 Yong Feng
Yong Feng Mengyun Jiang
Mengyun Jiang Weichang Yu
Weichang Yu Jiannan Zhou
Jiannan Zhou- 1Shenzhen Branch, Guangdong Laboratory of Lingnan Modern Agriculture, Genome Analysis Laboratory of the Ministry of Agriculture and Rural Affairs, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Shenzhen, China
- 2State Key Laboratory of Crop Stress Adaptation and Improvement, School of Life Sciences, Henan University, Kaifeng, China
- 3Guangdong Key Laboratory of Plant Epigenetics, College of Life Sciences and Oceanography, Shenzhen University, Shenzhen, China
- 4Liaoning Peanut Research Institute, Liaoning Academy of Agricultural Sciences, Fuxing, China
- 5Key Laboratory of Tropical Fruit Biology (Ministry of Agriculture), South Subtropical Crops Research Institute, Chinese Academy of Tropical Agricultural Sciences, Zhanjiang, China
The roles of short/small open reading frames (sORFs) have been increasingly recognized in recent years due to the rapidly growing number of sORFs identified in various organisms due to the development and application of the Ribo-Seq technique, which sequences the ribosome-protected footprints (RPFs) of the translating mRNAs. However, special attention should be paid to RPFs used to identify sORFs in plants due to their small size (~30 nt) and the high complexity and repetitiveness of the plant genome, particularly for polyploidy species. In this work, we compare different approaches to the identification of plant sORFs, discuss the advantages and disadvantages of each method, and provide a guide for choosing different methods in plant sORF studies.
Introduction
Short/small open reading frames (sORFs) with the capacity of encoding micropeptides shorter than 100 amino acids (aa) are widely distributed in plants, ranging from green algae (Xu et al., 2017a) to rice (Yang et al., 2021) and Arabidopsis (Hanada et al., 2007), and are engaged in various biological and molecular processes, such as plant growth, nitrogen response, symbiosis nitrogen fixation, stomatal closure, plant circadian clock, anther development, pollen tube growth, abiotic responses, plant disease resistance, morphogenesis, and growth regulation (Ong et al., 2022; Wu et al., 2022). sORFs are pervasive in plant genomes and have been detected beyond the known coding regions. According to their locations, sORFs can be classified into several groups, including uORF, dORF, lncRNA-sORFs, and intergenic-sORFs (Table 1). As well as in mammals and yeasts (Jeon et al., 2015; Libre et al., 2021), sORFs have also been reported in plants, and the translation of uORFs can potentially repress the translation of downstream major ORFs (mORFs) (David-Assael et al., 2005; Saul et al., 2009; Pajerowska-Mukhtar et al., 2012; Causier et al., 2022). Another function of sORFs is to mitigate the abundance of miRNAs, and therefore influence the translation of their target mRNAs. Lauressergues et al. (2015) reported two plant primary transcripts (pri-miRNAs) that contain sORFs encoding regulatory peptides, miPEP165a and miPEP171b, in A. thaliana and Medicago truncatula, respectively. Overexpressing miPEP171b in M. truncatula roots specifically improves the accumulation of endogenous mature miRNAs, resulting in a reduction in lateral root density to a similar extent as overexpression of the corresponding pri-miR171b. Furthermore, the peptides encoded by the sORFs, per se, can also be functional, involving various biological processes (reviewed in Ong et al., 2022). Given that sORFs can substantially regulate the translation of downstream ORFs and encode proteins with crucial functions, the mutation of many sORFs, particularly uORFs, would lead to dramatic phenotype changes in plants and crops. Therefore, the natural or artificial mutation of these sORFs can be used to improve vital plant processes and valuable crop traits (Xu et al., 2017b; Si et al., 2020).
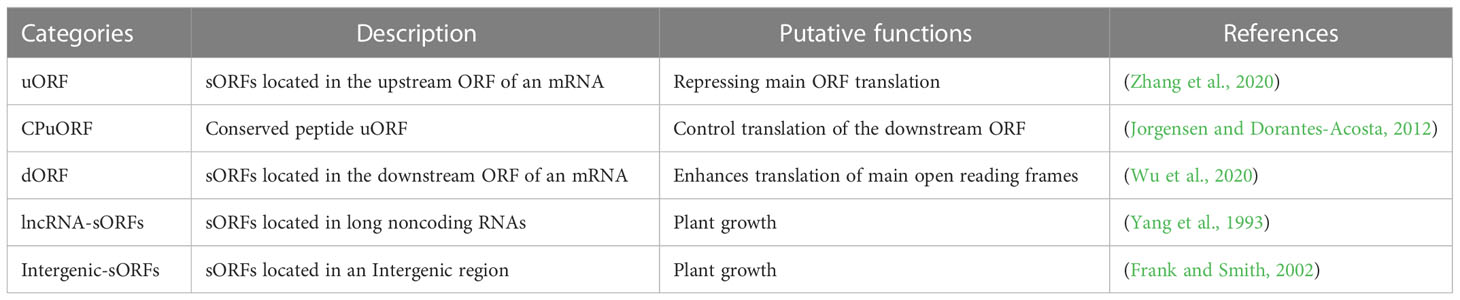
Table 1 Categories of sORFs.
It is noteworthy that although reports of plant sORFs are increasing dramatically, the identification of sORFs in plant genomes is still challenging. The recent advancement and application of Ribo-Seq technology have promoted the research of plant sORFs; however, the existing studies on plant sORFs are only focused on plants with simple genomes, including the model plants, Arabidopsis and rice, while investigations into sORFs in complex genomes are rare. In most, if not all, existing plant sORF studies, the methods and tools developed in mammals or yeasts were directly used to search for sORFs in plant genomes. Nevertheless, plant genomes are generally more repetitive, and many of them are polyploid or paleopolyploid. Special attention should be given to the studies of sORFs in plant genomes.
Despite the challenges and difficulties, many efforts have been made to identify sORFs from plant genomes. The first systematic study of sORFs in plants was conducted on Arabidopsis thaliana, where more than 7,000 sORFs were identified (Hanada et al., 2007), including 49 that induced visible phenotypic effects or that are associated with various morphological changes (Hanada et al., 2013). Subsequently, a total of 48,620 sORFs were identified in Oryza sativa through microarray analyses, and at least 36 were involved in Fe deficiency and excess (Bashir et al., 2014). Generally, plant sORFs can be identified using three different strategies: the conservation of coding sequences, ribosome-protected footprints, and nucleotide diversity in the natural population. In this review, we summarize the methods of sORF identification used in plant studies and discuss the challenges and caveats in plant sORF identification and possible solutions for future studies.
sORF identification through sequence conservation
As functional ORFs are conserved across genomes, early attempts at sORF identification were primarily based on sequence similarities using sequence alignment tools, such as BLAST (Altschul et al., 1997), coupled with ORF Finder (Wheeler et al., 2002), assuming that the functional sORFs would also have been preserved by natural selection (Figure 1A). For example, a total of 26 groups of conserved uORFs were identified in O. sativa and A. thaliana using uORF-Finder (Hayden and Jorgensen, 2007). In another study, sequence similarity was also observed at the amino acid level of uORFs across Arabidopsis and rice, 11 rice uORFs were conserved in Arabidopsis, and most of them were also conserved in other cereals (Tran et al., 2008). These conserved sORFs can be true; however, recent evidence has revealed varied degrees of sORF conservation. Although many sORFs are conserved, there are also many species- or lineage-specific sORFs (Hsu et al., 2016; Wang et al., 2021), which cannot be identified through sequence comparisons.
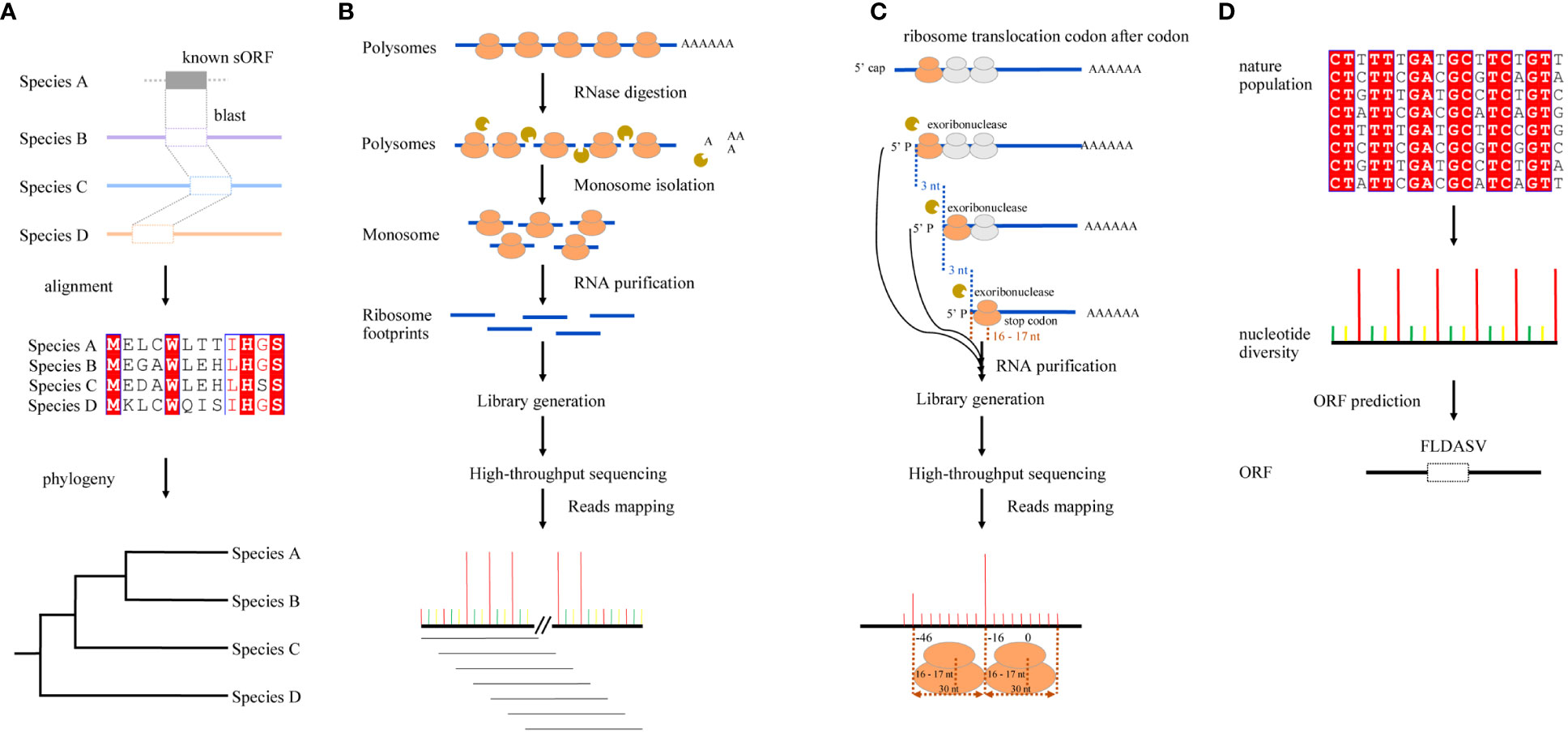
Figure 1 Graphic illustration for four different strategies of sORF identification. (A) Sequence conservation-based. It is assumed that functional sORFs are preserved by natural selection and are conserved across species; sORF identification could be based on sequence similarities using sequence alignment tools, such as BLAST. (B) RFP-based. The ribosome-associated mRNAs are digested by RNase and the fragments bound by ribosome are protected; they can then be isolated and sequenced using next-generation sequencing (NGS) technology. As ribosomes move along mRNA strands with a step of three nucleotides during the translation of mRNAs, the mapping loci of RPFs on mRNAs show a strong 3-nt periodicity. (C) Degradome sequencing-based. 5’-3’ exoribonuclease digests translating mRNAs chasing after the last translating ribosome, which translocates codon after codon on mRNAs, leaving truncated 5’ monophosphate mRNAs with a 3-nucleotide distance in length. After sequencing, this 3-nucleotide periodicity in the position of free 5’ mRNA ends can then be used for sORFs identification. (D) Natural nucleotide diversity-based. As only the nucleotide diversities in CDSs showed a significant 3-nt periodicity, the single-nucleotide polymorphism (SNP) datasets of natural populations can be used to predict sORFs.
sORF identification using ribosome-protected footprints
Ribo-seq is an emerging technology that enables the identification of sORFs (Ingolia, 2010). To date, most plant sORFs are identified using RPFs. Briefly, the ribosome-associated mRNAs are digested by RNase, and the fragments bound by ribosome are protected; they can then be isolated and sequenced using next-generation sequencing (NGS) technology. As ribosomes move along mRNA strands with a step of three nucleotides during the translation of mRNAs, the mapping loci of RPFs on mRNAs show a strong 3-nt periodicity, which provides information for the identification of translating frames on mRNAs (Figure 1B). To better analyze and mine the information in RPFs, many computation tools implementing different algorithms have been developed to predict sORFs or ORFs on noncoding RNAs (see review of Ong et al., 2022), including FLOSS (Ingolia et al., 2014), RiboTaper (Calviello et al., 2016), RiboCode (Xiao et al., 2018), ORFquant (Calviello et al., 2020), RiboNT (Song et al., 2021), and slORFfinder (Song et al., 2023). Among them, ORFScore and RiboTaper are the most frequently used tools in plant sORF studies. For example, Hsu et al. (2016) detected 187 uORFs, 10 dORFs, and 27 translated sORFs from annotated non-coding RNAs in Arabidopsis using RiboTaper (Hsu et al., 2016), and Wu et al. (2020) detected 1,406 and 1,153 dORFs in human cells and zebrafish embryos using ORFScore (Wu et al., 2020). As a caveat, it should be noted that most of these tools were originally developed in the study of mammals or yeasts, the genomes of which are much simpler than those of plants; the challenges in plant studies were not fully considered. sORF identification using RPFs is highly dependent on the accurate mapping of RPFs, and the Ribo-Seq strategy has some inherent shortcomings when it is applied in plant studies because of the short lengths of RPFs (~ 30 nt) and the complexity and repetitiveness of plant genomes. It is difficult to accurately map the short RPFs to the loci they are derived from in a complex and repetitive genome. In most of the existing studies, the solutions to this problem are either to remove the RPFs with multiple hits or to randomly retain only one of them (Hsu et al., 2016; Bazin et al., 2017; Erhard et al., 2018). However, these processes would certainly introduce errors in ORF identification, resulting in missing ORFs in the genome. Recently, a protocol profiling the footprints of two closely packed ribosomes (disomes) that can double the size of footprints was reported (Arpat et al., 2020). The RPFs of disomes (~60 nt) can somehow compromise the mapping problem caused by their short lengths; nevertheless, they are still too short to completely solve this problem, particularly in the study of polyploidy genomes. Furthermore, only ~10% of the ribosomes can be captured in disomes, with a significant bias towards rRNA and sequences encoding signal peptides; whether they can be used to identify sORFs genome-wide has not been tested. It is possible to increase the size of RPFs to ~ 90 nt by profiling the footprints of trisomes, but their representativeness should be evaluated before they can be used in sORF identification.
sORF identification using degradome sequencing
Degradome sequencing is a high-throughput method that was originally used for the identification of endogenous siRNA and miRNA targets by combining the modified rapid amplification of 5’ cDNA ends (5’-RACE) and NGS technology (Addo-Quaye et al., 2008). Briefly, 5’-3’ exoribonuclease cleaves the translating mRNAs at the last ribosomes that can be translocated codon by codon, leaving a set of truncated transcripts with both free 5’ monophosphates and 3-nt distance in length. After being sequenced with the NGS platform, this 3-nucleotide periodicity in the position of free 5’ mRNA ends could be revealed by mapping reads to mRNAs (Bertoni, 2016), which provides an alternative approach for sORF identification (Figure 1C) (Hou et al., 2016). Using genome-wide mapping of truncated mRNAs, Yu et al. (2016) discovered a 3-nt periodicity pattern throughout ORFs in Arabidopsis leaf samples, and the accumulation of cleavage events at 16 to 17 nucleotides upstream of the stop codons of both ORFs and uORFs was also observed because of ribosomal pausing during translation termination. These results, therefore, make it possible to search for potential sORFs (Yu et al., 2016). In another research, 3-nt periodicity was also observed, not only in Arabidopsis but also in Glycine max and Oryza sativa, and both novel and known uORFs were identified by searching the accumulation of 5’ RNA ends peaking upstream of the stop codons (Hou et al., 2016). While degradome-based ORF prediction relies on the 3-nt periodicity of the mapping positions of NGS reads on mRNAs, truncated RNA fragments are much longer than RPFs, making them beneficial for resolving the mapping problems in complex genomes caused by short lengths of RPFs (Carpentier et al., 2021). However, as degradome sequencing was originally developed to identify the target of sRNAs, the fact that the binding of sRNAs can result in the accumulation of degradome reads out of the translating frames might, therefore, introduce unexpected errors in the prediction of ORFs. Although ORF prediction from degradome reads is similar to that from RPFs, whether those RPF-based tools can also be used to predict ORFs from degradome reads has rarely been tested. To better utilize the degradome datasets, more tools need to be developed or tested for sORF prediction in the future.
sORF identification using nucleotide diversity
As the third nucleotides in codons are wobble nucleotides and are therefore subject to a more relaxed purification selection in nature (Hurst, 2002), resulting in higher nucleotide diversities every three nucleotides in the coding sequences (Jiang et al., 2022). This pattern resembles the 3-nt periodicity of RPFs on mRNAs and can therefore also be used to predict ORFs (Figure 1D). Jiang et al. (2022) recently developed a pipeline, OrfPP, to predict ORFs using the single-nucleotide polymorphisms (SNPs) datasets of natural populations and applied it to two polyploidy species: tetraploidy cotton (Gossypium hirsutum) and hexaploidy wheat (Triticum aestivum). As SNPs in most studies are usually called using 100 or 150 bp pair-ended reads, this strategy can overcome the troubles caused by the short lengths of RPFs in plant studies. Although SNP calling may also introduce several errors caused by the mismapping or multiple mapping of short reads, this problem can finally be solved by the future application of long reads in plant population studies. Indeed, long-read techniques have been used increasingly to detect genomic variants in natural populations of plants (Dorfner et al., 2022; Tang et al., 2022; Zhou et al., 2022). Another advantage of this method is the direct use of existing datasets (SNPs) requiring no extra experiments, such as the construction of Ribo-Seq libraries, which could be costly and technically challenging in some organisms or tissues, therefore allowing the large-scale identification of sORFs in plants with genome-wide SNP datasets. The SNP-based strategy can be used in complex genomes as long as their SNP datasets are available. Nevertheless, genome sequencing and assembly are also challenging and costly for these species, many of which do not have genome-wide SNP datasets either. The lack of SNPs prevents the application of an SNP-based approach in these species. Fortunately, for most polyploidy crops, such as oilseed, cotton, and wheat, the reference genome assembly and population resequencing have been completed, which can facilitate the identification of sORFs using the SNP-based approach.
Discussion
Although several approaches have been developed to identify sORFs, each has its own limitations, and caution should be taken in the application of different methods. Sequence conservation-based methods can only identify old and conserved sORFs but are powerless in identifying young sORFs. RPFs can only make use of short reads, thus resulting in incorrect mapping to genomes, and this problem would be even worse in a complex and repetitive genome. The degradome and SNP-based approaches take advantage of longer reads to produce high-quality unique mapping and should lead to better sORF prediction. Both the RPF and degradome-based identification are affected by the reads captured in the experiments, so the silent or lowly translated sORFs might have been missed in these data and the results can be substantially varied across different tissues or growth conditions. In contrast, the SNP-based strategy relies on the preparation of a high-quality library and can predict both active and inactive sORFs. Cross-identification using different approaches was also reported. Of the 89 conserved Arabidopsis sORFs, 39 were successfully identified by RPFs (Hsu et al., 2016). More than a quarter of the sORFs predicted from SNPs, which were actively translated, overlapped with those predicted using RPFs (Jiang et al., 2022). Thus, the SNP-based strategy is an effective approach to extending the study of sORFs, especially in complex genomes, but it requires the accumulation of nucleotide diversity in natural populations, and accuracy is also affected by the quality of the reference genome and SNPs datasets. In practice, these approaches are mutually complementary and can be chosen for different purposes. Overall, it has become increasingly clear that sORFs play important roles in various plant processes and are potential candidates for crop improvement. Although the application of Ribo-Seq in plant studies has substantially enhanced the understanding of plant sORFs, most of these studies were conducted in model species, such as Arabidopsis (Hsu et al., 2016; Mahboubi et al., 2021) and rice (Su et al., 2018; Xu et al., 2021), or crops with small and simple genomes, such as tomato (Wu et al., 2019); the sORFs in other plants, particularly polyploidy species, are poorly investigated. Given that approximately 47–70% of angiosperm species are polyploid (Masterson, 1994), more advanced techniques and algorithms are required in the future to enhance the understanding of plant sORFs.
Author contributions
JZ and WY conceived the idea. YF and MJ performed the literature search and data collection. YF and JZ wrote the manuscript. JZ and WY revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Natural Science Foundation of Hainan Province Youth Fund Project (320QN322) and Young Talent Start-up Fund of South Subtropical Crop Research Institution CATAS, the Shenzhen Commission of Science and Technology Innovation Projects (JCYJ20190808143207457, JCYJ20180305124101630, JCYJ20170818094958663), and Natural Science Foundation of Guangdong Province (2022A1515110240).
Acknowledgments
We thank Prof. Xiaoyu Yang of Shandong Agricultural University for his critical reading and comments.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Addo-Quaye, C., Eshoo, T. W., Bartel, D. P., Axtell, M. J. (2008). Endogenous siRNA and miRNA targets identified by sequencing of the arabidopsis degradome. Curr. Biol. 18, 758–762. doi: 10.1016/j.cub.2008.04.042
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Arpat, A. B., Liechti, A., De Matos, M., Dreos, R., Janich, P., Gatfield, D. (2020). Transcriptome-wide sites of collided ribosomes reveal principles of translational pausing. Genome Res. 30, 985–999. doi: 10.1101/gr.257741.119
Bashir, K., Hanada, K., Shimizu, M., Seki, M., Nakanishi, H., Nishizawa, N. K. (2014). Transcriptomic analysis of rice in response to iron deficiency and excess. Rice 7, 18. doi: 10.1186/s12284-014-0018-1
Bazin, J., Baerenfaller, K., Gosai, S. J., Gregory, B. D., Crespi, M., Bailey-Serres, J. (2017). Global analysis of ribosome-associated noncoding RNAs unveils new modes of translational regulation. Proc. Natl. Acad. Sci. 114, E10018–E10027. doi: 10.1073/pnas.1708433114
Bertoni, G. (2016). RNA Degradome studies give insights into ribosome dynamics. Plant Cell 28, 2348–2349. doi: 10.1105/tpc.16.00785
Calviello, L., Hirsekorn, A., Ohler, U. (2020). Quantification of translation uncovers the functions of the alternative transcriptome. Nat. Struct. Mol. Biol. 27, 717–725. doi: 10.1038/s41594-020-0450-4
Calviello, L., Mukherjee, N., Wyler, E., Zauber, H., Hirsekorn, A., Selbach, M., et al. (2016). Detecting actively translated open reading frames in ribosome profiling data. Nat. Methods 13, 165–170. doi: 10.1038/nmeth.3688
Carpentier, M.-C., Bousquet-Antonelli, C., Merret, R. (2021). Fast and efficient 5′P degradome library preparation for analysis of Co-translational decay in arabidopsis. Plants 10, 466.
Causier, B., Hopes, T., McKay, M., Paling, Z., Davies, B. (2022). Plants utilise ancient conserved peptide upstream open reading frames in stress-responsive translational regulation. Plant Cell Environ. 45, 1229–1241. doi: 10.1111/pce.14277
David-Assael, O., Saul, H., Saul, V., Mizrachy-Dagri, T., Berezin, I., Brook, E., et al. (2005). Expression of AtMHX, an arabidopsis vacuolar metal transporter, is repressed by the 5' untranslated region of its gene. J. Exp. Bot. 56, 1039–1047. doi: 10.1093/jxb/eri097
Dorfner, M., Ott, T., Ott, P., Oberprieler, C. (2022). Long-read genotyping with SLANG (Simple long-read loci assembly of nanopore data for genotyping). Appl. Plant Sci. 10, e11484. doi: 10.1002/aps3.11484
Erhard, F., Halenius, A., Zimmermann, C., L'Hernault, A., Kowalewski, D. J., Weekes, M. P., et al. (2018). Improved ribo-seq enables identification of cryptic translation events. Nat. Methods 15, 363–366. doi: 10.1038/nmeth.4631
Frank, M. J., Smith, L. G. (2002). A small, novel protein highly conserved in plants and animals promotes the polarized growth and division of maize leaf epidermal cells. Curr. Biol. 12, 849–853.
Hanada, K., Higuchi-Takeuchi, M., Okamoto, M., Yoshizumi, T., Shimizu, M., Nakaminami, K., et al. (2013). Small open reading frames associated with morphogenesis are hidden in plant genomes. Proc. Natl. Acad. Sci. 110, 2395–2400. doi: 10.1073/pnas.1213958110
Hanada, K., Zhang, X., Borevitz, J. O., Li, W.-H., Shiu, S.-H. (2007). A large number of novel coding small open reading frames in the intergenic regions of the Arabidopsis thaliana genome are transcribed and/or under purifying selection. Mol. Plant-Microbe Interact. 17, 632–640. doi: 10.1101/gr.5836207
Hayden, C. A., Jorgensen, R. A. (2007). Identification of novel conserved peptide uORF homology groups in Arabidopsis and rice reveals ancient eukaryotic origin of select groups and preferential association with transcription factor-encoding genes. BMC Biol. 5, 32. doi: 10.1186/1741-7007-5-32
Hou, C. Y., Lee, W. C., Chou, H. C., Chen, A. P., Chou, S. J., Chen, H. M. (2016). Global analysis of truncated RNA ends reveals new insights into ribosome stalling in plants. Plant Cell 28, 2398–2416. doi: 10.1105/tpc.16.00295
Hsu, P. Y., Calviello, L., Wu, H.-Y. L., Li, F.-W., Rothfels, C. J., Ohler, U., et al. (2016). Super-resolution ribosome profiling reveals unannotated translation events in arabidopsis. Proc. Natl. Acad. Sci. 113, E7126–E7135. doi: 10.1073/pnas.1614788113
Hurst, L. D. (2002). The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet. 18, 486–487. doi: 10.1016/S0168-9525(02)02722-1
Ingolia, N. T. (2010). Chapter 6 - genome-wide translational profiling by ribosome footprinting. Methods Enzymol. 470, 119–142. doi: 10.1016/S0076-6879(10)70006-9
Ingolia, N. T., Brar Gloria, A., Stern-Ginossar, N., Harris Michael, S., Talhouarne Gaëlle, J. S., Jackson Sarah, E., et al. (2014). Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep. 8, 1365–1379. doi: 10.1016/j.celrep.2014.07.045
Jeon, S., Lim, S., Ha, J., Kim, J. (2015). Identification of Psk2, Skp1, and Tub4 as trans-acting factors for uORF-containing ROK1 mRNA in saccharomyces cerevisiae. J. Microbiol. 53, 616–622. doi: 10.1007/s12275-015-5389-5
Jiang, M., Ning, W., Wu, S., Wang, X., Zhu, K., Li, A., et al. (2022). Three-nucleotide periodicity of nucleotide diversity in a population enables the identification of open reading frames. Briefings Bioinf. 23 (4), bbac210. doi: 10.1093/bib/bbac210
Jorgensen, R., Dorantes-Acosta, A. (2012). Conserved peptide upstream open reading frames are associated with regulatory genes in angiosperms, Vol. 2012. 3.
Lauressergues, D., Couzigou, J.-M., Clemente, H. S., Martinez, Y., Dunand, C., Bécard, G., et al. (2015). Primary transcripts of microRNAs encode regulatory peptides. Nature 520, 90–93.
Libre, C., Seissler, T., Guerrero, S., Batisse, J., Verriez, C., Stupfler, B., et al. (2021). Paillart JC. a conserved uORF regulates APOBEC3G translation and is targeted by HIV-1 vif protein to repress the antiviral factor. Biomedicines 10 (1). doi: 10.3390/biomedicines10010013
Mahboubi, A., Delhomme, N., Häggström, S., Hanson, J. (2021). Small-scale sequencing enables quality assessment of ribo-seq data: an example from arabidopsis cell culture. Plant Methods 17, 92. doi: 10.1186/s13007-021-00791-w
Masterson, J. (1994). Stomatal size in fossil plants: Evidence for polyploidy in majority of angiosperms. Science 264, 421–424. doi: 10.1126/science.264.5157.421
Ong, S. N., Tan, B. C., Al-Idrus, A., Teo, C. H. (2022). Small open reading frames in plant research: from prediction to functional characterization. 3 Biotech. 12, 76. doi: 10.1007/s13205-022-03147-w
Pajerowska-Mukhtar, K. M., Wang, W., Tada, Y., Oka, N., Tucker Chandra, L., Fonseca Jose, P., et al. (2012). The HSF-like transcription factor TBF1 is a major molecular switch for plant growth-to-Defense transition. Curr. Biol. 22, 103–112. doi: 10.1016/j.cub.2011.12.015
Saul, H., Elharrar, E., Gaash, R., Eliaz, D., Valenci, M., Akua, T., et al. (2009). The upstream open reading frame of the Arabidopsis AtMHX gene has a strong impact on transcript accumulation through the nonsense-mediated mRNA decay pathway. Plant J. 60, 1031–1042. doi: 10.1111/j.1365-313X.2009.04021.x
Si, X., Zhang, H., Wang, Y., Chen, K., Gao, C. (2020). Manipulating gene translation in plants by CRISPR–Cas9-mediated genome editing of upstream open reading frames. Nat. Protoc. 15, 338–363. doi: 10.1038/s41596-019-0238-3
Song, B., Jiang, M., Gao, L. (2021). RiboNT: A noise-tolerant predictor of open reading frames from ribosome-protected footprints. Life 11, 701. doi: 10.3390/life11070701
Song, B., Li, H., Jiang, M., Gao, Z., Wang, S., Gao, L., et al. (2023). slORFfinder: a tool to detect open reading frames resulting from trans-splicing of spliced leader sequences. Briefings Bioinf. 24 (1), bbac610. doi: 10.1093/bib/bbac610
Su, Z., Tang, Y., Ritchey, L. E., Tack, D. C., Zhu, M., Bevilacqua, P. C., et al. (2018). Genome-wide RNA structurome reprogramming by acute heat shock globally regulates mRNA abundance. Proc. Natl. Acad. Sci. 115, 12170–12175. doi: 10.1073/pnas.1807988115
Tang, D., Jia, Y., Zhang, J., Li, H., Cheng, L., Wang, P., et al. (2022). Genome evolution and diversity of wild and cultivated potatoes. Nature 606, 535–541. doi: 10.1038/s41586-022-04822-x
Tran, M. K., Schultz, C. J., Baumann, U. (2008). Conserved upstream open reading frames in higher plants. BMC Genomics 9, 361. doi: 10.1186/1471-2164-9-361
Wang, B., Hao, J., Pan, N., Wang, Z., Chen, Y., Wan, C. (2021). Identification and analysis of small proteins and short open reading frame encoded peptides in Hep3B cell. J. Proteomics 230, 103965. doi: 10.1016/j.jprot.2020.103965
Wheeler, D. L., Church, D. M., Lash, A. E., Leipe, D. D., Madden, T. L., Pontius, J. U., et al. (2002). Database resources of the national center for biotechnology information: 2002 update. Nucleic Acids Res. 30, 13–16. doi: 10.1093/nar/30.1.13
Wu, H.-W., Fajiculay, E., Wu, J.-F., Yan, C.-C. S., Hsu, C.-P., Wu, S.-H. (2022). Noise reduction by upstream open reading frames. Nat. Plants 8 (5), 474–480. doi: 10.1038/s41477-022-01136-8
Wu, H.-Y. L., Song, G., Walley, J. W., Hsu, P. Y. (2019). The tomato translational landscape revealed by transcriptome assembly and ribosome Profiling1 [OPEN]. Plant Physiol. 181, 367–380. doi: 10.1104/pp.19.00541
Wu, Q., Wright, M., Gogol, M. M., Bradford, W. D., Zhang, N., Bazzini, A. A. (2020). Translation of small downstream ORFs enhances translation of canonical main open reading frames. EMBO J. 39, e104763. doi: 10.15252/embj.2020104763
Xiao, Z., Huang, R., Xing, X., Chen, Y., Deng, H., Yang, X. (2018). De novo annotation and characterization of the translatome with ribosome profiling data. Nucleic Acids Res. 46, e61–e61. doi: 10.1093/nar/gky179
Xu, G., Greene, G. H., Yoo, H., Liu, L., Marqués, J., Motley, J., et al. (2017a). Global translational reprogramming is a fundamental layer of immune regulation in plants. Nature 545, 487–490. doi: 10.1038/nature22371
Xu, Q., Liu, Q., Chen, Z., Yue, Y., Liu, Y., Zhao, Y., et al. (2021). Histone deacetylases control lysine acetylation of ribosomal proteins in rice. Nucleic Acids Res. 49, 4613–4628. doi: 10.1093/nar/gkab244
Xu, G., Yuan, M., Ai, C., Liu, L., Zhuang, E., Karapetyan, S., et al. (2017b). uORF-mediated translation allows engineered plant disease resistance without fitness costs. Nature 545, 491–494. doi: 10.1038/nature22372
Yang, W. C., Katinakis, P., Hendriks, P., Smolders, A., de Vries, F. W., Spee, J. H., et al. (1993). HJJTPjfc, biology m. characterization of GmENOD40, a gene showing novel patterns of cell-specific expression during soybean nodule development. Plant J. 3 (4), 573–585.
Yang, X., Song, B., Cui, J., Wang, L., Wang, S., Luo, L., et al. (2021). Comparative ribosome profiling reveals distinct translational landscapes of salt-sensitive and -tolerant rice. BMC Genomics 22, 612. doi: 10.1186/s12864-021-07922-6
Yu, X., Willmann, M. R., Anderson, S. J., Gregory, B. D. (2016). Genome-wide mapping of uncapped and cleaved transcripts reveals a role for the nuclear mRNA cap-binding complex in cotranslational RNA decay in arabidopsis. Plant Cell 28, 2385–2397. doi: 10.1186/s12864-021-07922-6
Zhang, T., Wu, A., Yue, Y., Zhao, Y. (2021). uORFs: Important cis-regulatory elements in plants, Vol. 21. 6238.
Keywords: small open reading frame, sORFs, ribo-seq, plant, genome
Citation: Feng Y, Jiang M, Yu W and Zhou J (2023) Identification of short open reading frames in plant genomes. Front. Plant Sci. 14:1094715. doi: 10.3389/fpls.2023.1094715
Received: 17 November 2022; Accepted: 26 January 2023;
Published: 15 February 2023.
Edited by:
Julio Sáez-Vásquez, Université de Perpignan, FranceReviewed by:
Rémy Merret, UMR5096 Laboratoire Génome et développement des plantes, FranceThomas Blein, UMR9213 Institut des Sciences des Plantes de Paris Saclay (IPS2), France
Copyright © 2023 Feng, Jiang, Yu and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiannan Zhou, emhvdWppYW5uYW5AYWxpeXVuLmNvbQ==; Weichang Yu, d3l1QHN6dS5lZHUuY24=