
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 04 April 2023
Sec. Plant Systematics and Evolution
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1142147
This article is part of the Research Topic Crop Domestication and Selection: An Evolutionary View Affecting the Development of Agronomic Traits View all 8 articles
 Dayong Wei1†
Dayong Wei1† Nan Li1†Nan Zhang1Feng Liu1Jie Wu1Sa Zhao1Jinjuan Shen2
Nan Li1†Nan Zhang1Feng Liu1Jie Wu1Sa Zhao1Jinjuan Shen2 Zhimin Wang1Lisha Peng2Yonghong Fan2
Zhimin Wang1Lisha Peng2Yonghong Fan2 Jiaqin Mei3*Qinglin Tang1*
Jiaqin Mei3*Qinglin Tang1*The genus Brassica contains a diverse group of important vegetables and oilseed crops. Genome sequencing has been completed for the six species (B. rapa, B. oleracea, B. nigra, B. carinata, B. napus, and B. juncea) in U’s triangle model. The purpose of the study is to investigate whether positively and negatively selected genes (PSGs and NSGs) affect gene feature and function differentiation of Brassica tetraploids in their evolution and domestication. A total of 9,701 PSGs were found in the A, B and C subgenomes of the three tetraploids, of which, a higher number of PSGs were identified in the C subgenome as comparing to the A and B subgenomes. The PSGs of the three tetraploids had more tandem duplicated genes, higher single copy, lower multi-copy, shorter exon length and fewer exon number than the NSGs, suggesting that the selective modes affected the gene feature of Brassica tetraploids. The PSGs of all the three tetraploids enriched in a few common KEGG pathways relating to environmental adaption (such as Phenylpropanoid biosynthesis, Riboflavin metabolism, Isoflavonoid biosynthesis, Plant-pathogen interaction and Tropane, piperidine and pyridine alkaloid biosynthesis) and reproduction (Homologous recombination). Whereas, the NSGs of the three tetraploids significantly enriched in dozens of biologic processes and pathways without clear relationships with evolution. Moreover, the PSGs of B. carinata were found specifically enriched in lipid biosynthesis and metabolism which possibly contributed to the domestication of B. carinata as an oil crop. Our data suggest that selective modes affected the gene feature of Brassica tetraploids, and PSGs contributed in not only the evolution but also the domestication of Brassica tetraploids.
The genus Brassica includes several economically important species such as B. rapa (AA), B. nigra (BB), B. oleracea (CC), B. napus (AACC), B. juncea (AABB), and B. carinata (BBCC) which are widely grown around the world as fodder, vegetable, edible oil, biofuel, and condiment crops. Of the six Brassica species, the ancestors of three diploids (B. rapa, B. nigra, and B. oleracea) underwent hybridization events, giving rise to three allotetraploid species (B. napus, B. juncea, and B. carinata), forming the Triangle of U (Nagahara, 1935).
Many studies have illuminated the origin and evolution of A, B, and C genomes in Brassica. It was deduced that, before the divergence of A, B and C genomes, a common ancestor of Brassica was derived from Arabidopsis thaliana at 20-40 Mya (Beilstein et al., 2010) and experienced an extra whole-genome triplication (WGT) event at approximately 15.9–22.5 Mya (Beilstein et al., 2010; Liu et al., 2014). Consequently, the common ancestor diverged into the B genome (B. nigra) at about 11.5 Mya (Paritosh et al., 2021) and then the A (B. rapa) and C genomes (B. oleracea) at 4-6.8 Mya (Liu et al., 2014; Paritosh et al., 2021). Concerning to the tetraploid Brassica species, the A subgenome of B. napus might evolve from European turnip (B. rapa), and the C subgenome might evolve from the common ancestor of kohlrabi, cauliflower, broccoli, and Chinese kale (Lu et al., 2019), or from the B. oleracea cabbage-type lineage at ~2.63 Mya (Parkin et al., 2014; Yim et al., 2022). Comparison of the A subgenome in B. juncea with the A subgenome in B. napus indicated that the B. juncea A subgenome might derive from the ancestor of Asian B. rapa ssp. tricolaris and differ from the A subgenome in B. napus (Yang et al., 2016). In a recent study (Yim et al., 2022), the common ancestor of B. juncea and B. carinata diverged from the extant B genome of B. nigra at 6.61 Mya, and the B subgenome of B. carinata diverged from the B. juncea B subgenome at 5.44 Mya. The C subgenome of B. carinata is supposed to derive from the B. oleracea kale-type lineage at 2.88 Mya (Yim et al., 2022) thus appeared to have arisen from distinct B. oleracea lineage as compared with the B. napus C subgenome. These studies provided useful information on the origin and divergence of the A, B and C genomes of Brassica species, however, a more comprehensive and systematic understanding on the divergence of Brassica species is meaningful and available now since the release of reference genomes of all the six Brassica species.
Among the Brassicaceae species, A. thaliana was the first plant species with sequenced genome (Arabidopsis Genome, 2000) and thus is widely used as a model plant in genetic and genomic evolution studies. The six Brassica species in the triangle of U have also been sequenced. Among these, B. rapa accession Chiifu-401-42, a heading Chinese cabbage, was the first sequenced species (Wang et al., 2011), and the updated assembled version (v3.5) is now available (Zhang et al., 2022). A high-quality genome of B. nigra accession Ni100 v2 was recently released (Perumal et al., 2020). The genomes of two B. oleracea varieties, B. oleracea var. capitata line 02–12 and B. oleracea kale-like type TO1000 v2.1, have been assembled (Liu et al., 2014; Parkin et al., 2014). The genome of four B. juncea, i.e., var. tumida (T84−66 v1.0), T84−66 v2.0, var. varuna and a cultivar type (SY, yellow-seeded B. juncea) have also been released (Yang et al., 2016; Kang et al., 2021; Paritosh et al., 2021; Yang et al., 2021). For B. napus, the genome of a European winter oilseed cultivar ‘Darmor-bzh’ was assembled in 2014 (Chalhoub et al., 2014), and a high-quality genome assembly of version 10 was updated in 2020 (Rousseau-Gueutin et al., 2020). The high-quality genome of B. carinata was also published in 2021 and 2022 (Song et al., 2021; Yim et al., 2022). Owing to their agronomic importance and the availability in gaining their reference genomes, the six Brassica species composed an ideal model system for studying evolutionary changes in tetraploid species.
The release of reference genomes of all the six Brassica species not only provides the facility to investigate the divergence of Brassica species in depth, but also allows us to understand the evolution of Brassica species. In a study in B. rapa and B. oleracea (Guo et al., 2017), selective modes based on orthologous gene pairs (OGPs) between the two species, classified into positively selected genes (PSGs) and negatively selected genes (NSGs), were concerned as a determinant factor of evolutionary rates, gene compactness and expression patterns. In the present study, the divergence time (DT) of the Brassica genus was comprehensively and systematically estimated, and the PSGs and NSGs in Brassica tetraploids as comparing to corresponding diploid were investigated to understand the evolution of Brassica tetraploids.
The genome sequences for A. thaliana (Col_v10.1), B. rapa (Chiifu_v3.5), B. napus (Darmor-bzh_v10) and B. juncea (T84-66_v2.0) were obtained from Brassica genomics database (http://brassicadb.cn). The genome data of B. oleracea (TO1000_v2.1) and B. nigra (Ni100_v2) were downloaded from EnsemblPlants (https://plants.ensembl.org/Brassica_oleracea/Info/Index) and CGI (http://cruciferseq.ca/), respectively. The genome assembly and annotation for B. carinata (PGL_v1) is available from Comparative Genomics platform (CoGe) under id 63922.
To distinguish diverse subgenomes in Brassica species, we redesignated the subgenomes as follows: B. rapa genome as BraA, B. nigra genome as BniB, B. oleracea genome as BolC, B. juncea A and B subgenomes as BjuA and BjuB, respectively, B. napus A and C subgenomes as BnaA and BnaC, respectively, B. carinata B and C subgenomes as BcaB and BcaC, respectively. The interspecific all OGPs were determined using OrthoFinder2 (Emms and Kelly, 2018; Jing et al., 2023) platform (BLASTP all-vs-all, 10e-3, orthofinder -f./dataset -t 16 -a 8 -M msa -S blast -A mafft) in the combinations among different subgenome of Brassica species (BraA-BnaA, BraA-BjuA, BniB-BjuB, BniB-BcaB, BolC-BnaC and BolC-BcaC) and combinations between A. thaliana and Brassica species (Ath-BraA, Ath-BniB and Ath-BolC). The rates of nonsynonymous substitution (Ka) and the rates of synonymous substitution (Ks) values for each pair of homologous genes were estimated using ParaAT 2.0 (https://bigd.big.ac.cn/tools/). Genes with Ks > 1 were discarded, since such high Ks values may imply either potential sequence saturation or misalignment. The divergence time was calculated with the formula T = Ks/2λ, where λ is the neutral substitution rate of 8.22e–09 synonymous substitutions per site per year for Brassicaceae species (Beilstein et al., 2010).
Almost all the protein-coding genes can be classified into three categories: PSGs, NSGs, and neutral genes. To differentiate these genes, the Ka/Ks ratio was calculated for each gene, and Ka/Ks > 1, Ka/Ks = 1, and Ka/Ks < 1 were used as the indicators for positive selection, neutral evolution and negative selection (from small to large, the same number with PSGs) during gene sequence divergence, respectively. The physical location distribution of PSGs and NSGs were visualized by ‘CMplot’ R package (the widows size was 1 Mb).
Gene Ontology (GO) enrichment analysis was performed by ‘topGO v2.50.0’ package (https://bioconductor.org/packages/release/bioc/html/topGO.html) with R-4.1.2 software (significant genes≥5 and the Kolmogorov-Smirnov test ‘KS’<0.05) and TBtools v1.09876 software (GO level≥4, hits genes≥5 and P-value<0.01) (Chen et al., 2020). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway was analyzed using ‘clusterProfiler v4.2.2’ package (Wu et al., 2021) with R-4.1.2 software (gene count≥5 and P-value<0.05) and TBtools v1.09876 software (hits genes≥5 and P-value<0.05). These subgenomic OGPs of tetraploid Brassica species were used for background genes, and their function annotation were predicted by eggNOG-mapper v2 online service (http://eggnog-mapper.embl.de/) (Cantalapiedra et al., 2021).
The one-tailed test through a random sampling way was used to investigate whether PSGs affect the domestication of Brassica tetraploids. In details, for an interested tetraploid (for example B. carinata), the PSGs enriched in a given biologic process related to domestication was considered as gene set 1 (90 PSGs specifically enriched in lipid-related pathway in this study), and reported genes associating with the same biologic process was considered as gene set 2 (2,507 reported lipid-related genes). Firstly, the number of common genes overlapped between gene set 1 and 2 was found out and denoted by Ncom (21 intersecting genes). Then, a same number genes as gene set 1 (or gene set 2) were randomly chosen from all annotated genes of this tetraploid (90 or 2,507 genes randomly choose from 133,235 annotated gene), and then looked for the intersection with gene set 2 (or gene set 1). A total of 100,000 runs was carried out for the two algorithms, resulted in 50,000 intersections for each. The threshold of the right tail in the 99% confidence interval was calculated for each algorithm (50,000 intersections) as the mean gene number of corresponding 50,000 intersections plus 2.33*S.D (mean+2.33*S.D., 4.68). (Rosner, 2015). Finally, Ncom was compared with the threshold of the right tails calculated for the two algorithms (4.68). If Ncom falls outside of the 99% confidence intervals, the PSGs in gene set 1 will be considered to play a positive role in the domestication of the given biologic process. Other descriptive statistics were performed using Excel 2021. Analysis of variance (ANOVA) and Duncan’s multiple range test (DMRT) were carried out via IBM SPSS Statistics software v25 (https://www.ibm.com/products/spss-statistics).
A total of 561,662 OGPs was detected from the six combinations among Brassica species (BraA-BnaA, BraA-BjuA, BniB-BjuB, BniB-BcaB, BolC-BnaC and BolC-BcaC) and three combinations between A. thaliana and Brassica species (Ath-BraA, Ath-BniB and Ath-BolC). After discarding 112,332 genes with Ks>1 and Ka=0, the values of Ka, Ks and Ka/Ks were calculated for the remaining 449,330 gene pairs (Table 1). The Ka/Ks exhibited approximately normal distributions (single peak curves) in all the nine combinations (Figure S1). A significant positive correlation was found between Ka and Ks in each of the nine combinations (r=0.70~0.71, P<0.01). The Ka/Ks ratio was moderately correlated with Ka (r=0.12~0.26, P<0.01), while it showed a slight negative correlation with Ks (r=-0.14~-0.27, P<0.01), indicating that Ka is the determinant factor for Ka/Ks.
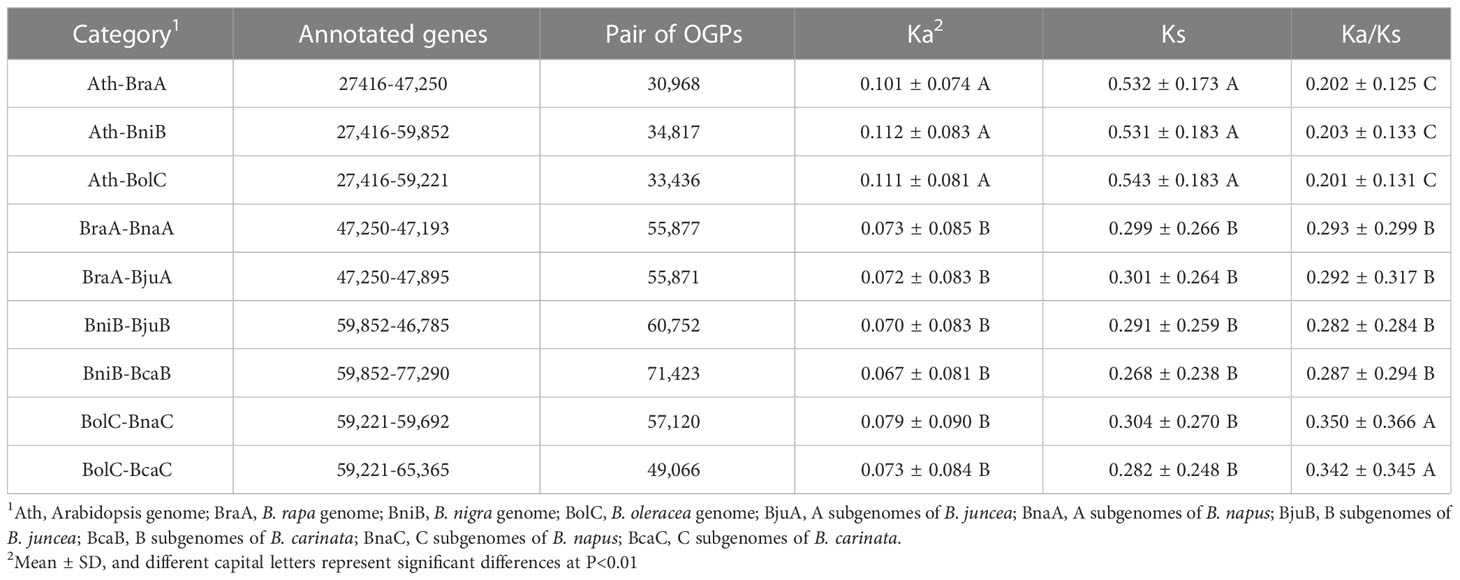
Table 1 Comparisons of Ka, Ks, and Ka/Ks.
Both the Ka and Ks of each A. thaliana–Brassica combination were significantly higher than those of the combinations of different Brassica species (DMRT, P<0.01). The mean Ka/Ks ratio of the A. thaliana–Brassica combinations was significantly lower than that of the within-Brassica combinations (Table 1). These results indicate that the A. thaliana genes possibly underwent a higher evolutionary rate and experienced more selective pressure than the Brassica genes.
A total of 9,701 PSGs were found in all combinations, with an average of 1,617 pairs in each combination. A higher number of PSGs were identified in the combination of C subgenome (average 2,028 pairs) than in A and B subgenomes (average 1373 and 1451 pairs) (Table 2). This indicates that more genes of B. oleracea (CC) were positively selected during the evolution of B. napus (AACC) and B. carinata (BBCC). Unevenly distribution of PSGs and NSGs were found in all chromosomes, and the NSGs are far from the centromere (Figure S2).
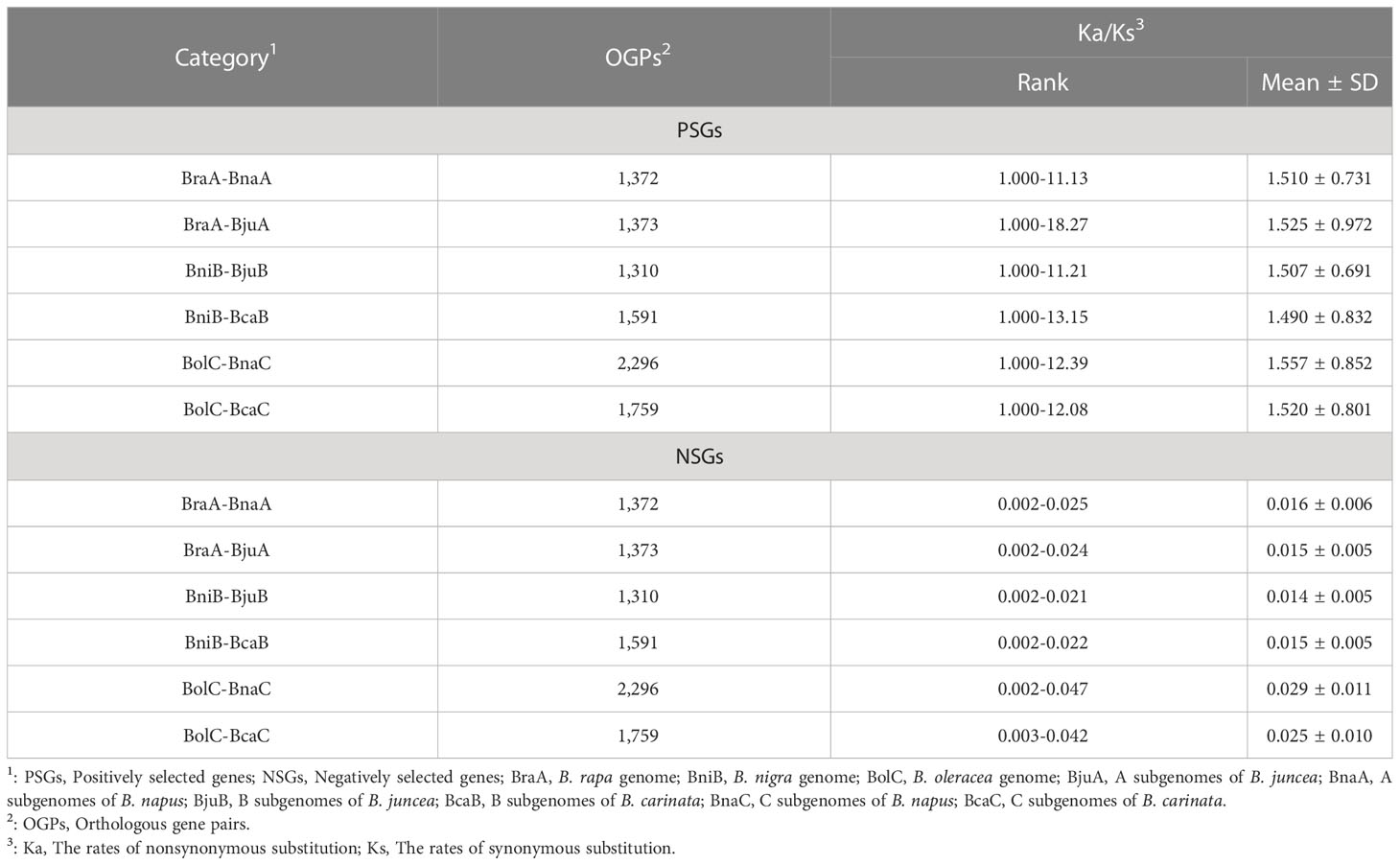
Table 2 The PSGs and NSGs in subgenome combinations among Brassica species.
To understand whether and how selective modes affect gene features, the tandem duplication, the copy number, the exon length and number of these PSGs and NSGs were characterized. It was found that PSGs had a significantly more tandem duplicated genes (average 5.47% vs. 1.68%), higher single copy (average 99.43% vs. 65.01%), lower multi-copy (average 0.28% vs. 17.50%), shorter exon length (average 1051 bp vs. 1798 bp) and fewer exon number (average 5.1 vs. 7.0) than the NSGs (Table 3), revealing obvious varied gene features between PSGs and NSGs. This finding indicated that selective modes may serve as an alternative indicator for gene compactness.
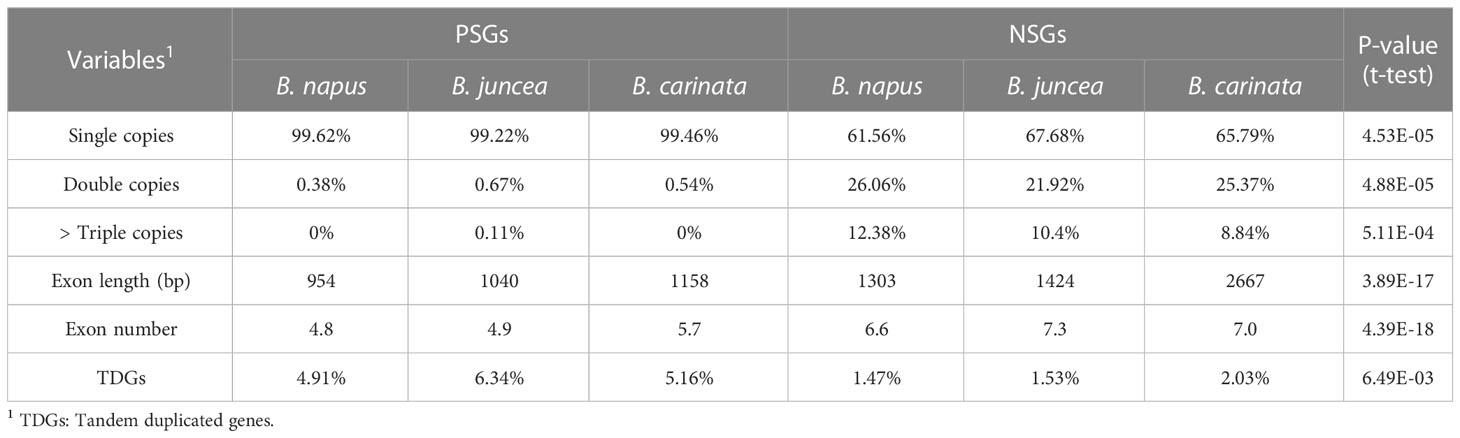
Table 3 Gene feature analysis between PSGs and NSGs.
To comprehensively and systematically estimate the divergence time of the Brassica species, we investigated the distribution of the Ks in nine combinations. The Ks peak between A. thaliana and diploid Brassica species (B. rapa, B. oleracea and B. nigra) was 0.408–0.424, indicating that the A. thaliana and diploid Brassica species separated at ~ 24.81–25.78 Mya. In similar, by estimating the peak Ks values in the A subgenomes (BraA-BnaA, BraA-BjuA), B subgenomes (BniB-BjuB, BniB-BcaB) and C subgenomes (BolC-BnaC, BolC-BcaC) combinations, the divergence time of subgenomes was speculated at ~1.27–1.96 Mya. Of which, significant differences were found between A and C subgenomes (average 1.77 vs 1.29 Mya, P=0.042), between B and C subgenomes (average 1.91 vs 1.29 Mya, P=0.0068), but no significant difference was detected between A and B subgenomes (average 1.77 vs 1.91 Mya, P=0.31) (Figure 1).
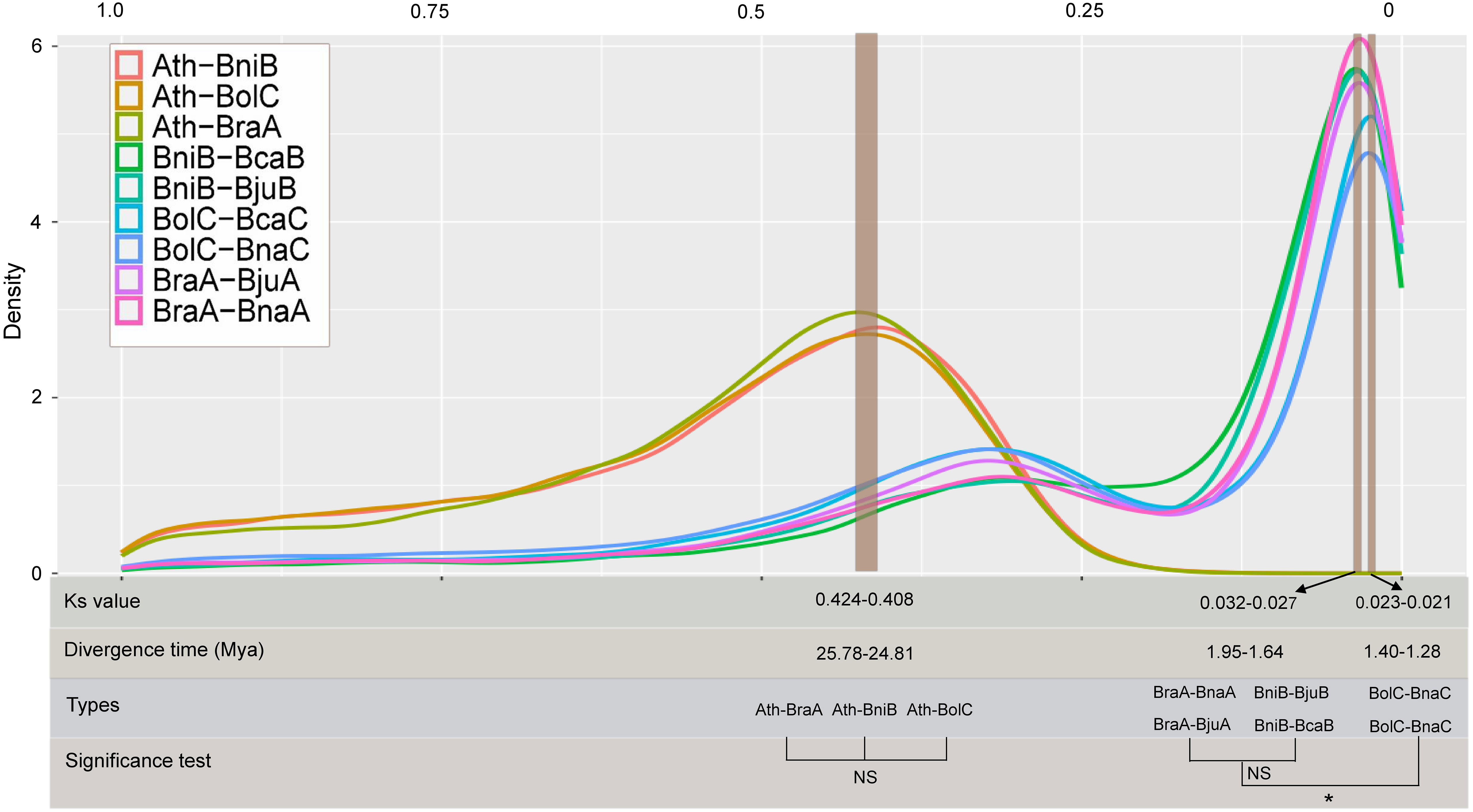
Figure 1 Divergence time estimation based on Ks distributions among different combinations. NS: no significance; *: significance level at 0.05. Ath, Arabidopsis genome; BraA, B rapa genome; BniB, B nigra genome; BolC, B oleracea genome; BjuA, A subgenome of B juncea; BnaA, A subgenome of B napus; BjuB, B subgenome of B juncea; BcaB, B subgenome of B carinata; BnaC, C subgenome of B napus; BcaC, C subgenome of B carinata.
To understand the functional differentiation of genes involved in the evolution from diploids to tetraploids, we performed functional enrichment analyses on the PSGs and NSGs in A, B and C subgenomes. By GO enrichment analyses through two methods, the PSGs of B. napus (BraA-BnaA and BolC-BnaC), B. juncea (BraA-BjuA and BniB-BjuB) and B. carinata (BniB-BcaB and BolC-BcaC) were significantly enriched in 42, 17 and 24 biological processes, respectively (Figure 2A). Among these terms, only two (GO:0006355, regulation of DNA-templated transcription and GO:2000112, regulation of cellular macromolecule biosynthetic process) were commonly enriched in all the three tetraploids (Figure 2B; Table S1-S3). In contrast, these NSGs were significantly enriched in 32 common terms among the three tetraploids, including five terms relating to transport and catabolism, nine associating with folding, sorting and degradation, two relating to response to metal ion, four relating to translation, seven associating with biosynthetic process and five unclassified terms (Figure 2C, D; Table S1-S3). In similar, the KEGG pathway analysis found that the PSGs were enriched in only one common pathway (B09124: Replication and repair) in the three tetraploids (Figure 2E, F; Table S4-S6), whereas, five common pathways (B09141: Transport and catabolism; B09123: Folding, sorting and degradation; B09102: Energy metabolism; B09101: Carbohydrate metabolism and B09121: Transcription) were significantly enriched by the NSGs of these tetraploids (Figure 2G, H; Table S4-S6). For each tetraploid Brassica, the PSGs enriched in less GO terms and KEGG pathways than the NSGs (average 85 vs. 237 GO terms, and 18 vs. 55 KEGG pathways) (Figure 3). The data may indicate that functional differentiation has occurred between PSGs and NSGs in the evolution of B. napus, B. juncea and B. carinata.
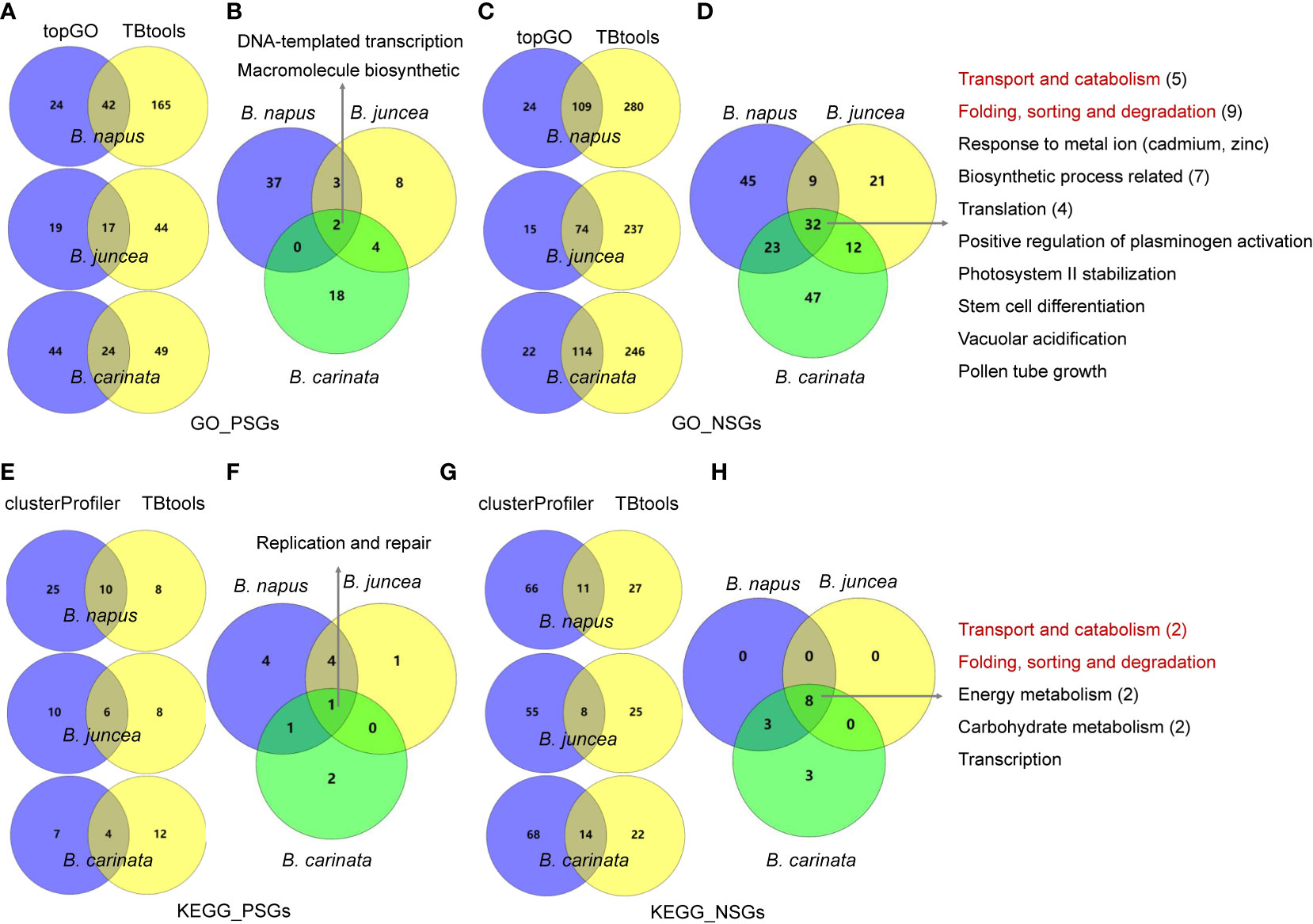
Figure 2 GO enrichment and KEGG pathway analysis for PSGs and NSGs in the evolution of tetraploid B napus, B juncea and B carinata. (A, C): GO enrichment analysis for PSGs and NSGs in three tetraploid Brassica using topGO and TBtools software, three intersecting terms were showed in middle; (B, D): two and 32 common terms were found in three tetraploid Brassica; (E, G): KEGG pathway analysis for PSGs and NSGs in three tetraploid Brassica by clusterProfiler and TBtools software, three intersecting terms were showed in middle; (F, H): one and eight common pathways were found in three tetraploid Brassica. The red font represent same terms between GO and KEGG.
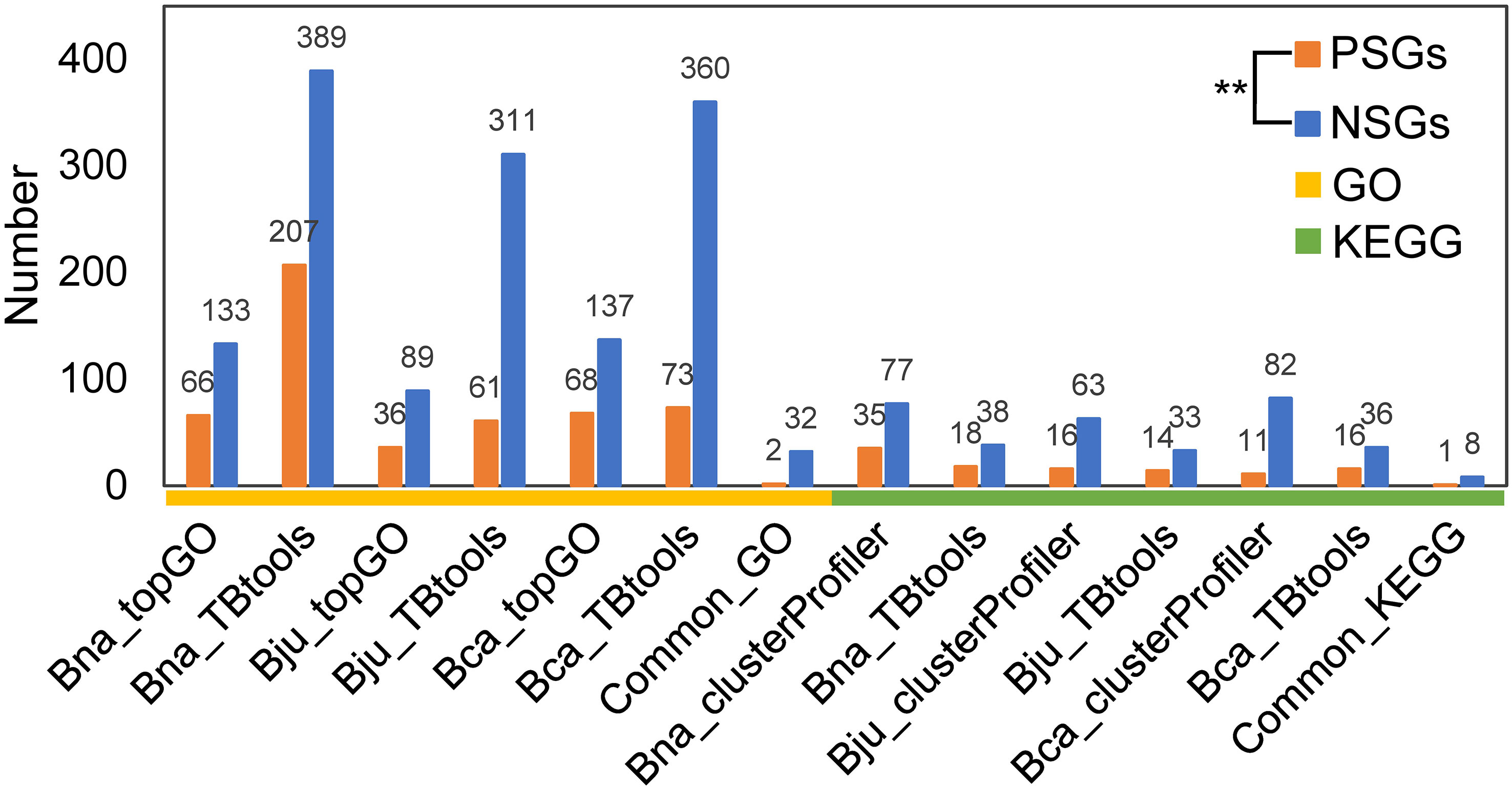
Figure 3 The number of GO terms and KEGG pathways for PSGs and NSGs. **significance level at 0.01. Bna, B.napus; Bju, B juncea; Bca, B carinata.
In total, ten, five and four KEGG pathways were enriched by the PSGs of B. napus, B. juncea and B. carinata, respectively (Figure 2F), with one common pathway “Homologous recombination” (ko03440) which is a secondary class of “Replication and repair” (B09124). This result indicates that genes associating with reproduction was positively selected in all the three tetraploids. On the other hand, four, one and two pathways were specifically enriched in B. napus, B. juncea and B. carinata respectively, however, most of these pathways were related to environmental adaptation. In detail, the PSGs of B. napus were specifically enriched in Starch and sucrose metabolism (ko00500), Cyanoamino acid metabolism (ko00460), Phenylpropanoid biosynthesis (ko00940) and Riboflavin metabolism (ko00740), of which the last two are related resistance to biotic or abiotic stresses. In similar, the PSGs of B. juncea were exclusively enriched in Isoflavonoid biosynthesis (ko00943), and Plant-pathogen interaction (ko04626) and Tropane, piperidine and pyridine alkaloid biosynthesis (ko00960) were specifically enriched in B. carinata. Our data suggest that PSGs play important roles in the evolution of tetraploids.
In addition, PSGs were also found to contribute to domestication. For example, B. carinata, an ancient crop from the Ethiopian highlands (Yim et al., 2022), is mainly domesticated as a crop with desirable seed fatty acid for biofuel production. In this study, 90 PSGs (gene set 1) of B. carinata were found specifically enriched in lipid biosynthesis (GO:0008610) and lipid metabolism (GO:0006629) (Table S7). In order to know whether PSGs affect lipid-related genes in B. carinata domestication, 2507 reported lipid-related genes of B. carinata (gene set 2) (Yim et al., 2022) were subjected to the one-tailed test as described in “Materials and Methods”. A total of 21 common genes (Ncom) was found between gene set 1 and gene set 2, which fell outside of the two 99% confidence intervals (the right tails were 4.684 and 4.678 for the two algorithms, respectively) (Figure 4). This suggests that lipid-related genes were positively selected in B. carinata domestication history.
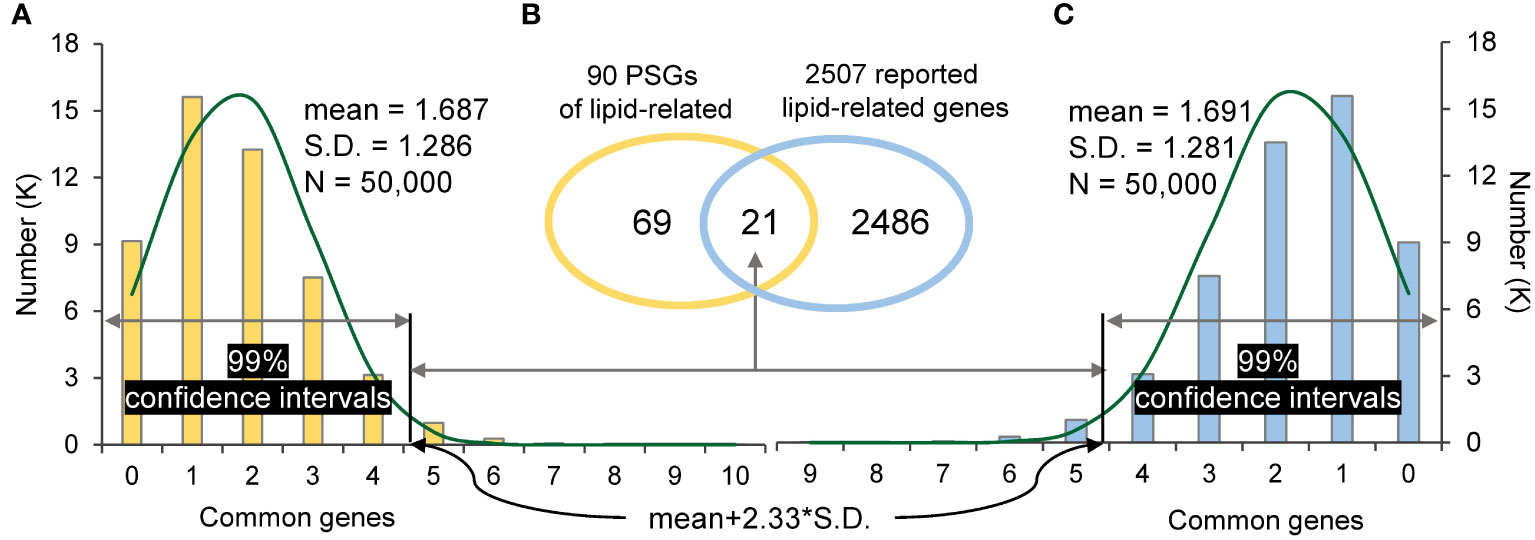
Figure 4 The random sampling distribution and significance test. (A): the distribution of common genes between 90 random sampling genes and 2507 reported lipid-related genes. (B): The common genes between 90 PSGs of lipid-related and 2507 reported lipid-related genes. (C): the distribution of common genes between 2507 random sampling genes and 90 PSGs of lipid-related. The threshold of the right tail is ‘mean+2.33*S.D.’ in the 99% confidence interval.
Combined with previous reports, a framework was provided here to illustrate the evolution and domestication of the Brassica genus (Figure 5). Based on the comprehensive and systematic estimation of the divergence time of the Brassica species according to Ks values, the Brassica species might be diverged from A. thaliana at about 24.81–25.78 Mya. This is in accordance with a previous study which found the Brassica lineage diverged from A. thaliana at about 29.5 Mya (Song et al., 2021). The divergence time was earlier than the hexaploidization event (23 Mya) and the known time of the WGT event (15.9–22.5 Mya) specific to Brassica (Beilstein et al., 2010; Liu et al., 2014). Based on the single-copy genes, we deduced that the divergence time of subgenomes at 1.27~1.96 Mya, which was significantly larger than that previously reports (0.043–0.076 Mya) using colinear genes (Song et al., 2021), however, consistent result was found that the formation time of B subgenomes was earlier than that of C subgenomes.
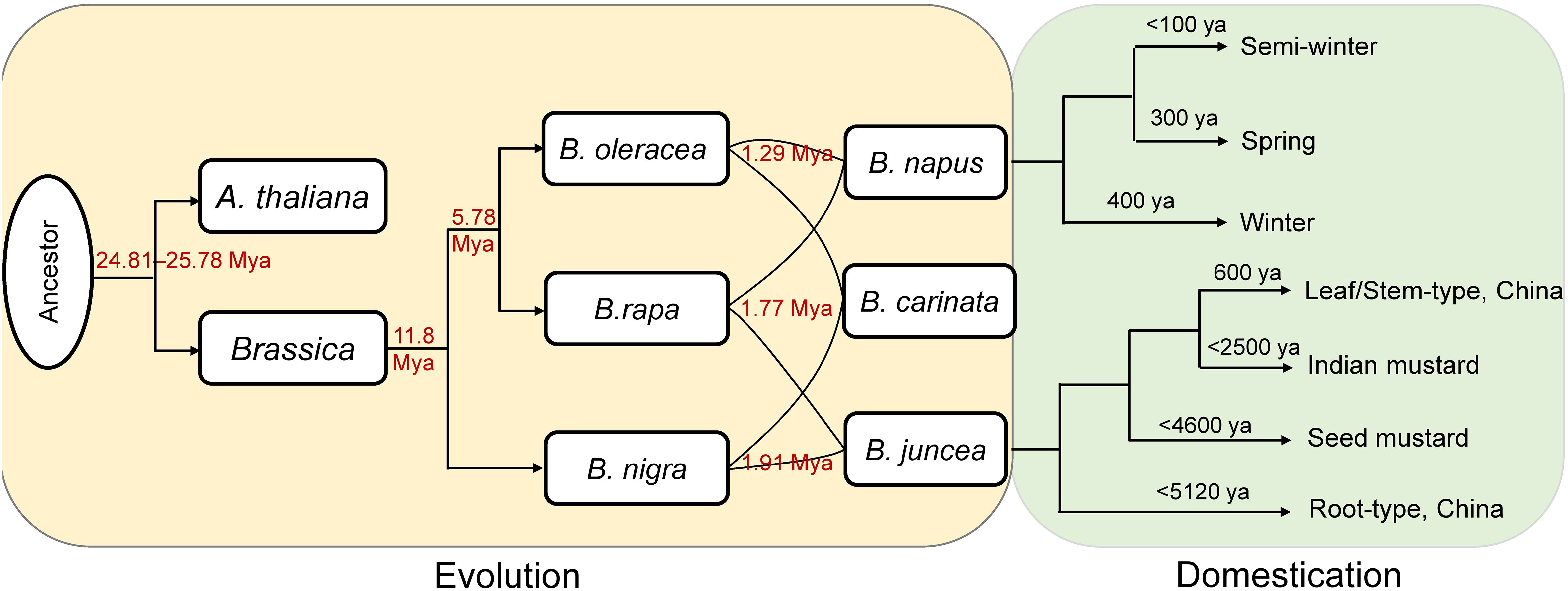
Figure 5 The evolution and domestication history. The red numbers were calculated in this study. ya, years ago.
Concerning to the diploid Brassica species, the divergence time among B. rapa, B. oleracea, and B. nigra estimated in the present study was generally consistent with other studies. For example, B. rapa and B. oleracea were proposed to have diverged from B. nigra at ~11.5 Mya (Parkin et al., 2014; Paritosh et al., 2021), and here we deduced that B. rapa and B. oleracea split from B. nigra at ~11.8 Mya (Figure 5). Likewise, the divergence time between B. rapa and B. oleracea was reported to be 4–6.8 Mya (Parkin et al., 2014; Paritosh et al., 2021), and we found the two species might diverge from each other at ~5.78 Mya (Figure 5).
Concerning to the tetraploid Brassica species, B. napus was found originate from the spontaneous hybridization between B. rapa and B. oleracea (Lu et al., 2019). The winter type (the original type) and the spring type of B. napus were formed at ~400 and ~300 ya, respectively, while the semi-winter type was domesticated more recently (<100 ya) (Qian et al., 2006; Bonnema, 2012; Wei et al., 2017). Recent research found that B. juncea most likely have a single origin from West Asia at about 8,000–14,000 ya (Kang et al., 2021). The earliest cultivation of B. juncea occurred at 2,500–5,120 ya in China as a vegetable crop (root type), followed by domestication of the seed mustard in West and Central Asia at 1,800–4,600 ya. The seed mustard spread from southern Afghanistan into the Indian subcontinent where it was domesticated into Indian mustard about 2,500 ya, and then spread further east into southwestern China at ~600 ya, formed a the broad-leaf/stem type B. juncea (Kang et al., 2021).
In this study, the number of PSGs was significantly greater in the C subgenome (average 2028) than in the A and B subgenomes (average 1412), and had no correlation with the subgenome size of Brassica (r=0.442, P=0.386). This result indicated that more genes of B. oleracea (CC) were positively selected during the evolution of B. napus (AACC) and B. carinata (BBCC). It is in consistence with previous studies that the C subgenome has undergone stronger selection in the evolution of B. napus and B. carinata (Wei et al., 2017; An et al., 2019; Yim et al., 2022). This could partially express the narrow genetic diversity of C subgenomes in B. napus and B. carinata, and therefore, introduce of novel genetic diversity into the C subgenomes is of great importance in broaden the genetic diversity of the two tetraploids. It is well known that B. oleracea has rich genetic variation (at least six cultivated and more than ten wild taxa) (Snogerup et al., 1990; Gladis and Hammer, 2001) and carries many favorable genes such as stress tolerance or disease resistance genes (Ellis et al., 1999; Mei et al., 2010; Wu et al., 2013; Wei et al., 2014). Although the two tetraploids and B. oleracea are cross-incompatibility, embryo rescue technique could solve this problem in a certain degree and it is possible to introduce genetic components from B. oleracea into B. napus or B. carinata via a hexaploid bridging strategy (Mei et al., 2015).
Positive selection and negative selection are considered as two major modes of natural selection (Xu et al., 2015). However, their influences on the evolutionary force of allotetraploids are still poorly understood. In the present study, we found obvious functional differentiation between PSGs and NSGs of tetraploid B. napus, B. juncea and B. carinata. It was found that PSGs of all the three tetraploids were significantly enriched in pathways associating with reproduction (Homologous recombination) and environmental adaptation. The role of homologous recombination in the evolution and domestication of Brassica allotetraploids has been well documented (Chalhoub et al., 2014), as well as in several other species (Bertioli et al., 2019; Edger et al., 2019; Zhang et al., 2020). The ability of environmental adaptation is a well-known factor in natural selection. Furthermore, we found lipid-related genes were positively selected in B. carinata. As we know, B. carinata is a crop with desirable seed fatty acid for biofuel production. Comparatively, this trait (seed fatty acid) seems more related to artificial selection rather than natural selection. Overall, our investigation suggests that positive selection is an important driving force in not only the evolution but also the domestication of Brassica tetraploids. Our study provides valuable insights into the selective mode and the evolution and domestication of Brassica tetraploid species.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
DW, JM and QT conceived and designed the experiments. NL, NZ and FL collected and analyzed the data. SZ, NZ, JW, LP, JS and ZW performed software analysis. YF and QT reviewed the manuscript. DW and NL wrote the manuscript; all authors reviewed and approved the final manuscript. All authors contributed to the article and approved the submitted version.
This study was performed under Key Project of Technological Innovation Application Development Plan of Chongqing (cstc2021jscx-gksbX0021), Fundamental Research Funds for the Central Universities (SWU-KT22054), National Natural Science Foundation of China (32172547), National Training Program of Innovation and Entrepreneurship for Undergraduates (202210635065), Ministry of Agriculture and Rural Affairs “National Characteristic Vegetable Industry Technology System Comprehensive Experimental Station in Yudongnan” (CARS-24-G-18). Regional Innovation Cooperation Project of Sichuan (2022YFQ0030).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1142147/full#supplementary-material
An, H., Qi, X. S., Gaynor, M. L., Hao, Y., Gebken, S., Mabry, M. E., et al. (2019). Transcriptome and organellar sequencing highlights the complex origin and diversification of allotetraploid Brassica napus. Nat. Commun. 10, 2878. doi: 10.1038/s41467-019-10757-1
Arabidopsis Genome (2000). Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815. doi: 10.1038/35048692
Beilstein, M. A., Nagalingum, N. S., Clements, M., Manchester, S. R., Mathews, S. (2010). Dated molecular phylogenies indicate a miocene origin for Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 107, 18724–18728. doi: 10.1073/pnas.0909766107
Bertioli, D. J., Jenkins, J. W., Clevenger, J. P., Dudchenko, O., Gao, D., Seijo, G., et al. (2019). The genome sequence of segmental allotetraploid peanut Arachis hypogaea. Nat. Genet. 51, 877–884. doi: 10.1038/s41588-019-0405-z
Bonnema, G. (2012). Genetics, genomics and breeding of oilseed brassicas. Eds. Edwards, D., Batley, J., Parkin, I., Kole, C. (Boca Raton: CRC press), 47–72.
Cantalapiedra, C. P., Hernández-Plaza, A., Letunić, I., Bork, P., Huerta-Cepas, J. (2021). eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829. doi: 10.1093/molbev/msab293
Chalhoub, B., Denoeud, F., Liu, S., Parkin, I. A., Tang, H., Wang, X., et al. (2014). Plant genetics. early allopolyploid evolution in the post-neolithic Brassica napus oilseed genome. Science 345, 950–953. doi: 10.1126/science.1253435
Chen, C. J., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y. H., et al. (2020). TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Edger, P. P., Poorten, T. J., VanBuren, R., Hardigan, M. A., Colle, M., McKain, M. R., et al. (2019). Origin and evolution of the octoploid strawberry genome. Nat. Genet. 51, 541–547. doi: 10.1038/s41588-019-0356-4
Ellis, P. R., Pink, D. A., Barber, N. E., Mead, A. (1999). Identification of high levels of resistance to cabbage root fly, Delia radicum, in wild Brassica species. Euphytica 110, 207–214. doi: 10.1023/A:1003752801143
Emms, D. M., Kelly, S. (2018). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol, 20. doi: 10.1186/s13059-019-1832-y
Gladis, T., Hammer, K. (2001). Nomenclatural note on the Brassica oleracea-group. Genet. Resour. Crop Evol. 48, 7–11. doi: 10.1023/A:1011201118844
Guo, Y., Liu, J., Zhang, J., Liu, S., Du, J. (2017). Selective modes determine evolutionary rates, gene compactness and expression patterns in Brassica. Plant J. 91, 34–44. doi: 10.1111/tpj.13541
Jing, D., Liu, X., He, Q., Dang, J., Hu, R., Xia, Y., et al. (2023). Genome assembly of wild loquat (Eriobotrya japonica) and resequencing provide new insights into the genomic evolution and fruit domestication in loquat. Hortic. Res. 10 (2), uhac265. doi: 10.1093/hr/uhac265
Kang, L., Qian, L., Zheng, M., Chen, L., Chen, H., Yang, L., et al. (2021). Genomic insights into the origin, domestication and diversification of Brassica juncea. Nat. Genet. 53, 1392–1402. doi: 10.1038/s41588-021-00922-y
Liu, S., Liu, Y. M., Yang, X. H., Tong, C. B., Edwards, D., Parkin, I. A. P., et al. (2014). The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 5, 3930. doi: 10.1038/ncomms4930
Lu, K., Wei, L., Li, X., Wang, Y., Wu, J., Liu, M., et al. (2019). Whole-genome resequencing reveals Brassica napus origin and genetic loci involved in its improvement. Nat. Commun. 10, 1154. doi: 10.1038/s41467-019-09134-9
Mei, J. Q., Liu, Y., Wei, D. Y., Wittkop, B., Ding, Y. J., Li, Q. F., et al. (2015). Transfer of sclerotinia resistance from wild relative of Brassica oleracea into Brassica napus using a hexaploidy step. Theor. Appl. Genet. 128, 639–644. doi: 10.1007/s00122-015-2459-3
Mei, J., Qian, L., Disi, J. O., Yang, X., Qian, W. (2010). Identification of resistant sources against sclerotinia sclerotiorum in Brassica species with emphasis on B. oleracea. Euphytica 177, 393–399. doi: 10.1007/s10681-010-0274-0
Nagahara, U. (1935). Genomic analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. Japan J. Bot. 7, 389–452.
Paritosh, K., Yadava, S. K., Singh, P., Bhayana, L., Mukhopadhyay, A., Gupta, V., et al. (2021). A chromosome-scale assembly of allotetraploid Brassica juncea (AABB) elucidates comparative architecture of the a and b genomes. Plant Biotech. J. 19, 602–614. doi: 10.1111/pbi.13492
Parkin, I. A., Koh, C., Tang, H., Robinson, S. J., Kagale, S., Clarke, W. E., et al. (2014). Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea. Genome Biol. 15, R77. doi: 10.1186/gb-2014-15-6-r77
Perumal, S., Koh, C. S., Jin, L., Buchwaldt, M., Higgins, E. E., Zheng, C., et al. (2020). A high-contiguity Brassica nigra genome localizes active centromeres and defines the ancestral Brassica genome. Nat. Plants. 6, 929–941. doi: 10.1038/s41477-020-0735-y
Qian, W., Meng, J., Li, M., Frauen, M., Sass, O., Noack, J., et al. (2006). Introgression of genomic components from Chinese Brassica rapa contributes to widening the genetic diversity in rapeseed (B. napus l.), with emphasis on the evolution of Chinese rapeseed. Theor. Appl. Genet. 113, 49–54. doi: 10.1007/s00122-006-0269-3
Rousseau-Gueutin, M., Belser, C., Da Silva, C., Richard, G., Istace, B., Cruaud, C., et al. (2020). Long-read assembly of the Brassica napus reference genome darmor-bzh. GigaScience 9 (12), giaa137. doi: 10.1093/gigascience/giaa137
Snogerup, S., Gustafsson, M., Von Bothmer, R. (1990). Brassica sect. Brassica (Brassicaceae) .I, taxonomy and variation. Willdenowia 19, 271–365. doi: 10.2307/3996645
Song, X., Wei, Y., Xiao, D., Gong, K., Sun, P., Ren, Y., et al. (2021). Brassica carinata genome characterization clarifies u's triangle model of evolution and polyploidy in Brassica. Plant Physiol. 186 (1), 388–406. doi: 10.1093/plphys/kiab048
Wang, X., Wang, H., Wang, J., Sun, R., Wu, J., Liu, S., et al. (2011). The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035–1039. doi: 10.1038/ng.919
Wei, D., Cui, Y., He, Y., Xiong, Q., Qian, L., Tong, C., et al. (2017). A genome-wide survey with different rapeseed ecotypes uncovers footprints of domestication and breeding. J. Exp. Bot. 68, 4791–4801. doi: 10.1093/jxb/erx311
Wei, D. Y., Mei, J. Q., Fu, Y., Disi, J. O., Li, J., Qian, W. (2014). Quantitative trait loci analyses for resistance to Sclerotinia sclerotiorum and flowering time in Brassica napus. Mol. Breed. 34, 1797–1804. doi: 10.1007/s11032-014-0139-7
Wu, J., Cai, G., Tu, J., Li, L., Liu, S., Luo, X., et al. (2013). Identification of QTLs for resistance to sclerotinia stem rot and BnaC.IGMT5.a as a candidate gene of the major resistant QTL SRC6 in Brassica napus. PloS One 8 (7), e67740. doi: 10.1371/journal.pone.0067740
Wu, T., Hu, E., Xu, S., Chen, M., Guo, P., Dai, Z., et al. (2021). clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb). 2 (3), 100141. doi: 10.1016/j.xinn.2021.100141
Xu, L., Bickhart, D. M., Cole, J. B., Schroeder, S. G., Song, J., Tassell, C. P., et al. (2015). Genomic signatures reveal new evidences for selection of important traits in domestic cattle. Mol. Biol. Evol. 32, 711–725. doi: 10.1093/molbev/msu333
Yang, J. H., Liu, D. Y., Wang, X. W., Ji, C. M., Cheng, F., Liu, B. N., et al. (2016). The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat. Genet. 48, 1225–1232. doi: 10.1038/ng.3657
Yang, J. H., Wang, J., Li, Z. P., Li, X. M., He, Z. S., Zhang, L. L., et al. (2021). Genomic signatures of vegetable and oilseed allopolyploid Brassica juncea and genetic loci controlling the accumulation of glucosinolates. Plant Biotechnol. J. 19, 2619–2628. doi: 10.1111/pbi.13687
Yim, W. C., Swain, M. L., Ma, D., An, H., Bird, K. A., Curdie, D. D., et al. (2022). The final piece of the triangle of U: Evolution of the tetraploid Brassica carinata genome. Plant Cell. 34, 4143–4172. doi: 10.1093/plcell/koac249
Zhang, Z., Gou, X., Xun, H., Bian, Y., Ma, X., Li, J., et al. (2020). Homoeologous exchanges occur through intragenic recombination generating novel transcripts and proteins in wheat and other polyploids. Proc. Natl. Acad. Sci. U.S.A. 117, 14561–14571. doi: 10.1073/pnas.2003505117
Keywords: Brassica tetraploid, evolution, domestication, functional differentiation, positively selected genes
Citation: Wei D, Li N, Zhang N, Liu F, Wu J, Zhao S, Shen J, Wang Z, Peng L, Fan Y, Mei J and Tang Q (2023) Selective modes affect gene feature and function differentiation of tetraploid Brassica species in their evolution and domestication. Front. Plant Sci. 14:1142147. doi: 10.3389/fpls.2023.1142147
Received: 11 January 2023; Accepted: 21 March 2023;
Published: 04 April 2023.
Edited by:
Francesco Mercati, National Research Council (CNR), ItalyReviewed by:
Chaobo Tong, Oil Crops Research Institute, Chinese Academy of Agricultural Sciences, Wuhan, ChinaCopyright © 2023 Wei, Li, Zhang, Liu, Wu, Zhao, Shen, Wang, Peng, Fan, Mei and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qinglin Tang, c3d1dHFsQDE2My5jb20=; Jiaqin Mei, amlhcWlubWF5QDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.