 Scott D. Anderson
Scott D. Anderson Cynthia A. Gleason
Cynthia A. Gleason
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 22 February 2023
Sec. Plant Pathogen Interactions
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1096239
This article is part of the Research TopicDetection, characterization, and management of plant pathogensView all 29 articles
Root-knot nematodes (Meloidogyne spp.) are major pests of many important crops around the world. In the Northwestern region of the United States of America (USA), Meloidogyne chitwoodi causes economic losses in potatoes because the nematodes can infect the tubers, which leads to potato galling and reductions in marketable yield. Meloidogyne chitwoodi is a quarantine pathogen in certain potato export markets, and there is little industry tolerance for the presence of this nematode. Recently, two Meloidogyne species that are not known to be present in agricultural fields in the USA were detected on golf turfgrasses in California and Washington. These species, M. fallax and M. minor, are morphologically similar to M. chitwoodi and can infect potatoes and cause tuber damage. Their detection in the USA means that they could potentially infest potato fields and become a problem in potato production. Additionally, M. fallax is a regulated plant pest in the USA, which makes the correct identification of potato-infecting root-knot nematodes important. Previously, there was no single-tube assay that could determine whether M. chitwoodi, M. fallax, and/or M. minor were present in a sample. Thus, a molecular beacon real-time PCR assay which can reliably detect M. chitwoodi, M. fallax, or M. minor from crude nematode extracts was designed and characterized.
Root-knot nematodes are obligate sedentary endoparasites of many important agricultural crops, causing upwards of $180 billion in global crop losses annually (Sasser and Freckman, 1987; Koenning et al., 1999). Root-knot nematodes are especially problematic in potato because the nematodes cause tuber galling and internal tuber defects (Lima et al., 2018; Bali et al., 2021a). In 2019, the United States had the 5th highest production of potatoes (FAO, 2017), with three states, Oregon, Washington, and Idaho, comprising over half of the country’s entire production (Zasada et al., 2018). These states have the root-knot nematodes M. chitwoodi and M. hapla. However, M. chitwoodi is a larger threat in potato production because it hatches at lower temperatures, which allows its populations to expand rapidly in a single growing season. Meloidogyne chitwoodi also causes more visible galling on potatoes compared to M. hapla (Pinkerton et al., 1991; Ingham et al., 2000). The galling causes the tuber surface to look bumpy, and necrotic dark spots form in the tuber flesh around the M. chitwoodi females. These visual defects can significantly decrease the potato market value due to the near zero tolerance to tuber blemishes in the processing market. Although M. chitwoodi is endemic to the western region of the United States, its regulated status means there is a zero- tolerance policy in potatoes destined for export to several world markets (Ingham et al., 2007). There is currently no genetic resistance against root-knot nematodes in commercial potato cultivars.
In Europe, root-knot nematodes that are commonly found in cooler climates include M. hapla, M. naasi, M. chitwoodi and M. fallax (Wesemael et al., 2011), and both M. chitwoodi and M. fallax are root-knot nematode species on the A2 EPPO alert list (Wesemael et al., 2011; Viaene, 2014; European and Mediterranean Plant Protection Organization [EPPO], 2022). Meloidogyne fallax was originally identified as M. chitwoodi, but subsequent morphological and biochemical investigations revealed that it was a distinct species (Karssen, 1996). Meloidogyne fallax and M. chitwoodi are difficult to distinguish morphologically. They also have some of the same hosts, such as potatoes and carrots, but they differ in their ability to infect some crops, such as corn (Karssen, 1996). Both M. chitwoodi and M. fallax can cause significant damage to potato tubers, and some data indicate that M. fallax is more aggressive on potato than M. chitwoodi (Van Meggelen et al., 1994; Van Der Beek et al., 1998; Suffert and Giltrap, 2012). Although M. fallax has not been reported on potato in the USA, it was detected during a survey of golf course greens in California (Nischwitz et al., 2013). Follow up surveys by APHIS in California did not detect M. fallax, so it is considered “not present” in the USA (Kantor et al., 2022), but there remains a threat of introduction.
In 2000 a new species of root-knot nematode called M. minor was found on heavily infected potato plants from a potato field in Zeijerveld, The Netherlands (Karssen et al., 2004). The potato tubers exhibited pimple-like galling similar to the symptoms caused by M. chitwoodi and M. fallax. Meloidogyne minor was subsequently found on golf courses in the Netherlands, Belgium, United Kingdom, and Ireland (Vandenbossche et al., 2011; Morris et al., 2013). Interestingly, M. minor infections did not significantly impact tuber yield or quality in two potato cultivars (cvs Astérix and Markies) in field plots in the Netherlands (Thoden et al., 2012). However, significant tuber damage was inflicted by M. minor on potatoes grown in greenhouses (Thoden et al., 2012), indicating that M. minor biology and pathogenicity at relatively warmer temperatures, such as those in greenhouses, are not fully understood. There is also limited information about M. minor infectivity on potato cultivars commonly used in the USA, such as the Russets. Recently M. minor was reported on turf grass in the USA (McClure et al., 2012; Nischwitz et al., 2013). It is possible that this nematode could be accidentally transmitted from golf courses to arable land via contaminated sports shoes or equipment (Morris et al., 2011; Morris et al., 2013). With the potential for M. minor to spread and cause damage to potatoes, it is prudent to monitor for M. minor to help mitigate the risk it poses to USA agriculture.
To ensure proper monitoring of these Meloidogyne species, a rapid and reliable molecular test to identify these three species is necessary. Many molecular techniques have been established for identifying Meloidogyne species (Baum et al., 1994; Vrain and Petersen, 1996; Blok et al., 1997; Petersen et al., 1997; Williamson et al., 1997; Zijlstra, 1997; Castagnone-Sereno et al., 1999; Castagnone-Sereno, 2000; Zeng et al., 2015; Zhang and Gleason, 2019). These molecular techniques were often based on PCR using species specific primers that target the mitochondrial DNA, or they target the intergenic region or the internal transcribed spacer (ITS) regions of the ribosomal DNA (Giorgi et al., 1994; Hadziavdic et al., 2014; Harris et al., 1990; Van Megen et al., 2009). The technique called PCR-RFLP, which is PCR followed by restriction digestion of the amplicons to create unique restriction fragment lengths, can be used to identify several Meloidogyne species (Harris et al., 1990; Vrain et al., 1992; Powers and Harris, 1993; Zijlstra et al., 1995; Powers et al., 1997; Orui, 1998; Waeyenberge et al., 2000; Han et al., 2004; Gamel et al., 2014). The technique was found to be sensitive enough to detect a single juvenile of M. fallax or M. chitwoodi, but PCR-RFLP results can be difficult to interpret when there are mixtures of species in the reaction (Gamel et al., 2014). Zijlstra (2000) showed that primers designed to amplify sequence-characterized amplified regions (SCARs) could be used in SCAR-PCR to identify M. hapla, M. chitwoodi and M. fallax. The drawback to the SCAR-PCR is that it is not as sensitive as PCR-RFLP, and it requires at least two juveniles of M. chitwoodi or one juvenile of M. fallax as template (Zijlstra, 2000). Moreover, the previously designed SCAR-PCR primers for M. fallax were shown to cross-react with M. minor allowing for false-positives (Nischwitz et al., 2013). A multiplex real-time PCR (TaqMan) using primers designed for the ITS region was developed for the simultaneous detection of M. chitwoodi and M. fallax (Zijlstra and Van Hoof, 2006). The TaqMan PCR was a breakthrough in root-knot nematode identification in multiplexed reactions, but it did not include a third nematode of interest, M. minor (Zijlstra and Van Hoof, 2006; Wesemael et al., 2014).
There are no molecular techniques that can identify and distinguish M. chitwoodi, M. fallax, and M. minor in a one tube assay. With this in mind, we have developed a new RT-PCR assay using molecular beacons to identify and distinguish M. chitwoodi, M. fallax, and M. minor in a single reaction. This assay can be used by regulatory agencies and diagnosticians to monitor for the presence of these potato-infecting nematodes, two of which are of regulatory importance, using as little as a single juvenile isolated from samples.
Primers and beacons were designed using Beacon Designer 8 (Premier Biosoft, Palo Alto, CA). Alignments of HSP90 gene sequences (Table 1) were used to find universally conserved primer sequences surrounding polymorphic regions in M. chitwoodi, M. fallax, and M. minor (Skantar and Carta, 2004). To ensure the beacon probes and primers were specific to their target organisms, the sequences were queried using the BLASTn search program and the non-redundant database (National Center for Biotechnology Information, NCBI). Primers and the beacons for the HSP90 region were synthesized by Sigma-Aldrich (St. Louis, Mo) and are described in Table 2. The M. chitwoodi beacon has 6-FAM, M. fallax beacon has HEX, and M. minor beacon has Cyan-5 as the reporters. For the quenchers, the Black Hole Quencher (BHQ-1 for 6-FAM and HEX, and BHQ- 3 for Cyan-5) was used.
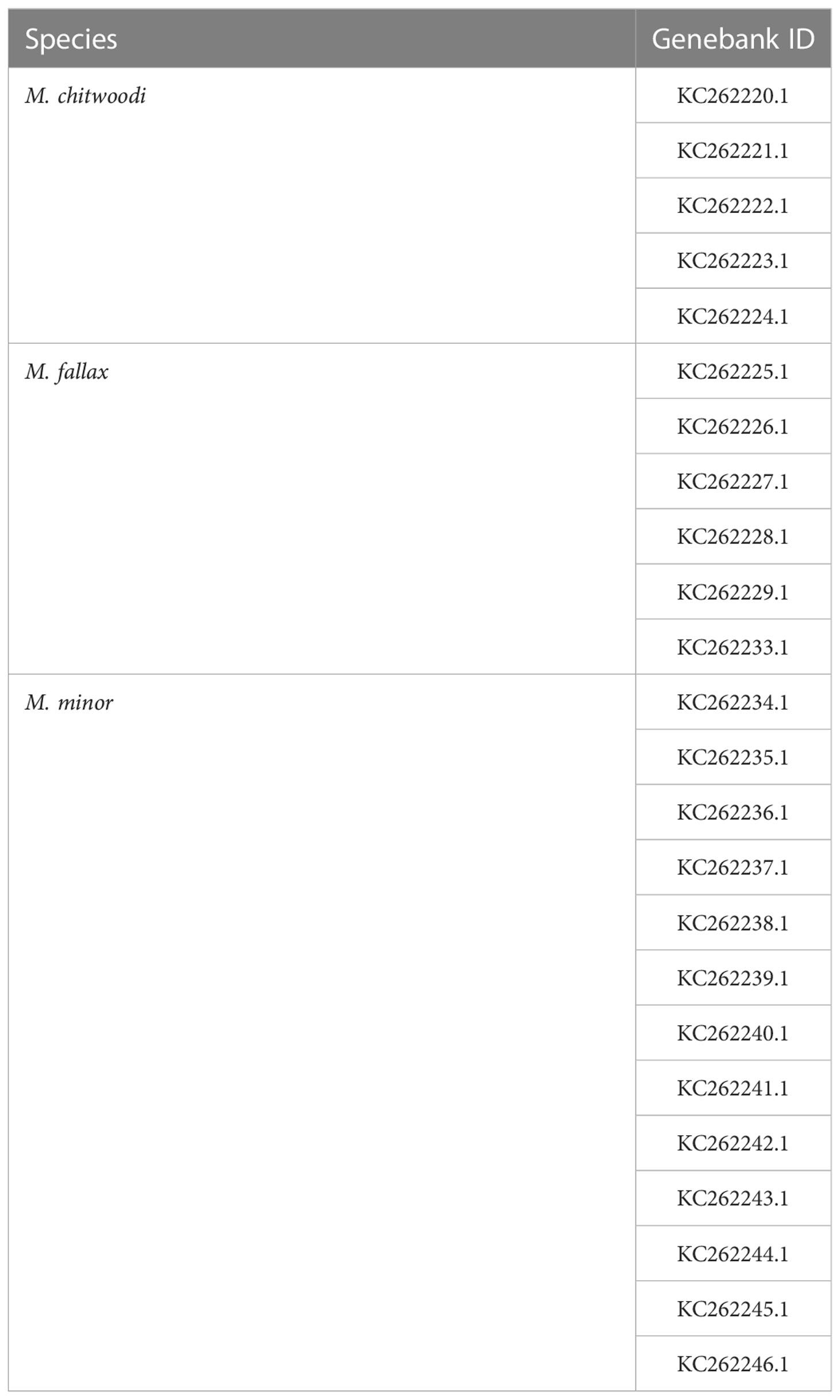
Table 1 Meloidogyne spp. HSP90 regions used for beacon designs.
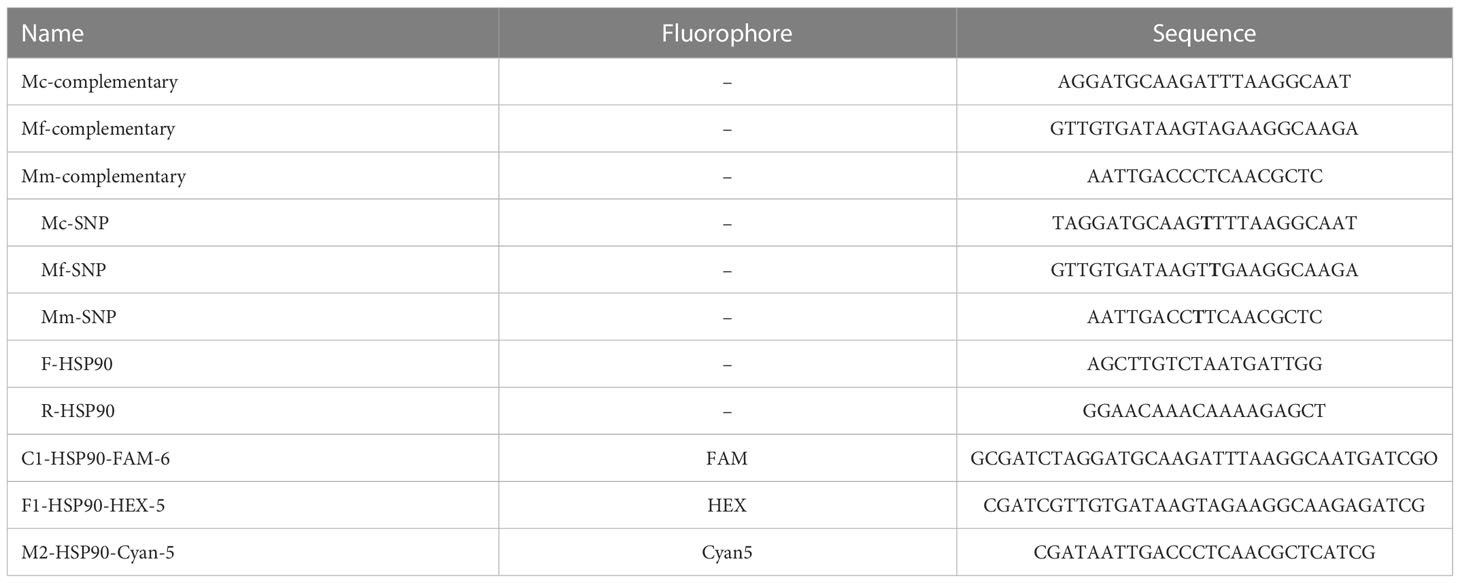
Table 2 Primers and beacon probes used in the molecular beacon RT-PCR assays.
Three synthetic oligos complementary to each beacon’s loop sequence and three off-target synthetic oligos containing a single nucleotide difference to the loop sequence were synthesized by Sigma-Aldrich (Table 2) and used for creating thermal denaturation curves. For each beacon, two 50 µL reactions containing 200 nM molecular beacon probe in 3 mM MgCl2 and 10 mM Tris- HCl, pH = 8.0 were prepared with 600 nM of either the complementary or off-target synthetic oligo. A third reaction for each beacon was run containing Milli-Q water in place of the synthetic oligo as a control. Reactions were prepared in a 96-well clear qPCR plate and run on a CFX96 Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA) starting at 80°C and decreasing to 30°C at a rate of 1°C/minute, with fluorescence measured every minute.
For genomic DNA templates, the DNA was extracted from M. chitwoodi (Race 1, Race 2, and Roza) (Bali et al., 2021b; Mojtahedi et al., 2007), M. incognita, M. javanica, M. arenaria, and M. hapla eggs using a phenol/chloroform extraction protocol as described by Gross and Williamson (2011). All three M. chitwoodi isolates (Race 1, Race 2, and Race 1 Roza) were originally provided by Dr. Charles Brown (USDA-ARS). The M. hapla isolate VW9, M. incognita isolate VW6, M. javanica isolate VW4, and M. arenaria isolate HarA were provided by Dr. Valerie Williamson (Table 3, UC-Davis). All Meloidogyne species were maintained on the susceptible tomato Solanum lycopersicum cv. Rutgers under greenhouse conditions. The identities of the M. chitwoodi, M. hapla, M. incognita, M. javanica and M. arenaria populations were confirmed using species-specific PCR (Powers and Harris, 1993; Zijlstra, 2000; Wishart et al., 2002).
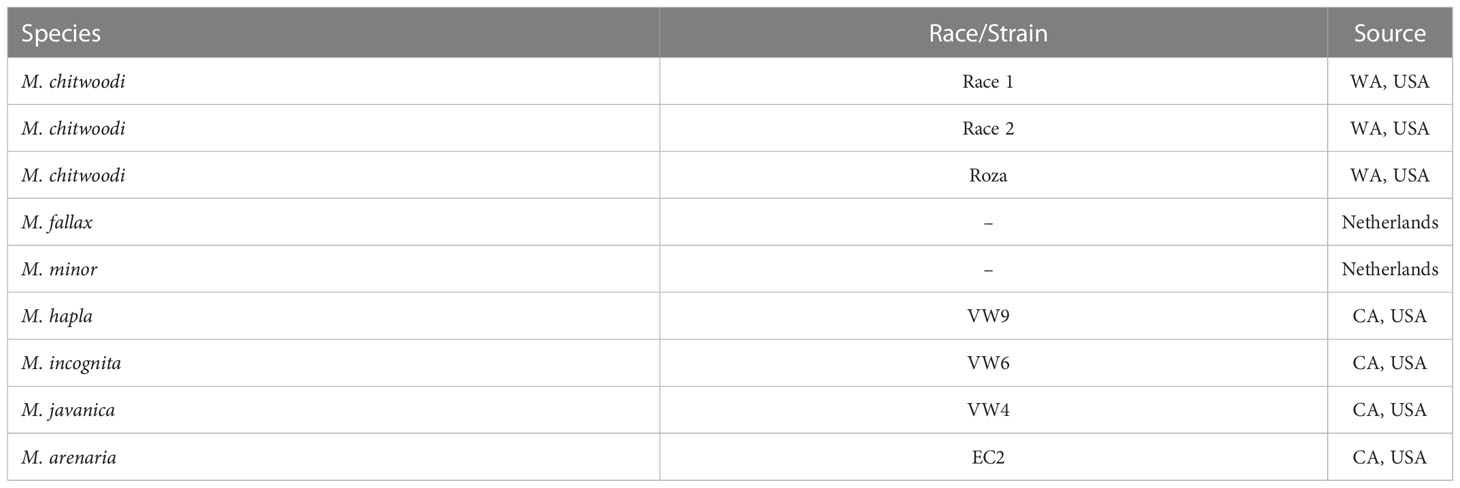
Table 3 Meloidogyne spp. used for testing molecular beacon RT-PCR specificity.
To amplify and clone the HSP90 amplicon from M. chitwoodi race 1, M. fallax and M. minor, F- HSP90 and R-HSP90 primers were used in a PCR with approximately 15 ng of M. chitwoodi, M. fallax, or M. minor DNA. Reactions were 50 µL total volume and contained 1x AmpliTaq Gold 360 buffer, 3 mM MgCl2, 200 µM dNTPs, 200 nM F-HSP90 and R-HSP90 primers, and 1.25 U of AmpliTaq Gold. The reactions were run on an Eppendorf Mastercycler Pro Thermal Cycler as follows: 94°C for 10 minutes, then 30 cycles of 94°C for 15 seconds, then 58°C for 30 seconds, and 72°C for 30 seconds, and then finally 72°C for 5 minutes. Reactions were pooled and cleaned up using a ThermoFisher GeneJET PCR purification kit (Thermo Fisher, Waltham, MA) and the purified PCR products were ligated into pGEM-T vector. The 10 µL ligation reaction contained 1x Rapid Ligation buffer, 3 Weiss Units of T4 ligase, purified PCR product, pGEM-T vector (Promega, Madison, WI), and Milli-Q water. Plasmids were cloned into TOP10 E. coli and extracted from the overnight bacterial cultures using a GenElute Plasmid Miniprep kit (Sigma-Aldrich, St. Louis, MO). The HSP90 amplicons from M. chitwoodi, M. minor, and M. fallax in the pGEM-T vector will be referred to as plasmid HSP90.
A standard curve using M. chitwoodi gDNA as template was prepared using 1:10 serial dilutions. Reactions were run at a volume of 50 µL on a CFX96 Real-Time PCR Detection System in a 96-well clear qPCR plate. Reaction mixes contained 1x AmpliTaq Gold 360 buffer, 3 mM MgCl2, 200 µM dNTPs, 200 nM F-HSP90 and R-HSP90 primers, 200 nM target beacon (C1-Hsp90-FAM-6) and 1.25 U of AmpliTaq Gold. The cycling conditions were as follows: 95°C for 10 minutes, then 55 cycles of 95°C for 15 seconds (sec), 54°C for 30 sec, and 72°C for 15 sec. The increase in fluorescent signal was registered during the annealing step of the reaction. The M. chitwoodi standard curve used 5 points, with template DNA concentrations ranging from 40 ng to 4 pg. Additional standard curves were made for M. chitwoodi, M. fallax (F1-Hsp90-HEX-5 beacon), and M. minor (M2-Hsp90-Cyan-5 beacon) using the same reaction conditions, but in which the plasmid HSP90 templates for the reactions were serial dilutions of 0.4 ng – 4 fg, 2 ng – 2 fg, and 0.4 ng – 0.04 fg respectively. Each concentration was run in triplicate. Non-template controls containing only water were included for all standard curves, and each curve was repeated with similar results. To calculate the PCR efficiency, the following equation was used
The second stage juveniles (J2s) of M. chitwoodi race 1 were hatched from eggs collected from Rutgers tomatoes grown in greenhouses at Washington State University. Meloidogyne fallax and M. minor J2s were provided by the Wageningen Nematode Collection (National Plant Protection Organization, the Netherlands) and stored in DESS at -20°C. Second stage juveniles were digested using the protocol described in Qiu et al. (2006). In brief, J2s were picked under a dissecting scope and transferred to 15 µL droplets of Milli-Q water. The nematodes were crushed using a pipet tip; 10 µL of the crushed nematode was transferred to a 10 µL solution containing 2 µL AmpliTaq Gold 360 buffer, 2 uL of 600 µg/mL Proteinase K, and 6 µL Milli-Q water. The reaction mixes were incubated at -20°C for 20 minutes, heated for 1 hour at 65°C, and then 95°C for 10 min before cooling to room temperature. The nematode samples were spun at 12,000 rpm for 2 min before storing at -20°C until used in PCR. Five µL of these J2 samples were used as template for molecular beacon RT-PCR, and each sample was run in duplicate or triplicate. The 50 µL reaction mixes contained 1x AmpliTaq Gold 360 buffer, 3 mM MgCl2, 200 µM dNTPs, 200 nM F-HSP90 and R-HSP90 primers, 200 nM target beacon (C1-Hsp90-FAM-6, F1-Hsp90-HEX-5, or M2-Hsp90-Cyan-5) and 1.25 U of AmpliTaq Gold. The reaction conditions were the same as those used for the standard curves. All molecular beacon RT-PCR assays with these three species were repeated at least twice with similar results.
To ensure the specificity of this assay 40 ng of gDNA from M. incognita, M. javanica, M. arenaria, or M. hapla was used as template for molecular beacon RT-PCR assay using C1-Hsp90-FAM-6, F1-Hsp90-HEX-5, and M2-Hsp90-Cyan-5 beacons. Non-template controls containing only Milli- Q water were included as well as positive controls using 4 pg of plasmid HSP90 for M. chitwoodi,
M. fallax, or M. minor. The 50 µL reaction mixes contained 1x AmpliTaq Gold 360 buffer, 3 mM MgCl2, 200 µM dNTPs, 200 nM F-HSP90 and R-HSP90 primers, 200 nM target beacon (C1- Hsp90-FAM-6, F1-Hsp90-HEX-5, or M2-Hsp90-Cyan-5) and 1.25 U of AmpliTaq Gold. The reaction conditions were the same as those used for the standard curves. The molecular beacon RT-PCR assays with non-target species were repeated twice with similar results.
All PCR products in this paper were visualized as follows: 10 µL of each PCR product was loaded onto a 1.5% agarose gel and separated for 45 min at 100 V before visualizing with ethidium bromide under UV light. The Invitrogen 1-kb plus DNA ladder was used as reference for size.
Initial multiplex molecular beacon RT-PCR assays were performed using equal template concentrations of each species. Each reaction tube contained the same amount of M. chitwoodi, M. minor and M. fallax template HSP90 plasmid DNA (20 pg of each species). To the template DNAs, the reaction mix was added. The reaction mix contained 1x AmpliTaq Gold 360 buffer, 3 mM MgCl2, 200 µM dNTPs, 200 nM F-HSP90 and R-HSP90 primers, 200 nM of each of the three target beacons (C1-Hsp90-FAM-6, F1-Hsp90-HEX-5, and M2-Hsp90-Cyan-5) and 1.25 U of AmpliTaq Gold polymerase. The amount of fluorescence for each reaction at different template concentrations with the three different probes was measured at each cycle on the CFX96 Real- Time PCR Detection System.
Varying ratios of plasmid HSP90 templates were used to test the validity of multiplexing. One template was kept at 20 pg while the other two were loaded ten times more concentrated at 200 pg each. Reaction mixes contained 1x AmpliTaq Gold 360 buffer, 3 mM MgCl2, 200 µM dNTPs, 200 nM F-HSP90 and R-HSP90 primers, 200 nM of each beacon (C1-Hsp90-FAM-6, F1-Hsp90-HEX-5, M2-Hsp90-Cyan-5) and 1.25 U of AmpliTaq Gold. The reaction conditions were the same as those used for the standard curves.
Multiplex with proteinase K digested J2s were tested with 1 J2 or 5 J2 of each species present in the reaction mix. A single J2 from M. chitwoodi, M. fallax, and M. minor were picked under a dissecting microscope and placed into a single 15 µL droplet of Milli-Q water and digested as described above and used as template for qPCR. Similarly, for the 5 J2 multiplexing, 5 J2s from M. chitwoodi, M. fallax, and M. minor were picked under a dissecting microscope and placed into a single 15 µL droplet of Milli-Q water and digested and used as template for qPCR. Reaction mixes contained 1x AmpliTaq Gold 360 buffer, 3 mM MgCl2, 200 µM dNTPs, 200 nM F-HSP90 and R-HSP90 primers, 200 nM of each beacon (C1-Hsp90-FAM-6, F1-Hsp90-HEX-5, M2- Hsp90-Cyan-5) and 1.25 U of AmpliTaq Gold. The reaction conditions were the same as those used for the standard curves.
All molecular beacon RT-PCR multiplex samples were run in duplicate, or triplicate and the assays were repeated at least once with similar results.
The molecular beacon probes were based on the heat shock protein 90 (HSP90) sequence information in Genbank for M. chitwoodi, M. fallax, and M. minor (Table 1). This gene was chosen because the sequence is polymorphic between the three species, and the gene had been previously used in a PCR-based nematode identification assay (Nischwitz et al., 2013; Skantar and Carta, 2004). Primers were designed that were specific for the conserved HSP90 sequence in M. chitwoodi, M. fallax, and M. minor, but span the polymorphic regions between these species within the gene (Figure 1). The HSP90 gene was amplified using the genomic DNA (gDNA) of M. chitwoodi race 1, M. fallax, and M. minor as the PCR templates. Sequencing the amplicons confirmed that the predicted polymorphic regions were present in the HSP90 sequence of each species.
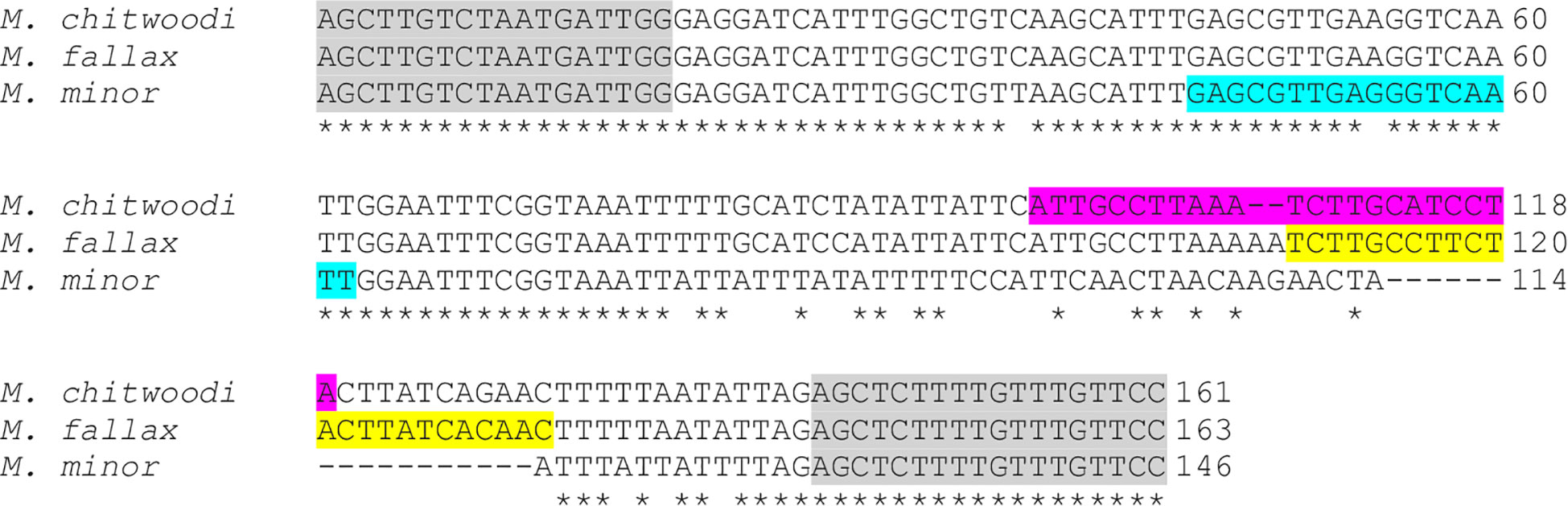
Figure 1 Alignment of partial HSP90 gene sequences for M. chitwoodi, M. fallax, and M. minor. Universal primer sequences are highlighted in gray. Variable regions targeted by the beacons are highlighted in teal for M. minor, magenta for M. chitwoodi, and yellow for M. fallax. Asterisks indicate conserved regions between all three species, gaps indicate missing nucleotides according to the alignment.
Molecular beacons were designed to target the polymorphic regions in each species, with the beacons hybridizing to the complementary sequences shown in Table 2. To determine the optimal annealing temperature for each molecular beacon probe, a melting curve analysis was performed using oligonucleotides complementary to the molecular beacon probe sequence and oligonucleotides with a single nucleotide substitution (Table 2, Figure 2). The optimal annealing temperature for M. chitwoodi, M. fallax, and M. minor molecular beacon probes was 54°C.
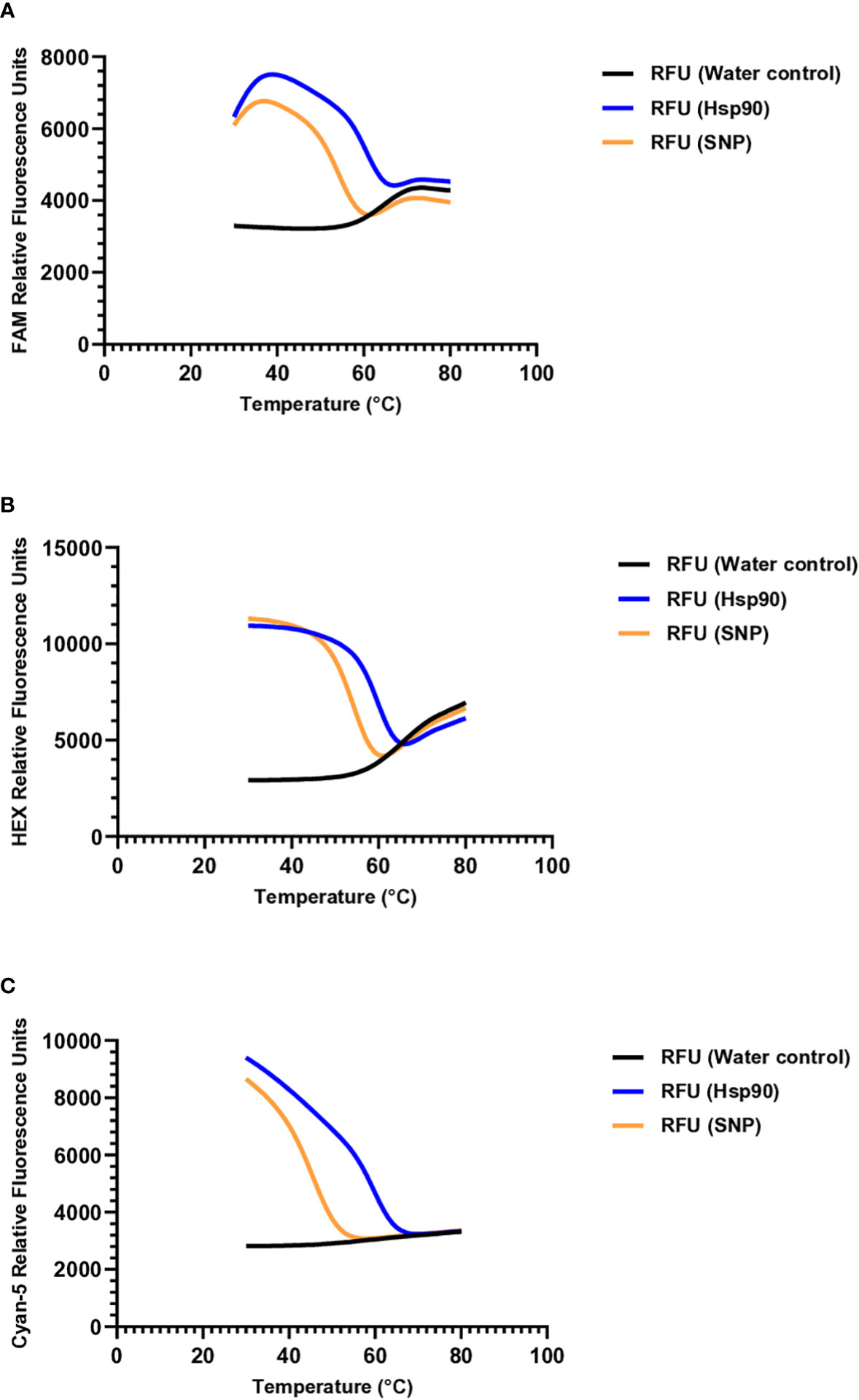
Figure 2 Denaturation curves of molecular beacon probes (A) M. chitwoodi beacon C1-Hsp90- FAM-6 in the presence of excess complementary template, SNP template, or no template (B) M. fallax beacon F1-Hsp90-HEX-5 in the presence of excess complementary template, SNP template, or no template. (C) M. minor beacon M2-Hsp90-Cyan-5 in the presence of excess complementary template, SNP template, or no template.
To measure the amplification efficiency of the molecular beacon RT-PCRs, the cycle threshold (Ct) values were obtained over a range of template concentrations for all three nematode species. This information was used to generate standard curves at an annealing temperature of 54°C. First, M. chitwoodi race 1 genomic DNA was used as the PCR template with DNA concentrations ranging from 40 ng to 4 pg. When using M. chitwoodi race 1 DNA template, the molecular beacon RT-PCR assay had an efficiency of 89% and an R2 = 0.9984 (Figure 3A). The commonly occurring M. chitwoodi Race 1 (Brown et al., 2009) was used as the template for subsequent PCRs and molecular beacon RT-PCR assays (Figure 3A). DNA from Race 2 and the pathotype Race 1 Roza of M. chitwoodi generated similar standard curves (Figure S1), indicating that the molecular beacon RT-PCR assay could detect M. chitwoodi, including isolates of the species common in Washington (Bali et al., 2021a).
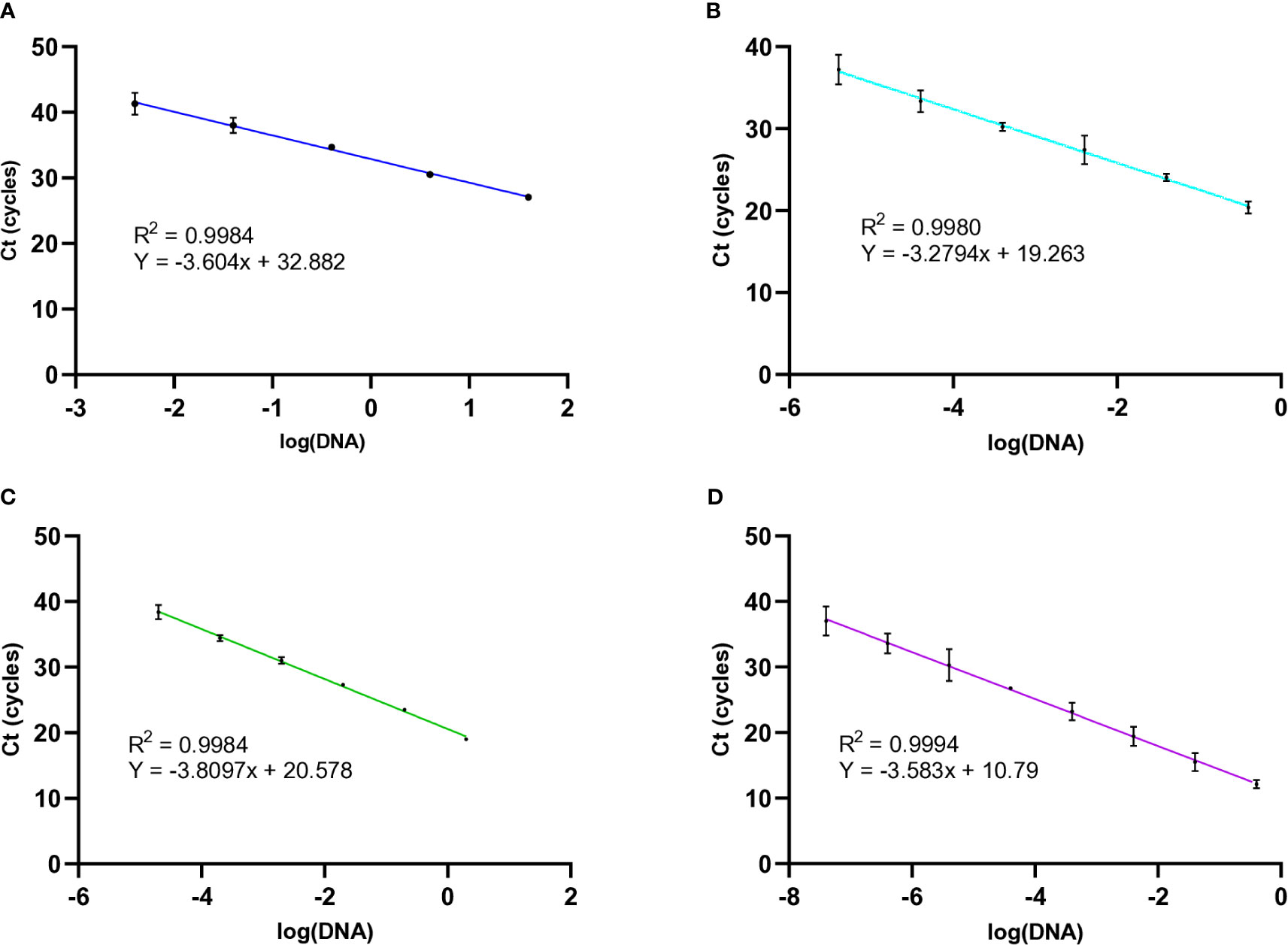
Figure 3 Standard curves of known concentrations of templates from M. chitwoodi, M, fallax, and M. minor with 95% confidence intervals (A) The relationship between Ct values and the natural log of M. chitwoodi race 1 gDNA from 40 ng to 4 pg (n = 3). (B) M. chitwoodi HSP90 plasmid DNA template from 2 ng to 20 fg (n = 3). (C) M. fallax HSP90 plasmid DNA template from 0.2 ng to 20 fg (n = 2). (D) M. minor HSP90 plasmid DNA template from 0.4 ng to 0.04 fg (n = 3).
Next, a fragment of the M. chitwoodi HSP90 gene was amplified using the F-HSP90 and R-HSP90 primers. This amplicon was cloned to generate the “M. chitwoodi HSP90 plasmid,” which was used as the PCR template for further reactions. The efficiency of the molecular beacon RT-PCR using the M. chitwoodi HSP90 plasmid as the PCR template was compared to using genomic DNA as the reaction template. By calculating the slope of the standard curve, the PCR efficiency for M. chitwoodi HSP90 plasmid was 101% with an R2 = 0.9980 (Figure 3B). This was better, but comparable to the reaction efficiencies using M. chitwoodi genomic DNA as the reaction template (efficiency of 89% and an R2 = 0.9984).
Cloned HSP90 fragments from M. fallax and M. minor were also used as template for the molecular beacon RT-PCR assays. The M. minor molecular beacon RT-PCR had an efficiency of 90% and an R2 = 0.9994 (Figure 3D), and the M. fallax molecular beacon RT-PCR had an efficiency of 83% with an R2 = 0.9984 (Figure 3C).
The M. chitwoodi molecular beacon probe could detect the M. chitwoodi amplicon, but not the amplicons from M. fallax and M. minor in a molecular beacon RT-PCR (Figure S2A). The same was true for the other molecular beacon probes; the M. fallax probe could only hybridize to the M. fallax amplicon (Figure S2B) and the M. minor probe could only hybridize to the M. minor amplicon (Figure S2C).
Meloidogyne hapla, M. incognita, M. javanica, and M. arenaria are the four major species of root- knot nematodes found worldwide. When the HSP90 sequences of M. hapla, M. incognita, M. javanica, or M. arenaria were aligned with the HSP90 sequences from M. chitwoodi, M. fallax and M. minor, the probes were specific to their respective nematode (Figure S3). When qPCR was run using gDNA from M. hapla, M. incognita, M. javanica, or M. arenaria none of the molecular beacon probes produced fluorescence (Figure S4), indicating that there is no off-target binding to these other root-knot nematode species.
To validate whether the molecular beacon RT-PCR assays could detect M. chitwoodi, M. fallax, or M. minor using DNA isolated directly from juveniles, crude extracts from 1 or 5 J2s of each species was used as the template for molecular beacon RT-PCR. The M. minor assay was the most sensitive with an average Ct of 35.7 (standard error of means (SEM) = 0.22, n = 7) for a single J2, and an average Ct of 32.7 for 5 J2s (SEM = 0.46, n = 2) (Figure 4C). The M. fallax assay was the least sensitive, with a single J2 sample having an average Ct of 43.6 (SEM = 1.16, n = 8) and 5 J2s having an average Ct of 39.8 (SEM = 1.04, n = 2) (Figure 4B). Finally, the molecular beacon RT- PCR for M. chitwoodi produced an average Ct for a single J2 of 38.9 (SEM = 0.64, n = 10) and a Ct of 34.7 for 5 J2s (SEM = 0.83, n = 2) (Figure 4A). All J2s digests were positive for all three species satisfying the 95% positive replicate criterion suggested by Forootan et al. (2017) for qPCR limit of detection. In the negative controls, no significant fluorescence was observed. These results demonstrate the sensitivity of the molecular beacon probes and their ability to detect the presence of M. chitwoodi, M. fallax, or M. minor from the crude extract of a single juvenile.
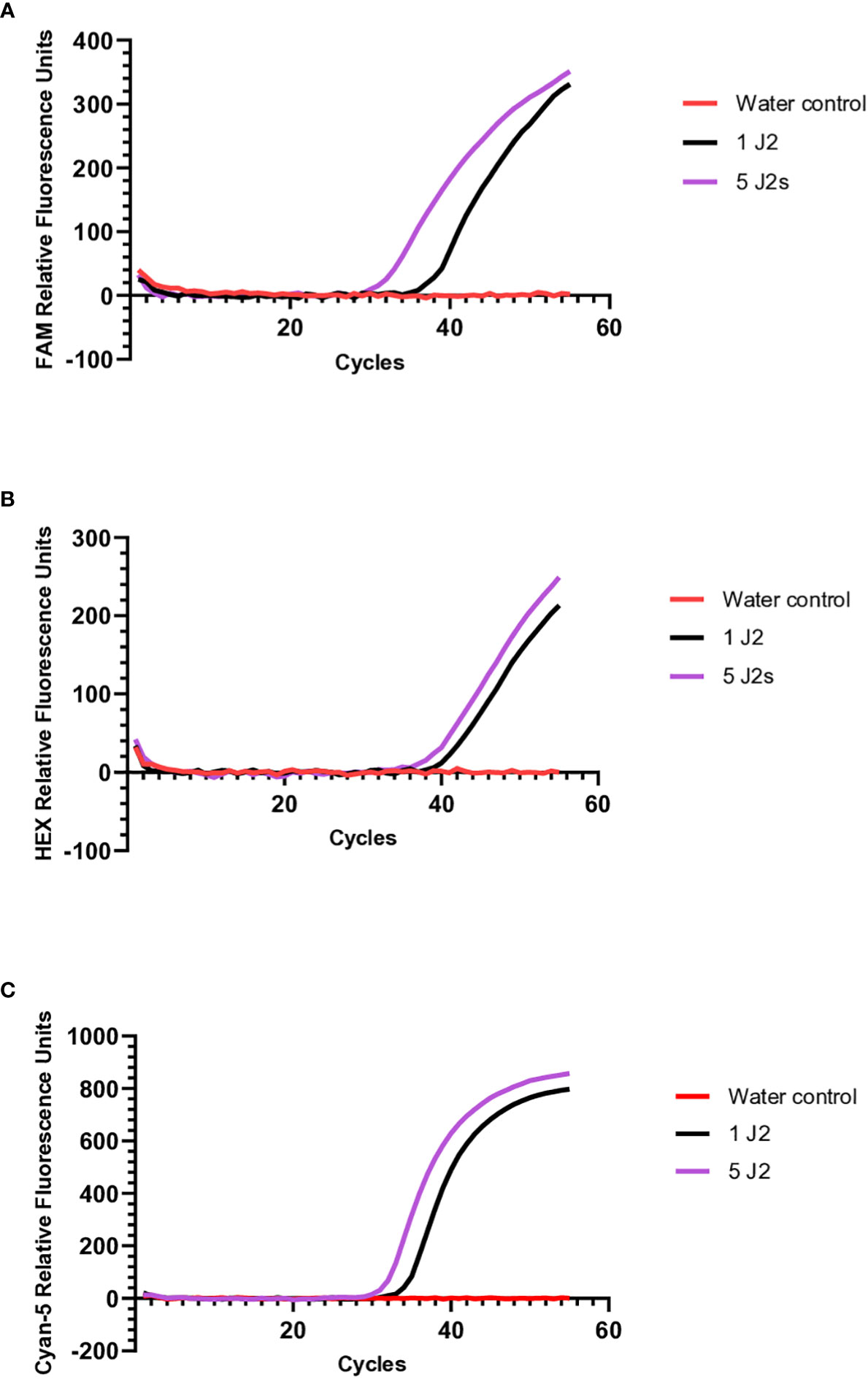
Figure 4 Molecular beacon RT-PCR amplification curves using crude extracts from 1 or 5 J2s of (A) M. chitwoodi (B) M. fallax, or (C) M. minor as the reaction template.
Further investigation was carried out to see if the molecular beacon RT-PCR assay could detect the nematode of interest when there is a mixture of nematodes in the reaction. When 20 pg of M. chitwoodi, M. fallax, and M. minor HSP90 templates were mixed in a single reaction containing all three beacon probes, it was possible to detect each species (Figure 5A). Detection of all three species was also possible using crude extracts from digested J2s when all 3 species were mixed, either as single J2s or as 5 J2s each (Figure 6).
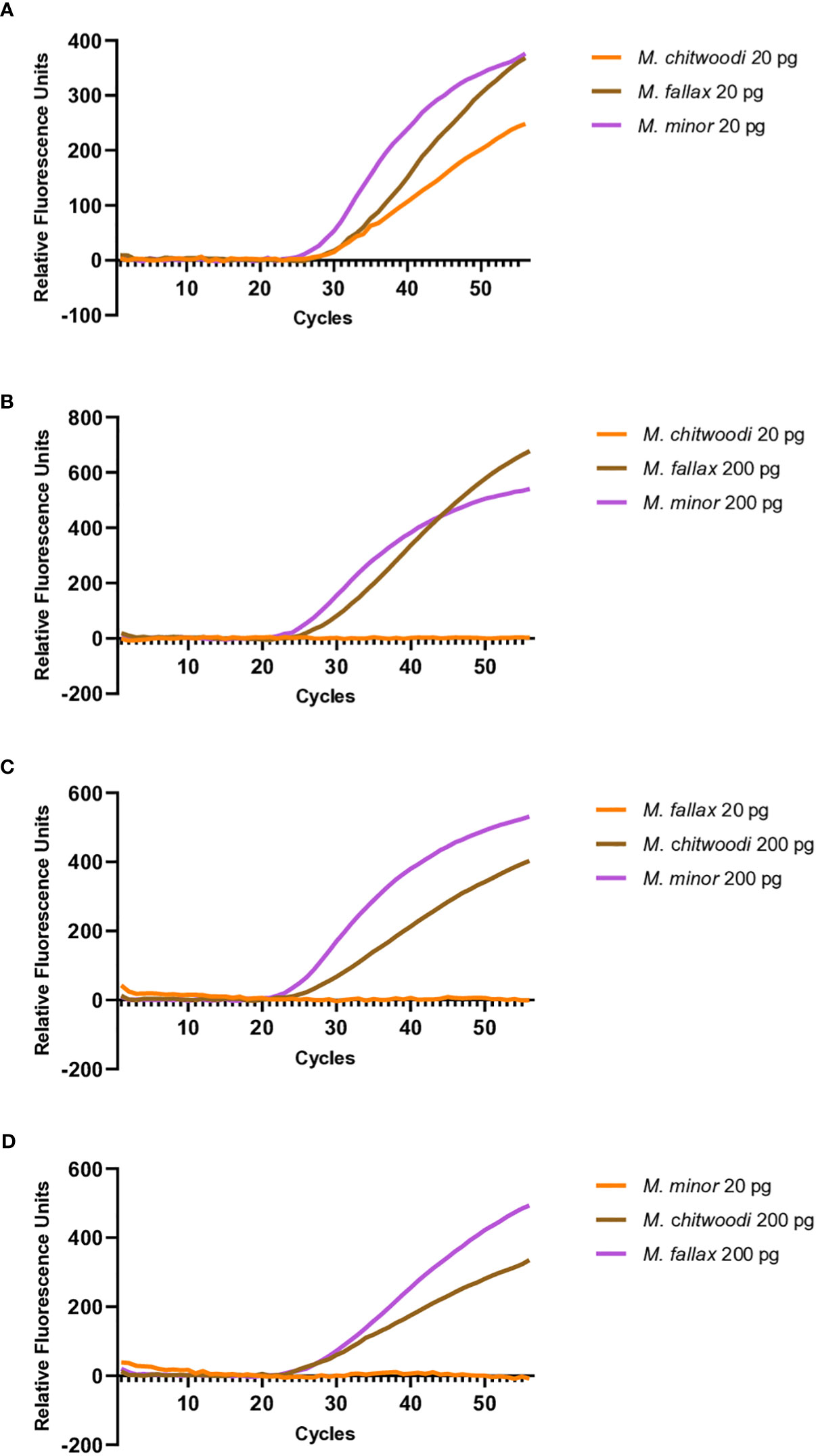
Figure 5 Multiplexed molecular beacon RT-PCR assays containing equal or unequal parts M. chitwoodi, M. fallax, and M. minor plasmid template. (A) Amplification curves from 20 pg of M. chitwoodi (average Ct = 31.3, SEM = 0.11, n = 6), M. fallax (average Ct = 30.9, SEM = 0.20, n = 6), and M. minor (average Ct = 30.1, SEM = 0.09, n = 6) in a single reaction (B) Amplification curves from 20 pg of M. chitwoodi (no amplification, n = 6), 200 pg M. fallax (average Ct = 26.8, SEM = 0.62, n = 6), and 200 pg M. minor (average Ct = 26.3, SEM = 0.39, n = 6) in a single reaction (C) Amplification curves from 20 pg of M. fallax (no amplification, n = 6), 200 pg M. chitwoodi (average Ct = 27.0, SEM = 0.50, n = 6), and 200 pg M. minor (average Ct = 25.1, SEM = 0.13, n = 6) in a single reaction. (D) Amplification curves from 20 pg of M. minor (no amplification, n = 6), 200 pg M. chitwoodi (average Ct = 26.6, SEM = 0.24, n = 6), and 200 pg M. fallax (average Ct = 26.6, SEM = 0.20, n = 6) in a single reaction.
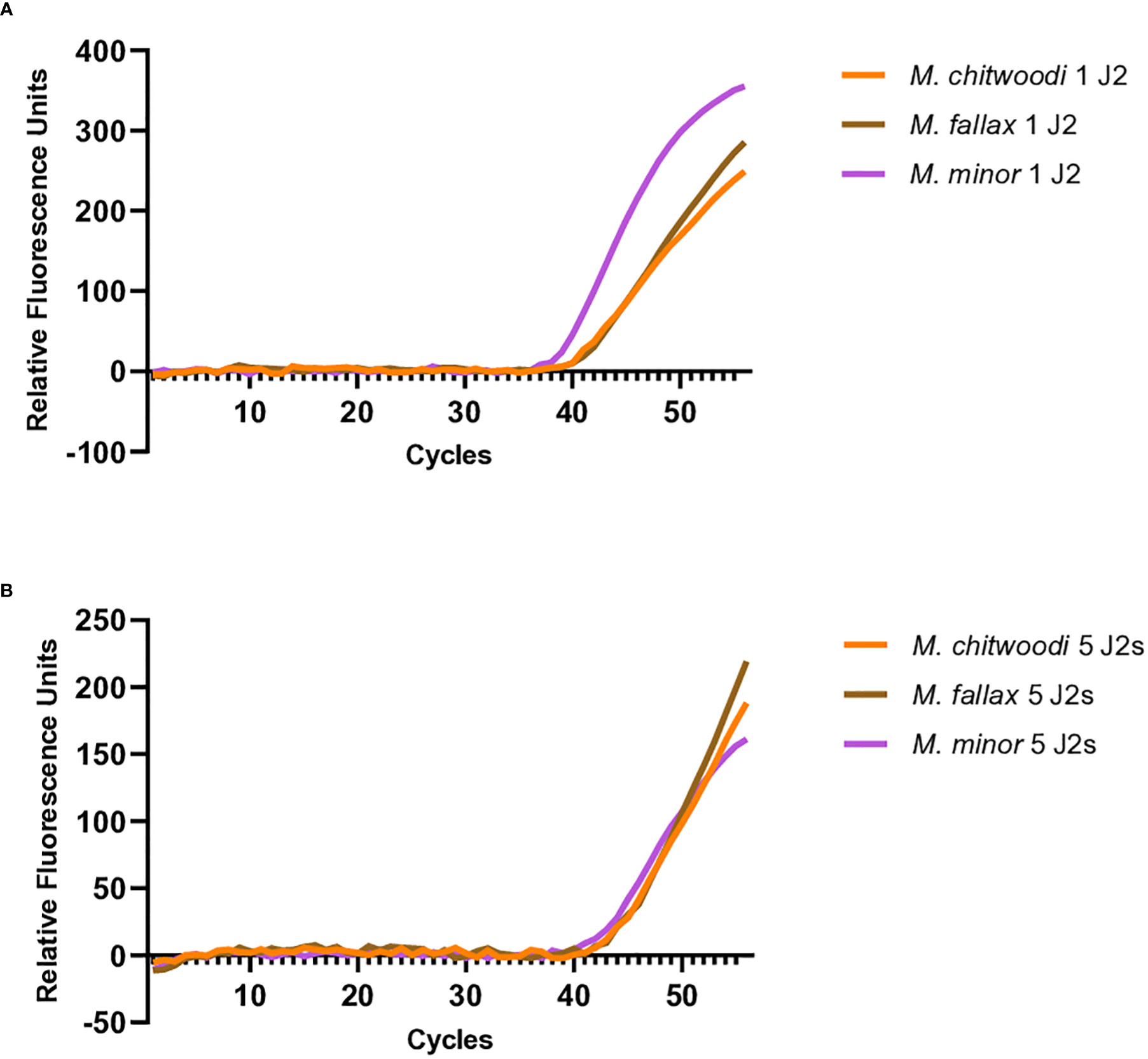
Figure 6 Multiplexed molecular beacon RT-PCR assays containing an equal mix of 1 or 5 J2s from M. chitwoodi, M. fallax, and M. minor as template. (A) Amplification curves from 1 J2 of M. chitwoodi (average Ct = 42.3, SEM = 0.43, n = 10), M. fallax (average Ct = 43.7, SEM = 0.78, n = 10), and M. minor (average Ct = 41.1, SEM = 0.69, n = 10) in a single reaction. (B) Amplification curves from 5 J2s of M. chitwoodi (average Ct = 42.4, SEM = 0.63, n = 4), M. fallax (average Ct = 42.7, SEM = 0.92, n = 4), and M. minor (average Ct = 42.7, SEM = 0.62, n = 4) in a single reaction.
To investigate how the assay performed when templates were not present in equal parts, mixes of M. chitwoodi, M. fallax, and M. minor HSP90 plasmid templates were made that had one species present at 20 pg, and the other two species present at 200 pg. The templates present at 200 pg were reliably detected, but the template at 20 pg could not be detected in these reactions (Figures 5B–D).
Overall, the data suggests that the molecular beacon RT-PCR assay can detect each species in a multiplexed reaction. However, an important caveat is that the sample detection is inhibited for the target species if the other two species are in relative excess in concentration.
Nematode identification is a critical component for biosecurity, particularly when dealing with regulated nematodes. Meloidogyne minor, M. chitwoodi and M. fallax juveniles can be difficult to distinguish from each other, making morphological-based identification challenging. We designed a molecular beacon RT-PCR assay to detect M. chitwoodi, M. minor, and M. fallax. The assay is species-specific, easy to perform, rapid, and reliable. It is also able to detect small amounts of DNA and could identify the species using a single J2 as the reaction template. This means it can be adapted for identifying J2s extracted from soil or from J2s hatched from eggs (Adam et al., 2007). This assay can be very useful because individual J2s can be picked and then used directly to identify if it is one of three species. If all three beacons are used on a single J2, a positive or negative identification of either M. chitwoodi, M. fallax, or M. minor can be obtained in a reaction time under 2 hours. This assay will help to determine the distribution of these nematodes and prevent their spread to new potato growing regions.
A previous comparison between TaqMan and molecular beacon probes for analyzing single nucleotide polymorphisms (SNPs) in human DNA found that molecular beacon probes are better in detecting sequence variants; in a panel of DNA samples, the molecular beacon probes were more reliable in detecting GC-rich targets compared to the TaqMan probes designed for that region (Täpp et al., 2000). In addition, the data showed that the molecular beacon probes could better detect minority sequence variants over a wider range of template concentrations compared to the TaqMan assay, suggesting that the molecular beacon probes offer advantages in sensitivity and robustness over TaqMan assays (Täpp et al., 2000). Our results indicate that the molecular beacon probes we developed for M. chitwoodi and M. fallax are more sensitive than the previously developed TaqMan assay for these two species (Zijlstra and Van Hoof, 2006). Our molecular beacon RT-PCR assays could detect DNA concentrations as low as 0.04 fg for M. minor and 4 fg and 2 fg for M. chitwoodi and M. fallax, respectively, which is better than or comparable to previous TaqMan molecular beacon RT-PCR assays (Braun-Kiewnick and Kiewnick, 2018; Zijlstra and Van Hoof, 2006; Weerdt et al., 2011). The M. chitwoodi and M. minor assays both had PCR efficiencies of ≥ 90%. This is an improvement in efficiency for M. minor (Weerdt et al., 2011) and a similar efficiency for M. chitwoodi compared to the previously published TaqMan assays (Zijlstra and Van Hoof, 2006). The M. fallax assay had the lowest PCR efficiency of the three at 83%. However, 83% is still a relatively good efficiency compared to previously published PCR assays for root-knot nematodes. For example, the TaqMan assay for M. minor had an efficiency of 62% (Weerdt et al., 2011). The performance of the molecular beacon RT-PCR assays indicate that the assay has a high sensitivity, specificity (the probes only detect the target species) and reproducibility over multiple experiments. While this assay is species-specific, standard curves using Race 1, Race 2, or Race 1 Roza gDNA from M. chitwoodi showed that this assay cannot distinguish between the M. chitwoodi isolates. Therefore, this the assay would be suitable for general M. chitwoodi detection despite the fact that the known races in Washington differ genetically (Bali et al., 2021b).
Multiplexing would allow for the simultaneous detection of the three species in one tube. The multiplexing experiments showed that we could detect each species of nematode when similar quantities of template of the three species were present. Because the PCR efficiencies were high and because the PCR primers are targeting the same sequences in all three species, when all three species are present in similar amounts, the amount of target amplicons produced from each species should be similar, resulting in detectable amplicons. However, we found that our ability to detect a species was compromised if its DNA template was present at relatively low ratios (≥1:10) compared to the other species. Although the sensitivity of multiplexing is limited by the relative quantities of DNA template from each of the three species, it is still a useful tool for the sensitive detection and identification of M. chitwoodi, M. fallax, and M. minor.
This assay provides a simple and rapid molecular protocol for distinguishing M. chitwoodi, M. fallax, and M. minor from one another using as little as a single J2 as the reaction template. The assays described here use DNA isolated from juvenile(s) since they are commonly found in soil samples, but DNA could be isolated from eggs and adult nematodes and used as the reaction template. This is the first molecular method that reliably identifies these three species of potato-infecting root-knot nematodes. It could be used by diagnosticians and extension agents as a tool for tracking the spread of M. chitwoodi, M. fallax and M. minor in the USA.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
SA performed the experiments and statistical analyses. SA and CG designed the experiments and wrote the manuscript text. Both authors contributed to the article and approved the submitted version.
Funding was provided by the Animal and Plant Health Inspection Service #AP18PPQS&T00C148. This work was supported in part by the USDA National Institute of Food and Agriculture, Hatch umbrella project #1015621 and Hatch project W4186.
The authors wish to thank Dr. Gerrit Karssen of the Wageningen Nematode Collection for providing the M. fallax and M. minor, and Dr. Sapinder Bali for help with nematode DNA extraction.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1096239/full#supplementary-material
Adam, M. A. M., Phillips, M. S., Blok, V. C. (2007). Molecular diagnostic key for identification of single juveniles of seven common and economically important species of root-knot nematode (Meloidogyne spp.). Plant Pathol. 56, 190–197. doi: 10.1111/j.1365-3059.2006.01455.x
Bali, S., Hu, S., Vining, K., Brown, C., Mojtahedi, H., Zhang, L., et al. (2021a). Nematode genome announcement: Draft genome of Meloidogyne chitwoodi, an economically important pest of potato in the pacific Northwest. Mol. Plant Microbe Interact. 34, 981–986. doi: 10.1094/MPMI-12-20-0337-A
Bali, S., Zhang, L., Franco, J., Gleason, C. (2021b). Biotechnological advances with applicability in potatoes for resistance against root-knot nematodes. Curr. Opin. Biotechnol. 70, 226–233. doi: 10.1016/j.copbio.2021.06.010
Baum, T. J., Gresshoff, P. M., Lewis, S. A., Dean, R. A. (1994). Characterization and phylogenetic analysis of four root-knot nematode species using DNA amplification fingerprinting and automated polyacrylamide gel electrophoresis. Mol. Plant Microbe Interact. 7, 39–47. doi: 10.1094/MPMI-7-0039
Blok, V. C., Phillips, M. S., Fargette, M. (1997). Comparison of sequences from the ribosomal DNA intergenic region of Meloidogyne mayaguensis and other major tropical root-knot nematodes. J. Nematol. 29, 16–22.
Braun-Kiewnick, A., Kiewnick, S. (2018). Real-time PCR, a great tool for fast identification, sensitive detection and quantification of important plant-parasitic nematodes. Eur. J. Plant Pathol. 152, 271–283. doi: 10.1007/s10658-018-1487-7
Brown, C. R., Mojtahedi, H., Zhang, L. H., Riga, E. (2009). Independent resistant reactions expressed in root and tuber of potato breeding lines with introgressed resistance to Meloidogyne chitwoodi. Phytopathology 99, 1085–1089. doi: 10.1094/PHYTO-99-9-1085
Castagnone-Sereno, P. (2000). Use of satellite DNA for specific diagnosis of the quarantine root- knot nematodes Meloidogyne chitwoodi and M. fallax. Bull. OEPP 30, 581–584. doi: 10.1111/j.1365-2338.2000.tb00951.x
Castagnone-Sereno, P., Leroy, F., Bongiovanni, M., Zijlstra, C., Abad, P. (1999). Specific diagnosis of two root-knot nematodes, Meloidogyne chitwoodi and M. fallax, with satellite DNA probes. Phytopathology 89, 380–384. doi: 10.1094/PHYTO.1999.89.5.380
European and Mediterranean Plant Protection Organization [EPPO] (2022) Meloidogyne fallax. EPPO datasheets on pests recommended for regulation. Available at: https://gd.eppo.int (Accessed December 23, 2022).
FAO. (2017). Crops and livestock products. Available at: https://www.fao.org/faostat/en/#data/QCL (Accessed 04-05-2021).
Forootan, A., Sjöback, R., Björkman, J., Sjögreen, B., Linz, L., Kubista, M. (2017). Methods to determine limit of detection and limit of quantification in quantitative real-time PCR (qPCR). Biomol. Detect. Quantif. 12, 1–6. doi: 10.1016/j.bdq.2017.04.001
Gamel, S., Huchet, E., Le Roux-Nio, A.-C., Anthoine, G. (2014). Assessment of PCR-based tools for the specific identification of some temperate Meloidogyne species including M. chitwoodi, M. fallax and M. minor. Eur. J. Plant Pathol. 138, 807–817. doi: 10.1007/s10658-013-0355-8
Giorgi, C. D., Sialer, M. F., Vito, M. D., Lamberti, F. (1994). Identification of plant-parasitic nematodes by PCR amplification of DNA fragments. Bull. OEPP 24, 447–451. doi: 10.1111/j.1365-2338.1994.tb01403.x
Gross, S. M., Williamson, V. M. (2011). Tm1: A mutator/foldback transposable element family in root-knot nematodes. PloS One 6, 9. doi: 10.1371/journal.pone.0024534
Hadziavdic, K., Lekang, K., Lanzen, A., Jonassen, I., Thompson, E. M., Troedsson, E. (2014). Characterization of the 18S rRNA gene for designing universal eukaryote specific primers. PLoS One 9, 2. doi: 10.1371/journal.pone.0087624
Han, H., Cho, M.-R., Jeon, H.-Y., Lim, C.-K., Jang, H.-I. (2004). PCR-RFLP identification of three major Meloidogyne species in Korea. J. Asia Pac. Entomol. 7, 171–175. doi: 10.1016/S1226-8615(08)60212-5
Harris, T. S., Sandall, L. J., Powers, T. O. (1990). Identification of single Meloidogyne juveniles by polymerase chain reaction amplification of mitochondrial DNA. J. Nematol. 22, 518–524.
Ingham, R. E., Hamm, P. B., Baune, M., David, N. L., Wade, N. M. (2007). Control of Meloidogyne chitwoodi in potato with shank-injected metam sodium and other nematicides. J. Nematol. 39, 161–168.
Ingham, R. E., Hamm, P. B., Williams, R. E., Swanson, W. H. (2000). Control of Meloidogyne chitwoodi in potato with fumigant and nonfumigant nematicides. J. Nematol. 32, 556–565.
Kantor, M., Handoo, Z., Kantor, C., Carta, L. (2022). Top ten most important U.S.-regulated and emerging plant-parasitic nematodes. Horticulturae 8, 208. doi: 10.3390/horticulturae8030208
Karssen, G. (1996). Description of Meloidogyne fallax n. sp. (Nematoda: Heteroderidae), a root- knot nematode from the Netherlands. Fundam. Appl. Nematol. 19, 593–599.
Karssen, G., Bolk, R. J., van Aelst, A. C., van den Beld, I., Kox, L. F. F., Korthals, G. W., et al. (2004). Description of Meloidogyne minor n. sp. (Nematoda: Meloidogynidae), a root-knot nematode associated with yellow patch disease in golf courses. Nematology 6, 59–72. doi: 10.1163/156854104323072937
Koenning, S. R., Overstreet, C., Noling, J. W., Donald, P. A., Becker, J. O., Fortnum, B. A. (1999). Survey of crop losses in response to phytoparasitic nematodes in the united states for 1994. J. Nematol. 31, 587–618.
Lima, F. S. O., Mattos, V. S., Silva, E. S., Carvalho, M. A. S., Teixeira, R. A., Silva, J. C., et al. (2018). “Nematodes affecting potato and sustainable practices for their management,” in Potato - from incas to all over the world. Ed. Yildiz, M. (London: IntechOpen). doi: 10.5772/intechopen.73056
Mojtahedi, H., Brown, C. R., Riga, E., Zhang, L. H. (2007). A new pathotype of Meloidogyne chitwoodi race 1 from Washington state. Plant Dis. 91, 1051.
McClure, M. A., Nischwitz, C., Skantar, A. M., Schmitt, M. E., Subbotin, S. A. (2012). Root- knot nematodes in golf course greens of the Western united states. Plant Dis. 96, 635–647. doi: 10.1094/PDIS-09-11-0808
Morris, K., Horgan, F., Downes, M., Griffin, C. (2011). The effect of temperature on hatch and activity of second-stage juveniles of the root-knot nematode, Meloidogyne minor, an emerging pest in north-west Europe. Nematology 13, 985–993. doi: 10.1163/138855411X571902
Morris, K., Horgan, F. G., Griffin, C. T. (2013). Spatial and temporal dynamics of Meloidogyne minor on creeping bentgrass in golf greens. Plant Pathol. 62, 1166–1172. doi: 10.1111/ppa.12025
Nischwitz, C., Skantar, A., Handoo, Z. A., Hult, M. N., Schmitt, M. E., McClure, M. A. (2013). Occurrence of Meloidogyne fallax in north America, and molecular characterization of M. fallax and M. minor from U.S. golf course greens. Plant Dis. 97, 1424–1430. doi: 10.1094/PDIS-03-13-0263-RE
Orui, Y. (1998). Identification of Japanese species of the genus Meloidogyne (Nematoda: Meloidogynidae) by PCR-RFLP analysis. Appl. Entomol. Zool. 33, 43–51. doi: 10.1303/aez.33.43
Petersen, D. J., Zijlstra, C., Wishart, J. (1997). Specific probes efficiently distinguish root-knot nematode species using signature sequences in the ribosomal intergenic spacer. Fundam. Appl. Nematol. 20, 619–626.
Pinkerton, J. N., Santo, G. S., Mojtahedi, H. (1991). Population dynamics of Meloidogyne chitwoodi on russet Burbank potatoes in relation to degree-day accumulation. J. Nematol. 23, 283–290.
Powers, T. O., Harris, T. S. (1993). A polymerase chain reaction method for identification of five major Meloidogyne species. J. Nematol. 25, 1–6.
Powers, T. O., Todd, T. C., Burnell, A. M., Murray, P. C. B., Fleming, C. C., Szalanski, A. L., et al. (1997). The rDNA internal transcribed spacer region as a taxonomic marker for nematodes. J. Nematol. 29, 441–450.
Qiu, J. J., Westerdahl, B. B., Anderson, C., Williamson, V. M. (2006). Sensitive PCR detection of Meloidogyne arenaria, M. incognita, and M. javanica extracted from soil. J. Nematol. 38, 434–441.
Sasser, J. N., Freckman, D. W. (1987). “A world perspective on nematology: The role of the society,” in Vistas on nematology. Eds. Veech, J. A., Dickson, D. W. (Hyattsville, MD: Society of Nematologists, Inc) 7–20.
Skantar, A. M., Carta, L. K. (2004). Molecular characterization and phylogenetic evaluation of the hsp90 gene from selected nematodes. J. Nematol. 36, 466–480.
Suffert, M., Giltrap, N. (2012). EPPO workshop on Meloidogyne chitwoodi and meloidogyne fallax, (2011-03-08): Importance for potato production and experience of management in EPPO countries. Bull. OEPP 42, 117–121. doi: 10.1111/j.1365-2338.2012.02528.x
Täpp, I., Malmberg, L., Rennel, E., Wik, M., Syvänen, A. C. (2000). Homogeneous scoring of single-nucleotide polymorphisms: Comparison of the 5’-nuclease TaqMan assay and molecular beacon probes. BioTechniques 28, 732–738. doi: 10.2144/00284rr02
Thoden, T. C., Korthals, G. W., Visser, J., van Gastel-Topper, W. (2012). A field study on the host status of different crops for Meloidogyne minor and its damage potential on potatoes. Nematology 14, 277–284. doi: 10.1163/156854111X594965
Van Der Beek, J. G., Vereijken, P. F. G., Poleij, L. M., Silfhout, C. H. V. (1998). Isolate-by- cultivar interaction in root-knot nematodes Meloidogyne hapla, M. chitwoodi, and M. fallax on potato. Can. J. Bot. 76, 75–82. doi: 10.1139/b97-161
Van Megen, H., van den Elsen, S., Holterman, M., Karssen, G., Mooyman, P., Bongers, T., et al. (2009). A phylogenetic tree of nematodes based on about 1200 full-length small subunit ribosomal DNA sequences. Nematology 11, 927–950. doi: 10.1163/156854109X456862
Van Meggelen, J. C., Karssen, G., Janssen, G. J. W., Verkerk-Bakker, B., Janssen, R. (1994). New race of Meloidogyne chitwoodi golden, O’Bannon, Santo & Finley 1980? Fundam. Appl. Nematol. 17, 93–96.
Vandenbossche, B., Viaene, N., de Sutter, N., Maes, M., Karssen, G., Bert, W. (2011). Diversity and incidence of plant-parasitic nematodes in Belgian turf grass. Nematology 13, 245–256. doi: 10.1163/138855410X517084
Viaene, N. (2014). Nematodes: An increasing threat for the potato crop in Europe? Potato Res. 57, 335–338. doi: 10.1007/s11540-015-9284-6
Vrain, T. C., Petersen, D. J. (1996). Rapid identification of Meloidogyne chitwoodi, M. hapla, and M. fallax using PCR primers to amplify their ribosomal intergenic spacer. Fundam. Appl. Nematol. 19, 601–605.
Vrain, T. C., Wakarchuk, D. A., Levesque, A. C., Hamilton, R. L. (1992). Intraspecific rDNA restriction fragment length polymorphism in the Xiphinema americanum group. Fundam. Appl. Nematol. 15, 563–573.
Waeyenberge, L., Ryss, A., Moens, M., Pinochet, J., Vrain, T. (2000). Molecular characterisation of 18 Pratylenchus species using rDNA restriction fragment length polymorphism. Nematology 2, 135–142. doi: 10.1163/156854100509024
Weerdt, M. D., Kox, L., Waeyenberge, L., Viaene, N., Zijlstra, C. (2011). A real-time PCR assay to identify Meloidogyne minor. J. Phytopathol. 159, 80–84. doi: 10.1111/j.1439-0434.2010.01717.x
Wesemael, W. M. L., Taning, L. M., Viaene, N., Moens, M. (2014). Life cycle and damage of the root-knot nematode Meloidogyne minor on potato, solanum tuberosum. Nematology 16, 185–192. doi: 10.1163/15685411-00002756
Wesemael, W., Viaene, N., Moens, M. (2011). Root-knot nematodes (Meloidogyne spp.) in Europe. Nematology 13, 3–16. doi: 10.1163/138855410X526831
Wishart, J., Phillips, M. S., Blok, V. C. (2002). Ribosomal intergenic spacer: a polymerase chain reaction diagnostic for Meloidogyne chitwoodi, M. fallax, and M. hapla. Phytopathology 92, 884–892. doi: 10.1094/PHYTO.2002.92.8.884
Williamson, V. M., Caswell-Chen, E. P., Westerdahl, B. B., Wu, F. F., Caryl, G. (1997). A PCR assay to identify and distinguish single juveniles of Meloidogyne hapla and M. chitwoodi. J. Nematol. 29, 9–15.
Zasada, I. A., Dandurand, L.-M., Gleason, C., Hagerty, C. H., Ingham, R. E. (2018). “Plant parasitic nematodes of the pacific Northwest: Idaho, Oregon and Washington,” in Plant parasitic nematodes in sustainable agriculture of north America. Eds. Subbotin, S. A., Chitambar, J. J. (Cham, Switzerland: Springer International Publishing), 211–239. doi: 10.1007/978-3-319-99585-4_8
Zeng, Y., Ye, W., Kerns, J., Tredway, L., Martin, S., Martin, M. (2015). Molecular characterization and phylogenetic relationships of plant-parasitic nematodes associated with turfgrasses in north Carolina and south Carolina, united states. Plant Dis. 99, 982–993. doi: 10.1094/PDIS-10-14-1060-RE
Zhang, L., Gleason, C. (2019). Loop-mediated isothermal amplification for the diagnostic detection of Meloidogyne chitwoodi and M. fallax. Plant Dis. 103, 12–18. doi: 10.1094/PDIS-01-18-0093-RE
Zijlstra, C. (1997). A fast PCR assay to identify Meloidogyne hapla, M. chitwoodi, and M. fallax, and to sensitively differentiate them from each other and from M. incognita in mixtures. Fundam. Appl. Nematol. 20, 505–511.
Zijlstra, C. (2000). Identification of Meloidogyne chitwoodi, M. fallax and M. hapla based on SCAR-PCR: A powerful way of enabling reliable identification of populations or individuals that share common traits. Eur. J. Plant Pathol. 106, 283–290. doi: 10.1023/A:1008765303364
Zijlstra, C., Van Hoof, R. A. (2006). A multiplex real-time polymerase chain reaction (TaqMan) assay for the simultaneous detection of Meloidogyne chitwoodi and M. fallax. Phytopathology 96, 1255–1262. doi: 10.1094/PHYTO-96-1255
Keywords: meloidogyne, nematode, diagnostic tool, RT-PCR, potato, pathogen detection
Citation: Anderson SD and Gleason CA (2023) A molecular beacon real-time polymerase chain reaction assay for the identification of M. chitwoodi, M. fallax, and M. minor. Front. Plant Sci. 14:1096239. doi: 10.3389/fpls.2023.1096239
Received: 11 November 2022; Accepted: 31 January 2023;
Published: 22 February 2023.
Edited by:
Brent Sipes, University of Hawaii at Manoa, United StatesReviewed by:
Tariq Mukhtar, Pir Mehr Ali Shah Arid Agriculture University, PakistanCopyright © 2023 Anderson and Gleason. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cynthia A. Gleason, Y3ludGhpYS5nbGVhc29uQHdzdS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.