 Yunus Emre Arvas
Yunus Emre Arvas Sevgi Marakli
Sevgi Marakli Yılmaz Kaya
Yılmaz Kaya Ruslan Kalendar
Ruslan Kalendar
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Plant Sci., 25 April 2023
Sec. Technical Advances in Plant Science
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1174339
This article is part of the Research TopicMobile Elements and Plant Genome Evolution, Comparative Analyses and Computational Tools, Volume IIView all 12 articles
The use of molecular markers has become an essential part of molecular genetics through their application in numerous fields, which includes identification of genes associated with targeted traits, operation of backcrossing programs, modern plant breeding, genetic characterization, and marker-assisted selection. Transposable elements are a core component of all eukaryotic genomes, making them suitable as molecular markers. Most of the large plant genomes consist primarily of transposable elements; variations in their abundance contribute to most of the variation in genome size. Retrotransposons are widely present throughout plant genomes, and replicative transposition enables them to insert into the genome without removing the original elements. Various applications of molecular markers have been developed that exploit the fact that these genetic elements are present everywhere and their ability to stably integrate into dispersed chromosomal localities that are polymorphic within a species. The ongoing development of molecular marker technologies is directly related to the deployment of high-throughput genotype sequencing platforms, and this research is of considerable significance. In this review, the practical application to molecular markers, which is a use of technology of interspersed repeats in the plant genome were examined using genomic sources from the past to the present. Prospects and possibilities are also presented.
The genomes of eukaryotic organisms mostly consist of interspersed repetitive sequences, in particular transposable elements (TEs). In most species studied, interspersed repeats are rather unevenly distributed, with some of them clustered around telomeres or centromeres (Kalendar et al., 2021a). Although TEs exhibit considerable sequence diversity, they can be divided into two well-defined classes according to their structure and propagation strategies. Retrotransposons are the most common type of TEs that belong to class I. Retrotransposons are retrovirus-related genetic elements that amplify through the process of reverse transcription use an RNA-mediated process to transpose, in contrast to class II transposons, which do not require an RNA intermediate (Papolu et al., 2022). Depending on their structure and transposition cycle, retrotransposons can be divided into two main subclasses, long terminal repeat (LTR) retrotransposons and non-LTR retrotransposons, which are based on the presence or absence of LTR at their ends (Wicker et al., 2007). Non-LTR retrotransposons can be further subdivided into long interspersed nucleotide elements (LINE) and short interspersed elements (SINE). Retrotransposons and retroviruses share similarities such as common structural features and basic life cycle stages (Figure 1). Retrovirus-like or LTR retrotransposons include endogenous pararetroviruses (Valli et al., 2023), which are remnants of previous rounds of germline virus infection that have lost their ability to reinfect and are fixed in the genome. Each group of transposons has corresponding non-autonomous forms, which are missing one or more genes necessary for transposition. In the case of class II transposons, these non-autonomous forms are known as miniature inverted-repeat transposable elements (MITE), while SINEs are non-autonomous forms of non-LTR retrotransposons. LTR retrotransposons have the following two types of non-autonomous forms: terminal-repeat retrotransposons in miniature (TRIM) and large retrotransposon derivatives (LARD) (Kalendar et al., 2004; Kalendar et al., 2008; Kalendar et al., 2020).
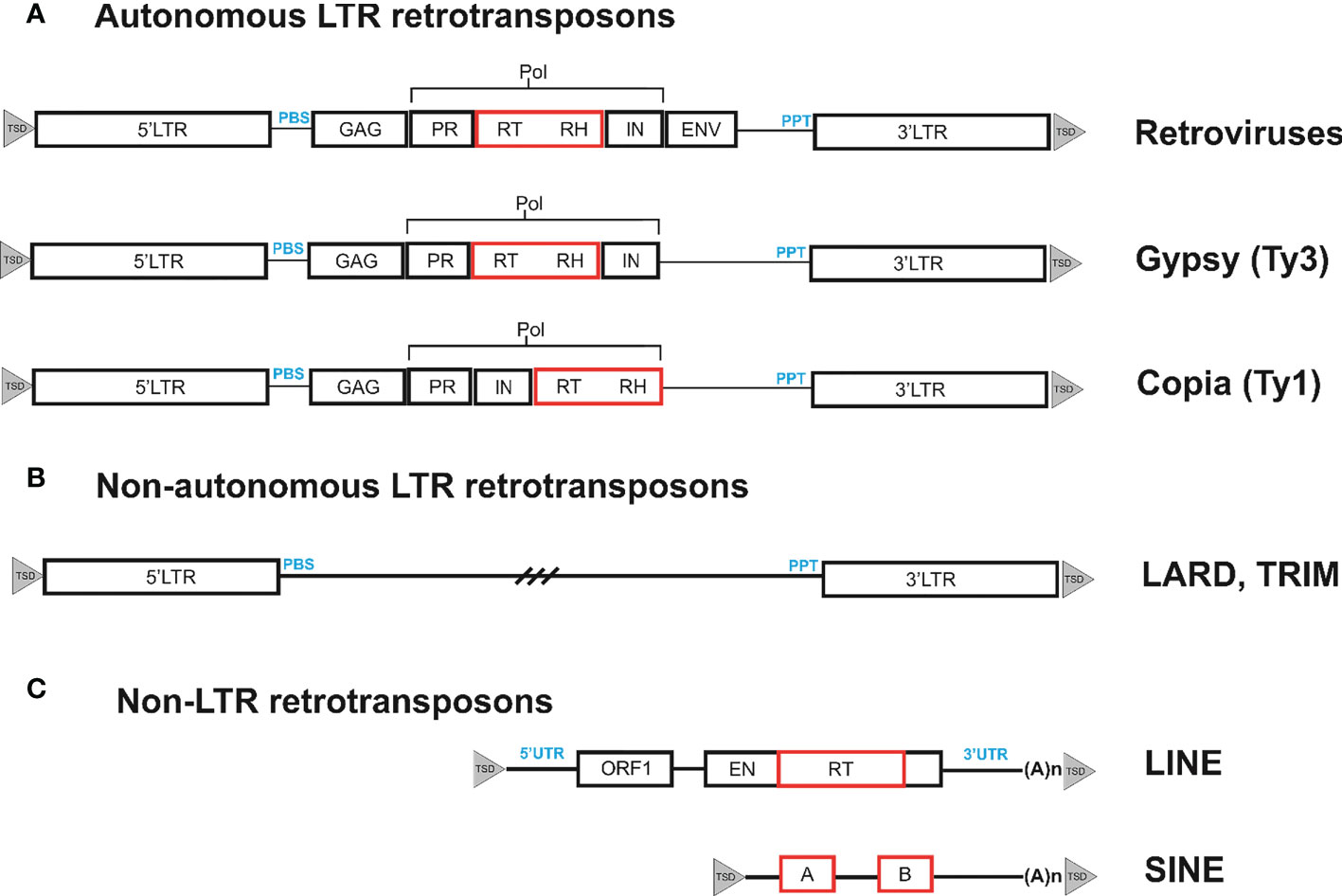
Figure 1 Organization of different types of retrotransposons. (A). Retrotransposons can be classified based on their structural features. One such type is bounded by long terminal repeats (LTRs), which contain the transcriptional promoter and terminator. These LTRs contain short inverted repeats at either end, shown as filled triangles. During reverse transcription, the primer binding site (PBS) and polypurine tract (PPT) domains prime the synthesis of the complementary DNA (cDNA) strand on the (−) and (+) strands, respectively. The internal region of the retrotransposon codes for the proteins necessary for the retrotransposon life cycle. These include the capsid protein (GAG), the aspartic proteinase (AP) that cleaves the polyprotein (AP), the integrase (IN) that inserts the cDNA copy into the genome, and the reverse transcriptase (RT) and RNaseH (RH) that together copy the transcript into cDNA. The internal region also contains evolutionarily conserved domains (noted below the element as black boxes) that are necessary for function and can be used to isolate retrotransposons from previously unstudied plant species. These conserved domains can be targeted for amplification and sequencing of retrotransposons from a variety of species. The LTRs are generally well-conserved within families and can serve as targets for primer design to generate DNA footprints. DNA footprints are useful for studying the evolutionary history and diversity of retrotransposons within and between species. (B). Non-autonomous LTR retrotransposons have two types of non-autonomous forms: Terminal-Repeat Retrotransposons in Miniature (TRIMs) and Large Retrotransposon Derivatives (LARDs). (C) Non-LTR retrotransposons, such as LINEs and SINEs, are terminated by a 3′ poly(A) stretch. LINEs have two open reading frames (ORFs) that encode a nucleocapsid protein (gag), an endonuclease (EN), and a reverse transcriptase (RT). The ORFs are flanked by untranslated regions (UTR), with the broken line indicating the 5′ truncations found in many LINEs. SINEs are composed of a tRNA-derived region, an unrelated DNA sequence. The two black boxes labeled A and B depict regions with homology to RNA polymerase III promoters.
The number of retrotransposons in an organism’s genome is directly proportional to the genome size (Hawkins et al., 2006; Holligan et al., 2006; Jaillon et al., 2007; Ming et al., 2008; Wang and Liu, 2008; Huang et al., 2009; Qiu et al., 2009; Wicker et al., 2009; Kovach et al., 2010; Schmutz et al., 2010; Argout et al., 2011; Shulaev et al., 2011). LTR retrotransposons are the most abundant subclass of retrotransposons in eukaryotic genomes (Bennetzen and Wang, 2014). Retrotransposons are believed to have a vital role in regulating chromosomal structure and structural genes due to their repetitive structure and presence of regulatory signals (Huang et al., 2012; Zervudacki et al., 2018). Eighty percent of the plant genomes of grasses (wheat, barley, and rye) consist of TEs and other repeats (Wicker et al., 2018). The corresponding value for the human genome is 45%; for Arabidopsis (Zhang and Wessler, 2004), retrotransposons form 14% of the genome (Turnbull et al., 2018; Akakpo et al., 2020).
Numerous studies have investigated the activities of LTR retrotransposons under stress conditions (Grandbastien, 1998; Pecinka et al., 2010; Tittel-Elmer et al., 2010; Ito et al., 2011; Butelli et al., 2012; Gaubert et al., 2017; Benoit et al., 2019; Cho et al., 2019; Papolu et al., 2021; Ramakrishnan et al., 2022). Mobile elements and endogenous retroviruses are important in the distribution of cis-regulatory elements, which contribute to genetic control in both the short (accumulation of hidden variability and processes of selection) and the long (adaptation and separation) term.
Transposons can insert into different positions within a genome; this leads to changes in the DNA sequence and consequently mutations (Mcclintock, 1950). Retrotransposons can also alter the amount of DNA in the genome by increasing the number of copies of the TE. Transposons have insertional, transcriptional, and translational properties, implying that transposon movements may change depending on the organism, environment, and even tissue specific. However, the movement mechanisms are not completely understood (Rebollo et al., 2011; Schrader and Schmitz, 2019).
TEs have long been considered as agents that give rise to mutations that disrupt gene function after inserting into coding or promoter sequences. These elements play a role in evolution but can also have negative effects on the host organism by disrupting normal gene function (Kalendar et al., 2020). This was demonstrated by Barbara McClintock, who showed that TEs can cause pigmentation loss in corn kernels (Mcclintock, 1950). There has been some opposition to this view; transposition may be beneficial to an organism by mediating epigenetic factors or by acting as cis-acting regulatory regions that exhibit alternative promoters that regulate gene expression (Mirouze and Vitte, 2014; Hirsch and Springer, 2017). Furthermore, amplification of TEs can drive significant biological novelty [i.e. placental pregnancy (Lynch et al., 2011) and innate immunity (Chuong et al., 2016)]; hence, transposition may drive eukaryotic evolution by reshaping gene networks that result in novel features. In crops, TEs have a role in variation of agronomic features, including tomato shape (Xiao et al., 2008) and red pigmentation of apples (Zhang et al., 2019). However, the degree that genetic variation caused by TE can be used for agronomic applications has not been fully verified for all crops (Ramakrishnan et al., 2023).
Molecular genetic markers are short DNA sequences that can be used to identify specific regions of the genome. They can be used in a variety of applications, including genome mapping, disease diagnosis, and classification of individuals or populations. The genetic marker might be either a gene or a sequence that possesses no known functions. Currently, genetic polymorphisms in DNA sequences are analyzed by several strategies, such as various PCR-based methods that detect polymorphisms and PCR-based genome profiling applications: various platforms for hybridization (NanoString Technologies, multiplex ligation-dependent probe amplification [MLPA], microarrays), and next-generation sequencing (NGS). Current analysis strategies have been developed depending on the main methods (Table 1).
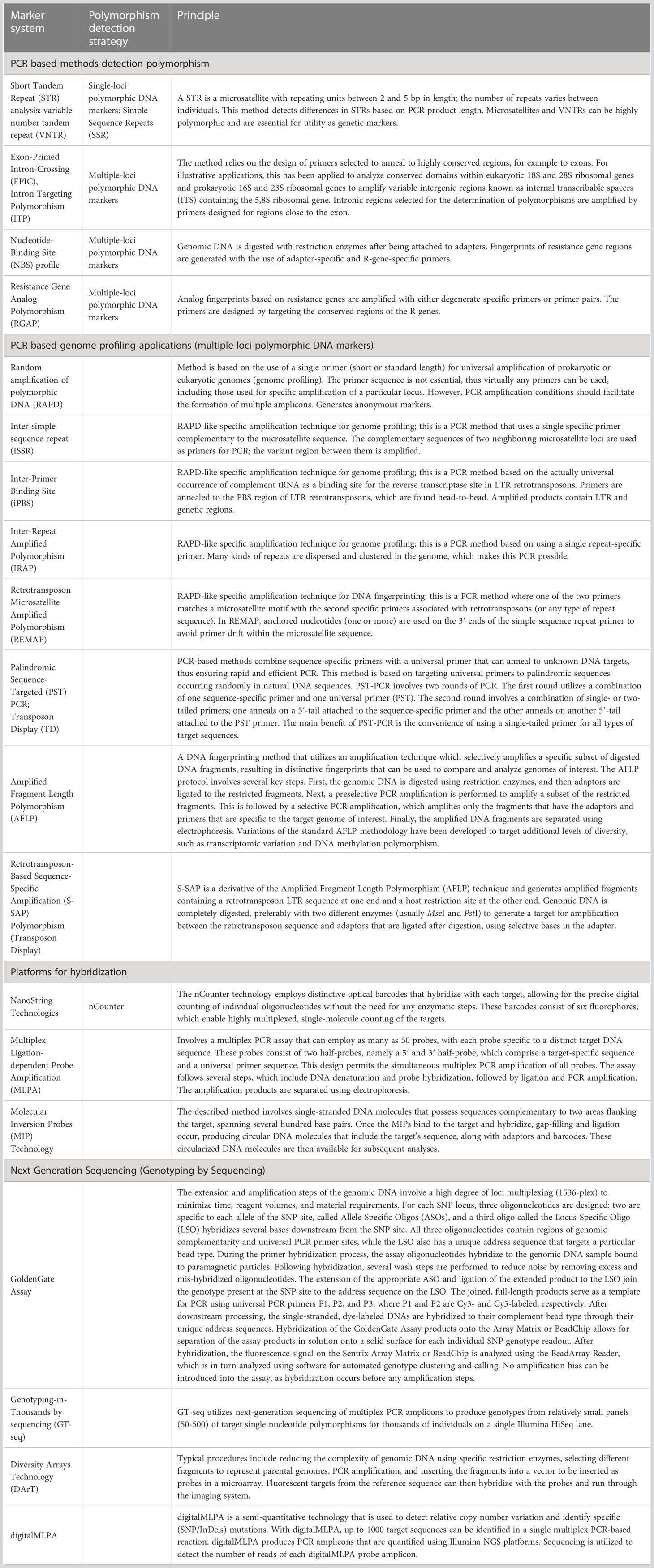
Table 1 Main strategies for detection of molecular genetic polymorphisms.
Molecular MAS is a method that improves the efficiency of plant breeding by relying on DNA markers to score for specific traits or characteristics. This method allows for earlier selection and reduces the population size of plants, thus saving time and effort. MAS can be used to screen for traits that are difficult or expensive to score phenotypically, such as disease resistance or fruit quality. The use of DNA markers also allows for detection of heredity patterns at the genomic level and the cloning of genes important for natural resistance to disease. This can result in more “green” and cost-effective solutions for disease control. MAS also has the potential to pyramid multiple desirable genes in a new plant variety. By increasing precision in the selection, unwanted side effects in future plant generations can be reduced.
The selection identified by MAS includes scoring in the absence or presence of a plant phenotype of interest that is based upon the DNA banding pattern of connected markers on an autoradiogram or a gel relying on the market framework. The rationale is that the banding pattern provides information about the parental source of the bands in segregants at a marker locus, which illustrates the absence or presence of a specifical chromosomal fragment harboring the allele of interest. The effectiveness of screening in breeding methods is improved in the following several regards. Segregants can be graded at the seedling phase for features that are expressed late in the progress of the plant; this involves features including photoperiod sensitivity, male sterility, and grain quality. It is likely that screening for features that are exceedingly time-consuming, difficult, or expensive to score and determine, such as resistance to biotypes of insects or diseases or certain types or nematodes, tolerance to root morphology, toxicity, mineral, salt deficiencies, and drought. Selection can be applied to certain features concurrently, which is difficult or is not possible via traditional means. Heterozygotes are readily defined and separated from homozygotes without referring to progeny testing, which saves effort and time. MAS is a promising choice to be used in improving many phenotypic features of interest in which the evaluation is usually unreliable or expensive. MAS may also increase the efficacy of selection by permitting earlier selection and decreasing the plant population size at the time of selection. Cultivators can rapidly detect heredity patterns at the genome level by directly analyzing the genetic makeup of empirical plants at the seedling stage. It may be useful for features that cannot be defined before plant maturity, such as fruit qualifications and for the features that are difficult to test (such as disease resistance). The selection of resistant plants is performed by using a DNA marker that is connected to the feature that controls the gene rather than turning the resistance of plants to disease into account. To achieve the most economical, environmentally safe, and effective outcomes in disease control, natural resistance genes should be marked in different plant varieties. This approach offers a “green” solution that eliminates the need for expensive chemicals used in disease control. To enhance precision in selection, a uniform practice for scoring involves determining which fragment of each chromosome belongs to each parent and identifying how many genes come from each parent. By increasing precision, undesired side effects can be reduced in the next generation of plants. Additionally, using MAS can help in pyramiding two or more desirable genes in a new plant variety. This approach can further enhance the efficacy of disease control in a cost-effective and environmentally friendly way (Das et al., 2017).
Interspersed repeats-based genome profiling is a range of different approaches that utilizes the polymorphic nature of TEs to study genetic variations in different plant populations. This approach involves identifying and analyzing the repetitive elements that are interspersed throughout the genome. As mentioned, TEs can insert into different positions within a genome, leading to changes in the DNA sequence and mutation. The emerging heterogeneity in the location of distinct TEs has been exploited for specific molecular marker methods focused on repetitive elements (Bennetzen and Wang, 2014). If integration occurs in the cell line from which pollen or eggs eventually originate, a new polymorphism is formed. These new integrated copies are useful for distinguishing breeding lines, varieties, or plant populations. Changes in the copy number of these repeats and internal rearrangements on both homologous chromosomes occur after the induction of recombination processes during prophase of meiosis (Belyayev et al., 2010). TEs create genomic variation in plants, which has been revealed by certain studies (Kalendar et al., 2000; Belyayev et al., 2010; Wicker et al., 2018; Kwolek et al., 2022). According to a comparative study on different plant genomes, most sequences associated with TEs come from modern insertions (El Baidouri and Panaud, 2013). This implies that the ancient TEs were removed from the genomes and, consequently, there must have been a force that balanced the expansion of the genome caused by TE. This was formulated previously in the “increase/decrease” model (Vitte and Panaud, 2005), which has recently been considered more explicitly with mathematical models (Dai et al., 2018).
Methods of detection of hidden (phenotypically invisible) genetic variations, such as the molecular marker system for genome profiling, were developed based on sequences of multiple families of complex interspersed genomic repeats (Figure 2). These genome-profiling PCR techniques for the study of genetic variability in eukaryotes that utilize multicopy and genomic diversity abundance of TEs and endogenous viruses can increase knowledge of genetic relationships and assess the genetic diversity of specific species. Interspersed repeats-based genome profiling applications are a simple PCR method and a cost-effective technique to study individual genetic polymorphisms. Genome profiling is essential as TEs, particularly LTR retrotransposons, are widely distributed throughout the genome and can facilitate recombination events during meiosis (Kent et al., 2017).
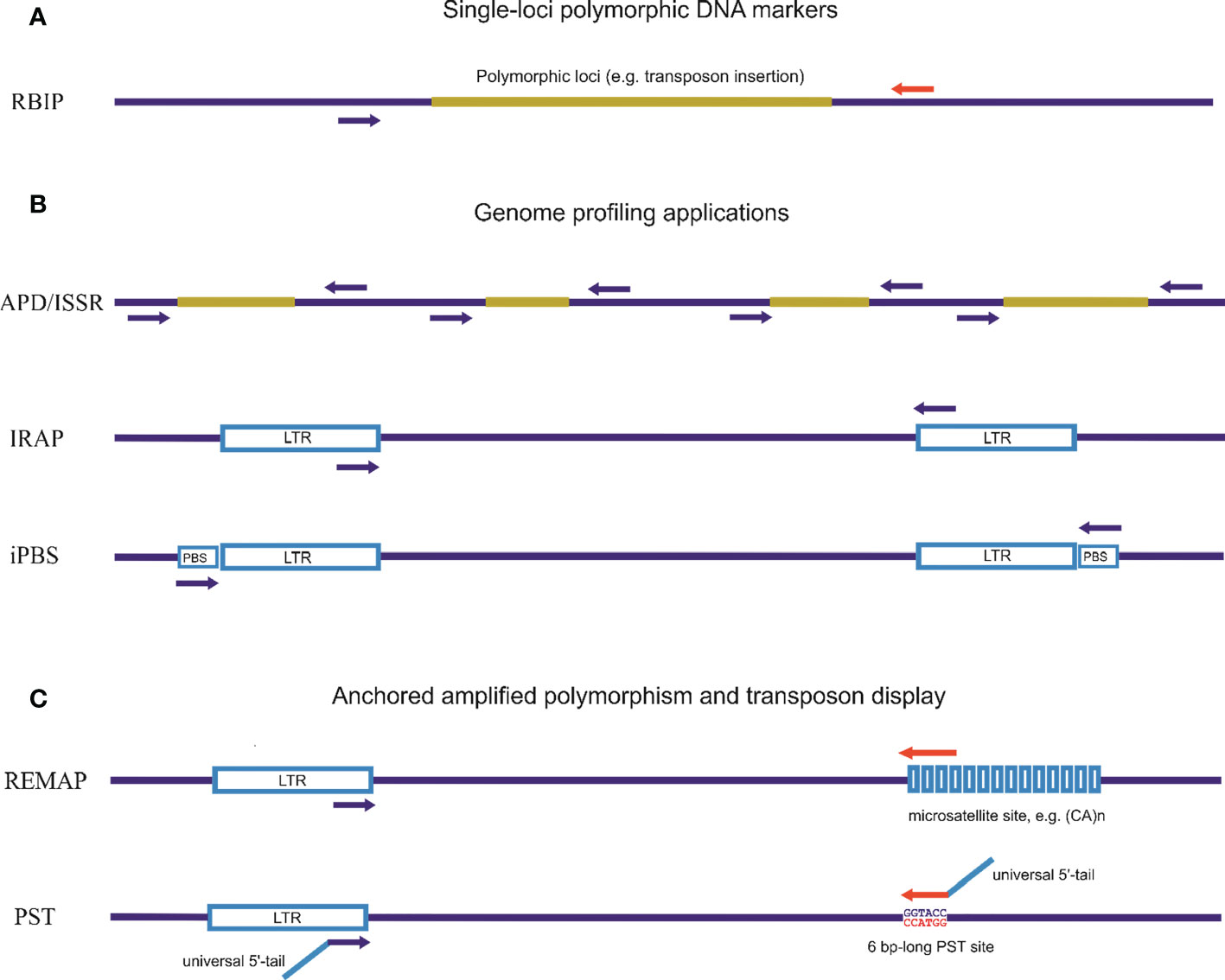
Figure 2 Different approaches for detecting inter-retrotransposon amplified polymorphisms using PCR techniques. (A). Single-loci polymorphic DNA markers. Retrotransposon-Based Insertional Polymorphism (RBIP) is a codominant marker system that uses PCR primers designed for flanking retrotransposon DNA to study insertional polymorphisms in individual retrotransposons. This method detects the presence of mobile element insertions or differences in tandem repeats based on PCR product length. (B). PCR-based genome profiling applications. Random amplification of polymorphic DNA (RAPD) is based on the use of a single primer (short or standard length) for amplification. Inter-simple sequence repeat (ISSR) is a PCR method for DNA fingerprinting using a single primer complementary to the microsatellite sequence. Inter-Repeat Amplified Polymorphism (IRAP) is a PCR method for DNA fingerprinting based on using a single repeat-specific primer. Inter-Primer Binding Site (iPBS) is a PCR method for DNA fingerprinting based on the actually universal occurrence of complement tRNA as a binding site for the reverse transcriptase site in LTR retrotransposons. (C). Anchored genome profiling and transposon display applications using one specific primer for repeat elements in combination with an anchored non-specific primer. Retrotransposon Microsatellite Amplified Polymorphism (REMAP) is a PCR method for DNA fingerprinting where one of the two primers matches a microsatellite motif with second primers associated with retrotransposons (or any type of repeat sequence). Palindromic Sequence-Targeted (PST) and Transposon Display (TD) are PCR-based methods that combine repeat-specific primers with a universal primer able to anneal to palindromic sequences. PST-PCR involves two rounds of PCR. The first round utilizes a combination of one sequence-specific primer and one universal primer (PST). The second round involves a combination of single- or two-tailed primers; one anneals to a 5′-tail attached to the sequence-specific primer and the other anneals to another 5′-tail attached to the PST primer.
The basic principle on which numerous PCR methods for genome profiling have been developed is the use of a single primer specific to the high-copy-number retrotransposon sequences or any other sequences for multiple families of complex interspersed genomic repeats. A second strategy is to use a specific primer to retrotransposon sequences in combination with an anchored primer that may be of varied origin, also including other sequences of interspersed genomic repeats (Figure 2).
Eukaryotic genomes harbor many retrotransposon elements, each with their own unique history and level of relatedness. As such, for closely related species, the sequences of a particular retrotransposon will be similar, which reflects the degree of relatedness between species. However, as species become more distantly related, the sequence of a particular retrotransposon will diverge, including the most conserved regions. In the case of closest species, such as within the grass families Triticum and Aegilops, the sequences of specific mobile elements are almost exactly the same. For species more distant from Triticum, such as Hordeum, sequences of mobile elements will differ but still retain more than 90% similarity. Therefore, the similarity of mobile elements can be used to study related but distinct species. PCR primers corresponding to the most conserved regions of these mobile elements can also be used (Kalendar et al., 2004; Kalendar et al., 2008; Moisy et al., 2014; Kalendar et al., 2020). One prominent example of such an application is PCR with a single specific primer corresponding to conserved sequences in LTR retrotransposons, which is used for interspersed repeats-based genome profiling. By comparing the sequences of mobile elements across different species, researchers elucidate the evolutionary relationships between those species and the processes that shaped their genomes. In particular, PCR approaches that rely on the identification of transposition element insertion or mutation site polymorphisms include Inter-retrotransposon amplified polymorphism (IRAP) (Kalendar and Schulman, 2006). IRAP is based on PCR amplification of the genomic region between two adjacent LTR retrotransposons oriented in opposite directions. This technique requires a single LTR primer for use in PCR. The PCR products are run on an agarose gel and polymorphisms among the samples are identified based on the banding pattern. IRAP is a technique that is straightforward and rapid. However, the sequences of LTR retrotransposons must be known for this method.
The Inter-Primer Binding Site (iPBS) amplification method is a powerful genomic profiling technique that does not require prior sequence identification of the retrotransposon. The iPBS amplification technique for DNA fingerprinting is a PCR method based on the universal presence of complement tRNA as a binding site for the reverse transcriptase site (PBS) in LTR retrotransposons (Kalendar et al., 2010). Primers are annealed to the PBS region of LTR retrotransposons, which are found head-to-head. Amplified products contain LTR and genetic regions. The LTR primers used in other marker methods are challenging to design, as retrotransposons have no conserved sequences in the LTR regions. On the contrary, many LTR retrotransposons include evolutionarily conserved PBS sequences. The most significant advantage of this method is that it does not need TE sequence information for primer design (Kalendar et al., 2022b). Many studies on TEs are on the determination of relationships using these marker techniques (Monden et al., 2014).
The second strategy for genome profiling and transposon display applications is based on using a specific primer for the repeat element in combination with an anchored non-specific primer. The Retrotransposon Microsatellite Amplified Polymorphism (REMAP) technique for genome profiling is a PCR method in which a specific primer (out of two primers) matches the LTR retrotransposon sequence (or any repeating sequence can be used) and the second anchored primer is annealed to the microsatellite sequence (Kalendar and Schulman, 2006). In REMAP, anchored nucleotides (one or more) are used on the 3′ ends of the microsatellite primer to avoid primer drift within the microsatellite sequence. REMAP and IRAP share similar working principles, as both of require retrotransposon-specific primers whose sequences are known (Hosid et al., 2012).
New methods for high-throughput targeted gene characterization and transposon display have been added to current methodologies, which may be modified to include high-throughput sequencing technologies, among other techniques. For example, Palindromic Sequence-Targeted (PST) PCR utilizes a pair of primers, one of which is complementary to 6-bp long palindromic sequence (PST site) and the other to conserved TE sequences (Kalendar et al., 2019; Kalendar et al., 2021b). The PST-PCR technique allows genome walking and profiling that can be used for the primary characterization of intraspecific and interspecific genetic variability and for screening lines and genotypes.
Retrotransposon-Based Insertional Polymorphism (RBIP) is primarily a codominant marker system that uses one or two pairs of PCR primers designed from combinations of sequences for the retrotransposon and its flanking DNA to study insertional polymorphisms in individual retrotransposons. RBIP is a marker method based on PCR and is used to determine the polymorphisms among two alleles. The comparison of two distinct PCRs permits determination of the polymorphisms. One of the PCRs uses primers that are specific to either retrotransposons or a genomic region near retrotransposons. At the end of the reaction, the interested transposon LTR region is amplified. In the other reaction, two primers specific to the genomic DNA surrounding the retrotransposon are used. The band profiles among the two reactions indicate whether a retrotransposon insertion occurred in the region of interest. RBIP requires sequence information for primer design (Flavell et al., 1998; Kalendar, 2022).
The retrotransposon-based Sequence-Specific Amplification Polymorphism (S-SAP), also known as transposon display, is a modification of the Amplified Fragment Length Polymorphism (AFLP) technique (Vos et al., 1995). This method involves complete digestion of genomic DNA with two different restriction enzymes, typically MseI and PstI, to generate a target for amplification between the retrotransposon sequence and adaptors that are ligated after digestion. The adaptors contain selective bases to facilitate amplification of specific regions. PCR is performed using two primers that are specific to the adapter sequence and the specific mobile element, allowing detection of variations in DNA flanking the mobile element insertion site. The technique uses a multiplex marker system to analyze band profiles among different samples and to detect polymorphisms (Queen et al., 2004). Compared to other retrotransposon marker techniques, S-SAP is costlier and is more difficult to perform. Additionally, the technique requires sequence information for primer generation, which must be designed specifically for the LTR site. However, S-SAP offers greater resolution and accuracy in detecting polymorphisms, making it a valuable tool for genetic analysis.
In plant genetics research and breeding practices, molecular techniques such as genetic characterization, genome profiling, genetic integrity, genetic mapping, feature mapping, MAS, and molecular breeding are widely used. Continuous improvements in molecular marker technology, such as high-throughput genotyping platforms, has led to development of new methods such as the GoldenGate assay, Genotyping-in-Thousands by sequencing (GT-seq) (Campbell et al., 2015), Diversity Arrays Technology (DArT) (Alheit et al., 2011), and NGS-based high-throughput hybridization platform systems (digitalMLPA [Multiplex Ligation-dependent Probe Amplification] and Molecular Inversion Probes [MIP]) (Table 1). These advancements have increased efficiency and reduced costs (Elbaidouri et al., 2013; Mir and Varshney, 2012). With such NGS-based high-throughput technological developments, low-throughput molecular markers, such as Kompetitive allele-specific PCR (KASP) (Makhoul et al., 2020), nevertheless remain indispensable for tracking specific genomic regions in molecular breeding programs when analyzing large numbers of samples (Kalendar et al., 2022a; Kalendar et al., 2022c). Therefore, single nucleotide polymorphism (SNP) markers continue to be the most preferred marker systems for development of high-throughput genotypic platforms for genome-wide marker scanning. However, detection of SNP-based markers alone drastically limits the potential to study diversity and genetic polymorphism. One type of polymorphism is the insertion/deletion polymorphism (InDel), which involves the addition or removal of a sequence of different lengths and origins. InDels can have important functional consequences for the chromosome and therefore studying them is crucial. Therefore, other techniques based on mobile element sequences should also be developed, as they can provide complementary information about genomic diversity and evolution.
Interspersed repeats-based genome profiling is a powerful tool for studying genetic polymorphisms and identifying markers for crop improvement programs. By analyzing the distribution and frequency of TEs within the genome, this method can provide valuable insights into the genetic diversity and evolution of plant populations. In addition to whole-genome sequencing, NGS platforms can also be used for targeted sequencing approaches that focus on specific regions of interest, such as the TEs interspersed throughout the genome. Practically all existing PCR approaches that involve identifying and analyzing repetitive elements can be adapted for use on modern NGS platforms (Figure 3). However, when designing primers for NGS-based analysis of repetitive elements, it is important to take into account the specificities of the platform being used. For example, the Illumina HiSeq platform requires the incorporation of adapter sequences in the 5′ tail structure of the primer to facilitate the attachment of the DNA fragments to the sequencing flowcell. One approach that has been successfully adapted for use on NGS platforms is the sequence-specific amplification polymorphism (SSAP) technique, which targets specific retrotransposon insertion sites. The SSAP method involves PCR amplification of the region between two specific primers, one of which is anchored within the retrotransposon and the other in the flanking genomic DNA. The resulting fragments can be sequenced on an NGS platform to identify polymorphisms. Another NGS-based approach for studying TEs is the retrotransposon insertion polymorphisms (RIPs) technique (Du et al., 2022), which uses PCR to amplify specific regions of the genome flanking retrotransposon insertions. The resulting fragments can be sequenced on an NGS platform to identify insertion and deletion events. Overall, the use of NGS platforms for studying TEs offers several advantages over traditional PCR-based methods. NGS allows for the simultaneous analysis of multiple samples and provides greater resolution and sensitivity in detecting polymorphisms. Additionally, NGS platforms can provide information on the structure and organization of TEs within the genome, which can aid in the identification of functional elements and the study of genome evolution.
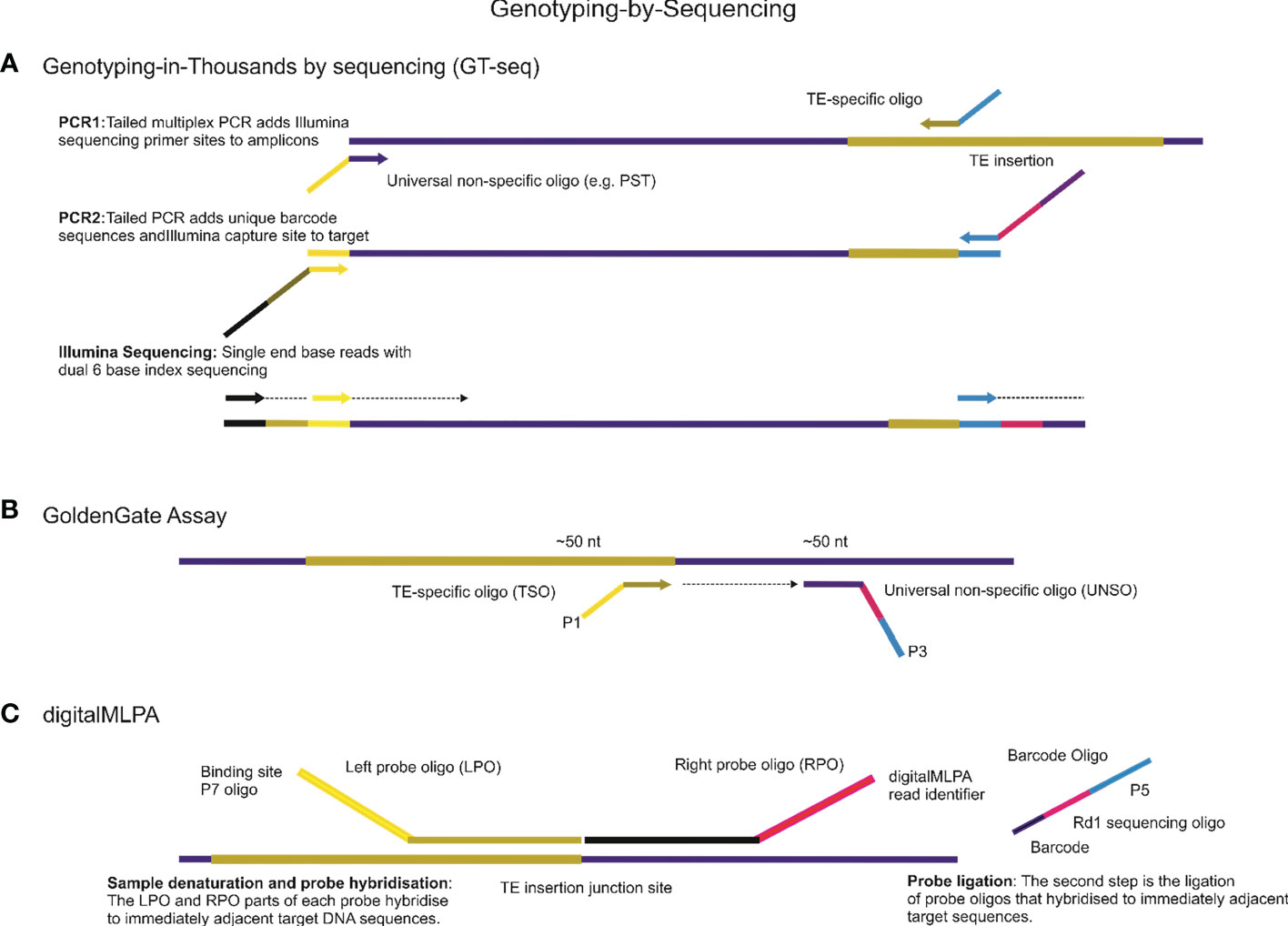
Figure 3 Genotyping-by-Sequencing (GBS) is a powerful technique for detecting genetic variations using Next-Generation Sequencing (NGS) platforms. This technique has several approaches, some of which are highlighted below. (A). One such approach is Genotyping-in-Thousands by sequencing (GT-seq), which uses NGS of multiplex PCR amplicons to produce genotypes from a small panel of mobile element insertions for thousands of individuals on Illumina NGS platforms. The basic principles and steps involved in this technology are also applicable to other NGS applications. (B) Another approach is the GoldenGate Assay, which can be designed for genotyping mobile element insertion sites involving a high degree of loci multiplexing (1536-plex). For all mobile element insertions sites, two oligonucleotides are designed, one is TE-specific to the mobile element site, called TE-Specific Oligos (TSO), and the second oligo, called the Universal Non-Specific Oligo (UNSO), for example, a universal primer able to anneal to palindromic sequences (PST), hybridizes downstream from the TE site. All oligonucleotides contain regions of genomic complementarity and universal PCR primer sites, while the UNSO also has a unique address sequence that targets a particular bead type. The extension of the appropriate TSO and ligation of the extended product to the UNSO join the genotype present at the TE site to the address sequence on the UNSO. (C) digitalMLPA is a semi-quantitative technology that is used to detect relative copy number variation and identify specific polymorphisms. With digitalMLPA, up to 1000 target sequences can be identified in a single multiplex PCR-based reaction.
NGS-based technologies rely on reduced representation sequencing (RRS) techniques (Andrews et al., 2016), including reduced-representation libraries (RRLs), complexity reduction of polymorphic sequences (CRoPS) (Van Orsouw et al., 2007), restriction site-associated DNA (RAD) sequencing (Baird et al., 2008), and low-coverage genotyping. The latter includes multiplexed shotgun genotyping (MSG) and genotyping by sequencing (GBS), which are innovative methods used in genomics (Elshire et al., 2011). These methods can provide informative results even when the reference genome is not available. The choice of method depends on the specific requirements of the study (Mir and Varshney, 2013). As more genome sequences become available, it will be necessary to develop new technologies that allow rapid exploration of the diversity of allelic gene variants in cultivated species that correspond to important plant physiological traits (Springer and Jackson, 2010). In conclusion, NGS-based analysis of repetitive elements is a powerful tool for genome profiling and can provide valuable insights into the genetic diversity and evolution of plant populations. However, careful consideration must be given to the specificities of the sequencing platform and the design of the primers used to ensure accurate and efficient sequencing.
Multiple NGS platforms and “omics” (i.e. genomics, proteomics, transcriptomics, epigenomics, and metabolomics) technologies offer many advantages and can therefore be used as high-throughput genotyping platforms. NGS platforms have revolutionized genomic approaches and have drastically reduced the time and cost required to obtain a DNA sequence. Markers can then be used for various applications, such as population genetics, association studies, and GWAS. The discovery of high-throughput genetic markers and the use of restriction enzymes for genotyping have several advantages and will become the methods of choice for marker research (Kalendar et al., 2022b). One of the advantages of these methods is that they can be used both for model organisms with high-quality reference genome sequences and for non-model species that do not have existing genomic data. Using these evolving technological methods, linkage mapping or (Quantitative Trait Locus) QTL may identify recombination breakpoints and genomic regions that are differentially expressed among populations for quantitative genetic research, genotype progenies for MAS, or resolve phylogeographies of wild populations.
Omics technologies that promise to detect tissue-specific changes with increased sensitivity and allow simultaneous analysis of thousands of genes, proteins, or metabolites (Kroeger, 2006) will increasingly provide sufficient data to create many digital platforms. The integration of new omics technologies with traditional breeding methods is important for seed production in the agriculture industry, as it helps plant growers make informed decisions based on genetic and molecular information. This combination of technologies will assist in meeting commercial criteria for seed production (Flavell, 2010; Li et al., 2018). Traditional plant breeding methods that rely exclusively on phenotypic mapping of desirable traits have limitations in defining gene-trait relationships (Flavell, 2010). The integration of modern omics technologies with traditional breeding can help identify specific gene functions related to seed development, leading to improved seed quality in economically important crops. The use of these cutting-edge technologies can benefit the modern seed industry by providing better tools for seed production (Toubiana and Fait, 2012). Studying the activity of genes at specific plant growth stages, such as grain filling or embryogenesis, may reveal critical components that regulate important metabolic processes that can be used to improve seed quality (Thompson et al., 2009).
TEs are a core component of all eukaryotic genomes, each with its own unique history and level of relatedness within the same species and between related species. Retrotransposons are widely present throughout the genome, and their replicative transposition allows them to insert themselves into the genome without removing the original elements. For closely related species, the sequences of a particular retrotransposon will be similar, reflecting the degree of relatedness between these species. Various applications of molecular markers have been developed to exploit the fact that these genetic elements are ubiquitous and their ability to be stably integrated into dispersed chromosomal localities that are polymorphic within a species. The ongoing development of molecular marker technologies is directly related to the deployment of high-throughput genotype sequencing platforms, and this research is of considerable significance. Digital NGS-based platforms can be used to study the transposition and site-specific recombination of TEs. Genotyping by sequencing includes a wide range of approaches for detecting genetic variations, and each of these approaches has unique advantages and limitations. Nonetheless, the advances in NGS technologies have greatly improved the ability to investigate the diverse nature of polymorphisms and their role in phenotypic variation. These platforms allow for high-throughput and cost-effective analysis of large amounts of genomic data, which can be used to identify and characterize TEs and their impact on the genome. This information can then be used for various applications, such as genetic characterization, genetic mapping, and marker-assisted selection.
RK, SM, YA, and YK wrote the manuscript. All authors reviewed the manuscript. All authors contributed to the article and approved the submitted version.
This work was funded by the Science Committee of the Ministry of Education and Science of the Republic of Kazakhstan (Grant No. AP14869076).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
TEs, Transposable Elements; LTR, Long Terminal Repeats; MAS, Marker-Assisted Selection; RFLP, Restriction Fragment Length Polymorphism; SSR, Simple Sequence Repeats; IRAP, Inter-Retrotransposon Amplified Polymorphism; iPBS, Inter-Primer Binding Site; RAPD, Random Amplification of Polymorphic DNA; TD, Transposon Display; AFLP, Amplified Fragment Length Polymorphism; S-SAP, Retrotransposon-Based Sequence-Specific Amplification; NGS, Next-Generation Sequencing; SNPs, single nucleotide polymorphisms; InDels,Insertion-deletion mutations.
Akakpo, R., Carpentier, M. C., Hsing, Y. I., Panaud, O. (2020). The impact of transposable elements on the structure, evolution and function of the rice genome. New Phytol. 226, 44–49. doi: 10.1111/nph.16356
Alheit, K. V., Reif, J. C., Maurer, H. P., Hahn, V., Weissmann, E. A., Miedaner, T., et al. (2011). Detection of segregation distortion loci in triticale (x triticosecale wittmack) based on a high-density DArT marker consensus genetic linkage map. BMC Genomics 12, 380. doi: 10.1186/1471-2164-12-380
Andrews, K. R., Good, J. M., Miller, M. R., Luikart, G., Hohenlohe, P. A. (2016). Harnessing the power of RADseq for ecological and evolutionary genomics. Nat. Rev. Genet. 17, 81–92. doi: 10.1038/nrg.2015.28
Argout, X., Salse, J., Aury, J. M., Guiltinan, M. J., Droc, G., Gouzy, J., et al. (2011). The genome of theobroma cacao. Nat. Genet. 43, 101–108. doi: 10.1038/ng.736
Baird, N. A., Etter, P. D., Atwood, T. S., Currey, M. C., Shiver, A. L., Lewis, Z. A., et al. (2008). Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One 3, e3376. doi: 10.1371/journal.pone.0003376
Belyayev, A., Kalendar, R., Brodsky, L., Nevo, E., Schulman, A. H., Raskina, O. (2010). Transposable elements in a marginal plant population: temporal fluctuations provide new insights into genome evolution of wild diploid wheat. Mobile DNA 1, 6. doi: 10.1186/1759-8753-1-6
Bennetzen, J. L., Wang, H. (2014). The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu. Rev. Plant Biol. 65, 505–530. doi: 10.1146/annurev-arplant-050213-035811
Benoit, M., Drost, H. G., Catoni, M., Gouil, Q., Lopez-Gomollon, S., Baulcombe, D., et al. (2019). Environmental and epigenetic regulation of rider retrotransposons in tomato. PLoS Genet. 15, e1008370. doi: 10.1371/journal.pgen.1008370
Butelli, E., Licciardello, C., Zhang, Y., Liu, J., Mackay, S., Bailey, P., et al. (2012). Retrotransposons control fruit-specific, cold-dependent accumulation of anthocyanins in blood oranges. Plant Cell 24, 1242–1255. doi: 10.1105/tpc.111.095232
Campbell, N. R., Harmon, S. A., Narum, S. R. (2015). Genotyping-in-Thousands by sequencing (GT-seq): a cost effective SNP genotyping method based on custom amplicon sequencing. Mol. Ecol. Resour 15, 855–867. doi: 10.1111/1755-0998.12357
Cho, J., Benoit, M., Catoni, M., Drost, H. G., Brestovitsky, A., Oosterbeek, M., et al. (2019). Sensitive detection of pre-integration intermediates of long terminal repeat retrotransposons in crop plants. Nat. Plants 5, 26–33. doi: 10.1038/s41477-018-0320-9
Chuong, E. B., Elde, N. C., Feschotte, C. (2016). Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 351, 1083–1087. doi: 10.1126/science.aad5497
Dai, X., Wang, H., Zhou, H., Wang, L., Dvorak, J., Bennetzen, J. L., et al. (2018). Birth and death of LTR-retrotransposons in aegilops tauschii. Genetics 210, 1039–1051. doi: 10.1534/genetics.118.301198
Das, G., Patra, J. K., Baek, K. H. (2017). Insight into MAS: a molecular tool for development of stress resistant and quality of rice through gene stacking. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.00985
Du, Z., D'alessandro, E., Zheng, Y., Wang, M., Chen, C., Wang, X., et al. (2022). Retrotransposon insertion polymorphisms (RIPs) in pig coat color candidate genes. Anim. (Basel) 12, 969. doi: 10.3390/ani12080969
Elbaidouri, M., Chaparro, C., Panaud, O. (2013). “Use of next generation sequencing (NGS) technologies for the genome-wide detection of transposition,” In: Peterson, T. (eds) Plant transposable elements (Springer) 1057, 265–274. doi: 10.1007/978-1-62703-568-2_19
El Baidouri, M., Panaud, O. (2013). Comparative genomic paleontology across plant kingdom reveals the dynamics of TE-driven genome evolution. Genome Biol. Evol. 5, 954–965. doi: 10.1093/gbe/evt025
Elshire, R. J., Glaubitz, J. C., Sun, Q., Poland, J. A., Kawamoto, K., Buckler, E. S., et al. (2011). A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6, e19379. doi: 10.1371/journal.pone.0019379
Flavell, R. (2010). From genomics to crop breeding. Nat. Biotechnol. 28, 144–145. doi: 10.1038/nbt0210-144
Flavell, A. J., Knox, M. R., Pearce, S. R., Ellis, T. H. (1998). Retrotransposon-based insertion polymorphisms (RBIP) for high throughput marker analysis. Plant J. 16, 643–650. doi: 10.1046/j.1365-313x.1998.00334.x
Gaubert, H., Sanchez, D. H., Drost, H. G., Paszkowski, J. (2017). Developmental restriction of retrotransposition activated in arabidopsis by environmental stress. Genetics 207, 813–821. doi: 10.1534/genetics.117.300103
Grandbastien, M. (1998). Activation of plant retrotransposons under stress conditions. Trends Plant Sci. 3, 181–187. doi: 10.1016/S1360-1385(98)01232-1
Hawkins, J. S., Kim, H., Nason, J. D., Wing, R. A., Wendel, J. F. (2006). Differential lineage-specific amplification of transposable elements is responsible for genome size variation in gossypium. Genome Res. 16, 1252–1261. doi: 10.1101/gr.5282906
Hirsch, C. D., Springer, N. M. (2017). Transposable element influences on gene expression in plants. Biochim. Biophys. Acta Gene Regul. Mech. 1860, 157–165. doi: 10.1016/j.bbagrm.2016.05.010
Holligan, D., Zhang, X., Jiang, N., Pritham, E. J., Wessler, S. R. (2006). The transposable element landscape of the model legume lotus japonicus. Genetics 174, 2215–2228. doi: 10.1534/genetics.106.062752
Hosid, E., Brodsky, L., Kalendar, R., Raskina, O., Belyayev, A. (2012). Diversity of long terminal repeat retrotransposon genome distribution in natural populations of the wild diploid wheat aegilops speltoides. Genetics 190, 263–274. doi: 10.1534/genetics.111.134643
Huang, C. R. L., Burns, K. H., Boeke, J. D. (2012). Active transposition in genomes. Annu. Rev. Genet. 46, 651–675. doi: 10.1146/annurev-genet-110711-155616
Huang, S., Li, R., Zhang, Z., Li, L., Gu, X., Fan, W., et al. (2009). The genome of the cucumber, cucumis sativus l. Nat. Genet. 41, 1275–1281. doi: 10.1038/ng.475
Ito, H., Gaubert, H., Bucher, E., Mirouze, M., Vaillant, I., Paszkowski, J. (2011). An siRNA pathway prevents transgenerational retrotransposition in plants subjected to stress. Nature 472, 115–119. doi: 10.1038/nature09861
Jaillon, O., Aury, J. M., Noel, B., Policriti, A., Clepet, C., Casagrande, A., et al. (2007). The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467. doi: 10.1038/nature06148
Kalendar, R. (2022). A guide to using FASTPCR software for PCR, in silico PCR, and oligonucleotide analysis. Methods Mol. Biol. 2392, 223–243. doi: 10.1007/978-1-0716-1799-1_16
Kalendar, R., Antonius, K., Smykal, P., Schulman, A. H. (2010). iPBS: a universal method for DNA fingerprinting and retrotransposon isolation. Theor. Appl. Genet. 121, 1419–1430. doi: 10.1007/s00122-010-1398-2
Kalendar, R., Baidyussen, A., Serikbay, D., Zotova, L., Khassanova, G., Kuzbakova, M., et al. (2022a). Modified “Allele-specific qPCR” method for SNP genotyping based on FRET. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.747886
Kalendar, R., Hunter, C., Orbovic, V. (2022b). Editorial: innovative applications of sequencing technologies in plant science. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1058347
Kalendar, R., Raskina, O., Belyayev, A., Schulman, A. H. (2020). Long tandem arrays of Cassandra retroelements and their role in genome dynamics in plants. Int. J. Mol. Sci. 21, 2931. doi: 10.3390/ijms21082931
Kalendar, R., Sabot, F., Rodriguez, F., Karlov, G. I., Natali, L., Alix, K. (2021a). Editorial: mobile elements and plant genome evolution, comparative analyzes and computational tools. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.735134
Kalendar, R., Schulman, A. (2006). IRAP and REMAP for retrotransposon-based genotyping and fingerprinting. Nat. Protoc. 1, 2478–2484. doi: 10.1038/nprot.2006.377
Kalendar, R., Shustov, A. V., Akhmetollayev, I., Kairov, U. (2022c). Designing allele-specific competitive-extension PCR-based assays for high-throughput genotyping and gene characterization. Front. Mol. Biosci. 9. doi: 10.3389/fmolb.2022.773956
Kalendar, R., Shustov, A., Schulman, A. (2021b). Palindromic sequence-targeted (PST) PCR, version 2: an advanced method for high-throughput targeted gene characterization and transposon display. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.691940
Kalendar, R., Shustov, A. V., Seppänen, M. M., Schulman, A. H., Stoddard, F. L. (2019). Palindromic sequence-targeted (PST) PCR: a rapid and efficient method for high-throughput gene characterization and genome walking. Sci. Rep. 9, 17707. doi: 10.1038/s41598-019-54168-0
Kalendar, R., Tanskanen, J., Chang, W., Antonius, K., Sela, H., Peleg, O., et al. (2008). Cassandra Retrotransposons carry independently transcribed 5S RNA. Proc. Natl. Acad. Sci. United States America 105, 5833–5838. doi: 10.1073/pnas.0709698105
Kalendar, R., Tanskanen, J., Immonen, S., Nevo, E., Schulman, A. H. (2000). Genome evolution of wild barley (Hordeum spontaneum) by BARE-1 retrotransposon dynamics in response to sharp microclimatic divergence. Proc. Natl. Acad. Sci. United States America 97, 6603–6607. doi: 10.1073/pnas.110587497
Kalendar, R., Vicient, C. M., Peleg, O., Anamthawat-Jonsson, K., Bolshoy, A., Schulman, A. H. (2004). Large Retrotransposon derivatives: abundant, conserved but nonautonomous retroelements of barley and related genomes. Genetics 166, 1437–1450. doi: 10.1534/genetics.166.3.1437
Kent, T. V., Uzunovic, J., Wright, S. I. (2017). Coevolution between transposable elements and recombination. Philos. Trans. R Soc. Lond B Biol. Sci. 372, 20160458. doi: 10.1098/rstb.2016.0458
Kovach, A., Wegrzyn, J. L., Parra, G., Holt, C., Bruening, G. E., Loopstra, C. A., et al. (2010). The pinus taeda genome is characterized by diverse and highly diverged repetitive sequences. BMC Genomics 11, 420. doi: 10.1186/1471-2164-11-420
Kroeger, M. (2006). How omics technologies can contribute to the ‘3R’principles by introducing new strategies in animal testing. Trends Biotechnol. 24, 343–346. doi: 10.1016/j.tibtech.2006.06.003
Kwolek, K., Kedzierska, P., Hankiewicz, M., Mirouze, M., Panaud, O., Grzebelus, D., et al. (2022). Diverse and mobile: eccDNA-based identification of carrot low-copy-number LTR retrotransposons active in callus cultures. Plant J. 110, 1811–1828. doi: 10.1111/tpj.15773
Li, Y., Xiao, J., Chen, L., Huang, X., Cheng, Z., Han, B., et al. (2018). Rice functional genomics research: past decade and future. Mol. Plant 11, 359–380. doi: 10.1016/j.molp.2018.01.007
Lynch, V. J., Leclerc, R. D., May, G., Wagner, G. P. (2011). Transposon-mediated rewiring of gene regulatory networks contributed to the evolution of pregnancy in mammals. Nat. Genet. 43, 1154. doi: 10.1038/ng.917
Makhoul, M., Rambla, C., Voss-Fels, K. P., Hickey, L. T., Snowdon, R. J., Obermeier, C. (2020). Overcoming polyploidy pitfalls: a user guide for effective SNP conversion into KASP markers in wheat. Theor. Appl. Genet. 133, 2413–2430. doi: 10.1007/s00122-020-03608-x
Mcclintock, B. (1950). The origin and behavior of mutable loci in maize. Proc. Natl. Acad. Sci. United States America 36, 344–355. doi: 10.1073/pnas.36.6.344
Ming, R., Hou, S., Feng, Y., Yu, Q., Dionne-Laporte, A., Saw, J. H., et al. (2008). The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 452, 991–996. doi: 10.1038/nature06856
Mir, R. R., Varshney, R. K. (2012). “Future prospects of molecular markers in plants,” in Molecular markers in plants. Ed. Henry, R. J. (Wiley-Blackwell Publication), 169–190. doi: 10.1002/9781118473023.ch10
Mirouze, M., Vitte, C. (2014). Transposable elements, a treasure trove to decipher epigenetic variation: insights from arabidopsis and crop epigenomes. J. Exp. Bot. 65, 2801–2812. doi: 10.1093/jxb/eru120
Moisy, C., Schulman, A. H., Kalendar, R., Buchmann, J. P., Pelsy, F. (2014). The Tvv1 retrotransposon family is conserved between plant genomes separated by over 100 million years. Theor. Appl. Genet. 127, 1223–1235. doi: 10.1007/s00122-014-2293-z
Monden, Y., Yamaguchi, K., Tahara, M. (2014). Application of iPBS in high-throughput sequencing for the development of retrotransposon-based molecular markers. Curr. Plant Biol. 1, 40–44. doi: 10.1016/j.cpb.2014.09.001
Papolu, P. K., Ramakrishnan, M., Mullasseri, S., Kalendar, R., Wei, Q., Zou, L. H., et al. (2022). Retrotransposons: how the continuous evolutionary front shapes plant genomes for response to heat stress. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1064847
Papolu, P. K., Ramakrishnan, M., Wei, Q., Vinod, K. K., Zou, L. H., Yrjala, K., et al. (2021). Long terminal repeats (LTR) and transcription factors regulate PHRE1 and PHRE2 activity in moso bamboo under heat stress. BMC Plant Biol. 21, 585. doi: 10.1186/s12870-021-03339-1
Pecinka, A., Dinh, H. Q., Baubec, T., Rosa, M., Lettner, N., Mittelsten Scheid, O. (2010). Epigenetic regulation of repetitive elements is attenuated by prolonged heat stress in arabidopsis. Plant Cell 22, 3118–3129. doi: 10.1105/tpc.110.078493
Qiu, D., Gao, M., Li, G., Quiros, C. (2009). Comparative sequence analysis for brassica oleracea with similar sequences in b. rapa and arabidopsis thaliana. Plant Cell Rep. 28, 649–661. doi: 10.1007/s00299-008-0661-3
Queen, R., Gribbon, B., James, C., Jack, P., Flavell, A. (2004). Retrotransposon-based molecular markers for linkage and genetic diversity analysis in wheat. Mol. Genet. Genomics 271, 91–97. doi: 10.1007/s00438-003-0960-x
Ramakrishnan, M., Papolu, P. K., Mullasseri, S., Zhou, M., Sharma, A., Ahmad, Z., et al. (2023). The role of LTR retrotransposons in plant genetic engineering: how to control their transposition in the genome. Plant Cell Rep. 42, 3–15. doi: 10.1007/s00299-022-02945-z
Ramakrishnan, M., Zhang, Z., Mullasseri, S., Kalendar, R., Ahmad, Z., Sharma, A., et al. (2022). How stress memory is regulated for cold and heat stress responses in plants: scope for future climate resilient agriculture. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1075279
Rebollo, R., Karimi, M. M., Bilenky, M., Gagnier, L., Miceli-Royer, K., Zhang, Y., et al. (2011). Retrotransposon-induced heterochromatin spreading in the mouse revealed by insertional polymorphisms. PLoS Genet. 7, e1002301. doi: 10.1371/journal.pgen.1002301
Schmutz, J., Cannon, S. B., Schlueter, J., Ma, J., Mitros, T., Nelson, W., et al. (2010). Genome sequence of the palaeopolyploid soybean. Nature 463, 178–183. doi: 10.1038/nature08670
Schrader, L., Schmitz, J. (2019). The impact of transposable elements in adaptive evolution. Mol. Ecol. 28, 1537–1549. doi: 10.1111/mec.14794
Shulaev, V., Sargent, D. J., Crowhurst, R. N., Mockler, T. C., Folkerts, O., Delcher, A. L., et al. (2011). The genome of woodland strawberry (Fragaria vesca). Nat. Genet. 43, 109–116. doi: 10.1038/ng.740
Springer, N. M., Jackson, S. A. (2010). Realizing the potential of genomics for crop improvement. Briefings Funct. Genomics 9, 93–94. doi: 10.1093/bfgp/elq006
Thompson, R., Burstin, J., Gallardo, K. (2009). Post-genomics studies of developmental processes in legume seeds. Plant Physiol. 151, 1023–1029. doi: 10.1104/pp.109.143966
Tittel-Elmer, M., Bucher, E., Broger, L., Mathieu, O., Paszkowski, J., Vaillant, I. (2010). Stress-induced activation of heterochromatic transcription. PLoS Genet. 6, e1001175. doi: 10.1371/journal.pgen.1001175
Toubiana, D., Fait, A. (2012). “Metabolomics-assisted crop breeding towards improvement in seed quality and yield,” in Seed development: OMICS technologies toward improvement of seed quality and crop yield (Dordrecht: Springer), 453–475. doi: 10.1007/978-94-007-4749-4_22
Turnbull, C., Scott, R. H., Thomas, E., Jones, L., Murugaesu, N., Pretty, F. B., et al. (2018). The 100 000 genomes project: bringing whole genome sequencing to the NHS. BMJ 361, k1687. doi: 10.1136/bmj.k1687
Valli, A. A., Gonzalo-Magro, I., Sanchez, D. H. (2023). Rearranged endogenized plant pararetroviruses as evidence of heritable RNA-based immunity. Mol. Biol. Evol. 40, msac240. doi: 10.1093/molbev/msac240
Van Orsouw, N. J., Hogers, R. C., Janssen, A., Yalcin, F., Snoeijers, S., Verstege, E., et al. (2007). Complexity reduction of polymorphic sequences (CRoPS): a novel approach for large-scale polymorphism discovery in complex genomes. PLoS One 2, e1172. doi: 10.1371/journal.pone.0001172
Vitte, C., Panaud, O. (2005). LTR Retrotransposons and flowering plant genome size: emergence of the increase/decrease model. Cytogenetic Genome Res. 110, 91–107. doi: 10.1159/000084941
Vos, P., Hogers, R., Bleeker, M., Reijans, M., Van De Lee, T., Hornes, M., et al. (1995). AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res. 23, 4407–4414. doi: 10.1093/nar/23.21.4407
Wang, H., Liu, J. S. (2008). LTR Retrotransposon landscape in medicago truncatula: more rapid removal than in rice. BMC Genomics 9, 382. doi: 10.1186/1471-2164-9-382
Wicker, T., Gundlach, H., Spannagl, M., Uauy, C., Borrill, P., Ramirez-Gonzalez, R. H., et al. (2018). Impact of transposable elements on genome structure and evolution in bread wheat. Genome Biol. 19, 103. doi: 10.1186/s13059-018-1479-0
Wicker, T., Sabot, F., Hua-Van, A., Bennetzen, J. L., Capy, P., Chalhoub, B., et al. (2007). A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 8, 973–982. doi: 10.1038/nrg2165
Wicker, T., Taudien, S., Houben, A., Keller, B., Graner, A., Platzer, M., et al. (2009). A whole-genome snapshot of 454 sequences exposes the composition of the barley genome and provides evidence for parallel evolution of genome size in wheat and barley. Plant J. 59, 712–722. doi: 10.1111/j.1365-313X.2009.03911.x
Xiao, H., Jiang, N., Schaffner, E., Stockinger, E. J., van der Knaap, E. (2008). A retrotransposon-mediated gene duplication underlies morphological variation of tomato fruit. Science 319, 1527–1530. doi: 10.1126/science.1153040
Zervudacki, J., Yu, A., Amesefe, D., Wang, J., Drouaud, J., Navarro, L., et al. (2018). Transcriptional control and exploitation of an immune-responsive family of plant retrotransposons. FEBS J. 37, e98482. doi: 10.15252/embj.201798482
Zhang, L., Hu, J., Han, X., Li, J., Gao, Y., Richards, C. M., et al. (2019). A high-quality apple genome assembly reveals the association of a retrotransposon and red fruit colour. Nat. Commun. 10, 1494. doi: 10.1038/s41467-019-09518-x
Keywords: molecular markers, interspersed repeats, amplification profiling, inter-retrotransposon amplified polymorphism, transposable elements, retrotransposon
Citation: Arvas YE, Marakli S, Kaya Y and Kalendar R (2023) The power of retrotransposons in high-throughput genotyping and sequencing. Front. Plant Sci. 14:1174339. doi: 10.3389/fpls.2023.1174339
Received: 26 February 2023; Accepted: 11 April 2023;
Published: 25 April 2023.
Edited by:
Diego H. Sanchez, Instituto de Investigaciones Fisiológicas y Ecologicas Vinculadas a la Agricultura (IFEVA) (CONICET), ArgentinaReviewed by:
Khalil Kashkush, Ben-Gurion University of the Negev, IsraelCopyright © 2023 Arvas, Marakli, Kaya and Kalendar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruslan Kalendar, cnVzbGFuLmthbGVuZGFyQGhlbHNpbmtpLmZp
†ORCID: Yunus Emre Arvas, orcid.org/0000-0001-6622-5083
Sevgi Marakli, orcid.org/0000-0001-5796-7819
Yılmaz Kaya, orcid.org/0000-0003-1506-7913
Ruslan Kalendar, orcid.org/0000-0003-3986-2460
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.