 Inom Juramurodov
Inom Juramurodov Dilmurod Makhmudjanov
Dilmurod Makhmudjanov Ziyoviddin Yusupov
Ziyoviddin Yusupov Komiljon Tojibaev
Komiljon Tojibaev
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 18 August 2023
Sec. Plant Systematics and Evolution
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1211247
This article is part of the Research Topic Economic Plant Genome and Database Construction and Research View all 10 articles
Hedysarum is one of the largest genera in the Fabaceae family, mainly distributed in the Northern Hemisphere. Despite numerous molecular studies on the genus Hedysarum, there is still a lack of research aimed at defining the specific characteristics of the chloroplast genome (cp genome) of the genus. Furthermore, the interrelationships between sections in the genus based on the cp genome have not yet been studied. In this study, comprehensive analyses of the complete cp genomes of six Hedysarum species, corresponding to sections Multicaulia, Hedysarum, and Stracheya were conducted. The complete cp genomes of H. drobovii, H. flavescens, and H. lehmannianum were sequenced for this study. The cp genomes of six Hedysarum species showed high similarity with regard to genome size (except for H. taipeicum), gene sequences, and gene classes, as well as the lacking IR region. The whole cp genomes of the six species were found to contain 110 genes ranging from 121,176 bp to 126,738 bp in length, including 76 protein-coding genes, 4 rRNA genes, and 30 tRNA genes. In addition, chloroplast SSRs and repetitive sequence regions were reported for each species. The six Hedysarum species shared 7 common SSRs and exhibited 14 unique SSRs. As well, three highly variable genes (clpP, accD, and atpF) with high Pi values were detected among protein-coding genes. Furthermore, we conducted phylogenetic analyses using the complete cp genomes and 76 protein-coding genes of 14 legume species, including the seven Hedysarum species. The results showed that the Hedysarum species form a monophyletic clade closely related to the genera Onobrychis and Alhagi. Furthermore, both of our phylogenetic reconstructions showed that section Stracheya is more closely related to section Hedysarum than to section Multicaulia. This study is the first comprehensive work to investigate the genome characteristics of the genus Hedysarum, which provides useful genetic information for further research on the genus, including evolutionary studies, phylogenetic relationships, population genetics, and species identification.
Hedysarum L. is one of the large genera in the Fabaceae family, containing more than 160 species (Lock, 2005). Plants belonging to the genus Hedysarum are distributed in Eurasia, North Africa, and North America (Lock, 2005; Liu et al., 2017). The species occur in meadows, clayey and stony places, deserts, steppes, forests, tundra, river valleys, and mountain slopes (Choi and Ohashi, 2003). The genus Hedysarum includes perennial herbs and rarely semi-shrubs, which differ from closely related genera in pod and pollen morphology (Fedtschenko, 1948; Choi and Ohashi, 1996). Previous studies have shown that many species of the genus Hedysarum have been employed in traditional Chinese medicine to strengthen the immune system and improve the energy of the body (Dong et al., 2013).
Recent molecular studies have proposed to divide this genus into three main clades, largely corresponding to the sections Hedysarum, Stracheya, and Multicaulia (Amirahmadi et al., 2014; Nafisi et al., 2019). In these molecular phylogenetic studies, species were divided into three sections that were well supported, but intersection relationships remained unresolved, especially in section Multicaulia. This could be due to the selection of regions with low variability in the cp genome. Therefore, it is necessary to identify regions with high nucleotide diversity in the cp genome as molecular markers for future molecular phylogenetic studies of Hedysarum. Furthermore, previous studies have reported conflicting results regarding the phylogenetic relationship of section Stracheya with the other two sections within the Hedysarum genus. Duan et al. (2015) found that section Stracheya was closely related to section Multicaulia based on both nrDNA ITS and some plastid markers. However, Liu et al. (2017) reported that section Stracheya was placed together with section Hedysarum based on nrDNA ITS and plastid markers. Conversely, Nafisi et al. (2019) supported a closer relationship between section Stracheya and section Multicaulia. Therefore, determining the phylogenetic position of section Stracheya in the genus Hedysarum based on the cp genome is necessary.
Chloroplasts are important intracellular organelles, having an independent genome with several genes responsible for the process of photosynthesis in green algae and plants (Nazareno et al., 2015; Smith and Keeling, 2015; Yin et al., 2017). Most complete cp genomes harbor a typical quadripartite structure including a long single copy (LSC) region, a small single copy (SSC) region, and two copies of inverted repeat (IR) regions (Bock, 2007). Hedysarum belonging to the IRLC (Inverted Repeat Lacking Clade) clade is described by the lack of one copy of the inverted repeat (IR) region in the whole cp genome (Wojciechowski et al., 2004; She et al., 2019). Species belonging to the IRLC clade are characterized by having a cp genome size of around 121,000–133,000 bp (Moghaddam et al., 2022; Yuan et al., 2022; Tian et al., 2021). To date, the size of the cp genomes of only four Hedysarum species are known, including H. petrovii, H. semenovii, H. polybotrys, and H. taipeicum, with genome sizes of 122,571 bp, 123,407 bp, 122,232 bp, and 126,699 bp, respectively.
Comparative genomics can be used to identify important structural sequences and detect evolutionary changes across genomes since the comprehensive analysis of the cp genome of genera belonging to the IRLC clade such as Astragalus, Onobrychis, Caragana, and Glycyrrhiza have been reported (Kang et al., 2018; Moghaddam et al., 2022; Yuan et al., 2022; Tian et al., 2021). A detailed characterization of these species’ cp genome, including size, gene content, structure repeats, and GC content, as well as information about highly variable nucleotide regions, was provided. However, comprehensive studies on the genome structure of the genus Hedysarum have not been conducted so far.
In the present investigation, we detailed an overview of the complete sequence of the six Hedysarum species cp genome. We sequenced the complete cp genome of H. drobovii, H. flavescens, and H. lehmannianum to explore the relationships among Hedysarum species. We obtained the other three species (H. petrovii, H. semenovii, and H. taipeicum) from the National Center for Biotechnology Information (NCBI). The following questions were addressed: (1) what are the features of the cp genome of selected Hedysarum species? (2) How many potential microsatellite markers can the cp genome provide? (3) Which regions in the cp genome can be used as candidate molecular markers for future molecular phylogenetic studies? (4) What is the phylogenetic placement of section Stracheya within the genus Hedysarum based on the cp genome data?
For the comparative genome analysis, species from each section of Hedysarum were selected in this study: Hedysarum drobovii and H. petrovii from the H. sect. Multicaulia; H. flavescens, H. semenovii, and H. taipeicum from the H. sect. Hedysarum; H. lehmaniannum from the H. sect. Stracheya. Fresh material for H. drobovii, H. flavescens, and H. lehmaniannum was collected from Uzbekistan (H. drobovii: Western Tien Shan, Chatkal Range, E. 70.1045, N. 41.560301, altitude: 970 m a.s.l., 06 June 2020, Dekhkonov, Ortiqov, Turdiev, Juramurodov 19062020117; H. flavescens: Western Tien Shan, Chatkal Range, E. 70.019246 N. 41.508587, altitude: 2290 m a.s.l., 06 June 2020, Dekhkonov, Ortiqov, Turdiev, Juramurodov 19062020089; H. lehmaniannum: Hisar Range, Boysun district, E. 67.163574, N. 38.337148, altitude: 2390 m a.s.l., 13 June 2021, Turginov, Rahmatov 13062021007), and their complete chloroplast (cp) genome sequences were generated (Figure 1). Herbarium materials of these three species were stored in the National Herbarium of Uzbekistan (TASH).

Figure 1 Three sequenced samples in this study. (A) H. flavescens, (B) H. lehmaniannum, and (C) H. drobovii. The photo of H. lehmaniannum was taken by O.Turginov.
Total genomic DNA was extracted from leaf material using DP305 Plant Genomic DNA kits (Tiangen, Beijing, China) following the manufacturer’s protocol. The sequencing library was generated using NEBNext® UltraTM DNA Library Prep Kit for Illumina (NEB, USA, Catalog: E7370L) following the manufacturer’s recommendations, and index codes were added to each sample. Briefly, the genomic DNA sample was fragmented by sonication to a size of 350 bp. Then DNA fragments were end polished, A-tailed, and ligated with the full-length adapter for Illumina sequencing, followed by further PCR amplification. After, PCR products were purified by the AMPure XP system (Beverly, USA). Subsequently, the library quality was assessed on the Agilent 5400 system(Agilent, USA) and quantified by QPCR (1.5 nM). The qualified libraries were pooled and sequenced on Illumina platforms with PE150 strategy in Novogene Bioinformatics Technology Co., Ltd (Beijing, China), according to effective library concentrations and data amount required.
The resulting clean reads were assembled using the GetOrganelle pipeline (Jin et al., 2020) with the optimized parameters “-F plant_cp -w 0.6 -o -R 20 -t 8 -k 75,95,115,127 and”. Gene annotation was performed in Geneious v.10.0.2 and H. polybotrys (unpublished, accession number: MZ322397) was set as the reference. Start and stop codons and intron/exon boundaries for protein-coding genes were checked manually (Kearse et al., 2012).
The chloroplast simple sequence repeats (SSRs) were identified using the MIcroSAtellite (MISA) web tool (Beier et al., 2017). The search conditions for SSRs were set to isolate perfect mono-, di-, tri-, tetra-, penta-, and hexa nucleotide motifs with a minimum of 10, 5, 4, 3, 3, and 3 repeats, respectively. The REPuter program (Kurtz et al., 2001) was used to identify repeats: forward, reverse, palindrome, and complement sequences in cp genomes. The following settings for repeat identification were used: (1) a hamming distance equal to three, (2) minimal repeat size set to 30 bp, and (3) maximum computed repeats set to 90 bp.
The cp genome was drawn using OGDRAWv1.1 (Lohse et al., 2007). Nucleotide variability (Pi) was calculated for the whole cp genome and protein-coding genes separately using DnaSP v. 6.12.03 software (Rozas et al., 2018). The window length was set to 800 bp and the step size was 200 bp. Furthermore, pairwise chloroplast genomic alignment among six species was compared by mVISTA in Shuffle-LAGAN mode (Frazer et al., 2004), and H. polybotrys (MZ322397) was used as a reference.
The three sequenced cp genomes of Hedysarum and 11 genomes from other species (including Onobrychis gaubae, O. viciifolia, Caragana jubata, C. kozlowii, Oxytropis aciphylla, and O. glabra as outgroups) retrieved from NCBI (Supplementary Table 1) were used to construct a phylogenetic tree. Phylogenetic tree reconstruction was performed using complete cp genomes and protein-coding sequences that were first aligned multiple times using MAFFT software (Katoh and Standley, 2013).
We reconstructed phylogenetic trees using Bayesian inference (BI), Maximum Parsimony (MP), and Maximum Likelihood (ML) methods. Nucleotide substitution models were selected statistically with the help of jModelTest2 on XSEDE (www.phylo.org) by considering the Akaike Information Criterion (AIC). The GTR+G model for the protein-coding sequences and the TVM+G model for the complete cp genomes were selected as the best model. For BI, we used MrBayes v. 3.2.7a (Ronquist et al., 2012) with 10 million generations with random trees sampled every 1000 generations. In the latter analysis, after discarding the first 25% of the trees as burn-in, a 50% majority-rule consensus tree was constructed from the remaining trees to estimate posterior probabilities (PP). The ML phylogeny was reconstructed using IQ-TREE 2.1.2 software (Minh et al., 2021) with 1000 bootstrap (BS) replicates to assess clade support (Nguyen et al., 2015). For MP analysis, we used PAUP* 4.0a169 (Swofford, 2002). The MP bootstrap analysis was performed with heuristic search, TBR branch-swapping, 1000 bootstrap replicates, random addition sequence with 10 replicates, and a maximum of 1000 trees saved per round.
The complete cp genomes of H. drobovii, H. flavescens, and H. lehmannianum were sequenced for this study. The sizes of the three newly sequenced species were 121,176 bp, 123,127 bp, and 123,586 bp, respectively. The H. petrovii, H. semenovii, and H. taipeicum species that were obtained from NCBI and the three newly sequenced species were without the typical quadripartite structure that contains a pair of IRs separated by LSC (large single-copy) and SSC (small single-copy) regions (Figure 2). The GC (guanine+cytosine) contents of the genomes of H. drobovii, H. petrovii, H. flavescens, H. semenovii, H. lehmannianum, and
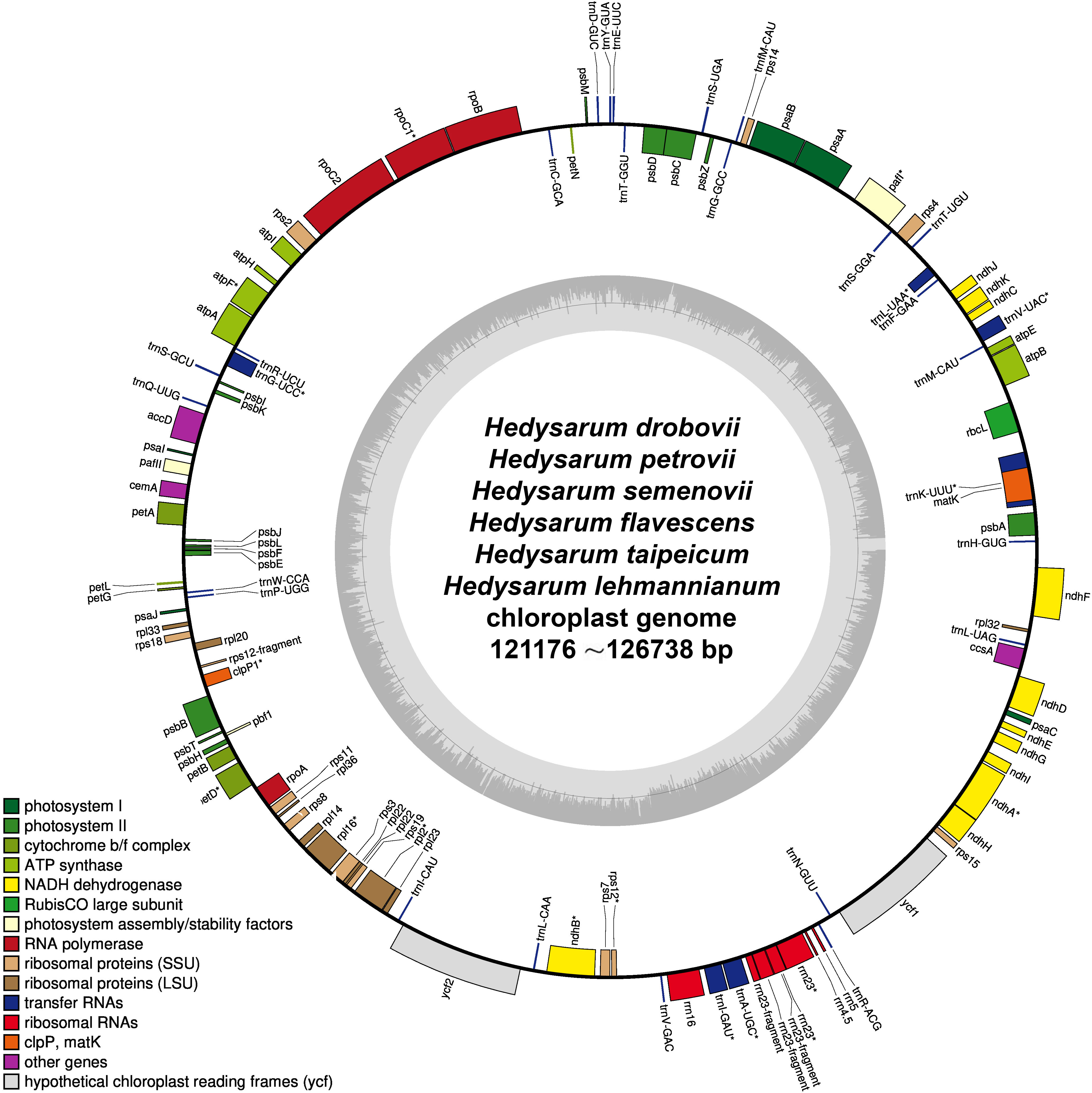
Figure 2 The chloroplast genome structure of six Hedysarum species. Genes shown outside the circles are transcribed clockwise, while those drawn inside are transcribed counterclockwise. Genes are color-coded according to their functional group.
H. taipeicum was 34.6%, 34.6%, 34.8%, 34.9%, 34.6%, and 35.1%, respectively. All six species’ genomes formed 110 genes including 76 protein-coding genes, 4 rRNA genes, and 30 tRNA genes (Table 1). A total of 16 genes in the cp genomes of six Hedysarum species consisted of introns, among which the genes trnK-UUU, trnC-ACA, trnL-UAA, rpoC1, atpF, trnG-UCC, clpP, petB, petD, rpl16, rpl2, ndhB, trnE-UUC, trnA-UGC, and ndhA each contained one intron, and only ycf3 gene contained two introns (Supplementary Table 2). The trnK-UUU gene contained the largest intron, from 2407 (H. petrovii) to 2503 (H. taipeicum). Additionally, the rps12 protein-coding gene is a trans-splicing gene that does not have introns in the 3’-end.
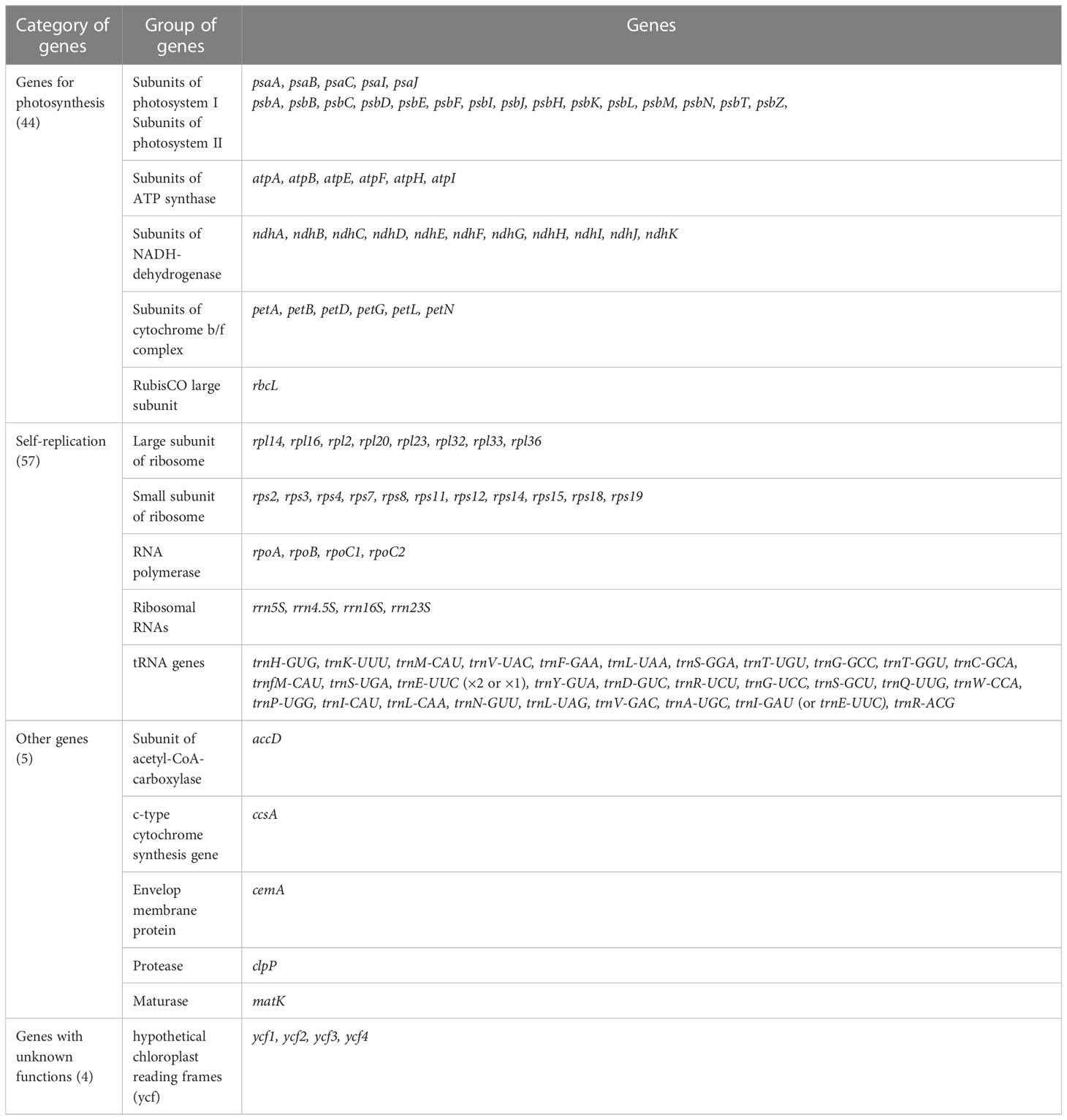
Table 1 List of genes annotated in the chloroplast genomes of six Hedysarum species in this study.
A total of 188 SSRs were detected using the MISA web tool in the cp genome of each H. drobovii, H. petrovii, and H. lehmannianum species, while H. flavescens, H. semenovii, and H. taipeicum had different SSRs of 184, 190, and 172, respectively. Among six Hedysarum cp genomes, the most abundant repeats were the mononucleotides from 145 (H. taipeicum) to 156 (H. lehmannianum), and the most dominant SSR was A/T (Figures 3A, B). Di-nucleotides (especially AT) were the second most predominant, varying from eight (H. taipeicum) to 21 (H. lehmannianum). A high number of trinucleotides was detected in H. semenovii (12), whereas a low number of trinucleotides was in H. lehmannianum (3). A total of 57 repeats of tetranucleotides, varying from seven (H. drobovii) to 12 (H. petrovii) were identified among the six Hedysarum cp genomes. Our analysis identified five pentanucleotide repeats in three Hedysarum species: H. drobovii (AAAAT, AAAGG, and AAGAC), H. petrovii (TTTCC), and H. taipeicum (AACCG), while the remaining three species did not exhibit any pentanucleotide repeats. Additionally, hexanucleotide repeats were detected only in H. flavescens (ATCAGT), H. semenovii (AAGACG, ATAGCT, and ATATTT), and H. taipeicum (AAGACG(× 2) and ATTCTT).
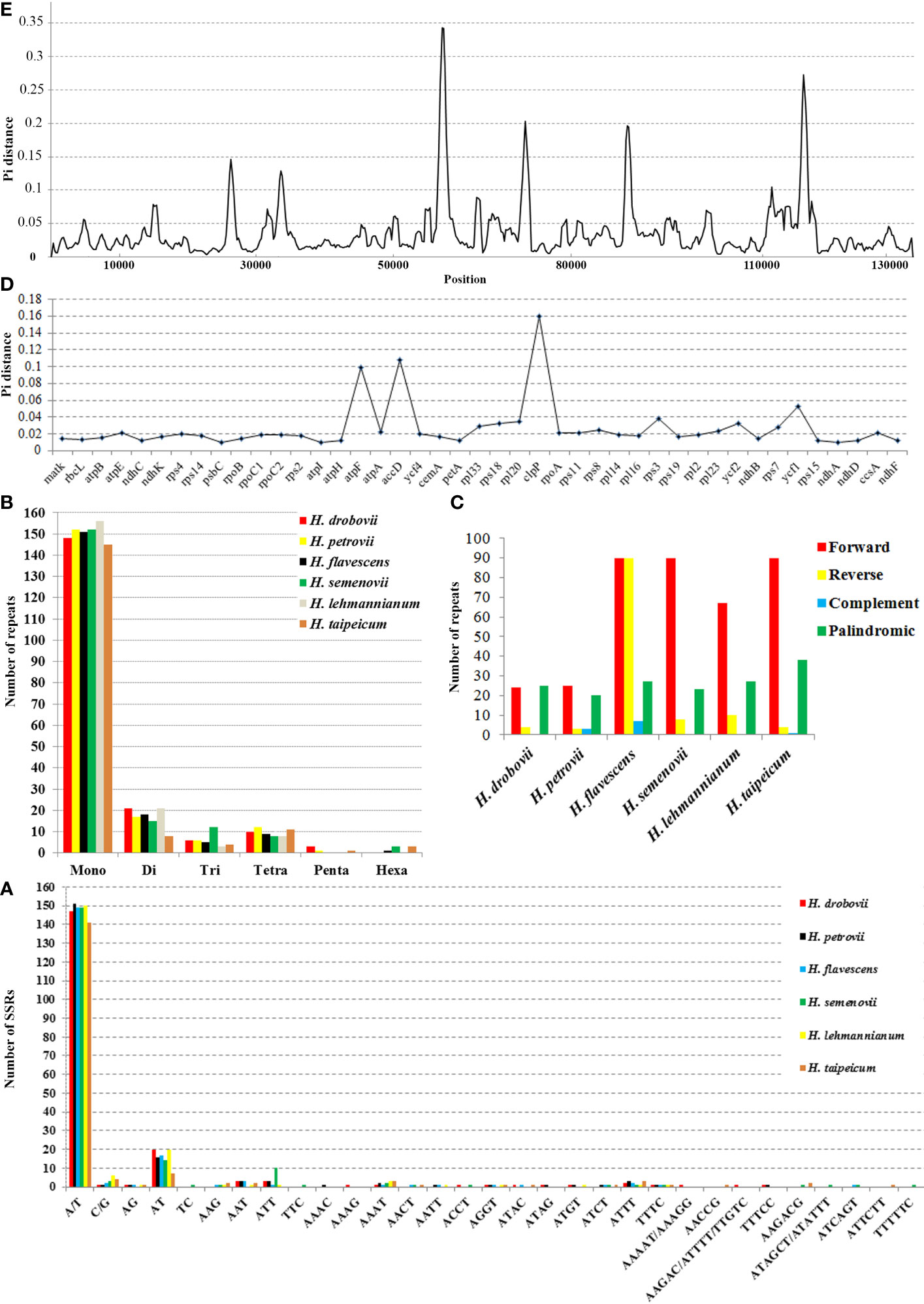
Figure 3 Chloroplast genome features of six Hedysarum species. Type of SSRs (A); long repetitive sequences (B); SSR distribution (C). Nucleotide diversity (Pi) in protein-coding genes (D) and whole chloroplast genomes (E). Among protein-coding genes, genes with nucleotide diversity < 0.01 are not shown.
In our study, we examined common and unique SSRs in six Hedysarum species (Supplementary Tables 3, 4), and we found that the majority of repeat units were composed of A and T, with rare occurrences of C or G, indicating that the SSRs of different species had an obvious bias in the base types of repeat units. Common SSRs included A, G, T, AT, AAAT, ATTT, and TTTC, which were present in all six species. We also identified 14 unique SSRs, including AAAG, AAAGG, AAAAT, AAGAC, ATTTT, and TTGTC in H. drobovii; TC, TTC, ATAGCT, and TTTTTC in H. semenovii; and AACCG and ATTCTT in H. taipeicum. Only one AAAC SSR was detected in H. petrovii, while no unique SSRs were identified in H. flavescens and H. lehmannianum.
In this study, we found many repeat regions including forward, reverse, palindromic, and complementary repeats (Figure 3C). Among the six studied Hedysarum species, the longest repetitive sequences were detected in the H. flavescens cp genome, which had 214 repetitive sequences with lengths of no more than 48 bp. On the contrary, the smallest repetitive sequences were found in H. drobovii and H. petrovii cp genomes, of which 53 and 51 scattered repetitive sequences with lengths of no more than 18 bp and 13 bp, respectively. The length of the largest forward and palindromic repeats were 62 bp and 36 bp in the H. taipeicum cp genome, respectively, whereas the largest reverse and complement repeat lengths were 48 bp and 7 bp in the H. flavescens cp genome, respectively. Equal numbers of forward repeats (90) were detected in H. flavescens, H. semenovii, and H. taipeicum. Additionally, the complement repeat was not found in the cp genomes of H. drobovii, H. semenovii, and H. lehmannianum.
We calculated the nucleotide diversity (Pi) values to estimate the divergence levels of the whole cp genome and protein-coding genes of the six Hedysarum species (Figures 3D, E). The most high-variation regions (Pi=0.3425) of the whole Hedysarum cp genomes were mainly concentrated between 55000 bp and 60000 bp. According to the Pi value results of protein-coding genes of six Hedysarum species, clpP (0.16), accD (0.108), and atpF (0.099) genes had the highest variability, while the psaC gene had a low nucleotide diversity (0.00136). In addition, the Pi values were less than 0.01 in 43.4% of the total protein-coding genes, while in 39.5% were 0.01–0.02. Only 26.3% of total protein-coding genes had Pi > 0.02 (Supplementary Table 5).
The cp genome sequences of six Hedysarum species were compared using the mVISTA software, and their alignments were visualized with annotation data (Figure 4). According to this visualization analysis, differences among sequences occurred in clpP, accD, and ycf1 genes from the coding regions and mainly in non-coding intergenic regions. Encoded gene classes and alignments of the main part of the coding regions among the six Hedysarum were highly congruous.
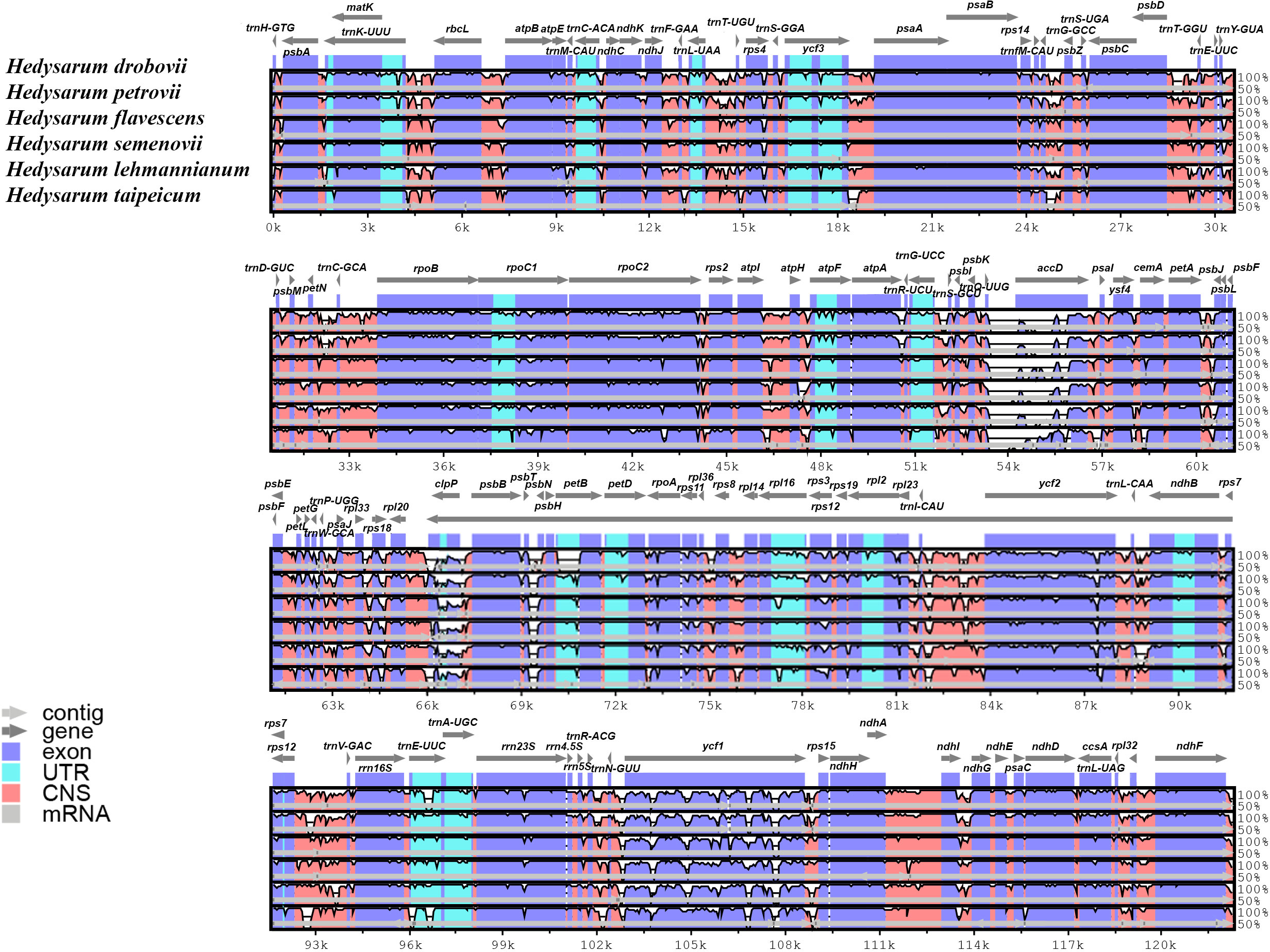
Figure 4 Alignment visualization of the chloroplast genome sequences of six Hedysarum species using mVISTA. Annotated genes are shown along the top. Genomic regions are color-coded to indicate protein-coding regions, exons, UTRs, and CNS. The similarity among the chloroplast genomes is shown on a vertical scale ranging from 50 to 100%.
Seven Hedysarum and seven related cp genome data were analyzed phylogenetically. Phylogenetic reconstructions based on the complete cp genome and protein-coding genes yielded similar results (Figures 5A, B). All clades in both trees were strongly supported by BI, ML, and MP analyses, with 1.00, 100%, and 100% bootstrap values, respectively. Additionally, the results of the phylogenetic analysis based on the complete cp genomes and 76 protein-coding genes showed that Hedysarum was monophyletic. The clade including species of the genus Onobrychis was sister to Hedysarum. The Hedysarum species used in this study were formed into two clades. One is a clade containing H. drobovii and H. petrovii species corresponding to section Multicaulia. The second clade was formed by five Hedysarum species. H. flavesens, H. semenovii, H. taipeicum, and H. polybotrys species were placed into the section Hedysarum, and H. lehmannianum belonged to the section Stracheya, which was sister to the section Hedysarum.
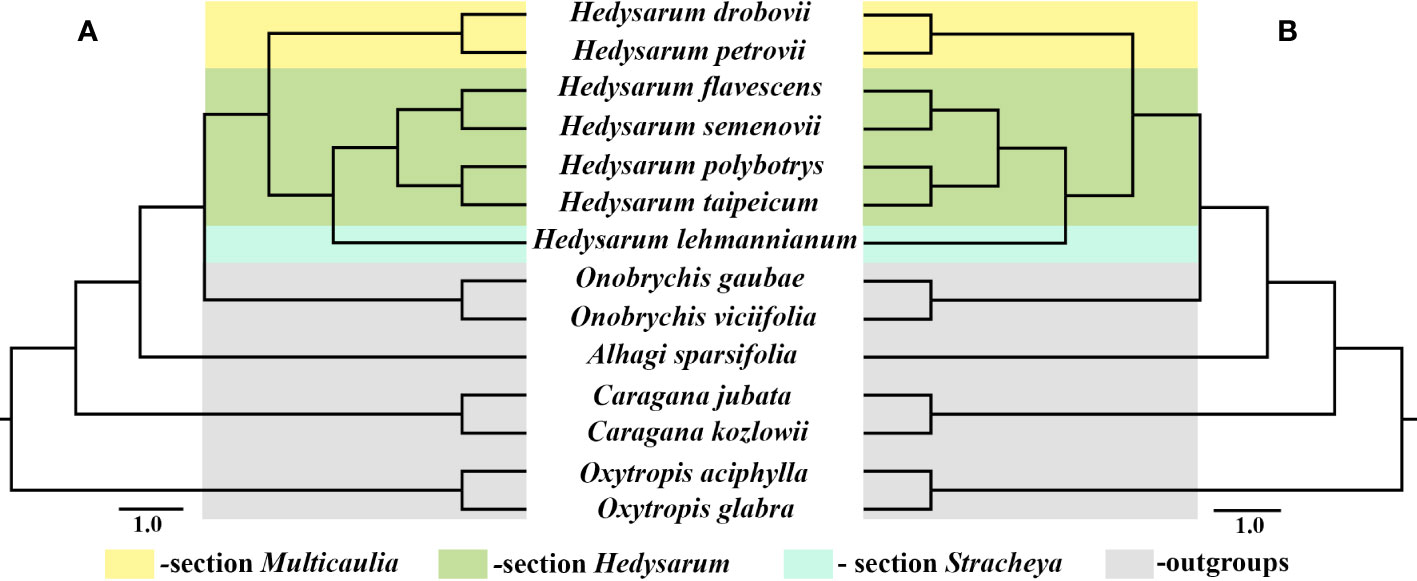
Figure 5 Phylogenetic tree of 14 species including seven Hedysarum species using BI, ML, and MP analyses based on complete cp genomes (A) and their 76 protein-coding genes (B). All branches were maximally supported by BI (1.00), ML (100%), and MP (100%) methods.
This study is the first to comprehensively examine the features of cp genomes in Hedysarum species. We compared the cp genomes of six species belonging to three sections that were distributed in different regions. We sequenced the cp genome of H. drobovii, H. flavescens, and H. lehmannianum for this study. The sizes of the six cp genomes ranged from 121,176 bp (H. drobovii) to 126,738 bp (H. taipeicum). It is worth noting that many related genera with similar cp genome sizes to Hedysarum have been reported in recent years (Tian et al., 2021; Bei et al., 2022; Moghaddam et al., 2022).
The cp genomes of Hedysarum species have 110 genes, including 76 protein-coding genes, 30 transfer RNA genes, and 4 ribosomal RNA genes. The structural composition of Hedysarum cp genomes revealed similarity with other IRLC clade species (Su et al., 2019; Bei et al., 2022; Moghaddam et al., 2022). The cp genomes of six Hedysarum species showed high similarity with regard to genome size (except for H. taipeicum which was 126,738 bp), gene sequences, gene classes, and the lacking IR region. All selected Hedysarum species were found to have lost one copy of the IR region, which was first identified in H. taipeicum by She et al. (2019). This loss of the IR region is common in most species belonging to the subfamily Papilionoideae in the family Fabaceae, forming a clade named the IR-lacking clade (IRLC) (Wojciechowski et al., 2004). The GC content of the six Hedysarum species in this study was highly similar, which is an important indicator of species affinity according to Tamura et al. (2011).
Introns are recognized as being central to the regulation of gene expression in plants and animals (Callis et al., 1987; Emami et al., 2013: Choi et al., 1991). In the present study, 15 genes with one intron and one gene (ycf3) with two introns were identified in each of the cp genomes of the six studied Hedysarum species. Most of the 16 identified genes have a high similarity in the structure of introns. However, a structural change was detected in the intron of the petB and clpP genes of H. drobovii and H. lehmannianum, respectively. The intron of the petB gene in the cp genome of H. drobovii is very short (9 bp); whereas, in the other five species, it ranges from 806 bp (H. flavescens) to 864 bp (H. taipeicum). Similarly, the intron of the clpP gene in the cp genome of H. lehmannianum is 6 bp long, while in other species it is from 159 bp (H flavescens) to 613 bp (H. taipeicum). However, the implications or link between gene expression and short or long introns for Hedysarum have not been studied. Further experimental work on the roles of introns in Hedysarum is therefore essential and should prove interesting. In consonance with previous studies, the trnK-UUU gene in the Hedysarum cp genome was observed to be harboring the largest intron (2407-2503 bp) which includes the matK gene.
The chloroplast SSRs were used in evolutionary studies, phylogenetic relationships, and plant population genetics and species identification as molecular markers (Olmstead and Palmer, 1994; Saski et al., 2005). A total of 172 SSRs (H. taipeicum) to 190 SSRs (H. semenovii) were found in the cp genome of six Hedysarum species. Several studies found that the mononucleotide repeats were dominant among SSRs in the cp genome, where A/T bases account for the majority (Ellegren, 2004; George et al., 2015; Ren et al., 2021). Likewise, A/T mononucleotide repeats were dominant among SSRs in the six Hedysarum cp genomes, ranging from 78.7% to 84.3%. Furthermore, the identified common and unique SSRs might play an important role in the analysis of the genetic diversity of the genus Hedysarum. In particular, the unique pentanucleotide SSRs identified in H. drobovii (AAAGG, AAAAT, AAGAC, ATTTT, and TTGTC) and H. taipeicum (AACCG), as well as the unique hexanucleotide SSRs identified in H. semenovii (ATAGCT and TTTTTC) and H. taipeicum (ATTCTT) may be effectively utilized in the future for species identification and assessment of genetic diversity in their populations. In addition, repeat sequences are known to play an important role in cp genome rearrangement, recombination, gene duplication, deletion, and gene expression (Gemayel et al., 2010; Do et al., 2014; Vieira et al., 2014; Li and Zheng, 2018),. They also have been reported to be responsible for substitutions and indels in the cp genome (Yi et al., 2013). We identified 51 (H. petrovii) to 214 (H. flavescens) repeat sequences among the six Hedysarum cp genomes analyzed, with forward repeats being the most common in H. petrovii, H. flavescens, H. semenovii, H. lehmannianum, and H. taipeicum, whereas palindromic and forward repeats were the most abundant in H. drobovii. Notably, H. drobovii and H. petrovii, both belonging to section Multicaulia, had significantly fewer repeat regions (53 and 51, respectively) compared to the other species (104–214). Further investigation into repeat sequences in section Multicaulia is necessary.
DNA barcodes with high variability are crucial for species identification, resource conservation, and phylogenetic analyses (Gregory, 2005; Bringloe and Saunders, 2019; Chen et al., 2019; Liu et al., 2019). Our comparison of Hedysarum species’ cp genomes revealed high similarity in gene content and gene order, with genome lengths ranging from 121,176 to 126,738 bp. However, mVISTA analyses indicated that sequence variation was higher in non-coding regions than in other regions. Nucleotide diversity analysis identified six highly variable regions in the whole cp genome of Hedysarum species, mainly located in non-coding regions. Three protein-coding genes, clpP, accD, and atpF, exhibited higher Pi values and were found to be highly variable regions. The variability of the clpP gene can be attributed to the large variation in its Exon I length between species, ranging from 3 bp (H. drobovii) to 219 bp (H. taipeicum and H. lehmannianum) (Supplementary Table 2). The clpP and accD genes have been reported to play an important role in counteracting biotic and abiotic stress (Singh et al., 2015; Sinha et al., 2018; Ali and Baek, 2020), while the atpF gene is involved in the synthesis of ATF during photosynthesis (Ghulam et al., 2012), which is greatly influenced by altitude conditions (Wang et al., 2017). The high Pi values observed in these genes may reflect adaptation to different environmental conditions. Moreover, these highly variable regions can serve as candidate molecular markers and a reference for identifying future Hedysarum species. The clpP, accD, and atpF gene exon regions have similarly been identified as some of the most highly variable hotspot regions in cp genomes of some species (Mo et al., 2020; Mascarello et al., 2021; Moghaddam et al., 2022; Long et al., 2023).
Our phylogenetic analysis based on complete cp genomes and protein-coding genes confirmed previous studies on cp genome data of IRLC clade species, determining the phylogenetic position of Hedysarum as a sister to Onobrychis (She et al., 2019; Jin et al., 2021; Moghaddam et al., 2022; Tian et al., 2021). Our study also confirms the monophyly of Hedysarum based on plastid DNA genes, which is consistent with previous studies by Duan et al. (2015); Liu et al. (2017), and Nafisi et al. (2019). Although a limited number of species were used in our study, the phylogenetic relationships among the three sections of Hedysarum were analyzed using complete cp genomes and 76 protein-coding genes for the first time. Our results suggested that sections of Hedysarum could be monophyletic based on both cp genome data. However, further studies with more species, particularly from section Stracheya, are necessary to confirm this outcome. Furthermore, both our phylogenetic reconstructions revealed a close relationship between section Stracheya and Hedysarum, which is consistent with previous findings by Liu et al. (2017), but incongruous with the outcomes of Duan et al. (2015) and Nafisi et al. (2019). Additionally, this relationship is supported by the shared morphological characteristics between species of section Stracheya and Hedysarum, including leaves with numerous leaflets (4-15 paired), wings longer than half of the keel, and pods lacking ribs, bristles, or spines.
Our study is the first research work to investigate the genome characteristics of the genus Hedysarum. We sequenced, assembled, and annotated the cp genome of H. drobovii, H. flavescens va H. lehmannianum using high-throughput technology. Our study is based on cp genome data from a total of six Hedysarum species, including three previously published species. The cp genomes of all six Hedysarum species analyzed contained 110 genes, including 76 protein-coding genes, 4 rRNA genes, and 30 tRNA genes. We identified between 172 and 190 microsatellites and 51 to 214 pairs of repeat sequences among the six Hedysarum species cp genomes. In addition, we identified seven common SSRs and 14 unique SSRs in the studied Hedysarum species. Furthermore, we detected highly variable regions in the clpP, accD, and atpF protein-coding genes. These repeat motifs and highly variable genes could be used for evolutionary studies, phylogenetic relationships, plant population genetics, and species identification. Our phylogenetic reconstructions using the complete cp genome and protein-coding genes confirmed the monophyly of Hedysarum. Additionally, we supported the close relationship between section Stracheya and section Hedysarum using all three BI, ML, and MP methods. However, future studies using more species will provide a better understanding of the relationships among Hedysarum sections.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
IJ: Conceptualization, methodology, data analysis, identification, visualization, writing, original draft preparation, reviewing, editing, and discussing. DM: methodology and data analysis. ZY: methodology and collection. KT: Supervision, investigation, identification, reviewing, editing, and discussing. All authors contributed to the article and approved the submitted version.
This study was supported by grants from the state research project “Taxonomic revision of polymorphic plant families of the flora of Uzbekistan” (FZ-20200929321) and the State Programs for 2021–2025 years “Grid mapping of the flora of Uzbekistan” and the “Tree of life: monocots of Uzbekistan” of the Institute of Botany of the Academy of Sciences of the Republic of Uzbekistan, the National Natural Science Foundation of China (32170215), the International Partnership Program of Chinese Academy of Sciences (151853KYSB20180009), Yunnan Young and Elite Talents Project (YNWRQNBJ-2019-033), the Ten Thousand Talents Program of Yunnan Province (202005AB160005), and the Chinese Academy of Sciences “Light of West China” Program.
The authors thank two reviewers for their comments and suggestions, which greatly improved the article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1211247/full#supplementary-material
Ali, M. S., Baek, K. H. (2020). Protective roles of cytosolic and plastidal proteasomes on abiotic stress and pathogen invasion. Plants 9 (7), 832. doi: 10.3390/plants9070832
Amirahmadi, A., Osaloo, S. K., Moein, F., Kaveh, A., Maassoumi, A. A. (2014). Molecular systematics of the tribe Hedysareae (Fabaceae) based on nrDNA ITS and plastid trnL-F and matK sequences. Plant System. Evol. 300 (4), 729–747. doi: 10.1007/s00606-013-0916-5
Bei, Z., Zhang, L., Tian, X. (2022). Characterization of the complete chloroplast genome of Oxytropis aciphylla Ledeb.(Leguminosae). Mitochondrial DNA Part B. 7 (9), 1756–1757. doi: 10.1080/23802359.2022.2124822
Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33 (16), 2583–2585. doi: 10.1093/bioinformatics/btx198
Bock, R. (2007). “Structure, function, and inheritance of plastid genomes,” in Cell and molecular biology of plastids (Berlin, Heidelberg: Springer Berlin Heidelberg). doi: 10.1007/4735_2007_0223
Bringloe, T. T., Saunders, G. W. (2019). DNA barcoding of the marine macroalgae from Nome, Alaska (Northern Bering Sea) reveals many trans-Arctic. Polar Biol. 42, 851–864. doi: 10.1007/s00300-019-02478-4
Callis, J., Fromm, M., Walbot, V. (1987). Introns increase gene expression in cultured maize cells. Genes Dev. 1, 1183–1200. doi: 10.1101/gad.1.10.1183
Chen, K. C., Zakaria, D., Altarawneh, H., Andrews, G. N., Ganesan, G. S., John, K. M., et al. (2019). DNA barcoding of fish species reveals low rate of package mislabeling in Qatar. Genome 62, 69–76. doi: 10.1139/gen-2018-0101
Choi, T., Huang, M., Gorman, C., Jaenisch, R. A. (1991). Generic intron increases gene expression in transgenic mice. Mol. Cell. Biol. 11, 3070–3074. doi: 10.1128/mcb.11.6.3070-3074.1991
Choi, B. H., Ohashi, H. (1996). Pollen morphology and taxonomy of Hedysarum and its related genera of the tribe Hedysareae (Legiminosae-Papilionoideae). J. Japanese Bot. 71, 191–213.
Choi, B. H., Ohashi, H. (2003). Generic criteria and an infrageneric system for Hedysarum and related genera (Papilionoideae-Leguminosae). Taxon 52, 567–576. doi: 10.2307/3647455
Do, H. D., Kim, J. S., Kim, J. H. (2014). A trnI_CAU triplication event in the complete chloroplast genome of Paris verticillata M. Bieb.(Melanthiaceae, Liliales). Genome Biol. Evol. 6 (7), 1699–1706. doi: 10.1093/gbe/evu138
Dong, Y., Tang, D., Zhang, N., Li, Y., Zhang, C., Li, L., et al. (2013). Phytochemicals and biological studies of plants in genus Hedysarum. Chem. Cent. J. 7 (1), 1–3. doi: 10.1186/1752-153X-7-124
Duan, L., Wen, J., Yang, X., Liu, P. L., Arslan, E., Ertuğrul, K., et al. (2015). Phylogeny of Hedysarum and tribe Hedysareae (Leguminosae: Papilionoideae) inferred from sequence data of ITS, matK, trnL-F and psbA-trnH. Taxon 64 (1), 49–64. doi: 10.12705/641.26
Ellegren, H. (2004). Microsatellites: Simple sequences with complex evolution. Nat. Rev. Genet. 5 (6), 435–445. doi: 10.1038/nrg1348
Emami, S., Arumainayagam, D., Korf, I., Rose, A. B. (2013). The efects of a stimulating intron on the expression of heterologous genes in Arabidopsis thaliana. Plant Biotechnol. J. 11, 555–563. doi: 10.1111/pbi.12043
Fedtschenko, B. A. (1948). Flora of URSS Vol. 13 (St. Petersburg: Academiae Scientiarum URSS, Mosqua-Leningrad Press), 259–319.
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., Dubchak, I. (2004). VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 32, 273–279. doi: 10.1093/nar/gkh458
Gemayel, R., Vinces, M. D., Legendre, M., Verstrepen, K. J. (2010). Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu. Rev. Genet. 1 (44), 445–477. doi: 10.1146/annurev-genet-072610-155046
George, B., Bhatt, B. S., Awasthi, M., George, B., Singh, A. K. (2015). Comparative analysis of microsatellites in chloroplast genomes of lower and higher plants. Curr. Genet. 61 (4), 665–677. doi: 10.1007/s00294-015-0495-9
Ghulam, M. M., Zghidi-Abouzid, O., Lambert, E., Lerbs-Mache, S., Merendino, L. (2012). Transcriptional organization of the large and the small ATP synthase operons, atpI/H/F/A and atpB/E, in Arabidopsis thaliana chloroplasts. Plant Mol. Biol. 79 (3), 259–272. doi: 10.1007/s11103-012-9910-5
Gregory, T. R. (2005). DNA barcoding does not compete with taxonomy. Nature 434, 1067. doi: 10.1038/4341067b
Jin, Z., Jiang, W., Yi, D., Pang, Y. (2021). The complete chloroplast genome sequence of Sainfoin (Onobrychis viciifolia). Mitochondrial DNA Part B. 6 (2), 496–498. doi: 10.1080/23802359.2020.1871439
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., DePamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 1–31. doi: 10.1186/s13059-020-02154-5
Kang, S. H., Lee, J. H., Lee, H. O., Ahn, B. O., Won, S. Y., Sohn, S. H., et al. (2018). Complete chloroplast genome and 45S nrDNA sequences of the medicinal plant species Glycyrrhiza glabra and Glycyrrhiza uralensis. Genes Genet. sys. 93 (3), 83–89. doi: 10.1266/ggs.17-00002
Katoh, K., Standley, D. M.. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30 (4), 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29 (22), 4633–4642. doi: 10.1093/nar/29.22.4633
Li, B., Zheng, Y. (2018). Dynamic evolution and phylogenomic analysis of the chloroplast genome in Schisandraceae. Sci. Rep. 8, 9285. doi: 10.1038/s41598-018-27453-7
Liu, X., Chang, E. M., Liu, J. F., Huang, Y. N., Wang, Y., Yao, N. (2019). Complete chloroplast genome sequence and phylogenetic analysis of Quercus bawanglingensis Huang, Li et Xing, a vulnerable oak tree in China. Forests 10 (7), 587. doi: 10.3390/f10070587
Liu, P. L., Wen, J., Duan, L., Arslan, E., Ertuðrul, K., Chang, Z. Y. (2017). Hedysarum L. (Fabaceae: Hedysareae) is not monophyletic – evidence from phylogenetic analyses based on five nuclear and five plastid sequences. PloS One 12, e0170596. doi: 10.1371/journal.pone.0170596
Lohse, M., Drechsel, O., Bock, R. (2007). OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52, 267–274. doi: 10.1007/s00294-007-0161-y
Long, L., Li, Y., Wang, S., Liu, Z., Wang, J., Yang, M. (2023). Complete chloroplast genomes and comparative analysis of Ligustrum species. Sci. Rep. 13, 212. doi: 10.1038/s41598-022-26884-7
Mascarello, M., Amalfi, M., Asselman, P., Smets, E., Hardy, O. J., Beeckman, H., et al. (2021). Genome skimming reveals novel plastid markers for the molecular identification of illegally logged African timber species. PloS One 16 (6), e0251655. doi: 10.1371/journal.pone.0251655
Minh, B. Q., Lanfear, R., Trifinopoulos, J., Schrempf, D., Schmidt, H. A. (2021) IQ-TREE version 2.1.2: Tutorials and manual phylogenomic software by Maximum Likelihood. Available at: http://www.iqtree.org/doc/iqtree-doc.pdf.
Mo, Z., Lou, W., Chen, Y., Jia, X., Zhai, M., Guo, Z., et al. (2020). The chloroplast genome of Carya illinoinensis: genome structure, adaptive evolution, and phylogenetic analysis. Forests 11 (2), 207. doi: 10.3390/f11020207
Moghaddam, M., Ohta, A., Shimizu, M., Terauchi, R., Kazempour-Osaloo, S. (2022). The complete chloroplast genome of Onobrychis gaubae (Fabaceae-Papilionoideae): comparative analysis with related IR-lacking clade species. BMC Plant Biol. 22 (1), 75. doi: 10.1186/s12870-022-03465-4
Nafisi, H., Kazempour-Osaloo, Sh., Mozaffarian, V., Schneeweiss, G. M. (2019). Molecular phylogeny and divergence times of Hedysarum (Fabaceae) with special reference to section Multicaulia in Southwest Asia. Plant System. Evol. 305 (10), 1001–1017. doi: 10.1007/s00606-019-01620-3
Nazareno, A. G., Carlsen, M., Lohmann, L. G. (2015). Complete chloroplast genome of tanaecium tetragonolobum: the first bignoniaceae plastome. PloS One 10, e0129930. doi: 10.1371/journal.pone.0129930
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32 (1), 268–274. doi: 10.1093/molbev/msu300
Olmstead, R. G., Palmer, J. D. (1994). Chloroplast DNA systematics: a review of methods and data analysis. Am. J. bot. 81 (9), 1205–1224. doi: 10.1002/j.1537-2197.1994.tb15615.x
Ren, F., Wang, L., Li, Y., Zhuo, W., Xu, Z., Guo, H., et al. (2021). Highly variable chloroplast genome from two endangered Papaveraceae lithophytes Corydalis tomentella and Corydalis saxicola. Ecol. Evol. 11 (9), 4158–4171. doi: 10.1002/ece3.7312
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. System. Biol. 61 (3), 539–542. doi: 10.1093/sysbio/sys029
Rozas, J., Ferrer-Mata, J. C., Sanchez-DelBarrio, P., Librado, P., Guirao-Rico, S. E. (2018) DnaSP version 6.12.03: A software for comprehensive analysis of DNA polymorphism data. Available at: http://www.ub.es/dnasp.
Saski, C., Lee, S. B., Daniell, H., Wood, T. C., Tomkins, J., Kim, H. G., et al. (2005). Complete chloroplast genome sequence of Glycine max and comparative analyses with other legume genomes. Plant Mol. Biol. 59, 309–322. doi: 10.1007/s11103-005-8882-0
She, R. X., Li, W. Q., Xie, X. M., Gao, X. X., Wang, L., Liu, P. L., et al. (2019). The complete chloroplast genome sequence of a threatened perennial herb species Taibai sweetvetch (Hedysarum taipeicum KT Fu). Mitochondrial DNA Part B. 4 (1), 1439–1440. doi: 10.1080/23802359.2019.1598817
Singh, R. P., Shelke, G. M., Kumar, A., Jha, P. N. (2015). Biochemistry and genetics of ACC deaminase: a weapon to “stress ethylene” produced in plants. Front. Microbiol. 6. doi: 10.3389/fmicb.2015.00937
Sinha, R., Pal, A. K., Singh, A. K. (2018). Physiological, biochemical and molecular responses of lentil (Lens culinaris Medik.) genotypes under drought stress. Indian J. Plant Physiol. 23, 772–784. doi: 10.1007/s40502-018-0411-7
Smith, D. R., Keeling, P. J. (2015). Mitochondrial and plastid genome architecture: reoccurring themes, but significant differences at the extremes. Proc. Natl. Acad. Sci. U.S.A. 112 (33), 10177–10184. doi: 10.1073/pnas.1422049112
Su, C., Liu, P. L., Chang, Z. Y., Wen, J. (2019). The complete chloroplast genome sequence of Oxytropis bicolor Bunge (Fabaceae). Mitochondrial DNA Part B. 4, 3762–3763. doi: 10.1080/23802359.2019.1682479
Swofford, D. L. (2002). PAUP*. Phylogenetic analysis using parsimony (* and other methods). Version. 4 (Sunderland: Sinauer Associates). doi: 10.1111/j.0014-3820.2002.tb00191.x
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. evol. 28 (10), 2731–2739. doi: 10.1093/molbev/msr121
Tian, C., Li, X., Wu, Z., Li, Z., Hou, X., Li, F. Y. (2021). Characterization and comparative analysis of complete chloroplast genomes of three species from the genus Astragalus (Leguminosae). Front. Genet. 12. doi: 10.3389/fgene.2021.705482
Vieira, L., Faoro, H., Rogalski, M., Fraga, H., Cardoso, R. L. A., de Souza, E. M., et al. (2014). The complete chloroplast genome sequence of Podocarpus lambertii: genome structure, evolutionary aspects, gene content and SSR detection. PloS One 9 (3), e90618. doi: 10.1371/journal.pone.0090618
Wang, H., Prentice, I. C., Davis, T. W., Keenan, T. F., Wright, I. J., Peng, C. (2017). Photosynthetic responses to altitude: an explanation based on optiMality principles. New Phytol. 213 (3), 976–982. doi: 10.1111/nph.14332
Wojciechowski, M. F., Lavin, M., Sanderson, M. J. (2004). A phylogeny of legumes (Leguminosae) based on analysis of the plastid gene resolves many well-supported subclades within the family. Amer J. Bot. 91, 1846–1862. doi: 10.3732/ajb.91.11.1846
Yi, X., Gao, L., Wang, B., Su, Y. J., Wang, T. (2013). The complete chloroplast genome sequence of Cephalotaxus oliveri (Cephalotaxaceae): evolutionary comparison of Cephalotaxus chloroplast DNAs and insights into the loss of inverted repeat copies in gymnosperms. Genome Biol. evol. 5 (4), 688–698. doi: 10.1093/gbe/evt042
Yin, D., Wang, Y., Zhang, X., Ma, X., He, X., Zhang, J. (2017). Development of chloroplast genome resources for peanut (Arachis hypogaea L.) and other species of Arachis. Sci. Rep. 7, 11649. doi: 10.1038/s41598-017-12026-x
Keywords: Hedysarum, chloroplast genome, comparative analysis, phylogeny, protein-coding genes
Citation: Juramurodov I, Makhmudjanov D, Yusupov Z and Tojibaev K (2023) First comparative analysis of complete chloroplast genomes among six Hedysarum (Fabaceae) species. Front. Plant Sci. 14:1211247. doi: 10.3389/fpls.2023.1211247
Received: 24 April 2023; Accepted: 20 July 2023;
Published: 18 August 2023.
Edited by:
Shuangyang Wu, Gregor Mendel Institute of Molecular Plant Biology (GMI), AustriaReviewed by:
Rufeng Wang, Shanghai University of Traditional Chinese Medicine, ChinaCopyright © 2023 Juramurodov, Makhmudjanov, Yusupov and Tojibaev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Inom Juramurodov, aWp1cmFtdXJvZG92QG1haWwucnU=; Komiljon Tojibaev, a3RvamliYWV2QG1haWwucnU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.