 Chan Zhang1,2
Chan Zhang1,2 Xiaofeng Liu
Xiaofeng Liu Jing Yu
Jing Yu Yougen Wu
Yougen Wu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 29 June 2023
Sec. Plant Metabolism and Chemodiversity
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1201486
Pogostemon cablin is a well-known protected species widely used in medicine and spices, however the underlying molecular mechanisms and metabolite dynamics of P. cablin flower development remain unclear due to the difficulty in achieving flowering in this species. A comparison of the transcriptome and widely targeted metabolome during P. cablin flower development was first performed in this study. Results showed that a total of 13,469 differentially expressed unigenes (DEGs) and 371 differentially accumulated metabolites (DAMs) were identified. Transcriptomic analysis revealed that the DEGs were associated with starch and sucrose metabolism, terpenoid biosynthesis and phenylpropanoid biosynthesis. Among these DEGs, 75 MIKC-MADS unigenes were associated with the development of floral organs. Gibberellins (GAs), auxin, and aging signaling might form a cross-regulatory network to regulate flower development in P. cablin. According to the metabolic profile, the predominant DAMs were amino acids, flavonoids, terpenes, phenols, and their derivatives. The accumulation patterns of these predominant DAMs were closely associated with the flower developmental stage. The integration analysis of DEGs and DAMs indicated that phenylpropanoids, flavonoids, and amino acids might be accumulated due to the activation of starch and sucrose metabolism. Our results provide some important insights for elucidating the reproductive process, floral organ, and color formation of P. cablin flowers at the molecular level. These results will improve our understanding of the molecular and genetic mechanisms involved in the floral development of P. cablin.
Pogostemon cablin Benth, a species of the Lamiaceae family, is a crucial aromatic plant famous for its volatile oils (Li et al., 2011; Swamy and Sinniah, 2016). It’s mainly cultivated in tropical and subtropical regions, such as Malaysia, China, and Philippines (Swamy and Sinniah, 2015). As a traditional Chinese medicine, P. cablin was listed in the first batch of “Lingnan Traditional Chinese Medicine Protected Species” in Guangdong Province, China, in 2017. The main active components of essential oil are sesquiterpene, including patchoulol, which decide the quality of P. cablin (Shen et al., 2022; Yan et al., 2022). In the biosynthesis of patchoulol, the isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP) are biosynthesized by the mevalonate (MVA), followed by the condensation to form farnesyl diphosphate (FPP). Finally, the FPP is conversed to patchoulol with the terpene synthases (TPSs), which is responsible for the production of patchouli oil (Yu et al., 2015; Shen et al., 2022). Besides leaves, the inflorescences of P. cablin also biosynthesize and accumulate large amounts of patchoulol, which are composed of 17 sesquiterpenes (97.7%) (Verma et al., 2019). However, a phenomenon of rare flowering accompanied by a lack of seeds has been observed in P. cablin in many countries, such as China, India and Cuba (Li et al., 2011; Swamy and Sinniah, 2016). Due to this phenomenon, the large-scale propagation and preservation of P. cablin on farms occurs mainly by cottage propagation (Swamy and Sinniah, 2016), resulting in the accumulation of viruses and root-knot nematodes, and decreases in the resistance and quality of P. cablin. This phenomenon seriously impedes the cultivation and development of P. cablin. Hence, an understanding of the flowering process of P. cablin is urgently needed to promote the breeding of this species and the production of patchouli oil.
Flowering involves the following two main steps: first, the shoot apex meristem changes from a vegetative state to a reproductive state driven by the perception and integration of floral induction signals; and second, the shoot apex differentiates into an inflorescence or flower (Satish and Manju, 2018). Floral induction is coordinated by six pathways, including the photoperiod, gibberellin (GA), autonomy, aging, temperature, and vernalization (Wellmer and Riechmann, 2010). In recent years, approximately 180 genes were identified to play an important role in flower development in Arabidopsis thaliana (Fornara et al., 2010; Ó’Maoiléidigh et al., 2014). According to the flowering process, these genes were divided into three main clades (Satish and Manju, 2018). The first clade comprises flower pathway integrators, including FLOWERING LOUCS T (FT) and SUPPRESSOR OF OVEREXPRESSION OF CONSTANS (SOC1); the second clade comprises floral meristem identity genes, such as APETALA1 (AP1) and FRUITFUL (FUL); and the third clade comprises floral organ identity genes, such as, APETALA2 (AP2), SEPALLATA (SEP) and AGAMOUS (AG) (Satish and Manju, 2018). For most flowering plants, ABCDE model play a key role in the flower development process. In this model, different organ developments are regulated by various genes, such as sepals (A + E), petals (A + B + E), stamens (B + C + E), carpels (C + E), and ovules (D + E). In addition, class A contains APETALA1 (AP1) and FRUITFULL (FUL); class B contains PISTILLATA (PI) and APETALA3 (AP3); class C contains AGAMOUS (AG); class D contains SEEDSTICK (STK) and class E contains SEPALLATA genes (SEP1, SEP2, SEP3, and SEP4). Interesting, most of these genes are belonging to MADS-box gene family (Satish and Manju, 2018; Ren et al., 2021). These genes work together to complete the transition of the shoot apex meristem into a flower. Then, it enters the bud stage and the full-bloom stage successively, followed by the fruit- or seed-formation stage.
P. cablin is also a flowering plant that naturally blooms after the beginning of spring, and the flowering period lasts around 30-42 days (Verma et al., 2019). P. cablin flowers are indeterminate inflorescences whose terminal inflorescence maintains the state of the meristem. Although the genomes of P. cablin have been published (He et al., 2018; Shen et al., 2022), limited studies have investigated P. cablin flowers due to their morphological structure (Li et al., 2011) and essential oil composition (Verma et al., 2019). However, the available information on the molecular mechanisms and metabolite dynamics of P. cablin flower development is scarce due to the flowering difficulties of this species. Herein, the transcriptomic and metabolic profiles during flower development in P. cablin were characterized using transcriptomic and widely targeted metabolomic technologies. This study had the following two objectives: (1) to verify the regulation pathways and genes of flower development and (2) to determine the dynamics of metabolites during flower development in P. cablin. This report provides the first description of the molecular genetic mechanisms of P. cablin flower development and provides a basis for further studies on P. cablin sexual reproduction and breeding.
P. cablin (Nan Xiang, grown in Hainan) was cultivated at the germplasm resource garden of Hainan University (20°05′78′′N, 110°31′90′′E). Twelve mixed samples were collected from 7-month-old plants at four different flower stages on April 3, 2020, including the inflorescence-bearing meristem stage (F1), flower bud stage (F2), full-bloom stage (F3) and withered flower stage (F4). Three replicates were collected at each flowering stage. The harvested samples were frozen immediately in liquid nitrogen. Half of the samples were stored at -80 °C for RNA extraction, and the others were freeze-dried for chemical component identification.
Total RNA was extracted using TRIzol®-Reagent according to the manufacturer’s instructions. After quality check, the high-quality RNA samples (OD260/280 = 1.8~2.2, RIN ≥ 8.0) were used to construct the RNA-seq library, according to the TruSeq™ instructions. First, the polyA selection method was performed to isolate mRNA, which was then segmented using lysis buffer. Second, cDNAs were obtained using a SuperScript Double-Stranded cDNA Synthesis Kit. Illumina adapters were then ligated to the cDNAs, and amplification fragments (200 bp) were obtained by PCR. After quantification with TBS380, the paired-end cDNA libraries were sequenced with a NovaSeq 6000 sequencer (Illumina) (2 × 150 bp read length). The raw data were submitted to the NCBI Short Read Archive database under accession number PRJNA769458.
Reads with a low quality (Q < 20) or containing more than 10% N bases were excluded using Trimmomatic-0.39.jar (Yan et al., 2022). The P. cablin genome sequence, including the coding DNA sequence (CDS), protein sequences and GFF3 files, was downloaded from the figshare database (https://figshare.com/). High-quality reads were mapped to the P. cablin genome using HISAT2 software (He et al., 2018). The NCBI RefSeq, Swiss-Prot, Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) and Pfam databases, were used for functional annotation via DIAMOND V0.9.24 (Buchfink et al., 2021).
The transcript levels of the unigenes were quantified based on the TPM reads system to identify the DEGs between the two flower periods. The transcript abundance was determined using RSEM (Li et al., 2011). DEGs (log2FC > 2 or < -2 and p value < 0.001) were screened using DESeq2 version 2.10 (Yan et al., 2022). Correlations were determined by calculating Pearson’s correlation coefficient (r). GO and KEGG enrichment analyses (p < 0.05 and FDR < 0.05) were performed using GOATOOLS and KOBAS software (version 2.0), respectively (Xie et al., 2011).
The sequences of the SRF (Pfam: PF00319) and K-box domains (Pfam: PF01486) served as queries to search the P. cablin genome and affirm MADS genes using HMMER (version 3.3.2). Phylogenetic relationships among MADS genes were aligned using Clustal Omega (https://www.ebi.ac.uk) and IQ-tree (http://iqtree.cibiv.univie.ac.at/) with the method of Maximum likelihood (ML), followed by visualizion with iTOL (https://itol.embl.de/).
Ten milligrams of sample were used to extract metabolites with the extract solution (methanol: water = 3:1) and then filtered. Metabolites in 2 μL were separated using an ExionLC system (AB SCIEX) equipped with a Waters UPLC HSS T3 column. The temperatures of the column and autosampler were 40 °C and 4 °C, respectively. In the analysis procedure, the gradient program (A, 0.1% formic acid: B, acetonitrile) was as follows: 98:2 (V/V) at 0-0.5 min, 50:50 (V/V) at 10.0 min, 5:95 (V/V) at 11.0-13.0 min, and 98:2 (V/V) at 13.1-15 min. The flow rate was 400 μL·min-1, and a SCIEX Q Trap 6500+ instrument (AB SCIEX Technologies) was used for the analysis.
SCIEX Analyst Work Station Software (1.6.3) was employed to clean the MRM data. Metabolite peak detection and annotation were performed using the R program. Principal component analysis (PCA), orthogonal partial least squares differential analysis (OPLS-DA) and hierarchical clustering analysis were performed using SIMCA Software (V16.0.2). KEGG enrichment analysis was conducted using the OmicShare platform (https://www.omicshare.com/), and pathways with p < 0.05 and FDR < 0.05 were identified as significantly different metabolic pathways. The heatmap was visualized using Morpheus (https://software.broadinstitute.org/).
To test the validation of transcriptomic data, 18 DEGs were chose and quantified at four flowering stages. qRT-PCR was performed using SYBR Green qPCR Mix in the Applied Biosystems device (Thermo Fisher). The relative transcript levels were computed with the 2−ΔΔCT method. All the primers used for qRT-PCR analysis are mentioned in Table S11.
A weighted gene correlation network analysis (WGCNA) was employed using a freely accessible R package (version 1.70-3) with the default parameters according to the protocol designed to identify coexpressed genes (Langfelder and Horvath, 2008). TOM similarity was also calculated, and correlation variables with TOM > 0.1 were considered correlated. A graphical representation of the coexpression network was constructed using Cytoscape v 3.8.1 (Mauceri et al., 2021).
A transcriptome and metabolome integrated correlation analysis was conducted by MetaboAnalyst 5.0 (Pang et al., 2021). Spearman’s rank correlation analysis was performed using the R package based on the DEGs and DAMs. Genes and metabolite network plots were constructed using Cytoscape v3.8.1. The correlation coefficients of DEGs and DAMs (value> 0.80 or < -0.80, and p < 0.05) was displayed in the network plot for clarity.
To identify the candidate genes connected with P. cablin flower development, 12 libraries were generated at the inflorescence-bearing meristem stage (F1), flower bud stage (F2), full-bloom stage (F3), and withered flower stage (F4) (Figure 1A). The transcriptomic data in each library were 7.67 G on average, with low average error rate (0.024%) and high Q30 (94.72%), indicating reliable databases (Table S1). A total of 109,498 unigenes were obtained from these databases. PCA displayed a similar gene expression pattern in the same flower stage, which were divided by PC1 (28.81%) and PC2 (22.41%) (Figure 1B). Venn diagrams showed that the overlap between F1 and F2 consisted of 1,554 (2.36%) individual unigenes, while only 250 (0.38%) unigenes were overlapped between F3 and F4 (Figure 1C). Approximately 75.59% of unigenes were shared during four stages. More stage-specific genes were identified in the F1 (2,197 unigenes) and F2 (2,090 unigenes) than in the F3 (864 unigenes) and F4 (876 unigenes) libraries (Figure 1C). A total of 13,469 DEGs were confirmed by DESeq2 (log2FC>2, P < 0.001) (Table S2). Notably, 4,520, 6,163, and 4,072 DEGs were identified in the F2 vs. F1, F3 vs. F1 and F4 vs. F1 comparison sets, respectively (Figure 1D).
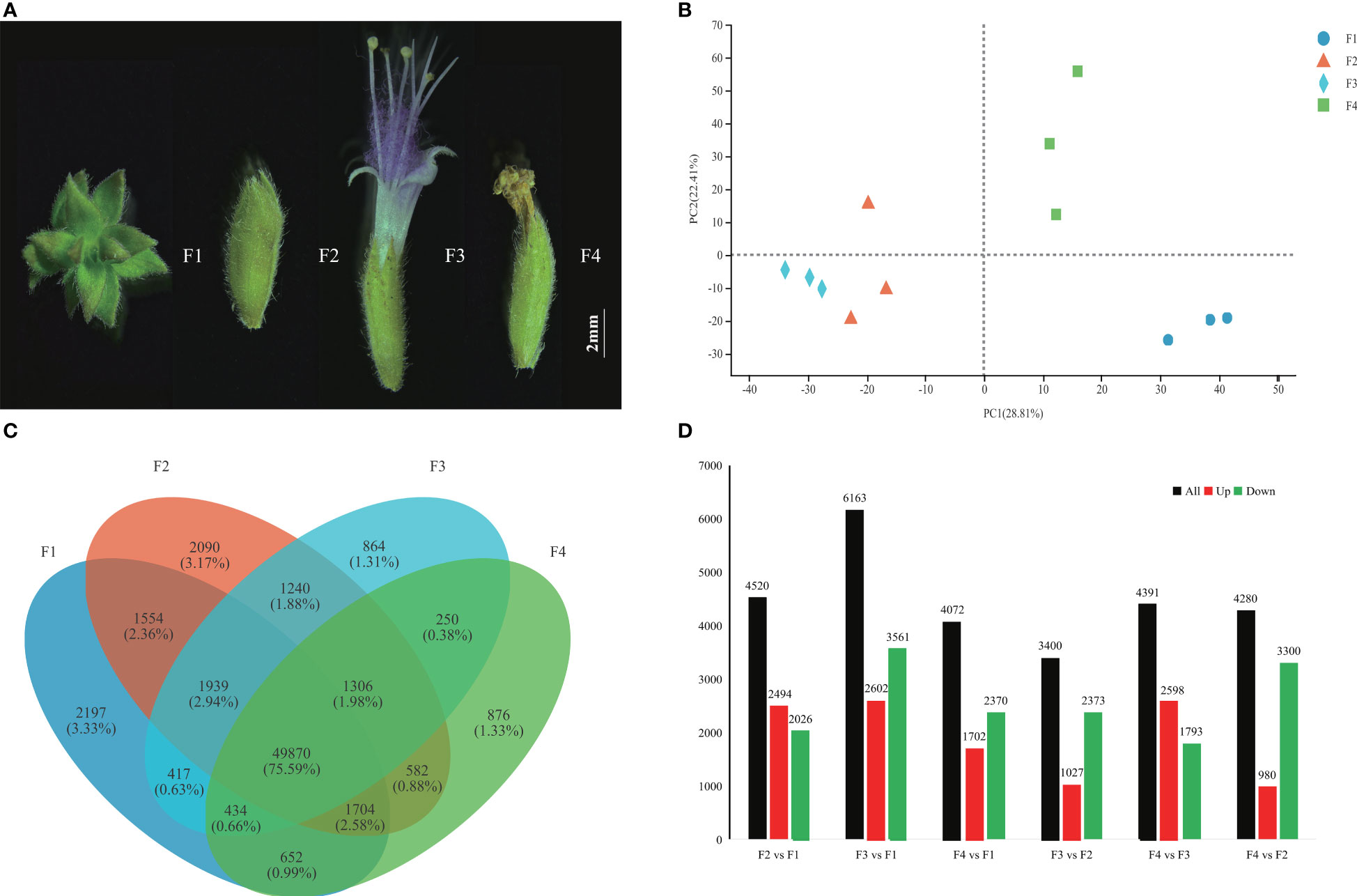
Figure 1 Transcriptome profiles of P. cablin flowers at four stages. (A) Four flower stages of P. cablin (from left to right): the inflorescence-bearing meristem stage (F1), flower bud stage (F2), full-bloom stage (F3), and withered flower stage (F4). The white line represents 2 mm. (B) PCA plot showing the gene expression patterns at the four flower stages. PC1 and PC2 represent the first principal component and the second principal component, respectively. (C) Venn diagram showing the expression profile of flower stage-specific unigenes. (D) Numbers of DEGs in different comparison sets, including F2 vs. F1, F3 vs. F1, F4 vs. F1, F3 vs. F2, F4 vs. F2, and F4 vs. F3. The black color indicates DEGs, the red color indicates upregulated unigenes, and the green color indicates downregulated unigenes.
GO and KEGG enrichment analyses showed the top 15 significantly different metabolic pathways (p < 0.05 and FDR < 0.05) in the Figures 2, S1. According to the GO functional analysis, 3,590, 4,927, and 3,227 DEGs from the three comparison sets were annotated to 4,139, 4,840, and 4,066 GO terms and were significantly enriched in 457, 667, and 510 GO terms, respectively (Table S3). Among them, the terpenoid biosynthesis process and anthocyanin-containing compound biosynthesis process were significantly more active from F2 to F4 (Figure S1). Moreover, the auxin-activated signaling pathway, pollen exine formation, and sporopollenin biosynthesis process were enriched at F2 (Figure S1). Two processes of carbohydrate metabolism, and four important processes involved in cell division and DNA replication, were enriched at F3 and F4, respectively (Figure S1). These results indicated that the development of P. cablin flowers is accompanied by the biosynthesis of terpenoids and anthocyanins.
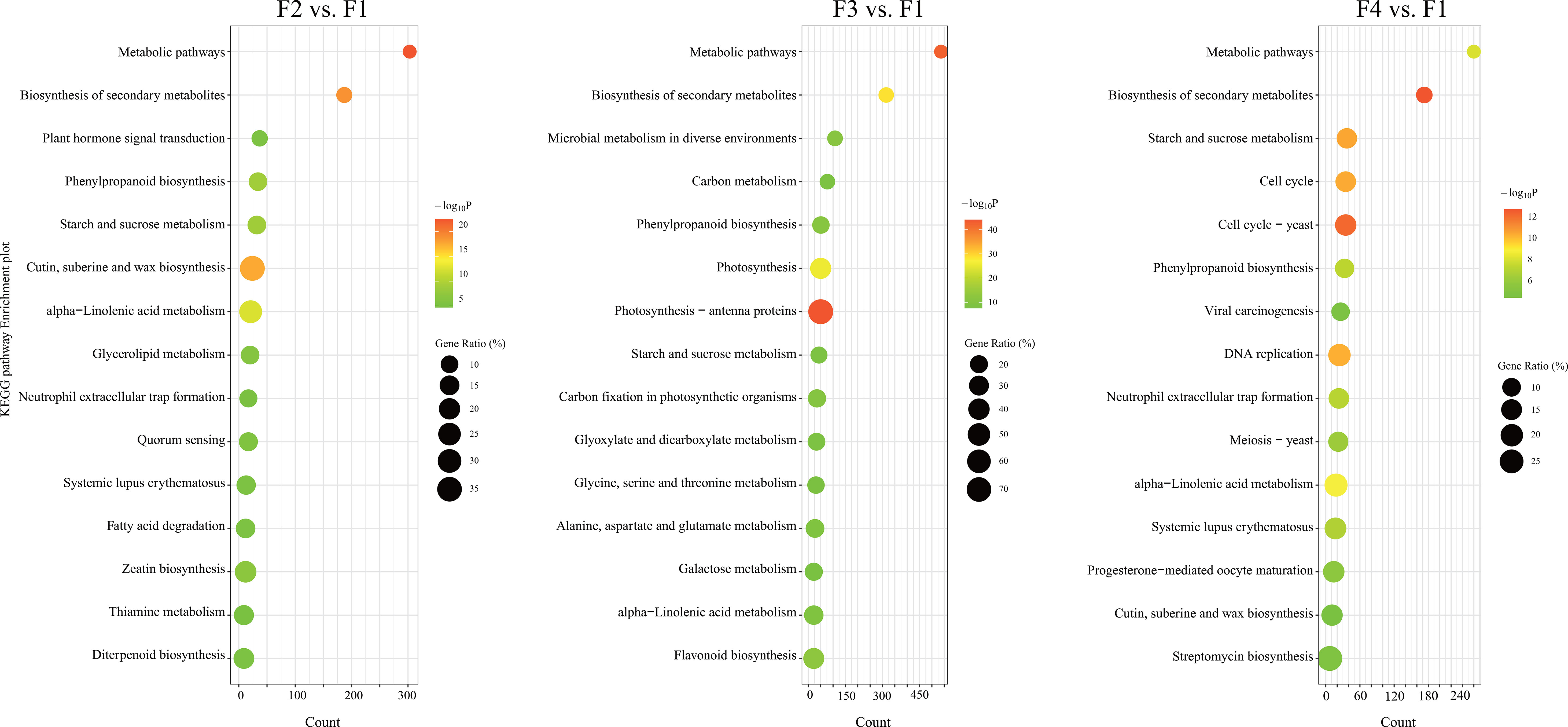
Figure 2 KEGG pathway enrichment analysis of DEGs at different flowering stages of P. cablin. KEGG pathway enrichment analysis of DEGs in three comparison sets: the flower bud stage (F2) vs. the inflorescence-bearing meristem stage (F1), the full-bloom stage (F3) vs. the inflorescence-bearing meristem stage (F1), and the withered flower stage (F4) vs. the inflorescence-bearing meristem stage (F1). The top fifteen significantly different metabolic pathways (p < 0.05, FDR < 0.05) are shown in the diagram. The percentage indicates the number of DEGs out of the total genes in the corresponding KEGG pathway. The size of the bubble represents the number of DEGs in the pathway; the color range indicates the significance of pathway enrichment.
The KEGG pathway enrichment analysis showed 1,786, 3,532, and 1,904 DEGs were annotated to 252, 297, and 254 KEGG pathways in the three comparison sets, respectively. Among them, 62, 85, and 86 KEGG pathways were significantly enriched in the F2 vs. F1, F3 vs. F1 and F4 vs. F1 comparison sets, respectively (Table S4). The phenylpropanoid biosynthesis, alpha-linolenic acid metabolism, and starch and sucrose metabolism pathways were all significantly enriched from F2 to F4, and the percentage of genes in these pathways was highest at F3 (Figure 2; Table S4). Furthermore, flavonoid biosynthesis and the plant hormone signal transduction pathway were also significantly enriched at F2 and F3 (Figure 2; Table S4), and the diterpenoid biosynthesis pathway was enriched at F2. DEGs involved in four pathways associated with carbohydrate metabolism were significantly enriched at F3. Similarly, four pathways associated with cell proliferation, including the cell cycle, meiosis and DNA replication were active at F4. All of pathways associated with growth, pigmentation and energy costs, displayed some distinctions in the stages of P. cablin flower development.
Hormone signaling pathways are important for flower development. The GO and KEGG enrichment results showed that auxin signal transduction was clearly enriched during P. cablin flower development (Figure S1; Table S4). Twenty-nine DEGs were related to the auxin signaling pathway (Figure S2). For instance, most genes including AUX/LAX, IAA, ARF, and SAUR, were upregulated from the F2 to F3 stage, while the transcript levels of GH3 genes decreased at the F2 and F3 stages (Figure S2). Thus, the activation of auxin signaling at the bud flower and full-bloom stages might favor P. cablin flower development.
As main active ingredients, terpenes are synthesized through methyl-erythritol phosphate and the mevalonate pathways (Figure 3A). KEGG analysis showed that 210 terpenoid synthesis-related genes were identified (Table S5), including 139 genes in the terpenoid backbone, 8 genes in monoterpenoids, 43 genes in diterpenoid biosynthesis, and 20 genes in sesquiterpenoid and triterpenoid biosynthesis (Table S5). We then identified 59 DEGs encoding 21 enzymes associated with terpenoid biosynthesis (Table S6). The p values and FDR values of these DEGs in the F2 vs. F1, F3 vs. F1 and F4 vs. F1 comparison sets are listed in Table S7.
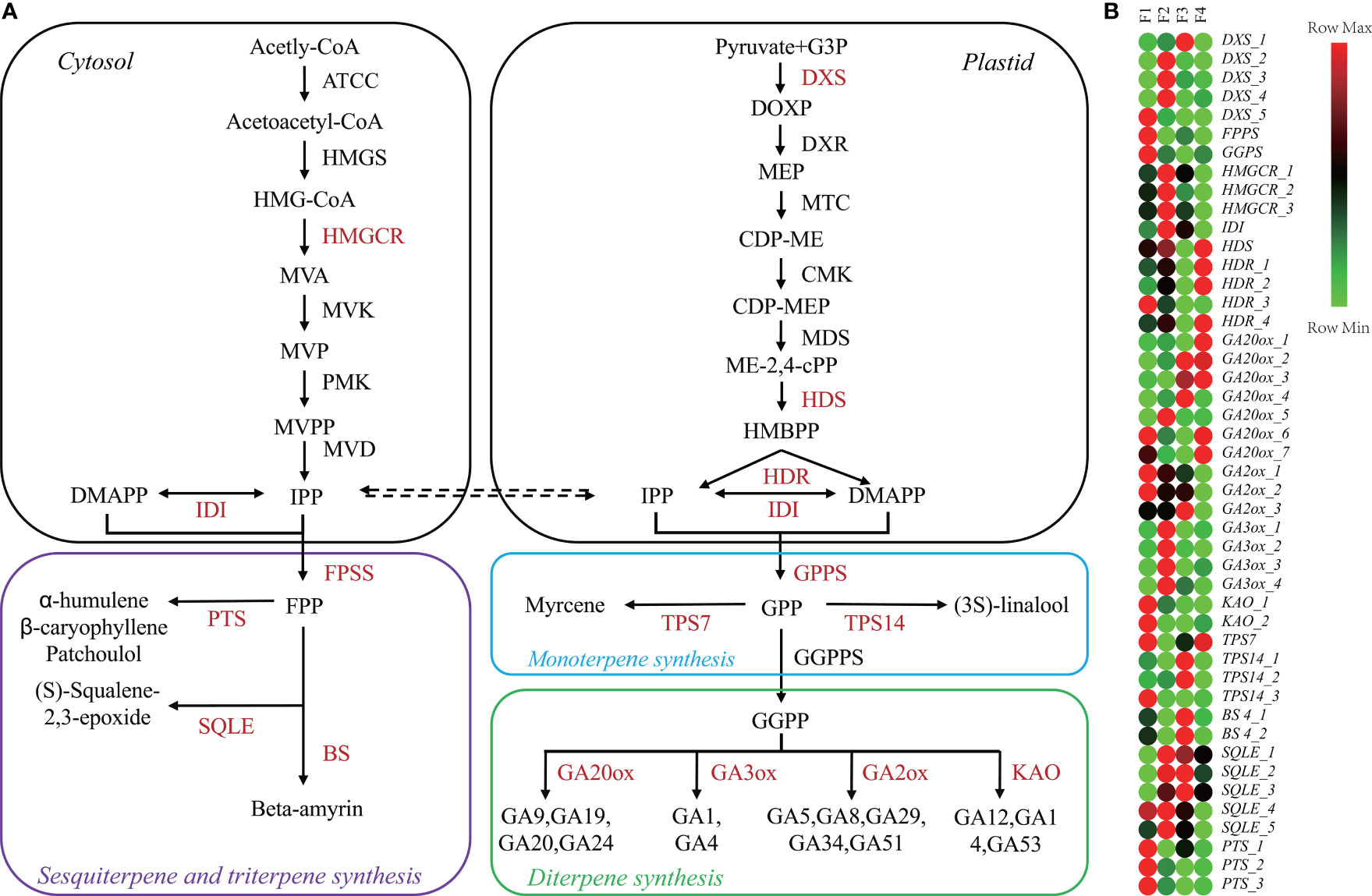
Figure 3 Changes in terpene metabolism-related genes during P. cablin flower development. (A) Diagram of the terpene biosynthesis pathway. (B) Heatmap showing the expression levels [log2(TPM+1)] of genes related to terpenoid biosynthesis during flower development. The pathway was redrawn based on ko0900, ko00902, ko00904, and ko00909 in the KEGG database (https://www.kegg.jp/). F1, inflorescence-bearing meristem stage; F2, flower bud stage; F3, full-bloom flower stage;and F4, withered flower stage.
A heatmap of 46 DEGs associated with terpenoid biosynthesis during the four flower stages was generated (Figure 3B). According to the heatmap, FPPS, GGPPS, KAO_1-2, GA2ox_1-2 and TPS7 were highly expressed at F1, while the expression levels of TPS14_1-2 and BS4_1-2 were increased at F3 (Figure 3B). The highest levels of HMGCR_1-3, IDI and GA3ox_1-4 transcripts were detected at F2. Patchoulol, a major component of P. cablin, is catalyzed by the enzyme patchoulol synthase (PTS). The PTS_1-3 genes were highly expressed at F1. Interestingly, 16 key GA-related genes such as KAO, GA2ox, GA3ox, and GA20ox, were differentially expressed throughout the process of flower development. Taken together, these results suggested that GA signaling might be involved in P. cablin flower development.
Two hundred thirty-seven DEGs related to flower development were characterized (Table S10). These DEGs were sorted into six clades based on flowering pathway, including 55 DEGs in the photoperiod/circadian pathway, 50 DEGs in the GA signaling, 29 DEGs in the auxin signaling, and 13 DEGs in the aging pathway. Nine and five DEGs were identified in the nutrient pathway and the vernalization pathway, respectively. Seventy-six DEGs were associated with flower pathway integration-related and floral organ identity genes, such as FT, SOC1, AP1, AG, PISTILLATA (PI) and SEP (Table S10).
Overall, 270 MADS genes, including 132 MIKC-type and 138 type I MADS genes, were identified (Figure S3). A phylogenetic analysis showed that MIKC-MADS genes were sorted into 12 classes such as SVP-like, AG-like, and AP1/FUL-like. Among them, 80 differentially expressed MADS unigenes (75 MIKC-type, 2 Mα and 3 Mδ MADS) were identified (Figure S3; Table S7). As expected, 69% of MIKC-MADS genes were involved in the ABCDE model: 12 genes in A-class (AP1-like), 13 genes in B-class (PI-like and AP3-like), 19 genes in C/D class (AG-like), and 8 genes in E-class (SEP-like) (Figure 4). Most genes of the AG-like, AP3-like, SEP-like, PI-like, and AP1-like clade were upregulated from F2 to F4 compared to F1. However, SVP-like and SOC1-like clade genes displayed downregulated expression from F2 to F4. Besides MIKC-MADS TF, 13 aging-related differentially expressed SPL genes were identified, and SPL1 and SPL8 exhibited upregulated expression at F2 and F3, respectively. Moreover, the expression levels of SPL3, SPL7, SPL13A, SPL15, and SPL12 were significantly upregulated at the inflorescence-bearing meristem stage (Figure S4). These results implied that MIKC-MADS and SPL genes might participate in the various phases of P. cablin floral organ formation.
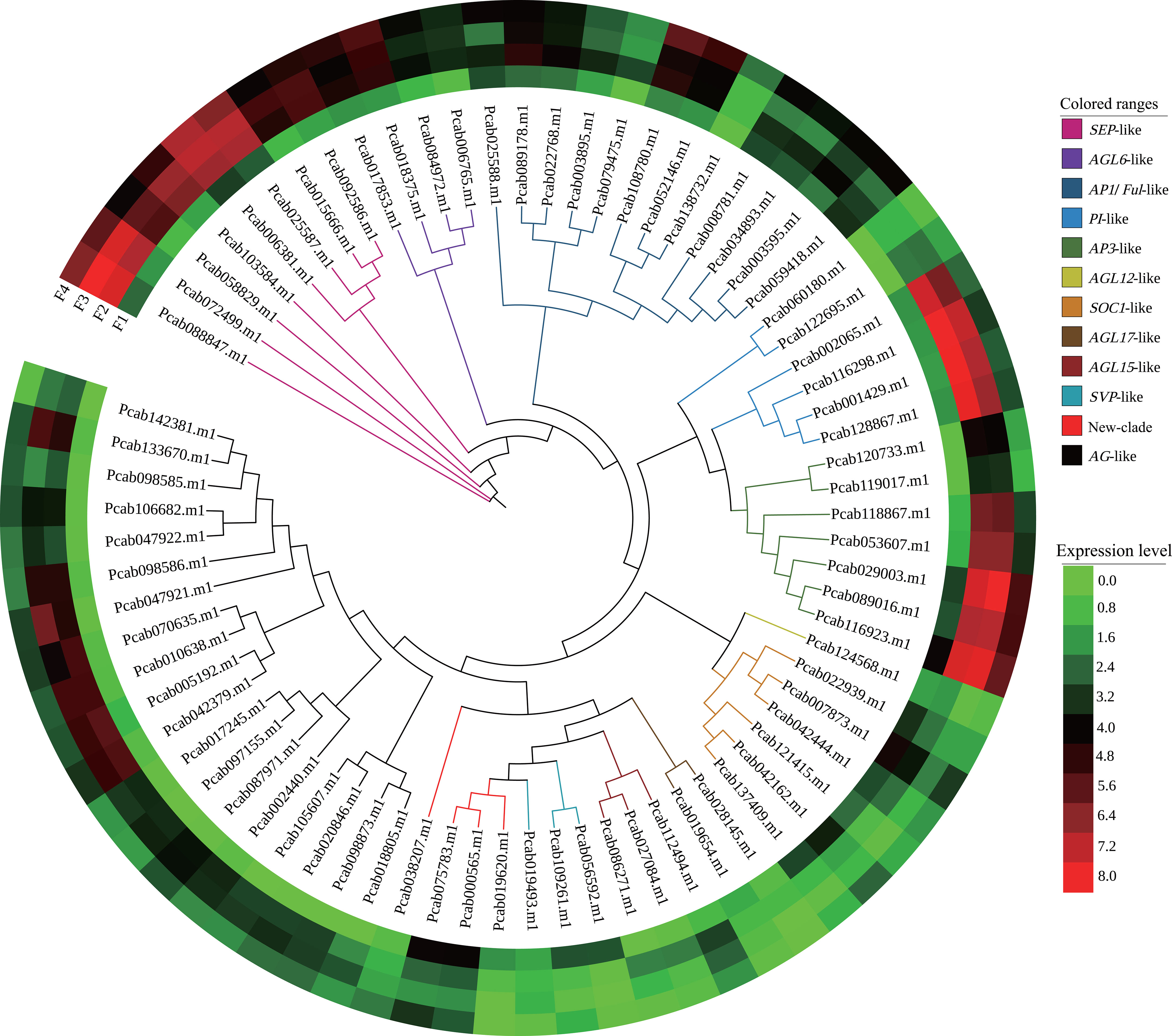
Figure 4 Phylogenetic and expression analyses of MIKC-MADS genes from P. cablin. Classification of differentially expressed MIKC-MADS genes identified in P. cablin. The phylogenetic tree was constructed using IQ-tree and EvolView. Heatmap showing the expression levels [log2(TPM+1)] of MIKC-MADS genes during flower development at the inflorescence-bearing meristem stage (F1), flower bud stage (F2), full-bloom stage (F3), and withered flower stage (F4).
To verify the validation of the transcriptomic data, the expression of 18 representative DEGs in the four flower stages were tested using qRT-PCR (Figure 5; Table S11). These DEGs were related to terpenoid biosynthesis and flower development. The relative expression levels of qRT-PCR and RNA-seq between the two flowering stages were logarithmically processed. Spearman correlation analysis was performed using the above logarithmic results. The Spearman correlation coefficient was 0.8562, indicating that the expression pattern of most transcripts revealed using qRT-PCR was consistent with the RNA-seq data (Figure 5).
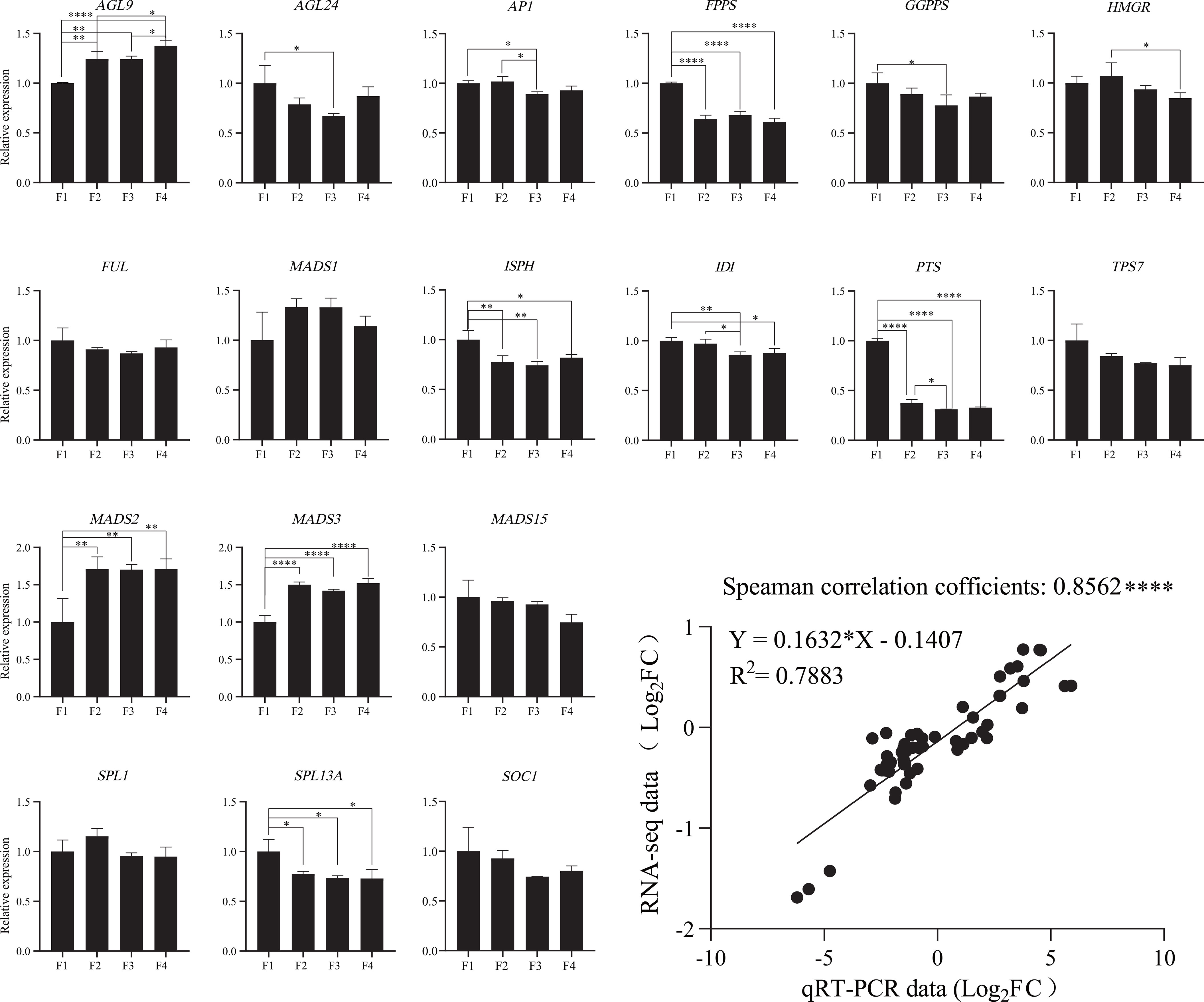
Figure 5 Validation of the expression of terpenoid biosynthesis and flowering-related genes in P. cablin. The expression levels of 18 flower development-related genes in four flower stages were validated using qRT-PCR. The relative expression levels of 18 DEGs were calculated using the 2−ΔΔCt method; 18S rRNA served as an internal reference gene. All these data were based on the analysis of three independent biological replicates. Linear regression analysis of the qRT-PCR data and RNA-seq data. “*” indicates significant differences, as determined by one-way ANOVA followed by Tukey’s test (P<0.05). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Metabolic analysis showed that 984 metabolites were identified across all flower samples (Table S8). KEGG analysis showed that 916 metabolites were classified into 14 known classe (Figure 6A). The percentages of primary and secondary metabolites were 32% and 61%, respectively. A total of 370 DAMs (VIP > 1 and p value < 0.05) were identified (Table S9). Amino acids, organic acids, and their derivatives were the major primary DAMs, while the most abundant secondary DAMs were flavonoids and their derivatives, followed by terpenes, alkaloids, phenols, and their derivatives (Figure 6A). PCA revealed that each group tended to cluster together, separated by PC1 (24.8%) and PC2 (12.9%) (Figure 6B). The composition of metabolites was different at the four flower development stages, proved by the Q2 values of the OPLS-DA (all greater than 0.8) (Figure 6C), and by hierarchical clustering analysis (Figure 6D). These results suggested a significant distinction in all three comparison sets.
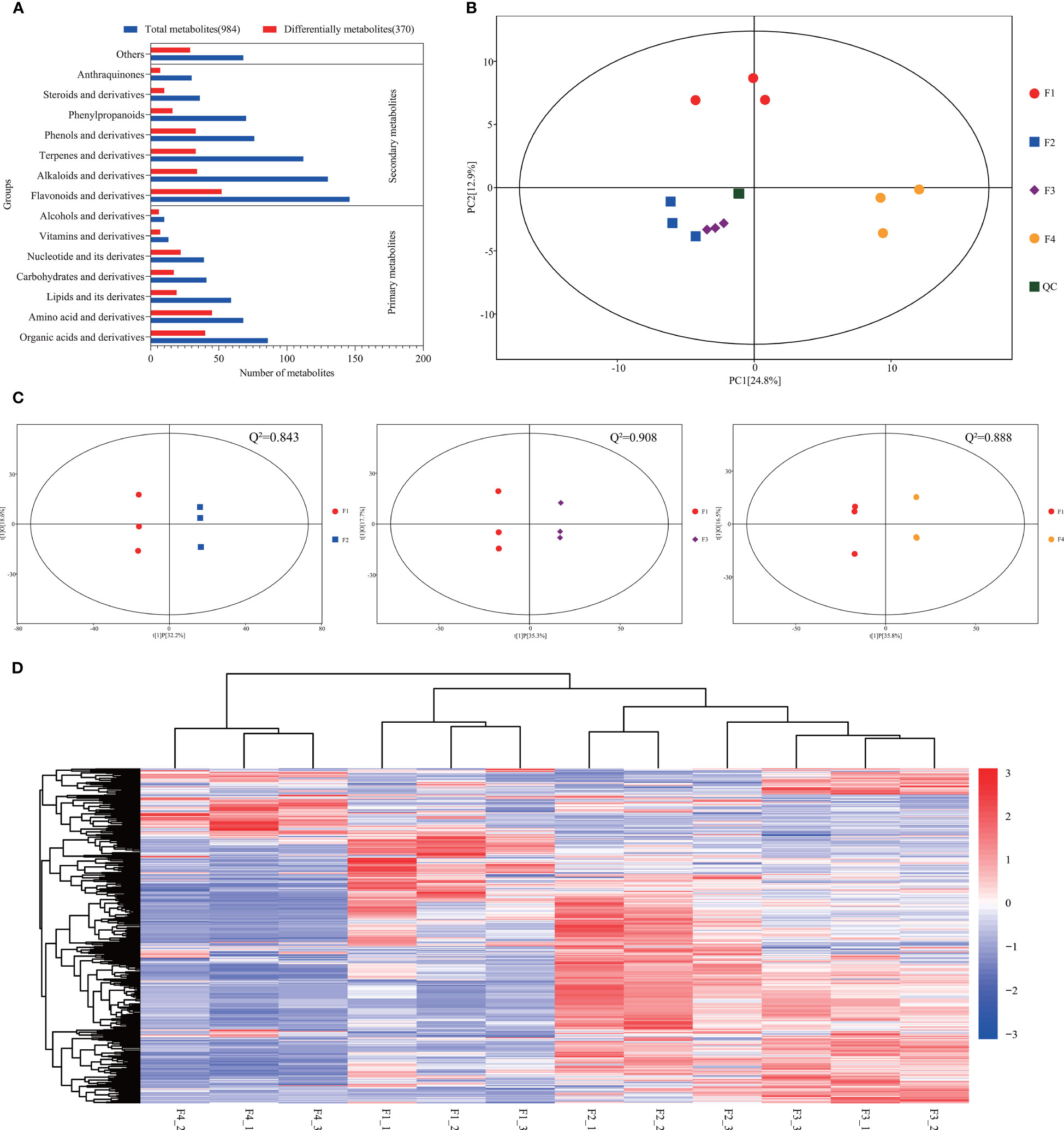
Figure 6 Metabolomic profiles during P. cablin flower development. (A) Classification of all the identified metabolites and DAMs. Red indicates the total identified metabolites, and blue indicates the DAMs. (B) PCA of the metabolite profiles of flower samples at four flowering stages. PC1 indicates the first principal component, and PC2 indicates the second principal component. (C) Score plot of the OPLS-DA of the metabolites at the four flowering stages. X axis, T score; Y axis, orthogonal T score. Q2 indicates the model predictability. (D) Bidirectional hierarchical clustering analysis of the DAMs and sample clustering analysis. The color palette represents the relative changes in metabolite contents among flower samples. The rows represent metabolites; the columns represent flower samples. Red and blue represent high gene expression and low gene expression, respectively. F1, inflorescence-bearing meristem stage; F2, flower bud stage; F3, full-bloom stage; F4, withered flower stage; and _1, _2, _3 represent three replicates.
KEGG pathway enrichment analysis showed that 62, 75, and 68 DAMs were annotated, and 9, 12, and 4 KEGG pathways were significantly enriched in the three comparison sets (p < 0.05 and FDR < 0.05) (Figure 7; Table S12). Among them, amino acids such as lysine, arginine, and alanine was constantly active during P. cablin flower development. DAMs in two carbohydrate metabolism processes were significantly enriched at F3 and F4, while two significantly different metabolic pathways (flavone and flavonol biosynthesis, and porphyrin and chlorophyll metabolism) were enriched in the flower bud stage compared with the inflorescence-bearing meristem stage (Figure 7). These pathways were associated with flower color formation. This result might be attributed to the coloration of petals during flower organ development, and was generally consistent with the results from the KEGG pathway enrichment analyses of DEGs.
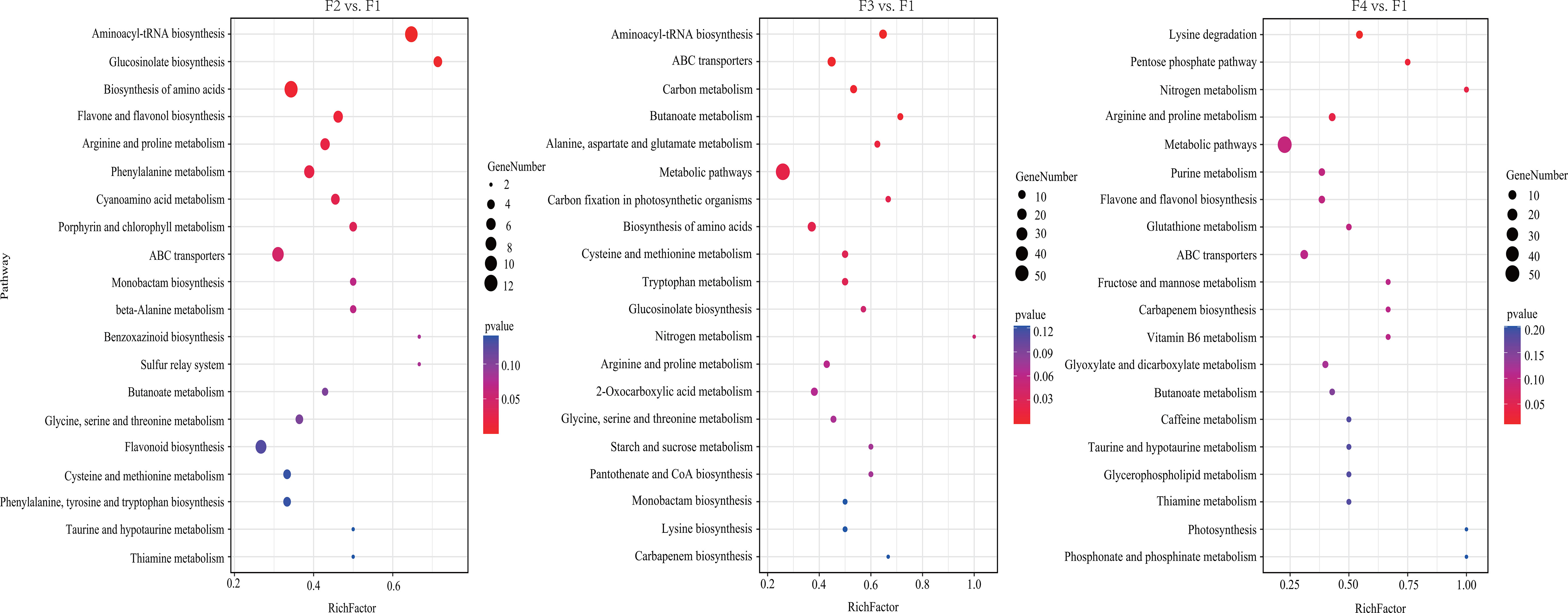
Figure 7 KEGG pathway enrichment analysis of DAMs at four flower stages of P. cablin. KEGG pathway enrichment analysis of DAMs in three comparison sets, including flower bud stage (F2) vs. inflorescence-bearing meristem stage (F1), full-bloom flower stage (F3) vs. inflorescence-bearing meristem stage (F1), and withered-flower stage (F4) vs. inflorescence meristem-stage (F1). The results are shown in the diagram with the twenty metabolic pathways along with the minimum p value. The bubble size represents the number of DAMs that were differentially accumulated in the pathway; the color range represents significant pathway enrichment. A p value < 0.05 and FDR < 0.05 were identified as significantly different metabolic pathways.
The accumulation patterns of the predominant DAMs (VIP>1 and p <0.05), including amino acids, flavonoids, terpenes, phenols, and their derivatives, were determined (Figure 8; Table S13). The Clustering analysis revealed that four predominant DAMs were clustered into three groups: group 1 (F1), group 2 (F2 and F3), and group 3 (F4) (Figure 8). These results were consistent with the results of PCA (Figure 6B). Twenty-two, four, and six amino acids were accumulated the highest at the F2 and F3 stages, the F4 stage, and the F2 stage, respectively (Figure 8A). Twenty-five, twenty-four, and three flavonoids and their derivatives exhibited the highest accumulation in group 1, group 2, and group 3, respectively (Figure 8B). In addition, twenty-one phenols and their derivatives exhibited the highest accumulation in group 2. Only five phenols and their derivatives and ten terpenes and their derivatives displayed the highest accumulation in group 3 (Figures 8C, D). Thus, the accumulation patterns of predominant DAMs were closely associated with flower development stage in P. cablin.
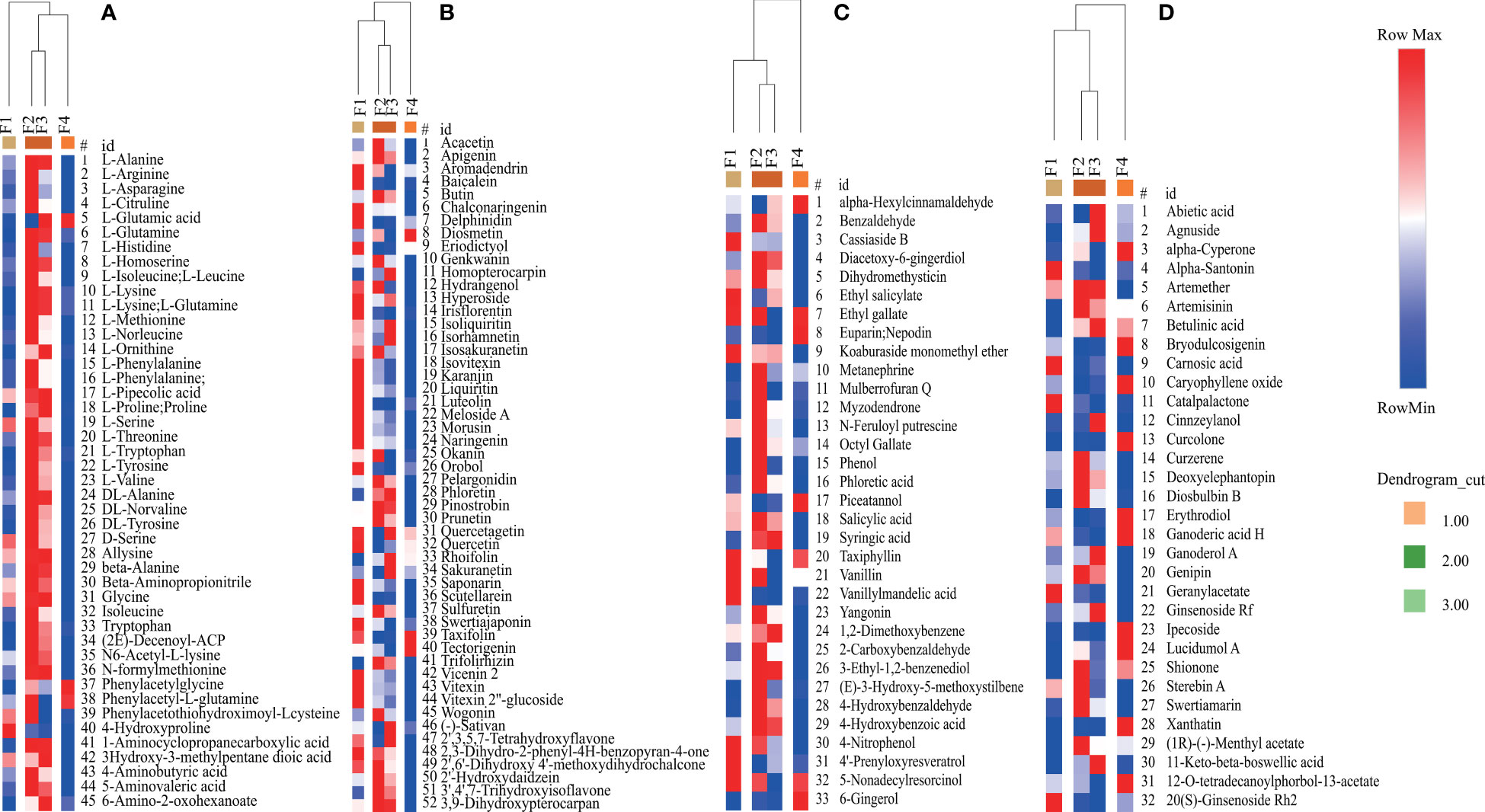
Figure 8 Relative changes in the contents of predominant DAMs during P. cablin flower development. (A) Relative changes in the contents of amino acids and their derivatives. (B) Relative changes in the contents of flavonoids and their derivatives. (C) Relative changes in the contents of phenols and their derivatives. (D) Relative changes in the contents of terpenes and their derivatives. The columns represent flower samples. Red and blue indicate the relative content and low relative content of metabolites, respectively. F1, inflorescence-bearing meristem stage; F2, flower bud stage; F3, full-bloom stage; and F4, withered-flower stage.
To understand the possible regulatory relationships of flower development-related genes, a coexpression analysis was performed using the auxin, GA, MIKC-MADS, and aging-related DEGs (Table S14). Two independent modules (blue and turquoise) with high correlation coefficients (R2 > 0.85), were defined as clusters of highly interconnected genes (Figure S5). The results showed that the blue and turquoise modules were each correlated with F1, and F2 and F3, and contained 42 genes and 101 genes, respectively (Figure S5C, Table S14). The blue module consisted of 13 MIKC-MADS genes, 1 FT gene, 1 FLK gene, 10 SPL genes, 5 auxin signaling-related genes and 12 GA signaling-related genes. However, in the turquoise module, MADS genes comprised the greatest percentage (52.47%) of the genes, followed by GA signaling-related genes (25.74%) and auxin signaling-related genes (19.80%).
The coexpression network derived from all modules showed that the possible regulatory relationship of MIKC-MADS genes with other genes was complicated (Figure 9). For example, SVP-like genes were correlated with SOC1, GA2ox, and KAO, and the TOM value ranged from 0.40 to 0.52 (Table S14). Two unknown MIKC-MADS genes (Pcab075783 and Pcab000565) were correlated with SOC1, GA2ox, IAA, and GH3. Moreover, AG (Pcab047921) was shown to be correlated with SPL8, IAA, GA2ox, GA20ox, and GASA, while FT was correlated with the SVP-like gene, SPL7 and KAO. SOC1 was strongly correlated with SVP-like genes, GH3, and GA2ox (Table S14).
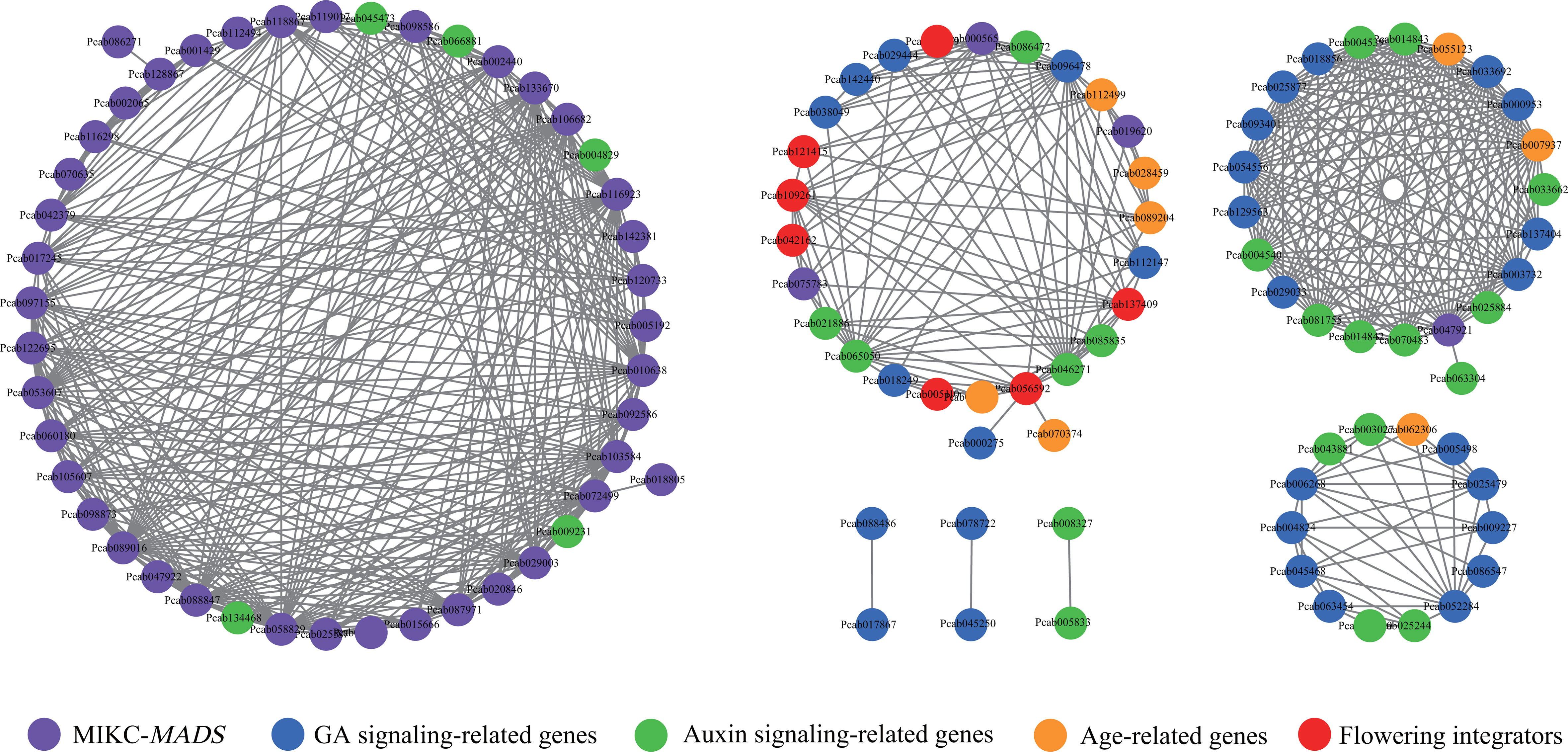
Figure 9 Coexpression network of flower development-related genes. The network was visualized using Cytoscapes. The purple, red, blue, green and orange round nodes represent MIKC-MADS genes, flowering integrators, GA signaling-related genes, auxin signaling-related genes, age-related genes, respectively. A TOM value > 0.4 is shown in the network plot.
The auxin and GA pathways are important hormone signaling pathways in flowering. Among auxin signaling-related genes, IAAs (Pcab004829 and Pcab045473) were strongly correlated with AP3, SEP, AG and AP3, respectively, while AUX (Pcab134468) was strongly correlated with SEP (Table S14). In the GA pathways, SVP-like genes (Pcab056592 and Pcab109261) were correlated with GA2ox and KAO. AG (Pcab047921) was correlated with GA2ox, GA20ox, and GASA. GID (Pcab137404) was strongly correlated with GASA. Notably, auxin signaling-related genes were strongly correlated with GA signaling-related genes. For example, ARF (Pcab043881) and AUX (Pcab003027) were strongly correlated with GASA. IAA and SAUR were strongly correlated with GA2ox, GA20ox, and GASA (Table S14). SPLs in aging pathway are important TFs involved in plant flowering. SPL12 was correlated with SOC1, SVP-like, GA2ox, GH3, and IAA. SPL8 was strongly correlated with IAA and GASA, while SPL7 was strongly correlated with SVP-like (Figure 9; Table S14).
These results indicated that the GA, auxin, and aging pathways might form a cross-network to regulate the development of P. cablin flowers, and auxin signaling might participate in P. cablin flower development by affecting GA signaling.
Spearman’s rank correlation analysis was performed to evaluate the correlation coefficients between DEGs and DAMs during flower development (Table S15; Figures S6, 10). Notably, 7801, 11245 and 10017 significantly correlated variables were involved in the biosynthesis and metabolism of primary and secondary DAMs at F2 vs. F1, F3 vs. F1 and F4 vs. F1, respectively (Table S15). The flavonoid biosynthesis (ko00941) and flavone and flavonol biosynthesis (ko00944) were the most significant metabolic pathways during flower development (Figure S7). Furthermore, the correlation network of DEGs and DAMs showed that many genes were positively correlated with flavonoids and phenylpropanoids (Figure S8; Table S15). For example, 4CL was positively correlated with 9 flavonoids and 1 phenylpropanoid (imperatorin), while GA3ox, GA20ox and GA2ox were negatively correlated with 27 flavonoids and 5 phenylpropanoids. IAA, ARF, AUX1/LAX, and SAUR were also negatively correlated with 33 flavonoids. These results indicated that these genes related to GA and auxin signaling might be involved in the metabolism and biosynthesis of flavonoids, phenylpropanoids and their derivatives.
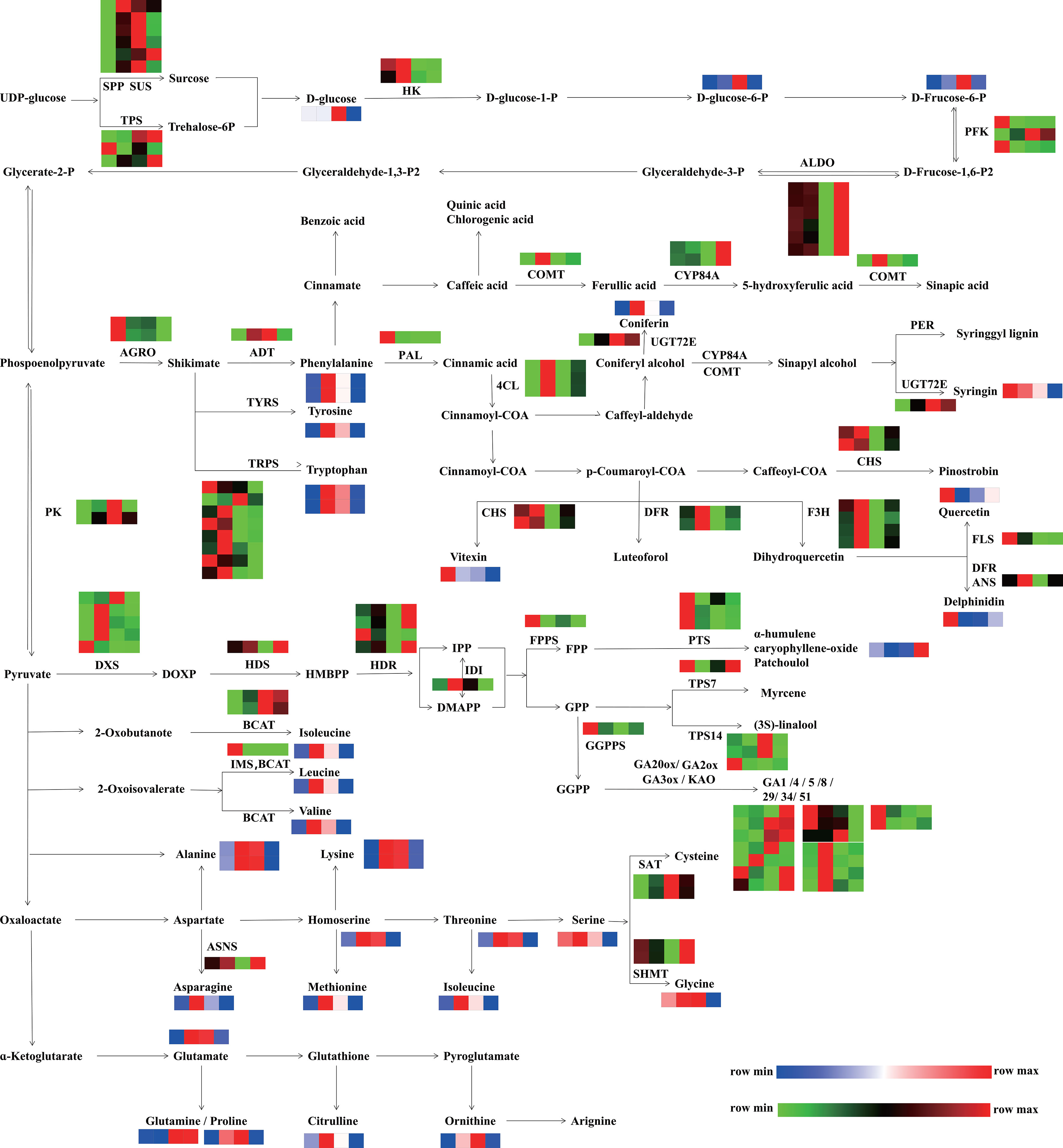
Figure 10 Integrated pathways of DEGs and DAMs involved in primary metabolism (starch and sucrose metabolism, carbon metabolism, biosynthesis of amino acids) and secondary metabolism (terpene biosynthesis, phenylpropanoid, phenols, and flavonoid biosynthesis). The pathway was redrawn based on the ko00500, ko01200, ko01230, ko00900, ko00904, ko00909, ko00940, ko00941, and ko00944 in the KEGG database (https://www.kegg.jp/). The red and green colors indicate upregulated unigenes and downregulated unigenes, respectively. The red and blue colors indicate the relative content and low relative content of metabolites, respectively. From left to right, the four squares represent the following stages: F1, inflorescence-bearing meristem stage; F2, flower bud stage; F3, full-bloom stage; and F4, withered-flower stage, respectively. SPP, sucrose-phosphatase; SUS, sucrose synthase; TPS, trehalose-phosphate phosphatase; HK, hexokinase; PFK, 6-phosphofructokinase 1; ALDO, fructose-bisphosphate aldolase, class I; PK, pyruvate kinase; AGRO, phosphoheptulonate synthase; PAL, phenylalanine ammonia-lyase; BCAT, branched-chain amino acid aminotransferase; TRPS, tryptophan synthase; 4CL, 4-coumarate CoA ligase; ASNS, aspartate-ammonia ligase; CYSE, serine O-acetyltransferase; ADT, arogenate dehydratase; IMS, 2-isopropylmalate synthase; UGT72E, coniferyl-alcohol glucosyltransferase; DFR, flavanone 4-reductase; ANS, anthocyanidin synthase; PER, peroxidase; CHS, Chalcone synthase; FLS, flavonol synthase; COMT, caffeic acid 3-O-methyltransferase; F3H, flavanone 3-dioxygenase; SAT, serine acetyltransferase; SHMT, glycine hydroxymethyl transferase; FPPS, farnesyl pyrophosphate synthase; GGPPS, geranylgeranyl diphosphate synthase; IDI, isopentenyl pyrophosphate isomerase, HDR, 1-hydroxy-2-methyl- 2-butenyl 4-diphosphate reductase; HDS, 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase; DXS, 1-deoxy-D-xylulose-5-phosphate synthase; PTS, patchoulol synthase; TPS7, trans-alpha-bergamotene synthase-like; TPS14, S-linalool synthase; KAO, ent-kaurenoic acid oxidase; GA3ox, GA 3-oxidase; GA2ox, GA 2-oxidase.
To illustrate the correlations of DEGs and DAMs related to carbohydrates, phenylpropanoids, flavonoids and phenols, and amino acids, an integrated pathway at four stages was constructed (Figure 10). In the starch and sucrose metabolism section, the accumulation of D-glucose, D-glucose-6-P, and D-fructose-6-P increased from the F1 to F3 stage and then decreased at the F4 stage, which was similar with the expression of the pyruvate kinase (PK). In the phenylpropanoids, flavonoid, and phenol biosynthesis pathway, ADT was highly expressed from the F2 to F3 stage, fitting the accumulation of phenylalanine. The expression levels of CHS, FLS and ANS were high at the F1 and F2 stages, while the transcription levels of 4CL, DFR, and F3H were highest at F3. The accumulation patterns of their products (vitexin, quercetin, delphinidin, and luteoforol) were highly consistent with the gene expression patterns. In amino acid biosynthesis, the expression pattern of ASNS was consistent with the accumulation of asparagine. The accumulation of alanine, lysine, homoserine, methionine, threonine, leucine, valine, isoleucine, glutamate, citrulline, and ornithine increased from the F2 to F3 stage. These results indicated that the active metabolism of starch and sucrose might be conducive to the biosynthesis of phenylpropanoids, flavonoids, and amino acids.
P. cablin has been widely cultivated due to the high demand for its essential oils, a crucial industrial ingredient in medicine, perfumes and cosmetics. Besides leaves, the inflorescences of P. cablin also accumulate large amounts of oil, which are composed of 17 sesquiterpenes (97.7%), the main active ingredient of patchoulol (Verma et al., 2019). In our results, 111 terpenoids and their derivatives were identified across the four flower stages and the relative content of sesquiterpenoids was highest (82.77%-88.80%) (Table S16). Recent studies of lavender have shown that terpenoid contents fluctuated with flower development stage (Li et al., 2019). Similarly, our results showed that 32 differentially accumulated terpenoids and their derivatives were identified, and their accumulation patterns were related to the flower development stage. We found that, patchoulol, a key ingredient in assessing the quality of P. cablin, was the predominant sesquiterpene present at each flower stage, and its relative content changed little during flower development (Figure S9). In addition, the relative content of patchoulol in the leaves of flowering and nonflowering plants also changed slightly (Figure S9).
As flower development progresses, flowers shifts from autotrophy to heterotrophy, accompanied with a progressively decreased photosynthesis rate and the emergence of anther secondary metabolism (Müller et al., 2010; Muhlemann et al., 2012). As expected, the pathways involved in the biosynthesis of secondary metabolites were significantly enriched from the bud stage to the withered flower stage (Figure 2). The accumulation of secondary metabolites were related to color, scent and taste (Ruan et al., 2010; Shan et al., 2019), depended on the pollinator, precursors and energy generated from the metabolism of primary metabolites (Payyavula et al., 2013; Borghi and Fernie, 2017). Starch and sucrose metabolism is generally active (Figure 2) (Jing et al., 2020; Lu et al., 2020; Cai et al., 2021), and participates in the biosynthesis of flavonoids and phenylpropanoids during flower development (Ferri et al., 2011; Payyavula et al., 2013). Sucrose, a major type of carbohydrates, is translocated from photosynthetically active tissues to nonphotosynthetic sinks (flowers) (Eveland and Jackson, 2012; Borghi and Fernie, 2017), and is degraded rapidly to provide carbon skeleton and energy for secondary metabolites (Eveland and Jackson, 2012). According to the KEGG enrichment results, a number of genes involved in phenylpropanoids, alpha-linolenic acid metabolism, and starch and sucrose metabolism, significantly increased from the F2 to F3 stage and then decreased at the F4 stage (Figure 2, Table S4). Alpha-linolenic acid metabolism is closely linked to JA (jasmonic acid) synthesis, which influence the various metabolites production, such as terpenoids and pyrethrin via the MEP pathway (Ghorbel et al., 2021; Zeng et al., 2022). Moreover, in the integration pathway of DEGs and DAMs (Figure 10), the starch and sucrose might be important energy sources for P. cablin reproduction, and phenylpropanoids, flavonoids, and amino acids might accumulate due to activated starch and sucrose metabolism. These results were similar to those from a previous study of Lonicera japonica Thunb. flower development (Yang et al., 2019).
In flower development, amino acids play a crucial role in enzyme synthesis, osmotic regulation, and providing nitrogen and energy for pollen and ovule maturation (Biancucci et al., 2015; Gaufichon et al., 2017). Recently, researchers discovered that the contents of amino acids increased in Agave amica flowers at the bud and full-bloom stages (Kutty et al., 2021). Interestingly, we also detected increased accumulation of 53 differentially accumulated amino acids at the bud and full-bloom stages, followed by a decrease at the withered flower stages. Flower opening requires osmotic oscillations including amino acids, which is followed by an influx of water into cells (Forterre, 2013; van Doorn et al., 2013). This finding might explain why amino acids accumulated mainly at the bud and full-bloom stages. In addition, our data showed that many amino acid metabolism and degradation pathways were enriched at the withered flower stage. The reason might be decomposition and translocation of proteins from senescent flowers to phloem (van Doorn, 2004). Therefore, these results suggested that the energy and carbon sources consumed during P. cablin reproduction might be derived from sucrose and starch metabolism, whereas amino acids are used more as structural components to maintain the proper status and shape of flowers. In addition, phenylpropanoids, flavonoids and amino acids might accumulate due to activated starch and sucrose metabolism.
Chlorophyll, carotenoids, and anthocyanins contributes to various flowers color, which are also affected by copigmentation, vacuolar pH, metal chelation and other processes (Trouillas et al., 2016; Stavenga et al., 2021). In our research, anthocyanin-containing compound biosynthesis processes were significantly more active from F2 to F4, indicating that anthocyanins are related to the color formation of P. cablin flowers. Copigmentation is a natural approach to stabilize anthocyanins, by forming noncovalent complexes (Stavenga et al., 2021). Flavonoids and phenolic acids, stabilizing anthocyanins and intensify color, are the most effective copigments (Klisurova et al., 2019). These two classes of metabolites are synthesized from the phenylpropanoid pathway (Buer et al., 2010). Previous research has found that flavonoids and phenylpropanoids-related genes tend to be highly expressed during the flower color formation stage (Zheng et al., 2019). In our study, the phenylpropanoid and flavonoid biosynthesis pathways were also enriched from the bud to withered flower stages in P. cablin (Figure 2; Table S4). Our results were generally consistent with the studies on Rosmarinus officinalis flowers (del Baño et al., 2003).
Previous research has shown that the contents of phenolic compounds and flavonoids change during flower development in some rose species (Elmastaş et al., 2017; Chamani et al., 2020). In the present study, flavonoid and phenolic acid contents also changed during the development of P. cablin flowers, indicating similar accumulation patterns between the bud and full-bloom stages. The spikelets of P. cablin have whorls of flowers ranging in color from white to lilac, purplish-white petals, purple stamen filaments, yellow anthers, and a green calyx and ovaries. Buds and full-bloom flowers contain similar floral organs, including purplish-white petals, purple stamen filaments, and yellow anthers, which are different from those at other stages. This finding may explain why the accumulation of metabolites at the bud and full-bloom stages was similar. Taken together, our results suggested that flavonoids and phenolic acids might contribute to pigment formation in P. cablin.
Plant hormones, such as GAs, auxin, and cytokinin, have important roles in flowering (Conti, 2017). Among them, GAs play key roles in plant flowering (Mutasa-Gottgens and Hedden, 2009; Conti, 2017). First, GAs can accelerate floral induction in several species, such as A. thaliana (Sven Eriksson et al., 2006) and Eriobotrya japonica (Jing et al., 2020). Second, GAs can promote the growth and maturation of floral organs, such as petals (Plackett et al., 2012), stamens (Plackett et al., 2011), ovaries (Lange and Lange, 2016), anthers and pistils (Hu et al., 2008). GAs are synthesized in anthers and pistils, and transfer to other floral organs for the development of them (Pimenta Lange et al., 2012; Binenbaum et al., 2018). The biosynthesis of active GAs involves 12 steps, regulated by a series of enzymes (Figure 3A) (Fan et al., 2017). As the starting and final enzymes, GA3ox and GA20ox are particularly important for controlling bioactive GA levels. In our study, the highest expression of GA3ox was detected at the flower bud stage while GA20ox was mainly upregulated from the full-bloom to the withered flower stages (Figure 3B). A recent survey found that VpGA3ox and VpGA20ox participate in the initiation of floral organ formation in Viola philippica, proved by upregulated expressions in the primordia of pistils and stamens (Li et al., 2021). Therefore, GA signaling might participate in floral organ formation and maturation during P. cablin flower development.
GA signaling exerted opposite effects on different flower development stages (Yamaguchi et al., 2014), since the concentration of GAs is relatively high during the flower bud stage and then decreases after anthesis (Cheng et al., 2004; Serrani et al., 2007; Iftikhar et al., 2020). A recent study of Petunia hybrida also found that GA1 and GA4 were detected at negligible amounts when the phenylpropanoid volatiles were actively produced and emitted after anthesis, implying a negative regulation of GAs on the synthesis of phenylpropanoids (Ravid et al., 2017). Similarly, no corresponding products were detected in our results, although several key enzyme genes (KAO, GA2ox, GA3ox, and GA20ox) were active at the flower bud and full-bloom stages. In A. thaliana flowers, two potential sites (stamens and receptacles) for bioactive GA synthesis were identified, suggesting that GAs might be dynamically transported from these organs to other parts during flower development (Hu et al., 2008). Thus, we proposed that GAs may have been transferred to other parts of the inflorescence to produce scent-associated metabolites during the development of P. cablin flowers. These results also suggested that GA signaling might be a regulatory switch between flower developmental and flower-related scent metabolism in P. cablin.
Auxin is another important hormone in the flowering process, including inflorescence and floral organ initiation (Cucinotta et al., 2021). Auxin signal transduction involves four core components: AUX/IAA, AUX/LAX, transport inhibitor response 1/auxin signaling F-box protein (TIR1/AFB), and ARF family member proteins (Swarup and Péret, 2012; Lavy and Estelle, 2016). As shown in our results, auxin signal transduction was clearly enriched at the bud flower stage (Figure S1). Interestingly, all AUX/LAXs, major auxin inflow carriers, were mainly upregulated from the bud to withered flower stages (Figure S2), indicating that high concentration of auxin is required for P. cablin flower development. In nature, IAA presents in active free-state or inactive combined-state, controlled by GH3 (Ludwig-Müller, 2011). In our study, expressions of most IAA genes were upregulated from the bud stage to the full-bloom stage, however the transcript levels of GH3 genes decreased at same stages (Figure S2), suggesting that more active auxin are needed for the continuous stimulation of P. cablin flower development.
Research has shown that auxin participates in flower closing-opening activity, proved by upregulated expressions of IAA and ARF during flower opening but downregulated expressions during closing (Ke et al., 2018). Similarly, our transcriptomic data showed that many homologs of IAA, ARF, and SAUR were maintained high transcript levels from the bud to the full-bloom stage (Figure S2). According to these results, the activation of auxin signaling at the full-bloom stage might be associated with flower opening and the maintenance of blooms. In addition, auxin signaling has many functions in the growth of stamens, petals, and pedicels (Cheng et al., 2006; van Doorn et al., 2013) and accelerates the maturation and development of pollen, anthers and ovules (Cecchetti et al., 2008; Galbiati et al., 2013). According to our observations, the floral organs within most bud samples of P. cablin were incomplete and still developing. Therefore, the activation of auxin signaling at the bud flower stage might favor floral organ formation.
In summary, GA and auxin signaling might be involved in floral organ development in P. cablin. In addition, GA signaling might be a regulatory switch between flower development and flower-related scent/color metabolism, while auxin signaling might be beneficial for flower opening.
Three categories of genes are responsible for flower development, including floral pathway integrators (FT and SOC1), floral meristem identity genes (AP1 and FUL) and floral organ identity genes (AP2, AP3, PI, SEP and AG) (Satish and Manju, 2018; Cao et al., 2021). Floral organ identity genes are regulated by flower pathway integrators. For example, AtFT can positively regulate the expression of AtAP1 and AtFUL, and AtSOC1 upregulates the transcription of AtAP2, AtPI, and AtAG via AtLFY (Blázquez, 2000). As expected, our results showed that FT was correlated with SVP-like, SPL7, and KAO, whereas SOC1 were strongly correlated with the SVP-like and two new MIKC-MADS genes. Another function of flowering integrators is to integrate the environmental and internal flower induction signals. For example, AtSPL3/4/5 and AtSPL2/9/10/11/13/15 can directly bind to AtFT and AtSOC1 in the shoot apex meristem, and then induce the expression of AtAP1, AtLFY, and AtFUL (Xu et al., 2016). EjSPL3/4/5 can significantly upregulate the expression levels of EjSOC1-1, EjAP1, and EjLFY-1 in E. japonicat (Jiang et al., 2019). PvSPL also directly upregulates the expression levels of PvSEP3 and PvAP1/FUL to promote Panicum virgatum flowering (Yamaguchi et al., 2009; Gou et al., 2019). According to our results, SPL12 was correlated with SOC1, and SVP-like gene, whereas SPL7, SPL3, and SPL8 were strongly correlated with SVP-like, new MIKC-MADS gene (Pcab019620), and AG genes. This result suggested that the transcription of SOC1, FT, SVP, AG and new MICK-MADS genes might be regulated by aging-related genes (SPL) in P. cablin.
Hormone signaling pathways are important for flower development. Previous reporters also revealed that GAs regulate the transcription of AtSVP and AtSOC1 via an AtDELLA-mediated signaling pathway (Li et al., 2008). Interestingly, AtSVP negatively regulates GA signaling by repressing the transcription of GA20ox (Leijten et al., 2018). In our results, SVP-like genes were correlated with GA2ox and KAO, while new MIKC-MADS genes and AG were correlated with GA2ox, respectively. Auxin signaling appears to be a positive regulator of the flowering process (Renau-Morata et al., 2021). AtARF5 is presumed to be closely associated with the initiation of flower primordia and organs, and significantly upregulates the expression level of AtLFY (Krizek and Eaddy, 2012; Yamaguchi et al., 2016). In our results, IAA was strongly correlated with AP3, SEP, AG, and GH3, while AUX was strongly correlated with SEP. These results also indicated that auxin signaling might regulate P. cablin flower development by affecting the expression of these flower development-related genes. In addition, auxin can promote flowering by regulating the GA content and promoting DELLA degradation (O’Neill and Ross, 2002; Fu and Harberd, 2003; Frigerio et al., 2006). Similarly, our results also showed that auxin signaling-related genes were strongly correlated with GA signaling-related genes (Figure 9). These results implied that GA and auxin signaling might regulate P. cablin flower development in a cross-linked manner.
The flower induction regulation pathway typically forms a regulatory network integrating flower stimulation and triggering the transition from vegetative to reproductive stages. Previous research found that the aging-related AtSPL3/9/15 gene interacts with AtDELLA and impairs the activation of flower-related genes (such as AtSOC1), suggesting that SPLs are key targets of GA signaling (Yu et al., 2012; Hyun et al., 2017; Li et al., 2018). In our results, SPL12 was correlated with GA2ox, GH3, and IAA, while SPL8 was strongly correlated with IAA and GASA (Figure 9). These results suggested crosstalk between GA, auxin, and aging pathways during flower development in P.cablin. In summary, the GA, auxin, and aging pathways might form a cross-regulatory network to regulate P. cablin flowering.
Plants also produce volatiles and pigments, including phenylpropanoids and flavonoids, for pollinators during flowering. Those natural products are regulated by core proteins, including PAL, 4CL, C4H, and PAAS. Hormone signals are an important regulatory link in floral color/scent during flower development (Ravid et al., 2017; Lu et al., 2020). In our results, the phenylpropanoid and flavonoid biosynthesis pathways were also enriched from the bud to withered flower stages in P. cablin (Figure 2; Table S4), and GA signaling-related genes (GA3ox, GA20ox and GA2ox) were negatively correlated with 27 flavonoids, 5 phenylpropanoids and 8 terpenes. Auxin signaling-related genes (IAA, ARF, AUX1, LAX, and SAUR) were also negatively correlated with 33 flavonoids, 7 phenylpropanoids, and 15 terpenes (Figure S8). These results suggested GA and auxin signaling might be involved in the metabolism and biosynthesis of flavonoids, phenylpropanoids, terpenes and their derivatives. In addition, DELLA proteins can bind with a variety of transcription factors (i.e., AtSPL15, AtMYC2, and AtMYB23) and prevent them from binding to their target genes (Qi et al., 2014; Davière and Achard, 2016). EOBI, EOBII and ODO1 in the MYB family, were proven to regulate the expression of some phenylpropanoid-related genes (CS, CM, and PAL) (Verdonk et al., 2005; Spitzer-Rimon et al., 2010). What’s more, DELLA can also bind with AtMYC2 to inhibit the expression of sesquiterpene synthase genes (AtTPS11 and AtTSP22) (Hong et al., 2012). Similarly, PatMYC2b1/PatMYC2b2 can directly regulate the expression of PatPTS in P. cablin (Wang et al., 2019). Thus, we propose that DELLA might bind to some MYB and MYC TFs to regulate some genes related to the phenylpropanoid, flavonoid and terpene biosynthesis pathways.
Based on our results, a proposed model of the molecular and genetic networks regulating P. cablin flower development is shown in Figure 11. GA signaling acts as an important regulatory switch connecting flower development and pollinator attraction. DELLAs are an important inhibitor of GA signaling and are degraded after interacting with the GA receptor GID1 when GA accumulates at high levels. IAAs can induce the expression of GA2ox, GA3ox and GA20ox to increase the biosynthesis of GAs, which results in DELLA degradation to promote flower development. SPL induces the expression of SOC1, FT, SEP, AG, AP1/FUL and new MIKC-MADS to promote flower development in P. cablin, and is inhibited by DELLA. In addition, the floral repressor SVP, another target of DELLA, negatively regulates GA signaling by repressing the transcription of GA2ox, GA3ox and GA20ox. Meanwhile, GA also negatively regulates the transcription of genes (PAL and 4CL, PTS and TPS) involved in phenylpropanoid, flavonoid and terpene production via DELLA-mediated signaling pathway.
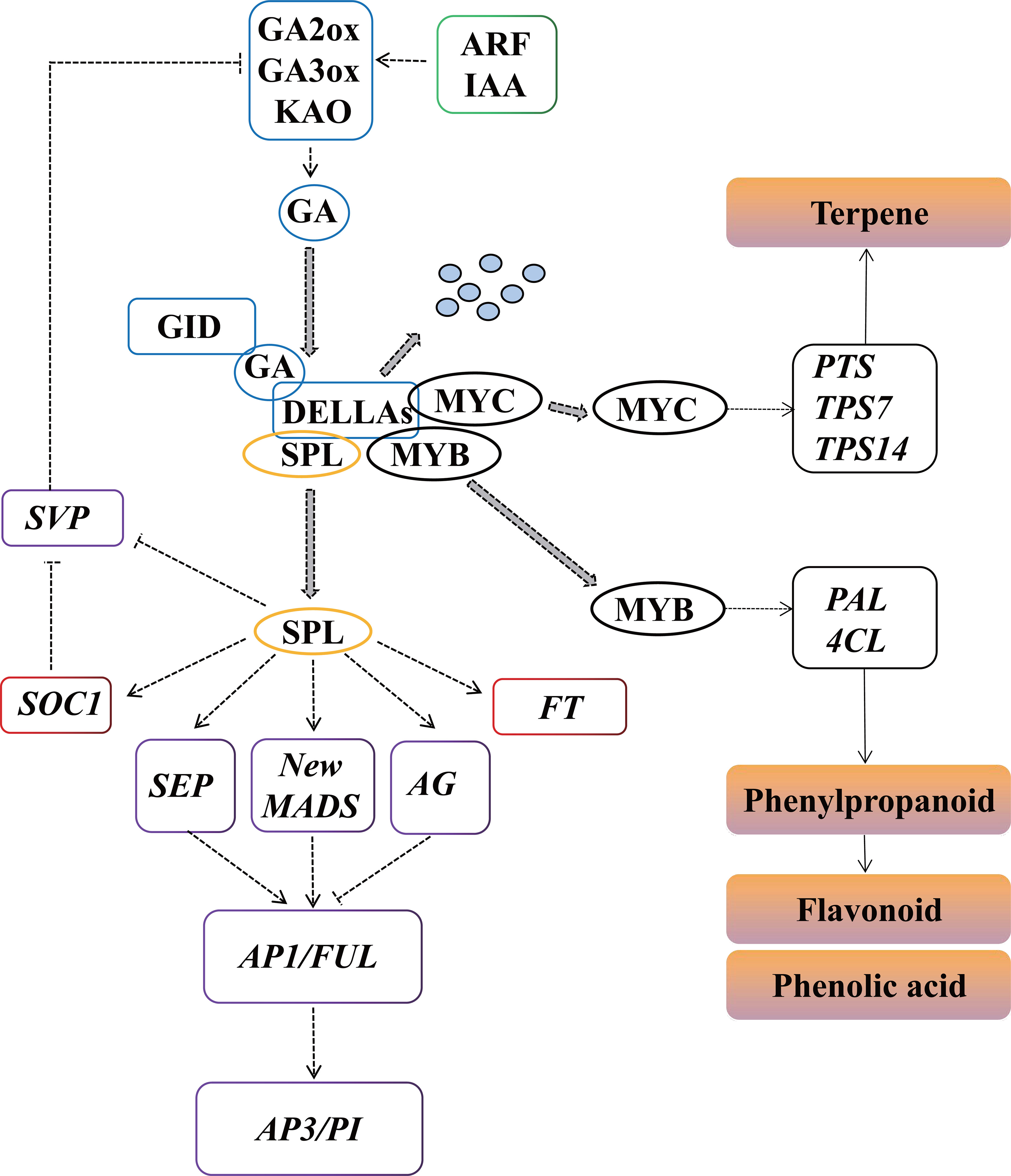
Figure 11 Predicted regulatory model involved in P. cablin flower development. KAO, ent-kaurenoic acid oxidase; GA3ox, GA 3-oxidase; GA2ox, GA 2-oxidase; GID1, gibberellin receptor; IAA, indole-3-acetic acid; ARF, auxin response factor; GH3, Gretchen Hagen3; SPL, squamosa promoter binding protein-like; FT, FLOWERING LOCUS T; SOC1, SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1; AP1, APETALA1; FUL, FRUITFUL; PI, PISTILLATA; AP3, APETALA3; SEP, SEPALLATA; AG, AGAMOUS, PAL, phenylalanine ammonia-lyase; 4CL, 4-coumarate CoA ligase, PTS, patchoulol synthase; TPS7, trans-alpha-bergamotene synthase-like; TPS14, S-linalool synthase.
P. cablin is an important medicinal plant species known for its essential oil. However, due to the difficulty in flowering, it is mainly cultivated and preserved though cottage propagation. Cottage propagation tends to result in virus accumulation and quality losses, hindering the development and utilization of P. cablin. Flowering, the beginning of seed formation, is regulated by precise molecular and genetic networks involved in a variety of signaling pathways. A model of the molecular and genetic regulatory networks of P. cablin flower development is proposed in Figure 11. In this model, three signaling pathways (GA, auxin and aging) can regulate the transcript levels of floral organ identity genes (AP1, AG, FUL, PI and SEP, etc.) to promote the development of floral organs and anthesis. Moreover, GA also negatively regulate the transcription of some genes involved in floral color/scent (phenylpropanoid and flavonoids) production via the DELLA-mediated signaling pathway. GA signaling acts as an important regulatory switch connecting flower morphology and pollinator attraction, to maintain the flowers in the best pollination state for successful reproduction. These results will provide a basis for future studies on the sexual reproduction and breeding of P. cablin.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
YW conceived, designed, and supervised the research project. CZ, XL and YL performed the experiments. CZ, JY and GY analyzed the data. CZ and YL wrote the manuscript. HY and DY provided input on the data presentation and critically revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was financially supported by the National Natural Science Foundation of China (No. 82260737), the Key Research and Development Program of Hainan Province (No. ZDYF2021SHFZ075), and the Startup Funding from Hainan University (No. KYQD(ZR)23018).
We would like to thank Dr. Mingxun Ren for his guidance on the experiments and HLZ, CJ and YFH from SHANGHAI BIOTREE BIOMEDICAL TECHNOLOGY Co. Ltd. (Shanghai, China), Majorbio Biotech Co. Ltd. (Shanghai, China) and PlantTech Biotech Co. Ltd. (Beijing) for their help with data processing, respectively. We would also like to thank AJE (https://www.aje.cn/) for providing English language editing.
Author CZ was employed by the company Guangdong VTR BioTech Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1201486/full#supplementary-material
Biancucci, M., Mattioli, R., Forlani, G., Funck, D., Costantino, P., Trovato, M. (2015). Role of proline and GABA in sexual reproduction of angiosperms. Front. Plant Sci. 6. doi: 10.3389/fpls.2015.00680
Binenbaum, J., Weinstain, R., Shani, E. (2018). Gibberellin localization and transport in plants. Trends Plant Sci. 23, 410–421. doi: 10.1016/j.tplants.2018.02.005
Blázquez, M. A. (2000). Flower development pathways. J. Cell Sci. 113, 3547–3548. doi: 10.1242/jcs.113.20.3547
Borghi, M., Fernie, A. R. (2017). Floral metabolism of sugars and amino acids: implications for pollinators’ preferences and seed and fruit set. Plant Physiol. 175, 1510–1524. doi: 10.1104/pp.17.01164
Buchfink, B., Reuter, K., Drost, H. G. (2021). Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368. doi: 10.1038/s41592-021-01101-x
Buer, C. S., Imin, N., Djordjevic, M. A. (2010). Flavonoids: new roles for old molecules. J. Integr. Plant Biol. 52, 98–111. doi: 10.1111/j.1744-7909.2010.00905.x
Cai, Z., Yang, C., Liao, J., Song, H., Zhang, S. (2021). Sex-biased genes and metabolites explain morphologically sexual dimorphism and reproductive costs in Salix paraplesia catkins. Hortic. Res. 8, 125. doi: 10.1038/s41438-021-00566-3
Cao, S., Luo, X., Xu, D., Tian, X., Song, J., Xia, X., et al. (2021). Genetic architecture underlying light and temperature mediated flowering in Arabidopsis, rice, and temperate cereals. New Phytol. 230, 1731–1745. doi: 10.1111/nph.17276
Cecchetti, V., Altamura, M. M., Falasca, G., Costantino, P., Cardarelli, M. (2008). Auxin regulates arabidopsis anther dehiscence, pollen maturation, and filament elongation. Plant Cell. 20, 1760–1774. doi: 10.1105/tpc.107.057570
Chamani, E., Wagstaff, C., Kanani, M. (2020). Phenolics pattern of cut H3O rose flowers during floral development. Sci. Hortic. 271, 109460. doi: 10.1016/j.scienta.2020.109460
Cheng, Y., Dai, X., Zhao, Y. (2006). Auxin biosynthesis by the YUCCA flavin monooxygenases controls the formation of floral organs and vascular tissues in Arabidopsis. Genes Dev. 20, 1790–1799. doi: 10.1101/gad.1415106
Cheng, H., Qin, L., Lee, S., Fu, X., Richards, D. E., Cao, D., et al. (2004). Gibberellin regulates Arabidopsis floral development via suppression of DELLA protein function. Development 131, 1055–1064. doi: 10.1242/dev.00992
Conti, L. (2017). Hormonal control of the floral transition: can one catch them all? Dev. Biol. 430, 288–301. doi: 10.1016/j.ydbio.2017.03.024
Cucinotta, M., Cavalleri, A., Chandler, J. W., Colombo, L. (2021). Auxin and flower development: a blossoming field. Cold Spring Harb. Perspect. Biol. 13, a039974. doi: 10.1101/cshperspect.a039974
Davière, J.-M., Achard, P. (2016). A pivotal role of DELLAs in regulating multiple hormone signals. Mol. Plant 9, 10–20. doi: 10.1016/j.molp.2015.09.011
del Baño, M. J., Lorente, J., Castillo, J., Benavente-García, O., del Río, J. A., Ortuño, A., et al. (2003). Phenolic diterpenes, flavones, and rosmarinic acid distribution during the development of leaves, flowers, stems, and roots of Rosmarinus officinalis. antioxidant activity. J. Agric. Food Chem. 51, 4247–4253. doi: 10.1021/jf0300745
Elmastaş, M., Demir, A., Genç, N., Dölek, Ü., Güneş, M. (2017). Changes in flavonoid and phenolic acid contents in some Rosa species during ripening. Food Chem. 235, 154–159. doi: 10.1016/j.foodchem.2017.05.004
Eveland, A. L., Jackson, D. P. (2012). Sugars, signalling, and plant development. J. Exp. Bot. 63, 3367–3377. doi: 10.1093/jxb/err379
Fan, X., Yuan, D., Tian, X., Zhu, Z., Liu, M., Cao, H. (2017). Comprehensive transcriptome analysis of phytohormone biosynthesis and signaling genes in the flowers of chinese chinquapin (Castanea henryi). J. Agric. Food Chem. 65, 10332–10349. doi: 10.1021/acs.jafc.7b03755
Ferri, M., Righetti, L., Tassoni, A. (2011). Increasing sucrose concentrations promote phenylpropanoid biosynthesis in grapevine cell cultures. J. Plant Physiol. 168, 189–195. doi: 10.1016/j.jplph.2010.06.027
Fornara, F., de Montaigu, A., Coupland, G. (2010). SnapShot: control of flowering in Arabidopsis. Cell 141, 550–550.e2. doi: 10.1016/j.cell.2010.04.024
Forterre, Y. (2013). Slow, fast and furious: understanding the physics of plant movements. J. Exp. Bot. 64, 4745–4760. doi: 10.1093/jxb/ert230
Frigerio, M., Alabadí, D., Pérez-Gómez, J., García-Cárcel, L., Phillips, A. L., Hedden, P., et al. (2006). Transcriptional regulation of gibberellin metabolism genes by auxin signaling in Arabidopsis. Plant Physiol. 142, 553–563. doi: 10.1104/pp.106.084871
Fu, X., Harberd, N. P. (2003). Auxin promotes Arabidopsis root growth by modulating gibberellin response. Nature 421, 740–743. doi: 10.1038/nature01387
Galbiati, F., Sinha Roy, D., Simonini, S., Cucinotta, M., Ceccato, L., Cuesta, C., et al. (2013). An integrative model of the control of ovule primordia formation. Plant J. 76, 446–455. doi: 10.1111/tpj.12309
Gaufichon, L., Marmagne, A., Belcram, K., Yoneyama, T., Sakakibara, Y., Hase, T., et al. (2017). ASN1-encoded asparagine synthetase in floral organs contributes to nitrogen filling in Arabidopsis seeds. Plant J. 91, 371–393. doi: 10.1111/tpj.13567
Ghorbel, M., Brini, F., Sharma, A., Landi, M. (2021). Role of jasmonic acid in plants: the molecular point of view. Plant Cell Rep. 40, 1471–1494. doi: 10.1007/s00299-021-02687-4
Gou, J., Tang, C., Chen, N., Wang, H., Debnath, S., Sun, L., et al. (2019). SPL7 and SPL8 represent a novel flowering regulation mechanism in switchgrass. New Phytol. 222, 1610–1623. doi: 10.1111/nph.15712
He, Y., Peng, F., Deng, C., Xiong, L., Huang, Z., Zhang, R., et al. (2018). Building an octaploid genome and transcriptome of the medicinal plant Pogostemon cablin from lamiales. Sci. Data 5, 180274. doi: 10.1038/sdata.2018.274
Hong, G. J., Xue, X. Y., Mao, Y. B., Wang, L. J., Chen, X. Y. (2012). Arabidopsis MYC2 interacts with DELLA proteins in regulating sesquiterpene synthase gene expression. Plant Cell 24, 2635–2648. doi: 10.1105/tpc.112.098749
Hu, J., Mitchum, M. G., Barnaby, N., Ayele, B. T., Ogawa, M., Nam, E., et al. (2008). Potential sites of bioactive gibberellin production during reproductive growth in Arabidopsis. Plant Cell 20, 320–336. doi: 10.1105/tpc.107.057752
Hyun, Y., Richter, R., Coupland, G. (2017). Competence to flower: age-controlled sensitivity to environmental cues. Plant Physiol. 173, 36–46. doi: 10.1104/pp.16.01523
Iftikhar, J., Lyu, M., Liu, Z., Mehmood, N., Munir, N., Ahmed, M. A. A., et al. (2020). Sugar and hormone dynamics and the expression profiles of SUT/SUC and SWEET sweet sugar transporters during flower development in Petunia axillaris. Plants (Basel) 9, E1770. doi: 10.3390/plants9121770
Jiang, Y., Peng, J., Wang, M., Su, W., Gan, X., Jing, Y., et al. (2019). The role of EjSPL3, EjSPL4, EjSPL5, and EjSPL9 in regulating flowering in loquat (Eriobotrya japonica lindl.). Int. J. Mol. Sci. 21, E248. doi: 10.3390/ijms21010248
Jing, D., Chen, W., Hu, R., Zhang, Y., Xia, Y., Wang, S., et al. (2020). An integrative analysis of transcriptome, proteome and hormones reveals key differentially expressed genes and metabolic pathways involved in flower development in loquat. Int. J. Mol. Sci. 21, 5107. doi: 10.3390/ijms21145107
Ke, M., Gao, Z., Chen, J., Qiu, Y., Zhang, L., Chen, X. (2018). Auxin controls circadian flower opening and closure in the waterlily. BMC Plant Biol. 18, 143. doi: 10.1186/s12870-018-1357-7
Klisurova, D., Petrova, I., Ognyanov, M., Georgiev, Y., Kratchanova, M., Denev, P. (2019). Co-Pigmentation of black chokeberry (Aronia melanocarpa) anthocyanins with phenolic co-pigments and herbal extracts. Food Chem. 279, 162–170. doi: 10.1016/j.foodchem.2018.11.125
Krizek, B. A., Eaddy, M. (2012). AINTEGUMENTA-like6 regulates cellular differentiation in flowers. Plant Mol. Biol. 78, 199–209. doi: 10.1007/s11103-011-9844-3
Kutty, N. N., Ghissing, U., Mitra, A. (2021). Revealing floral metabolite network in tuberose that underpins scent volatiles synthesis, storage and emission. Plant Mol. Biol. 106, 533–554. doi: 10.1007/s11103-021-01171-7
Lange, M. J. P., Lange, T. (2016). Ovary-derived precursor gibberellin A9 is essential for female flower development in cucumber. Development 143, 4425. doi: 10.1242/dev.135947
Langfelder, P., Horvath, S. (2008). WGCNA: an r package for weighted correlation network analysis. BMC Bioinf. 9, 559. doi: 10.1186/1471-2105-9-559
Lavy, M., Estelle, M. (2016). Mechanisms of auxin signaling. Development 143, 3226–3229. doi: 10.1242/dev.131870
Leijten, W., Koes, R., Roobeek, I., Frugis, G. (2018). Translating flowering time from Arabidopsis thaliana to brassicaceae and asteraceae crop species. Plants (Basel) 7 (4), 111. doi: 10.3390/plants7040111
Li, H., Li, J., Dong, Y., Hao, H., Ling, Z., Bai, H., et al. (2019). Time-series transcriptome provides insights into the gene regulation network involved in the volatile terpenoid metabolism during the flower development of lavender. BMC Plant Biol. 19, 313. doi: 10.1186/s12870-019-1908-6
Li, Q., Li, J., Zhang, L., Pan, C., Yang, N., Sun, K., et al. (2021). Gibberellins are required for dimorphic flower development in Viola philippica. Plant Sci. 303, 110749. doi: 10.1016/j.plantsci.2020.110749
Li, X. Y., Lin, E. P., Huang, H. H., Niu, M. Y., Tong, Z. K., Zhang, J. H. (2018). Molecular characterization of SQUAMOSA PROMOTER BINDING PROTEIN-like (SPL) gene family in betula luminifera. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00608
Li, D., Liu, C., Shen, L., Wu, Y., Chen, H., Robertson, M., et al. (2008). A repressor complex governs the integration of flowering signals in Arabidopsis. Dev. Cell 15, 110–120. doi: 10.1016/j.devcel.2008.05.002
Li, C., Wu, Y., Guo, Q. (2011). Floral and pollen morphology of Pogostemon cablin (Lamiaceae) from different habitats and its taxonomic significance. Proc. Eng. 18, 295–300. doi: 10.1016/j.proeng.2011.11.046
Lu, B., Chen, L., Hao, J., Zhang, Y., Huang, J. (2020). Comparative transcription profiles reveal that carbohydrates and hormone signalling pathways mediate flower induction in Juglans sigillata after girdling. Ind. Crops Prod. 153, 112556. doi: 10.1016/j.indcrop.2020.112556
Ludwig-Müller, J. (2011). Auxin conjugates: their role for plant development and in the evolution of land plants. J. Exp. Bot. 62, 1757–1773. doi: 10.1093/jxb/erq412
Mauceri, A., Abenavoli, M. R., Toppino, L., Panda, S., Mercati, F., Aci, M. M., et al. (2021). Transcriptomics reveal new insights into molecular regulation of nitrogen use efficiency in solanum melongena. J. Exp. Bot. 72, 4237–4253. doi: 10.1093/jxb/erab121
Muhlemann, J. K., Maeda, H., Chang, C. Y., San Miguel, P., Baxter, I., Cooper, B., et al. (2012). Developmental changes in the metabolic network of snapdragon flowers. PloS One 7, e40381. doi: 10.1371/journal.pone.0040381
Müller, G. L., Drincovich, M. F., Andreo, C. S., Lara, M. V. (2010). Role of photosynthesis and analysis of key enzymes involved in primary metabolism throughout the lifespan of the tobacco flower. J. Exp. Bot. 61, 3675–3688. doi: 10.1093/jxb/erq187
Mutasa-Gottgens, E., Hedden, P. (2009). Gibberellin as a factor in floral regulatory networks. J. Exp. Bot. 60, 1979–1989. doi: 10.1093/jxb/erp040
Ó’Maoiléidigh, D. S., Graciet, E., Wellmer, F. (2014). Gene networks controlling Arabidopsis thaliana flower development. New Phytol. 201, 16–30. doi: 10.1111/nph.12444
O’Neill, D. P., Ross, J. J. (2002). Auxin regulation of the gibberellin pathway in pea. Plant Physiol. 130, 1974–1982. doi: 10.1104/pp.010587
Pang, Z., Chong, J., Zhou, G., de Lima Morais, D. A., Chang, L., Barrette, M., et al. (2021). MetaboAnalyst 5.0: narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 49, W388–W396. doi: 10.1093/nar/gkab382
Payyavula, R. S., Singh, R. K., Navarre, D. A. (2013). Transcription factors, sucrose, and sucrose metabolic genes interact to regulate potato phenylpropanoid metabolism. J. Exp. Bot. 64, 5115–5131. doi: 10.1093/jxb/ert303
Pimenta Lange, M. J., Knop, N., Lange, T. (2012). Stamen-derived bioactive gibberellin is essential for male flower development of Cucurbita maxima l. J. Exp. Bot. 63, 2681–2691. doi: 10.1093/jxb/err448
Plackett, A. R. G., Powers, S. J., Fernandez-Garcia, N., Urbanova, T., Takebayashi, Y., Seo, M., et al. (2012). Analysis of the developmental roles of the arabidopsis gibberellin 20-oxidases demonstrates that GA20ox1, -2, and -3 are the dominant paralogs. Plant Cell 24, 941–960. doi: 10.1105/tpc.111.095109
Plackett, A. R. G., Thomas, S. G., Wilson, Z. A., Hedden, P. (2011). Gibberellin control of stamen development: a fertile field. Trends Plant Sci. 16, 568–578. doi: 10.1016/j.tplants.2011.06.007
Qi, T., Huang, H., Wu, D., Yan, J., Qi, Y., Song, S., et al. (2014). Arabidopsis DELLA and JAZ proteins bind the WD-Repeat/bHLH/MYB complex to modulate gibberellin and jasmonate signaling synergy. Plant Cell 26, 1118–1133. doi: 10.1105/tpc.113.121731
Ravid, J., Spitzer-Rimon, B., Takebayashi, Y., Seo, M., Cna’ani, A., Aravena-Calvo, J., et al. (2017). GA as a regulatory link between the showy floral traits color and scent. New Phytol. 215, 411–422. doi: 10.1111/nph.14504
Ren, L., Sun, H., Dai, S., Feng, S., Qiao, K., Wang, J. A., et al. (2021). Identification and characterization of MIKC(c)-type MADS-box genes in the flower organs of Adonis amurensis. Int. J. Mol. Sci. 22 (17), 9362. doi: 10.3390/ijms22179362
Renau-Morata, B., Nebauer, S. G., García-Carpintero, V., Cañizares, J., Gómez Minguet, E., de los Mozos, M., et al. (2021). Flower induction and development in saffron: timing and hormone signalling pathways. Ind. Crops Prod. 164, 113370. doi: 10.1016/j.indcrop.2021.113370
Ruan, Y. L., Jin, Y., Yang, Y. J., Li, G. J., Boyer, J. S. (2010). Sugar input, metabolism, and signaling mediated by invertase: roles in development, yield potential, and response to drought and heat. Mol. Plant 3, 942–955. doi: 10.1093/mp/ssq044
Satish, C. B., Manju, A. L. (2018). Plant physiology, development and metabolism: physiology of flowering (Singapore: Springer Singapore).
Serrani, J. C., Sanjuán, R., Ruiz-Rivero, O., Fos, M., García-Martínez, J. L. (2007). Gibberellin regulation of fruit set and growth in tomato. Plant Physiol. 145, 246–257. doi: 10.1104/pp.107.098335
Shan, H., Cheng, J., Zhang, R., Yao, X., Kong, H. (2019). Developmental mechanisms involved in the diversification of flowers. Nat. Plants 5, 917–923. doi: 10.1038/s41477-019-0498-5
Shen, Y., Li, W., Zeng, Y., Li, Z., Chen, Y., Zhang, J., et al. (2022). Chromosome-level and haplotype-resolved genome provides insight into the tetraploid hybrid origin of patchouli. Nat. Commun. 13, 3511. doi: 10.1038/s41467-022-31121-w
Spitzer-Rimon, B., Marhevka, E., Barkai, O., Marton, I., Edelbaum, O., Masci, T., et al. (2010). EOBII, a gene encoding a flower-specific regulator of phenylpropanoid volatiles’ biosynthesis in petunia. Plant Cell 22, 1961–1976. doi: 10.1105/tpc.109.067280
Stavenga, D. G., Leertouwer, H. L., Dudek, B., van der Kooi, C. J. (2021). Coloration of flowers by flavonoids and consequences of pH dependent absorption. Front. Plant Sci. 11. doi: 10.3389/fpls.2020.600124
Sven Eriksson, H. B., Moritz, T., Nilsson, O. (2006). GA4 is the active gibberellin in the regulation of LEAFY transcription and arabidopsis floral initiation. Plant Cell 18, 2172–2181. doi: 10.1105/Ftpc.106.042317
Swamy, M. K., Sinniah, U. R. (2015). A comprehensive review on the phytochemical constituents and pharmacological activities of Pogostemon cablin benth.: an aromatic medicinal plant of industrial importance. Molecules 20, 8521–8547. doi: 10.3390/molecules20058521
Swamy, M. K., Sinniah, U. R. (2016). Patchouli (Pogostemon cablin benth.): botany, agrotechnology and biotechnological aspects. Ind. Crops Prod 87, 161–176. doi: 10.1016/j.indcrop.2016.04.032
Swarup, R., Péret, B. (2012). AUX/LAX family of auxin influx carriers-an overview. Front. Plant Sci. 3. doi: 10.3389/fpls.2012.00225
Trouillas, P., Sancho-García, J. C., De Freitas, V., Gierschner, J., Otyepka, M., Dangles, O. (2016). Stabilizing and modulating color by copigmentation: insights from theory and experiment. Chem. Rev. 116, 4937–4982. doi: 10.1021/acs.chemrev.5b00507
van Doorn, W. G. (2004). Is petal senescence due to sugar starvation? Plant Physiol. 134, 35–42. doi: 10.1104/pp.103.033084
van Doorn, W. G., Dole, I., Çelikel, F. G., Harkema, H. (2013). Opening of iris flowers is regulated by endogenous auxins. J. Plant Physiol. 170, 161–164. doi: 10.1016/j.jplph.2012.09.014
Verdonk, J. C., Haring, M. A., van Tunen, A. J., Schuurink, R. C. (2005). ODORANT1 regulates fragrance biosynthesis in petunia flowers. Plant Cell 17, 1612–1624. doi: 10.1105/tpc.104.028837
Verma, R. S., Padalia, R. C., Chauhan, A., Singh, V. R. (2019). Chemical composition of leaves, inflorescence, whole aerial-parts and root essential oils of patchouli Pogostemon cablin (Blanco) benth. J. Essent. Oil Res. 31, 319–325. doi: 10.1080/10412905.2019.1566100
Wang, X., Chen, X., Zhong, L., Zhou, X., Tang, Y., Liu, Y., et al. (2019). PatJAZ6 acts as a repressor regulating ja-induced biosynthesis of patchouli alcohol in Pogostemon cablin. IJMS 20, 6038. doi: 10.3390/ijms20236038
Wellmer, F., Riechmann, J. L. (2010). Gene networks controlling the initiation of flower development. Trends Genet. 26, 519–527. doi: 10.1016/j.tig.2010.09.001
Xie, C., Mao, X., Huang, J., Ding, Y., Wu, J., Dong, S., et al. (2011). KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, W316–W322. doi: 10.1093/nar/gkr483
Xu, M., Hu, T., Zhao, J., Park, M. Y., Earley, K. W., Wu, G., et al. (2016). Developmental functions of miR156-regulated SQUAMOSA PROMOTER BINDING PROTEIN-like (SPL) genes in Arabidopsis thaliana. PloS Genet. 12, e1006263. doi: 10.1371/journal.pgen.1006263
Yamaguchi, N., Jeong, C. W., Nole-Wilson, S., Krizek, B. A., Wagner, D. (2016). AINTEGUMENTA and AINTEGUMENTA-like6/PLETHORA3 induce LEAFY expression in response to auxin to promote the onset of flower formation in Arabidopsis. Plant Physiol. 170, 283–293. doi: 10.1104/pp.15.00969
Yamaguchi, N., Winter, C. M., Wu, M. F., Kanno, Y., Yamaguchi, A., Seo, M., et al. (2014). Gibberellin acts positively then negatively to control onset of flower formation in Arabidopsis. Science 344, 638–641. doi: 10.1126/science.1250498
Yamaguchi, A., Wu, M. F., Yang, L., Wu, G., Poethig, R. S., Wagner, D. (2009). The microRNA regulated SBP-box transcription factor SPL3 is a direct upstream activator of LEAFY, FRUITFULL, and APETALA1. Dev. Cell 17, 268–278. doi: 10.1016/j.devcel.2009.06.007
Yan, W., Cao, S., Liu, X., Yao, G., Yu, J., Zhang, J., et al. (2022). Combined physiological and transcriptome analysis revealed the response mechanism of Pogostemon cablin roots to p-hydroxybenzoic acid. Front. Plant Sci. 13, 980745. doi: 10.3389/fpls.2022.980745
Yang, B., Zhong, Z., Wang, T., Ou, Y., Tian, J., Komatsu, S., et al. (2019). Integrative omics of Lonicera japonica thunb. flower development unravels molecular changes regulating secondary metabolites. J. Proteomics 208, 103470. doi: 10.1016/j.jprot.2019.103470
Yu, S., GalvO, V. C., Zhang, Y. C., Horrer, D., Zhang, T. Q., Hao, Y. H., et al. (2012). Gibberellin regulates the Arabidopsis floral transition through miR156-targeted SQUAMOSA PROMOTER BINDING-like transcription factors. Plant Cell 24, 3320–3332. doi: 10.2307/41692803
Yu, Z.-X., Wang, L.-J., Zhao, B., Shan, C. M., Zhang, Y. H., Chen, D. F., et al. (2015). Progressive regulation of sesquiterpene biosynthesis in arabidopsis and patchouli (Pogostemon cablin) by the miR156-targeted SPL transcription factors. Mol. Plant 8, 98–110. doi: 10.1016/j.molp.2014.11.002
Zeng, T., Li, J., Xu, Z., Zhou, L., Li, J., Yu, Q., et al. (2022). TcMYC2 regulates pyrethrin biosynthesis in Tanacetum cinerariifolium. Hortic. Res. 9, uhac178. doi: 10.1093/hr/uhac178
Keywords: Pogostemon cablin, flower development, GA and auxin signaling, MIKC-MADS, coexpression network, correlation analysis
Citation: Zhang C, Liu X, Liu Y, Yu J, Yao G, Yang H, Yang D and Wu Y (2023) An integrated transcriptome and metabolome analysis reveals the gene network regulating flower development in Pogostemon cablin. Front. Plant Sci. 14:1201486. doi: 10.3389/fpls.2023.1201486
Received: 06 April 2023; Accepted: 09 June 2023;
Published: 29 June 2023.
Edited by:
Xiang Gao, Northeast Normal University, ChinaReviewed by:
Caiyun Wang, Huazhong Agricultural University, ChinaCopyright © 2023 Zhang, Liu, Liu, Yu, Yao, Yang, Yang and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yougen Wu, d3lnZW5nMjAwM0AxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.