
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 02 June 2023
Sec. Plant Biotechnology
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1166728
This article is part of the Research TopicGenomic and Biotechnological Interventions for the Concurrent Improvement of Stress Resilience and Seed-Associated Traits in CropsView all 8 articles
 Ankit Saroha1
Ankit Saroha1 Sunil S. Gomashe2
Sunil S. Gomashe2 Vikender Kaur3Deepa Pal1Shraddha Ujjainwal1
Vikender Kaur3Deepa Pal1Shraddha Ujjainwal1 J. Aravind4
J. Aravind4 Mamta Singh3
Mamta Singh3 S. Rajkumar1
S. Rajkumar1 Kuldeep Singh5†
Kuldeep Singh5† Ashok Kumar3
Ashok Kumar3 Dhammaprakash Pandhari Wankhede1*
Dhammaprakash Pandhari Wankhede1*Flaxseed/linseed is an important oilseed crop having applications in the food, nutraceutical, and paint industry. Seed weight is one of the most crucial determinants of seed yield in linseed. Here, quantitative trait nucleotides (QTNs) associated with thousand-seed weight (TSW) have been identified using multi-locus genome-wide association study (ML-GWAS). Field evaluation was carried out in five environments in multi-year-location trials. SNP genotyping information of the AM panel of 131 accessions comprising 68,925 SNPs was employed for ML-GWAS. From the six ML-GWAS methods employed, five methods helped identify a total of 84 unique significant QTNs for TSW. QTNs identified in ≥ 2 methods/environments were designated as stable QTNs. Accordingly, 30 stable QTNs have been identified for TSW accounting up to 38.65% trait variation. Alleles with positive effect on trait were analyzed for 12 strong QTNs with r2 ≥ 10.00%, which showed significant association of specific alleles with higher trait value in three or more environments. A total of 23 candidate genes have been identified for TSW, which included B3 domain-containing transcription factor, SUMO-activating enzyme, protein SCARECROW, shaggy-related protein kinase/BIN2, ANTIAUXIN-RESISTANT 3, RING-type E3 ubiquitin transferase E4, auxin response factors, WRKY transcription factor, and CBS domain-containing protein. In silico expression analysis of candidate genes was performed to validate their possible role in different stages of seed development process. The results from this study provide significant insight and elevate our understanding on genetic architecture of TSW trait in linseed.
Linseed/flaxseed (Linum usitatissimum L.) is an important crop in several countries across the world and has myriad of food, nutritional, and industrial applications. It is a self-pollinated diploid crop species (2n = 2x = 30) with genome size of ~368–373 Mb (Ragupathy et al., 2011; Wang et al., 2012). Linseed is considered to be originated in the Middle East or Indian regions (Vavilov, 1951; Green et al., 2008). There are two morphotypes of L. usitatissimum, “flax type” and “linseed type” (Diederichsen and Ulrich, 2009; Kaur et al., 2017). The linseed type is short statured, branched with high number of capsules bearing larger seeds.
Although, linseed is among the earliest domesticated crop and has been under cultivation since ancient times, in recent years, there is a surge of renewed interest and popularity largely due to the health benefits it offers. It is one of the richest plant-based sources of omega-3 fatty acid, alpha linolenic acid, and has an impressive nutritional profile with high amounts of proteins, dietary fibers, vitamin B1, and lignans, especially secoisolariciresinol diglucoside (SDG) (Yadav et al., 2022). The nutritional value of linseed extends to a wide range of health benefits in cardiovascular disease, cancer, neurological disorder, and other lifestyle related diseases/conditions such as diabetes, atherosclerosis, hypertension, and obesity (Pan et al., 2009; Katare et al., 2012; Goyal et al., 2014; Kajla et al., 2015; Mohammadi-Sartang et al., 2017). Accordingly, the higher demand has led to an upward trend in the global production of linseed in the last decade (FAOSTAT, 2022; Yadav et al., 2022). India, which ranks seventh in the world in terms of linseed production, however, has productivity substantially lower (0.543 tonne/ha) than the world average (1.053 tonne/ha) (Kaur et al., 2017).
Thousand-seed weight (TSW) (1,000 seed weight) is one of the major components of seed yield in linseed. Seed weight is widely recognized as a complex trait controlled by many genetic and environmental factors (Kraft et al., 1963; Mian et al., 1996). The synergistic growth of embryo, endosperm, and ovule contributes to seed development, wherein several signaling pathways and regulatory networks coupled with spatiotemporal disposition of an array of phytohormones play critical roles (Cao et al., 2020). Signaling pathways such as G-protein signaling, IKU (HAIKU), ubiquitin-proteosome pathway, and phytohormones especially auxins and associated factors have a role in the regulation of seed development and seed weight (Locascio et al., 2014; Cao et al., 2020). Several genes encoding Indole-3-acetic acid (IAA)–glucose hydrolase, GLYCOGEN SYNTHASE KINASE 3/SHAGGY-like, GRAS transcription factor, AUXIN RESPONSE FACTOR (ARF), WRKY transcription factor, and cytochrome P450 enzyme have been identified crucial in seed development and seed weight in other plants (Ishimaru et al., 2013; Chen et al., 2015; Liu et al., 2015a; Zhu et al., 2015; Ge et al., 2016; Hirano et al., 2017; Hu et al., 2018; Cao et al., 2020).
In linseed, a few research groups have attempted genetic dissection of seed weight trait using biparental linkage mapping and association studies in natural population and identified 44 unique quantitative trait loci (QTLs) (Soto-Cerda et al., 2014; Kumar et al., 2015; Xie et al., 2018a; Xie et al., 2018b; Guo et al., 2020; You and Cloutier, 2020). Among the earliest studies to understand the genetic architecture of TSW in linseed, Soto-Cerda et al. (2014) using association mapping identified five SSR markers associated with TSW, explaining 30% of the trait variation in the Canadian flax core collection. Kumar et al. (2015) using biparental mapping population (RILs) developed an SSR-SNP genetic map and identified a minor QTL for TSW encompassing important genes encoding leucine-rich receptor-like protein kinase, cytochrome P450, WRKY, and GRAS family transcription factors. Further using genome-wide association study (GWAS) and GWAS combined with LD heatmap in flax core and cultivated accessions, important candidate genes for TSW have been identified, namely, SPX & EXS domain-containing protein (PHO1), ubiquitin-proteosome pathway genes, RING/U box protein, cytochrome P450, Auxin canalization (AC), and 26S proteosome regulatory subunit (RPN) (Xie et al., 2018a; Guo et al., 2020).
As known from plethora of studies, seed weight is considered a complex trait governed by several genes. In rice, for example, over 400 QTLs have been reported for seed weight and shape traits (Huang et al., 2013). In linseed, in spite of the 44 reported QTLs/quantitative trait nucleotides (QTNs)/markers and underlying candidate genes, more light is certainly required for a better understanding of the genetic architecture of TSW.
GWAS has been a preferred strategy in recent times for genetic dissection of complex traits including seed weight/size in a number of crop plants such as wheat, soybean, rice, maize, and oilseed rape (Li et al., 2014; Lu et al., 2017; Kumar et al., 2020; Qi et al., 2020; Tao et al., 2020; Zhang et al., 2020; Chaurasia et al., 2021; Niu et al., 2021). In linseed, GWAS has been used for genetic dissection of flowering time traits, days to maturity, plant height, fiber-related traits, seed weight and size, pasmo resistance, mucilage content, and important agronomic traits (Soto-Cerda et al.,2018; Xie et al., 2018a; Xie et al., 2018b; He et al., 2019; Singh et al., 2019; Guo et al., 2020; Soto-Cerda et al., 2021; Saroha et al., 2022a; Yadav et al., 2022). For genotyping, reduced representation sequencing approach such as Genotyping by Sequencing (GBS) and SLAF has been successfully used for genotyping and further deployed in GWAS studies in different crop plants (Elshire et al., 2011; Hirsch et al., 2014; Kujur et al., 2015; Xie et al., 2018a; Xie et al., 2018b; Milner et al., 2019).
In present study, genetic dissection of TSW trait in linseed was undertaken using multi-locus GWAS methods in a panel of diverse germplasm accessions conserved in Indian National Genebank (INGB) to identify quantitative traits nucleotides (QTNs) and associated candidate genes.
Association mapping panel of 131 germplasm accessions (Supplementary Table S1) from a total of 220 accessions grown in five environments in three consecutive years (Saroha et al., 2022a; Saroha et al., 2022b) were used in this study. Observations on TSW were recorded on three representative plants from each accession in all five environments at two locations including Delhi (28°38’53.7”N 77°09’05.4”E) for 3 years (2017-18: DL17-18; 2018-19: DL18-19 and 2019-20: DL19-20) and Akola (20°42’03.2”N 77°01’53.6”E) for 2 years (2018-19: AK18-19 and 2019-20: AK19-20). Data analysis for each individual environment was done and adjusted means were generated in “R” with the package augmentedRCBD version 0.1.5 (Aravind et al., 2021), and the homogeneity of variances was tested by Bartlett’s chi-square test. Adjusted means were used for the calculation of descriptive statistics, frequency distribution plots, and GWAS.
SNP data on the association mapping panel (131 accessions) was previously generated using genotyping by sequencing approach (Saroha et al., 2022a). Total of 68,925 SNPs distributed on 15 chromosomes were employed for multi-locus genome-wide association study (ML-GWAS) for the five independent environments using mrMLM package version 4.0.2 (Wen et al., 2018) of “R”. All the six multi-locus models: mrMLM (Wang et al., 2016), FASTmrMLM (Tamba and Zhang, 2018), FASTmrEMMA (Wen et al., 2018), pKWmEB (Ren et al., 2018), pLARmEB (Zhang et al., 2017), and ISIS EM-BLASSO (Tamba et al., 2017) were deployed for analysis using default values for all the parameters. A threshold of logarithm of odds (LOD) score ≥ 3.0 was used to consider the QTNs statistically significant for the trait to balance the high-power and low false positive rate for QTN detection using ML-GWAS methods (Zhang et al., 2019). For eliminating the model and environment biasness, QTNs identified either by ≥ 2 models or in ≥ 2 environments were considered as stable QTNs.
Genes around 30 kb region (30 kb upstream and 30 kb downstream, total 60 kb) of the stable QTNs were considered as potential candidate genes. Annotated genes as per the flax genome annotation (You et al., 2018a; You and Cloutier, 2020) were aligned against the Swiss-Prot sequences (reviewed and manually curated) of the Plants taxonomic division at the Uniprot database (https://www.uniprot.org/). Functional annotation of the potential candidate genes was performed as per the best hit.
In silico gene expression of the candidate genes was studied using publicly available transcriptome data of the rice variety Nipponbare available at Rice Genome Annotation Project database (http://rice.uga.edu/index.shtml) at four developmental stages: Embryo - 25 Days After Pollination (DAP) (SRX100753), Endosperm - 25 DAP (SRX100754) and Seed - 5 DAP (SRX100749), and 10 DAP (SRX100755). Candidate genes were aligned against the Nopponbare protein sequences and heatmap plots of the best hit were generated by the ComplexHeatmap package version 2.10.0 (Gu et al., 2016) of “R”.
A wide range of phenotypic variation was observed for TSW in AM panel under all the five environments (Table 1 and Figure 1). The heterogeneity of the error variances for TSW was high for the studied environments and, therefore, analysis was performed independently for each of the environments. Among all the five environments, the lowest and highest TSW value was 1.76 g and 12.52 g, respectively. The mean TSW was significantly lower at Akola location than at Delhi location as the former comes under Zone-3 of linseed growing areas of India and the later under Zone-2. At Akola, mean TSW for the year 2018-19 and 2019-20 was 5.85 g and 5.92 g with CV of 29.8 and 27.57, respectively. At the Delhi location for the year 2017-18, 2018-19, and 2019-20 mean TSW was 6.73, 8.09, and 8.93 with CV of 30.10, 24.86, and 18.94, respectively. The variation within each single environment was statistically significant as evident in the analysis of variance (ANOVA) (Supplementary Table S2).
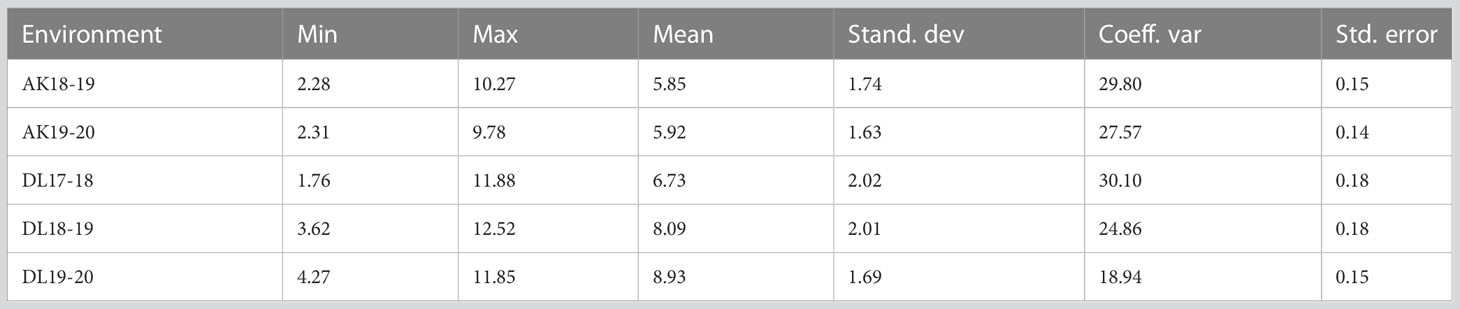
Table 1 Descriptive statistics of thousand seed weight (TSW) in association mapping panel in five environments at Akola (AK) and Delhi (DL).
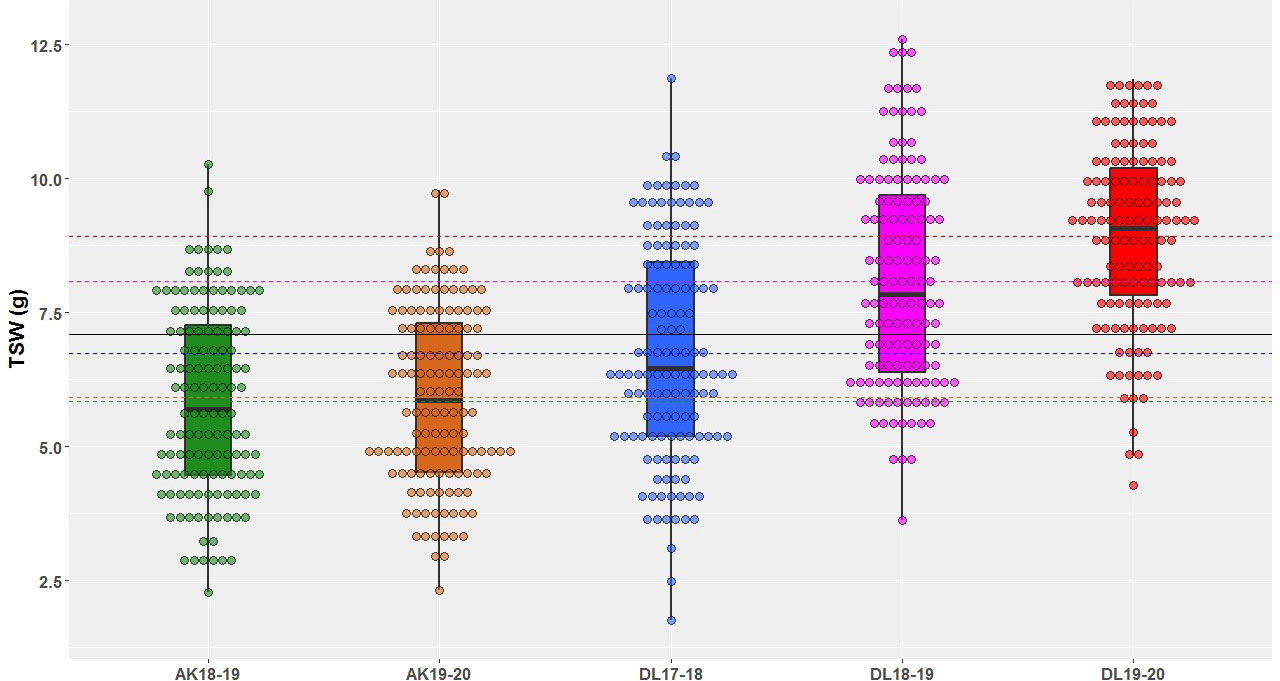
Figure 1 Thousand-seed weight phenotypic variation illustrated in box & dot plots in association mapping panel of linseed germplasm accessions grown at Akola during years 2018-19 (AK18-19) and 2019-20 (AK19-20) and Delhi during 2017-18 (DL17-18), 2018-19 (DL18-19), and 2019-20 (DL19-20).
For five environments, there were total 137 statistically significant QTNs identified with a threshold LOD score of ≥ 3.0 using five of the six ML-GWAS methods, of which 84 QTNs were unique across methods and environments (Table 2). Among the six ML-GWAS methods across the five environments, pLARmEB identified maximum of 39 unique QTNs followed by 27 QTNs each by mrMLM and ISIS EM-BLASSO. FASTmrMLM identified 24 unique QTNs, whereas the least number of seven QTNs were identified by FASTmrEMMA. There was no significant QTN identified by pKWmEB in any of the environments for TSW. With respect to the five environments, across five methods maximum unique QTNs were identified for Delhi 2018-19, followed by Delhi 2017-18 and Akola 2019-20. The five ML-GWAS methods combined Manhattan and Quantile-Quantile (QQ) plots for TSW trait for five environments depicting the significant QTNs have been shown (Figure 2).
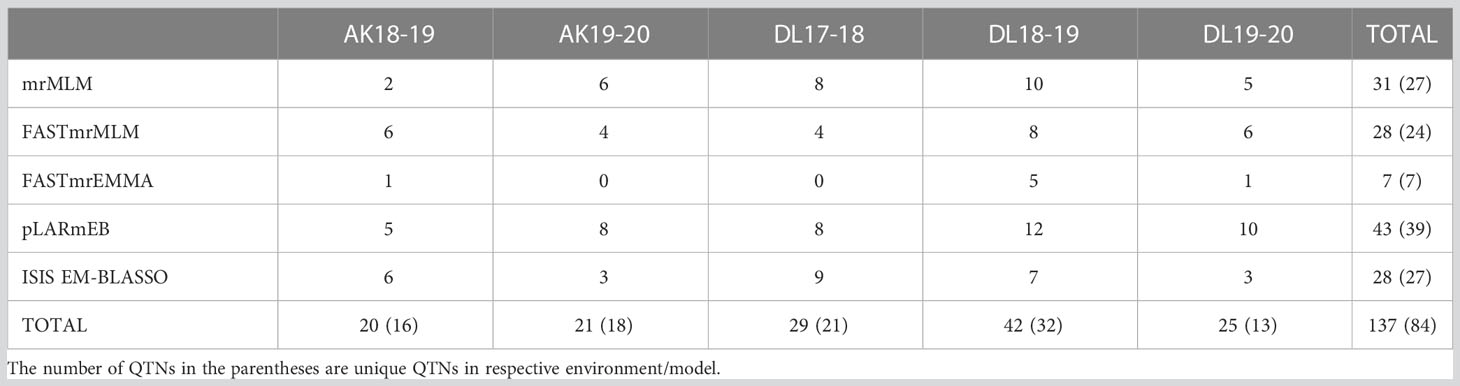
Table 2 Distribution of QTNs identified for TSW in linseed across five environments and methods.
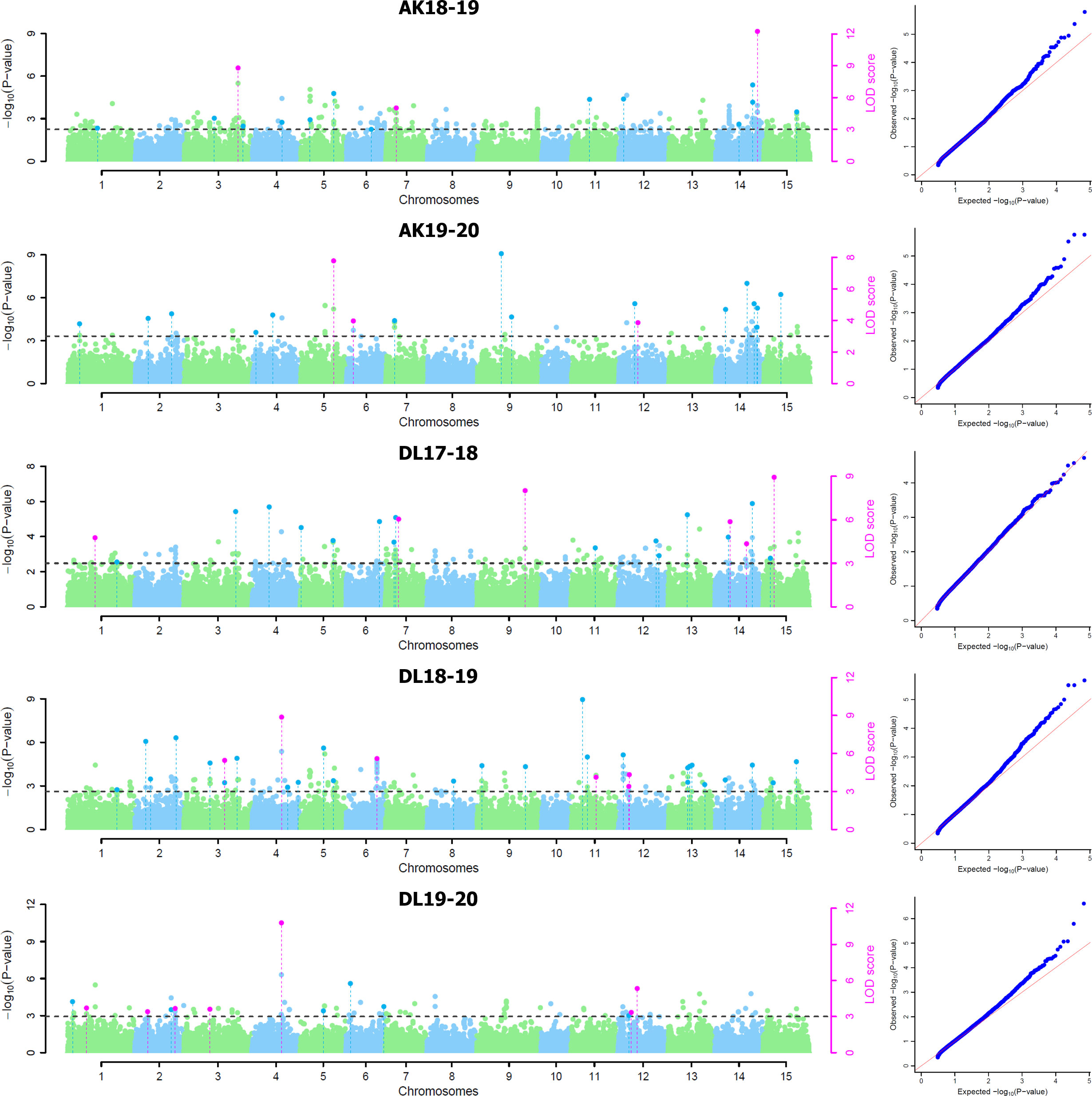
Figure 2 Combined Manhattan plots and respective quantile–quantile plots of five ML-GWAS methods for thousand-seed weight trait in five environments at Akola during 2018-19 (AK18-19) and 2019-20 (AK19-20) and New Delhi during 2017-18 (DL17-18), 2018-19 (DL18-19), and 2019-20 (DL19-20). LOD threshold score of 3.0 is indicated as dashed line in Manhattan plots. Dots above the threshold line represent significant QTNs, whereas QTNs identified in ≥ 2 methods are depicted in pink color.
In order to identify the stable QTNs by removing method and/or environment biasness, QTNs identified by ≥ 2 models or in ≥ 2 environments were designated as stable QTNs. Accordingly, 30 stable QTNs have been identified for TSW (Table 3), and the rest were considered as potential QTNs (Supplementary Table S3). The LOD score and –log10(p) value for the stable QTNs ranged from 3.02 to 12.57 and 3.72 to 13.56, respectively, which explained up to 38.65% of the variation in the TSW trait across environments (Table 3). The genomic locations of all the identified stable QTNs for the TSW trait have been depicted on 15 chromosomes of linseed (Figure 3).
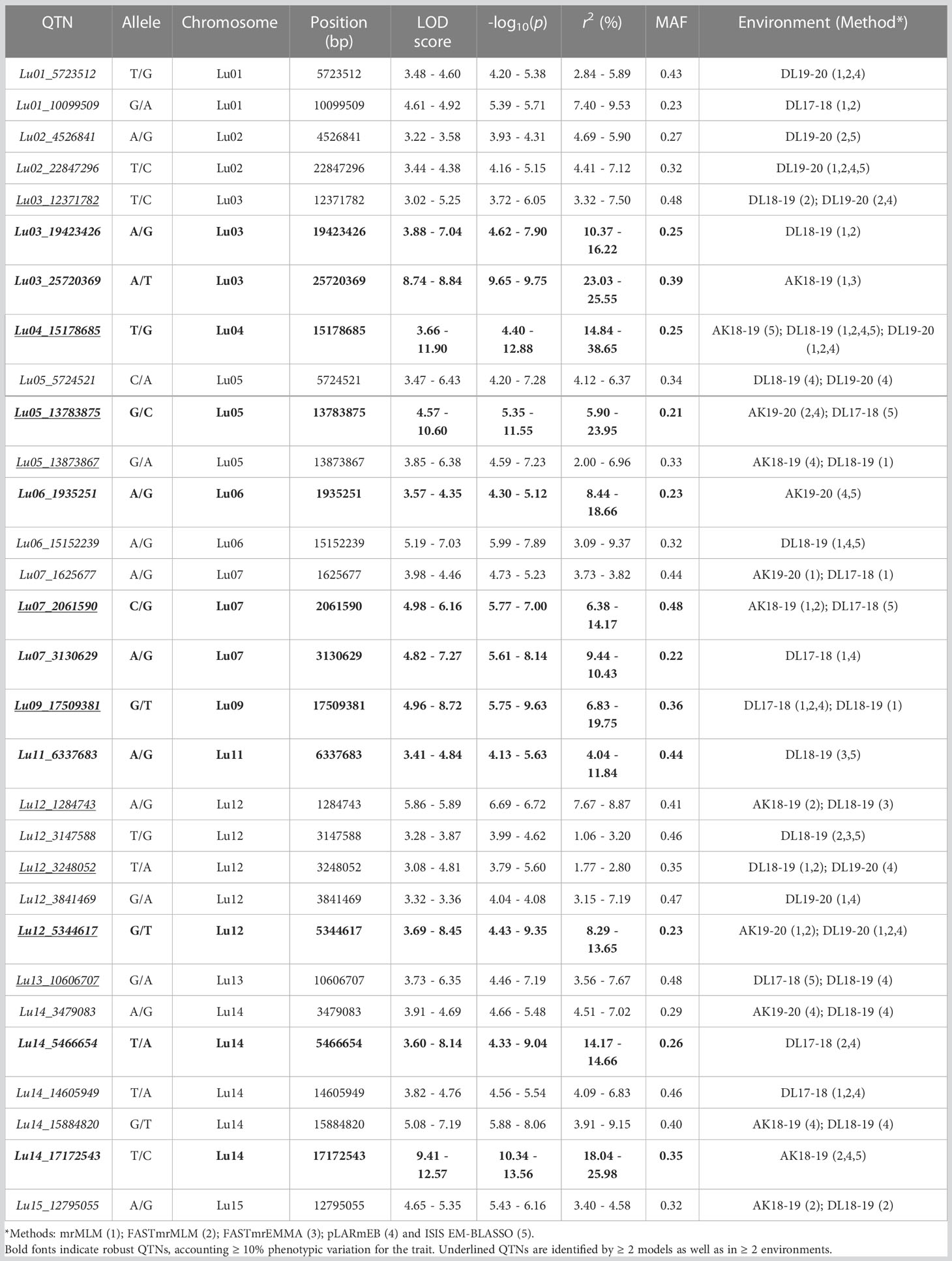
Table 3 List of stable QTNs identified for TSW in linseed.
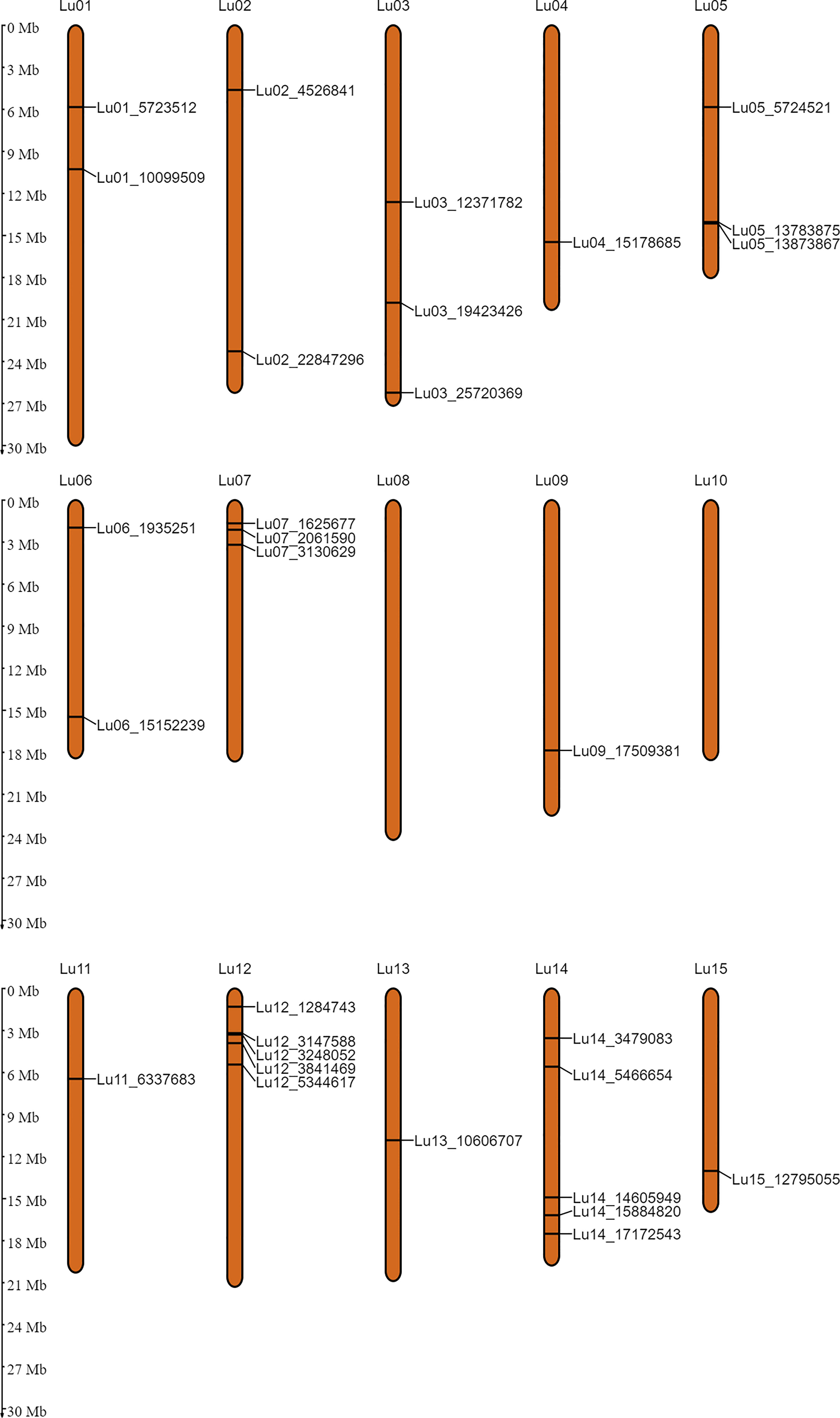
Figure 3 Genomic positions of the stable QTNs for thousand-seed weight on 15 chromosomes of linseed.
To study whether specific alleles of the QTNs carry any positive effect on the trait value in different environments, the robust QTNs having r2 ≥ 10.0% in at least one of the identified environments/models were selected (Table 3). For each of the 12 selected robust QTN loci, the AM panel was divided into two groups based on the allele type they carried and the significance of the phenotypic difference between the two groups studied. For eight of the 12 studied QTNs, there was a statistically significant difference in TSW value between two groups carrying distinct alleles in at least three environments (Figure 4). For example, in the case of Lu03_25720369, accessions carrying allele “Thymine” (T) showed a higher five environment mean TSW 8.21 g compare with the accession group with another allele “Adenine” (A) having mean TSW 6.46 g. Four QTNs, Lu03_25720369, Lu07_2061590, Lu11_6337683, and Lu14_17172543 showed a positive effect of the respective alleles on TSW consistent across the five environments.
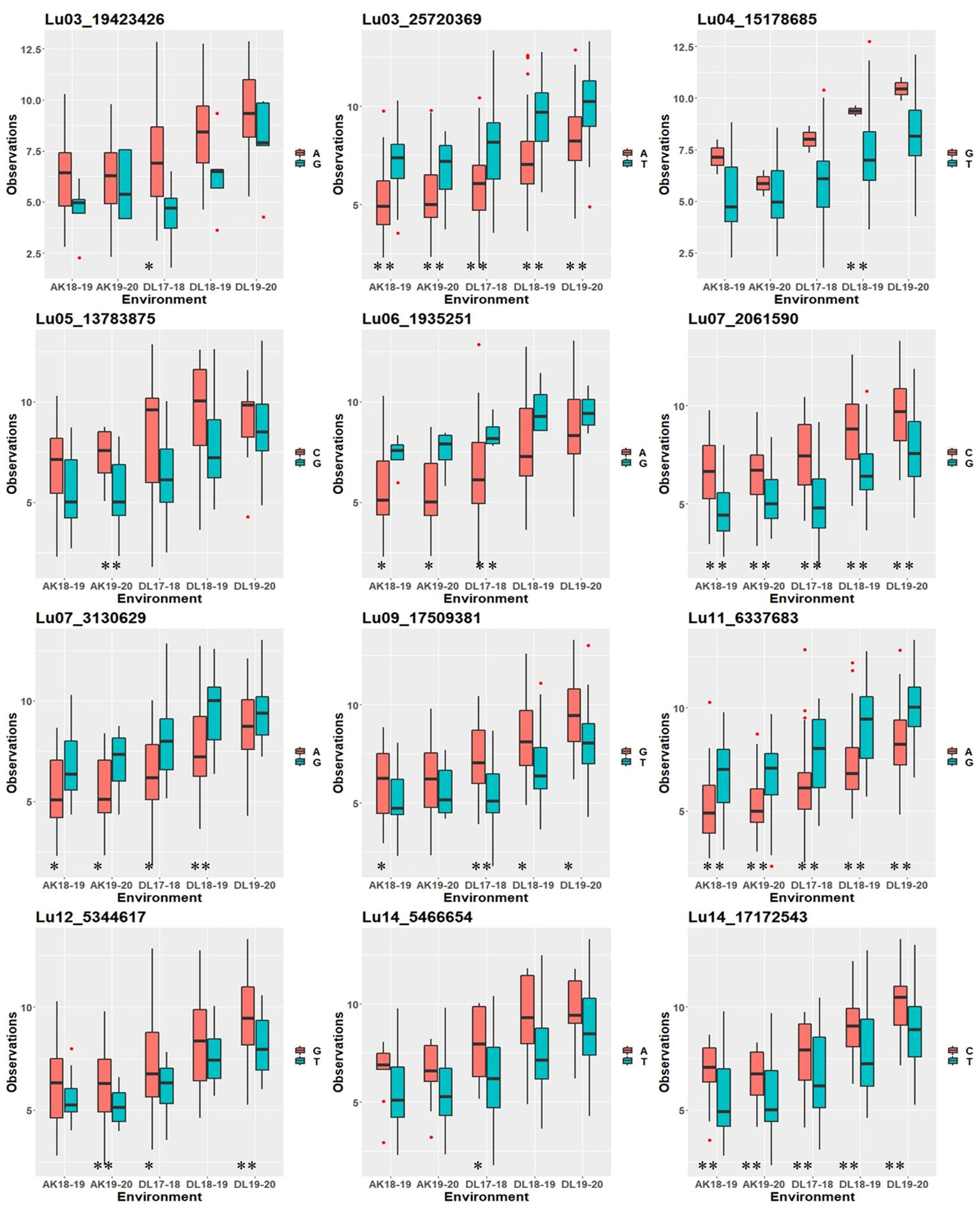
Figure 4 Box plot showing positive effect of alleles of the robust QTNs (r2 ≥ 10.0%) on TSW trait values in five environments at Akola (2018-19 and 2019-20) and Delhi (2017-18, 2018-19, and 2019-20). Statistical significance at 0.01 and 0.05 were indicated with ** and *, respectively.
Genes around the 30 kb region (30 kb upstream and downstream, total 60 kb) of the stable QTNs were extracted as per the linseed genome assembly (You et al., 2018a; You and Cloutier, 2020). A total of 333 genes were found around the stable QTNs. The list of the potential candidate genes for 30 stable QTNs of TSW and their functional annotation has been provided (Supplementary Table S4). Based on the functional annotation, 23 genes were shortlisted as candidate genes for TSW trait. Among the notable candidate genes included were Lus10012572 and Lus10012573 (B3 domain-containing transcription factor, FUS3), Lus10015274 (Shaggy-related protein kinase/Protein BRASSINOSTEROID INSENSITIVE 2), Lus10017640 and Lus10017641 (Auxin response factor 6), Lus10012582 (Cytochrome P450 84A/Ferulate-5-hydroxylase), Lus10030685 (ANTIAUXIN-RESISTANT 3), Lus10030697 (Probable ubiquitin conjugation factor E4/RING-type E3 ubiquitin transferase E4), Lus10037500 (Protein pleiotropic regulatory locus 1), Lus10003792 (E3 ubiquitin-protein ligase UPL5/Ubiquitin-protein ligase 5), Lus10042538 (WRKY transcription factor), Lus10015277 (MALE DISCOVERER 1/LRR receptor-like serine/threonine-protein kinase), Lus10015279 (Non-specific lipid-transfer protein, NLTP), Lus10023553 (Glucose-1-phosphate adenylyltransferase), Lus10028804 (SUMO-activating enzyme), Lus10033912 (Protein SCARECROW), Lus10037503 and Lus10037504 (Sucrose nonfermenting 4-LIKE protein/CBS domain-containing protein CBSCBS3) (Table 4).

Table 4 List of candidate genes for TSW in linseed.
In order to validate the identified candidate genes and have the supporting evidence of their possible function in seed weight trait in model plants, in silico expression analysis was performed with rice ortholog of selected candidate genes using rice RNA-seq data as a basis. All but one (Lus10010348) candidate genes showed significant hit and found ortholog in the rice genome. For the expression study, rice RNA-seq data of seeds at 5 and 10 DAP, the embryo (25 DAP), and endosperm (25 DAP) tissues were used. All 22 genes showed good expression in either developing seeds, embryo or endosperm tissues (Figure 5).
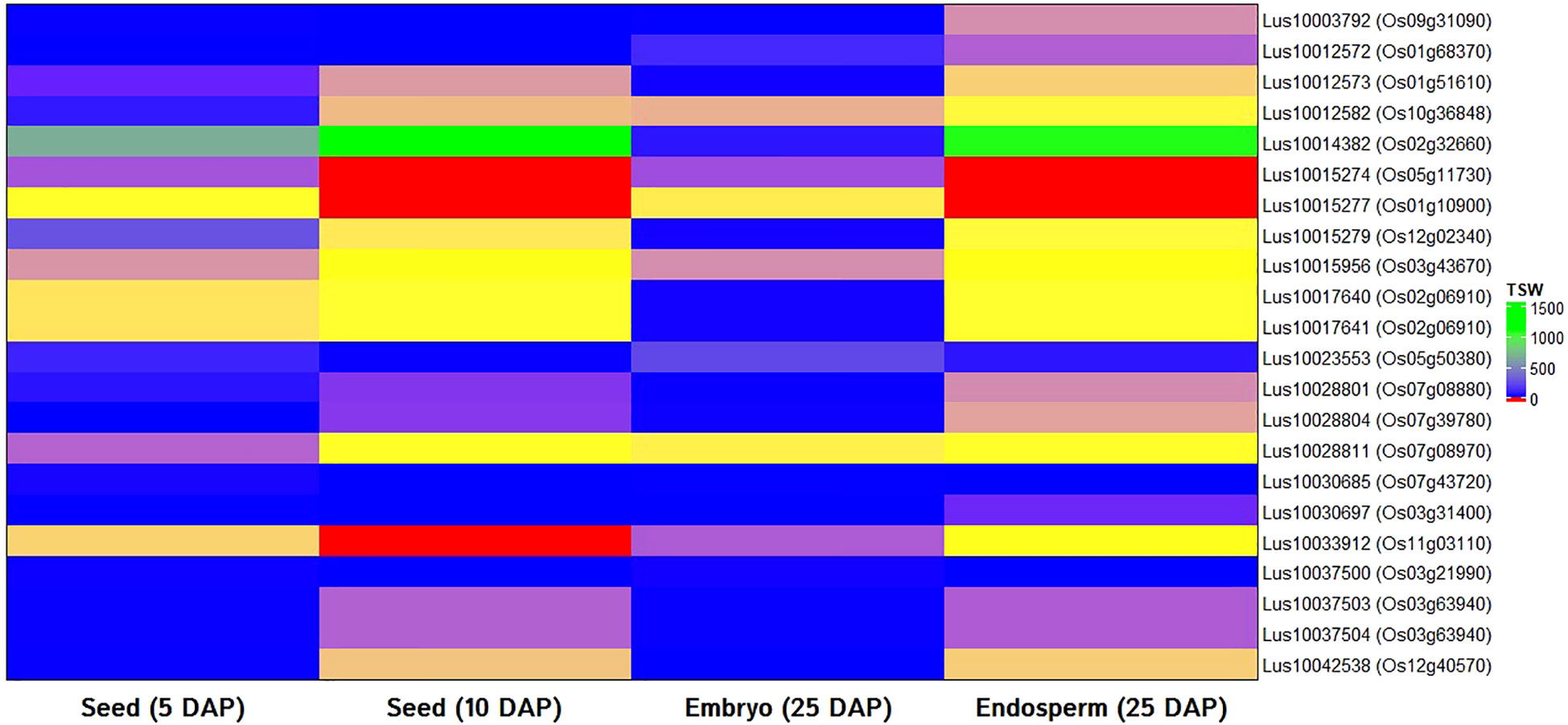
Figure 5 In silico gene expression of orthologs of candidate genes for thousand-seed weight in linseed using RNA-seq data of rice seeds (5 and 10 days after pollination: 5 DAP; 10 DAP), embryo (25 DAP), and endosperms (25 DAP). Transcriptome data of the rice variety Nipponbare available at Rice Genome Annotation Project database was used.
Substantial variation for TSW trait was observed in the AM panel for all the studied environments (Figure 1 and Table 1). Owing to the high-error variance (environmental factor), especially across geographical areas, statistical analyses were performed independently for each individual environment. GWAS studies performed in linseed for important agronomic and flowering time traits also followed a similar approach (Xie et al., 2018b; Soto-Cerda et al., 2021). There was a significant difference in the trait expression between the two geographical locations. Linseed accessions displayed relatively lower seed weight in Akola compared with Delhi. Akola and Delhi fall under the 2nd and 3rd linseed growing zones in India and have significant environmental/temperature differences due to latitudinal locations and thus have shorter growing seasons at the former location. The differences in trait expression at the two geographical locations were also expected, as the higher temperature is known to negatively affect plant growth and development and, thereby, the seed yields (Li et al., 2014; Čeh et al., 2020). Significant location effect on TSW trait in linseed was also observed in GWAS studies using Canadian flax core collection (Soto-Cerda et al., 2014).
The positive effect of alleles of 12 robust QTNs on TSW was observed in this study (Figure 4). Alleles with a positive effect on the trait would be helpful for trait improvement, as it would facilitate combining positive alleles in the genetic background of popular varieties (Zhang et al., 2012). These alleles would also help in the selection of parents for pyramiding in the desired genetic background. Favorable SSR alleles for TSW were reported earlier in Canadian flax core collection (Soto-Cerda et al., 2014). Combining the positive alleles of important yield components would make a pertinent strategy for yield improvement through its components (Shi et al., 2009; Soto-Cerda et al., 2014).
For TSW, a total of 44 QTLs have been identified so far from the earlier studies (Soto-Cerda et al., 2014; Wu et al., 2018; Xie et al., 2018a; Xie et al., 2018b, Guo et al., 2020, You and Cloutier, 2020). The 30 QTNs identified in this study were checked for co-location with earlier known QTLs/markers for TSW in linseed.
It was observed that a few of the stable QTNs were in close physical proximity of at least five earlier known markers associated with TSW on the linseed pseudomolecules (You et al., 2018a; You and Cloutier, 2020). SSR markers Lu58a and Lu2555 were among the first reported markers associated with TSW (Soto-Cerda et al., 2014). QTNs Lu12_3841469, Lu12_3248052, Lu12_3147588, Lu12_5344617 and Lu12_1284743 were 0.04, 0.55, 0.66, 1.54, and 2.52 Mb close, respectively, to Lu58a on chromosome 12 (You and Cloutier, 2020). QTN Lu06_15152239 was co-located 0.89 Mb close to Lu2555 on chromosome 6. The same QTN was located just 0.2 Mb away from marker “scaffold1491_58878” for TSW (Xie et al., 2018a; You and Cloutier, 2020). Another QTN Lu02_22847296 was also found in the proximity of 0.44 and 0.86 Mb to markers “scaffold43-1111162” and “scaffold107-300735”, respectively on chromosome 2 (You and Cloutier, 2020). Co-locating these QTLs in the present work and other independent studies (Soto-Cerda et al., 2014; Xie et al., 2018a) highlights their importance in the regulation of TSW in linseed. This also underpins the power of ML-GWAS methods in the genetic dissection of complex traits. The QTNs/markers identified for TSW in this study as well as the earlier identified QTNs/markers for TSW, oil and seed yield (Soto-Cerda et al., 2014; Kumar et al., 2015; You et al., 2018b; You and Cloutier, 2020) have been depicted on linseed chromosomes (Supplementary Figure S1) to facilitate identification of QTL/QTN rich regions associated with these economically important traits.
In addition, the QTNs near the known QTLs/markers, other QTNs identified in this study are novel and hitherto unknown QTNs/genomic regions for TSW in linseed (Table 3).
“Priori knowledge of biology” is one of the most important corner stones in identifying candidate genes in GWAS studies (Burghardt et al., 2017). As per the LD Decay (Saroha et al., 2022a), 23 candidate genes have been identified from 30 kb regions (total 60 kb up- and downstream) around the stable QTNs. Earlier studies in linseed reported important candidate genes for seed weight including leucine-rich receptor-like protein kinase, cytochrome P450 family protein, WRKY genes, GRAS family transcription factors, Pho1 (SPX & EXS domain-containing protein), auxin canalization, kinase family, RING/U-box protein and, a few genes related to ubiquitin proteosome pathway (Kumar et al., 2015; Xie et al., 2018a; Guo et al., 2020). Indeed, in the present work, candidate genes such as cytochrome P450 proteins (Lus10012582, Lus10030687), WRKY (Lus10042538), Probable LRR receptor-like serine/threonine-protein kinase (Lus10015277), SCARECROW/GRAS family protein (Lus10033912), RING-type E3 ubiquitin transferase/ubiquitin conjugation factor E4 (Lus10030697), and E3 ubiquitin-protein ligase (Lus10003792) have been identified underpinning their importance in the regulation of thousand seed weight in linseed.
In other plants, involvement of cytochrome P450 proteins in seed weight and yield is known (Huang et al., 2013; Ayalew et al., 2022). The crucial roles of ubiquitin-proteasome pathways in regulating seed size have been shown in several recent studies (Xia et al., 2013; Kelley, 2018; Xu and Xue, 2019; Wang et al., 2020). In flaxseed, six of the 13 identified candidate genes through GWAS were related to the ubiquitin-proteasome pathway (Guo et al., 2020), which highlighted the important role of deubiquitination in seed size regulation. In congruence, the present study identified seven candidates/potential candidate genes encoding RING-type E3 ubiquitin transferase for TSW (Table 4 and Supplementary Table S4), which further substantiates the possible involvement of deubiquitination/ubiquitination in seed weight regulation in linseed.
Two promising candidate genes Lus10017640 and Lus10017641 for TSW showed similarity to Auxin response factor 6 of Arabidopsis and rice with putative function in modulating early auxin response genes expression (Table 4). Role of auxin as a key regulator of seed and seed weight development is well established (Cao et al., 2020). A number of genes involved in auxin biosynthesis, supply, transport, metabolism, and auxin–BR signaling have been identified to play crucial role in varied aspect of seed development in different plant species (Dharmasiri et al., 2005; Bernardi et al., 2012; Ishimaru et al., 2013; Liu et al., 2015a; Liu et al., 2015b; Hirano et al., 2017; Shi et al., 2019; Cao et al., 2020).
One of the most important candidates identified for TSW in this study is shaggy-related protein kinase/protein BRASSINOSTEROID INSENSITIVE 2 (BIN2) (Lus10015274). Role of brassinosteroid hormone in normal growth and development of plants as well as regulating seed size/weight is well known (Wu et al., 2016; Song, 2017; Xiong et al., 2021). In rice, for grain width, a major QTL (GW5/qSW5/GSE5) was identified harboring a domestication related gene GW5, which encodes a protein with two IQ calmodulin-binding motifs (Shomura et al., 2008; Weng et al., 2008; Duan et al., 2017; Liu et al., 2017; Song, 2017). GW5 was shown to impart a negative effect on grain width as its reduced and upregulated expression gave wider and narrower grains, respectively (Duan et al., 2017; Liu et al., 2017). GLYCOGEN SYNTHASE KINASE 2 (GSK2), an ortholog of the Arabidopsis GSK3/SHAGGY-like kinase BIN2 was identified as a viable interacting partner of GW5 (Liu et al., 2017; Song, 2017). GSK2 is known negative regulator of brassinosteroid signaling and inhibit stomatal development by phosphorylation mediated inhibition of MAPKK kinase, YODA, and the MAPK kinases MKK4 and MKK5 (Li and Nam, 2002; Kim et al., 2012; Khan et al., 2013). In Arabidopsis, SHAGGY-like kinase BIN2 also promote and restrict vital process of asymmetric cell division (ACD) by phosphorylating MAPK-signaling components and the downstream transcription factor SPEECHLESS (SPCH), respectively (Houbaert et al., 2018). Therefore, Lus10015274, a shaggy-related protein kinase/protein BIN2 is a promising candidate gene for TSW in linseed.
WRKY transcription factor gene (Lus10042538) and LRR receptor-like serine/threonine-protein kinase (Lus10015277) are other important candidates identified for TSW (Table 4). Genes encoding both WRKY and LRR were also earlier identified as candidate gene for TSW in linseed (Kumar et al., 2015). In Arabidopsis, WRKY and leucine-rich repeat (LRR) KINASE family genes are known to regulate seed size (Luo et al., 2005; Gu et al., 2017). It is interesting to note that WRKY46, WRKY54, and WRKY70 were shown to exert positive roles in brassinosteroid-regulated plant growth and negative role in drought stress response. Shaggy-related protein kinase/protein BIN2 reportedly destabilizes WRKY46, WRKY54, and WRKY70 by phosphorylation (Chen et al., 2017). It is further astonishing to know that candidate gene (Lus10010348) encoding VQ motif-containing protein 9 has been identified for TSW in this study. AtVQ9 in Arabidopsis act as a negative regulator of salt stress and repressor of WRKY (Hu et al., 2013). Another promising candidate gene (Lus10003792) encodes E3 ubiquitin-protein ligase 5, whose gene ontology function showed regulation of leaf senescence through ubiquitination and subsequent degradation of WRKY53 (Miao and Zentgraf, 2010).
Candidate gene Lus10033912 encoding GRAS family protein 20/SCARECROW was identified for TSW, whose Arabidopsis counterpart was shown to be involved in ACD, specification of quiescent center and maintenance of surrounding stem cells toward radial pattern formation in roots. The GRAS gene was also found essential for cell division, shoot gravitropism, and regulation of radial organization of shoot axial organs (Heidstra et al. 2004; Wysocka-Diller et al., 2000; Sabatini et al., 2003; Sena et al., 2004). Role of SCARECROW-LIKE 15 in repressing the seed maturation by interaction with HISTONE DEACETYLASE19 is known (Gao et al., 2015).
Among others, an important gene Lus10015279 (non-specific lipid-transfer protein, NLTP) showed possible involvement in multiple seed development-related processes such as pollen development, pollen tube adhesion-growth, seed coat development, seed maturation, germination, and fruit ripening (Missaoui et al., 2022). NLTPs are also known to play role in biosynthesis and storage of lipids in seeds. In sesame, two members of the NLTP gene family, SiLTPI.23 and SiLTPI.28, have been identified for high oil content in seeds (Song et al., 2021). Another important gene Lus10014382 (1,4-alpha-glucan-branching enzyme 1, chloroplastic/amyloplastic or Starch branching enzyme I, SBEI) showed function in glycogen and starch biosynthesis. Absence of the SBEI enzyme caused reduced amylopectin and starch, while increase in sucrose in the developing seeds, which resulted in increased osmosis and thereby larger cell size. In maturing seeds, loss of water leads to the cell shrinking and wrinkled seed phenotype (Bhattacharyya et al., 1993).
Orthologs of genes Lus10012572 and Lus10012573 (B3 domain–containing transcription factor FUS3) have been reported to play diverse roles in plant life cycle, such as seed germination, dormancy, embryo formation, seed and fruit development, and seed maturation (Santos-Mendoza et al., 2008). In Arabidopsis, FUS3 positively regulates seed filling by suppressing expression of TRANSPARENT TESTA GLABRA1 (TTG1) gene. TTG1 negatively regulates accumulation/synthesis of the seed storage reserves such as storage proteins and fatty acids during embryogenesis. TTG1 also indirectly suppresses the expression of a group of genes that either act as master regulators of seed development or are involved in synthesis/modification of fatty acids during seed development (Chen et al., 2015). FUS3, together with the ABI3, LEC1, and LEC2, also regulates biosynthesis of fatty acids in Arabidopsis (Mu et al., 2008; Roscoe et al., 2015). Recently, Ahmad et al. (2022) reported that, in tomato, overexpression of VvFUS3 gene resulted in a reduction in seed number and seed weight, however, with no effect on size of the fruits. Another gene Lus10023553 (Glucose-1-phosphate adenylyltransferase or ADP-glucose synthase or ADP-glucose pyrophosphorylase, AGPase) is possibly involved in the biosynthesis of glycogen and starch. In oilseeds, accumulation of starch during seed development is temporary and almost absent in mature seeds. Suppression of AGPase activity in Camelina seeds significantly increased seed size and weight (Na et al., 2018).
To study the possible role of the identified candidate genes in seed development process, in silico gene expression of orthologs of 22 candidate genes were studied using RNA-seq data of different seed developmental stages and tissues of rice (Figure 5). Orthology and in silico expression of candidate genes have been considered important to extrapolate the underlying gene function from model to other plant species (Das et al., 2016). In silico gene expression approach has earlier been employed in identification and successful validation of candidate genes for important traits including seed weight, seed development, seed yield, flowering time, agronomic traits in other plants, and flaxseed as well (Ngu et al., 2014; Soto-Cerda et al., 2021; Ayalew et al., 2022; Raboanatahiry et al., 2022).
Present study reports genetic dissection of TSW trait in linseed using ML-GWAS approach. The study identified 84 unique significant QTNs for TSW, of which 30 QTNs were designated as “stable” QTNs as have been identified in two or more methods/environments. The stable QTNs accounted up to 38.65% of the trait variation. Several important candidate genes have also been identified including shaggy-related protein kinase/BRASSINOSTEROID INSENSITIVE 2 (BIN2), ANTIAUXIN-RESISTANT 3, SCARECROW, RING-type E3 ubiquitin transferase E4, Auxin response factor, and WRKY transcription factor. The study not only reported novel QTNs and candidate genes but also collocated a few QTNs near to the established QTLs/markers for TSW in linseed. Unravelling QTNs/markers/candidate genes associated with TSW in independent studies underpin the importance of these genomic regions/QTLs in regulation of TSW. With more light shed on different possible attributes of TSW, this study gives a novel insight in the genetic architecture of complex TSW trait in this ancient crop.
The datasets used in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, PRJNA706105.
DW, VK, and AK conceptualized the project and designed experiments. AS, DP, SG, and SU have done the investigation, data recording, and curation. AS, DW, JA, and SR have done statistical and GWAS analyses. JA provided germplasm resources. DW provided resources. DW, VK, AK, and KS facilitated funding acquisition. AS and DW wrote the original draft. KS, AK, MS, and DW reviewed the manuscript. All authors contributed to the article and approved the submitted version.
This research was funded by Science and Engineering Research Board (SERB), Government of India in the form of Early Career Research Award (ECR/2017/001280) and from Department of Biotechnology (DBT), Government of India for project (No. BT/Ag/Network/Linseed/2019-20).
Authors acknowledge funding to project (ECR/2017/001280) from Science & Engineering Research Board (SERB), and Department of Biotechnology (DBT), Government of India for project (No. BT/Ag/Network/Linseed/2019-20). Authors thank Director, ICAR-NBPGR and Heads, DGR, DGE for research facilities and facilitation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1166728/full#supplementary-material
Supplementary Figure 1 | Chromosomal positions of QTNs/QTLs for TSW, oil, and seed yield based on earlier and present study. QTL/QTN/markers from earlier published work (Soto-Cerda et al., 2014; Kumar et al., 2015; You et al., 2018b; You and Cloutier, 2020) within 3.0 Mb proximality of QTNs identified in this study have been depicted on linseed chromosomes (You et al., 2018a; You et al., 2018b; You and Cloutier, 2020).
Ahmad, B., Zhang, S., Yao, J., Chai, S., Yadav, V., Athar, H. R., et al. (2022). Ectopic expression of VvFUS3, B3-domain transcription factor, in tomato influences seed development via affecting endoreduplication and hormones. Hortic. Plant J. 8 (3), 351–360. doi: 10.1016/j.hpj.2020.12.009
Aravind, J., Mukesh, S. S., Wankhede, D. P., Kaur, V. (2021). augmentedRCBD: analysis of augmented randomised complete block designs. R package version 0.1.5 (R package version 0.1.5). Available at: https://cran.r-project.org/package=augmentedRCBD.
Ayalew, H., Schapaugh, W., Vuong, T., Nguyen, H. T. (2022). Genome-wide association analysis identified consistent QTL for seed yield in a soybean diversity panel tested across multiple environments. Plant Genome 15, 4, e20268. doi: 10.1002/tpg2.20268
Bernardi, J., Lanubile, A., Li, Q. B., Kumar, D., Kladnik, A., Cook, S. D., et al. (2012). Impaired auxin biosynthesis in the defective endosperm18 mutant is due to mutational loss of expression in the ZmYuc1 gene encoding endosperm-specific YUCCA1 protein in maize. Plant Physiol. 160 (3), 1318–1328. doi: 10.1104/pp.112.204743
Bhattacharyya, M., Martin, C., Smith, A. (1993). The importance of starch biosynthesis in the wrinkled seed shape character of peas studied by Mendel. Plant Mol. Biol. 22 (3), 525–531. doi: 10.1007/BF00015981
Burghardt, L. T., Young, N. D., Tiffin, P. (2017). A guide to genome-wide association mapping in plants. Curr. Protoc. Plant Biol. 2, 22–38. doi: 10.1002/cppb.20041
Cao, J., Li, G., Qu, D., Li, X., Wang, Y. (2020). Into the seed: auxin controls seed development and grain yield. Int. J. Mol. Sci. 21 (5), 1662. doi: 10.3390/ijms21051662
Čeh, B., Štraus, S., Hladnik, A., Kušar, A. (2020). Impact of linseed variety, location and production year on seed yield, oil content and its composition. Agronomy 10, 1770. doi: 10.3390/agronomy10111770
Chaurasia, S., Singh, A. K., Kumar, A., Songachan, L. S., Yadav, M. C., Kumar, S., et al. (2021). Genome-wide association mapping reveals key genomic regions for physiological and yield-related traits under salinity stress in wheat (Triticum aestivum l.). Genomics 113 (5), 3198–3215. doi: 10.1016/j.ygeno.2021.07.014
Chen, J., Nolan, T. M., Ye, H., Zhang, M., Tong, H., Xin, P., et al. (2017). Arabidopsis WRKY46, WRKY54, and WRKY70 transcription factors are involved in brassinosteroid-regulated plant growth and drought responses. Plant Cell. 29 (6), 1425–1439. doi: 10.1105/tpc.17.00364
Chen, M., Zhang, B., Li, C., Kulaveerasingam, H., Chew, F. T., Yu, H. (2015). TRANSPARENT TESTA GLABRA1 regulates the accumulation of seed storage reserves in arabidopsis. Plant Physiol. 169, 391–402. doi: 10.1104/pp.15.00943
Das, M., Haberer, G., Panda, A., Das Laha, S., Ghosh, T. C., Schäffner, A. R. (2016). Expression pattern similarities support the prediction of orthologs retaining common functions after gene duplication events. Plant Physiol. 171 (4), 2343–2357. doi: 10.1104/pp.15.01207
Dharmasiri, N., Dharmasiri, S., Weijers, D., Lechner, E., Yamada, M., Hobbie, L., et al. (2005). Plant development is regulated by a family of auxin receptor f box proteins. Dev. Cell. 9, 1:109–1:119. doi: 10.1016/j.devcel.2005.05.014
Diederichsen, A., Ulrich, A. (2009). Variability in stem fibre content and its association with other characteristics in 1177 flax (Linum usitatissimum L.) genebank accessions. Ind. Crops Prod. 30, 33–39. doi: 10.1016/j.indcrop.2009.01.002
Duan, P., Xu, J., Zeng, D., Zhang, B., Geng, M., Zhang, G., et al. (2017). Natural variation in the promoter of GSE5 contributes to grain size diversity in rice. Mol. Plant 10 (5), 685–694. doi: 10.1016/j.molp.2017.03.009
Elshire, R. J., Glaubitz, J. C., Sun, Q., Poland, J. A., Kawamoto, K., Buckler, E. S., et al. (2011). A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PloS One 6, e19379. doi: 10.1371/journal.pone.0019379
FAOSTAT (2022). Available at: https://www.fao.org/faostat/en/#data/QCL (Accessed June 5, 2022).
Gao, M. J., Li, X., Huang, J., Gropp, G. M., Gjetvaj, B., Lindsay, D. L., et al. (2015). SCARECROW-LIKE15 interacts with HISTONE DEACETYLASE19 and is essential for repressing the seed maturation programme. Nat. Commun. 6, 7243. doi: 10.1038/ncomms8243
Ge, L., Yu, J., Wang, H., Luth, D., Bai, G., Wang, K., et al. (2016). Increasing seed size and quality by manipulating BIG SEEDS1 in legume species. Proc. Natl. Acad. Sci. (USA) 113, 12414–12419. doi: 10.1073/pnas.1611763113
Goyal, A., Sharma, V., Upadhyay, N., Gill, S., Sihag, M. (2014). Flax and flaxseed oil: an ancient medicine & modern functional food. J. Food Sci. Technol. 51 (9), 1633–1653. doi: 10.1007/s13197-013-1247-9
Green, A. G., Singh, S. P., Chen, Y., Dribnenki, J. C. P. (2008). “Flax,” in Compendium transgenic crop plants: transgenic oilseed crops. Eds. Kole, C., Hall, T. C. (Oxford, UK: Blackwell Publishing Ltd), 199–226. doi: 10.1002/9781405181099.k0205
Gu, Z., Eils, R., Schlesner, M. (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 18:2847–9. doi: 10.1093/bioinformatics/btw313
Gu, Y., Li, W., Jiang, H., Wang, Y., Wang, Y., Gao, H., Liu, M., et al. (2017). Differential expression of a WRKY gene between wild and cultivated soybeans correlates to seed size. J. Exp. Bot. 68, 11:2717–2729. doi: 10.1093/jxb/erx147
Guo, D., Jiang, H., Yan, W., Yang, L., Ye, J., Wang, Y., et al. (2020). Resequencing 200 flax cultivated accessions identifies candidate genes related to seed size and weight and reveals signatures of artificial selection. Front. Plant Sci. 10, 1682. doi: 10.3389/fpls.2019.01682
He, L., Xiao, J., Rashid, K. Y., Yao, Z., Li, P., Jia, G., et al. (2018). Genome-wide association studies for pasmo resistance in flax (Linum usitatissimum L.). Front. Plant Sci. 9, 1982. doi: 10.3389/fpls.2018.01982
Heidstra, R., Welch, D., Scheres, B. (2004). Mosaic analyses using marked activation and deletion clones dissect arabidopsis SCARECROW action in asymmetric cell division. Genes Dev. 18 16, 1964–1969. doi: 10.1101/gad.305504
Hirano, Y., Nakagawa, M., Suyama, T., Murase, K., Shirakawa, M., Takayama, S., et al. (2017). Structure of the SHR–SCR heterodimer bound to the BIRD/IDD transcriptional factor JKD. Nat. Plants 3, 17010. doi: 10.1038/nplants.2017.10
Hirsch, C. D., Evans, J., Buell, C. R., Hirsch, C. N. (2014). Reduced representation approaches to interrogate genome diversity in large repetitive plant genomes. Brief Funct. Genomics 134, 257–267. doi: 10.1093/bfgp/elt051
Houbaert, A., Zhang, C., Tiwari, M., Wang, K., de Marcos Serrano, A., Savatin, D.V., et al. (2018). POLAR-guided signalling complex assembly and localization drive asymmetric cell division. Nature 563, 574–578. doi: 10.1038/s41586-018-0714-x
Hu, Y., Chen, L., Wang, H., Zhang, L., Wang, F., Yu, D. (2013). Arabidopsis transcription factor WRKY8 functions antagonistically with its interacting partner VQ9 to modulate salinity stress tolerance. Plant J. 74 (5), 730–745. doi: 10.1111/tpj.12159
Hu, Z., Lu, S. J., Wang, M. J., He, H., Sun, L., Wang, H., et al. (2018). A novel QTL qTGW3 encodes the GSK3/SHAGGY-like kinase OsGSK5/OsSK41 that interacts with OsARF4 to negatively regulate grain size and weight in rice. Mol. Plant 11, 5:736–5:749. doi: 10.1016/j.molp.2018.03.005
Huang, R., Jiang, L., Zheng, J., Wang, T., Wang, H., Huang, Y., et al. (2013). Genetic bases of rice grain shape: so many genes, so little known. Trends Plant Sci. 18, 4:218–4:226. doi: 10.1016/j.tplants.2012.11.001
Ishimaru, K., Hirotsu, N., Madoka, Y., Murakami, N., Hara, N., Onodera, H., et al. (2013). Loss of function of the IAA-glucose hydrolase gene TGW6 enhances rice grain weight and increases yield. Nat. Genet. 45 (6), 707–711. doi: 10.1038/ng.2612
Kajla, P., Sharma, A., Sood, D. R. (2015). Flaxseed-a potential functional food source. J. Food Sci. Technol. 52 (4), 1857–1871. doi: 10.1007/s13197-014-1293-y
Katare, C., Saxena, S., Agrawal, S., Prasad, G. B., Bisen, P. S. (2012). Flax seed: a potential medicinal food. J. Nutr. Food Sci. 2, 1:120–1:127. doi: 10.4172/2155-9600.1000120
Kaur, V., Yadav, R., Wankhede, D. P. (2017). Linseed genetic resources for climate change intervention and future breeding. J. Appl. Nat. Sci. 9, 1112–1118. doi: 10.31018/jans.v9i2.1331
Kelley, D. R. (2018). E3 ubiquitin ligases: key regulators of hormone signaling in plants. Mol. Cell Proteomics. 17 (6), 1047–1054. doi: 10.1074/mcp.MR117.000476
Khan, M., Rozhon, W., Bigeard, J., Pflieger, D., Husar, S., Pitzschke, A., et al. (2013). Brassinosteroid-regulated GSK3/Shaggy-like kinases phosphorylate mitogen-activated protein (MAP) kinase kinases, which control stomata development in Arabidopsis thaliana. J. Biol. Chem. 288, 11:7519–7527. doi: 10.1074/jbc.M112.384453
Kim, T. W., Michniewicz, M., Bergmann, D. C., Wang, Z. Y. (2012). Brassinosteroid regulates stomatal development by GSK3-mediated inhibition of a MAPK pathway. Nature 482, 7385:419–22. doi: 10.1038/nature10794
Kraft, J. M., Kommedahl, T., Linck, A. L. (1963). Histological study of malformation in flaxseed after exposure to 31°CC. Bot. Gaz 125, 367–371. doi: 10.1086/336221
Kujur, A., Bajaj, D., Upadhyaya, H., Das, S., Ranjan, R., Shree, T., et al. (2015). A genome-wide SNP scan accelerates trait-regulatory genomic loci identification in chickpea. Sci. Rep. 5, 11166. doi: 10.1038/srep11166
Kumar, S., Kumari, J., Bhusal, N., Pradhan, A. K., Budhlakoti, N., Mishra, D. C., et al. (2020). Genome-wide association study reveals genomic regions associated with ten agronomical traits in wheat under late-sown conditions. Front. Plant Sci. 11. doi: 10.3389/fpls.2020.549743
Kumar, S., You, F. M., Duguid, S., Booker, H., Rowland, G., Cloutier, S. (2015). QTL for fatty acid composition and yield in linseed (Linum usitatissimum L.). Theor. Appl. Genet. 128, 965–984. doi: 10.1007/s00122-015-2483-3
Li, J., Nam, K. H. (2002). Regulation of brassinosteroid signaling by a GSK3/SHAGGY-like kinase. Science 295, 5558:1299–301. doi: 10.1126/science.1065769
Li, S. Z., Sun, L. L., Ma, Y. P., Ma, Y. P., Xu, Y. D., You-Hao, E. (2014). Effects of climate change on flax development and yield in guyuan of ningxia, Northwest China. Ying Yong Sheng Tai Xue Bao. 25 (10), 2892–2900.
Liu, J., Chen, J., Zheng, X., Wu, F., Lin, Q., Heng, Y., et al. (2017). GW5 acts in the brassinosteroid signaling pathway to regulate grain width and weight in rice. Nat. Plants 3, 17043. doi: 10.1038/nplants.2017.43
Liu, J., Hua, W., Hu, Z., Yang, H., Zhang, L., Li, R., et al. (2015a). Natural variation in ARF18 gene simultaneously affects seed weight and silique length in polyploid rapeseed. Proc. Natl. Acad. Sci. U.S.A. 112 37, E5123–E5132. doi: 10.1073/pnas.1502160112
Liu, L., Tong, H., Xiao, Y., Che, R., Xu, F., Hu, B., et al. (2015b). Activation of big Grain1 significantly improves grain size by regulating auxin transport in rice. Proc. Natl. Acad. Sci. U.S.A. 112, 11102–11107. doi: 10.1073/pnas.1512748112
Locascio, A., Roig-Villanova, I., Bernardi, J., Varotto, S. (2014). Current perspectives on the hormonal control of seed development in arabidopsis and maize: a focus on auxin. Front. Plant Sci. 5, 412. doi: 10.3389/fpls.2014.00412
Lu, K., Peng, L., Zhang, C., Lu, J., Lu, J., Yang, B., Xiao, Z., et al. (2017). Genome-wide association and transcriptome analyses reveal candidate genes underlying yield-determining traits in brassica napus. Front. Plant Sci. 8, 206. doi: 10.3389/fpls.2017.00206
Luo, M., Dennis, E. S., Berger, F., et al. (2005). MINISEED3 (MINI3), a WRKY family gene, and HAIKU2 (IKU2), a leucine-rich repeat (LRR) KINASE gene, are regulators of seed size in arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 102, 17531–17536. doi: 10.1073/pnas.0508418102
Mian, M., Bailey, M., Tamulonis, J., Shipe, E.R., Carter, T.E., Jr, Parrott, W.A., et al. (1996). Molecular markers associated with seed weight in two soybean populations. Theor. Appl. Genet. 93, 7:1011–7:1016. doi: 10.1007/BF00230118
Miao, Y., Zentgraf, U. (2010). A HECT E3 ubiquitin ligase negatively regulates arabidopsis leaf senescence through degradation of the transcription factor WRKY53. Plant J. 63, 2:179–2:188. doi: 10.1111/j.1365-313X.2010.04233.x
Milner, S. G., Jost, M., Taketa, S., Mazón, E.R., Himmelbach, A., Oppermann, M., et al. (2019). Genebank genomics highlights the diversity of a global barley collection. Nat. Genet. 51, 319–326. doi: 10.1038/s41588-018-0266-x
Missaoui, K., Gonzalez-Klein, Z., Pazos-Castro, D., Hernandez-Ramirez, G., Garrido-Arandia, M., Brini, F., et al. (2022). Plant non-specific lipid transfer proteins: an overview. Plant Physiol. Biochem. 171, 115–127. doi: 10.1016/j.plaphy.2021.12.026
Mohammadi-Sartang, M., Mazloom, Z., Raeisi-Dehkordi, H., Barati-Boldaji, R., Bellissimo, N., Totosy de Zepetnek, J.O., et al. (2017). The effect of flaxseed supplementation on body weight and body composition: a systematic review and meta-analysis of 45 randomized placebo-controlled trials. Obes. Rev. 18, 9:1096–9:1107. doi: 10.1111/obr.12550
Mu, J., Tan, H., Zheng, Q., Fu, F., Chen, K., Shen, C., Chen, H., et al. (2008). LEAFY COTYLEDON1 is a key regulator of fatty acid biosynthesis in arabidopsis. Plant Physiol. 148 (2), 1042–1054. doi: 10.1104/pp.108.126342
Na, G., Aryal, N., Fatihi, A., Kang, J., Lu, C. (2018). Seed-specific suppression of ADP-glucose pyrophosphorylase in Camelina sativa increases seed size and weight. Biotechnol. Biofuels. 11, 330. doi: 10.1186/s13068-018-1334-2
Ngu, M. S., Thomson, M. J., Bhuiyan, M. A., Ho, C., Wickneswari, R. (2014). Fine mapping of a grain weight quantitative trait locus, qGW6, using near isogenic lines derived from Oryza rufipogon IRGC105491 and Oryza sativa cultivar MR219. Genet. Mol. Res. 13, 4:9477–4:9488. doi: 10.4238/2014
Niu, Y., Chen, T., Wang, C., Chen, K., Shen, C., Chen, H., et al. (2021). Identification and allele mining of new candidate genes underlying rice grain weight and grain shape by genome-wide association study. BMC Genomics 22, 1:602. doi: 10.1186/s12864-021-07901-x
Pan, A., Yu, D., Demark-Wahnefried, W., Franco, O. H., Lin, X. (2009). Meta-analysis of the effects of flaxseed interventions on blood lipids. Am. J. Clin. Nutr. 90 (2), 288–297. doi: 10.3945/ajcn.2009.27469
Qi, Z., Song, J., Zhang, K., Liu, S., Liu, S., Tian, X., Wang, Y., et al. (2020). Identification of QTNs controlling 100-seed weight in soybean using multilocus genome-wide association studies. Front. Genet. 11, 689. doi: 10.3389/fgene.2020.00689
Raboanatahiry, N., Chao, H., He, J., Li, H., Yin, Y., Li, M. (2022). Construction of a quantitative genomic map, identification and expression analysis of candidate genes for agronomic and disease-related traits in Brassica napus. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.862363
Ragupathy, R., Rathinavelu, R., Cloutier, S. (2011). Physical mapping and BAC-end sequence analysis provide initial insights into the flax (Linum usitatissimum L.) genome. BMC Genom. 12, 217. doi: 10.1186/1471-2164-12-217
Ren, W. L., Wen, Y. J., Dunwell, J. M., Zhang, Y. M. (2018). pKWmEB: integration of kruskal-Wallis test with empirical bayes under polygenic background control for multi-locus genome-wide association study. Heredity 120, 208–218. doi: 10.1038/s41437-017-0007-4
Roscoe, T. T., Guilleminot, J., Bessoule, J. J., Berger, F., Devic, M. (2015). Complementation of seed maturation phenotypes by ectopic expression of ABSCISIC ACID INSENSITIVE3, FUSCA3 and LEAFY COTYLEDON2 in arabidopsis. Plant Cell Physiol. 56, 1215–1228. doi: 10.1093/pcp/pcv049
Sabatini, S., Heidstra, R., Wildwater, M., Scheres, B. (2003). SCARECROW is involved in positioning the stem cell niche in the arabidopsis root meristem. Genes Dev. 17, 3:354–3:358. doi: 10.1101/gad.252503
Santos-Mendoza, M., Dubreucq, B., Baud, S., Parcy, F., Lepiniec, L. (2008). Deciphering gene regulatory networks that control seed development and maturation in arabidopsis. Plant J. 54, 608–620. doi: 10.1111/j.1365-313X.2008.03461.x
Saroha, A., Pal, D., Gomashe, S. S., Akash, Kaur, V., Ujjainwal, S., et al. (2022a). Identification of QTNs associated with flowering time, maturity, and plant height traits in Linum usitatissimum L. using genome-wide association study. Front. Genet. 13. doi: 10.3389/fgene.2022.811924
Saroha, A., Pal, D., Kaur, V., Kumar, S., Bartwal, A., Aravind, J., et al. (2022b). Agro-morphological variability and genetic diversity in linseed (Linum usitatissimum L.) germplasm accessions with emphasis on flowering and maturity time. Genet. Resour Crop Evol. 69, 315–333. doi: 10.1007/s10722-021-01231-3
Sena, G., Jung, J. W., Benfey, P. N. (2004). A broad competence to respond to SHORT ROOT revealed by tissue-specific ectopic expression. Development 131, 12:2817–26. doi: 10.1242/dev.01144
Shi, J., Li, R., Qiu, D., Jiang, C., Long, Y., Morgan, C., et al. (2009). Unraveling the complex trait of crop yield with quantitative trait loci mapping in Brassica napus. Genetics 182, 851–861. doi: 10.1534/genetics.109.101642
Shi, L. L., Song, J. R., Guo, C. C., Wang, B., Guan, Z., Yang, P., et al. (2019). A CACTA-like transposable element in the upstream region of BnaA9.CYP78A9 acts as an enhancer to increase silique length and seed weight in rapeseed. Plant J. 98, 524–539. doi: 10.1111/tpj.14236
Shomura, A., Izawa, T., Ebana, K., Ebitani, T., Kanegae, H., Konishi, S., et al. (2008). Deletion in a gene associated with grain size increased yields during rice domestication. Nat. Genet. 40, 1023–1028. doi: 10.1038/ng.169
Singh, N., Agarwal, N., Yadav, H. K. (2019). Genome-wide SNP-based diversity analysis and association mapping in linseed (Linum usitatissimum L.). Euphytica 215, 139. doi: 10.1007/s10681-019-2462-x
Song, X. J. (2017). Crop seed size: BR matters. Mol. Plant 10, 5:668–5:669. doi: 10.1016/j.molp.2017.04.007
Song, S., You, J., Shi, L., Sheng, C., Zhou, W., Dossou, S. S. K., et al. (2021). Genome-wide analysis of nsLTP gene family and identification of SiLTPs contributing to high oil accumulation in sesame (Sesamum indicum l.). Int. J. Mol. Sci. 22 (10), 5291. doi: 10.3390/ijms22105291
Soto-Cerda, B. J., Aravena, G., Cloutier, S. (2021). Genetic dissection of flowering time in flax (Linum usitatissimum L.) through single and multi-locus genome-wide association studies. Mol. Genet. Genomics 296 (4), 877–891. doi: 10.1007/s00438-021-01785-y
Soto-Cerda, B. J., Cloutier, S., Quian, R., Gajardo, H. A., Olivos, M., You, F. M. (2018). Genome-wide association analysis of mucilage and hull content in flax (Linum usitatissimum L.) seeds. Int. J. Mol. Sci. 19, 2870. doi: 10.3390/ijms19102870
Soto-Cerda, B. J., Duguid, S., Booker, H., Rowland, G., Diederichsen, A., Cloutier, S. (2014). Genomic regions underlying agronomic traits in linseed (Linum usitatissimum L.) as revealed by association mapping. J. Integr. Plant Biol. 56, 75–87. doi: 10.1111/jipb.12118
Tamba, C. L., Ni, Y. L., Zhang, Y. M. (2017). Iterative sure independence screening EM-Bayesian LASSO algorithm for multilocus genome-wide association studies. PloS ComputBiol. 13, e1005357. doi: 10.1371/journal.pcbi.1005357
Tamba, C. L., Zhang, Y. M. (2018). A fast mrMLM algorithm for multi-locus genome-wide association studies. bioRxiv, p.341784. doi: 10.1101/341784
Tao, Y., Zhao, X., Wang, X., Hathorn, A., Hunt, C., Cruickshank, A.W., et al. (2020). Large-Scale GWAS in sorghum reveals common genetic control of grain size among cereals. Plant Biotechnol. J. 18 (4), 1093–1105. doi: 10.1101/710459
Vavilov, N. I. (1951). The origin, variation, immunity and breeding of cultivated plants. Chronica Botanica 13, 1–366. doi: 10.2134/agronj1952.00021962004400020016x
Wang, S. B., Feng, J. Y., Ren, W. L., Huang, B., Zhou, L., Wen, Y.J., et al. (2016). Improving power and accuracy of genome-wide association studies via multi-locus mixed linear model methodology. Sci. Rep. 6, 19444. doi: 10.1038/srep19444
Wang, Z., Hobson, N., Galindo, L., Zhu, S., Shi, D., McDill, J., et al. (2012). The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. Plant J. 72, 461–473. doi: 10.1111/j.1365-313X.2012.05093.x
Wang, J., Wang, R., Mao, X., Zhang, J., Liu, Y., Xie, Q., et al. (2020). RING finger ubiquitin E3 ligase gene TaSDIR1-4A contributes to determination of grain size in common wheat. J. Exp. Bot. 19, 5377–5388. doi: 10.1093/jxb/eraa271
Wen, Y. J., Zhang, H., Ni, Y. L., Huang, B., Zhang, J., Feng, J. Y., et al. (2018). Methodological implementation of mixed linear models in multi-locus genome-wide association studies. Brief Bioinform. 19, 700–712. doi: 10.1093/bib/bbw145
Weng, J., Gu, S., Wan, X., Gao, H., Guo, T., Su, N., et al. (2008). Isolation and initial characterization of GW5, a major QTL associated with rice grain width and weight. Cell Res. 18, 1199–1209. doi: 10.1038/cr.2008.307
Wu, Y., Fu, Y., Zhao, S., Gu, P., Zhu, Z., Sun, C., et al. (2016). CLUSTERED PRIMARY BRANCH1, a new allele of DWARF11, controls panicle architecture and seed size in rice. Plant Biotechnol. J. 14, 377–386. doi: 10.1111/pbi.12391
Wu, J., Zhao, Q., Zhang, L., Li, S., Ma, Y., Pan, L., et al. (2018). QTL mapping of fiber-related traits based on a high-density genetic map in flax (Linum usitatissimum L.). Front. Plant Sci. 9, 885. doi: 10.3389/fpls.2018.00885
Wysocka-Diller, J. W., Helariutta, Y., Fukaki, H., Malamy, J. E., Benfey, P. N. (2000). Molecular analysis of SCARECROW function reveals a radial patterning mechanism common to root and shoot. Development 127, 3:595–3:603. doi: 10.1242/dev.127.3.595
Xia, T., Li, N., Dumenil, J., Li, J., Kamenski, A., Bevan, M. W., et al. (2013). The ubiquitin receptor DA1 interacts with the E3 ubiquitin ligase DA2 to regulate seed and organ size in arabidopsis. Plant Cell 25, 3347–3359. doi: 10.1105/tpc.113.115063
Xie, D., Dai, Z., Yang, Z., Sun, J., Zhao, D., Yang, X. (2018a). Genome-wide association study identifying candidate genes influencing important agronomic traits of flax (Linum usitatissimum L.) using SLAF-seq. Front. Plant Sci. 8, 2232. doi: 10.3389/fpls.2017.02232
Xie, D., Dai, Z., Yang, Z., Tang, Q., Sun, J., Yang, X., et al. (2018b). Genomic variations and association study of agronomic traits in flax. BMC Genom 19, 512. doi: 10.1186/s12864-018-4899-z
Xiong, M., Chu, L., Li, Q., Yu, J., Yang, Y., Zhou, P., et al. (2021). Brassinosteroid and gibberellin coordinate rice seed germination and embryo growth by regulating glutelin mobilization. Crop J. 9(5), 1039–1048. doi: 10.1016/j.cj.2020.11.006
Xu, F. Q., Xue, H. W. (2019). The ubiquitin-proteasome system in plant responses to environments. Plant Cell Environ. 42 (10), 2931–2944. doi: 10.1111/pce.13633
Yadav, B., Kaur, V., Narayan, O. P., Yadav, S. K., Kumar, A., Wankhede, D. P. (2022). Integrated omics approaches for flax improvement under abiotic and biotic stress: current status and future prospects. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.931275
You, F. M., Cloutier, S. (2020). Mapping quantitative trait loci onto chromosome-scale pseudomolecules in flax. Methods Protoc. 3, 28. doi: 10.3390/mps3020028
You, F. M., Xiao, J., Li, P., Yao, Z., Jia, G., He, L., et al. (2018a). Chromosome-scale pseudomolecules refined by optical, physical, and genetic maps in flax. Plant J. 95, 371–384. doi: 10.1111/tpj.13944
You, F. M., Xiao, J., Li, P., Yao, Z., Jia, G., He, L., et al. (2018b). Genome-wide association study and selection signatures detect genomic regions associated with seed yield and oil quality in flax. Int. J. Mol. Sci. 19, 2303. doi: 10.3390/ijms19082303
Zhang, J., Feng, J. Y., Ni, Y. L., Wen, Y.J., Niu, Y., Tamba, C.L., et al. (2017). pLARmEB: integration of least angle regression with empirical bayes for multilocus genome-wide association studies. Heredity 118, 517–524. doi: 10.1038/hdy.2017.8
Zhang, X., Guan, Z., Wang, L., Fu, J., Zhang, Y., Li, Z., et al. (2020). Combined GWAS and QTL analysis for dissecting the genetic architecture of kernel test weight in maize. Mol. Genet. Genomics 295 (2), 409–420. doi: 10.1007/s00438-019-01631-2
Zhang, D., Hao, C., Wang, L., Zhang, X. (2012). Identifying loci influencing grain number by microsatellite screening in bread wheat (Triticum aestivum l.). Planta 236, 1507–1517. doi: 10.1007/s00425-012-1708-9
Zhang, Y. M., Jia, Z., Dunwell, J. M. (2019). Editorial: the applications of new multi-locus GWAS methodologies in the genetic dissection of complex traits. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.00100
Keywords: linseed, flaxseed, seed weight, genome-wide association studies, quantitative trait nucleotides, candidate genes
Citation: Saroha A, Gomashe SS, Kaur V, Pal D, Ujjainwal S, Aravind J, Singh M, Rajkumar S, Singh K, Kumar A and Wankhede DP (2023) Genetic dissection of thousand-seed weight in linseed (Linum usitatissimum L.) using multi-locus genome-wide association study. Front. Plant Sci. 14:1166728. doi: 10.3389/fpls.2023.1166728
Received: 15 February 2023; Accepted: 08 May 2023;
Published: 02 June 2023.
Edited by:
Harmeet Kaur, Indian Council of Agricultural Research, IndiaReviewed by:
Mahendra Vikram Singh Rajawat, Prabhat Fertilizers & Chemical Works, IndiaCopyright © 2023 Saroha, Gomashe, Kaur, Pal, Ujjainwal, Aravind, Singh, Rajkumar, Singh, Kumar and Wankhede. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dhammaprakash Pandhari Wankhede, ZC53YW5raGVkZUBpY2FyLmdvdi5pbg==
†Present address: Kuldeep Singh, International Crops Research Institute for the Semi-Arid Tropics (ICRISAT), Patancheru, Hyderabad, Telangana, India
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.