 Min Wang1,2†
Min Wang1,2† Songguang Yang1,2†
Songguang Yang1,2† Wei Liu1,2Zhenqiang Cao1,2Lin Chen1,2Wenrui Liu1,2Dasen Xie1,2
Wei Liu1,2Zhenqiang Cao1,2Lin Chen1,2Wenrui Liu1,2Dasen Xie1,2 Jinqiang Yan1,2
Jinqiang Yan1,2 Biao Jiang1,2*Qingwu Peng1*
Biao Jiang1,2*Qingwu Peng1*- 1Vegetable Research Institute, Guangdong Academy of Agricultural Sciences, Guangzhou, China
- 2Guangdong Key Laboratory for New Technology Research of Vegetables, Guangdong Academy of Agricultural Sciences, Guangzhou, China
Gynoecy demonstrates an earlier production of hybrids and a higher yield and improves the efficiency of hybrid seed production. Therefore, the utilization of gynoecy is beneficial for the genetic breeding of chieh-qua. However, little knowledge of gynoecious-related genes in chieh-qua has been reported until now. Here, we used an F2 population from the cross between the gynoecious line ‘A36’ and the monoecious line ‘SX’ for genetic mapping and revealed that chieh-qua gynoecy was regulated by a single recessive gene. We fine-mapped it into a 530-kb region flanked by the markers Indel-3 and KASP145 on Chr.8, which harbors eight candidate genes. One of the candidate genes, Bhi08G000345, encoding networked protein 4 (CqNET4), contained a non-synonymous SNP resulting in the amino acid substitution of isoleucine (ATA; I) to methionine (ATG; M). CqNET4 was prominently expressed in the female flower, and only three genes related to ethylene synthesis were significantly expressed between ‘A36’ and ‘SX.’ The results presented here provide support for the CqNET4 as the most likely candidate gene for chieh-qua gynoecy, which differed from the reported gynoecious genes.
Introduction
Sex differentiation in higher plants is often determined by sex chromosomes and sex-determining genes (Lai et al., 2017), and the regulation of unisexual flower development has been the focus of plant sex determination (Bai et al., 2004). According to the distribution or proportion of three flower types (female flowers, male flowers, and complete flowers) in a plant, the cucurbit plant can be classified into six phenotypes, namely, gynoecy, monoecy, subgynoecy, androecy, andromonoecy, and hermaphrodites (Li et al., 2019). Gynoecy has been the focus of research on the sexual type of cucurbits as it directly affects the yield and hybrid seed purity (Boualem et al., 2009; Boualem et al., 2015; Zhang et al., 2021). Melon and cucumber have been excellent models for studying the molecular mechanisms of gynoecism development. The recessive gene CmWIP1 (encoding the C2H2-type zinc finger protein transcription factor) (Boualem et al., 2015) and ethylene synthesis-related genes (Zhang et al., 2021) are involved in gynoecism regulation.
The expression of the CmWIP1 gene in melon could inhibit carpel development and the expression of downstream CmACS7, thus promoting the development of the male flower (Martin et al., 2009; Boualem et al., 2015; Boualem et al., 2016). Gene editing of CsWIP1 in cucumber exerts a gynoecious phenotype (Chen et al., 2016; Hu et al., 2017). The chromosomal translocation of the ClWIP1 gene in watermelon leads to an insertion mutation and results in gynoecy (Zhang et al., 2020). A recent study demonstrates that CmWIP1 in melon interacts with the co-suppressor protein TPL and binds to the promoter of CRC (carpel development gene CRABS CLAW), which deacetylates the CRC genomic protein to suppress its expression, finally interfering with the floral meristem determination in the carpel primordium and promoting the male flower development. However, mutation of CmWIP1 can promote the expression of CRC and also promote the development of the carpel and the expression of the CmACS7 gene, thus producing female flowers (Zhang et al., 2021).
Except for the negative regulation of the CsWIP1 gene in cucumber, there is also a dominant F gene. A recent study identifies that the F gene is ACS1G rather than the MYB transcription factor in the repeat sequence. Due to the structural variation of the genome, ACS1G acquired a promoter and a new expression pattern, which is different from that of ACS1 in monoecious materials (Li et al., 2020; Zhang et al., 2021). In addition, CsACS1G acts upstream of CsWIP1 and inhibits its expression by producing ethylene, resulting in a gynoecy phenotype (Zhang et al., 2021).
Chieh-qua (Benincasa hispida Cogn. var. chieh-qua How), a wax gourd variety, is an important fruit vegetable widely cultivated in South China and Southeast Asia that is rich in vitamins, propanol diacid, and other nutrients (He et al., 2007). Like other cucurbit crops, chieh-qua also has several sexual flower types including gynoecism, which is important in breeding and hybrid seed production. However, the genetic studies on gynoecy in chieh-qua are very limited. Since the release of the reference genome of wax gourd (Xie et al., 2019), it has provided us with a great opportunity to explore the genetic basis of chieh-qua, such as the trait of gynoecy.
In this study, we fine-mapped the gynoecy trait using an F2 population derived from the cross between ‘A36’ and ‘SX.’ Candidate genes in the 530-kb interval were sequenced and further assessed by the qRT-PCR assay. Combining sequencing and expression analysis, we found that the candidate gene related to chieh-qua gynoecy was different from the homologous gynoecious genes WIP1 and ACS1G. Collectively, our findings not only provided practical markers for molecular gynoecious breeding of chieh-qua but also enriched the cucurbit sex differentiation theory.
Materials and methods
Plant materials
Two chieh-qua inbred lines, ‘A36’ and ‘SX,’ were used for phenotypic characterization and segregating populations. ‘A36’ with a gynoecious phenotype was a homozygous inbred line derived from the native variety ‘Jiangxinjie’ in Guangdong Province. ‘SX’ with a monoecious trait was also a high-generation inbred line, derived from a cross of ‘Cuiyu’ and ‘Guiyou’ chieh-qua. F1 plants were obtained by crossing ‘A36’ (P1) and ‘SX’ (P2), and the F2 population was obtained by self-crossing F1 plants. Parents and F1 individuals were planted in the spring of 2020. A total of 308 and 196 F2 individuals were planted in the spring and autumn of 2020, respectively, and a total of 2,120 F2 individuals were planted in the autumn of 2021. For phenotyping, individuals producing only female flowers in the main stem were considered gynoecious, while individuals producing both male and female flowers were monoecious. All plants were grown in the Baiyun study base of Guangdong Academy of Agricultural Sciences (Guangzhou, China) at 23.4°N (latitude) and 113.4°E (longitude).
Generation of the whole genome resequencing data
Young leaves of parents and F2 individuals were used to extract genomic DNAs using cetyltrimethylammonium bromide (CTAB) (Healey et al., 2014). We constructed the gynoecium pool (G-pool) and the monoecism pool (M-pool) by mixing an equal amount of DNAs from 30 gynoecious and 30 monecious F2 plants, respectively. Then, the DNA of parents and two DNA pools were sequenced using the Illumina sequencing platform by Genedenovo Biotechnology Co., Ltd. (Guangzhou, China). To ensure the correctness of the resequencing results, the depth of sequencing data in the parent pool was larger than 10×, and that in the G-bulk and M-bulk was larger than 50×.
Alignment and analysis of BSA data
In order to align clean reads from each sample, we used the software Burrows-Wheeler Aligner (BWA) (Li and Durbin, 2010; Abuín et al., 2015) against the public reference genome (Xie et al., 2019), and alignment files were converted to SAM/BAM files using SAMtools (Li et al., 2009). Variant calling was carried out for all the samples using GATK’s UnifiedGenotyper (McKenna et al., 2010). Single-nucleotide polymorphisms (SNPs) and indels were filtered using GATK’s Variant Filtration with proper standards (-Window 4, -filter “QD < 4.0||FS > 60.0|| MQ < 40.0”, -G_filter “GQ < 20”). Next, to determine the physical positions of each obtained variant, we used the software tool ANNOVAR (Wang et al., 2010a) to align and annotate SNPs or indels, and the SNP index and delta (SNP index) were calculated and analyzed to identify the candidate region for gynoecium in chieh-qua (Takagi et al., 2013; Lu et al., 2014). Furthermore,the original sequencing data are being uploaded to the NCBI database with the BioProject ID:PRJNA941947.
Development of molecular markers for fine mapping
Based on the primary mapping of BSA-seq, simple sequence repeat (SSR) markers were used for gene linkage analysis. New indel and Kompetitive Allele-Specific PCR (KASP) markers were developed based on the DNA sequence polymorphisms from the parents’ resequencing data and B227 reference genome (http://cucurbitgenomics.org/organism/22). The primer design was performed using the Primer Premier 5 (Premier Biosoft, Palo Alto, CA, USA). All primers were synthesized commercially, and all newly developed markers were firstly screened for polymorphism between the two parental lines. The primer information is listed in Supplemental Table 1. The obtained polymorphic markers were applied to the 2,120 F2 individuals. Based on the confirmed recombination events and sex phenotype of the detected recombinants, the initial mapping region was further narrowed down.
Amplification of candidate genes
Total RNA was extracted from the terminal buds of A36 and SX. The cDNA was synthesized from total RNA using the PrimeScript RT reagent kit and gDNA Eraser (TaKaRa, Dalian, China) according to the manufacturer’s instructions. The coding sequences of eight candidate genes (Bhi08G000338–Bhi08G000345) in the interval were amplified using the PCR primers listed in Table S2. The PCR amplifications were then performed using the PrimeSTAR® Max DNA Polymerase (Takara Bio, Shiga, Japan) and respectively ligated into pEASY®-Blunt Zero Cloning Vector (TransGen Biotech, Beijing, China) according to the manufacturer’s instructions. We identified the positive clones using colony PCR and analyzed the Sanger sequencing results using DNAMAN software to confirm the candidate gene.
Phylogenetic analysis
In order to analyze the evolutionary relationship between CqNET4 and its homologs, we performed a Basic Local Alignment Search Tool (BLAST) (https://blast.ncbi.nlm.nih.gov/Blast.cgi) to explore the homolog sequences from the NCBI database using the CqNET4 protein sequence as the query. The obtained amino acid sequences were aligned using ClustalW, and a phylogenetic tree was built using the neighbor-joining method with 1,000 bootstrap replications in the MEGA X software.
Subcellular localization
The full-length coding sequence (CDS) of CqNET4 was cloned into pCAMBIA1305 vector to create a 35S:GFP-CqNET4 fusion construct, which was subsequently transformed into Agrobacterium tumefaciens strain EHA105. The constructs were infiltrated into Nicotiana benthamiana leaves, and the mesophyll protoplasts were obtained by polyethylene glycol-mediated infiltration. Fluorescence signals were visualized using an LSM880 laser scanning confocal microscope (Carl Zeiss, Jena, Germany).
qRT-PCR analysis
RNA was prepared according to the instructions of the TRIzol® reagent (TaKaRa, Japan) and purified and concentrated using an RNeasy MinElute Cleanup Kit (Qiagen, Germany). Quantitative real-time PCR (qRT-PCR) (20 μl) was performed with 0.5 μl of cDNA, 0.2 μM of primer mix, and SYBR Premix Ex Taq Kit (TaKaRa, Japan). We used the 7500 Real-Time PCR System (Applied Biosystems, America) to monitor the gene expression by fluorescence at the end of each cycle. For each plate, three technical replicates and three biological replicates were generated. The candidate gene expression was evaluated by the 2−ΔΔCt method (Livak and Schmittgen, 2001). The significant differences were detected by IBM SPSS Statistics 20 (Student’s t-test). The primer sequences are listed in Supplemental Table 3.
Results
Genetic inheritance of gynoecious trait
To determine the inheritance pattern of the gynoecious trait, the gynoecious parent A36 was crossed to the monoecious parent SX to construct a segregating population. When the individual plants of parents and the F1 and F2 populations start to bear flowers, the sexual flower type (female or male flower) on every node was determined. The images of typical flower types for the two parental lines are shown in Figure 1. Generally, every node in the main stem of A36 was a female flower, whereas both female and male flowers existed in the SX main stem (Figure 1A). Furthermore, the color of the stigma of female flowers differed, with A36 having white stigmas and SX having green stigmas (Figures 1B–D). Like the phenotype of SX, all F1 individuals produced both female and male flowers. The segregation in the F2 population was 235 plants with monoecious phenotype and 73 with gynoecious phenotype (χ23:1 = 0.28; P = 0.6) in the spring of 2020 and 144 monoecious plants and 52 gynoecious plants (χ23:1 = 0.24; P = 0.62) in the autumn of 2020 (Table 1), indicating that the chieh-qua gynoecious trait was caused by a single recessive gene.

Figure 1 Characteristics of chieh-qua flower types. (A) The typical images of flower types and fruits of A36 and SX. (B, C) The female flowers of gynoecious (A36) and monoecious (SX) lines. (D) The stigma color of A36 and SX female flowers. ♀ represents a female flower; ♂ represents a male flower.
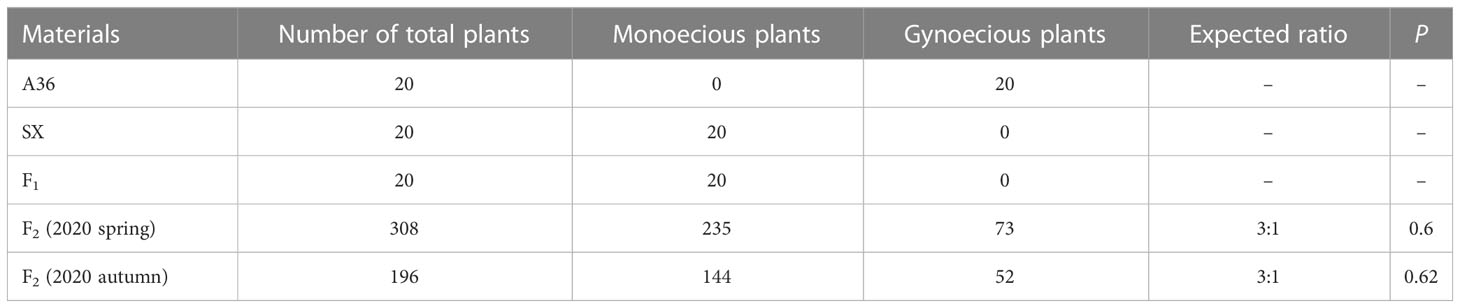
Table 1 Genetic analysis of chieh-qua gynoecy using the F2 population derived from A36 and SX.
Fine mapping of the candidate gene
To determine the chromosomal location of the gene controlling the gynoecious trait, DNA pools from 30 homozygous gynoecious (G-pool) and 30 homozygous monoecious (M-pool) F2 plants were created and genotyped using the Illumina high-throughput sequencing platform. We finally obtained 207,126,558 and 198,796,506 reads from the G-pool and the M-pool, respectively. These reads were aligned to the B227 reference genome (Xie et al., 2019), and the average paired mapped reads were 175,114,378 with 93.8% align ratio (Supplemental Table 4). Next, the SNP index graphs of the G-pool and the M-pool (Supplemental Figure 1) were constructed by computing the average SNP index, and the delta SNP index was computed and plotted against the genome locations based on the information of the SNP index of these two extreme pools. The results showed that the region on Chr.8 from 13.08 to 16.06 Mb, with a total length of 2.98 Mb (Supplemental Figure 1; Figure 2A), was the most likely interval, indicating the presence of a significant locus governing the related gynoecy characteristic.
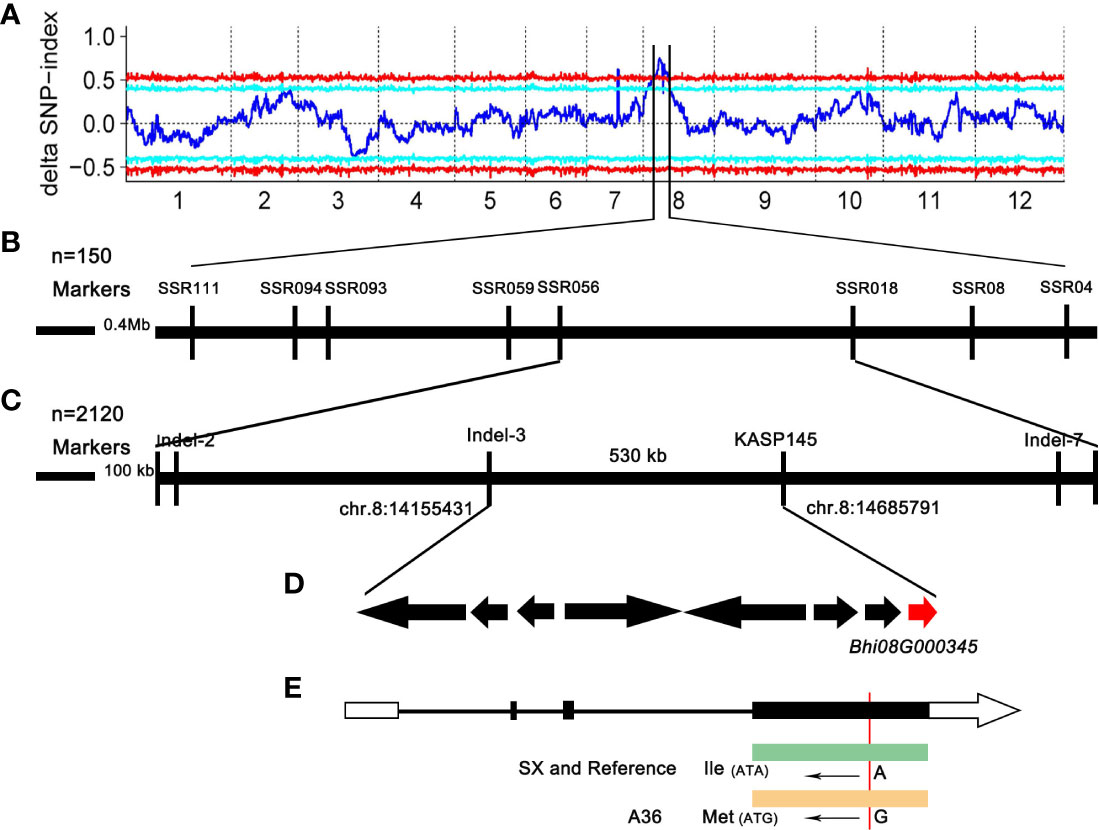
Figure 2 Genetic mapping of the gynoecious gene in chieh-qua. (A) Delta SNP index distribution on chieh-qua chromosomes. The abscissa is the chromosome name, the blue line represents the fitted ED value, and the red line represents the significance association threshold of ±0.5. The higher the ED value, the better the association effect of this point. (B, C) Fine mapping of the gynoecious gene. (D) Candidate genes in the fine mapping region. (E) The candidate gene structure and comparison of the base sequence in ‘A36’ and ‘SX.’ The white boxes, black boxes, and solid line represent 5′ and 3′ UTR, exons, and introns, respectively.
Based on the reference genome, we designed 45 SSR markers in the candidate region for screening the polymorphism between A36 and SX. In the candidate interval, eight polymorphic SSR markers were identified and mapped. Combining the linkage analysis of 150 F2 individuals revealed that SSR056 and SSR018 were the two closest flanking markers with 1.5 Mb physical length (Figure 2B). In order to fine map the candidate locus, 2,120 F2 plants were firstly screened using the major flanking SSR056 and SSR018 markers to obtain 25 recombinant plants. In addition, 20 indel and 18 KASP markers were further developed to genotype the recombinant plants. After genotyping the F2 plants, only three indel and one KASP markers were polymorphic with a low polymorphism rate. Finally, the candidate gene was positioned between 14.15 and 14.68Mb in an approximately 530-kb region bordered by the markers Indel-3 and KASP145 (Figure 2C).
According to the reference genome sequence (http://cucurbitgenomics.org/organism/22), the genomic candidate area contained eight protein-coding genes (Bhi08G000338–Bhi08G000345) (Figure 2D), which encoded the MYB transcription factor, secretory protein HlyD family protein, NETWORKED 4 (NET4) protein, and so on (Figure 2D; Table 2). In order to compare the eight annotated genes in the candidate interval, these candidate genes from parents were amplified by PCR. Sequencing data showed that except Bhi08G000345, CDS sequence alignment showed no nucleotide differences in the other genes between ‘A36’ and ‘SX,’ indicating that the other seven genes were not associated with gynoecy. Indeed, sequence analysis of Bhi08G000345 exerted an SNP base substitution (A→G) in the third exon, causing an L (SX: Lle) to M (A36: Met) amino acid alternation (Figure 2E). Moreover, we found that Bhi08G000345 shared a sequence homology with Arabidopsis thaliana (AT5G58320), which encoded the protein NETWORKED 4. Therefore, Bhi08G000345 (denoted CqNET4) is the most possible gene governing gynoecy in chieh-qua.
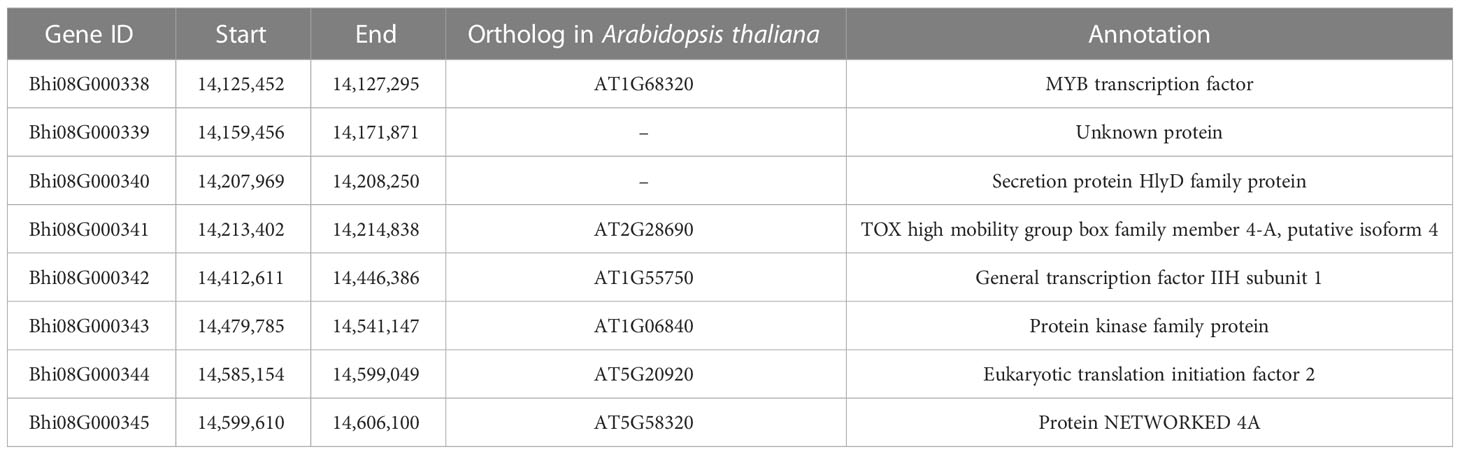
Table 2 The candidate genes in the mapping interval.
Development of molecular markers
In order to identify the molecular markers linked to chieh-qua gynoecy, the DNA of 13 F2 plants and 10 monoecious varieties was amplified using Indel-3 primers. We found that the accuracy of the marker was over 93% (Figure 3A). Next, we used the KASP145 marker to analyze the DNA of 50 germplasm chieh-qua materials consisting of 5 gynoecious plants and 45 monoecious plants. The genotype of gynoecy was T:T, the monoecious genotype was C:C, and the heterozygous genotype was T:C. Results showed that except for the phenotypes of two materials (AW2 and AW10) inconsistent with the genotypes, the detection accuracy of KASP145 was 96.0% (Table 3). In addition, we analyzed the variation of CqNET4 in 25 chieh-qua materials and found that base 548 of the exon region of this gene was ‘G’ in the gynoecious materials, while ‘A’ was found in the monoecious materials (Figure 3B).
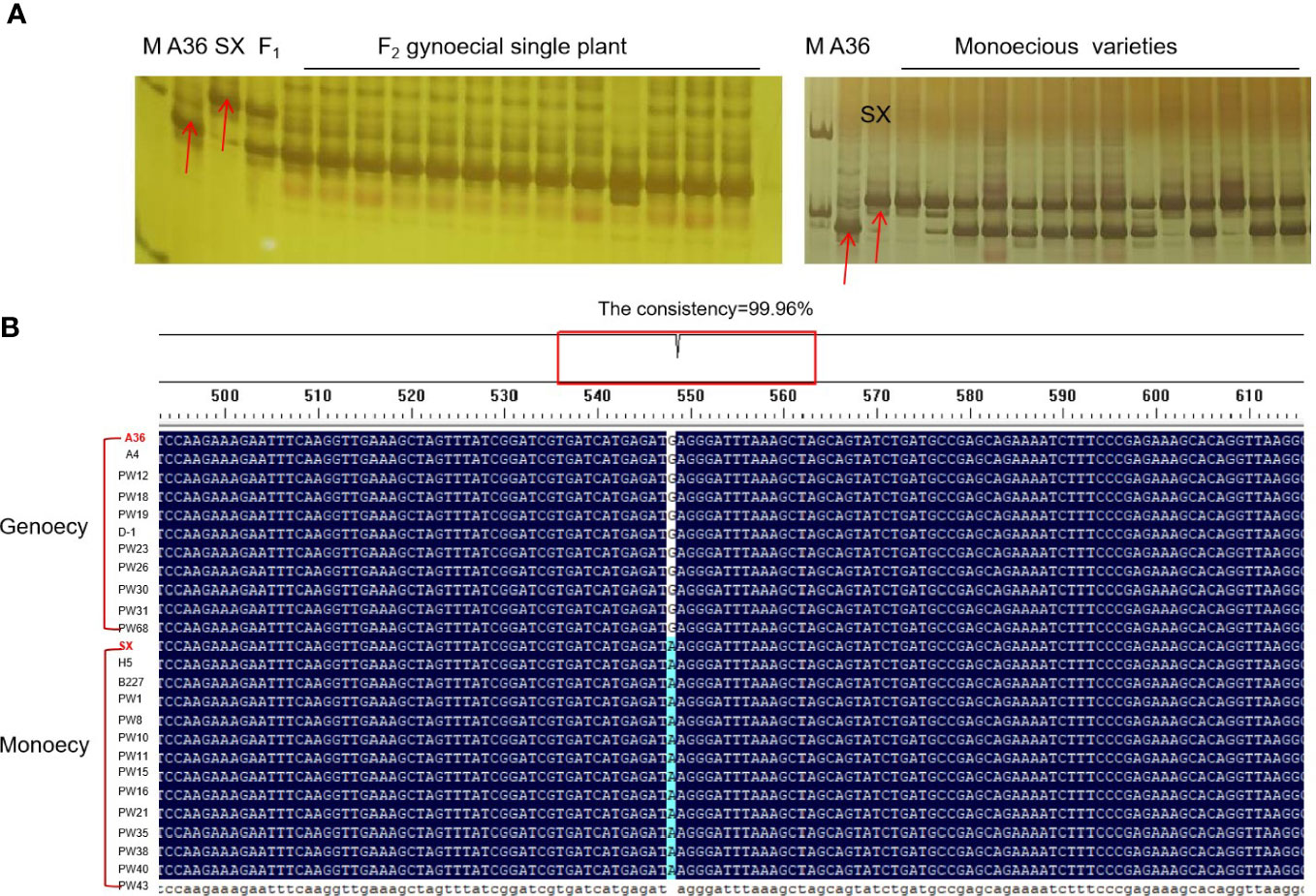
Figure 3 Validation of molecular markers closely linked to gynoecy trait. (A) The detection of Indel-3 in F2 individuals and monoecious plant. (B) Analysis of variation of CqNET4 in 25 chieh-qua materials.
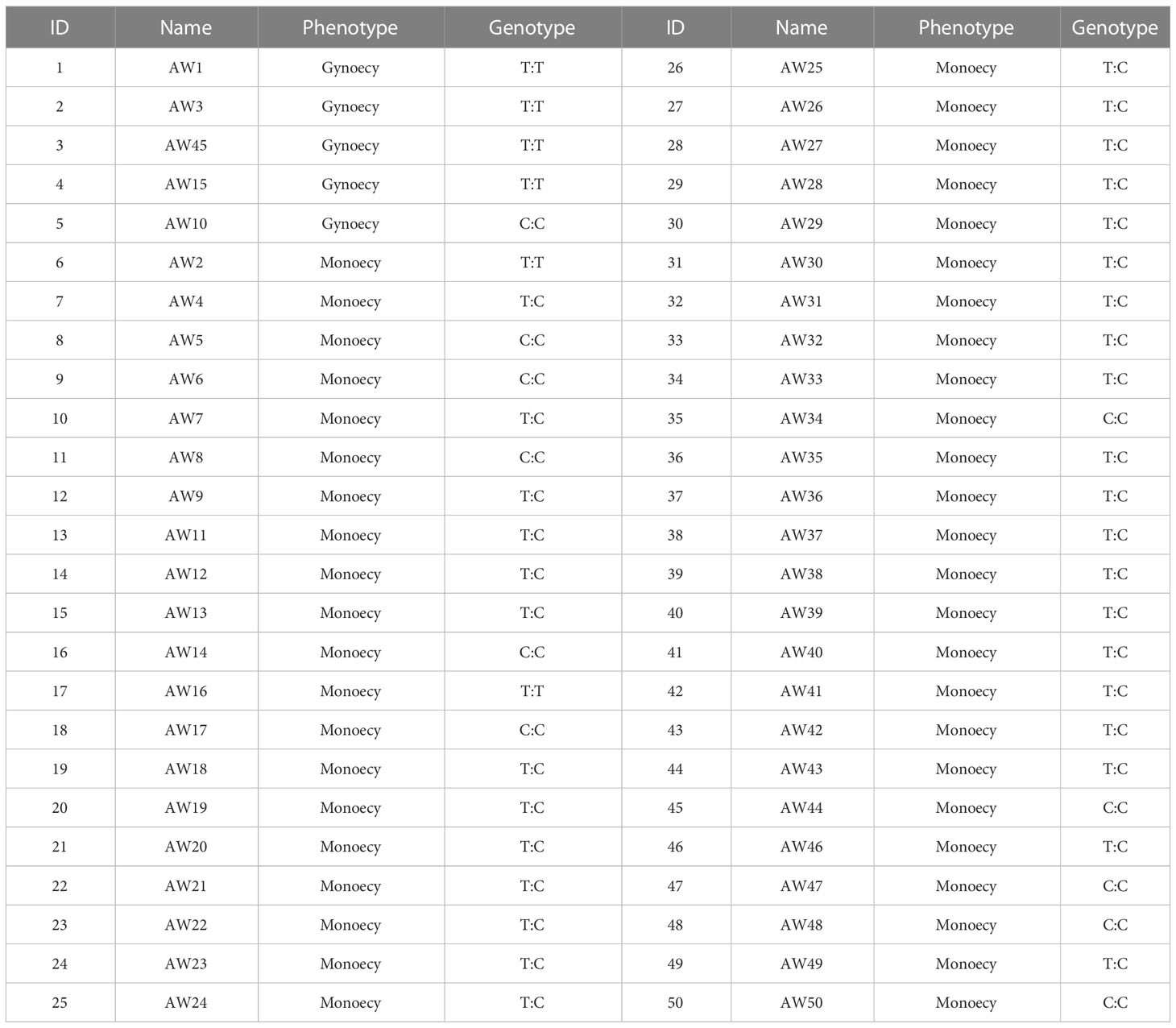
Table 3 Identification of KASP145 in 50 chieh-qua germplasm resources.
Expression analysis of genes in the candidate region
In order to further detect the expression of candidate genes in the fine mapping interval, the qRT-PCR assay was carried out using terminal buds of ‘A36’ and ‘SX.’ The expression of Bhi08G000338–Bhi08G000343 showed no significant difference between ‘A36’ and ‘SX,’ while the expression of Bhi08G000344 and Bhi08G000345 was significantly downregulated in ‘SX’ than in ‘A36’ (Supplemental Figure 2).
Phylogenic analysis of CqNET4
To explore the relationship between CqNET4 and its homologous sequences in other species, we obtained the homolog sequences of CqNET4 using NCBI and TBtools (Chen et al., 2020). The neighbor-joining tree showed that the amino acids in the motif of CqNET4 in plant species were highly conserved (Supplemental Figure 3). Moreover, CqNET4 had a close phylogenetic relationship with Cucurbitaceae, including Cucumis melo (CmNET4), Cucumis sativus (CsNET4), Cucurbita maxima (CmaNET4), and Cucurbita moschata (CmoNET4) (Figure 4). In addition, we found that the NET4 sequences in ‘A36’ and ‘SX’ were longer than those in wax gourd because of a Motif 5 existing in chieh-qua (Figure 4).
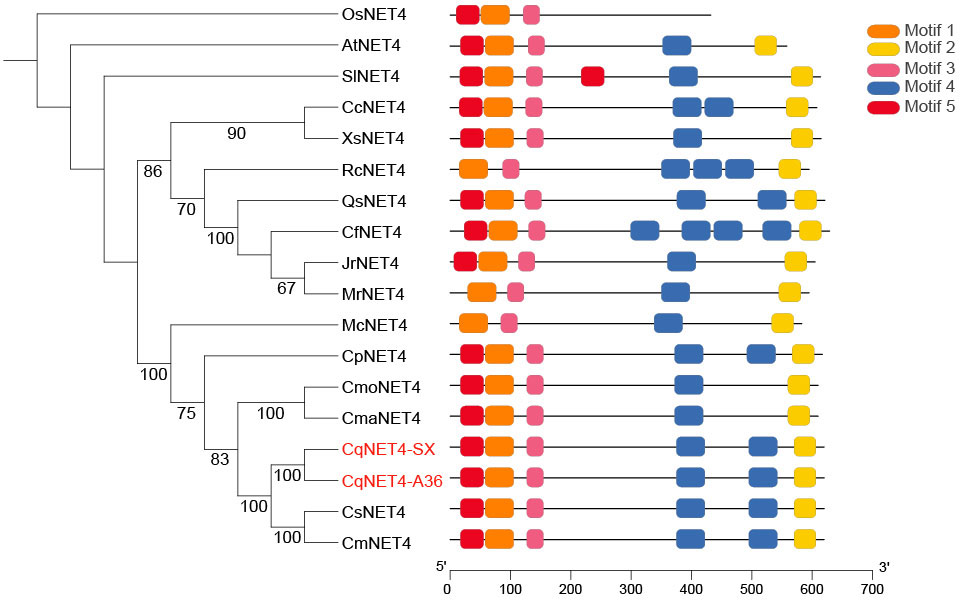
Figure 4 Phylogenetic and motif analyses of NET4A in chieh-qua and other species. The phylogenetic tree was constructed using MEGA-5.2 with 1,000 bootstrap replications. Numbers at the tree forks indicated bootstrap values. The motif distribution of different NET4 from Oryza sativa (Os), Arabidopsis (At), Solanum lycopersicum (Sl), Cucurbita maxima (Cma), Cucurbita pepo (Cp), Myrica rubra (Mr), Quercus variabilis (Qs), Xanthoceras sorbifolia (Xs), Carpinus fangiana (Cf), Rosa chinensis (Rc), Clement pomelo (Cp), Cucumis melo (Cm), Cucumis sativus (Cs), Momordica charantia (Mc), and Cucurbita moschata (Cmo) was shown.
Expression analysis of CqNET4
To further understand the CqNET4 expression pattern in different tissues, we performed the qRT-PCR assay using six chieh-qua tissues, namely, leaves, stems, tendrils, terminal buds, female flowers, and male flowers from SX. Results showed that CqNET4 was broadly expressed in various tissues, with a high expression in female flowers (Figure 5A), implying its crucial role in regulating flower development. Next, to determine the subcellular localization of CqNET4, the full-length CDS sequence without stop codon was cloned and inserted into the pCAMBIA1305 vector. Then, the obtained 35S::CqNET4-GFP fusion construct and 35S::GFP were transiently transformed in the protoplasts of N. benthamiana leaf cells. Compared with the GFP control, whose fluorescence signal was distributed throughout the cell, the CqNET4-GFPs fusion proteins of A36 (CqNET4G-GFP) and SX (CqNET4A-GFP) were both observed exclusively in the nuclei of N. benthamiana leaf cell protoplasts (Figure 5B), indicating CqNET4 is a nuclear-localized protein and the amino acid change did not influence its subcellular localization.
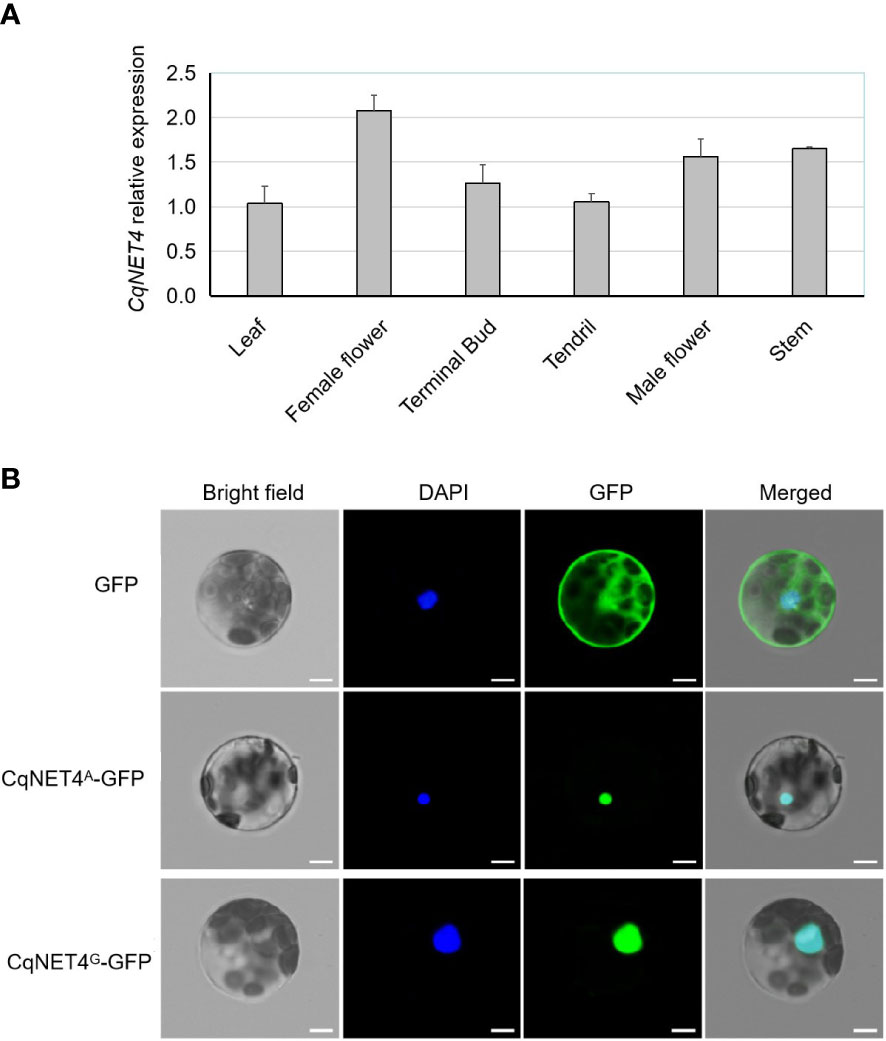
Figure 5 The expression pattern and subcellular localization of CqNET4. (A) The relative expression pattern of CqNET4 in different tissues of chieh-qua. Data are the means ± SE of three independent replicates. (B) Subcellular localization of CqNET4A and CqNET4G proteins. Scale bars = 5 µm.
Gene expression related to ethylene synthesis
WIP1 and ACS1G were identified to control gynoecy development in melon and cucumber, respectively (Boualem et al., 2015; Zhang et al., 2021). To identify whether there existed sequence and expression differences of WIP1 and ACS1G between ‘A36’ and ‘SX,’ we carried out PCR amplification and qRT-PCR assays. Results showed that no differences on the gene sequence of WIP1 and ACS1G were detected between A36 and SX (Figures 6A, B). Nevertheless, WIP1 was not expressed differently between A36 and SX in the terminal bud and leaf tissues (Figure 6C), similar to ACS1G (Figure 6D). Previous studies reported that ethylene played crucial roles in the early stages of floral meristem development of Cucurbitaceae (Boualem et al., 2015; Li et al., 2020; Zhang et al., 2021). Thus, the ethylene synthesis-related genes were chosen for qPCR analysis between the ‘A36’ and ‘SX’ terminal buds. By detecting 18 ethylene synthesis-related genes, only three genes were found significantly changed between ‘A36’ and ‘SX.’ Bhi12G001445 encoding ACC (1-aminocyclopropane-1-carboxylate) oxidase (ACO) was prominently upregulated in ‘SX’ in comparison with ‘A36.’ Both Bhi01G001789 and Bhi02G001590 encoding ACC synthase (ACS) were expressed more highly in ‘A36’ than in ‘SX’ (Figure 6E).
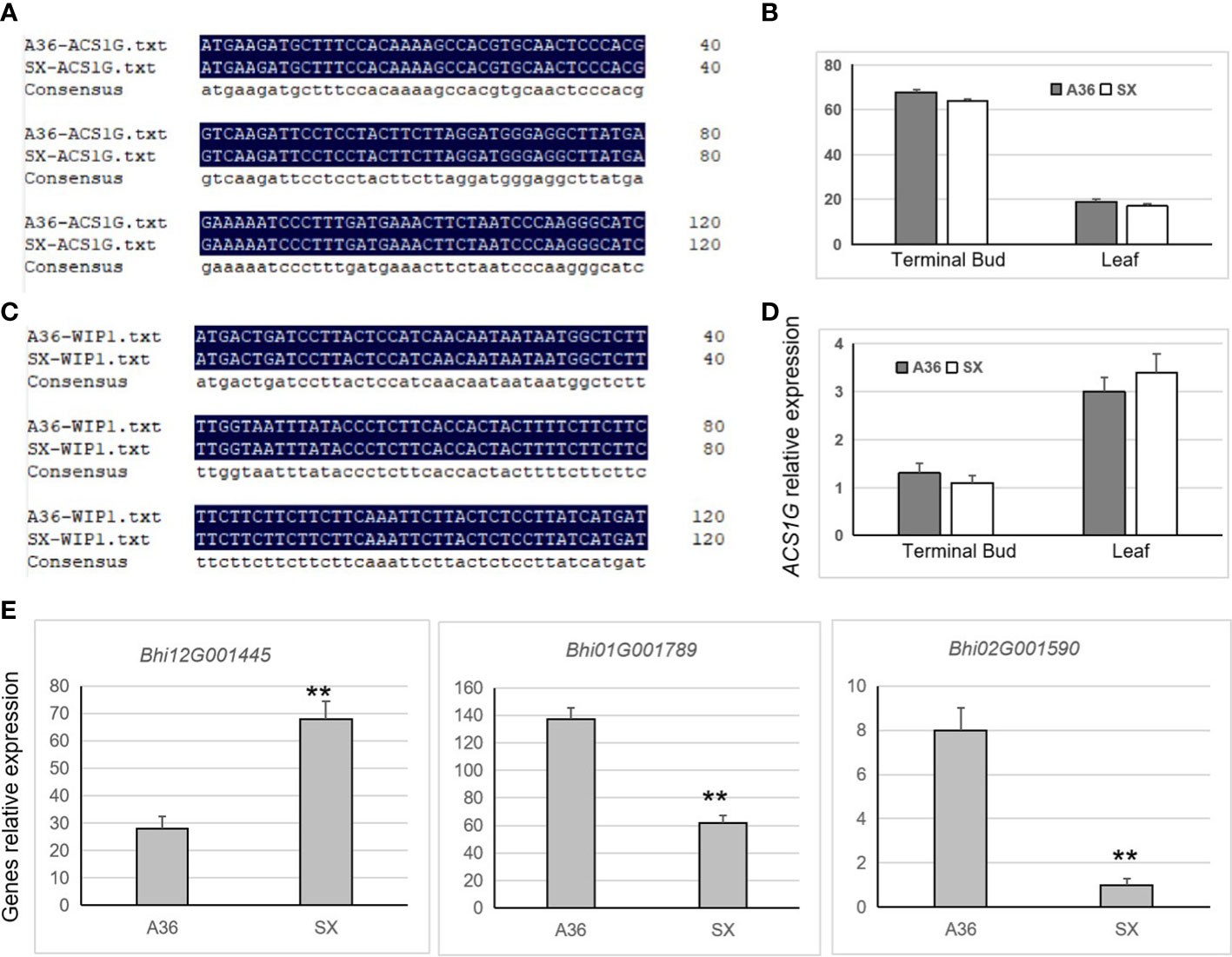
Figure 6 Partial sequence alignment and expression analysis of the homologous genes WIP1 and ACS1G and ethylene synthesis. (A, B) Partial sequence alignment results of the homologous genes WIP1 and ACS1G in melon ‘A36’ and ‘SX.’ (C, D) Expression levels of CqACS1 and CqWIP1 in the leaves and terminal buds of ‘A36’ and ‘SX.’ (E) Expression detection of genes related to ethylene synthase. The asterisks indicate significant differences (Student’s t-test): *Padj < 0.05, **Padj < 0.01.
Discussion
Gynoecy could not only improve fruit yield but also reduce labor cost during crossing seed production (Robinson, 1999). Previous studies reported that the gynoecy of bitter gourd was controlled by a single recessive gene (Behera et al., 2009; Cui et al., 2018), as well as that of watermelon (Li et al., 2019) and melon (Boualem et al., 2016). However, in cucumber, some reports showed that its gynoecy was controlled by a dominant gene (Pati et al., 2015) or multiple genes as a quantitative trait (Li et al., 2019). However, as far as we know, no genetic research related to chieh-qua gynoecy has been reported. In this study, by constructing the F2 population using gynoecious and monoecious lines, we found that the gynoecy of chieh-qua was controlled by a single recessive locus (Table 1), which was consistent with most other cucurbits.
In melon and cucumber, the genetic control for sex determination or female development has been clarified with several crucial genes. Moreover, WIP1 and ACS1G were the crucial genes controlling gyneocy for melon and cucumber, respectively (Boualem et al., 2015; Zhang et al., 2021). Our results showed that Bhi08G000345 (CqNET4) was the most likely possible candidate gene involved in gynoecy in chieh-qua (Figure 2) with a higher expression in female flower (Figure 4A). Furthermore, we found that CqNET4G was specifically enriched in gynoecious plants, and CqNET4A was in the reference genome B227 and monoecious materials (Figure 3C). Thus, we predicted that the chieh-qua gyneocy determination gene was different from other cucurbits.
Microfilament, as part of the cytoskeleton, is involved in important biological processes, including cytoplasmic morphogenesis, signal transduction, and pollen germination and tube growth (Thomas et al., 2009; Blanchoin et al., 2014). NET4s are plant microfilament-specific binding proteins, which potentially couple different membranes to the actin cytoskeleton in plant cells (Deeks et al., 2012). Overexpression of NET4A leads to a shorter root length in A. thaliana (Kaiser et al., 2019). NET2 can interact with pollen tube receptor kinase to co-regulate pollen tube development (Duckney et al., 2017). The dual mutation of NET3B and NET3C could result in a lethal phenotype of Arabidopsis gametophytes (Wang et al., 2014). Overexpression of AtNET3B could enhance the association between ER (endoplasmic reticulum) and the actin cytoskeleton (Wang and Hussey, 2017). A further study indicated that NET4 is localized to highly constricted regions of the vacuolar membrane in A. thaliana (Kaiser et al., 2019). However, our study found that CqNET4A and CqNET4G proteins were located in the cell nucleus, which was different from previous studies. Thus, we predicted that the CqNET4 protein might have homology with other species, while it possessed a novel function during the chieh-qua sex determination.
Previous studies demonstrated that the phytohormone ethylene played critical functions in controlling female or male flower development (Wang et al., 2010b; Manzano et al., 2014; Boualem et al., 2015; Martínez and Jamilena, 2021; Zhang et al., 2021). The gyneocy crucial gene WIP1 functions in a pathway with other ethylene-related genes such as CmACS11 and CmACS-7 in melon (Boualem et al., 2015). CsACS1G (a vital enzyme for ethylene synthesis) is the critical gene responsible for the formation of female flowers, rather than CsACS1 or other genes in the F locus (Zhang et al., 2021). Our results showed that the sequence and expression level of homologous WIP1 and ACS1G between A36 and SX showed no difference (Figures 6A–D). In addition, the ethylene synthesis-related genes encoding ACS were significantly upregulated in gyneocious plants (Figure 6E). Therefore, we propose that CqNET4 might regulate chieh-qua gynoecy by affecting ethylene synthesis. However, the biological function of CqNET4 should be elucidated by combining efficient genetic transformation systems such as genome editing or overexpression technology in the future.
Conclusion
Our study found that the gynoecy of chieh-qua was controlled by a single recessive gene, and it was fine-mapped to a 530-kb region between Indel-3 and KASP145 on Chr.8. Combining the mapped cloning, sequence, and expression results, we predicted Bhi08G000345 (CqNET4) as the candidate gene of gynoecy, which differed from the homology of gynoecious genes, such as WIP1 or ACS1G in melon and cucumber.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
Conceived and designed all the experiments: MW, SY, and QP. Performed part of the experiments: WeiL, ZC, and JY. Analyzed the data: LC, WenL, and DX. Wrote the manuscript: MW. Edited the manuscript: BJ. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (32002038), Guangzhou Science and Technology Project (202102020750), Special Fund for Scientific Innovation Strategy-Construction of High Level Academy of Agricultural Science (R2021PY-QF008), and Agricultural Competitive Industry Discipline Team Building Project of Guangdong Academy of Agricultural Sciences (202114TD, 202103TD).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1158735/full#supplementary-material
References
Abuín, J. M., Pichel, J. C., Pena, T. F., Amigo, J. (2015). BigBWA: approaching the burrows–wheeler aligner to big data technologies. Bioinformatics 31, 4003–4005. doi: 10.1093/bioinformatics/btv506
Bai, S. L., Peng, Y. B., Cui, J. X., Gu, H. T., Xu, L. Y., Li, Y. Q., et al. (2004). Developmental analyses reveal early arrests of the spore-bearing parts of reproductive organs in unisexual flowers of cucumber (Cucumis sativus l.). Planta 220, 230–240. doi: 10.1007/s00425-004-1342-2
Behera, T. K., Dey, S., Munshi, A. D., Gaikwad, A. B., Pal, A., Singh, I. (2009). Sex inheritance and development of gynoecious hybrids in bitter gourd (Momordica charantia l.). Scientia Hortic. 120, 130–133. doi: 10.1016/j.scienta.2008.09.006
Blanchoin, L., Boujemaa-Paterski, R., Sykes, C., Plastinoet, J. (2014). Actin dynamics, architecture, and mechanics in cell motility. Physiol. Rev. 94, 235–263. doi: 10.1152/physrev.00018.2013
Boualem, A., Lemhemdi, A., Sari, M. A., Pignoly, S., Troadec, C., Abou Choucha, F., et al. (2016). The andromonoecious sex determination gene predates the separation of cucumis and citrullus genera. PLoS One 11, e0155444. doi: 10.1371/journal.pone.0155444
Boualem, A., Troadec, C., Camps, C., Lemhemdi, A., Morin, H., Sari, M.-A., et al. (2015). A cucurbit androecy gene reveals how unisexual flowers develop and dioecy emerges. Science 350, 688–691. doi: 10.1126/science.aac8370
Boualem, A., Troadec, C., Kovalski, I., Sari, M.-A., Perl-Treves, R., Bendahmane, A. (2009). A conserved ethylene biosynthesis enzyme leads to andromonoecy in two cucumis species. PLoS One 4, e6144. doi: 10.1371/journal.pone.0006144
Chen, C. J., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y. H., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chen, H. M., Sun, J. J., Li, S., Cui, Q. Q., Zhang, H., Xin, F. J., et al. (2016). An ACC oxidase gene essential for cucumber carpel development. Mol. Plant 9, 1315–1327. doi: 10.1016/j.molp.2016.06.018
Cui, J. J., Luo, S. B., Niu, Y., Huang, R. K., Wen, Q. F., Su, J. W., et al. (2018). A RAD-based genetic map for anchoring scaffold sequences and identifying QTLs in bitter gourd (Momordica charantia). Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00477
Deeks, M. J., Calcutt, J. R., Ingle, E. K. S., Hawkins, T. J., Chapman, S., Richardson, A. C., et al. (2012). A superfamily of actin-binding proteins at the actin-membrane nexus of higher plants. Curr. Biol. 22, 1595–1600. doi: 10.1016/j.cub.2012.06.041
Duckney, P., Deeks, M., Dixon, M., Kroon, J., Hawkins, T., Hussey, P. (2017). Actin-membrane interactions mediated by NETWORKED2 in arabidopsis pollen tubes through associations with pollen receptor-like kinase 4 and 5. New Phytol. 216, 1170–11180. doi: 10.1111/nph.14745
He, X. M., Xie, D. S., Chen, Q., Peng, Q. (2007). Chieh-qua biotechnology, progress and prospects. Asia Aust. J. Sci. Biotechnol. 1, 19–22.
Healey, A., Furtado, A., Cooper, T., Henry, R. J. (2014). Protocol: a simple method for extracting next-generation sequencing quality genomic DNA from recalcitrant plant species. Plant Methods 10, 1–8. doi: 10.1186/1746-4811-10-21
Hu, B. W., Li, D. W., Liu, X., Qi, J. J., Gao, D. L., Zhao, S. Q., et al. (2017). Engineering non-transgenic gynoecious cucumber using an improved transformation protocol and optimized CRISPR/Cas9 system. Mol. Plant 10, 1575–1578. doi: 10.1016/j.molp.2017.09.005
Kaiser, S., Eisa, A., Kleine-Vehn, J., Scheuring, D. (2019). NET4 modulates the compactness of vacuoles in arabidopsis thaliana. Int. J. Mol. Sci. 20, 4752. doi: 10.3390/ijms20194752
Lai, Y., Zhang, X. H., Zhang, W., Shen, D., Wang, H. P., Xia, Y., et al. (2017). The association of changes in DNA methylation with temperature-dependent sex determination in cucumber. J. Exp. Bot. 68, 2899–2912. doi: 10.1093/jxb/erx144
Li, H., Durbin, R. (2010). Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, Z., Han, Y. H., Niu, H. H., Wang, Y. H., Jiang, B., Weng, Y. Q. (2020). Gynoecy instability in cucumber (Cucumis sativus l.) is due to unequal crossover at the copy number variation-dependent femaleness (F) locus. Horticulture Res. 7, 32. doi: 10.1038/s41438-020-0251-2
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, D. D., Sheng, Y. Y., Niu, H. H., Li, Z. (2019). Gene interactions regulating sex determination in cucurbits. Front. Plant Sci. 10, 1231. doi: 10.3389/fpls.2019.01231
Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lu, H. F., Lin, T., Klein, J., Wang, S. H., Qi, J. J., Zhou, Q., et al. (2014). QTL-seq identifies an early flowering QTL located near flowering locus T in cucumber. Theor. Appl. Genet. 127, 1491–1499. doi: 10.1007/s00122-014-2313-z
Manzano, S., Martínez, C., García, J. M., Megías, Z., Jamilena, M. (2014). Involvement of ethylene in sex expression and female flower development in watermelon (Citrullus lanatus). Plant Physiol. Biochem. 85, 96–104. doi: 10.1016/j.plaphy.2014.11.004
Martin, A, Troadec, C, Boualem, A, Rajab, M, Fernandez, R, Morin, H, et al. (2009). A transposon-induced epigenetic change leads to sex determination in melon. Nature 461, 1135–1138. doi: 10.1038/nature08498
Martínez, C., Jamilena, M. (2021). To be a male or a female flower, a question of ethylene in cucurbits. Curr. Opin. Plant Biol. 59, 101981. doi: 10.1016/j.pbi.2020.101981
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Pati, K., Munshi, D. A., Behera, K. T. (2015). Inheritance of gynoecism in cucumber (Cucumis sativus l.) using genotype GBS-1 as gynoecious parent. Genetika 47, 349–356. doi: 10.2298/GENSR1501349P
Robinson, R. W. (1999). Rationale and methods for producing hybrid cucurbit seed. J. New Seeds 1, 1–47. doi: 10.1300/J153v01n03-01
Takagi, H., Abe, A., Yoshida, K., Kosugi, S., Natsume, S., Mitsuoka, C., et al. (2013). QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 74, 174–183. doi: 10.1111/tpj.12105
Thomas, C., Tholl, S., Moes, D., Dieterle, M., Papuga, J., Moreau, F., et al. (2009). Actin bundling in plants. Cell Motil. Cytoskeleton 66, 940–957. doi: 10.1002/cm.20389
Wang, P., Hussey, P. J. (2017). NETWORKED 3B: a novel protein in the actin cytoskeleton-endoplasmic reticulum interaction. J. Exp. Bot. 68, 1441–1450. doi: 10.1093/jxb/erx047
Wang, P, Hawkins, TJ, Richardson, C, Cummins, I, Deeks, MJ, Sparkes, I, et al. (2014). The plant cytoskeleton, NET3C, and VAP27 mediate the link between the plasma membrane and endoplasmic reticulum. Curr Biol 16, 1397–1405. doi: 10.1016/j.cub.2014.05.003
Wang, D. H., Li, F., Duan, Q. H., Han, T., Xu, Z. H., Bai, S. N. (2010b). Ethylene perception is involved in female cucumber flower development. Plant J. 61, 862–872. doi: 10.1111/j.1365-313X.2009.04114.x
Wang, K., Li, M. Y., Hakonarson, H. (2010a). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164–e164. doi: 10.1093/nar/gkq603
Xie, D. S., Xu, Y. C., Wang, J. P., Liu, W. R., Zhou, Q., Luo, S. B., et al. (2019). The wax gourd genomes offer insights into the genetic diversity and ancestral cucurbit karyotype. Nat. Commun. 10, 5158. doi: 10.1038/s41467-019-13185-3
Zhang, J., Guo, S., Ji, G. J., Zhao, H., Sun, H. H., Ren, Y., et al. (2020). A unique chromosome translocation disrupting ClWIP1 leads to gynoecy in watermelon. Plant J. 101, 265–277. doi: 10.1111/tpj.14537
Keywords: chieh-qua, gynoecy, gene mapping, CqNET4, ethylene synthesis related genes
Citation: Wang M, Yang S, Liu W, Cao Z, Chen L, Liu W, Xie D, Yan J, Jiang B and Peng Q (2023) Fine mapping and candidate gene analysis of gynoecy trait in chieh-qua (Benincasa hispida Cogn. var. chieh-qua How). Front. Plant Sci. 14:1158735. doi: 10.3389/fpls.2023.1158735
Received: 04 February 2023; Accepted: 23 March 2023;
Published: 20 April 2023.
Edited by:
Rafael Lozano, University of Almeria, SpainReviewed by:
Yong Xu, Beijing Academy of Agriculture and Forestry Sciences, ChinaChengjun Zhang, Zhejiang Agriculture and Forestry University, China
Copyright © 2023 Wang, Yang, Liu, Cao, Chen, Liu, Xie, Yan, Jiang and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingwu Peng, cGVuZ3Fpbmd3dUBnZGFhcy5jbg==; Biao Jiang, amlhbmdiaWFvQGdkYWFzLmNu
†These authors have contributed equally to this work