
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 24 October 2023
Sec. Plant Systematics and Evolution
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1270052
 He Su1,2†
He Su1,2† Xiaoxia Ding1†
Xiaoxia Ding1† Baosheng Liao1Danchun Zhang1
Baosheng Liao1Danchun Zhang1 Juan Huang1,2
Juan Huang1,2 Junqi Bai1,2Subing Xu1
Junqi Bai1,2Subing Xu1 Jing Zhang1,2Wen Xu1,2Xiaohui Qiu1,2*
Jing Zhang1,2Wen Xu1,2Xiaohui Qiu1,2* Lu Gong1,2*
Lu Gong1,2* Zhihai Huang1,2*
Zhihai Huang1,2*Generally, chloroplast genomes of angiosperms are always highly conserved but carry a certain number of variation among species. In this study, chloroplast genomes of 13 species from Datureae tribe that are of importance both in ornamental gardening and medicinal usage were studied. In addition, seven chloroplast genomes from Datureae together with two from Solanaceae species retrieved from the National Center for Biotechnology Information (NCBI) were integrated into this study. The chloroplast genomes ranged in size from 154,686 to 155,979 and from 155,497 to 155,919 bp for species of Datura and Brugmansia, respectively. As to Datura and Brugmansia, a total of 128 and 132 genes were identified, in which 83 and 87 protein coding genes were identified, respectively; Furthermore, 37 tRNA genes and 8 rRNA genes were both identified in Datura and Brugmansia. Repeats analysis indicated that the number and type varied among species for Simple sequence repeat (SSR), long repeats, and tandem repeats ranged in number from 53 to 59, 98 to 99, and 22 to 30, respectively. Phylogenetic analysis based on the plastid genomes supported the monophyletic relationship among Datura and Brugmansia and Trompettia, and a refined phylogenic relationships among each individual was resolved. In addition, a species-specific marker was designed based on variation spot that resulted from a comparative analysis of chloroplast genomes and verified as effective maker for identification of D. stramonium and D. stramonium var. inermis. Interestingly, we found that 31 genes were likely to be under positive selection, including genes encoding ATP protein subunits, photosystem protein subunit, ribosome protein subunits, NAD(P)H dehydrogenase complex subunits, and clpP, petB, rbcL, rpoCl, ycf4, and cemA genes. These genes may function as key roles in the adaption to diverse environment during evolution. The diversification of Datureae members was dated back to the late Oligocene periods. These chloroplast genomes are useful genetic resources for taxonomy, phylogeny, and evolution for Datureae.
Datureae, a tribe belonging to Solanaceae, widely distributed around the world, consists of three clades: Datura L., Brugmansia Pers., and Trompettia gen. nov. It is of importance both in ornamental gardening for its charismatic large flowers (Figure 1) and in medicinal usage for its therapeutic effects in inflammations, skin infections, and rheumatic arthritis (Zhengping et al., 2003; Pigatto et al., 2015; Algradi et al., 2021). Datureae species are widely distributed around the world, while the geographical distribution varied among its members. For example, Datura are found in the southwest U.S.A. and Mexico and parts of central America (Dupin and Smith, 2018), while species of Brugmansia distribute in the Andes and southern portions of the Atlantic forest in Brazil (Chellemi et al., 2011; Kim et al., 2020). In addition, expanding the distribution of several species is also attributed to human activities. The phylogenic analysis for Datureae clade have been addressed and made it easily distinguished through a suite of morphological features. For example, Datura and Brungmansia are distinguished by phenotypes such as fruit type, fruit shape, seed shape, and seed margin (Bye and Sosa, 2013). However, revision of taxonomy and phylogeny relationships continues to be updated; a new monotypic genus, Trompettia, a species previously described in Iochrom Benth, was incorporated in Datureae that owns contorted-conduplicate corolla aestivation characteristics, leading Trompettia to be a new member of Datureae (Smith and Baum, 2006; Olmstead et al., 2008). In addition, the phylogeny relationships among those genus have been verified by a study combining information of morphological and three nuclear markers (Dupin and Smith, 2018). However, the reality that the majority of classification characteristics for Datureae depends on characteristics of flowers and seeds have made it hard to distinguish or identify before flowering period, leading to unpredictable disasters. For instance, some species of Datura and Brugmansia, abundant in tropane alkaloids varying in species and in dose, have been used as phytomedicines in the treatment of inflammations, skin infections, rheumatic arthritis, etc. (Berkov et al., 2006; Benítez et al., 2018; Petricevich et al., 2020; Cinelli and Jones, 2021). However, tropane alkaloids also result in insidious toxicity and may cause hallucinations and poisoning outbreaks and even death in high dose (Doan et al., 2019; Mutebi et al., 2022). Due to similar appearance, the Chinese crude drugs of these species are frequently misidentified; for example, the dried flowers of D. metel listed in the Chinese Pharmacopeia are always mixed with those of D. stramonium, D. inoxia, and even B. arborea during sale process (Han et al., 2011; Wu et al., 2015).

Figure 1 Collected species of Datureae in this study. (A) D. stramonium; (B) D. stramonium var. inermis; (C) D. stramonium var. tatula; (D) D. inoxia; (E) D. metel; (F) B. arborea; (G) B. aurea. Photo taken by Chongjian Zhou.
Although the monophyly of Datureae is not contested, relationships within and among genera are still under-addressed. For example, the phylogenetic relationships between Datura and other two genera are still unclear, although the phylogeny of Datura have been extensively studied (Luna-Cavazos et al., 2009). Some researchers regarded T. cardenasiana as sister to Brugmansia (Särkinen et al., 2013) (Särkinen et al., 2013), while others thought it as sister to Datura and Brugmansia (Ng and Smith, 2016; Dupin and Smith, 2018). High similarity in morphological characteristics among species for the sub-genus of Datureae have made it a challenge for its classification and led to confusion in commercial herbal market. Previous studies have conducted the molecular classification and identification analysis on those genus. Han et al. employed four universal DNA barcodes (ITS2, psbA-trnH, matK, and rbcL) to identify D. metel and its adulterants (Han et al., 2011). Wu conducted the identification study of Datura by using the ITS2 barcode (Wu et al., 2015). However, limited resolution of Sanger sequencing has reduced the capacity of variation identification in Datureae, which limited its application in identification and phylogenetic studies of Datureae. In addition, it was found that the origin of Datureae tribe was approximately 35 million years ago, when the beginning of the Andean uplift based on three nuclear markers (ITS+5.8S, waxy and lfy) (Dupin and Smith, 2018). The uniparental inherited chloroplast (cp) genome, a functional important organelle conducting photosynthesis that converts solar energy into chemical energy and release oxygen, owing to >100 kb genome in size and is highly conserved both in genome structure and nucleotide substitution rates during evolution, has been proven to be a potential tool in species identification and evolution trajectory estimation (Brunkard et al., 2015; Li et al., 2015). For instance, Cui reported the cp genome of Zingiber officinale and constructed a robust phylogenetic relationship among species in the family Zingiberaceae (Cui et al., 2019). Fan identified the sequence differentiation and adaptive variation in cp genomes among the order Dipsacales and provided much useful genetic information not only for species identification but also for the understanding of evolution trajectory for Dipsacales (Fan et al., 2018). Chen showed inconsistencies between the molecular phylogeny and traditional taxonomy for subg. Seriphidium and found out cp genome could be used as super barcode in resolving interspecific relationships as in authentic for subg. Seriphidium (Chen et al., 2023). Gong et al. reported that the cp genome of Amomum villosum, which provided significantly higher resolution in species identification than that of ITS2 and the union of those information, gave power for the detection of hybridization event between A. villosum and A. longiligulare (Gong et al., 2022).
However, to the best of our understanding, only two species of cp genomes belonging to Datureae have been reported; more detailed studies for this tribe are still needed to be carried out (Yang et al., 2014; De-la-Cruz and Núñez-Farfán, 2020). In this study, a total of 13 complete cp genomes of Datureae were sequenced and annotated. Together with seven online available cp genomes in Datureae, we aimed to (1) study the comprehensive characteristics of Datureae cp genomes, (2) reveal the phylogenic relationships and evolutionary history among Datureae species based on cp genomes, and (3) develop the molecular markers on cp genomes for Datureae species identification. This study will facilitate the studies of genetics and evolution of Datureae species.
A total of 13 individuals from seven Datureae taxa were identified by Chongjian Zhou, an engineer in HuBei Guizhenyuan Chinese Herbal Medicine Co., Ltd; Huagu Ye, a professor in South China Botanical Garden, Chinese Academy of Sciences; and Jizhu Liu, a professor in Guangdong Pharmaceutical University (Figure 1; Supplementary Table S1). Fresh leaves were packaged in thin foil, then frozen by liquid nitrogen, and stored in a −80°C fridge for high throughput sequencing. Furthermore, 44 fresh samples of 13 Datureae species were used to test and verify the molecular markers (five universal DNA barcodes) (Supplementary Tables S1, S2). Genomic DNA was extracted using a DNA easy Plant Mini Kit (Qiagen Co., Hilden, Gemany) following the manufactures’ instructions. NanoDrop2000C spectrophotometry and electrophoresis in 1% (w/v) agarose gel were used to detect the concentration and integration of the total DNA, respectively.
Extracted DNA was fragmented to an average size of approximately 400 bp using CovarisM220 (Gene Company Limited, China) for paired-end library construction. Paired-end library was constructed using NEXTFLEX® Rapid DNA-Seq (Bioo Scientific, Austin, TX, USA). Adapters containing the full complement of sequencing primer hybridization sites were ligated to the blunt end of fragments. Paired-end sequencing was performed on Illumina NovaSeq platform (Illumina Inc., San Diego, CA, USA) at Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China). More than 4-Gb raw reads per sample were generated (Supplementary Table S2). Raw reads were quality controlled with Trimmomatic and Fast QC software (https://www.bioinformatics.babraham.ac.uk/projects/fastqc). The assembly strategy of cp genome referred to Zhou’s (Zhou et al., 2017). Cp-like reads were extracted by mapping clean reads against the collection of cp genomes retrieved from the NCBI nucleotide database on the basis of their coverage and similarity. Cp contigs were assembled based on cp-like reads using SOAPdenovo2 (Luo et al., 2015), then scaffolded by SSPACE (Boetzer et al., 2011). Finally, gaps were filled with clean reads using Gap Filler package (Nadalin et al., 2012). The cp genomes generated from this study are available at GPGD (http://www.gpgenome.com/species/) under species IDs 296, 297, 8797, 16984, 16994, 16995, and 62347 (Liao et al., 2022).
The cp genomes were annotated using CPGAVAS2 (Shi et al., 2019) with default settings, except for taking 2,544 rather than 43 plastomes as the reference dataset. Predicted protein-coding genes were extracted and blasted against Swiss-Prot database and then manually corrected in Apollo software (Lewis et al., 2002), from which the latest GFF3 file was used to update the original CPGAVAS2’s prediction. Additionally, tRNA genes were identified by tRNA scan-SE (Lowe and Chan, 2016). The cp genome structure was visualized using Chloroplot software (Zheng et al., 2020). The GC content of the cp genome was calculated by custom R scripts with functions in seqinr (Charif et al., 2005). The distribution of codon usage was investigated using the software CodonW (Sharp and Li, 1986) with RSCU ratios. SSRs (mono-, di-, tri-, tetra-, penta-, and hexanu-cleotide repeats) were detected by MISA (Beier et al., 2017) by setting mono-, di-, tri-, tetra-, penta-, and hexane-nucleotide SSRs to 10, 5, 4, 3, 3, and 3 repeat units, respectively. The long dispersed repeats: including forward (F), palindromic (P), reverse (R), and complement (C) repeats were identified using the online tool REPuter (Kurtz et al., 2001) with default settings. In addition, the tandem repeats (>10 bp in length) were identified using Tandem Repeats Finder program (Benson, 1999), in which the alignment parameters referred to Fan’s (Fan et al., 2018).
The complete cp genomes of Datureae were aligned using MAFFT (Katoh et al., 2019) and compared at cp genome-level by mVISTA (Frazer et al., 2004). The nucleotide diversity (Pi) was calculated by DnaSP with sliding window analysis by setting step size to 200 bp and window length to 800 bp (Rozas et al., 2017). In addition, IRscope was used to evaluate the IR expansion and contraction with the GenBank files (Amiryousefi et al., 2018).
We extracted shared non-redundant gene CDS and among 22 cp genomes from Datureae species, and extracted each gene’s CDS pair of one-by-one species combination and aligned them with MAFFT (Katoh et al., 2019). The rate of non-synonymous (Ka) and synonymous (Ks) substitutions and Ka/Ks were then calculated by ParaAT2.0 (Zhang et al., 2012), which is called KaKs_Calculator2.0 (Zhang et al., 2006) with “MA” model. The command that we applied is as follows: “ParaAT.pl -c 11 -h homologs.txt -n CDS -a PEP -p proc -o OUT -k -f axt -m mafft -v”. In addition to 13 cp genomes of Datureae sequenced in this study (Supplementary Table S1), nine published cp genomes from NCBI were retrieved for further phylogeny analysis. In details, Nicandra physalodes and Atropa belladonna were used as an outgroup to construct the phylogenetic tree (Supplementary Table S4), among which four more cp genomes (D. stramonium var. tatula-2, D. stramonium var. tatula-3, B. arborea-2, and B. aurea-3) sequenced in this study were added to obtain more reliable phylogenetic relationships. Maximum likelihood (ML) method in RAxML-ng (Kozlov et al., 2019) was used by providing multiple sequence alignment file generated from whole cp genomes with MAFFT (Katoh et al., 2019). For RAxML-ng, the model parameter was set to “GTR+I+G4,” which was chosen by model test planted in RAxML-ng; bs-metric was set to “fbp, tbe;” 100 starting trees (50 random and 50 parsimony-based) were used to pick the best-scoring topology; and bootstrap replicates were set to 1,000. In addition, MrBayes (Huelsenbeck and Ronquist, 2001) was used for Bayesian inference of phylogeny by setting MCMC simulations to 10,000,000 generations, sampling frequency to one out of every 1,000 generations, and heating coefficient to 0.07. The first 25% of the trees were regarded as burn-ins. Two-runs-two-independent analyses starting from different random trees was used to calculate convergence diagnostics on the fly.
The divergence times between lineages were estimated by clocks module in MEGA11 (Tamura et al., 2021), where we applied the RelTime method (Tamura et al., 2012). Theoretical foundation of the RelTime method for estimating divergence times from variable evolutionary rates to the user-supplied phylogenetic tree using the ML method generated by RAxML-ng and general time reversible substitution model with four categories was chosen. Three calibration constraints queried from TimeTree (Kumar et al., 2022) with normal distribution were set, with node Bar_1-Bau_1 (mean=10.1, sd=0.2), Dino-Dme_2 (mean=9.5, sd=0.2), and Dine-Dst_1 (mean=1.71, sd=0.1), respectively; in addition, Abe-Nph node was set as outgroup.
First, five universal plant DNA barcodes (ITS, ITS2, matK, rbcL, and psbA-trnH) were used for the testing of the identification for Datureae species. Second, specific primer was designed based on highly variable regions of D. stramonium and D. stramonium var. inermis and used for further validation. The PCR reaction system was performed in a total volume of 25 μl that contained 2× Taq PCR Mix of 12.5 µL, forward primer (10.0 µM) of 1.0 µL, reverse primer (10.0 µM) of 1.0 µL, genomic DNA of 2.0 µL (30–100 ng), and added up to 25 µL with ddH2O. The primers and conditions for PCR are listed in Supplementary Table S3. All the PCR products were sent to Sangon Biotech Guangzhou branch office for sequencing. The bi-directionally sequenced peaks of DNA markers were assembled using the CondonCode Aligner v8.0.1 software (https://www.codoncode.com/aligner). Neighbor-joining (NJ) trees were constructed with 1,000 bootstrap replicates for each marker in MEGA (Tamura et al., 2021) to evaluate its discrimination power, using 50% as a cutoff value for the condensed tree.
Fresh leaves of seven Datureae taxa were extracted with total DNA and subjected to NGS with Illumina NovaSeq paired-end sequencing. Cp-like sequences were extracted from clean Illumina reads by BLAST searches against an in-house constructed chloroplast database. As a result, the cp genomes showed typical quadripartite structures, which were divided into LSC region and SSC region by IRa/IRb regions with genome size ranging from 154 bp, 686 bp to 155 bp, 979 bp (Figure 2). The overall GC content for these seven taxa was nearly identical (~37.8%) but was unevenly distributed in the cp genomes (Table 1). In details, the GC content was the highest in IR regions (approximately 43.0%) while the lowest in SSC regions (approximately 32.0%), and 35.80%–36.02% for LSC regions in both genera. After annotation, 128–132 genes including 83–87 protein-coding genes, 8 rRNA, and 37 tRNA genes were predicted in Datureae species (Supplememtary Table S5). Among these genes, 18 duplicated genes were found in the IR region, including eight protein-coding, six tRNA, and four rRNA genes (Table S5). We found that D. metel and D. stramonium var. tatula have one more copy of rps19 gene than other species, while there is a lack of a copy of ycf1 gene in D. metel-1. Furthermore, petB and petD genes in D. stramonium, D. stramonium var. inmeris, and two samples of D. metel contained one intron for each. Moreover, petB in D. inoxia also contained one intron. However, there is no intron in petB or petD genes of Brugmansia species (Supplementary Table S5).
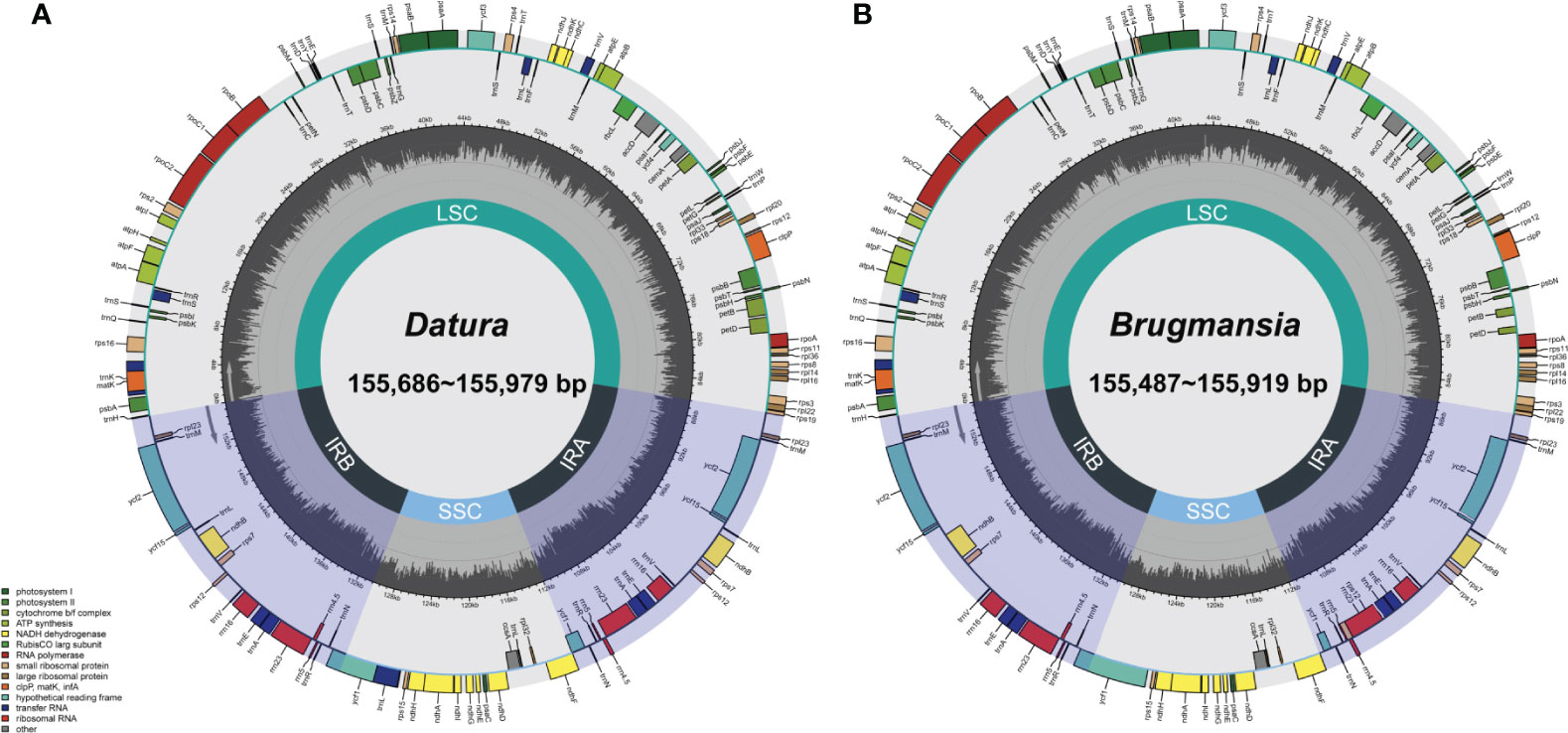
Figure 2 Datura (A) and Brugmansia (B) cp genome maps. Genes drawn within the circle are transcribed clockwise; genes drawn outside are transcribed counterclockwise. Genes in different functional groups are shown in different colors. Dark bold lines indicate the extent of IR regions that separate the genomes into SSC and LSC regions.
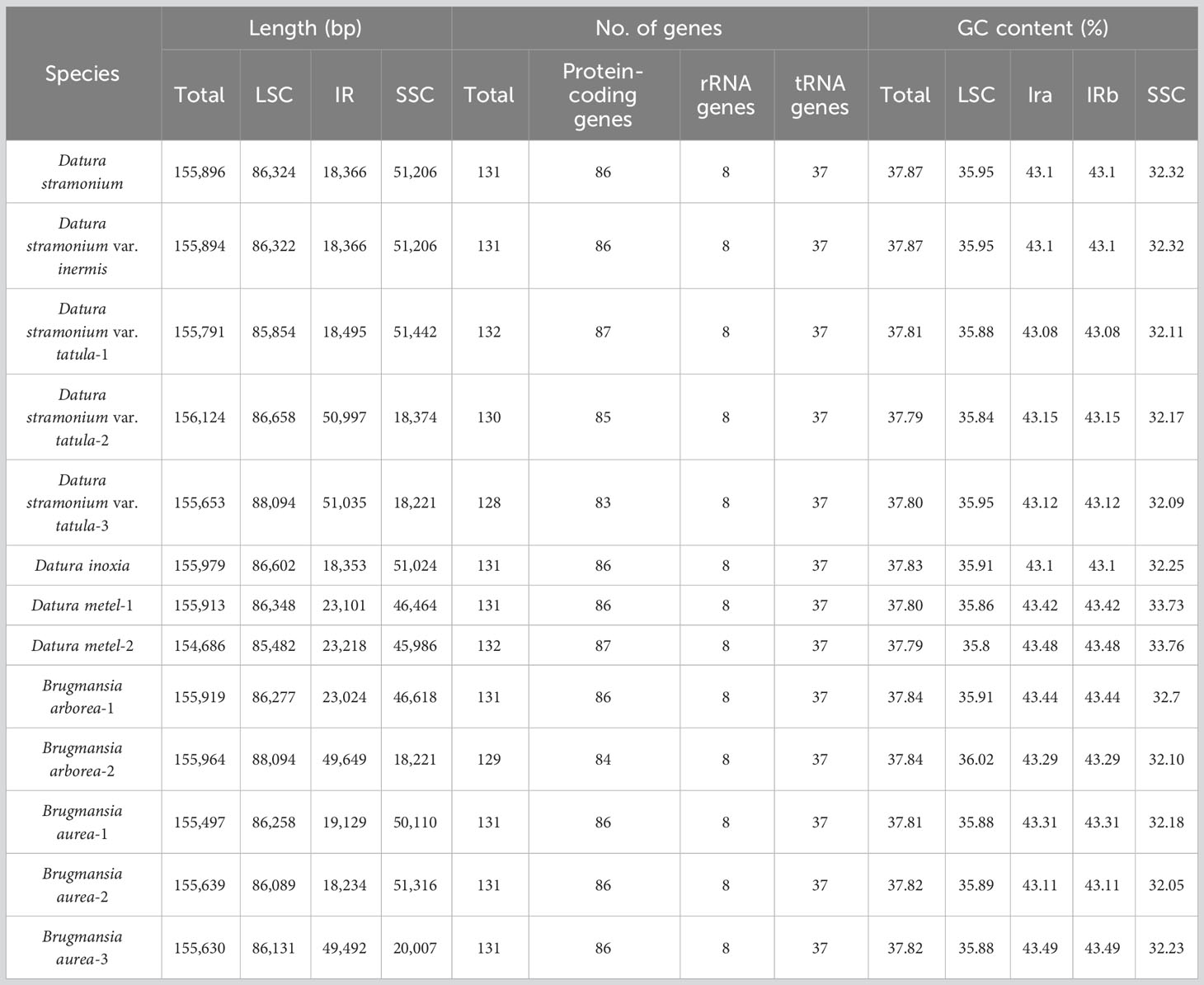
Table 1 Chloroplast genome features of seven Datureae taxa.
Codon usage patterns and nucleotide composition help to lay a theoretical foundation for genetic modifications of the cp genome (Romero et al., 2000; Angellotti et al., 2007). Amino acids frequency and codon usage were determined in this study, a range of 26, 207–26, and 358 codons were included in the protein-coding genes wherein leucine was the most abundant (10.6%) while cysteine was the least (1.2%) (Supplementary Figure S1).
Simple sequence repeats (SSRs) are tandem repeats of DNA sequences with 1–6 bp in length that have been widely applied as molecular markers in species authentication (Powell et al., 1995; Song et al., 2014). In total, 53–59 SSRs were detected among nine cp genomes, among which, mono-nucleotide repeats were the most frequent, followed by di-, tetra-, and tri-repeats. Noteworthy, penta-repeats were only found in B. arborea and B. aurea, while hexa-repeats were only found in D. stramonium var. tatula, D. inoxia, and D. metel with the least numbers (Supplementary Figure S2A). Among these SSRs, A/T repeats were the most frequent, followed by AT/AT repeats (Supplementary Figure S2B), which were consistent with the majority reported angiosperms (Gao et al., 2019; Li et al., 2019). Except for SSRs, a total of 98–99 long dispersed repeats were detected in nine cp genomes by REPuter (Kurtz et al., 2001), among which the forward and palindrome repeats were found to be the most abundant repeats while complement repeats were the least (Supplementary Figure S3A) and repeats with 11–20 bp were most frequent and 41–51 bp were the least among nine cp genomes (Supplementary Figure S3B). Additionally, 22–30 tandem repeats were identified in nine cp genomes using Tandem Repeats Finder program (Benson, 1999), in which most of the repeat units were 11–30 bp in length (Supplementary Figure S3C).
The expansion and contraction of IR regions are common phenomena during the evolution and the main reason for variations in cp genome length. Therefore, the IRs and SC borders were detected among nine cp genomes. The border of LSC/IRb junction (JLB) located in rps19 gene in D. metel_1, D. stramonium, D. stramonium var. inermis, B. arborea_1, and B. aurea_2, but varied 64–112 bp in length overlapping with IRb. In B. aurea_1, D. metel_2, and D. inoxia, rps19 gene located in LSC region with 14–332 bp from LSC/IRb border, while in D. stramonium var. tatula, rps19 gene completely contracted into IRb region. To the border of SSC/IRa junction (JSA), except for B. arborea_1, D. metel_1, and D. metel_2, the IRa of other species expanded into ycf1 gene. The border of SSC/IRb junction (JSB) was located between ycf1 and ndhF genes, and JSB of B. aurea_2 was located in the overlap of these two genes, while in B. arborea_1, two ycf1 genes were located in IRb and SSC, respectively. Gene trnH located on the border of LSC/IRa was 4–157 bp away from IRa region (Figure 3).
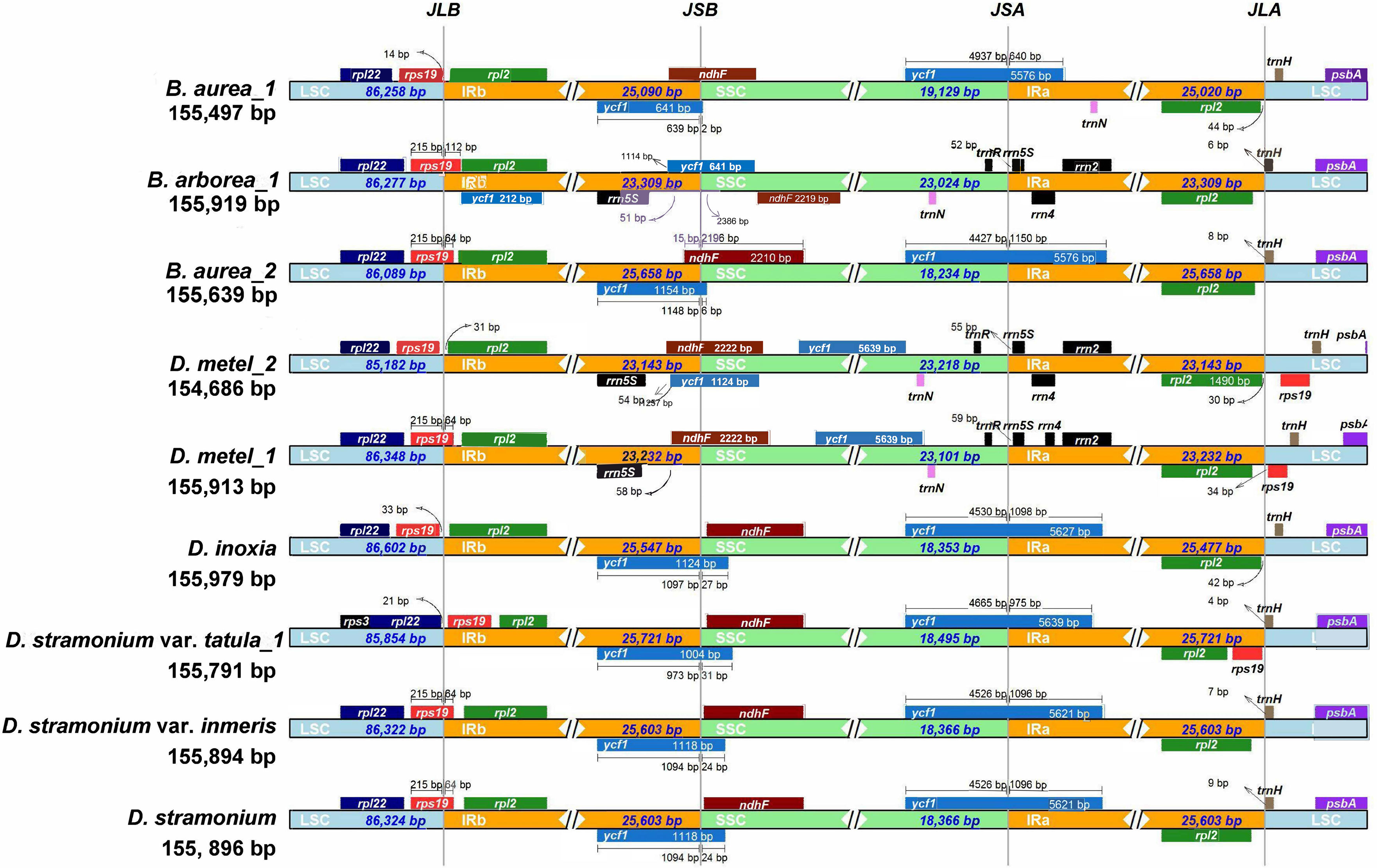
Figure 3 Contraction and expansion of IR borders of Datureae cp genomes.
Multiple alignments of cp genomes were conducted taking D. stramonium as a reference. The results indicated that gene or sequence order and organization were conserved among these cp genomes. Sequences in non-coding regions were more divergent than coding regions (Figure 4). In non-coding regions, the most divergent regions were mainly located in the intergenic spacers. Furthermore, the nucleotide diversity (Pi) values of cp genomes were calculated using DnaSP. Based on DNA polymorphisms, five highly diverged regions were identified, including ycf1_trnN-GUU, ccsA-ndhD, rps16-psbK, atpH-atpI, and ycf1, and the Pi value of these regions ranged from 0.1673 (atpH-atpI) to 0.02932 (ycf1_trnN-GUU), which might be potential candidates for Datureae identification (Supplementary Figure S4).
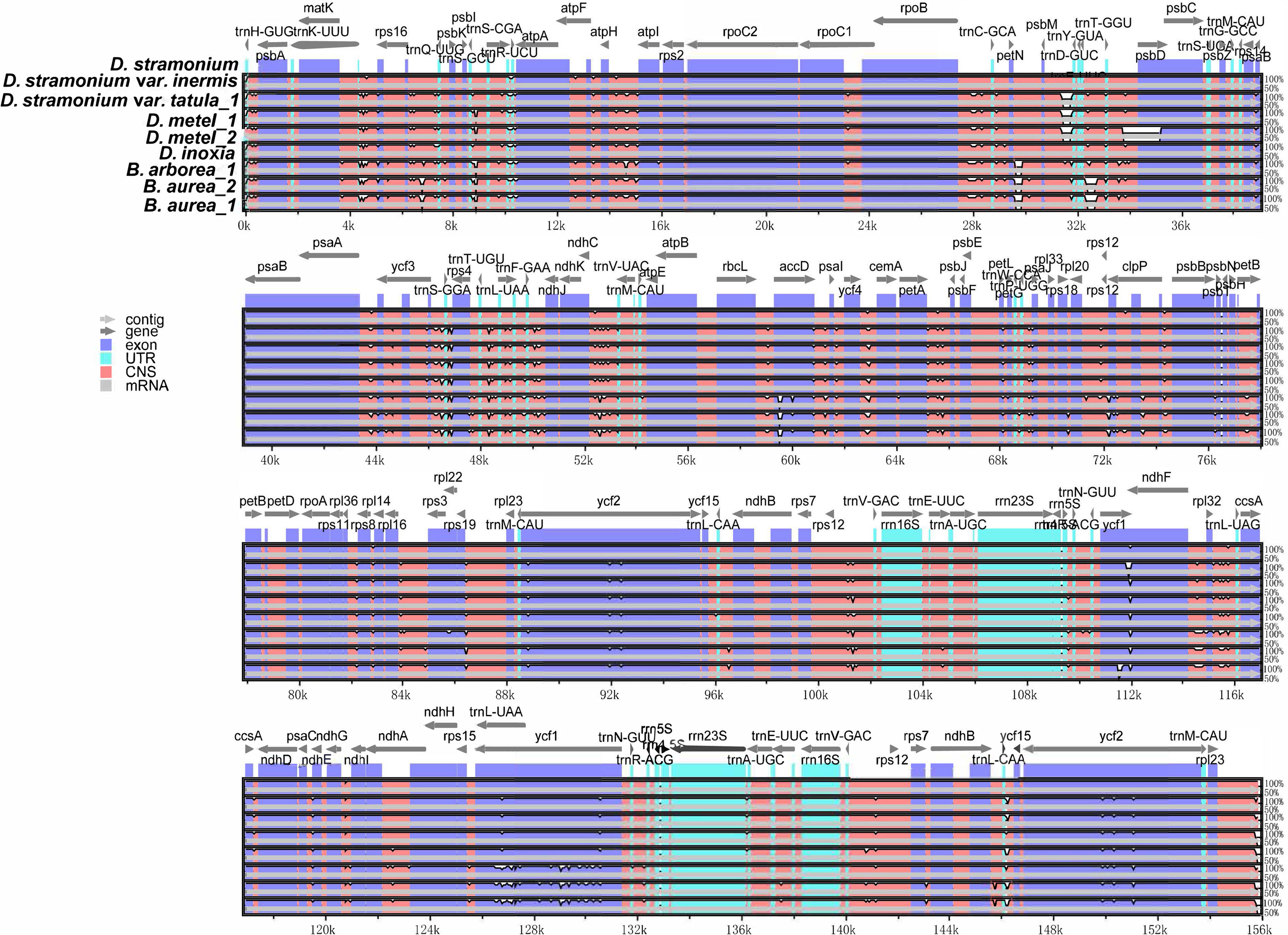
Figure 4 Sequence alignments of Datureae cp genomes using mVISTA with D. stramonium as a reference. Gray arrows and thick black lines above the alignment indicate genes with their orientation and the position of the inverted repeats (IRs), respectively. A cutoff of 70% identity was used for the plots, and the Y-scale represents the percent identity ranging from 50% to 100%.
To unravel the phylogenetic relationships among Datureae species, the phylogenetic tree was re-constructed based on complete cp genomes and super gene of protein coding genes from the 22 Solanaceae species using ML and Bayesian inference (BI) methods, respectively (Supplementary Tables S1, S4). The topologies of these trees were nearly consistent (Figure 5; Supplementary Figure S5). The phylogenetic tree of available Datureae species presented two main clades. One clade comprised T. cardenasiana, while the other clade was further divided into two subclades, in which one subclade contained species from Brugmansia and another subclade contained species from Datura, which revealed the monophyly of these three genera. In Brugmansia subclade, B. aurea and B. suaveolens clustered to two branches separately with strong support (bootstrap value=100%). The Datura subclade was separated into two branches, one contained D. stramonium and D. stramonium var. inermis and the other contained D. inoxia, D. metel, and D. stramonium var. tatula. In detail, different accessories of one species were always clustered together except for those of D. stramonium and D. stramonium var. inermis, indicating close relationships between these two taxa. Molecular divergence dates of the eight Datureae taxa were computed based on the shared unique cp protein-coding gene sequences (Supplementary Figure S6). The diversification of Datureae members could be dated back to approximately 27.08 mya, which is in the late Oligocene periods; the divergence between Datura and Brugmansia occurred at 24.9 mya, which is in the early Neogene periods, suggesting a late Oligocen origin for these two genera within the tribe.
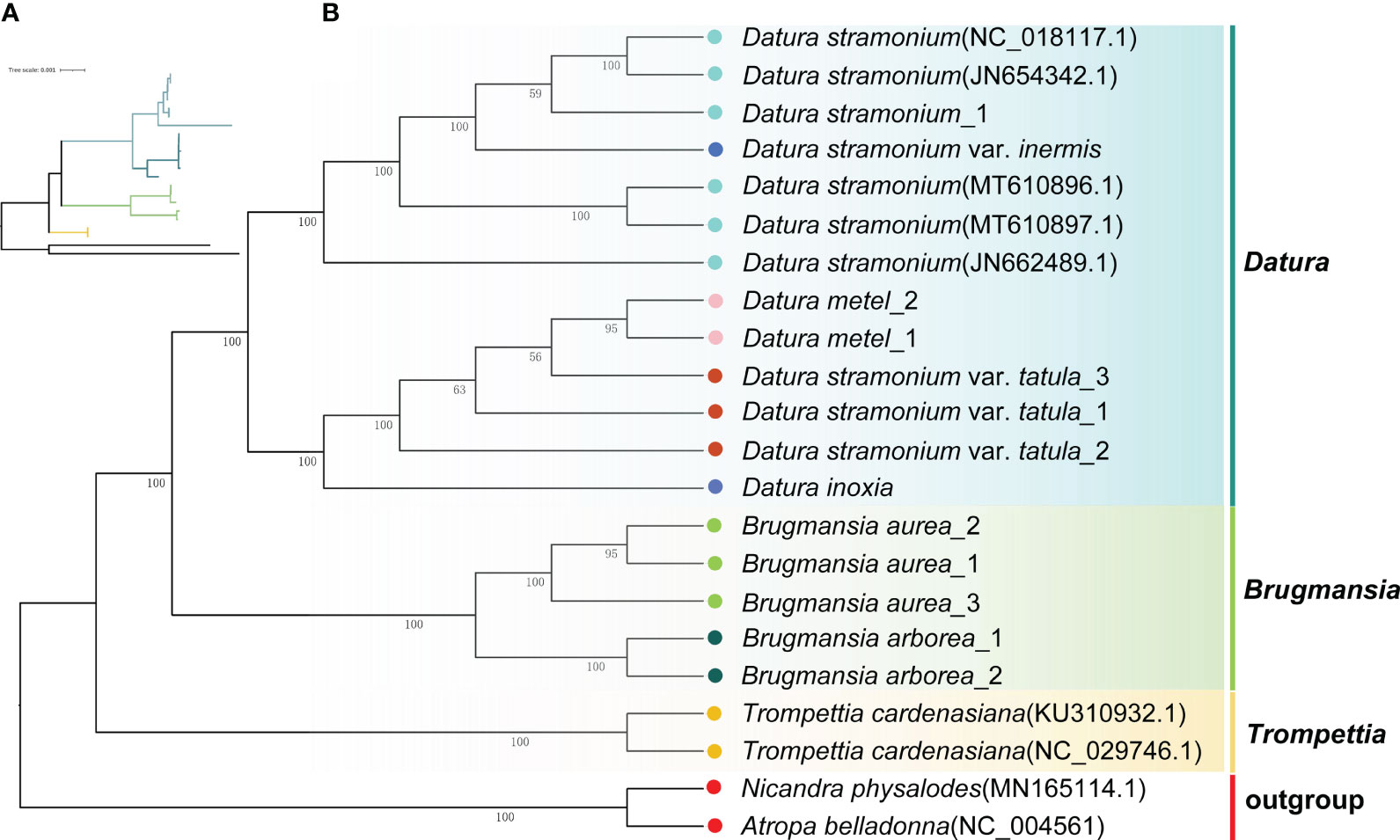
Figure 5 Phylogenetic tree inferred from ML analysis. (A) The tree topology of Datureae species. (B) Complete ML tree using the cp genomes of 20 Datureae species. The number above the lines indicates the ML bootstrap values.
In addition, we calculated the non-synonymous (Ka) and synonymous (Ks) substitution ratios (Ka/Ks) for all the shared unique 69 protein coding genes of cp genomes from 20 Datureae and 2 outgroup species from Solanaceae family, respectively, with KaKs_calculator by “MA” model and statistically tested by Fisher’s exact test. As a result, 31 genes were identified to be positively selected genes along the lineage; in detail, three genes included in ATP subunits (atpB, atpE, and atpH), nine genes in photosystem subunit (psaA, psaI, psaJ, psbB, psbC, psbF, psbJ, psbK, and psbT), four genes in ribosome large subunit (rpl14, rpl16, rpl32, and rpl33), six genes in ribosome small subunit (rps3, rps4, rps11, rps15, rps16, and rps18), three genes in NAD(P)H dehydrogenase complex (ndhE, ndhG, and ndhI), and clpP, petB, rbcL, rpoC1, ycf4, and cemA genes. However, only one substitution for those genes were detected in the multiple sequence alignment (MSA) files except for cemA (chloroplast envelope membrane protein), which owned >20 substitutions in the MSA file. It is noteworthy to mention that there are only ~9.4% of gene pairs, and ~10.0% (after excluding genes with one substitution) are with Ka/Ks >1. Considering that the essence that cp genes are functionally conserved genes and non-synonymous variation is not preferable, we just visualized Ka/Ks for genes with >1 substitutions (Figure 6). Overall, Ka/Ks values were <0.5 for the majority genes, suggesting that cp genes of the Datureae species are conserved and mainly under a purifying selection during the evolution process, which is reasonable for necessary functions played by the chloroplast genes and is in accordance with previous studies (Liu et al., 2020).

Figure 6 Pairwise Ka/Ks for shared non-redundance from Datureae and genes with >1 substitutions and significant different Ka and Ks values, examined by Fisher’s exact test by KaKs_calculator, were plotted. Genes like rps16 only have approximately 20 Ka/Ks values indicating that most of them among species are too conserved to calculate out the Ka/Ks values.
Five universal DNA barcodes (ITS, ITS2, psbA-trnH, matK, and rbcL) were amplified and sequenced to assess the identification ability and reveal the phylogenetic relationships among 44 samples of Datureae species (Figure 7A; only Supplementary Figure S7). The result suggested that universal barcodes were limited in variation detection, and closely related species such as D. stramonium and D. stramonium var. inermis cannot be distinguished from each other. Based on the phylogeny relationship among those species, comparative analysis for genomes sequence was conducted between D. stramonium and its variant D. stramonium var. inermis using sliding window method. As a result, a species-specific site with two mutation sites (TT-AA) was screened out, which were located at 79 bp, 749–79 bp, and 750 bp in the multiple alignment sequence (MSA) file; specific primers for the 330 bp of cp genome region spanned by the mutation site had been tested and validated by PCR amplification and Sanger sequencing (Figure 7B; Supplementary Figure S8).
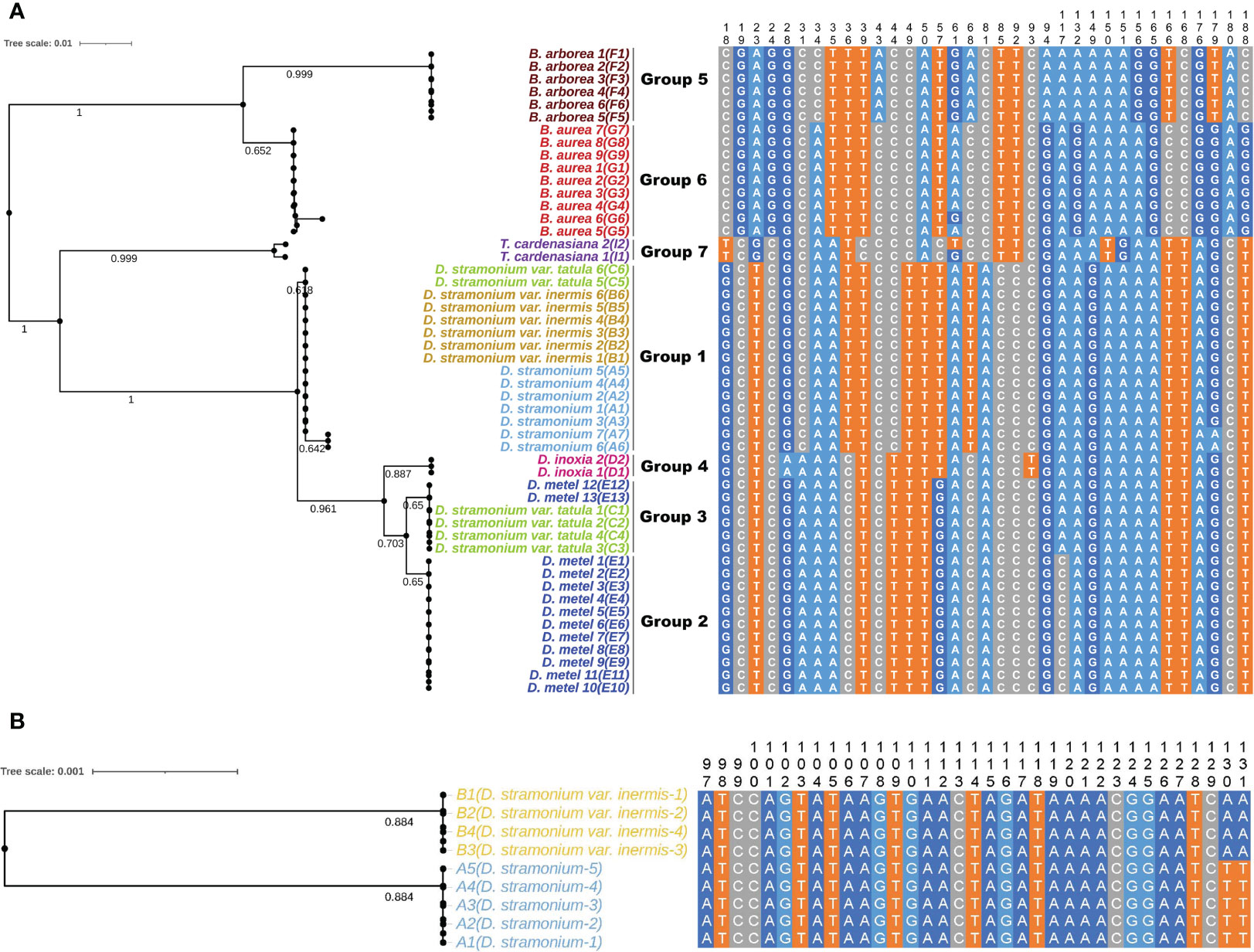
Figure 7 The NJ trees and sequence alignment based on ITS2 (A) and the mined marker (B) for Datureae species.
The cp genomes ranged in size from 154 bp, 686 bp to 155 bp, 979 bp and from 155 bp, 497 bp to 155 bp, 919 bp for species of Datura and Brugmansia, respectively. It is a common evolutionary phenomenon in cp genome of plants that the expansion and contraction events of four IR boundaries make the whole cp genome size differ in the same plant population or among different plant populations. As a result, we think that the size variation for these cp genomes may be attributed to the expansion and contraction of the border positions between IR and SC regions (Fan et al., 2018). In addition, repeats composition is another reason responsible for variations in genome size (Chumová et al., 2021; Schley et al., 2022). However, the composition varied little among Datureae species, indicating that expansion and contraction of IR boundaries contribute to the most for cp genome size variation. In most flowering plants, the rearrangement and variation in cp genome sequence are closely related to the frequent variation of repeat regions, mainly caused by unconventional recombination and mismatch (Park et al., 2017). This study analyzed four types of repeat sequences in Datureae species. We found that repeats are similar both in amount and category among Datureae where species clustered in the same branch of the phylogenetic tree have the most similar SSR type and length distribution; the phenomenon is consistent with previous studies for flowering plants (Sugita and Sugiura, 1996). Gene loss-and-gain events is present in some species, despite that cp genomes of land plants are considered to be highly conserved (Daniell et al., 2016). In this study, we found that the number of protein coding gene ranges from 83 to 87 and 84 to 86 in Datura and Brugmansia, respectively, indicating that gene loss-and-gain event might occurred during the evolutionary process. Furthermore, the Datureae cp genomes have quadripartite structures and conserved gene contents and arrangements, while GC content for IRs is higher than that of LSC and SSC regions. GC content varied among regions, which might attribute to the location of four rRNAs on IRs because rRNA is lacking of AT nucleotides from previous reports (Qian et al., 2013).
Synonymous and non-synonymous nucleotide substitution patterns are important markers for gene evolution studies. In most genes, synonymous nucleotide substitutions have occurred more frequently than non-synonymous ones (Ogawa et al., 1999). Thus, a ratio of Ka/Ks < 1 indicates purifying selection, Ka/Ks > 1 denotes probable positive selection, and Ka/Ks values close to one indicate neutral evolution. In this study, the majority of the protein-coding genes of Datureae species were found to be under purifying selection by Ka/Ks analysis, which was conservative in plastid genomes of most angiosperms (Gong et al., 2022). However, we found that 31 cp genes might be under positive selection during the evolution process; these genes are connected to proteins from ATP subunit, photosystem subunit, ribosome large subunit, NAD(P)H dehydrogenase complex, and other genes such as clpP, petB, rbcL, rpoC1, ycf4, and cemA. These positive selected genes might explain the higher fitness to diverse environment situation for Datureae species. For example, cemA plays an important role in protein sorting signals, and it is also be found to undergo adaptation evolution in Anisodus tanguticus of Solanaceae and Gossypium (Zhou et al., 2022). psbT encodes a small hydrophobic polypeptide, which is functional essential to optimize the electron acceptor complex of the acceptor side of PS II (Fagerlund et al., 2020).
The phylogenetic trees constructed with cp genomes indicates that all Datureae species converge into a monophyletic branch, which is divided into three branches with high support for Datura, Brugmansia, and Trompettia. Trompettia was previously thought as a sister genus of Brugmansia (Särkinen et al., 2013), but in recent years, it was regarded as a common sister genus of Brugmansia and Datura (Ng and Smith, 2016; Dupin and Smith, 2018) The result was convinced by our study by introducing both universal DNA barcodes and cp genomes. In Datura, there are two major sections defined: sect. Datura and sect. Dutra (Bye and Sosa, 2013). Two branches of this genus in our phylogenetic trees match well with this definition. The similar topologies obtained based on various analyses, including these obtained in this study, indicate the clear phylogenetic relationships in Datureae. In FRPS and Flora of China, only one genus Datura of this tribe was included. There are four Datura species listed in FRPS; they are D. arborea, D. innoxia, D. metel, and D. stramonium. The monophyly of Datura and Brugmansia was once disputed based on morphological characters (Persoon, 1805; Bernhardi, 1833; Safford, 1921; Barclay, 1959), and B. arborea was listed as D. arborea in FRPS. However, multiple phylogenetic studies supported the separating genera (Bye and Sosa, 2013; Särkinen et al., 2013; Ng and Smith, 2016). Thus, in flora of China, D. arborea was removed, and only three species were left. We support this revision according to this and previous phylogenetic studies. However, the opinion described in FRPS that the variation in flower color and seed texture among D. inermis, D. tatula, and D. stramonium might attribute to genetically unstable and evolutionarily meaningless dominant and recessive genes was not fully supported in our study. We supported the hypothesis that D. inermis might be regarded as D. stramonium, but D. tatula was different from D. stramonium. In addition, we have estimated the divergence times of eight Datureae taxa based on the protein-coding sequences in the complete cp genomes. The origin time of Datureae tribe was estimated approximately 27.08 mya in Oligocene, which is consistent with the time queried from TimeTree database (a median of 28.6 mya). However, the divergence time estimated in in this study differed from that reported by Dupin and Smith (2018) slightly. Future phylogenetic work in Datureae would benefit from more extensive sampling with cp genomes analysis to resolve uncovered regions of the phylogeny and to provide a robust test for the diversification of the genus.
The accurate identification of plant species is important not only for taxonomy but also in agriculture and pharmaceuticals. Generally, ITS2 is an efficient DNA barcode for species identification and have been applied to authenticate for herb extensively (Chen et al., 2023); our study for species identification with ITS2 and other four universal DNA barcodes failed to resolve the identification issues for several closely related Datureae species even with our custom database, which contains 1,276 barcodes (Gong et al., 2018).
Using cp genomes as super barcodes, different accessories of one species clustered together with higher supports than that of ITS2, suggesting that cp genome could be a super barcode with potential in the authentication of Datureae species. However, D. stramonium and D. stramonium var. inermis still could not be discriminated between each other, indicating close relationships between these two taxa. Intergenic regions play important roles in gene expression regulation and can accumulate more mutations than protein-coding regions, and therefore, they can be used to develop molecular markers for reconstruction of phylogeny (Parenteau and Abou Elela, 2019). Based on the hotspots of variation between D. stramonium and D. stramonium var. inermis, we have developed a molecular marker and succeeded in distinguishing these closely related species. However, it is worth mentioning that we found that D. stramonium var. tatula, supposed to be relatives to D. stramonium, was closer to D. metel, which is inconsistent with a previous report (Wu et al., 2015). Taking the uniparental inheritance characteristics of cp genome and similarity in morphological traits, we hypothesized that D. stramonium var. tatula might be a hybrid of D. stramonium and D. metel; however, substantial evidence still needs to be provided.
In this study, we have sequenced, de novo assembled and annotated 13 complete cp genomes of seven taxa from Datureae tribe. Variations in genome size, repeat composition, gene composition, selective pressure, and evolution trajectory among them were studied. A total of 31 genes identified as positive-selected genes during evolution might play important function roles in the adaptive process. In addition, we succeeded in distinguishing D. stramonium from its closely related species D. stramonium var. inermis by screening out molecular markers based on the high variation hotspots that were resulted from comparative analysis at cp genomic level. These cp genomes are useful genetic resources for taxonomy, phylogeny, and evolution for Datureae.
The data presented in the study are deposited in the Global Pharmacopoeia Genome Database (http://www.gpgenome.com/species/) under species IDs 296, 297, 8797, 16984, 16994, 16995 and 62347.
HS: Methodology, Software, Writing – original draft. XD: Writing – original draft, Data curation, Formal Analysis. BL: Data curation, Formal Analysis, Visualization, Writing – original draft. DZ: Visualization, Writing – original draft. JH: Supervision, Writing – original draft. JB: Supervision, Writing – review & editing. SX: Validation, Writing – review & editing. JZ: Project administration, Writing – review & editing. WX: Writing – review & editing, Project administration. XQ: Investigation, Validation, Writing – review & editing. LG: Investigation, Validation, Writing – review & editing. ZH: Project administration, Writing – review & editing, Conceptualization, Funding acquisition, Resources.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from Guangdong Provincial Medical Products Administration of China (2019KT1261 and 2020ZDB25) and Project for Automatic Analyzer of DNA barcodes based on Sanger Sequencing and DNA barcodes database construction for medicinal plants (YN2019QJ05).
We sincerely thank Dr. Jiang Xu from Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences for giving us important instructions and guidance about this work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1270052/full#supplementary-material
Cp, chloroplast; LSC, large single-copy region; IR, inverted repeat region; SSC, small single-copy region; CDS, coding DNA sequence; tRNAs, transport RNAs; rRNAs, ribosomal RNAs; AT1, AT content in first codon positions of protein coding genes; AT2, AT content in second codon positions of protein coding genes; AT3, AT content in third codon positions of protein coding genes; RSCU, relative synonymous codon usage; SSRs, simple sequence repeats; Pi, nucleotide diversity; Ka/Ks, the rate of non-synonymous substitutions to the rate of synonymous substitutions.
Algradi, A. M., Liu, Y., Yang, B. Y., Kuang, H. X. (2021). Review on the genus Brugmansia: Traditional usage, phytochemistry, pharmacology, and toxicity. J. Ethnopharmacol. 279, 113910. doi: 10.1016/j.jep.2021.113910
Amiryousefi, A., Hyvönen, J., Poczai, P. (2018). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics. 34 (17), 3030–3031. doi: 10.1093/bioinformatics/bty220
Angellotti, M. C., Bhuiyan, S. B., Chen, G., Wan, X. F. (2007). CodonO: codon usage bias analysis within and across genomes. Nucleic Acids Res. 35 (Web Server issue), W132–W136. doi: 10.1093/nar/gkm392
Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics. 33 (16), 2583–2585. doi: 10.1093/bioinformatics/btx198
Benítez, G., March-Salas, M., Villa-Kamel, A., Cháves-Jiménez, U., Hernández, J., Montes-Osuna, N., et al. (2018). The genus Datura L. (Solanaceae) in Mexico and Spain - Ethnobotanical perspective at the interface of medical and illicit uses. J. Ethnopharmacol. 219, 133–151. doi: 10.1016/j.jep.2018.03.007
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27 (2), 573–580. doi: 10.1093/nar/27.2.573
Berkov, S., Zayed, R., Doncheva, T. (2006). Alkaloid patterns in some varieties of Datura stramonium. Fitoterapia. 77 (3), 179–182. doi: 10.1016/j.fitote.2006.01.002
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D., Pirovano, W. (2011). Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 27 (4), 578–579. doi: 10.1093/bioinformatics/btq683
Brunkard, J. O., Runkel, A. M., Zambryski, P. C. (2015). Chloroplasts extend stromules independently and in response to internal redox signals. Proc. Natl. Acad. Sci. U S A. 112 (32), 10044–10049. doi: 10.1073/pnas.1511570112
Bye, R., Sosa, V. (2013). Molecular phylogeny of the jimsonweed genus datura (Solanaceae). Systematic Botany. 38 (3), 818–829. doi: 10.1600/036364413X670278
Charif, D., Thioulouse, J., Lobry, J. R., Perrière, G. (2005). Online synonymous codon usage analyses with the ade4 and seqinR packages. Bioinformatics. 21 (4), 545–547. doi: 10.1093/bioinformatics/bti037
Chellemi, D. O., Webster, C. G., Baker, C. A., Annamalai, M., Achor, D., Adkins, S. (2011). Widespread Occurrence and Low Genetic Diversity of Colombian datura virus in Brugmansia Suggest an Anthropogenic Role in Virus Selection and Spread. Plant Dis. 95 (6), 755–761. doi: 10.1094/pdis-09-10-0654
Chen, S., Yin, X., Han, J., Sun, W., Yao, H., Song, J., et al. (2023). DNA barcoding in herbal medicine: Retrospective and prospective. J. Pharm. Anal. 13 (5), 431–441. doi: 10.1016/j.jpha.2023.03.008
Chumová, Z., Záveská, E., Hloušková, P., Ponert, J., Schmidt, P. A., Čertner, M., et al. (2021). Repeat proliferation and partial endoreplication jointly shape the patterns of genome size evolution in orchids. Plant J. 107 (2), 511–524. doi: 10.1111/tpj.15306
Cinelli, M. A., Jones, A. D. (2021). Alkaloids of the genus datura: review of a rich resource for natural product discovery. Molecules 26 (9), 2629. doi: 10.3390/molecules26092629
Cui, Y., Chen, X., Nie, L., Sun, W., Hu, H., Lin, Y., et al. (2019). Comparison and phylogenetic analysis of chloroplast genomes of three medicinal and edible amomum species. Int. J. Mol. Sci. 20 (16), 4040. doi: 10.3390/ijms20164040
Daniell, H., Lin, C. S., Yu, M., Chang, W. J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17 (1), 134. doi: 10.1186/s13059-016-1004-2
De-la-Cruz, I. M., Núñez-Farfán, J. (2020). The complete chloroplast genomes of two Mexican plants of the annual herb Datura stramonium (Solanaceae). Mitochondrial DNA B Resour. 5 (3), 2823–2825. doi: 10.1080/23802359.2020.1789516
Doan, U. V., Wu, M. L., Phua, D. H., Mendez Rojas, B., Yang, C. C. (2019). Datura and Brugmansia plants related antimuscarinic toxicity: an analysis of poisoning cases reported to the Taiwan poison control center. Clin. Toxicol. (Phila). 57 (4), 246–253. doi: 10.1080/15563650.2018.1513527
Dupin, J., Smith, S. D. (2018). Phylogenetics of Datureae (Solanaceae), including description of the new genus Trompettia and re-circumscription of the tribe. Taxon. 67 (2), 359–375. doi: 10.12705/672.6
Fagerlund, R. D., Forsman, J. A., Biswas, S., Vass, I., Davies, F. K., Summerfield, T. C., et al. (2020). Stabilization of Photosystem II by the PsbT protein impacts photodamage, repair and biogenesis. Biochim. Biophys. Acta Bioenerg. 1861 (10), 148234. doi: 10.1016/j.bbabio.2020.148234
Fan, W. B., Wu, Y., Yang, J., Shahzad, K., Li, Z. H. (2018). Comparative chloroplast genomics of dipsacales species: insights into sequence variation, adaptive evolution, and phylogenetic relationships. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00689
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32 (Web Server issue), W273–W279. doi: 10.1093/nar/gkh458
Gao, C., Deng, Y., Wang, J. (2019). The complete chloroplast genomes of Echinacanthus species (acanthaceae): phylogenetic relationships, adaptive evolution, and screening of molecular markers. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.01989
Gong, L., Ding, X., Guan, W., Zhang, D., Zhang, J., Bai, J., et al. (2022). Comparative chloroplast genome analyses of Amomum: insights into evolutionary history and species identification. BMC Plant Biol. 22 (1), 520. doi: 10.1186/s12870-022-03898-x
Han, J. P., Li, M. N., Luo, K., Liu, M. Z., Chen, X. C., Chen, S. L. (2011). Identification of Daturae flos and its adulterants based on DNA barcoding technique. Yao Xue Xue Bao. 46 (11), 1408–1412. doi: 10.16438/j.0513-4870.2011.11.009
Huelsenbeck, J. P., Ronquist, F. (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 17 (8), 754–755. doi: 10.1093/bioinformatics/17.8.754
Katoh, K., Rozewicki, J., Yamada, K. D. (2019). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 20 (4), 1160–1166. doi: 10.1093/bib/bbx108
Kim, H. G., Jang, D., Jung, Y. S., Oh, H. J., Oh, S. M., Lee, Y. G., et al. (2020). Anti-inflammatory effect of flavonoids from brugmansia arborea L. Flowers. J. Microbiol. Biotechnol. 30 (2), 163–171. doi: 10.4014/jmb.1907.07058
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B., Stamatakis, A. (2019). RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics. 35 (21), 4453–4455. doi: 10.1093/bioinformatics/btz305
Kumar, S., Suleski, M., Craig, J. M., Kasprowicz, A. E., Sanderford, M., Li, M., et al. (2022). TimeTree 5: an expanded resource for species divergence times. Mol. Biol. Evol. 39 (8). doi: 10.1093/molbev/msac174
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29 (22), 4633–4642. doi: 10.1093/nar/29.22.4633
Lewis, S. E., Searle, S. M., Harris, N., Gibson, M., Lyer, V., Richter, J., et al. (2002). Apollo: a sequence annotation editor. Genome Biol. 3 (12), Research0082. doi: 10.1186/gb-2002-3-12-research0082
Li, D. M., Zhao, C. Y., Liu, X. F. (2019). Complete chloroplast genome sequences of kaempferia galanga and kaempferia elegans: molecular structures and comparative analysis. Molecules. 24 (3), 474. doi: 10.3390/molecules24030474
Li, X., Yang, Y., Henry, R. J., Rossetto, M., Wang, Y., Chen, S. (2015). Plant DNA barcoding: from gene to genome. Biol. Rev. Camb Philos. Soc 90 (1), 157–166. doi: 10.1111/brv.12104
Liao, B., Hu, H., Xiao, S., Zhou, G., Sun, W., Chu, Y., et al. (2022). Global Pharmacopoeia Genome Database is an integrated and mineable genomic database for traditional medicines derived from eight international pharmacopoeias. Sci. China Life Sci. 65 (4), 809–817. doi: 10.1007/s11427-021-1968-7
Liu, Q., Li, X., Li, M., Xu, W., Schwarzacher, T., Heslop-Harrison, J. S. (2020). Comparative chloroplast genome analyses of Avena: insights into evolutionary dynamics and phylogeny. BMC Plant Biol. 20 (1), 406. doi: 10.1186/s12870-020-02621-y
Lowe, T. M., Chan, P. P. (2016). tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44 (W1), W54–W57. doi: 10.1093/nar/gkw413
Luna-Cavazos, M., Bye, R., Jiao, M. (2009). The origin of Datura metel (Solanaceae): genetic and phylogenetic evidence. Genet. Resour. Crop Evolution. 56 (2), 263–275. doi: 10.1007/s10722-008-9363-5
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J., et al. (2015). Erratum: SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 4, 30. doi: 10.1186/s13742-015-0069-2
Mutebi, R. R., Ario, A. R., Nabatanzi, M., Kyamwine, I. B., Wibabara, Y., Muwereza, P., et al. (2022). Large outbreak of Jimsonweed (Datura stramonium) poisoning due to consumption of contaminated humanitarian relief food: Uganda, March-April 2019. BMC Public Health 22 (1), 623. doi: 10.1186/s12889-022-12854-1
Nadalin, F., Vezzi, F., Policriti, A. (2012). GapFiller: a de novo assembly approach to fill the gap within paired reads. BMC Bioinf. 13 Suppl 14 (Suppl 14), S8. doi: 10.1186/1471-2105-13-s14-s8
Ng, J., Smith, S. D. (2016). Widespread flower color convergence in Solanaceae via alternate biochemical pathways. New Phytol. 209 (1), 407–417. doi: 10.1111/nph.13576
Ogawa, T., Ishii, C., Kagawa, D., Muramoto, K., Kamiya, H. (1999). Accelerated evolution in the protein-coding region of galectin cDNAs, congerin I and congerin II, from skin mucus of conger eel (Conger myriaster). Biosci. Biotechnol. Biochem. 63 (7), 1203–1208. doi: 10.1271/bbb.63.1203
Olmstead, R. G., Bohs, L., Migid, H. A., Santiago-Valentin, E., Garcia, V. F., Collier, S. M., et al (2008). A molecular phylogeny of the Solanaceae. TAXON 57 (4), 1159–1181. doi: 10.1002/tax.574010
Parenteau, J., Abou Elela, S. (2019). Introns: good day junk is bad day treasure. Trends Genet. 35 (12), 923–934. doi: 10.1016/j.tig.2019.09.010
Park, I., Yang, S., Choi, G., Kim, W. J., Moon, B. C. (2017). The complete chloroplast genome sequences of Aconitum pseudolaeve and Aconitum longecassidatum, and development of molecular markers for distinguishing species in the Aconitum subgenus Lycoctonum. Molecules. 22 (11), 2012. doi: 10.3390/molecules22112012
Petricevich, V. L., Salinas-Sánchez, D. O., Avilés-Montes, D., Sotelo-Leyva, C., Abarca-Vargas, R. (2020). Chemical compounds, pharmacological and toxicological activity of brugmansia suaveolens: A review. Plants (Basel) 9 (9), 1161. doi: 10.3390/plants9091161
Pigatto, A. G., Blanco, C. C., Mentz, L. A., Soares, G. L. (2015). Tropane alkaloids and calystegines as chemotaxonomic markers in the Solanaceae. Acad. Bras. Cienc. 87 (4), 2139–2149. doi: 10.1590/0001-3765201520140231
Powell, W., Morgante, M., McDevitt, R., Vendramin, G. G., Rafalski, J. A. (1995). Polymorphic simple sequence repeat regions in chloroplast genomes: applications to the population genetics of pines. Proc. Natl. Acad. Sci. U.S.A. 92 (17), 7759–7763. doi: 10.1073/pnas.92.17.7759
Qian, J., Song, J., Gao, H., Zhu, Y., Xu, J., Pang, X., et al. (2013). The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PloS One 8 (2), e57607. doi: 10.1371/journal.pone.0057607
Romero, H., Zavala, A., Musto, H. (2000). Codon usage in Chlamydia trachomatis is the result of strand-specific mutational biases and a complex pattern of selective forces. Nucleic Acids Res. 28 (10), 2084–2090. doi: 10.1093/nar/28.10.2084
Rozas, J., Ferrer-Mata, A., Sánchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 34 (12), 3299–3302. doi: 10.1093/molbev/msx248
Särkinen, T., Bohs, L., Olmstead, R. G., Knapp, S. (2013). A phylogenetic framework for evolutionary study of the nightshades (Solanaceae): a dated 1000-tip tree. BMC Evol. Biol. 13, 214. doi: 10.1186/1471-2148-13-214
Schley, R. J., Pellicer, J., Ge, X. J., Barrett, C., Bellot, S., Guignard, M. S., et al. (2022). The ecology of palm genomes: repeat-associated genome size expansion is constrained by aridity. New Phytol. 236 (2), 433–446. doi: 10.1111/nph.18323
Sharp, P. M., Li, W. H. (1986). An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 24 (1-2), 28–38. doi: 10.1007/bf02099948
Shi, L., Chen, H., Jiang, M., Wang, L., Wu, X., Huang, L., et al. (2019). CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47 (W1), W65–w73. doi: 10.1093/nar/gkz345
Smith, S. D., Baum, D. A. (2006). Phylogenetics of the florally diverse Andean clade Iochrominae (Solanaceae). Am. J. Bot. 93 (8), 1140–1153. doi: 10.3732/ajb.93.8.1140
Song, S. L., Lim, P. E., Phang, S. M., Lee, W. W., Hong, D. D., Prathep, A. (2014). Development of chloroplast simple sequence repeats (cpSSRs) for the intraspecific study of Gracilaria tenuistipitata (Gracilariales, Rhodophyta) from different populations. BMC Res. Notes. 7, 77. doi: 10.1186/1756-0500-7-77
Sugita, M., Sugiura, M. (1996). Regulation of gene expression in chloroplasts of higher plants. Plant Mol. Biol. 32 (1-2), 315–326. doi: 10.1007/bf00039388
Tamura, K., Battistuzzi, F. U., Billing-Ross, P., Murillo, O., Filipski, A., Kumar, S. (2012). Estimating divergence times in large molecular phylogenies. Proc. Natl. Acad. Sci. U S A. 109 (47), 19333–19338. doi: 10.1073/pnas.1213199109
Tamura, K., Stecher, G., Kumar, S. (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38 (7), 3022–3027. doi: 10.1093/molbev/msab120
Wu, Y. N., Xu, L., Chen, L., Wang, B., Zhao, R. (2015). DNA molecular identification of datura medicinal plants using ITS2 barcode sequence. Zhong Yao Cai. 38 (9), 1852–1857. doi: 10.13863/j.issn1001-4454.2015.09.014
Yang, Y., Dang, Y., Li, Q., Lu, J., Li, X., Wang, Y. (2014). Complete chloroplast genome sequence of poisonous and medicinal plant Datura stramonium: organizations and implications for genetic engineering. PloS One 9 (11), e110656. doi: 10.1371/journal.pone.0110656
Zhang, Z., Li, J., Zhao, X. Q., Wang, J., Wong, G. K., Yu, J. (2006). KaKs_Calculator: calculating Ka and Ks through model selection and model averaging. Genomics Proteomics Bioinf. 4 (4), 259–263. doi: 10.1016/s1672-0229(07)60007-2
Zhang, Z., Xiao, J., Wu, J., Zhang, H., Liu, G., Wang, X., et al. (2012). ParaAT: a parallel tool for constructing multiple protein-coding DNA alignments. Biochem. Biophys. Res. Commun. 419 (4), 779–781. doi: 10.1016/j.bbrc.2012.02.101
Zheng, S., Poczai, P., Hyvönen, J., Tang, J., Amiryousefi, A. (2020). Chloroplot: an online program for the versatile plotting of organelle genomes. Front. Genet. 11. doi: 10.3389/fgene.2020.576124
Zhengping, N., Jiali, X., Yuxi, L. (2003). The study of the anti-epileptic function of datarametel L. Chin. J. Integr. Med. Cardio-/Cerebrovascular Dis (04), 193-195.
Zhou, J., Chen, X., Cui, Y., Sun, W., Li, Y., Wang, Y., et al. (2017). Molecular structure and phylogenetic analyses of complete chloroplast genomes of two aristolochia medicinal species. Int. J. Mol. Sci. 18 (9). doi: 10.3390/ijms18091839
Keywords: Datureae, chloroplast genome, comparative analysis, species identification markers, evolutionary relationship
Citation: Su H, Ding X, Liao B, Zhang D, Huang J, Bai J, Xu S, Zhang J, Xu W, Qiu X, Gong L and Huang Z (2023) Comparative chloroplast genomes provided insights into the evolution and species identification on the Datureae plants. Front. Plant Sci. 14:1270052. doi: 10.3389/fpls.2023.1270052
Received: 31 July 2023; Accepted: 05 October 2023;
Published: 24 October 2023.
Edited by:
Xiaohua Jin, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Zhi Chao, Southern Medical University, ChinaCopyright © 2023 Su, Ding, Liao, Zhang, Huang, Bai, Xu, Zhang, Xu, Qiu, Gong and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaohui Qiu, cWl1eGlhb2h1aUBnenVjbS5lZHUuY24=; Lu Gong, Z29uZ2x1MDkwNEBnenVjbS5lZHUuY24=; Zhihai Huang, emhodWFuZzczMDhAMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.