 Lungowe Mulozi1†
Lungowe Mulozi1† Amaranatha R. Vennapusa1†
Amaranatha R. Vennapusa1† Sathya Elavarthi1*
Sathya Elavarthi1* Oluwatomi E. Jacobs1
Oluwatomi E. Jacobs1 Krishnanand P. Kulkarni1
Krishnanand P. Kulkarni1 Purushothaman Natarajan2
Purushothaman Natarajan2 Umesh K. Reddy2
Umesh K. Reddy2 Kalpalatha Melmaiee1*
Kalpalatha Melmaiee1*- 1Department of Agriculture and Natural Resources, Delaware State University, Dover, DE, United States
- 2Department of Biology and Gus R. Douglass Institute, West Virginia State University, Institute, WV, United States
Sugar maple (Acer saccharum Marshall) is a temperate tree species in the northeastern parts of the United States and is economically important for its hardwood and syrup production. Sugar maple trees are highly vulnerable to changing climatic conditions, especially drought, so understanding the physiological, biochemical, and molecular responses is critical. The sugar maple saplings were subjected to drought stress for 7, 14, and 21 days and physiological data collected at 7, 14, and 21 days after stress (DAS) showed significantly reduced chlorophyll and Normalized Difference Vegetation Index with increasing drought stress time. The drought stress-induced biochemical changes revealed a higher accumulation of malondialdehyde, proline, and peroxidase activity in response to drought stress. Transcriptome analysis identified a total of 14,099 differentially expressed genes (DEGs); 328 were common among all stress periods. Among the DEGs, transcription factors (including NAC, HSF, ZFPs, GRFs, and ERF), chloroplast-related and stress-responsive genes such as peroxidases, membrane transporters, kinases, and protein detoxifiers were predominant. GO enrichment and KEGG pathway analysis revealed significantly enriched processes related to protein phosphorylation, transmembrane transport, nucleic acids, and metabolic, secondary metabolite biosynthesis pathways, circadian rhythm-plant, and carotenoid biosynthesis in response to drought stress. Time-series transcriptomic analysis revealed changes in gene regulation patterns in eight different clusters, and pathway analysis by individual clusters revealed a hub of stress-responsive pathways. In addition, qRT-PCR validation of selected DEGs revealed that the expression patterns were consistent with transcriptome analysis. The results from this study provide insights into the dynamics of physiological, biochemical, and gene responses to progressive drought stress and reveal the important stress-adaptive mechanisms of sugar maple saplings in response to drought stress.
Introduction
Sugar maple (Acer saccharum Marshall) is a keystone tree species abundantly distributed in the northeastern forests of the United States and eastern Canada (Horsley et al., 2002). It is broadly known for its vibrant fall colors, syrup, and hardwood, and plays a vital role in its native ecosystems (Godman et al., 1990; Lucash et al., 2012). The maple industry contributed $147 million to the US economy in 2016 (Snyder et al., 2019). The United States was the second major producer of maple syrup worldwide, with a total production of ~4.24 million gallons in 2019. The state of Vermont produced more than 1.5 million gallons in 2021 and was the top maple syrup producer state in the United States. New York was the second leading producer, with a production of about 647 thousand gallons in 2021 (Shahbandeh, 2021). The global market value for maple syrup was at an all-time high of $1.24 billion in 2018 and is anticipated to increase by 2023 with a compound annual growth rate of 7% to $1.7 billion. Even though the United States is the second highest producer of maple syrup, it imports more than it produces each year. The import rate has increased over the past 40 years, from 607,000 in 1975 to 6.1 million in 2018. In 2018, the United States represented 50% ($182 million) of the world’s import value, with 100% of the imports coming from Canada1. The current demand for maple products worldwide and in the United States is increasing steadily (Farrel, 2021).
Drought being the inevitable factor, significantly affects the plant morphological, physiological, biochemical, and molecular features with an adverse influence on growth, photosynthetic capacity, membrane fluidity, and antioxidant machinery. At cellular and molecular levels, a significant effect of drought stress is seen on turgor pressure maintenance, membrane stability, sugar metabolism, respiration, interruption of metabolic and biochemical pathways, protein degradation, enzyme dysfunctions, induction of stress signaling, and sensitive genes (Farooq et al., 2009; McDowell, 2011). Plants cope with drought stress through various mechanisms, such as morphological by water mining with profound root systems, conservation by waxes, reduced leaf size, stay green nature, growth and development, and physiological mechanisms by maintaining the photosynthetic machinery, assimilate partitioning, stomatal regulation, turgor maintenance (Anjum et al., 2011; Sajeevan et al., 2017; Seleiman et al., 2021; Venkatesh et al., 2022). Biochemical mechanisms involved osmolyte and secondary metabolite production, enzymatic functions, maintenance of metabolic, protein, and lipid biosynthetic pathways, and molecular mechanisms involved in the maintenance of RNA and DNA synthesis and stress signaling, cascade, and regulatory genes expression, especially the stress-induced transcription factors, and stress-responsive downstream genes (Mickelbart et al., 2015; Kiranmai et al., 2018; Lokesh et al., 2019; Zhang et al., 2022).
Due to global climate change, the frequency and intensity of drought have continuously increased (Dai, 2013; Lima et al., 2015; Gugger et al., 2017). In plants, the ability to respond to increasing drought conditions will be particularly important if the current climate scenario continues (Gugger et al., 2017). Episodes of sugar maple decline have arisen sporadically since the early 1900s due to climate change. The major factors affecting decline include defoliation by insects, intense drought, elevated seasonal temperatures, winter freezing injury, and early autumn frosts (Horsley et al., 2000; Wong et al., 2009; Rogers et al., 2017; Oswald et al., 2018; Rogers, 2020). Severe and recurrent droughts are a significant threat to the productivity of sugar maple and account for unusual mortality in several states in the United States (Horsley et al., 2002; Oswald et al., 2018). Drought stress-induced changes were reported in crown growth, soluble sugars, net photosynthesis, leaf color, nutrient limitation during the autumn, and anthocyanin production in sugar maple plants (Schaberg et al., 2003; Wong et al., 2009). The drought stress in sugar maple trees reduces the chlorophyll content, and growth and induces early abscission of leaves (Carlson, 2009).
Although observational studies and reports have indicated that drought is one of the major factors responsible for sugar maple decline, no comprehensive physiological and genetic studies have been conducted to substantiate this theory (Harmon et al., 2017). Understanding the mechanisms involved in plants coping with drought stress has been a central topic in plant physiology for decades (Bray, 1997: Gugger et al., 2017; Hauer et al., 2021). Despite knowing the significant effect of drought stress on sugar maple survival, plant growth, and development, limited information is available on plant physiological processes, and the molecular mechanisms in response to drought stress remain unexplored. Hence, investigating the molecular mechanisms in response to drought stress in sugar maple offers a possibility for understanding stress adaptation mechanisms.
Next-generation sequencing has become a powerful platform to elucidate molecular mechanisms in many research fields due to its rapidity and cost-effectiveness (Gao et al., 2018; Callwood et al., 2021; McEvoy et al., 2022). The comprehensive data generated facilitates the development of genetic analyses and functional genomics studies between plant species, specifically for non-model plants and tree species (Kulkarni et al., 2021). Transcriptome studies are a powerful tool to provide valuable molecular and genetic information, including the discovery of molecular markers and novel candidate genes; insight into genetic networks, transcriptional and post-transcriptional regulation; and pathways involved in drought stress tolerance in many plant species (Aranda et al., 2012; Zhang et al., 2020; Song et al., 2022). Additionally, a meta-analysis of large transcriptomic datasets can reveal the key genetic resources responsible for complex tolerant traits, which are useful in crop improvement programs (Baldoni, 2022). In particular, global transcriptomic analysis has been widely used to understand drought stress response in various tree species, such as oak (Spieß et al., 2012; Gugger et al., 2017), pine (Fox et al., 2018), and beech (Müller et al., 2017).
The present study investigated the physiological, biochemical, and transcriptome changes of sugar maple to drought stress. Potential responsive genes were identified using RNAseq. This study describes the first genome-wide expression profiles of sugar maple in response to drought stress. The results from this study provide a comprehensive understanding of the molecular, physiological, and biochemical mechanisms of sugar maple plants in response to drought stress and help identify potential candidate genes for drought resistance, which can be used in sugar maple breeding programs for improving drought stress tolerance.
Materials and methods
Drought-stress experiments and sample collection
Sugar maple saplings were obtained from the Cold Stream Farms Nursery (Free Soil, MI, USA). The saplings were transplanted into medium-sized pots (15 cm deep) filled with peat moss and perlite. They were maintained under ambient environmental conditions, regularly watered, and fertilized for 12 months to establish and obtain sufficient biomass before undergoing drought stress. Then 28 uniform-sized saplings were transferred into the greenhouse and kept for one month for acclimatization. The conditions in the greenhouse were an average temperature of 28 ± 2°C with an average photoperiod of 14 h/day provided by natural sunlight. The plants were randomly divided into four groups: one control and three different drought stress treatments. Each group contained seven plants that were tagged with four different colored tags to indicate the four different treatments. Drought stress was imposed in May 2020. The control treatment plants were regularly irrigated every day with 500 ml water. For drought stress, a group of plants was left unwatered for 21 days (21 days after stress imposition [21DAS]), another group for 14 days (14DAS), and another group for 7 days (7DAS). The stress was imposed ahead of the corresponding number of days of treatment so that the experiments could be ended on the same day. Physiological measurements were taken, and leaf samples were collected at the same time at the end of the experiments. This sampling was based on the fact that all plants will undergo the same environmental and growth changes except for the water stress they receive. There were seven plants for each treatment, among them, three plants were utilized for biochemical analysis and four plant for RNAseq analysis for each treatment as biological replicates. The topmost fully expanded young fresh leaf samples were collected from the selected plants and were frozen in liquid nitrogen and stored at -80°C.
Soil volumetric water content measurements
The volumetric water content of the soil (moisture content in peat moss) was measured by using a hand-held FieldScout Time Domain Reflectometer (TDR) 100 Soil Moisture Meter (Spectrum Technologies, Aurora, IL, USA). Moisture content readings were taken in three different spots in the pot, and the final readings were the average of the three readings. The readings were taken from both control and drought stress pots (7, 14, and 21DAS) (Gebre & Earl, 2021).
Physiological and biochemical measurements
Physiological parameters were measured on three randomly selected leaves from each plant (one on the top, one in the center, and one at the lower base). For each leaf, measurements were taken three times on different spots of the leaf to obtain an average reading. These leaves were labeled, and the Normalized Difference Vegetation Index (NDVI) was measured similarly by using a handheld PolyPen RP 410 device (Qubit systems, Canada) (Wu et al., 2019). The chlorophyll index was measured by using the Soil and Plant Analysis Development (SPAD) metric (Konica Minolta, Japan) (Vennapusa et al., 2021).
Proline content was estimated according to the modified method (Vemanna et al., 2017; Siddique et al., 2018; Bissiwu et al., 2022) from Bates et al. (1973). The frozen leaf samples were ground by adding 500 μL of 3% sulfosalicylic acid in microcentrifuge tubes using a Fisher scientific bead mill 24. The samples were centrifuged for 5 min at 14,000 rpm by using a centrifuge (5417R, Eppendorf, Hamburg, Germany). Then, 100 μL supernatant was collected and added to the reaction mixture containing 100 μL of 3% sulfosalicylic acid, 200 μL glacial acetic acid, and 200 μL acidic ninhydrin and incubated at 96°C for 60 min. The reaction was terminated by keeping the samples on ice for 5 min; after cooling with 600 μL toluene added to the incubated samples and brief vortexing for 5 min for separating the samples, the upper aqueous phase of the 200-μL sample was used for spectrophotometer analysis. The absorbance was measured at 520 nm by using a 96-well microplate reader. The proline content in samples was determined with a standard curve using L-proline and calculated on a fresh weight basis and expressed as μg/g fresh weight (FW).
Malondialdehyde (MDA) content was estimated by using Lipid Peroxidation (MDA) Assay Kit (Munich, Germany). A homogenate of leaf samples (50 mg) was prepared in 500 μL MDA lysis buffer containing 3 μL butylated hydroxytoluene (BHT 100x). Samples were centrifuged at 13,000 rpm for 10 min to separate solid particles from the supernatant. A clear supernatant of 200 μL was added to 600 μL of the Thiobarbituric acid (TBA, 20%) solution and incubated at 95°C for 60 min to form the MDA-TBA adducts. Then the samples were cooled to room temperature in an ice bath for 10 min. Finally, 200 μL of the reaction mixture from plant samples, blank, and standards was transferred to 96-well plates for analysis, and absorbance of the supernatant was measured at 532 nm. Both standard and sample values were corrected by subtracting the blank value from all readings. The MDA concentration in treatment samples was estimated in µmoles/g FW with a standard curve plot according to guidelines given in the protocol.
Total RNA isolation, library preparation, poly-A selection, and HiSeq sequencing
Total RNA was isolated from leaf tissues of four biological replicates for each treatment by using Qiagen RNeasy plus a universal mini kit following the manufacturer’s instructions (Qiagen, Hilden, Germany). Total RNA was quantified using NanoDrop 2000C (Thermo Scientific, Waltham, MA, USA), and quality was checked using the Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA, USA). The integrity (RIN) was studied using Bioanalyzer, Agilent TapeStation 4200 (Agilent Technologies, Palo Alto, CA, USA). RNAseq libraries were prepared using the NEBNext Ultra RNA Library Prep Kit for Illumina with the manufacturer’s guidelines (NEB, Ipswich, MA, USA). Briefly, mRNAs were initially enriched with Oligod (T) beads. Enriched mRNAs were fragmented for 15 min at 94°C. First- and second-strand cDNA was synthesized. cDNA fragments were end-repaired and adenylated at 3’ends, and universal adapters were ligated to cDNA fragments, followed by index addition and library enrichment by PCR with limited cycles. The sequencing library was validated on the Agilent TapeStation (Agilent Technologies, Palo Alto, CA, USA) and quantified by using the Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA, USA) as well as by quantitative PCR (KAPA Biosystems, Wilmington, MA, USA). The sequencing libraries were clustered on three lanes of a flow cell, and the flow cell was loaded on the Illumina HiSeq instrument (4000) according to the manufacturer’s guidelines (Illumina. San Diego, CA, USA). The samples were sequenced by using a 2x150-bp paired-end configuration. Image analysis and base calling involved using HiSeq Control Software (HCS). Raw sequence data (.bcl files) generated from Illumina HiSeq were converted into fastq files using Illumina’s bcl2fastq 2.17 software (Illumina, USA).
De novo transcriptome assembly, read mapping, and sequence annotation
The adapters and low-quality sequencing reads (Q <30) were trimmed using Trimmomatic v.0.39 (Bolger and Giorgi, 2014; Natarajan et al., 2021). The quality-filtered reads were mapped to the reference de novo transcriptome generated with Trinity assembler and clustering with CD-HIT-EST using default parameters. The completeness and integrity of the de novo transcriptome assembly were assessed using Benchmarking Universal Single-Copy Orthologs (BUSCO) analysis and embryophyta_odb10 dataset (Simão et al., 2015). The read count table was created by using RSEM and Bowtie2. The differentially expressed genes (DEGs) among pair-wise stress treatment combinations were identified using the R package DESEq2 (Love et al., 2014). The minimum log 2-fold change = 1 and false discovery rate (FDR) cutoff 0.05 were used to filter the DEGs. Gene Annotation and Gene Ontology (GO) enrichment analyses were performed using the Omicsbox tool (https://www.biobam.com/omicsbox/). The unigenes were annotated against databases of NR, InterPro (UniProtKB/Swiss-Prot), COG, KOG, and CDD) Pathway mapping involved using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Natarajan et al., 2021) and the Pathview web tool (Luo et al., 2017). Time-series gene expression and co-expressed gene clusters with similar expression patterns across conditions were analyzed using maSigPro (Wan et al., 2017).
Quantitative real-time PCR
Quantitative real-time PCR (RT-qPCR) analysis was performed to validate the expression of DEGs identified in the RNAseq analysis. Total RNA for sugar maple leaves from all drought stress treatments and control samples was extracted using the Spectrum Plant Total RNA Kit (Sigma-Aldrich, St Louis, MO, USA). The RevertAid First Strand cDNA Synthesis Kit was used for cDNA synthesis (Thermo Scientific, Waltham, MA, USA). Primers for the selected genes were designed by Primer 3 software and custom-synthesized from IDT (Integrated DNA Technologies, Coralville, IA, USA). A list of the primers used for the validation is in Table S1. Quantitative gene expression analysis used the RT-qPCR system (Step Plus one, Applied Biosystems, Waltham, MA, USA). Four biological and three technical replicates were used for each treatment. The RT-qPCR reaction mixture contained 8 μL of Power track SYBR Green master mix (Applied Biosystems, Waltham, MA, USA), 2 μL of each of the forward and reverse primer (5 μM), and 20 ng template cDNA (2 μL) for a final volume of 14 μL. The RT-qPCR program was set at 95°C for 5 min, followed by 35 cycles of 95°C for 30 s and 60°C for 30 s. Relative gene expression was quantified by the 2-ΔΔCt method, and fold change in gene expression under stress conditions was calculated relative to control samples (Livak and Schmittgen, 2001; Vennapusa et al., 2020). The gene expression was normalized to the expression of B-tubulin and actin as the reference genes.
Estimation of peroxidase activity
The peroxidase activity (POD) was measured using the guaiacol peroxidase protocol (Cavalcanti et al., 2004). The frozen leaf material (1.0 g) was homogenized in 3 ml of ice-cold 100 mM potassium phosphate buffer with pH 6.8 containing 0.1 mM EDTA using mortar and pestle. The homogenate samples were allowed for 5 min and filtered through cheesecloth to remove the plant debris and centrifuged at 16 000 g for 15 min at 4˚C, and all the steps were carried out on the ice. The supernatant was used as a crude enzyme extract for the assay. The activity of the guaiacol peroxidase (POD) enzyme was determined by adding 25 µl of the crude enzyme extract to 2 ml of an assay buffer containing 50 mM K-phosphate buffer with pH 6.8, 20 mM guaiacol, and 20 mM H202. The assay samples were incubated at 30˚C for 10 min, and the reaction was stopped by adding 0.5 ml of 5% (v/v) H2SO4, and the absorbance was measured at 480 nm. One unit of POD was expressed as the change of 1.0 absorbance unit per ml enzymatic extract and defined as units of enzyme activity per g fresh weight per min (UA g-1 FW min-1).
Statistical analysis
One-way ANOVA was used to estimate the significant differences between drought stress treatments on various physiological and biochemical traits and gene expression studies (Pandian et al., 2020). The R package ggplot2 was used to generate bar graphs (Wickham and Wickham, 2007; Vennapusa et al., 2022). Fisher’s LSD test was used to separate the mean of significant treatments at P ≤ 0.05 by using the R package agricolae (de Mendiburu, 2020; Pandian et al., 2021).
Results
We investigated the physiological, biochemical, and molecular responses of sugar maple to drought stress. Transcriptome analysis was performed, and a de novo reference genome was assembled to identify DEGs between control and drought-stressed samples.
Morpho-physiological responses of sugar maple to drought stress
The drought stress effect on sugar maple was assessed by imposing drought stress for 7, 14, and 21 days. The control plant pots (watered every day) maintained a soil volumetric water content of 45%; 7-day stressed plant pots maintained a soil moisture content of about 29%, and 14- and 21-day stressed plant pots maintained a soil moisture content of about 17% (Figure S1). The control plants grew normally and maintained the stay-green nature, with the regeneration of young leaves (Figure S2). The 7-day drought-stressed plants showed relatively fewer yellow symptoms on leaves as compared with 14- and 21-day stressed plants. The plants exposed to 14 days of stress showed distinct yellowish symptoms and some necrotic spots as compared with 7-day stressed plants. The plants exposed to 21 days of stress showed severe yellow and necrotic symptoms with wilting and shoot meristem burning at the tips as compared with all other stress treatments (Figure S2).
The drought stress effect on chlorophyll content and photosynthesis in sugar maple was assessed with SPAD and NDVI measurements. All leaf-level physiological variables showed significant variation (p ≤ 0.05) with different times of drought stress exposure. The chlorophyll content represented by SPAD units showed a significant effect of drought stress. The control plants maintained a higher chlorophyll index, and a higher reduction was observed at 21DAS (22.7) compared to other stress treatments (7DAS [24.7] and 14DAS [23.7]; Figure 1A). The NDVI in the drought-subjected leaf samples was significantly affected by drought stress. The control plants maintained higher NDVI (0.55), and a decreasing trend was observed with increasing stress times (7DAS [0.50], 14DAS [0.48], and 21DAS [0.44]). Reduction in NDVI was significantly greater with 21 days of drought stress than with other stress times and in control plants (Figure 1B).
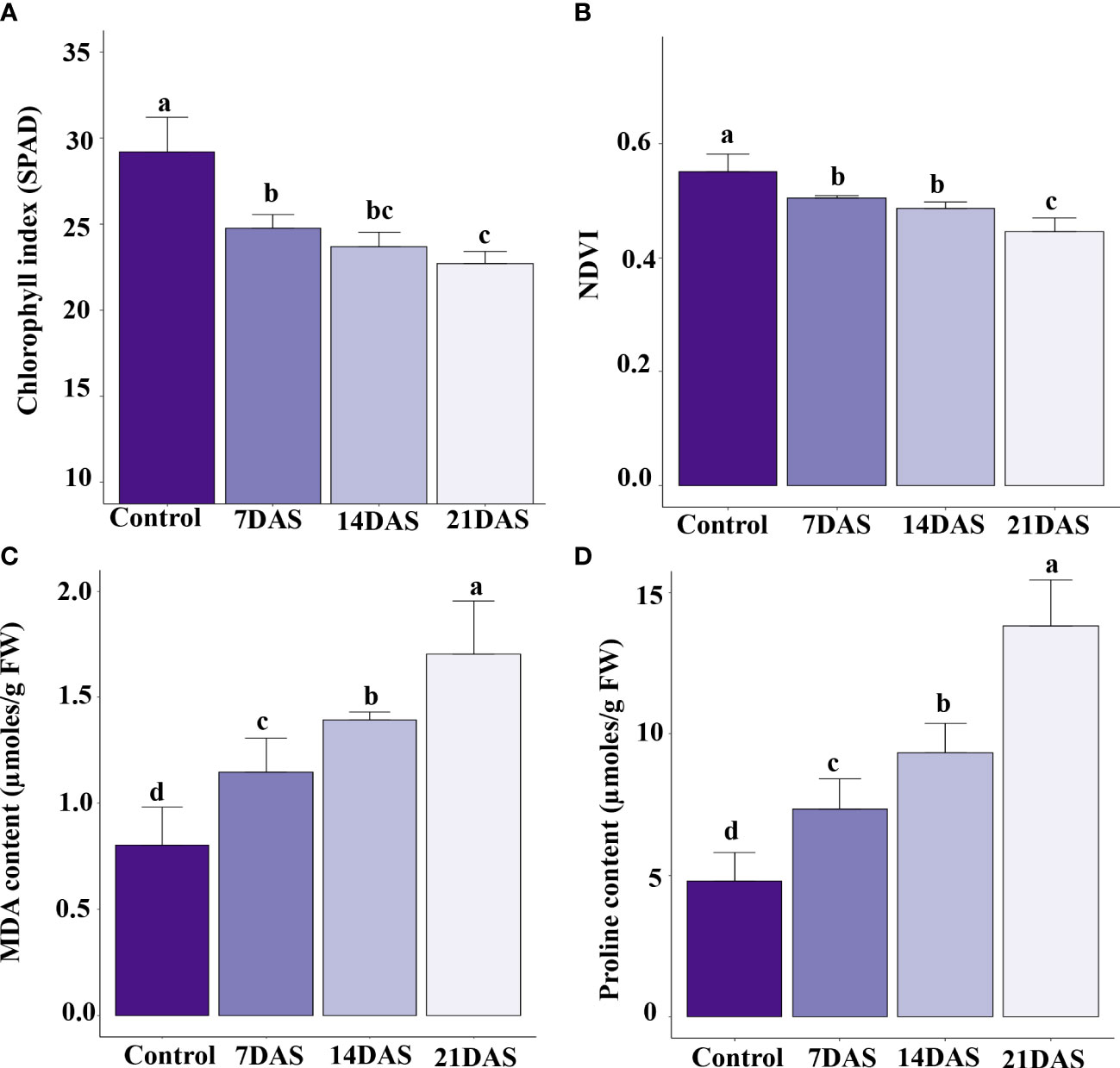
Figure 1 Physiological and biochemical changes in sugar maple saplings in response to drought stress: Sugar maple plants were subjected to drought stress for 7, 14, and 21 days and data were analyzed by comparison with the control samples. (A) Chlorophyll index (SPAD), (B) NDVI, (C) MDA, and (D) proline content. Different alphabetic letters on top of the error bar graphs indicate statistically significant differences between the stress periods (p ≤ 0.05). SPAD: Soil and Plant Analysis Development; NDVI: Normalized Difference Vegetation Index; MDA: malondialdehyde.
Biochemical changes with drought stress
Biochemical profiling of leaves with drought stress treatments helps understand the stress responses and induced protective mechanisms in plants. We observed enhanced production of MDA in sugar maple with increasing drought stress time. The MDA content was significantly induced at 7DAS (1.14 µmoles/g FW) and showed an increasing trend with increasing stress time: 14DAS and 21DAS (1.39 and 1.71 µmoles/g FW, respectively) (Figure 1C). Among all the time points, control samples showed less MDA content (0.8 µmoles/g FW).
Therefore, we studied the accumulation of proline in sugar maple leaves in response to drought stress. Proline content showed an increasing trend with prolonged stress in sugar maple leaves. It was significantly higher at 21DAS (13.8 µmoles/g FW) than at other stress times (9.34 µmoles/g FW at 14DAS, and 7.35 µmoles/g FW at 7DAS) and control samples (4.79 µmoles/g FW) (Figure 1D).
De novo transcriptome assembly and annotation
The quality filtered paired-end reads from the control and treatment samples (7, 14, and 21DAS) were assembled de novo using Trinity assembler. The de novo assembly and further clustering produced 310, 981 transcripts which included 218, 885 unique longest open reading frame (ORF) transcripts. The average contig size was 1,177, and the contig N50 was 2115 (Table 1). The resulting unigenes were used as a reference transcriptome for further analysis of RNAseq data. The completeness and integrity of the de novo transcriptome assembly from sugar maple were assessed using BUSCO analysis and the embryophyta_odb10 dataset. Of the 1614 BUSCOs from the embryophyta orthologue groups, 89% were identified as complete BUSCOs in the sugar maple transcriptome assembly, indicating the high-quality reference transcriptome data set comparable with transcriptome assembly from land plants (Gault et al., 2018). The assembly also contained 81.8% of the single-copy BUSCOs and only 5.8% of the missing BUSCOs (Table 2). A total of 55,955 unigenes were successfully annotated using the NR database at NCBI and including InterPro (UniProtKB/Swiss-Prot), COG, KOG, and CDD databases provided high confidence annotation to a total of 75,959 unigenes (Table S2; Figure 2).

Table 1 Summary of de novo transcriptome assembly.
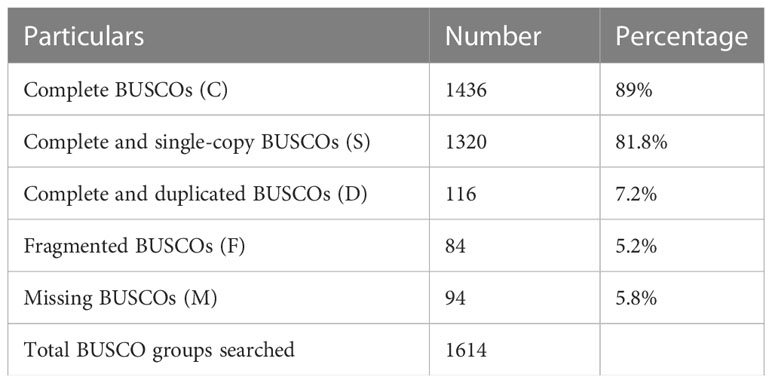
Table 2 Summary of BUSCO analysis.
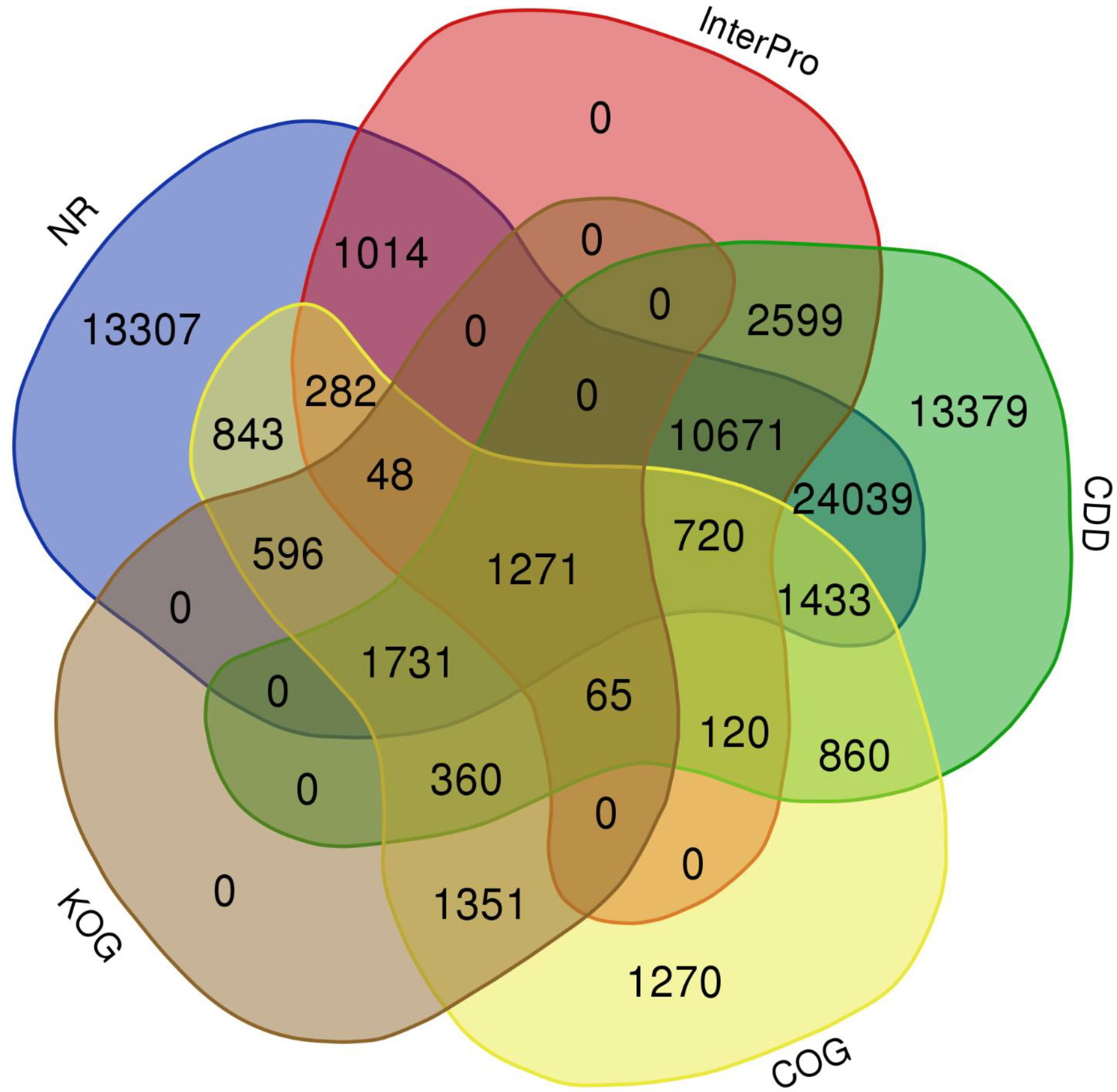
Figure 2 Venn diagram shows the annotation shared among NR, InterPro, CDD, COG, and KOG databases.
Summary of RNAseq and mapping
RNAseq was performed under drought stress to assess the global transcriptome profile of sugar maple leaf samples. RNAseq of 16 samples with the four treatments and four biological replicates under “irrigated control” or “drought stress treatment (7, 14, and 21DAS)” generated a total of 395,240,587 raw reads; 365,445,423 were identified as filtered reads (Table S3). These samples were clustered based on the gene expression and presented with a principal component analysis (PCA) plot in Figure S3. The filtered reads were aligned and mapped to the reference de novo transcriptome with an average mapping percentage of 92%. The summary of RNAseq reads and genome mapping are given in Table S3.
DEGs in response to drought stress
DEGs were determined between the control and the three different drought stress treatments in sugar maple leaf samples. The number of DEGs between the control and 7, 14, and 21DAS samples are shown in Table S4 and were filtered using log2 fold-change cutoff ≥ 1 and FDR <0.05. The number of total DEGs was greater between 7DAS and the control (9,936); most were downregulated (9,520). The number of total DEGs was lowest among 14DAS and the control (2,374), and more were upregulated (1,223) than downregulated genes (1,151). The total number of DEGs between 21DAS and the control was 4,456: 2,978 were upregulated and 1,478 downregulated (Table S4). The overall differential gene expression pattern and their variation under specific drought stress regimes are visualized with a PCA plots and volcano plot in Figures S3 and S4.
We used a pair-wise comparison of the DEGs from three different drought stress conditions to explore unique as well as common DEGs in response to drought stress in sugar maple (Figure 3). A total of 14,099 DEGs were identified during drought stress against the control samples: 328 genes were common between all three drought stress times, and 8480, 639, and 2,587 were unique at 7, 14, and 21DAS, respectively. The gene number shared between 7DAS and 14DAS was 470, and between 7DAS and 21DAS was 658. The number shared between 14DAS, and 21DAS was 883. Lists of significantly up- or downregulated DEGs among all drought stress treatments and their annotations are in Tables S5-S8.
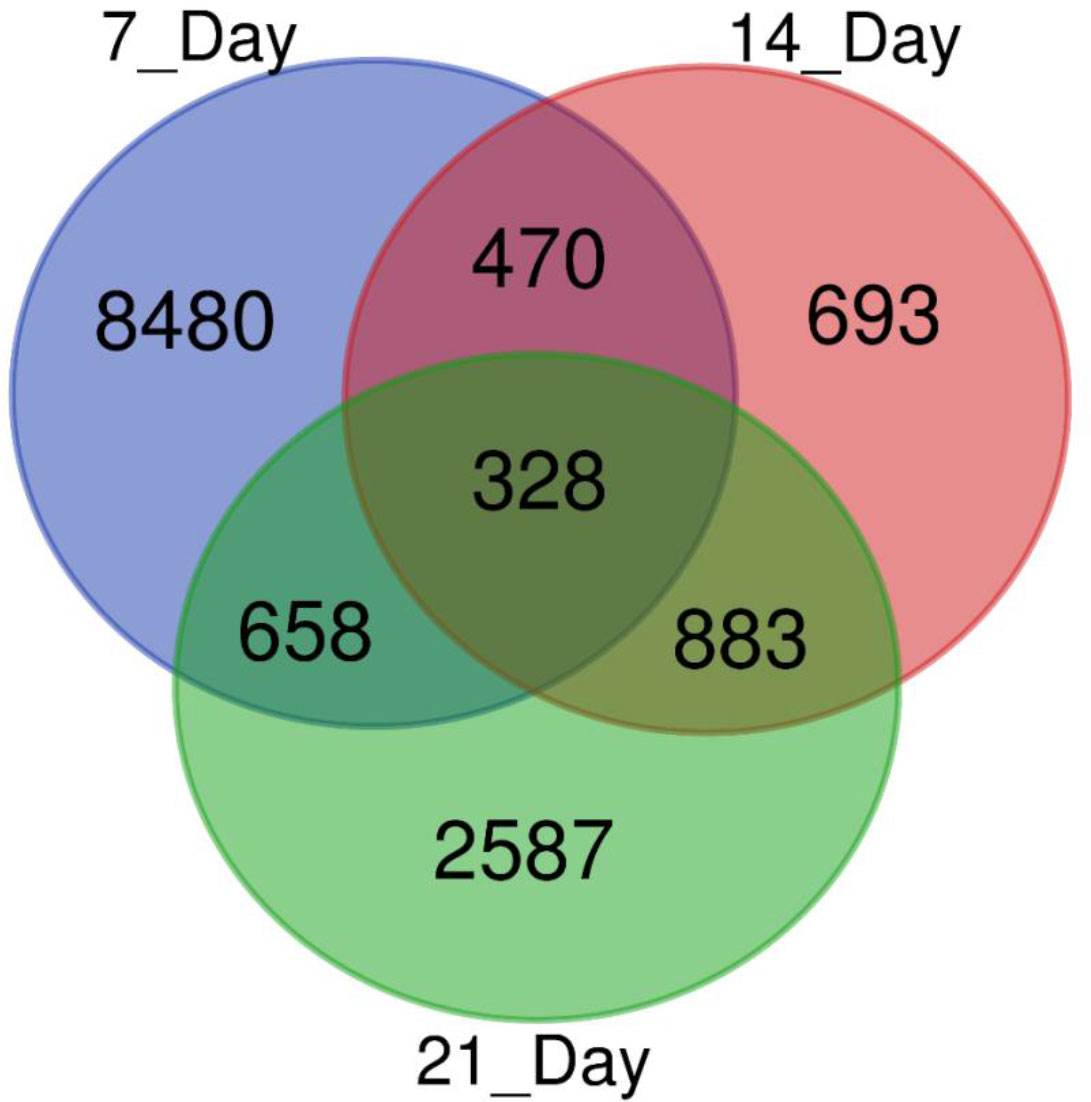
Figure 3 Venn diagram showing the number of DEGs obtained from different drought stress imposition periods in sugar maple.
Among the 328 DEGs common to all stress times, a large number were related to the chloroplast, protein kinases, membrane transporters, and protein detoxification. Many genes encoded stress-responsive transcription factors, such as NAC, HSF, ZFPs, RING-finger, YTH, HD-Zip, GRFs, and ERF, which are involved in alleviating drought stress in plants (Joshi et al., 2016). In addition, DEGs included stress-responsive genes involved in antioxidant activity (peroxidases, dehydrogenases, reductases, cytochrome P450, PAL), sugar metabolism, splicing, ubiquitination, hormonal, and signaling (Table S8).
GO assignments for functional classification of DEGs
We used GO enrichment analysis of 14,099 genes to identify the enriched biological processes and functions in the DEGs. GO enrichment analysis is used for understanding the regulation of DEGs under the three essential categories: biological process (BP), molecular function (MF), and cellular component (CC). The top 20 enriched GO terms under each category (BP, MF, and CC) at different stress periods are in Figure 4. GO enrichment analysis of DEGs from the three different drought stress periods revealed the functional categories activated under drought stress treatments. Under BP, the highest percentage of DEGs commonly enriched with all three drought stress treatments included protein phosphorylation (2.5-3.9%) and transmembrane transport (2.4-3.1%). Among MF, the highest percentage of DEGs commonly enriched was ATP (7-10%), DNA (2-2.4%), RNA (2.2-2.8%), metal ion (4.8-5.9%), and zinc ion binding (1.9-2.6%). Among CC, the highest percentage of DEGs commonly enriched was an integral component of the membrane (19.3-22.8%), nucleus (10.1-12.9%), cytoplasm (6-6.4%), and membrane (5-6.3%) processes. The overall list of the GO enrichment process is in Table S9.
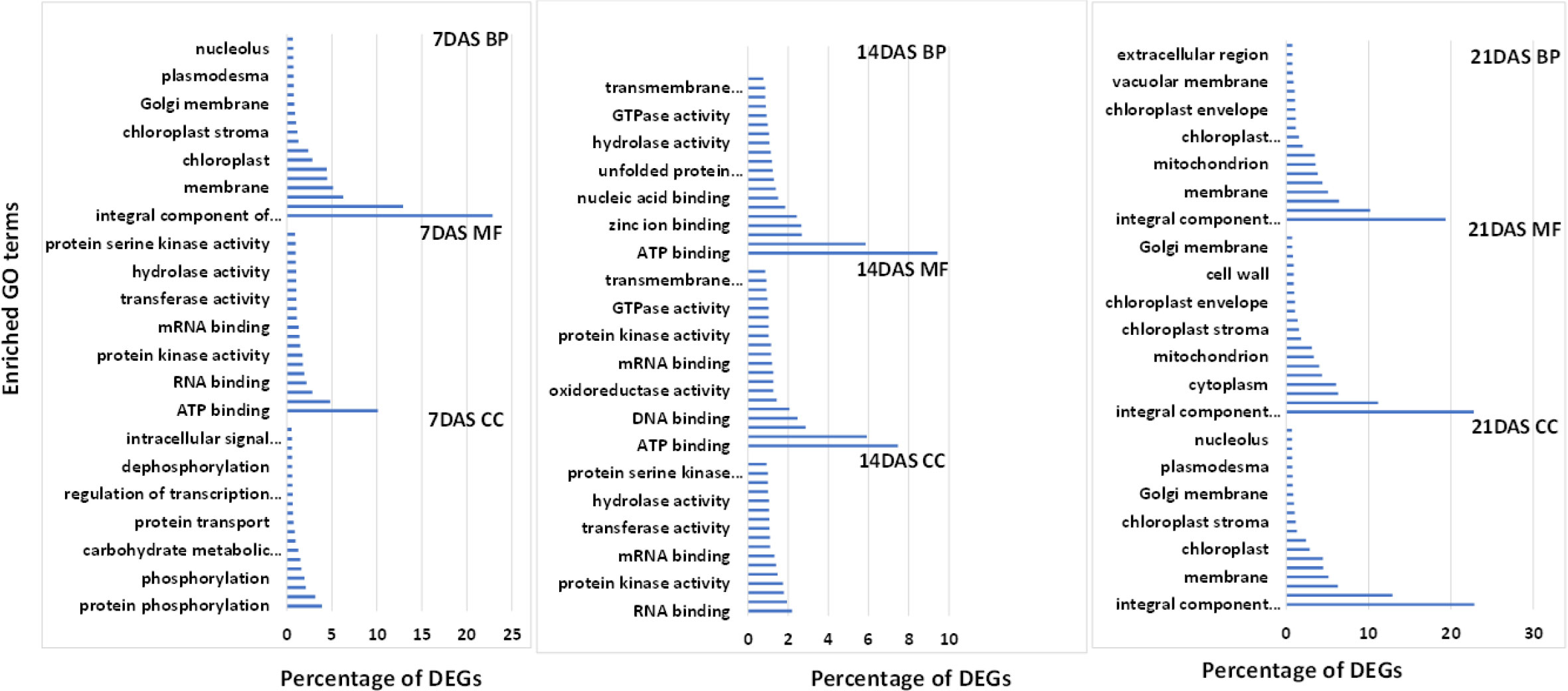
Figure 4 Top 20 enriched GO terms for the common DEGs in sugar maple leaf samples imposed to different drought stress periods (7, 14, and 21 days of drought stress). BP: biological process, CC: cellular component, and MF: molecular functions.
KEGG pathway enrichment analysis of DEGs
A total of 249 KEGG terms were assigned to the 14,099 DEGs from all drought stress experiments. Overall, 56 KEGG pathways were significantly enriched (FDR-adjusted P < 0.05) with different drought stress periods (7, 14, and 21 days). Both up- and downregulated DEGs were involved in the metabolic pathways and biosynthesis of secondary metabolites pathway under all drought stress regimes (Table 3). The number of pathways in upregulated DEGs was highest at 21DAS and lowest at 7DAS. The number of pathways in downregulated DEGs was highest at 7DAS and lowest at 14DAS. The other most enriched pathways in both up- and downregulated DEGs included carbon metabolism, protein processing in the endoplasmic reticulum, circadian rhythm-plant, carotenoid biosynthesis, carbon fixation in photosynthetic organisms, fatty acid degradation glycerolipid metabolism, biosynthesis of amino acids, and flavonoid biosynthesis pathways (Tables 3, S10).
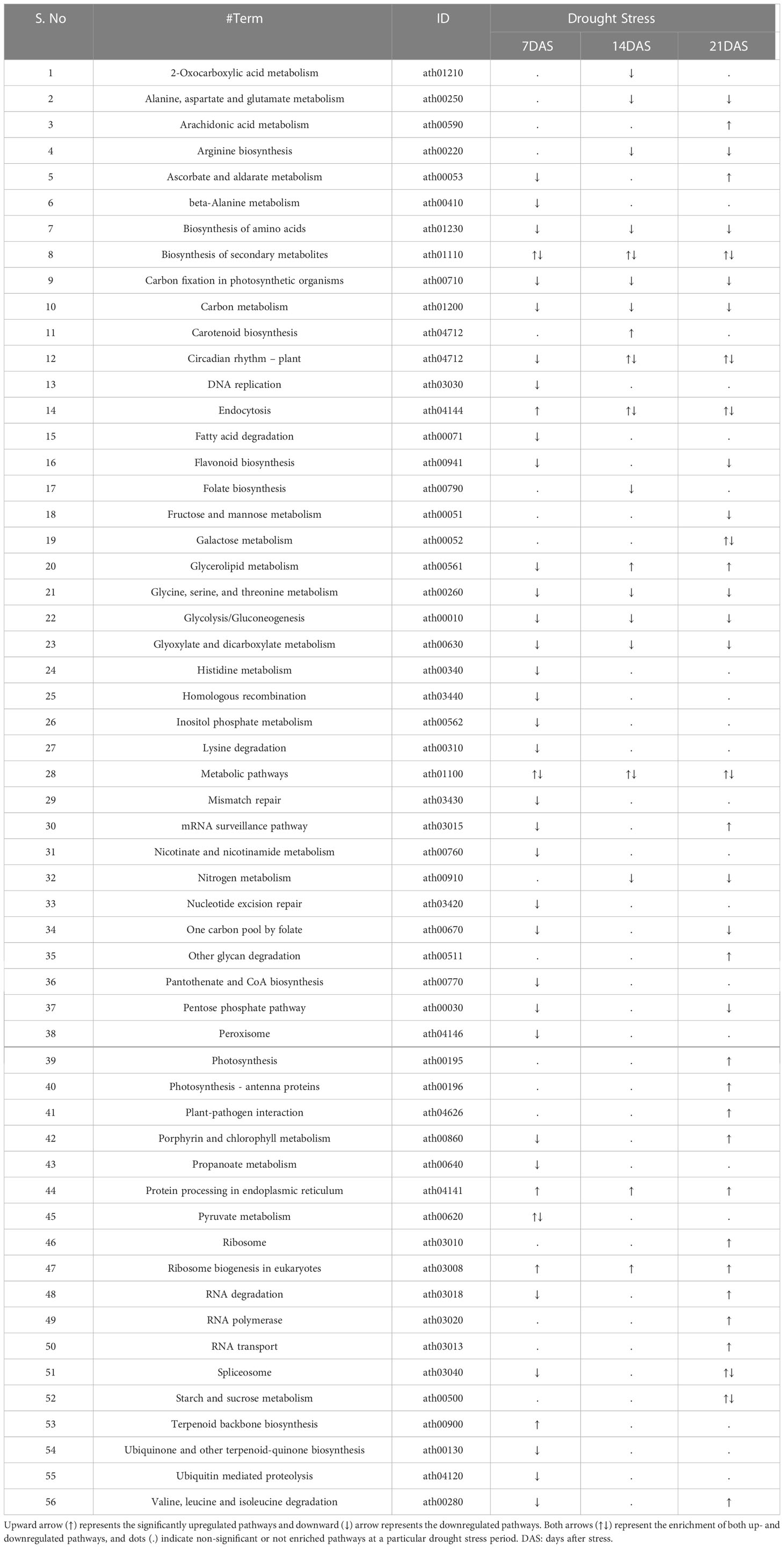
Table 3 KEGG pathway enrichment analysis of differentially expressed genes in response to drought stress in sugar maple.
Time series analysis of gene expression upon drought stress
The genes regulated under drought stress were clustered according to the expression ratios of drought stress samples to the control samples at each time point, which resulted in eight clusters based on their regulation pattern (Figure 5). The gene regulation in clusters 1 and 4 showed a downregulation pattern with increasing drought stress time, and the genes from four major clusters (2, 3, 5, and 7) showed an upregulation pattern. In cluster 6, the gene expression showed an increasing pattern at 7DAS, and with increasing stress periods at 14 and 21DAS, the expression showed a marked decrease. In cluster 8, the pattern of gene regulation was decreased at 7 to 14DAS and recovered with the increased regulation pattern at 21DAS. Eight major clusters, consisting of 1,317 genes, were classified into three groups: genes showing the upregulated pattern on drought stress (upregulated DEGs: clusters 2, 3, 5, and 7) and a downregulated pattern (downregulated DEGs: clusters 1 and 4) and a differential pattern (clusters 6 and 8), which contained 445,824 and 48 genes, respectively (Figure 5). We performed pathway enrichment analysis to identify significantly enriched stress pathway hubs individually from each of the five clusters. We identified 1, 4, and 9 significant KEGG pathways with FDR cutoff, P < 0.05 for clusters 2, 3, and 5 in the upregulated pattern and 12 and 18 pathways in clusters 1 and 4 in downregulated pattern, respectively. The significantly enriched pathways in clusters 2, 3, and 5 with the upregulated pattern are RNA degradation and transport, metabolic pathways, biosynthesis of secondary metabolites, Ascorbate and aldarate metabolism, porphyrin, and chlorophyll metabolism, circadian rhythm-plant, fatty acid degradation, and glycerolipid metabolism. The downregulated pattern that occurred in the cluster 1 and 4 showed the pathways such as carbon metabolism, carbon fixation in photosynthetic organisms, alanine, aspartate, and glutamate metabolism, metabolic pathways, biosynthesis of amino acids, biosynthesis of secondary metabolites, arginine biosynthesis, and glyoxylate and dicarboxylate metabolism (Table S11). Detailed cluster-wise information for all genes is in Table S11.
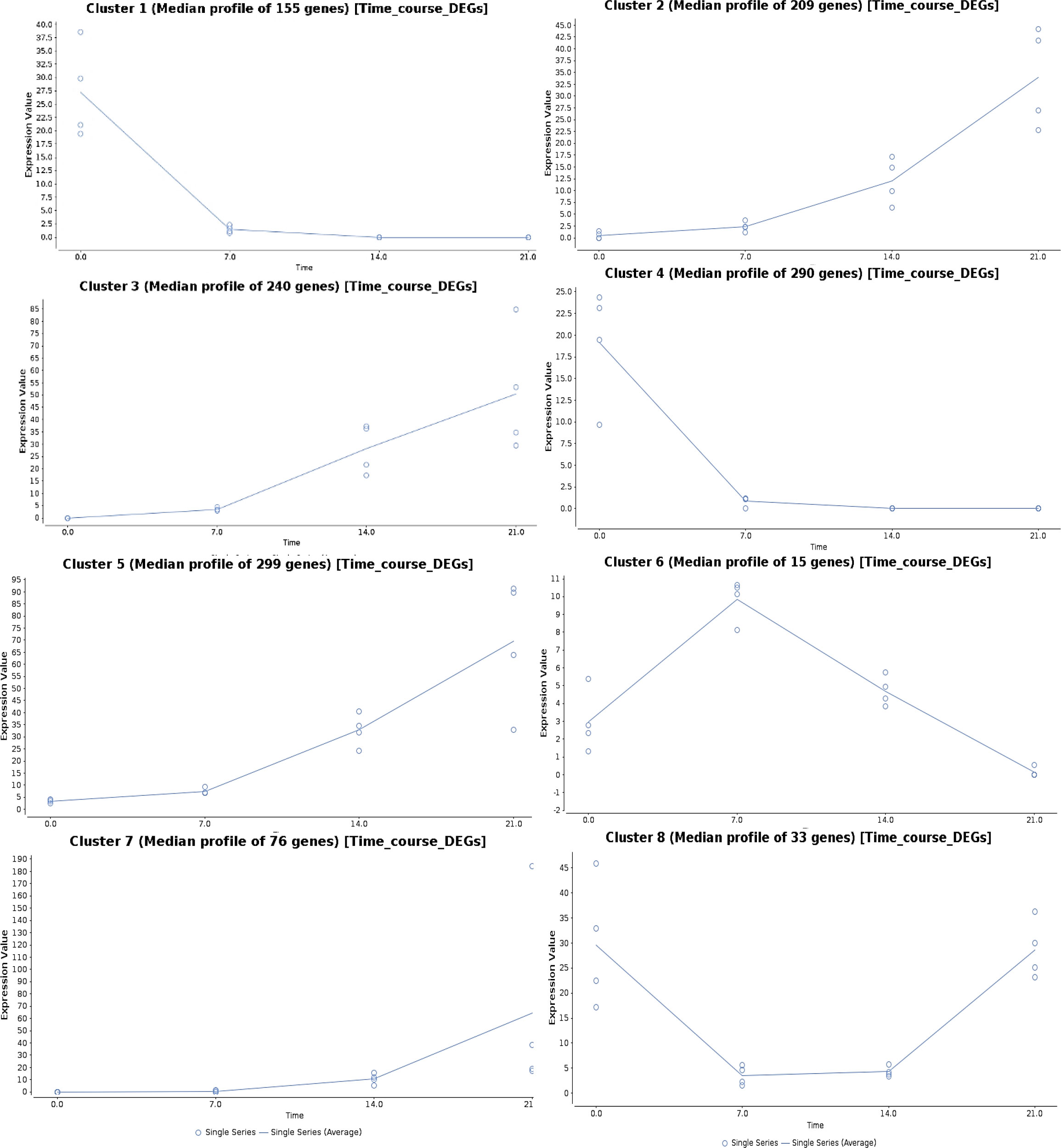
Figure 5 Time-series gene expression analysis showing the expression pattern in eight different clusters upon different drought stress periods in sugar maple.
Validation of DEGs expression by qRT-PCR and enzyme assay
To confirm the reliability and validity of the RNAseq results in sugar maple, we randomly selected four genes (TRINITY_DN1230_c0_g1_i11, TRINITY_DN287_c0_g2_i28, TRINITY_DN172_c0_g2_i1, and TRINITY_DN1259_c0_g1_i3) from DEGs for the 7, 14, and 21DAS samples for qRT-PCR analysis. The expression of the selected genes was positively correlated with the RNAseq results. Even though the detected fold change expression do not exactly match, the selected genes showed the same trends of expression patterns. For example, the upregulation of the peroxidase P7-like gene (TRINITY_DN1230_c0_g1_i11) and downregulation of the 4-alpha-glucanotransferase, chloroplastic/amyloplastic gene (TRINITY_DN287_c0_g2_i28) was confirmed by both RNAseq and qRT-PCR. Overall, the qRT-PCR expression followed a similar expression pattern of DEGs, thus confirming the consistency of the transcriptome sequencing analysis (Figure 6).
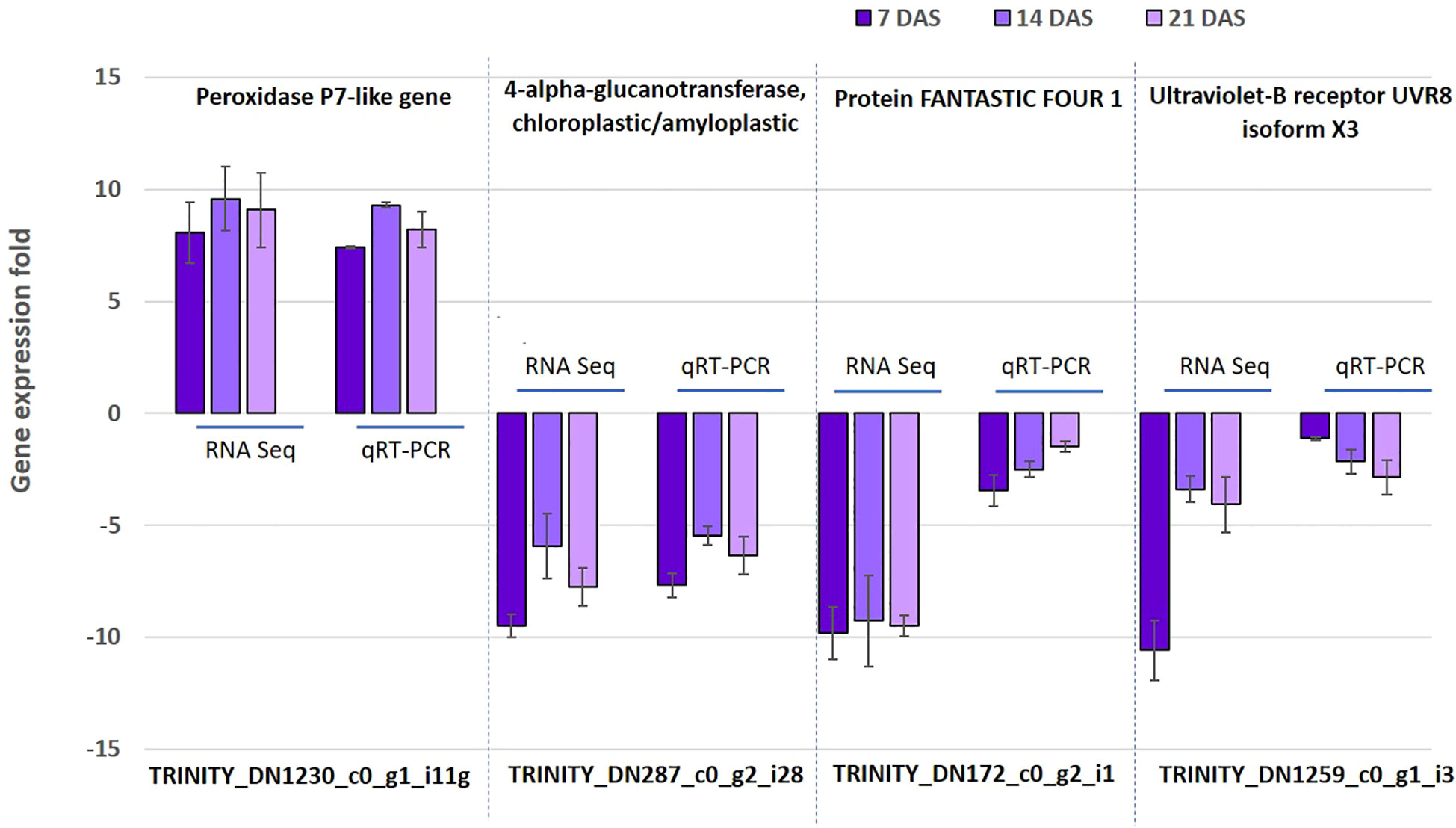
Figure 6 Validation of RNAseq data using quantitative real-time PCR. The expression of four selected genes was studied using qRT-PCR. Data are mean (SE) expressions from three biological replicates of sugar maple at each drought stress time. The β-tubulin and actin gene were used as an internal control for normalizing gene expression and fold change was calculated over the control.
In order to assess the protein level expression of one of the top 10 upregulated DEGs (peroxidase P7-like gene), the peroxidase enzyme assay was performed. The increase in enzyme activity with increasing stress periods showed the activity of enzyme results is directly proportional to the gene expression data. However, no significant differences were observed in the activity of enzymes between the 14 and 21 DAS (Figures 6; S5).
Discussion
Considering the increased demand for sugar maple and its multiple roles in providing ecosystem services across its range, understanding how sugar maple plants will be affected by climate change signifies the importance of handling our natural resources within the Anthropocene (Wolkovich and Cleland, 2014). This study describes the molecular mechanisms involved in the physiological and biochemical responses of sugar maple saplings to drought stress.
Morpho-physiological signatures in response to drought stress
Perennial trees signal their water deficit with several symptoms. The most common alterations in appearance are lighter green to yellow-green foliage, scorches around the leaf margins, leaf wilting, necrosis, and drooping (Li et al., 2015; Ball, 2021; Ribeyre et al., 2022). We observed similar symptoms in sugar maple upon drought stress in the present study; severe symptoms occurred with increased drought stress time (Figure S2). The physiological indexes of reduced chlorophyll and NDVI observed in sugar maple under drought stress were consistent with the results of other drought stress studies of crop and tree species (Silber et al., 2003; Hassan and Ali, 2014; Li et al., 2015; Kargar et al., 2017; Basal and Szabo, 2020; Bissiwu et al., 2022) (Figures 1A, B).
Biochemical responses of sugar maple to drought stress
MDA is an oxidative stress-induced product that acts as a biomolecular marker for cell membrane damage caused by lipid peroxidation injury in plants (Nisarga et al., 2017; Demirel et al., 2020). Its accumulation varies in response to drought stress depending on the length of stress exposure and stress intensity (Davey et al., 2005; Singh et al., 2017). Membrane lipid peroxidation also serves as an indicator of the prevalence of reactive oxygen species (ROS) and indicates the impact of stress on plant cell damage (Patel and Hemantaranjan, 2012; Nisarga et al., 2017; Zhang et al., 2021). We observed a marked increase in MDA level with increasing stress time in our sugar maple leaf samples (Figure 1C), which suggests that drought stress-induced oxidative stress caused lipid membrane damage in corroboration with prior reports.
The production of osmolytes like proline stabilizes membrane and protein structural damage under low leaf water potential conditions upon drought stress (Kiranmai et al., 2018). Osmolyte production varies according to stress severity, the length of stress exposure, and plant species (Anjum et al., 2011). Moreover, proline accumulation is a well-reported compatible osmolyte in plants in response to drought stress (Demirel et al., 2020). Our results showed higher levels of proline accumulation under drought stress conditions with prolonged stress in sugar maple (Figure 1D). Higher accumulation of proline in plants was involved in reducing photo-oxidative damage in chloroplast thylakoid membranes by scavenging and/or reducing ROS production (Verbruggen and Hermans, 2008; Kiranmai et al., 2018; Lokesh et al., 2019). Also, proline has been reported to stabilize and protect ROS scavenging enzymes and activate alternate detoxification pathways in plants upon exposure to various abiotic stresses (Ramu et al., 2016; Patykowski et al., 2018; Kijowska-Oberc et al., 2020; Nareshkumar et al., 2020). Therefore, induced proline content with increasing drought stress possibly acts as a direct antioxidant via the osmo-protecting mechanism as well as activating other antioxidant mechanisms.
Molecular mechanisms in response to drought stress
Elucidating the gene-level stress adaptive features to overcome adverse environmental conditions is vital to understand the molecular mechanisms in response to drought stress in sugar maple. In this study, we identified 14,099 potential DEGs in response to drought stress; 328 were commonly expressed during all three drought-stress periods (Figure 3; Table S5-S8).
A number of DEGs common to all three stress periods that were related to chloroplast and photosynthesis were more frequently downregulated than upregulated in sugar maple, and similar patterns of DEGs were reported in other plant species in response to drought stress (Haider et al., 2017). Also, these results agree with reduced photosynthetic parameters (Chaves et al., 2009; Li et al., 2020). Many common DEGs encode protein kinases, which play crucial roles in stress sensing and signal transduction (Priya et al., 2019; Yang et al., 2019; Chen et al., 2021). Membrane transporter genes were differentially expressed in common DEGs; these genes play a key role in response to drought stress by transporting signaling molecules, ions, or osmolytes (Jarzyniak and Jasiński, 2014). We found the downregulation of protein detoxification genes (multidrug and toxic compound extrusion [MATE] transporters). These enzymes regulate plant growth and development processes by transporting secondary metabolites, toxic cell compounds, and hormone regulation (Du et al., 2021; Wang et al., 2022).
Transcription factors play an important role in signal transduction pathways in plants during stress by regulating the expression of downstream target genes via specific binding to cis-acting elements. NAC, bZIP, MYB, HSF, GRFs and ERF, WRKY, DREB, and ERFs are well-known transcription factor families that are responsive to drought stress (Kiranmai et al., 2018; Lokesh et al., 2019; Yang et al., 2019). In this study, transcription factor genes including NAC, HSF, ZFPs, YTH, HD-Zip, GRFs, and ERF were commonly expressed under drought stress (Table S5-S8). Among the transcription factor genes identified as being differentially expressed in the present study, many are known to play a key role in drought tolerance (Alves et al., 2011; Omidbakhshfard et al., 2015; Zhu et al., 2019; Sun et al., 2020; Yao et al., 2021). Furthermore, a plethora of drought stress tolerance genes differentially expressed in sugar maple included peroxidases, ubiquitin, cytochrome P450, hormone, lipid, antioxidant, and sugar metabolism-related and other stress-responsive genes (Table S8).
The unavailability of water induces a wide range of physiological and biochemical changes in plants. These responses include regulation of the cellular process, metabolic functions, and maintenance of photosynthesis, respiration, and oxidative damage of the cellular components under stress conditions in plants (Shinozaki & Yamaguchi-Shinozaki, 2007; Gao et al., 2018). Similar responses were observed in the present study. We identified a number of DEGs involved in pathways such as protein phosphorylation, photosynthesis, chloroplast organization, and their integral parts of stroma and thylakoid membrane. This observation shows the effect of drought stress on chlorophyll synthesis or photosynthetic activities and transcript level responses as evidenced by the reduced physiological indices like chlorophyll index and NDVI with increasing drought stress. The higher percentage of DEGs from transmembrane transport, ATP binding, metal and zinc ion binding, and integral component of membrane pathways suggest the drought-induced effect on membrane damage and gene-level response to overcome the effects of stress. This finding is also evidenced by the increased lipid peroxidation of the membrane in sugar maple upon drought stress (Figure 1C and Table S8).
A high number of DEGs involved in protein phosphorylation, carbohydrate metabolic process, oxidoreductase activity, and cytoplasm suggests the cellular level response of the sugar maple to overcome the oxidative stress induced by increasing drought stress, which is also corroborated by the enhanced production of osmolytes (Figure 1D). GO terms enriched in processes such as ATP, DNA, and RNA binding, nucleus, DNA-binding transcription factor activity, nucleotide binding, nucleic acid binding, and mRNA binding suggests the stress-induced gene signaling and molecular level response in sugar maple to drought stress (Figure 4 and Table S9). Similar GO terms in response to drought stress were reported in other plant species as well as the expression of a high percentage of DEGs involved in photosynthesis, respiration, and oxidative damage (Estravis-Barcala et al., 2020; Sobreiro et al., 2021; Tahmasebi and Niazi, 2021).
Metabolic pathways and biosynthesis of secondary metabolites were the most commonly expressed pathways at 7, 14, and 21 days of drought stress and were significantly enriched both in up- and down-regulated DEGs. Other pathways such as carbon metabolism, circadian rhythm-plant, carotenoid biosynthesis, glycerolipid metabolism, and protein processing in the endoplasmic reticulum were significantly enriched, which suggests that these pathways were activated under drought stress. Induced and downregulated DEGs involved in metabolic pathways, carotenoid biosynthesis, circadian rhythm, carbon metabolism, porphyrin and chlorophyll metabolism, photosynthesis-antenna proteins, photosynthesis, and carbon fixation in photosynthetic organism pathways suggesting that the photosynthetic response copes with stress-induced damage on chlorophylls and other pigments, as evidenced by the reduced SPAD metric and NDVI in sugar maple (Table 3, Table S10). Significantly enriched DEGs in metabolic pathways, glycerolipid metabolism, fatty acid degradation, glycolysis/gluconeogenesis, and protein processing in the endoplasmic reticulum were upregulated possibly to protect the lipid membrane protein damage caused by the induced lipid peroxidation upon drought stress. Significantly enriched DEGs in metabolic pathways, biosynthesis of amino acids, biosynthesis of secondary metabolites, pentose phosphate pathway, starch and sucrose metabolism, fructose, and mannose metabolism pathways may induce osmolyte production for protecting cellular level damage in sugar maple in response to drought stress. DEGs enriched in flavonoid biosynthesis, folate biosynthesis, beta-alanine metabolism, and biosynthesis of secondary metabolite pathways suggest the activation of antioxidant machinery under drought stress in sugar maple.
In addition to nonenzymatic antioxidative machinery plants have developed various enzymatic antioxidative detoxification mechanisms in adoption to drought stress. Antioxidant enzymes such as peroxidase, ascorbate peroxidase, superoxide dismutase, and catalase play major roles in ROS scavenging molecules and minimize their levels under drought stress (Sharma and Zheng, 2019; Wei et al., 2019; Li et al., 2023). In our study, many DEGs related to antioxidative activity, peroxidases, and other detoxification enzymes like cytochrome P450 suggest the important role of enzymes in ROS scavenging in response to drought stress (Table S8). Significantly enhanced expression of peroxidase at both gene and protein (enzyme activity) levels suggests the substantial role of sugar maple in coping with drought stress (Table S8 and Figure S5). We found a gene (TRINITY_DN1230_c0_g1_i11g) that encodes peroxidase-like protein was significantly upregulated at all the drought stress periods (7, 14, and 21 DAS) and maintained enhanced enzyme activity suggesting that ROS scavenging system plays a key role in protecting the sugar maple from drought stress.
Time-series transcriptomic analysis allows for investigating the dynamic gene responses to drought stress and characterizing drought-responsive genes with respect to increasing drought stress and relevance to drought tolerance. Time-series gene expression analysis revealed different functional groups of genes regulated at 7, 14, and 21 days of drought (Table S11). Time series analysis was used in previous studies to understand the gene regulation patterns in response to drought stress in plants (He et al., 2020; Yi et al., 2022). Some plant physiological functions were suppressed, and a series of responses were activated to overcome the drought stress effects by complex gene regulatory and networks, as evidenced by global transcriptomic changes in eight clusters of regulation patterns we found in sugar maple (Table S11). Our results indicate that drought stress remarkably altered the expression of chlorophyll/photosynthesis-related and plant cell membrane and osmotic adjustment related genes to avoid cell damage in sugar maple. Overall, the results suggest that drought stress-induced changes in photosynthesis and cell membrane damage are possibly encountered by both enzymatic (peroxidase) and nonenzymatic antioxidant (proline) mechanisms in sugar maple saplings.
Conclusion
The present study reports for the first time the characterization of the global transcriptome of sugar maple in response to drought stress. A set of physiological and biochemical traits examined in this study showed distinct signatures upon drought stress, suggesting that changes in chlorophyll biosynthesis, lipid membrane damage, and osmolyte production play a pivotal role in determining the sugar maple response to the severity of drought stress. The 328 common DEGs identified under all three drought stress periods in sugar maple will be a potential source for understanding the drought-tolerant mechanisms. Genes related to chlorophyll and carotenoid biosynthesis, membrane, oxidative stress response, osmolytes, and secondary metabolite production related to antioxidant activity pathways were significantly enriched on GO and KEGG analyses. These genes may play important roles in sugar maple adaptation or tolerance to the oxidative stresses caused by drought. In addition, we demonstrated the time-series expression profiling of global genes in response to drought stress for three time periods and identified the stress-responsive pathways. Overall, the study results help in understanding the drought stress-responsive molecular mechanisms complemented by physiological and biochemical responses of sugar maple and expand this knowledge in other tree species.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/bioproject/895722.
Author contributions
KM and SE conceived and designed the experiments. LM conducted the major experiments. AV, OJ and KK performed qRT-PCR. AV conducted a statistical analysis and summarized the transcriptome data. AV and LM wrote the manuscript. UR and PN performed RNAseq analysis. KM, KK, and SE revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This project was funded by McIntire Stennis Cooperative Forestry Research Program (NI19MSCFRXXXG045) to Delaware State University and the USDA-NIFA grants (2020-38821-31083 and 2018-38821-27744) to KM.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1150204/full#supplementary-material
Supplementary Table 3 | Summary of RNAseq and mapping analysis.
Supplementary Table 9 | GO enrichment analysis.
Footnotes
- ^ https://agriculture.vermont.gov/sites/agriculture/files/documents/AgDevReports/Maple%20Syrup%20Market%20Research%20Report.pdf.
References
Alves, M. S., Fontes, E. P., Fietto, L. G. (2011). Early responsive to dehydration 15, a new transcription factor that integrates stress signaling pathways. Plant Signal. Behav. 6, 1993–1996. doi: 10.4161/psb.6.12.18268
Anjum, S. A., Xie, X. Y., Wang, L. C., Saleem, M. F., Man, C., Lei, W. (2011). Morphological, physiological and biochemical responses of plants to drought stress. Afr. J. Agric. Res. 6, 2026–2032. doi: 10.5897/AJAR10.027
Aranda, I., Gil-Pelegrín, E., Gascó, A., Guevara, M. A., Cano, J. F., Miguel, M., et al. (2012). “Drought response in forest trees: from the species to the gene,” in Plant responses to drought stress. Ed. Aroca, R. (Heidelberg: Springer Berlin Heidelberg), 293–333. doi: 10.1007/978-3-642-32653-0_12
Baldoni, E. (2022). Improving drought tolerance: Can comparative transcriptomics support strategic rice breeding? Plant Stress 3, 100058. doi: 10.1016/j.stress.2022.100058
Ball, J. (2021) Trees & drought stress. Available at: https://extension.sdstate.edu/trees-drought-stress.
Basal, O., Szabo, A. (2020). Physiomorphology of soybean as affected by drought stress and nitrogen application. Scientifica 2020, 1–7. doi: 10.1155/2020/6093836
Bates, L. S., Waldren, R. P., Teare, I. D. (1973). Rapid determination of free proline for water-stress studies. Plant Soil. 39, 205–207. doi: 10.1007/BF00018060
Bissiwu, P., Kulkarni, K. P., Melmaiee, K., Elavarthi, S. (2022). Physiological and molecular responses of red maple (Acer rubrum l.) cultivars to drought stress. Plant Breed. Biotechnol. 10, 62–74. doi: 10.9787/PBB.2022.10.1.62
Bolger, A., Giorgi, F. (2014). Trimmomatic: a flexible read trimming tool for illumina NGS data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bray, E. A. (1997). Plant responses to water deficit. Trends Plant Sci. 2, 48–54. doi: 10.1016/S1360-1385(97)82562-9
Callwood, J., Melmaiee, K., Kulkarni, K. P., Vennapusa, A. R., Aicha, D., Moore, M., et al. (2021). Differential morpho-physiological and transcriptomic responses to heat stress in two blueberry species. Int. J. Mol. Sci. 22, 2481. doi: 10.3390/ijms22052481
Carlson, M. (2009). An assessment of stress in acer saccharum as a possible response to climate change (Durham, NH, USA: University of New Hampshire). Retrieved from: http://gradworks.umi.com/14/72/1472053.html.
Cavalcanti, F. R., Oliveira, J. T. A., Martins-Miranda, A. S., Viégas, R. A., Silveira, J. A. G. (2004). Superoxide dismutase, catalase, and peroxidase activities do not confer protection against oxidative damage in salt-stressed cowpea leaves. New Phytol. 163, 563–571. doi: 10.1093/aob/mcn125
Chaves, M. M., Flexas, J., Pinheiro, C. (2009). Photosynthesis under drought and salt stress: regulation mechanisms from whole plant to cell. Ann. Bot. 103, 551–560. doi: 10.1093/aob/mcn125
Chen, X., Ding, Y., Yang, Y., Song, C., Wang, B., Yang, S., et al. (2021). Protein kinases in plant responses to drought, salt, and cold stress. J. Integr. Plant Biol. 63, 53–78. doi: 10.1111/jipb.13061
Dai, A. (2013). Increasing drought under global warming in observations and models. Nat. Clim. Change 3, 52–58. doi: 10.1038/nclimate1633
Davey, M. W., Stals, E., Panis, B., Keulemans, J., Swennen, R. L. (2005). High-throughput determination of malondialdehyde in plant tissues. Anal. Biochem. 347, 201–207. doi: 10.1016/j.ab.2005.09.041
de Mendiburu (2020) Agricolae: Statistical procedures for agricultural research. r packageVersion1.3-2. Available at: https://cran.r-project.org/web/packages/agricolae/index.html.
Demirel, U., Morris, W. L., Ducreux, L. J., Yavuz, C., Asim, A., Tindas, I., et al. (2020). Physiological, biochemical, and transcriptional responses to single and combined abiotic stress in stress-tolerant and stress-sensitive potato genotypes. Front. Plant Sci. 11. doi: 10.3389/fpls.2020.00169
Du, Z., Su, Q., Wu, Z., Huang, Z., Bao, J., Li, J., et al. (2021). Genome-wide characterization of MATE gene family and expression profiles in response to abiotic stresses in rice (Oryza sativa). BMC Ecol. Evol. 21, 1–14. doi: 10.1186/s12862-021-01873-y
Estravis-Barcala, M., Mattera, M. G., Soliani, C., Bellora, N., Opgenoorth, L., Heer, K., et al. (2020). Molecular bases of responses to abiotic stress in trees. J. Exp. Bot. 71, 3765–3779. doi: 10.1093/jxb/erz532
Farooq, M., Wahid, A., Kobayashi, N., Fujita, D., Basra, S. M. A. (2009). Plant drought stress: effects, mechanisms and management. Agron. Sustain. Dev. 29, 185–212. doi: 10.1051/agro:2008021
Farrel, M. (2021) The future of the maple sugar industry in the united states: Assessing the growth potential based on ecologic, economic, demographic, and public policy factors. Available at: https://nnyagdev.org/index.php/mapleforest/maple/the-future-of-the-maple-sugar-industry-in-the-us/.
Fox, H., Doron-Faigenboim, A., Kelly, G., Bourstein, R., Attia, Z., Zhou, J., et al. (2018). Transcriptome analysis of Pinus halepensis under drought stress and during recovery. Tree Physiol. 38, 423–441. doi: 10.1093/treephys/tpx137
Gao, C., Cheng, C., Zhao, L., Yu, Y., Tang, Q., Xin, P., et al. (2018). Genome-wide expression profiles of hemp (Cannabis sativa l.) in response to drought stress. Int. J. Genomics 2018, 1–13. doi: 10.1155/2018/3057272
Gault, C. M., Kremling, K. A., Buckler, E. S. (2018). Tripsacum de novo transcriptome assemblies reveal parallel gene evolution with maize after ancient polyploidy. Plant Genome 11, 180012. doi: 10.3835/plantgenome2018.02.0012
Gebre, M. G., Earl, H. J. (2021). Soil water deficit and fertilizer placement effects on root biomass distribution, soil water extraction, water use, yield, and yield components of soybean [Glycine max (L.) merr.] grown in 1-m rooting columns. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.581127
Godman, R. M., Yawney, H. W., Tubbs, C. H. (1990). ““Acer saccharum marsh. sugar maple,” in Silvics of north America: Hardwoods, vol. Vol. 2 . Eds. Burns, R. M., Honkala, B. H. (Washington, DC: USDA Forest Service), 194–215.
Gugger, P. F., Peñaloza-Ramírez, J. M., Wright, J. W., Sork, V. L. (2017). Whole-transcriptome response to water stress in a California endemic oak, Quercus lobata. Tree Physiol. 37, 632–644. doi: 10.1093/treephys/tpw122
Haider, M. S., Zhang, C., Kurjogi, M. M., Pervaiz, T., Zheng, T., Zhang, C., et al. (2017). Insights into grapevine defense response against drought as revealed by biochemical, physiological and RNAseq analysis. Sci. Rep. 7, 1–15. doi: 10.1038/s41598-017-13464-3
Harmon, M., Lane, T., Staton, M., Coggeshall, M. V., Best, T., Chen, C. C., et al. (2017). Development of novel genic microsatellite markers from transcriptome sequencing in sugar maple (Acer saccharum marsh.). BMC Res. Notes 10, 1–7. doi: 10.1186/s13104-017-2653-2
Hassan, F. A. S., Ali, E. F. (2014). Impact of different water regimes based on class-a pan on growth, yield and oil content of Coriandrum sativum l. plant. J. Saudi. Soc Agric. Sci. 13, 155–161. doi: 10.1016/j.jssas.2013.05.001
Hauer, R. J., Wei, H., Koeser, A. K., Dawson, J. O. (2021). Gas exchange, water use efficiency, and biomass partitioning among geographic sources of Acer saccharum subsp. saccharum and subsp. nigrum seedlings in response to water stress. Plants 10, 742. doi: 10.3390/plants10040742
He, C., Du, Y., Fu, J., Zeng, E., Park, S., White, F., et al. (2020). Early drought-responsive genes are variable and relevant to drought tolerance. G3.: Genes Genomes Genet. 10, 1657–1670. doi: 10.1534/g3.120.401199
Horsley, S. B., Long, R. P., Bailey, S. W., Hallett, R. A., Hall, T. J. (2000). Factors associated with the decline disease of sugar maple on the Allegheny plateau. Can. J. For. Res. 30, 1365–1378. doi: 10.1139/x00-057
Horsley, S. B., Long, R. P., Bailey, S. W., Hallett, R. A., Wargo, P. M. (2002). Health of eastern north American sugar maple forests and factors affecting decline. North. J. Appl. For. 19, 34–44. doi: 10.1093/njaf/19.1.34
Jarzyniak, K. M., Jasiński, M. (2014). Membrane transporters and drought resistance – a complex issue. Front. Plant Sci. 5. doi: 10.3389/fpls.2014.00687
Joshi, R., Wani, S. H., Singh, B., Bohra, A., Dar, Z. A., Lone, A. A., et al. (2016). Transcription factors and plants response to drought stress: current understanding and future directions. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.01029
Kargar, M., Suresh, R., Legrand, M., Jutras, P., Clark, O. G., Prasher, S. O. (2017). Reduction in water stress for tree saplings using hydrogels in soil. J. Environ. Prot. Sci. 5, 27. doi: 10.4236/gep.2017.51002
Kijowska-Oberc, J., Staszak, A. M., Wawrzyniak, M. K., Ratajczak, E. (2020). Changes in proline levels during seed development of orthodox and recalcitrant seeds of genus Acer in a climate change scenario. Forests 11, 1362. doi: 10.3390/f11121362
Kiranmai, K., Lokanadha Rao, G., Pandurangaiah, M., Nareshkumar, A., Amaranatha Reddy, V., Lokesh, U., et al. (2018). A novel WRKY transcription factor, MuWRKY3 (Macrotyloma uniflorum lam. verdc.) enhances drought stress tolerance in transgenic groundnut (Arachis hypogaea l.) plants. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00346
Kulkarni, K. P., Vorsa, N., Natarajan, P., Elavarthi, S., Iorizzo, M., Reddy, U. K., et al. (2021). Admixture analysis using genotyping-by-sequencing reveals genetic relatedness and parental lineage distribution in highbush blueberry genotypes and cross derivatives. Int. J. Mol. Sci. 22, 163. doi: 10.3390/ijms22010163
Li, L., Liu, Y., Liu, Y., He, B., Wang, M., Yu, C., et al. (2015). Physiological response and resistance of three cultivars of Acer rubrum l. @ to continuous drought stress. Acta Ecol. Sin. 35, 196–202. doi: 10.1016/j.chnaes.2015.09.006
Li, Q., Shen, C., Zhang, Y., Zhou, Y., Niu, M., Wang, H. L., et al. (2023). PePYL4 enhances drought tolerance by modulating water-use efficiency and ROS scavenging in Populus. Tree Physio. 43 (1), 102–117. doi: 10.1093/treephys/tpac106
Li, T., Wang, R., Zhao, D., Tao, J. (2020). Effects of drought stress on physiological responses and gene expression changes in herbaceous peony (Paeonia lactiflora pall.). Plant Signal. Behav. 15, 1746034. doi: 10.1080/15592324.2020.1746034
Lima, J. M., Nath, M., Dokku, P., Raman, K. V., Kulkarni, K. P., Vishwakarma, C., et al. (2015). Physiological, anatomical and transcriptional alterations in a rice mutant leading to enhanced water stress tolerance. AoB. Plants 7, plv023. doi: 10.1093/aobpla/plv023
Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2– ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lokesh, U., Venkatesh, B., Kiranmai, K., Nareshkumar, A., Amarnathareddy, V., Rao, G. L., et al. (2019). Overexpression of ß-ketoacyl Co-a Synthase1 gene improves tolerance of drought susceptible groundnut (Arachis hypogaea l.) cultivar K-6 by increased leaf epicuticular wax accumulation. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.01869
Love, M., Anders, S., Huber, W. (2014). Differential analysis of count data-the DESeq2 package. Genome Biol. 15, 550. doi: 10.1186/s13059-014-0550-8
Lucash, M. S., Yanai, R. D., Blum, J. D., Park, B. B. (2012). Foliar nutrient concentrations related to soil sources across a range of sites in the northeastern united states. Soil Sci. Soc Am. J. 76, 674–683. doi: 10.2136/sssaj2011.0160
Luo, W., Pant, G., Bhavnasi, Y. K., Blanchard, S. G., Jr., Brouwer, C. (2017). Pathview web: user friendly pathway visualization and data integration. Nucleic Acids Res. 45, W501–W508. doi: 10.1093/nar/gkx372
McDowell, N. G. (2011). Mechanisms linking drought, hydraulics, carbon metabolism, and vegetation mortality. Plant Physiol. 155, 1051–1059. doi: 10.1104/pp.110.170704
McEvoy, S. L., Sezen, U. U., Trouern-Trend, A., McMahon, S. M., Schaberg, P. G., Yang, J. (2022). Strategies of tolerance reflected in two north American maple genomes. Plant J. 109, 1591–1613. doi: 10.1111/tpj.15657
Mickelbart, M. V., Hasegawa, P. M., Bailey-Serres, J. (2015). Genetic mechanisms of abiotic stress tolerance that translate to crop yield stability. Nat. Rev. Genet. 16, 237–251. doi: 10.1038/nrg3901
Müller, M., Seifert, S., Lübbe, T., Leuschner, C., Finkeldey, R. (2017). De novo transcriptome assembly and analysis of differential gene expression in response to drought in European beech. PloS One 12, e0184167. doi: 10.1371/journal.pone.0184167
Nareshkumar, A., Subbarao, S., Vennapusa, A. R., Ashwin, V., Banarjee, R., Kulkarni, M. J., et al. (2020). Enzymatic and non-enzymatic detoxification of reactive carbonyl compounds improves the oxidative stress tolerance in cucumber, tobacco and rice seedlings. J. Plant Growth Regul. 39, 1359–1372. doi: 10.1007/s00344-020-10072-w
Natarajan, P., Ahn, E., Reddy, U. K., Perumal, R., Prom, L. K., Magill, C. (2021). RNAsequencing in resistant (QL3) and susceptible (Theis) sorghum cultivars inoculated with johnsongrass isolates of Colletotrichum sublineola. Front. Genet. 1476. doi: 10.3389/fgene.2021.722519
Nisarga, K. N., Vemanna, R. S., Kodekallu Chandrashekar, B., Rao, H., Vennapusa, A. R., Narasimaha, A., et al. (2017). Aldo-ketoreductase 1 (AKR1) improves seed longevity in tobacco and rice by detoxifying reactive cytotoxic compounds generated during ageing. Rice 10, 1–12. doi: 10.1186/s12284-017-0148-3
Omidbakhshfard, M. A., Proost, S., Fujikura, U., Mueller-Roeber, B. (2015). Growth-regulating factors (GRFs): A small transcription factor family with important functions in plant biology. Mol. Plant 8, 998–1010. doi: 10.1016/j.molp.2015.01.013
Oswald, E. M., Pontius, J., Rayback, S. A., Schaberg, P. G., Wilmot, S. H., Dupigny-Giroux, L. A. (2018). The complex relationship between climate and sugar maple health: Climate change implications in Vermont for a key northern hardwood species. For. Ecol. Manage. 422, 303–312. doi: 10.1016/j.foreco.2018.04.014
Pandian, B. A., Sathishraj, R., Prasad, P. V., Jugulam, M. (2021). A single gene inherited trait confers metabolic resistance to chlorsulfuron in grain sorghum (Sorghum bicolor). Planta 253, 1–12. doi: 10.1007/s00425-020-03563-3
Pandian, B. A., Varanasi, A., Vennapusa, A. R., Sathishraj, R., Lin, G., Zhao, M., et al. (2020). Characterization, genetic analyses, and identification of QTLs conferring metabolic resistance to a 4-hydroxyphenylpyruvate dioxygenase inhibitor in sorghum (Sorghum bicolor). Front. Plant Sci. 11. doi: 10.3389/fpls.2020.596581
Patel, P. K., Hemantaranjan, A. (2012). Salicylic acid induced alteration in dry matter partitioning, antioxidant defence system and yield in chickpea (Cicer arietinum l.) under drought stress. Asian J. Crop Sci. 4, 86–102. doi: 10.3923/ajcs.2012.86.102
Patykowski, J., Kołodziejek, J., Wala, M. (2018). Biochemical and growth responses of silver maple (Acer saccharinum l.) to sodium chloride and calcium chloride. PeerJ 6, e5958. doi: 10.7717/peerj.5958
Priya, M., Dhanker, O., Siddique, K. H. M., HanumanthaRao, B., Nair, R., Pandey, S., et al. (2019). Drought and heat stress-related proteins: an update about their functional relevance in imparting stress tolerance in agricultural crops. Theor. Appl. Genet. 132, 1607–1638. doi: 10.1007/s00122-019-03331-2
Ramu, V. S., Swetha, T. N., Sheela, S. H., Babitha, C. K., Rohini, S., Reddy, M. K., et al. (2016). Simultaneous expression of regulatory genes associated with specific drought-adaptive traits improves drought adaptation in peanut. Plant Biotechnol. J. 14, 1008–1020. doi: 10.1111/pbi.12461
Ribeyre, Z., Messier, C., Nolet, P. (2022). No stress memory pattern was detected in sugar maple and white spruce seedlings subjected to experimental droughts. Ecosphere 13, e4332. doi: 10.1002/ecs2.4332
Rogers, N. S. (2020). Long-term dynamics and silviculture of northern hardwood forests in the northeast united states (Burlington, VT, USA: The University of Vermont and State Agricultural College). Available at: https://www.proquest.com/docview/2321858563?pq-origsite=gscholar&fromopenview=true.
Rogers, B. M., Jantz, P., Goetz, S. J. (2017). Vulnerability of eastern US tree species to climate change. Glob. Change Biol. 23, 3302–3320. doi: 10.1111/gcb.13585
Sajeevan, R. S., Parvathi, M. S., Nataraja, K. N. (2017). Leaf wax trait in crops for drought and biotic stress tolerance: regulators of epicuticular wax synthesis and role of small RNAs. Indian J. Plant Physiol. 22, 434–447. doi: 10.1093/treephys/23.5.325
Schaberg, P. G., Van den Berg, A. K., Murakami, P. F., Shane, J. B., Donnelly, J. R. (2003). Factors influencing red expression in autumn foliage of sugar maple trees. Tree Physiol. 23, 325–333. doi: 10.1093/treephys/23.5.325
Seleiman, M. F., Al-Suhaibani, N., Ali, N., Akmal, M., Alotaibi, M., Refay, Y., et al. (2021). Drought stress impacts on plants and different approaches to alleviate its adverse effects. Plants 10, 1–25. doi: 10.3390/plants10020259
Shahbandeh, M. (2021) Maple syrup production in the united states in 2021, by state. Available at: https://www.statista.com/statistics/372013/us-maple-syrup-production-by-state/.
Sharma, A., Zheng, B. (2019). Melatonin mediated regulation of drought stress: physiological and molecular aspects. Plants 8, 190. doi: 10.3390/plants8070190
Shinozaki, K., Yamaguchi-Shinozaki, K. (2007). Gene networks involved in drought stress response and tolerance. J. Exp. Bot. 58, 221–227. doi: 10.1093/jxb/erl164
Siddique, A., Kandpal, G., Kumar, P. (2018). Proline accumulation and its defensive role under diverse stress condition in plants: An overview. J. Pure. Appl. Microbiol. 12, 1655–1659. doi: 10.22207/JPAM.12.3.73
Silber, A., Xu, G., Wallach, R. (2003). High irrigation frequency: The effect on plant growth and on uptake of water and nutrients. Acta Hortic. 627, 89–96. doi: 10.17660/ActaHortic.2003.627.10
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Singh, D., Singh, C. K., Taunk, J., Tomar, R. S. S., Chaturvedi, A. K., Gaikwad, K., et al. (2017). Transcriptome analysis of lentil (Lens culinaris medikus) in response to seedling drought stress. BMC Genom. 18, 1–20. doi: 10.1186/s12864-017-3596-7
Snyder, S. A., Kilgore, M. A., Emery, M. R., Schmitz, M. (2019). Maple syrup producers of the lake states, USA: Attitudes towards and adaptation to social, ecological, and climate conditions. Environ. Manage. 63, 185–199. doi: 10.1007/s00267-018-1121-7
Sobreiro, M. B., Collevatti, R. G., Dos Santos, Y. L., Bandeira, L. F., Lopes, F. J., Novaes, E. (2021). RNAseq reveals different responses to drought in Neotropical trees from savannas and seasonally dry forests. BMC Plant Biol. 21, 1–17. doi: 10.1186/s12870-021-03244-7
Song, F., Zhou, J., Quan, M., Xiao, L., Lu, W., Qin, S., et al. (2022). Transcriptome and association mapping revealed functional genes respond to drought stress in populus. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.829888
Spieß, N., Oufir, M., Matušíková, I., Stierschneider, M., Kopecky, D., Homolka, A., et al. (2012). Ecophysiological and transcriptomic responses of oak (Quercus robur) to long-term drought exposure and rewatering. Environ. Exp. Bot. 77, 117–126. doi: 10.1016/j.envexpbot.2011.11.010
Sun, J., Bie, X. M., Wang, N., Zhang, X. S., Gao, X. Q. (2020). Genome-wide identification and expression analysis of YTH domain-containing RNA-binding protein family in common wheat. BMC Plant Biol. 20, 1–14. doi: 10.1186/s12870-020-02505-1
Tahmasebi, A., Niazi, A. (2021). Comparison of transcriptional response of C3 and C4 plants to drought stress using meta-analysis and systems biology approach. Front. Plant Sci. 12.1295. doi: 10.3389/fpls.2021.668736
Vemanna, R. S., Babitha, K. C., Solanki, J. K., Reddy, V. A., Sarangi, S. K., Udayakumar, M. (2017). Aldo-keto reductase-1 (AKR1) protect cellular enzymes from salt stress by detoxifying reactive cytotoxic compounds. Plant Physiol. Biochem. 113, 177–186. doi: 10.1016/j.plaphy.2017.02.012
Venkatesh, B., Vennapusa, A. R., Kumar, N. J., Jayamma, N., Reddy, B. M., Johnson, A. M. A., et al. (2022). Co-Expression of stress-responsive regulatory genes, MuNAC4, MuWRKY3 and MuMYB96 associated with resistant-traits improves drought adaptation in transgenic groundnut (Arachis hypogaea l.) plants. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1055851
Vennapusa, A. R., Agarwal, S., Hm, H. R., Aarthy, T., Babitha, K. C., Thulasiram, H. V., et al. (2022). Stacking herbicide detoxification and resistant genes improves glyphosate tolerance and reduces phytotoxicity in tobacco (Nicotiana tabacum l.) and rice (Oryza sativa l.). Plant Physiol. Biochem. 189, 126–138. doi: 10.1016/j.plaphy.2022.08.025
Vennapusa, A. R., Assefa, Y., Sebela, D., Somayanda, I., Perumal, R., Riechers, D. E., et al. (2021). Safeners improve early-stage chilling-stress tolerance in sorghum. J. Agron. Crop Sci. 207, 705–716. doi: 10.1111/jac.12503
Vennapusa, A. R., Somayanda, I. M., Doherty, C. J., Jagadish, S. K. (2020). A universal method for high-quality RNA extraction from plant tissues rich in starch, proteins and fiber. Sci. Rep. 10, 1–13. doi: 10.1038/s41598-020-73958-5
Verbruggen, N., Hermans, C. (2008). Proline accumulation in plants: a review. Amino Acids 35, 753–759. doi: 10.1007/s00726-008-0061-6
Wan, H., Cui, Y., Ding, Y., Mei, J., Dong, H., Zhang, W., et al. (2017). Time-series analyses of transcriptomes and proteomes reveal molecular networks underlying oil accumulation in canola. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.02007
Wang, S., Chen, K., Zhang, J., Wang, J., Li, H., Yang, X., et al. (2022). Genome-wide characterization of MATE family members in cucumis melo l. and their expression profiles in response to abiotic and biotic stress. Hortic. Plant J. 8, 474–488. doi: 10.1016/j.hpj.2022.05.004
Wei, T., Wang, Y., Xie, Z., Guo, D., Chen, C., Fan, Q., et al. (2019). Enhanced ROS scavenging and sugar accumulation contribute to drought tolerance of naturally occurring autotetraploids in Poncirus trifoliata. Plant Biotechnol. J. 17, 1394–1407. doi: 10.1111/pbi.13064
Wickham, H., Wickham, M. H. (2007). The ggplot Package. Available at: http://ftp.uni-bayreuth.de/math/statlib/R/CRAN/doc/packages/ggplot.pdf (accessed June 6, 2021).
Wolkovich, E. M., Cleland, E. E. (2014). Phenological niches and the future of invaded ecosystems with climate change. AoB. Plants 6, 1–16. doi: 10.1093/aobpla/plu013
Wong, B. L., Baggett, K. L., Rye, A. H. (2009). Cold-season patterns of reserve and soluble carbohydrates in sugar maple and ice-damaged trees of two age classes following drought. Botany 87, 293–305. doi: 10.1139/B08-123
Wu, L. B., Holtkamp, F., Wairich, A., Frei, M. (2019). Potassium ion channel gene OsAKT1 affects iron translocation in rice plants exposed to iron toxicity. Front. Plant Sci. 10, 579. doi: 10.3389/fpls.2019.00579
Yang, X., Liu, J., Xu, J., Duan, S., Wang, Q., Li, G., et al. (2019). Transcriptome profiling reveals effects of drought stress on gene expression in diploid potato genotype P3-198. Int. J. Mol. Sci. 20, 852. doi: 10.3390/ijms20040852
Yao, T., Zhang, J., Xie, M., Yuan, G., Tschaplinski, T. J., Muchero, W., et al. (2021). Transcriptional regulation of drought response in arabidopsis and woody plants. Front. Plant Sci. 11. doi: 10.3389/fpls.2020.572137
Yi, F., Huo, M., Li, J., Yu, J. (2022). Time-series transcriptomics reveals a drought-responsive temporal network and crosstalk between drought stress and the circadian clock in foxtail millet. Plant J. 110, 1213–1228. doi: 10.1111/tpj.15725
Zhang, J., Gao, Y., Feng, M., Cui, Y., Li, S., Liu, L., et al. (2022). Genome-wide identification of the HD-ZIP III subfamily in upland cotton reveals the involvement of GhHB8-5D in the biosynthesis of secondary wall in fiber and drought resistance. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.806195
Zhang, T., Gao, G., Liu, J., Yang, G., Lv, Z., Zhang, J., et al. (2020). Transcripts and ABA-dependent signaling in response to drought stress in Hippophae rhamnoides l. Trees 34, 1033–1045. doi: 10.1007/s00468-020-01979-8
Zhang, J., Huang, D., Zhao, X., Zhang, M. (2021). Evaluation of drought resistance and transcriptome analysis for the identification of drought-responsive genes in Iris germanica. Sci. Rep. 11, 1–21. doi: 10.1038/s41598-021-95633-z
Keywords: Acer saccharum, sugar maple, transcriptome, drought, stress, physiology, gene expression
Citation: Mulozi L, Vennapusa AR, Elavarthi S, Jacobs OE, Kulkarni KP, Natarajan P, Reddy UK and Melmaiee K (2023) Transcriptome profiling, physiological, and biochemical analyses provide new insights towards drought stress response in sugar maple (Acer saccharum Marshall) saplings. Front. Plant Sci. 14:1150204. doi: 10.3389/fpls.2023.1150204
Received: 23 January 2023; Accepted: 30 March 2023;
Published: 19 April 2023.
Edited by:
Marcelo Menossi Menossi, State University of Campinas, BrazilReviewed by:
Amber Gupta, Maharaja Sayajirao University of Baroda, IndiaRadha Sivarajan Sajeevan, Swedish University of Agricultural Sciences, Sweden
Copyright © 2023 Mulozi, Vennapusa, Elavarthi, Jacobs, Kulkarni, Natarajan, Reddy and Melmaiee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kalpalatha Melmaiee, a21lbG1haWVlQGRlc3UuZWR1; Sathya Elavarthi, c2VsYXZhcnRoaUBkZXN1LmVkdQ==
†These authors have contributed equally to this work and share first authorship