 Jimei Lu1†
Jimei Lu1† Liangping Zha
Liangping Zha- 1College of Pharmacy, Anhui University of Chinese Medicine, Hefei, China
- 2Anhui Institute for Food and Drug Control, Hefei, China
Polysaccharides and saponins are the main active components of Polygonati Rhizoma. Studying the molecular mechanism of their synthesis pathway is helpful in improving the content of active components at the molecular level. At present, transcriptome analysis of three Polygonatum species (Polygonatum sibiricum Red., Polygonatum cyrtonema Hua, Polygonatum kingianum Coll. et Hemsl.) has been reported, but no comparative study has been found on the transcriptome data of the three species. Transcriptome sequencing was performed on the rhizomes of three Polygonatum species based on high-throughput sequencing technology, and all transcripts were assembled. A total of 168,108 unigenes were generated after the removal of redundancy, of which 121,642 were annotated in seven databases. Through differential analysis and expression analysis of key enzyme genes in the synthesis pathway of three Polygonatum polysaccharides and steroidal saponins, 135 differentially expressed genes encoding 18 enzymes and 128 differentially expressed genes encoding 28 enzymes were identified, respectively. Numerous transcription factors are involved in the carbohydrate synthesis pathway. Quantitative real-time PCR was used to further verify the gene expression level. In this paper, we present a public transcriptome dataset of three medicinal plants of the genus Polygonatum, and analyze the key enzyme genes of polysaccharide and steroidal saponins synthesis pathway, which lays a foundation for improving the active component content of Polygonati Rhizoma by molecular means.
Introduction
Polygonati Rhizoma is a traditional Chinese herb used in both food and medicine, and has a medicinal history of more than 2000 years (Hu et al., 2022; Li et al., 2023). In China, there are about 39 species of the genus Polygonatum. Among them, Polygonatum cyrtonema Hua (P. cyrtonema), Polygonatum kingianum Coll. et Hemsl. (P. kingianum) and Polygonatum sibiricum Red. (P. sibiricum) were included in Chinese Pharmacopoeia (2020 edition) as the original plants of Polygonati Rhizoma. Modern pharmacological studies have shown that polysaccharide and saponin, the main active components of Polygonati Rhizoma, have a series of important functions such as anti-aging, immune regulation, anti-tumor, hypoglycemic (Long et al., 2018; Zhao et al., 2018; Shen et al., 2021; Shen et al., 2022).
Polygonatum polysaccharide is a class of biological macromolecules composed of a variety of monosaccharides in different proportions. The researchers revealed that there are differences in polysaccharide structure, monosaccharide type, and content in different types and origins of Polygonati Rhizoma, which means that its biological activities also have certain differences (Wang et al., 2022). Comparative studies of polysaccharides from P. cyrtonema (PCP), P. kingianum (PKP), and P. sibiricum (PSP) showed that their physicochemical properties, antioxidant activity in vitro, and the regulation of macrophage polarization were different (Bai et al., 2021). Previous studies have shown that UDP-glucose is the basis for the production of multiple NDP-monosaccharides in the biosynthesis pathway of polysaccharides, and is also the main form of activated sugar in higher plants. It is directly generated by sucrose under the action of sucrose synthase (SUS) or is catalyzed by UDP-glucose pyrophosphorylase (UGPase) to produce using glucose 1-phosphate as the substrate (Decker et al., 2017; Wang et al., 2017). UDP-glucose generates a series of NDP-sugar under the action of UDP-glucose 4-epimerase (GALE), UDP-glucose dehydrogenase (UGDH), and other enzymes. At the same time, sucrose is catalyzed by enzymes such as hexokinase (HK), fructokinase (scrK), mannose-6-phosphate isomerase (MPI), and phosphomannose isomerase (PMM) to generate GDP-mannose and GDP-fucose. These monosaccharides are finally incorporated into polysaccharide polymers by glycosyl transferases (GTs) to form characteristic polysaccharides. At present, research on the synthesis pathway of polysaccharides has been reported in medicinal plants such as Panax ginseng (Fang et al., 2022), Dendrobium moniliform (Yuan et al., 2019), and Codonopsis pilosula (Gao et al., 2015).
Saponins include steroid saponins and triterpenoid saponins, which are composed of one or more hydrophilic sugar residues and hydrophobic sapogenin. Steroidal saponins are mainly present in plants such as the Liliaceae (Luo et al., 2018), Smilaxaceae (Tian et al., 2017), Asparagaceae, etc.(Sidana et al., 2016), and their anti-tumor activity has been widely studied (Mimaki et al., 1994; Lu et al., 2009; Han et al., 2018). The biosynthesis process of steroidal saponins is divided into three stages, and their upstream synthesis includes the cytoplasmic mevalerate (MVA) pathway and the plastid methyl-ethrolitol 4-phosphate (MEP) pathway, which eventually produces mutually transferable isopentenyl pyrophosphate (IPP) and dimethylallyl diphosphate (DMAPP). In the action of geranyl diphosphate synthase (GPS), farnesyl diphosphate synthase (FPS), squalene synthase (SQS), and squalene epoxidase (SE), IPP and DMAPP generated 2,3-oxidosqualene. Cycloartenol synthase (CAS) catalyzes 2,3-oxidosqualene to cycloartenol, the precursor of steroidal sapogenin, which undergoes a series of reactions including oxidation, hydroxylation, and glycosylation to ultimately generate steroidal saponins (Upadhyay et al., 2018; Guo et al., 2021). Some studies have shown that lanosterol can participate in the synthesis of plant sterols and steroid glycoalkaloids. However, the current theory is more accepted that cycloartenol is converted to sitosterol, and then steroidal saponins are produced by hydroxylase and glycosyltransferase (Chen et al., 2021). At present, the genes related to plant steroidal saponins biosynthesis need to be further studied. It has been reported that the expression levels of key regulatory and rate-limiting enzyme 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGR) in the MVA pathway can affect the content of monoglycosylated saponins in Medicago truncatula (Moses et al., 2014). Therefore, identifying the key genes in the synthesis pathway of steroidal saponins is of great significance for understanding the synthesis mechanism of steroidal saponins.
Transcriptomics takes all transcripts in the sample as the research object. It plays an important role in revealing the molecular mechanism of biological processes and mining functional genes. It is the most widely used and rapidly developing omics technology (Lowe et al., 2017). Since RNA sequencing technology was proposed, it has gradually become the first choice for gene expression analysis due to its advantages such as high sensitivity, high accuracy, high repeatability, and not limited by the completeness of genome data of species (Su et al., 2014; Imadi et al., 2015). It has been widely used in transcriptome analysis of species such as Panax japonicus (Rai et al., 2016), Carthamus tinctorius L. (Huang et al., 2012), and Atractylodes lancea (Ahmed et al., 2016). Based on the transcriptome data mining of P. cyrtonema, P. kingianum, and P. sibiricum, scholars have obtained multiple key enzyme genes related to the synthesis of polysaccharides, saponins, and flavonoids, which is conducive to improving the content of effective components of polygonati rhizoma through genetic engineering (Wang et al., 2019; Yang et al., 2019; Han et al., 2023). However, at present, comparative studies on the biological activities and synthesis pathways of three Polygonatum polysaccharides and steroidal saponins are relatively lacking. In this paper, through sequencing and analysis of the transcriptome of P. cyrtonema, P. kingianum, and P. sibiricum, the key enzyme genes affecting the content differences of Polygonatum polysaccharides and steroidal saponins were explored, laying a foundation for the regulation of the genes of Polygonatum polysaccharide and steroidal saponins synthesis pathway.
Materials and methods
Plant materials and total RNA extraction
P. cyrtonema and P. sibiricum were collected from Guoziyuan Township, Jinzhai County, Lu’an City, Anhui Province, and P. kingianum was collected from Zhangshan Village, Xinghua Township, Hong’an County, Huanggang City, Hubei Province. Three plants of each variety were taken as replicates. The test materials were identified by associate researcher Liangping Zha (Anhui University of Chinese Medicine) as Liliaceae family plants P. cyrtonema, P. sibiricum, and P. kingianum. The rhizomes of the plants were collected, cleaned with sterilized water, dried on filter paper, snap-frozen in liquid nitrogen, and stored at -80°C. According to the manufacturer’s instructions, the sample was first ground into a powder with liquid nitrogen, and total RNA was extracted using CTAB-PBIOZOL and the ethanol precipitation protocol. The RNA concentration, 28S/18S, and RNA integrity number (RIN) were detected using the Standard Sensitivity RNA Analysis Kit, which was performed by the Fragment Analyzer.
Extraction and determination of total polysaccharide and saponins
Rhizomes of three Polygonatum plants were dried in an oven at 60°C to constant weight, ground into powder, and passed through an 80 mesh sieve. The extraction method of polysaccharides was as described in Chinese Pharmacopoeia. The absorbance was measured at 574 nm, D-glucose was used as a reference, and total polysaccharide was determined using the anthrone-sulfuric acid method. The extraction method of Polygonatum saponin refers to the method of Hu et al. and saponin content was analyzed using the 5% vanillin-glacial acetic acid–perchloric acid method, diosgenin as the reference, the wavelength was measured at 530 nm (Hu et al., 2022).
RNA−Seq library construction and sequencing
The mRNA with polyA of the three varieties of polygonati rhizoma (three replicates) was enriched using Oligo (dT) magnetic beads, cleaved into short fragments, and cDNA strands were obtained by reverse transcription with random N6 primers. The double-stranded cDNA ends were repaired, the 5’ end was phosphorylated and a single nucleotide A was added at the 3’ end. Then, cDNA was ligated to the sequencing adapters, and the ligated products were amplified by PCR to construct a cDNA library and sequenced on the DNBSEQ platform (Beijing Genomics Institute, Shenzhen, China).
The SOAPnuke filtering software removed reads containing adapters, unknown nucleotides content greater than 5%, and low-quality (more than 20% of bases with quality <10). De novo assembly and redundant sequence removal were performed in Trinity and Tgicl, respectively.
Functional annotation
To obtain more comprehensive information on gene function, the assembled unigenes were annotated to seven databases, including the NCBI non-redundant nucleotide sequence (NT, ftp://ftp.ncbi.nlm.nih.gov/blast/db), NCBI non-redundant protein sequences (NR, ftp://ftp.ncbi.nlm.nih.gov/blast/db), Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.genome.jp/kegg), Clusters of euKary-otic Orthologous Groups (KOG, http://www.ncbi.nlm.nih.gov/KOG), SwissProt (http://ftp.ebi.ac.uk/pub/databases/swissprot), Gene Ontology (GO, http://geneontology.org) and Pfam (http://pfam.xfam.org). Blast2GO software (version 2.5.0) was used to further perform GO annotation and functional analysis of unigenes annotated to the NR database, and the annotation results were divided into three parts: molecular function, cellular component, and biological process.
Identification of differentially expressed genes
Clean reads were matched to reference gene sequences by Bowtie2 software (version 2.2.5), and then gene expression levels were calculated for each sample using RSEM software (version 1.2.8). Transcripts per kilobase per million fragments (FPKM) were used to normalize read counts of transcripts. DEGseq2 (version 1.4.5) software was applied to the analysis of DEGs. Genes with Q value ≤ 0.001 and |log2FC|>1 were defined as DEGs. DEGs enrichment in GO terms and KEGG pathways were determined using the phyper function with Q value ≤0.05.
Analysis of genes encoding transcription factors
TFs can regulate the transcription level of genes by binding to promoters. In order to analyze the TF family of the transcriptome of three Polygonatum plants, getorf software (EMBOSS:6.5.7.0) was used to detect the open reading frame (ORF) of Unigene, and hmmsearch (version 3.0) searched for proteins with certain gene family domains in candidate sequences by comparing ORFs. Then the genes encoding TFs were identified based on PlantTFDB.
Bioinformatics analysis of UDP-apiose/xylose synthase genes
DNAMAN software was used for homologous amino acid alignment, and SWISS MODEL (http://swissmodel.expasy.org/) and PyMOL software to predict the tertiary structure of the proteins. The phylogenetic tree was constructed in MEGA7.0 software using the neighbor-joining method.
Quantitative real−time PCR validation
Total RNA from the three Polygonatum plants was reverse transcribed using the All-in-One First-Strand Synthesis MasterMix kit (LABLEAD, China). The qRT-PCR was performed using the Bio-Rad CFX96 platform (Bio-Rad, USA) according to the instructions of the 2×Realab Green PCR Fast mixture kit. The mRNA abundance of the target genes was normalized to the mRNA abundance of the internal reference gene EF-1α2 (Yang et al., 2020), and the relative gene expression analysis was performed using the 2-ΔΔCt method. The specific primers were designed using the Primer software (version 5.0).
Results
Total polysaccharide content of Polygonatum samples
We used the anthrone-sulfuric acid method to determine the polysaccharide content of three polygonatum species, with three replicates per sample, expressed as the mean value with standard deviation. The results showed that the polysaccharide content of P. sibiricum and P. cyrtonema were almost the same, both significantly higher than that of P. kingianum, 1.66 and 1.70 times that of P. kingianum, respectively (Figure 1A).
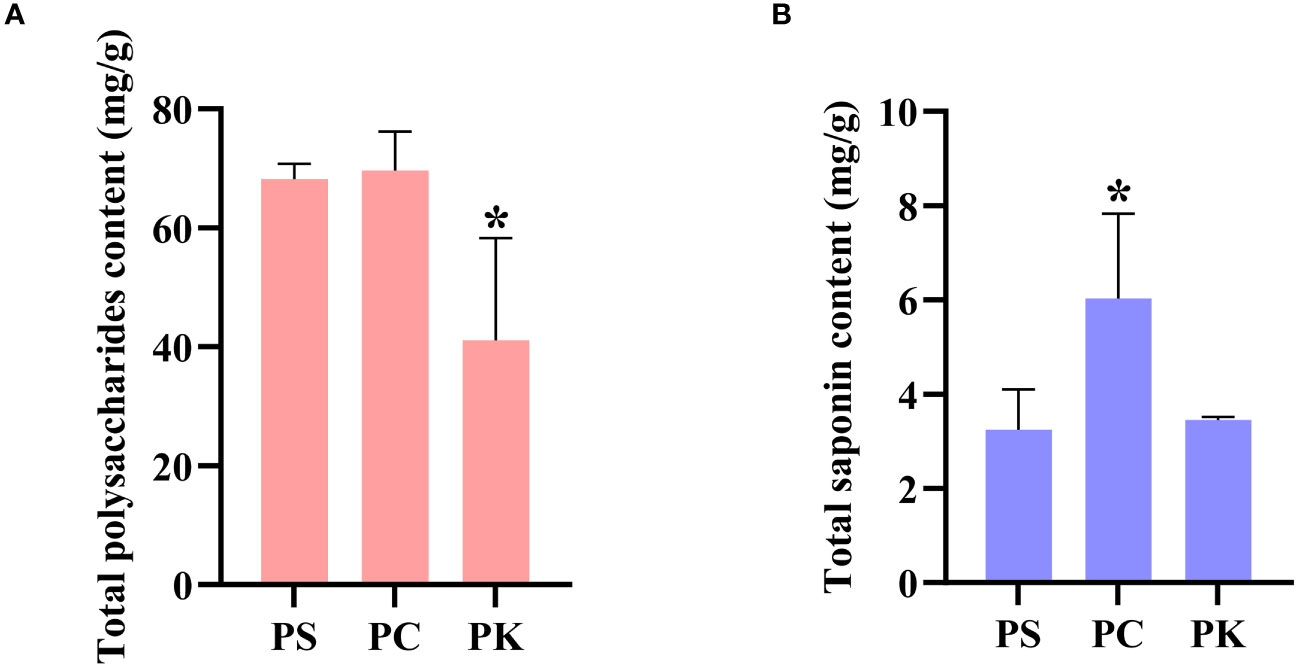
Figure 1 Contents of polysaccharides and saponins in P. sibiricum, P. cyrtonema, and P. kingianum (Mean values ± SD, n = 3). (A) Contents of polysaccharides. (B) Contents of saponins. *data compared with the P. sibiricum, P < 0.05. PS, PC, and PK represent P. sibiricum, P. cyrtonema, and P. kingianum, respectively.
Total saponin content of Polygonatum samples
As the main active secondary metabolite of polygonati rhizoma, total saponin has attracted much attention. The vanillin-glacial acetic acid method was used to determine the total saponin content of three botanical origins of polygonati rhizoma. The results showed that the total saponin content of P. cyrtonema was significantly higher than that of P. sibiricum and P. kingianum, which were 1.86 and 1.75 times higher, respectively. The total saponin content of P. kingianum was slightly higher than that of P. sibiricum, which was 1.07 times that of P. sibiricum (Figure 1B).
Transcriptome analysis based on RNA−Seq
Nine samples of P. sibiricum, P. cyrtonema, and P. kingianum (three replicates for each species) were sequenced on the DNBSEQ platform, and the raw reads were filtered by the filtering software SOAPnuke to obtain clean reads of 128.07 M, 127.72 M, and 128.11 M respectively. Each sample yielded more than 6.32 Gb of clean data. The Q30 percentages were more than 92.52%. Using Tgicl to cluster the transcripts to remove redundancy, a total of 168,108 unigenes were obtained, a mean length of 1,236 base pairs (bp) and a mean N50 of 1,839 bp, and the average GC content was 43.61%. Among them, 70.57% (118,640) and 46.95% (78,927) unigenes were longer than 500 bp and 1000 bp, respectively (Table S1; Figure S1).
Functional annotation and expression overview of unigenes
The assembled unigenes were compared with NR, NT, Swissprot, KEGG, KOG, Pfam, and GO databases. 27.34% of the unigenes were mapped simultaneously in the seven databases, and 72.36% of the unigenes were mapped to at least one public database (Table 1). Among the six databases of NR, NT, SwissProt, Pfam, KOG, and KEGG, there were 3196, 2870, 104, 1299, 33, and 250 unique unigenes, respectively, and unique genes were not annotated in the GO database (Figure 2A).
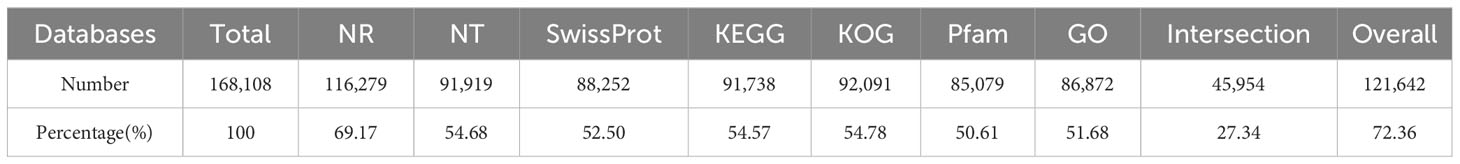
Table 1 Functional annotation results from seven public databases.
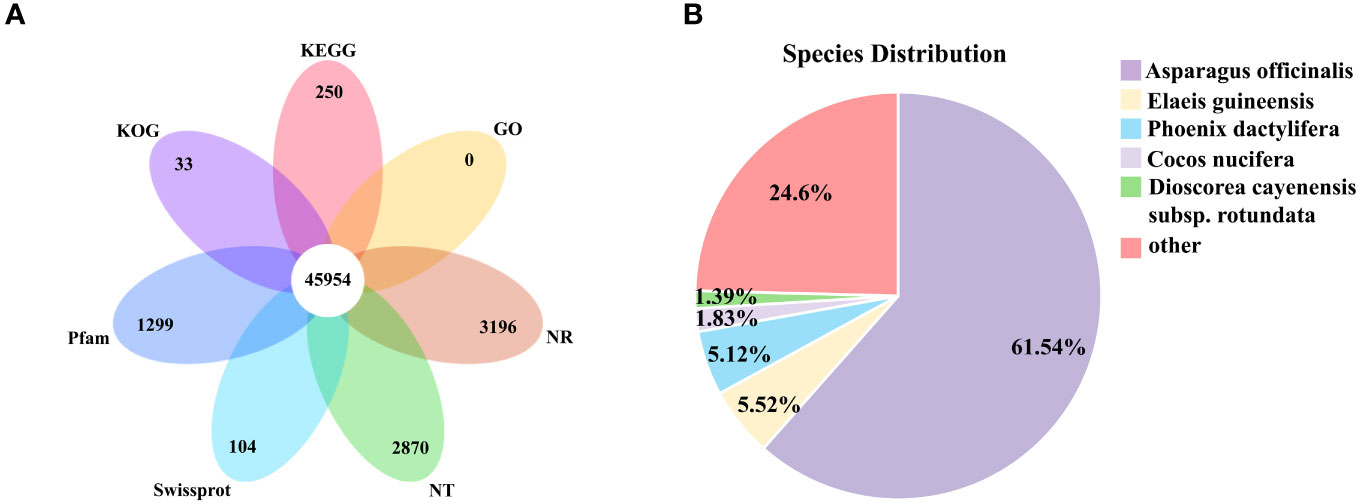
Figure 2 Transcriptome annotation. (A) Venn diagram of functional annotation of seven public protein databases (NR, NT, SwissProt, Pfam, KOG, KEGG and GO). (B) Species distribution of homologous sequences against Nr database.
The 116,279 unigenes annotated in NR database were subjected to homology analysis, among which the highest homology was Asparagus officinalis (61.54%). Followed by Elaeis guineensis (5.52%), Phoenix dactylifera (5.12%), Cocos nucifera (1.83%), Dioscorea cayenensis subsp. Rotundata (1.39%) (Figure 2B).
In order to further explore the interactions and biological functions of genes in polygonati rhizoma, a total of 91,738 unigenes were mapped to the KEGG database and assigned to five categories: genetic information processing, environmental information processing, cellular processes, metabolism, and organismal systems. It contains 19 subcategories such as “carbohydrate metabolism”, “biosynthesis of other secondary metabolites”, “transport and catabolism”, among which 11 subcategories belong to the “metabolism” category (Figure 3A; Table S2). The results of KEGG enrichment showed that the “carbohydrate metabolism” subcategory involved 15 pathways. The “pentose and glucuronate interconversions” pathway contains the largest number of unigenes (2,179). Eighteen pathways were assigned to the “biosynthesis of other secondary metabolites” subcategory, and “phenylpropanoid biosynthesis” was the most abundant pathway containing unigenes. Nine pathways were involved in “metabolism of terpenoids and polyketides” subcategory, most unigenes (426) were enriched in “terpenoid backbone biosynthesis”, 211 unigenes were enriched in “Sesquiterpenoid and triterpenoid biosynthesis” pathway (Figure S2).
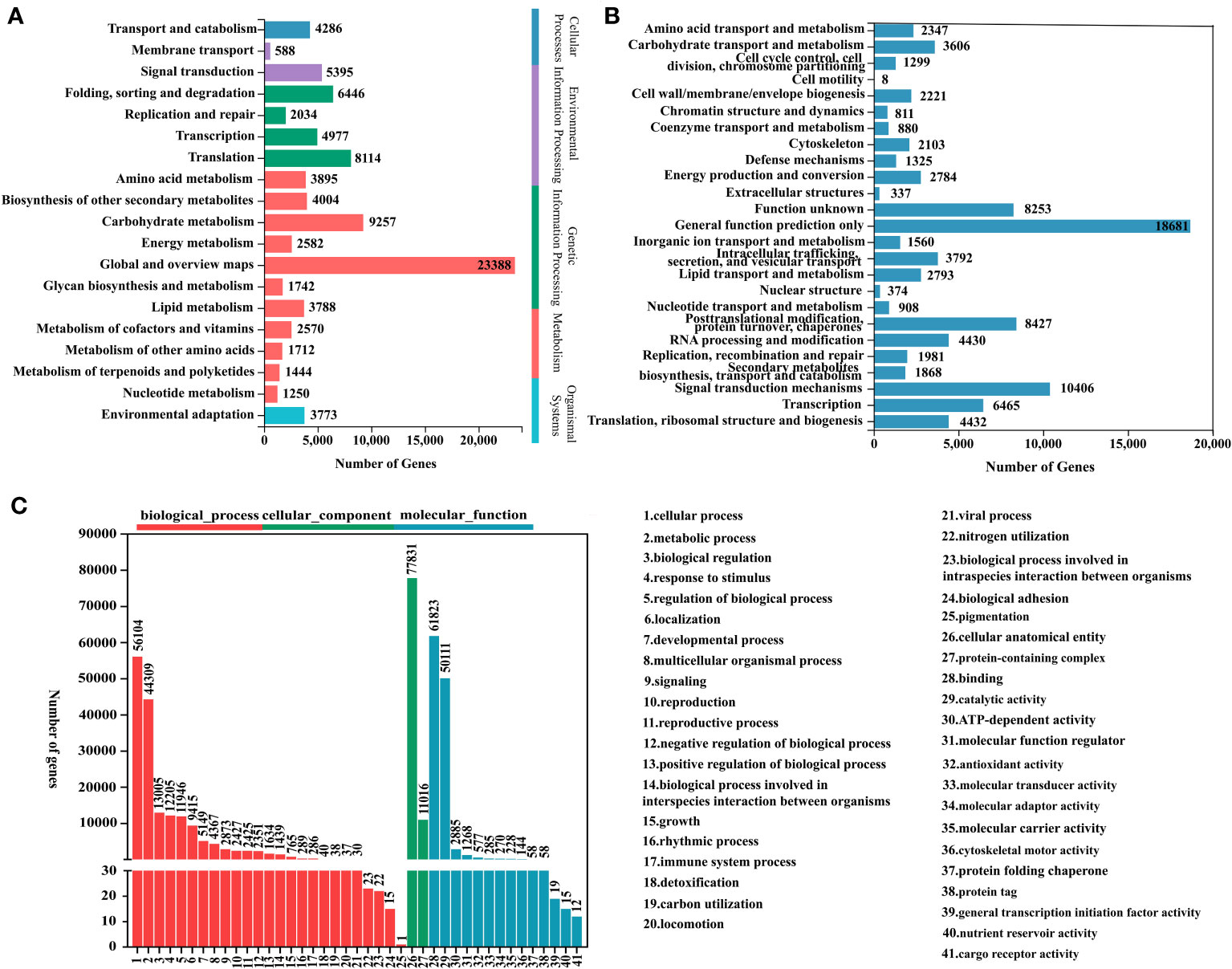
Figure 3 Functional annotations for three Polygonatum plants. (A) KEGG pathway classification. (B) KOG functional classification. (C) GO functional classification.
Based on the NR database annotation results, the 86,872 transcripts mapped to the GO database were classified into three categories: biological process, molecular function, and cellular component by the Blast2GO software. Among them, biological process was the largest functional category, and the main terms were “cellular process” (GO:0009987; 56,104 genes) and “metabolic process” (GO:0008152; 44,309 genes). In the cell component and molecular function categories, “cellular anatomical entity” (GO:0110165; 77,831 genes) and “binding” (GO:0005488; 61,823 genes) were the richest terms, respectively (Figure 3C).
The KOG database alignment showed that 92,091 unigenes were divided into 25 functional clusters, and that the “general function prediction only” had the largest number of unigenes (18,681), followed by “signal transduction mechanisms”, including 10,406 unigenes (Figure 3B).
Unigenes with FPKM>1 were analyzed. The results showed that 48,397, 55,259 and 53,099 unigenes were expressed in P. sibiricum, P. cyrtonema, and P. kingianum, respectively (Figure S3A). The boxplot showed that the expression level of P. kingianum was slightly higher than that of P. sibiricum and P. cyrtonema (Figure S3B).
Identification of differentially expressed genes
The screening of DEGs is helpful in analyzing the important functional genes of polygonati rhizoma. Taking Q ≤ 0.001 as the screening condition of DEGs, 47,858 DEGs were selected from the P. cyrtonema vs P. kingianum analysis, including 23,596 up-regulated genes and 24,262 down-regulated genes. 38,308 DEGs were detected by comparing the expression levels of P. sibiricum and P. cyrtonema, with 19,915 up-regulated genes and 18,393 down-regulated genes. The comparison of expression levels between P. sibiricum and P. kingianum identified 50,210 DEGs, including 25,380 up-regulated genes and 24,830 down-regulated genes (Figure 4A). The analysis of DEGs showed that there were 7444 specific DEGs in P. sibiricum vs P. kingianum, and then there were 5089 and 3467 specific DEGs in P. cyrtonema vs P. kingianum and P. sibiricum vs P. cyrtonema, respectively. In addition, a total of 3868 DEGs were detected among the three comparison groups (Figure 4B).
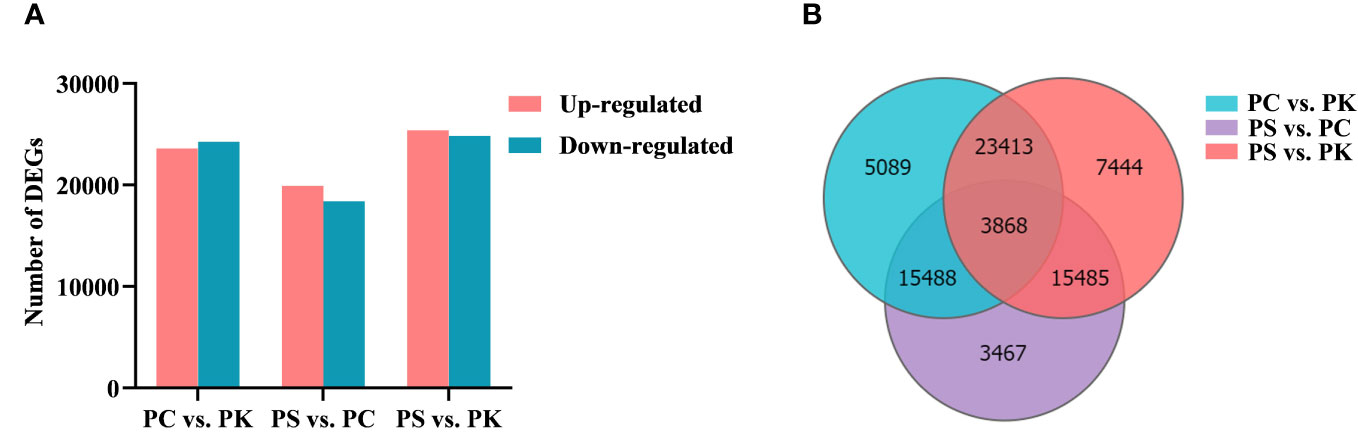
Figure 4 Analysis of DEGs. (A) Upregulated and downregulated DEGs in different samples. (B) Venn diagram of DEGs in diverse comparison groups.
In order to further analyze the DEGs, KEGG and GO enrichment were performed using phyper function in R software. In KEGG analysis, P. cyrtonema vs P. kingianum, P. sibiricum vs P. kingianum, P. sibiricum vs P. cyrtonema identified 20,258, 21,180, and 16,154 DEGs, respectively. In the P. cyrtonema vs P. kingianum, there were 220, 368, 757 and 677 DEGs enriched to fructose and mannose metabolism (ko00051) pathway, galactose metabolism (ko00052) pathway, starch and sucrose metabolism (ko00500) pathway, amino sugar and nucleotide sugar metabolism (ko00520) pathway related to polysaccharide biosynthesis, respectively. Among them, the fructose and mannose metabolism (ko00051) pathway and galactose metabolism (ko00052) pathway were included in the top 20 pathways. In P. sibiricum vs P. kingianum, 256, 378, 828, and 691 DEGs were enriched to ko00051, ko00052, ko00500, and ko00520 pathways, respectively, and only fructose and mannose metabolism (ko00051) pathway was included in the top 20 pathways. 174, 278, 601, and 526 DEGs in P. sibiricum vs P. cyrtonema were enriched in the above four polysaccharide synthesis pathways, respectively. In conclusion, the largest number of DEGs enriched to starch and sucrose metabolism (ko00500) pathway, however, the higher enrichment of fructose and mannose metabolism (ko00051) pathway in both P. cyrtonema vs P. kingianum and P. sibiricum vs P. kingianum, this pathway can be focused on in subsequent studies of polysaccharide metabolism. Among the three pathways of terpenoid backbone biosynthesis (ko00900), sesquiterpenoid and triterpenoid biosynthesis (ko00909) and steroid biosynthesis (ko00100) associated with the synthesis of steroidal saponins, the largest number of DEGs were enriched to sesquiterpenoid and triterpenoid biosynthesis (ko00909) pathway. The most enriched pathway among the three comparison groups was “ribosome”. In GO enrichment analysis, the most expansive GO term among the different comparison groups was “RNA binding” (Figure S4).
Putative DEGs involved in polysaccharide biosynthesis
Polysaccharide is the main active component of polygonati rhizoma. Based on the KEGG enrichment results, we identified the genes encoding key enzymes on the starch and sucrose metabolism and amino sugar and nucleotide sugar metabolism pathways. After removing the sequences with FPKM<1, incomplete sequences, and completely consistent sequences, we ultimately obtained 163 genes encoding 18 key enzymes for polygonatum polysaccharide biosynthesis, of which 135 were further identified as DEGs, including mannose-1-phosphate guanylyltransferase (GMPP, 16 unigenes), scrK (16 unigenes), UDP-glucuronate 4-epimerase (UGE, 14 unigenes), HK (15 unigenes) et al. (Table 2; Figure 5). Heat maps were drawn based on FPKM values to reflect the expression levels of these genes in P. cyrtonema, P. kingianum and P. sibiricum (Figure 6). The expression levels of 28 genes encoding key enzymes were higher in P. sibiricum and P. cyrtonema, while the expression levels were the lowest in P. kingianum. Among them, the expression levels of 13 genes encoding 8 key enzymes-AXS (Unigene15181), GMPP (CL15301.Contig2, CL4107.Contig6, Unigene16104, CL1098.Contig4), MPI (CL10643.Contig2), scrK (CL10568.Contig2, CL5289.Contig3), GALE (CL8435.Contig2, Unigene20863), GMDS(CL14382.Contig7), GPI (CL12106.Contig4) and SacA (CL787.Contig8) were the highest in P. cyrtonema, suggesting that they play an important role in the polysaccharide synthesis pathway of polygonati rhizoma.
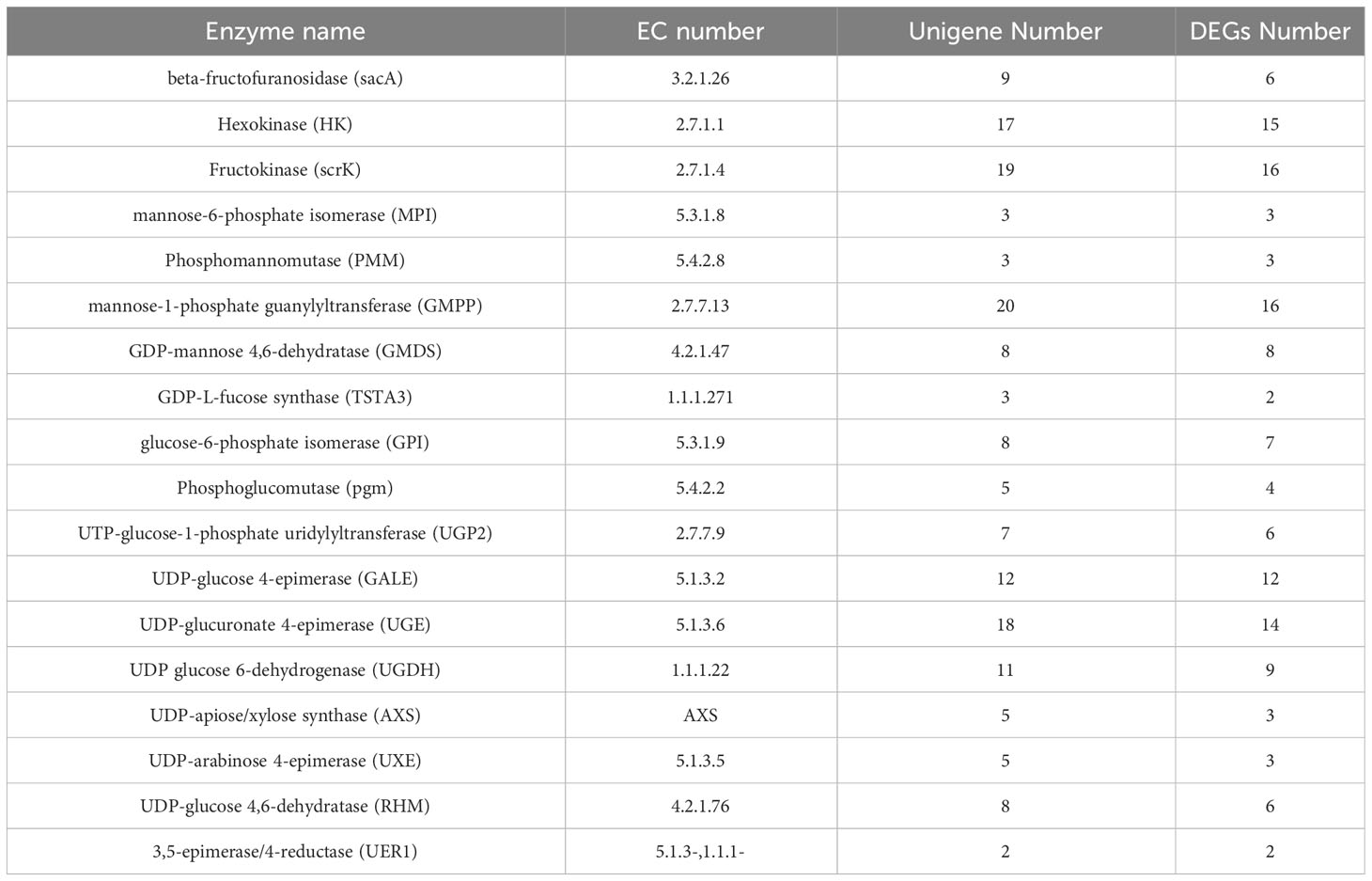
Table 2 Number of unigenes encoding key enzymes involved in polysaccharide biosynthesis in three Polygonatum plants.
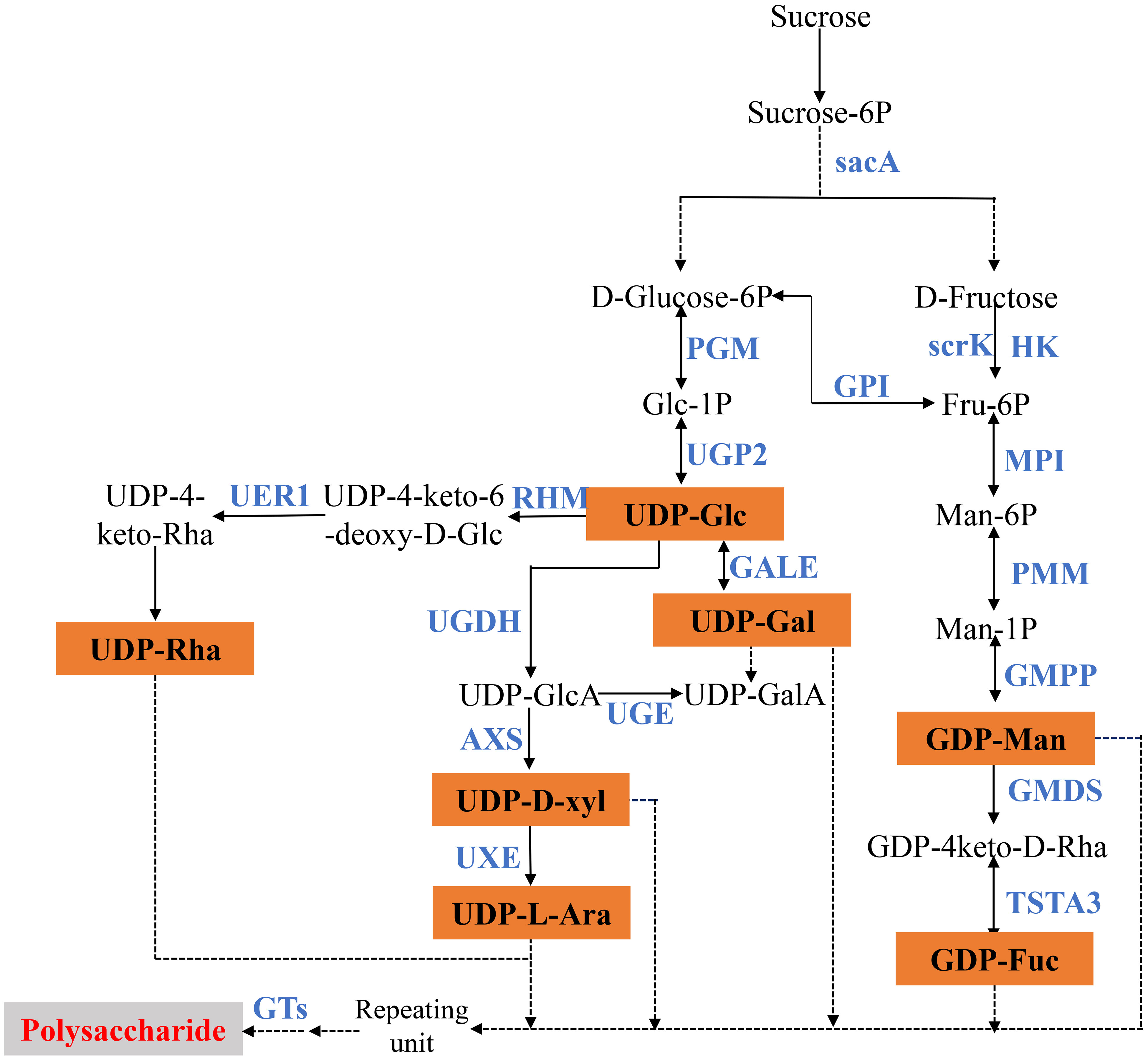
Figure 5 Proposed pathways for polysaccharide biosynthesis in three Polygonatum plants.
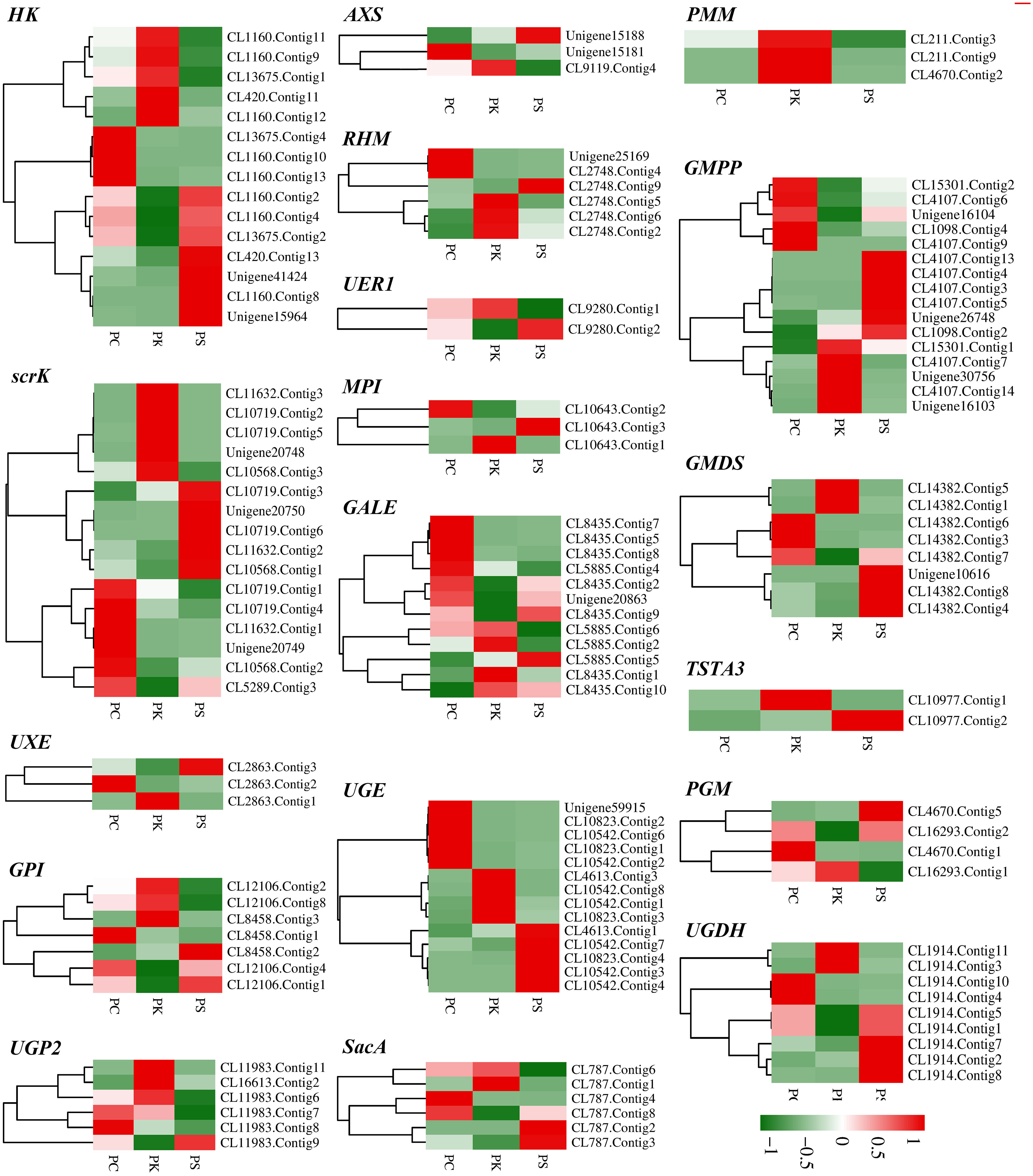
Figure 6 Relative expression patterns of unigenes encoding enzymes involved in polysaccharide biosynthesis.
Putative DEGs involved in steroidal saponins biosynthesis
The upstream pathway of steroidal saponins synthesis is the same as that of terpenoid skeleton synthesis, including MVA and MEP pathways. The downstream pathway includes the biosynthesis of steroid sapogenin precursor sterols and the oxidation and modification of steroidal saponins. Terpenoid backbone biosynthesis (ko00900), sesquiterpenoid and triterpenoid biosynthesis (ko00909), steroid biosynthesis (ko00100) were the three metabolic pathways of steroidal saponins synthesis. After screening FPKM>1 and complete sequences, 155 unigenes encoding key enzymes of steroidal saponins were obtained, of which 128 were further identified as DEGs (Table 3).
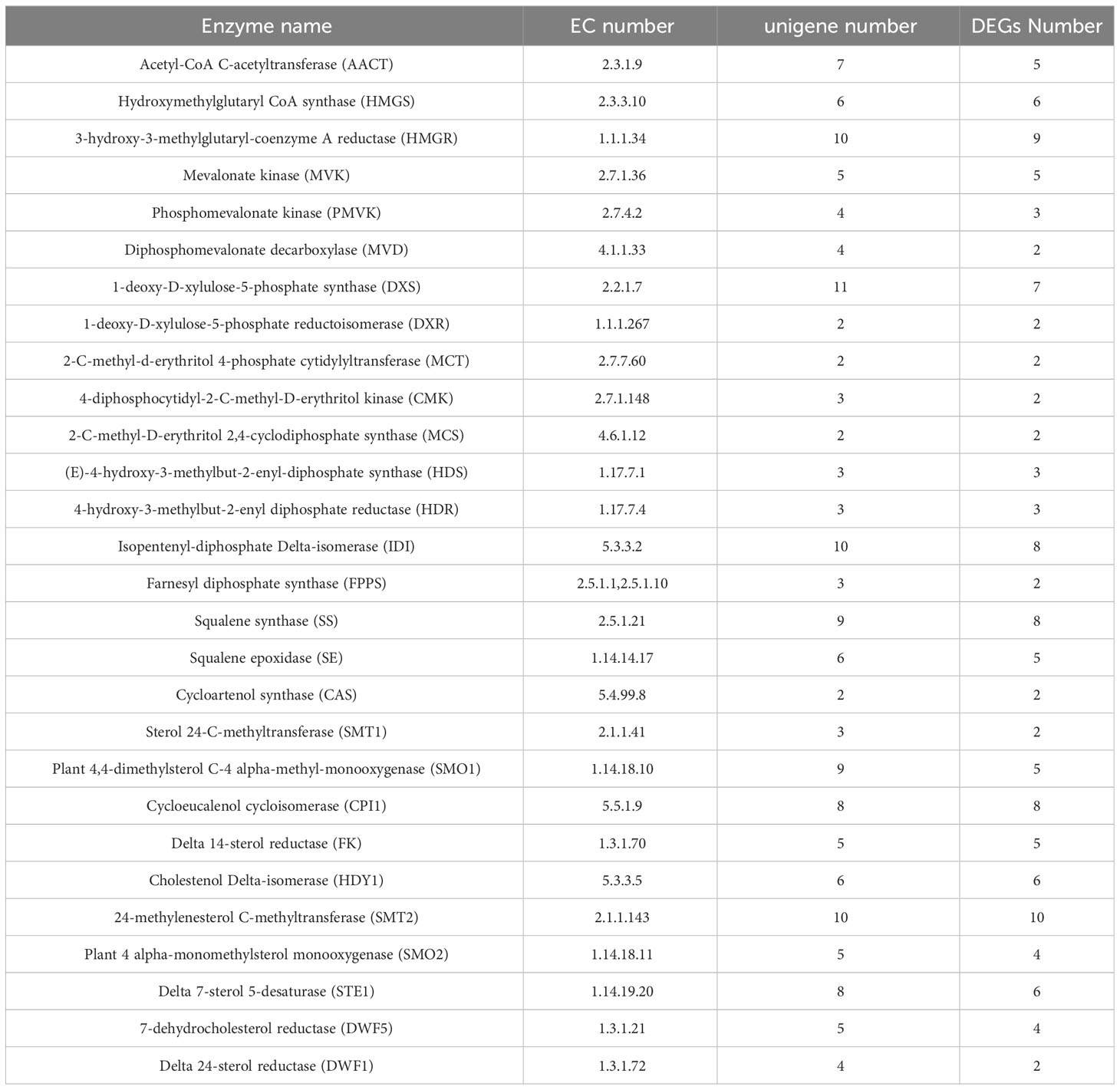
Table 3 Number of unigenes encoding key enzymes involved in steroidal saponins biosynthesis in three Polygonatum plants.
In the MVA pathway, 30 DEGs encoding six key enzymes including Acetyl-CoA C-acetyltransferase (AACT, 5 unigenes), Hydroxymethylglutaryl CoA synthase (HMGS, 6 unigenes), HMGR (9 unigenes), Mevalonate kinase (MVK, 5 unigenes), Phosphomevalonate kinase (PMVK, 3 unigenes) and Diphosphomevalonate decarboxylase (MVD, 2 unigenes) were identified. In the MEP pathway, 21 DEGs encoding seven key enzymes were identified, namely 1-deoxy-D-xylulose-5-phosphate synthase (DXS, 7 unigenes), 1-deoxy-D-xylulose-5-phosphate reductoisomerase (DXR, 2unigenes), 2-C-methyl-d-erythritol 4-phosphate cytidylyltransferase (MCT, 2 unigenes), 4-diphosphocytidyl-2-C-methyl-D-erythritol kinase (CMK, 2 unigenes), 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase (MCS, 2 unigenes), (E)-4-hydroxy-3-methylbut-2-enyl-diphosphate synthase (HDS, 3 unigenes) and 4-hydroxy-3-methylbut-2-enyl diphosphate reductase (HDR, 3 unigenes). IPP and DMAPP can be converted into each other under the catalysis of Isopentenyl-diphosphate Delta-isomerase (IDI), and then they generate the precursor substance FPP of squalene under the action of GPPS and FPPS. We identified eight DEGs encoding IDI and two DEGs encoding FPPS. In the sesquiterpenoid and triterpenoid biosynthesis and steroid biosynthesis pathways, we obtained 67 DEGs, which encoded 13 key enzymes including SS (8 unigenes), SE (5 unigenes), Sterol 24-C-methyltransferase (SMT1, 2 unigenes), Cycloeucalenol cycloisomerase (CPI1, 8 unigenes), Delta 14-sterol reductase (FK, 5 unigenes), Cholestenol Delta-isomerase (HDY1, 6 unigenes), etc. Notably, two CAS genes that catalyzed the formation of Cycloartenol from 2,3-Oxidosqualene were highly expressed in P. cyrtonema, while genes encoding SMT1 and DWF1 were expressed at low levels in P. cyrtonema. In addition, we found that the gene encoding DWF5 was highly expressed in P. kingianum (Figures 7, 8).
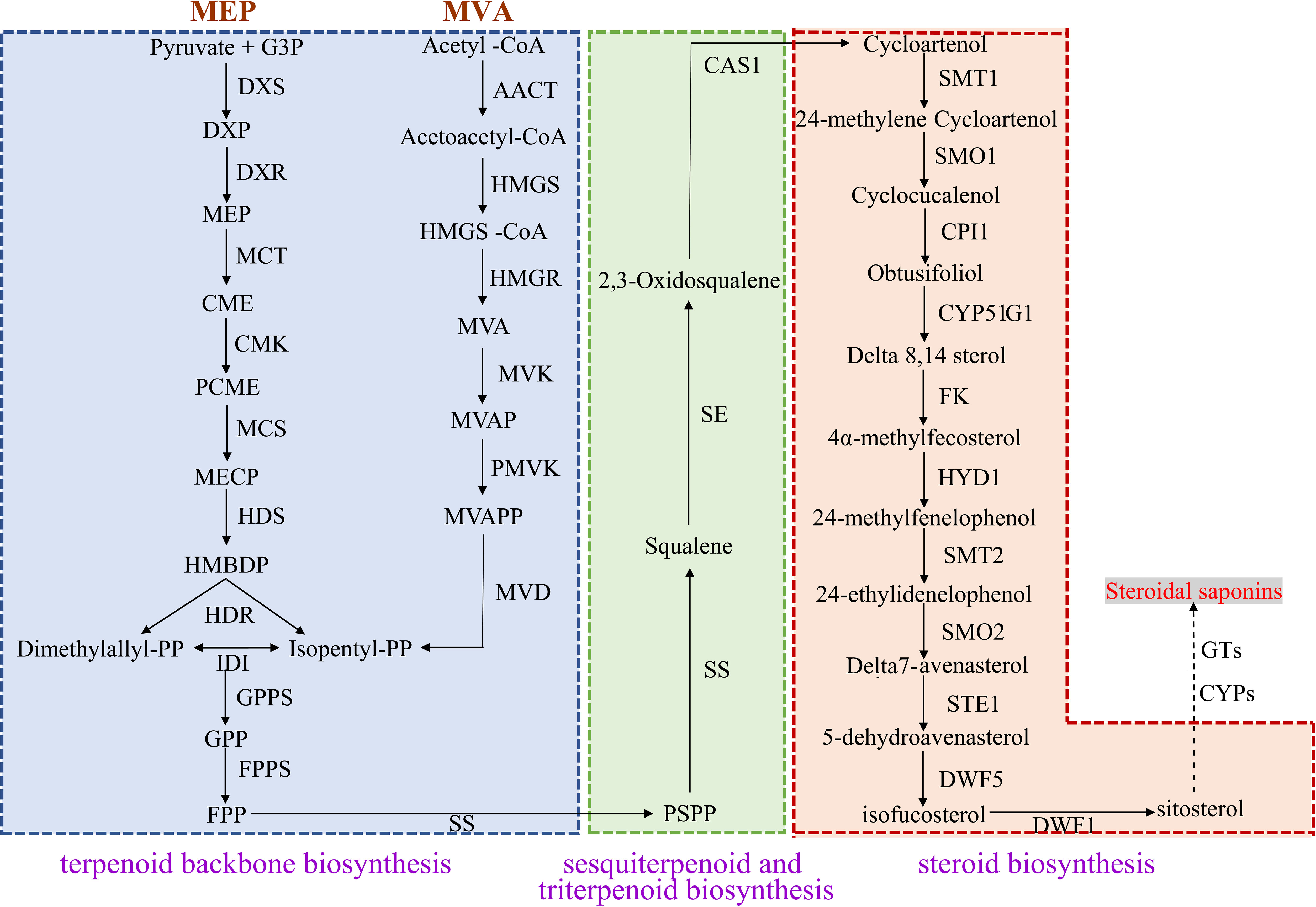
Figure 7 Pathway for steroidal saponins biosynthesis in three Polygonatum plants.
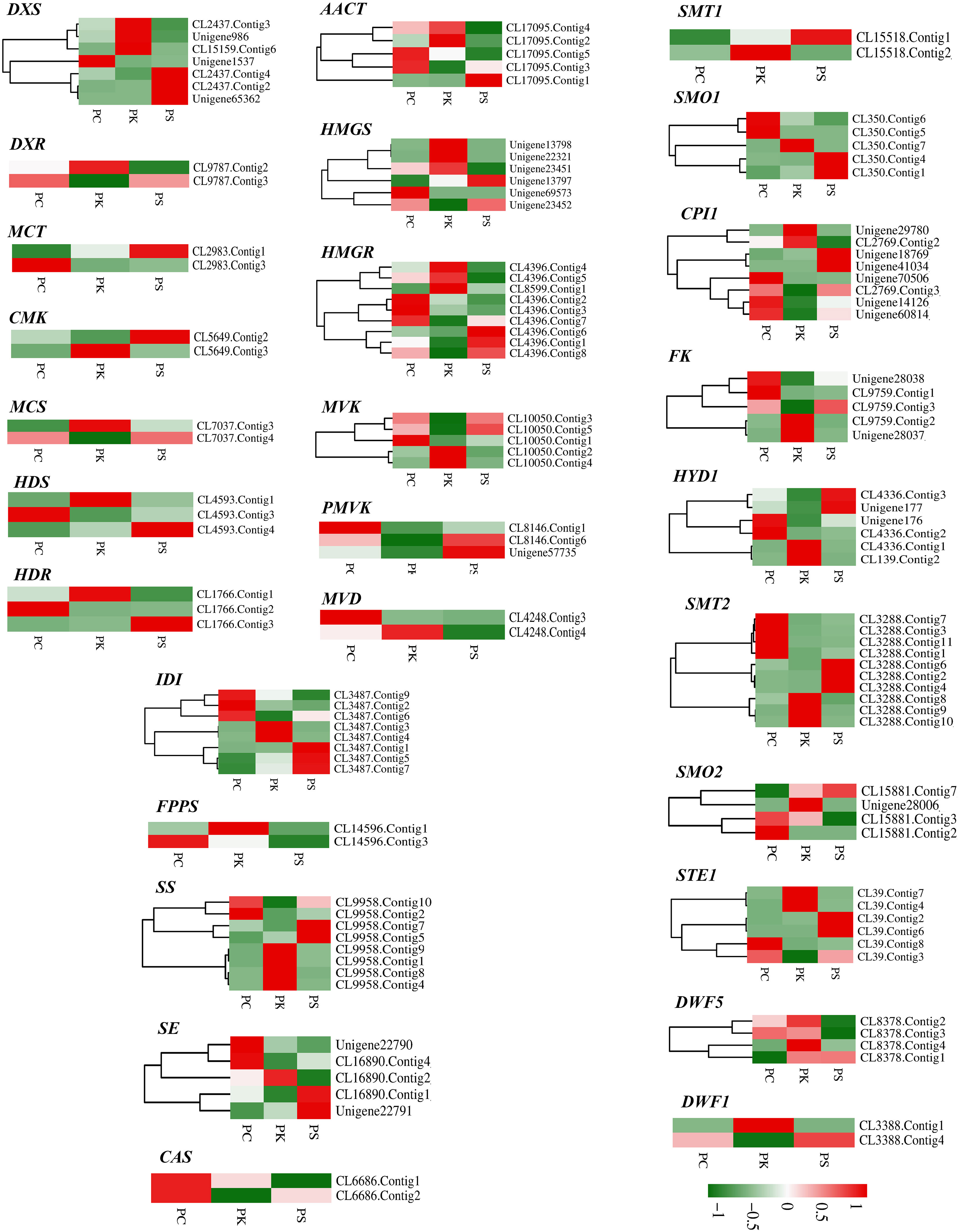
Figure 8 Relative expression patterns of unigenes encoding enzymes involved in steroidal saponins biosynthesis.
Identification of TFs involved in the biosynthesis of polysaccharides and other secondary metabolites
TFs can regulate the expression of genes in metabolic pathways and are important regulatory factors for plant development, stress response, and other activities (Riaño-Pachón et al., 2007). A total of 4454 TFs belonging to 57 TF families were identified in P. sibiricum, P. cyrtonema, and P. kingianum. With P. cyrtonema as the control group, 767 up-regulated TFs and 847 down-regulated TFs were identified in P. sibiricum, and 1010 up-regulated TFs and 1043 down-regulated TFs were identified in P. kingianum. In this study, 510 TFs were annotated to MYB TF family, accounting for the largest proportion, followed by AP2-EREBP, C3H, bHLH, and WRKY TF families (Table 4). Meanwhile, annotation results showed that 197 TFs from 20 TF families were involved in the carbohydrate metabolism pathway, and 164 TFs from 16 TF families were involved in the biosynthesis of other secondary metabolites (Figure 9). By KEGG enrichment analysis, there were 3, 85, 32 and 38 unigenes encoding TFs assigned to fructose and mannose metabolism (ko00051) pathway, galactose metabolism (ko00052) pathway, starch and sucrose metabolism (ko00500) pathway, amino sugar and nucleotide sugar metabolism (ko00520) pathway related to polysaccharide biosynthesis, respectively.
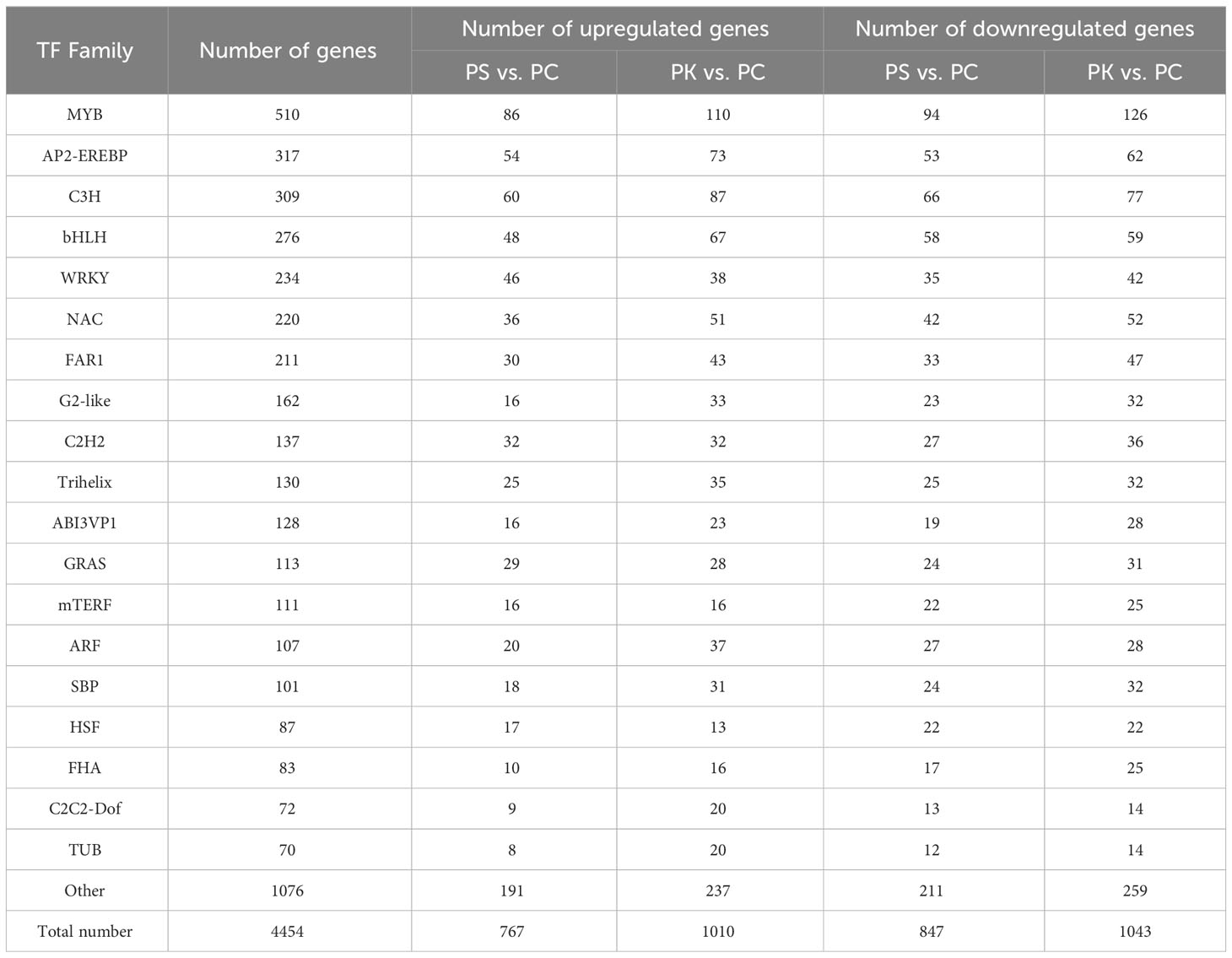
Table 4 Classification and number of TF families identified in the DEG database.
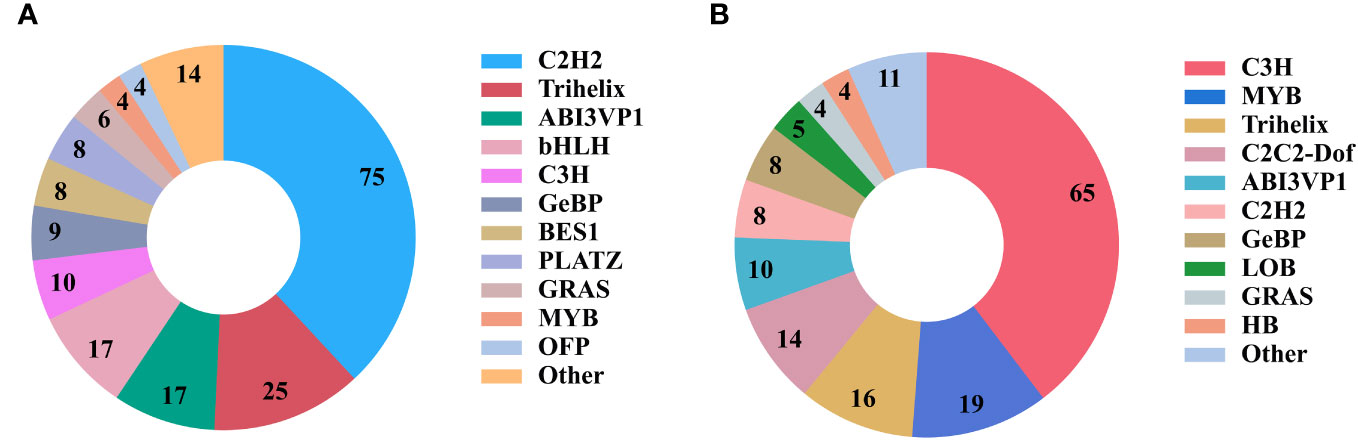
Figure 9 Transcription factors (TFs) involved in metabolic pathways. (A) TF families involved in carbohydrate metabolism. (B) TF families involved in the metabolism of other secondary metabolites.
Bioinformatics analysis of UDP-apiose/xylose synthase genes
AXS catalyzes the ring contraction and closure of UDP-D-glucuronic acid (UDP-GlcA) to produce UDP-Aal and UDP-Xyl. D-apiose (Api) is essential for the development of plant cell walls (Zhao et al., 2020b), and D-xyl is a component of P. cyrtonema, P. sibiricum, and P. kingianum polysaccharides (Zhao et al., 2020a). We identified three DEGs encoding AXS in the transcriptome data.
Multiple sequence alignment showed that each of the three AXS genes had a GxxGxxG motif at the N-terminal, which was thought to be related to cofactor NAD+ binding. In addition, sequences contained conserved ST motifs and YxxxK motifs, and YxxxK motifs may play catalytic roles (Thoden et al., 1997). AXS (Unigene15181) gene was further selected to create a 3D construction model (Figures 10, S5; Table S3).
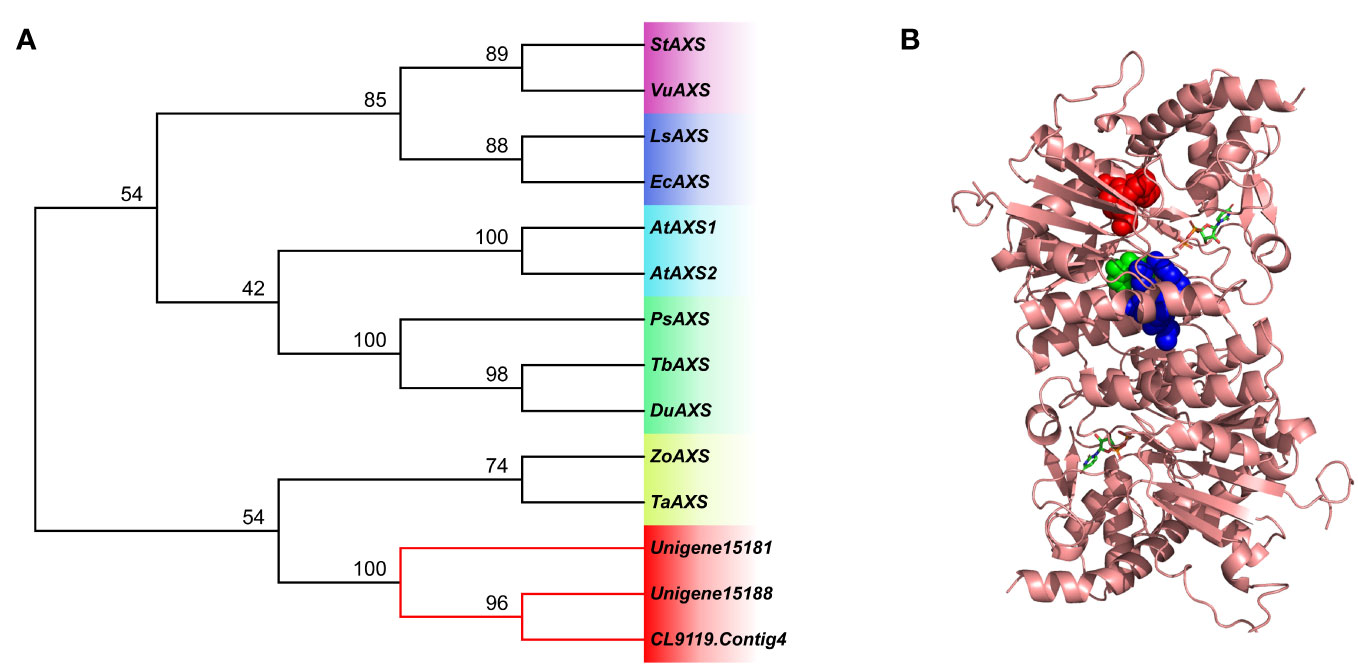
Figure 10 (A) Phylogenetic tree showing the three unigenes of AXS. The neighbor-joining phylogenetic trees were constructed using the bootstrap method in MEGA 7.0, and the number of bootstrap replications was 1000. (B) Spatial structure models and domain analysis of AXS (Unigene15181) gene. Conserved regions, GxxGxxG, ST, and YxxxK, are separately depicted as spheres in red, green, and blue.
Validation and expression analysis of genes encoding key enzymes
Using EF-1α2 as the reference gene, 9 DEGs involved in polysaccharide and steroidal saponins synthesis were verified by qRT-PCR, all primers used were listed in Table S4. The results showed that the trend of RNA-seq and qRT-PCR data was consistent, indicating that the data of RNA-seq was reliable (Figure 11).
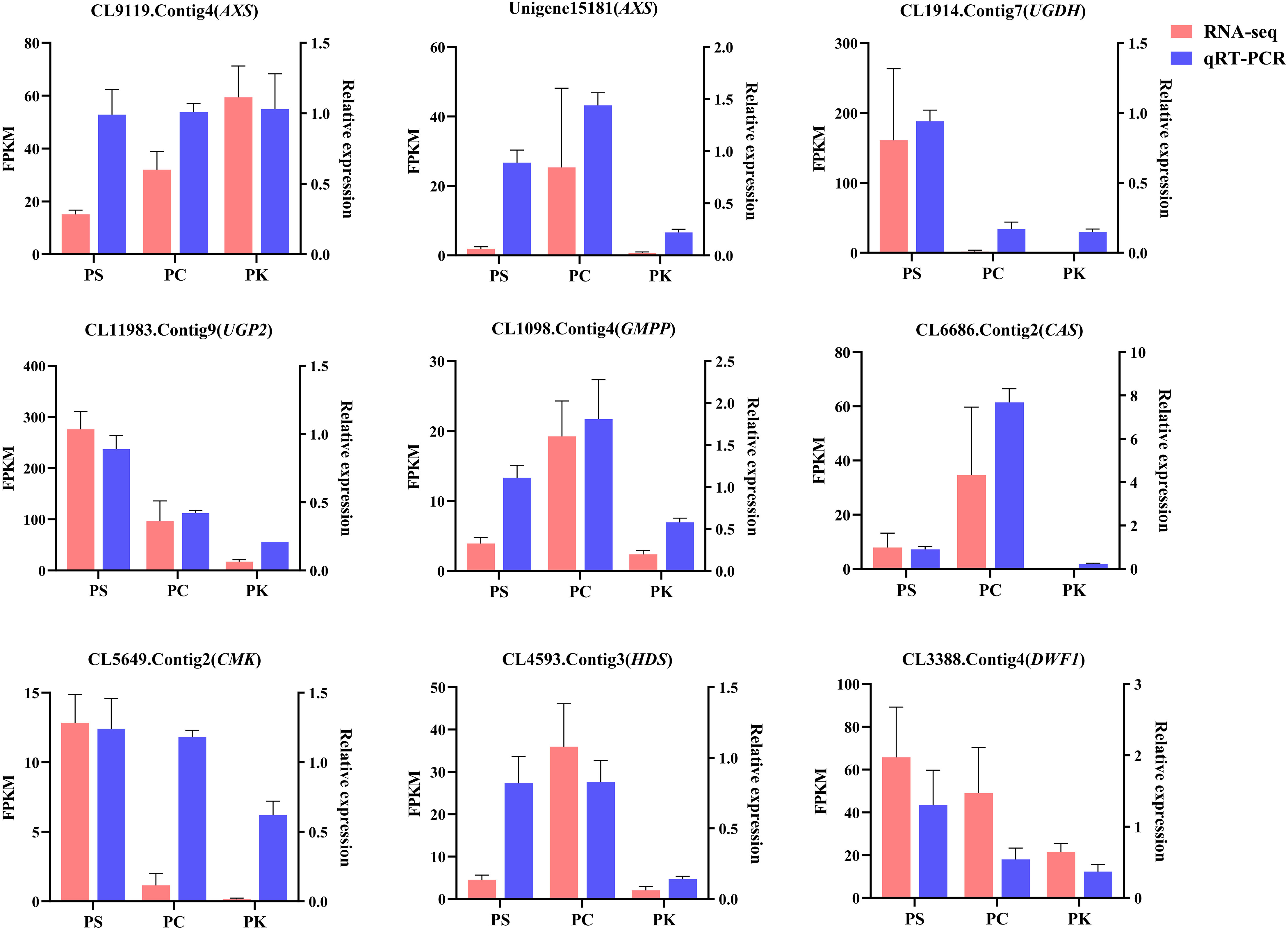
Figure 11 qRT-PCR validation of selected genes. The red bars represent the FPKM values of genes from RNA-seq, and blue bars represent the relative expression determined by qRT-PCR. Data represent mean ± standard error of three replicates. The left Y-axis denotes the FPKM values, and the right Y-axis denotes relative expression levels.
Discussion
The traditional Chinese medicine Huangjing is based on the rhizome of P. sibiricum, P. cyrtonema, and P. kingianum. It is listed as the top grade in the Ming Yi Bie Lu, and can be used as food materials. It is a good tonic medicine for both medicine and food. At present, there are no reports on the genome of P. sibiricum, P. cyrtonema, and P. kingianum at home and abroad. High-throughput transcriptome sequencing technology has become an effective means to study the metabolic pathways of active components of polygonati rhizoma and to mine key enzyme genes due to its advantages of convenience and efficiency. The biosynthetic pathways of polysaccharides and steroidal saponins of polygonati rhizoma have been gradually analyzed, but the differences in the expression of key genes in the metabolic pathways of polysaccharides and steroidal saponins of these three plants have not been reported. Therefore, the transcriptome data of these three plants were analyzed in this paper to study the similarities and differences in their biosynthetic pathways, laying a foundation for improving the content of effective components by molecular means.
β-fructofuranosidase belongs to the 32 family of glycoside hydrolase J clan and is the first key enzyme in the synthesis of polygonatum polysaccharide (Lammens et al., 2009; Naumoff, 2011). Scholars based on transcriptome data analysis, combined with the polysaccharide content determination results, found that the expression pattern of β-fructofuranosidase and polysaccharide content is positively correlated, in the genus Polygonatum another plant Polygonatum odoratum also found that most unigenes expression pattern of encoding β-fructofuranosidase is consistent with the content changes (Wang et al., 2017; Zhang et al., 2020; Feng et al., 2022). After antisense inhibition of GMPP, the content of mannose in transgenic potato decreased by 30-50% compared with wild type (Keller et al., 1999). The mannose content of DoGMP1 isolated from Dendrobium officinale was significantly increased after overexpression, and transgenic plants showed better growth under salt stress, suggesting that GMPP gene may have potential as a candidate gene to improve plant abiotic stress tolerance (He et al., 2017a). In addition to catalyzing the interconversion of UDP-galactose and UDP-glucose, GalE can also catalyze the interconversion of D-galactose and D-glucose and other free monosaccharides, providing a new scheme for the preparation of rare free monosaccharides (Kim et al., 2011). The analysis of transcriptome data indicates that SacA, GMPP, GALE, and others play important roles in the polysaccharide synthesis pathway of Polygonatum plants (Wang et al., 2017; Li et al., 2022). We found that 13 genes encoding SacA, scrK, GMPP, MPI, GMDS, GPI, GALE, and AXS were highly expressed in P. cyrtonema, while their expression levels were low in P. kingianum, suggesting that they might be putative genes affecting polysaccharide synthesis in Polygonati Rhizoma. DoPMM overexpression resulted in 77%, 22%, and 39% increases in polysaccharide content of Arabidopsis thaliana transgenic lines #1, #2, and #5 compared to wild type (WT) levels (He et al., 2017b). Ganoderma and Pseudoalteromonas agarivorans Hao also confirmed that PMM expression level was positively correlated with polysaccharide content (Ju et al., 2022; Zhao et al., 2022). However, the results of this study showed that the expression level of PMM was the highest in P. kingianum.
AXS and UDP-xylose synthase (UXS) has the function of converting UDP-D-GlcA to UDP-D-xyl, and AXS can also catalyze UDP-D-GlcA to UDP-D-Api through decarboxylation and rearrangement of the carbon skeleton. The functional expression of Arabidopsis AtAXS1 in Escherichia coli confirmed that it is a bifunction enzyme (Mølhøj et al., 2003). VIGS of a Nicotiana benthamiana homolog of the AXS genes resulted in cell death in leaves, obstructed synthesis of RG-II, and abnormal cell wall (Ahn et al., 2006). Many studies have provided evidence that AXS is a key regulator of polysaccharide production. The polysaccharide content of rice root cell walls increased under vanadium stress, and the polysaccharide level of tolerant varieties was higher than that of sensitive varieties. Transcriptome analysis showed that genes encoding AXS were also highly expressed in tolerant varieties (Yuan et al., 2022). The polysaccharide content of P. cyrtonema at different growth years was positively correlated with the AXS gene expression pattern (Li et al., 2022). We identified three differentially expressed AXS genes, and multiple sequence alignments showed that they had conserved motifs GxxGxxG, ST, and YxxxK. The expression pattern of one AXS gene (Unigene15181) was consistent with that of polysaccharide content, and its expression level was verified by qRT-PCR.
Some enzymes related to the synthesis of steroidal saponins have been identified, but exploring the enzymes in the pathway of steroidal saponins biosynthesis and the signaling molecules involved in regulation are still the key research areas that need attention (Upadhyay et al., 2018). In this study, 155 genes encoding key enzymes during steroidal saponins synthesis were identified, and 128 DEGs were screened out. Among them, 30 DEGs were associated with MVA pathway, and 21 DEGs were associated with MEP pathway. MVA and MEP pathways may be all involved in the synthesis of saponins (Sun et al., 2017; Upadhyay et al., 2018). The transcriptome analysis of P. kingianum, Asparagus racemosus, and Trillium govanianum showed that monocotyledons seem to produce steroidal saponins through the cycloatenol pathway (Upadhyay et al., 2014; Singh et al., 2017b; Yang et al., 2019). CAS with lupeol synthase (LS), lanosterol synthase (LAS), α-amyrin synthase (AAS), β -amyrin synthase (BAS) belongs to the oxidosqualene cyclases (OCSs) gene family, it can catalyze 2,3-oxidosqualene to produce cycloartenol, the precursor of steroid saponins, sterols, and steroid alkaloids, through chair-boat-chair conformation (Chen et al., 2022). The important functions of CAS gene have been identified in a variety of plants. Overexpression of FPPS and RNA interference of CAS in Panax notoginseng cells finally led to increased total triterpene saponins content and decreased phytosterols content, and inhibition of CAS expression in Tobacco BY-2 cell suspensions also caused a significant decrease in phytosterol accumulation. We obtained two CAS genes in the transcriptome data, which were highly expressed in P. cyrtonema, consistent with the high content of saponin than P. sibiricum and P. kingianum.
TFs can be combined with specific sequences of DNA, inhibit or enhance the expression of target genes, a TF can regulate the expression of multiple genes, in improving the active ingredient content has more advantages than adjusting the single enzyme (Yuan and Grotewold, 2015). In our study, a total of 4454 TFs belonging to 57 TF families were found, among which the MYB family contained the most TFs, followed by AP2-EREBP, C3H, bHLH, WRKY, etc. MYB is one of the largest TF families in eukaryotes and plays an important role in many physiological processes such as secondary metabolism regulation and stress response in plants. It has been reported that overexpression of the candidate gene DOMYB75 in Arabidopsis thaliana can increase the water-soluble polysaccharide content of seeds by about 14% (He et al., 2019). AtMYB46 can directly bind to the CSLA9 promoter sequence, and its overexpression can significantly increase mannose content (Kim et al., 2014). A total of 510 MYB TFs were identified, 94 and 126 TFs were up-regulated in P. cyrtonema compared with P. sibiricum and P. kingianum, respectively. These up-regulated genes may be related to the content of polysaccharides and saponins in Polygonati Rhizoma. Yeast one-hybrid assay and electrophoretic mobility shift assay showed that SMBHLH2 and SMWRKY27 might play important roles in sapindite triterpenoid saponin accumulation by directly regulating the transcription of SMCYP71D-3 (Xu et al., 2022). Panax notoginseng PnbHLH1 TF positively regulates the synthesis of triterpenoid saponins (Zhang et al., 2017). Previous studies have shown that TF family members such as MYB (Li and Lu, 2014; Hao et al., 2020), WRKY (Ma et al., 2009; Singh et al., 2017a) and bHLH (Mertens et al., 2016; Jiang et al., 2021) are involved in the regulation of various secondary metabolites in plants; therefore, the TFs identified in this study that were upregulated in P. cyrtonema need to be further investigated.
Conclusion
Comparative transcriptome analysis was conducted using the RNA-seq method on P. sibiricum, P. cyrtonema, and P. kingianum. The key enzymes involved in the biosynthesis pathway of polysaccharide and steroidal saponins of Polygonati Rhizoma were discovered and the DEGs were further analyzed. In this paper, we reported the transcriptome sequencing data of three medicinal plants of the genus Polygonatum, and verified the accuracy of the sequencing results by qRT-PCR. The acquired transcriptome data are helpful for the molecular study of polysaccharide and steroidal saponins synthesis pathways in Polygonatum SPP.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA974718.
Author contributions
JML: Writing – original draft. JY: Writing – original draft. JP: Writing – original draft. DW: Writing – original draft. JLL: Writing – original draft. YZ: Writing – review & editing. LZ: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Science Research Project at the Universities of Anhui Province for Distinguished Young Scholars (2023AH020036); Excellent Young Scholars Project of Natural Science Foundation of Anhui Province in China (grant numbers 2208085Y30); the NMPA Key Laboratory for Quality Research and Evaluation of Traditional Chinese Medicine (AHYJ-KFKY-202102); Young Elite Scientists Sponsorship Program by CACM (CACM-2023-QNRC2-B23); Key Project Foundation of Support Program for the Excellent Young Faculties in Universities of Anhui Province in China (grant numbers gxyqZD2022051); Traditional Chinese Medicine high-level key discipline construction project of National Administration of Traditional Chinese Medicine - Science of Chinese medicinal material resources (pharmaceutical botany) (zyyzdxk-2023095).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1293411/full#supplementary-material
References
Ahmed, S., Zhan, C., Yang, Y., Wang, X., Yang, T., Zhao, Z., et al. (2016). The transcript profile of a traditional chinese medicine, atractylodes lancea, revealing its sesquiterpenoid biosynthesis of the major active components. PloS One 11, e0151975. doi: 10.1371/journal.pone.0151975
Ahn, J. W., Verma, R., Kim, M., Lee, J. Y., Kim, Y. K., Bang, J. W., et al. (2006). Depletion of UDP-D-apiose/UDP-D-xylose synthases results in rhamnogalacturonan-II deficiency, cell wall thickening, and cell death in higher plants. J. Biol. Chem. 281, 13708–13716. doi: 10.1074/jbc.M512403200
Bai, J. B., Ge, J. C., Zhang, W. J., Liu, W., Luo, J. P., Xu, F. Q., et al. (2021). Physicochemical, morpho-structural, and biological characterization of polysaccharides from three Polygonatum spp. RSC Adv. 11, 37952–37965. doi: 10.1039/d1ra07214e
Chen, C., Pang, Y., Chen, Q., Li, C., Lü, B. (2022). Oxidosqualene cyclases in triterpenoids biosynthesis: a review. Chin. J. Biotechnol. 38, 443–459. doi: 10.13345/j.cjb.210169
Chen, Y., Wu, J., Yu, D., Du, X. (2021). Advances in steroidal saponins biosynthesis. Planta 254, 91. doi: 10.1007/s00425-021-03732-y
Decker, D., Öberg, C., Kleczkowski, L. A. (2017). Identification and characterization of inhibitors of UDP-glucose and UDP-sugar pyrophosphorylases for in vivo studies. Plant J. 90, 1093–1107. doi: 10.1111/tpj.13531
Fang, X., Wang, H., Zhou, X., Zhang, J., Xiao, H. (2022). Transcriptome reveals insights into biosynthesis of ginseng polysaccharides. BMC Plant Biol. 22, 594. doi: 10.1186/s12870-022-03995-x
Feng, T., Jiang, Y., Jia, Q., Han, R., Wang, D., Zhang, X., et al. (2022). Transcriptome analysis of different sections of rhizome in polygonatum sibiricum red. and mining putative genes participate in polysaccharide biosynthesis. Biochem. Genet. 60, 1547–1566. doi: 10.1007/s10528-022-10183-x
Gao, J. P., Wang, D., Cao, L. Y., Sun, H. F. (2015). Transcriptome sequencing of Codonopsis pilosula and identification of candidate genes involved in polysaccharide biosynthesis. PloS One 10, e0117342. doi: 10.1371/journal.pone.0117342
Guo, S. Y., Yin, Y., Lei, T., Shi, Y. H., Gao, W., Zhang, X. N., et al. (2021). A cycloartenol synthase from the steroidal saponin biosynthesis pathway of Paris polyphylla. J. Asian. Nat. Prod. Res. 23, 353–362. doi: 10.1080/10286020.2020.1730331
Han, F. Y., Song, X. Y., Chen, J. J., Yao, G. D. (2018). and song, S Timosaponin AIII: A novel potential anti-tumor compound from Anemarrhena asphodeloides. J.Steroids. 140, 125–130. doi: 10.1016/j.steroids.2018.09.014
Han, Z., Gong, Q., Huang, S., Meng, X., Xu, Y., Li, L., et al. (2023). Machine learning uncovers accumulation mechanism of flavonoid compounds in Polygonatum cyrtonema Hua. Plant Physiol. Biochem. 201, 107839. doi: 10.1016/j.plaphy.2023.107839
Hao, X., Pu, Z., Cao, G., You, D., Zhou, Y., Deng, C., et al. (2020). Tanshinone and salvianolic acid biosynthesis are regulated by SmMYB98 in Salvia miltiorrhiza hairy roots. J. Adv. Res. 23, 1–12. doi: 10.1016/j.jare.2020.01.012
He, C., Teixeira da Silva, J. A., Wang, H., Si, C., Zhang, M., Zhang, X., et al. (2019). Mining MYB transcription factors from the genomes of orchids (Phalaenopsis and Dendrobium) and characterization of an orchid R2R3-MYB gene involved in water-soluble polysaccharide biosynthesis. Sci. Rep. 9, 13818. doi: 10.1038/s41598-019-49812-8
He, C., Yu, Z., Teixeira da Silva, J. A., Zhang, J., Liu, X., Wang, X., et al. (2017a). DoGMP1 from Dendrobium officinale contributes to mannose content of water-soluble polysaccharides and plays a role in salt stress response. Sci. Rep. 7, 41010. doi: 10.1038/srep41010
He, C., Zeng, S., Teixeira da Silva, J. A., Yu, Z., Tan, J., Duan, J. (2017b). Molecular cloning and functional analysis of the phosphomannomutase (PMM) gene from Dendrobium officinale and evidence for the involvement of an abiotic stress response during germination. Protoplasma 254, 1693–1704. doi: 10.1007/s00709-016-1044-1
Hu, Y., Yin, M., Bai, Y., Chu, S., Zhang, L., Yang, M., et al. (2022). An evaluation of traits, nutritional, and medicinal component quality of Polygonatum cyrtonema hua and P. sibiricum red. Front. Plant Sci. 13, 891775. doi: 10.3389/fpls.2022.891775
Huang, L. L. ,. Y., X., S. ,. P., Tong, W., Hu, S. Q. (2012). The first Illumina-based de novo transcriptome sequencing and analysis of safflower flowers. PloS One 7, e38653. doi: 10.1371/journal.pone.0038653
Imadi, S. R., Kazi, A. G., Ahanger, M. A., Gucel, S., Ahmad, P. (2015). Plant transcriptomics and responses to environmental stress: an overview. J. Genet. 94, 525–537. doi: 10.1007/s12041-015-0545-6
Jiang, L., Yu, Y. L., Jiang, M., Cui, X. M., Liu, D. Q., Ge, F. (2021). Synergistic effect on biosynthesis of Panax notoginseng saponins by overexpressing a transcription factor PnbHLH and RNA interference of cycloartenol synthase gene. Chin. J. Chin. Mater. Med. 46, 94–102. doi: 10.19540/j.cnki.cjcmm.20201011.101
Ju, Y., Shan, K., Liu, W., Xi, C., Zhang, Y., Wang, W., et al. (2022). Effect of Different Initial Fermentation pH on Exopolysaccharides Produced by Pseudoalteromonas agarivorans Hao 2018 and Identification of Key Genes Involved in Exopolysaccharide Synthesis via Transcriptome Analysis. Mar. Drugs 20, 89. doi: 10.3390/md20020089
Keller, R., Renz, F. S., Kossmann, J. (1999). Antisense inhibition of the GDP-mannose pyrophosphorylase reduces the ascorbate content in transgenic plants leading to developmental changes during senescence. Plant J. 19, 131–141. doi: 10.1046/j.1365-313x.1999.00507.x
Kim, H. J., Kang, S. Y., Park, J. J., Kim, P. (2011). Novel activity of UDP-galactose-4-epimerase for free monosaccharide and activity improvement by active site-saturation mutagenesis. Appl. Biochem. Biotechnol. 163, 444–451. doi: 10.1007/s12010-010-9052-7
Kim, W. C., Reca, I. B., Kim, Y., Park, S., Thomashow, M. F., Keegstra, K., et al. (2014). Transcription factors that directly regulate the expression of CSLA9 encoding mannan synthase in Arabidopsis thaliana. Plant Mol. Biol. 84, 577–587. doi: 10.1007/s11103-013-0154-9
Lammens, W., Le Roy, K., Schroeven, L., Van Laere, A., Rabijns, A., Van den Ende, W. (2009). Structural insights into glycoside hydrolase family 32 and 68 enzymes: functional implications. J. Exp. Bot. 60, 727–740. doi: 10.1093/jxb/ern333
Li, C., Lu, S. (2014). Genome-wide characterization and comparative analysis of R2R3-MYB transcription factors shows the complexity of MYB-associated regulatory networks in Salvia miltiorrhiza. BMC Genomics 15, 277. doi: 10.1186/1471-2164-15-277
Li, D., Wang, Q., Chen, S., Liu, H., Pan, K., Li, J., et al. (2022). De novo assembly and analysis of Polygonatum cyrtonema Hua and identification of genes involved in polysaccharide and saponin biosynthesis. BMC Genomics 23, 195. doi: 10.1186/s12864-022-08421-y
Li, X. L., Ma, R. H., Zhang, F., Ni, Z. J., Thakur, K., Wang, S., et al. (2023). Evolutionary research trend of Polygonatum species: a comprehensive account of their transformation from traditional medicines to functional foods. Crit. Rev. Food Sci. Nutr. 63, 3803–3820. doi: 10.1080/10408398.2021.1993783
Long, T., Liu, Z., Shang, J., Zhou, X., Yu, S., Tian, H., et al. (2018). Polygonatum sibiricum polysaccharides play anti-cancer effect through TLR4-MAPK/NF-κB signaling pathways. Int. J. Biol. Macromol. 111, 813–821. doi: 10.1016/j.ijbiomac.2018.01.070
Lowe, R., Shirley, N., Bleackley, M., Dolan, S., Shafee, T. (2017). Transcriptomics technologies. PloS Comput. Biol. 13, e1005457. doi: 10.1371/journal.pcbi.1005457
Lu, Y., Luo, J., Huang, X., Kong, L. (2009). Four new steroidal glycosides from Solanum torvum and their cytotoxic activities. Steroids. 74, 95–101. doi: 10.1016/j.steroids.2008.09.011
Luo, L. M., Qin, L., Pei, G., Huang, S. G., Zhou, X. J., Chen, N. H. (2018). Study on structural conversion of dihydrochelerythrine in different solvents. China J. Chin. materia medica. 43 (7), 1416–1426. doi: 10.19540/j.cnki.cjcmm.2018.0055
Ma, D., Pu, G., Lei, C., Ma, L., Wang, H., Guo, Y., et al. (2009). Isolation and characterization of AaWRKY1, an Artemisia annua transcription factor that regulates the amorpha-4,11-diene synthase gene, a key gene of artemisinin biosynthesis. Plant Cell Physiol. 50, 2146–2161. doi: 10.1093/pcp/pcp149
Mertens, J., Pollier, J., Vanden Bossche, R., Lopez-Vidriero, I., Franco-Zorrilla, J. M., Goossens, A. (2016). The bHLH Transcription Factors TSAR1 and TSAR2 Regulate Triterpene Saponin Biosynthesis in Medicago truncatula. Plant Physiol. 170, 194–210. doi: 10.1104/pp.15.01645
Mimaki, Y., Nakamura, O., Sashida, Y., Satomi, Y., Nishino, A., Nishino, H. (1994). Steroidal saponins from the bulbs of Lilium longiflorum and their antitumour-promoter activity. Phytochemistry. 37, 227–232. doi: 10.1016/0031-9422(94)85030-5
Mølhøj, M., Verma, R., Reiter, W. D. (2003). The biosynthesis of the branched-chain sugar d-apiose in plants: functional cloning and characterization of a UDP-d-apiose/UDP-d-xylose synthase from Arabidopsis. Plant J. 35, 693–703. doi: 10.1046/j.1365-313x.2003.01841.x
Moses, T., Papadopoulou, K. K., Osbourn, A. (2014). Metabolic and functional diversity of saponins, biosynthetic intermediates and semi-synthetic derivatives. Crit. Rev. Biochem. Mol. Biol. 49, 439–462. doi: 10.3109/10409238.2014.953628
Naumoff, D. G. (2011). Hierarchical classification of glycoside hydrolases. Biochem. (Mosc) 76, 622–635. doi: 10.1134/S0006297911060022
Rai, A., Yamazaki, M., Takahashi, H., Nakamura, M., Kojoma, M., Suzuki, H., et al. (2016). RNA-seq transcriptome analysis of panax japonicus, and its comparison with other panax species to identify potential genes involved in the saponins biosynthesis. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.00481
Riaño-Pachón, D. M., Ruzicic, S., Dreyer, I., Mueller-Roeber, B. (2007). PlnTFDB: an integrative plant transcription factor database. BMC Bioinf. 8, 42. doi: 10.1186/1471-2105-8-42
Shen, W. D., Li, X. Y., Deng, Y. Y., Zha, X. Q., Pan, L. H., Li, Q. M., et al. (2021). Polygonatum cyrtonema Hua polysaccharide exhibits anti-fatigue activity via regulating osteocalcin signaling. Int. J. Biol. Macromol. 175, 235–241. doi: 10.1016/j.ijbiomac.2021.01.200
Shen, F., Xie, P., Li, C., Bian, Z., Wang, X., Peng, D., et al. (2022). Polysaccharides from polygonatum cyrtonema hua reduce depression-like behavior in mice by inhibiting oxidative stress-calpain-1-NLRP3 signaling axis. Oxid. Med. Cell. Longev. 2022, 2566917. doi: 10.1155/2022/2566917
Sidana, J., Singh, B., Sharma, O. P. (2016). Saponins of agave: chemistry and bioactivity. Phytochemistry 130, 22–46. doi: 10.1016/j.phytochem.2016.06.010
Singh, A. K., Kumar, S. R., Dwivedi, V., Rai, A., Pal, S., Shasany, A. K., et al. (2017a). A WRKY transcription factor from Withania somnifera regulates triterpenoid withanolide accumulation and biotic stress tolerance through modulation of phytosterol and defense pathways. New. Phytol. 215, 1115–1131. doi: 10.1111/nph.14663
Singh, P., Singh, G., Bhandawat, A., Singh, G., Parmar, R., Seth, R., et al. (2017b). Spatial transcriptome analysis provides insights of key gene(s) involved in steroidal saponin biosynthesis in medicinally important herb Trillium govanianum. Sci. Rep. 7, 45295. doi: 10.1038/srep45295
Su, Z., Fang, H., Hong, H., Shi, L., Zhang, W., Zhang, W., et al. (2014). An investigation of biomarkers derived from legacy microarray data for their utility in the RNA-seq era. Genome Biol. 15, 523. doi: 10.1186/s13059-014-0523-y
Sun, H., Li, F., Xu, Z., Sun, M., Cong, H., Qiao, F., et al. (2017). De novo leaf and root transcriptome analysis to identify putative genes involved in triterpenoid saponins biosynthesis in Hedera helix L. PloS One 12, e0182243. doi: 10.1371/journal.pone.0182243
Thoden, J. B., Hegeman, A. D., Wesenberg, G., Chapeau, M. C., Frey, P. A., Holden, H. M. (1997). Structural analysis of UDP-sugar binding to UDP-galactose 4-epimerase from Escherichia coli. Biochemistry 36, 6294–6304. doi: 10.1021/bi970025j
Tian, L. W., Zhang, Z., Long, H. L., Zhang, Y. J. (2017). Steroidal saponins from the genus smilax and their biological activities. Nat. Prod Bioprospect. 7, 283–298. doi: 10.1007/s13659-017-0139-5
Upadhyay, S., Jeena, G. S., Shikha, Shukla, R. K. (2018). Recent advances in steroidal saponins biosynthesis and in vitro production. Planta 248, 519–544. doi: 10.1007/s00425-018-2911-0
Upadhyay, S., Phukan, U. J., Mishra, S., Shukla, R. K. (2014). De novo leaf and root transcriptome analysis identified novel genes involved in steroidal sapogenin biosynthesis in Asparagus racemosus. BMC Genomics 15, 746. doi: 10.1186/1471-2164-15-746
Wang, C., Peng, D., Zhu, J., Zhao, D., Shi, Y., Zhang, S., et al. (2019). Transcriptome analysis of Polygonatum cyrtonema Hua: identification of genes involved in polysaccharide biosynthesis. Plant Methods 15, 65. doi: 10.1186/s13007-019-0441-9
Wang, F., Jiang, Y., Jin, S., Wang, D., Wu, K., Yang, Q., et al. (2022). Structure characterization and bioactivity of neutral polysaccharides from different sources of Polygonatum Mill. Biopolymers 113, e23490. doi: 10.1002/bip.23490
Wang, S., Wang, B., Hua, W., Niu, J., Dang, K., Qiang, Y., et al. (2017). De novo assembly and analysis of polygonatum sibiricum transcriptome and identification of genes involved in polysaccharide biosynthesis. Int. J. Mol. Sci. 18, 1950. doi: 10.3390/ijms18091950
Xu, Y., Zhao, G., Ji, X., Liu, J., Zhao, T., Gao, Y., et al. (2022). Metabolome and transcriptome analysis reveals the transcriptional regulatory mechanism of triterpenoid saponin biosynthesis in soapberry (Sapindus mukorossi gaertn.). J. Agric. Food Chem. 70, 7095–7109. doi: 10.1021/acs.jafc.2c01672
Yang, Y., Ye, B. H., Song, Q. Y., Chen, Y. W., Hu, C. J., DU, G. J., et al. (2020). Selection and validation of internal reference genes for qPCR in Polygonatum cyrtonema tubers at different development stages and in response to abiotic stress. China J. Chin. Materia Med. 45, 5967–5975. doi: 10.19540/j.cnki.cjcmm.20200927.101
Yang, Z., Yang, L., Liu, C., Qin, X., Liu, H., Chen, J., et al. (2019) Transcriptome analyses of Paris polyphylla var. chinensis, Ypsilandra thibetica, and Polygonatum kingianum characterize their steroidal saponin biosynthesis pathway. Fitoterapia 135, 52–63. doi: 10.1016/j.fitote.2019.04.008
Yuan, L., Grotewold, E. (2015). Metabolic engineering to enhance the value of plants as green factories. Metab. Eng. 27, 83–91. doi: 10.1016/j.ymben.2014.11.005
Yuan, Y., Imtiaz, M., Rizwan, M., Dai, Z., Hossain, M. M., Zhang, Y., et al. (2022). The role and its transcriptome mechanisms of cell wall polysaccharides in vanadium detoxication of rice. J. Hazard. Mater. 425, 127966. doi: 10.1016/j.jhazmat.2021.127966
Yuan, Y., Zhang, J., Kallman, J., Liu, X., Meng, M., Lin, J. (2019). Polysaccharide biosynthetic pathway profiling and putative gene mining of Dendrobium moniliforme using RNA-Seq in different tissues. BMC Plant Biol. 19, 521. doi: 10.1186/s12870-019-2138-7
Zhang, S., Shi, Y., Huang, L., Wang, C., Zhao, D., Ma, K., et al. (2020). Comparative transcriptomic analysis of rhizomes, stems, and leaves of Polygonatum odoratum (Mill.) Druce reveals candidate genes associated with polysaccharide synthesis. Gene 744, 144626. doi: 10.1016/j.gene.2020.144626
Zhang, X., Ge, F., Deng, B., Shah, T., Huang, Z., Liu, D., et al. (2017). Molecular cloning and characterization of pnbHLH1 transcription factor in panax notoginseng. Molecules (Basel Switzerland) 22, 1268. doi: 10.3390/molecules22081268
Zhao, L. N., Cao, Y. B., Luo, Q., Xu, Y. L., Li, N., Wang, C. X., et al. (2022). Overexpression of phosphomannomutase increases the production and bioactivities of Ganoderma exopolysaccharides. Carbohydr. Polym. 294, 119828. doi: 10.1016/j.carbpol.2022.119828
Zhao, P., Zhao, C., Li, X., Gao, Q., Huang, L., Xiao, P., et al. (2018). The genus Polygonatum: A review of ethnopharmacology, phytochemistry and pharmacology. J. Ethnopharmacol. 214, 274–291. doi: 10.1016/j.jep.2017.12.006
Zhao, P., Li, X., Wang, Y., Yan, L., Guo, L., Huang, L., et al. (2020a). Characterisation and saccharide mapping of polysaccharides from four common Polygonatum spp. Carbohydr Polym. 233, 115836. doi: 10.1016/j.carbpol.2020.115836
Keywords: Polygonati Rhizoma, transcriptome, polysaccharides biosynthesis, steroidal saponins biosynthesis, transcription factor
Citation: Lu J, Yao J, Pu J, Wang D, Liu J, Zhang Y and Zha L (2023) Transcriptome analysis of three medicinal plants of the genus Polygonatum: identification of genes involved in polysaccharide and steroidal saponins biosynthesis. Front. Plant Sci. 14:1293411. doi: 10.3389/fpls.2023.1293411
Received: 13 September 2023; Accepted: 30 October 2023;
Published: 17 November 2023.
Edited by:
Wei Sun, China Academy of Chinese Medical Sciences, ChinaReviewed by:
Huan Zhao, Capital Medical University, ChinaXiaohui Ma, Yunnan University of Traditional Chinese Medicine, China
Copyright © 2023 Lu, Yao, Pu, Wang, Liu, Zhang and Zha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liangping Zha, emxwX2FodGNtQDEyNi5jb20=; Yazhong Zhang, MjgyNDgzNTA3QHFxLmNvbQ==
†These authors have contributed equally to this work