 Jinyang Liu
Jinyang Liu Jingbin Chen
Jingbin Chen Qiang Yan
Qiang Yan Ranran Wu
Ranran Wu Xin Chen
Xin Chen Xingxing Yuan
Xingxing Yuan- Institute of Industrial Crops, Jiangsu Academy of Agricultural Sciences/Jiangsu Key Laboratory for Horticultural Crop Genetic Improvement, Nanjing, China
Although mungbean (Vigna radiata (L.) R. Wilczek) is an important legume crop, its seed yield is relatively low. To address this issue, here 196 accessions with 3,607,508 SNP markers were used to identify quantitative trait nucleotides (QTNs), QTN-by-environment interactions (QEIs), and their candidate genes for seed length (SL), seed width, and 100-seed weight (HSW) in two environments. As a result, 98 QTNs and 20 QEIs were identified using 3VmrMLM, while 95, >10,000, and 15 QTNs were identified using EMMAX, GEMMA, and CMLM, respectively. Among 809 genes around these QTNs, 12 were homologous to known seed-development genes in rice and Arabidopsis thaliana, in which 10, 2, 1, and 0 genes were found, respectively, by the above four methods to be associated with the three traits, such as VrEmp24/25 for SL and VrKIX8 for HSW. Eight of the 12 genes were significantly differentially expressed between two large-seed and two small-seed accessions, and VrKIX8, VrPAT14, VrEmp24/25, VrIAR1, VrBEE3, VrSUC4, and Vrflo2 were further verified by RT-qPCR. Among 65 genes around these QEIs, VrFATB, VrGSO1, VrLACS2, and VrPAT14 were homologous to known seed-development genes in A. thaliana, although new experiments are necessary to explore these novel GEI-trait associations. In addition, 54 genes were identified in comparative genomics analysis to be associated with seed development pathway, in which VrKIX8, VrABA2, VrABI5, VrSHB1, and VrIKU2 were also identified in genome-wide association studies. This result provided a reliable approach for identifying seed-size-related genes in mungbean and a solid foundation for further molecular biology research on seed-size-related genes.
Background
Mungbean (Vigna radiata (L.) R. Wilczek) is a basic source of protein and carbohydrate, as it contains approximately 20% protein and 75% carbohydrate, and is a traditional and important legume in Asia (Somta et al., 2007). Due to its short life cycle (60–75 days), relative drought tolerance, and the ability to restore atmospheric nitrogen in association with Rhizobium/Bradyrhizobium bacteria, mungbean plays a crucial role in cropping systems and soil improvement (Somta et al., 2007; Alam et al., 2014).
The crop is generally grown as a cash crop in cereal-based farming systems. However, the major constraint in mungbean production is low seed yield. The average seed yield of mungbean is only approximately 700 kg per ha (Islam et al., 2015). Therefore, improving seed yield is the main goal in mungbean breeding. Understanding the genetic basis underlying seed-size-related traits is critical for the genetic improvement of mungbeans. In mungbeans, the ideotype of high-yielding cultivars are generally characterized by a large seed size, a short and synchronous maturity, a low sensitivity or insensitivity to day length, and the resistances to insects and disease (Fernandez et al., 1988). However, the knowledge on genes related to seed size has been limited. Moreover, the genes involved in the pathway of seed developments are not yet fully known.
Seed weight is the most important yield component and directly proportional to seed yield per plant in mungbean. To date, there have been seven studies of QTLs for seed weight in mungbean. Most of these studies are based on bi-parental segregation populations derived from interspecific crosses between cultivated and wild (V. radiata var. sublobata) mungbeans, and only two studies have evaluated seed size in more than one environments. The number of QTLs identified in those studies ranged from 3 to 11. Humphry et al. (2010) reported 11 loci for seed weight using SSR-marks, and Mei et al. (2009) identified a major QTL associated with both bruchid resistance and seed mass. Nonetheless, no candidate gene was identified for this trait.
Although many genes for seed weight have been reported in Arabidopsis (Plackett et al., 2012; Ge et al., 2016; Lu et al., 2016; Cheng et al., 2018; Zhang et al., 2020), soybeans, and rice (Luo et al., 2013; Ge et al., 2016; Liu et al., 2020a; Hao et al., 2021; Nguyen et al., 2021), few genes were reported in mungbean. In Arabidopsis, FATB (Bonaventure et al., 2003) was involved in the synthesis of short-chain fatty acids and influenced seed development. Although GA20OX regulated Arabidopsis in late floral development (Plackett et al., 2012), the overexpression of GmGA20OX in Arabidopsis enhanced seed size and weight. KIX8 controlled seed size in Arabidopsis and soybeans (Liu et al., 2020a; Nguyen et al., 2021). BES1 suppressed the cell elongation and increased seed size in legume species (Ge et al., 2016). ERG2 promoted early seed development and influenced the length of mature siliques (Cheng et al., 2018). In soybeans, GA20OX (Lu et al., 2016), GmFAD3 (Singh et al., 2011), GmLEC2 (Manan et al., 2017), GmPDAT (Liu et al., 2020c), GmKIX8-1 (Nguyen et al., 2021), and GmGA3ox1 (Hu et al., 2022) were found to influence seed size by regulating lipid accumulation or increasing cell proliferation. In rice, D1 (Sun et al., 2018), D2 (Fang et al., 2016), flo2 (She et al., 2010), GS3 (Sun et al., 2018), OsBZR1 (Liu et al., 2021), GW2 (Hao et al., 2021), D11 (Wu et al., 2016), and OsHT (Guo et al., 2020) were found to control seed weight by regulating rice grain size or starch quality.
Knowledge regarding seed development pathway is also a valuable source for transgenic strategies to improve crop production. As reported, there are several signaling pathways that control seed size, including the G-protein signaling, ubiquitin proteasome pathways, mitogen-activated protein kinase (MAPK) signaling, auxin pathways, and some transcriptional regulators (Li et al., 2019). In Arabidopsis, GPA1, AGB, and AGG3 were involved in G-protein-signaling pathways. DA1, DA2, SOD2, UBP15, EOD1, and SAMBA were involved in ubiquitin proteasome pathways. In addition, ABA2, ABI5, SHB1, MINI3, IKU2, and CKX were involved in the HAIKU (IKU) pathway. Additional genes were found to be related to seed size developments, but their pathways are uncertain, such as KIX8, BES1, MES1, and KLU (Orozco-Arroyo et al., 2015; Li et al., 2019). However, some reports have been focused on genetic foundation and molecular mechanism of seed developments in mungbean.
Genome-wide association studies (GWASs), along with multi-omics analysis, have been frequently used to mine candidate genes for most important agronomic traits in crops. Integrating GWAS with comparative genomics, transcriptome analysis, and molecular experiments, genes have been identified to be associated with complex traits (Liu et al., 2020c). For example, Gong et al. (2022) conducted a GWAS with high-quality single nucleotide polymorphism (SNP) data and seed-size traits, and found that Cla97C05G104360 and Cla97C05G104380, which are involved in abscisic acid metabolism, played important role in regulating the seed size in watermelon. Duan et al. (2022) identified GmST05 to be associated with soybean seed size through the GWAS of 1800 soybean germplasm resources, and GmST05 differed significantly at the transcriptional level. Liu et al., 2022a,c used GWASs and biological experiments to identify a pleiotropic gene GmPDAT for seed size- and oil-related traits in soybean, and a salt-stress-tolerance gene VrFRO8 in mungbean. Nonetheless, the related genes responsible for seed-size-related traits remained unknown in mungbean.
To address the above issues, 196 mungbean accessions with 3,607,508 SNP markers were used to conduct GWAS for seed length (SL), seed width (SW), 100-seed weight (HSW) using 3VmrMLM (Li et al., 2022b), efficient mixed-model association expedited (EMMAX) (Kang et al., 2010), genome-wide efficient mixed-model association (GEMMA) (Zhou and Stephens, 2012), and compressed mixed linear model (CMLM) (Zhang et al., 2010) methods. Candidate genes around quantitative trait nucleotides (QTNs) and QTN-by-environment interactions (QEIs) for the three traits were predicted by transcriptomics and comparative genomics. Key candidate genes were verified by RT-PCR analysis. Moreover, genes in seed-development-regulation pathway were also mined by comparative genomics. It should be noted that VrEmp24/25 and VrKIX8 were found to be associated with SL and HSW, and a major gene VrPAT14 (LOD = 61.95, r2 = 5.80%) was identified in QEI detection via 3VmrMLM.
Materials and methods
Plant materials and treatments
A diverse set of 196 mungbean accessions including 20 wild and 176 cultivated accessions from 23 countries, were used in this study (Supplementary Data Set 1). All the accessions were planted in a randomized complete block design with two replicates in an experimental field of Kasetsart University, Kamphaeng Saen Campus, Nakhon Pathom, Thailand in 2018 and 2020. In each replicate, each accession was planted in a single row 2.5 m long with 12.5 cm intra-row spacing (ca. 20 plants/row) and 50 cm inter-row spacing. Cultural practices were performed according to Park (1978). SW (mm), SL (mm), and HSW (g) were measured. At maturity. The SL and SW traits for each accession were averaged based on 20 seeds and 100SW for each accession was averaged based on three replicates.
Whole-genome resequencing
The young leaves of the above 196 mungbean accessions were collected 1 week after planting. The DNA was extracted in 2018, using the CTAB method (Smith et al., 2005). Short reads sequenced by an Illumina HiSeq 4000 platform (Illumina, San Diego, CA, United States), and mapped to scaffolds using Burrows-Wheeler-Alignment Tool (BWA) (Version 0.7.15)1 (Li and Durbin, 2009). Genome Analysis Toolkit (GATK) was used to select SNP and indel2 (McKenna et al., 2010). Sulv 1 genome was selected as the reference genome in the GATK analysis (Yan et al., 2020). High-quality SNPs and Indel variations were obtained as the following steps. (a) Retaining concordant sites both identified by GATK and VCFtools were retained (Danecek et al., 2011). (b) Filtering out SNP with quality value below 30, removing SNPs with an average coverage depth < 8× and with minor allele frequency (MAF) less than 5%. (c) Deleting insertions and deletions (InDels) with length less than10 bp were deleted. A total of 3,607,508 SNPs were identified.
As described in Liu et al. (2022a), the number of subpopulations was five (K = 5), and the population structure (Q matrix) was calculated using ADMIXTURE software (version is 1.3.0).3 The K matrix was calculated using the above CMLM (GAPIT version 3),4 EMMAX (GAPIT),5 GEMMA (Version 0.94.1)6, and 3VmrMLM programs (IIIVmrMLM)7 (Supplementary Data Set 2; Li et al., 2022a).
Genome-wide association study for seed width, seed length, and 100-seed weight
Only the SNPs with MAF ≥ 0.05 and missing rate < 10% were used in GWAS (Pongpanich et al., 2010). The lines with more than 95% missing for trait were filtered out (Liaw and Wiener, 2002). SW, SL, and HSW, and the above SNP markers in 196 mungbean accessions were used to conduct GWAS using four different methods, including 3VmrMLM (Li et al., 2022b) via software IIIVmrMLM (Li et al., 2022a), EMMAX (Kang et al., 2010), GEMMA (Zhou and Stephens, 2012), and CMLM (Zhang et al., 2010). The probability threshold for significant QTNs was set at 1/m = 2.77e-07 (m = 3,607,508) for all the four methods (Xu et al., 2018; Zhang Y. M. et al., 2019; Zhang Y. M. et al., 2019), and the LOD score threshold for suggested QTNs was set at LOD ≥ 3.0 for 3VmrMLM (Li et al., 2022b). Heatmaps of the linkage disequilibrium was generated by LDheatmap package (Shin et al., 2006), haplotype analysis was conducted by LDheatmap package (Barrett et al., 2005). The averages for those traits measured in 2018 and 2020 were used in GWAS.
Candidate gene identification
Candidate genes for salt tolerance were mined in the follow steps. (a) All the genes between the 30 Kb around regions for each of the significantly QTN were mined, where the LD-value was about 20 Kb in mungbean, (b) mined the Arabidopsis, rice and soybean homologous genes of those candidate genes, which were reported related to seed developments, seed production, phytohormone signaling pathways and carbohydrate metabolism pathways, etc. (Li et al., 2019), as the candidate genes. (c) The selected genes showing different expression between two groups of mungbean accessions contrasting in seed size (large seed vs. small seed) (see below) were considered as candidate genes.
Differentially expressed gene based on RNA-sequenced data
Two large-seeded accessions [G141 and G143; 19.32 ± 7.09 (g)] and two small-seeded accessions [G169 and G171; 11.58 ± 5.93 (g)] were selected for RNA sequencing (RNA-seq) analysis. Data in seed set were collected at three seed development stages (10, 15, and 25 DAF) for RNA extraction in 2021. Total RNA was extracted using RNAprep Pure Plant Kit (DP441) according to the manufacturer’s instructions. 1 μg high-quality RNA samples (OD260/280 = 1.8∼2.2; OD260/230 ≥ 2.0; RIN ≥ 6.5; 28S:18S ≥ 1.0 and >10 μg) were used to construct the sequencing library (G9691B, Agilent). The RNA were analyzed in an Illumina Novaseq Sequencer. Raw reads were cleaned by trimmomatic8 (Bolger et al., 2014), and clean reads were mapped to reference sequences using Hisat2 (Pertea et al., 2016). The gene expression level was calculated by using RPKM method by Subread package (Mortazavi et al., 2008).
In the key candidate gene identification, the extracted RNA in two large-seeded accessions at 10 and 25 DAF were treated with RNase-free DNase I (Promega, Madison, WI, United States). After reverse transcription, the cDNA was used as a template for RT-qPCR using the Takara Bio TB Green Premix Ex Taq (Tli RNase H Plus). The detail progress was described by Liu et al. (2022b). Reactions were run on a Bio-Rad CFX96 system. EVM0007380 (homologous of At3g18780) was used as the CK in this experiment. Primers were designed by NCBI and tested by RCR of tubulin. The t-test was adopted in the hypothesis testing, P < 0.05, P < 0.01, and P < 0.001 indicated significant probability levels at 0.05, 0.01, and 0.001, respectively. Information of the primers used is presented in Supplementary Table 1.
Protein–protein interaction
The protein–protein interactions (PPIs) were detected used the online tools STRING9 (Jensen et al., 2009). The mungbean (V. radiata (L.) R. Wilczek) protein database was used as the protein library.
Results
Phenotypic variation for mungbean seed-size-related traits
100-seed weight, SW, and SL in 196 mungbean accessions were measured in 2018 and 2020. The average-plus-standard deviations for the three traits across the 2 years were 5.05 ± 1.91 (g), 3.48 ± 0.51 (mm), and 4.64 ± 0.99 (mm), respectively, and their average coefficients of variation (CV) across the 2 years were 38.5, 14.5, and 16.5 (%), respectively (Supplementary Table 2). Although the trends for those traits in the 2 years were similar (Figures 1A–C), HSW (38.5%) had much larger phenotypic variation than SW (14.5%) and SL (16.5%), indicating their large phenotypic variation and typical quantitative traits. In general, the wild mungbeans showed low seed weight (1.68 ± 0.61) as well as short SW (2.45 ± 0.401) and SL (3.12 ± 0.43), while the cultivated mungbeans had high seed weights (5.29 ± 1.68) as well as long SW (3.56 ± 0.41) and SL (4.76 ± 0.92) (Supplementary Table 2). Moreover, significant difference for each trait between the 2 years was observed (P < 0.001), and these traits had significant correlations with each other (r > 0.87, P < 0.001) (Figure 1D), indicating the existence of common QTNs among these traits (Liu et al., 2020b).
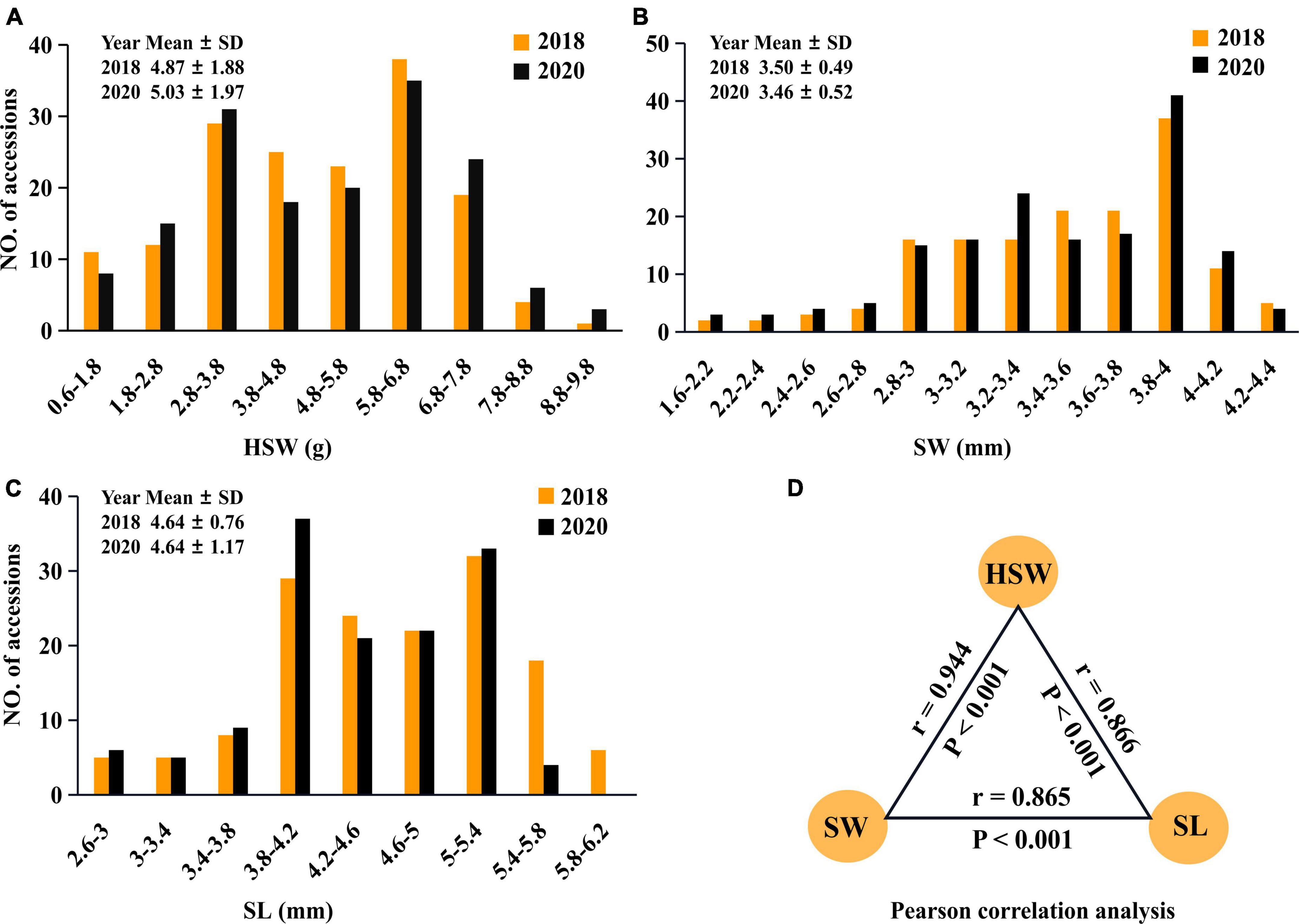
Figure 1. The frequency distributions of seed-size-related traits. Frequency distributions of HSW (A) (g), SL (B) (mm), and SW (C) (mm) in 196 mungbean accessions, which were measured in 2018 (brown bar) and 2020 (black bar). SD, standard deviation. The associations of HSW with SW and SL, the average dates of those traits measured in 2018 and 2020 were used in the partial correlation analysis (D).
Genome-wide association studies for seed-size-related traits in mungbean
Detection of main-effect quantitative trait nucleotides for seed-size-related traits in each environment
After removing the SNPs with an average coverage depth < 8× and with a MAF less than 5%, we identified more than 3.6 million SNP markers. In the single-environment analysis, the phenotypic observations for each trait in 196 accessions measured in 2018 and 2020 were used to associate with 3,607,508 SNPs using 3VmrMLM, EMMAX, GEMMA, and CMLM under the situations of five subpopulations and polygenic background control (kinship matrix) (Supplementary Data Set 3). As more than 10,000 QTNs were identified by GEMMA for HSW in 2018, the relevant results were not used in the subsequent analysis. As a result, 208 significant QTNs were identified for the above traits. Thirteen significant QTNs were simultaneously identified in two environments by two GWAS methods (Supplementary Table 3; Supplementary Data Set 4), some significant QTNs are presented in Figure 2. For example, Chr10-25206533-25223155 (LOD = 15.40∼37.89, P = 3.16E-08∼5.15E-09) was detected in 2018 and 2020 by MLM, EMMAX, and 3VmrMLM to be associated with HSW, SW, and SL (Table 1; Figures 2A–F), and the Q-Q plot in the Supplementary Figures 1A–D, which was corresponding to the GWAS results in Figure 2, except 3VmrMLM. And Chr1-71543546 (LOD = 7.70∼12.44) was detected in 2018 and 2020 by 3VmrMLM to be associated with SW (Supplementary Table 3). These QTNs were distributed on chromosomes 1–4, and 10 (≥20 QTNs for each chromosome) and had a 1.15% average proportion of their total phenotypic variation explained by each QTN, and there were 47, 115, and 46 QTNs, respectively, for HSW, SL, and SW (Supplementary Data Set 4).
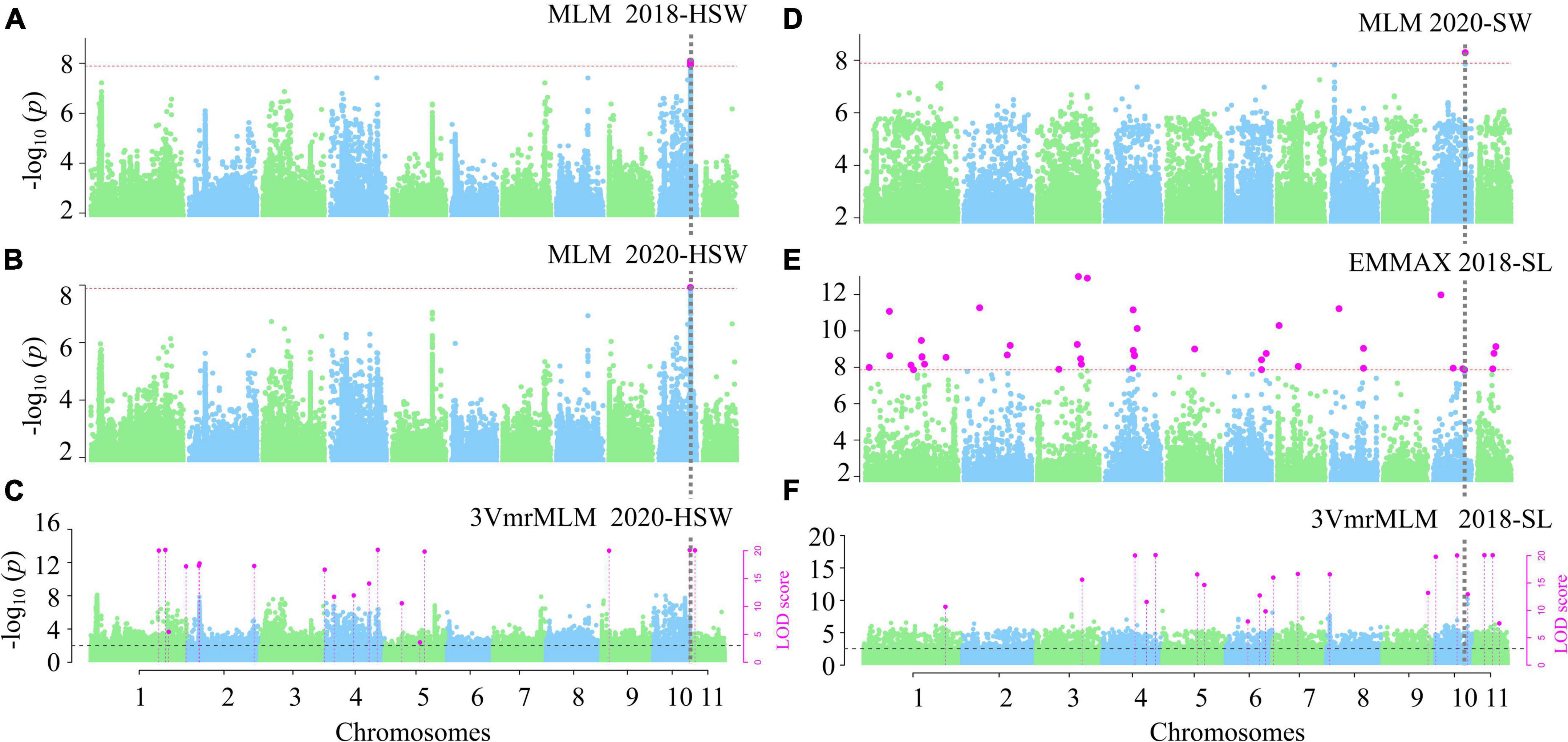
Figure 2. Manhattan plots for the GWAS for seed-yield-related traits. GWAS for HSW (A–C), SW (D) and SL (E,F). Significant QTN in phenotypic GWAS was set at P-value ≤ 0.05/m = 1.39e-08 (m = 3607508), ≤2.77e-09 for CMLM and EMMAX (A,B,D,E); and LOD ≥ 3.0 for the 3VmrMLM as the significant QTN, and all the critical values were marked by horizontal lines, Y-axis on the left side reports –log10 P-values of SNP, while Y-axis on the right side reports LOD scores, and LOD scores are shown in points with straight lines.
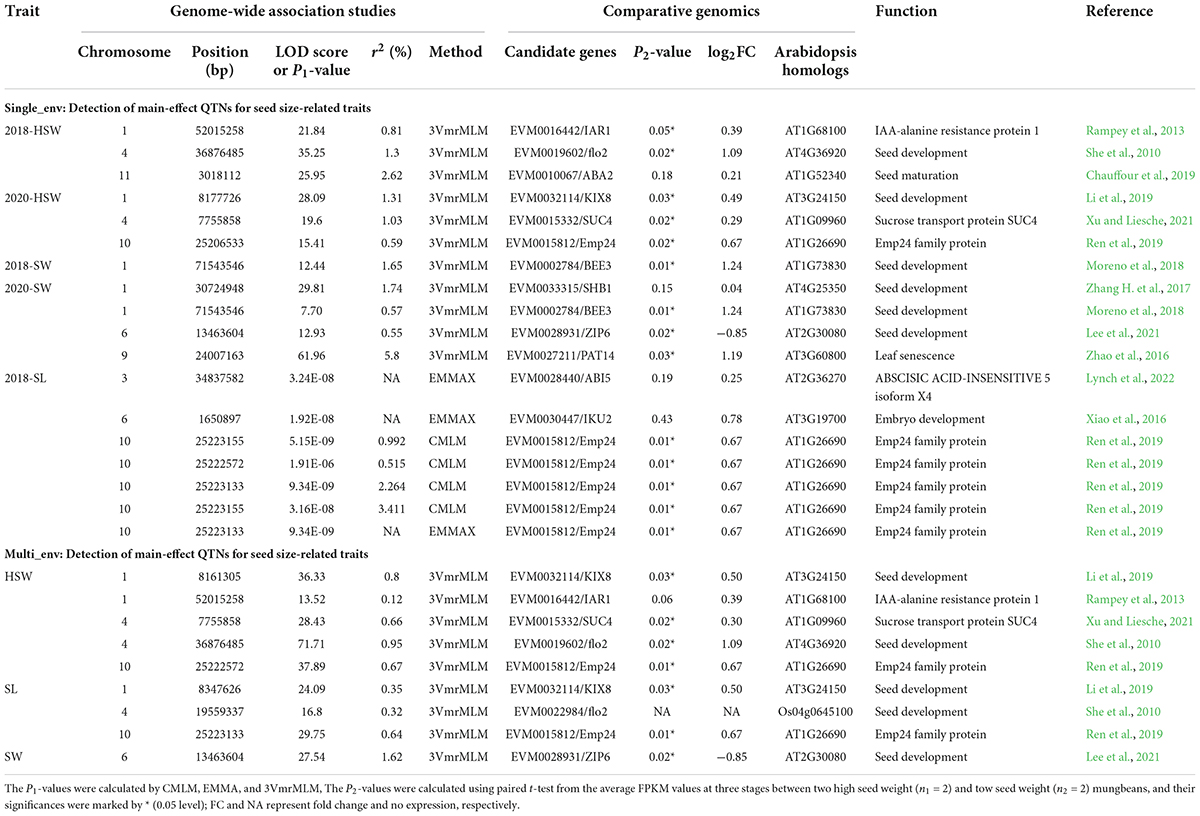
Table 1. Eight key candidate genes derived from genome-wide association studies for seed-related traits.
Detection of quantitative trait nucleotides for seed-size-related traits in multiple environments
To detect more stable QTNs, three seed-size-related traits of 196 mungbean accessions measured in 2018 and 2020 were used to associate with 3607508 SNP markers using two-environment 3VmrMLM joint analysis. As a result, 32, 33, and 18 significant QTNs were identified for HSW, SL, and SW, respectively (Supplementary Table 3), and had a 1.08% average proportion of total phenotypic variation explained by each QTN. Moreover, eight significant QTNs were identified (Supplementary Table 4). For example, Chr1-8161305-8347626 (LOD = 24.09∼36.33) and Chr10-25222572-25223133 loci (LOD = 29.75∼37.89) were detected to be associated with HSW and SL, respectively (Supplementary Tables 3, 4).
Based on all the above main-effect QTNs in single- and multiple-environment analysis, five stable QTNs across various methods and/or two environments were found (Supplementary Table 5), including Chr1-8161305-8347626 (LOD = 24.09∼36.33), Chr2-12602704 (LOD = 17.71∼38.08), Chr4-10069367 (LOD = 17.72∼34.19), Chr5-10834954 (LOD = 9.53∼30.03), and Chr10-Chr10-25222572-25223133 (LOD = 29.75∼37.89), especially, Chr1-8161305-8347626 and Chr10-25222572-25223133 were simultaneously identified across methods and two environments.
Detection of quantitative trait nucleotide-by-environment interactions for seed-size-related traits in multiple environments
All the above datasets in GWAS were used to detect QEIs using 3VmrMLM. As a result, 5, 10, and 5 significant QEIs were found to be associated with HSW, SL, and SW, respectively (Supplementary Figure 2; Table 2). Among these QEIs, 5 had zero dominant-by-environment interaction effects, and 7 had zero additive-by-environment interaction effects. For example, the two loci Chr4-26262890 and Chr4-31677341 for HSW had only additive-by-environment interaction effects of 0.12 (Supplementary Figures 2A–C, LOD = 12.70; r2 = 0.26) and 0.08 (Supplementary Figures 2A–C, LOD = 12.65; r2 = 0.27), respectively.
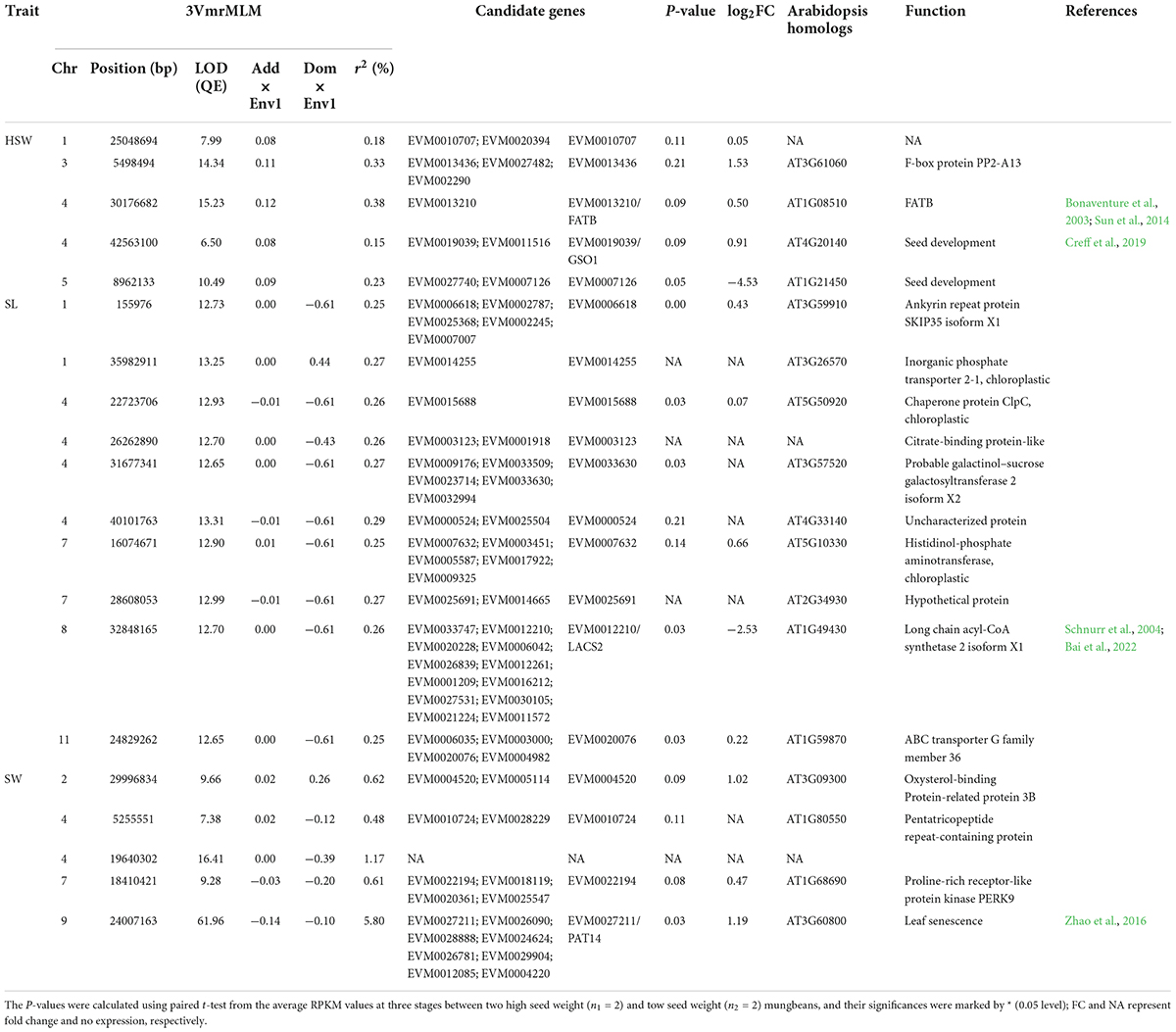
Table 2. Twenty significant QTN-by-environment interactions for seed-size-related traits under multi-environments.
The two loci Chr1-155976 and Chr1-3598291 for HSW had only dominant-by-environment interaction effects of −0.61 (LOD = 12.73; r2 = 0.25) and 0.44 (LOD = 13.25; r2 = 0.27), respectively. Among the 20 QEIs, the loci Chr4-5255551 and Chr7-16074671 had inconsistent directions between additive- and dominant-by-environment interaction effects.
In addition, among these QEIs, the QEI locus Chr9-24007163 for SW had large effect, and r2 was 5.8% (Supplementary Figure 2B, LOD = 61.95). The additive and dominant effects in environment 1 were −0.14 and −0.098, respectively.
Candidate genes for seed-size-related traits
A total of 6912 DEGs were identified between two high-seed-weight and low-seed-weight mungbeans (FDR ≤ 0.05) (Supplementary Figures 3A,B; Supplementary Data Set 6). These DEGs were intersected with 809 genes around significant QTNs for HSW, SL, and SW (Supplementary Tables 3, 4; Supplementary Data Sets 4, 5). As a result, 53 out of 809 genes were differentially expressed (P ≤ 0.05, Log2FC ≥ 0.5). Using comparative genomics analysis, 12 out of 53 DEGs were homologous to previously reported seed development related genes in rice and Arabidopsis thaliana, in which KIX8, PAT14, Emp24/25, IAR1, BEE3, SUC4, flo2, and Zip6 had been confirmed via functional analysis in rice and A. thaliana (Table 1), such as VrKIX8 (LOD = 24.09∼36.33), VrEmp24/25 (LOD = 15.40∼37.89, P = 3.16E-08∼5.15E-09), VrPAT14 (LOD = 61.96), and VrZIP6 (LOD = 27.54). Among the eight genes, VrKIX8, VrEmp24/25, VrIAR1, VrBEE3, VrSUC4, and Vrflo2 were significantly upregulated in high-HSW accessions, VrPAT14 was significantly downregulated, and VrZIP6 had no significant difference (Figure 3A), as compared to those in low-HSW accessions using the transcriptome data at 10, 15, and 25 DAF (Supplementary Data Set 4). We conducted RT-qPCR analysis to further confirm the eight key candidate genes. The results showed that seven genes were confirmed, except VrZIP6, a transcription factor related to seed development. All the seven genes had higher expression levels in the early stage of seed development (10 DAF) than in the late maturation stage of seed development (25 DAF) (Figure 3B; Supplementary Data Set 7), indicating their essential roles at early stage of seed development.
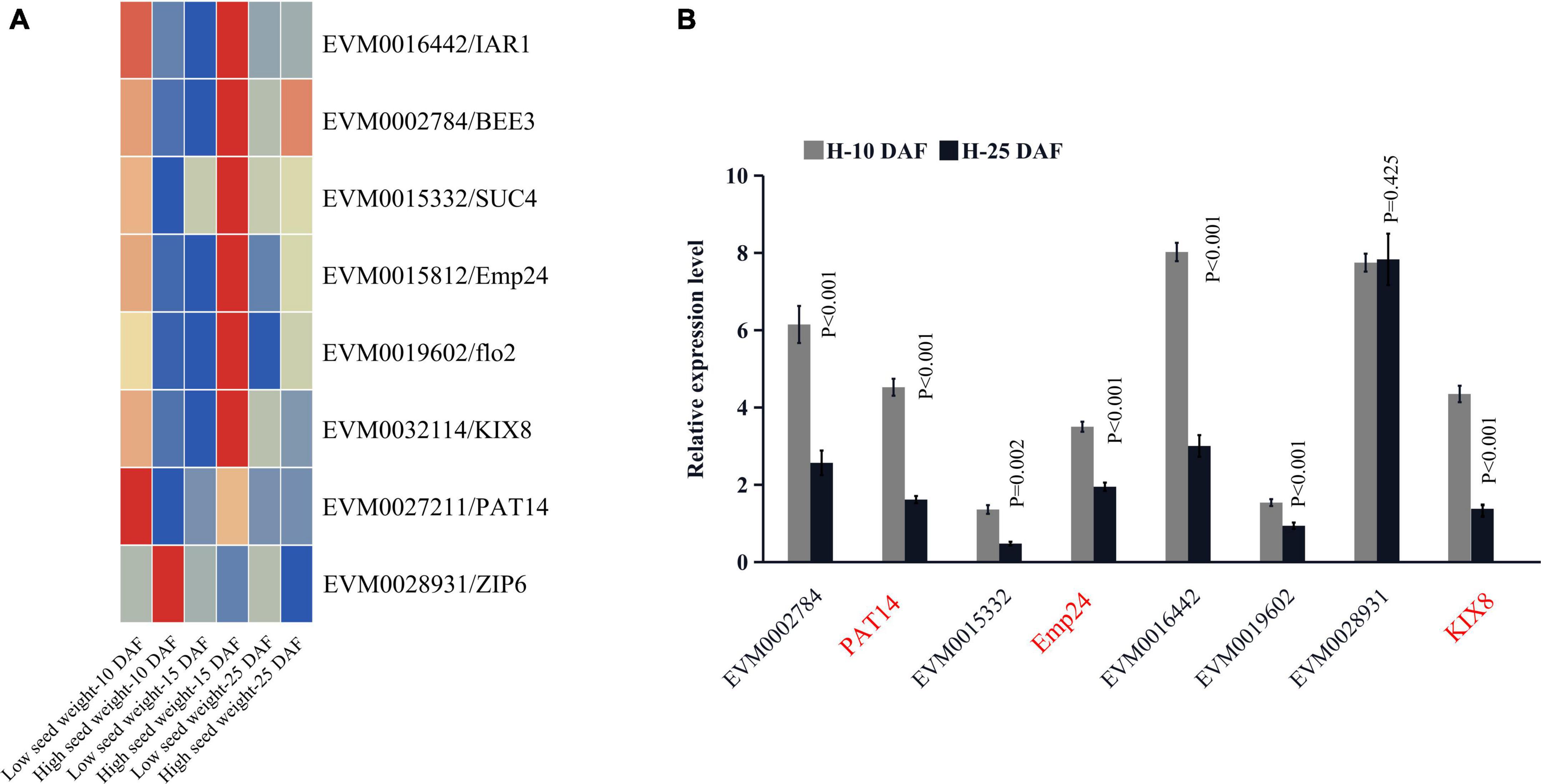
Figure 3. The expression of eight key candidate genes. The expression profiling of eight key candidate genes significantly associated with seed-size-related traits. The expression profiling of eight key candidate genes between two high-seed-weight and two low-seed-weight mungbeans (A). Real-time PCR analysis of the eight key candidate genes; the t-test was used to test the significant differences of genes expression between two high-seed-weight mungbeans at 10 DAF and 25 DAF (B). DAF, days after flowering.
Using the same approach described above, among 65 genes around 20 QEIs, four were homologous to previously reported seed development related genes in rice and A. thaliana (Table 2), although new experiments are necessary to explore these novel GEI-trait associations. The four genes were described as below. VrFATB was linked to the locus Chr4-30176682 (Supplementary Figure 2A). As described in Bonaventure et al. (2003) and Sun et al. (2014), FATB is a major determinant of saturated fatty-acid synthesis, and increases FATB activity at low temperature during seedling establishment caused high saturated fatty-acid content in plant. VrGSO1 was linked to the locus Chr4-42563100 (Supplementary Figure 2A). As observed in Creff et al. (2019), GSO1 was a stress signal-pathway-related gene, and stress-associated MPK6 protein acted downstream of GSO1 in developing embryo. VrPAT14 was linked to the locus Chr9-24007163 (Supplementary Figure 2B). In Zhao et al. (2016), PAT14 was involved with NPR1-dependent salicylic-acid signaling. VrLACS2 was linked to the locus Chr8-32848165 (Supplementary Figure 2C), in which VrLACS2 was essential for normal cuticle development in Arabidopsis (Schnurr et al., 2004) and CrLACS2 suppression resulted in 50% less oil, yet with a higher amount of chloroplast lipids under N-deprivation (Bai et al., 2022).
Haplotype analysis of the main candidate genes
Two DEGs, VrEmp24/25 and VrKIX8, were detected in the single- and multi-environment analyses (Figures 4A,B), and verified by RT-qPCR. Their haplotypic analyses were described as below.
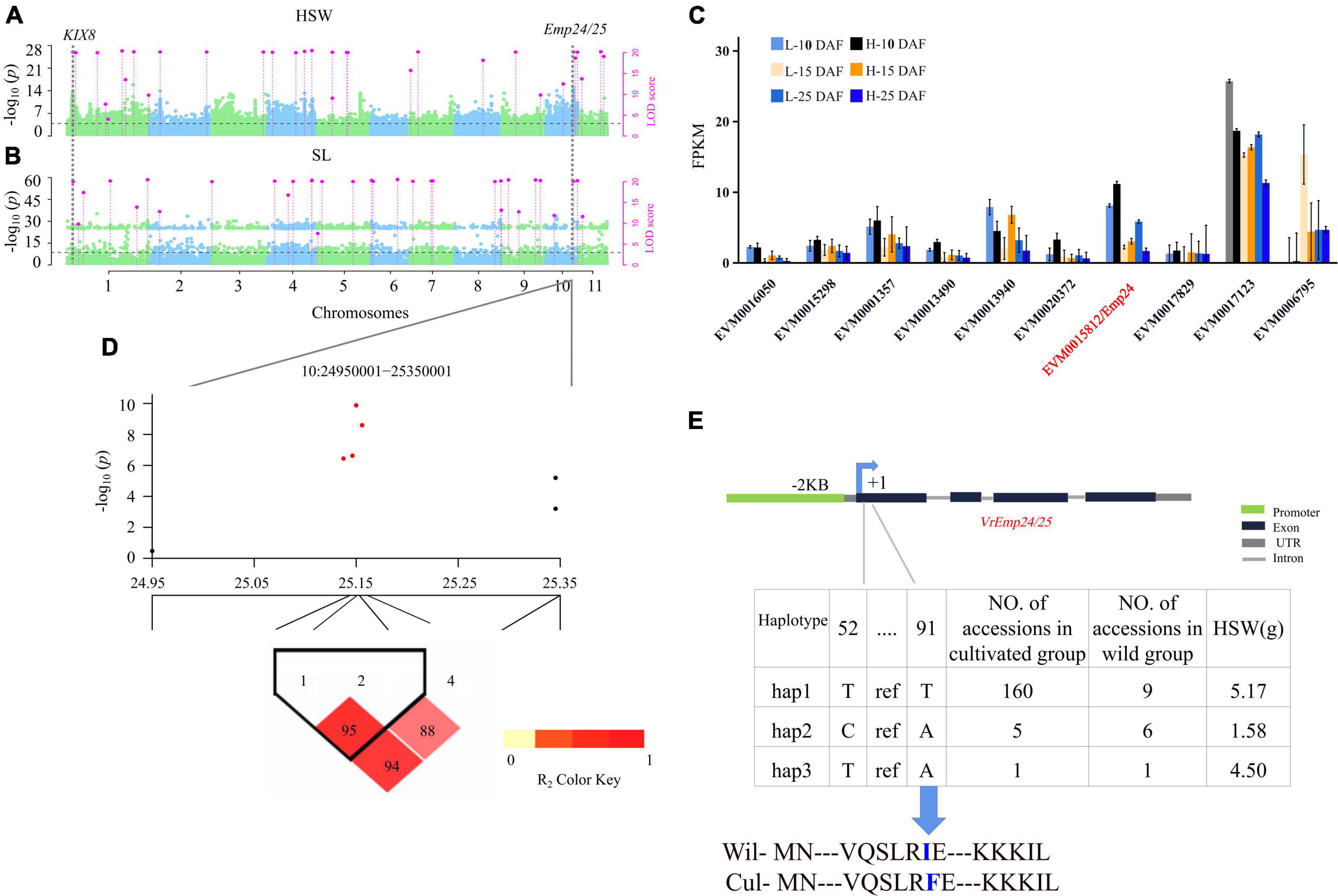
Figure 4. Genetic analysis of VrEmp24/25. Local Manhattan plots for HSW under multi-environments. LOD ≥ 3.0 for the 3VmrMLM as the significant QTN (A,B). The expression profiling of 10 candidate genes for HSW identified at 30 Kb around Chr10-25222572-25223133 loci in the seed between two high-seed-weight and two low-seed-weight mungbeans (C). LD heatmaps surrounding Chr10-25222572-25223133 loci (D). Haplotype analysis of VrEmp24/25 (E), the thirtieth amino acid of VrEmp24/25 changed from ATT (Ile, I) to TTT (Phe, F). DAF, days after flowering. Wil, the wild accessions. Cul, the cultivated accessions.
In the haplotype analysis of VrEmp24/25, five SNP markers were found to be within VrEmp24/25 and the promoter region (Supplementary Data Set 8), and the two SNP markers in VrEmp24/25 were used to consist of three haplotypes (Figure 4D). Among the three haplotypes, hap 1 (5.17 g) had significantly higher HSW than hap 2 (1.58 g) and hap 3 (4.50 g; P = 2.11E-29) (Supplementary Table 7). Thus, hap 1 is elite haplotype. And the elite haplotypes TT made up more than 90.9% (160/176) in the cultivated mungbeans. VrEmp24/25 with elite haplotype frequencies less than 45% in wild mungbeans (Supplementary Table 7; Figure 4) can be exploited for the improvement of mungbean cultivars.
Around the significant QTN Chr1-8161305-8347626 (Figure 5A; Supplementary Data Set 8), eight genes were found distributed in the region (Figure 5B). And six polymorphic loci, i.e., Chr1_8243935, Chr1_8243938, Chr1_8243939, Chr1_8243940, Chr1_8243945, and Chr1_8244001 were found in VrKIX8 and the promoter region. All the six SNP were used to conduct the haplotype analysis (Figure 5C). Among the three haplotypes, hap 1 (5.09 g) had significantly higher HSW than hap 2 (4.56 g), hap 3 (3.47 g), and hap 4 (3.86 g) (Supplementary Table 7). Thus, hap 1 is elite haplotype. The elite haplotypes ATCGAA made up more than 73.2% (129/176) in the cultivated mungbeans, while the haplotype frequencies of CGAGT and CTAGGA were more than 25% (5/20) in wild mungbeans. Though Chr1_8243945 and Chr1_8244001 were located within the 5′ UTR of VrKIX8, and the amino acid sequence had not changed between cultivated mungbeans and wild mungbeans (Figure 5D). The SNP in 5′ UTRs could influence the translation efficiency of VrKIX8 (Evfratov et al., 2017). The HSW in hap 1 (5.16 g) was significantly higher than that in hap 2 to hap 4 (3.50–4.66 g; P = 1.19E-21).
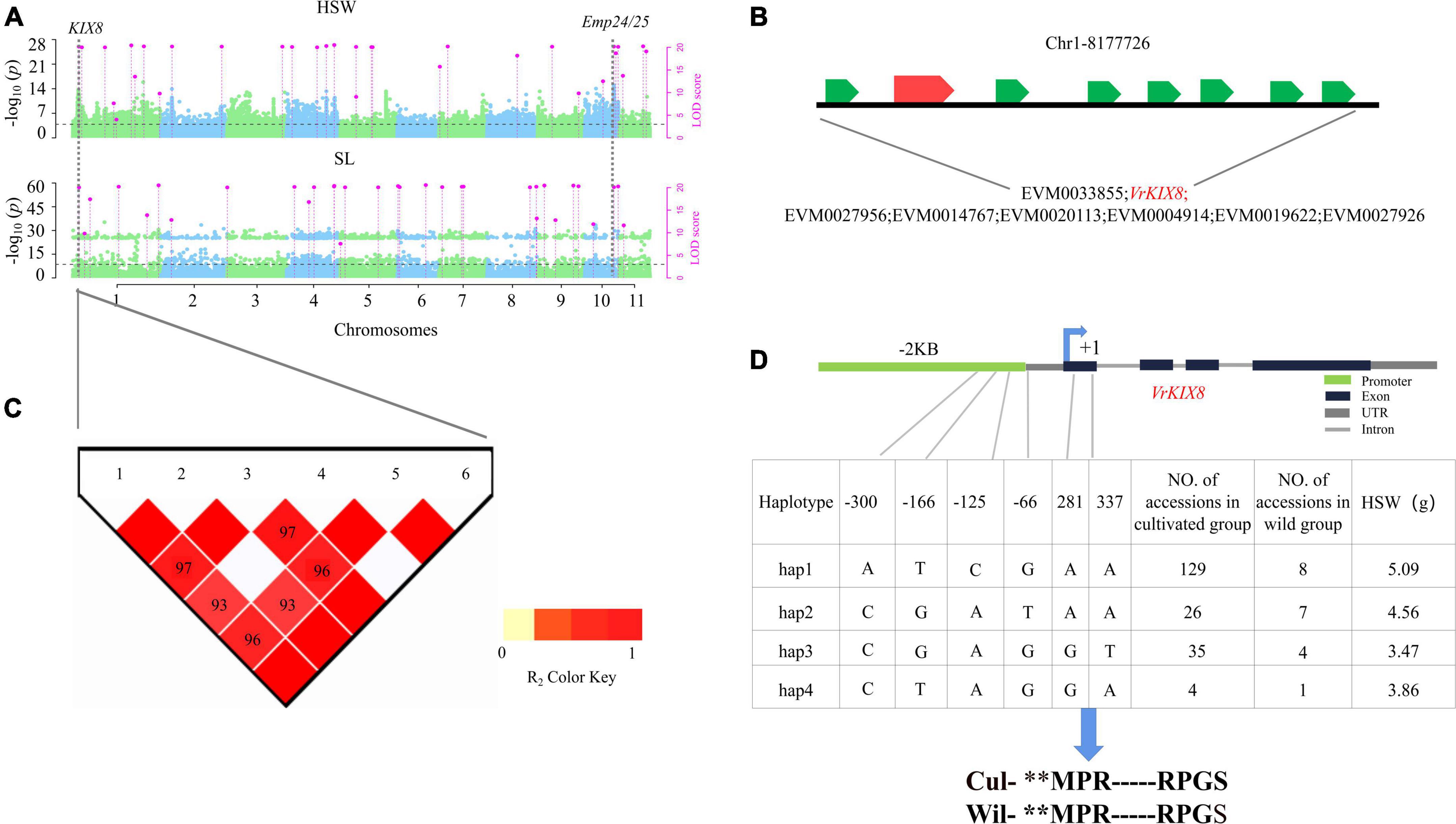
Figure 5. Genetic analysis of VrKIX8. Local Manhattan plots for HSW in multi-environments. LOD ≥ 3.0 for the 3VmrMLM as the significant QTN (A). LD heatmaps surrounding Chr1-8161305-8347626 loci (B). Genes around the significant QTN region, shown at the bottom (C). Haplotype analysis of VrKIX8 (D). Wil, the wild accessions. Cul, the cultivated accessions. symbol “**” means omit the same sequence part.
Based on these results, we deduced that these two SNP and six SNP cause the difference expression of the VrEmp24/25 and VrKIX8 gene, respectively. The discovery of VrEmp24/25 and VrKIX8 two domestication/improvement genes can accelerate breeding selections and facilitate ideal crop designs.
Expression patterns of seed development pathway genes in mungbean
As seed development pathway genes were largely unknown in mungbean, we mined seed development pathway genes by comparative genomics and transcriptomics analysis. As a result, 54 genes in seed-development pathway were identified in this study (Figure 6; Supplementary Data Set 9), such as two GPA1, one AGB, and one AGG3. In the ubiquitin proteasome pathways, two DA1, one DA2, one SOD2, one EOD1, and one UBP15 rather than SAMBA were identified. In the auxin pathways, two ABA2, one ABI5, three SHB1, five IKU2, and three CKX2 rather than IKU1 and MINI3 were identified (Figure 6A). Five transcription factors including three BES1, and two SOD7 were identified. Moreover, 16 genes for seed size developments were found to be with uncertain pathways, including three KIX8, five MES1, and one KLU (Figure 6A; Supplementary Data Set 9). Among the 54 genes, 13 genes were significantly differentially expressed (P-value < 0.05, t-test) between two low-seed-weight (nos. G169 and G171) and two high-seed-weight (no. G141 and G143) accessions in the 196 mungbean accessions using the transcriptome data at 10, 15, and 25 DAF (Figure 6B; Supplementary Data Set 8). Moreover, almost 90% of the 54 genes (48/54) had higher expressions in the early stage of seed development (10 and 15 DAF) than in the late maturation stage (25 DAF), including VrKIX8 (EVM0032114), which was commonly identified in the GWAS by 3VmrMLM for HSW and SL. And EVM0010067/VrABA2, EVM0033315/VrSHB1, EVM0028440/VrABI5, and EVM0030447/VrIKU2 were also identified in the GWAS by 3VmrMLM, within 100 Kb region of significant QTNs (Table 1).
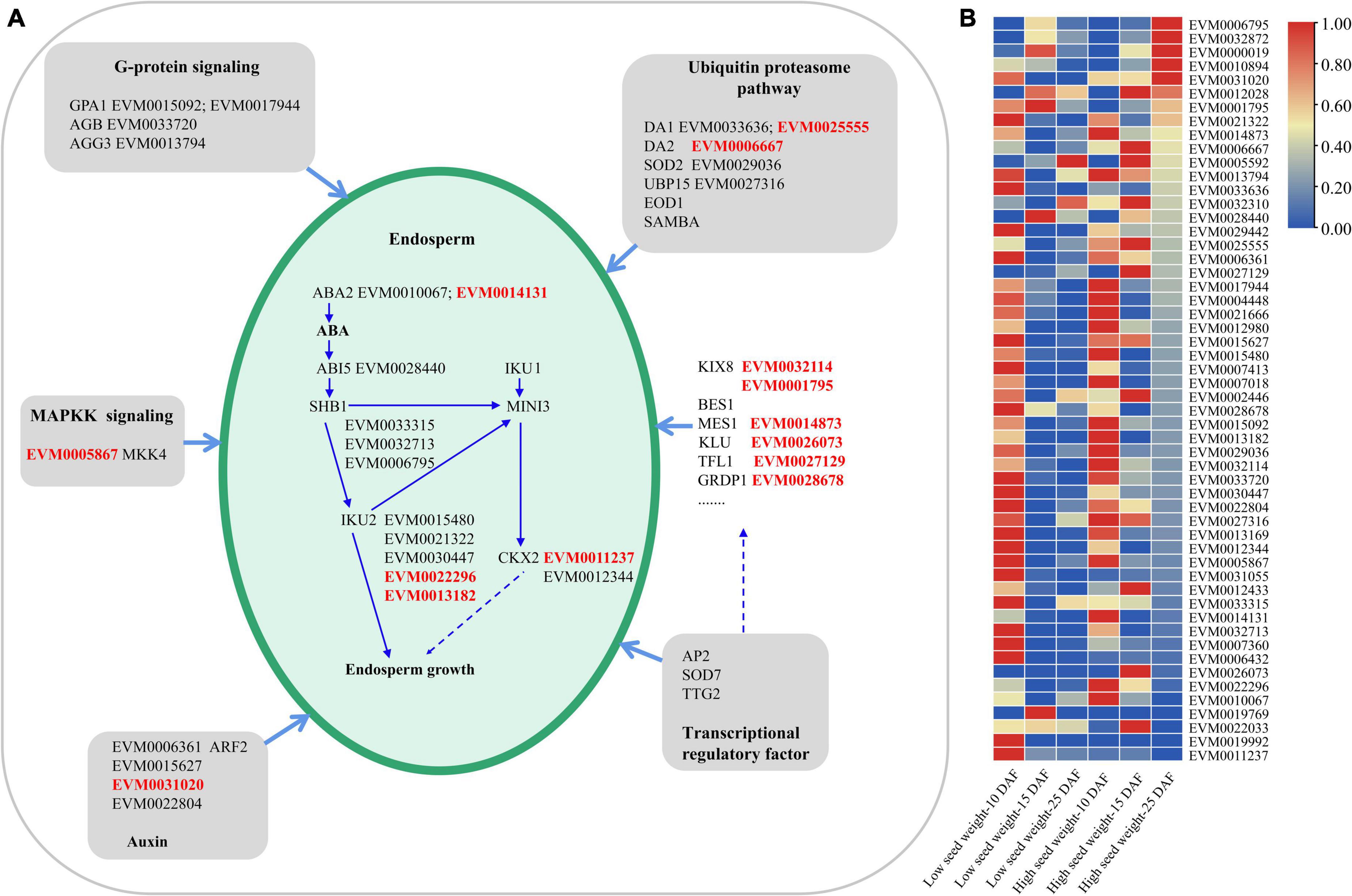
Figure 6. The seed development pathway in mungbean (A) and the expression profiling of 54 candidate genes predicted by comparative genomics, identified in this study (B). GPA1, G PROTEIN ALPHA SUBUNIT 1; AGB, heterotrimeric G-protein beta subunit; AGG3, heterotrimeric G-protein gamma-subunit; DA1/DA2, a ubiquitin-activated peptides; SOD2, chloroplastic copper/zinc superoxide dismutase CSD2; EOD1, E3 ubiquitin ligase; UBP15, ubiquitin-specific protease; SAMBA, plant-specific negative regulator of the APC/C complex; MKK4, mitogen-activated map kinase; ABA2, ABA DEFICIENT 2; ABI5, ABA INSENSITIVE 5; SHB1, a nuclear and cytosolic protein; IKU1/IKU2, leucine rich repeat (LRR) kinase; CKX2, CYTOKININ OXIDASE 2; MINI3, MINISEED 3; ARF2, auxin response factor; AP2, AP2/EREBP (ethylene-responsive element-binding protein) class of transcription factors (Figure 5A). We also identified five transcription factors, three BES1 and two SOD7, transcription repressor involved in regulation of inflorescence architecture; KIX8, KINASE-INDUCIBLE DOMAIN INTERACTING8; MES1, METHYL ESTERASE 1; KLU, cytochrome P450 CYP78A5 monooxygenase. DAF, days after flowering; ABA, abscisic acid; DAF, days after flowering.
We also did the PPI analysis among the seed development pathway genes, and found five pairs of PPIs were larger than the medium confidence value of 0.40 (Supplementary Table 7), indicating the existence of significant PPIs, i.e., EVM0013794.1 (VrAGG3) and EVM0006667.1 (VrDA2) (0.478), EVM0033720.1 (VrAGB) and EV944.1 (VrGPA1-1) (0.995), as well as EVM0033720.1 (VrAGB) and EVM0015092.1 (VrGPA1-2) (0.995).
Discussion
The high-yield and efficiency breeding progress of mungbeans have been limited by the lack of ideal yield-related genes. At present, few QTNs or QTLs of yield-related traits in mungbeans have been reported (Kang et al., 2014). This study provided a genetic analysis of seed-size-related traits in mungbeans, to improve the accuracy of significant QTNs, we used multiple genome-wide M0017 association studies combined with multi-omics analysis to mine candidate genes associated with yield-related traits. Firstly, a total of 98 QTNs and 20 QEIs were identified using 3VmrMLM, while 95 and 15 QTNs were identified using EMMAX, and CMLM, respectively. Then, in the identification of candidate genes, 12 key candidate genes were mined, and seven of them including VrKIX8, VrEmp24/25, and VrPAT14 were evidenced by transcriptome analysis and RT-qPCR analysis. Lastly, through haplotype analysis, the thirtieth amino acid of VrEmp24/25 in the elite haplotype was changed from Ile to Phe. And there were six SNP in the promoter and 5′ UTRs of VrKIX8, however, the amino acid sequence of VrKIX8 in the elite haplotype was not changed. The results provided the theoretical basis for both the functional identification of seed-size-related genes and for quality improvements in mungbean breeding.
Multiple genome-wide association studies methods combined with multi-omics analysis in mining candidate genes
In the GWAS, how to identify candidate genes around significant QTNs has been a challenge. Liu et al. (2020c), Zhang et al. (2021), and Gong et al. (2022) selected the 100-kb interval upstream and downstream of the significant QTN as the candidate interval in watermelon and soybeans. Usually, the interval has been chosen according to the LD decay values.
In order to determine stable QTNs and key candidate genes for seed-size-related traits, we adopted the following analyses. Firstly, we used CMLM, EMMAX, GEMMA, and 3VmrMLM to identify stable QTNs, as a result, five stable QTNs for seed-size-related traits were detected in single- and multiple-environments (Supplementary Table 5), i.e., Chr1-8161305-8347626 (LOD = 24.09∼36.33), and Chr10-25222572-25223133 loci (LOD = 29.75∼37.89).
Second, in the identification of candidate genes, we conducted issue expression analysis, and comparative genomics analysis. 53 out of the 809 candidate genes were significantly differentially expressed between high and low HSW accessions (P ≤ 0.05, Log2FC ≥ 0.5). Among the 53 DEGs, Arabidopsis homologous genes of the 12 key candidate genes had certain molecular functions. Notably, 10 of those genes were identified by 3VmrMLM (Table 1). Seven key candidate genes (VrKIX8, VrEmp24/25, VrIAR1, VrBEE3, VrSUC4, VrPAT14, and Vrflo2) were significantly differentially expressed between the low-seed-weight and high-seed-weight accessions, and further verified by RT-qPCR analysis (Table 1; Figure 4). VrKIX8 (Chr1-8161305-8347626) and VrEmp24/25 (Chr10-25222572-25223133) may be main genes in controlling seed-size-related traits.
Notably, 3VmrMLM showed more powerful ability in the detection of significant QTN than GEMMA, EMMAX, and CMLM, as it found more differentially expressed key candidate genes than other methods. The combination of 3VmrMLM and multi-omics analysis in the genetic analysis of complex traits was helpful.
Genome-wide association study provided potential genes VrEmp24/25 and VrKIX8 for mungbean seed-size-related traits
VrEmp24/25 was an important seed-size traits related gene, the evidence was as below: Firstly, Chr10-25206533-25223155 locus for seed size traits was detected in 2018 and 2020 by CMLM, EMMAX, and 3VmrMLM (Figure 2), and there were 10 genes in its interval (Figure 4C). Secondly, among the 10 genes, only VrEmp24/25 (EVM0015812) (P = 0.014, Log2FC = 0.67) had deferentially expressed across different phenotype accessions (Figure 4C; Supplementary Data Set 4). Besides, in maize, the loss function of EMP24 and Emp25 would impair embryo and endosperm development (Xiu et al., 2020). EMP24 was required for the splicing of nad4 (Ren et al., 2019), and the lack of either Nad4 or Nad5 blocked the assembly of complex I holoenzyme in Arabidopsis (Ligas et al., 2019). The loss of the steady-state level of mitochondrial nad5 mature mRNA blocked the assembly of complex I and caused an arrest in endosperm development (Zhang Y. F. et al., 2017). Lastly, the elite haplotypes of VrEmp24/25 (TT) made up the main proportion of more than 90.9% in cultivated mungbeans, 45% in wild mungbeans (Figure 4E). The HSW in hap 1 haplotypes accessions was significantly higher than that in hap 2 and hap 3 (P = 2.11E-29). It was reported that a single amino acid completely prevented the appearance of the enzyme in the medium, and we inferred that the related variation could lead to the change in enzyme activity (East et al., 1990; Alfson et al., 2018).
There have four evidences to take VrKIX8 as another important seed-size trait gene. Firstly, VrKIX8 associated with Chr1-8161305-8347626 (LOD = 24.09∼36.33) for HSW and SL were detected in multi-environment by 3VmrMLM (Figure 5A; Supplementary Table 5). Secondly, VrKIX8 (LOD = 24.09∼36.33) had significantly differentially expressed between high- and low-HSW accessions (Figure 3A). Then, in Arabidopsis, the disruption of KIX8/9 and PPD1/2 could cause large seeds due to increased cell proliferation and cell elongation in the integuments (Liu et al., 2020a). In soybeans, the loss of the function GmKIX8-1 showed a significant increase in the size of seeds and leaves. In addition, the increase in organ size was due to the increased cell proliferation, rather than cell expansion. GmKIX8-1 showed negatively regulated cell proliferation in plants (Nguyen et al., 2021). Lastly, the elite haplotypes of VrKIX8 (ATCGAA) made up the main proportion of more than 73% in cultivated mungbeans, 40% in wild mungbeans. Moreover, there are four SNPs in the promoter and of VrKIX8, and two SNPs in the CDS region, however the amino acid sequence did not change between the elite haplotypes and the other haplotypes (Figure 5C). The HSW in hap 1 haplotypes accessions was higher than that in hap 2 to hap 4 (P = 1.19E-21). We supposed that the mutations may have influenced the translation efficiency of VrKIX8 and caused low expression in cultivated accessions during mungbean domestication.
Genes participate in seed development progress
The genes controlling seed development progress in mungbean are largely unknown (Ha et al., 2021). In this study, we identified fifty-four candidate genes in the seed-development pathways, i.e., aba2 (Cheng et al., 2014; Chauffour et al., 2019), ABI5 (Lynch et al., 2022), SHB1, MINI3, and IKU2 (Garcia et al., 2003; Xiao et al., 2016; Zhang H. et al., 2017), mutants of those genes induced abnormal seed development in Arabidopsis. And, five genes were also commonly identified via GWAS (Table 1). Those five genes (VrKIX8, VrABA2, VrSHB1, VrABI5, and VrIKU2) are more likely to be reliable, especially for VrKIX8, as described above.
We also analyze the possible correlation between the main seed development pathways. Among the 54 genes, five genes (VrAGG, VrDA2, VrAGB, VrGPA1-1, and VrGPA1-2) consisted of five pairs of significant PPIs. Interestingly, four pairs PPIs were found to be in the G-protein-signaling pathway, and one pair of PPIs was found to be in the G-protein-signaling and the ubiquitin proteasome pathways (Figure 6; Supplementary Table 6). Ubiquitin proteasome pathway is an important pathway for the selective degradation of proteins and seed development (Smalle and Vierstra, 2004), and the G-protein-signaling pathway is a ubiquitous cell transmembrane signal transduction pathway in eukaryotes (Huang et al., 2006). Moreover, mutations in GPA1 or AGB1 could cause short flowers (Lease et al., 2001; Ullah et al., 2001). The overexpression of AGG3 promoted seed and organ growth by increasing cell proliferation, and loss-of-function mutations in AGG3 caused small seeds and organs (Chakravorty et al., 2011; Li et al., 2012). The ubiquitin receptor DA1 could control seed size by restricting cell proliferation in maternal integuments (Li et al., 2008). DA1 functioned synergistically with DA2 to restrict seed growth, and DA2 physically interacted with DA1 in vitro and in vivo (Song et al., 2007; Xia et al., 2013). This interaction could mediate the interactions between the G-protein-signaling pathway and the ubiquitin proteasome pathway, which might offer an important clue in the mechanism analysis of seed development.
In addition, 48 genes had higher expressions in the early stage of seed development than in the late maturation stage of seed development, indicating that seed-development-related genes function primarily in the early stages of seed development, which was consistent with the findings of Zuo et al. (2022) in soybean.
Conclusion
This study conducted GWAS for seed-size-related traits in mungbeans. 98 QTNs and 20 QEIs were identified using 3VmrMLM, while 95, >10,000, and 15 QTNs were identified using EMMAX, GEMMA, and CMLM, respectively. A total of 12 key candidate genes were mined, which were homologous to known seed-development genes in rice and A. thaliana. VrEmp24/25 and VrKIX8 were identified as main candidate genes around two stable QTNs, the two candidate genes were further confirmed by RT-qPCR and haplotype analysis, and prevalent haplotypes of VrEmp24/25 and VrKIX8 may be useful in mungbean breeding.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: The WGS sequencing data of 196 mungbean accessions was uploaded to NGDC, with subCRA011538, subSAM100395, and PRJCA010704 ID.
Author contributions
JL, XY, and XC conceived of the project and its components. JL, JC, and YL performed the field experiments. JL, QY, CX, and RW performed the bioinformatics analysis and real data analysis. JL, XC, and XY wrote and revised the manuscript. All authors reviewed the manuscript.
Funding
This work was supported by Natural Science Foundation of Jiangsu Province (BK20190257), National Natural Science Foundation of China (31871696), China Agriculture Research System-Food Legumes (CARS-08), Jiangsu Seed Industry Revitalization Project (JBGS[2021]004), and Jiangsu Planned Projects for Postdoctoral Research Funds (2021K393C).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.997988/full#supplementary-material
Abbreviations
GWAS, genome-wide association study; HSW, 100-seed weight; FPKM, Fragments Reads Per Kilobases per Million reads; PPI, protein–protein interaction; RNA-seq, RNA sequencing; QEIs, QTN-by-environment interactions; GEMMA, genome-wide efficient mixed-model association; CMLMs, compressed mixed linear models; EMMAX, efficient mixed-model association expedited; KIX8, KINASE-INDUCIBLE DOMAIN INTERACTING8; Emp24/25, emp24/gp25L/p24 family; QTNs, quantitative trait nucleotides; SNP, single nucleotide polymorphism; SW, seed width; SL, seed length.
Footnotes
- ^ http://bio-bwa.sourceforge.net/bwa.shtml
- ^ https://gitee.com/mirrors/GATK
- ^ http://dalexander.github.io/admixture/download.html
- ^ http://zzlab.net/GAPIT
- ^ http://csg.sph.umich.edu//kang/emmax/download/index.html
- ^ https://github.com/genetics-statistics/GEMMA
- ^ https://github.com/YuanmingZhang65/IIIVmrMLM
- ^ http://www.usadellab.org/cms/index.php?page=trimmomatic
- ^ https://string-db.org//
References
Alam, M. K., Islam, M. M., Salahin, N., and Hasanuzzaman, M. (2014). Effect of tillage practices on soil properties and crop productivity in wheat-mungbean-rice cropping system under subtropical climatic conditions. Sci. World J. 2014:437283. doi: 10.1155/2014/437283
Alfson, K. J., Avena, L. E., Delgado, J., Beadles, M. W., Patterson, J. L., and Carrion, R. Jr., et al. (2018). A single amino acid change in the Marburg virus glycoprotein arises during serial cell culture passages and attenuates the virus in a macaque model of disease. mSphere 3:e00401-17. doi: 10.1128/mSphere.00401-17
Bai, F., Yu, L., Shi, J., Li-Beisson, Y., and Liu, J. (2022). Long-chain acyl-CoA synthetases activate fatty acids for lipid synthesis, remodeling and energy production in Chlamydomonas. New Phytol. 233, 823–837. doi: 10.1111/nph.17813
Barrett, J. C., Fry, B., Maller, J., and Daly, M. J. (2005). Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265. doi: 10.1093/bioinformatics/bth457
Bolger, A. M., Marc, L., and Bjoern, U. (2014). Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120.
Bonaventure, G., Salas, J. J., Pollard, M. R., and Ohlrogge, J. B. (2003). Disruption of the FATB gene in Arabidopsis demonstrates an essential role of saturated fatty acids in plant growth. Plant Cell 15, 1020–1033. doi: 10.1105/tpc.008946
Chakravorty, D., Trusov, Y., Zhang, W., Acharya, B. R., Sheahan, M. B., McCurdy, D. W., et al. (2011). An atypical heterotrimeric G-protein γ-subunit is involved in guard cell K+-channel regulation and morphological development in Arabidopsis thaliana. Plant J. 67, 840–851. doi: 10.1111/j.1365-313X.2011.04638.x
Chauffour, F., Bailly, M., Perreau, F., Cueff, G., Suzuki, H., Collet, B., et al. (2019). Multi-omics analysis reveals sequential roles for ABA during seed maturation. Plant Physiol. 180, 1198–1218. doi: 10.1104/pp.19.00338
Cheng, P., Li, H., Yuan, L., Li, H., Xi, L., Zhang, J., et al. (2018). The ERA-related GTPase AtERG2 associated with mitochondria 18S RNA is essential for early embryo development in Arabidopsis. Front. Plant Sci. 9:182. doi: 10.3389/fpls.2018.00182
Cheng, Z. J., Zhao, X. Y., Shao, X. X., Wang, F., Zhou, C., Liu, Y. G., et al. (2014). Abscisic acid regulates early seed development in Arabidopsis by ABI5-mediated transcription of SHORT HYPOCOTYL UNDER BLUE1. Plant Cell 26, 1053–1068. doi: 10.1105/tpc.113.121566
Creff, A., Brocard, L., Joubès, J., Taconnat, L., Doll, N. M., Marsollier, A. C., et al. (2019). A stress-response-related inter-compartmental signalling pathway regulates embryonic cuticle integrity in Arabidopsis. PLoS Genet. 15:e1007847. doi: 10.1371/journal.pgen.1007847
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158.
Duan, Z., Zhang, M., Zhang, Z., Liang, S., Fan, L., Yang, X., et al. (2022). Natural allelic variation of GmST05 controlling seed size and quality in soybean. Plant Biotechnol. J. 20, 1807–1818. doi: 10.1111/pbi.13865
East, A. K., Curnock, S. P., and Dyke, K. G. (1990). Change of a single amino acid in the leader peptide of a staphylococcal beta-lactamase prevents the appearance of the enzyme in the medium. FEMS Microbiol. Lett. 57, 249–254. doi: 10.1016/0378-1097(90)90075-2
Evfratov, S. A., Osterman, I. A., Komarova, E. S., Pogorelskaya, A. M., Rubtsova, M. P., Zatsepin, T. S., et al. (2017). Application of sorting and next generation sequencing to study 5′-UTR influence on translation efficiency in Escherichia coli. Nucleic Acids Res. 45, 3487–3502.
Fang, N., Xu, R., Huang, L., Zhang, B., Duan, P., Li, N., et al. (2016). SMALL GRAIN 11 controls grain size, grain number and grain yield in rice. Rice 9:64.
Fernandez, G., Shanmugasundaram, S., Shanmugasundaram, S., and Mclean, B. T. (1988). The AVRDC mungbean improvement program: The past, present and future. Shanhua: AVRDC.
Garcia, D., Saingery, V., Chambrier, P., Mayer, U., Jürgens, G., and Berger, F. (2003). Arabidopsis haiku mutants reveal new controls of seed size by endosperm. Plant Physiol. 131, 1661–1670. doi: 10.1104/pp.102.018762
Ge, L., Yu, J., Wang, H., Luth, D., Bai, G., Wang, K., et al. (2016). Increasing seed size and quality by manipulating BIG SEEDS1 in legume species. Proc. Natl. Acad. Sci. U.S.A. 113, 12414–12419. doi: 10.1073/pnas.1611763113
Gong, C., Zhao, S., Yang, D., Lu, X., Anees, M., He, N., et al. (2022). Genome-wide association analysis provides molecular insights into the natural variation of watermelon seed size. Hortic. Res. 9:uhab074. doi: 10.1093/hr/uhab074
Guo, N., Gu, M., Hu, J., Qu, H., and Xu, G. (2020). Rice OsLHT1 functions in leaf-to-panicle nitrogen allocation for grain yield and quality. Front. Plant Sci. 11:1150. doi: 10.3389/fpls.2020.01150
Ha, J., Satyawan, D., Jeong, H., Lee, E., Cho, K. H., Kim, M. Y., et al. (2021). A near-complete genome sequence of mungbean (Vigna radiata L.) provides key insights into the modern breeding program. Plant Genome 14:e20121. doi: 10.1002/tpg2.20121
Hao, J., Wang, D., Wu, Y., Huang, K., Duan, P., Li, N., et al. (2021). The GW2-WG1-OsbZIP47 pathway controls grain size and weight in rice. Mol. Plant 14, 1266–1280. doi: 10.1016/j.molp.2021.04.011
Hu, D., Li, X., Yang, Z., Liu, S., Hao, D., Chao, M., et al. (2022). Downregulation of a gibberellin 3β-hydroxylase enhances photosynthesis and increases seed yield in soybean. New Phytol. 235, 502–517. doi: 10.1111/nph.18153
Huang, J., Taylor, J. P., Chen, J. G., Uhrig, J. F., Schnell, D. J., Nakagawa, T., et al. (2006). The plastid protein THYLAKOID FORMATION1 and the plasma membrane G-protein GPA1 interact in a novel sugar-signaling mechanism in Arabidopsis. Plant Cell 18, 1226–1238. doi: 10.1105/tpc.105.037259
Humphry, M. E., Lambrides, C. J., Chapman, S. C., Aitken, E., and Liu, C. J. (2010). Relationships between hard-seededness and seed weight in mungbean (Vigna radiata) assessed by QTL analysis. Plant Breed. 124, 292–298.
Islam, M. A., Islam, M. R., Haque, M. E., Yeasmin, F., and Hossain, M. A. (2015). Impacts of famers’ participation in upscaling technologies on mungbean (Vigna radiata l.) production in the south-western region of Bangladesh. Agriculturists 12, 39–47. doi: 10.3329/agric.v12i2.21730
Jensen, L. J., Kuhn, M., Stark, M., Chaffron, S., Creevey, C., Muller, J., et al. (2009). STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 37, D412–D416.
Kang, H. M., Sul, J. H., Service, S. K., Zaitlen, N. A., Kong, S. Y., Freimer, N. B., et al. (2010). Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42, 348–354.
Kang, Y. J., Kim, S. K., Kim, M. Y., Lestari, P., Kim, K. H., Ha, B. K., et al. (2014). Genome sequence of mungbean and insights into evolution within Vigna species. Nat. Commun. 5:5443. doi: 10.1038/ncomms6443
Lease, K. A., Wen, J., Li, J., Doke, J. T., Liscum, E., and Walker, J. C. (2001). A mutant Arabidopsis heterotrimeric G-protein beta subunit affects leaf, flower, and fruit development. Plant Cell 13, 2631–2641. doi: 10.1105/tpc.010315
Lee, S., Lee, J., Ricachenevsky, F. K., Punshon, T., Tappero, R., Salt, D. E., et al. (2021). Redundant roles of four ZIP family members in zinc homeostasis and seed development in Arabidopsis thaliana. Plant J. 108, 1162–1173. doi: 10.1111/tpj.15506
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760.
Li, M., Zhang, Y. W., Xiang, Y., Liu, M. H., and Zhang, Y. M. (2022a). IIIVmrMLM: The R and C++ tools associated with 3VmrMLM, a comprehensive GWAS method for dissecting quantitative traits. Mol. Plant 15, 1251–1253. doi: 10.1016/j.molp.2022.06.002
Li, M., Zhang, Y. W., Zhang, Z. C., Xiang, Y., Liu, M. H., Zhou, Y. H., et al. (2022b). A compressed variance component mixed model for detecting QTNs, and QTN-by-environment and QTN-by-QTN interactions in genome-wide association studies. Mol. Plant 15, 630–650. doi: 10.1016/j.molp.2022.02.012
Li, N., Xu, R., and Li, Y. (2019). Molecular networks of seed size control in plants. Annu. Rev. Plant Biol. 70, 435–463.
Li, S., Liu, Y., Zheng, L., Chen, L., Li, N., Corke, F., et al. (2012). The plant-specific G protein γ subunit AGG3 influences organ size and shape in Arabidopsis thaliana. New Phytol. 194, 690–703. doi: 10.1111/j.1469-8137.2012.04083.x
Li, Y., Zheng, L., Corke, F., Smith, C., and Bevan, M. W. (2008). Control of final seed and organ size by the DA1 gene family in Arabidopsis thaliana. Genes Dev. 22, 1331–1336. doi: 10.1101/gad.463608
Ligas, J., Pineau, E., Bock, R., Huynen, M. A., and Meyer, E. H. (2019). The assembly pathway of complex I in Arabidopsis thaliana. Plant J. 97, 447–459.
Liu, D., Yu, Z., Zhang, G., Yin, W., Li, L., Niu, M., et al. (2021). Diversification of plant agronomic traits by genome editing of brassinosteroid signaling family genes in rice. Plant Physiol. 187, 2563–2576. doi: 10.1093/plphys/kiab394
Liu, J. Y., Chen, J. B., Anochar, K., Lin, Y., Xue, C. C., Wu, R. R., et al. (2022a). High-quality genome assembly and genome-wide association studies provide genetic insights into natural variation in yield-related traits in mungbean.
Liu, J. Y., Lin, Y., Chen, J. B., Xue, C. C., Wu, R. R., Yan, Q., et al. (2022b). Identification and clarification of VrCYCA1: A key genic male sterility-related gene in mungbean by multi-omics analysis. Agriculture 12:686.
Liu, J. Y., Xue, C. C., Lin, Y., Yan, Q., Chen, J. B., Wu, R. R., et al. (2022c). Genetic analysis and identification of VrFRO8, a salt tolerance-related gene in mungbean. Gene 836:146658. doi: 10.1016/j.gene.2022.146658
Liu, Z., Li, N., Zhang, Y., and Li, Y. (2020a). Transcriptional repression of GIF1 by the KIX-PPD-MYC repressor complex controls seed size in Arabidopsis. Nat. Commun. 11:1846. doi: 10.1038/s41467-020-15603-3
Liu, J. Y., Zhang, Y. W., Han, X., Zuo, J. F., Zhang, Z., Shang, H., et al. (2020b). An evolutionary population structure model reveals pleiotropic effects of GmPDAT for traits related to seed size and oil content in soybean. J. Exp. Bot. 71, 6988–7002. doi: 10.1093/jxb/eraa426
Liu, J. Y., Li, P., Zhang, Y. W., Zuo, J. F., Li, G., Han, X., et al. (2020c). Three-dimensional genetic networks among seed oil-related traits, metabolites and genes reveal the genetic foundations of oil synthesis in soybean. Plant J. 103, 1103–1124. doi: 10.1111/tpj.14788
Lu, X., Li, Q. T., Xiong, Q., Li, W., Bi, Y. D., Lai, Y. C., et al. (2016). The transcriptomic signature of developing soybean seeds reveals the genetic basis of seed trait adaptation during domestication. Plant J. 86, 530–544. doi: 10.1111/tpj.13181
Luo, J., Liu, H., Zhou, T., Gu, B., Huang, X., Shangguan, Y., et al. (2013). An-1 encodes a basic helix-loop-helix protein that regulates awn development, grain size, and grain number in rice. Plant Cell 25, 3360–3376. doi: 10.1105/tpc.113.113589
Lynch, T., Née, G., Chu, A., Krüger, T., Finkemeier, I., and Finkelstein, R. R. (2022). ABI5 binding protein2 inhibits ABA responses during germination without ABA-INSENSITIVE5 degradation. Plant Physiol. 189, 666–678. doi: 10.1093/plphys/kiac096
Manan, S., Ahmad, M. Z., Zhang, G., Chen, B., Haq, B. U., Yang, J., et al. (2017). Soybean LEC2 regulates subsets of genes involved in controlling the biosynthesis and catabolism of seed storage substances and seed development. Front. Plant Sci. 8:1604. doi: 10.3389/fpls.2017.01604
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Mei, L., Cheng, X. Z., Wang, S. H., Wang, L. X., and Liu, C. J. (2009). Relationship between bruchid resistance and seed mass in mungbean based on QTL analysis. Genome 52, 589–596. doi: 10.1139/G09-031
Moreno, J. E., Moreno-Piovano, G., and Chan, R. L. (2018). The antagonistic basic helix-loop-helix partners BEE and IBH1 contribute to control plant tolerance to abiotic stress. Plant Sci. 271, 143–150. doi: 10.1016/j.plantsci.2018.03.024
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628.
Nguyen, C. X., Paddock, K. J., Zhang, Z., and Stacey, M. G. (2021). GmKIX8-1 regulates organ size in soybean and is the causative gene for the major seed weight QTL qSw17-1. New Phytol. 229, 920–934. doi: 10.1111/nph.16928
Orozco-Arroyo, G., Paolo, D., Ezquer, I., and Colombo, L. (2015). Networks controlling seed size in Arabidopsis. Plant Reprod. 28, 17–32. doi: 10.1007/s00497-015-0255-5
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T., and Salzberg, S. L. (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667. doi: 10.1038/nprot.2016.095
Plackett, A. R., Powers, S. J., Fernandez-Garcia, N., Urbanova, T., Takebayashi, Y., Seo, M., et al. (2012). Analysis of the developmental roles of the Arabidopsis gibberellin 20-oxidases demonstrates that GA20ox1, -2, and -3 are the dominant paralogs. Plant Cell 24, 941–960. doi: 10.1105/tpc.111.095109
Pongpanich, M., Sullivan, P. F., and Tzeng, J. Y. (2010). A quality control algorithm for filtering SNPs in genome-wide association studies. Bioinformatics 26, 1731–1737.
Rampey, R. A., Baldridge, M. T., Farrow, D. C., Bay, S. N., and Bartel, B. (2013). Compensatory mutations in predicted metal transporters modulate auxin conjugate responsiveness in Arabidopsis. G3 3, 131–141. doi: 10.1534/g3.112.004655
Ren, Z., Fan, K., Fang, T., Zhang, J., Yang, L., Wang, J., et al. (2019). Maize empty pericarp602 encodes a P-type PPR protein that is essential for seed development. Plant Cell Physiol. 60, 1734–1746. doi: 10.1093/pcp/pcz083
Schnurr, J., Shockey, J., and Browse, J. (2004). The acyl-CoA synthetase encoded by LACS2 is essential for normal cuticle development in Arabidopsis. Plant Cell 16, 629–642. doi: 10.1105/tpc.017608
She, K. C., Kusano, H., Koizumi, K., Yamakawa, H., and Shimada, H. (2010). A novel factor floury endosperm2 is involved in regulation of rice grain size and starch quality. Plant Cell 22, 3280–3294. doi: 10.1105/tpc.109.070821
Shin, J. H., Blay, S., Mcneney, B., and Graham, J. (2006). LDheatmap: An R function for graphical display of pairwise linkage disequilibria between single nucleotide polymorphisms. J. Stat. Softw. 16, 1–9.
Singh, A. K., Fu, D. Q., El-Habbak, M., Navarre, D., Ghabrial, S., and Kachroo, A. (2011). Silencing genes encoding omega-3 fatty acid desaturase alters seed size and accumulation of bean pod mottle virus in soybean. Mol. Plant Microbe Interact. 24, 506–515. doi: 10.1094/MPMI-09-10-0201
Smalle, J., and Vierstra, R. D. (2004). The ubiquitin 26S proteasome proteolytic pathway. Annu. Rev. Plant Biol. 55, 555–590.
Smith, D. S., Maxwell, P. W., and De Boer, S. H. (2005). Comparison of several methods for the extraction of DNA from potatoes and potato-derived products. J. Agric. Food Chem. 53, 9848–9859. doi: 10.1021/jf051201v
Somta, P., Ammaranan, C., Ooi, P. A.-C., and Srinives, P. (2007). Inheritance of seed resistance to bruchids in cultivated mungbean (Vigna radiata, L. Wilczek). Euphytica 155, 47–55. doi: 10.1007/s10681-006-9299-9
Song, X. J., Huang, W., Shi, M., Zhu, M. Z., and Lin, H. X. (2007). A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat. Genet. 39, 623–630. doi: 10.1038/ng2014
Sun, J. Y., Hammerlindl, J., Forseille, L., Zhang, H., and Smith, M. A. (2014). Simultaneous over-expressing of an acyl-ACP thioesterase (FatB) and silencing of acyl-acyl carrier protein desaturase by artificial microRNAs increases saturated fatty acid levels in Brassica napus seeds. Plant Biotechnol. J. 12, 624–637.
Sun, S., Wang, L., Mao, H., Shao, L., Li, X., Xiao, J., et al. (2018). A G-protein pathway determines grain size in rice. Nat. Commun. 9:851. doi: 10.1038/s41467-018-03141-y
Ullah, H., Chen, J. G., Young, J. C., Im, K. H., Sussman, M. R., and Jones, A. M. (2001). Modulation of cell proliferation by heterotrimeric G protein in Arabidopsis. Science 292, 2066–2069.
Wu, Y., Fu, Y., Zhao, S., Gu, P., Zhu, Z., Sun, C., et al. (2016). CLUSTERED PRIMARY BRANCH 1, a new allele of DWARF11, controls panicle architecture and seed size in rice. Plant Biotechnol. J. 14, 377–386. doi: 10.1111/pbi.12391
Xia, T., Li, N., Dumenil, J., Li, J., Kamenski, A., Bevan, M. W., et al. (2013). The ubiquitin receptor DA1 interacts with the E3 ubiquitin ligase DA2 to regulate seed and organ size in Arabidopsis. Plant Cell 25, 3347–3359. doi: 10.1105/tpc.113.115063
Xiao, Y. G., Sun, Q. B., Kang, X. J., Chen, C. B., and Ni, M. (2016). SHORT HYPOCOTYL UNDER BLUE1 or HAIKU2 mixepression alters canola and Arabidopsis seed development. New Phytol. 209, 636–649.
Xiu, Z., Peng, L., Wang, Y., Yang, H., Sun, F., Wang, X., et al. (2020). Empty Pericarp24 and empty Pericarp25 are required for the splicing of mitochondrial introns, complex I assembly, and seed development in maize. Front. Plant Sci. 11:608550. doi: 10.3389/fpls.2020.608550
Xu, Q., and Liesche, J. (2021). Sugar export from Arabidopsis leaves: Actors and regulatory strategies. J. Exp. Bot. 72, 5275–5284. doi: 10.1093/jxb/erab241
Xu, Y., Yang, T., Zhou, Y., Yin, S., Li, P., Liu, J., et al. (2018). Genome-wide association mapping of starch pasting properties in maize using single-locus and multi-locus models. Front. Plant Sci. 9:1311. doi: 10.3389/fpls.2018.01311
Yan, Q., Wang, Q., Cheng, X., Wang, L., and Chen, X. (2020). High-quality genome assembly, annotation and evolutionary analysis of the mungbean (Vigna radiata) genome. Hoboken, NJ: Authorea. doi: 10.22541/au.160587196.63922177/v1
Zhang, B., Li, C., Li, Y., and Yu, H. (2020). Mobile terminal flower1 determines seed size in Arabidopsis. Nat. Plants 6, 1146–1157. doi: 10.1038/s41477-020-0749-5
Zhang, H., Cheng, F., Xiao, Y., Kang, X., Wang, X., Kuang, R., et al. (2017). Global analysis of canola genes targeted by SHORT HYPOCOTYL UNDER BLUE 1 during endosperm and embryo development. Plant J. 91, 158–171. doi: 10.1111/tpj.13542
Zhang, W., Xu, W., Zhang, H., Liu, X., Cui, X., Li, S., et al. (2021). Comparative selective signature analysis and high-resolution GWAS reveal a new candidate gene controlling seed weight in soybean. Theor. Appl. Genet. 134, 1329–1341. doi: 10.1007/s00122-021-03774-6
Zhang, Y. F., Suzuki, M., Sun, F., and Tan, B. C. (2017). The mitochondrion-targeted PENTATRICOPEPTIDE REPEAT78 protein is required for nad5 mature mRNA stability and seed development in maize. Mol. Plant 10, 1321–1333. doi: 10.1016/j.molp.2017.09.009
Zhang, Y. M., Jia, Z., and Dunwell, J. M. (2019). Editorial: The applications of new multi-locus GWAS methodologies in the genetic dissection of complex traits. Front. Plant Sci. 10:100. doi: 10.3389/fpls.2019.00100
Zhang, Z., Ersoz, E., Lai, C. Q., Todhunter, R. J., Tiwari, H. K., Gore, M. A., et al. (2010). Mixed linear model approach adapted for genome-wide association studies. Nat. Genet. 42, 355–360.
Zhang, Z., Zhao, H., Huang, F., Long, J., Song, G., and Lin, W. (2019). The 14-3-3 protein GF14f negatively affects grain filling of inferior spikelets of rice (Oryza sativa L.). Plant J. 99, 344–358. doi: 10.1111/tpj.14329
Zhao, X. Y., Wang, J. G., Song, S. J., Wang, Q., Kang, H., Zhang, Y., et al. (2016). Precocious leaf senescence by functional loss of PROTEIN S-ACYL TRANSFERASE14 involves the NPR1-dependent salicylic acid signaling. Sci. Rep. 6:20309. doi: 10.1038/srep20309
Zhou, X., and Stephens, M. (2012). Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 44, 821–824.
Zuo, J. F., Ikram, M., Liu, J. Y., Han, C. Y., Niu, Y., Dunwell, J. M., et al. (2022). Domestication and improvement genes reveal the differences of seed size- and oil-related traits in soybean domestication and improvement. Comput. Struct. Biotechnol. J. 20, 2951–2964. doi: 10.1016/j.csbj.2022.06.014
Keywords: multiple genome-wide association studies, QTN-by-environment interactions, VrEmp24/25, multi-omics analysis, RT-qPCR
Citation: Liu J, Lin Y, Chen J, Yan Q, Xue C, Wu R, Chen X and Yuan X (2022) Genome-wide association studies provide genetic insights into natural variation of seed-size-related traits in mungbean. Front. Plant Sci. 13:997988. doi: 10.3389/fpls.2022.997988
Received: 19 July 2022; Accepted: 15 August 2022;
Published: 13 October 2022.
Edited by:
Zhenyu Jia, University of California, Riverside, United StatesReviewed by:
Jia Wen, The University of North Carolina at Chapel Hill, United StatesYoulu Yuan, Cotton Research Institute (CAAS), China
Copyright © 2022 Liu, Lin, Chen, Yan, Xue, Wu, Chen and Yuan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin Chen, Y3hAamFhcy5hYy5jbg==; Xingxing Yuan, eXh4QGphYXMuYWMuY24=