 Jin Li
Jin Li Han Tang2
Han Tang2 Lizheng Xiao
Lizheng Xiao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 19 January 2023
Sec. Plant Biotechnology
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1117002
Camellia sinensis var. Assamica cv. Duntsa (C.duntsa), a valuable Theaceae from Hunan Province, has been looked at as a precious tea resource by local farmers because of its economic and ecological value. Genomics study on C.duntsa is essential for the domestication and enhancement of tea tree varieties. In the present study, we used a hybrid approach based on Illumina and PacBio data to sequence and assemble the mitochondrial genome of C.duntsa. The mitochondrial genome of C.duntsa was estimated to be 1,081,996 base pairs (bp) and eighty-one genes consisting of one pseudogene, three ribosomal RNA (rRNA) genes, thirty transfer RNA (tRNA) genes, and forty-seven protein-coding genes (PCGs). Tetramer repetitions made up 43.90% of simple sequence repeats (SSRs). The codon usage bias of the Theaceae mitochondrial gene atp9 was altered by mutation, but the codon usage of other genes was shaped by natural selection. Besides, there are eighteen gene-containing homologous regions between the chloroplast and mitochondrial genomes of C. duntsa.Some genomes including atp8, cox1, cox3, nad7, nad9, rpl16, rpl2, rps19, rps4, and sdh4 are absent in the mitochondrial genome of several Theaceae plant. However, C. duntsa maintains these genes integrity and functionality. Another gene, rps16, is either lacking from the mitochondrial genome of C. duntsa or is present as a pseudogene. C. duntsa and C. sinensis (OM809792) are very similar, as shown by a collinear match across four species of Theaceae; the most conservative genes are nad5, atp9, cox2, rps3, trnA-TGC, trnI-GAT, rrn18, trnV-GAC, and ccmFN. Similarly, the genome’s phylogenetic trees revealed that C. duntsa was the sister species to C. sinensis. The results confirmed that the C. duntsa and C. sinensis (OM809792) mitochondrial genome underwent gene rearrangement.In general, our results shows that genomic information from organelles can help us understand plant phylogeny and can also be used to make molecular markers and study how genetic traits change over time. Our research will contribute to the population genetics and evolution of tea plant.
The basic function of mitochondria in living cells is to transform biomass energy into chemical energy (Birky, 1995).Most seed plants inherit mitochondrial genetic information from the maternal line (Cusimano and Wicke, 2016).The size, sequence alignment, number of repeats, and structure of plant mitochondrial genomes are all very complex and different (Mower et al., 2012). Higher plants have mitochondrial genomes that are between 200 and 2000 kb in size (Fauron et al., 1995). Some recently sequenced plant mitochondrial genomes are bigger than this range. For example, the flowering plant Silene conica has a mitochondrial genome of 11.3 Mb (Sloan et al., 2012).
In the plant mitochondrial genome, there are many repeats, including huge repeats (>1 kb), tiny repeats (<100 nt), and tandem repeats.Most changes in the structure of mitochondrial DNA are thought to be caused by repeat-mediated duplication (Maréchal and Brisson, 2010). Even though plant mitochondrial genomes are usually big, their gene pools (24 core genes and 17 variant genes) are generally small. This is because, during the evolution of angiosperms, many genes were lost or moved to the nucleus, but the coding sequences of the remaining genes are very stable (Adams et al., 2002).
As a result, the mitochondrial genome can be used to create molecular markers and figure out how the mitochondrial genome evolved (Zubaer et al., 2018). When comparing the mitochondrial genomes of closely related species, new things can be learned about how evolution works and how mitochondrial genomes change over time. It can also help with the classification of species (Zubaer et al., 2018).
With the Illumina Hiseq Platform, the entire DNA of plant organelles can be sequenced. But, Illumina read lengths frequently do not span larger repetitions, which leads to an inadequate assembly of these areas and, as a result, affects both the size and the content of the genome. Third-generation sequencing approaches, which include long-read Oxford Nanopore and PacBio sequencing, can enhance the coverage and assembly accuracy of unassembled genomic areas. This is a useful tool for understanding the information contained in the plant organelle genome (Shearman et al., 2016).
As new technologies come out, more and more organelle genomic data is being made available and analyzed, and gene transfer between chloroplasts and mitochondria is thought to be a part of long-term evolution (Nguyen et al., 2020). In the past, scientists have focused on gene transfer in the DNA of organelles in angiosperms (Park et al., 2014). Because of this, more research needs to be done on the mitochondrial and chloroplast genomic information of Theaceae species.
Camellia sinensis var. Assamica cv.Duntsa(C.duntsa), also known as “Cheng Bu Dong Cha “in Chinese. It is a tea plant native to the Chengbu Miao Autonomous County, Hunan Province, China (Supplementary Figure 1), with a cultivation history of over 200 years (Xinghui et al., 1998).
In botanical morphology, they are primarily small trees with large soft leaves, uplifted and glossy leaves, fuzz buds and leaves, and unique fragrances, which have the value of variety breeding and promotion (Guoben, 1987; Xinghui et al., 1998).
C.duntsa is often called “the Hunan province local characteristics tea plant resources”.The primary and secondary metabolites found in the fresh leaves of C.duntsa, such as polyphenols, amino acids, soluble sugars, and volatile organic compounds (Graham, 1992; Zhang et al., 2019b), have been linked to a lower risk of diabetes and several cancers (McCarty et al., 2020).
Nevertheless, there are currently no reports of any C. duntsa mitochondrial genomes. The NCBI Genbank database only has three mitochondrial genomes of tea plant species. Because of this, more research needs to be done on the mitochondrial genomic information of the C. duntsa species to understand how it evolved and how to classify it as a species.
Herein, the full mitochondrial genome of C.duntsa were identified using a combination of third-generation sequencing and second-generation sequencing techniques. After assembling and annotating the entire mitochondrial genome of C.duntsa, we performed an analysis of the content, organization, and phylogenetic tree of the genome. We carried out a comparative investigation of the mitochondrial of tea plant species in order to locate areas of the genome that vary, are conserved, and have been rearranged. In addition to this, we investigated the movement of genes between the chloroplast and mitochondrial genomes of C. duntsa. The information contained in a plant’s mitochondrial genome can be utilized to develop molecular markers and for genetic engineering, and it can also explain the phylogenetic and evolutionary links among plant species.
C. duntsa was cultivated at Chengbu Miao Autonomous County, Hunan Province, China(26° 05’ 52” N, 110° 28’ 78” E). Liquid nitrogen frozen fresh leaves at 80°C. The Nanopore (2000cUV-Vis) sequencing device sequenced C. duntsa mitochondrial DNA from the sample. Fastp (v0.20.0, https://github.htm ) was used to filter the raw data, removing primer sequences and sequencing junctions, discarding reads with quality scores below Q5, and removing N values greater than 5.The Perl script Filling v0.2.1 tallied and filtered sequence data from three different sources.
Using the Canu assembly program (Koren et al., 2017), we combined data from the first three generations to generate contig sequences.Then, we check the sequences against the plant mitochondrial gene database using BLAST v2.6 (https://blast.ncbi.nlm.nih.gov/Blast.Cgi). The mitochondrial gene contig that was compared served as the seed sequence, and the original data were used to loop and expand the sequence, ultimately revealing its primary structure (or daughter ring).Newest version of Next Polish is 1.3.1(available at https://github.com/Nextomics/NextPolish ). Assembling is done with data from the second and third generations. Errors were addressed and final results from the assembling process were achieved by human inspection.
Methods used in mitochondrial. Annotation BLAST were used to search for and find matches to previously reported plant mitochondrial sequences, and then additional changes were done by hand for closely related species based on the encoded proteins and rRNA. The tRNA was annotated with the help of the program tRNA scanSE (Chan and Lowe, 2019)(http://lowel.ab.ucsc.edu/tRNAs.can-SE/ ).The program Open Reading Frame Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) is utilized in the process of annotating Open Reading Frames (ORF). For the assembly of the mitochondrial genome, the program OrganellarGenomeDRAW was utilized (https://chlor.obox.mpimp-golm.mpg.de/OGDraw.html).
To determine whether sequences are duplicates, we use the DupFinder Perl script in conjunction with the vmatch v2.3.0 software (http://www.vmatch.de/). Forward, backward, reverse, and complementary sequences of at least 30 bp in length are all recognized. For the purpose of doing simple repeat sequence analysis, we made use of MISA (https://webblast.ipk-gatersleben.de/misa/ ), a web-based tool. In this research, we found a total of six instances of one-base, two-base, three-base, four-base, five-base, and six-base repeats. SSRs have a minimum separation of 100 bp. The Tandem Repeats Finder v4.09 software (http://tandem.bu.edu/trf/trf.submit.options.html) was utilized in order to recognize tandem repeats with lengths of more than six base pairs and more than ninety-five percent of matched repeats.
We extracted C.duntsa’s chloroplast genome sequence from the NCBI Organelle Genome Resource database (OL450397). Utilizing BLAST v2.10.1, we were able to identify comparable sequences in the mitochondrial and chloroplast genomes. Define the selection criteria that will result in a successful pairing rate of seventy percent. The editing sites of the mitochondrial RNA were located by a process that involved comparing the mitochondrial RNA of C. duntsa to the protein that was encoded by the mitochondrial gene. The Plant Predictive RNA Editor (PREP) tool (http://prep.unl.edu/) was utilized in order to do an analysis on the data collected for this investigation.
We determined the frequencies of non-synonymous (Ka) and synonymous (Ks) substitution for each PCG between C.duntsa and four other plant species: C. sinensis (NC 043914), C. sinensis (OM809792), A. thaliana (JF729201), and G. biloba (KM672373).The PCG sequence was aligned using Mafft version 7.427, and the Ka/Ks analysis was performed using the MLWL-based Ka/Ks Calculator version 2.0 (Wang et al., 2010).
Utilizing MAFFT, the mitochondrial genomes of nineteen distinct species that belong to a variety of families were compared and aligned. After the sequences were aligned end-to-end, trimming was carried out using trimAl (v1.4.rev15) with the parameter -gt 0.7, then jmodeltest-2.1.10 was used to produce a prediction on the GTR status of the model. Employing the RAxML (Stamatakis, 2014)program, the GTRGAMMA model, and a bootstrap size of one thousand in order to construct an evolutionary tree with the highest probability achievable. An iterative analytic method known as Markov Chain Monte Carlo (MCMC) was used in version 3.2.7 of MrBayes to simulate a population for a total of 1,000,000 generations, and samples were taken at intervals of 100 generations. The simulation was run for a total of 1,000,000 generations. After the first 25% of the phylogenetic tree has been burnt in,a tree is produced that is consistent the majority of the time.
The codon usage bias parameter RSCU and the Effective number of codons (ENC) were calculated using an online cloud platform (cloud.genepioneer.com.).ENC was used to analyze the effect of gene base composition on codon usage preference. These two parameters are used to describe the pattern of codon usage preference. Four mitochondrial genomes from the tea plant species, including C.duntsa, C.sinensis(NC043914), C.sinensis(OM809792), and C. sinensis var. Assamica, calculate their codon preference parameters.
The C. duntsa mitochondrial genome was used as a reference and compared with three other mitochondrial genomes of the tea plant species, C.sinensis(NC043914), C.sinensis(OM809792), and C. sinensis var. Assamica. The mitochondrial genomes of C. duntsa and C. sinensis (OM809792) were analyzed for covariance using Dottup software. Whole-gene covariance comparisons were performed using LASTZ version 1.02.00 to detect covariance regions.The parameters were set as follows: step = 20 and seed pattern = 12 of 19.
The C.duntsa genomic sequence generated in this study is now archived in GenBank (accession number OL989850). In this study, the average read coverage of the assembled mitochondrial genome was more significant than 268×. Whole mitochondrial genome sequencing delivered 26,855,210 clean reads from the second-generation sequencing platform, whereas the third-generation sequencing platform generated 1,034,825 clear reads (SRA accession number SRR17287593). The sequencing and assembly statistics are shown in Table Supplemental 1.
The mitochondrial genome of C.duntsa consists of 1,081,996bp of closed-loop DNA molecules (Figure 1). C.duntsa mitochondrial has a nucleotide makeup of 27.03% A, 27.35% T, 22.78% G, 22.84% C, and a GC content of 45.62%. PCGs, tRNAs, and rRNAs made up 3.62%, 0.21%, and 0.52% of the total mitochondrial genome, respectively. The mitochondrial genome of C.duntsa has eighty-one annotated genes, including forty-seven PCGs, thirty tRNAs, three rRNAs, and one pseudogene (Table 1).
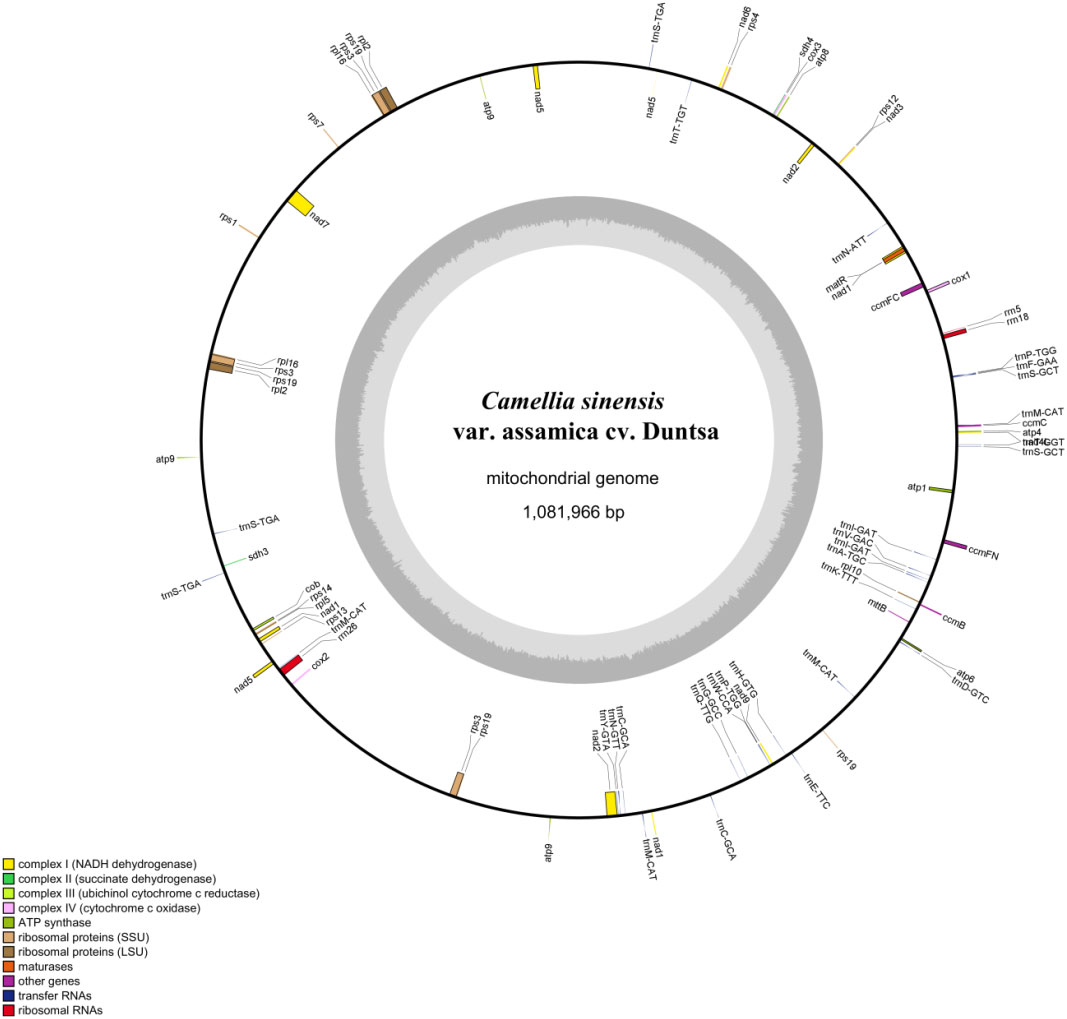
Figure 1 C.duntsa mitochondrial genome circular map.
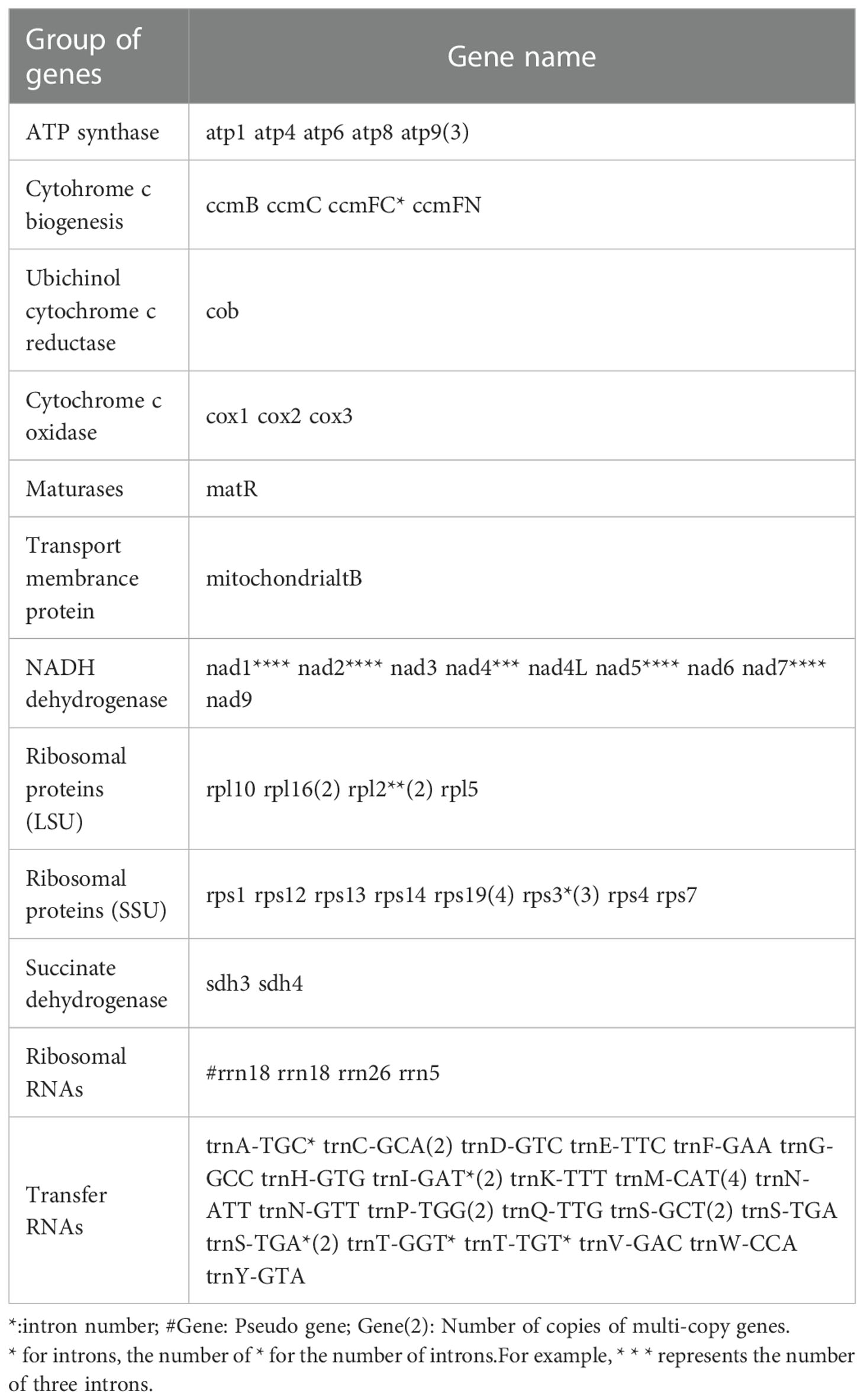
Table 1 Gene profile and organization of the C.duntsa mitochondrial genome.
Most terrestrial plants contain three rRNA genes (Cavalier-tsmith, 1975; Archibald, 2011). The rrn26 (3595 bp), rrn18(1923 bp), and rrn5(121bp), were annotated within the C.duntsa. In addition, the number of introns contained in the mitochondrial genomes of terrestrial plants might vary considerably. Eighteen of the annotated genes in C.duntsa contained introns, and eleven of those genes (CCMFc,rps3(3), trnA-TGC, trnI-GAT(2), trnS-TGA(2), trnT-GGT, and trnT-TGT) included an intron (Table 1).
C. duntsa PCGs have a length of 39,126 bp. All PCGs used ATG as the starting codon, while the terminating codons TAG, TGA, and TAA were utilized at 19.15%, 34.04%, and 46.81%, respectively (Supplementary Table 2). Leucine (Leu) (7.32–11.12%), Serine (Ser) (8.64–10.30%), and Aspartic Acid (Arg) (6.09–7.82%) were found to be the most frequently occurring amino acids compared to other species (Figure 2). In contrast, Cysteine (Cys) (1.21–1.49%) and Tryptophan (Trp) (1.17–1.54%) were identified only frequently. The Relative Synonymous Codon Usage (RSCU) of the C. duntsa mitochondrial genome was another aspect that we investigated. To do this, we analyzed 10,237 codons out of 47 PCGs(excluding terminated codons). 6,469 codons (63.02%) had RSCU values over 1.0, showing that they were used more than synonymous codons(Supplementary Table 3).
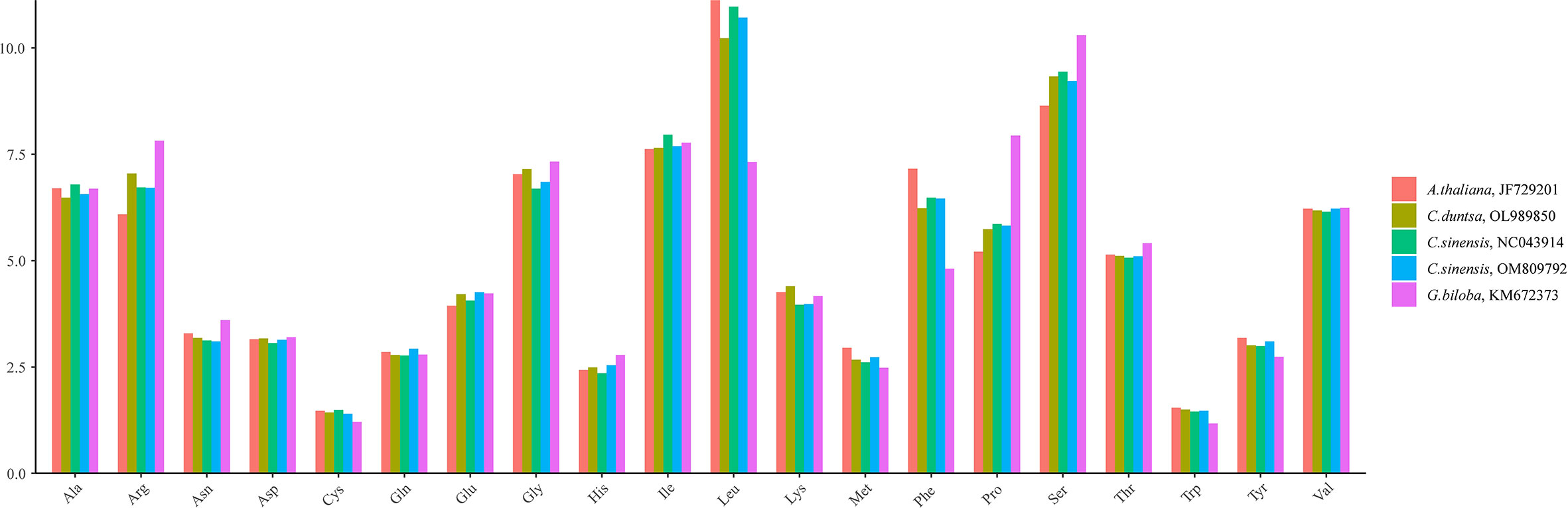
Figure 2 C.duntsa mitochondrial codon usage pattern compared to C.sinensis(NC043914), C.sinensis(OM809792), A.thaliana(JF729201),and G.biloba(KM672373).
According to Supplemental Table 3, which displays the codon usage frequency of four mitochondrial genomes from tea plant species, these four species contain between 4631 and 6351 codons with an RSCU greater than 1. The most prevalent codons among these four species were GCU (Ala), UAU (Tyr), and CAU (His) (RSCU > 1.5), whereas CAC (His) was the least common (RSCU< 0.5). The number of genes with ENC values< 35 in the mitochondrial genomes of C.duntsa, C. sinensis (NC043914), C. sinensis (OM809792), and C. sinensis var. Assamica is 0, 1 (atp9), 0 and 0 correspondingly (Supplementary Table 4). Four genes, including atp4, rps12, rps7, and nad9, are positioned on or above the standard curve line, while the remaining genes are located below the standard curve line (Figure 3 and Table 2).
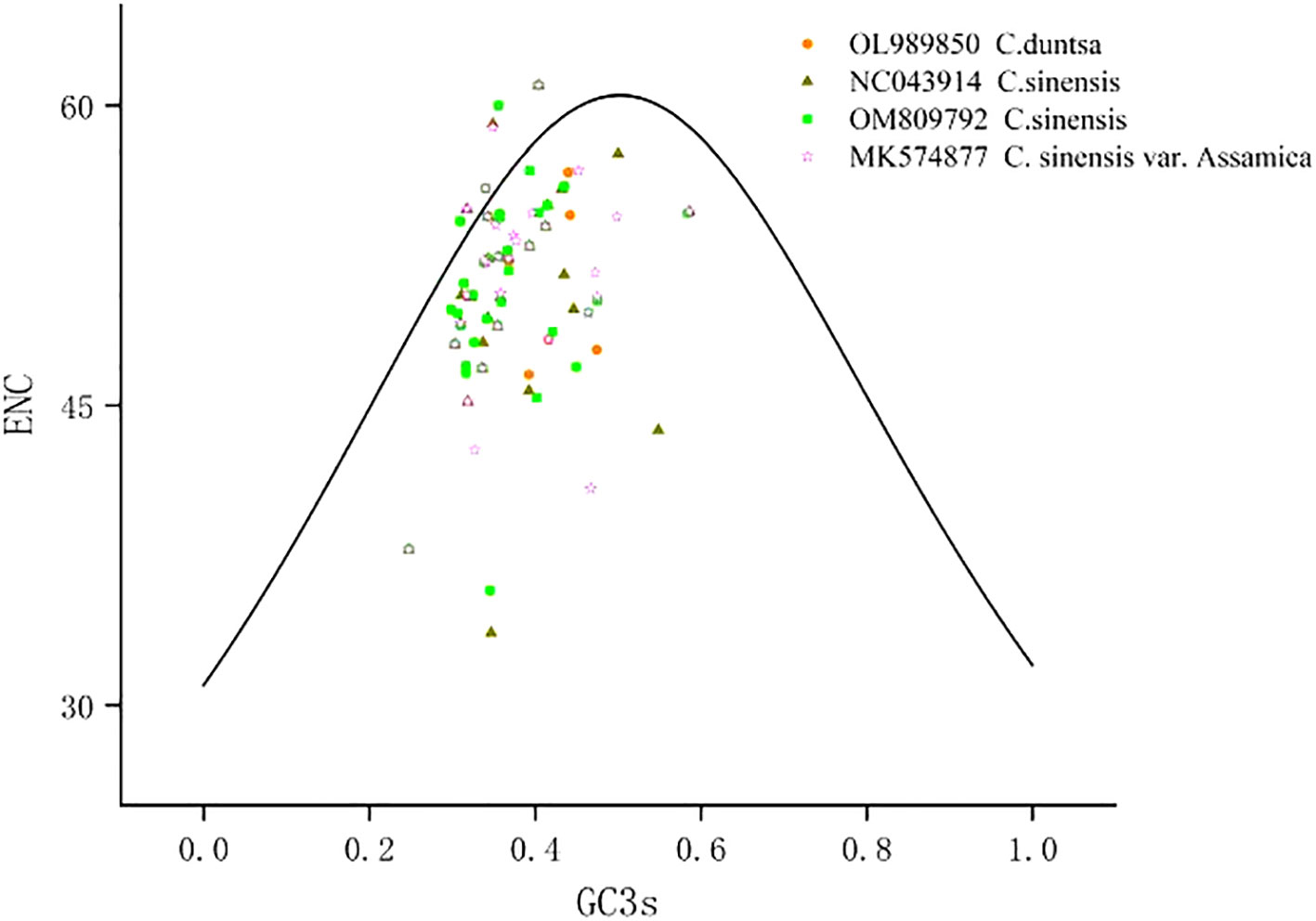
Figure 3 ENC plot of four Theaceae mitochondrial genomes.Standard curve line is calculated as follows: ENC = 2 + GC3 + 29/(GC32 + (1-GC3)2).
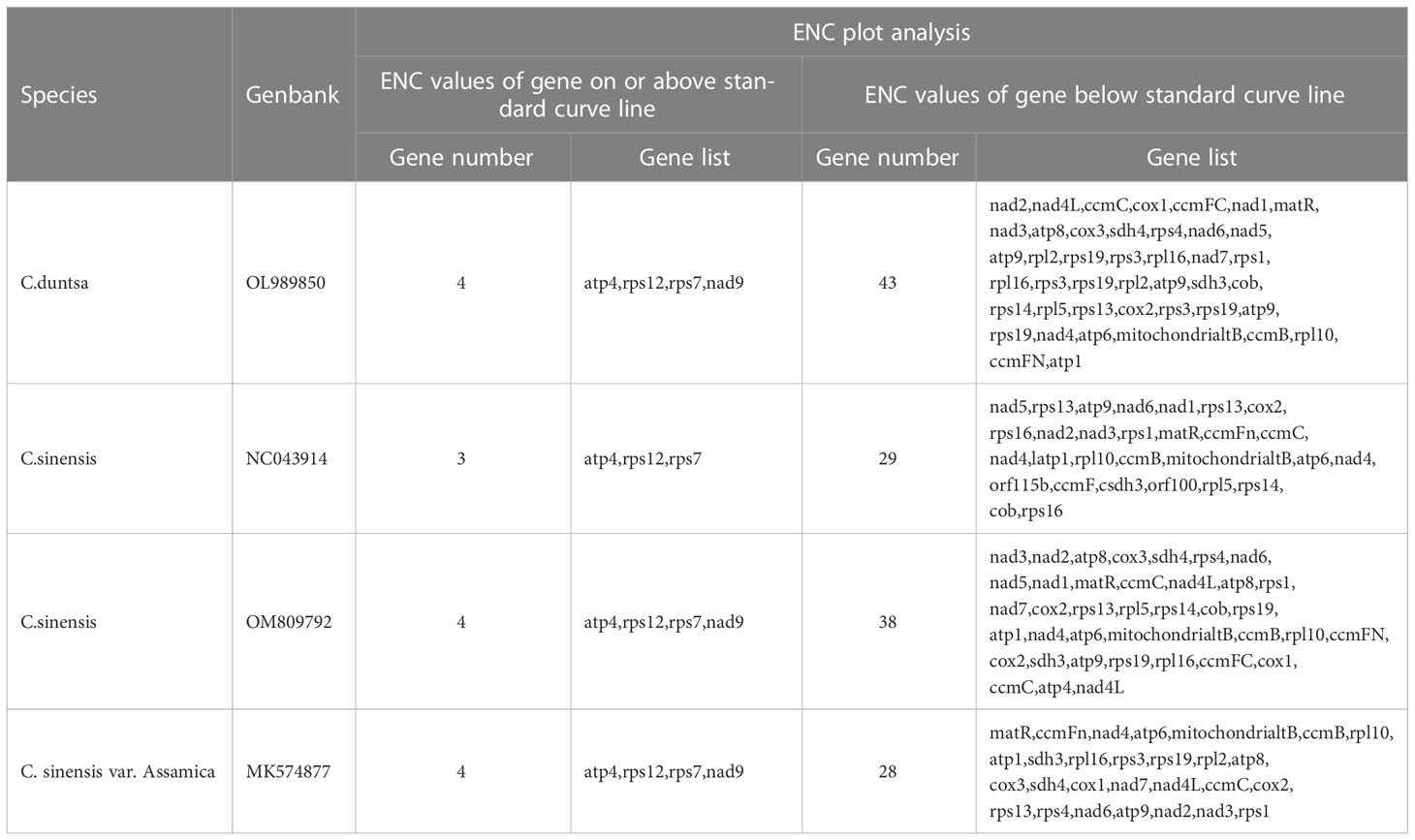
Table 2 Plot of ENC of genes in 4 Theaceae species mitochondrial genome.
Calculating non-synonymous substitutions (Ka) and synonymous substitutions (Ks) is essential for reconstructing phylogenies and studying the evolution of protein-coding sequences among closely related species. Positive selection (Ka/Ks >1), neutral selection (Ka/Ks =1), and negative selection (Ka/Ks<1) are all possible outcomes. Here, we analyzed the Ka, Ks ratio of thirty-eight PCGs present in the mitochondrial genomes of C. duntsa, C. sinensis (NC043914), C. sinensis (OM809792), A. thaliana (JF729201), and G. biloba (KM672340) (Figure 4). Ka/Ks values for the thirty-eight shared PCGs between C. duntsa and C. sinensis (OM809792) were zero. For the thirty-five shared PCGs between C. duntsa and C. sinensis (NC043914), the Ka/Ks values were also zero. In contrast, eleven of the thirty-eight genes shared by all four species have Ka/Ks values that are larger than 1, indicating that they were positively chosen during the process of evolution. These genes include nad2, ccmB, atp4, nad6, atp9, rps7, rps19, rps14, ccmB, nad3 and nad1.
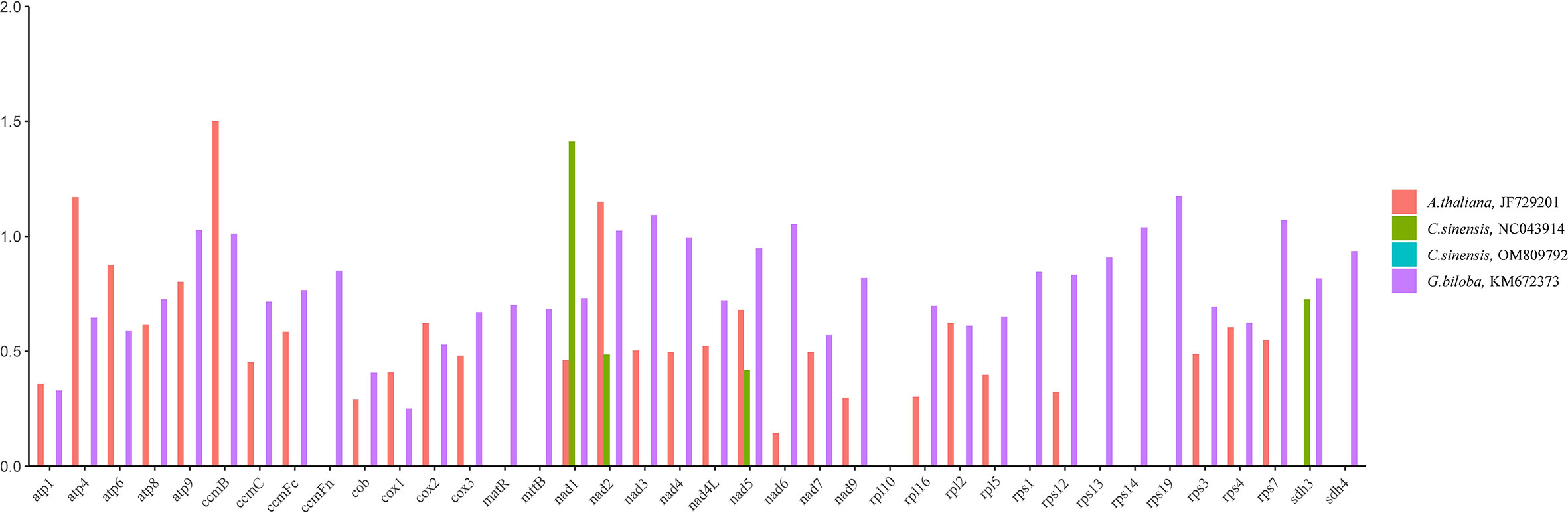
Figure 4 Ka/Ks ratios of thirty-eight PCGs in C.duntsa,C.sinensis(NC043914),C.sinensis(OM809792), A.thaliana(JF729201),and G.biloba(KM672373).
Simple sequence repeats (SSRs) are sequence-repeating units ranging in length from one to six base pairs (Qiu et al., 2010).SSRs are useful due to their polymorphism, ease of PCR detection, codominant inheritance, and extensive genome coverage (Powell et al., 1996).The C.duntsa mitochondrial genome SSRs were identified using the Tandem Repeats Finder program. There were 316 SSRs discovered, and Figure 5 displays the proportion of each kind. According to weighted SSRs, tetramer repeats comprised 43.90% of the total, followed by dimer repeats at 26.00%. AGAA repetitions were the most common form of tetramer SSRs, accounting for 7.10%, whereas CT repeats were the most common form of dimer SSRs, accounting for 17.80%. The C.duntsa mitochondrial genome has only four hexamers SSRs, located between atp9 and rpl2, rpl2-2 and atp9-2, cox2 and rps3-3, and rps19-3 and atp9-3. In Supplemental Table 5, the positions of the pentamer and hexamer are displayed.
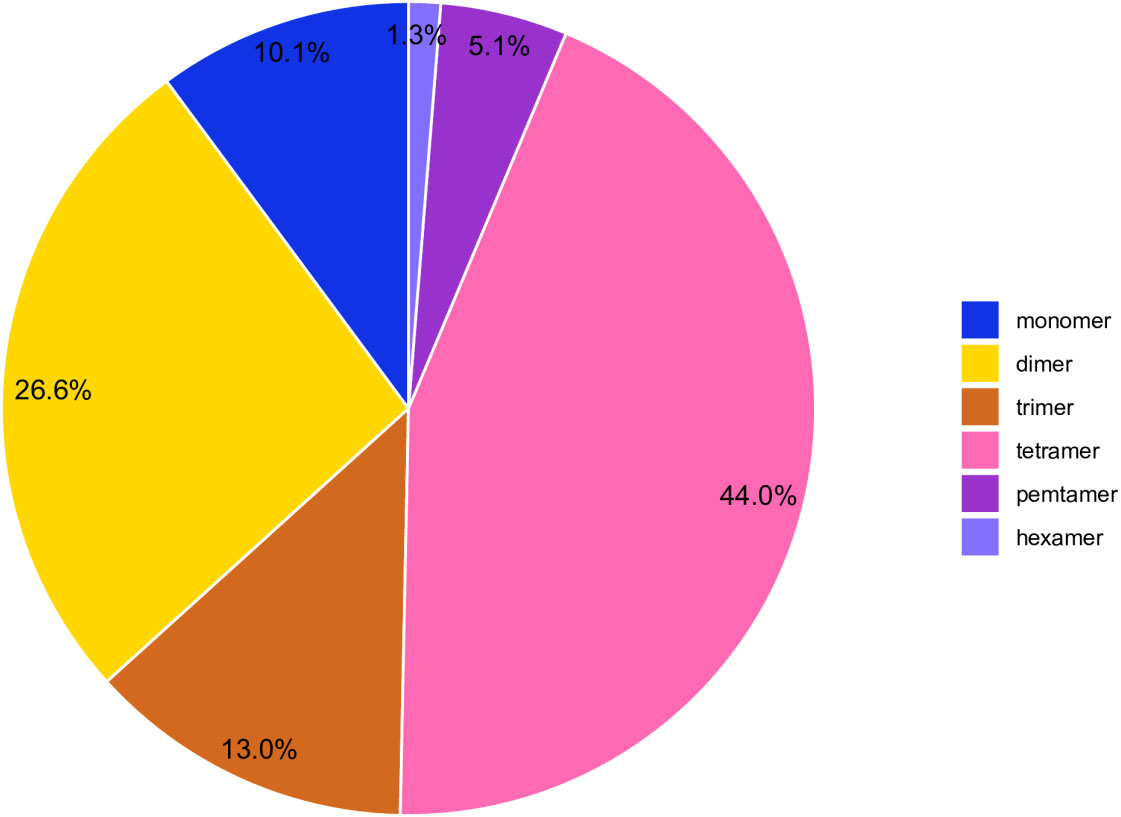
Figure 5 C.duntsa SSR distribution. Different hues denote SSRs. The graphic shows SSR percentages.
Tandem repetitions refer to the approximately 1-200 base repeat units repeated repeatedly. They are widespread in eukaryotic and certain prokaryotic genomes (Huan and Jie, 2005). The mitochondrial genome of C. duntsa has thirteen sequence repetitions with a significance level greater than 95%, ranging in length from 12 to 39 bp, according to Supplemental Table 6. The REPuter program (Kurtz et al., 2001) identified the C. duntsa mitochondrial genome’s interspersed repeat sequence. The longest forward repetition was 92,293bp while the longest palindromic repeat was 88,112bp. Both types of repeats are mostly made up of 30–39 bp repeats (Figure 6).
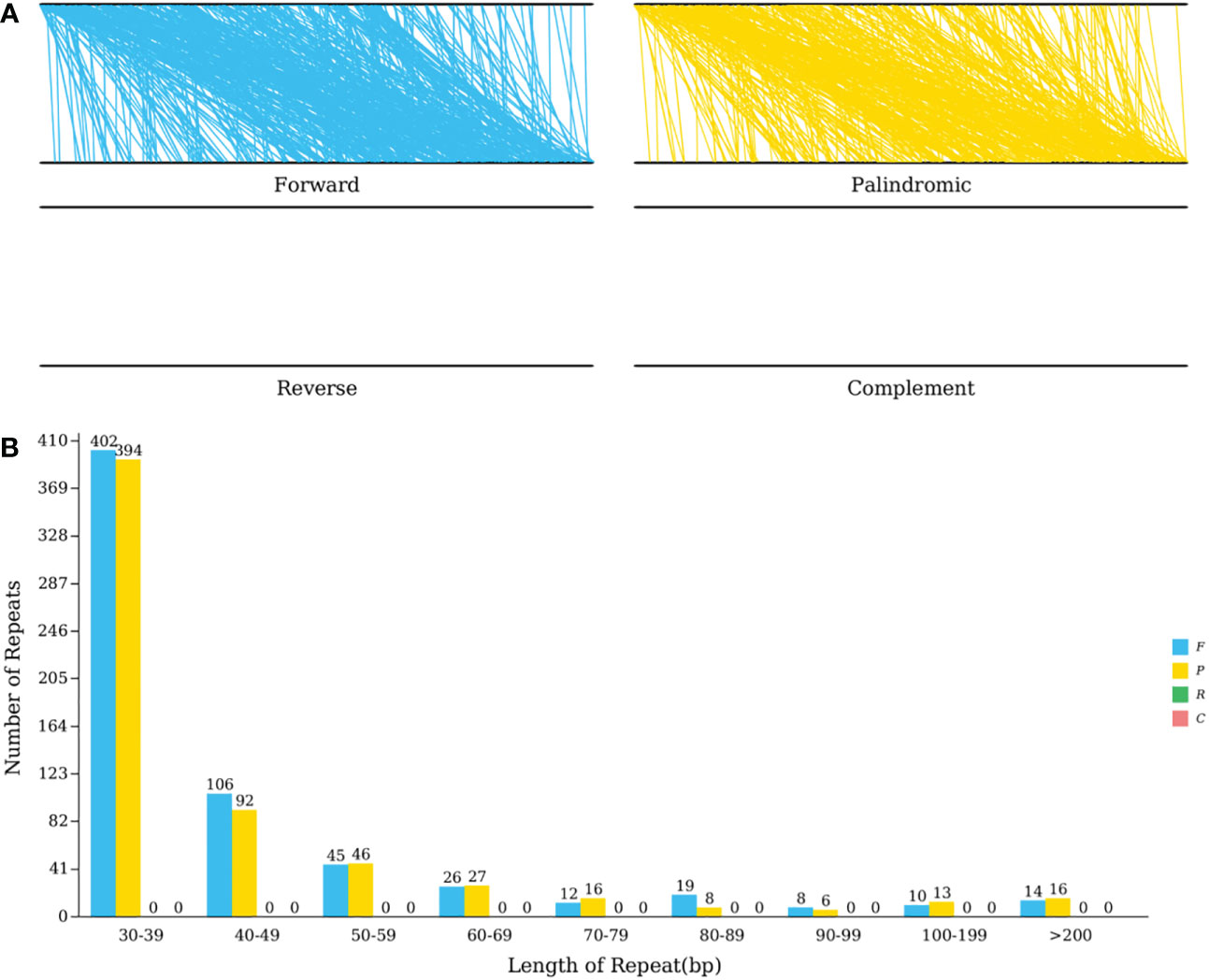
Figure 6 C.duntsa mitochondrial repeats. (A) The mitochondrial genome and its forward copy both contain forward repeats.Palindromic repeats in mitochondrial genome and its reverse complement. (B) C.duntsa mitochondrial genome repeat length distribution. Histograms display the repeat number of given lengths.
RNA editing, the insertion, deletion, or replacement of nucleotides in a transcribed RNA’s coding region, is widespread in land plants other than mosses. (Malek et al., 1996; Brennicke et al., 1999).This work predicted the RNA editing sites of forty-one PCGs conserved among five angiosperms to search for similarities. The fact that G.biloba (KM672373;1315) contained more RNA editing sites than A. thaliana (JF729201), C. duntsa (OL989850), C. sinensis (NC043914), and C. sinensis (OM809792) (351, 536, 436, and 522) implies that these positions are highly conserved in tea plant PCGs (Figure 7).
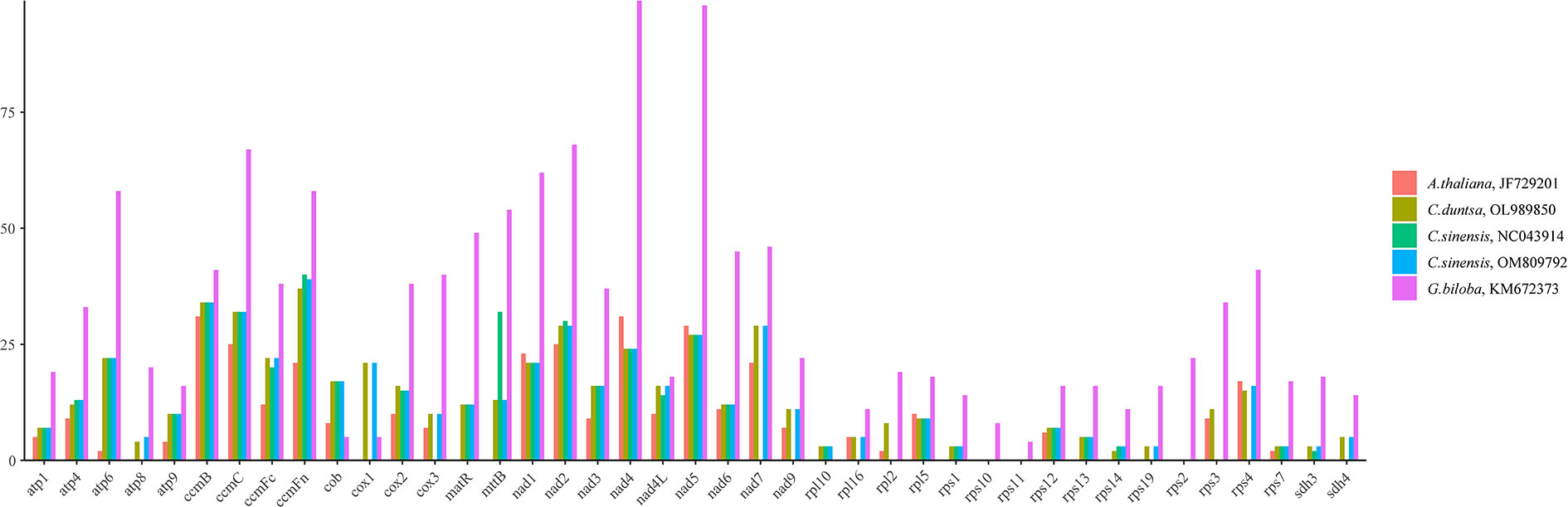
Figure 7 The distribution of RNA editing sites across the PCGs of the mitochondrial genomes of five different angiosperms.
Among the RNA editing sites in C.duntsa PCGs, ccmFn contains the greatest number, 37, accounting for 6.90% of all editing sites.The rps14 has minor editing sites, 2, accounting for 0.3%. The first position of triplet codes contained 32.07% of these 536 RNA editing sites, whereas the second position had 67.93%.(Supplementary Table 7).The water-repelling properties of 43.74 percent of the amino acids did not change, 7.29% became hydrophilic, and 48.22% became hydrophobic.Atp6, atp9, cox2, and ccmFc of the C.duntsa mitochondrial genome are prematurely terminated due to the presence of RNA editing. Our research also indicated that following RNA editing, leucine was changed to 45.61% of the edited amino acids, which suggests that leucine was the predominant amino acid among the projected editing codons(Supplementary Table 7).
It is generally accepted that the size of the genome and the proportion of GCs it contains are two of the most critical factors determining the genomes of plant organelles. The mitochondrial genomes of twenty-three other plant species were analyzed, and their sizes and percentages of GC content were compared to the C. duntsa mitochondrial genome. Of the twenty-three species, one was a gymnosperm, two were monocots, and the remaining twenty were dicotyledons. Supplementary Table 8 lists these plants’ species abbreviations and mitochondrial genome access numbers. There was a wide range in plant mitochondrial genome size, from 219,736 Kb (Brassica rapa) to 1,325,823 Kb (Hevea brasiliensis). Although there is a wide range in plant mitochondrial genome size, the GC content (often approximately 44%) shows that the mitochondrial genome has remained generally stable throughout evolution (Figure 8).
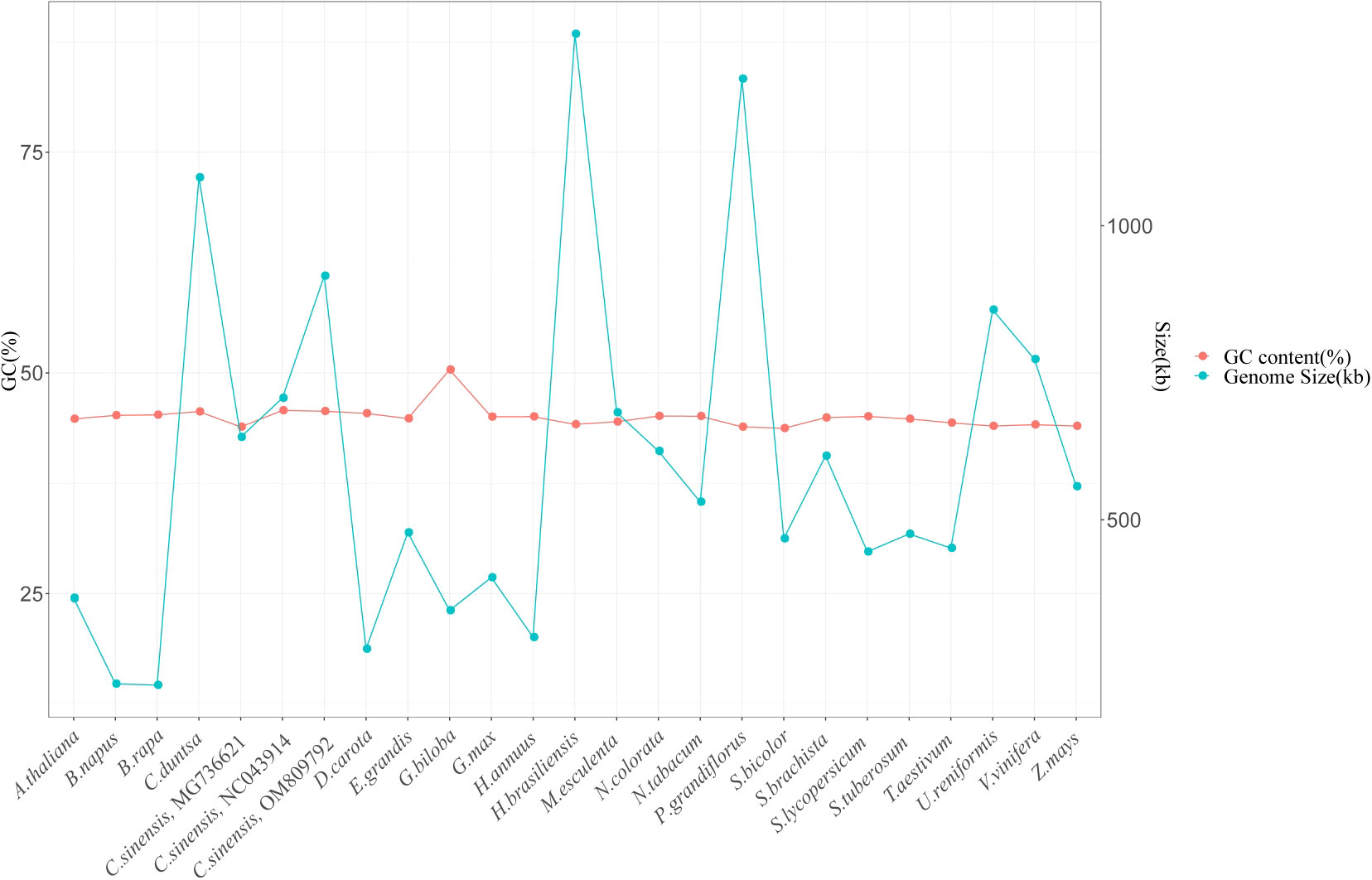
Figure 8 Gene content (GC) and genome size (kb) of twenty-four plant mitochondrial genomes.
As displayed in Figure 9, the C. duntsa mitochondrial genome served as a standard for evaluating the outcomes of a whole-genome correlation match performed on four different tea plant taxa. In the gene region of the mitochondrial genomes of these related species, there was a higher level of similarity with C. duntsa; the most conserved genes were nad5,atp9,cox2,rps3,trnA-TGC,trnI-GAT, rrn18, trnV-GAC, and ccmFN. Compared to those of the other two tea plant species, the mitochondrial genomes of C. duntsa and C. sinensis (OM809792) were more similar to one another. In addition, the arrangement of the mitochondrial genomes of C. duntsa and C. sinensis (OM809792) shown that substantial variation still existed (Supplementary Figure 3).
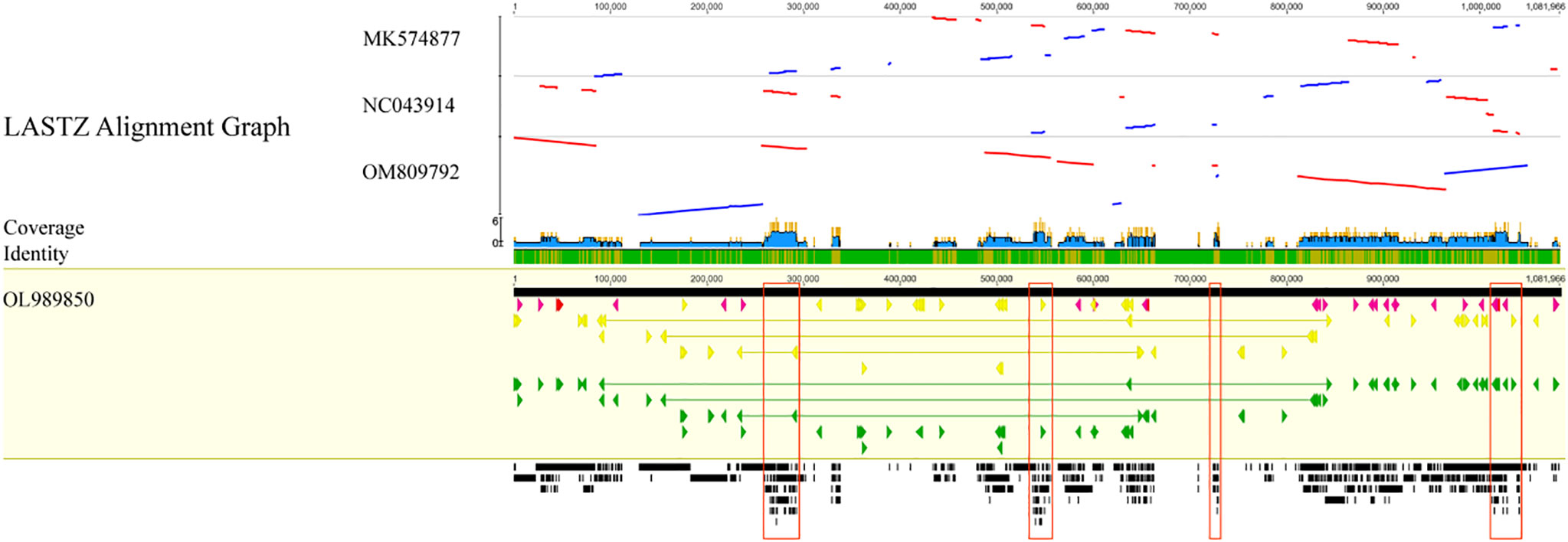
Figure 9 Four Theaceae mitochondrial genomes aligned collinearly.High-scoring segment co-direction pairings are shown by blue bars, and reversed pairs are shown by red bars. Arrows in yellow indicate coding sequences, red ribosomal RNA genes, purple transfer RNA genes, green protein-coding genes, and gray exonic areas. In this case, the larger the black squares, the more genetic material the two species share.Conservative regions are red. These sections comprise nad5, atp9, cox 2, rps 3, trnA-TGC, trnI-GAT, rrn18, trnV-GAC, and ccmFN.
In order to evaluate mitochondrial genome rearrangement and collinearity, the mitochondrial genome of C. duntsa and C.sinensis (OM809792) were compared using the Mauve and Lastz programs.C. duntsa and C.sinensis mitochondrial genome (OM809792) were split into fourteen locally collinear blocks (LCBs). Both the size of these LCBs and their position in relation to one another varied greatly between the two tea plant species that were researched (Figure 10). In addition, the majority of LCBs contained gene sequences, with a total of thirty-one genes involved.The examination of the rearrangement revealed that the mitochondrial genomes of C. duntsa and C. sinensis(OM809792)had undergone a significant amount of rearrangement (Supplementary Table 9).
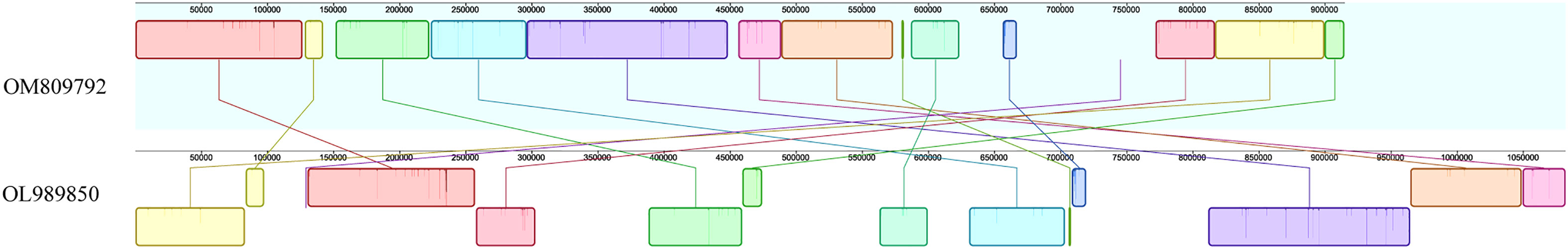
Figure 10 Mauve comparison between C.duntsa and C.sinensis genomes (OM809792).
With sequencing technology advancing rapidly, the entire plant mitochondrial genome has been constructed and reported, enabling comparative research of mitochondrial genome features across plant species (Wei et al., 2016). Four mitochondrial genomes from this family can now be accessed: C.duntsa, C.sinensis(NC043914), C.sinensis(OM809792), and C. sinensis var. Assamica. These four mitochondrial genomes each include a total of 47, 30, 42, and 44 protein-coding genes, respectively(Supplementary Table 10).
In distinct species, gene duplication and gene loss have been documented. For instance,rpl16, rpl2, atp9, rps3, and rrn18 were replicated in the C.duntsa genome, rps13 and rps16 replicated in the C.sinensis (NC043914) genome, atp4, atp8, ccmC, cox2, and nad4L replicated in the C.sinensis (OM809792) genome, and atp9, nad1, nad2, nad9, rps19, sdh3 replicated in the C. sinensis var. Assamica.
C.duntsa and C. sinensis var. Assamica has the complete mitochondrial genome, with only one gene (rps16) being lost, rpl2, rps16, and rps3 from C.sinensis (OM809792) being lost, and C.sinensis (NC043914) losing most of eleven genes(Supplementary Table 10).
To better understand the evolution of the C. duntsa genome, a taxonomic study was carried out on the mitochondrial genome of the C. duntsa species, in addition to nineteen other published plant mitochondrial genomes.The phylogenetic trees were constructed through a combination of the maximum likelihood method and the Bayesian method. The species that were taken into consideration are listed, together with their respective NCBI accession numbers, in Supplemental Table 11.In the study of phylogenetic relationships,Sorghum bicolor, Triticum aestivum, Zea mays, and Ginkgo biloba are employed as outgroups. Both analysis approaches produce mutually consistent results for classifying the phylogenetic tree. This evolutionary tree shows that gymnosperms and dicots are distinct from monocots and dicots, respectively (Figure 11). The phylogenetic tree’s clustering matches these species’ family and genus affiliations, confirming mitochondrial genome-based clustering. The phylogenetic tree also demonstrated that C.duntsa, C.sinensis(NC 043914), and C.sinensis(OM809792) are linked. Our mitochondrial genome study provides a solid foundation for future tea plant relatedness studies.
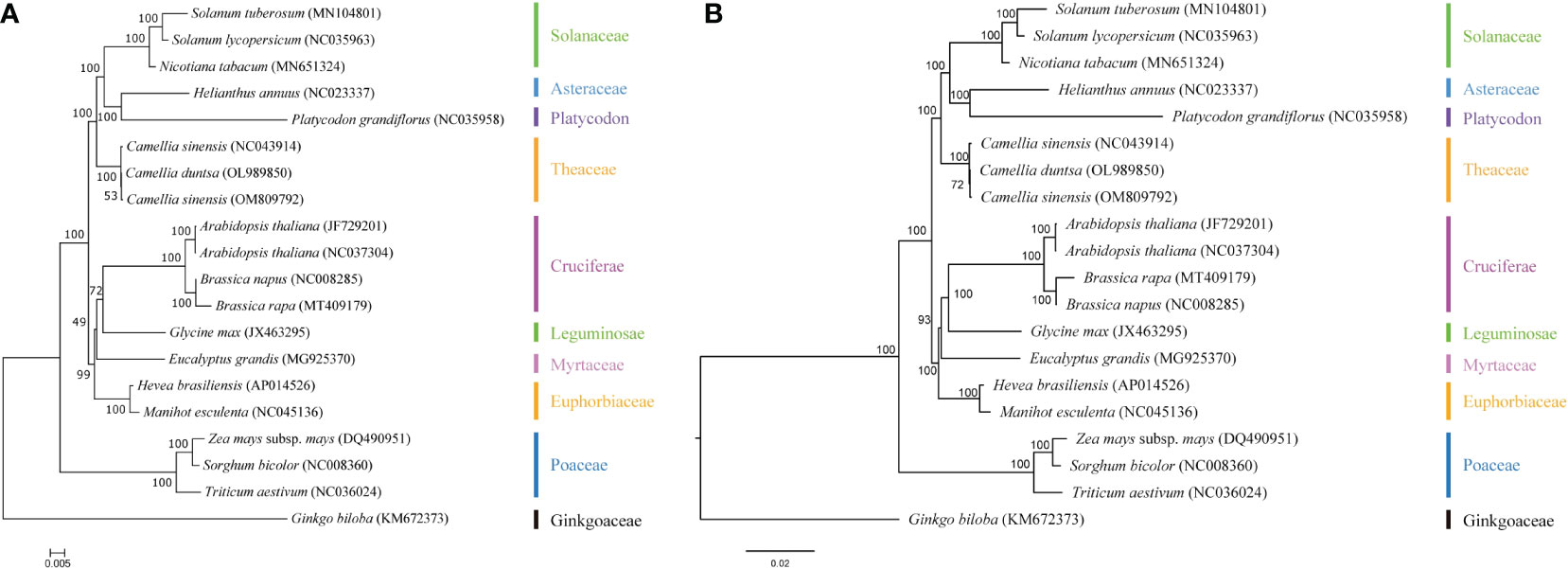
Figure 11 C.duntsa’s nineteen plant phylogenetic connections. Outgroup: Triticum aestivum, Sorghum bicolor, Ginkgo biloba, Zea mays. (A) Maximum-likelihood phylogenies. (B) Bayesian phylogeny.
Using BLAST v2.10.1, we performed homologous fragment analysis on C.duntsa mitochondrial and chloroplast genome segments with > 70% similarity (Supplementary Figure 4). It was determined that the C.duntsa mitochondrial genome sequence (1,081,966 bp) is roughly 6.9 times longer than the chloroplast genome (157,026 bp). eighteen gene-containing homologous segments were discovered between the chloroplast and mitochondrial genomes of C.duntsa (Table 3). These fragments comprised 1.56 and 10.7 percent of the mitochondrial and chloroplast genomes, respectively. The following twelve mitochondrial genes were identified: trnV-GAC,trnP-TGG,trnW-CCA,trnI-GAU,trnA-UGC,trnD-GTC,trnN-GTT,trnM-CAT,trnI-CAU,rrn16,rrn23,rrn18,and rpl2 (Table 3).
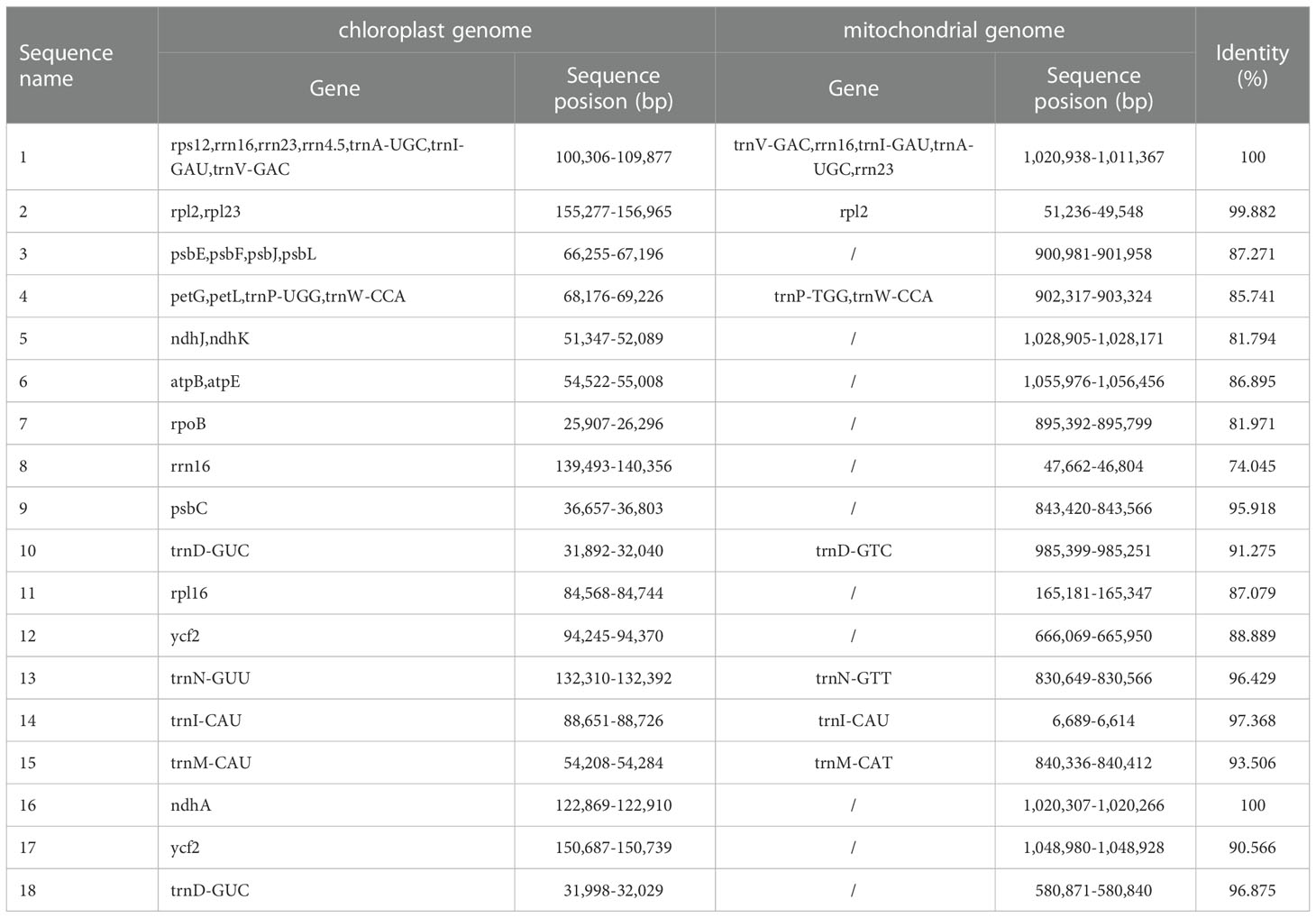
Table 3 Plot of ENC of genes in 4 Theaceae species mitochondrialgenomes.
Mitochondria give plants the energy they need to live. Plant mitochondrial genomes are more complicated than animal ones because they are bigger and have more repeated sequences (Kozik et al., 2019). The key features of the Chinese C.duntsa mitochondrial genome are described in this article. Black tea made from its tender buds is a rare resource with a rich fragrance, mellow taste, and convergence. Many mitochondrial genomes are circular, but some are linear, like those found in Polytomella parva (Zhang et al., 2018). The circular C.duntsa mitochondrial genome that was published here is 1,081,966 bp long, and it has a GC content of 45.62%. The GC content was comparable to that of other sequenced plant mitochondrial genomes, for example, B. chinense, 45.68% (Qiao et al., 2022);B. juncea, 45.24% (Chang et al., 2011).
The mitochondrial genome contains many types of repeats, including tandem, short, and large repeats (Gualberto et al., 2014; Guo et al., 2017). Repeated sequences have been shown to be crucial to intermolecular recombination in previous studies of mitochondria. As a result, repetitive sequences play a crucial role in the assembly of the mitochondrial genome (Dong et al., 2018). SSRs, longer tandem repeats, and non-tandem repeats were the primary targets of this study (Figure 5-6). Numerous repetitive sequences have been discovered in the mitochondrial genome of C.duntsa, which may be evidence of frequent intermolecular recombination that has dynamically altered the structure and conformation of the mitochondrial genome over the course of evolution. We also compared the genome of C.duntsa to those of other terrestrial plants to learn more about its structure and organization. In conclusion, C.duntsa’s mitochondrial genome shares features that are common among other land-dwelling green plants.
Mitochondrial genomes for the four tea plant species ranged in size from 701,719 to 1,081,966bp. From the shape and size of the mitochondrial genome in tea plant species, it might be possible to figure out phylogenetic relationships and the rate at which species change.The size and structure of the mitochondrial genome are very similar in closely related species,such as R. simsii and R. pulchrum (Xu et al., 2021),the mitochondrial genome may be an important tool for understanding plant evolution and defining taxonomic taxonomy.
C.duntsa has forty-seven PCGs, thirty tRNAs, three rRNAs, and one pseudogene in its mitochondrial genome, it is the tea plant species with the longest genome that has been identified to this point.In the mitochondrial genomes of four distinct species of tea plant, researchers discovered several copies of the same gene.For example, genome atp9 has three copies in the mitochondrial genome of C.duntsa and two copies in the mitochondrial genome of C. sinensis var. Assamica. In addition, the mitochondrial genome of the C.duntsa species had a greater number of annotated genes than the mitochondrial genome of the C.sinensis (NC043914) species. These genes included atp8, cox1, cox3, nad7, nad9, rpl16, rpl2, rps19, rps3, rps4, and sdh4.Therefore, plant mitochondrial genomes are vital for future research, as they will reveal information on the evolution of genomes that has not been discovered before.
The results of the mitochondrial phylogenetic trees of three tea plant species and seventeen other plant species were coherent with the taxonomic information linked with those species. These findings illustrate the possibility of employing information acquired from organelle genomes in plant phylogeny studies. (Wu and Ge, 2012).The authors of this study made use of classification trees to show that C. sinensis and C. duntsa are closely related to one another (Figure 11).
The mitochondrial genome of C. duntsa was used as a reference in this study. According to a whole-genome correlation match, three tea plant species share more genomic areas with C.duntsa than interval sections. This demonstrates that segments of the mitochondrial genome containing genes are more conserved throughout tea plant species than are segments containing intervals.
This study found nine conservative genes: nad5, atp9, cox2, rps3, trnA-TGC, trnI-GAT, rrn18, trnV-GAC, and ccmFN. These genes will help researchers understand more about evolution and classify tea plant species (Li et al., 2021). Fourteen LCBs were found in the mitochondrial genomes of C.duntsa and C.sinensis (OM809792). These LCBs vary greatly in size and location (Figure 10).
Moreover, the majority of LCBs contain DNA sequences. Many genes are rearranged between the mitochondrial genome of C.duntsa and C.sinensis (OM809792), their mitochondrial genome has extremely complicated variations that have the potential to result in hybridization and duplication.These findings are consistent with a prior study that found extensive diversity and rearrangement in the plant mitochondrial genome (Xu et al., 2021). Furthermore, the order of genes in the mitochondrial genome of plants varies widely,the structure of mitochondrial DNA is useful for tracing common ancestry among diverse species (Xiao et al., 2017).
RNA editing is a process that occurs following transcription. It occurs in the mitochondrial genomes of higher plants and aids in the folding of proteins. The study of RNA editing sites aids in the comprehension of plant mitochondrial gene expression. Arabidopsis contains 441 RNA editing sites for 36 genes (Unseld et al., 1997), while rice has 491 RNA editing sites for 34 genes(A et al., 2019), according to previous study. This investigation uncovered 536 RNA editing sites within 41 genes.After RNA editing, the water-repelling properties of 43.74 percent of the amino acids did not change, 7.29% became hydrophilic, and 48.22% became hydrophobic. There are consistent results in the mitochondrial genome of B. chinense (Qiao et al., 2022).
Prior research has found that editing at the second codon location accounts for almost half of all RNA editing (Cheng et al., 2021). Consistent with earlier research, 67.93% of editing sites in the C.duntsa mitochondrial genome were found at the second base of the triplet codon. The identification of RNA editing sites gives crucial information for predicting the function of genes containing new codons.
Ka/Ks analysis of mitochondrial genome from C.duntsa, C.sinensis(NC043914), C.sinensis(OM809792), A.thaliana, and G.biloba shows that the PCGs of the C.duntsa mitochondrial gene are generally preserved. However, we also found genes in which the Ka/Ks >1, such as nad2, ccmB, atp4, nad6, atp9, rps7, rps19, rps14, nad3, and nad1. This shows that positive selection has worked on these coding genes over the course of their evolutionary history. In studies of gene selection and evolution in the Theaceae family, high Ka/Ks gene ratios are very important.
Besides, we investigated the amount of GC found in the mitochondrial genomes of C.duntsa and other other green plants. The findings provide support for the theory that the GC content of higher plants maintains a high degree of consistency across time (Cheng et al., 2021).
On animals and insects, phylogenetic and evolutionary analyses based on codon use bias are frequently undertaken, however less studies have been conducted on plants (Sun et al., 2009).The low ENC mitochondrial genes (ENC< 35; Supplementary Tables 4) may be prone to mutation, while the codon usage bias observed in the remaining genes(ENC>35)is most likely due to natural selection or another cause (Wright, 1990). Our research reveals that just one mitochondrial gene (atp9,ENC=33.62) in the Theaceae family has had its codon use bias altered by mutation, whereas the other genes’ codon usage bias was altered by natural or artificial selection. These results suggested that NEC analysis of the mitochondrial genome would be valuable for studying the evolutionary history of plants belonging to the tea plant family.
Sequencing analyses of the nuclear, mitochondrial, and chloroplast genomes have shown that genes can move between different genomes inside a cell (Nguyen et al., 2020).There has been an upsurge in the publication and analysis of data relevant to organelle genomes as a result of the introduction of novel methods (Park et al., 2014). It is thought that plant long-term evolution includes gene transfer between chloroplast and mitochondria (Nguyen et al., 2020).There are eighteen gene-containing homologous regions that are shared between the chloroplast and mitochondrial genomes of C.duntsa (Table 3), which points to the possibility of horizontal transfer of genes among organelle genomes.From the chloroplast, nine tRNA genes were moved to the mitochondria. It is frequent in angiosperm for tRNA genes to be transferred from the genome of the chloroplast to the genome of the mitochondria (Bi et al., 2019).
Although their homologous fragments have been found, twenty-two chloroplast genes in the mitochondrial genome of C.duntsa remain unannotated. These include one tRNA (trnD-GUC), three ribosomal genes (rrn16, rrn16, and rrn4.5), one photosynthetic gene (psbC), and three NADH dehydrogenase gene (ndhJ, ndhK, and ndhA)(Table 3). The ribosome complex, which is partially generated by the ribosomal genes, is responsible for the synthesis of the proteins that are necessary for the correct operation of the cell. During the process of photosynthesis, genes that code for photosynthetic processes and genes that code for NADH dehydrogenase work together to produce the photosynthetic system II and the chloroplast NADH dehydrogenase complex, respectively (Sazanov et al., 1998).It is hypothesized that psbC, a photosynthesis-related gene, evolved in the mitochondria of C.duntsa due to the transfer of chloroplast genes to mitochondria. By studying the genomes of plant chloroplast and mitochondria, additional insights will be acquired into the evolution of phylogenetic analyses and molecular markers of these specially arranged genomes.
This research mapped out the complete mitochondrial genome of C.duntsa. Various aspects of the genome have been investigated, including its organization, genome comparisons with those of closely related species, and horizontal gene transfer between the mitochondrial and chloroplast genomes. Several tea plant plants lack the atp8, cox1, cox3, nad7, nad9, rpl16, rpl2, rps19, rps4, and sdh4 genomes in their mitochondrial genomes. However, C. duntsa maintains the integrity of these genes. Those findings can be utilized in the development of molecular markers.
Phylogenetic relationships, patterns of biased codon usage, and evolutionary history were investigated and studied.Our research identified nine genes associated with conservatism: nad5, atp9, cox2, rps3, trnA-TGC, trnI-GAT, rrn18, trnV-GAC, and ccmFN. These genes will aid scientists in understanding evolution and classifying tea plant species.
The complexity of the organelle genome variation among the tea plant species is demonstrated by this study. The findings would be helpful in understanding the mechanisms of evolution as well as the taxonomy of species within the Camellia family.
The original contributions presented in the study are included in the article/Supplementary material. Further inquiries can be directed to the corresponding author.
JL and LX: design; validation; resources; database gathering; writing; preparation; analysis; and editing of initial drafts. HT and HL: methods. HL, NZ, and JT are all types of software. LX: finance acquisition. All authors contributed to the article and approved the submitted version.
This work was supported by the Modern Tea Industry Technology System of Hunan Province (Xiang Cai Nong Index [2020] No. 65).
The authors thank Hunan Baiyun Lake Ecological Tea Co., LTD for their help in sample collection.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1117002/full#supplementary-material
Adams, K. L., Qiu, Y.-L., Stoutemyer, M., Palmer, J. D. (2002). Punctuated evolution of mitochondrial gene content: High and variable rates of mitochondrial gene loss and transfer to the nucleus during angiosperm evolution. Proc. Natl. Acad. Sci. 99, 9905–9912. doi: 10.1073/pnas.042694899
Archibald, J. M. (2011). Origin of eukaryotic cells: 40 years on. Symbiosis 54, 69–86. doi: 10.1007/s13199-011-0129-z
Bi, C., Paterson, A. H., Wang, X., Xu, Y., Ye, N. (2019). Corrigendum to “Analysis of the complete mitochondrial genome sequence of the diploid cotton gossypium raimondii by comparative genomics approaches”. BioMed. Res. Int. 2019, 1–2. doi: 10.1155/2019/9691253
Birky, C. W. (1995). Uniparental inheritance of mitochondrial and chloroplast genes: mechanisms and evolution. Proc. Natl. Acad. Sci. U. S. A. 92, 11331–11338. doi: 10.1073/pnas.92.25.11331
Brennicke, A., Marchfelder, A., Binder, S. (1999). RNA Editing. FEMS Microbiol. Rev. 23, 297–316. doi: 10.1111/j.1574-6976.1999.tb00401.x
Cavalier-tsmith, T. (1975). The origin of nuclei and of eukaryotic cells. Nature 256, 463–468. doi: 10.1038/256463a0
Chang, S., Yang, T., Du, T., Huang, Y., Chen, J., Yan, J., et al. (2011). Mitochondrial genome sequencing helps show the evolutionary mechanism of mitochondrial genome formation in brassica. BMC Genomics 12, 497. doi: 10.1186/1471-2164-12-497
Chan, P. P., Lowe, T. M. (2019). tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol. Biol. Clifton NJ 1962, 1–14. doi: 10.1007/978-1-4939-9173-0_1
Cheng, Y., He, X., Priyadarshani, S. V. G. N., Wang, Y., Ye, L., Shi, C., et al. (2021). Assembly and comparative analysis of the complete mitochondrial genome of suaeda glauca. BMC Genomics 22, 167. doi: 10.1186/s12864-021-07490-9
Cusimano, N., Wicke, S. (2016). Massive intracellular gene transfer during plastid genome reduction in nongreen orobanchaceae. New Phytol. 210, 680–693. doi: 10.1111/nph.13784
Dong, S., Zhao, C., Chen, F., Liu, Y., Zhang, S., Wu, H., et al. (2018). The complete mitochondrial genome of the early flowering plant nymphaea colorata is highly repetitive with low recombination. BMC Genomics 19, 614. doi: 10.1186/s12864-018-4991-4
Fauron, C., Casper, M., Gao, Y., Moore, B. (1995). The maize mitochondrial genome: dynamic, yet functional. Trends Genet. 11, 228–235. doi: 10.1016/S0168-9525(00)89056-3
Graham, H. N. (1992). Green tea composition, consumption, and polyphenol chemistry. Prev. Med. 21, 334–350. doi: 10.1016/0091-7435(92)90041-f
Guoben, C. (1987). A discussion on the genetic relationship and taxonomic position of tea resources in Hunan. J. Hunan Agri. Univ. 1987 (03), 71–79. doi: 10.13331/j.cnki.jhau.1987.03.013
Gualberto, J. M., Mileshina, D., Wallet, C., Niazi, A. K., Weber-Lotfi, F., Dietrich, A. (2014). The plant mitochondrial genome: dynamics and maintenance. Biochimie 100, 107–120. doi: 10.1016/j.biochi.2013.09.016
Guo, W., Zhu, A., Fan, W., Mower, J. P. (2017). Complete mitochondrial genomes from the ferns ophioglossum californicum and psilotum nudum are highly repetitive with the largest organellar introns. New Phytol. 213, 391–403. doi: 10.1111/nph.14135
Huan, G., Jie, K. (2005). Distribution characteristics and biological function of tandem repeat sequences in the genomes of different organisms. Zool. Res. 26, 555–564. doi: 10.3321/j.issn:0254-5853.2005.05.017
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Kozik, A., Rowan, B. A., Lavelle, D., Berke, L., Schranz, M. E., Michelmore, R. W., et al. (2019). The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PloS Genet. 15, e1008373. doi: 10.1371/journal.pgen.1008373
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Li, J., Lv, Q., Zhang, X.-M., Han, H.-L., Zhang, A.-B. (2021). Characterization and phylogenetic analysis of the complete mitochondrial genome of laelia suffusa (Lepidoptera: Erebidae, lymantriinae). J. Insect Sci. Online 21, 5. doi: 10.1093/jisesa/ieaa138
Malek, O., Lättig, K., Hiesel, R., Brennicke, A., Knoop, V. (1996). RNA Editing in bryophytes and a molecular phylogeny of land plants. EMBO J. 15, 1403–1411. doi: 10.1002/j.1460-2075.1996.tb00482.x
Maréchal, A., Brisson, N. (2010). Recombination and the maintenance of plant organelle genome stability. New Phytol. 186, 299–317. doi: 10.1111/j.1469-8137.2010.03195.x
McCarty, M. F., Assanga, S. I., Lujan, L. L. (2020). Flavones and flavonols may have clinical potential as CK2 inhibitors in cancer therapy. Med. Hypotheses 141, 109723. doi: 10.1016/j.mehy.2020.109723
Mower, J. P., Sloan, D. B., Alverson, A. J. (2012). “Plant mitochondrial genome diversity: The genomics revolution,” in Plant genome diversity volume 1: Plant genomes, their residents, and their evolutionary dynamics. Eds. Wendel, J. F., Greilhuber, J., Dolezel, J., Leitch, I. J. (Vienna: Springer), 123–144. doi: 10.1007/978-3-7091-1130-7_9
Nguyen, V. B., Linh Giang, V. N., Waminal, N. E., Park, H.-S., Kim, N.-H., Jang, W., et al. (2020). Comprehensive comparative analysis of chloroplast genomes from seven panax species and development of an authentication system based on species-unique single nucleotide polymorphism markers. J. Ginseng Res. 44, 135–144. doi: 10.1016/j.jgr.2018.06.003
Park, S., Ruhlman, T. A., Sabir, J. S. M., Mutwakil, M. H. Z., Baeshen, M. N., Sabir, M. J., et al. (2014). Complete sequences of organelle genomes from the medicinal plant rhazya stricta (Apocynaceae) and contrasting patterns of mitochondrial genome evolution across asterids. BMC Genomics 15, 405. doi: 10.1186/1471-2164-15-405
Powell, W., Machray, G. C., Provan, J. (1996). Polymorphism revealed by simple sequence repeats. Trends Plant Sci. 1, 215–222. doi: 10.1016/1360-1385(96)86898-1
Qiao, Y., Zhang, X., Li, Z., Song, Y., Sun, Z. (2022). Assembly and comparative analysis of the complete mitochondrial genome of bupleurum chinense DC. BMC Genomics 23, 664. doi: 10.1186/s12864-022-08892-z
Qiu, L., Yang, C., Tian, B., Yang, J.-B., Liu, A. (2010). Exploiting EST databases for the development and characterization of EST-SSR markers in castor bean (Ricinus communis l.). BMC Plant Biol. 10, 278. doi: 10.1186/1471-2229-10-278
Sazanov, L. A., Burrows, P. A., Nixon, P. J. (1998). The plastid ndh genes code for an NADH-specific dehydrogenase: Isolation of a complex I analogue from pea thylakoid membranes. Proc. Natl. Acad. Sci. 95, 1319–1324. doi: 10.1073/pnas.95.3.1319
Shearman, J. R., Sonthirod, C., Naktang, C., Pootakham, W., Yoocha, T., Sangsrakru, D., et al. (2016). The two chromosomes of the mitochondrial genome of a sugarcane cultivar: assembly and recombination analysis using long PacBio reads. Sci. Rep. 6, 31533. doi: 10.1038/srep31533
Sloan, D. B., Alverson, A. J., Chuckalovcak, J. P., Wu, M., McCauley, D. E., Palmer, J. D., et al. (2012). Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PloS Biol. 10, e1001241. doi: 10.1371/journal.pbio.1001241
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinforma. Oxf. Engl. 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Sun, Z., Wan, D.-G., Murphy, R. W., Ma, L., Zhang, X.-S., Huang, D.-W. (2009). Comparison of base composition and codon usage in insect mitochondrial genomes. Genes Genomics 31, 65–71. doi: 10.1007/BF03191139
Unseld, M., Marienfeld, J. R., Brandt, P., Brennicke, A. (1997). The mitochondrial genome of arabidopsis thaliana contains 57 genes in 366,924 nucleotides. Nat. Genet. 15, 57–61. doi: 10.1038/ng0197-57
Wang, D., Zhang, Y., Zhang, Z., Zhu, J., Yu, J. (2010). KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinf. 8, 77–80. doi: 10.1016/S1672-0229(10)60008-3
Wei, S., Wang, X., Bi, C., Xu, Y., Wu, D., Ye, N. (2016). Assembly and analysis of the complete salix purpurea l. (Salicaceae) mitochondrial genome sequence. SpringerPlus 5, 1894. doi: 10.1186/s40064-016-3521-6
Wright, F. (1990). The “effective number of codons” used in a gene. Gene 87, 23–29. doi: 10.1016/0378-1119(90)90491-9
Wu, Z.-Q., Ge, S. (2012). The phylogeny of the BEP clade in grasses revisited: Evidence from the whole-genome sequences of chloroplasts. Mol. Phylogenet. Evol. 62, 573–578. doi: 10.1016/j.ympev.2011.10.019
Xiao, L., Xu, X., Zhang, F., Wang, M., Xu, Y., Tang, D., et al. (2017). The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 11, 297–311. doi: 10.1016/j.redox.2016.12.022
Xinghui, L., Gesheng, X., Xingyan, C., Benqing, S., Dehua, L., Junwu, L. (1998). Evaluation of tea plant resources in Hunan [J]. Guangxi Plants 1998 (02), 14–19.
Xu, J., Luo, H., Nie, S., Zhang, R.-G., Mao, J.-F. (2021). The complete mitochondrial and plastid genomes of rhododendron simsii, an important parent of widely cultivated azaleas. Mitochondrial DNA Part B Resour. 6, 1197–1199. doi: 10.1080/23802359.2021.1903352
Zhang, L., Ho, C.-T., Zhou, J., Santos, J. S., Armstrong, L., Granato, D. (2019). Chemistry and biological activities of processed camellia sinensis teas: A comprehensive review. Compr. Rev. Food Sci. Food Saf. 18, 1474–1495. doi: 10.1111/1541-4337.12479
Zhang, W., Li, L., Li, G. (2018). Characterization of the complete chloroplast genome of shrubby sophora (Sophora flavescens ait.). Mitochondrial DNA Part B Resour. 3, 1282–1283. doi: 10.1080/23802359.2018.1532839
Keywords: Camellia sinensis var. Assamica cv. Duntsa, horizontal transfer, repeat sequence, genome size variation, mitochondrial genome, evolution, phylogenetic
Citation: Li J, Tang H, Luo H, Tang J, Zhong N and Xiao L (2023) Complete mitochondrial genome assembly and comparison of Camellia sinensis var. Assamica cv. Duntsa. Front. Plant Sci. 14:1117002. doi: 10.3389/fpls.2023.1117002
Received: 06 December 2022; Accepted: 04 January 2023;
Published: 19 January 2023.
Edited by:
Pankaj Bhatt, Purdue University, United StatesReviewed by:
Zefa Liu, Hunan University of Humanities, Science and Technology, ChinaCopyright © 2023 Li, Tang, Luo, Tang, Zhong and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lizheng Xiao, ZW1haWxAdW5pLmVkdQ==; MTM2OTk0OTA1NkBxcS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.