 Jianli Wang
Jianli Wang Zhenfei Yan2
Zhenfei Yan2 Zhongbao Shen
Zhongbao Shen Guofeng Yang
Guofeng Yang Lichao Ma
Lichao Ma
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 07 September 2022
Sec. Plant Bioinformatics
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.998863
This article is part of the Research Topic Multi-omics and Computational Biology in Horticultural Plants: From Genotype to Phenotype View all 32 articles
There is currently international interest in applying DNA barcoding as a tool for plant species discrimination and identification. In this study, we evaluated the utility of four candidate plant DNA barcoding regions [rbcL, matK, trnL-F, and internal transcribed spacer (ITS)] in seven genera of Gramineae including Agropyron, Bromus, Elymus, Elytrigia, Festuca, Leymus, and Lolium. Fourteen accessions were analyzed, and matK and ITS showed the highest species, subspecies, and variety discriminatory power, each resolving 11 accessions. Species discrimination using rbcL and trnL-F was lower, resolving 7 and 8 accessions, respectively. Subspecies and variety discrimination using rbcL and trnL-F could not identify 4 accessions of Agropyron. A technical system can be established using the proposed DNA barcode to rapidly and reliably identify the seven genera of Gramineae. This study serves as a “useful reference” for identifying the genetic diversity of grass germplasm resources. DNA barcoding can be utilized to uncover the relatives of different species within the same family or between different families. It can also be used to determine the related groups of important herbage, turfgrass, and crops and provide crucial background information for discovering excellent genes and improving existing crop varieties.
Canadian taxonomist Paul Hebert first proposed the concept of DNA barcoding in 2003 (Hebert et al., 2003a). It involves using one or several standard and universal DNA fragments of the genome to identify species. Because of its rapid, simple, and accurate features, DNA barcoding has been adopted worldwide to facilitate DNA recognition and species identification (Hebert et al., 2003b; Kress et al., 2005; Hollingsworth et al., 2011; Miller, 2011). Species identification is an important foundation in taxonomy, diversity management, conservation biology, and other fields. The traditional species identification methods mainly relied on morphological characterization. However, morphological identification of species is time-consuming, laborious, and prone to error. Thus, the emergence of DNA barcoding technology was a breakthrough in species identification as it can be used to precisely identify an organism at the species level (Savolainen et al., 2005).
Research on DNA barcoding for animals has seen tremendous progress since its inception. Paul Hebert utilized mitochondrial gene COI to characterize 11 animal phylum and identified 13,320 species based on sequence analysis. Thus, COI gene was selected as the general DNA barcode for identifying animal species. So far, several new animal species have been identified using the COI barcode, including butterflies (Astraptes fulgerator) (Hebert et al., 2004), Mactra spp. (Chetoui et al., 2022), and Protaphorura spp. (Sun et al., 2017). Notably, mitochondrial genes evolve at a relatively slow rate in land plants and thus are not suitable for DNA barcoding in plants. Several studies have been conducted to find ideal DNA barcodes from chloroplast and nuclear genomes of plants (Cho et al., 2004; Chase et al., 2005). At the Third International Conference on DNA Barcoding held in Mexico in 2009, participants agreed that the chloroplast genome fragments rbcL and matK would serve as the core barcode for plant DNA barcoding, and the chloroplast genome fragment trnH-psbA and nuclear gene fragment ITS would serve as the supplementary barcode for plant DNA barcoding.
Plant species identification is the basis of botanical research and application. In plant taxonomy, applying plant DNA barcoding can aid in the identification of some cryptic species, as well as new species (Besse et al., 2021). Liu et al. (2011) analyzed Taxus from Eurasia using four chloroplast gene fragments and one nuclear gene fragment and identified 11 species and four new taxa. DNA barcoding has also been extensively applied to identify plant germplasm resources. For example, three DNA barcodes were used to identify six species and seven easily confused plants of the genus Sabia, and the sequence difference rate between the Sabia species and the easily confused plants was as high as 24.5% (Sui et al., 2011). For instance, by analyzing the DNA barcodes of 274 plant species belonging to 87 genera, 77 plant species were found to be misidentified (China Plant BOL Group et al., 2011).
Grassland is an important green ecological barrier on China’s land, and a vital source of livelihood for farmers and herders. It is also a primary foundation for high-quality development of pastoral areas, which occupies the largest area in the northern grassland, accounting for 40.72% of the country’s total grassland area (Dong et al., 2015). Gramineae grasses are the dominant and constructive species in China’s northern grasslands. The grasses are rich in genetic information, but with the enhancement of human activities, their genetic diversity has been gradually declining; therefore, protecting the grass germplasm resources is essential (Liu et al., 2021). Species identification is the prerequisite and basis for the protection of grass germplasm resources. DNA barcoding is a potential and effective method for identification and it gets rid of the obstacle that traditional morphological identification methods rely on long-term experience (Yang et al., 2022). The application of DNA barcoding to the identification of grass species will be an innovation in the methodology of grass resource identification. Thus, DNA barcoding can be utilized for grass germplasm identification, which is critical for the protection of the diversity of grass germplasm resources. In this study, we established a DNA barcode database and uncovered the genetic relationship of 14 accessions of gramineous grasses. The aim of the present study was to determine the best DNA barcode sequences for grass accessions that are common in China’s northern grasslands and our findings provide a “useful reference” for identifying genetic diversity of grass germplasm resources.
Seeds of 14 accessions of gramineous forage grass used in this study were provided by the Pratacultural Science Institute, Heilongjiang academy of agricultural sciences, Harbin, China (Table 1). The plant samples were grown in the greenhouse under 16 h of light (390 μE m–2 S–1) and 8 h of darkness per day at 25°C (Figure 1). Four weeks later, the shoots of three plants (representing one sample) were harvested. Subsequently, total genomic DNA was extracted from each sample using the CTAB method (Doyle, 1987). Four pairs of primers were designed using DNAMAN, according to the sequences of three chloroplast genes (matK, rbcL, and trnL-F) and one nuclear region (ITS) of Agropyron, Bromus, Elymus, Elytrigia, Festuca, Leymus, and Lolium.

Table 1. Details of the 14 grass accessions used in this study.

Figure 1. The seven genera of Gramineae.
The DNA sequences of the chloroplast and nuclear regions of the various grasses were obtained from the Gene Bank (Table 2). The primers were synthesized by Sangon Biotech (Shanghai) Co., Ltd. Polymerase chain reactions were then conducted in a 25-μL tube containing 1 μL genomic DNA (100 ng/μL), 1 μL of each primer (10 μmol/μL), 10 μL Takara Taq DNA polymerase master mix and water to a final volume of 20 μL. ITS gene was amplified under the PCR conditions of 95°C for 3 min (initial denaturation), followed by 30 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 15 s, and extension at 72°C for 1 min, and a final extension at 72°C for 10 min. Meanwhile, the three chloroplast genes were amplified at 95°C for 3 min, then 30 cycles at 95°C for 30 s, annealing at 53°C for 15 s, extension at 72°C for 30 s, and a final extension step at 72°C for 5 min (Di et al., 2015). Subsequently, sequencing reactions were performed by Sangon Biotech (Shanghai) Co., Ltd.

Table 2. Primer sequences.
Haplotype analysis of the 11 species was performed by comparing the sequence matrices of the inland, coastal, and total samples using MEGA X to obtain a K2P genetic distance matrix (Kumar et al., 2018). Haplotype analysis was performed using DNAsp v6.12.03 (Rozas et al., 2017) for the sequences of ITS, matK, rbcL, and trnL-F, respectively. The number of haplotypes showed included ITS, matK, rbcL, and trnL-F. The species delimitation was based on the sequence length and haplotype of DNA barcodes.
Evolutionary analysis of the plants belonging to seven genera of Gramineae was conducted using MEGA X. Sequence alignment was initially performed using Clustal W (Kumar et al., 2018). The evolutionary history was inferred using the Neighbor-Joining method (Saitou and Nei, 1987). The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) are shown next to the branches (Felsenstein, 1985). The evolutionary distances were computed using the number of differences method (Nei and Kumar, 2000) and expressed as the number of base differences per sequence. The rate variation among sites was modeled with a gamma distribution (shape parameter = 1). The proportion of sites where at least 1 unambiguous base is present in at least 1 sequence for each descendent clade is shown next to each internal node in the tree. All ambiguous positions were removed for each sequence pair (pairwise deletion option). The evolutionary analyses involved 8, 8, and 11 nucleotide sequences, as well as 706, 1,479, and 2,201 positions in the ITS, cpDNAs, and 4-DNA dataset, respectively.
The primer sequences of ITS, matK, trnL-F, and rbcL were designed to allow amplification within the target regions and did not need any modification (Table 2). A total of 56 sequences were obtained from the 14 processed specimens (14 from each target gene). The size of the sequences ranged from 682 to 701 bp for ITS, 395 to 408 bp for matK, 444 to 473 bp for trnL-F, and 572 bp for rbcL (Tables 3–5). Haplotype analysis showed that 8 haplotypes were included in ITS, matK, and rbcL of 8 grass genera, and 7 haplotypes were included in trnL-F of 8 grass genera (Table 3). Between different species or subspecies of the same genus, 4 haplotypes were included in ITS and matK, 1 haplotype was included in rbcL, and 2 haplotypes were included in trnL-F (Table 4). Between the different varieties of Lolium perenne, 1 haplotype was included in ITS, matK, rbcL, and trnL-F (Table 5).
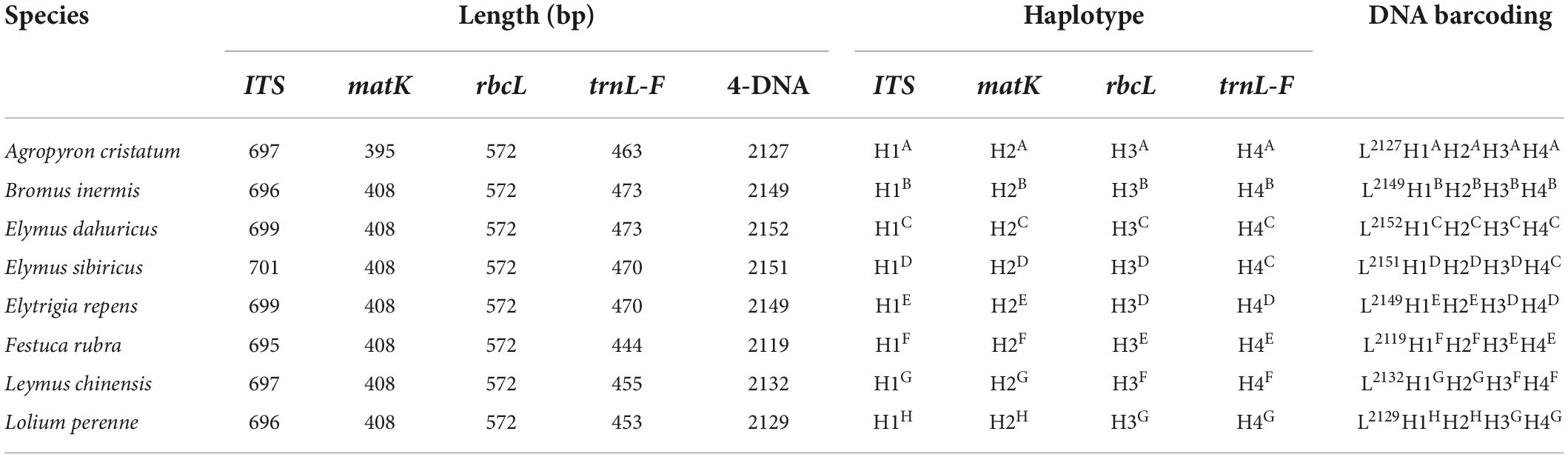
Table 3. Database of DNA barcoding for eight species grasses.
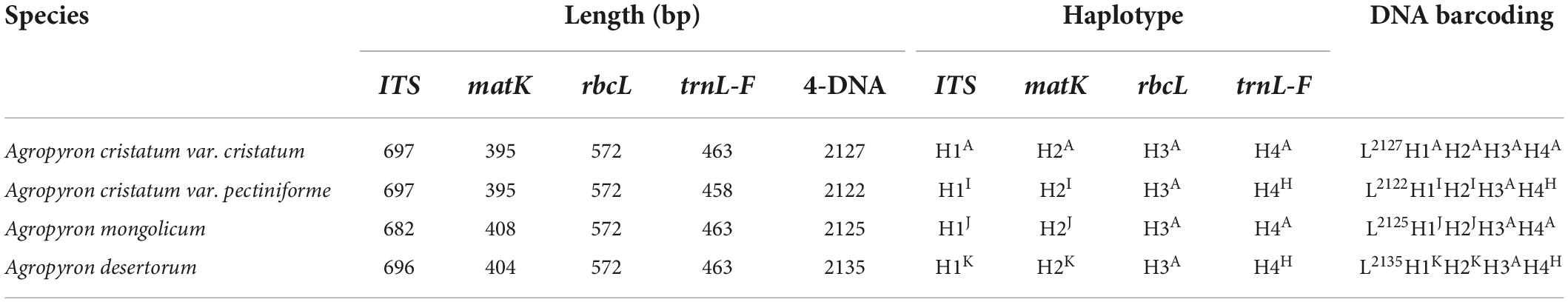
Table 4. Database of DNA barcoding for four samples of Agropyron species.
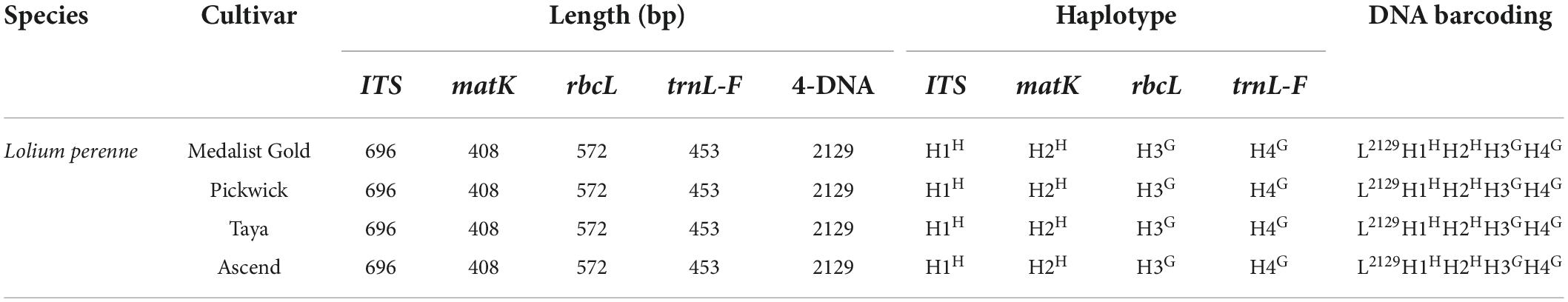
Table 5. Database of DNA barcoding for four varieties of Lolium perenne.
A DNA barcoding database was generated based on the differences in base number and haplotype of 4 gene fragments of ITS, matK, trnL-F, and rbcL in 8 grass varieties. The results showed that DNA barcodes of different species of the same genus were different (Table 3). Also, DNA barcodes of different subspecies of the same species were different (Table 4). However, the DNA barcodes of different varieties of the same species were the same (Table 5). Notably, DNA barcoding did not reveal any differences within species, but large differences among species were observed, enabling identification.
The ITS-based phylogenetic tree of the 8 grass varieties on the 706-bp alignment is shown in Figure 2A. The optimal tree with the sum of branch length being equal to the 170.97 was generated. The results showed that all the 8 grass varieties formed a monophyletic clade with a high bootstrap value. Elymus sibiricus and Elytrigia repens clustered within the same subclade, suggesting that they may have closer genetic relationships than E. sibiricus and E. dahuricus. Festuca rubra and Lolium perenne also clustered within the same subclade and were separated from the other six species, suggesting that they are more closely related in evolution.
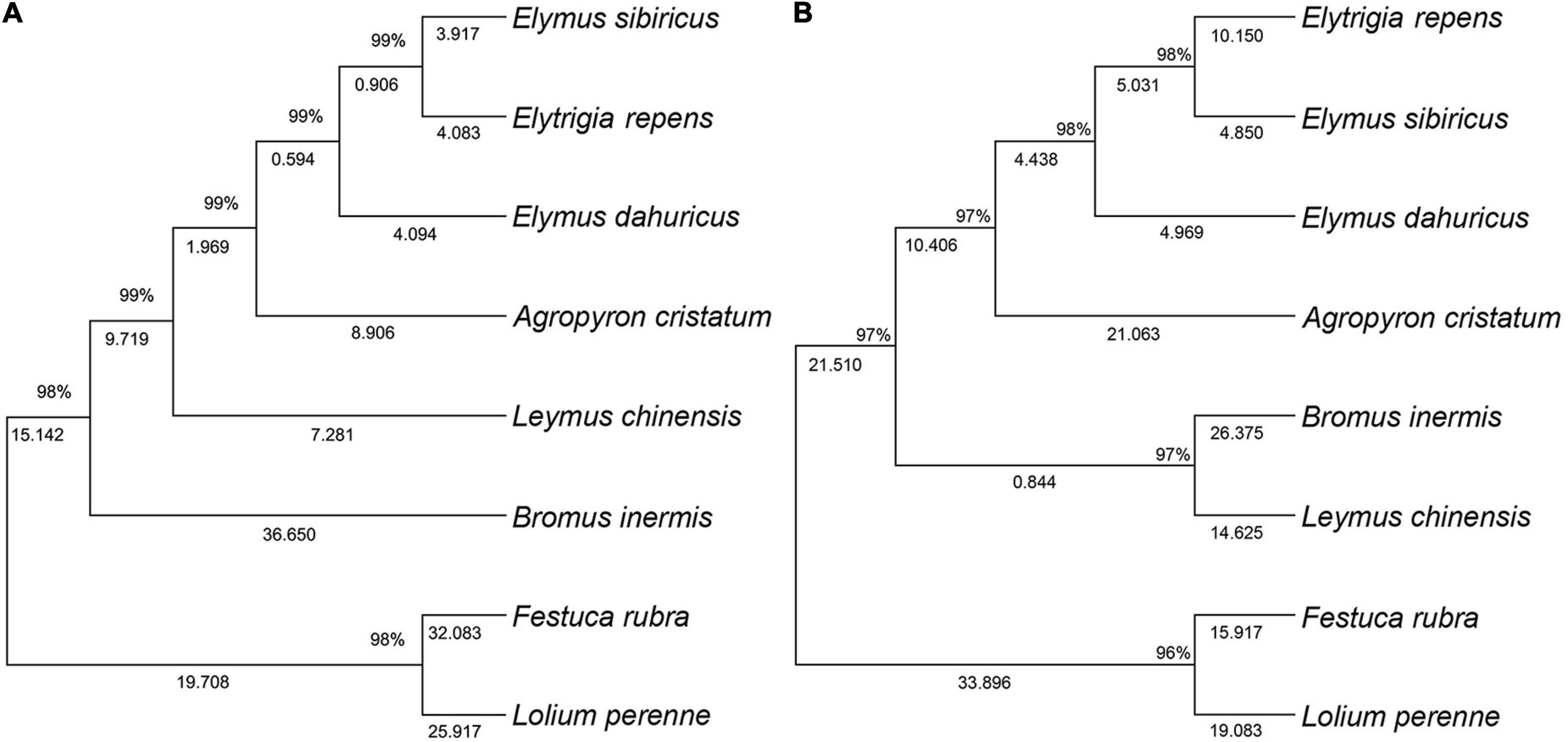
Figure 2. Topology resulting from Neighbor-Joining analysis of ITS and 3-cpDNA in the seven genera of Gramineae. One ITS dataset (A), 3-cpDNA dataset (matK, rbcL, and trnL-F) (B).
The 3-cpDNA tree of Melilotus based on 1,479-bp of concatenated plastid sequences (rbcL, matK, and trnL-F) is shown in Figure 2B. Similar to the ITS tree for the 8 grass varieties, the 3-cpDNA tree indicated that all the 8 grass varieties formed a monophyletic clade with a high bootstrap value. E. sibiricus and E. repens also clustered within the same subclade, confirming that they may have closer genetic relationships than E. sibiricus and E. dahuricus. Meanwhile, F. rubra and L. perenne clustered within the same subclade with a single clade. The only difference compared to the ITS tree is a single subclade of Bromus inermis and Leymus chinensis, suggesting that the two are more closely related.
The 4-gene tree of 11 forage species yielded 2,998 bp of four concatenated genes (rbcL, matK, trnL-F, and ITS) (Figure 3). The results showed that the two variants of Agropyron clustered in the same subclade, and the 4 species of Agropyron clustered together. Notably, the evolutionary relationship of the other grasses was similar to that of the ITS tree and the 3-cpDNA tree.
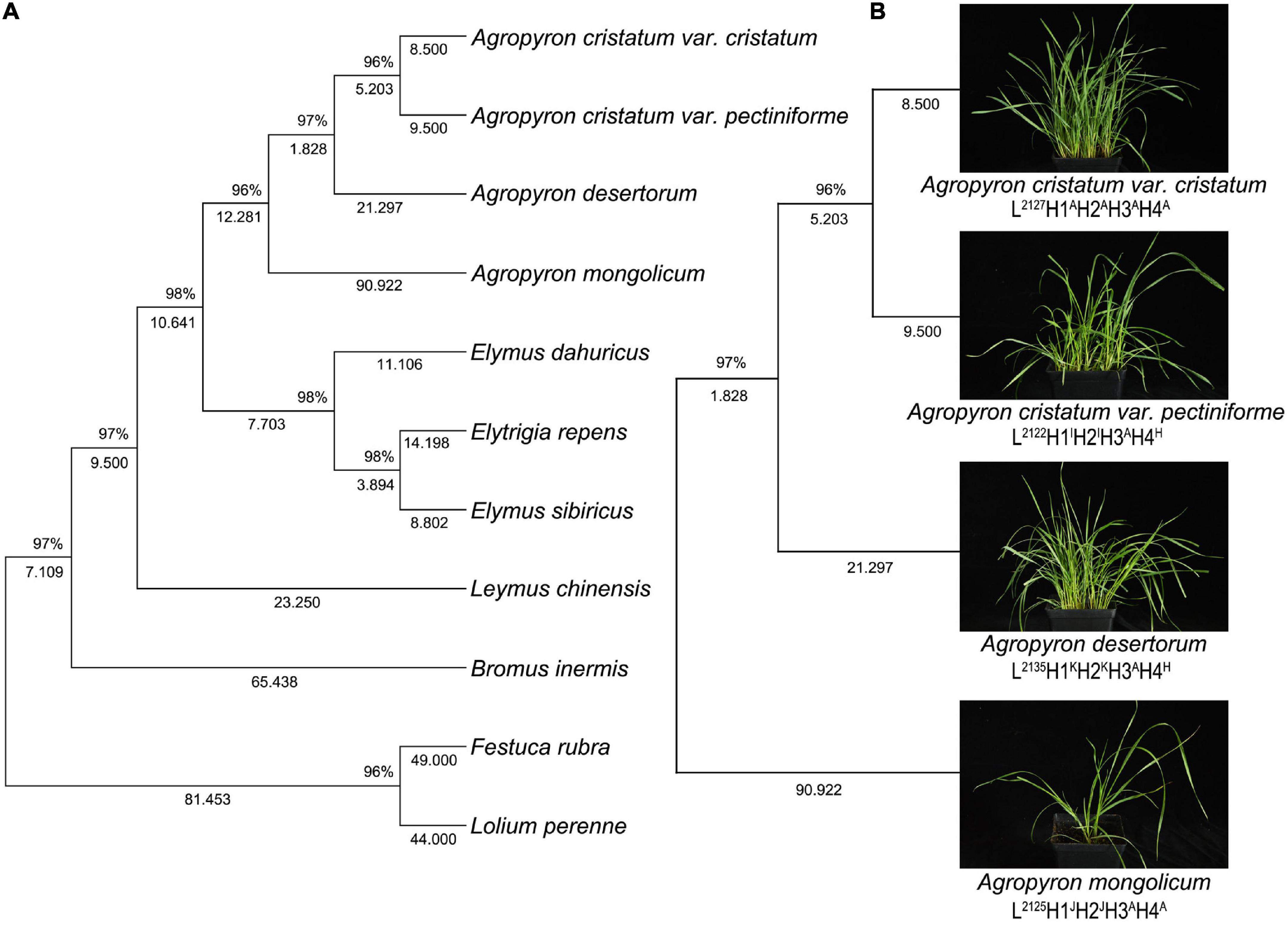
Figure 3. Topology resulting from Neighbor-Joining analysis of one 4-gene dataset using MEGA X. Neighbor-Joining analysis of one 4-gene dataset of 14 samples of grass belonging to Gramineae (A), neighbor-Joining analysis of one 4-gene dataset in 4 samples of Agropyron species (B).
Comparison of the genetic diversity of grass germplasm resources in different regions can reveal the distribution rules of different taxa, determine the center of diversity distribution, and provide guidance for the collection and conservation of grass germplasm resources. Chloroplast genes matK and rbcL were proposed as the candidate sequences for plant DNA barcoding by The Plant Working Group of the Consortium for the Barcode of Life (CBOL) (Kress and Erickson, 2007; CBOL Plant Working Group, 2009). However, in large scale studies, matK and rbcL provide a discriminatory efficiency at the species level of 72 and 49.7%, respectively, and they often fail to differentiate closely related species (Kress and Erickson, 2007; Ferri et al., 2009; Li et al., 2009; China Plant BOL Group et al., 2011; Hollingsworth et al., 2011; Liu et al., 2011; Peterson et al., 2014).
matK is characterized by rapid evolution and a high ability of interspecific identification, but the primer is not universal (Arca et al., 2012). Meanwhile, rbcL has high generality, easy amplification, and comparability, but its discriminatory efficiency at the species is not efficient. As a result, other chloroplast regions such as trnH-psbA, trnL, trnL-F (Fazekas et al., 2008; Arca et al., 2012) and the nuclear ribosomal Internal Transcribed Spacer (ITS) region are routinely used as supplementary barcodes alongside matK and rbcL (Sass et al., 2007; Fazekas et al., 2008).
Diversity is key in the protection of grass germplasm resources. Evaluation of genetic diversity is crucial for the protection of grass germplasm resources and plays a guiding role in formulating the next protection objects and methods. Current genetic diversity evaluations mainly focus on understanding the genetic diversity within species by analyzing DNA markers such as RFLP (Restriction Fragment Length Polymorphism), AFLP (Amplified Fragment Length Polymorphism), RAPD (Random Amplification Polymorphic DNA), and SSR (Simple Sequence Repeats) (Brummer et al., 1995). However, a unified appraisal evaluation system has not been formulated because of the diversification of detection methods and the lack of universality of inter-species data.
Plant DNA barcodes are suitable for classification levels above species. However, significantly different populations within species can also be identified with high versatility in some taxa. Analyzing the genetic diversity of grass germplasm resources using DNA barcodes can reveal the genetic relationship between different species within a family and between different families. Moreover, DNA barcoding can be used to determine the relative groups of important pastures, turfgrass, and crops and uncover important background information for the discovery of excellent genes and superior varieties. Comparing the genetic diversity of grass germplasm resources in different regions can reveal the distribution rules of different groups, determine the diversified distribution center, and provide guidance for collecting and protecting grass germplasm resources.
Previous studies showed that the barcodes matK and rbcL had about 50% correct assignment rate (CAR) in grasses (Li et al., 2015; Loera-Sánchez et al., 2020). The low CARs for grass DNA barcodes could be due to various factors. Some grass species, such as Poa spp., are notoriously hard to discriminate morphologically and their phylogeny is subject to controversy. This could have resulted in misidentified reference sequences (Loera-Sánchez et al., 2020). Another factor is the high genetic similarity between some grass taxa. This may result in a higher proportion of incorrect taxonomic assignments for such grass species (Meyer and Paulay, 2005; Loera-Sánchez et al., 2020). Our results showed that the highest CAR for grasses was 100% with matK followed by rbcL (87.5%; Table 6). ITS, matK, rbcL, and trnL-F genes were make for good candidate for large-scale DNA barcoding of some grasses. However, further work is needed to produce reference sequences in more grass species of Gramineae.
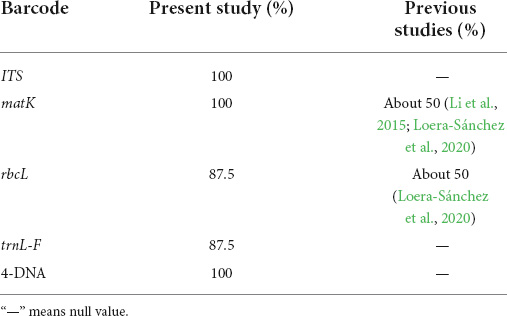
Table 6. Species-level assignment success by barcode.
In this study, we utilized highly conserved universal primers to obtain ideal DNA barcoding sequences. ITS, matK, rbcL, and trnL-F genes were selected and used in combination to identify gramineous forages. The bases of the four gene fragments and the haplotype combination of the marker sites constituted the DNA identification code. Each forage has its own specific DNA. The identification success rate at the genus and species levels was 100%. However, this combination method could not identify different varieties of the same grass species. For example, the four perennial ryegrass varieties, including Medalist Gold, Pickwick, Taya, and Ascend have a common DNA identification code.
In this study, eight forage species were identified through polymorphic locus analysis, haplotype delineation, and different haplotype combinations of marker loci. The K2P model was used to construct a phylogenetic tree, which classified the eight forage species into different clades. Combining ITS, matK, rbcL, and trnL-F had a significantly higher identification effect than using a single fragment. The monophyly of each species of Gramineae was verified based on auxiliary analysis of the phylogenetic tree. Our results meet the requirements of DNA barcoding to locate species in a taxonomic system (family, genus, etc.) with sufficient phylogenetic information.
The original contributions presented in this study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
JW and ZY conducted the experiments. PZ and ZS give advice and assistance in this research. GY revised the manuscript. LM designed the experiments and wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Foundation Project of Shandong Natural Science Foundation (ZR2020MC172), the Fundamental Research Funds for the Universities (6631119040), and “First Class Grassland Science Discipline” programme of Shandong Province.
We thank GY for his comments and guidance on the article and thank the National Foundation and the Key Laboratory of the Yellow River Delta Grassland Resources and Ecology of the Chinese Forestry and Grassland Administration for the support of this project.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Arca, M., Hinsinger, D. D., Cruaud, C., Tillier, A., Bousquet, J., and Frascaria-Lacoste, N. (2012). Deciduous trees and the application of universal DNA barcodes: A case study on the circumpolar Fraxinus. PLoS One 7:e34089. doi: 10.1371/journal.pone.0034089
Besse, P., Da Silva, D., and Grisoni, M. (2021). Plant DNA barcoding principles and limits: A case study in the genus Vanilla. Methods Mol. Biol. 2222, 131–148. doi: 10.1007/978-1-0716-0997-2_8
Brummer, E. C., Bouton, J. H., and Kochert, G. (1995). Analysi s of annual medicargo species using RAPD markers. Genome 38, 362–367. doi: 10.1139/g95-047
CBOL Plant Working Group (2009). A DNA barcode for land plants. Proc. Natl. Acad. Sci. U.S.A. 106, 12794–12797. doi: 10.1073/pnas.0905845106
Chase, M. W., Salamin, N., Wilkinson, M., Dunwell, J. M., Kesanakurthi, R. P., Haider, N., et al. (2005). Land plants and DNA barcodes: Short-term and long-term goals. Proc. Biol. Sci. 360, 1889–1895. doi: 10.1098/rstb.2005.1720
Chetoui, I., Baraket, G., Tir, M., Lekired, A., Boussaid, M., Cafsi, M. E., et al. (2022). Molecular signature of phylogenetic relationships and demographic history of Tunisian Mactra stultorum: Evidence from mitochondrial and nuclear DNA data. Zoology 151:125989. doi: 10.1016/j.zool.2021.125989
China Plant BOL Group, Li, D. Z., Gao, L. M., Li, H. T., Wang, H., Ge, X. J., et al. (2011). Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants. Proc. Natl. Acad. Sci. U.S.A. 108, 19641–19646. doi: 10.1073/pnas.1104551108
Cho, Y., Mower, J. P., Qiu, Y. L., and Palmer, J. D. (2004). Mitochondrial substitution rates are extraordinarily elevated and variable in a genus of flowering plants. Proc. Natl. Acad. Sci. U.S.A. 101, 17741–17446. doi: 10.1073/pnas.0408302101
Di, H., Duan, Z., Luo, K., Zhang, D., Wu, F., Zhang, J., et al. (2015). Interspecific phylogenic relationships within genus Melilotus based on nuclear and chloroplast DNA. PLoS One 10:e0132596. doi: 10.1371/journal.pone.0132596
Dong, X. B., Dai, G. S., Ulgiati, S., Na, R., Zhang, X. S., Kang, M. Y., et al. (2015). On the relationship between economic development, environmental integrity and well-being: The point of view of herdsmen in Northern China grassland. PLoS One 10:e0134786. doi: 10.1371/journal.pone.0134786
Doyle, J. J. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15.
Fazekas, A. J., Burgess, K. S., Kesanakurti, P. R., Graham, S. W., Newmaster, S. G., Husband, B. C., et al. (2008). Multiple multilocus DNA barcodes from the plastid genome discriminate plant species equally well. PLoS One 3:e2802. doi: 10.1371/journal.pone.0002802
Felsenstein, J. (1985). Confidence limits on phylogenies: An approach using the bootstrap. Evolution 39, 783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x
Ferri, G., Alù, M., Corradini, B., and Beduschi, G. (2009). Forensic botany: Species identification of botanical trace evidence using a multigene barcoding approach. Int. J. Legal Med. 123, 395–401. doi: 10.1007/s00414-009-0356-5
Hebert, P. D. N., Cywinska, A., Ball, S. L., and deWaard, J. R. (2003a). Biological identifications through DNA barcodes. Proc. Biol. Sci. 270, 313–321. doi: 10.1098/rspb.2002.2218
Hebert, P. D. N., Ratnasingham, S., and deWaard, J. R. (2003b). Barcoding animal life: Cytochrome c oxidase subunit 1 divergences among closely related species. Proc. Biol. Sci. 270(Suppl. 1) S96–S99. doi: 10.1098/rsbl.2003.0025
Hebert, P. D. N., Penton, E. H., Burns, J. M., Janzen, D. H., and Hallwachs, W. (2004). Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc. Natl. Acad. Sci. U.S.A. 101, 14812–14817. doi: 10.1073/pnas.0406166101
Hollingsworth, P. M., Graham, S. W., and Little, D. P. (2011). Choosing and using a plant DNA barcode. PLoS One 6:e19254. doi: 10.1371/journal.pone.0019254
Kress, W. J., and Erickson, D. L. (2007). A two-locus global DNA barcode for land plants: The coding rbcL gene complements the non-coding trnH-psbA spacer region. PLoS One 2:e508. doi: 10.1371/journal.pone.0000508
Kress, W. J., Wurdack, K. J., Zimmer, E. A., Weigt, L. A., and Janzen, D. H. (2005). Use of DNA barcodes to identify flowering plants. Proc. Natl. Acad. Sci. U.S.A. 102, 8369–8374. doi: 10.1073/pnas.0503123102
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Li, F. W., Tan, B. C., Buchbender, V., Moran, R. C., Rouhan, G., Wang, C. N., et al. (2009). Identifying a mysterious aquatic fern gametophyte. Plant Syst. Evol. 281, 77–86. doi: 10.1007/s00606-009-0188-2
Li, X. W., Yang, Y., Henry, R. J., Rossetto, M., Wang, Y. T., and Chen, S. L. (2015). Plant DNA barcoding: From gene to genome. Biol. Rev. Camb. Philos. Soc. 90, 157–166. doi: 10.1111/brv.12104
Liu, J., Möller, M., Gao, L. M., Zhang, D. Q., and Li, D. Z. (2011). DNA barcoding for the discrimination of Eurasian yews (Taxus L., Taxaceae) and the discovery of cryptic species. Mol. Ecol. Resour. 11, 89–100. doi: 10.1111/j.1755-0998.2010.02907.x
Liu, Z. P., Zhou, Q., Liu, W. X., Zhang, J. Y., Xie, W. G., Fang, L. F., et al. (2021). Some scientific issues of forage breeding in China. Acta Pratacult. Sin. 30, 184–193. doi: 10.11686/cyxb2021314
Loera-Sánchez, M., Studer, B., and Kölliker, R. (2020). DNA barcode trnH-psbA is a promising candidate for efficient identification of forage legumes and grasses. BMC Res. Notes 13:35. doi: 10.1186/s13104-020-4897-5
Meyer, C. P., and Paulay, G. (2005). DNA barcoding: Error rates based on comprehen-sive sampling. PLoS Biol. 3:e422. doi: 10.1371/journal.pbio.0030422
Miller, S. E. (2011). DNA barcoding and the renaissance of taxonomy. Proc. Natl. Acad. Sci. U.S.A. 104, 4775–4776. doi: 10.1073/pnas.0700466104
Nei, M., and Kumar, S. (2000). Molecular Evolution and Phylogenetics. New York, NY: Oxford University Press.
Peterson, P. M., Romaschenko, K., and Soreng, R. J. (2014). A laboratory guide for generating DNA barcodes in grasses: A case study of Leptochloa s.l. (Poaceae: Chloridoideae). Webbia. 69, 1–12. doi: 10.1080/00837792.2014.927555
Rozas, J., Ferrer-Mata, A., Sánchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Saitou, N., and Nei, M. (1987). The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Sass, C., Little, D. P., Stevenson, D. W., and Specht, C. D. (2007). DNA barcoding in the cycadales: Testing the potential of proposed barcoding markers for species identification of cycads. PLoS One 2:e1154. doi: 10.1371/journal.pone.0001154
Savolainen, V., Cowan, R. S., Vogler, A. P., Roderick, G. K., and Lane, R. (2005). Towards writing the encyclopedia of life: An introduction to DNA barcoding. Proc. Biol. Sci. 360, 1805–1811. doi: 10.1098/rstb.2005.1730
Sui, X. Y., Huang, Y., Tan, Y., Guo, Y., Guo, Y., and Long, C. L. (2011). Molecular authentication of the ethnomedicinal plant Sabia parviflora and its adulterants by DNA barcoding technique. Planta Med. 77, 492–496. doi: 10.1055/s-0030-1250468
Sun, X., Zhang, F., Ding, Y., Davies, T. W. L. Y., and Wu, D. H. (2017). Delimiting species of Protaphorura (Collembola: Onychiuridae): Integrative evidence based on morphology, DNA sequences and geography. Sci. Rep. 7:8261. doi: 10.1038/s41598-017-08381-4
Keywords: DNA barcoding, haplotype, rbcL, matK, trnL-F, ITS
Citation: Wang J, Yan Z, Zhong P, Shen Z, Yang G and Ma L (2022) Screening of universal DNA barcodes for identifying grass species of Gramineae. Front. Plant Sci. 13:998863. doi: 10.3389/fpls.2022.998863
Received: 20 July 2022; Accepted: 18 August 2022;
Published: 07 September 2022.
Edited by:
Yunpeng Cao, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Rui Dong, Guizhou University, ChinaCopyright © 2022 Wang, Yan, Zhong, Shen, Yang and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guofeng Yang, eWFuZ2dmQHFhdS5lZHUuY24=; Lichao Ma, bWEtbGljaGFvQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.