
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 17 July 2023
Sec. Functional and Applied Plant Genomics
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1210513
This article is part of the Research Topic Omics and Bioinformatics Approaches to Decipher Plant-Microbe Interactions View all 5 articles
 Hongyuan Zhang1
Hongyuan Zhang1 Hao Chen1
Hao Chen1 Jie Tan1Shuping Huang1
Jie Tan1Shuping Huang1 Xia Chen1Hongxia Dong1
Xia Chen1Hongxia Dong1 Ru Zhang2Yikui Wang3Benqi Wang1
Ru Zhang2Yikui Wang3Benqi Wang1 Xueqiong Xiao4
Xueqiong Xiao4 Zonglie Hong5
Zonglie Hong5 Junhong Zhang6*
Junhong Zhang6* Jihong Hu2*Min Zhang1*
Jihong Hu2*Min Zhang1*Solanum torvum (Swartz) (2n = 24) is a wild Solanaceae plant with high economic value that is used as a rootstock in grafting for Solanaceae plants to improve the resistance to a soil-borne disease caused by root-knot nematodes (RKNs). However, the lack of a high-quality reference genome of S. torvum hinders research on the genetic basis for disease resistance and application in horticulture. Herein, we present a chromosome-level assembly of genomic sequences for S. torvum combining PacBio long reads (HiFi reads), Illumina short reads and Hi-C scaffolding technology. The assembled genome size is ~1.25 Gb with a contig N50 and scaffold N50 of 38.65 Mb and 103.02 Mb, respectively as well as a BUSCO estimate of 98%. GO enrichment and KEGG pathway analysis of the unique S. torvum genes, including NLR and ABC transporters, revealed that they were involved in disease resistance processes. RNA-seq data also confirmed that 48 NLR genes were highly expressed in roots and fibrous roots and that three homologous NLR genes (Sto0288260.1, Sto0201960.1 and Sto0265490.1) in S. torvum were significantly upregulated after RKN infection. Two ABC transporters, ABCB9 and ABCB11 were identified as the hub genes in response to RKN infection. The chromosome-scale reference genome of the S. torvum will provide insights into RKN resistance.
Solanum torvum (Swartz) (2n = 24) is a wild Solanaceae plant with high economic value, and its roots, leaves, stems and fruits can be used as medicine for the relief of swelling and pain (Govindaraju et al., 2010). S. torvum, commonly known as turkey berry, is distributed in most pantropical areas, including Pakistan, India, China, and tropical America. Eggplant (Solanum melongena L.) is a vegetable crop species belonging to the Solanaceae family, which includes economically important species such as tomato (S. lycopersicum L.), potato (S. tuberosum L.), pepper (Capsicum annuum L.), and tobacco (Nicotiana tabacum L.) (Hirakawa et al., 2014). Root-knot nematodes (RKNs, Meloidogyne spp.) are destructive endoparasites that mainly parasitize a variety of cultivated plants. In many countries, the production of eggplant is seriously threatened by RKNs. Once infected with RKNs, large knots form in the roots of the host plant. They eventually lead to giant cells that are multinucleated and hypertrophic cells that are a source of nutrients for nematodes to grow, so that the surrounding root tissue cannot obtain enough nutrients for them to grow (Ling et al., 2017). There is evidence that S. torvum, is resistant to serious soil borne diseases caused by bacteria, fungal wilt and RKNs (Bagnaresi et al., 2013; Zhou et al., 2018). Therefore, S. torvum is often used as rootstock in the grafting of solanaceous plants to improve the resistance to soil borne diseases caused by RKNs (Gousset et al., 2005).
The ban on highly toxic nematicides makes it urgent to need environmentally friendly and pesticide free control methods, and the breeding of RKNs resistant varieties seems to be a promising alternative. Therefore, the identification of nematode resistance (Nem-R) may be of great importance for eggplant cultivation. Using genome-specific satellite markers and chromosome breakpoint analysis, the Hs1 (pro-1) gene with resistance to the beet cyst nematode (Heterodera schachtii Schmidt) was cloned from sugar beet (Beta vulgaris L.) (Cai et al., 1997). Nem-R genes cloned from tomato or potato relatives, including Mi-1, Hero A, Gpa2 and Gro1-4, are very similar to known plant R-genes in their domains and belong to NBS-LRR like R-genes (Williamson and Kumar, 2006). With regard to the subclass of RKN R-genes (RKN-R), almost no resistance genes were found, and only Mi-1 from tomato and Ma from Myrobalan plum were cloned. The Mi-1 gene with resistance to RKN was cloned from tomato by bacterial artificial chromosome cloning and isolation. This gene encodes a protein sharing structural characteristics with leucine rich repetitive plant resistance genes (Milligan et al., 1998; Vos et al., 1998). The Ma gene of RKN resistance has full spectrum and heat stable resistance to Meloidogvne. In contrast to Mi-I of tomato, the latter has more limited spectrum and lower efficiency under high temperature (Claverie et al., 2004). Pepper resistance to RKNs is controlled by the Me gene, which is thought to play an independent role in gene interactions (Djian-Caporalino et al., 2001; Gisbert et al., 2013). To identify the defense mechanism of RKNs in Solanaceae, some studies compared the transcriptional characteristics of eggplant and S. torvum, and found that many plant hormone related genes and transcription factors, such as MYB, WRKY and NAC, were differentially expressed at different time points after infection with RKNs (Zhang et al., 2021).
NLR (nucleotide-binding and leucine rich repeat) gene belongs to one of the largest gene families in plants. Most of the plant resistance genes (R-genes) are characterized by molecules belonging to the NLR receptor family, and are prone to duplication and translocation, with high sequence diversity (Chovelon et al., 2021). Most NLRs share a common biological structural basis, including a highly conserved nucleic acid binding domain at the N-terminal (nucleotide binding, NB) and a highly variable leucine rich repeat at the C-terminal (leucine rich repeat, LRR) (Zheng et al., 2022). These large and abundant proteins are mainly involved in the detection of various pathogens and many NLR genes only show activated expression under pathogen invasion, although some NLR members still show tissue/space specific expression in wild type plants (McHale et al., 2006). In wild eggplant Solanum aculeatissimum, a nucleotide binding site rich repeat (NBS-LRR) resistance gene, SacMi was isolated and characterized. Silencing of SacMi enhanced the susceptibility of S. aculatissimum to RKNs, indicating that SacMi may participate in the resistance to RKNs (Zhou et al., 2018).
Although it is very important for other solanaceous crops, especially eggplant, to identify the resistance of S. torvum to RKN, there is little research on the resistance mechanism of S. torvum at present. The main reason for this phenomenon is the lack of high-quality S. torvum genome. Combined genomic and transcriptome analysis could elucidate the possible mechanism underlying the RKN disease resistance (Hu et al., 2022; Zhang et al., 2022; Zhao et al., 2022). Therefore, it is necessary to obtain a high-quality chromosome-scale genome of S. torvum and study the mechanism for RKN resistance of S. torvum at the whole-genome level.
In this study, we sequenced and assembled the whole genome of S. torvum using Illumina paired-end reads, PacBio sequencing, and Hi-C technology. We structurally and functionally annotated the genome with clustered gene families related to diverse biological processes. Our work provides a valuable resource for revealing the evolution of S. torvum and for understanding the mechanism of resistance to RKNs.
Solanum torvum (Swartz) was planted in the field of the Wuhan Vegetable Research Institute, Wuhan Academy of Agricultural Sciences (30.82°N, 114.37°E), Wuhan, China. The tender leaves at the seedling stage were collected for further experiments. Leaves, roots, stems, fibrous roots, buds, flowers and fruits were harvested at the corresponding stage, immediately frozen in liquid nitrogen, and stored at –80°C until use.
High-quality genomic DNA was extracted from tender leaves of S. torvum using a Qiagen DNeasy Plant Mini Kit. For third-generation genome sequencing, the SMRT Bell library was prepared using the SMRTbell Express Template Prep Kit 2.0, which was performed using SMRT sequencing on a PacBio Sequel system following the manufacturer’s standard protocol. To estimate genome size, the Illumina DNA libraries were constructed using the Next Ultra DNA library prep kit (NEB) and sequenced on the HiSeq X Ten platform (Illumina, San Diego, USA).
Before genome assembly, the read information obtained by sequencing to estimate genome features was subjected to 17-mer analysis to estimate the genome size. We assembled the S. torvum genome based on PacBio subreads, Illumina short reads, and Hi-C data using a hierarchical method. The longer PacBio subreads were de novo assembled by Canu v1.5 (Koren et al., 2017). All the PacBio reads were then mapped to the previously assembled contigs using minimap2 (Li, 2016) and further corrected with arrow software. To increase the accuracy of assembly, all the filtered Illumina reads were mapped to the corrected contigs with BWA-mem (Li, 2013) and further corrected by Pilon (Walker et al., 2014) to correct indel errors associated with homopolymer repeats in the PacBio data. Hi-C is a technology derived from chromosome conformation capture technology that utilizes high-throughput data and is mainly used to assist in genome assembly. To anchor scaffolds onto pseudochromosomes, HiCUP v0.6.1 (Wingett et al., 2015) was used to map and process the reads from the Hi-C library. For Hi-C analysis, the raw reads were trimmed to obtain clean Hi-C reads. Then, the obtained clean reads were compared with the preassembled contigs using Juicer (Durand et al., 2016). After filtering the results and removing the misaligned reads, 3D-DNA software was used to preliminarily cluster, sequence, and direct the pseudochromosomes (Dudchenko et al., 2017). Juicer-box was used to adjust, reset, and cluster the pseudochromosomes to improve the chromosome assembly quality. For the evaluation of the Hi-C assembly results, the final pseudochromosome assemblies were divided into 500 kb bins of equal lengths, and a heatmap was used to visualize the interaction signals generated by the valid mapped read pairs between each bin.
To assess genome assembly quality, the Benchmarking Universal Single-Copy Orthologs v3 (BUSCO) tool (http://busco.ezlab.org/) (Simão et al., 2015) was used with single-copy orthologous genes. Genome integrity is assessed using Long Terminal Repeat through LTR_FINDER and LTR_retriever software, which is finally represented by the LAI index.
Repetitive sequences are an important part of a genome and are divided into two types, namely, tandem repeats and interspersed repeats. Two methods, do novo prediction and homology-based search, were used to annotate repeat sequences in the genome. Tandem repeats in genome sequences were found by TRF (Benson, 1999). RepeatMasker v4.1.3 (Tarailo-Graovac and Chen, 2009) and RepeatProteinMask v4.1.3 (http://www.repeatmasker.org) were used to identify repetitive sequences based on the Repbase database (Jurka et al., 2005). For de novo prediction, the final sequence file obtained by RepeatModeler v2.0.3 and LTR-FINDER (Xu and Wang, 2007) was used as the library, and the genome sequence was annotated through RepeatMaker software to obtain repetitive sequences. Finally, after integrating the above methods and removing redundancy, it will be used as the result of the last repetitive sequences.
The annotation of high-quality protein-coding genes was carried out by integrating de novo, homology-based and transcriptome-based predictions. AUGUSTUS v2.4 (Nachtweide and Stanke, 2019) and GlimmerHMM v3.04 (Majoros et al., 2004) was used for ab initio prediction. For homology-based prediction, protein sequences from six species (S. torvum, S.pennellii, S.tuberosum, S.lycopersicum, C.annuum, N.sylvestris) were analyzed using tblastn with a cutoffe-value of 1e-5, and aligned against the contents of Swiss-Prot, TrEMBL (http://www.UniProt.org/), NR and so on. The RNA-seq data of S. torvum were mapped to genome sequences through HISAT2 v2.0.4 (Kim et al., 2015) and StringTie v2.0.4 (Pertea et al., 2015). Finally, the MAKER package v2.31.878 (Cantarel et al., 2008) was used to annotate and integrate the results generated by the above methods.
For non-coding RNAs annotations, tRNAscan-SE (Lowe and Eddy, 1997) was used to annotate transfer RNA (tRNA) sequences. BLASTN (Kent, 2002) was used to search for ribosomal RNAs (rRNAs) and microRNAs (miRNAs), and snRNA sequences were predicted by Infernal 1.1 (http://eddylab.org/infernal/) against the Rfam database (Gardner et al., 2011) using default parameters.
Homological relationships among the protein sequences derived from 10 plants (S.torvum, S.melongena, S.tuberosum, S.aethiopicum, C.annuum, S.lycopersicum, A.thaliana, N.tabacum, P.inflata, O.sativa) were determined by BLAST analysis. The distribution of orthologous gene families from the 10 different species was identified using the OrthoMCL package v2.0.9 (Li et al., 2003). After gene family clustering, we aligned all high-quality single-copy orthologous gene protein sequences using MUSCLE v3.8.31 (Edgar, 2004) and constructed a phylogenetic tree using PhyML v3.1 (http://www.atgc-montpellier.fr/phyml/versions.php). CAFE v4.2 (Han et al., 2013) was used to calculate the number of gene families amplified and contracted on each phylogenetic tree branch based on divergence time and gene family clustering.
We used the synonymous substitution rate (Ks) to infer the occurrence of WGD events. First, BLASTP was used to search for putative paralogous and orthologous genes by aligning the genomes of S. lycopersicum, S. torvum, and S. melongena. Then, syntenic blocks within and among species were identified using MCScanX (Wang et al., 2012). Subsequently, Ks values of each gene pair were calculated with KaKs_Calculator 2.0 (Wang et al., 2010), and four-fold synonymous third-codon transversion (4DTv) rate was determined using a Perl script (https://github.com/JinfengChen/Scripts). The WGD events in the S. torvum genome were determined by plotting the distribution frequency of the 4DTv values.
Total RNA was isolated from different tissues (leaf, stem, bud, flower, fruit, fibrous root and root) of S. torvum (Swartz) with three biological replicates using TRIzol reagent (Tiangen, Beijing, China). Approximately 2 μg of high-quality RNA per sample was used for sequencing library construction as previous described (Zhang et al., 2021). The 150 bp paired-end sequencing was performed on the Illumina HiSeq 4000 platform (Illumina, San Diego, USA). The transcriptome sequencing data were also used for genomic gene prediction and annotation. We used fastp (Chen et al., 2018) to remove adaptors from raw reads and filter out low-quality reads. Clean reads were compared to our assembled S.torvum genome using HISAT2, and transcripts were analyzed using Stringtie v2.0.4. After comparing the existing annotations with cuffcompare v2.2.1, new transcripts were obtained, and new transcripts with protein coding potential confirmed by CPC2 (Kang et al., 2017) were added to the gene set. Finally, the gene set was quantified through RSEM. The differentially expressed genes (DEGs) were identified with the DESeq2 R package v3.11 (Love et al., 2014) based on |log2 (fold-change)|≥1 and adjusted P value (P < 0.05).
A weighted gene co-expression network of differentially expressed genes was constructed using the WGCNA package in R (Langfelder and Horvath, 2008). An unsupervised co-expression relationship was built based on the adjacency matrix which represents the network connection strength between gene pairs. The one-step network construction option with a soft-thresholding power value of 10, min module size = 30 and merge cut height = 0.25 were used. The other parameters were set to default values. Highly similar modules were subsequently identified by clustering and then merged together to new modules on the bias of eigengenes. The correlation of each module was also analyzed and visualized by a heatmap. Then, the co-expression network was visualized by Cytoscape software (Smoot et al., 2011).
Solanum torvum is a shrub and its roots can resist to RKNs (Figure 1A).Using k-mer analysis (k = 17), we estimated that the S. torvum genome size is approximately 1, 185 Mb (Supplementary Figure S1). A total of 148.60 Gb of PacBio single-molecule long reads and 118.95 Gb of Illumina paired-end clean reads were generated for initial assembly. The original sketch length of the S. torvum genome sequence is 1.25 Gb with 309 contigs. To anchor the scaffolds to chromosomes, we constructed high-throughput chromosome conformation capture (Hi-C) libraries of S. torvum, generating 136.27 Gb clean Hi-C reads (Figure 1B). After Hi-C assisted genome assembly, the genome sequence and direction of S. torvum were finally determined. The genome was assembled, and approximately 98.80% of the contigs were anchored into the 12 pseudo-chromosomes, generating chromosome-level sequences of 1.25 Gb with a contig N50 and scaffold N50 of 38.65 Mb and 103.02 Mb, respectively, which significant higher than those of eggplant (Table 1; Supplementary Tables 1, S2).
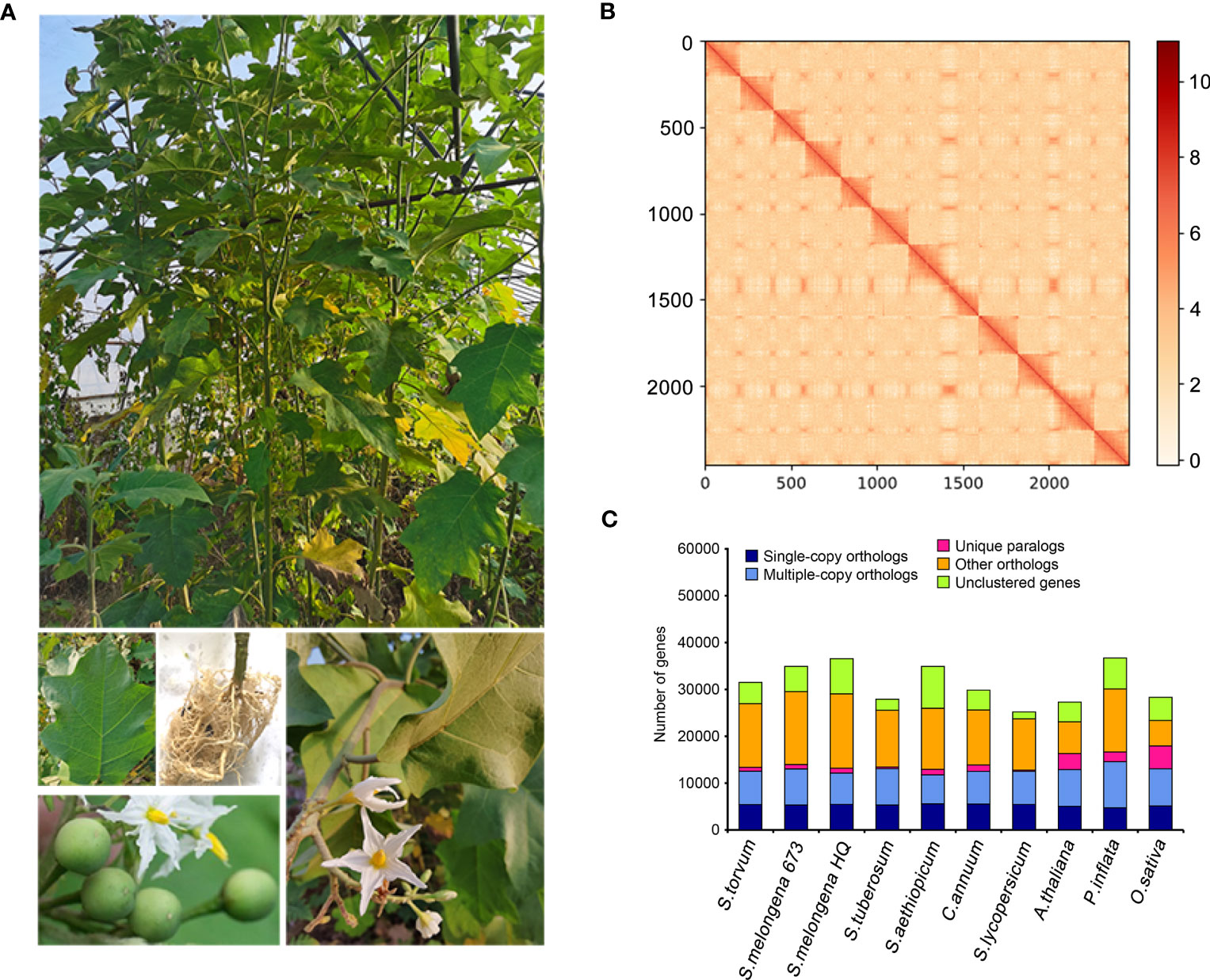
Figure 1 (A) Morphological characteristics of S. torvum. (B) Interchromosomal Hi-C contact map of S. torvum. (C) The distribution of orthologous genes in different species.
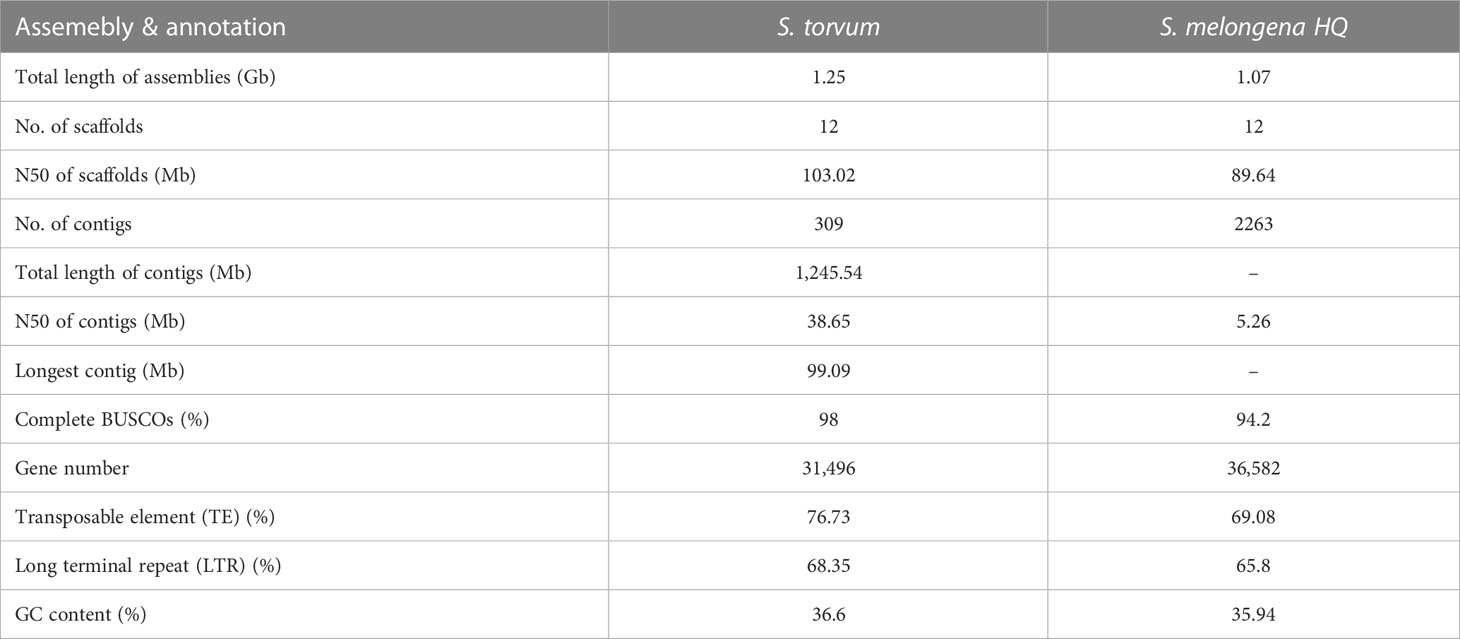
Table 1 Global statistics of the Solanum torvum genome assembly and annotation compared with Solanum melongena HQ.
BUSCO analysis showed that the completeness of the S. torvum genome was 98% with low redundancy (3.3% complete and duplicated BUSCOs) and the long terminal repeat (LTR) assembly index (LAI) had a high score of 10.47 (Supplementary Table S3). Moreover, to evaluate the sequence consistency, the software BWA was used to compare the Illumina paired-end clean reads to the assembled genome, and the paired mapping rate and coverage reached 98.60% and 99.68%, respectively. In addition, 17,714 gene families were constructed with 517 single-copy genes identified among the 11 species (Figure 1C). Therefore, these results indicated that the genome assembly is of high quality with high coverage (Figure 2).
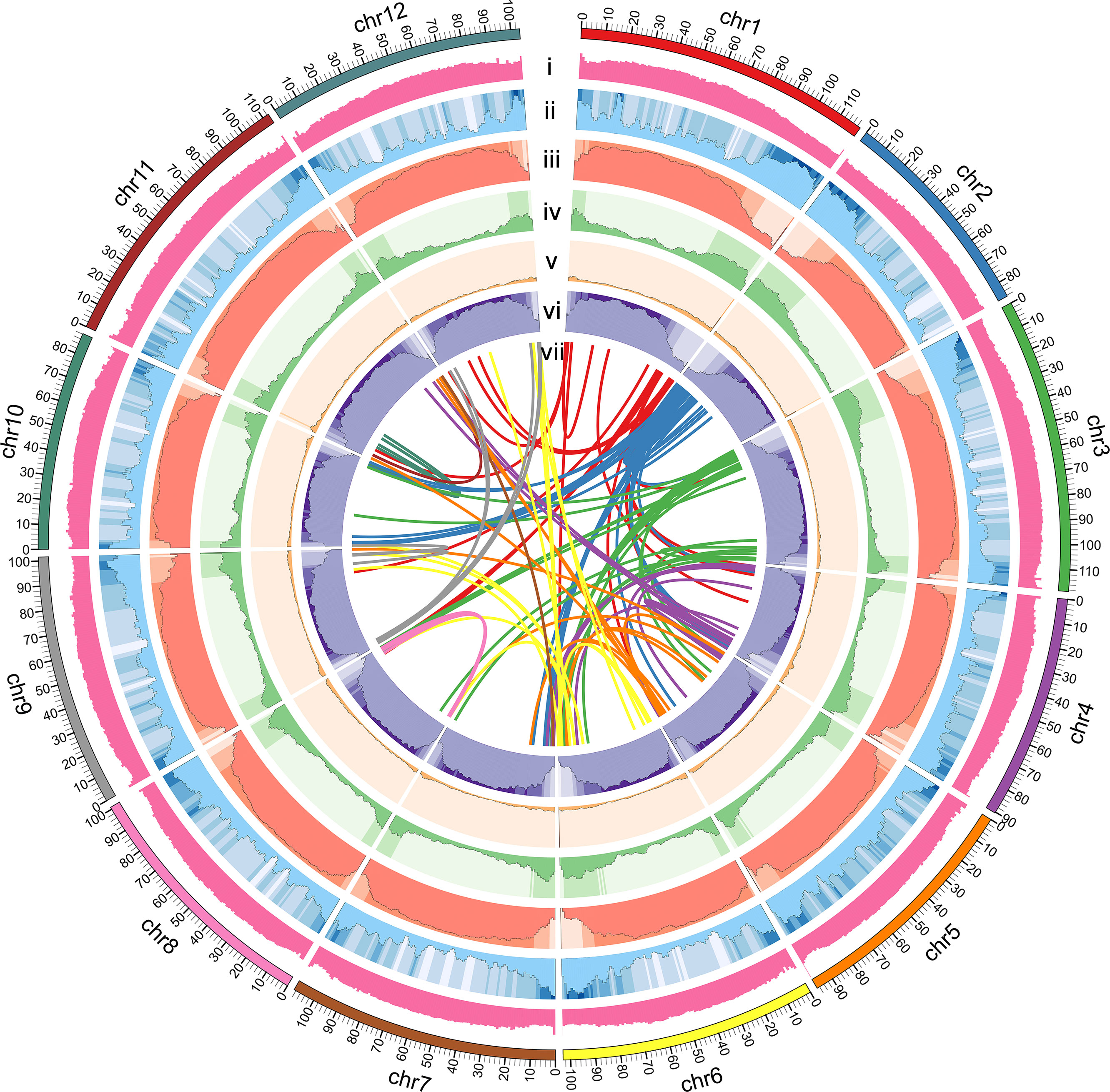
Figure 2 Circular map of S. torvum genome. (i) GC content, (ii) gene density, (iii) repeat content, (iv) DNA Transposon, (v) LINE, (vi) LTR content, and (vii) synteny relations.
The repetitive sequences of the S. torvum genome were annotated by combining de novo prediction and homology-based searches at the DNA and protein levels, identifying 955.75 Mb of repeated sequences accounting for 76.73% of the assembled genome, which is slightly higher than that of eggplant (Li et al., 2021) (Supplementary Figure S2 and Table S4). Transposable elements (TEs) are the largest single component of most eukaryotic genetic materials, and play an important role in shaping eukaryotic genomes and promoting their evolution (Feschotte et al., 2002). In S. torvum, TEs accounted for 76.73% of the genome size, with long-terminal repeat (LTR) retrotransposons accounting for 68.35% of the genome as the most common type of transposon elements (Figure 2 and Supplementary Table S4). As another major class of transposable elements, DNA transposons account for 5.17% (Supplementary Figure S2 and Table S4). The proportions of TEs and LTRs were slightly higher than those in eggplant (Table 1) (Wei et al., 2020; Li et al., 2021).
A total of 31,496 high-quality protein-coding genes were annotated in the S. torvum genome, of which 27,719 (88.01%) were further detected by RNA sequencing (RNA-seq) in seven tissues (Supplementary Tables S5, S6). The number of protein-coding genes was comparable with that of eggplant (36,582 genes), tomato (35,768 genes), potato (39,028 genes) and pepper (34,903 genes), indicating that the number of genes in these related species is similar (Xu et al., 2011; Sato et al., 2012; Kim et al., 2014; Wei et al., 2020) (Table 1).
Comparative analysis of the genomes among the five (S. aethiopicum, S. torvum, S. melongena, S. melongena and S. tuberosum) and four (S. torvum, S. melongena, S. melongena and S. tuberosum) Solanum species identified a total of 12,856 and 13,958 gene orthologs, respectively (Supplementary Figure S3). We further assessed the quality of these annotated genes by comparing them with several closely related species (S. pennellii, S. tuberosum, S. lycopersicum, C. annuum and N. sylvestris) (Supplementary Figure S4). The accuracy of these predicted genes assessed by comparing gene features including the distribution of mRNA length, CDS length, exon length, intron length and the exon number, showed similar distribution patterns, revealing high confidence gene models (Supplementary Figure S4). Analysis of gene functional annotations using the InterPro, GO, KEGG, SwissProt, TrEMBL, and NR databases revealed that most of the genes of S. torvum have close homologs from other organisms in the public databases (Supplementary Table S6). Noncoding RNAs of S. torvum, including 192 microRNAs and 1561 tRNAs, were also identified (Supplementary Table S7).
The expanded and contracted gene families of S. torvum were compared with those of nine other plant species with O. sativa as an outgroup. Phylogenetic analysis showed that S. torvum diverged from the common ancestor of S. torvum, S. melongena and S. aethiopicum for 33.6 million years ago (Mya), with a confidence interval ranging from 20.8 to 48.3 Mya (Figure 3). Moreover, 368 gene families in S. torvum underwent expansion, including the ATP-binding cassette (ABC) transporter and nucleotide-binding leucine-rich repeat (NLR) gene families, while 707 gene families were contracted (Figure 4A). GO enrichment analysis of these unique S. torvum gene families showed that they participate in a series of biological processes, such as purine nucleotide metabolic processes and chitin metabolic processes, which may explain why they are resistant to RKNs (Zhou et al., 2020). KEGG pathway analysis revealed that these expanded genes were involved in plant-pathogen interaction, diterpenoid biosynthesis, monoterpenoid biosynthesis and phenylpropanoid biosynthesis (Supplementary Table S8). These pathways were reported in response to pathogen infection, suggesting their possible roles in the resistance to RKNs in S. torvum (Kyndt et al., 2012). Comparative genome analysis revealed that 26 genes were positively selected in the S. torvum genome (Supplementary Table S9).
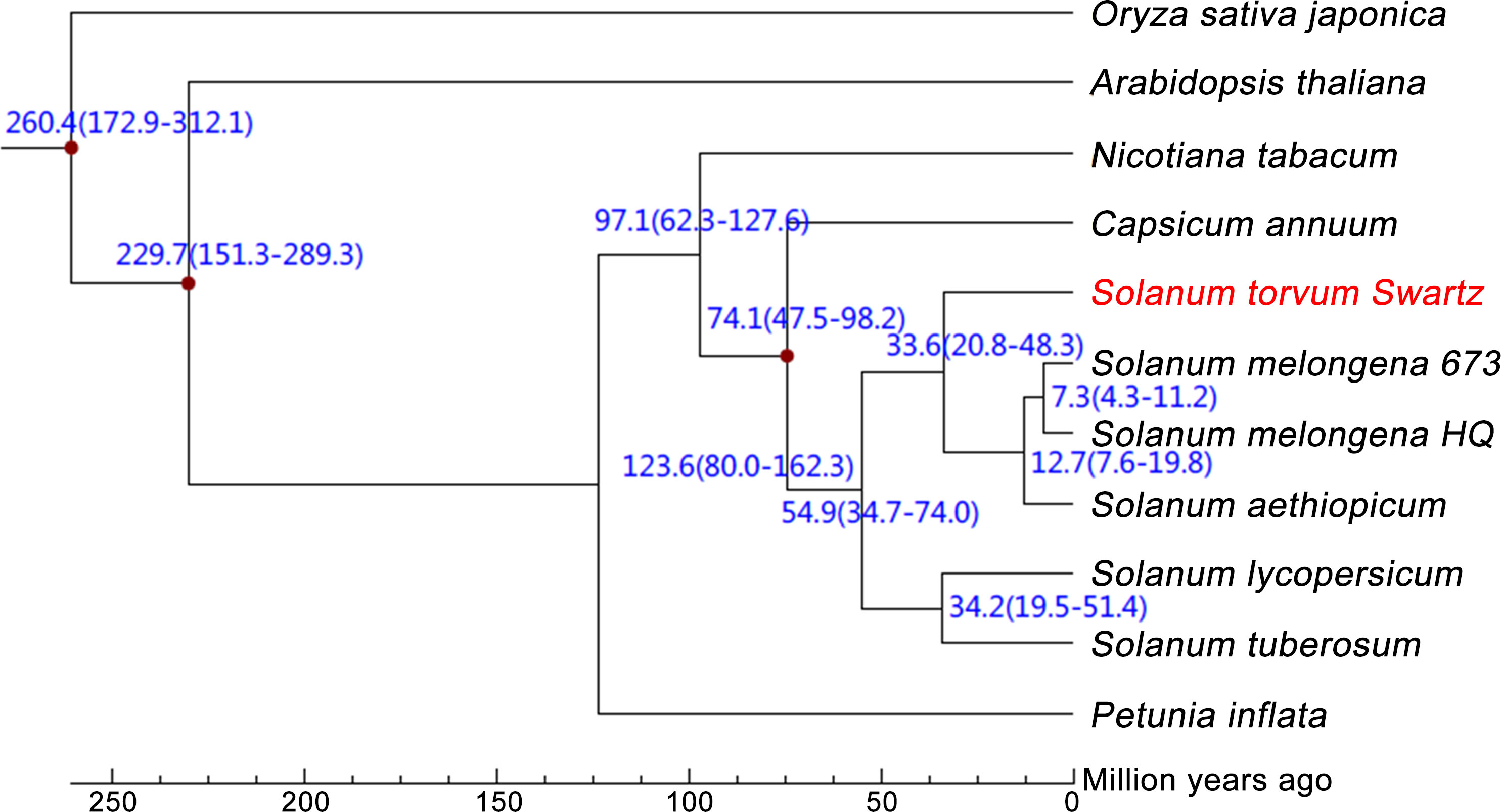
Figure 3 Phylogenetic tree showing divergence times based on ten sequenced plant genomes with O. sativa as an outgroup.
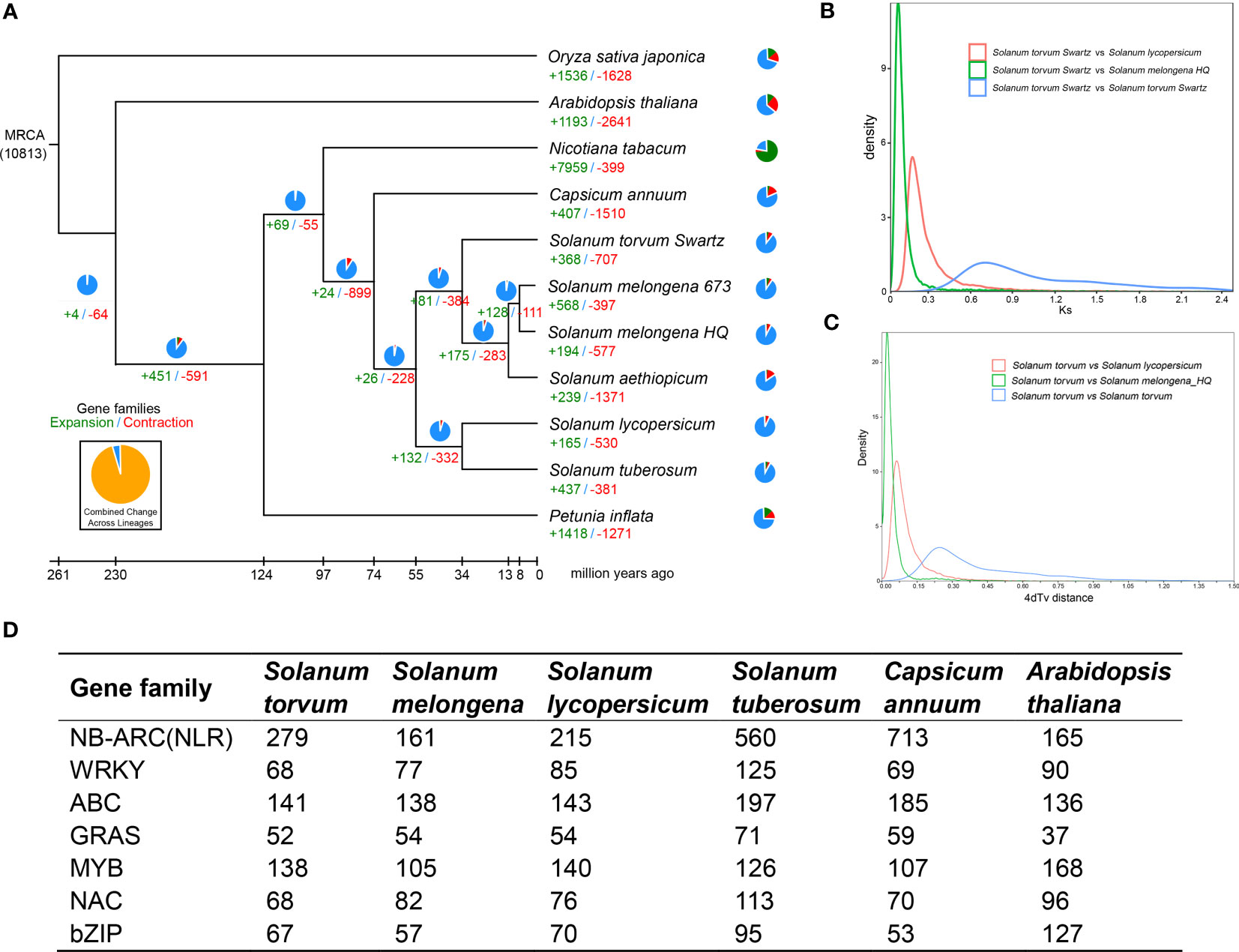
Figure 4 (A) Phylogenetic tree showing divergence times and the evolution of gene families in S. torvum. (B) Density distribution of Ks for paralogous gene pairs of the three Solanaceae genomes. (C) Age distribution of 4dTv values for the homologous gene pairs between the Solanaceae genomes. (D) Comparison of the number of different gene families between S. torvum and other species.
Gene replication provides evolutionary potential for generating new functions. WGD doubles are strongly supported by the evidence common in many species-rich lineages of eukaryotes, so they are considered as the main driving force of species diversity (Song et al., 2012; Ren et al., 2018). To determine the occurrence of WGD events in S. torvum, the distributions of synonymous substitution rates (Ks) and distance-transversion rates (4DTv) of paralogous gene pairs were estimated to shape a WGD event in S. torvum (Figures 4B, C). The divergence of these three species is consistent with the phylogenetic analysis (Figure 4A), confirming the accuracy of the present results (Li et al., 2021). Comparison of the gene families among different species in Solanaceae revealed some genes were expanded in S. torvum (Figure 4D). Population history analysis showed that the effective population size of S. torvum expanded along with that of S. melongena and then declined to a stable level, suggesting a possible concerted evolution (Supplementary Figure S5). Genome collinearity analysis of the three Solanum species indicated that some chromosomes were conserved, but chromosomes 3, 4, 5 and chromosomes 10, 11, 12 contained a large percentage of collinear regions among S. lycopersicum, S. torvum and S. melongena (Figure 5).
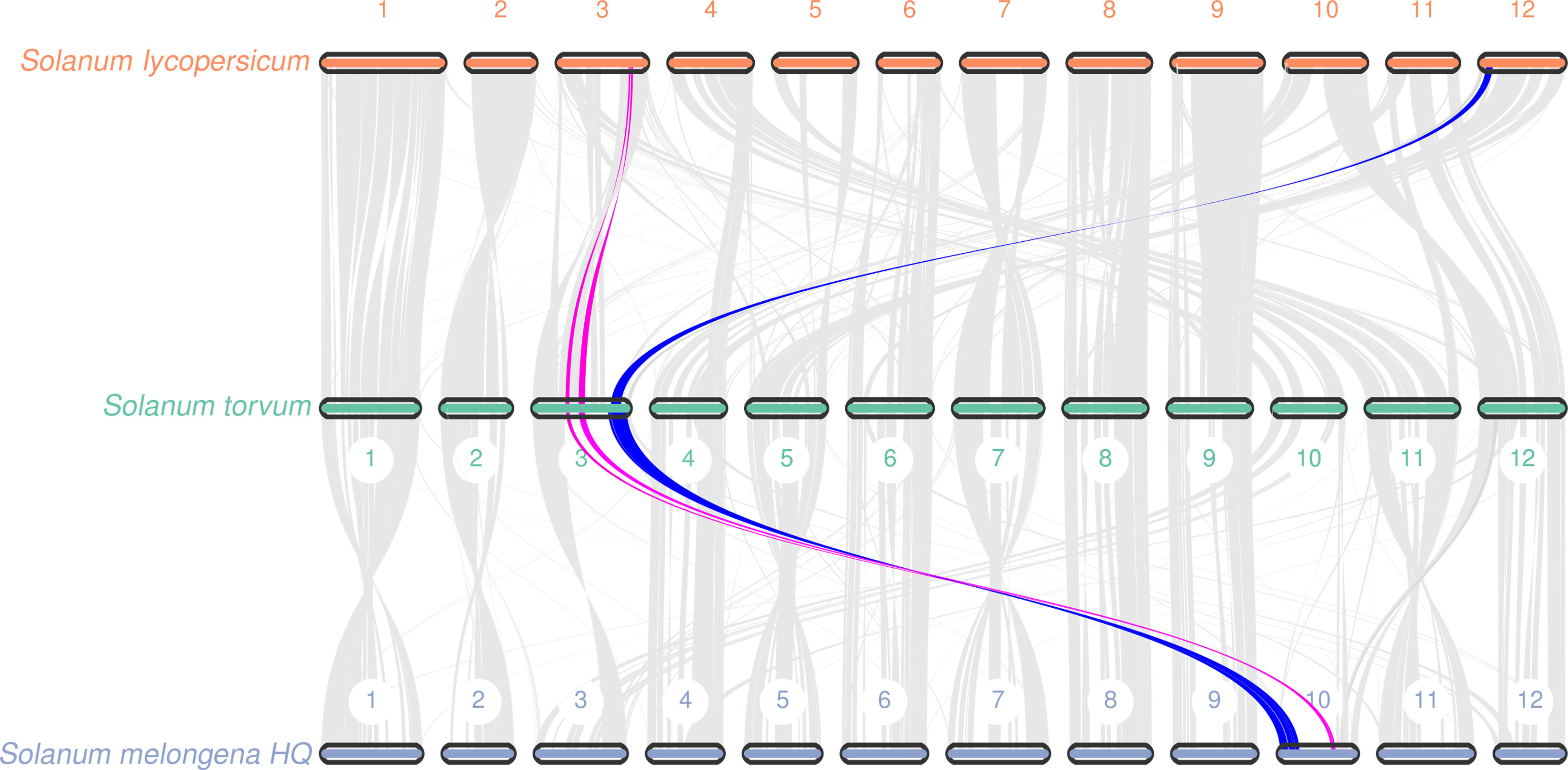
Figure 5 Collinear relationship at the chromosome level among S. torvum, S. lycopersicum and S. melongena. The red and blue lines showed the collinearity of genomic sequences on the chromosome 3 with S. lycopersicum and S. melongena.
Many RKN resistance genes in tomato, pepper and other plants have been identified to as NLR genes containing NB-ARC domains (Milligan et al., 1998). In this study, we detected dramatic expansions of 279 NLR genes within 26 gene clusters and 68 WRKY transcription factors, compared with those in S. melongena (Figure 4D; Supplementary Figures S6-S9 and Tables S10, S11), suggesting their roles in disease resistance. The NLR gene was mainly located at both ends of the S. torvum chromosome, and most of them were tandem repeats, which indicates that NLR has undergone genetic amplification during evolution, which is consistent with previous research results (Zhang et al., 2014). RNA-seq data also confirmed that all the three subgroups (TNL, CNL and PN) of 48 NLR genes were highly expressed in roots and fibrous roots (Figure 6A and Supplementary Figure S9), indicating that the NLR genes may be involved in the resistance to RKNs in S. torvum. Compared with eggplant in response to RKN infection (Zhang et al., 2021), three homologous NLR genes (Sto0288260.1, Sto0201960.1 and Sto0265490.1) in S. torvum were significantly upregulated after RKN infection (Figure 6B).
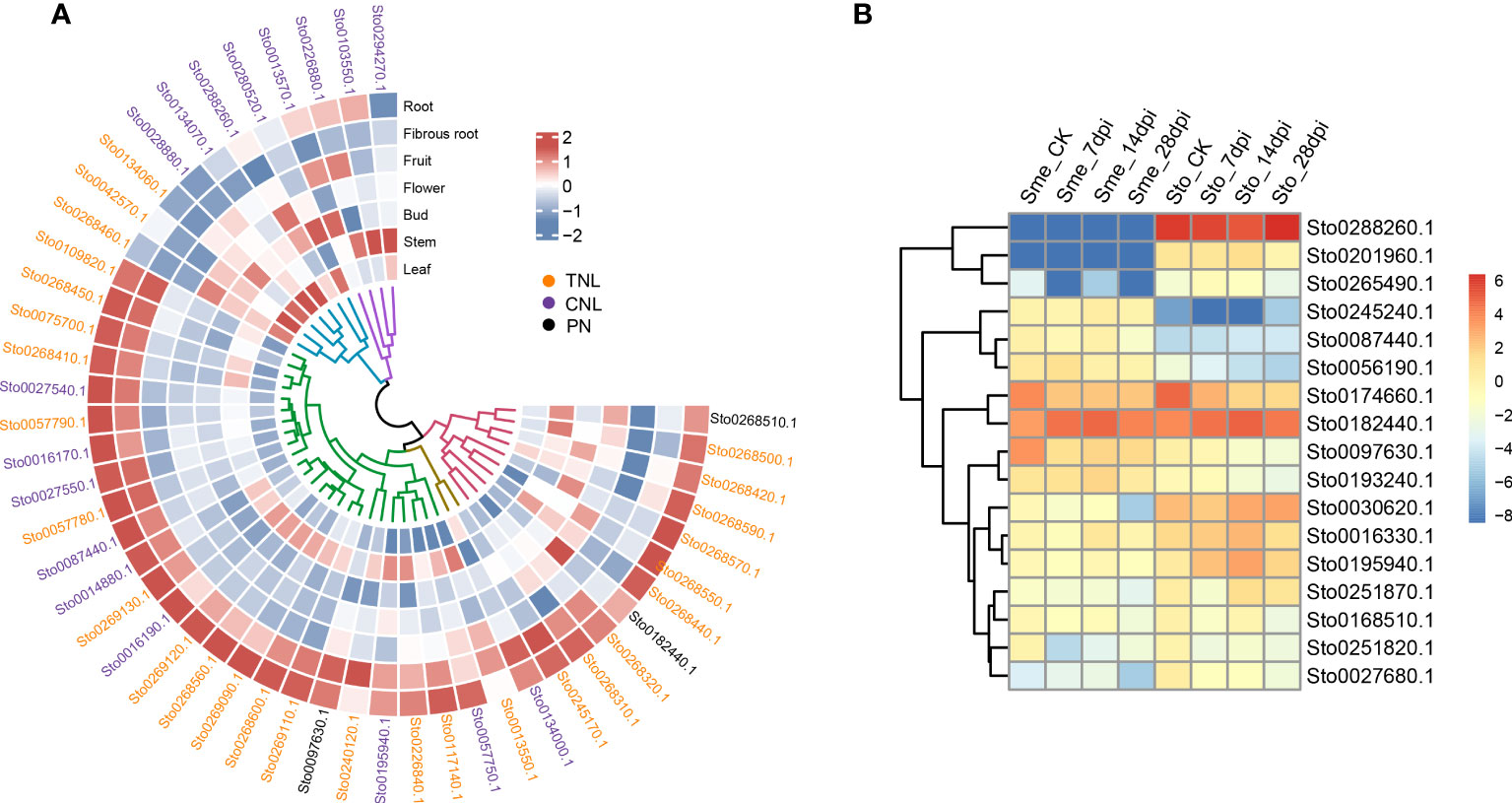
Figure 6 (A) Heatmap showing the expression patterns of 48 NLR genes. Expression levels with log2(FPKM). (B) The expression patterns of NLR genes in S. torvum (Sto) and their homologous genes in S. melongena (Sme) in response to root-knot nematodes (RKNs).
A weighted gene co-expression network based on differentially expressed genes (DEGs) from RNA-seq data revealed that the genes in three modules (“blue”, “midnightblue” and “saddlebrown”) were significantly associated with RKN infection (Figure 7A). In our study, the S. torvum unique expansion genes contained 25 ABC transporter genes (ABCB subfamily), which may participate in RKN resistance (Figure 7B and Supplementary Table S8). In the “saddlebrown” module at 28 days post-inoculation (dpi), two ABCB genes, ABCB9 (Sto0189000.1) and ABCB11 (Sto0286710.1) were identified as the hub genes that interact with several disease resistance genes in response to RKN infection (Figure 7C). Particularly, ABCB11 was one of the unique ABC transporter genes identified in S. torvum, implying its important role in RKN resistance (Figures 7B, C). The expression patterns of the expanded genes in different pathways were also showed that some of these genes were highly expressed in the root, including the 25 unique ABC transporter genes (Figure 8). In addition, the expression levels of the homologous genes in eggplant and S. torvum indicated WRKY and ABC gene families related to RKN resistance (Figure 9). Thus, some metabolite synthesis pathways and the unique NLR genes and ABC transporters may be involved in the resistance to RKNs in S. torvum.
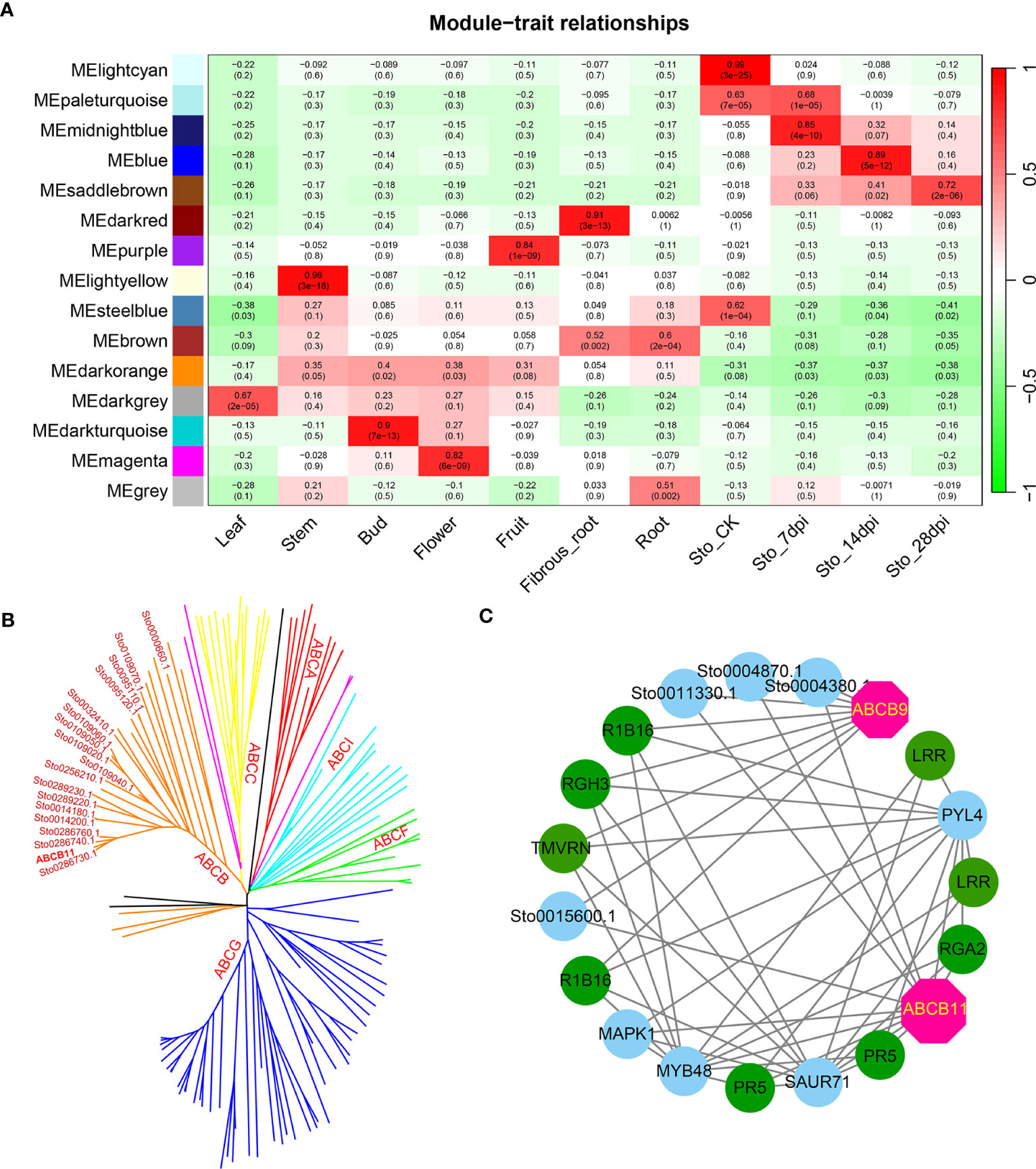
Figure 7 Co-expression network in different tissues and roots after RKN infection in S. torvum constructed by WGCNA. (A) The module-trait relationships showing the modules correlated with RKN infection. (B) Phylogenetic relationship of 139 ABC transporters in S. torvum. The unique expansion genes are labeled in red. (C) The subnetwork of the “saddlebrown” module with the hub genes in pink.
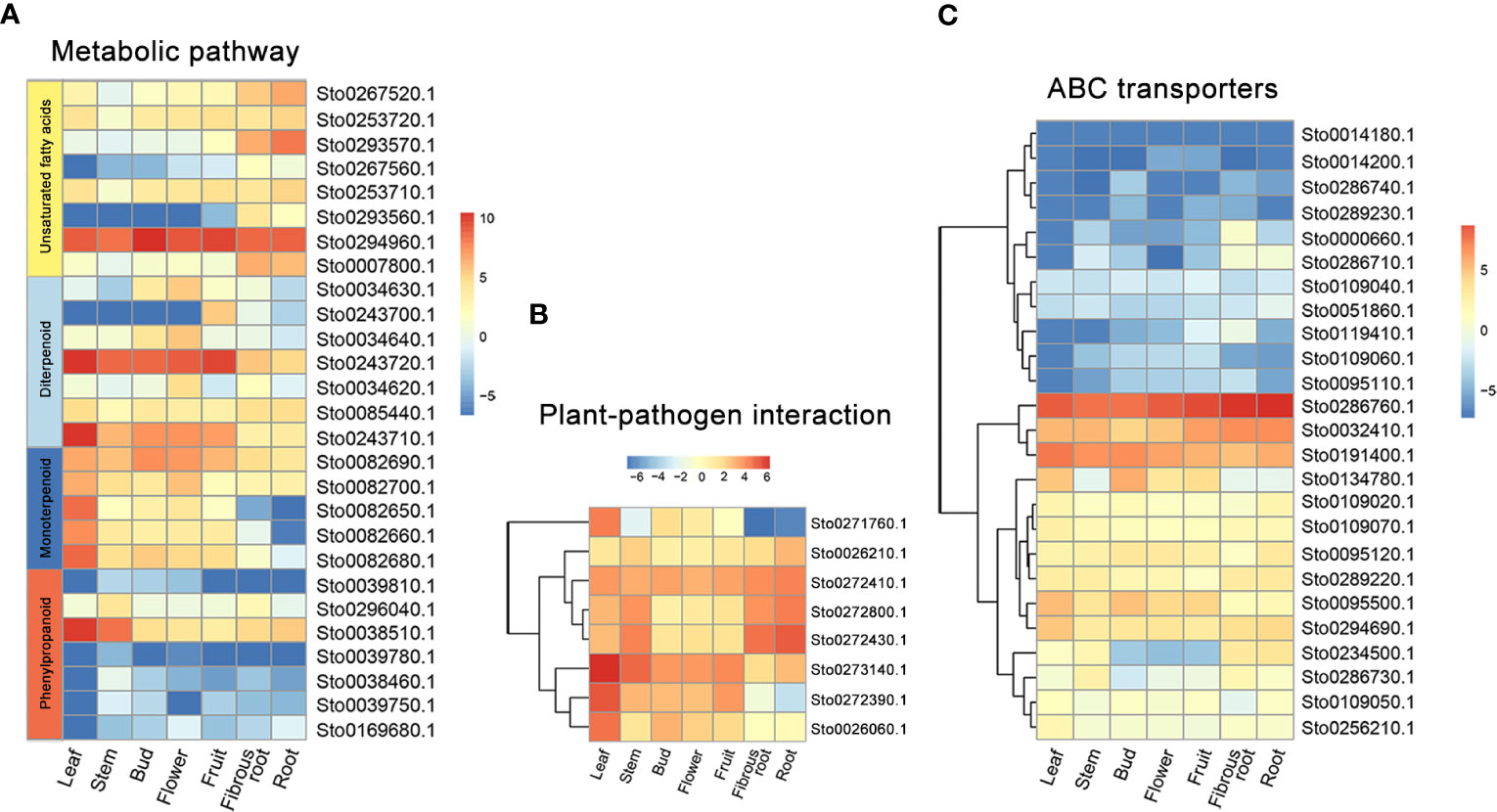
Figure 8 The expression patterns of specific expanded genes in different tissues of S torvum. (A) Genes involved in metabolic pathway. (B) Genes for plant-pathogen interaction. (C) ABC transporter genes.
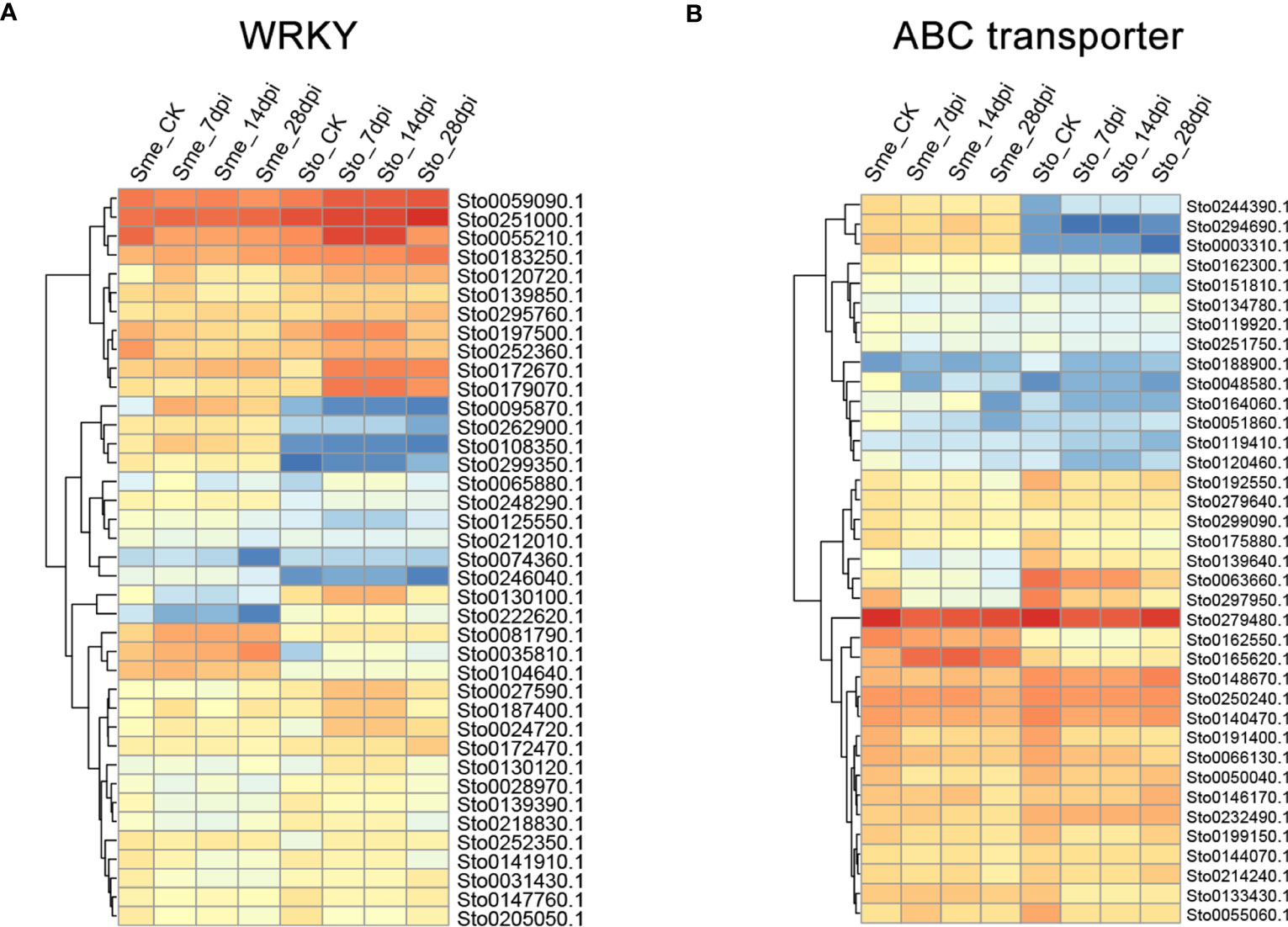
Figure 9 The expression patterns of WRKY (A) and ABC transporter genes (B) in S. torvum (Sto) and their homologous genes in S. melongena (Sme) in response to root-knot nematodes (RKNs).
Chlorogenic acid (CGA) is the phenolic metabolite of the phenylpropanoid pathway in several solanaceous plants, and plays an important role in plant defense (Plazas et al., 2013). In this study, some of the expanded genes in S. torvum were involved in phenylpropanoid biosynthesis, and genes associated with CGA biosynthesis were highly expressed in root tissues (Figure 10), which is consistent with other solanaceous plants, including eggplant and tomato (Li et al., 2021). However, the expression patterns of these genes were higher in eggplant than those in S. torvum after RKN infection (Supplementary Figure S10 and Table S12). Thus, CGA might not be involved in the resistance of S. torvum to RKNs.
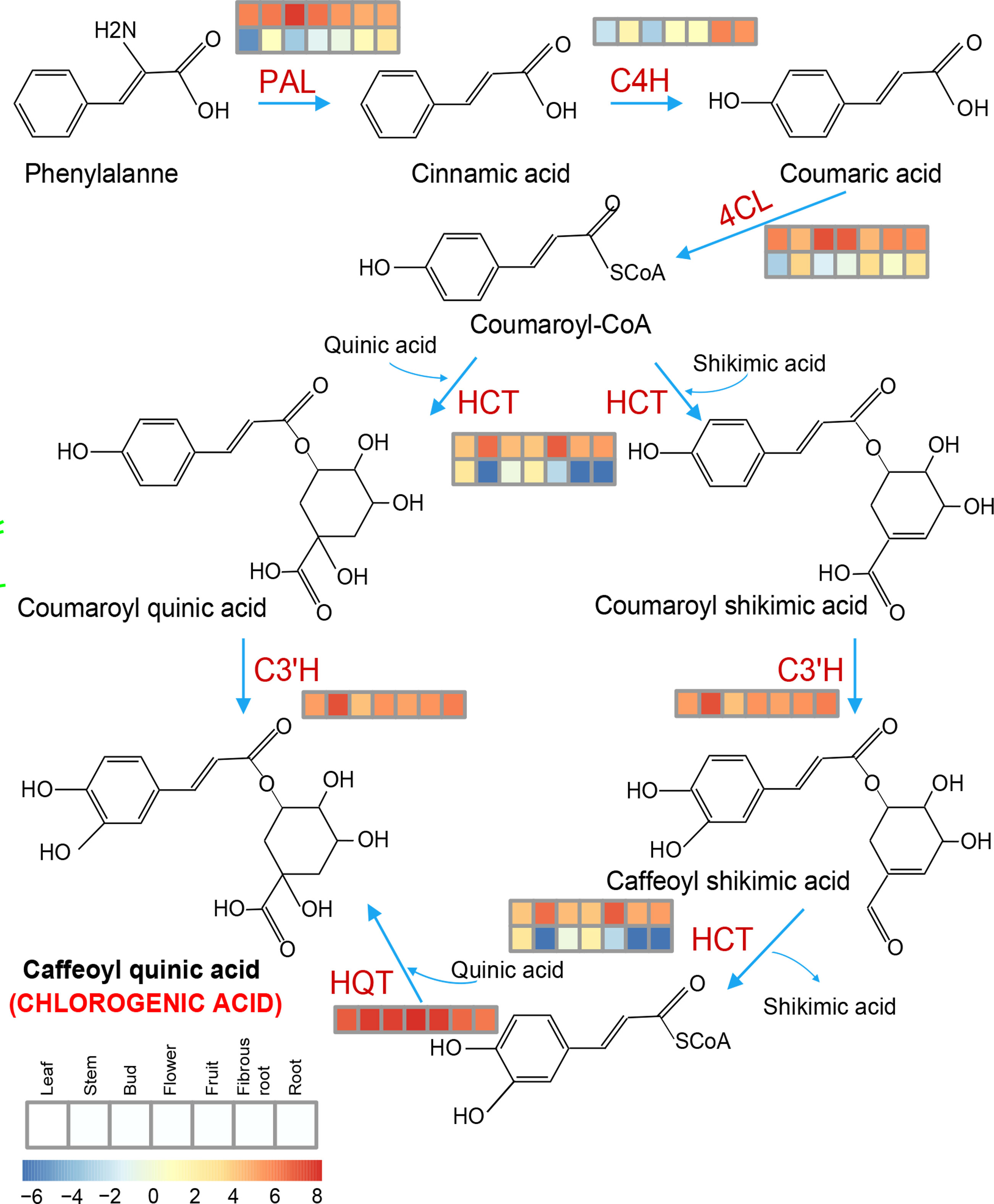
Figure 10 Key genes involved in the chlorogenic acid (CGA) synthesis pathway and their expression patterns in the different tissues of S. torvum. Expression levels with log2(FPKM).
Solanum torvum (Turkey berry) is widely used as an important folk medicinal plant in tropical and subtropical countries and is also used as a rootstock in grafting for Solanaceae plants to improve the resistance to a soil-borne disease caused by RKNs (Gousset et al., 2005). RKN is one of the common pathogens for eggplant (Solanum melongena) and utilizes water and nutrients from the host eggplant, resulting in root and shoot growth retardation. Thus, Turkey berry can provide a valuable genetic resource for eggplant improvement (Sato et al., 2021). Herein, we present a chromosome-level assembly of genomic sequences for S. torvum, which will provide insights into understanding RKN resistance in Solanaceae plants.
The genome of S. torvum was assembled with the three different datasets of genomic DNA sequences and approximately 98.80% of the contigs were anchored into the 12 pseudo-chromosomes with an LAI of 10.47, indicating the high quality of the assembly (Figure 2 and Supplementary Table S3). The annotated genes in S. torvum were comparable to those in other related species, indicating the high quality of prediction (Supplementary Figure S4). Genome collinearity analysis also revealed the conservation of genome sequences among the related species in Solanum genus. These results indicated that the assembled chromosome-level genome is highly complete and contiguous. Furthermore, comparison of the expanded and contracted gene families in S. torvum revealed that 368 gene families have been underwent expansion, including the NLR and ABC transporter gene families (Figure 4A). GO enrichment and KEGG pathway analysis of the S. torvum unique gene families suggested that they were involved in purine nucleotide metabolic process, chitin metabolic process and plant-pathogen interaction (Kyndt et al., 2012).
RKNs are one of the major pathogens of solanaceous crops and decrease root and shoot growth, resulting in considerable economic damage to crop production. In solanaceous crops, R-genes have been identified to confer resistance to RKNs and nearly all the R-genes reported were mapped to a collinear cluster of NLR genes within solanaceous genomes (Rutter et al., 2022). The tomato Mi-1 gene that belongs to CNL type NLR gene, confers resistance to both RKNs and potato aphids (Vos et al., 1998). And Mi1.2 gene that encode an NLR from tomato has been reported to confer resistance to M. incognita and M. arenaria (Milligan et al., 1998). In Prunus, a NLR gene Ma with TNL structure was reported for complete-spectrum resistance to Meloidogyne (Claverie et al., 2011). In our study, a total of 279 NLR genes along to the three subgroups (TNL, CNL and PN) were detected with 45 of TNL type, which is more number than eggplant (Supplementary Figure S7). Compared with the eggplant in response to RKN infection (Zhang et al., 2021), three homologous NLR genes (Sto0288260.1, Sto0201960.1 and Sto0265490.1) in S. torvum were significantly highly expressed after RKN infection (Figure 6B). And Sto0288260.1 is a CNL type NLR gene with significantly up-regulated in after RKN infection (Figure 6B), indicating the important role for RKN resistance. Furthermore, 48 NLR genes were highly expressed in roots and fibrous roots (Figure 6), indicating that the NLR genes play important roles in the resistance to RKNs in S. torvum. Therefore, further study of these NLR genes in S. torvum or eggplant might be elucidate their roles in resistance to RKNs.
In plants, ABC transporters have been reported to modulate resistance genes by improving the disease resistance of RKNs (Fujimoto et al., 2015). In this study, 25 ABCB genes were uniquely identified in S. torvum, indicating their roles in RKN resistance (Figure 7 and Supplementary Table S8). Furthermore, the WGCNA revealed that two ABCB genes, ABCB9 (Sto0189000.1) and ABCB11 (Sto0286710.1) were identified as hub genes in response to RKN infection (Figure 7C). In rice, the ABC transporter OsPDR1 has been reported to regulate plant growth and pathogen resistance via affecting jasmonate biosynthesis (Zhang et al., 2020). And FgABCC9 is required for fungicide resistance and pathogenicity toward wheat (Qi et al., 2018). In particular, knockdown of ABC-C6 can inhibit egg hatching of Meloidogyne incognita in tomato (Cox et al., 2019). Thus, combining genome and transcriptome analyses revealed that the ABCB11 gene which is a unique expansion gene of the S. torvum, plays an important role in the RKN resistance.
Chlorogenic acid (CGA) is a phenolic metabolite that is involved in plant defense (Plazas et al., 2013). In this study, the genes associated with CGA biosynthesis were highly expressed in root tissues, but the expression patterns of these genes were higher in eggplant than those in turkey berry after RKN infection, indicating that CGA might not be involved in resistance to RKNs in S. torvum (Figure 10 and Supplementary S12). Thus, the induced resistance of S. torvum to RKNs could be caused via increasing secondary metabolite synthesis and the expression levels of unique NLR genes and ABC transporters.
In this study, the high-quality genome assembly of S. torvum provides some new insights into the nematode resistance and lays a foundation for better elucidating the evolution and diversification of Solanaceae. Through gene prediction and annotation, 31,496 high-quality protein genes were annotated, and 27,719 (88.01%) of them were further detected by RNA-seq of seven tissues. A total of 368 gene families underwent expansion, including the NLR and ABC transporter gene families, and 707 gene families were contracted in the S. torvum. Integration of genomic and transcriptomic analyses revealed that secondary metabolite synthesis and the expression levels of unique NLR genes and ABC transporters may be caused to RKN resistance in S. torvum.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
MZ, JH and JZ conceived and supervised the research. HZ, HC, MZ, XX, JT, SH, XC, HD and BW performed the experiments. HZ, JH, RZ, HD, and YW performed bioinformatics analysis. HZ, JH, MZ, ZH and JZ wrote and revise the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Wuhan Major Project of Key Technologies in Biological Breeding and New Variety Cultivation (2022021302024852), Key research and development project of Hubei Province Department of Science and Technology (2022BBA0060), and Wuhan Academy of Agricultural Sciences Innovation Project (XKCX202301-4).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1210513/full#supplementary-material
Bagnaresi, P., Sala, T., Irdani, T., Scotto, C., Lamontanara, A., Beretta, M., et al. (2013). Solanum torvum responses to the root-knot nematode meloidogyne incognita. BMC Genomics 14, 540. doi: 10.1186/1471-2164-14-540
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Cai, D., Kleine, M., Kifle, S., Harloff, H. J., Sandal, N. N., Marcker, K. A., et al. (1997). Positional cloning of a gene for nematode resistance in sugar beet. Science 275, 832–834. doi: 10.1126/science.275.5301.832
Cantarel, B. L., Korf, I., Robb, S. M., Parra, G., Ross, E. J., Moore, B., et al. (2008). MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196. doi: 10.1101/gr.6743907
Chen, S., Zhou, Y., Chen, Y., Gu, J. (2018). Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Chovelon, V., Feriche-Linares, R., Barreau, G., Chadoeuf, J., Callot, C., Gautier, V., et al. (2021). Building a cluster of NLR genes conferring resistance to pests and pathogens: the story of the vat gene cluster in cucurbits. Hortic. Res. 8, 72. doi: 10.1038/s41438-021-00507-0
Claverie, M., Dirlewanger, E., Bosselut, N., Ghelder, C., Voisin, R., Kleinhentz, M., et al. (2011). The Ma gene for complete-spectrum resistance to Meloidogyne species in Prunus is a TNL with a huge repeated c-terminal post-LRR region. Plant Physiol. 156, 779–792. doi: 10.1104/pp.111.176230
Claverie, M., Dirlewanger, E., Cosson, P., Bosselut, N., Lecouls, A. C., Voisin, R., et al. (2004). High-resolution mapping and chromosome landing at the root-know nematode resistance locus ma from myrobalan plum using a large-insert BAC DNA library. Theor. Appl. Genet. 109, 1318–1327. doi: 10.1007/s00122-004-1749-y
Cox, D. E., Dyer, S., Weir, R., Cheseto, X., Sturrock, M., Coyne, D., et al. (2019). ABC transporter genes ABC-C6 and ABC-G33 alter plant-microbe-parasite interactions in the rhizosphere. Sci. Rep. 9, 19899. doi: 10.1038/s41598-019-56493-w
Djian-Caporalino, C., Pijarowski, L., Fazari, A., Samson, M., Gaveau, L., O’Byrne, C., et al. (2001). High-resolution genetic mapping of the pepper (Capsicum annuum l.) resistance loci Me3 and Me4 conferring heat-stable resistance to root-knot nematodes (Meloidogyne spp.). TAG Theor. Appl. Genet. 103, 592–600. doi: 10.1007/pl00002914
Dudchenko, O., Batra, S. S., Omer, A. D., Nyquist, S. K., Hoeger, M., Durand, N. C., et al. (2017). De novo assembly of the aedes aegypti genome using Hi-c yields chromosome-length scaffolds. Science 356, 92–95. doi: 10.1126/science.aal3327
Durand, N. C., Shamim, M. S., Machol, I., Rao, S. S., Huntley, M. H., Lander, E. S., et al. (2016). Juicer provides a one-click system for analyzing loop-resolution Hi-c experiments. Cell Syst. 3, 95–98. doi: 10.1016/j.cels.2016.07.002
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Feschotte, C., Jiang, N., Wessler, S. R. (2002). Plant transposable elements: where genetics meets genomics. Nat. Rev. Genet. 3, 329–341. doi: 10.1038/nrg793
Fujimoto, T., Mizukubo, T., Abe, H., Seo, S. (2015). Sclareol induces plant resistance to root-knot nematode partially through ethylene-dependent enhancement of lignin accumulation. Mol. Plant Microbe Interact. 28, 398–407. doi: 10.1094/MPMI-10-14-0320-R
Gardner, P. P., Daub, J., Tate, J., Moore, B. L., Osuch, I. H., Griffiths-Jones, S., et al. (2011). Rfam: Wikipedia, clans and the "decimal" release. Nucleic Acids Res. 39, D141–D145. doi: 10.1093/nar/gkq1129
Gisbert, C., Trujillo-Moya, C., Sánchez-Torres, P., Sifres, A., Sánchez-Castro, E., Nuez, F. (2013). Resistance of pepper germplasm to meloidogyne incognita. Ann. Appl. Biol. 162, 110–118. doi: 10.1111/aab.12006
Gousset, C., Collonnier, C., Mulya, K., Mariska, I., Rotino, G. L., Besse, P., et al. (2005). Solanum torvum, as a useful source of resistance against bacterial and fungal diseases for improvement of eggplant (S. melongena l.). Plant Sci. 168, 319–327. doi: 10.1016/j.plantsci.2004.07.034
Govindaraju, K., Tamilselvan, S., Kiruthiga, V., Singaravelu, G. (2010). Biogenic silver nanoparticles by Solanum torvum and their promising antimicrobial activity. J. Biopestic 3, 394–399.
Han, M. V., Thomas, G. W., Lugo-Martinez, J., Hahn, M. W. (2013). Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 30, 1987–1997. doi: 10.1093/molbev/mst100
Hirakawa, H., Shirasawa, K., Miyatake, K., Nunome, T., Negoro, S., Ohyama, A., et al. (2014). Draft genome sequence of eggplant (Solanum melongena l.): the representative solanum species indigenous to the old world. DNA Res. 21, 649–660. doi: 10.1093/dnares/dsu027
Hu, J. H., Chen, B. Y., Zhao, J., Zhang, F. G., Xie, T., Xu, K., et al. (2022). Genomic selection and genetic architecture of agronomic traits during modern rapeseed breeding. Nat. Genet. 54, 694–704. doi: 10.1038/s41588-022-01055-6
Jurka, J., Kapitonov, V. V., Pavlicek, A., Klonowski, P., Kohany, O., Walichiewicz, J. (2005). Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467. doi: 10.1159/000084979
Kang, Y. J., Yang, D. C., Kong, L., Hou, M., Meng, Y. Q., Wei, L., et al. (2017). CPC2: a fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 45, W12–W16. doi: 10.1093/nar/gkx428
Kent, W. J. (2002). BLAT–the BLAST-like alignment tool. Genome Res. 12, 656–664. doi: 10.1101/gr.229202
Kim, D., Langmead, B., Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Kim, S., Park, M., Yeom, S. I., Kim, Y. M., Lee, J. M., Lee, H. A., et al. (2014). Genome sequence of the hot pepper provides insights into the evolution of pungency in capsicum species. Nat. Genet. 46, 270–278. doi: 10.1038/ng.2877
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087
Kyndt, T., Denil, S., Haegeman, A., Trooskens, G., Bauters, L., Van Criekinge, W., et al. (2012). Transcriptional reprogramming by root knot and migratory nematode infection in rice. New Phytol. 196, 887–900. doi: 10.1111/j.1469-8137.2012.04311.x
Langfelder, P., Horvath, S. (2008). WGCNA: an r package for weighted correlation network analysis. BMC Bioinf. 9, 559. doi: 10.1186/1471-2105-9-559
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv: Genomics, 1303. doi: 10.48550/arXiv.1303.3997
Li, H. (2016). Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32, 2103–2110. doi: 10.1093/bioinformatics/btw152
Li, D., Qian, J., Li, W., Yu, N., Gan, G., Jiang, Y., et al. (2021). A high-quality genome assembly of the eggplant provides insights into the molecular basis of disease resistance and chlorogenic acid synthesis. Mol. Eol Res. 21, 1274–1286. doi: 10.1111/1755-0998.13321
Li, L., Stoeckert, C. J., Jr, Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Ling, J., Mao, Z., Zhai, M., Zeng, F., Yang, Y., Xie, B. (2017). Transcriptome profiling of cucumis metuliferus infected by meloidogyne incognita provides new insights into putative defense regulatory network in cucurbitaceae. Sci. Rep. 7, 3544. doi: 10.1038/s41598-017-03563-6
Love, M. I., Huber, W., Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi: 10.1186/s13059-014-0550-8
Lowe, T. M., Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection oftransfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Majoros, W. H., Pertea, M., Salzberg, S. L. (2004). TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879. doi: 10.1093/bioinformatics/bth315
McHale, L., Tan, X., Koehl, P., Michelmore, R. W. (2006). Plant NBS-LRR proteins: adaptable guards. Genome Biol. 7, 212. doi: 10.1186/gb-2006-7-4-212
Milligan, S. B., Bodeau, J., Yaghoobi, J., Kaloshian, I., Zabel, P., Williamson, V. M. (1998). The root knot nematode resistance gene mi from tomato is a member of the leucine zipper, nucleotide binding, leucine-rich repeat family of plant genes. Plant Cell. 10, 1307–1319. doi: 10.1105/tpc.10.8.1307
Nachtweide, S., Stanke, M. (2019). Multi-genome annotation with AUGUSTUS. Methods Mol. Biol. 1962, 139–160. doi: 10.1007/978-1-4939-9173-0_8
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Plazas, M., Andujar, I., Vilanova, S., Hurtado, M., Gramazio, P., Herraiz, F. J., et al. (2013). Breeding for chlorogenic acid content in eggplant: interest and prospects. Notulae Botanicae Horti Agrobotanici Cluj-Napoca 41, 26–35. doi: 10.15835/nbha4119036
Qi, P. F., Zhang, Y. Z., Liu, C. H., Zhu, J., Chen, Q., Guo, Z. R., et al. (2018). Fusarium graminearum ATP-binding cassette transporter gene FgABCC9 is required for its transportation of salicylic acid, fungicide resistance, mycelial growth and pathogenicity towards wheat. Int. J. Mol. Sci. 19 (8), 2351. doi: 10.3390/ijms19082351
Ren, R., Wang, H., Guo, C., Zhang, N., Zeng, L., Chen, Y., et al. (2018). Widespread whole genome duplications contribute to genome complexity and species diversity in angiosperms. Mol. Plant 11, 414–428. doi: 10.1016/j.molp.2018.01.002
Rutter, W. B., Franco, J., Gleason, C. (2022). Rooting out the mechanisms of root-knot nematode-plant interactions. Ann. Rev. Phytopathol. 60, 43–76. doi: 10.1146/annurev-phyto-021621-120943
Sato, S., Tabata, S., Mueller, L. A., Huang, S., Du, Y., Li, C., et al. (2012). The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641. doi: 10.1038/nature11119
Sato, K., Uehara, T., Holbein, J., Sasaki-Sekimoto, Y., Gan, P., Bino, T., et al. (2021). Transcriptomic analysis of resistant and susceptible responses in a new model root-knot nematode infection system using Solanum torvum and Meloidogyne arenaria. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.680151
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Smoot, M. E., Ono, K., Ruscheinski, J., Wang, P. L., Ideker, T. (2011). Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432. doi: 10.1093/bioinformatics/btq675
Song, C., Guo, J., Sun, W., Wang, Y. (2012). Whole genome duplication of intra- and inter-chromosomes in the tomato genome. J. Genet. Genomics 39, 361–368. doi: 10.1016/j.jgg.2012.06.002
Tarailo-Graovac, M., Chen, N. (2009). Using repeatMasker to identify repetitive elements in genomic sequencesV. Curr. Protoc. Bioinf. 4, 4.10.1-4.10.14. doi: 10.1002/0471250953.bi0410s25
Vos, P., Simons, G., Jesse, T., Wijbrandi, J., Heinen, L., Hogers, R., et al. (1998). The tomato mi-1 gene confers resistance to both root-knot nematodes and potato aphids. Nat. Biotechnol. 16, 1365–1369. doi: 10.1038/4350
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963. doi: 10.1371/journal.pone.0112963
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49. doi: 10.1093/nar/gkr1293
Wang, D. P., Zhang, Y. B., Zhang, Z., Zhu, J., Yu, J. (2010). KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinf. 8, 77–80. doi: 10.1016/S1672-0229(10)60008-3
Wei, Q., Wang, J., Wang, W., Hu, T. H., Hu, H. J., Bao, C. L. (2020). A high-quality chromosome-level genome assembly reveals genetics for important traits in eggplant. Hortic. Res. 7, 153. doi: 10.1038/s41438-020-00391-0
Williamson, V. M., Kumar, A. (2006). Nematode resistance in plants: the battle underground. Trends Genet. 22, 396–403. doi: 10.1016/j.tig.2006.05.003
Wingett, S., Ewels, P., Furlan-Magaril, M., Nagano, T., Schoenfelder, S., Fraser, P., et al. (2015). HiCUP: pipeline for mapping and processing Hi-c data. F1000Res 4, 1310. doi: 10.12688/f1000research.7334.1
Xu, X., Pan, S., Cheng, S., Zhang, B., Mu, D., Ni, P., et al. (2011). Genome sequence and analysis of the tuber crop potato. Nature 475, 189–195. doi: 10.1038/nature10158
Xu, Z., Wang, H. (2007). LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268. doi: 10.1093/nar/gkm286
Zhang, H., Jing, W., Zheng, J., Jin, Y., Wu, D., Cao, C., et al. (2020). The ATP-binding cassette transporter OsPDR1 regulates plant growth and pathogen resistance by affecting jasmonates biosynthesis in rice. Plant Sci. 298, 110582. doi: 10.1016/j.plantsci.2020.110582
Zhang, R., Murat, F., Pont, C., Langin, T., Salse, J. (2014). Paleo-evolutionary plasticity of plant disease resistance genes. BMC Genomics 15, 187. doi: 10.1186/1471-2164-15-187
Zhang, M., Zhang, H., Tan, J., Huang, S., Chen, X., Jiang, D., et al. (2021). Transcriptome analysis of eggplant root in response to root-knot nematode infection. Pathogens 10, 470. doi: 10.3390/pathogens10040470
Zhang, R., Zhang, C., Yu, C., Dong, J., Hu, J. (2022). Integration of multi-omics technologies for crop improvement: status and prospects. Front. Bioinform. 2. doi: 10.3389/fbinf.2022.1027457
Zhao, D. X., Zhang, Y., Lu, Y. Z., Fan, L. Q., Zhang, Z. B., Zheng, J., et al. (2022). Genome sequence and transcriptome of Sorbus pohuashanensis provide insights into population evolution and leaf sunburn response. J. Genet. Genomics 49, 547–558. doi: 10.1016/j.jgg.2021.12.009
Zheng, X., Zhou, Z., Gong, Z., Hu, M., Ahn, Y., Zhang, X., et al. (2022). Two plant NLR proteins confer strain-specific resistance conditioned by an effector from Pseudomonas syringae pv actinidiae. J. Genet. Genomics 49, 823–832. doi: 10.1016/j.jgg.2022.06.006
Zhou, X. H., Liu, J., Bao, S. Y., Yang, Y., Zhuang, Y. (2018). Molecular cloning and characterization of a wild eggplant Solanum aculeatissimum NBS-LRR gene, involved in plant resistance to meloidogyne incognita. Int. J. Mol. Sci. 19, 583. doi: 10.3390/ijms19020583
Keywords: Solanum torvum, genome, transcriptome, root-knot nematode, ABC transporter
Citation: Zhang H, Chen H, Tan J, Huang S, Chen X, Dong H, Zhang R, Wang Y, Wang B, Xiao X, Hong Z, Zhang J, Hu J and Zhang M (2023) The chromosome-scale reference genome and transcriptome analysis of Solanum torvum provides insights into resistance to root-knot nematodes. Front. Plant Sci. 14:1210513. doi: 10.3389/fpls.2023.1210513
Received: 22 April 2023; Accepted: 26 June 2023;
Published: 17 July 2023.
Edited by:
Feng Cheng, Insititute of Vegetables and Flowers (CAAS), ChinaCopyright © 2023 Zhang, Chen, Tan, Huang, Chen, Dong, Zhang, Wang, Wang, Xiao, Hong, Zhang, Hu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Zhang, emhhbmdtaW5Ad3VoYW5hZ3JpLmNvbQ==; Jihong Hu, aHVqaDA1QG53YWZ1LmVkdS5jbg==; Junhong Zhang, emhhbmdqdW5obmdAbWFpbC5oemF1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.