 Emmanuel Nyongesa Waswa1,2,3
Emmanuel Nyongesa Waswa1,2,3 Elijah Mbandi Mkala1,2,3
Elijah Mbandi Mkala1,2,3 Wyclif Ochieng Odago1,2,3
Wyclif Ochieng Odago1,2,3 Sara Getachew Amenu1,2,3
Sara Getachew Amenu1,2,3 Elizabeth Syowai Mutinda1,2,3
Elizabeth Syowai Mutinda1,2,3 Samuel Wamburu Muthui1,2,3
Samuel Wamburu Muthui1,2,3 Shi-Xiong Ding1,2,3
Shi-Xiong Ding1,2,3 Guang-Wan Hu1,2,3*
Guang-Wan Hu1,2,3* Qing-Feng Wang1,2,3
Qing-Feng Wang1,2,3- 1Key Laboratory of Plant Germplasm Enhancement and Specialty Agriculture, Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan, China
- 2Sino-Africa Joint Research Center, Chinese Academy of Sciences, Wuhan, China
- 3Botany Department, University of Chinese Academy of Sciences, Beijing, China
Sambucus L. is found in the family Viburnaceae (syn. Adoxaceae) and encompasses approximately 29 accepted species. The complex morphology of these species has caused continued confusion concerning their nomenclature, classification, and identification. Despite previous attempts to resolve taxonomic complexities in the Sambucus genus, there are still unclear phylogenetic relationships among several species. In this study, the newly obtained plastome of Sambucus williamsii Hance. as well as the populations of Sambucus canadensis L., Sambucus javanica Blume, and Sambucus adnata Wall. ex DC were sequenced, and their sizes, structural similarity, gene order, gene number, and guanine–cytosine (GC) contents were analyzed. The phylogenetic analyses were conducted using the whole chloroplast genomes and protein-coding genes (PCGs). The findings revealed that the chloroplast genomes of Sambucus species exhibited typical quadripartite double-stranded DNA molecules. Their lengths ranged from 158,012 base pairs (bp) (S. javanica) to 158,716 bp (S. canadensis L). Each genome comprised a pair of inverted repeats (IRs), which separated the large single-copy (LSC) and small single-copy (SSC) regions. In addition, the plastomes contained 132 genes, encompassing 87 protein-coding, 37 tRNA, and four rRNA genes. In the simple sequence repeat (SSR) analysis, A/T mononucleotides had the highest proportion, with the most repetitive sequences observed in S. williamsii. The comparative genome analyses showed high similarities in structure, order, and gene contents. The hypervariable regions in the studied chloroplast genomes were trnT-GGU, trnF-GAA, psaJ, trnL-UAG, ndhF, and ndhE, which may be used as candidate barcodes for species discrimination in Sambucus genus. Phylogenetic analyses supported the monophyly of Sambucus and revealed the separation of S. javanica and S. adnata populations. Sambucus chinensis Lindl. was nested within S. javanica in the same clade, collaborating their conspecific treatment. These outcomes indicate that the chloroplast genome of Sambucus plants is a valuable genetic resource for resolving taxonomic discrepancies at the lower taxonomic levels and can be applied in molecular evolutionary studies.
1 Introduction
Chloroplasts (cp), the key organelles for photosynthesis and carbon fixation in green plants, are believed to have originated from ancestral cyanobacteria via endosymbiosis (Dyall et al., 2004). They are among the many distinguishing characteristic organelles in plant cells and possess genomes whose genetic information is maternally inherited from generation to generation (Birky, 1995). This genetic information contains enzymatic machinery essential for gene expression and encodes for many vital proteins that usually participate in photosynthesis and other metabolic processes (Green, 2011; Allen, 2015). The cp genomes are organized into large clusters of polycistronic transcribed genes that are highly conserved and comprise a single circular molecule with a quadripartite arrangement (Shen et al., 2018). A typical tetrad structure of the cp genome contains two copies of inverted repeat (IRa and IRb) regions and small single-copy (SSC) and large single-copy (LSC) regions in most plants (Wu et al., 2009; Kong and Yang, 2015). Plastomes’ IRs usually separate the LSC and SSC regions (Lee et al., 2007). The number of genes encoded by a circular cp genome is commonly 110–130, consisting of about 79 proteins, 30 transfer RNAs, and four ribosomal RNAse (Daniell et al., 2016).
Advances in high- throughput sequencing technologies have made large-scale cp genome sequence acquisition possible (Kyalo et al., 2020). Therefore, the cp genomes have highly been used as essential tools for comparative phylogenetic studies (Shen et al., 2018). For instance, numerical improvement of cp genome sequenced plants has been enhanced by the emergence and implementation of technical developments in DNA sequences such as next-generation sequencing (NGS) technologies. These approaches are cost-effective and time-efficient, enabling the exploration of high numbers of plant genomes at molecular levels (Raveendar et al., 2015). The cp genome analyses substantially contribute to the evolutionary and phylogenetic studies (Liu et al., 2013; Huang et al., 2014; Zhang et al., 2016; He et al., 2017) and have been applied at lower taxonomic levels to resolve close taxonomic relationships (Dong et al., 2012).
The family Viburnaceae as currently conceived encompasses the variously formerly classified genera in Caprifoliaceae, Sambucaceae, and Adoxaceae (Willis et al., 1973). Previous phylogenetic analyses within the family using morphological characters and rbcL sequences indicated Adoxa to be nested within Sambucus and inferred strong support for Sambucus monophyly (Chase et al., 1993; Olmstead et al., 1993). The internal transcribed spacer (ITS) regions of nuclear ribosomal DNA and morphological characters revealed Adoxa as a sister group to Sambucus (Eriksson and Donoghue, 1997). The close relatedness between Sinadoxa, Tetradoxa, and Adoxa was revealed by rbcL sequences and ITS regions (Eriksson and Donoghue, 1997; Jacobs et al., 2010). Moreover, a study on the phylogenetic relationship of Viburnaceae using the complete cp genomes confirmed a sister relationship between Sambucus and Adoxa–Tetradoxa–Sinadoxa groups (Ran et al., 2020).
Sambucus L. is a relatively small genus in the family Viburnaceae consisting of small trees, deciduous shrubs, and/or perennial herbs (Amini et al., 2019). They are highly distributed in the temperate and subtropical regions of the Northern Hemisphere with some species extending toward the Southern Hemisphere, surrounding both the high and low latitudes areas of North America, Asia, Europe, Northern Africa, West Indies, Eastern and South-eastern Australia, and the Andean region of South America (Figure 1). The species of this genus are commonly known as elders or elderberries and are characterized by compound leaves (5–30 cm long) with serrated margins and five to nine pinnate to ovate-lanceolate or ovate-elliptic leaflets (Hummer et al., 2012). They form dense cymes of flowers that are white-yellowish or sometimes pinkish-purple. At maturity, elders bear brown-black, blue, red, orange, or yellow berry-like drupes (4–7 mm in diameter) (Fu et al., 2020).
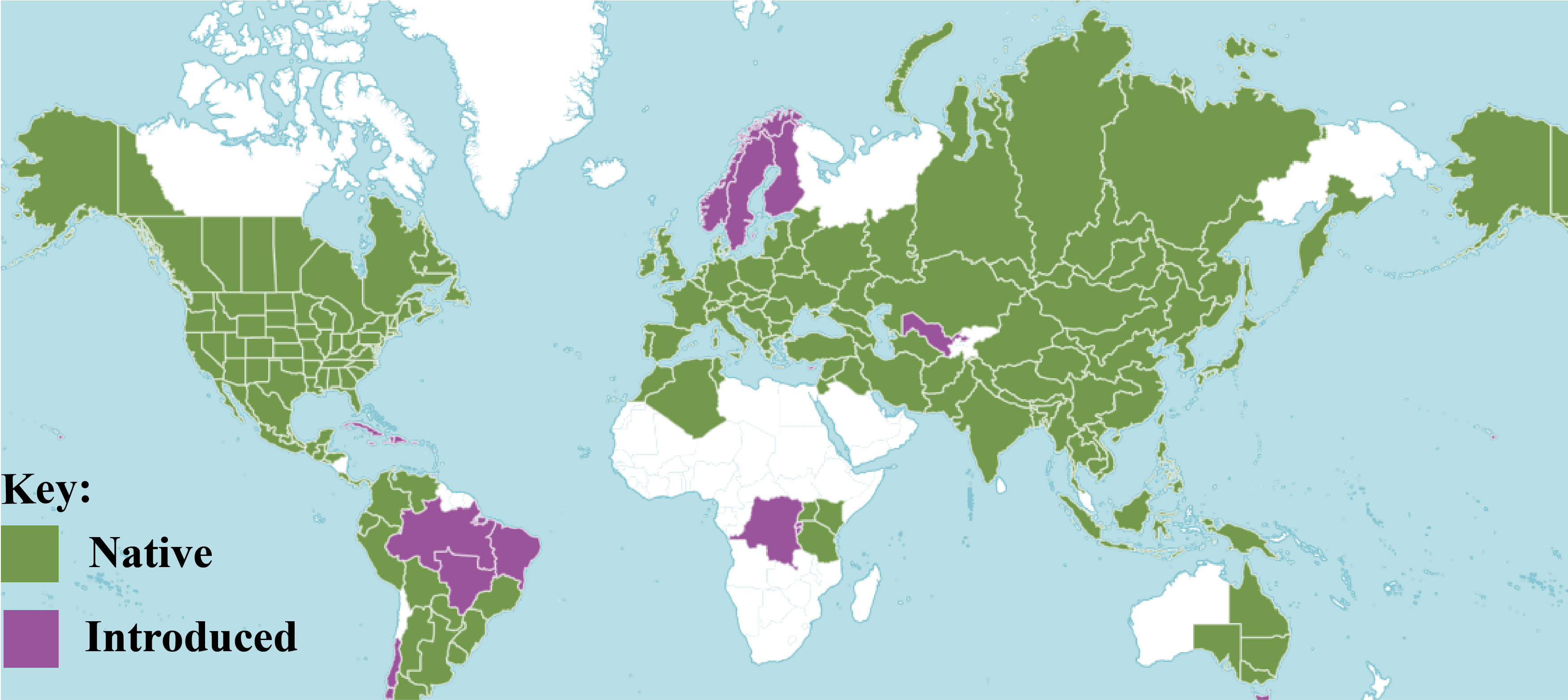
Figure 1 Distribution of Sambucus species (POWO, 2023).
Sambucus species have long been traditionally employed in the treatment of various ailments including bone fractures, diabetes, rheumatism, respiratory and pulmonary disorders, diarrhea, skin diseases, wounds, and inflammatory illnesses (Waswa et al., 2022). The species Sambucus adnata and Sambucus williamsii are by far the most popular Chinese traditional medicines extensively used by the Yi people of Liangshan in Sichuan province to treat bone fractures and rheumatism, whereas Sambucus javanica (syn. Sambucus chinensis) is medicinally employed to cure wounds and relieve throbbing pains (Wang et al., 2020; Otsuka et al., 2021). The medicinal properties of Sambucus plants are associated with the isolated bioactive metabolites such as phenolic compounds, terpenoids, fatty acids, and organic acids (Hearst et al., 2010; Do Nascimento et al., 2014; Przybylska-Balcerek et al., 2021). In addition, pharmacological investigations show that Sambucus plants are outstanding sources of antioxidants, antimicrobial, antidiabetic, anti-inflammatory, antidepressant, and anti-glycation activities, among others (Waswa et al., 2022).
Linnaeus was the first to provide the taxonomic description of the genus Sambucus, hence its botanical authority. The taxonomy of this genus is sophisticated due to plastic morphological characters induced by considerable diversity within species with vast geographical ranges and possible interspecific hybridizations (Applequist, 2015). von Schwerin (1920) recognized 28 species and several varieties, which relied heavily on important diagnostic morphological characters. At present, the most comprehensive taxonomic study was achieved by Bolli (1994), who included nine species in the genus by primarily focusing on morphological resemblances within the groups. Nevertheless, Bolli’s work lacked a molecular basis, and several researchers were hesitant to adopt it (Yatskievych, 2006; Applequist, 2015). In Bolli’s treatments, the two economically important members of the genus Sambucus nigra L. and Sambucus canadensis L. were given the subspecies status and recognized as S. nigra ssp. nigra (L.) R. Bolli and S. nigra ssp. canadensis (L.) R. Bolli. Conversely, molecular analysis of the microsatellites revealed their separation as distinct species (Clarke and Tobutt, 2006). Jacobs et al. (2009) assessed the phylogeny of Sambucus using the ITS, trnK, and matK molecular sets. The molecular datasets showed discrimination between the closely related Sambucus racemosa and Sambucus ebulus. Moreover, Amini et al. (2019) established delimitation of S. nigra and S. ebulus populations in the molecular investigation using micro-morphological characters and nuclear (nrDNA ITS) markers. Furthermore, the results from the complete cp genomes dataset displayed divergence between the populations of S. nigra and S. williamsii (Ran et al., 2020). Presently, the genus Sambucus encompasses 30 accepted taxa (22 species, four subspecies, and four varieties), 68 synonyms, and 73 ambiguous taxa (Table S1) (The Plant List, 2013; IPNI, 2023; POWO, 2023; WFO, 2023).
Representatives of the genus Sambucus usually show high variability within individual species with wide geographical ranges, and thus artificial classification based solely on morphological trait-based systems is undependable and controversial. Thus, the outcome of this classification cannot be outrightly adopted and require molecular-based studies to confirm their taxonomic treatments. Elucidating the phylogenetic relationships of Sambucus plants using complete plastomes is essential in understanding their taxonomic treatments. In our fieldwork, several species of Sambucus were collected from China including populations of S. adnata and S. javanica that were observed to share high morphological similarities, including bladelike stipule leaves, stems with white pith, pedunculate inflorescences, calyx urceolate, and red fruits (http://www.efloras.org/). They exhibited limited phenotypic variations (Figure 2), necessitating investigations of their molecular affinities and phylogenetic relationships. The results obtained by Ran et al. (2020) using a complete chloroplast genome sequence dataset indicated that S. javanica Blume is a close relative of S. adnata Wall. ex DC. However, the molecular investigation attempts to assess species limits between the closely related S. adnata and S. javanica are lacking. The present study aimed to I) characterize and compare the cp genomes of S. williamsii and the populations of S. adnata, S. javanica, and S. canadensis; II) examine the phylogenetic relationships of the main clades of Viburnaceae, with a particular focus on the generic status of Sambucus; III) perform comparative analyses of Sambucus for future species identification and phylogeographic studies; IV) e xplore the taxonomic treatment of S. adnata Wall. ex DC and S. javanica Blume.

Figure 2 Morphological characters of Sambucus species. (1) Sambucus adnata Wall. ex DC. (2) Sambucus javanica Blume. (3) Sambucus nigra L. (A) Compound odd-pinnate leaves with serrated margins, (B) compound polychasial cyme, and (C) small rounded berry-like drupes.
2 Materials and methods
We performed the comparative genomics and phylogenetic relationships of various Sambucus species obtained from Africa and Asia. In this investigation, the complete chloroplast genomes of S. canadensis were sequenced and reported for the first time.
2.1 Plant material collections
Fresh leaf materials of Sambucus plants were obtained from several areas in China and Kenya (Table S2). The samples that did not show any obvious disease symptoms were washed and dried in silica gel for preservation until DNA extraction. Voucher specimens were deposited and stored in the Herbarium of Wuhan Botanical Garden (HIB) at −80°C pending chloroplast DNA extraction.
2.2 Plant DNA extraction, sequencing, and assembly
The total genomic DNA of each specimen was extracted from 100 mg of the leaves using improved cetyltrimethylammonium bromide (CTAB) protocol (Doyle, 1990). The NanoDrop spectrometer (Beckman Coulter, Krefeld, Germany) and gel electrophoresis (Beijing Liuyi Instrument Factory, Beijing, China) were used to determine the quantity and quality of the isolated DNA material. The other 30 species from the family Viburnaceae and two outgroups were downloaded from the NCBI (https://www.ncbi.nlm.nih.gov/) (Table S2). The cp genomes were sequenced using the Illumina platform at Novo Gene Company (Beijing, China). High-quality reads were used for de novo assembly to reconstruct the Sambucus chloroplast genome using GetOrganelle v.1.7.2 with a word size of 150 and K-mer size of 105 using plastome data of S. chinensis (MW455170) as a reference. The resulting scaffolds and their connectivity were visualized using Bandage 0.7.1 software (Wick et al., 2015) to authenticate the produced plastid genomes. Finally, the quality of the assembled plastomes was confirmed based on the reading level by aligning the trimmed raw reads to the de novo assemblies using Geneious Prime 2021 (Kearse et al., 2012), with medium-to low-sensitivity option and iteration up to five times (Hourahine et al., 2020). File S1 contains the text generated following the assembly of the complete cp genome of Sambucus, while its depiction is illustrated in Figure S1.
2.3 Annotation of the chloroplast genomes
The complete cp genome sequences were annotated using CPGAVAS2 software (Shi et al., 2019), with the gene boundaries (intron/exon) and codons (start/stop) checked and modified manually using the reference genome. tRNAscan-SE v1.21 software (Meier-Kolthoff et al., 2013) was used with its default settings to confirm the tRNAs. The web application (https://irscope.shinyapps.io/chloroplot/) was used to create the gene maps of Sambucus cp genomes. The complete cp genomes were deposited at the GenBank with the following accession numbers: S. javanica (OM868260, ON006397, ON006398, and ON006402), S. williamsii (OM937121), S. canadensis (OM937119 and OM937120), and S. adnata (ON006399, ON006400, and ON006401).
2.4 Repeats and codon usage analysis
The whole cp genomes of Sambucus species were aligned in GENEIOUS v11.1.4 (Kearse et al., 2012), using MAFFT multiple aligner v7. The Perl script MISA (Beier et al., 2017) was used to identify the simple sequence repeat (SSR) loci (i.e., mono-, di-, tri-, tetra-, penta-, and hexa-) nucleotide repeats with the following thresholds: 10 repeats for mononucleotides, five repeats for dinucleotides, four repeats for trinucleotides, and three repeats each for tetranucleotides, pentanucleotides, and hexa nucleotides. In addition, the number of positions of repeat elements was estimated using the program REputer (Kurtz et al., 2001), including forward, palindromic, complementary, and reverse repeats with a minimum size of 30 bp and sequencing identity of not less than 90% with the hamming distance of 3.
2.5 Comparative complete chloroplast genome analysis
The boundary shifts of Sambucus plastomes at the IR borders were examined using the IRscope online program (https://irscope.shinyapps.io/irapp/). For expansion/contraction analysis, 10 plastomes were compared with the S. chinensis_MW455170 plastome using Geneious v11.1.4 (Kearse et al., 2012). Afterward, the disparities of the gene position at the plastome boundary (IR-SC) of the four junctions (JLB–LSC/IRB, JSB–IRB/SSC, JSA–SSC/IRA, and JLA–IRA/LSC) were assessed. The 10 plastomes and the reference were aligned using the progressive Mauve algorithm in default settings (Jin et al., 2020) to detect the gene inversions. The parameters were set to automatically calculate the seed weight (15) and locally collinear blocks (LCBs) with the minimum LCB score of 30,000 (Darling et al., 2004). The cp genomes of the studied species were constructed using the mVISTA program (Frazer et al., 2004) in Shuffle-LAGAN mode, with S. chinensis_MW455170 as a reference to assess divergence in the genomic structures. Sliding windows analysis in Dnasp v5.10 (Librado and Rozas, 2009) was used to calculate the nucleotide sequence variability. The window length was calibrated at 600 bp with a 200- bp step size.
2.6 Synonymous and non-synonymous substitution rates
We also assessed the substitutions of synonymous (Ka) and non-synonymous (Ks) ratios (Ka/Ks) using the KaKs calculator (Zhang et al., 2006). In this analysis, the protein-coding sequences were extracted using PhyloSuite (Zhang et al., 2020), and sequence alignment was performed using MAFFT v7 software (Katoh and Standley, 2013). Unrealistic Ka/Ks ratios were excluded to ensure precise screening of conserved and divergent genes. Thus, we adopted a more accurate threshold screening: Ka/Ks < 0.01 treated as qualified conserved genes, Ka/Ks = 1 for neutral selection, and Ka/Ks >1 (greater than 1) as positively selected orthologs based on Abdullah et al. (2020) and Ka/Ks < 1 (less than 1) for purifying selection.
2.7 Phylogeny
The phylogenies within the studied family were examined using the maximum-likelihood (ML), Bayesian inference (BI), and Neighbor-Net (NN) methods. To infer phylogenetic relationships in the family Viburnaceae, 42 species were examined that incorporated the newly obtained plastomes of Sambucus species, including S. williamsii (OM937121) and populations of S. javanica (OM868260, ON006397, ON006398, and ON006402), S. adnata (ON006399 and ON006400), and S. canadensis (OM937119 and OM937120) (Table S2). Additionally, the complete cp genome sequences of five other Sambucus species, together with 22 Viburnum and three Adoxa species (including Adoxa moschatellina, Adoxa corydalifolia (Sinadoxa corydalifolia), and Adoxa omeiensis (Tetradoxa omeiensis)) and two outgroups (Panax ginseng (MH049735) and Eleutherococcus gracilistylus (KT153020)) from Araliaceae family were downloaded from GenBank (Table S2).
The downloaded plastomes that contained errors were re-annotated. Protein-coding genes were extracted using PhyloSuite v 1.2.2 software (Zhang et al., 2020), and the “–auto” strategy and normal alignment mode were used to align the sequences in MAFFT v 7 (Katoh and Standley, 2013). The “automated1” command was set to omit gap sites using trimAl (Capella-Gutiérrez et al., 2009), while the fragments that were aligned ambiguously were eliminated by default parameter settings in Gblocks (Talavera and Castresana, 2007). The cleaned sequences were concatenated into different formats for other analyses. By applying the corrected Akaike information criterion (AICc) criterion and all algorithms, pre-defined partitions were chosen by the best partitioning arrangement and evolutionary models in PartitionFinder2 (Lanfear et al., 2017). Based on our obtained dataset of 58 protein-coding genes that were common in all species, we used ML phylogenies embedded in IQtree (Nguyen et al., 2015) with the GTR+R2+F model for 5000 ultrafast and the BI by Bayes (Ronquist and Huelsenbeck, 2003) to perform the phylogenetic analysis. The maximum-likelihood phylogeny was inferred based on the Minh et al. (2013) model and the likelihood-ratio test defined by Guindon et al. (2010). The complete cp genome data were aligned using clustal format by MAFFT (v7.504). In this study, we used the term “clade” to refer to clusters for the recovered phylogenies. Moreover, we adopted the name “Adoxoideae” to represent the Sambucus–Adoxa–Tetradoxa–Sinadoxa group and “Adoxina” for the Adoxa–Tetradoxa–Sinadoxa (Adoxina) group based on Jacobs et al. (2010). The distance-based network construction method NN incorporated in the SplitsTree4 v.4.14.4 (Huson and Bryant, 2001) was used to reconstruct phylogenetic split networks (Levy and Pachter, 2011). We used the term “lineage” to denote a group of specimens in the split graph (Neighbor-Net diagram).
3 Results
3.1 General characterization of chloroplast genomes
The assembled and annotated genomes varied in size (158,012–158,716 bp) and displayed a quadripartite conformation. They contained two copies of IR regions ranging from 26,149 to 26,269 bp. The IRs were segregated by the SSC and LSC regions, which varied in size from 18,920 to 19,099 and 86,624 to 87,376 bp, respectively (Figure 3; Table 1). The genomes showed high similarities in guanine–cytosine (GC) contents of 38.0%, with the LSC, SSC, and IR regions exhibiting respective values of 36.3%, 31.7%, and 43.0% (Table 1).
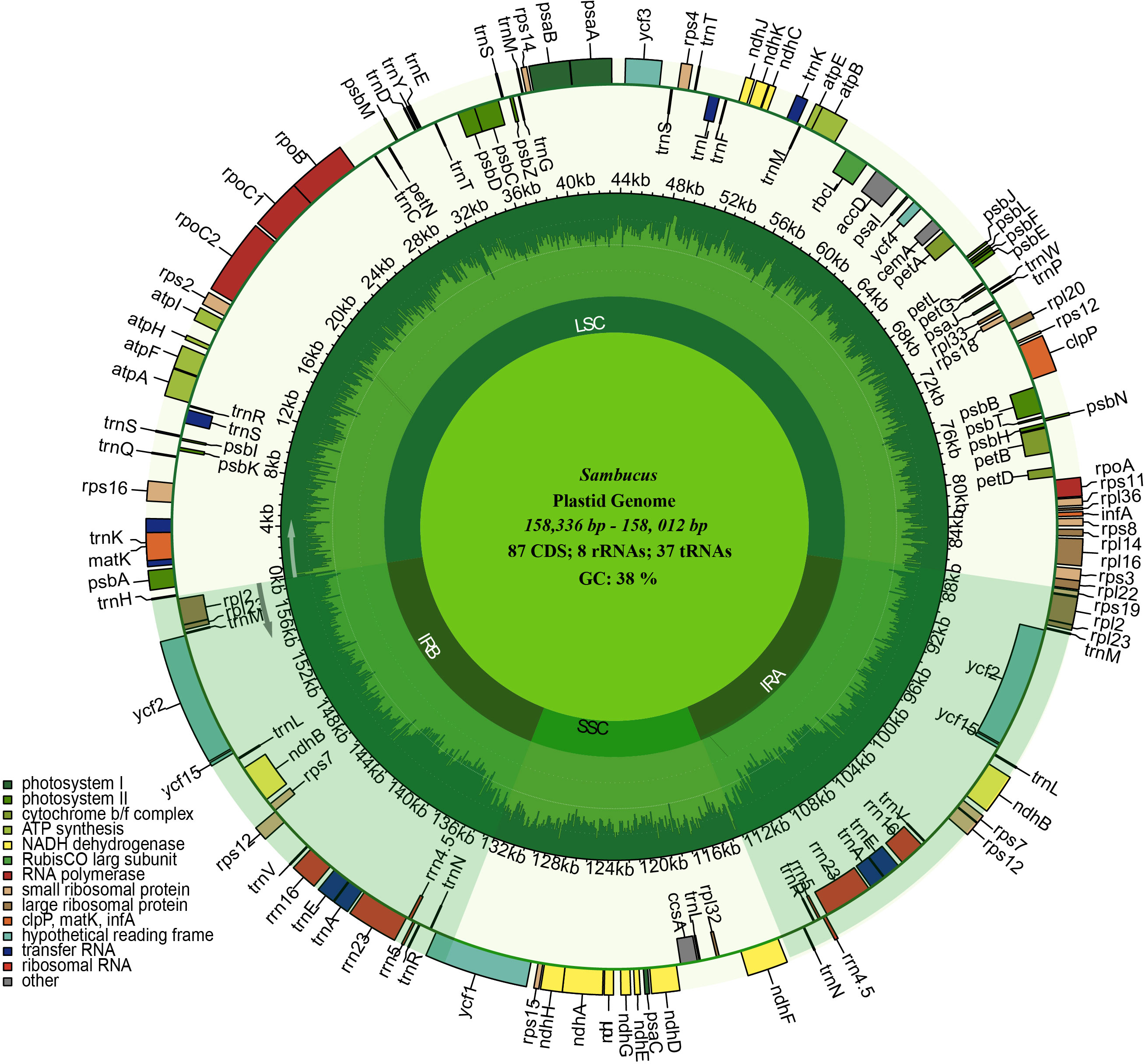
Figure 3 Circular maps of the chloroplast genomes of three Sambucus species. Genes outside the circle are transcribed clockwise, while the genes inside the circle are transcribed counterclockwise. The dark gray plot in the inner circle corresponds to the guanine–cytosine (GC) content. LSC, SSC, and IRs (IRA and IRB) denote large single- copy, small single- copy, and inverted repeat regions, respectively.
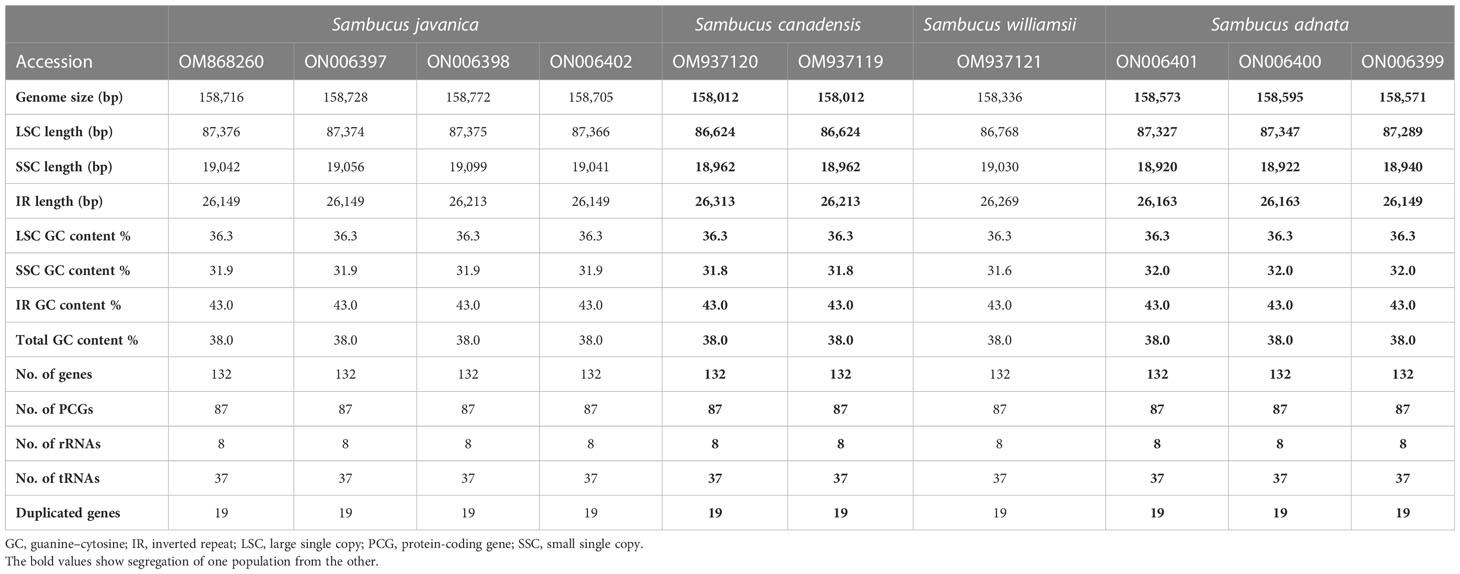
Table 1 Detailed comparison of the complete chloroplast genomes of Sambucus species.
We detected 132 functional genes encompassing 87 protein-coding, 37 tRNA, and eight rRNA genes. Among them, 19 genes were replicated in the IR regions, comprising seven protein-coding genes (PCGs) (ycf1, rpl2, rps7, ndhB, ycf2, rpl23, and rps12), four rRNA genes (rrn5, rrn4.5, rrn23, and rrn16), and eight tRNA genes (trnN-GUU, trnR-ACG, trnA-UGC, trnL-GAU, trnV-GAC, trnL-CAA, trnI- CAU, and trnH-GUG) (Figure 3). rps12, clpP, and ycf3 genes were identified to have two introns, whereas 15 genes including trnK-UUU, trnG-UCC, trnL-UAA, trnV-UAC, trnl-GAU, trnA-UGC, petB, petD, atpF, ndhB, ndhA, rpl1, rpl2, rps16, and rpoC1 contained one intron. ycf1 genes were detected as pseudogenes in the studied Sambucus species with a trans-splicing event, whereby the 5′ end of rps12 gene located in the LSC part and the 3′ situated at the IRa/IRb region were observed. Comparison between the studied and other selected cp genomes showed no significant gene order rearrangements or inversions. The genes encoded by Sambucus cp genomes are listed in Table 2.
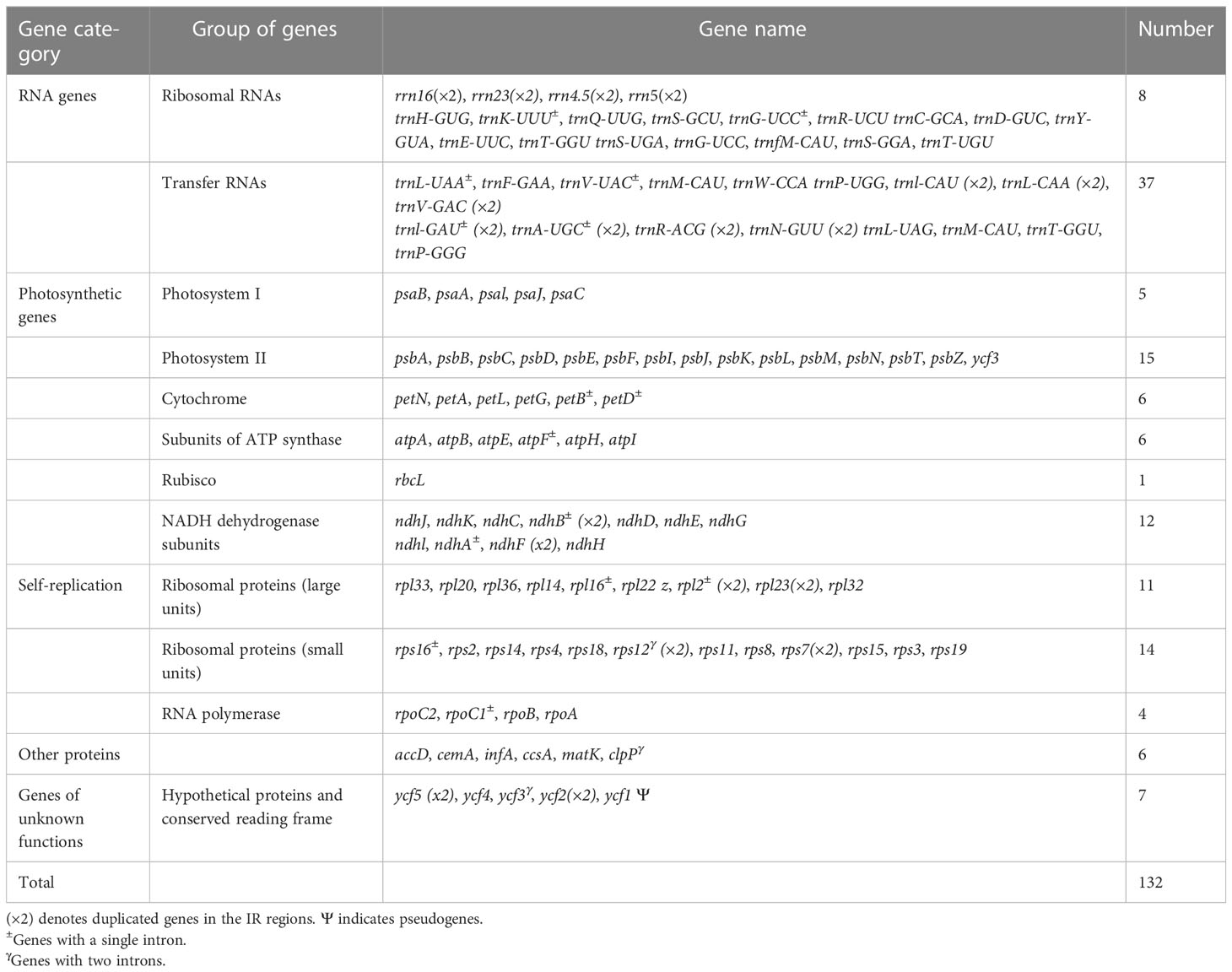
Table 2 Genes existing in the chloroplast genome of Sambucus.
3.2 Repeat sequence characterization
In the repeat structure analysis, four types of long repeats including palindromic, forward, complementary, and reverse elements were identified in the studied Sambucus genomes (Figure 4). Moreover, analysis of the microsatellites showed the presence of mononucleotide, dinucleotide, trinucleotide, tetranucleotide, and hexanucleotide SSRs in Sambucus cp genomes (Table S3). S. williamsii (OM937121) had the highest number of repeats [68 SSRs], followed by S. canadensis [64 (OM937119 and OM937120) SSRs] and S. adnata [53 (ON006399) and 52 (ON006400 and ON006401) SSRs], while 49 (ON006398), 50 (OM868260), and 51 (ON006397) were found in S. javanica. Additionally, the variety of S. javanica and S. chinensis var. pinnatilobatus had a total of 50 SSRs. The most abundant nucleotide contents in all studied species were mononucleotide repeats (SSR loci A/T), followed by dinucleotides, tetranucleotides, trinucleotides, and pentanucleotides, whereas hexanucleotides exhibited the least number of SSRs. S. williamsii encompassed 48 mononucleotide, seven dinucleotide, three trinucleotide, eight tetranucleotide, and nine hexanucleotide repeats. Additionally, pentanucleotide repeats were absent in S. williamsii but were detected in the rest of the studied Sambucus species. Hexanucleotide repeats were present in S. williamsii but absent in other species (Figure 5). The distribution pattern of SSRs between S. adnata and S. javanica exhibited high resemblances, and the hexanucleotide repeats were absent in both species (Figure 5; Table S3).
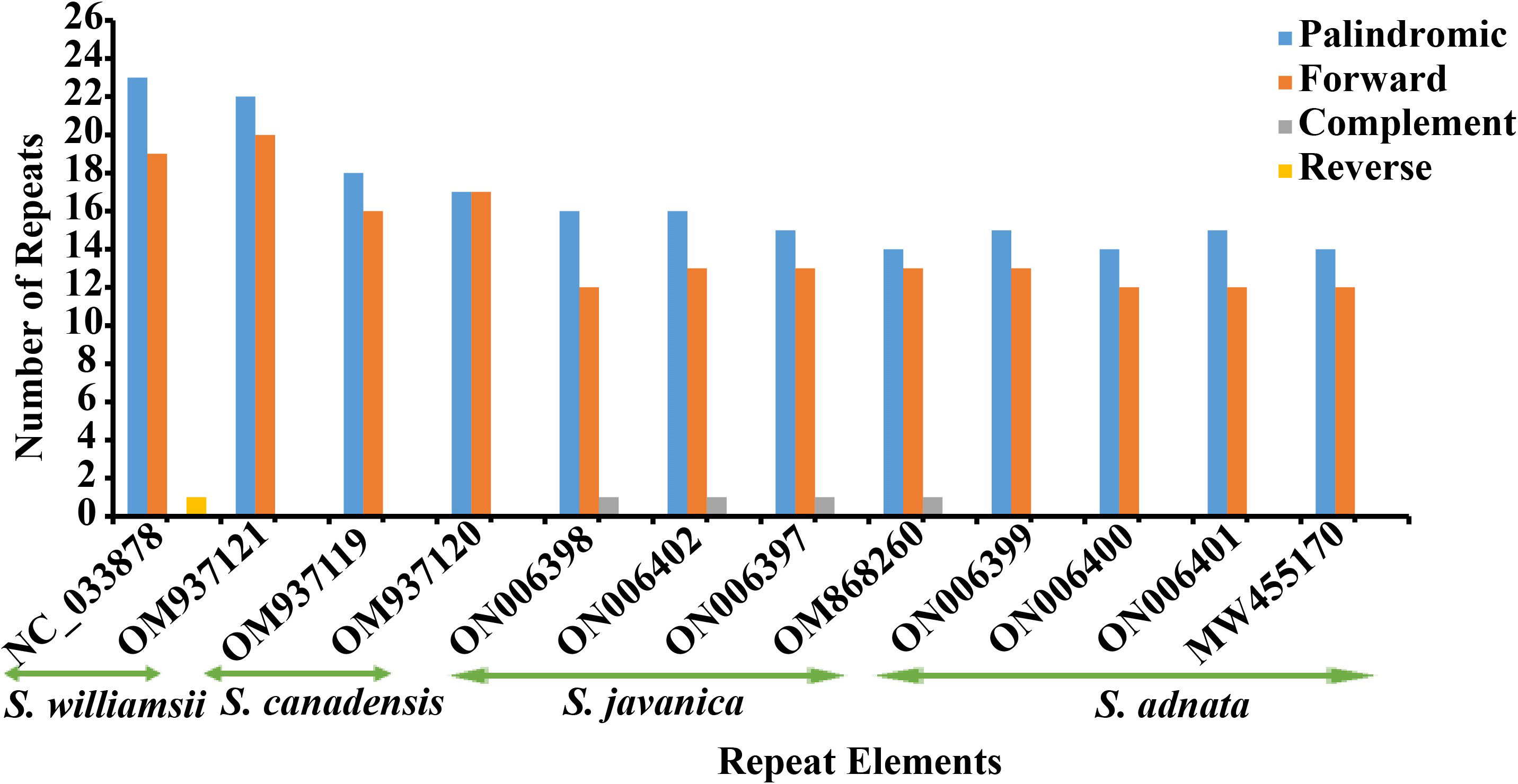
Figure 4 Long repeat elements in Sambucus species.
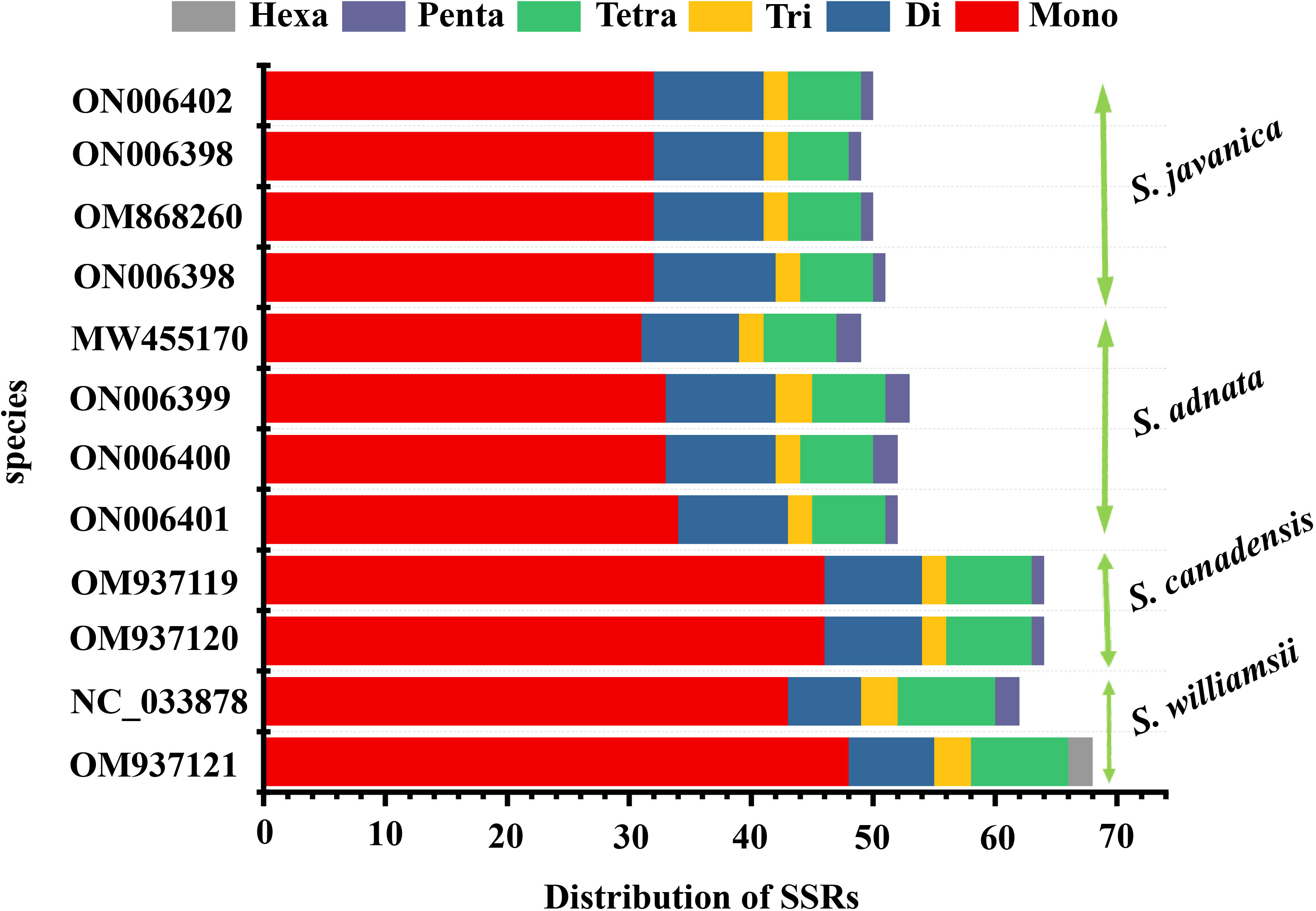
Figure 5 Analysis of SSRs in Sambucus chloroplast genomes. SSRs, simple sequence repeats.
3.3 Expansion and contraction of inverted repeat regions
We performed comparisons between IR border regions of the four closely related Sambucus plastomes and six congeners. The detailed IR shift comparison was executed at the LSC/IRa, LSC/IRb, SSC/IRa, and SSC/IRb junction sites and the adjacent genes. Expansion/contraction of IRs was examined using S. chinensis (syn. S. javanica)_MW455170 as a reference genome. Our findings showed that LSC, IR, and SSC areas are slightly variable among Sambucus genomes (Figures 6, 7). Moreover, the sizes of S. javanica IR regions were identical (26, 149 bp), congruent to that of S. chinensis var. pinnatilobatus, a variety of S. chinensis (S. javanica). Furthermore, the populations of S. canadensis and S. adnata exhibited IR sizes of 26,213 and 26,163 bp, respectively, except for S. adnata_ON006399 (26,171 bp). ndhF gene was entirely situated within SSC and partially expanded in the range of 201 bp in S. javanica_ON006398 to 93 bp in S. canadensis (OM937120). The junction SSC/IRA (JSA) largely lies within ycf1 gene that ranges from 4,586 bp in S. williamsii_MN937121 to 4,568 bp in S. canadensis_OM937120, whereas in S. adnata_ON006401, ycf1 gene is located far from 414 bp in the SSC. ycf1 pseudogene and rps19 gene were present in all Sambucus species. In addition, trnH-GUG gene at the LSCs was far from the border JLA in the range of 1–12 bp (Figure 6). rpl2 gene was correspondingly positioned around IRb/LSC border, whereas rps19 was located at the LSC/IRb junction site. The LSC/IRB (JSB) junction located within rps19 gene largely lies in the range of 166 bp in S. williamsii _MN937121 to 39 bp in S. canadensis_MN937120.
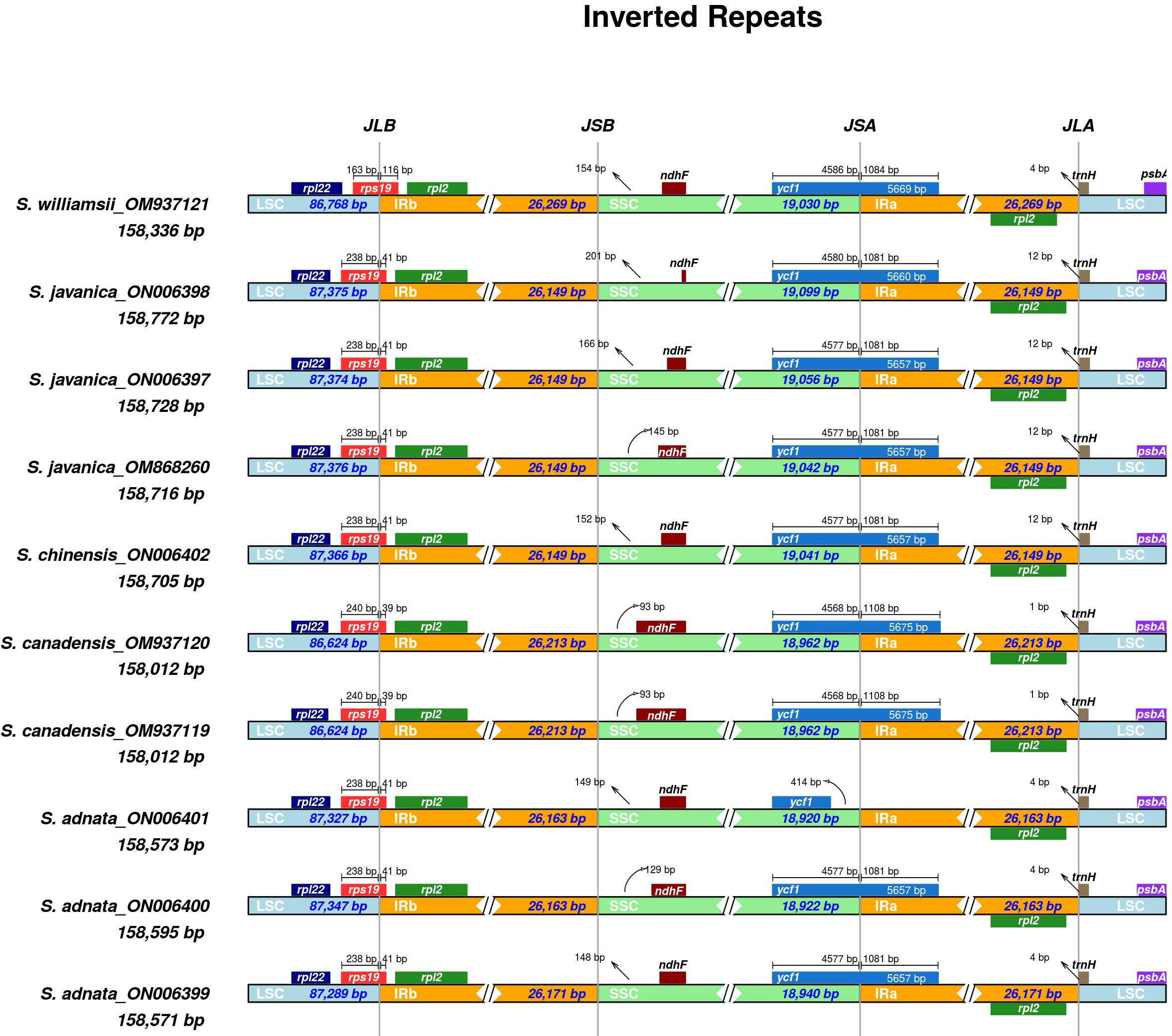
Figure 6 Comparison of the borders of LSC, SSC, and IR regions among Sambucus plastomes. LSC, large single copy; SSC, small single copy; IR, inverted repeat.

Figure 7 Structural genome comparison of the Sambucus species using Mauve program. The aligned DNA sequences positioned above the line are indicated in the clockwise direction while counterclockwise is presented below the line.
3.4 Comparative analysis of chloroplast genomes
Disparities in cp genome divergence between the four Sambucus species (S. adnata (ON006400), S. canadensis (OM937119), S. javanica (OM868260), and S. williamsii (OM937121)) were analyzed using the mVISTA program with the annotated sequence of S. nigra_NC_045061 as a reference genome (Figure 8). The overall sequence identity of the plastomes indicated high consistency in the arrangement of genes, and the single-copy regions were more diversified and variable compared to the IR regions. The results showed high conservation in gene number, orientation, and order. Despite slight variations at the IR/SSC border, the conservative nature of IRs was evident. rRNA genes rrn16, rrn23, rrn4.5, and rrn5 located at IRa and IRb were highly conserved. With the minority showing long repeats of more than 60 bp, most plastomes exhibited repeat lengths between 20 and 58 bp. The intergenic regions were more divergent, especially atpH-atpA, petN-psbM, psbL-petG, trnV-rps12, rps16-psbK, and trn-GUU-ccsA regions. Furthermore, the protein-coding regions including ndhD, ndhH, ndhF, ycf4, psbB, psbL, psbP, rps3, ycf1, and ycf2 were highly divergent.
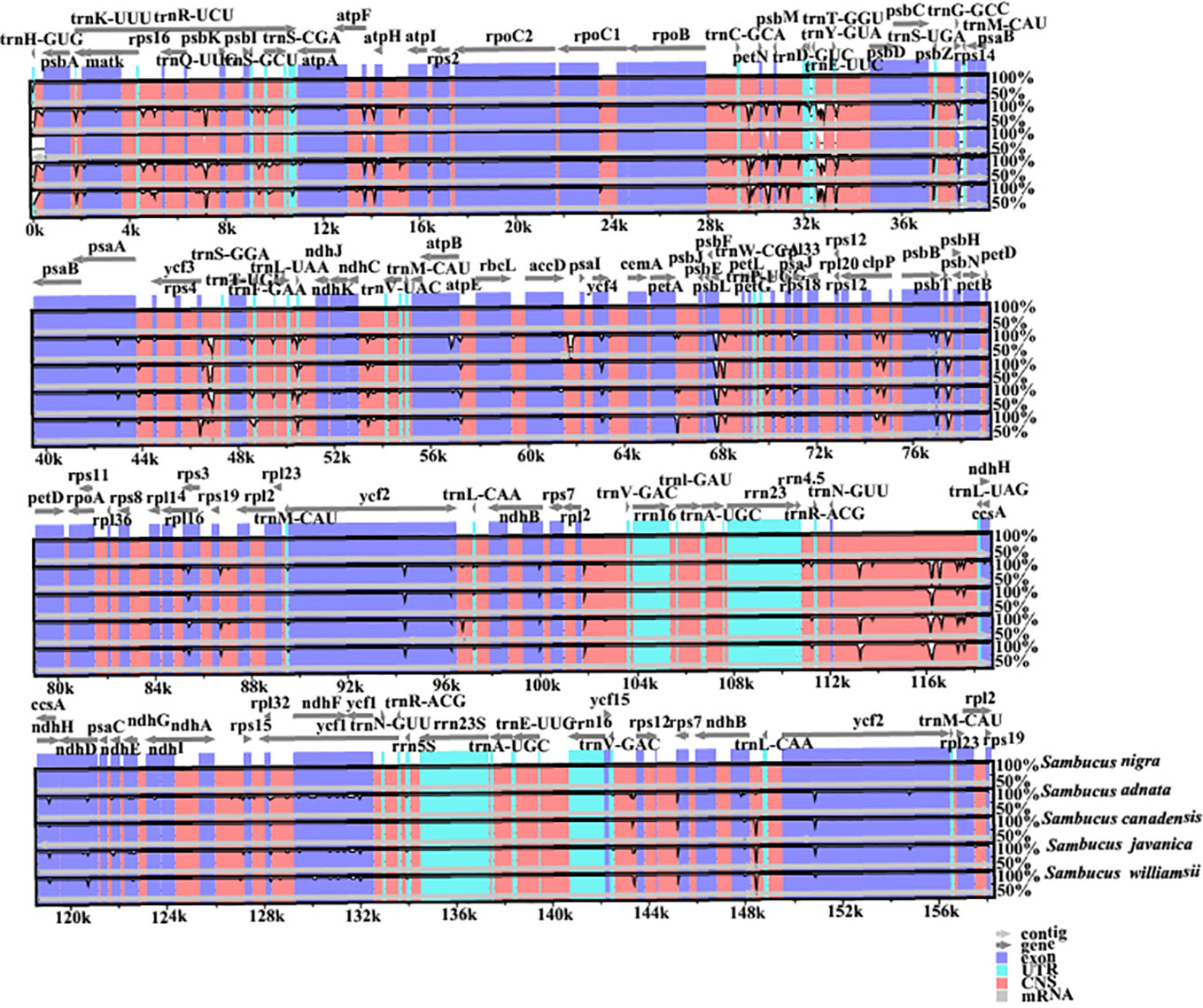
Figure 8 Visualized alignments of the four Sambucus species cp genomes. The sequence identity plot was made using mVISTA, with Sambucus nigra as a reference genome. The y-axis represents identity ranging from 50% to 100%.
Additionally, the nucleotide diversities of the cp genomes of five Sambucus species were compared using DNASP (Librado and Rozas, 2009), and the sequence variability (Pi) values were plotted using a reference annotation of S. chinensis (MW455170) (Figure 9). The most divergent hotspots regions were trnT-GGU, trnF-GAA, psaJ, ndhF, trnL-UAG, and ndhE genes, situated in the coding and non-coding areas with the values of Pi values between 0 and 0.09. The LSC region exhibited higher divergence at trnT-GGU and trnF-GAA regions and psaJ genes (>0.04) compared to SSC (ndhF, trnL-UAG, and ndhE) regions. The SC regions displayed more variations than IR regions (Figure 9). The nucleotide comparison between S. javanica and S. chinensis revealed negligible nucleotide variance (Pi = 0.00018) (Table S4).
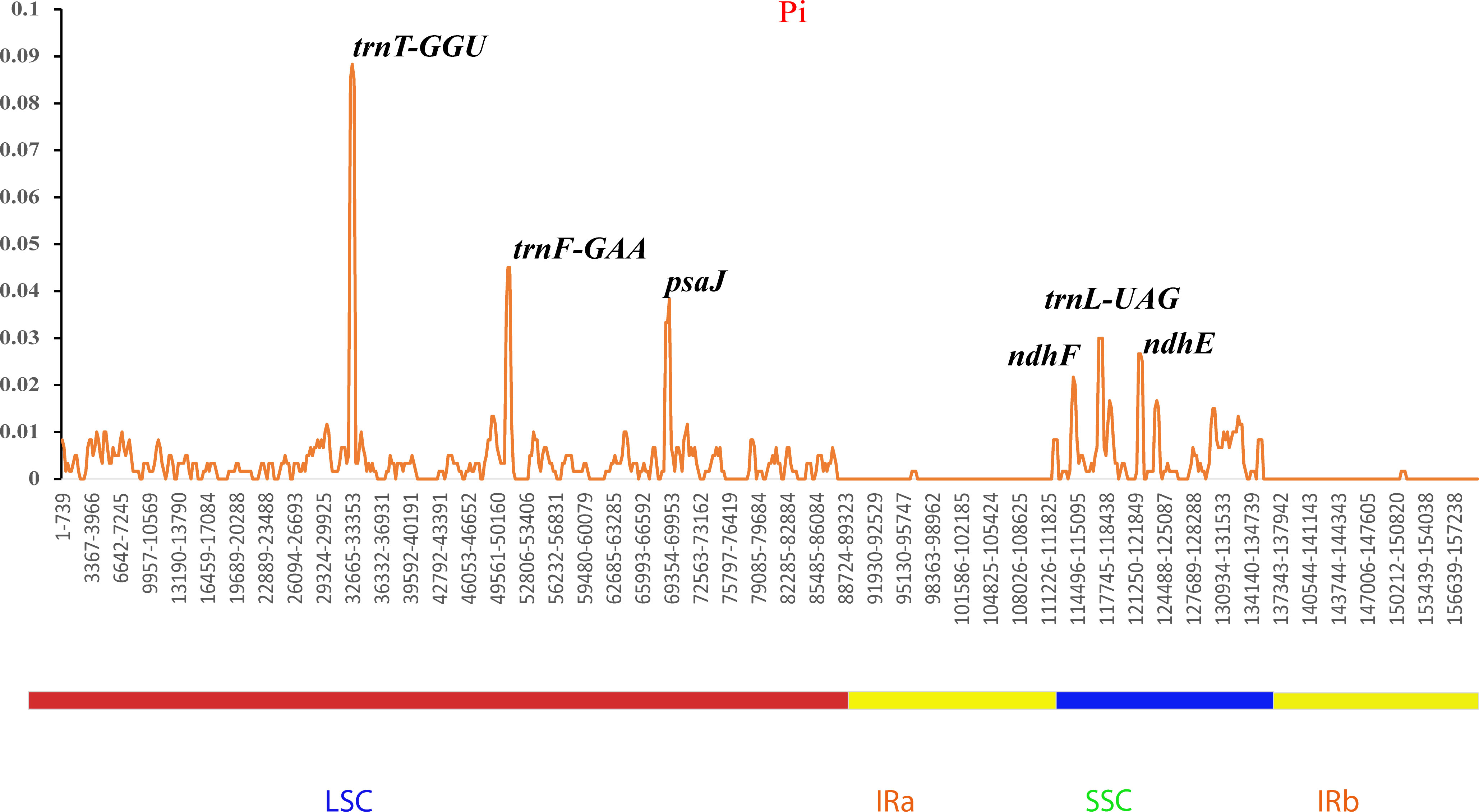
Figure 9 Sliding window analyses of the complete chloroplast genomes of five Sambucus species. The x-axis represents the position of the window, while the y-axis represents the nucleotide diversity (Pi) of each window (window length, 600 bp; step size, 200 bp).
The nucleotide diversity of protein-coding genes extracted from five Sambucus species indicated several genes including atpE, ccsA, ndhD, ndhF, petD, psaJ, psbJ, and rpl33 as highly variable (Figure 10). Conversely, S. javanica_OM868260 and S. chinensis_MW455170 (reference genome) exhibited zero nucleotide variability (Pi = 0).
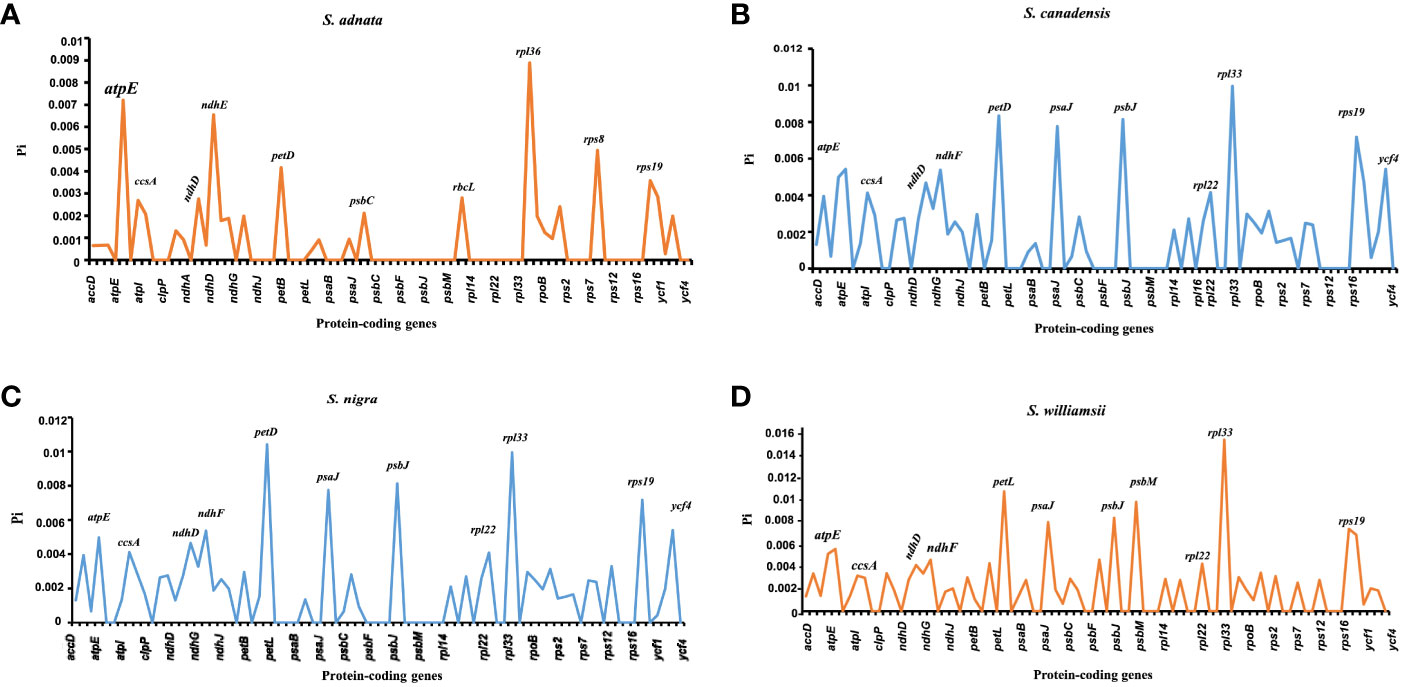
Figure 10 Nucleotide variability of the protein-coding genes of Sambucus plastomes. (A) The most divergent regions in Sambucus adnata. (B) The most divergent regions in Sambucus canadensis. (C) The most divergent regions in Sambucus nigra. (D) The most divergent regions in Sambucus williamsii.
3.5 Codon usage analysis
A comparison was made between selected populations of Sambucus species including S. javanica (OM868260), S. williamsii (OM937121), S. canadensis (OM937119), and S. adnata (ON006399). The number of codons ranged from 52,670 to 52,909, and both species exhibited 64 different types of codons (Table S5). We found 20 different amino acids (AAs) in all studied species (Figure 11), which revealed high similarities in the distribution patterns. Leucine, encoded by six codons, was the most abundant AA and attributed to an average of 9.83%–10.19%. Encoded UUA exhibited the highest Relative Synonymous Codon Usage (RSCU) values, and the ATG codon was found to highly occur as a start codon. The absence of bias was exhibited in methionine and tryptophan, which were encoded for ATG and UGG, respectively (RSCU = 1.00). In addition, the codon bias of most AAs had high preferences (RSCU > 1). Moreover, higher encoding of serine was also exhibited, while the rarest encoding was revealed in cysteine. The RSCU between S. javanica and S. adnata showed high resemblances in the encoding pattern.
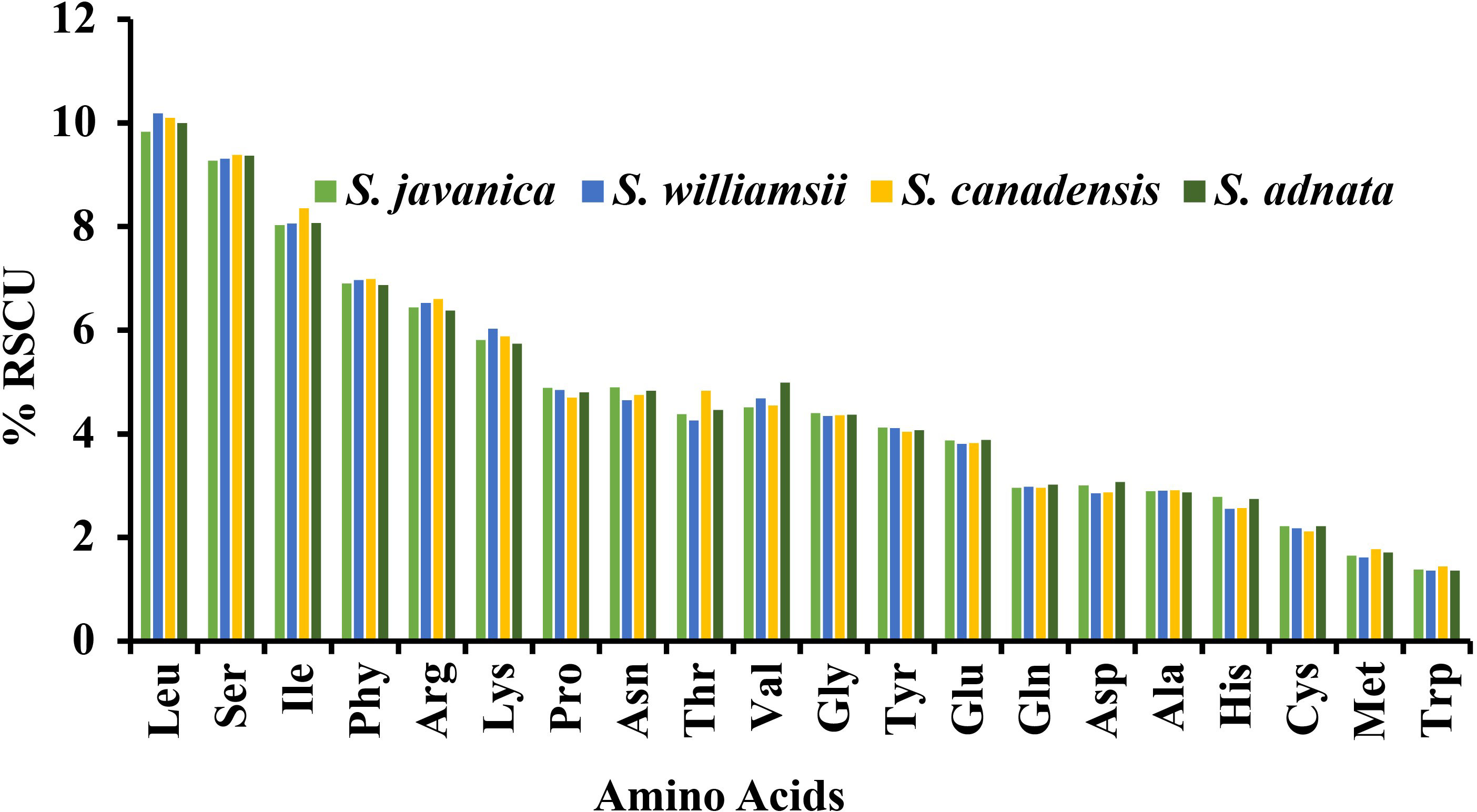
Figure 11 Percentage RSCU of amino acids in Sambucus species.
3.6 Adaptive evolution analysis
Ka/Ks ratios indicate selection pressures and are significant in determining whether a mutation is beneficial, purifying, or neutral. Eighty-one protein-coding genes were extracted from S. williamsii (OM937121), and the values of Ka and Ks were evaluated using S. chinensis (MW455170) as a reference genome. Eighteen photosynthetic genes (atpB, matK, ndhB, ndhC, ndhF, petA, psaA, psbB, psbZ, rpl14, rpl20, rpl32, rpl33, rps2, rps3, rps14, rps15, rps16, and rps19) exhibited zero Ka, Ks, and Ks/Ks substitutions (Ka = 0, Ks = 0, and Ka/Ks = 0). The Ka and Ks values and the substitution rates (Ka/Ks) of the remaining 60 protein-coding genes were calculated (Figure 12; Table S6). The findings showed that a higher average was recorded in Ks (1.068651) than in Ka (0.985531). Twenty-one positively selected genes encompassing atpF, atpH, atpI, clpP, ndhD, petG, petN, psaB, psaC, psaI, psbA, psbC, psbF, psbI, psbK, psbL, rps11, rpl22, rpoB, ycf1, and ycf15 were obtained. Averagely, low rates of Ka/Ks substitutions were displayed by most genes (0.970625), and the high Ka/Ks substitution rates were observed in the LSC and SSC regions compared to the IRs (Figure 13).
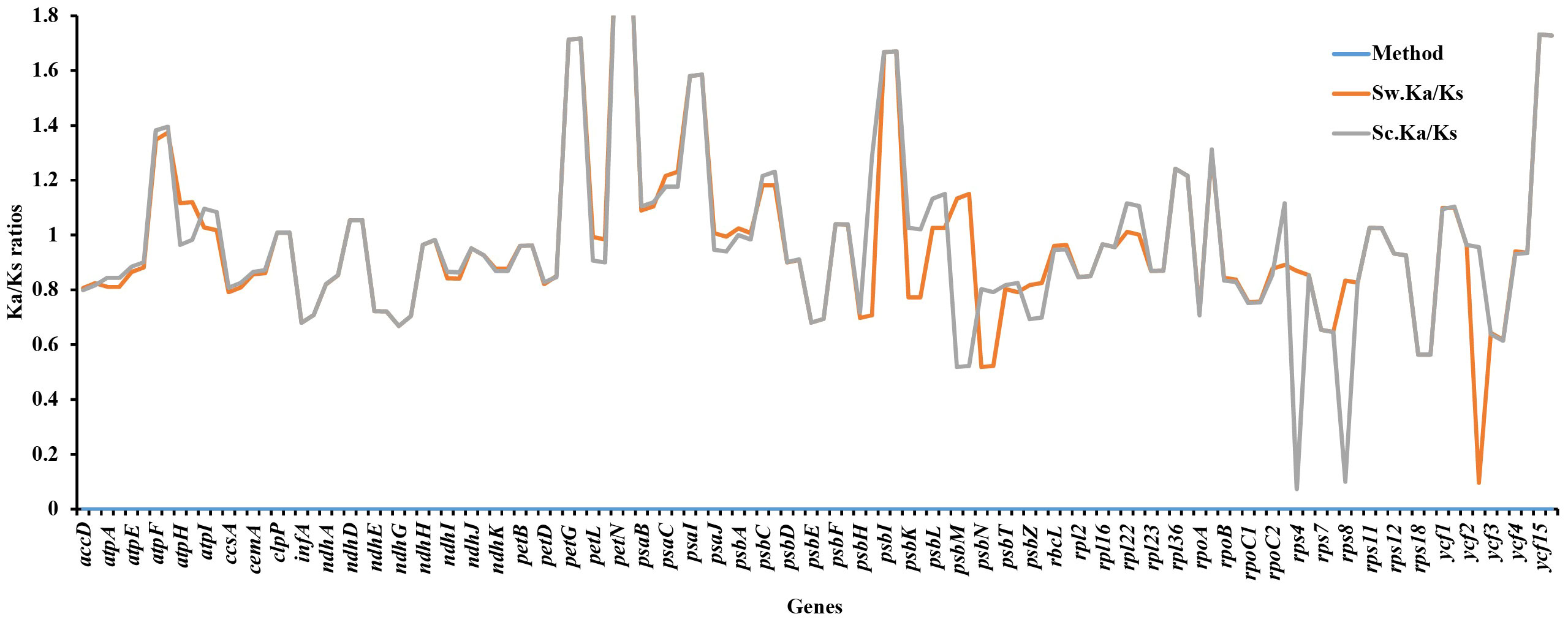
Figure 12 Synonymous and non-synonymous substitution rates of Sambucus. Note: Sc represents Sambucus chinensis, while Sw denotes Sambucus williamsii..

Figure 13 Ka/Ks substitution ratios in the LSC, SSC, and IRs. Sc represents Sambucus chinensis, while Sw denotes Sambucus williamsii. LSC, large single copy; SSC, small single copy; IRs, inverted repeats.
3.7 Phylogenetic analysis
In the present study, the whole plastomes as well as 58 shared protein-coding genes extracted from complete cp genomes of 40 Viburnaceae species and two outgroups from Araliaceae were selected to reconstruct phylogenetic trees. Similar tree topologies were yielded in all datasets by the ML and BI methods and confirmed the monophyly of Sambucus (Figures 14; S2). The phylogenetic relationship within the family recovered two major groups encompassing the Viburnum (larger group) and Sambucus–Adoxa–Tetradoxa–Sinadoxa genera (Adoxoideae) (smaller group) and was significantly supported with bootstrap values greater than 78/100 (Figure 14). In the Adoxoideae group, Adoxa–Tetradoxa–Sinadoxa (Adoxina) was closely related to Sambucus. The taxa S. williamsii, S. nigra, S. adnata, and S. javanica clustered into distinct clades. In this study, the population encompassing S. javanica was treated as clade I, while clade II included the population of S. adnata. Individuals of S. chinensis and S. javanica were clustered together in the same clade in ML, BI, and NN analyses; thus, S. chinensis was treated as a conspecific/synonym to S. javanica. The population of S. nigra clustered together as monophyletic with inconsistent nesting of one individual of S. canadensis (OM937119) in the same clade, while the other (S. canadensis_OM937120) separated from the latter in both datasets. Therefore, S. canadensis might be closely related to S. nigra. Moreover, S. williamsii clustered into a separate monophyletic clade.
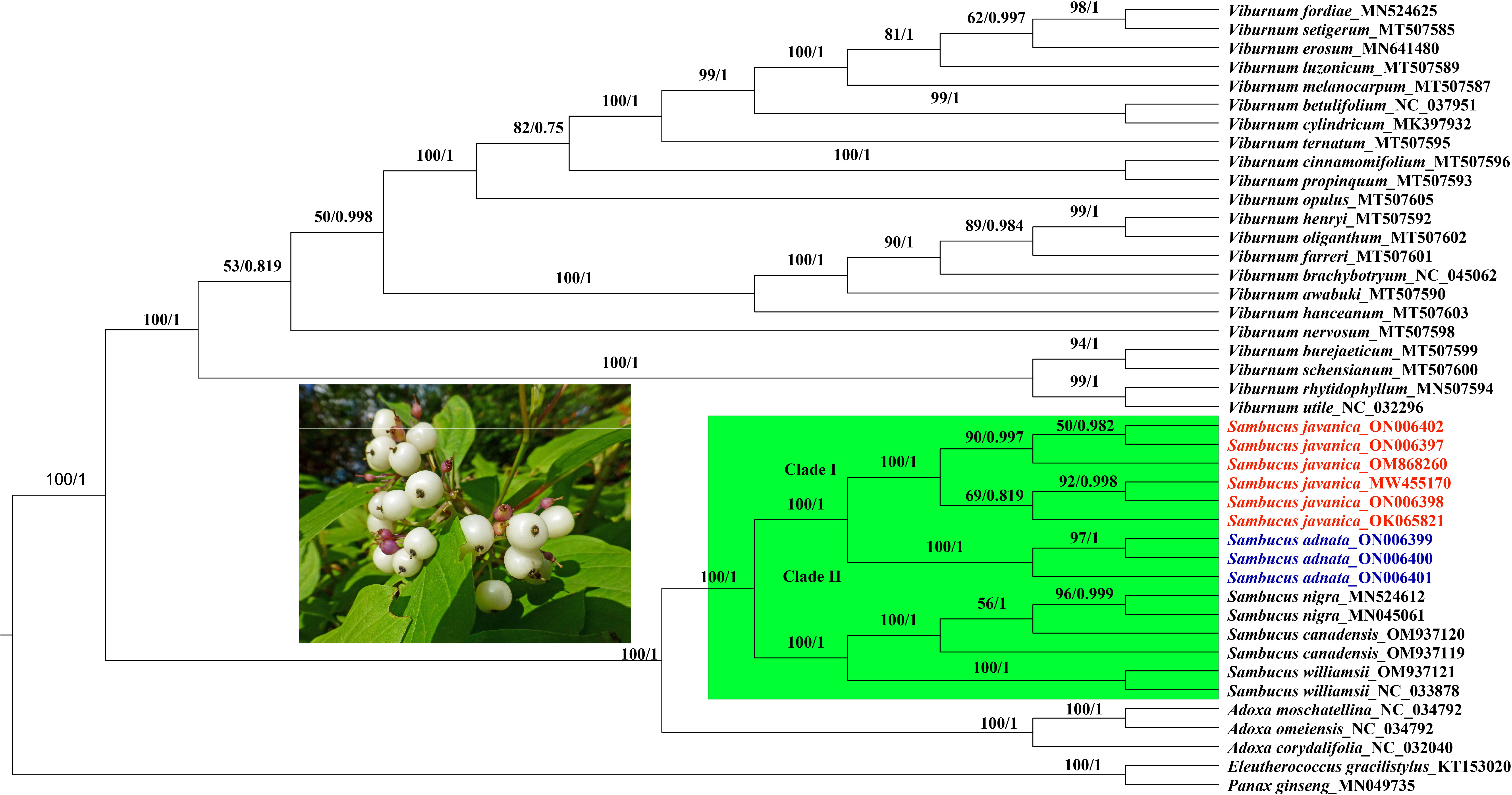
Figure 14 Phylogenetic tree reconstruction based on 58 shared protein-coding genes of 42 Viburnaceae species. The numbers indicate bootstrap values from the ML (left) and BI (right). Eleutherococcus gracilistylus and Panax ginseng were used as outgroups. ML, maximum likelihood; BI, Bayesian inference.
NN analysis was used to determine the phylogenetic resolution between the two closely related S. adnata and S. javanica. In the NN diagram, the term “lineage” was used to denote clustered populations. Individuals of S. javanica (1, 2, 3, 4, 5, and 6) (trivially lineage 1) were distinct and remained isolated from the rest of the population (7, 8, and 9) encompassing S. adnata (lineage 1) (Figure 15). The CDS split graph showed two major groups, and lineage 1 corresponds to clade I, while the populations of S. javanica (lineage 1) correlate to clade II in Figures 9; S1. Nonetheless, the population comprising lineage 1 and clade I appears in close proximity to each other, exhibiting a great genetic affinity.
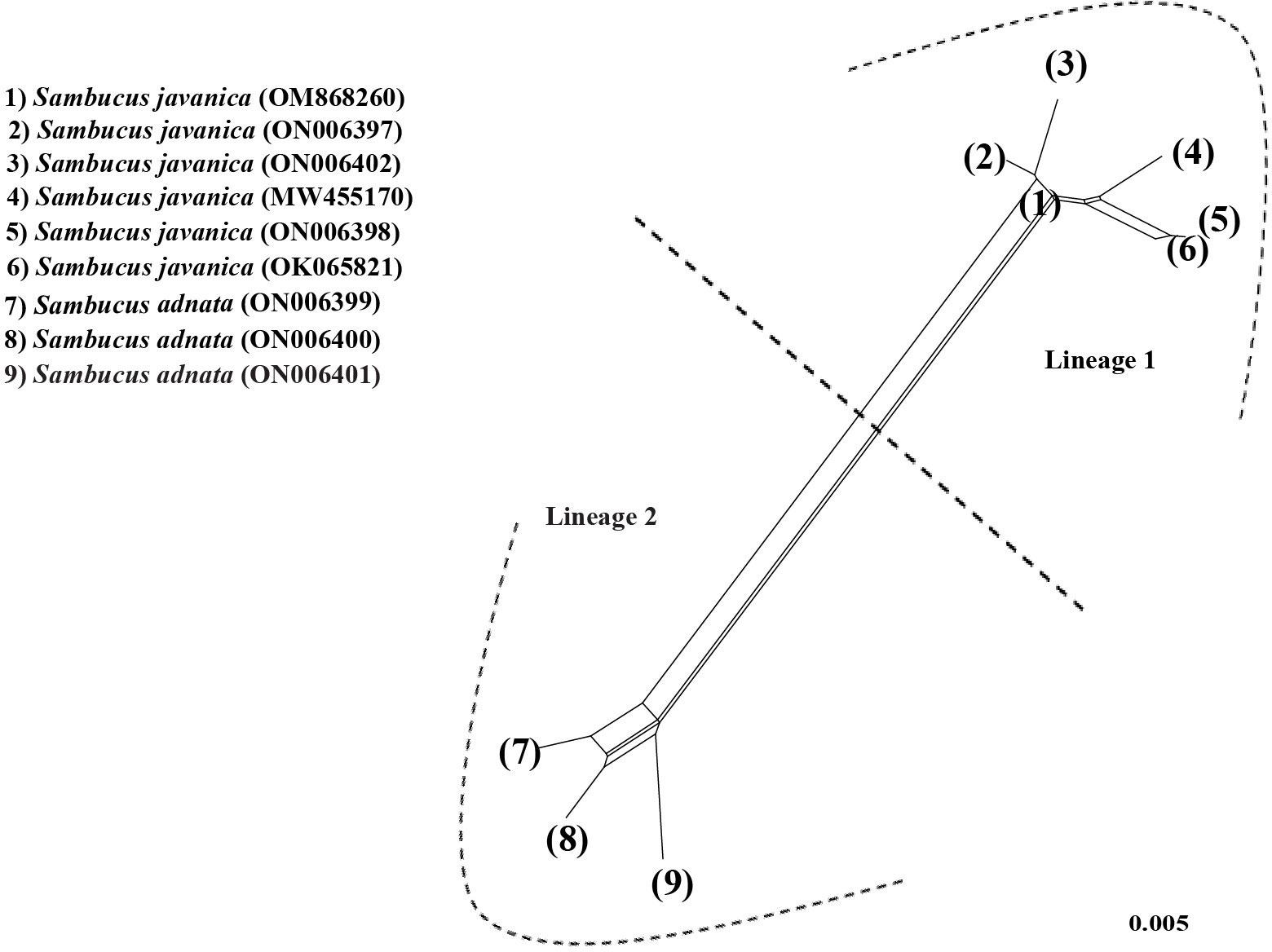
Figure 15 Neighbor-Net for protein-coding sequences of Sambucus.
4 Discussion
4.1 Organization and comparison of genomic features
In the present study, we sequenced the cp genomes of four Sambucus species, annotated and assembled the cp genomes, identified the SSRs within the genomes, analyzed the codon usage and adaptive evolution, and performed comparative phylogenetic analysis within the family Viburnaceae. The findings showed that all the cp genomes presented a typical quadripartite conformation congruent to vast angiosperms (Park et al., 2019; Ran et al., 2020). They were highly conserved in terms of structure, organization, and gene content, with only a few differences in their sizes primarily caused by evolutionary events (IR contraction and expansion) at the border regions (Shaw et al., 2014; Rabah et al., 2019; Abdullah et al., 2020).
The inclusive GC of 38% was detected in the cp genomes of all Sambucus species and collaborated with the previously reported values in the family Viburnaceae (Fan et al., 2018). Additionally, IR regions exhibited higher estimated GC contents when compared to the LSC and SSC regions. As previously reported, the high content of GC nucleotides in several genes in IR regions could be possibly linked to the higher GC percentages (Curci et al., 2015; Raman et al., 2017; Fu et al., 2022).
Based on the gene contents, 132 genes were found in all Sambucus cp genomes, encompassing 37 tRNA, eight rRNA, and 87 CDs, and their arrangements were largely consistent with the majority of the previously reported cp genomes in the genus (Fan et al., 2018; Ran et al., 2020). The evolution of plant lineages is a dynamic process in IR contraction/expansion at the boundaries of the cp genomes and plays a vital role in revealing evolutionary events (Oyebanji et al., 2020; Bai et al., 2021). In our findings, it was evident that IR contraction/expansion varies at the boundaries, and this character is a synapomorphy of the 10 studied plastomes, consistent with several angiosperms (Shaw et al., 2014; Amenu et al., 2022). Sambucus genomes reported no gene loss, and the presence of ycf1 pseudogene might be due to evolutionary events at the IRs (Ran et al., 2020). Interestingly, rps12 gene had a unique trans-splicing with LSC incorporating the 5′ end of the exon and 3′ end positioned in the IRs, a phenomenon highly detected in most plants (Hildebrand et al., 1988; Munyao et al., 2020). infA gene usually participates in the initiation of the translation process (Gichira et al., 2017; Chen et al., 2019). In the present study, this gene was found to be non-functional, congruent to the previous related investigations on other plants (Ewels et al., 2016; Shen et al., 2016; Amiryousefi et al., 2018). Nonetheless, it is suggested to be translocated into the nuclear plastome genome in an active form that is functional or sometimes its copy (Piot et al., 2018; Shahzadi et al., 2019).
4.2 Analysis of repeats and codon usage
Dispersed repeat sequences are essential in the rearrangement of genomes and thus form a basis for resolving the phylogenetic complexities among different taxa (Raman et al., 2017). They are normally used as commendable molecular markers in plant taxonomy (Gu et al., 2019). Our present study assessed the dispersion of repetitive sequences in several Sambucus cp genomes, which showed similarities in SSR motif distribution, congruent to previous findings (Abdullah et al., 2020; Mehmood et al., 2020a). Palindromic repeats were the richest among studied species, similar to previous outcomes in the Sambucus genus (Fan et al., 2018; Ran et al., 2020). Oligonucleotide repeats (SSRs) usually generate mutations in genomes and thus are regarded as proxies to identify mutational hotspots (Ahmed et al., 2012; Abdullah et al., 2020; Mehmood et al., 2020b). Our results indicated that most SSRs are positioned at the non-coding spacers (NCSs) and thus could be potential DNA markers for species identification (Fan et al., 2018; Ran et al., 2020).
Codon usage refers to how an organism encodes amino acids in its protein genes using identical codons (Munyao et al., 2020). However, several previous studies showed variabilities in codon usage among different species (Li et al., 2016; Srivastava and Shanker, 2016). Codons link proteins and nucleic acids, thus acting as significant transmitters of genetic information used in plastome evolution (Liu et al., 2005). Each gene in an organism has its favorite amino acid codon, which is referred to as codon use bias (Wu et al., 2007), which is greatly influenced by natural selection (Liu and Xue, 2005). Our findings revealed high RSCU values for the codons with A/T at the 3′ end in place of C/G, similar to previous observations in several cp genomes of land plants (Abdullah et al., 2020; Mehmood et al., 2020a), and it is presumably influenced by enormous AT content in the plastid genomes. The present study showed high proportions of leucine and isoleucine, while cysteine exhibited the least AA. The cp genomes of Sambucus species established similarities in codon usage, and the codons ending in A/U had higher encoding efficacy than those ending in C/G. Previous studies indicated that close phylogenetic relationships among closely related species may be due to similarities in codon selection strategies (Qian et al., 2013). Cui et al., 2019; Fu et al., 2022 In this study, the codon bias of most AAs was highly preferred (RSCU ≥ 1), suggesting that codon usage happens more often than expected. RSCU ≥ 1 was notably displayed by the majority of codons ending with A/U. The codon usage is less often than expected when values of less than one (RSCU < 1) are exhibited (Li et al., 2017). RSCU < 1 was exhibited for most codons ending with C/G in this study. Moreover, the codon usage of methionine and tryptophan was not preferred (RSCU = 1).
4.3 Comparative genome characterization analysis
The various regions of the chloroplast DNAs (cpDNAs) were reported as possible markers to explore the phylogenetic relationships of the closely related species (Gu et al., 2019). Some of the hypervariable regions in the cpDNAs including ycf1, trnF-ndhJ, rpl33, rps2-rpoC2, rps18-rpl20, rps16, trnG-trnR, atpE, trnM-psbD, ccsA, trnN-ndhF, clpP, ycf4-cemA, ycf4, ndhG-ndhI, ndhF, rpl32trnL, ndhD, atpI-rps2, and rpl16 were stated as significant markers in studying the phylogenetic relationships of Sambucus species (Fan et al., 2018; Ran et al., 2020). In the present study, the most divergent regions were trnT-GGU, trnF-GAA, and trnL-UAG intergenic spacers as well as atpE, ccsA, ndhD, ndhF, petD, psaJ, psbJ, and rpl33 genes. The proportion of variable sites in non-coding regions was higher than in the coding regions, consistent with the previous findings by Fu et al. (2022). Hence, they can be employed as candidate DNA barcodes for phylogenetic and phylogeographic studies. The SC regions were more variable than IR regions and thus less conserved. In this study, the most informative plastome region was trnT-GGU and thus could be a useful marker in phylogenetic resolution at lower-level phylogenetic studies.
The protein-coding genes have been widely used to determine selection pressures (Wang et al., 2021). Under positive selection in the Ka/Ks analysis, 21 identified genes were correlated with adaptive evolution. High evolution rates were observed in functional genes with high Ka/Ks ratios, whereas the genes that were linked to photosynthesis exhibited slow evolutionary rates, agreeing with the results obtained by Song et al. (2022). The latter exhibited no obvious nucleotide changes (Ka and Ks = 0), indicating that Sambucus plastomes were relatively conserved. Most genes exhibited Ka/Ks values of less than one, suggesting that they may have experienced extensive purifying selection and could remove deleterious mutations (Wu et al., 2020).
4.4 Phylogenetic analysis
Phylogenetic relationships within Viburnaceae were established and resolved by several authors in the past few decades (Chase et al., 1993; Eriksson and Donoghue, 1997; Donoghue et al., 2001; Jacobs et al., 2010). The complete cp genomes and protein-coding genes dataset have been previously utilized to reconstruct phylogenies (Blazier et al., 2016; Hou et al., 2016; Namgung et al., 2021). In this study, we used complete cp genomes and protein-coding genes to examine the phylogenetic relationships between several Sambucus species by BI, ML, and NN methods.
Our findings revealed significantly supported clades within Viburnaceae and confirmed the monophyly of Sambucus, similar to previous findings (Fan et al., 2018; Ran et al., 2020). The obtained topologies based on different analyses exhibited similar branching patterns, consistent with prior studies within the family (Lens et al., 2016; Ran et al., 2020). In the present work, we adopted the most recent taxonomic treatments of Adoxa, Tetradoxa, and Sinadoxa. Based on this treatment, Tetradoxa L. and Sinadoxa L. are circumscribed within Adoxa L., which is regarded as an accepted genus encompassing four recognized species including A. corydalifolia (C.Y.Wu, Z.L.Wu & R.F.Huang) christenh. & Byng (S. corydalifolia), A. moschatellina L., A. omeiensis H. Hara (T. omeiensis (H. Hara) C.Y.Wu), and Adoxa xizangensis G.Yao (POWO, 2023). Therefore, Tetradoxa C.Y.Wu, and Sinadoxa C.Y.Wu, Z.L.Wu & R.F.Huang are treated as synonyms of Adoxa L.
Previous morphological and molecular studies showed a close relationship between S. adnata and S. javanica, which were clustered in the same clade (Eriksson and Donoghue, 1997; Lens et al., 2016). The findings by Ran et al. (2020) inferred by the complete chloroplast genome sequence dataset indicated that S. javanica Blume is a close relative of S. adnata Wall. ex DC. The latter are morphologically united by valvate corolla lobes, yellow anthers, bladelike stipules, umbellate cymes, three-lobed stigma, and urceolate calyx (Eriksson and Donoghue, 1997; Silalahi and Wakhidah, 2021); thus, difficulties may arise during the discrimination process. In this study, molecular analysis showed amplification of primers in S. adnata that were distinct from those amplified by S. javanica and segregated into different clades. The segregation between S. javanica and S. adnata is supported by several morphological characteristics such as inconspicuous stem lenticels, two to three pairs of leaflets, flower filaments connate at base, and fruits characterized by verrucate pyrenes, which distinguish S. javanica from S. adnata (http://www.efloras.org/).
Populations of the same species tend to cluster together within the tree, and possible reticulation events can be evidenced by the NN phylogenetic networks (Bryant and Moulton, 2002). The phylogenetic relationships indicated by edges revealed a complete divergence between the populations of S. javanica and S. adnata. Although S. adnata appears closely related to S. javanica, we treat them as distinct species due to existing morphological differences and their discrimination into different clusters and/or lineages observed in ML, BI, and NN analyses. The results obtained in both analyses indicate similarities in the retention of distinct gene pools between S. javanica and S. adnata and thus should be treated as discrete taxonomic entities. Moreover, negligible nucleotide variability between S. javanica and S. chinensis was observed in the current study. Thus, we confirm S. chinensis as a synonym for S. javanica, congruent to previous findings (Kern and van Steenis, 1948; Ohashi, 2015; WFO, 2023). Representatives of the Sambucus genus frequently show slight variations within individual species (Elkiran, 2021). Consequently, the infraspecific classification by Ohashi (2015) regarded S. chinensis as a form of S. javanica due to the sole distinctive morphological attribute of fruit color (black or blackish purple), distinguishing the latter from S. javanica (red). However, S. chinensis was considered a synonym of S. javanica subsp. chinensis (Lindl) Fukuoka and conspecific to S. javanica (POWO, 2023). The present findings support the conspecific nature of S. chinensis and S. javanica. Our results indicate the close relatedness of the medicinally significant S. nigra and S. canadensis, consistent with previous findings (Eriksson and Donoghue, 1997; Jacobs et al., 2010). Therefore, more specimens will be required to further analyze their taxonomic treatment in the future.
5 Conclusion
In the present study, the newly sequenced cp genomes of the populations of S. javanica, S. adnata, S. canadensis, and S. williamsii were reported and combined with 16 others to perform the comparative molecular analysis of 40 Viburnaceae cp genomes. A comparative phylogenetic analysis of the studied species was concluded. The structure, order, and gene content of the cp genomes of Sambucus species exhibited striking similarities, revealing their highly conservative nature. This study showed that trnT-GGU, trnF-GAA, psaJ, ndhF, trnL-UAG, and ndhE genes as the most divergent hotspot regions. The analyzed hypervariable regions, SSRs, and repeat sequences could be utilized as possible markers for molecular phylogeographic and genetic studies. Moreover, the phylogenetic analysis showed complete segregation of Viburnaceae species into two major branches representing Viburnum, Sambucus, and Adoxa (Adoxoideae) groups. Adoxa L. encompassed the Adoxa–Tetradoxa–Sinadoxa (Adoxina) group. Populations of different Sambucus species clustered separately, with the exception of S. canadensis, while individuals of the same species clustered clearly in the same clade. Further, the classical taxonomy of Sambucus based on protein-coding genes and whole cp genomes supported the separation of S. javanica Blume and S. adnata Wall. ex DC. Therefore, the results presented in this work demonstrate that the complete cp genomes and protein-coding genes could be used to discriminate species. The findings of this study provide rich genetic data for phylogenetic investigations, which might be useful for future research within Viburnaceae.
Data availability statement
The data presented in this study are deposited in the Genbank repository. The accession number(s) can be found in the article/Supplementary material.
Author contributions
EW, G-WH, and Q-FW participated in the study de sign and carried out experiments. EW and G-WH collected plant specimens. WO, SA, EMM, and ESM contributed to the drafting of the manuscript and data analysis. SM and S-XD revised the draft manuscript. All authors read and approved the final version of the manuscript.
Funding
This work was supported by grants from the National Natural Science Foundation of China (31961143026, 31970211), National Science and Technology Fundamental Resources Investigation Program of China (2019FY101800), International Partnership Program of Chinese Academy of Sciences (151853KYSB20190027), and Sino-Africa Joint Research Center, CAS (SAJC202101).
Acknowledgments
We thank Consolata Nanjala for her insightful suggestions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1179510/full#supplementary-material
References
Abdullah, H., Mehmood, F., Shahzadi, I., Ali, Z., Waheed, M. T., Croat, T. B., et al. (2020). Comparison among the first representative chloroplast genomes of Orontium, lasia, Zamioculcas, and Stylochaeton of the plant family araceae: inverted repeat dynamics are not linked to phylogenetic signalling. bioRxiv 2020-04. doi: 10.1101/2020.04.07.029389
Ahmed, I., Biggs, P. J., Matthews, P. J., Collins, L. J., Hendy, M. D., Lockhart, P. J. (2012). Mutational dynamics of aroid chloroplast genomes. Genome Biol. Evol. 4 (12), 1316–1323. doi: 10.1093/gbe/evs110
Allen, J. F. (2015). Why chloroplasts and mitochondria retain their own genomes and genetic systems: colocation for redox regulation of gene expression. Proc. Natl. Acad. Sci. United States America 112 (33), 10231–10238. doi: 10.1073/pnas.1500012112
Amenu, S. G., Wei, N., Wu, L., Oyebanji, O., Hu, G., Zhou, Y., et al. (2022). Phylogenomic and comparative analyses of coffeeae alliance (Rubiaceae): deep insights into phylogenetic relationships and plastome evolution. BMC Plant Biol. 22 (1), 1–13. doi: 10.1186/s12870-022-03480-5
Amini, E., Nasrollahi, F., Sattarian, A., Habibi, M. (2019). Systematic and molecular biological study of Sambucus L. (Caprifoliaceae) Iran. Thaiszia J. Bot. 29, 133–150. doi: 10.33542/TJB2019-2-02
Amiryousefi, A., Hyvo, J., Poczai, P. (2018). Genome analysis IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34 (17), 3030–3031. doi: 10.1093/bioinformatics/bty220
Applequist, W. L. (2015). A brief review of recent controversies in the taxonomy and nomenclature of Sambucus nigra sensu lato. In Acta Hortic. 1061 (1994), 25–34). doi: 10.17660/actahortic.2015.1061.1
Bai, H.-R., Oyebanji, O., Zhang, R., Yi, T.-S. (2021). Plastid phylogenomic insights into the evolution of subfamily dialioideae (Leguminosae). Plant Diversity 43 (1), 27–34. doi: 10.1016/j.pld.2020.06.008
Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33 (16), 2583–2585. doi: 10.1093/bioinformatics/btx198
Birky, C. W. (1995). Uniparental inheritance of mitochondrial and chloroplast genes: mechanisms and evolution. Proc. Natl. Acad. Sci. United States America 92 (25), 11331–11338. doi: 10.1073/pnas.92.25.11331
Blazier, J. C., Jansen, R. K., Mower, J. P., Govindu, M., Zhang, J., Weng, M. L., et al. (2016). Variable presence of the inverted repeat and plastome stability in erodium. Ann. Bot. 117 (7), 1209–1220. doi: 10.1093/aob/mcw065
Bryant, D., Moulton, V. (2002). NeighborNet: an agglomerative method for the construction of planar phylogenetic networks. International Workshop on Algorithms in Bioinformatics (Rome, Italy: Springer Berlin Heidelberg) 2, 375–391.
Capella-Gutiérrez, S., Silla-Martínez, J. M., Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25 (15), 1972–1973. doi: 10.1093/bioinformatics/btp348
Chase, M. W., Soltis, D. E., Olmstead, R. G., Morgan, D., Les, D. H., Mishler, B. D., et al. (1993). Phylogenetics of seed plants: an analysis of nucleotide sequences from the plastid gene rbcL. Ann. Missouri Botanical Garden 80 (3), 528–580. doi: 10.2307/2399846
Chen, Y., Hu, N., Wu, H. (2019). Analyzing and characterizing the chloroplast genome of Salix wilsonii. BioMed. Res. Int. 2019, 1–14. doi: 10.1155/2019/5190425
Clarke, J. B., Tobutt, K. R. (2006). Development of microsatellite primers and two multiplex polymerase chain reactions for the common elder (Sambucus nigra). Mol. Ecol. Notes 6 (2), 453–455. doi: 10.1111/j.1471-8286.2006.01270.x
Cui, Y., Nie, L., Sun, W., Xu, Z., Wang, Y., Yu, J., et al. (2019). Comparative and phylogenetic analyses of ginger (Zingiber officinale) in the family zingiberaceae based on the complete chloroplast genome. Plants 8 (8), 283. doi: 10.3390/plants8080283
Curci, P. L., De Paola, D., Danzi, D., Vendramin, G. G., Sonnante, G. (2015). Complete chloroplast genome of the multifunctional crop globe artichoke and comparison with other asteraceae. PloS One 10 (3), e0120589. doi: 10.1371/journal.pone.0120589
Daniell, H., Lin, C. S., Yu, M., Chang, W. J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17 (1), 1–29. doi: 10.1186/s13059016-1004-2
Darling, A. C. E., Mau, B., Blattner, F. R., Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14 (7), 1394–1403. doi: 10.1101/gr.2289704
Do Nascimento, P. G. G., Lemos, T. L. G., Bizerra, A. M. C., Arriaga, A. M. C., Ferreira, D. A., Santiago, G. M. P., et al. (2014). Antibacterial and antioxidant activities of ursolic acid and derivatives. Molecules 19 (1), 1317–1327. doi: 10.3390/molecules19011317
Dong, W., Liu, J., Yu, J., Wang, L., Zhou, S. (2012). Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PloS One 7 (4), 1–9. doi: 10.1371/journal.pone.0035071
Donoghue, M. J., Eriksson, T., Reeves, P. A., Olmstead, R. G. (2001). Phylogeny and phylogenetic taxonomy of dipsacales, with special reference to Sinadoxa and Tetradoxa (Adoxaceae). Harvard Papers Bot. 6 (2), 459–479. Available at: http://www.jstor.org/stable/41761757.
Dyall, S. D., Brown, M. T., Johnson, P. J. (2004). Ancient invasions: from endosymbionts to organelles. Science 304 (5668), 253–257. doi: 10.1126/science.1094884
Elkiran, O. (2021). Comparative Anatomical Study in The Genus Sambucus L. (Adoxaceae) Distributed in Turkey. Avrupa Bilim ve Teknoloji Dergisi 31, 670–674. doi: 10.31590/ejosat.1028351
Eriksson, T., Donoghue, M. J. (1997). Phylogenetic relationships of Sambucus and Adoxa (Adoxoideae, adoxaceae) based on nuclear ribosomal ITS sequences and preliminary morphological data. Systemat. Bot. 22 (3), 555–573. doi: 10.2307/2419828
Ewels, P., Magnusson, M., Lundin, S., Käller, M. (2016). MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32 (19), 3047–3048. doi: 10.1093/bioinformatics/btw354
Fan, W. B., Wu, Y., Yang, J., Shahzad, K., Li, Z. H. (2018). Comparative chloroplast genomics of dipsacales species: insights into sequence variation, adaptive evolution, and phylogenetic relationships. Front. Plant Sci. 9 (May). doi: 10.3389/fpls.2018.00689
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32 (Web Server ISS.), 273–279. doi: 10.1093/nar/gkh458
Fu, G., Liu, Y., Caraballo-Ortiz, M. A., Zheng, C., Liu, T., Xu, Y., et al. (2022). Characterization of the complete chloroplast genome of the dragonhead herb, Dracocephalum heterophyllum (Lamiaceae), and comparative analyses with related species. Diversity 14 (2), 110. doi: 10.3390/d14020110
Fu, F., Xu, M., Li, W. (2020). Antiviral phytomedicine elderberry (Sambucus) inhibition of 2019nCoV. Authorea. doi: doi.10.22541/au.158168950.03798827
Gichira, A. W., Li, Z., Saina, J. K., Long, Z., Hu, G., Gituru, R. W., et al. (2017). The complete chloroplast genome sequence of an endemic monotypic genus Hagenia (Rosaceae): structural comparative analysis, gene content and microsatellite detection. PeerJ 5, e2846. doi: 10.7717/peerj.2846
Green, B. R. (2011). Chloroplast genomes of photosynthetic eukaryotes. Plant J. 66 (1), 34–44. doi: 10.1111/j.1365-313X.2011.04541.x
Gu, C., Ma, L., Wu, Z., Chen, K., Wang, Y. (2019). Comparative analyses of chloroplast genomes from 22 Lythraceae species: inferences for phylogenetic relationships and genome evolution within Myrtales. BMC Plant Biology 19 (1), 1–19. doi: 10.1186/s12870-019-1870-3
Guindon, S., Dufayard, J.-F., Lefort, V., Anisimova, M., Hordijk, W., Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Systemat. Biol. 59 (3), 307–321. doi: 10.1093/sysbio/syq010
He, L., Qian, J., Li, X., Sun, Z., Xu, X., Chen, S., et al. (2017). Complete chloroplast genome of medicinal plant Lonicera japonica: genome rearrangement, intron gain and loss, and implications for phylogenetic studies. Molecules 22 (2), 1–12. doi: 10.3390/molecules22020249
Hearst, C., McCollum, G., Nelson, D., Ballard, L. M., Millar, B. C., Goldsmith, C. E., et al. (2010). Antibacterial activity of elder (Sambucus nigra l.) flower or berry against hospital pathogens. J. Medicinal Plants Res. 4 (17), 1805–1809. doi: 10.5897/JMPR10.147
Hildebrand, M., Hallick, R. B., Passavant, C. W., Bourque, D. P. (1988). Trans-splicing in chloroplasts: the rps 12 loci of Nicotiana tabacum. Proc. Natl. Acad. Sci. 85 (2), 372–376. doi: 10.1073/pnas.85.2.372
Hou, C., Wikström, N., Strijk, J. S., Rydin, C. (2016). Resolving phylogenetic relationships and species delimitations in closely related gymnosperms using high-throughput NGS, Sanger sequencing and morphology. Plant Systemat. Evol. 302 (9), 1345–1365. doi: 10.1007/s00606-016-1335-1
Hourahine, B., Aradi, B., Blum, V., Bonafé, F., Buccheri, A., Camacho, C., et al. (2020). DFTB+, a software package for efficient approximate density functional theory based atomistic simulations. J. Chem. Phys. 152 (12), 124101. doi: 10.1063/1.5143190
Huang, D. I., Hefer, C. A., Kolosova, N., Douglas, C. J., Cronk, Q. C. B. (2014). Whole plastome sequencing reveals deep plastid divergence and cytonuclear discordance between closely related Balsam poplars, Populus balsamifera, and P. trichocarpa (S alicaceae). New Phytologist 204 (3), 693–703. doi: 10.1111/nph.12956.
Hummer, K. E., Pomper, K. W., Postman, J., Graham, C. J., Stover, E., Mercure, E. W., et al. (2012). Emerging Fruit Crops. In: Badenes, M., Byrne, D. (eds) Fruit Breeding. Handbook of Plant Breeding (Boston, MA: Springer) 8. doi: 10.1007/978-1-4419-0763-9_4
Huson, D. H., Bryant, D. (2001). Application of phylogenetic networks in evolutionary studies. Mole. Bio. Evol. 23 (2), 254–267. doi: 10.1093/molbev/msj030
IPNI (2023). International plant names index. Available at: https://www.ipni.org/. (Accessed February 15, 2023).
Jacobs, B., Huysmans, S., Smets, E. (2010). Evolution and systematic value of fruit and seed characters in adoxaceae (Dipsacales). Taxon 59 (3), 850–866. doi: 10.1002/tax.593014
Jacobs, B., Lens, F., Smets, E. (2009). Evolution of fruit and seed characters in the Diervilla and Lonicera clades (Caprifoliaceae, Dipsacales). Ann. Bot. 104 (2), 253–276. doi: 10.1093/aob/mcp131
Jin, D. M., Wicke, S., Gan, L., Yang, J. B., Jin, J. J., Yi, T. S. (2020). The loss of the inverted repeat in the putranjivoid clade of malpighiales. Front. Plant Sci. 11 (June). doi: 10.3389/fpls.2020.00942
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30 (4), 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28 (12), 1647–1649. doi: 10.1093/bioinformatics/bts199
Kern, J. H., van Steenis, C. G. G. J. (1948). Caprifoliaceae. Flora Malesiana-Series 1 Spermatophyta 4 (1), 175–194.
Kong, W., Yang, J. (2015). The complete chloroplast genome sequence of Morus mongolica and a comparative analysis within the fabidae clade. Curr. Genet. 62, 165–172. doi: 10.1007/s00294-0150507-9
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29 (22), 4633–4642. doi: 10.1093/nar/29.22.4633
Kyalo, C. M., Li, Z.-Z., Mkala, E. M., Malombe, I., Hu, G.-W., Wang, Q.-F. (2020). The first glimpse of Streptocarpus ionanthus (Gesneriaceae) phylogenomics: analysis of five subspecies’ chloroplast genomes. Plants 9 (4), 456. doi: 10.3390/plants9040456
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T., Calcott, B. (2017). PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34 (3), 772–773. doi: 10.1093/molbev/msw260
Lee, H., Jansen, R. K., Chumley, T. W., Kim, K. (2007). Gene relocations within chloroplast genomes of Jasminum and Menodora (Oleaceae) are due to multiple, overlapping inversions. Mole. Bio. Evol. 24 (5), 1161–1180. doi: 10.1093/molbev/msm036
Lens, F., Vos, R. A., Charrier, G., van der Niet, T., Merckx, V., Baas, P., et al. (2016). Scalariformto-simple transition in vessel perforation plates triggered by differences in climate during the evolution of adoxaceae. Ann. Bot. 118 (5), 1043–1056. doi: 10.1093/aob/mcw151
Levy, D., Pachter, L. (2011). Advances in applied mathematics the neighbor-net algorithm. Adv. Appl. Math. 47 (2), 240–258. doi: 10.1016/j.aam.2010.09.002
Li, Y., Kuang, X.-J., Zhu, X.-X., Zhu, Y.-J., Sun, C. (2016). Codon usage bias of Catharanthus roseus. Zhongguo Zhong Yao Za Zhi= Zhongguo Zhongyao Zazhi= China J. Chin. Materia Med. 41 (22), 4165–4168. doi: 10.4268/cjcmm20162213
Li, B., Lin, F., Huang, P., Guo, W., Zheng, Y. (2017). Complete chloroplast genome sequence of decaisnea insignis: genome organization, genomic resources and comparative analysis. Sci. Rep. 7 (1), 1–10. doi: 10.1038/s41598-017-10409-8
Librado, P., Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25 (11), 1451–1452. doi: 10.1093/bioinformatics/btp187
Liu, Q., Dou, S., Ji, Z., Xue, Q. (2005). Synonymous codon usage and gene function are strongly related in Oryza sativa. Biosystems 80 (2), 123–131. doi: 10.1016/j.biosystems.2004.10.008
Liu, Y., Huo, N., Dong, L., Wang, Y., Zhang, S., Young, H. A., et al. (2013). Complete chloroplast genome sequences of Mongolia medicine Artemisia frigida and phylogenetic relationships with other plants. PloS One 8 (2). doi: 10.1371/journal.pone.0057533
Liu, Q., Xue, Q. (2005). Comparative studies on codon usage pattern of chloroplasts and their host nuclear genes in four plant species. J. Genet. 84 (1), 55–62. doi: 10.1007/BF02715890
Mehmood, F., Shahzadi, I., Ahmed, I., Waheed, M. T., Mirza, B. (2020a). Characterization of Withania somnifera chloroplast genome and its comparison with other selected species of solanaceae. Genomics 112 (2), 1522–1530. doi: 10.1016/j.ygeno.2019.08.024
Mehmood, F., Shahzadi, I., Waseem, S., Mirza, B., Ahmed, I., Waheed, M. T. (2020b). Chloroplast genome of Hibiscus rosa-sinensis (Malvaceae): comparative analyses and identification of mutational hotspots. Genomics 112 (1), 581–591. doi: 10.1016/j.ygeno.2019.04.010
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H.-P., Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinf. 14 (1), 1–14. doi: 10.1186/1471-2105-14-60
Minh, B. Q., Nguyen, M. A. T., von Haeseler, A. (2013). Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30 (5), 1188–1195. doi: 10.1093/molbev/mst024
Munyao, J. N., Dong, X., Yang, J. X., Mbandi, E. M., Wanga, V. O., Oulo, M. A., et al. (2020). Complete chloroplast genomes of Chlorophytum comosum and Chlorophytum gallabatense: genome structures, comparative and phylogenetic analysis. Plants 9 (3), 1–19. doi: 10.3390/plants9030296
Namgung, J., Dang, H., Do, K., Kim, C., Choi, H. J., Kim, J. H. (2021). Complete chloroplast genomes shed light on phylogenetic relationships, divergence time, and biogeography of allioideae (Amaryllidaceae). Sci. Rep. 11 (1), 1–13. doi: 10.1038/s41598-021-82692-5
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32 (1), 268–274. doi: 10.1093/molbev/msu300
Olmstead, R. G., Bremer, B., Scott, K. M., Palmer, J. D. (1993). A parsimony analysis of the asteridae sensu lato based on rbcL sequences. Ann. Missouri Botanical Garden 80, 700–722. doi: 10.2307/2399855
Otsuka, H., Shitamoto, J., Sueyoshi, E., Matsunami, K., Takeda, Y. (2021). A megastigmane glucoside from sambucus chinensis. J. Medicinal Plants 9 (1), 29–32.
Oyebanji, O., Zhang, R., Chen, S.-Y., Yi, T.-S. (2020). New insights into the plastome evolution of the Millettioid/Phaseoloid clade (Papilionoideae, leguminosae). Front. Plant Sci. 11, 151. doi: 10.3389/fpls.2020.00151
Park, J., Choi, Y. G., Yun, N., Xi, H., Min, J., Oh, S. (2019). The complete chloroplast genome sequence of Viburnum erosum (Adoxaceae). Mitochondrial DNA Part B 4 (2), 3278–3279. doi: 10.1080/23802359.2019.1667919
Piot, A., Hackel, J., Christin, P.-A., Besnard, G. (2018). One-third of the plastid genes evolved under positive selection in PACMAD grasses. Planta 247 (1), 255–266. doi: 10.1007/s00425-017-2781-x
POWO (2023) Plants of the world online. kew: facilitated by the royal botanic gardens (Accessed February 15, 2023).
Przybylska-Balcerek, A., Szablewski, T., Szwajkowska-Michałek, L., Świerk, D., CegielskaRadziejewska, R., Krejpcio, Z., et al. (2021). Sambucus nigra extracts–natural antioxidants and antimicrobial compounds. Molecules 26 (10), 2910. doi: 10.3390/molecules26102910
Qian, J., Song, J., Gao, H., Zhu, Y., Xu, J., Pang, X., et al (2013). The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PloS One 8 (2). doi: 10.1371/journal.pone.0057607
Rabah, S. O., Shrestha, B., Hajrah, N. H., Sabir, M. J., Alharby, H. F., Sabir, M. J., et al. (2019). Passiflora plastome sequencing reveals widespread genomic rearrangements. J. Systemat. Evol. 57 (1), 1–14. doi: 10.1111/jse.12425
Raman, G., Park, V., Kwak, M., Lee, B., Park, S. (2017). Characterization of the complete chloroplast genome of Arabis stellari and comparisons with related species. PloS One 12 (8), e0183197. doi: 10.1371/journal.pone.0183197
Ran, H., Liu, Y., Wu, C., Cao, Y. (2020). Phylogenetic and comparative analyses of complete chloroplast genomes of Chinese viburnum and Sambucus (Adoxaceae). Plants 9 (9), 1–17. doi: 10.3390/plants9091143
Raveendar, S., Na, Y. W., Lee, J. R., Shim, D., Ma, K. H., Lee, S. Y., et al. (2015). The complete chloroplast genome of Capsicum annuum var. glabriusculum using illumina sequencing. Molecules 20 (7), 13080–13088. doi: 10.3390/molecules200713080
Ronquist, F., Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19 (12), 1572–1574. doi: 10.1093/bioinformatics/btg180
Shahzadi, I., Mehmood, F., Ali, Z., Malik, M. S., Waseem, S., Mirza, B., et al. (2019). Comparative analyses of chloroplast genomes among three Firmiana species: identification of mutational hotspots and phylogenetic relationship with other species of malvaceae. Plant Gene 19, 100199. doi: 10.1016/j.plgene.2019.100199
Shaw, J., Shafer, H. L., Rayne, L. O., Kovach, M. J., Schorr, M., Morris, A. B. (2014). Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: the tortoise and the hare IV. Am. J. Bot. 101 (11), 1987–2004. doi: 10.3732/ajb.1400398
Shen, L., Guan, Q., Amin, A., Zhu, W., Li, M., Li, X., et al. (2016). Complete plastid genome of Eriobotrya japonica (Thunb.) lindl and comparative analysis in rosaceae. SpringerPlus 5 (1), 1–14. doi: 10.1186/s40064-016-3702-3
Shen, X., Guo, S., Yin, Y., Zhang, J., Yin, X., Liang, C. (2018). Complete chloroplast genome sequence and phylogenetic analysis of Aster tataricus. Molecules 23 (10), 2426. doi: 10.3390/molecules23102426
Shi, L., Chen, H., Jiang, M., Wang, L., Wu, X., Huang, L., et al. (2019). CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47 (W1), W65–W73. doi: 10.1093/nar/gkz345
Silalahi, M., Wakhidah, A. Z. (2021). Sambucus javanica Reinw. ex Blume Viburnaceae. Ethnobotany of the Mountain Regions of Southeast Asia, 955–961. doi: 10.1007/978-3-030-38389-3_140
Song, W., Chen, Z., He, L., Feng, Q., Zhang, H., Du, G., et al. (2022). Comparative chloroplast genome analysis of wax gourd (Benincasa hispida) with three benincaseae species, revealing evolutionary dynamic patterns and phylogenetic implications. Genes 13 (3), 461. doi: 10.3390/genes13030461
Srivastava, D., Shanker, A. (2016). Identification of simple sequence repeats in chloroplast genomes of magnoliids through bioinformatics approach. Interdiscip. Sciences: Comput. Life Sci. 8 (4), 327–336. doi: 10.1007/s12539-015-0129-4
Talavera, G., Castresana, J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Systemat. Biol. 56 (4), 564–577. doi: 10.1080/10635150701472164
The Plant List. (2013). Available at: http://www.theplantlist.org/. (Accessed February, 2023).
Wang, J., Seyler, B. C., Ticktin, T., Zeng, Y., Ayu, K. (2020). An ethnobotanical survey of wild edible plants used by the yi people of liangshan prefecture, sichuan province, China. J. Ethnobiol. Ethnomed. 16 (1), 1–27. doi: 10.1186/s13002-019-0349-5
Wang, J., Sheng, J., Zhu, J., Hu, Z., Diao, Y. (2021). Comparative transcriptome analysis and identification of candidate adaptive evolution genes of Miscanthus lutarioriparius and Miscanthus sacchariflorus. Physiol. Mol. Biol. Plants 27 (7), 1499–1512. doi: 10.1007/s12298-021-01030-1
Waswa, E. N., Li, J., Mkala, E. M., Wanga, V. O., Mutinda, E. S., Nanjala, C., et al. (2022). Ethnobotany, phytochemistry, pharmacology, and toxicology of the genus Sambucus L.(Viburnaceae). J. Ethnopharmacol. 292, 115102. doi: 10.1016/j.jep.2022.115102
WFO. (2023). World flora online. Available at: http://www.worldfloraonline.org/. (Accessed February 15, 2023).
Wick, R. R., Schultz, M. B., Zobel, J., Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31 (20), 3350–3352. doi: 10.1093/bioinformatics/btv383
Willis, J. C. (1973). A dictionary of the flowering plants and ferns, Rev. (Cambridge: HK Airy Shaw. Univ. Press).
Wu, C., Lai, Y., Lin, C., Wang, Y., Chaw, S. (2009). Molecular phylogenetics and evolution of reduced and compact chloroplast genomes (cpDNA) in gnetophytes: selection toward a lower-cost strategy. Mol. Phylogenet. Evol. 52 (1), 115–124. doi: 10.1016/j.ympev.2008.12.026
Wu, Z., Liao, R., Yang, T., Dong, X., Lan, D., Qin, R., et al. (2020). Analysis of six chloroplast genomes provides insight into the evolution of Chrysosplenium (Saxifragaceae). BMC Genomics 21 (1), 1–14. doi: 10.1186/s12864-020-07045-4
Wu, X.-M., Wu, S.-F., Ren, D.-M., Zhu, Y.-P., He, F.-C. (2007). The analysis method and progress in the study of codon bias. Yi Chuan= Hereditas 29 (4), 420–426. doi: 10.1360/yc-007-0420
Yatskievych, G. (2006). Steyermark’s flora of Missouri, vol. 2 (revised ed.) (St. Louis, MO: Missouri Botanical Garden Press).
Zhang, Y., Du, L., Liu, A., Chen, J., Wu, L., Hu, W., et al. (2016). The complete chloroplast genome sequences of five Epimedium species: lights into phylogenetic and taxonomic analyses. Front. Plant Sci. 7, 306. doi: 10.3389/fpls.2016.00306
Zhang, D., Gao, F., Jakovlić, I., Zou, H., Zhang, J., Li, W. X., et al. (2020). PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20 (1), 348–355. doi: 10.1111/17550998.13096
Keywords: chloroplast, genomes, phylogeny, taxonomy, Sambucus, Viburnaceae
Citation: Waswa EN, Mkala EM, Odago WO, Amenu SG, Mutinda ES, Muthui SW, Ding S-X, Hu G-W and Wang Q-F (2023) Comparative chloroplast genome analysis of Sambucus L. (Viburnaceae): inference for phylogenetic relationships among the closely related Sambucus adnata Wall. ex DC Sambucus javanica Blume. Front. Plant Sci. 14:1179510. doi: 10.3389/fpls.2023.1179510
Received: 04 March 2023; Accepted: 31 May 2023;
Published: 16 June 2023.
Edited by:
Rongrong Zhai, Zhejiang Academy of Agricultural Sciences, ChinaReviewed by:
Kumar Paritosh, University of Delhi, IndiaCássio Van Den Berg, State University of Feira de Santana, Brazil
Copyright © 2023 Waswa, Mkala, Odago, Amenu, Mutinda, Muthui, Ding, Hu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guang-Wan Hu, Z3Vhbmd3YW5odUB3YmdjYXMuY24=