
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 25 October 2023
Sec. Functional and Applied Plant Genomics
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1269995
 Muhammad Zeshan Haider1
Muhammad Zeshan Haider1 Adnan Sami1
Adnan Sami1 Muhammad Shafiq2*
Muhammad Shafiq2* Waheed Anwar3
Waheed Anwar3 Sajid Ali4
Sajid Ali4 Qurban Ali1*
Qurban Ali1* Sher Muhammad5Irfan Manzoor5
Sher Muhammad5Irfan Manzoor5 Muhammad Adnan Shahid6
Muhammad Adnan Shahid6 Daoud Ali7
Daoud Ali7 Saud Alarifi7
Saud Alarifi7Rice constitutes a foundational cereal and plays a vital role in the culinary sector. However, the detriments of abiotic stress on rice quality and productivity are noteworthy. Carotenoid cleavage oxygenases (CCO) hold vital importance as they enable the particular breakdown of carotenoids and significantly contribute towards the growth and response to abiotic stress in rice. Due to the insufficient information regarding rice CCOs and their potential role in abiotic stress, their utilization in stress-resistant genetic breeding remains limited. The current research identified 16 CCO genes within the Oryza sativa japonica group. These OsCCO genes can be bifurcated into three categories based on their conserved sequences: NCEDs (9-Cis-epoxycarotenoid dioxygenases), CCDs (Carotenoid cleavage dioxygenases) and CCD-like (Carotenoid cleavage dioxygenases-like). Conserved motifs were found in the OsCCO gene sequence via MEME analysis and multiple sequence alignment. Stress-related cis-elements were detected in the promoter regions of OsCCOs genes, indicating their involvement in stress response. Additionally, the promoters of these genes had various components related to plant light, development, and hormone responsiveness, suggesting they may be responsive to plant hormones and involved in developmental processes. MicroRNAs play a pivotal role in the regulation of these 16 genes, underscoring their significance in rice gene regulation. Transcriptome data analysis suggests a tissue-specific expression pattern for rice CCOs. Only OsNCED6 and OsNCED10 significantly up-regulated during salt stress, as per RNA seq analyses. CCD7 and CCD8 levels were also higher in the CCD group during the inflorescence growth stage. This provides insight into the function of rice CCOs in abiotic stress response and identifies possible genes that could be beneficial for stress-resistant breeding.
Carotenoids, these fascinating isoprenoid compounds, have captivated scientific curiosity ever since their serendipitous discovery in the 19th century (Stafsnes et al., 2010). Their enigmatic diversity unfolds through the analysis and identification of more than 700 distinct types over the years, revealing a rich tapestry of potential benefits waiting to be unraveled (Gitelson and Merzlyak, 1998). These compounds have various biological functions across different organisms, such as bacteria and plants (Misawa, 2011). Carotenoids serve as important secondary pigments in photosynthetic organisms, protecting them from photooxidation and helping to capture light more efficiently (Hashimoto et al., 2016). Additionally, certain apocarotenoids, produced through the cleavage of carotenoids, aid in regulating plant growth and development, as well as their response to biotic and abiotic stresses (Felemban et al., 2019). The carotenoid cleavage oxygenase’s (CCOs) are a series of enzymes involved in the breakdown of carotenoids which are naturally occurring pigments that contribute color to many fruits, vegetables, and flowers (Liang et al., 2018). CCOs comprise two types of enzymes carotenoid cleavage dioxygenases (CCDs) and 9-cis-epoxycarotenoid dioxygenases (NCEDs). These enzymes are essential for turning carotenoids into apocarotenoids, which have degraded (Yue et al., 2022). CCOs use oxygen to split the carotenoid molecule and produce apocarotenoids, which have a variety of physiological roles such as serving as plant hormones, enticing pollinators, and giving defense against environmental stress (Shi et al., 2023).
9-Cis-epoxycarotenoid dioxygenase (NCED) is a major enzyme in the ABA production pathway found in numerous plant species (Sun et al., 2012). NCED is evolutionarily conserved throughout plant lineages, with little divergence within its subfamily and excellent exon conservation (Liu et al., 2023). This demonstrates the significance of NCED in preserving the structural integrity of the protein for its function. NCED genes change somewhat slowly, which may contribute to the functional divergence of NCED subfamilies across tissues (Wan et al., 2021). Various expression patterns and activity levels of NCED subfamilies in various plant tissues may contribute to controlling ABA biosynthesis and plant responses to environmental signals (Pu et al., 2020).
The identification of the NCED gene marked a significant milestone in advancing our comprehension of ABA production and its role in plant growth and development. The NCED gene was initially cloned in the maize ABA-deletion mutant Vp14, leading to a better understanding of NCED’s involvement in ABA synthesis (Fei et al., 2023). Further research in Arabidopsis revealed nine CCOs genes, five of which (AtNCED2, AtNCED3, AtNCED5, AtNCED6, and AtNCED9) are involved in ABA synthesis (Hu et al., 2021). Manipulation of AtNCED3 increased drought tolerance by increasing endogenous ABA accumulation (Seki et al., 2007). Identifying NCED genes in additional plant species, including Tamatim, cowpea, and rice, increased our understanding of NCED evolution and function across plant taxa (Wang and Guo, 2023). These findings show that NCED is a conserved and essential component of the ABA biosynthesis pathway that governs plant responses to environmental stress and developmental stimuli (Tan et al., 2003). The NCED gene family has been identified and investigated in many plant species, such as Arabidopsis (Tan et al., 2003), citrus (Xian et al., 2014), cotton (Li et al., 2021), cucumber (Zhang et al., 2022), wheat, and tobacco (Zhang et al., 2014). However, the function of NCED genes in rice has not been clearly defined. Carotenoid cleavage dioxygenases (CCDs) are an enzyme that uses molecular oxygen to cleave carotenoids, a group of pigments commonly found in plants, animals, and microorganisms (Daruwalla and Kiser, 2020). CCDs are responsible for various biological functions such as vision, stress response, and reproduction, producing apocarotenoids with multiple biological activities (Wei et al., 2022). Arabidopsis now has nine CCO gene members, five NCED genes, and four CCD genes. The CCD gene family is divided into five subfamilies based on differences in cleavage sites and substrates: CCD1, CCD4, CCD7, CCD8, and NCED (Cheng et al., 2023). For the first time, CCD-like (CCDL), a new member of the CCD family, was found in tomatoes (Cheng et al., 2022). The retinal pigment epithelium membrane protein (RPE65) domain is found in all CCD genes (Kiser, 2022), which is typical of enzymes involved in carotenoid cleavage (Kim et al., 2016). The CCD gene family has been found and studied in several plant species, including Arabidopsis (Chernys and Zeevaart, 2000). CCD gene family is described in grass species maize (Zea mays), rice (O. sativa), and sorghum (Sorghum bicolor) (Zhou et al., 2020). CCD research has also been conducted in wheat (Takezawa, 2000), watermelon (Cheng et al., 2022), pepper (Yao et al., 2022), tobacco (Zhou et al., 2019), rapeseed (Zhou et al., 2020), and maize (Sun et al., 2022). However, the function of CCD genes in rice has not been precisely defined. Extensive research on CCO genes has been conducted on Arabidopsis thaliana. Still, due to the absence of CCDL proteins, it is not an ideal candidate for investigating CCD enzymes and their functions. Fortunately, CCDL genes have been discovered in Citrullus lanatus and Cucumis melo, providing opportunities for further exploration (Xu et al., 2023). Carotenoids provide vibrant colors to fruits and vegetables, and their production and degradation are facilitated by CCD enzyme family (Kiokias et al., 2016). C. lanatus and C. melo have CCDL genes, making them good candidates for future research into CCD enzymes and their functions.
Rice (O. sativa) is a cereal crop grown in many parts of the world (Sami et al., 2023). It is a staple food for over half of the world’s population and is Asia’s most important food crop (Fukagawa and Ziska, 2019). The International Rice Genome Sequencing Project (IRGSP), a cooperation of research institutes from ten nations, sequenced the genome of rice (O. sativa) for the first time in 2002 (Project and Sasaki, 2005). However, research on rice CCO genes is sparse (He et al., 2022). The functional role of the CCO family in rice development remains unclear. To address this issue, we performed an extensive bioinformatics analysis of the complete rice genome in order to identify all potential members of the OsCCO gene family. We discovered several genes involved in the change in ABA content during rice seedling growth and development under environmental stress. The RNA-seq data analysis has identified promising candidates in OsNCED6 and OsNCED10 genes, offering potential avenues for the development of salt stress-resistant rice varieties to enhance crop yield in saline conditions. Meanwhile, OsCCD7 and OsCCD8 genes display higher activity in inflorescences, implying their potential involvement in grain development, although further investigations, such as gene cloning and functional analysis, are essential to validate their roles in diverse physiological and biological processes. This in-silico analysis expands our comprehensive understanding of the rice OsCCO gene family across the genome. The results of this study shed new light on the functional diversity and evolutionary aspects of the CCO gene family in plants. Our findings will serve as a valuable resource for further investigations, enabling the functional analysis and cloning of these genes through the comprehensive genome-wide identification and characterization conducted in this study.
The amino acid sequence data for O. sativa was obtained from the Phytozome v13 database (https://phytozome-next.jgi.doe.gov), and the particular domain PF03055 (RPE65) linked to the gene for carotenoid cleavage dioxygenase 1 (CCD1) was retrieved. The BLAST-P (Basic Local Alignment Search Tool for Protein Sequences) program was used to find the CCD1 genes in the O. sativa genome database at Phytozome v13 using the PF03055 domain as a query. Using the default parameters, the obtained amino acid sequences were then cross-checked against NCBI’s Conserved Domain Database (CDD) (https://www.ncbi.nlm.nih.gov/genome/) (Zhang et al., 2016).
Information on 16 protein was collected from Protparam (https://web.expasy.org/protparam/) and Phytozome. The Phytozome database provided crucial details such as chromosome number, location, and direction, mRNA length (CDS), and peptide length, while the Protparam database provided additional information including the theoretical pI (isoelectric point), molecular weight, GRAVY (Grand Average of Hydropathy) and stability index of the proteins. To identify the subcellular localization of the rice proteins, the WoLF PSORT database (https://wolfpsort.hgc.jp/) was utilized. This database employs protein sequences as input to generate a list of potential locations and their associated prediction scores. This investigation aimed to determine the most probable location of the proteins within the cell (Kavas et al., 2023). The upstream 1000bp promotor regions were extracted from phytozomeV3 (https://phytozome-next.jgi.doe.gov) for cis-regulatory elements extraction using a web tool PlantCARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/). Putative cis-elements for the promotor region were retrieved from 5 to 20 bp. The results were visualized in a heatmap with the help of TBtools (Bülow and Hehl, 2016).
The default values for the optimum matching length parameter were used in the Multiple Expectation Maximization for Motif Elicitation (MEME) program, which can be found online at (http://meme.sdsc.edu/meme/website/intro.html). Next, using the CDD search function, which discovers and curates domains identified by NCBI, the amino acid sequences were added to the Conserved Domain Database (CDD) (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). In analyzing the distribution of exons and introns, the OsCCO gene family’s genomic and CDS sequences were subjected to the web tool Gene Structure Display Server (GSDS) at (http://gsds.cbi.pku.edu.cn/) (Barre et al., 2009).
CCO proteins’ amino acid (AA) sequences including O. sativa, A. thaliana, S. lycopersicum, Citrullus lanatus, and Cucumis melo were aligned to generate a phylogenetic tree. The tree was constructed using the neighbor-joining (NJ) method in the MEGA 11 software with 1000 replications for bootstrapping. The tree was further visualized and modified using the Interactive Tree of Life (iTOL) program (https://itol.embl.de/), which provided a user-friendly interface for exploring and annotating the tree (Rehman et al., 2022).
The PmiREN website (https://www.pmiren.com/) was used to identify the target site of rice’s 16 CCO gene family. The PsRNA (https://www.zhaolab.org/psRNATarget/) online server tool was used to compare the CDS sequences of the genes with the mature miRNA sequences, and the default settings were utilized. The Cytoscape program was used to visualize the connections between target genes and the predicted miRNA (Mazhar et al., 2023).
The evolution of CCO genes in rice was examined using duplication and synteny analysis. The genes’ divergence period was estimated using Ka/Ks values. The protein sequences were aligned using the MUSCLE program, and for the calculation of Ka/Ks substitution rates, Tbtools 1.108 with default parameters was employed. This study calculated the molecular evolution rate for each gene pair by determining the Ka/Ks ratios of paralogous genes. To estimate the time of divergence between the paralogous, we used the Ks value in the T = Ks/2 equation, where λ equals 6.5 × 10−9. Gene duplication events were identified using MCScanX v1.0 with default settings. This analysis allowed us to determine the frequency of gene duplications and their potential impact on the molecular evolution of the paralogous genes. Tbtools were used to create syntenic and dual syntenic maps to ascertain the synteny link between Arabidopsis and tomato’s orthologous genes and rice’s paralogous genes (Mazhar et al., 2023).
The gene structure was analyzed using the gene structure display server (GSDS v2.0) (http://gsds.cbi.pku.edu.cn/) to identify intron-exon arrangements. PlantCare database (http://bioinformatics.psb.ugent.be/webtools/plantcare/html) was used to identify the cis-regulatory elements (Abdulla et al., 2023).
Using GO annotations, a GO enrichment analysis was carried out to evaluate the roles of CCO genes in rice. Information on the molecular activities of the CCO genes and various biological processes was accessed from the Uniprot online database (https://www.uniprot.org/), which was consulted for this purpose (Langenbacher et al., 2020). The GO word enrichment analysis was then carried out using the CCO gene sequences entered into the ShinyGo v0.741 web tool, a resource accessible on (http://bioinformatics.sdstate.edu/go/) (Mazhar et al., 2023).
The activities of the CCO genes were further validated using GO annotations by gene ontology (GO) term enrichment study. The online program called ShinyGo v0.741.(https://string-db.org/cgi/input?sessionId=b16GBNb2GGmA) was used for better understand of CCO genes function in cucumber. For visualization of biological, molecular and cellular function, an online database shiny Go was used.
For this research, two types of rice plants (CSR28 and IR28) were selected to investigate their response to salt. Their data was retrieved from the NCBI GEO (https://www.ncbi.nlm.nih.gov/geo/) (GSE133480) database to explore the salt stress expression profile of the CCO gene family. The expression profiles of rice genotype were sub-categories into G1, G2, G3, G4, G5, G6, G7, and G8. Salt was applied to the roots and shoots of even genotypes for 6 hours. The second application of salt to the roots and shoots of genotypes with odd numbers for 54 hours. The Statistix 8.1 pairwise comparison tool was used to improve comprehension of up/down-regulated gene expression by highlighting significant differences between circumstances, assisting in the identification of possible targets for further research (Mazhar et al., 2023).
Wild-type (WT) and Osgl1/2 mutant genotypes were analyzed for their inflorescence development stage in a study. Data from the NCBI GEO database (https://www.ncbi.nlm.nih.gov/geo/) (GSE227706) were used to evaluate the gene expression profiles of the CCO gene family. Statistix 8.1 pairwise comparison tool was used to determine significant differences in gene expression between the two genotypes, paving the way for further investigation into potential targets (Mazhar et al., 2023).
The gene expression profiles of the CCO gene family were assessed from the Expression Atlas database (https://www.ebi.ac.uk/gxa/experiments?experimentType=differential/). To gain insights into the expression patterns of these genes, RNA sequencing data were analyzed with the TBtools, which provides visual representations of gene expression levels.
A total of 16 CCO genes have been identified in rice, with the encoding protein lengths ranging from 173 to 801 and molecular weights varying from 19,432.02 to 88,580.62 kDa. The smallest protein was OsNCED4, and the longest was OsCCD9 (Table 1). Proteins were anticipated to be stable if their value was below 40 and unstable if it is above 40, according to the instability index. The genes OsCCDL3, OsNCED4, OsCCD10, OsCCD13, OsCCD14, and OsNCED16 were predicted to be stable, while the remaining ten proteins, including OsCCD1, OsCCDL2, OsCCD5, OsNCED6, OsCCD7, OsCCD8, OsCCD9, OsCCD11, OsCCDL12, and OsCCD15, ware predicted to be unstable. The grand average of hydropathy (GRAVY) value suggests that most genes are predominantly hydrophobic, with OsCCD1 and OsNCED4 showing a slight hydrophobic nature. The pI values for these proteins vary between 4.66 and 9.04, suggesting that they will carry no overall electrical charge within this pH range. Of the identified genes, 56.25% were in the forward direction, while the remaining 43.75% were in the reverse direction (Table 1).
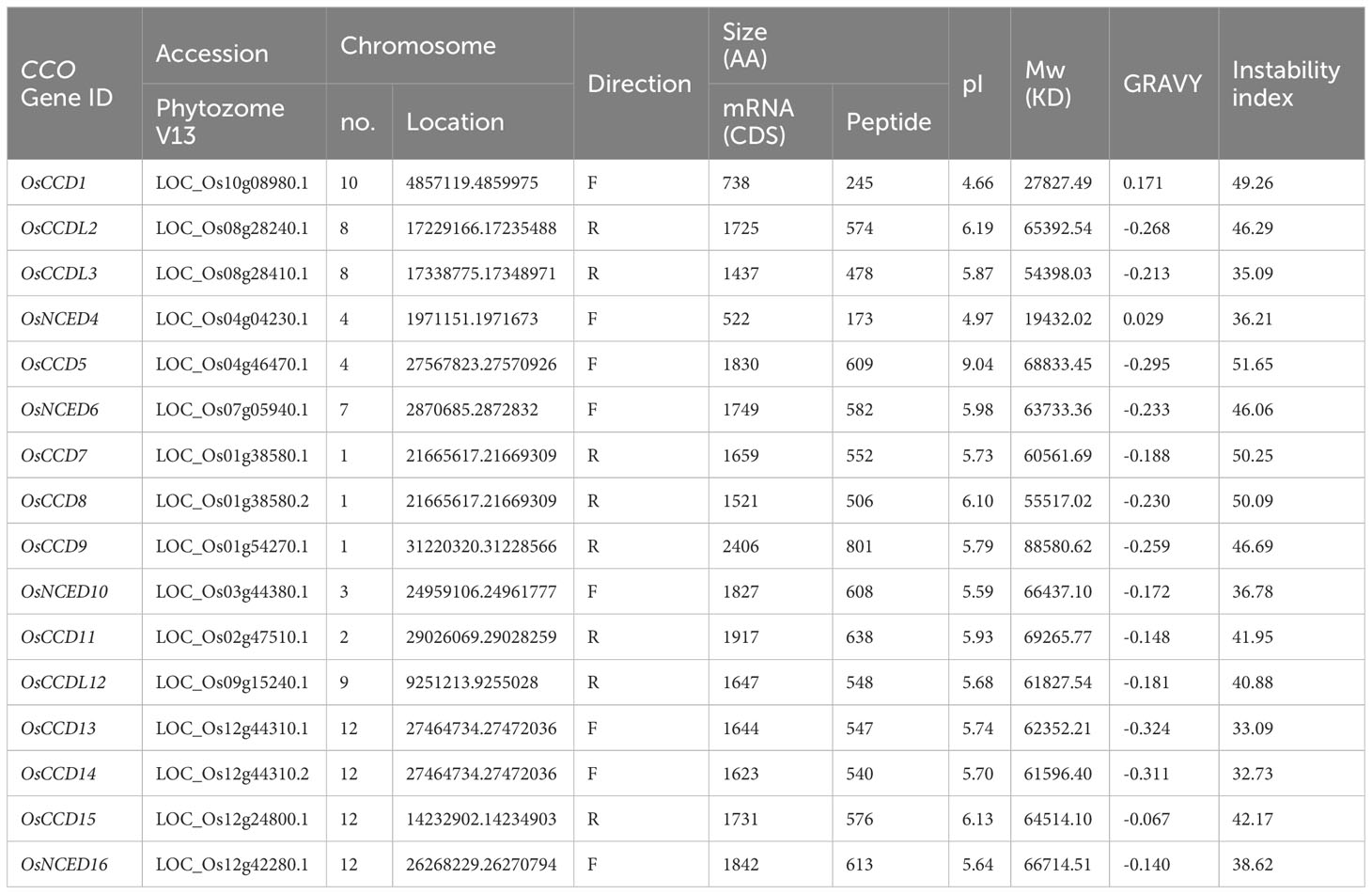
Table 1 Physiochemical properties of CCO gene family.
The sub-cellular localization of these genes shows that OsCCD1 was localized in both the cytoplasm and nucleus, while OsCCDL2 was located in the chloroplast, mitochondria, and extracellular space.OsCCDL3 and OsNCED4 are located in the cytoplasm, while OsNCED5, OsNCED6, OsNCED7, OsNCED8, OsNCED9, OsCCD11, OsCCD13, OsCCD14, and OsCCD15 were localized in the chloroplast. OsNCED16 is also located in the chloroplast, while OsNCED10 and OsCCDL12 were localized in both the chloroplast and mitochondria (Figure 1; Table 1).
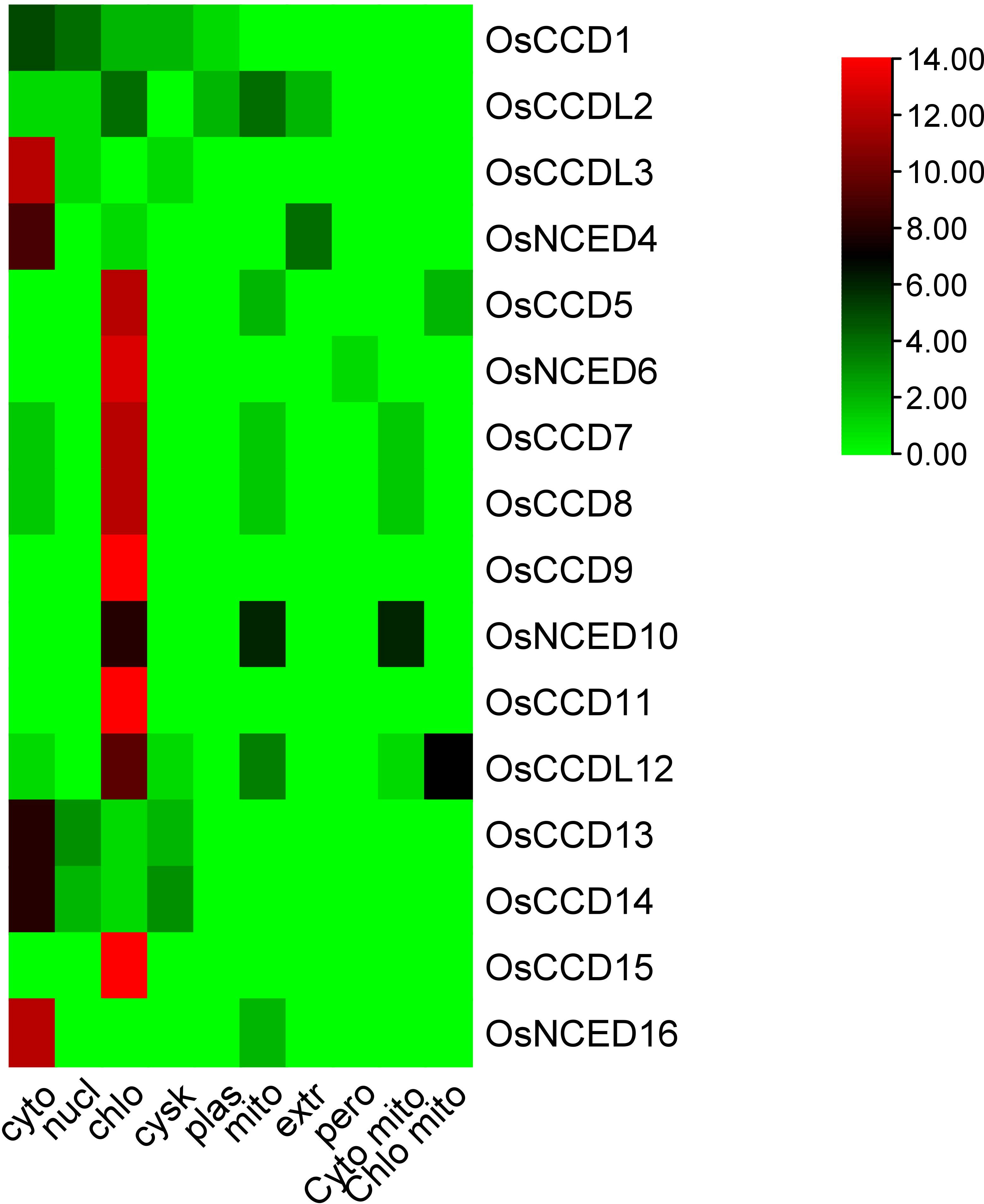
Figure 1 The subcellular localization prediction analysis of OsCCO proteins indicated their predominant localization in chloroplasts and cytoplasm. The analysis highlighted that the maximum number of proteins showed a red color, representing their localization in these cellular compartments.
The analysis of OsCCO gene promoter regions identified 74 cis-elements, excluding common elements such as the TATA-box and CAAT-box, as well as unidentified functional elements. OsCCO genes in rice were subjected to cis-element analysis, and a variety of responsive elements were discovered, including 15 light-responsive elements, 28 stress-responsive elements, 19 development and metabolism-responsive elements, and 15 hormone-responsive elements. The light sensitivity of about 20.27% of the elements suggests that OsCCO genes may be involved in the response to light stress. The discovery that 37.83% of the cis-elements are linked to the stress response also suggests a likely role for OsCCO genes in stress-related activities. The study also identified 25.67% of the cis-elements associated with plant growth and metabolism, indicating that OsCCOgenes may be involved in the growth and development of rice plants. The hormone-responsive cis-elements discovered (20.27%) include auxin, MeJA, and ABA. These elements may be interesting targets for study in order to understand how hormones behave under stress. All 16 CCO genes were found to contain most of the hormone-related regions. By employing molecular breeding techniques, the analysis provides valuable insights into the potential roles of CCO genes in rice, facilitating the creation of improved rice varieties (Figure 2; Supplementary Table 1).
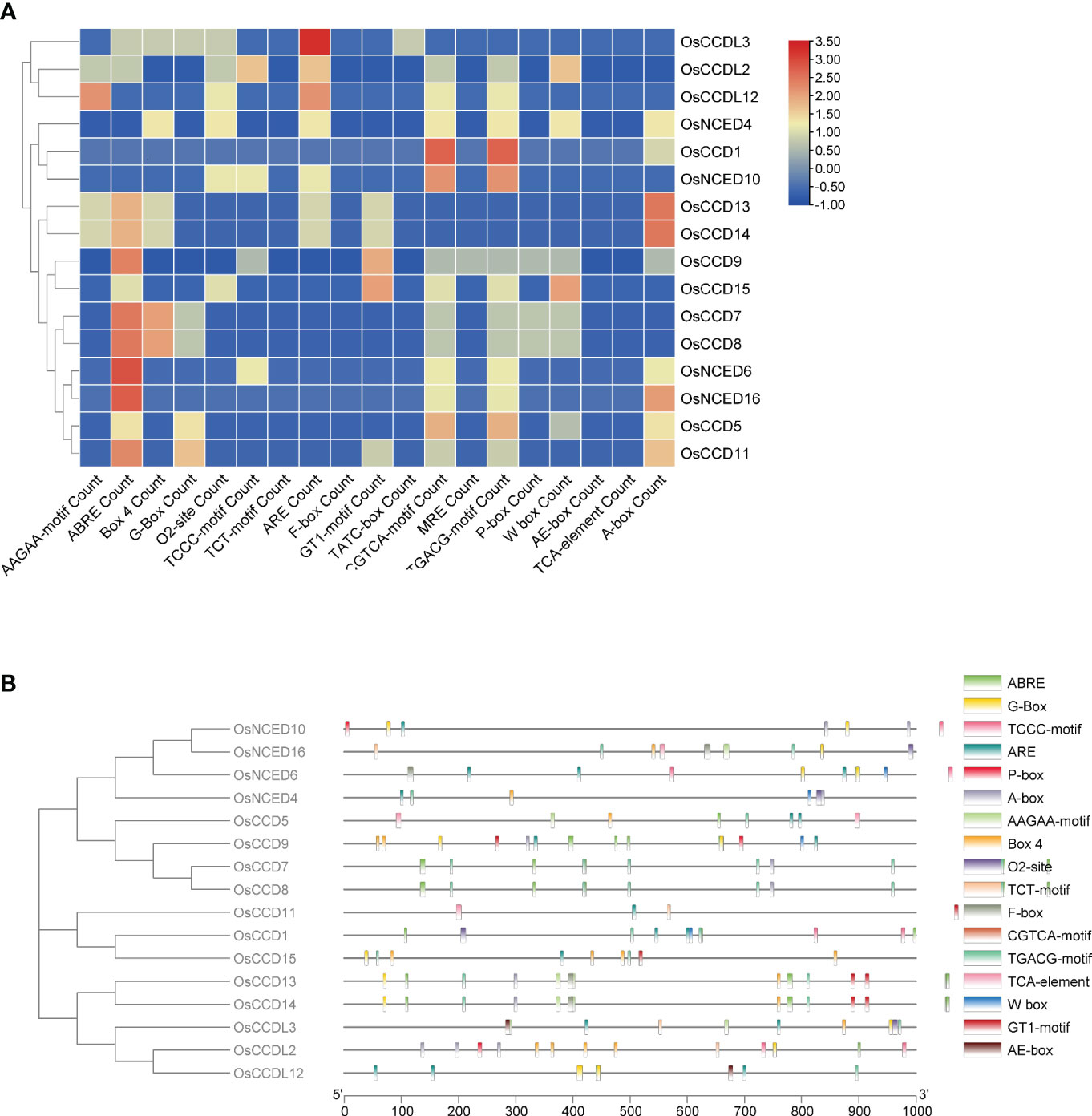
Figure 2 The analysis of cis-regulatory elements in the promoters of OsCCO genes in Rice revealed their association with various plant developmental processes. (A) The analysis provided statistics on the presence of cis-regulatory elements in each OsCCO gene. (B) The distribution of these cis-regulatory elements across the promoters was also investigated, shedding light on their spatial arrangement.
The investigation carried out by the MEME tool on rice OsCCO looked into the discovery and distribution of 20 distinct motifs. Among all the OsCCO proteins, RPE65 superfamily, RPE65, PLN02258, PLN02969 domains were present (Supplementary Figure 1; Supplementary Table 2). During conserved domain investigation the OsCCD1, OsNCED4, OsCCD11, and OsCCD15 protein were categorized under the RPE65 superfamily. Within the same RPE65 superfamily the RPE65 were consisted of OsCCDL2, OsCCDL3, OsCCD7, OsCCD8 and OsCCDL12. Notably, OsCCD9 was associated with both the RPE65 and RT_LTR subfamilies’ While OsNCED6, OsNCED10 and OsNCED16 were grouped under the PLN02258 classification. In a separate classification, OsCCD5 was linked to the PLN02969 subfamily. OsCCD13 and OsCCD14 were both members of the PLN02491 subfamily. So it was cleared that domain was conserved throughout all proteins.While analyzing the OsCCO gene family, it was discovered that genes within the same group shared similar motifs. This suggests that conserved motifs might play a role in specific activities within a particular group or subgroup. During conserved motif investigation, the motif (2, 8, 13, 15, and 16) were conserved in the RPE65 superfamily. While RPE65 had a conserved motif of motif (1, 3, 5, 6, 7, 10, 12 and 13). While motif (1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 15, 16, 18, 19 and 20) were conserved in the PLN02258. The motif (1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 15, 16, 18 and 19) were conserved in the PLN02491 subfamily (Supplementary Figure 2). Notably, RPE65 and RT_LTR subfamilies’ were consist of motif (1, 2, 3, 5, 6, 8, 9, 10, 12, 13, 14, and 17), while PLN02969 subfamily was consist of motif (1, 2, 3, 5, 7, 8, and 10) (Supplementary Figure 2; Supplementary Table 3). The presence of similar motifs across different members of the OsCCO gene family hints that gene expansion might have played a role in the evolution of these genes. Unquestionably, OsNCED4, OsCCD5, and OsCCD1 had fewer motifs like 6, 7, and 7, respectively. This study discovered that none of the 16 genes analyzed had any conserved motifs. However, 15 genes have a conserved motif, indicating a possible functional significance (Figure 3).
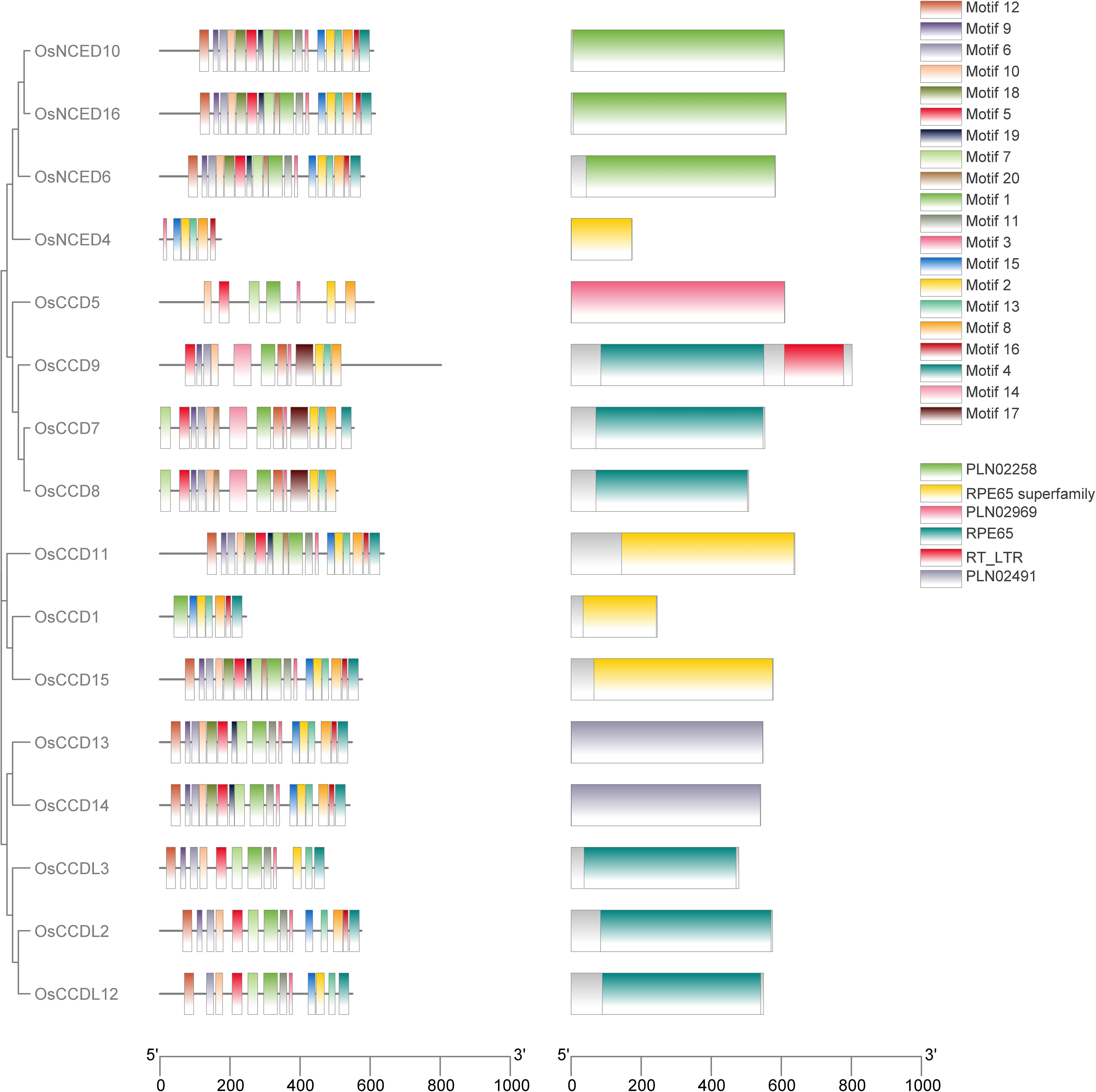
Figure 3 A bar graph with color-coded bars was generated to depict the motif distribution analysis of rice OsCCO proteins using MEME version 5.5.2. The analysis identified a total of 20 distinct motifs. To better understand the relationship between the OsCCO proteins and motif distribution, the graph was connected to a phylogenetic tree, providing additional insights into the evolutionary patterns and functional relationships among the OsCCO proteins.
According to gene structure predictions, 6 OsCCO genes (37.5%) were found to have only one exon and no introns. This indicates that these genes have a simple structure with no gaps in their coding sequence. Three genes (18.75%) had 13 exons and 12 introns, while 2 genes (12.5%) had 12 exons and 11 introns. There are 19 cis-elements found in the CCO gene (Figure 4).
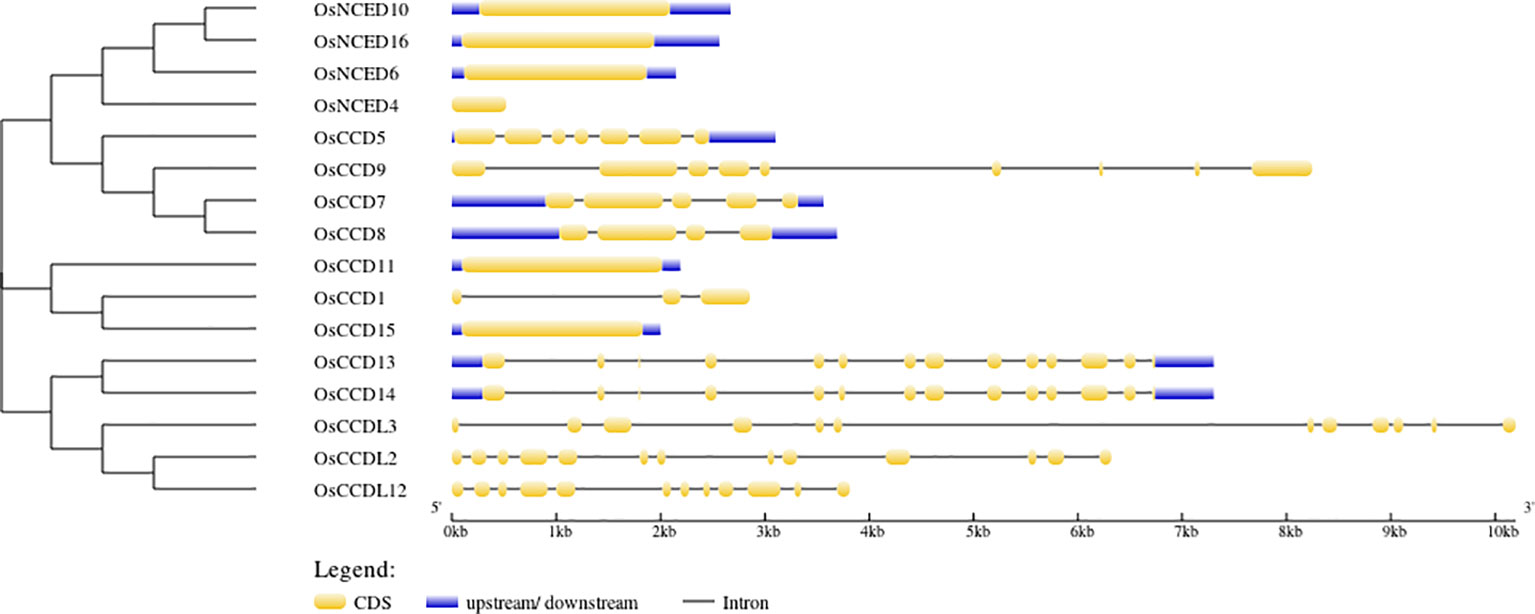
Figure 4 The intron-exon structures were illustrated using yellow and black shapes, representing exons within the genes.
The phylogenetic analysis revealed that the CCO genes from five plant species were classified into three subgroups, denoted as subfamilies I-III. Specifically, the study analyzed 37 CCO genes, including 16 from O. sativa, 11 from S. lycopersicum, 8 from Arabidopsis thaliana, and 1 from and Cucumis melo. To better understand the properties of subfamily III, represented by CCDL, sequences from C. lanatus and C. melo were added to the analysis. CCDL is noticeably absent in Arabidopsis thaliana, so genomes from several additional plant species were inserted. In the resulting phylogenetic tree, O. sativa CCO genes were marked by blue dots, and subfamilies were differentiated by color coding, with subfamily I represented by green, subfamily II by blue, and subfamily III by pink (Figure 5).
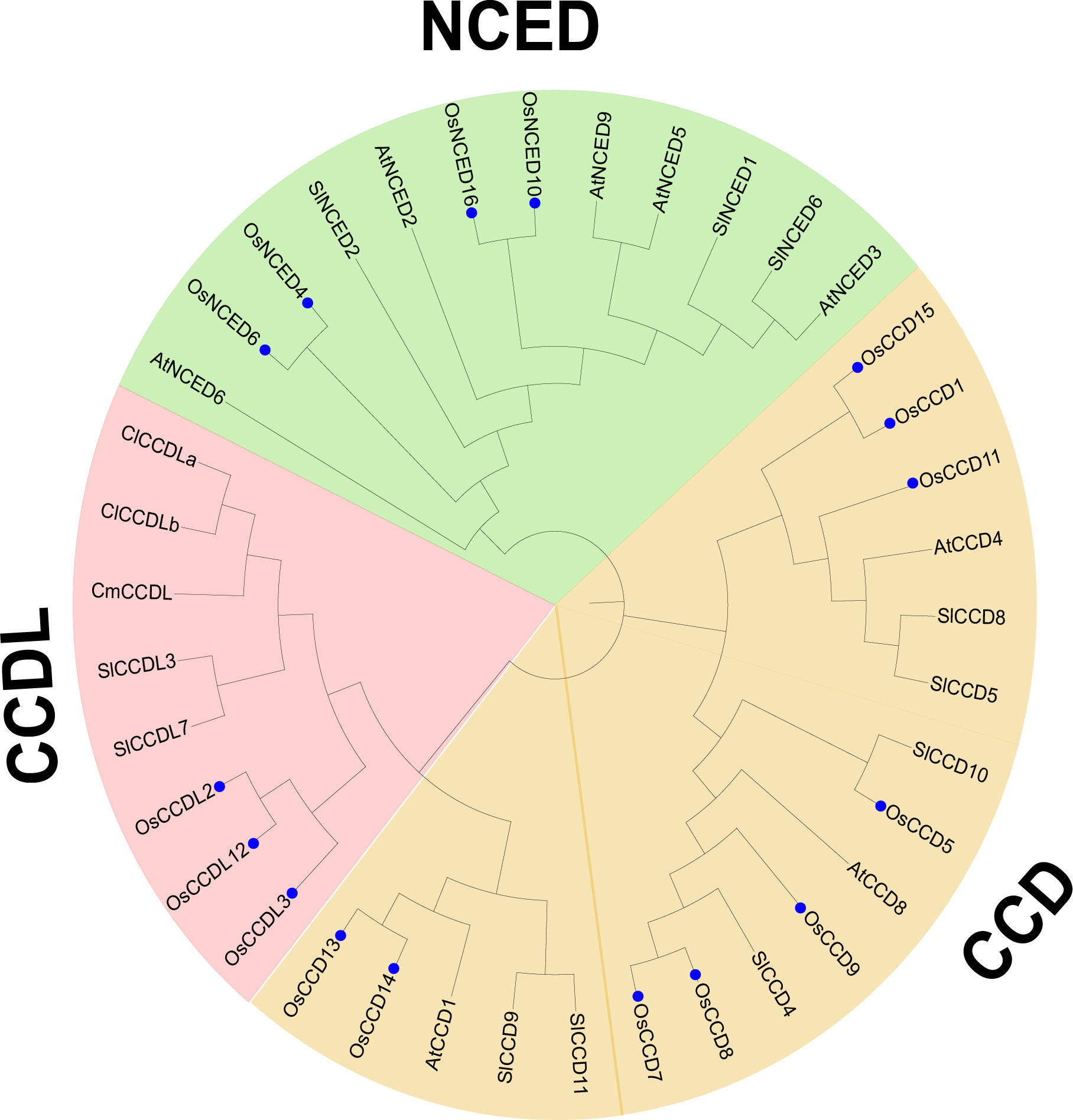
Figure 5 Evolutionary analysis was conducted to investigate the phylogenetic relationship among the CCO gene family members of O. sativa, Citrullus lanatus, Cucumis melo, Arabidopsis thaliana, and Solanum lycopersicum. The O. sativa genes were specifically identified using blue stars. The analyses were performed using MEGA 11 software, allowing for comprehensive insights into the evolutionary patterns and relationships among these species’.
A total of 153 miRNAs were identified that target all 16 genes. These 153 miRNAs belong to 45 groups of miRNAs. The length of these miRNAs ranges from 20 to 24 amino acids. OsCCDL12 (Osa-miR169, Osa-miR5802, Osa-miR1430, Osa-miRN2366, Osa-miR1432, Osa-miRN2322) is targeted by 6 groups of miRNAs, which is the highest number of targets, while OsCCD1 (Osa-miRN2275) is targeted by only 1 miRNA. OsCCD7 and OsCCD8 are targeted by 4 and 2 groups of miRNAs, respectively. OsCCDL3 and OsNCED6 are targeted by 4 and 1 group of miRNAs, respectively. OsCCD13 and OsCCD14 are targeted by 3 and 2 groups of miRNAs, respectively. OsCCD11, OsCCD15, OsCCD5, OsCCD9, OsNCED10, and OsNCED16 are targeted by 4, 3, 3, 4, 3, and 1 group of miRNAs, respectively. The miRNAs are specific and target only one gene, but multiple miRNAs can target a single gene. Most of the miRNAs inhibit cleavage, while others inhibit the translation of their respective targeted genes. In total, 49 groups consist of 12 translation-inhibited miRNAs and 37 cleavage-inhibited miRNAs found in the results. Osa-miR169, Osa-miR827, Osa-miR394, Osa-miR395, Osa-miR396 and Osa-miR397 were involved during salt in rice (Wang et al., 2013) (Supplementary Table 4).
Chromosomal location anticipated that they were dispersed on distinct chromosomes. CCO genes were found on chromosomes 1, 2, 3, 4, 7, 8, 9, 10, and 12. No CCO genes exist on chromosomes 5, 11, 13, 14, or 16. OsCCDL2 and OsCCDL3 are found on chromosome 8. OsCCD7, OsCCD8, and OsCCD9 were also found on one chromosome. OsNCED4 and OsCCD5 were discovered on four chromosomes. OsCCD13, OsCCD14, OsCCD15, and OsNCED16 are all found on chromosome 12 (Figure 6).

Figure 6 The chromosomal distribution of the OsCCO gene family in rice is illustrated in the figure. The figure’s blue bars represent rice’s individual chromosomes, while the red lines indicate the functional relationships or co-regulation between OsCCO genes. This analysis provides insights into the spatial arrangement and potential interactions among OsCCO genes within the rice genome.
For the gene pair OsCCD7_OsCCD8, the Ka and Ks values are relatively low (0.0037 and 0.0073, respectively), indicating that the genes have experienced relatively few substitutions. The MYA value of 0.56 suggests that the gene divergence occurred relatively recently. While for the gene pair OsNCED4_OsCCD15, the Ka and Ks values are much higher (0.72 and 4.18, respectively), indicating that these genes have experienced many substitutions over time. The MYA value of 321.65 suggests that these genes diverged from a common ancestor long ago. The two gene pairs have undergone different levels of evolutionary change, with OsNCED4_OsCCD15 experiencing more divergence over time than OsCCD7_OsCCD8 (Figure 7).
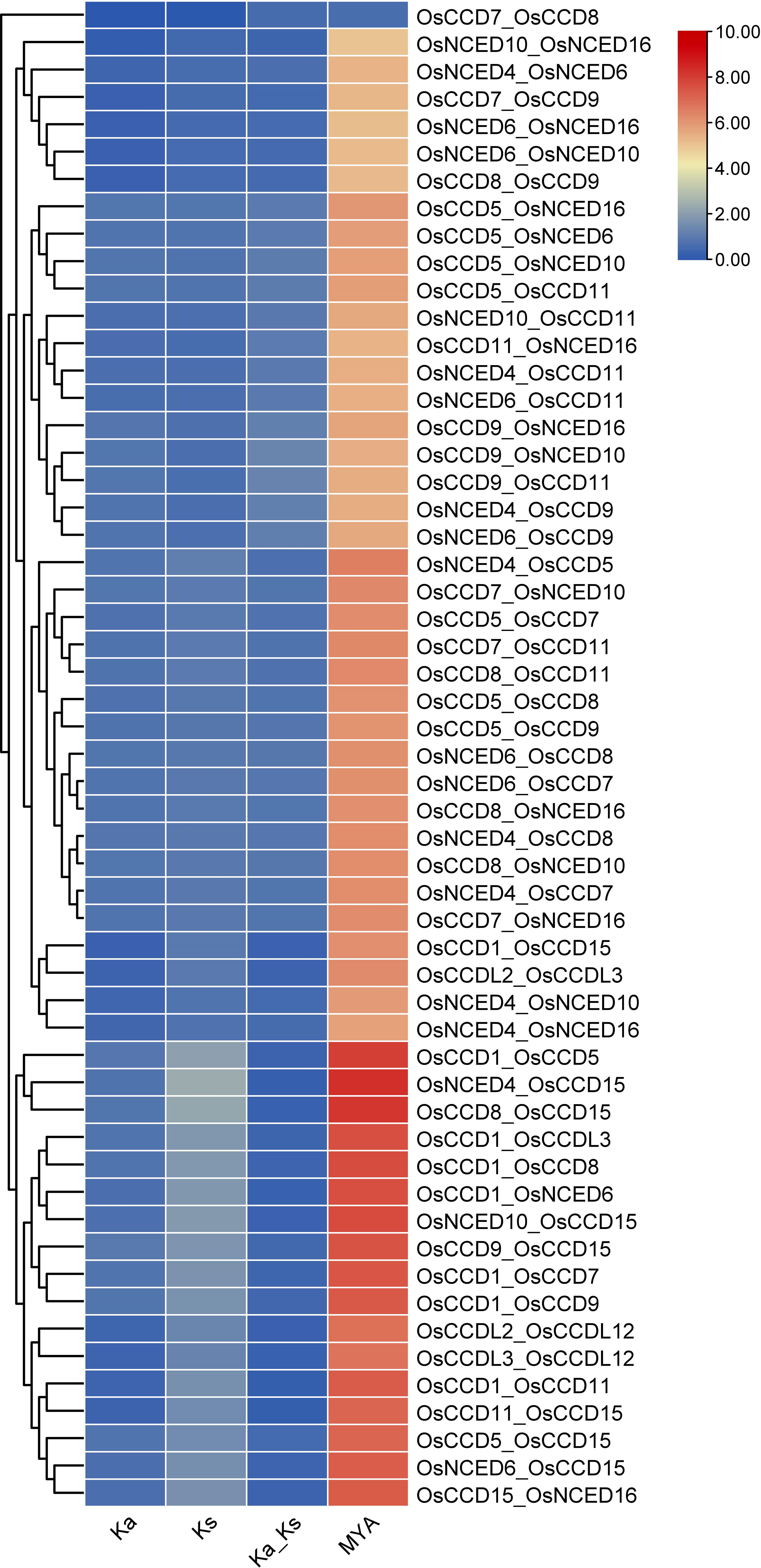
Figure 7 The Ks (synonymous substitution rate) and Ka (nonsynonymous substitution rate) were estimated using TBTools. The rectangular-like rate (λ) for rice was calculated to be 6.5 × 10−9. The date of the duplication event was determined using the formula T = Ks/2λ. This analysis provides insights into the timing and evolutionary dynamics of gene duplications in rice.
During synteny analysis, 56 paralogous genes were found, but the OsNCED10 gene was a singleton, meaning no copies were discovered (Figure 8A). One gene was duplicated in Arabidopsis, however two duplicate genes were found in rice and tomato, according to the dual synteny study (Figures 8A, B).

Figure 8 (A) Displays the distribution of OsCCO genes on rice chromosomes, where potential gene duplications are indicated by lines connecting genes on different chromosomes. Panel (B) shows the chromosomal distribution and intrachromosomal linkages of CCO genes between rice-Arabidopsis and rice-Tomato. Red lines represent duplicate CCO gene pairs, while gray lines represent synteny blocks in the rice genome. The chromosome number is indicated at the top of each chromosome, highlighting that segmental duplication of genes is more prevalent than tandem duplication within the OsCCO gene family. This analysis provides insights into the evolutionary history and genomic organization of OsCCO genes.
The Go enrichment analysis conducted in this study revealed valuable insights into the functional roles of OsCCO genes. These genes were found to be primarily involved in carotene metabolic processes (GO:0016119) and terpene catabolic processes (GO:0046247) within the biological process category (Rothenberg et al., 2019). In terms of molecular functions, they were prominently associated with carotenoid dioxygenase activity (GO:0010436). Furthermore, the analysis indicated a high enrichment of OsCCOs proteins in the chloroplast stroma (GO:0009570), suggesting their diverse roles in cellular metabolism (Figure 9; Supplementary Table 5).
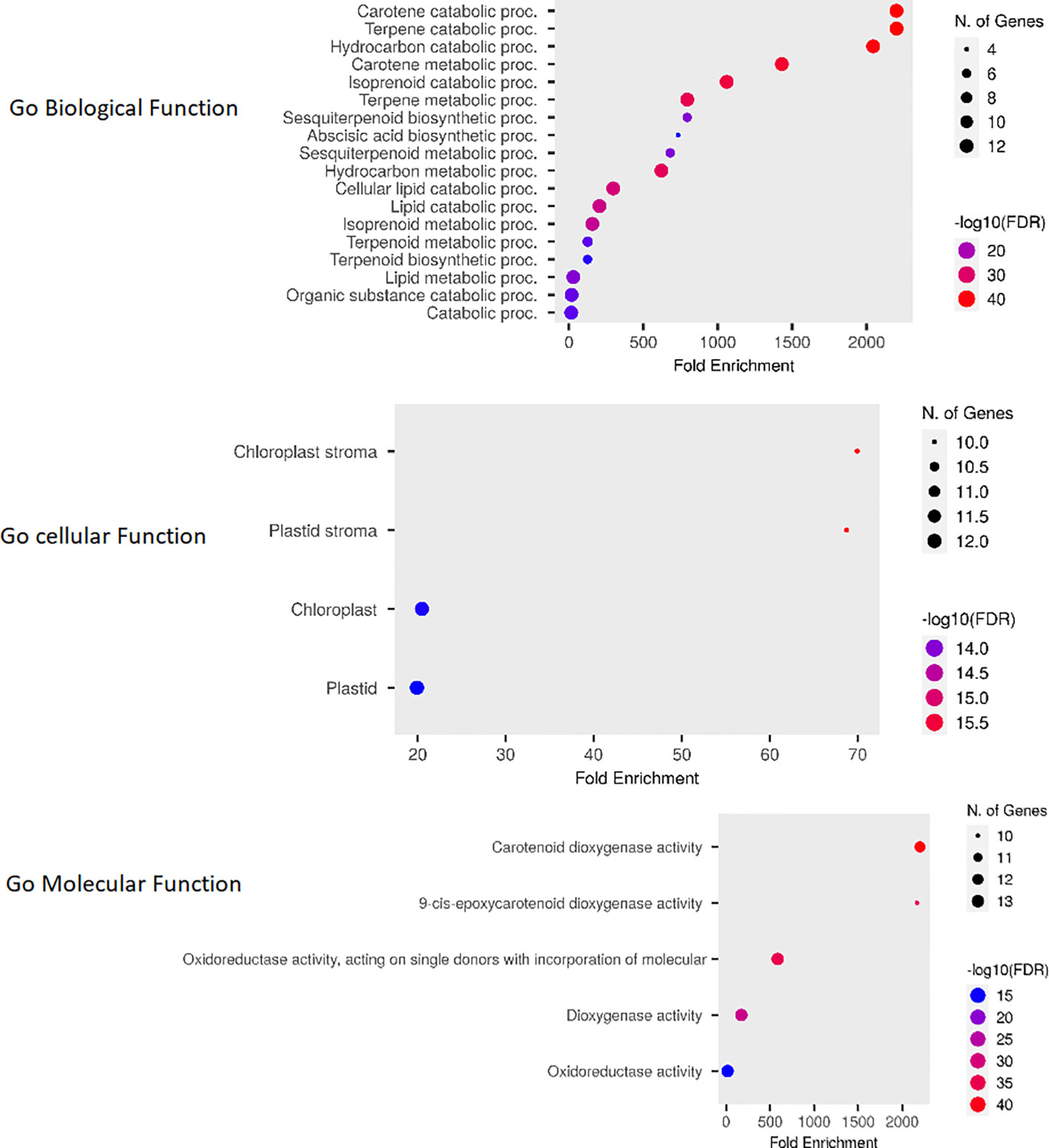
Figure 9 The Fold Enrichment chart visually represents the overlapping functions of OsCCO genes. The chart uses dot plots, where red dots indicate a higher number of genes associated with a particular process, while smaller blue dots represent a lower number of genes. This chart provides a quick overview of the functional distribution among OsCCO genes and highlights the processes in which they are predominantly involved.
The OsCCD11 gene exhibits the highest number of associations in the protein-protein interaction network. Notably, OsCCD11, OsNCED4, OsNCED6, OsNCED16, and OsCCD1 establish distinctive connections among themselves. Conversely, OsCCDL12 and OsCCDL3 engage with a diverse set of proteins in the dataset, indicating their broad interaction profiles. OsCCD11 appears to participate in various connections and play roles in multiple cellular processes. Meanwhile, OsCCD8, OsCCD9, and OsCCD5 form a sub-network within the larger protein-protein interaction network, suggesting a specific biological process or coordinated functional relationship among these proteins (Figure 10).
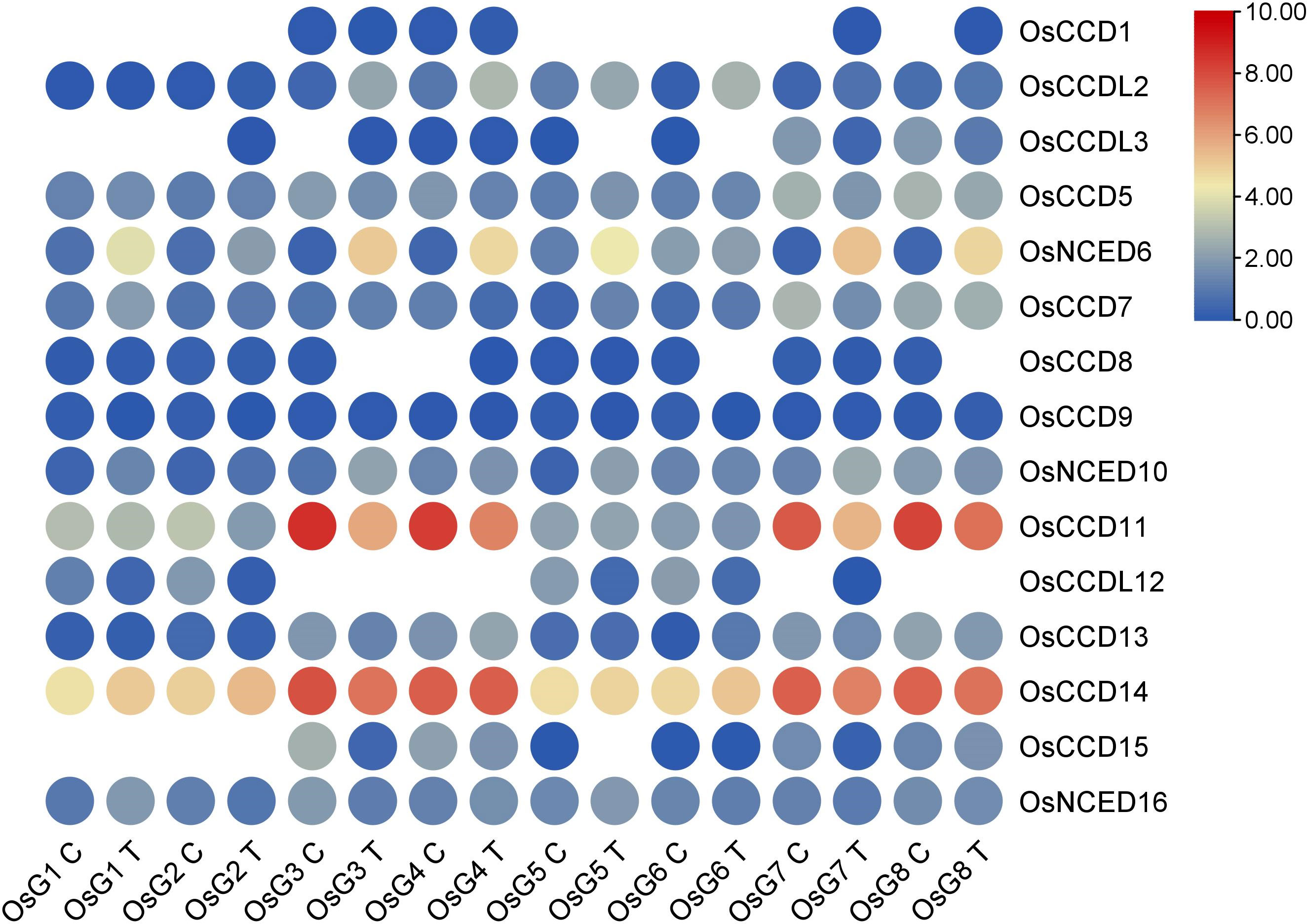
Figure 10 Protein-protein interactions involve various connections with distinct genes.
Based on the RNA-seq analysis, it was observed that OsNCED10 and OsNCED6 showed high expression levels and upregulation, indicating their potential role in abscisic acid (ABA) production. This suggests these genes regulate ABA synthesis in rice plants, especially in response to stress conditions. However, based on their expression patterns, the remaining genes did not exhibit significant roles in salt stress (Figure 11).
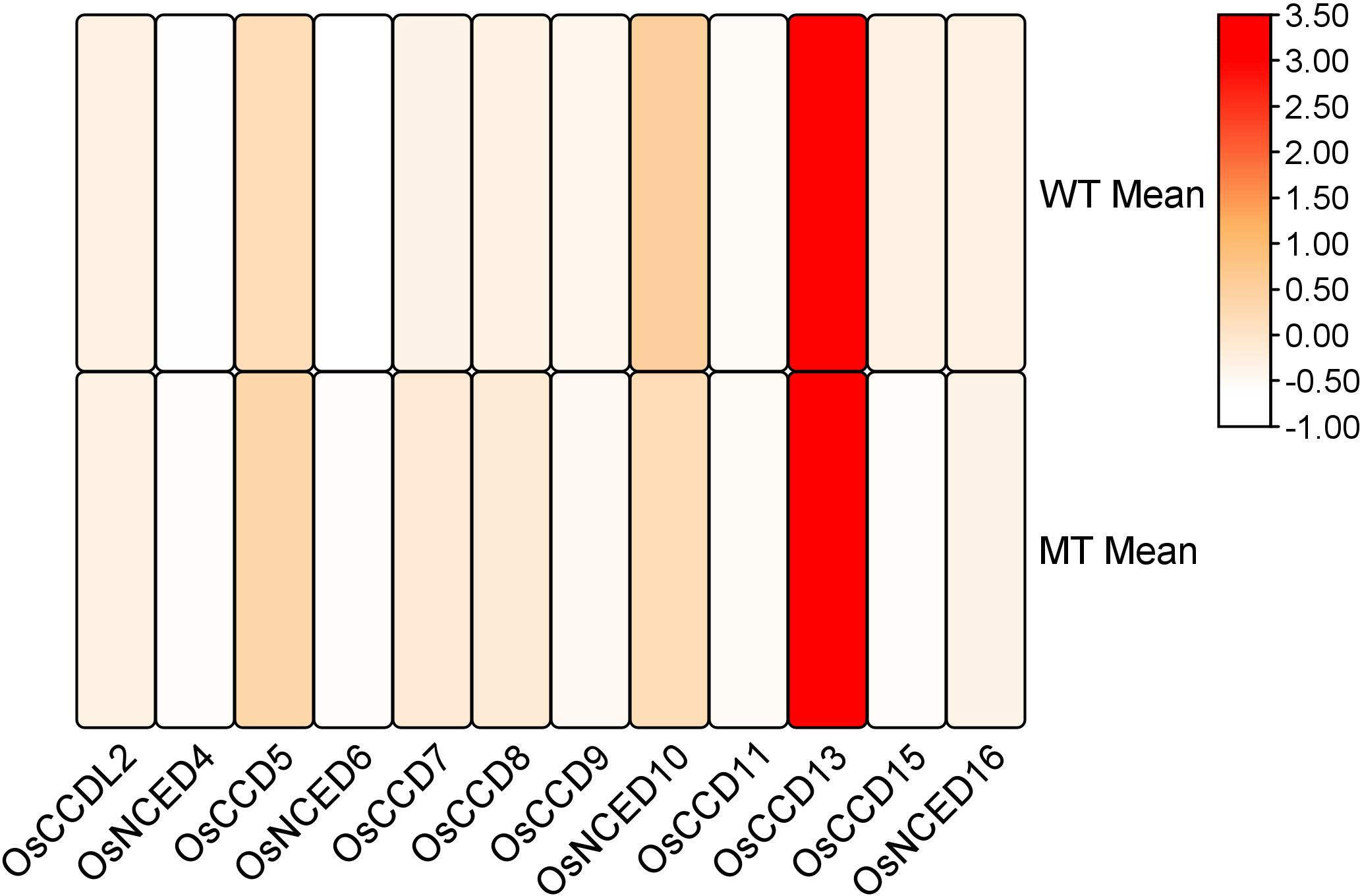
Figure 11 Heat map of transcriptomic expression profile of the OsCCO genes of rice under salt conditions.
RNA-seq analysis revealed that during the inflorescence development stage, the mutant genotype exhibited high expression levels and upregulation of two genes, CCD7 and CCD8. These genes showed significant involvement in the developmental process. However, the remaining genes were also expressed during the inflorescence development stage but did not display a significant role (Figure 12).

Figure 12 Heat map of transcriptomic expression profile of the OsCCO genes during inflorescences development.
RNA-seq analysis revealed distinct expression patterns of various genes. OsCCDL2 was predominantly expressed in seed, while OsCCDL3 showed expression specifically in carpel tissue. OsNCED4 exhibited high expression levels in anther tissue, while OsCCD5 was highly expressed in shoot tissue. OsNCED6 showed upregulation in carpel tissue, whereas OsCCD7 displayed upregulation in seed and expression in carpel and shoot tissues. OsCCD8 was expressed in carpel tissue, and OsCCD9 showed expression in anther and seed tissues.
Furthermore, OsNCED10 was highly upregulated in carpel tissue, indicating its potential role in this tissue-specific process. OsCCD11 exhibited high upregulation in shoot and seed tissues. OsCCDL12 was expressed in carpel tissue, while OsCCD13 showed high upregulation in shoot, seed, and carpel tissues. OsCCD14 displayed significant upregulation in anther tissue, and OsCCD15 showed expression specifically in shoot tissue. Lastly, OsNCED16 exhibited high upregulation in seed and shoot tissues (Figure 13).
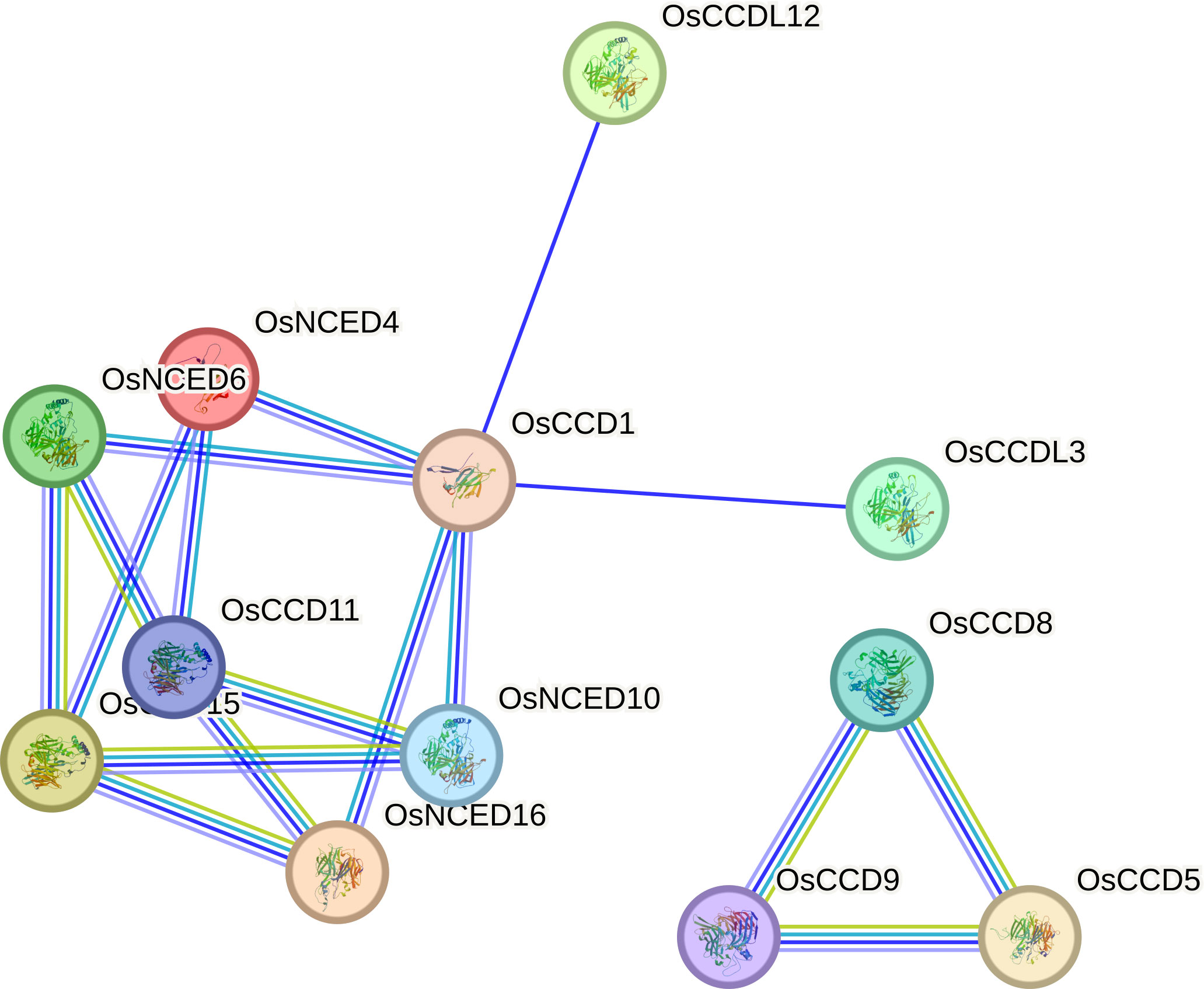
Figure 13 The heat map visually displays the transcriptomic expression patterns of OsCCO genes in different organs of rice. It uses colors to represent the varying levels of gene expression.
Rice is a major crop for many poor countries, but because of many environmental stressors, both abiotic and biotic, it is vulnerable to yield reductions (Seah et al., 2023). To help minimize these losses, transcription factors (TFs) play a vital role in regulating plant responses to these stresses. The investigation of OsCCOs gene family is essential in plant research particularly in relation to its role in environmental stress such as drought, seed dormancy, salinity, cold and heat stress. The role of the OsCCOs gene family in rice is yet to be fully comprehended. Knowledge of CCO genes’ functions and their association with these activities opens the door for novel approaches to improve crop production and increase the resilience of rice plants against environmental stressors (Dhankher and Foyer, 2018).
A genome-wide investigation was carried out to find and understand the CCO genes in rice (Griffiths et al., 2003). The physicochemical properties of sixteen OsCCO genes in the rice genome were investigated to observe their distinctions within a clade of proteins. It was determined that all identified OsCCO proteins exhibited hydrophilic characteristics as evidenced by negative GRAVY values, reflecting a tendency to interact with water and possess electrical charges dependent on pH levels (Wilkins et al., 1998). Upon examination using the instability index, it was observed that ten distinct proteins possessed features suggestive of instability. The examination of subcellular localization demonstrated that OsCCO proteins were dispersed among multiple organelles, such as the chloroplast, mitochondria, cytoplasm, cytosol, endoplasmic reticulum nucleus and plasma membrane (Tan et al., 2003). It is significant to observe that more than half of the proteins, precisely 47.76% and 23.98%, were correspondingly identified in the chloroplast and cytoplasm. This suggests that the OsCCO proteins could serve vital roles within these particular organelles.
By examining the genome of diverse species, substantial insight can be gained regarding the history and arrangement of genes. Moreover, such analyses can enable the transfer of genomic data from a well-researched taxonomic group to one that is not as extensively studied (Auldridge et al., 2006). In the current research, 56 paralogous genes were detected within OsCCO, indicating genes replicating via gene duplication (Xue et al., 2023). This occurrence of duplication grants particularly valuable knowledge into the expansion of gene families, a phenomenon frequently observed in the realm of plants as a result of both tandem and segmental duplications (Fatima et al., 2023).
Comparative analysis of members of similar subgroups can provide significant insights into their functional behavior. This study, 37 CCO proteins were discovered and classified into three subfamilies based on their sequence structures and evolutionary relationships (Xue et al., 2023). Twelve OsNCED proteins, seventeen OsCCD proteins, and eight OsCCDL proteins were discovered using phylogenetic analysis. These findings imply that the OsNCED and OsCCD proteins in their respective subgroups may perform roles similar to AtNCED and AtCCD in Arabidopsis. In contrast, the OsCCDL proteins may perform functions similar to ClCCDLa, ClCCDLb, and CmCCDL proteins (Zhao et al., 2022).
Previous studies have suggested that positioning exons and introns within gene families is important for evolution (Zhao et al., 2022). This study’s investigation of gene structure and motifs revealed that the distribution of exons, introns, and motifs among members of the same population and clade was compatible with the shape of the phylogenetic tree (Wei et al., 2022). Every CCO gene contained exons and introns (Figure 4). Notably, in keeping with the traits of plants, the motifs of the NCED subfamily were found to be more maintained than those of the CCD subfamily.
Cis-regulatory elements are frequently located in the promoter region of genes and are essential for controlling gene expression at the transcriptional level (Zhou et al., 2019). Research on cis-regulatory elements revealed that a significant portion of the largest group, constituting 28 elements (equivalent to 37.833%), had a stress response and contained motifs such as ABRE, MYB, STRE, TGACG-motif, and WRE3. Meanwhile, the second-largest group, composed of 19 elements (or 25.67%), focused on metabolism and development and contained motifs such as A-box, Box 4, and TCCC-motif (Zhou et al., 2019). In addition, OsCCO contains various motifs responsible for different responses, such as CGTCA-motif and TGACG-motif for the MeJA response, TCA-element for the SA response, GARE-motif, TATC-box, and P-box for the GA response, and the ABRE TGA-element for the Auxin response.
Plants can undergo transpiration and photosynthetic processes, which can be affected by drought and salinity, resulting in reduced crop yields. The role of stomata is crucial in this process (Ahluwalia et al., 2021). Rice plants under salt stress were examined for the functions of various genes, including OsCCO genes, using transcriptome data (Liang et al., 2021). Interestingly, while CCD genes did not show any impact during salt stress, OsNCED6 and OsNCED10 genes were identified as possible candidates for developing salt-resistant rice varieties based on the RNA-seq data analysis. By producing ABA hormone, these genes facilitate stomata closure, resulting in reduced water loss due to transpiration and improved water-use efficiency under salt-stress conditions, thereby impacting rice Physiology and development.
The Go enrichment analysis of OsCCO genes highlights their significant involvement in carotene metabolic and terpene catabolic processes (Bao et al., 2021), pointing to their roles in pigment synthesis, vitamin A production and plant defense mechanisms. Additionally, their association with carotenoid dioxygenase activity underscores their potential in carotenoid cleavage reactions. The high enrichment of OsCCO gene in the chloroplast stroma suggests their crucial participation in diverse metabolic processes within this organelle, particularly related to photosynthesis and pigment biosynthesis (Chengcheng et al., 2022).
OsCCD11 stands out with the highest number of associations in our network analysis. This protein, along with OsNCED4, OsNCED6, OsNCED16, and OsCCD1 forms a distinct cluster of interconnected proteins, implying their potential involvement in shared cellular processes. Conversely, OsCCDL12 and OsCCDL3 exhibit a broader range of interactions, suggesting their roles in diverse biological contexts. These results hint at a specific functional relationship or concerted involvement in a particular biological process within the larger interaction network (Shimada et al., 2017).
Through a comprehensive transcriptome analysis utilizing RNA sequencing data from the NCBI GEO database (GSE227706), the importance of the CCD group in rice has been elucidated (Zhao et al., 2021). An in-depth investigation unveiled that CCD7 and CCD8 exhibited elevated expression levels during the critical floral development phase in rice. This significant discovery not only opens doors for further research aimed at enhancing rice productivity and quality but also establishes a potential foundation for the development of novel rice varieties with enhanced traits (Yue et al., 2022).
The analysis of RNA-seq data of various part of rice gene expression investigations (available at https://www.ebi.ac.uk/gxa/experiments?experimentType=differential/) has revealed important information about the expression patterns of OsCCOs gene in rice (Priya et al., 2019). The results show that most CCD genes are expressed in seed and shoot tissues, implying that they are involved in important activities associated to these organs. Furthermore, the CCDL group expresses specifically in the seed and carpel, whereas the NCED group expresses specifically in the anther, carpel, and seed. This extensive analysis of gene expression data demonstrates the importance of the OsCCOs gene family in rice crop improvement. The discovery of their tissue-specific expression patterns opens the door to future breeding programs to produce new rice varieties with improved characteristics and attributes.
MicroRNAs (miRNAs) are crucial regulatory molecules in plants that play a significant role in almost all biological processes, including plant growth, development, and responses to biotic and abiotic stress (Ding et al., 2020). They are highly conserved and exhibit specific functions (Millar et al., 2019). The investigation showed that the OsCCOs has a variety of miRNAs with various roles, including by focusing on genes involved in nitrogen uptake and assimilation, Osa-miR169 controls how effectively the plant uses nitrogen (Peng et al., 2020). It is believed that Osa-miR5802 regulates rice grain yield and size (Kumar and Sanan-Mishra, 2022). By targeting genes associated with stress signaling and cellular homeostasis, Osa-miR1430 aids rice plants in surviving drought stress. Osa-miRN2366 is thought to be involved in controlling the growth and development of rice plants, while its exact role is still unclear (61). By focusing on genes involved in stress signaling and ion transport, Osa-miR1432 controls how rice plants react to salt stress (Zhai et al., 2020). Osa-miR2275 is an OsCCD1 microRNA that has been connected to the production of siRNAs, small interfering RNAs that are unique to the inflorescence of rice (Fei et al., 2021). The possible implication from the observed results conveys that miRNAs could potentially govern the post-transcriptional control relating to OsCCO genes in rice progression.
This study’s outcomes provide fresh insights into the functional diversity and evolutionary dimensions of the CCO gene family in plants. These discoveries will serve as a valuable asset for future research, facilitating the functional examination and cloning of these genes. This comprehensive genome-wide identification and characterization, as undertaken in this study, will enable further investigations.
Sixteen OsCCO genes were discovered in O.sativa in this study. Based on structural analyses, the number of introns in OsCCO genes ranged from one to twelve. The presence of cis-regulatory elements related to light responsiveness, development-related response, developmental and hormone responsiveness, and certain abiotic stress in the promoter of OsCCO genes suggested their function in the abiotic stress of rice. OsNCED6 and OsNCED10 genes identified by RNA-seq data analysis can potentially be utilized to develop salt stress-resistant rice varieties for enhanced crop yield under salt conditions. The OsCCD7 and OsCCD8 genes are more active in inflorescences than in other organs, which suggests that they might play a role in grain development. However, additional research, including gene cloning and functional analysis, is required to confirm the significance of these genes in various physiological and biological processes. This in-silico analysis reveals the extensive genome-wide knowledge of rice OsCCO genes.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
MZ: Data curation, Formal Analysis, Writing – original draft. AS: Conceptualization, Data curation, Methodology, Writing – original draft. MS: Data curation, Formal Analysis, Software, Supervision, Writing – original draft. WA: Data curation, Investigation, Methodology, Writing – review & editing. SAli: Funding acquisition, Methodology, Resources, Writing – original draft. QA: Formal Analysis, Funding acquisition, Resources, Software, Writing – review & editing. SM: Writing – review & editing, Methodology, Resources. MS: Data curation, Funding acquisition, Investigation, Writing – review & editing. DA: Formal Analysis, Funding acquisition, Project administration, Supervision, Visualization, Writing – review & editing. SAla: Data curation, Funding acquisition, Methodology, Project administration, Resources, Writing – review & editing. IM: Writing – review & editing, Methodology, Formal Analysis, Software.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was funded by Researchers Supporting Project number (RSP-2023R27), King Saud University Riyadh, Saudi Arabia.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1269995/full#supplementary-material
Abdulla, M. F., Mostafa, K., AYDIN, A., Kavas, M. (2023). GATA transcription factor in common bean: a comprehensive genome-wide functional characterization, identification, and abiotic stress response evaluation. bioRxiv. 2023.2004. 2008.536124. doi: 10.1101/2023.04.08.536124
Ahluwalia, O., Singh, P. C., Bhatia, R. A. (2021). review on drought stress in plants: Implications, mitigation and the role of plant growth promoting rhizobacteria. Resources Environ. Sustainability 5, 100032. doi: 10.1016/j.resenv.2021.100032
Auldridge, M. E., Block, A., Vogel, J. T., Dabney-Smith, C., Mila, I., Bouzayen, M., et al. (2006). Characterization of three members of the Arabidopsis carotenoid cleavage dioxygenase family demonstrates the divergent roles of this multifunctional enzyme family. Plant J. 45, 982–993. doi: 10.1111/j.1365-313X.2006.02666.x
Bao, Q., Zhang, X., Bao, P., Liang, C., Guo, X., Chu, M., et al. (2021). Using weighted gene co-expression network analysis (WGCNA) to identify the hub genes related to hypoxic adaptation in yak (Bos grunniens). Genes Genomics 43, 1231–1246. doi: 10.1007/s13258-021-01137-5
Barre, A., Culerrier, R., Granier, C., Selman, L., Peumans, W. J., Van Damme, E. J., et al. (2009). Mapping of IgE-binding epitopes on the major latex allergen Hev b 2 and the cross-reacting 1, 3β-glucanase fruit allergens as a molecular basis for the latex-fruit syndrome. Mol. Immunol. 46, 1595–1604. doi: 10.1016/j.molimm.2008.12.007
Bülow, L., Hehl, R. (2016). Bioinformatic identification of conserved cis-sequences in coregulated genes. Plant Synthetic Promoters: Methods Protoc., 233–245. doi: 10.1007/978-1-4939-6396-6_15
Cheng, D., Wang, Z., Li, S., Zhao, J., Wei, C., Zhang, Y. (2022). Genome-wide identification of CCD gene family in six Cucurbitaceae species and its expression profiles in melon. Genes 13, 262. doi: 10.3390/genes13020262
Cheng, C., Yang, R., Yin, L., Zhang, J., Gao, L., Lu, R., et al. (2023). Characterization of carotenoid cleavage oxygenase genes in Cerasus humilis and functional analysis of ChCCD1. Plants 12, 2114. doi: 10.3390/plants12112114
Chengcheng, L., Raza, S. H. A., Shengchen, Y., Mohammedsaleh, Z. M., Shater, A. F., Saleh, F. M., et al. (2022). Bioinformatics role of the WGCNA analysis and co-expression network identifies of prognostic marker in lung cancer. Saudi J. Biol. Sci. 29, 3519–3527. doi: 10.1016/j.sjbs.2022.02.016
Chernys, J. T., Zeevaart, J. A. (2000). Characterization of the 9-cis-epoxycarotenoid dioxygenase gene family and the regulation of Abscisic acid biosynthesis in avocado. Plant Physiol. 124, 343–354. doi: 10.1104/pp.124.1.343
Daruwalla, A., Kiser, P. D. (2020). Structural and mechanistic aspects of carotenoid cleavage dioxygenases (CCDs). Biochim. Biophys. Acta (BBA)-Molecular Cell Biol. Lipids 1865, 158590. doi: 10.1016/j.bbalip.2019.158590
Dhankher, O. P., Foyer, C. H. (2018). Climate resilient crops for improving global food security and safety. Plant Cell Environ. 41, 877–884. doi: 10.1111/pce.13207
Ding, Y., Ding, L., Xia, Y., Wang, F., Zhu, C. (2020). Emerging roles of microRNAs in plant heavy metal tolerance and homeostasis. J. Agric. Food Chem. 68, 1958–1965. doi: 10.1021/acs.jafc.9b07468
Fatima, S., Cheema, K., Shafiq, M., Manzoor, M., Ali, Q., Haider, M., et al. (2023). The genome-wide bioinformatics analysis of 1-aminocyclopropane-1-carboxylate synthase (acs), 1-aminocyclopropane-1-carboxylate oxidase (aco) and ethylene overproducer 1 (eto1) gene family of fragaria vesca (woodland strawberry). Bull. Biol. Allied Sci. Res. 2023, 38–38. doi: 10.54112/bbasr.v2023i1.38
Fei, Y., Feng, J., Wang, R., Zhang, B., Zhang, H., Huang, J. (2021). PhasiRNAnalyzer: an integrated analyser for plant phased siRNAs. RNA Biol. 18, 1622–1629. doi: 10.1080/15476286.2021.1879543
Fei, R., Guan, S., Duan, S., Ge, J., Sun, T., Sun, X. (2023). Elucidating biological functions of 9-cis-epoxycarotenoid dioxygenase genes involved in seed dormancy in Paeonia lactiflora. Plants 12, 710. doi: 10.3390/plants12040710
Felemban, A., Braguy, J., Zurbriggen, M. D., Al-Babili, S. (2019). Apocarotenoids involved in plant development and stress response. Front. Plant Sci. 10, 1168. doi: 10.3389/fpls.2019.01168
Fukagawa, N. K., Ziska, L. H. (2019). Rice: Importance for global nutrition. J. Nutr. Sci. vitaminology 65, S2–S3. doi: 10.3177/jnsv.65.S2
Gitelson, A. A., Merzlyak, M. N. (1998). Remote sensing of chlorophyll concentration in higher plant leaves. Adv. Space Res. 22, 689–692. doi: 10.1016/S0273-1177(97)01133-2
Griffiths, S., Dunford, R. P., Coupland, G., Laurie, D. A. (2003). The evolution of CONSTANS-like gene families in barley, rice, and Arabidopsis. Plant Physiol. 131, 1855–1867. doi: 10.1104/pp.102.016188
Hashimoto, H., Uragami, C., Cogdell, R. J. (2016). Carotenoids and photosynthesis. Carotenoids nature: biosynthesis Regul. Funct., 111–139. doi: 10.1007/978-3-319-39126-7_4
He, Y., Chen, W., Tan, J., Luo, X., Zhou, Y., Gong, X., et al. (2022). Rice CENTRORADIALIS 2 regulates seed germination and salt tolerance via ABA-mediated pathway. Theor. Appl. Genet. 135, 4245–4259. doi: 10.1007/s00122-022-04215-8
Hu, Q., Ao, C., Wang, X., Wu, Y., Du, X. (2021). GhWRKY1-like, a WRKY transcription factor, mediates drought tolerance in Arabidopsis via modulating ABA biosynthesis. BMC Plant Biol. 21, 1–13. doi: 10.1186/s12870-021-03238-5
Kavas, M., Mostafa, K., Secgin, Z., Yerlikaya, B. A., Yıldırım, K., Gökdemir, G. (2023). Genome-wide analysis of duf221 domain-containing gene family in common bean and identification of its role on abiotic and phytohormone stress response. Genet. Resour. Crop Evol. 70, 169–188. doi: 10.1007/s10722-022-01421-7
Kim, Y., Hwang, I., Jung, H.-J., Park, J.-I., Kang, J.-G., Nou, I.-S. (2016). Genome-wide classification and abiotic stress-responsive expression profiling of carotenoid oxygenase genes in Brassica rapa and Brassica oleracea. J. Plant Growth Regul. 35, 202–214. doi: 10.1007/s00344-015-9520-y
Kiokias, S., Proestos, C., Varzakas, T. (2016). A review of the structure, biosynthesis, absorption of carotenoids-analysis and properties of their common natural extracts. Curr. Res. Nutr. Food Sci. J. 4, 25–37. doi: 10.12944/CRNFSJ.4.Special-Issue1.03
Kiser, P. D. (2022). Retinal pigment epithelium 65 kDa protein (RPE65): An update. Prog. retinal eye Res. 88, 101013. doi: 10.1016/j.preteyeres.2021.101013
Kumar, S., Sanan-Mishra, N. (2022). Mapping the expression profiles of grain yield QTL associated miRNAs in rice varieties with different grain size. Am. J. Plant Sci. 13, 1261–1281. doi: 10.4236/ajps.2022.139086
Langenbacher, A. D., Shimizu, H., Hsu, W., Zhao, Y., Borges, A., Koehler, C., et al. (2020). Mitochondrial calcium uniporter deficiency in zebrafish causes cardiomyopathy with arrhythmia. Front. Physiol. 11, 617492. doi: 10.3389/fphys.2020.617492
Li, Q., Yu, X., Chen, L., Zhao, G., Li, S., Zhou, H., et al. (2021). Genome-wide identification and expression analysis of the NCED family in cotton (Gossypium hirsutum L.). PLoS One 16, e0246021. doi: 10.1371/journal.pone.0246021
Liang, M.-H., He, Y.-J., Liu, D.-M., Jiang, J.-G. (2021). Regulation of carotenoid degradation and production of apocarotenoids in natural and engineered organisms. Crit. Rev. Biotechnol. 41, 513–534. doi: 10.1080/07388551.2021.1873242
Liang, M.-H., Zhu, J., Jiang, J.-G. (2018). Carotenoids biosynthesis and cleavage related genes from bacteria to plants. Crit. Rev. Food Sci. Nutr. 58, 2314–2333. doi: 10.1080/10408398.2017.1322552
Liu, X., Li, X., Yang, H., Yang, R., Zhang, D. (2023). Genome-wide characterization and expression profiling of ABA biosynthesis genes in a desert moss Syntrichia caninervis. Plants 12, 1114. doi: 10.3390/plants12051114
Mazhar, H.S.-U.-D., Shafiq, M., Ali, H., Ashfaq, M., Anwar, A., Tabassum, J., et al. (2023). Genome-wide identification, and in-silico expression analysis of YABBY gene family in response to biotic and abiotic stresses in potato (Solanum tuberosum). Genes 14, 824. doi: 10.3390/genes14040824
Millar, A. A., Lohe, A., Wong, G. (2019). Biology and function of miR159 in plants. Plants 8, 255. doi: 10.3390/plants8080255
Misawa, N. (2011). Pathway engineering for functional isoprenoids. Curr. Opin. Biotechnol. 22, 627–633. doi: 10.1016/j.copbio.2011.01.002
Peng, Y., Zhang, X., Liu, Y., Chen, X. (2020). Exploring heat-response mechanisms of microRNAs based on microarray data of rice post-meiosis panicle. Int. J. Genomics 2020. doi: 10.1155/2020/7582612
Priya, R., Sneha, P., Dass, J. F. P., Manickavasagam, M., Siva, R. (2019). Exploring the codon patterns between CCD and NCED genes among different plant species. Comput. Biol. Med. 114, 103449. doi: 10.1016/j.compbiomed.2019.103449
Project, I. R. G. S., Sasaki, T. (2005). The map-based sequence of the rice genome. Nature 436, 793–800. doi: 10.1038/nature03895
Pu, X., Li, Z., Tian, Y., Gao, R., Hao, L., Hu, Y., et al. (2020). The honeysuckle genome provides insight into the molecular mechanism of carotenoid metabolism underlying dynamic flower coloration. New Phytol. 227, 930–943. doi: 10.1111/nph.16552
Rehman, O. U., Uzair, M., Chao, H., Fiaz, S., Khan, M. R., Chen, M. (2022). Role of the type-B authentic response regulator gene family in fragrant rice under alkaline salt stress. Physiologia plantarum 174, e13696. doi: 10.1111/ppl.13696
Rothenberg, D. O. N., Yang, H., Chen, M., Zhang, W., Zhang, L. (2019). Metabolome and transcriptome sequencing analysis reveals anthocyanin metabolism in pink flowers of anthocyanin-rich tea (Camellia sinensis). Molecules 24, 1064. doi: 10.3390/molecules24061064
Sami, A., Haider, M., Imran, M., Abbas, A., Javed, M. (2023). SYNERGIZING FOOD SAFETY, QUALITY AND GENETIC IMPROVEMENT: THE INTERSECTION OF FOOD MICROBIOLOGY AND PROCESSING. Bull. Biol. Allied Sci. Res. 2023, 44–44. doi: 10.54112/bbasr.v2023i1.44
Seah, C. C., Tan, C. H., Arifin, N., Hafriz, R., Salmiaton, A., Nomanbhay, S., et al. (2023). Co-pyrolysis of biomass and plastic: Circularity of wastes and comprehensive review of synergistic mechanism. Results Eng., 100989. doi: 10.1016/j.rineng.2023.100989
Seki, M., Umezawa, T., Urano, K., Shinozaki, K. (2007). Regulatory metabolic networks in drought stress responses. Curr. Opin. Plant Biol. 10, 296–302. doi: 10.1016/j.pbi.2007.04.014
Shi, L., Chang, L., Yu, Y., Zhang, D., Zhao, X., Wang, W., et al. (2023). Recent advancements and biotechnological implications of carotenoid metabolism of brassica. Plants 12, 1117.
Shimada, S., Shinzawa-Itoh, K., Baba, J., Aoe, S., Shimada, A., Yamashita, E., et al. (2017). Complex structure of cytochrome c–cytochrome c oxidase reveals a novel protein–protein interaction mode. EMBO J. 36, 291–300.
Stafsnes, M. H., Josefsen, K. D., Kildahl-Andersen, G., Valla, S., Ellingsen, T. E., Bruheim, P. (2010). Isolation and characterization of marine pigmented bacteria from Norwegian coastal waters and screening for carotenoids with UVA-blue light absorbing properties. J. Microbiol. 48, 16–23.
Sun, Q., Gu, X., Chen, L., Xu, X., Wei, Z., Pan, Y., et al. (2022). Monitoring maize canopy chlorophyll density under lodging stress based on UAV hyperspectral imagery. Comput. Electron. Agric. 193, 106671.
Sun, L., Sun, Y., Zhang, M., Wang, L., Ren, J., Cui, M., et al. (2012). Suppression of 9-cis-epoxycarotenoid dioxygenase, which encodes a key enzyme in abscisic acid biosynthesis, alters fruit texture in transgenic tomato. Plant Physiol. 158, 283–298.
Takezawa, D. (2000). A rapid induction by elicitors of the mRNA encoding CCD-1, a 14 kDa Ca2+-binding protein in wheat cultured cells. Plant Mol. Biol. 42, 807–817. doi: 10.1023/A:1006431724090
Tan, B. C., Joseph, L. M., Deng, W. T., Liu, L., Li, Q. B., Cline, K., et al. (2003). Molecular characterization of the Arabidopsis 9-cis epoxycarotenoid dioxygenase gene family. Plant J. 35, 44–56. doi: 10.1046/j.1365-313X.2003.01786.x
Wan, T., Liu, Z., Leitch, I. J., Xin, H., Maggs-Kölling, G., Gong, Y., et al. (2021). The Welwitschia genome reveals a unique biology underpinning extreme longevity in deserts. Nat. Commun. 12, 4247. doi: 10.1038/s41467-021-24528-4
Wang, H., Guo, J. (2023). Molecular cloning and functional characterization of Jatropha curcas NCED3 involved in cold resistance. Plant Biotechnol. Rep. 17, 89–99.
Wang, M., Wang, Q., Zhang, B. (2013). Response of miRNAs and their targets to salt and drought stresses in cotton (Gossypium hirsutum L.). Gene 530, 26–32. doi: 10.1016/j.gene.2013.08.009
Wei, H., Movahedi, A., Liu, G., Li, Y., Liu, S., Yu, C., et al. (2022). Comprehensive analysis of carotenoid cleavage dioxygenases gene family and its expression in response to abiotic stress in poplar. Int. J. Mol. Sci. 23, 1418. doi: 10.3390/ijms23031418
Wilkins, M. R., Gasteiger, E., Sanchez, J. C., Bairoch, A., Hochstrasser, D. F. (1998). Two-dimensional gel electrophoresis for proteome projects: The effects of protein hydrophobicity and copy number. Electrophoresis 19, 1501–1505. doi: 10.1002/elps.1150190847
Xian, L., Sun, P., Hu, S., Wu, J., Liu, J.-H. (2014). Molecular cloning and characterization of CrNCED1, a gene encoding 9-cis-epoxycarotenoid dioxygenase in Citrus reshni, with functions in tolerance to multiple abiotic stresses. Planta 239, 61–77. doi: 10.1007/s00425-013-1963-4
Xu, M., Wang, Y., Zhang, M., Chen, M., Ni, Y., Xu, X., et al. (2023). Genome-wide identification of BES1 gene family in six Cucurbitaceae species and its expression analysis in Cucurbita moschata. Int. J. Mol. Sci. 24, 2287. doi: 10.3390/ijms24032287
Xue, G., Hu, L., Zhu, L., Chen, Y., Qiu, C., Fan, R., et al. (2023). Genome-wide identification and expression analysis of CCO gene family in liriodendron Chinense. Plants 12, 1975. doi: 10.3390/plants12101975
Yao, Y., Jia, L., Cheng, Y., Ruan, M., Ye, Q., Wang, R., et al. (2022). Evolutionary origin of the carotenoid cleavage oxygenase family in plants and expression of pepper genes in response to abiotic stresses. Front. Plant Sci. 12, 3169. doi: 10.3389/fpls.2021.792832
Yue, X.-Q., Zhang, Y., Yang, C.-K., Li, J.-G., Rui, X., Ding, F., et al. (2022). Genome-wide identification and expression analysis of carotenoid cleavage oxygenase genes in Litchi (Litchi chinensis Sonn.). BMC Plant Biol. 22, 394. doi: 10.1186/s12870-022-03772-w
Zhai, R., Ye, S., Zhu, G., Lu, Y., Ye, J., Yu, F., et al. (2020). Identification and integrated analysis of glyphosate stress-responsive microRNAs, lncRNAs, and mRNAs in rice using genome-wide high-throughput sequencing. BMC Genomics 21, 1–15. doi: 10.1186/s12864-020-6637-6
Zhang, Y., Fu, X., Feng, Y., Zhang, X., Bi, H., Ai, X. (2022). Abscisic acid mediates salicylic acid induced chilling tolerance of grafted cucumber by activating H2O2 biosynthesis and accumulation. Int. J. Mol. Sci. 23, 16057. doi: 10.3390/ijms232416057
Zhang, X., Liu, H., Guo, Q., Zheng, C., Li, C., Xiang, X., et al. (2016). Genome-wide identification, phylogenetic relationships, and expression analysis of the carotenoid cleavage oxygenase gene family in pepper. Genet. Mol. Res. 15, 15048695. doi: 10.4238/gmr.15048695
Zhang, S., Song, G., Li, Y., Gao, J., Liu, J., Fan, Q., et al. (2014). Cloning of 9-cis-epoxycarotenoid dioxygenase gene (TaNCED1) from wheat and its heterologous expression in tobacco. Biol. plantarum 58, 89–98. doi: 10.1007/s10535-013-0373-6
Zhao, J., Li, J., Zhang, J., Chen, D., Zhang, H., Liu, C., et al. (2021). Genome-wide identification and expression analysis of the carotenoid cleavage oxygenase gene family in five rosaceae species. Plant Mol. Biol. Rep., 1–13. doi: 10.1007/s11105-021-01284-9
Zhao, X.-L., Yang, Y.-L., Xia, H.-X., Li, Y. (2022). Genome-wide analysis of the carotenoid cleavage dioxygenases gene family in Forsythia suspensa: Expression profile and cold and drought stress responses. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.998911
Zhou, X.-T., Jia, L.-D., Duan, M.-Z., Chen, X., Qiao, C.-L., Ma, J.-Q., et al. (2020). Genome-wide identification and expression profiling of the carotenoid cleavage dioxygenase (CCD) gene family in Brassica napus L. PLoS One 15, e0238179. doi: 10.1371/journal.pone.0238179
Keywords: carotenoid cleavage oxygenases, apocarotenoids, RNA seq analysis, 9-cisepoxycarotenoid dioxygenases, Oryza sativa, O. sativa, abscisic acid, transcriptome
Citation: Haider MZ, Sami A, Shafiq M, Anwar W, Ali S, Ali Q, Muhammad S, Manzoor I, Shahid MA, Ali D and Alarifi S (2023) Genome-wide identification and in-silico expression analysis of carotenoid cleavage oxygenases gene family in Oryza sativa (rice) in response to abiotic stress. Front. Plant Sci. 14:1269995. doi: 10.3389/fpls.2023.1269995
Received: 31 July 2023; Accepted: 06 October 2023;
Published: 25 October 2023.
Edited by:
Syed Adeel Zafar, University of California, Riverside, United StatesReviewed by:
Irfan Ali Sabir, Shanghai Jiao Tong University, ChinaCopyright © 2023 Haider, Sami, Shafiq, Anwar, Ali, Ali, Muhammad, Manzoor, Shahid, Ali and Alarifi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Muhammad Shafiq, c2hhZmlxLmlhZ3NAcHUuZWR1LnBr; Qurban Ali, c2FpbTE2OTJAZ2FtaWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.