 Jian Zhong1,2
Jian Zhong1,2 Junjie Cui3
Junjie Cui3 Mingjun Miao2Fang Hu4
Mingjun Miao2Fang Hu4 Jichi Dong1Jia Liu1Chunfeng Zhong1
Jichi Dong1Jia Liu1Chunfeng Zhong1 Jiaowen Cheng1*
Jiaowen Cheng1* Kailin Hu1*
Kailin Hu1*- 1Key Laboratory of Biology and Genetic Improvement of Horticultural Crops (South China), College of Horticulture, South China Agricultural University, Guangzhou, China
- 2Horticulture Research Institute, Sichuan Academy of Agricultural Sciences, Chengdu, Sichuan, China
- 3Department of Horticulture, Foshan University, Foshan, China
- 4Henry Fok School of Biology and Agricultural, Shaoguan University, Shaoguan, China
In Cucurbitaceae crops, the first flower node (FFN) is an important agronomic trait which can impact the onset of maturity, the production of female flowers, and yield. However, the gene responsible for regulating FFN in bitter gourd is unknown. Here, we used a gynoecious line (S156G) with low FFN as the female parent and a monoecious line (K8-201) with high FFN as the male parent to obtain F1 and F2 generations. Genetic analysis indicated that the low FFN trait was incompletely dominant over the high FFN trait. A major quantitative trait locus (QTL)-Mcffn and four minor effect QTLs-Mcffn1.1, Mcffn1.2, Mcffn1.3, and Mcffn1.4 were detected by whole-genome re-sequencing-based QTL mapping in the S156G×K8-201 F2 population (n=234) cultivated in autumn 2019. The Mcffn locus was further supported by molecular marker-based QTL mapping in three S156G×K8-201 F2 populations planted in autumn 2019 (n=234), autumn 2020 (n=192), and spring 2022 (n=205). Then, the Mcffn locus was fine-mapped into a 77.98-kb physical region on pseudochromosome MC06 using a large S156G×K8-201 F2 population (n=2,402). MC06g1112, which is a homolog of FLOWERING LOCUS T (FT), was considered as the most likely Mcffn candidate gene according to both expression and sequence variation analyses between parental lines. A point mutation (C277T) in MC06g1112, which results in a P93S amino acid mutation between parental lines, may be responsible for decreasing FFN in bitter gourd. Our findings provide a helpful resource for the molecular marker-assisted selective breeding of bitter gourd.
Introduction
The appearance of the first flower is a signal of the pivotal transition from vegetative to reproductive growth in flowering plants (Pnueli et al., 1998; Zahid et al., 2021). Both the time of first flowering and the first flower node (FFN) are useful for the evaluation of crop maturity, and are thus considered important agronomic traits in crop improvement endeavors (Yuan et al., 2008; Zhang et al., 2018; Zhang et al., 2019). In the model plant Arabidopsis thaliana, FLOWERING LOCUS T (FT) is an important regulator gene in determining the flowering time (Corbesier et al., 2007; Turck et al., 2008). The function of the FT gene has also been characterized in several Cucurbitaceae crops. In cucumber (Cucumis sativus), the CsFT gene has been reported to explain 52.3% of the phenotypic variation in flowering time, and is theorized to have been crucial to the spread of this species from its origin in the tropics to higher latitudes (Lu et al., 2014; Wang et al., 2020). The overexpression of CsFT, as well as Cm-FTL1 and Cm-FTL2 from squash (Cucurbita maxima) and CmFT from melon (Cucumis melo), in Arabidopsis promotes early flowering (Lin et al., 2007; Yang et al., 2022). Furthermore, the overexpression of Arabidopsis-derived AtFT in squash also results in early flowering (Lin et al., 2007).
Several studies have reported that other genes are also associated with the regulation of flowering time in Cucurbitaceae crops. For example, in cucumber, silencing CsGL2-LIKE results in delayed male flowering through inhibition of CsFT expression (Cai et al. (2020). Yi et al. (2020), utilizing haplotype analysis, report that ClGA2/KS is associated with flowering time in watermelon (Citrullus lanatus). The overexpression of cucumber-derived CsTFL1b, a homolog of TERMINAL FLOWER 1 (TFL1), results in later flowering in transgenic Arabidopsis (Zhao et al., 2018; Cai et al., 2020). Contrarily, the overexpression of cucumber-derived CsBCAT (CsBCAT2, CsBCAT3, and CsBCAT7) and CsMADS02 has been shown to accelerate flowering in transgenic Arabidopsis (Lee et al., 2019; Zhou et al., 2019). Although the function of these genes has not been universally verified, these initial reports provide clues for the further dissection of the regulation of flowering time in Cucurbitaceae crops. In addition, several quantitative trait loci (QTLs) have been reported to be associated with flowering time in cucurbits. Pan et al. (2017) and Sheng et al. (2020) identified three and two QTLs associated with flowering time in cucumber, respectively. McGregor et al. (2014) identified a major QTL associated with male flowering time in watermelon, which was later verified by Gimode et al. (2020).
Bitter gourd (Momordica charantia), so named because of its characteristically bitter taste, is an edible and medicinal cucurbit that has been used to treat hypertension, cancer, diabetes, infection, hyperlipidemia, and obesity (Akihisa et al., 2007; Zhang et al., 2012; Wang et al., 2017). Bitter gourd originated in Africa (Schaefer et al., 2009; Schaefer and Renner, 2010) and has become an important crop across Asia, Africa, the Caribbean, and South America, among other regions (Basch et al., 2003). In bitter gourd, low FFN or early flowering is usually considered as an important indicator of the early maturity trait. To date, genetic mapping studies have revealed at least 21 QTLs associated with female flowering time and 12 QTLs associated with male flowering time in bitter gourd (Wang and Xiang, 2013; Cui et al., 2018; Gangadhara Rao et al., 2018; Kaur et al., 2022). However, there are currently no research reports about genetic mapping of the FFN trait in bitter gourd.
Thanks to the completion of the fully sequenced and assembled bitter gourd genome (Urasaki et al., 2017; Cui et al., 2020; Matsumura et al., 2020), the mapping and cloning of genes controlling important agronomic traits has become easier. Like typical cucurbits species such as cucumber or melon, there are many types of sexual plants in bitter gourd, of which monoecy that carries both unisexual male and female flowers and gynoecy that harbors only female flower have been reported (Kole, 2020; Zhong et al., 2023). Here, we used a segregating F2 populations crossing from a gynoecious female parent and a monoecious male parent to elucidate the molecular mechanism of FFN regulation in bitter gourd. We first performed a whole-genome re-sequencing-based QTL mapping to rapidly identify FFN-associated genetic loci. Next, we conducted a molecular marker-based classical QTL mapping to confirm the stability of the major effected QTL in three F2 population cultivated three different environments, respectively. Finally, we fine-mapped the identified candidate gene. Both expression and sequence variation analyses suggest that the candidate gene MC06g1112 regulates FFN in bitter gourd. The results of this study will be invaluable for breeding improved bigger gourds, and further our understanding of the regulation of floral timing in cucurbits.
Materials and methods
Plant materials
A gynoecious, low-FFN (7-10th nodes) inbred line (S156G, P1) (Figure 1A) and a monecious, high-FFN (16-19th nodes) inbred line (K8-201, P2) (Figure 1B) were used as the female and male parents, respectively, to construct the F1 generation, which was then self-crossed to generate the F2 population. Both of the parental lines (S156G and K8-201) had been previously whole-genome re-sequenced (Zhong et al., 2022). All plants, representing four generations (P1, P2, F1, and F2), were cultivated across three quarters (autumn 2019, autumn 2020, and spring 2022) at the experimental field of the SCAU Teaching & Research Base in Zengcheng District, Guangzhou, China (23.24N, 113.64E), under standard agronomic management. Plants from the S156G×K8-201 F2 population (n=234), which were cultivated in autumn 2019, were used to preliminarily map FFN-associated genetic loci by whole-genome re-sequencing-based QTL mapping. Plants from three S156G×K8-201 F2 populations, cultivated in autumn 2019 (n=234), autumn 2020 (n=192), and spring 2022 (n=205), were employed for molecular marker-based QTL mapping to confirm the stability of the major effected QTL. Finally, a large S156G×K8-201 F2 population (n=2,402) was used to fine-map the candidate region associated with FFN. The number of nodes from the node with the first alternate leaf to the node carrying the first flower was used to quantify FFN in bitter gourd.
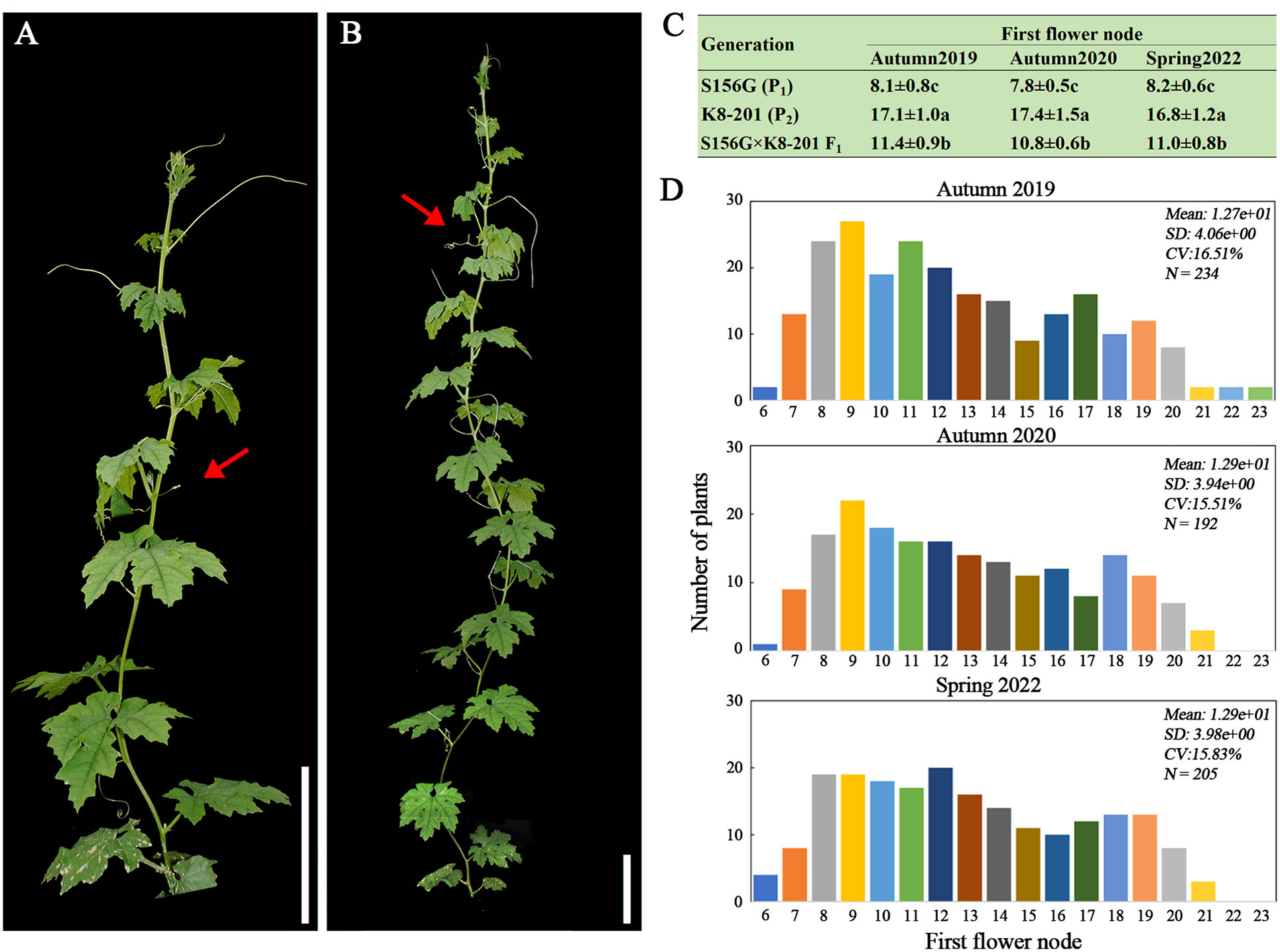
Figure 1 Phenotypic evaluation of the FFN trait. (A) Low-FFN (7-10th nodes) inbred line S156G (P1). Bar=10 cm. (B) High-FFN (16-19th nodes) inbred line K8-201(P2). Bar=10 cm. The red arrows in (A, B) indicate FFNs. (C) Average ( ± SD) FFN values of the S156G, K8-201, and S156G×K8-201 F1 generations recorded from autumn 2019, autumn 2020, and spring 2022. Different lowercase letters indicate statistical significance at the 0.01 level. (D) Phenotypic distribution of the FFN trait from the three S156G×K8-201 F2 populations cultivated in autumn 2019, autumn 2020, and spring 2022.
DNA library preparation for whole-genome re-sequencing
The CTAB method (Porebski et al., 1997) was used to isolate genomic DNA (gDNA) from young leaves, and each sample was stored at -20°C prior to analysis. gDNA isolated from the S156G×K8-201 F2 population (n=234) was utilized to construct DNA sequencing libraries with a KAPA-Hyper Plus Kit (KAPA Biosystems, MA, USA). Briefly, DNA was fragmented by ultrasonication to sizes of 250-350 bp, which were utilized for end-repairing and 3’ adenylation. After the adapters were ligated to the ends of these 3’-adenylated fragments, the products were purified by gel recovery. The recovered products were amplified by polymerase chain reaction (PCR) to construct the DNA sequencing libraries. The quality of the DNA sequencing libraries was evaluated using an Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA) and a Real-Time PCR (qPCR) System (Bio-Rad, CA, USA). Finally, the qualified DNA sequencing libraries were sequenced on an Illumina Nova-Seq platform (Illumina, CA, USA).
Whole-genome re-sequencing-based QTL mapping
Quality control of raw whole-genome re-sequencing data, including removal of adapter sequences and low-quality reads, was conducted with Fastp (Chen et al., 2018). Clean reads were aligned to the Dali-11 reference genome with BMA-MEM2 (Vasimuddin et al., 2019; Cui et al., 2020), and the alignment results were evaluated with Qualimap2 (Okonechnikov et al., 2016). Both single nucleotide polymorphisms (SNPs) and insertions and deletions (InDels) were called with BCTtools (Li, 2011), and all variations were annotated with ANNOVAR (Wang et al., 2010). SNPs with a minor allele frequency <0.05 or a missing call frequency >0.1 were removed with VCFtools (Danecek et al., 2011). High quality SNPs were further used to QTL mapping using QTL package in R language. First, the multiple imputation method was used to calculate the LOD value by QTL scanning, and then the significant threshold of LOD value was obtained by 1000 permutation test. Finally, the confidence interval of the selected QTL was identified by LOD support intervals evaluation method.
Molecular marker-based QTL mapping
The variation in SNPs and InDels between the two parental lines (S156G and K8-201) was obtained by aligning the clean re-sequencing data to the Dali-11 reference genome using SOAP2 (Li et al., 2009). Primers for SNP and InDel molecular markers within the whole-genome re-sequencing-based QTL mapping-delimited candidate region were designed with Primer3 Plus (https://www.primer3plus.com), and SNPs were converted to cleaved amplified polymorphic sequences (CAPS) or derived CAPS (dCAPS) markers. Primer sequences are listed in Supplementary Table 1. PCR was carried out in a 10 µL of reaction volume consisting of 0.2 µL of forward and reverse primers (10 µmol/L), 50-100 ng of DNA template, 5 µL of Green Taq Mix (Vazyme, Nanjing, China), and 3.6 µL of nuclease-free water. The PCR procedure was as follows: initial denaturation at 94 °C for 3 min; 34 cycles of denaturation at 94 °C for 15 s, annealing at 55 °C for 15 s, and extension at 72 °C for 30 s; and final extension at 72 °C for 5 min. The InDel primer-amplified PCR products were directly visualized with 6% polyacrylamide gel electrophoresis (PAGE). The CAPS- or dCAPS-amplified PCR products were first digested with corresponding restriction endonucleases (Supplementary Table 1) at a stationary temperature of 37 °C for 30 min, and the digested products were then visualized with 6% PAGE.
Polymorphic markers from within the molecular marker-based QTL mapping-delimited candidate region were utilized to genotype the three F2 populations cultivated between autumn 2019, autumn 2020, and spring 2022. Genetic distances of those polymorphic markers were calculated with JoinMap 4.0 (Van Ooijen, 2006). Based on marker genotypes and FFN phenotypes of the three F2 populations, FFN-associated QTL mapping was conducted with MapQTL 6.0 using the multiple QTL model (MQM mapping) procedure (Van Ooijen, 2009).
Fine-mapping
Recombinant and non-recombinant members of the three F2 populations were identified using two markers flanking the candidate region identified by molecular marker-based QTL mapping. Non-recombinant plants were divided into three groups (dominant homozygote, recessive homozygote, and heterozygote) depending on whether both flanking markers were identical to S156G, K8-201, or S156G×K8-201 F1, respectively. During the process of fine-mapping, the average FFN values of the recessive homozygote and heterozygote groups were used as a reference to evaluate the FFN phenotype of the recombinant plant. Recombinant plants were divided into two groups: group one plants contained a recombination of the dominant homozygote and heterozygote genotypes, and group two plants contained a recombination of the recessive homozygote and heterozygote genotypes. Only group two plants were utilized for further genotyping with six newly-developed markers from within the flanked region. By using a combination of FFN phenotype data and genotype markers obtained from the group two plants, we identified a more accurate candidate region and two new flanking markers. These two new flanking markers were used to screen the S156G×K8-201 F2 population (n=2,402) for plants containing a recombination of the recessive homozygote and heterozygote genotypes. The selected recombinant plants were then grown in the field to evaluate their FFN phenotypes, and genotyped using nine markers from within the newly-identified flanked region. Finally, by using a combination of FFN phenotype data and genotype markers obtained from these recombinant plants, we delimited the FFN-associated fine-mapping interval.
Expression analysis and cloning of the candidate genes
Prior to RNA extraction, tissue samples, including roots, leaves, petioles, female flowers, sarcocarps, and stems (including the 5th, 10th, 15th, 20th, and 25th node [shoot tip, ST]), were collected from parental plants at the 25-leaf stage and frozen in liquid nitrogen. Three biological replicates were used for all analyses. Total RNA was extracted with an Eastep Super Total RNA Extraction kit (Promega, Shanghai, China), and first-strand cDNA was synthesized with an Eastep RT Master Mix kit (Promega, Shanghai, China), according to the manufacturer’s instructions. Quantitative real-time PCR (qRT-PCR) was performed using a TB Green Premix Ex TaqTM II kit (Takara Bio, Shiga, Japan) on a CFX384 Real-Time System (Bio-Rad, CA, USA). All primers are listed in Supplementary Table 1. Six categories of tissue samples, including roots, stems (15th node), leaves, petioles, female flowers, and sarcocarps, were utilized to perform qRT-PCR for the genes annotated within the fine-mapped interval. The five categories of stem samples were utilized to perform qRT-PCR for the FNN-associated candidate gene. Three technical replicates were used for all assays. The relative expression level of each gene was normalized using the bitter gourd beta-actin gene (MC02g1395) and quantified using the delta-delta Ct method (2-ΔΔCt) (Livak and Schmittgen, 2001).
The primer sequences used to clone the full-length cDNA of the FNN-associated candidate gene were designed according to the gene annotation of Dali-11 reference genome (Cui et al., 2020) (Supplementary Table 1). PCR amplifications of cDNA collected from parental stem (15th node) samples were performed with Phanta Max Super-Fidelity DNA Polymerase (Vazyme, Nanjing, China), according to the manufacturer’s instructions. The PCR products were purified and then ligated into the pMD19-T vector (Takara, Shiga, Japan). At least three positive colonies per amplicon were selected for Sanger sequencing, and the generated sequences were assembled with ContigExpress (Lu and Moriyama, 2004). Both nucleotide and amino acid sequences were aligned with ESPript 3.0 (https://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi).
Results
FFN phenotypic characteristics across four generations
We evaluated the FFN phenotype of each plant across four generations, including P1 (S156G), P2 (K8-201), F1, and F2, planted respectively in autumn 2019, autumn 2020, and spring 2022. Across all three quarters, the FFN of the P1 (S156G) was significantly lower than that of the P2 (K8-201) generation, with the FFN of the P1 (S156G) generation ranging from the 7th to the 9th node (average of ~8th node) and the FFN of the P2 (K8-201) generation ranging from the 15th to the 19th node (average of ~17th node) (Figures 1A-C). The FFN of the F1 generation was significantly higher than the P1 (S156G) generation and lower than the P2 (K8-201) generation, ranging from the 9th to the 13th node (average of ~11th node) (Figure 1C), indicating that the low FFN trait is incompletely dominant over the high FFN trait. The FFN of plants in the three F2 populations was highly stratified but tended toward the low FFN of the P1 (S156G) generation, ranging from the 6th to the 23th node (Figure 1D; Supplementary Table 2).
Whole-genome re-sequencing-based QTL mapping detects FFN-associated genetic locus
Whole-genome re-sequencing of the 234 F2 individuals planted in autumn 2019 resulted in the generation of 557.9 Gb of raw data, and 533.9 Gb of clean data was obtained after filtering (Supplementary Table 3). The clean data exhibited a Q20 of 97.3% and an average sequencing depth of 7.6×, indicating that the data was of high quality enough for subsequent bioinformatics analysis. Approximately 98.0% of the clean reads were aligned to Dali-11 reference genome, with a sample-specific genomic coverage of 88.6% (Supplementary Table 4). After aligning the clean reads to the Dali-11 reference genome (Cui et al., 2020), a total of 175,019 high-quality SNPs were obtained (Supplementary Figure 1). A QTL mapping combining FFN phenotype and SNP data from the 234 F2 individuals identified one ~3.77 Mb FFN-associated major effected QTL designated as the Mcffn locus located between 9.26 Mb and 13.03 Mb on pseudochromosome MC06 (hereafter referred to as MC06), and four minor effected QTLs named Mcffn1.1, Mcffn1.2, Mcffn1.3, and Mcffn1.4 located in pseudochromosome MC01, MC02, MC03, and MC08, respectively. (Figure 2A, Supplementary Table 5).
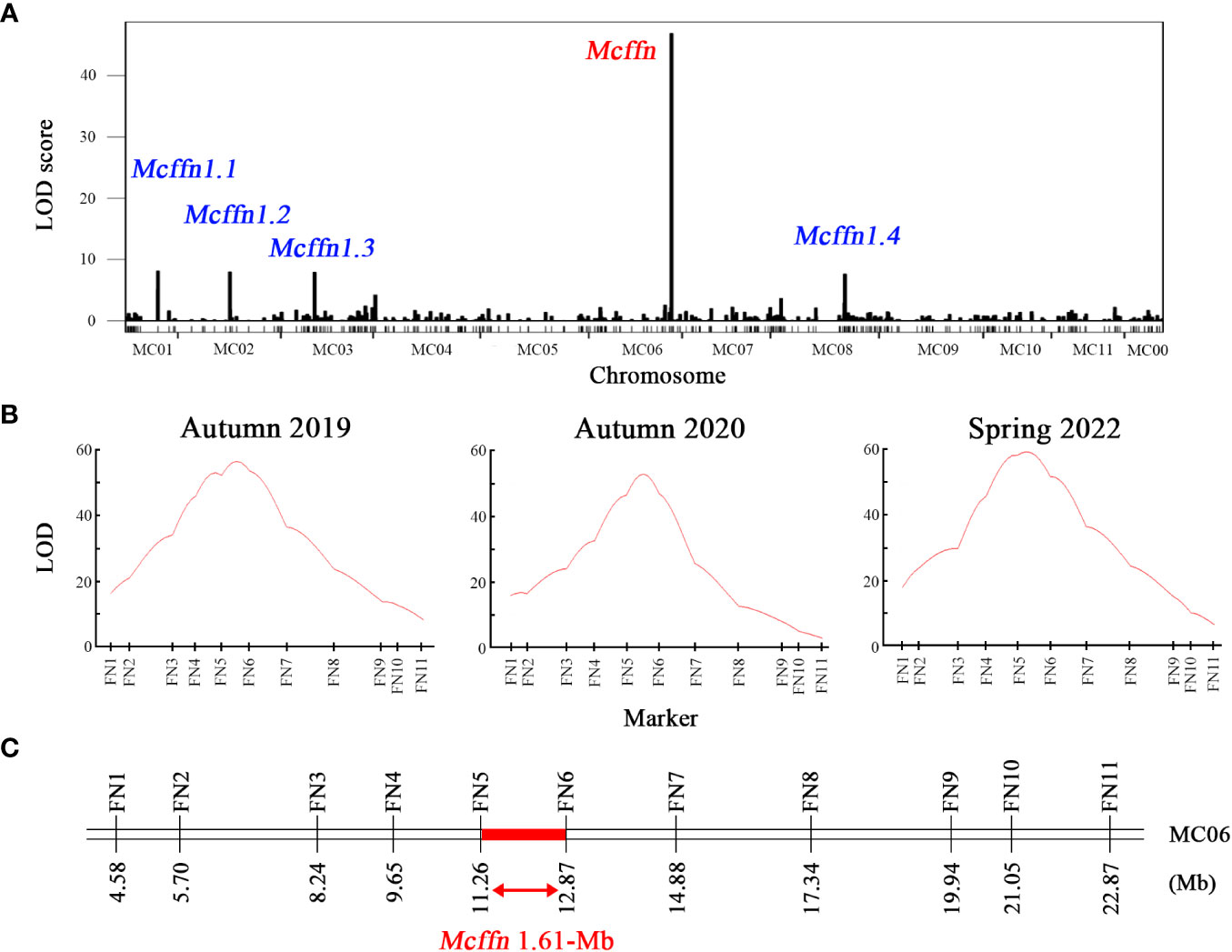
Figure 2 Preliminary mapping of the Mcffn locus. (A) Whole-genome re-sequencing-based QTL mapping in the S156G×K8-201 F2 mapping population (n=234) planted in autumn 2019. (B) Molecular marker-based QTL mapping in the Mcffn locus in three S156G×K8-201 F2 mapping populations planted in autumn 2019, autumn 2020, and spring 2022. (C) Physical map of the molecular markers used for molecular marker-based QTL mapping. The numbers under the bar correspond to the physical positions (Mb). The red bar represents the region of the Mcffn locus delimited by QTL analysis.
The Mcffn locus is narrowed into a 1.61-Mb interval by molecular marker-based QTL mapping
Eleven polymorphic InDel markers (FN1-FN11) were developed within the ~3.77 Mb candidate region (Supplementary Table 1) and used to genotype 631 F2 individuals cultivated in autumn 2019 (n=234), autumn 2020 (n=192), and spring 2022 (n=205). QTL mapping combining FFN phenotype and marker genotype data revealed that the 11 polymorphic InDel markers exhibited different LOD values between the three quarters: 8.50-53.81 in autumn 2019, 3.27-47.01 in autumn 2020, and 6.90-58.70 in spring 2022 (Figure 2B). These results suggested that all of the 11 InDel markers were linked to the FFN phenotypes. It is worth noting that the maximum LOD values of 56.73, 52.87, and 59.76, which explained 67.3%, 71.9%, and 73.9% of the variation in the FFN phenotype in the three F2 populations planted in autumn 2019, autumn 2020, and spring 2022, respectively, were all located between two markers, FN5 and FN6 (Figure 2B). Accordingly, we suggested that the Mcffn locus was located within a 1.61-Mb physical interval between the FN5 (11,262,463 bp) and FN6 (12,873,591 bp) markers on MC06 (Figure 2C).
The Mcffn locus is fine-mapped into a 77.98-kb interval
Based on genotyping with the two flanking markers (FN5 and FN6), the 631 F2 individuals were divided into 579 non-recombinant plants and 52 recombinant plants. In order to study the relationship between Mcffn genotypes and FFN phenotypes, the 579 non-recombinant plants were divided into three groups: 142 dominant homozygotes, 128 recessive homozygotes, and 309 heterozygotes. The FFN phenotype was significantly different between groups across all three quarters, while within-group differences were not significant (Table 1), suggesting that the FFN trait is genetically, rather than environmentally, determined. To reduce the possibility of errors when categorizing plants as either dominant homozygotes or heterozygotes during the fine-mapping process, we used the FFN value of 11.6 ± 2.6 for the heterozygote genotype and 18.3 ± 2.0 for the recessive homozygote genotype (autumn 2019) as reference criteria (Table 1).
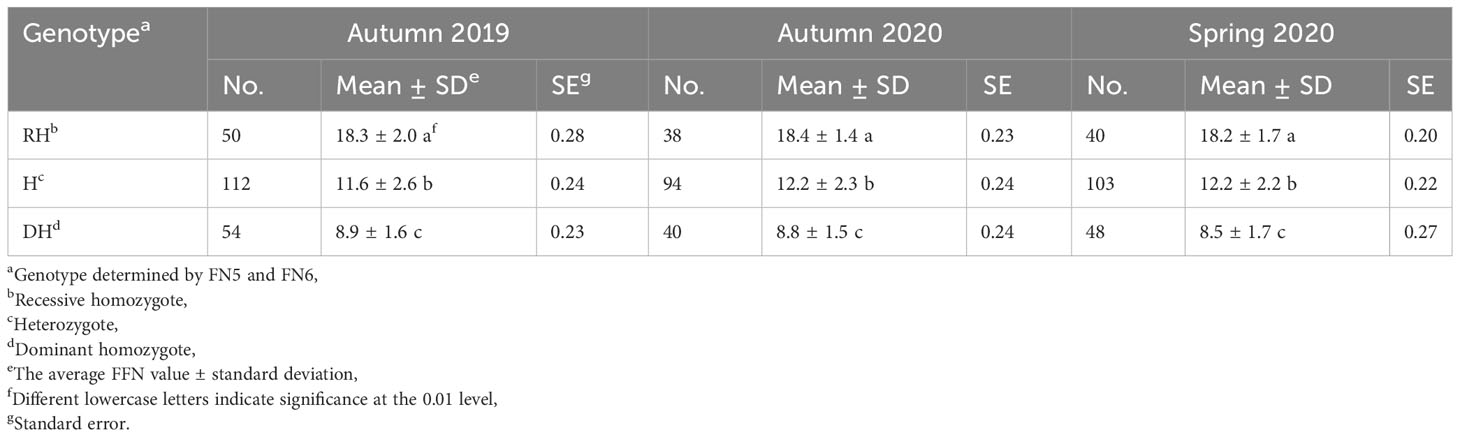
Table 1 Statistical analysis of FFN trait across three genotypes.
The 52 recombinant plants were divided into two groups: group one plants (n=30) contained a recombination of the dominant homozygote and heterozygote genotypes, and group two plants (n=22) contained a recombination of the recessive homozygote and heterozygote genotypes. The group two plants were further divided into nine haplotypes using six newly-developed markers (FN12-FN17) (Figure 3A). By utilizing the FFN phenotype and marker genotype data of the group two plants, as well as the FFN as the reference, the Mcffn locus was further mapped into a 463.01-kb physical interval between the FN13 (11,585,385 bp) and FN16 (12,048,398 bp) markers on MC06 (Figures 3A, B).
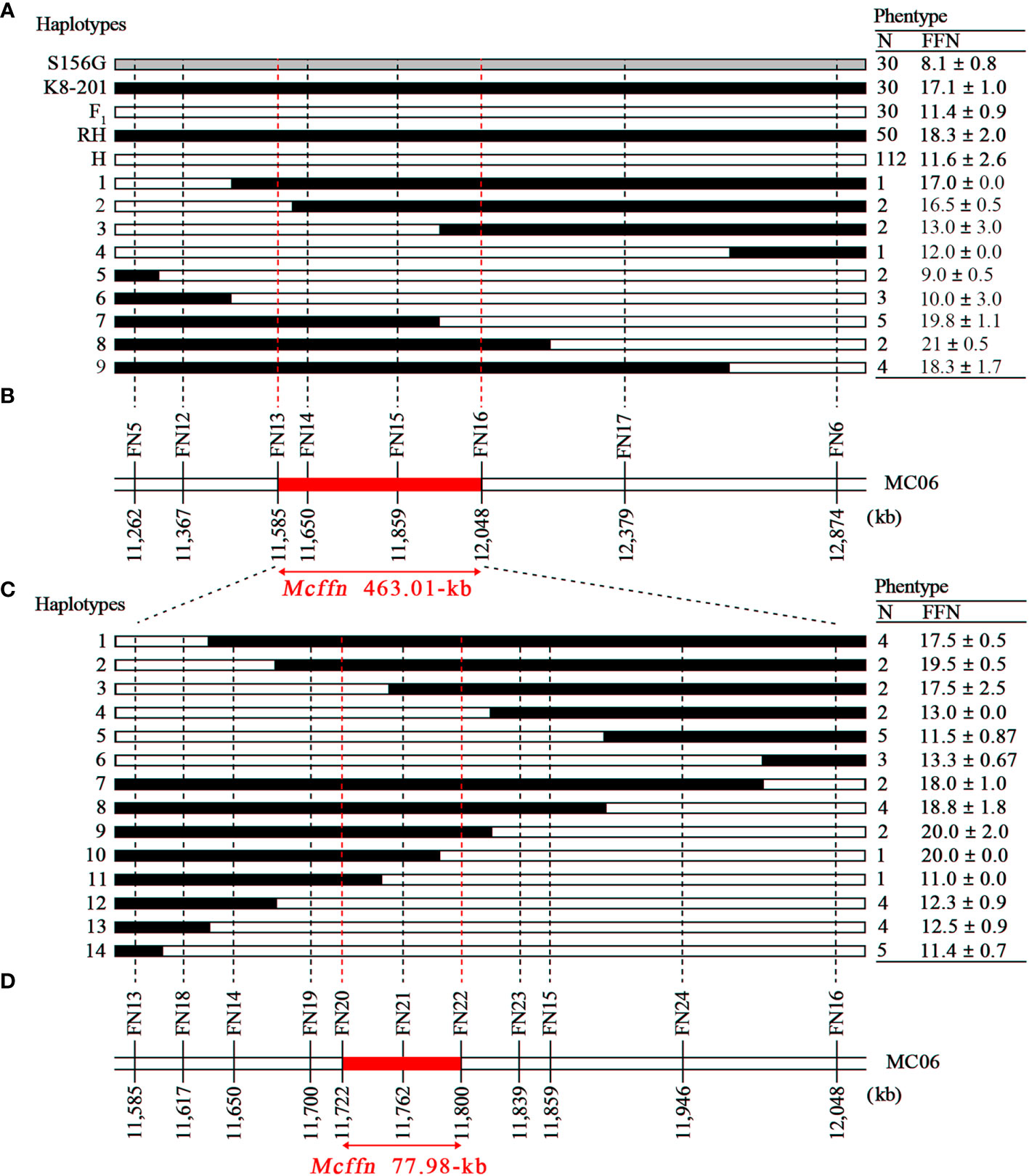
Figure 3 Fine-mapping of the Mcffn locus. (A) Nine haplotypes representing the 22 recombinant plants screened (with flanking markers FN5 and FN6) from the three F2 populations planted in autumn 2019, autumn 2020, and spring 2022. The dotted red lines indicate the boundaries of the Mcffn locus. RH, recessive homozygote. H, heterozygote. N, number. FFN, first flower node. (B) Physical map of the molecular markers used to genotype the 22 recombinant plants. The red bar represents the Mcffn locus. (C) Fourteen haplotypes representing the 41 recombinant plants screened (with flanking markers FN13 and FN16) from the large F2 population (n=2,402). (D) Physical map of the molecular markers used to genotype the 41 recombinant plants (fine-mapping).
To determine a more precise region for the Mcffn locus, the S156G×K8-201 F2 population (n=2,402) was genotyped using the two new flanking markers (FN13 and FN16). Of these, 41 plants containing a recombination of the recessive homozygote and heterozygote genotypes were obtained. Using the FN14 and FN15 markers, and seven newly-developed markers (FN18-FN24), these recombinant plants were divided into 14 haplotypes (Figure 3C and Supplementary Table 1). By utilizing the FFN phenotype and marker genotype data of the 41 recombinant plants, as well as the FFN reference criteria, the Mcffn locus was finally fine-mapped into a 77.98-kb physical interval between the FN20 (11,722,144 bp) and FN22 (11,800,118 bp) markers on MC06 (Figures 3C, D).
Differential expression reveals MC06g1112 as the Mcffn candidate gene
By examining the annotation of the Dali-11 reference genome (Cui et al., 2020), we identified four annotated genes (MC06g1110, MC06g1111, MC06g1112, and MC06g_new0263) within the 77.98-kb fine-mapping interval. We considered MC06g1111 a pseudogene because its predicted cDNA is only a 72-bp short nucleotide fragment lacking a complete gene structure and because no transcripts were detected in any sampled tissues (Cui et al., 2020). Only minimal (Cq>35) expression was detected for MC06g_new0263 across tissues in both parental lines, and no significant differences in the relative expression of MC06g1110 were detected across tissues between parental lines (Figure 4A). However, MC06g1112 exhibited significantly different relative expression across tissues between parental lines (Figure 4B). Furthermore, MC06g1112 exhibited different relative expression across the five different stem categories, increasing from the 5th to the 15th node, and decreasing from the 15th to the 25th node, with almost no expression at the ST (Figure 4C). Additionally, MC06g1112 exhibited significantly higher expression in the stems of P1 (S156G) plants than in the stems of P2 (K8-201) plants at all nodes from the 5th to the 20th (Figure 4C). Accordingly, we proposed that MC06g1112 was the FFN-associated Mcffn candidate gene.
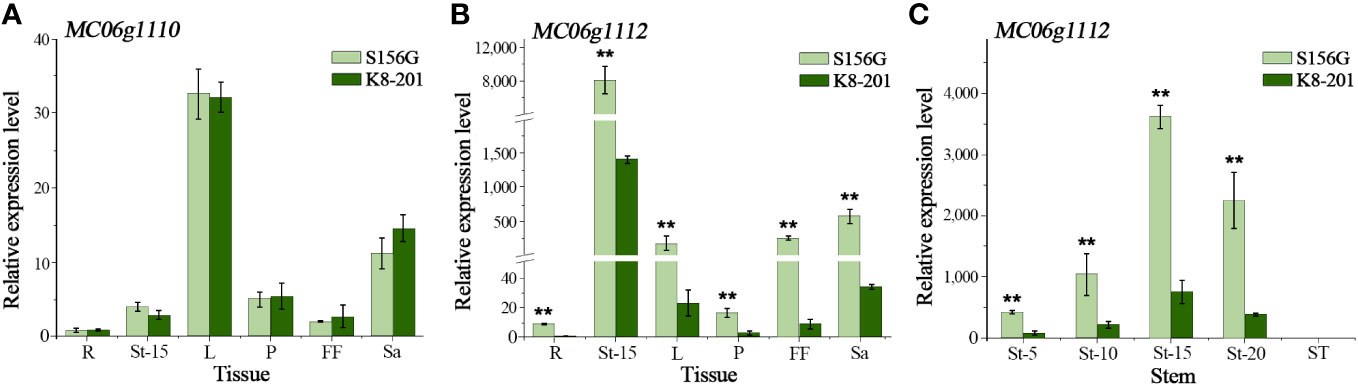
Figure 4 Relative expression of candidate genes. (A) The relative expression of MC06g1110 in different tissues at the 25-leaf stage. (B) The relative expression of MC06g1112 in different tissues at the 25-leaf stage. (C) The relative expression of MC06g1112 at different stem node at the 25-leaf stage. R, root. L, leaf. P, petiole. FF, female flower. Sa, sarcocarp. St-5, stem at 5th node. St-10, stem at 10th node. St-15, stem at 15th node. St-20, stem at 20th node. ST, stem at 25th node. The expression levels are presented as the mean ± SD (n=3). ** represents significance at the 0.01 level (Student’s t test).
A point mutation of MC06g1112 may decrease the FFN
By comparing the genomic sequences of the parental lines, we identified seven single-nucleotide variations (SNV-1~SNV-7) within the 77.98-kb fine-mapping interval (Supplementary Table 6). Of these, only SNV-2 (11,775,926 bp) was located within the MC06g1112 coding region, while the other six SNVs were located in the intergenic spacer region (Supplementary Table 6). The full-length MC06g1112 cDNA sequences of parental lines were cloned and compared, as a result, MC06g1112 gene consisted of 540 base pairs, which is a homolog of the FT gene encoding a phosphatidylethanolamine-binding protein (PEBP), and therefore was called McFT; additionally, we identified a point mutation (C>T) located 277 bp away from the MC06g1112 start codon, which leaded proline (P) of K8-201 to serine (S) of S156G (hereinafter referred to as P93S) (Figure 5).

Figure 5 Alignment of full-length cDNA and amino acid sequences of MC06g1112 between S156G and K8-201. Black boxes represent cDNA sequences and red boxes represent amino acid sequences.
To further verify the association of SNV-2 (C277T) and FFN, we designed a dCPAS marker to target SNV-2 by introducing a mismatched base (C) at the end of forward primer to create a Msp I restriction enzyme site, which can theoretically produce a 184-bp single fragment with the DNA template of K8-201, a 205-bp single fragment with the DNA template of S156G, and double fragments of 184-bp and 205-bp with the DNA template of S156G×K8-201 F1 generation (Supplementary Figure 2). Actually, however, the results of marker genotyping showed that both S156G and S156G×K8-201 F1 generation displayed double fragments of 184-bp and 205-bp, and K8-201 displayed a single 184-bp fragment; in 234 S156G×K8-201 F2 individuals (autumn 2019), all dominant homozygous and heterozygous plants exhibited double fragments of 184-bp and 205-bp, and all recessive homozygous plants exhibited a single 184-bp fragment (Supplementary Figure 3). Therefore, we conducted Sanger sequencing targeting SNV-2, which indicated that the SNV-2 locus in both DNA and cDNA of K8-201 were recessive homozygous genotype (C), in cDNA of S156G was dominant homozygous genotype (T), while in DNA of S156G was heterozygous genotype (C/T) (Supplementary Figure 4), which implied that the region where SNV-2 is located might have two or multiple copies on the bitter gourd genome.
The McFT proteins of the two parental lines were compared with previously-characterized FT from C. sativus (CsFT), C. melo (CmFT), C. lanatus (ClFT), Benincasa hispida (BhiFT), Lagenaria siceraria (LsiFT), Cucurbita maxima (Cm-FTL1 and Cm-FTL2), Cucurbita moschata (Cmo-FTL1 and Cmo-FTL2), Nicotiana tabacum (NtFT), Oryza sativa (OsFT/Hd3a), and Arabidopsis thaliana (AtFT). Sequence alignment and phylogenetic analysis indicated that the McFT proteins from S156G and K8-201 were highly homologous with these previously-characterized FTs, especially FTs of cucurbits species, sharing between 73.63 and 97.21% sequence identity (Figure 6 and Supplementary Figure 5). Additionally, P93 was a strictly-conserved amino acid across all of the examined species, with only one mutant S93 identified in S156G (Figure 6). We speculated that the P93S mutation might be responsible for the decreased FFN exhibited by S156G, since the FFN of S156G was significantly lower than that of K8-201.
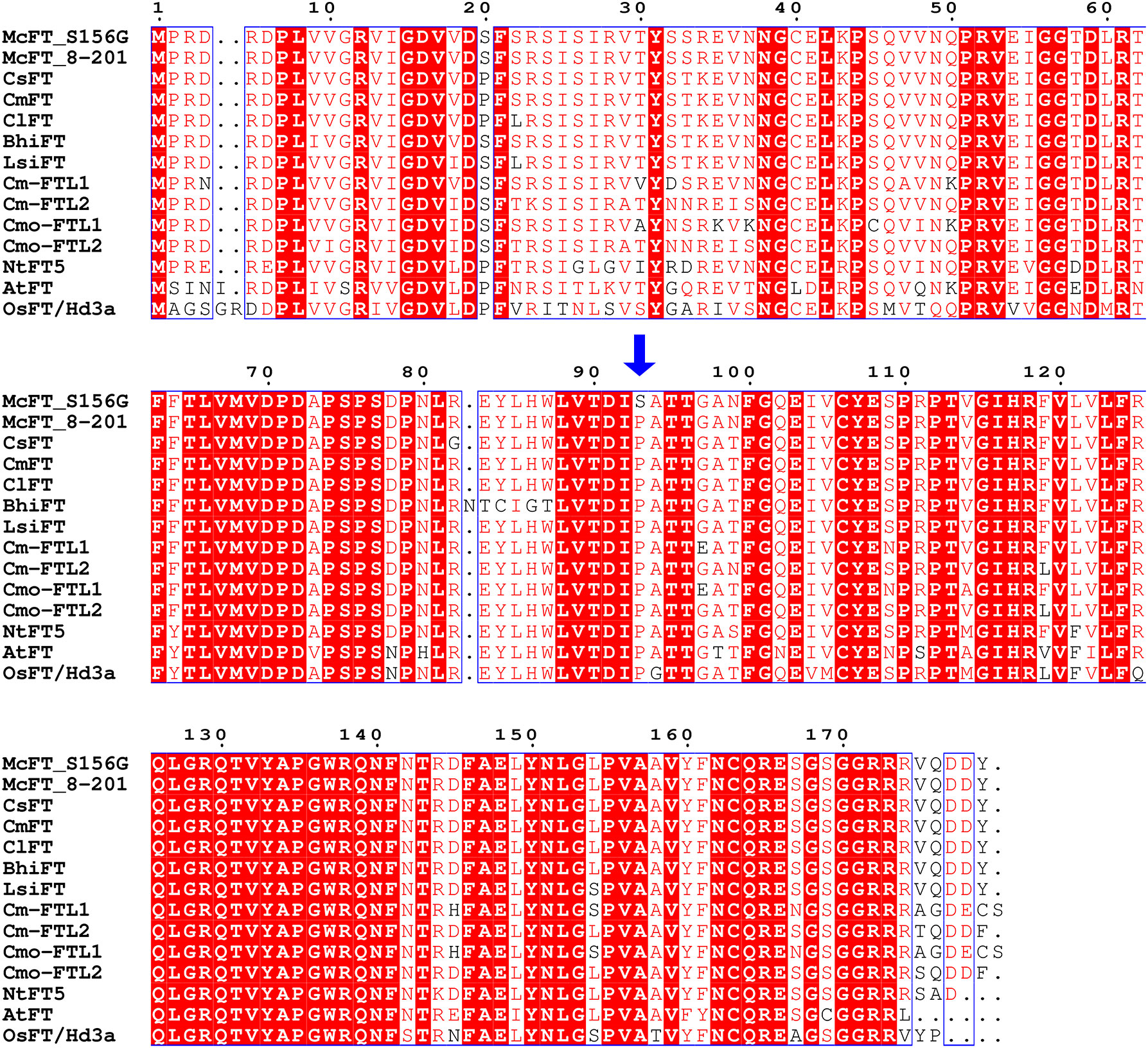
Figure 6 Alignment of amino acid sequences of FT proteins across different species of flowering plants. McFT, Momordica charantia; CsFT, Cucumis sativus; CmFT, Cucumis melo; ClFT, Citrullus lanatus; BhiFT, Benincasa hispida; LsiFT, Lagenaria siceraria; NtFT5, Nicotiana tabacum; OsFT/Hd3a, Oryza sativa; AtFT, Arabidopsis thaliana; Cm-FTL1 and Cm-FTL2, Cucurbita maxima; Cmo-FTL1 and Cmo-FTL2, Cucurbita moschata. The blue arrow indicates the P93S amino acid mutation. The source of each FT protein sequence is listed in Supplementary Table 7.
Discussion
The onset of flowering, which signals the transition from vegetative to reproduction growth, is a particularly important agronomic trait in Cucurbitaceae crops, as this trait can influence the onset of maturity, the production of female flowers, and yield (Lu et al., 2014; Zhao et al., 2018; Wen et al., 2019). Previous research on the timing of flowering in bitter gourd has primarily focused on the time of onset of flowering, either female or male, from sowing, and has led to the identification of flowering-associated QTLs through genetic mapping (Wang and Xiang, 2013; Cui et al., 2018; Gangadhara Rao et al., 2018; Kaur et al., 2022). However, the gene responsible for regulating flowering time in bitter gourd remained unidentified.
As one of the model plants for research on sex differentiation, Cucurbitaceae species harbor all three basic types of flower sexes, namely female, male, and hermaphroditic flowers (Dellaporta and Calderon-Urrea, 1993; Schaefer and Renner, 2011). All these three basic types carry both pistil and stamen primordia at early development stage of flower bud, and the formation of female and male flowers are resulted by the arrest of stamen and pistil development, respectively (Bai et al., 2004). Previous studies have revealed that the “arrest” processes are genetically controlled, such as the loss of function of CmWIP1, CsWIP1, and ClWIP1 lead to gynoecious lines in melon, cucumber, and watermelon, respectively (Martin et al., 2009; Hu et al., 2017; Zhang et al., 2020). Also, our previous works using S156G×K8-201 F2 population have confirmed that the gene locus responsible for gynoecy in bitter gourd is located at the end of MC01 (Zhong et al., 2023). In addition, our results of phenotype investigation showed that there was no direct relationship between gynoecy and FFN in the three S156G×K8-201 F2 populations (Supplementary Table 2). Hence, we speculate that gynoecy and FFN are independently inherited in bitter gourd.
Here, we used FFN as a proxy for flowering time in bitter gourd and detected a main effect QTL (Mcffn) associated with FFN via whole-genome re-sequencing-based QTL mapping (Figure 2A). Then molecular marker-based QTL mapping indicated that Mcffn could explain 67.3-73.9% of the variation in the FFN phenotype (Figure 2B), which is higher than the explanatory power reported for QTLs related to either female or male flowering time in bitter gourd (Wang and Xiang, 2013; Cui et al., 2018; Gangadhara Rao et al., 2018; Kaur et al., 2022). Furthermore, the consecutive variation in FFN exhibited by the segregating F2 populations (Figure 1D) implies that there may be multiple QTLs associated with flowering time in bitter gourd, which is consistent with our QTL mapping results (Figure 2A). Based on fine-mapping, gene expression, and sequence comparison analyses, the MC06g1112 (McFT) gene was identified as the most likely FFN-associated Mcffn candidate gene (Figures 3–5). In cucumber, CsFT has been found to explain 52.3% of the variation in flower time, and two large structural variations upstream of CsFT are associated with earlier flowering (Lu et al., 2014; Zhao et al., 2018; Gimode et al., 2020). Previous comparative genome analyses have shown that most of genomic sequences of pseudochromosome MC06 of bitter gourd, including the genomic fragment where McFT is located, are mapped to the chromosome 1 of cucumber (Cui et al., 2020). It is worth mentioning that CsFT is just in this collinear genomic interval (Lu et al., 2014), which may imply that bitter gourd and cucumber evolved from the same ancestor and the molecular mechanism regulating FFN or flowering time is highly similar in bitter gourd and cucumber. In squash, the ectopic expression of Arabidopsis-derived AtFT, which is responsive to inductive short-day (SD) photoperiods, has highly effective in mediating floral induction under long-day (LD) treatment (Lin et al., 2007). These evidences indicate that FT genes of cucurbit species may be conserved, and thus its development and application are beneficial to early maturity breeding for cucurbit crops.
Several previous studies have confirmed that the FT gene is the downstream target of many transcription factors (TFs) associated with flowering time, such as CONSTANS (CO), PHYTOCHROME INTERACTING FACTOR4 (PIF4), FLOWERING LOCUS C (FLC), and PHYTOCHROME AND FLOWERING TIME 1 (PFT1), among others (Putterill et al., 1995; Cerdán and Chory, 2003; Crevillén and Dean, 2011; Kumar et al., 2012), and hence plays a vital role in regulating flowering time across diverse flowering plants, including Arabidopsis, rice (O. sativa), and winter oilseed rape (Brassica napus), among others (Komiya et al., 2008; Ho and Weigel, 2014; Vollrath et al., 2021). In Arabidopsis, the FT mRNA is expressed in the vasculature of cotyledons and leaves while the FT protein interacts with a bZIP TF (FD) in the shoot apex to promote floral transition and initiate floral development (Abe et al., 2005; Wigge et al., 2005). Lin et al. (2007), using squash as a model system, report that the FT protein is translocated long distances through the phloem to the shoot apical meristem, where it induces flowering. Because of this, FT is generally considered a long-distance signal, or a leaf-to-apex communicator, for the induction of flowering (Corbesier et al., 2007; Lin et al., 2007). In this study, we detected almost no expression of McFT in STs of both parental lines (Figure 4C), suggesting that the functional mechanism of McFT in bitter gourd may be similar with previously-reported squash (Lin et al., 2007). In addition, previous studies have focused on FT expression in cucumber mainly using leaf tissues (Lu et al., 2014; Wang et al., 2020; Yang et al., 2022). Unlike these previous studies, we examined FT expression in several bitter gourd tissues and found that this gene was expressed in all tissues (with the exception of STs), and the expression was particularly high in stem tissues (Figure 4B). Our results suggest that the stem tissue may have the greatest impact on flowering time, although the precise regulatory mechanism underlying McFT expression requires further study.
In general, FT is a highly conserved protein which is robust to a wide range of mutations and plays a similar functional role in many species (Lin et al., 2007; Ho and Weigel, 2014; Putterill and Varkonyi-Gasic, 2016). However, Ho and Weigel (2014) reported that the P93 mutation of the FT protein may alter flowering time in Arabidopsis, with the P93A and P93T mutations resulting in early flowering and the P93H mutation resulting in delayed flowering. In the present study, we displayed the conservatism of P93 of FT protein across cucurbit species and some other flowering plant species, and identified a P93S amino acid mutation of the McFT protein in bitter gourd (Figure 6). We speculate that the P93S mutation may be responsible for the decreased FFN exhibited by S156G (Table 1). Furthermore, the results of genotypes and Sanger sequencing targeting SNV-2 (C277T) suggested that the region where SNV-2 is located might have two or multiple copies on the bitter gourd genome (Supplementary Figure 3 and 4). The genome replication events may still be the cause of the change of FFN, such as CsACS1G, which is a copy of CsACS1 and leads to gynoecy in cucumber (Mibus and Tatlioglu, 2004; Li et al., 2020). Overall, our results suggest that the FT genes may be highly conserved across cucurbits, and thus they should be considered targets for the molecular breeding of early-maturing Cucurbitaceae crops.
Conclusions
FFN of bitter gourd is regulated by a major effect QTL named Mcffn, with the low FFN is incompletely dominant over the high FFN. The Mcffn locus was fine-mapped into a 77.98-kb physical region on MC06. MC06g1112, a homolog of FT, was considered as the most likely Mcffn candidate gene according to expression and sequence variation analyses. A point mutation (C277T) in MC06g1112, which results in a P93S amino acid mutation between parental lines, may be responsible for decreasing FFN in bitter gourd.
Data availability statement
The data presented in the study are deposited in the Genome Sequence Archive in National Genomics Data Center, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences repository, accession number CRA009976.
Author contributions
KH and JWC conceived and designed the research. JZ performed most of the experiments and wrote the manuscript. JL, CFZ and MJM performed statistical analysis. JJC, FH and JCD provided helpful discussions. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Science and Technology Planning Project of Guangdong Province (2022B0202160015, 2019A050520002 and 2022-NPY-00-027) and the Guangzhou Science and Technology Plan Project (202206010170 and SL2024A04J01673).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1153208/full#supplementary-material
References
Abe, M., Kobayashi, Y., Yamamoto, S., Daimon, Y., Yamaguchi, A., Ikeda, Y., et al. (2005). FD, a bZIP protein mediating signals from the floral pathway integrator FT at the shoot apex. Science 309, 1052–1056. doi: 10.1126/science.1115983
Akihisa, T., Higo, N., Tokuda, H., Ukiya, M., Akazawa, H., Tochigi, Y., et al. (2007). Cucurbitane-type triterpenoids from the fruits of Momordica charantia and their cancer chemopreventive effects. J. Nat. Prod. 70, 1233–1239. doi: 10.1021/np068075p
Bai, S. L., Peng, Y. B., Cui, J. X., Gu, H. T., Xu, L. Y., Li, Y. Q., et al. (2004). Developmental analyses reveal early arrests of the spore-bearing parts of reproductive organs in unisexual flowers of cucumber (Cucumis sativus L.). Planta 220, 230–240. doi: 10.1007/s00425-004-1342-2
Basch, E., Gabardi, S., Ulbricht, C. (2003). Bitter melon (Momordica charantia): a review of efficacy and safety. Am. J. Health Syst. Pharm. 60, 356–359. doi: 10.1093/ajhp/60.4.356
Cai, Y., Bartholomew, E. S., Dong, M., Zhai, X., Yin, S., Zhang, Y., et al. (2020). The HD-ZIP IV transcription factor GL2-LIKE regulates male flowering time and fertility in cucumber. J. Exp. Bot. 71, 5425–5437. doi: 10.1093/jxb/eraa251
Cerdán, P. D., Chory, J. (2003). Regulation of flowering time by light quality. Nature 423, 881–885. doi: 10.1038/nature01636
Chen, S., Zhou, Y., Chen, Y., Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Corbesier, L., Vincent, C., Jang, S., Fornara, F., Fan, Q., Searle, I., et al. (2007). FT protein movement contributes to long-distance signaling in floral induction of Arabidopsis. Science 316, 1030–1033. doi: 10.1126/science.1141752
Crevillén, P., Dean, C. (2011). Regulation of the floral repressor gene FLC: the complexity of transcription in a chromatin context. Curr. Opin. Plant Biol. 14, 38–44. doi: 10.1016/j.pbi.2010.08.015
Cui, J., Luo, S., Niu, Y., Huang, R., Wen, Q., Su, J., et al. (2018). A RAD-based genetic map for anchoring scaffold sequences and identifying QTLs in bitter gourd (Momordica charantia). Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00477
Cui, J., Yang, Y., Luo, S., Wang, L., Huang, R., Wen, Q., et al. (2020). Whole-genome sequencing provides insights into the genetic diversity and domestication of bitter gourd (Momordica spp.). Hortic. Res. 7, 85. doi: 10.1038/s41438-020-0305-5
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Dellaporta, S. L., Calderon-Urrea, A. (1993). Sex determination in flowering plants. Plant Cell 5, 1241–1251. doi: 10.1105/tpc.5.10.1241
Gangadhara Rao, P., Behera, T. K., Gaikwad, A. B., Munshi, A. D., Jat, G. S., Boopalakrishnan, G. (2018). Mapping and QTL analysis of gynoecy and earliness in bitter gourd (Momordica charantia L.) using genotyping-by-sequencing (GBS) technology. Front. Plant Sci. 9 1555. doi: 10.3389/fpls.2018.01555
Gimode, W., Clevenger, J., McGregor, C. (2020). Fine-mapping of a major quantitative trait locus Qdff3-1 controlling flowering time in watermelon. Mol. Breed. 40, 1–12. doi: 10.1007/s11032-019-1087-z
Ho, W. W., Weigel, D. (2014). Structural features determining flower-promoting activity of Arabidopsis FLOWERING LOCUS T. Plant Cell 26, 552–564. doi: 10.1105/tpc.113.115220
Hu, B., Li, D., Liu, X., Qi, J., Gao, D., Zhao, S., et al. (2017). Engineering non-transgenic gynoecious cucumber using an improved transformation protocol and optimized CRISPR/cas9 system. Mol. Plant 10, 1575–1578. doi: 10.1016/j.molp.2017.09.005
Kaur, G., Pathak, M., Singla, D., Chhabra, G., Chhuneja, P., Kaur Sarao, N. (2022). Quantitative trait loci mapping for earliness, fruit, and seed related traits using high density genotyping-by-sequencing-based genetic map in bitter gourd (Momordica charantia L.). Front. Plant Sci. 12. doi: 10.3389/fpls.2021.799932
Komiya, R., Ikegami, A., Tamaki, S., Yokoi, S., Shimamoto, K. (2008). Hd3a and RFT1 are essential for flowering in rice. Development 135, 767–774. doi: 10.1242/dev.008631
Kumar, S. V., Lucyshyn, D., Jaeger, K. E., Alós, E., Alvey, E., Harberd, N. P., et al. (2012). Transcription factor PIF4 controls the thermosensory activation of flowering. Nature 484, 242–245. doi: 10.1038/nature10928
Lee, J. H., Kim, Y. C., Jung, Y., Han, J. H., Zhang, C., Yun, C. W., et al. (2019). The overexpression of cucumber (Cucumis sativus L.) genes that encode the branched-chain amino acid transferase modulate flowering time in Arabidopsis thaliana. Plant Cell Rep. 38, 25–35. doi: 10.1007/s00299-018-2346-x
Li, H. (2011). Improving SNP discovery by base alignment quality. Bioinformatics 27, 1157–1158. doi: 10.1093/bioinformatics/btr076
Li, R., Yu, C., Li, Y., Lam, T. W., Yiu, S. M., Kristiansen, K., et al. (2009). SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25, 1966–1967. doi: 10.1093/bioinformatics/btp336
Li, Z., Han, Y., Niu, H., Wang, Y., Jiang, B., Weng, Y. (2020). Gynoecy instability in cucumber (Cucumis sativus L.) is due to unequal crossover at the copy number variation-dependent Femaleness (F) locus. Hortic. Res. 7, 32. doi: 10.1038/s41438-020-0251-2
Lin, M. K., Belanger, H., Lee, Y. J., Varkonyi-Gasic, E., Taoka, K., Miura, E., et al. (2007). FLOWERING LOCUS T protein may act as the long-distance florigenic signal in the cucurbits. Plant Cell 19, 1488–1506. doi: 10.1105/tpc.107.051920
Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lu, H., Lin, T., Klein, J., Wang, S., Qi, J., Zhou, Q., et al. (2014). QTL-seq identifies an early flowering QTL located near Flowering Locus T in cucumber. Theor. Appl. Genet. 127, 1491–1499. doi: 10.1007/s00122-014-2313-z
Lu, G., Moriyama, E. N. (2004). Vector NTI, a balanced all-in-one sequence analysis suite. Brief. Bioinform. 5, 378–388. doi: 10.1093/bib/5.4.378
Martin, A., Troadec, C., Boualem, A., Rajab, M., Fernandez, R., Morin, H., et al. (2009). A transposon-induced epigenetic change leads to sex determination in melon. Nature 461, 1135–1138. doi: 10.1038/nature08498
Matsumura, H., Hsiao, M. C., Lin, Y. P., Toyoda, A., Taniai, N., Tarora, K., et al. (2020). Long-read bitter gourd (Momordica charantia) genome and the genomic architecture of nonclassic domestication. Proc. Natl. Acad. Sci. U. S. A. 117, 14543–14551. doi: 10.1073/pnas.1921016117
McGregor, C. E., Waters, V., Vashisth, T., Abdel-Haleem, H. (2014). Flowering time in watermelon is associated with a major quantitative trait locus on chromosome 3. J. Amer. Soc Hortic. Sci. 139, 48–53. doi: 10.21273/JASHS.139.1.48
Mibus, H., Tatlioglu, T. (2004). Molecular characterization and isolation of the F/f gene for femaleness in cucumber (Cucumis sativus L.). Theor. Appl. Genet. 109, 1669–1676. doi: 10.1007/s00122-004-1793-7
Okonechnikov, K., Conesa, A., García-Alcalde, F. (2016). Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32, 292–294. doi: 10.1093/bioinformatics/btv566
Pan, Y., Qu, S., Bo, K., Gao, M., Haider, K. R., Weng, Y. (2017). QTL mapping of domestication and diversifying selection related traits in round-fruited semi-wild Xishuangbanna cucumber (Cucumis sativus L. var. xishuangbannanesis). Theor. Appl. Genet. 130, 1531–1548. doi: 10.1007/s00122-017-2908-2
Pnueli, L., Carmel-Goren, L., Hareven, D., Gutfinger, T., Alvarez, J., Ganal, M., et al. (1998). The SELF-PRUNING gene of tomato regulates vegetative to reproductive switching of sympodial meristems and is the ortholog of CEN and TFL1. Development 125, 1979–1989. doi: 10.1242/dev.125.11.1979
Porebski, S., Bailey, L. G., Baum, B. R. (1997). Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 15, 8–15. doi: 10.1007/BF02772108
Putterill, J., Robson, F., Lee, K., Simon, R., Coupland, G. (1995). The CONSTANS gene of Arabidopsis promotes flowering and encodes a protein showing similarities to zinc finger transcription factors. Cell 80, 847–857. doi: 10.1016/0092-8674(95)90288-0
Putterill, J., Varkonyi-Gasic, E. (2016). FT and florigen long-distance flowering control in plants. Curr. Opin. Plant Biol. 33, 77–82. doi: 10.1016/j.pbi.2016.06.008
Schaefer, H., Heibl, C., Renner, S. S. (2009). Gourds afloat: a dated phylogeny reveals an Asian origin of the gourd family (Cucurbitaceae) and numerous oversea dispersal events. Proc. Biol. Sci. 276, 843–851. doi: 10.1098/rspb.2008.1447
Schaefer, H., Renner, S. S. (2010). A three-genome phylogeny of Momordica (Cucurbitaceae) suggests seven returns from dioecy to monoecy and recent long-distance dispersal to Asia. Mol. Phylogenet. Evol. 54, 553–560. doi: 10.1016/j.ympev.2009.08.006
Schaefer, H., Renner, S. S. (2011). “Cucurbitaceae. In: kubitzki K (ed) flowering plants eudicots: sapindales, cucurbitales, myrtaceae,” in The families and genera of vascular plants. (Berlin, Heidelberg: Springer Berlin Heidelberg), 112–174. doi: 10.1007/978-3-642-14397-7_10
Sheng, Y., Pan, Y., Li, Y., Yang, L., Weng, Y. (2020). Quantitative trait loci for fruit size and flowering time-related traits under domestication and diversifying selection in cucumber (Cucumis sativus). Plant Breed. 139, 176–191. doi: 10.1111/pbr.12754
Turck, F., Fornara, F., Coupland, G. (2008). Regulation and identity of florigen: FLOWERING LOCUS T moves center stage. Annu. Rev. Plant Biol. 59, 573–594. doi: 10.1146/annurev.arplant.59.032607.092755
Urasaki, N., Takagi, H., Natsume, S., Uemura, A., Taniai, N., Miyagi, N., et al. (2017). Draft genome sequence of bitter gourd (Momordica charantia), a vegetable and medicinal plant in tropical and subtropical regions. DNA Res. 24, 51–58. doi: 10.1093/dnares/dsw047
Van Ooijen, J. W. (2006). Joinmap®4, software for calculation of genetic linkage maps in experimental populations (Wageningen: Kyazma B.V).
Van Ooijen, J. W. (2009). MapQTL®6: software for the mapping of quantitative trait loci in experimental populations of diploid species (Wageningen: Kyazma B.V).
Vasimuddin, M., Misra, S., Li, H., Aluru, S. (2019). “Efficient architecture-aware acceleration of BWA-MEM for multicore systems,” in 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS). (Rio de Janeiro, Brazil: IEEE), 314–324. doi: 10.1109/IPDPS.2019.00041
Vollrath, P., Chawla, H. S., Schiessl, S. V., Gabur, I., Lee, H., Snowdon, R. J., et al. (2021). A novel deletion in FLOWERING LOCUS T modulates flowering time in winter oilseed rape. Theor. Appl. Genet. 134, 1217–1231. doi: 10.1007/s00122-021-03768-4
Wang, K., Li, M., Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic. Acids Res. 38, e164. doi: 10.1093/nar/gkq603
Wang, S., Li, H., Li, Y., Li, Z., Qi, J., Lin, T., et al. (2020). FLOWERING LOCUS T improves cucumber adaptation to higher latitudes. Plant Physiol. 182, 908–918. doi: 10.1104/pp.19.01215
Wang, S., Li, Z., Yang, G., Ho, C. T., Li, S. (2017). Momordica charantia: a popular health-promoting vegetable with multifunctionality. Food Funct. 8, 1749–1762. doi: 10.1039/c6fo01812b
Wang, Z., Xiang, C. (2013). Genetic mapping of QTLs for horticulture traits in a F2-3 population of bitter gourd (Momordica charantia L.). Euphytica 193, 235–250. doi: 10.1007/s10681-013-0932-0
Wen, C., Zhao, W., Liu, W., Yang, L., Wang, Y., Liu, X., et al. (2019). CsTFL1 inhibits determinate growth and terminal flower formation through interaction with CsNOT2a in cucumber. Development 146, dev180166. doi: 10.1242/dev.180166
Wigge, P. A., Kim, M. C., Jaeger, K. E., Busch, W., Schmid, M., Lohmann, J. U., et al. (2005). Integration of spatial and temporal information during floral induction in Arabidopsis. Science 309, 1056–1059. doi: 10.1126/science.1114358
Yang, A., Xu, Q., Hong, Z., Wang, X., Zeng, K., Yan, L., et al. (2022). Modified photoperiod response of CsFT promotes day neutrality and early flowering in cultivated cucumber. Theor. Appl. Genet. 135, 2735–2746. doi: 10.1007/s00122-022-04146-4
Yi, L., Wang, Y., Huang, X., Gong, Y., Wang, S., Dai, Z. (2020). Genome-wide identification of flowering time genes in cucurbit plants and revealed a gene ClGA2/KS associate with adaption and flowering of watermelon. Mol. Biol. Rep. 47, 1057–1065. doi: 10.1007/s11033-019-05200-z
Yuan, X. J., Pan, J. S., Cai, R., Guan, Y., Liu, L. Z., Zhang, W. W., et al. (2008). Genetic mapping and QTL analysis of fruit and flower related traits in cucumber (Cucumis sativus L.) using recombinant inbred lines. Euphytica 164, 473–491. doi: 10.1007/s10681-008-9722-5
Zahid, N., Maqbool, M., Hamid, A., Shehzad, M., Tahir, M. M., Mubeen, K., et al. (2021). Changes in Vegetative and Reproductive Growth and Quality Parameters of Strawberry (Fragaria × ananassa Duch.) cv. Chandler Grown at Different Substrates. J. Food Qual. 2021, 9. doi: 10.1155/2021/9996073
Zhang, J., Guo, S., Ji, G., Zhao, H., Sun, H., Ren, Y., et al. (2020). A unique chromosome translocation disrupting ClWIP1 leads to gynoecy in watermelon. Plant J. 101, 265–277. doi: 10.1111/tpj.14537
Zhang, J., Huang, Y., Kikuchi, T., Tokuda, H., Suzuki, N., Inafuku, K., et al. (2012). Cucurbitane triterpenoids from the leaves of Momordica charantia, and their cancer chemopreventive effects and cytotoxicities. Chem. Biodivers. 9, 428–440. doi: 10.1002/cbdv.201100142
Zhang, X., Wang, G., Chen, B., Du, H., Zhang, F., Zhang, H., et al. (2018). Candidate genes for first flower node identified in pepper using combined SLAF-seq and BSA. PloS One 13, e0194071. doi: 10.1371/journal.pone.0194071
Zhang, X. F., Wang, G. Y., Dong, T. T., Chen, B., Du, H. S., Li, C. B., et al. (2019). High-density genetic map construction and QTL mapping of first flower node in pepper (Capsicum annuum L.). BMC Plant Biol. 19, 167. doi: 10.1186/s12870-019-1753-7
Zhao, W., Gu, R., Che, G., Cheng, Z., Zhang, X. (2018). CsTFL1b may regulate the flowering time and inflorescence architecture in cucumber (Cucumis sativus L.). Biochem. Biophys. Res. Commun. 499, 307–313. doi: 10.1016/j.bbrc.2018.03.153
Zhong, J., Cheng, J., Cui, J., Hu, F., Dong, J., Liu, J., et al. (2022). MC03g0810, an important candidate gene controlling black seed coat color in bitter gourd (Momordica spp.). Front. Plant Sci. 13. doi: 10.3389/fpls.2022.875631
Zhong, J., Cui, J., Liu, J., Zhong, C., Hu, F., Dong, J., et al. (2023). Fine-mapping and candidate gene analysis of the Mcgy1 locus responsible for gynoecy in bitter gourd (Momordica spp.). Theor. Appl. Genet. 136, 81. doi: 10.1007/s00122-023-04314-0
Keywords: bitter gourd, first flower node, quantitative trait locus, fine-mapping, FLOWERING LOCUS T
Citation: Zhong J, Cui J, Miao M, Hu F, Dong J, Liu J, Zhong C, Cheng J and Hu K (2023) A point mutation in MC06g1112 encoding FLOWERING LOCUS T decreases the first flower node in bitter gourd (Momordica charantia L.). Front. Plant Sci. 14:1153208. doi: 10.3389/fpls.2023.1153208
Received: 29 January 2023; Accepted: 25 September 2023;
Published: 10 October 2023.
Edited by:
Baohua Wang, Nantong University, ChinaReviewed by:
Zhansheng Li, Chinese Academy of Agricultural Sciences, ChinaQiusheng Kong, Huazhong Agricultural University, China
Jianbin Hu, Henan Agricultural University, China
Copyright © 2023 Zhong, Cui, Miao, Hu, Dong, Liu, Zhong, Cheng and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiaowen Cheng, amlhb2xvbmcxMDE1QDEyNi5jb20=; Kailin Hu, aHVrYWlsaW5Ac2NhdS5lZHUuY24=