 Liya Zhang
Liya Zhang Yanyan Zhang
Yanyan Zhang Bin Liu
Bin Liu Tao Zhao
Tao Zhao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 04 April 2023
Sec. Plant Abiotic Stress
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1153840
This article is part of the Research Topic Light, Clock, Flowering, and Hormone Pathways in Attaining Abiotic Stress Tolerance View all 6 articles
N6-methyladenosine (m6A) modification of messenger RNA (mRNA) is the most prevalent and abundant modification in eukaryotic mRNA and posttranscriptionally modulates the transcriptome at almost all stages of mRNA metabolism. In plants, m6A is crucial for embryonic-phase growth, flowering time control, microspore generation and fruit maturation. However, the role of m6A in plant responses to light, the most important environmental stimulus, remains unexplored. Here, we profile the m6A transcriptome of Williams 82, a soybean cultivar, and reveal that m6A is highly conserved and plays an important role in the response to light stimuli in soybean. Similar to the case in Arabidopsis, m6A in soybean is enriched not only around the stop codon and within the 3’UTR but also around the start codon. Moreover, genes with methylation occurring in the 3’UTR have higher expression levels and are more prone to alternative splicing. The core genes in the light signaling pathway, GmSPA1a, GmPRR5e and GmBIC2b, undergo changes in methylation modification and transcription levels in response to light. KEGG pathway analysis revealed that differentially expressed genes with differential m6A peaks were involved in the “photosynthesis” and “circadian rhythm” pathways. Our results highlight the important role played by epitranscriptomic mRNA methylation in the light response in soybean and provide a solid basis for determining the functional role of light on RNA m6A modification in this plant.
More than 150 RNA modifications have been identified on various types of RNAs, including mRNA, tRNA, rRNA, lncRNA, and microRNA (Cantara et al., 2011; Meyer and Jaffrey, 2014; Boccaletto et al., 2018). Common mRNA modifications include N6-methyladenosine (m6A), 5-methylcytidine (m5C), N1-methyladenosine (m1A), and pseudouridine (Ψ) (Roundtree et al., 2017). m6A is the most abundant methylation modification in eukaryotic mRNA, and it is also the most thoroughly studied type of RNA modification (Shi et al., 2017). m6A methylation is a reversible process that is dynamically coordinated by writers, readers and erasers. RNA m6A methylation is a unique molecular mechanism for influencing gene expression (Meyer and Jaffrey, 2014). Studies have shown that m6A plays a functional role in mRNA, altering properties such as stability (Frye et al., 2018), decay (Yang et al., 2018), splicing (Haussmann et al., 2016; Xiao et al., 2016), translation efficiency (Meyer et al., 2015; Luo et al., 2020), nuclear export (Zheng et al., 2013) and localization (Hartmann et al., 1999; Yang et al., 2018).
In plants and other eukaryotes, m6A modification occurs at a highly conserved RRACH (R = G or A; H: U>A>C) motif (Shen et al., 2016). Unlike that in animals, m6A methylation modification in plants is present not only in stop codons and 3’untranslated regions (3’UTR) (Dominissini et al., 2012; Meyer et al., 2012; Schwartz et al., 2014; Ke et al., 2015), but also in start codons (Slobodin et al., 2017). m6A modification can affect many aspects of plant biology, including embryo development, shoot apical meristem development, apical dominance, organ size, root development, viral infection, floral transition, leaf and trichome morphogenesis, flowering time, microspore generation and fruit ripening (Shen et al., 2019). There is increasing evidence that m6A is also involved in regulating responses to environmental stresses (Wei et al., 2018; Yue et al., 2019; Huong et al., 2020). For example, in Arabidopsis, m6A methylation plays an important role in salt stress tolerance (Hu et al., 2021). The m6A peaks in Pak-choi and rice under heat stress and cadmium stress were significantly reduced compared with those in controls (Liu et al., 2020; Cheng et al., 2021). The methyltransferase PtrMTA enhances drought tolerance by regulating the development of trichomes and roots (Lu et al., 2020). Disruption of RNA demethylase AtALKBH10B resulted in the suppression of heat-activated genes and greatly reduced plant fertility (Wang et al., 2022). AtMTA can regulate the methylation level and translation efficiency of DGAT1, which preliminarily revealed the molecular mechanism of mRNA m6A modification in chilling responses in Arabidopsis (Wang et al., 2023). In addition, Under high light conditions, m6A writer VIRILIZER (VIR) positively regulates photosynthesis, and its mutation significantly reduces photosynthetic activity and photosystem protein abundance (Zhang et al., 2022). These findings clearly indicate that m6A is essential for plant growth and development. However, the significance and cellular role of m6A RNA modification in the plant response to environmental stimuli remain largely unknown.
Light is essential for plants, and perception of the light environment determines plant growth, morphology, and developmental changes. Light acts as an important developmental switch, affecting photomorphogenesis, germination, flowering time and circadian rhythms (Hoecker, 2017). Soybean is a typical short-day plant. Compared with other crops, soybean is particularly dependent on specific light conditions for its growth and development, which restricts soybean cultivation to particular areas (Li et al., 2020). Its yield at a given latitude depends mainly on the appropriate photoperiod (Lu et al., 2017). However, it is possible to genetically alter soybeans to reduce this photoperiod sensitivity, creating strains that can be grown in a wide range of locations and agricultural settings, to increase yields. Core genes in the light signaling pathway, such as Suppressor of phytochrome A (SPA), Blue light inhibitors of cryptochromes (BIC) and Pseudoresponse regulator 5 (PRR5), sense changes in the light environment, which in turn affects plant growth and development (Wang et al., 2017; Hwang et al., 2021; Ponnu and Hoecker, 2021). For example, GmBICs regulate soybean stem elongation under low levels of blue light (Mu et al., 2022). The Arabidopsis COP1/SPA complex acts as a cullin4-based E3 ubiquitin ligase to suppress photomorphogenesis in darkness (Kerner et al., 2021). PRR5 is one of the core circadian clock components, and PRR5 mutants in Arabidopsis display a shorter circadian rhythm, later flowering and a red light-sensitive phenotype (Eriksson et al., 2003; Michael et al., 2003; Yamamoto et al., 2003). A recent study showed that CRY2 can interact with the m6A writers MTA, MTB and FIP37 and modulate m6A writer activity, mRNA methylation and abundance and circadian rhythms in plants (Wang et al., 2021), establishing a preliminary link between light and m6A modification. However, to date, there has been very little research on the transcriptome-wide N6-methyladenosine methylome profile response to light in soybean or other plants.
Thus, in this study, to further explore whether light affects m6A methylation modifications in soybean, we performed MeRIP-seq and RNA-seq analysis on soybean seedlings under dark and continuous light conditions. Here, we obtain the first known m6A map of the transcriptome range in soybean. We also compared and analyzed the patterns of m6A distribution between dark and continuous light conditions to obtain differentially methylated peaks and then analyzed the potential impacts of m6A on gene expression regulation under different light conditions. Collectively, our data constitute a comprehensive picture of m6A methylation in mRNA in soybean seedlings and provide a basis for future studies of its function and biological significance in soybean.
Soybean varieties TL1 (Tianlong 1) and Williams 82 were grown under continuous light or dark conditions in a growth chamber at 25°C. Aboveground tissue was harvested on day 4 after germination and flash frozen in liquid nitrogen. Total RNA was extracted using TRIzol reagent (Invitrogen, Cat. No 15596026).
Total RNA from each sample was isolated using TRIzol reagent (Invitrogen) and then quantified using a NanoDrop ND-1000 instrument and gel electrophoresis. Polyadenylated RNA was extracted using VAHTS mRNA Capture Beads (VAHTS, Cat. No. N401-01/02), fragmented to 100 nt-200 nt using 20 mM ZnCl2 at 95°C for 5 min-10 min and placed on ice. A portion of the purified RNA was kept as input. The remainder of the purified RNA was incubated with m6A antibody in IP buffer for 2 h at 4°C. Then, the immunoprecipitation reaction was incubated with protein A/G magnetic beads (Thermo Scientific, Cat. No. 88845) for 2 h at 4°C. The beads were then washed two times with 1× IP buffer, and the RNA was eluted with phenol–chloroform. Then, the sample was dissolved in RNase-free H2O. The m6A -seq libraries were constructed with a KC-Digital™ stranded mRNA library kit (Seqhealth, Cat. No. DR08502) according to the provided instructions.
RNA-seq libraries were prepared as follows: high-quality mRNA was obtained and fragmented using fragmentation buffer. Using the fragmented mRNA as a template, six-base random primers were used to synthesize single-stranded cDNA, and then buffer, dNTPs and DNA polymerase I were added to synthesize double-stranded cDNA. The purified double-stranded cDNA was eluted for end repair and poly(A) tailing; then, a UID adapter was added as sequencing adapter. Magnetic beads were used to recover the target fragment, and PCR amplification was conducted. Library quality was verified using gel electrophoresis and Qubit 2.0. Finally, the samples were sequenced using an Illumina gene sequencer.
After the data were downloaded, the quality of the reads was evaluated with FastQC (version 0.11.5), and Trimmomatic (version 0.36) was used for data quality control, mainly removal of the linker sequence from the reads and elimination of low-quality reads. Using STAR (version 2.5.3a) software, we mapped the clean data from the input and IP-sequenced samples to the soybean reference genome. Following previous studies, we used RSeQC (version 2.6) to evaluate the mapped read distribution, coverage uniformity, strand specificity and other parameters. Next, based on the background information (input RNA-seq data), we used the R package exomePeak2 (Version 3.8) to obtain robust and high-resolution RIP-Seq peak predictions with two biological replicates. Bedtools (Version v2.25.0) was used to annotate m6A peaks in the soybean annotation file, characterize the distribution of peaks on chromosomes, and count the peaks in the transcript regions. The m6A site differential algorithm was used for the identification of differentially methylated peaks between the dark and white light samples with criteria of P value <0.05 and log2 (fold change) ≥1. The conserved sequences (RRACH and DRACH) of identified m6A peaks were found by HOMER (Version v4.10). According to past research, we used deepTools (Version 2.4.1) to study the coverage mapping of reads and peaks in the CDSs and UTRs of all genes and generate a coverage heatmap.
To further explore the essential role of m6A mRNA modification in soybean, the identified m6A peaks and differentially expressed genes (DEGs) were subjected to network analyses. Here, we used reads per kilobase per million reads (RPKM) as a measure of gene expression levels with a cut off of log2FC ≥ 1 and P value < 0.05 (Mortazavi A. et al., 2008). RPKM was calculated as total exon reads/mapped reads (millions) x exon length (KB). GO and KEGG analyses of the differentially methylated genes and differentially expressed genes were conducted using the Gene Ontology Database (http://www.geneontology.org/) and Kyoto Encyclopedia of Genes and Genomes database (http://www.genome.jp/kegg/). We used Replicate Multivariate Analysis of Transcript Splicing (rMATS) software for differential alternative splicing (AS) analysis. Related data are presented in Table S1.
The aboveground tissue was quickly frozen with liquid nitrogen and ground into a powder with a mortar. Total RNA was extracted using TRIzol reagent. First-strand cDNA was synthesized from 3 μg of total RNA in a 20 μL volume using an Oligo(dT)18 primer with TransScript II One-Step gDNA Removal (TransGen Biotech. Cat. No. AT311-03) and cDNA Synthesis SuperMix time PCR was performed on Biometra Tone 96 G according to a two-step method using the ChamQ Universal SYBR qPCR Master kit (Vazyme. Cat. No. Q711-02). Briefly, each 10 μL qRT–PCR contained 4.6 μL of 20-fold diluted cDNA, 0.2 μL of each primer and 5 μL ChamQ Universal SYBR Qpcr Master Mix. The reaction procedure was predenaturated at 95 °C for 30s, followed by a 40-cycle program (95°C 10 s and 60°C 30s). GmActin (Glyma.18G290800) was used as an internal control to calculate the transcript levels of selected genes, which were quantified by the cycle threshold 2-ΔCt method. △Ct=Ct (selected gene)-Ct (GmActin). Each set of experiments was performed with at least three biological replicates and two technical replicates. Related genes and primer lists are presented in Table S1.
60 μg of total RNA per sample was used, fragmented with 0.272 g ZnCl2 (0.1 M) at 94°C for 100 s and then incubated with beads for at least 3 h or overnight. 30 μL of fragmented mRNA was used as the input control. m6A-IP was performed using the magna RIP kit (Millipore, 17-700). 50 μL of beads was used and then immunoprecipitated with 5 μg anti-m6A antibody (Synaptic Systems, 202003) at room temperature for 30 min-1 h. Reverse transcription was performed with random primers or specific primers. PCR amplification was performed by qRT-PCR. Each set of experiments was performed with at least three biological replicates and three technical replicates.
The effect of light on soybean germination and growth were has been studied extensively. Our study showed that soybean seedlings grown in continuous dark conditions had a slightly lower germination percentage than those grown under continuous white light (96.18% vs. 100%) (Supplementary Figure 1A). However, the hypocotyl length (8.085 cm vs. 3.74 cm) (Figure 1A) and root length (11.4 cm vs. 9.475 cm) (Supplementary Figure 1B) of the seedlings grown in the dark were significantly higher than those grown in continuous white light. These results suggest that continuous dark conditions may be beneficial for soybean seedling growth in terms of hypocotyl and root length. To further investigate the physiological differences between these two growth conditions, we performed m6A MeRIP-seq of aboveground tissue on four-day soybean seedlings subjected to continuous dark and continuous light growth conditions. After quality control, high-quality sequencing data were uniquely mapped into the soybean reference genome using STAR (version 2.5.3a) software (Supplementary Figure 2). A total of 123386 m6A peaks with an average length of 200 bp were detected in the total set of soybean samples, of which 61737 m6A enrichment peaks represented the transcripts of 31225 genes expressed in the dark conditions, and 61646 m6A enrichment peaks represented 31600 genes expressed under continuous light. Among them, 29295 genes were detected under both dark and continuous light conditions (Figure 1B). The soybean genome (wm82.a4. v1) contains 52872 protein-coding genes, of which 33530 exhibit m6A methylation, indicating that m6A methylation modification is very common in this plant. Our results suggest that m6A methylation is an important factor in regulating the physiological response of soybean seedlings to different light conditions.
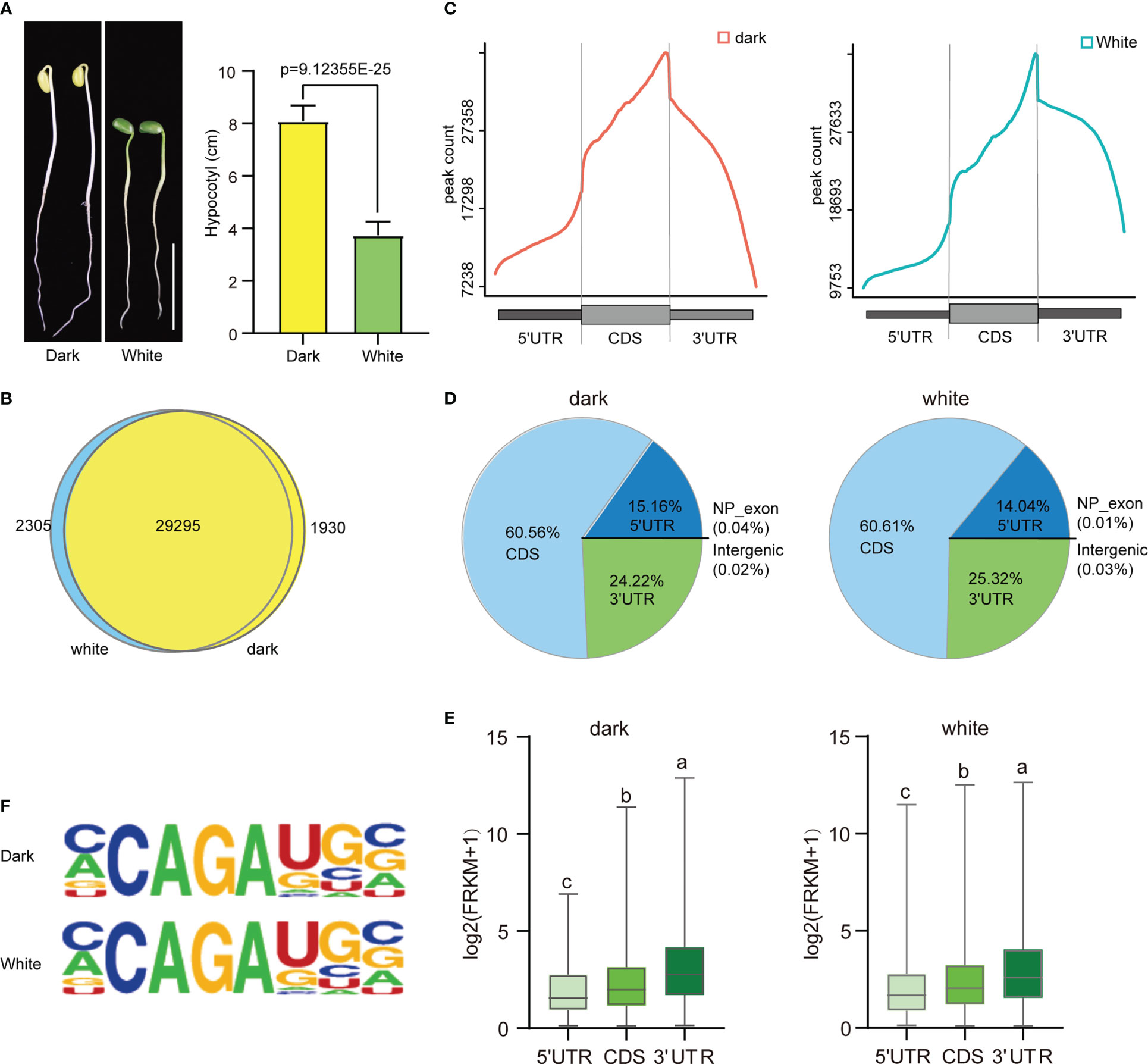
Figure 1 Overview of the m6A methylome in soybean. (A) The phenotype and hypocotyl statistics of Williams 82 grown in the dark and with continuous white light (100 μmol m-2 s-1) for 4 days. Scale bars correspond to 5 cm.The comparisons were performed using Student’s t-tests. Data are shown as means ± SD (n = 15). (B) Venn diagram showing the overlap of m6A peak-containing genes in two independent m6A-seq experiments for wild type plants grown under dark and continuous white light conditions. (C) Distribution of m6A peaks in transcript segments. (D) Pie charts showing the distribution of m6A peaks plants grown under dark and continuous white light conditions. (E) Gene expression (log2(FRKM+1)) of m6A peaks within three transcript segments. (F) Most representative motifs called for each group (dark and white light). The letters above the bars indicate significant differences (P < 0.05) as determined by one-way ANOVA.
Next, we analyzed the distribution of m6A peaks along the 5’UTR, CDS and 3’UTR of genes and found that the m6A peaks were enriched around the stop codon in all the samples (Figure 1C and Supplementary Figure 3), similar to the distributions observed in other animals and plants. Intriguingly, we found that there was a slight difference in the abundance of m6A peaks at different positions under light and dark conditions, with a slight increase within 5′UTRs (15.16% vs. 14.04%) and a slight decrease within CDSs (60.56% vs. 60.61%) and 3′UTRs (24.22% vs. 25.32%) (Figure 1D).
To study the effect of the distribution of the m6A peak on gene expression, we analyzed the expression levels of genes containing only one peak, and the results indicated that m6A peaks in the 3’UTR correlated with the highest levels of gene expression, followed by peaks in the CDS and 5’UTR (Figure 1E).
To determine whether the m6A peaks share the common sequence elements previously characterized in plants, we used homer to search for consensus motifs in the dark and continuous light groups. Conserved motifs, such as CCAGAUGC (dark: p=1e-157; white: p=1e-143) were all enriched in both groups (Figure 1F). Thus, the same sequence motif observed in mammalian and yeast mRNA appears to be necessary for m6A methylation in plant mRNA.
We also analyzed the distribution of m6A on chromosomes and revealed a lower density of m6A peaks at the centromere regions than at other regions (Supplementary Figure 4). Further analysis showed that the average density was the highest on chromosome 13 under both conditions (Supplementary Figure 4).
For AS analysis, we used replicate multivariate analysis of transcript splicing (rMATS) software for differential AS analysis. AS events (ASE) can be grouped into five categories, namely, alternative 3’ splice site (A3SS), alternative 5’ splice site (A5SS), mutually exclusive exon (MXE), skipped exon (SE) and retained intron (RI) (Figure 2A). There are two situations in which reads are aligned to exons: reads on target and junction reads. We calculated the expression level of alternatively spliced transcripts based on the number of junction reads. We screened ASEs with the criteria p value<0.05 and |Δψ|>0.05. The results revealed that 386 differentially spliced genes were detected, with 179 genes were up-regulated and 207 genes down-regulated when exposed to continuous white light compared to dark conditions (Figure 2B). Furthermore, the analysis demonstrated that A3SS and SE were the most frequent alternative splicing events, followed by A5SS, RI, and MXE (Figure 2C). A comparison of the number of genes up-regulated and down-regulated under each splicing event, A3SS (62 vs.75), A5SS (23 vs. 24), MEX (5 vs. 7), RI (20 vs. 20) and SE (69 vs. 81), revealed that the number of genes down-regulated was slightly higher than those up-regulated (Figure 2C). Moreover, the analysis of genes containing only one m6A peak indicated that an m6A peak in the 3’UTR had the greatest effect on alternative splicing in both dark and continuous white light conditions, followed by peaks in the CDS and 5’UTR (Figure 2D). These results suggest that m6A methylation in different transcript regions may affect alternative mRNA splicing in soybean.
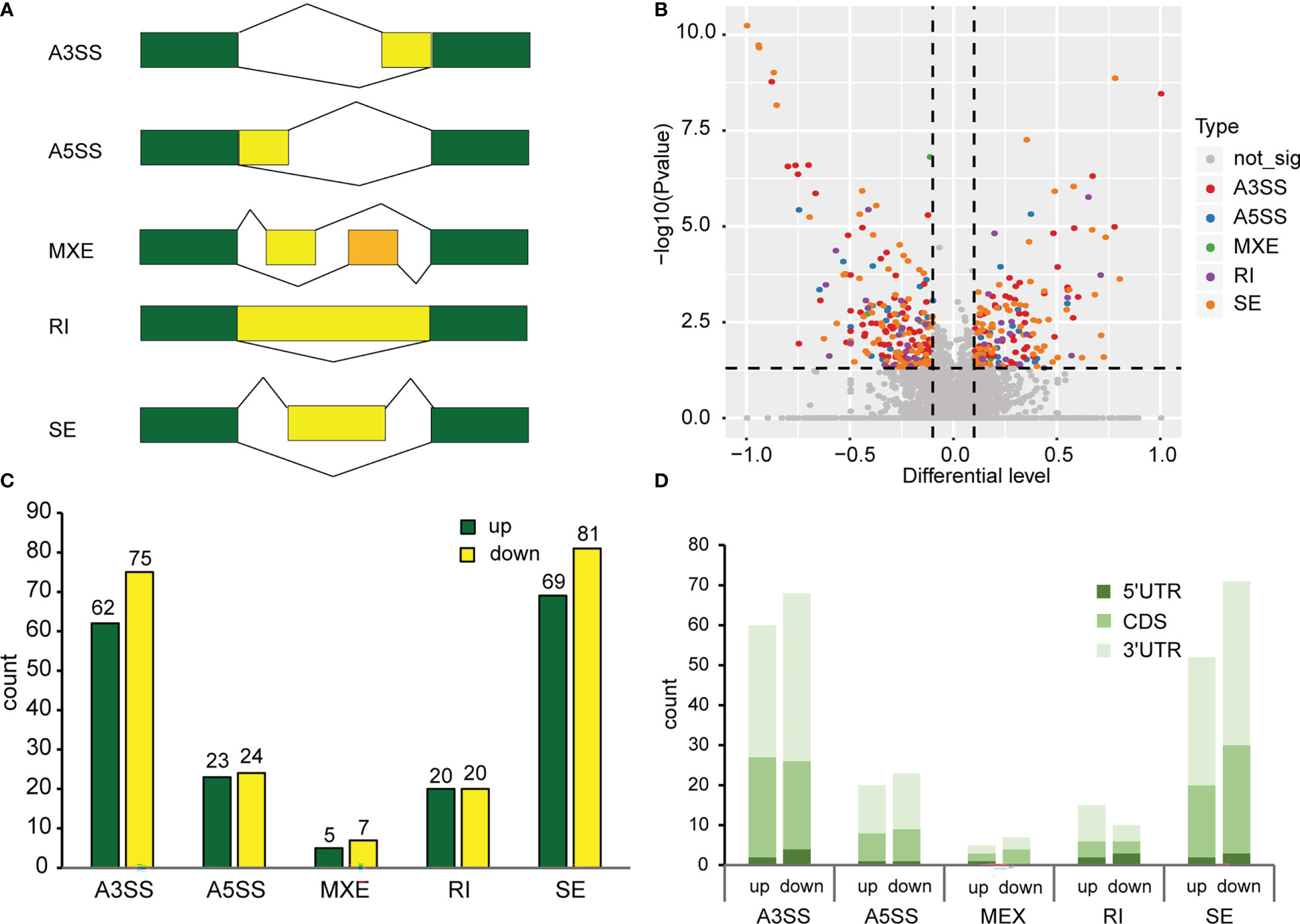
Figure 2 Alternative splicing events in methylated genes. (A) Types of alternative splicing events. A3SS, Alternative 3’ splice site; A5SS, Alternative 5’ splice site; MXE, Mutually exclusive exon; RI, Retained intron; SE, Skipped exon. Green boxes represent constitutive exons. Yellow boxes and orange boxes represent alternatively spliced exons. The broken lines represent the way exons are connected. (B) Analysis of differential alternative splicing. p value<0.05, |Δψ|>0.05. (C) Number of different types of alternative splicing events. (D) The effect of methylation of different transcript regions on alternative splicing.
To study the biological significance of m6A modification in soybean, GO and KEGG pathway analyses of the mRNAs methylated in dark and continuous light conditions were conducted with the p value <0.05 and log2 fold change ≥1. Gene Ontology (GO) enrichment analysis of the genes methylated in the dark showed that “ubiquitin-dependent protein catabolic process”, “organelle membrane” and “modification-dependent protein catabolic process” terms were enriched (Figure 3A). However, Gene Ontology (GO) enrichment analysis of the genes methylated under continuous light showed that “ribonucleoprotein complex assembly”, “cullin-RING ubiquitin ligase complex” and “ATP binding” terms were enriched (Figure 3B). For KEGG pathway analysis, genes methylated in dark and continuous light were both enriched in the “spliceosome”, “ribosome” and “plant hormone signal transduction” pathways (Figures 3C, D).
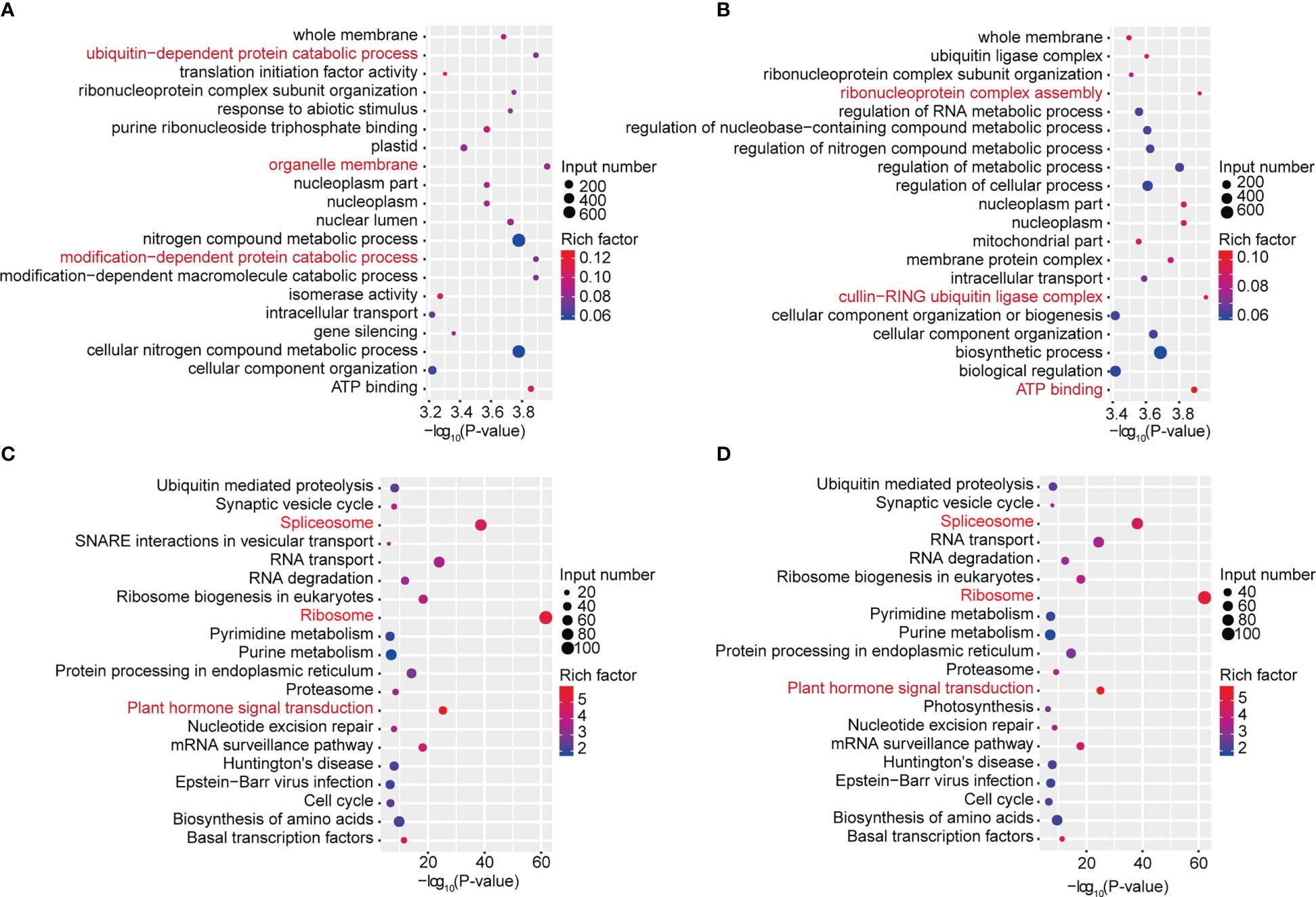
Figure 3 GO and KEGG pathway analyses of all methylated mRNAs. (A) The top 20 GO terms among genes modified by m6A in the dark. (B) The top 20 GO terms among genes modified by m6A under continuous white light. (C) The top 20 KEGG pathways among genes modified by m6A in the dark. (D) The top 20 KEGG pathways among genes modified by m6A under continuous white light.
To explore the roles of m6A methylation in the soybean response to light, we compared the modification profiles of seedlings grown in the dark and under continuous light treatment. Using RNA-seq, we characterized the transcriptome profiles of soybean seedlings from the two groups. Differentially expressed genes were detected. Compared to samples grown under continuous light, samples grown in the dark had 1,533 significantly upregulated genes and 1,438 significantly downregulated genes (Figures 4A, B). Gene Ontology (GO) enrichment analysis of upregulated genes revealed that terms related to “RNA biosynthetic process”, “nucleic acid-templated transcription” and “biological regulation” were significantly enriched (Supplementary Figure 5A). Similarly, GO enrichment analysis of downregulated genes demonstrated that terms related to “single-organism metabolic process”, “single-organism biosynthetic process” and “catalytic activity” were significantly enriched (Supplementary Figure 5B). KEGG pathway analysis revealed that upregulated genes were enriched in “Plant hormone signal transduction”, “Phenylpropanoid biosynthesis” and “Biosynthesis of secondary metabolites” pathways (Supplementary Figure 5C), while downregulated genes were enriched in “Photosynthesis”, “Photosynthesis - antenna proteins” and “Carbon fixation in photosynthetic organisms” pathways (Supplementary Figure 5D). Next, using MeRIP-seq analysis, we found 1,211 upregulated m6A peak-related genes and 3569 downregulated m6A peak-related genes. We then performed codifferential analysis to further associate m6A modification and gene expression. As a result, all differentially methylated m6A peaks with differential mRNA levels (418) were identified by conjoint analysis of the MeRIP-seq and RNA-seq data. Finally, we found that 111 genes exhibited both a significant upregulation of m6A and a significant upregulation of expression, 29 genes exhibited both a significant upregulation of m6A and a significantly downregulation of expression, 90 genes exhibited both a significant downregulation of m6A and a significant upregulation of expression, and 188 differential genes exhibited both a significant downregulation of m6A and a significant downregulation of expression (Figure 4B). We randomly selected a number of these 418 genes and used m6A IP qPCR and qRT–PCR assays to detect their methylation modification levels and transcription levels, respectively (Figure 4C; Supplementary Figure 6). The results were consistent with the MeRIP-seq and RNA-seq results, further verifying the accuracy of the sequencing results. We also examined the changes in the methylation modification levels and transcription levels of several of these genes in Tianlong 1, another soybean cultivar, and the results were consistent with the sequencing results (Supplementary Figure 7), thus verifying the conservation of m6A methylation modification among soybean varieties.
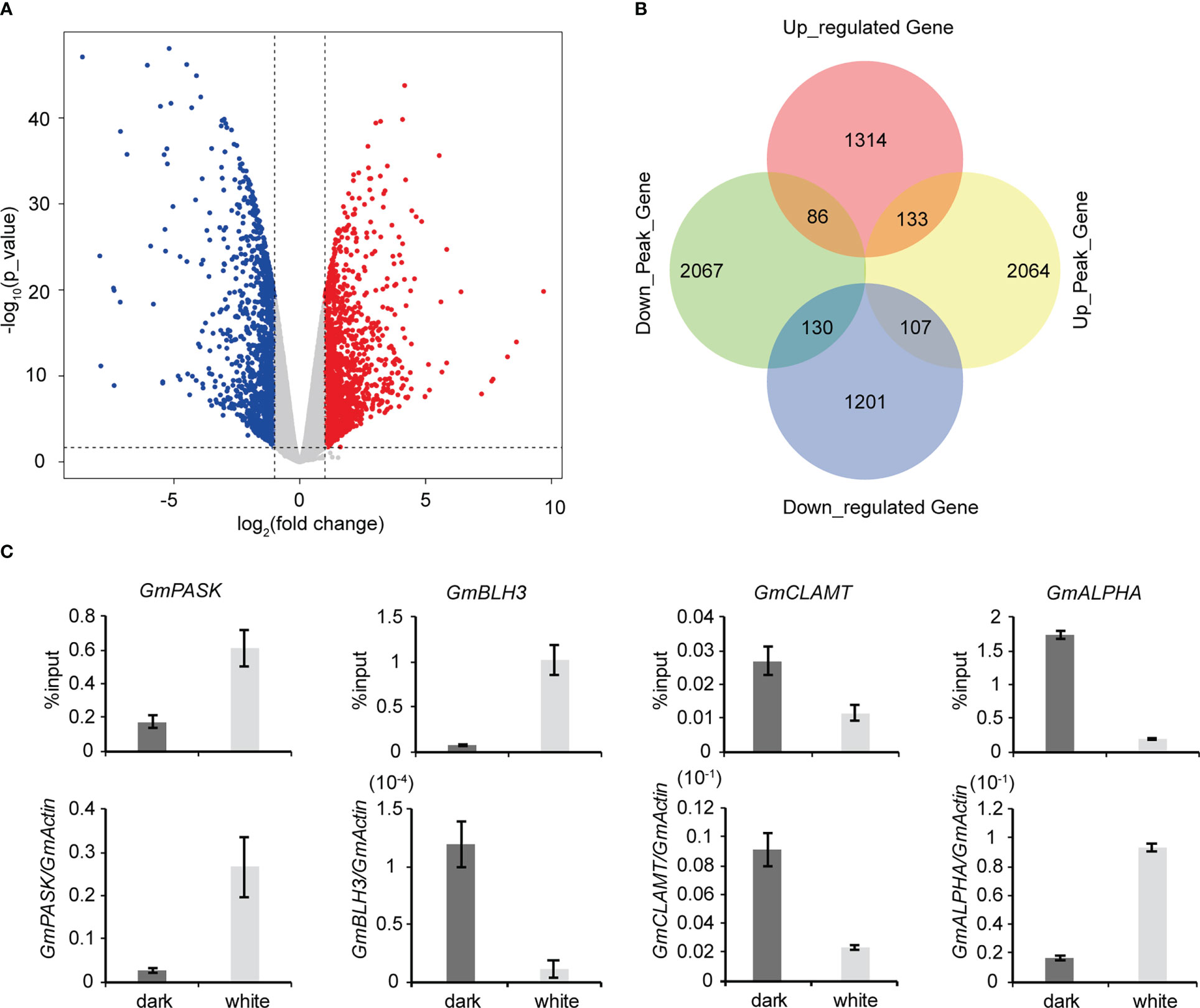
Figure 4 Conjoint analysis of MeRIP-seq and RNA-seq data. (A) Scatter plots showing the differential expression of genes under dark and continuous white light conditions. |logFC|>1, FC= RPKM (dark)/RPKM (white), p value<0.05. (B) Venn diagram showing the distribution of genes with significant changes in both the m6A peaks and mRNA levels. (C) Validation of differential m6A modification and differential gene expression. The values represent the means ± SD (n=3).
To further characterize the important pathways involved in differentially expressed genes with differential m6A peaks, we performed GO and KEGG pathway analyses to investigate the biological significance of those 418 genes with synchronous differential expression and differential m6A peaks. Gene Ontology (GO) enrichment analysis showed that the biological processes “transferase activity, transferring acyl-groups other than amino-acyl groups”, “transferase activity, transferring acyl groups” and “fatty acid biosynthetic process” were significantly enriched (Figure 5A). Moreover, KEGG pathway analysis revealed that in addition to “starch and sucrose metabolism”, “plant hormone signal transduction” and “phenylpropanoid biosynthesis”, the three most significant pathways, the “photosynthesis” and “circadian rhythm” pathways were also associated with differentially expressed genes with differential m6A peaks (Figure 5B). Next, we used qRT–PCR assays to detect related genes involved in circadian rhythm (Supplementary Figure 8A), plant hormone signal transduction (Supplementary Figure 8B) and photosynthesis (Supplementary Figure 8C) pathways. The results indicated that were consistent with the RNA-seq results. Taken together, these results suggested that differentially methylated m6A peaks with differential mRNA levels were involved in important signaling pathways.
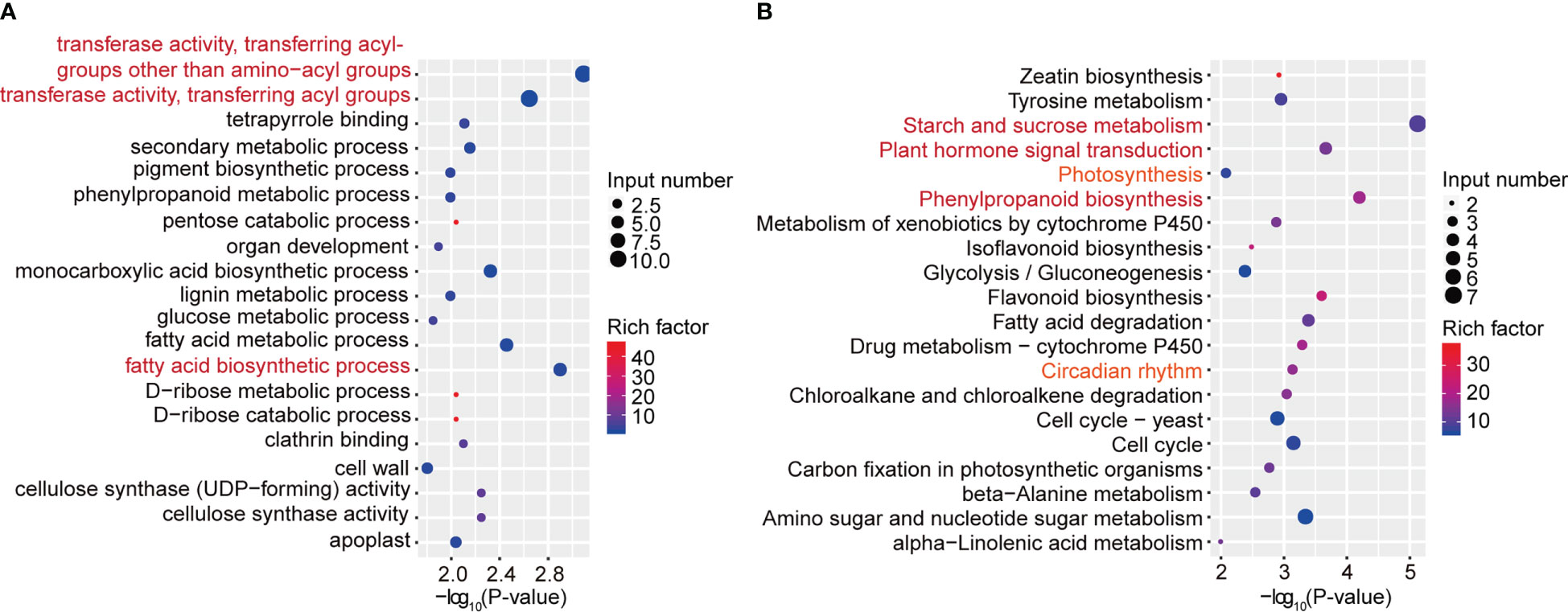
Figure 5 GO and KEGG pathway analyses of differentially expressed genes with differential m6A peaks. (A) The top 20 GO terms among differentially expressed genes with differentially m6A peaks. (B) The top 20 KEGG pathways among differentially expressed genes with differentially m6A peaks.
To further explore the effect of m6A methylation on the soybean growth response to light, methylation modification levels and transcription levels of core genes in the light signaling pathway were detected and compared between the light and dark groups. Among the 418 differentially expressed genes with differential m6A peaks, we found three core light signaling pathway genes, namely, GmPRR5e (Glyma.19G260400), GmBIC2b (Glyma.13G316500) and GmSPA1a (Glyma.16G027200). GmPRR5e and GmBIC2b were upregulated in the light group and exhibited an m6A peak in their CDS regions (Figures 6A, B). GmSPA1a was also upregulated in the light group but the m6A peak was observed in its 3’UTR region (Figure 6C). To validate these findings, we used m6A IP qPCR and qRT–PCR assays to detect the methylation modification levels and transcription levels of these genes, respectively (Figure 6D). The results were consistent with the sequencing results, indicating that the transcription levels of these three genes were significantly increased in response to light. Therefore, the changes in the m6A levels and expression levels of light signaling pathway-related genes in soybean under light conditions may contribute to the phenotypic differences observed in soybean in response to light. Our findings suggest that light affects the levels of methylation modification in soybean seedlings, and that changes in the levels of methylation modification may be essential for soybean growth.
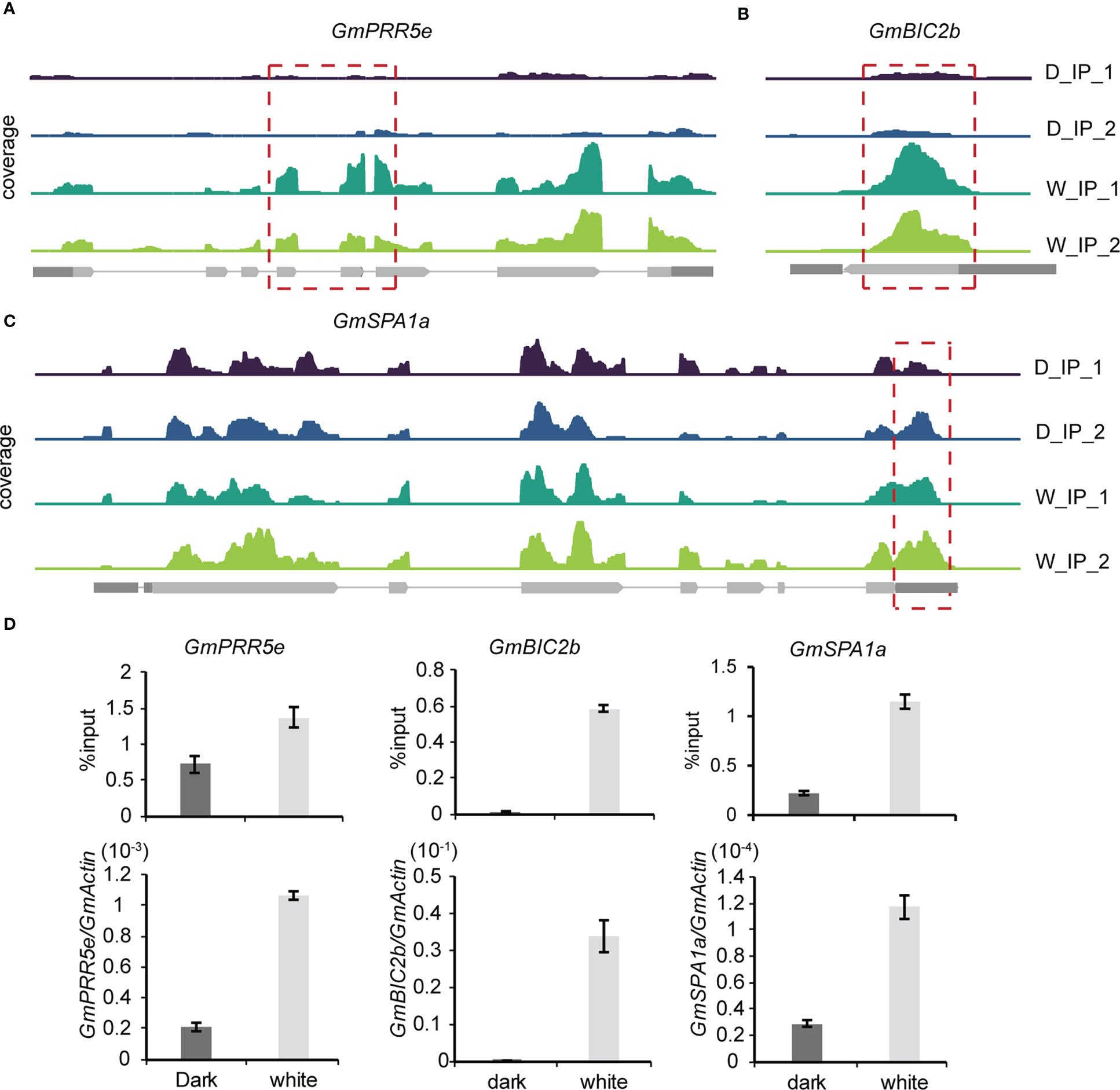
Figure 6 Gene expression-regulated m6A modification correlates with light signaling. (A–C), Genomic visualization of m6A density maps of representative genes of the light signaling pathway (GmPRR5e, GmBIC2b and GmSPA1a). (D) The m6A abundance of individual sites of selected genes and their expression levels were analyzed by m6A-IP-qPCR and RT–qPCR. The values represent the means ± SD (n=3).
Recent studies have demonstrated that m6A writers, erasers and readers are essential for normal plant development and are involved in the plant response to abiotic stresses (Zhong et al., 2008; Bodi et al., 2012; Hu et al., 2019). However, the role of m6A methylation in light mediated responses in soybean remains largely unexplored. While it is known that light and circadian cycles can affect the stability and translation of specific plant transcripts (Tang et al., 2003; Juntawong and Bailey-Serres, 2012), further research is needed to investigate the effects of light and circadian rhythm on soybean m6A methylation and its associated mechanisms.
In this study, we used soybean as a material to investigate the dynamic modification of m6A under light and dark conditions. Our findings showed that the distribution characteristics of m6A in soybean are similar to those observed in Arabidopsis and other plants, with m6A being significantly enriched at stop codons and 3’UTRs, and the conserved CCAGAUGC motifs being found in yeast, plants, and mammals (Dominissini et al., 2012; Schwartz et al., 2013). Furthermore, we observed that the genes with m6A modifications in the 3’UTR had higher expression levels, which is contrary to the findings in mammalian systems, where m6A modification around stop codons and at 3’UTRs is negatively correlated with gene expression (Liu et al., 2014; Wang X. et al., 2014; Wang Y. et al., 2014). In addition, we found that in soybean, the genes with m6A in the 3’UTR are more prone to alternative splicing. Studies in animals have demonstrated that YTHDC1/SRSF3 and SRSF10 differentially regulate AS in a m6A-dependent manner, and the binding regions of YTHDC1, SRSF3 and SRSF10 were significantly enriched in coding sequences and 3’UTRs, concurrent with the m6A distribution pattern (Adhikari et al., 2016). This indicates that the role of m6A modification in plants may be distinct from that in animals.
Soybean is a photoperiod-sensitive crop, which means it is essential to investigate the influence of light on soybean growth and development in order to maximize its adaptability and productivity. Studies have demonstrated that photoperiodic responses in soybean are largely modulated by specific photoreceptors, such as phytochromes, cryptochromes, and phototropins, which enable the plant to sense the daylength and respond accordingly (Liu et al., 2016). Several genes in the light signaling pathway have been identified, such as the COP1/SPA complex, which can regulate photomorphogenic responses appropriate for the light environment (Podolec and Ulm, 2018). BIC1 and BIC2 have been shown to inhibit cryptochrome function and regulate hypocotyl elongation in Arabidopsis (Wang et al., 2017). PRR5 has been found to play a crucial role in the regulation of flowering and the circadian clock in plants (Uehara et al., 2019; Nakamichi et al., 2020). Our conjoint analysis of MeRIP-seq and RNA-seq data revealed 418 differentially expressed genes with differential m6A peaks, including GmSPA1a, GmPRR5e and GmBIC2b, providing evidence for a link between soybean light signaling and m6A methylation. KEGG pathway analysis indicated that these differentially expressed genes with differential m6A peaks were involved in the “photosynthesis” and “circadian rhythm” pathways, suggesting that m6A methylation may be involved in the regulation of plant photosynthesis and the circadian clock in soybean. This comprehensive map of light-regulated m6A modification in soybean lays a solid foundation for further research into the functional role of light on RNA m6A modification in soybean.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author. The original high-throughput m6A-seq and RNA-seq data had been deposited in GSA (https://ngdc.cncb.ac.cn/gsa/). The accession No is PRJCA015274.
Contributions TZ designed the studies. LZ, YZ, JL, HL, and BL performed the experiments. LZ conducted the bioinformatic analyses. TZ wrote the manuscript. All authors contributed to the article and approved the submitted version.
This research was funded by the National Natural Science Foundation of China (32072091), the National Natural Science Foundation of China (32171965).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1153840/full#supplementary-material
Adhikari, S., Xiao, W., Zhao, Y. L., Yang, Y. G. (2016). m(6)A: Signaling for mRNA splicing. RNA Biol. 13 (9), 756–759. doi: 10.1080/15476286.2016.1201628
Boccaletto, P., Machnicka, M. A., Purta, E., Piatkowski, P., Baginski, B., Wirecki, T. K., et al. (2018). MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 46 (D1), D303–d307. doi: 10.1093/nar/gkx1030
Bodi, Z., Zhong, S., Mehra, S., Song, J., Graham, N., Li, H., et al. (2012). Adenosine methylation in Arabidopsis mRNA is associated with the 3' end and reduced levels cause developmental defects. Front. Plant Sci. 3. doi: 10.3389/fpls.2012.00048
Cantara, W. A., Crain, P. F., Rozenski, J., McCloskey, J. A., Harris, K. A., Zhang, X., et al. (2011). The RNA modification database, RNAMDB: 2011 update. Nucleic Acids Res. 39 (Database issue), D195–D201. doi: 10.1093/nar/gkq1028
Cheng, Q., Wang, P., Wu, G., Wang, Y., Tan, J., Li, C., et al. (2021). Coordination of m(6)A mRNA methylation and gene transcriptome in rice response to cadmium stress. Rice (N Y) 14 (1), 62. doi: 10.1186/s12284-021-00502-y
Dominissini, D., Moshitch-Moshkovitz, S., Schwartz, S., Salmon-Divon, M., Ungar, L., Osenberg, S., et al. (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485 (7397), 201–206. doi: 10.1038/nature11112
Eriksson, M. E., Hanano, S., Southern, M. M., Hall, A., Millar, A. J. (2003). Response regulator homologues have complementary, light-dependent functions in the Arabidopsis circadian clock. Planta 218 (1), 159–162. doi: 10.1007/s00425-003-1106-4
Frye, M., Harada, B. T., Behm, M., He, C. (2018). RNA Modifications modulate gene expression during development. Science 361 (6409), 1346–1349. doi: 10.1126/science.aau1646
Hartmann, A. M., Nayler, O., Schwaiger, F. W., Obermeier, A., Stamm, S. (1999). The interaction and colocalization of Sam68 with the splicing-associated factor YT521-b in nuclear dots is regulated by the src family kinase p59(fyn). Mol. Biol. Cell 10 (11), 3909–3926. doi: 10.1091/mbc.10.11.3909
Haussmann, I. U., Bodi, Z., Sanchez-Moran, E., Mongan, N. P., Archer, N., Fray, R. G., et al. (2016). m(6)A potentiates sxl alternative pre-mRNA splicing for robust drosophila sex determination. Nature 540 (7632), 301–304. doi: 10.1038/nature20577
Hoecker, U. (2017). The activities of the E3 ubiquitin ligase COP1/SPA, a key repressor in light signaling. Curr. Opin. Plant Biol. 37, 63–69. doi: 10.1016/j.pbi.2017.03.015
Hu, J., Cai, J., Park, S. J., Lee, K., Li, Y., Chen, Y., et al. (2021). N(6) -methyladenosine mRNA methylation is important for salt stress tolerance in Arabidopsis. Plant J. 106 (6), 1759–1775. doi: 10.1111/tpj.15270
Hu, J., Manduzio, S., Kang, H. (2019). Epitranscriptomic RNA methylation in plant development and abiotic stress responses. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.00500
Huong, T.T., Ngoc, L.N.T., Kang, H. (2020). Functional Characterization of a Putative RNA Demethylase ALKBH6 in Arabidopsis Growth and Abiotic Stress Responses. Int. J. Mol. Sci. 21 (18), 6707. doi: 10.3390/ijms21186707
Hwang, G., Park, J., Kim, S., Park, J., Seo, D., Oh, E. (2021). Overexpression of BBX18 promotes thermomorphogenesis through the PRR5-PIF4 pathway. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.782352
Juntawong, P., Bailey-Serres, J. (2012). Dynamic light regulation of translation status in Arabidopsis thaliana. Front. Plant Sci. 3. doi: 10.3389/fpls.2012.00066
Ke, S., Alemu, E. A., Mertens, C., Gantman, E. C., Fak, J. J., Mele, A., et al. (2015). A majority of m6A residues are in the last exons, allowing the potential for 3' UTR regulation. Genes Dev. 29 (19), 2037–2053. doi: 10.1101/gad.269415.115
Kerner, K., Nagano, S., Lübbe, A., Hoecker, U. (2021). Functional comparison of the WD-repeat domains of SPA1 and COP1 in suppression of photomorphogenesis. Plant Cell Environ. 44 (10), 3273–3282. doi: 10.1111/pce.14148
Li, C., Li, Y. H., Li, Y., Lu, H., Hong, H., Tian, Y., et al. (2020). A domestication-associated gene GmPRR3b regulates the circadian clock and flowering time in soybean. Mol. Plant 13 (5), 745–759. doi: 10.1016/j.molp.2020.01.014
Liu, G., Wang, J., Hou, X. (2020). Transcriptome-Wide N(6)-Methyladenosine (m(6)A) Methylome Profiling of Heat Stress in Pak-choi (Brassica rapa ssp. chinensis). Plants (Basel) 9 (9), 1080. doi: 10.3390/plants9091080
Liu, B., Yang, Z., Gomez, A., Liu, B., Lin, C., Oka, Y. (2016). Signaling mechanisms of plant cryptochromes in Arabidopsis thaliana. J. Plant Res. 129 (2), 137–148. doi: 10.1007/s10265-015-0782-z
Liu, J., Yue, Y., Han, D., Wang, X., Fu, Y., Zhang, L., et al. (2014). A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 10 (2), 93–95. doi: 10.1038/nchembio.1432
Lu, L., Zhang, Y., He, Q., Qi, Z., Zhang, G., Xu, W., et al. (2020). MTA, an RNA m(6)A methyltransferase, enhances drought tolerance by regulating the development of trichomes and roots in poplar. Int. J. Mol. Sci. 21 (7), 2462. doi: 10.3390/ijms21072462
Lu, S., Zhao, X., Hu, Y., Liu, S., Nan, H., Li, X., et al. (2017). Natural variation at the soybean J locus improves adaptation to the tropics and enhances yield. Nat. Genet. 49 (5), 773–779. doi: 10.1038/ng.3819
Luo, J. H., Wang, Y., Wang, M., Zhang, L. Y., Peng, H. R., Zhou, Y. Y., et al. (2020). Natural variation in RNA m(6)A methylation and its relationship with translational status. Plant Physiol. 182 (1), 332–344. doi: 10.1104/pp.19.00987
Meyer, K. D., Jaffrey, S. R. (2014). The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat. Rev. Mol. Cell Biol. 15 (5), 313–326. doi: 10.1038/nrm3785
Meyer, K. D., Patil, D. P., Zhou, J., Zinoviev, A., Skabkin, M. A., Elemento, O., et al. (2015). 5' UTR m(6)A promotes cap-independent translation. Cell 163 (4), 999–1010. doi: 10.1016/j.cell.2015.10.012
Meyer, K. D., Saletore, Y., Zumbo, P., Elemento, O., Mason, C. E., Jaffrey, S. R. (2012). Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell 149 (7), 1635–1646. doi: 10.1016/j.cell.2012.05.003
Michael, T. P., Salomé, P. A., Yu, H. J., Spencer, T. R., Sharp, E. L., McPeek, M. A., et al. (2003). Enhanced fitness conferred by naturally occurring variation in the circadian clock. Science 302 (5647), 1049–1053. doi: 10.1126/science.1082971
Mortazavi, A., Williams, B.A., McCue, K., Schaeffer, L., Wold, B.. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5(7), 621–628. doi: 10.1038/nmeth.1226
Mu, R., Lyu, X., Ji, R., Liu, J., Zhao, T., Li, H., et al. (2022). GmBICs modulate low blue light-induced stem elongation in soybean. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.803122
Nakamichi, N., Kudo, T., Makita, N., Kiba, T., Kinoshita, T., Sakakibara, H. (2020). Flowering time control in rice by introducing Arabidopsis clock-associated PSEUDO-RESPONSE REGULATOR 5. Biosci. Biotechnol. Biochem. 84 (5), 970–979. doi: 10.1080/09168451.2020.1719822
Podolec, R., Ulm, R. (2018). Photoreceptor-mediated regulation of the COP1/SPA E3 ubiquitin ligase. Curr. Opin. Plant Biol. 45 (Pt A), 18–25. doi: 10.1016/j.pbi.2018.04.018
Ponnu, J., Hoecker, U. (2021). Illuminating the COP1/SPA ubiquitin ligase: Fresh insights into its structure and functions during plant photomorphogenesis. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.662793
Roundtree, I. A., Evans, M. E., Pan, T., He, C. (2017). Dynamic RNA modifications in gene expression regulation. Cell 169 (7), 1187–1200. doi: 10.1016/j.cell.2017.05.045
Schwartz, S., Agarwala, S. D., Mumbach, M. R., Jovanovic, M., Mertins, P., Shishkin, A., et al. (2013). High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell 155 (6), 1409–1421. doi: 10.1016/j.cell.2013.10.047
Schwartz, S., Mumbach, M. R., Jovanovic, M., Wang, T., Maciag, K., Bushkin, G. G., et al. (2014). Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5' sites. Cell Rep. 8 (1), 284–296. doi: 10.1016/j.celrep.2014.05.048
Shen, L., Liang, Z., Gu, X., Chen, Y., Teo, Z. W., Hou, X., et al. (2016). N(6)-methyladenosine RNA modification regulates shoot stem cell fate in Arabidopsis. Dev. Cell 38 (2), 186–200. doi: 10.1016/j.devcel.2016.06.008
Shen, L., Liang, Z., Wong, C. E., Yu, H. (2019). Messenger RNA modifications in plants. Trends Plant Sci. 24 (4), 328–341. doi: 10.1016/j.tplants.2019.01.005
Shi, H., Wang, X., Lu, Z., Zhao, B. S., Ma, H., Hsu, P. J., et al. (2017). YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 27 (3), 315–328. doi: 10.1038/cr.2017.15
Slobodin, B., Han, R., Calderone, V., Vrielink, J., Loayza-Puch, F., Elkon, R., et al. (2017). Transcription impacts the efficiency of mRNA translation via Co-transcriptional N6-adenosine methylation. Cell 169 (2), 326-337 e312. doi: 10.1016/j.cell.2017.03.031
Tang, L., Bhat, S., Petracek, M. E. (2003). Light control of nuclear gene mRNA abundance and translation in tobacco. Plant Physiol. 133 (4), 1979–1990. doi: 10.1104/pp.103.029686
Uehara, T. N., Mizutani, Y., Kuwata, K., Hirota, T., Sato, A., Mizoi, J., et al. (2019). Casein kinase 1 family regulates PRR5 and TOC1 in the Arabidopsis circadian clock. Proc. Natl. Acad. Sci. U.S.A. 116 (23), 11528–11536. doi: 10.1073/pnas.1903357116
Wang, X., Jiang, B., Gu, L., Chen, Y., Mora, M., Zhu, M., et al. (2021). A photoregulatory mechanism of the circadian clock in Arabidopsis. Nat. Plants 7 (10), 1397–1408. doi: 10.1038/s41477-021-01002-z
Wang, Y., Li, Y., Toth, J. I., Petroski, M. D., Zhang, Z., Zhao, J. C. (2014). N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 16 (2), 191–198. doi: 10.1038/ncb2902
Wang, X., Lu, Z., Gomez, A., Hon, G. C., Yue, Y., Han, D., et al. (2014). N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505 (7481), 117–120. doi: 10.1038/nature12730
Wang, X., Wang, Q., Han, Y. J., Liu, Q., Gu, L., Yang, Z., et al. (2017). A CRY-BIC negative-feedback circuitry regulating blue light sensitivity of Arabidopsis. Plant J. 92 (3), 426–436. doi: 10.1111/tpj.13664
Wang, S., Wang, H., Xu, Z., Jiang, S., Shi, Y., Xie, H., et al. (2023). m6A mRNA modification promotes chilling tolerance and modulates gene translation efficiency in Arabidopsis. Plant Physiol. doi: 10.1093/plphys/kiad112
Wang, L., Zhuang, H., Fan, W., Zhang, X., Dong, H., Yang, H., et al. (2022). m(6)A RNA methylation impairs gene expression variability and reproductive thermotolerance in Arabidopsis. Genome Biol. 23 (1), 244. doi: 10.1186/s13059-022-02814-8
Wei, L. H., Song, P., Wang, Y., Lu, Z., Tang, Q., Yu, Q., et al. (2018). The m(6)A reader ECT2 controls trichome morphology by affecting mRNA stability in Arabidopsis. Plant Cell 30 (5), 968–985. doi: 10.1105/tpc.17.00934
Xiao, W., Adhikari, S., Dahal, U., Chen, Y. S., Hao, Y. J., Sun, B. F., et al. (2016). Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol. Cell 61 (4), 507–519. doi: 10.1016/j.molcel.2016.01.012
Yamamoto, Y., Sato, E., Shimizu, T., Nakamich, N., Sato, S., Kato, T., et al. (2003). Comparative genetic studies on the APRR5 and APRR7 genes belonging to the APRR1/TOC1 quintet implicated in circadian rhythm, control of flowering time, and early photomorphogenesis. Plant Cell Physiol. 44 (11), 1119–1130. doi: 10.1093/pcp/pcg148
Yang, Y., Hsu, P. J., Chen, Y. S., Yang, Y. G. (2018). Dynamic transcriptomic m(6)A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. 28 (6), 616–624. doi: 10.1038/s41422-018-0040-8
Yue, H., Nie, X., Yan, Z., Weining, S. (2019). N6-methyladenosine regulatory machinery in plants: composition, function and evolution. Plant Biotechnol. J. 17 (7), 1194–1208. doi: 10.1111/pbi.13149
Zhang, M., Zeng, Y., Peng, R., Dong, J., Lan, Y., Duan, S., et al. (2022). N(6)-methyladenosine RNA modification regulates photosynthesis during photodamage in plants. Nat. Commun. 13 (1), 7441. doi: 10.1038/s41467-022-35146-z
Zheng, G., Dahl, J. A., Niu, Y., Fedorcsak, P., Huang, C. M., Li, C. J., et al. (2013). ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 49 (1), 18–29. doi: 10.1016/j.molcel.2012.10.015
Keywords: soybean, M6A, light response, signaling pathway, epitranscriptomic modification
Citation: Zhang L, Zhang Y, Liu J, Li H, Liu B and Zhao T (2023) N6-methyladenosine mRNA methylation is important for the light response in soybean. Front. Plant Sci. 14:1153840. doi: 10.3389/fpls.2023.1153840
Received: 30 January 2023; Accepted: 20 March 2023;
Published: 04 April 2023.
Edited by:
Madhusmita Panigrahy, National Institute of Science Education and Research (NISER), IndiaCopyright © 2023 Zhang, Zhang, Liu, Li, Liu and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Zhao, emhhb3RhbzAyQGNhYXMuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.