 Hao Guo†
Hao Guo† Yiqin Wang
Yiqin Wang Suqin Zhang
Suqin Zhang- College of Agriculture, Guizhou University, Guiyang, Guizhou, China
Drought stress is one of the main factors influencing the growth and development of an organism. Auricularia fibrillifera has strong dessication resistance. In A. fibrillifera under dessication-stress, the melanin content of fruiting bodies elevated significantly by >10-fold compared with the control. Folate content also increased sharply but decreased significantly after rehydration, and amino acid and biotin levels increased by 40.11 and 22.14%, respectively. In proteomic analysis, 1,572 and 21 differentially abundant proteins (DAPs) were identified under dessication-stress and rehydration, respectively. A large number of DAPs were annotated in “amino acid metabolism,” “carbohydrate metabolism,” and “translation” pathways, and the DAPs related to osmotic regulation and antioxidant enzymes were significantly increased in abundance. Transcriptome-proteome association analysis showed that most DAPs (30) were annotated in the “biosynthesis of antibiotics” pathway. DAPs and corresponding differentially expressed genes were all up-regulated in the “biotin biosynthesis” pathway and associated with “folate biosynthesis” and “phenylalanine, tyrosine, and tryptophan biosynthesis.” In the analysis of protein–protein interactions, the DAPs annotated in the “phenylalanine, tyrosine, and tryptophan biosynthesis” pathway had the strongest interactions with other DAPs. These enriched pathways could enhance amino acid, folate, biotin, and melanin levels during desiccation stress, which is consistent with the physiological data (amino acid, folate, biotin, and melanin contents). In addition, many DAPs related to the cytoskeleton were significantly increased in abundance under dessication-stress. Physiological and transcriptome data were in agreement with proteomic results. This work provides valuable insight into the dessication-tolerant mechanisms of A. fibrillifera.
Introduction
Auricularia fibrillifera is a common traditional Chinese food and medicine, which is the third most important cultivated mushroom worldwide (Yuan et al., 2019). It has a pleasant taste and many health-associated characteristics such as antioxidant, anticoagulant, antitumor, immunomodulatory, and cholesterol-lowering properties (Sekara et al., 2015). It is expected that the duration and severity of droughts will increase, resulting in adverse effects on agriculture and causing significant declines in crop production on a global scale (Lesk et al., 2016). Thus, it is important to improve drought tolerance in organisms for global food security and necessary to clarify the physiological and molecular mechanisms of dessication tolerance.
Some mechanisms of drought response are similar between A. fibrillifera and plant species (Wang et al., 2016, 2022; Zandalinas et al., 2018; Mahmood et al., 2019). The physiological effects induced by drought stress include altered cell wall elasticity, increased oxidative stress (Caruso et al., 2009), toxic metabolite generation, and extensive cellular damage in plants (Ahuja et al., 2010; Zandalinas et al., 2018). Plants have also evolved various drought tolerance mechanisms to adapt to drought stress. The drought resistance of plants is quite complex (Wang et al., 2015). Osmotic adjustment, hormonal regulation, antioxidant systems, and signal transduction play vital roles in drought tolerance (Zandalinas et al., 2018; Mahmood et al., 2019). Amino acids have crucial roles in osmotic adjustment. Melanins exist in fungi, plants, and microorganisms, and have the functions of free-radical scavenging, antioxidant activity, and radiation protection (Burmasova et al., 2019; Cordero and Casadevall, 2020; Cao et al., 2021). Biotin and biotinylation might be involved in energy management to cope with drought and flooding in the early stage of soybean-root tip (Wang et al., 2016). Foliar application of folate was found to be not only suitable for drought stress alleviation in Coriandrum sativum L. but also beneficial for improvement in growth and yield under water-deficit circumstances (Khan et al., 2022).
A loss of more than 10% of plant fresh weight can lead to water stress, which can induce the synthesis of some specific ones (including S-like RNase homolog, actin depolymerizing factor, rubisco activase, and translational initiation factor EF-Tu), maintain others, and decrease the levels of some plant proteins (such as isoflavone reductase-like protein and chloroplast Rieske Fe-S protein) (Hsiao, 1973; Salekdeh et al., 2002). Investigating the proteome profiles under drought stress can provide detailed information regarding the specific protein changes associated with drought responses (Koh et al., 2015). Under drought stress response/tolerance, numerous proteins related to metabolism, photosynthesis, stress, and defense were identified by a comparative proteome analysis in Brassica napus seedlings (Koh et al., 2015). Proteomic studies on post-drought recovery have clarified the mechanisms of plants in response to drought stress (Khan and Komatsu, 2016). Four novel drought-responsive proteins were identified during drought stress and recovery in rice leaves by proteomic analysis (Salekdeh et al., 2002). Aldehyde dehydrogenase and peroxidase are known to decrease aldehydes and toxic reactive oxygen species from soybean roots and help in the recovery from drought stress (Khan and Komatsu, 2016). Under drought stress, the abundance of most protein changes may be associated with gene transcription. There was a positive correlation between protein expression and gene transcription in B. napus, although different patterns between proteins and transcripts were detected at various time points (Koh et al., 2015). These reports provide valuable information for investigating the molecular mechanisms of dessication tolerance in A. fibrillifera.
For general organisms (the majority of terrestrial plants and mushrooms), the vegetative bodies will dry up or even die under severe drought stress. Selaginella lepidophylla is a desiccation-tolerant plant capable of surviving complete vegetative tissue dehydration and reviving under water conditions (Pampurova and Van Dijck, 2014). A candidate basic helix-loop-helix (bHLH) transcription factor was observed to be highly expressed at 4% relative water content in S. lepidophylla (SlbHLH), and its overexpression significantly increased integrated water-use efficiency and green cotyledon emergence rates under water-deficit stress in Arabidopsis (Ariyarathne and Wone, 2022). The fruit bodies of Auricularia dries out and enters dormancy under dessication conditions. The dormancy may be broken once watered. Hence, the dessication tolerance and rehydration capability of A. fibrillifera make it a suitable model to elucidate its adaptive mechanism against dessication-stress (Ma et al., 2014). Currently, there are limited reports on the molecular mechanisms of dessication tolerance in A. fibrillifera. In this study, protein markers and pathways were investigated under dessication-stress in A. fibrillifera by data-independent acquisition (DIA) proteomic profiling to explore the dessication-tolerant mechanism of A. fibrillifera and provide novel information for dessication-tolerant breeding for A. fibrillifera.
Materials and methods
Materials and treatments
A dessication-tolerant A. fibrillifera cultivar “CSLZ” was used for this study. The strain was maintained in a culture medium to generate fruiting bodies (Wang et al., 2022). Upon reaching full mycelial colonization, polyethylene bags were removed, and the substrate was cultured at 25 ± 1°C under a 15:9 h-light/dark cycle. The substrate-containing bags were routinely sprayed with 15 mL water/bag 8 times daily. When the diameters of the fruit bodies reached 2-3 cm, dessication-stress treatment was initiated, and the fruit bodies on the substrate naturally dehydrated. The regularly watered fruit bodies on the substrate served as a parallel control. When the water loss rate of fruit bodies achieved 60% (desiccation-stress, DS) compared to the CK1 parallel control, uniformly sized fruit bodies were harvested as the first samples. The fruit bodies were rewatered. The next sampling process was conducted when the water loss rate of fruit bodies was 50% [after rehydration (RE) for 1 h] compared to the CK2 parallel control. Each sample pool included 15 individual fruit bodies, and the experiment was performed in triplicate. All the specimens were immediately frozen in liquid nitrogen and kept at -80°C for further analysis.
Physiological analysis
The amino acid (Cas No.: BC1575, Solarbio, Beijing, China), biotin (Cas No.: BC4804, Solarbio), and folate (Cas No.: BC4834, Solarbio) contents were determined according to the kit instructions. In brief, the α-amino group of amino acids can react with hydrated ninhydrin to produce a blue-violet compound with an absorption peak at 570 nm, which was detected with a microplate reader (Thermo-Fisher-Scientific, San Jose, CA, United States). Both biotin and folate have ultraviolet absorption at 210 nm, and their contents were determined by high performance liquid chromatography (HPLC) (Shimadzu, Kyoto, Japan) and ultraviolet detector (Shimadzu). Melanin was extracted using a method by Wang et al. (2022). Shortly, 1.0 g fruit body was ground in 50 mL of 1 M NaOH. The samples were treated in an ultrasonic cleaner (300 W) for 2 h at 60°C. The supernatant (pH adjusted to 1.5) was immersed in a boiling water bath for 10 h, and then centrifuged at 9,156 × g for 15 min. After air-drying the precipitation, the melanin content was calculated.
Protein extraction
Total proteins of fruit bodies were isolated according to the phenol method (Isaacson et al., 2006) with slight modifications: briefly, 0.5 g fruit bodies were ground into a fine powder in a lysis buffer containing 877 mM sucrose, 100 mM EDTA, 20 mM Tris-HCL (pH = 8.0), 1 mM dithiothreitol (DTT), 2% (v:v) β-mercaptoethanol, 1% (v:v) Triton X-100, and 0.1 × Cocktail (Roche, Switzerland). Subsequently, 2 × volume of tris-saturated phenol (pH = 7.5) was added and centrifuged at 25,000 × g for 15 min at 4°C. After collecting the supernatant, 5 × volume of precooled precipitate solution, containing 0.1 M ammonium acetate in methanol and 10 mM DTT, was added to the protein mixture. Every sample was maintained for 2 h at -20°C. Then, the samples were centrifuged at 25,000 g for 15 min at 4°C, and the supernatant was removed. The pellets were further washed with 1 mL of precooled acetone [precooled acetone:sample = 5:1 (v/v)] with centrifugation at 25,000 × g for 15 min at 4°C. After air-drying the pellets, 200 μL of L3 lysis buffer containing 7 M urea, 2 M thiourea, 20 mM Tris, 10 mM DTT, and 1 × Cock-tail (Roche) were added, ground (60 Hz, 2 min), and centrifuged at 25,000 × g for 15 min at 4°C. DTT (10 mM) was added to the supernatant and it was kept in a water bath at 56°C for 1 h. Subsequently, iodoacetamide (55 mM) was added and kept in the dark for 45 min and, after adding 1 mL cold acetone, all samples were maintained 2 h at -20°C. All samples were centrifuged at 25,000 × g for 15 min at 4°C. After removing the supernatants, the pellets were air-dried and dissolved in 200 μL L3 lysis buffer. Trypsin [protein:trypsin = 40:1 (w/w)] was added for enzymolysis, and then desalinated, vacuum-dried and redissoluted. Nanodrop ND-1000 (Thermo-Fisher-Scientific) was used to measure protein concentrations.
Liquid chromatography-tandem mass spectrometry analysis
As an internal standard for quantification, 2 mL of mixed extract solution (100 μg/mL) were used. After HPLC (Shimadzu) fractionation, the eluents were combined into 10 fractions for LC-tandem mass spectrometry (MS/MS). The freeze-dried peptides were dissolved in Solvent A (2% acetonitrile and 0.1% formic acid), and the peptide specimens were separated using an UltiMate 3000 UHPLC (Thermo-Fisher-Scientific). The analytical conditions were as follows: LC column, C18 (150 μm × 35 cm, 1.8 μm, 100 Å); gradient program, 5% B (98% acetonitrile and 0.1% formic acid) for 5.0 min, 5%–25% B for 115 min, 25%–35% B for 40 min, 35%–80% B for 10 min, 80% B for 5 min, and 5% B for 5 min; and flow rate, 300 nL/min. The peptide specimens were ionized by a nanoESI, and then put into a Q-Exactive HF tandem mass spectrometer (Thermo-Fisher-Scientific) for data-dependent acquisition mode detection (Tsou et al., 2015). The spectra of first-grade MS (MS1) were acquired in the scan range of 350–1,500 m/z with spray voltage of 1.6 kV, resolution of 120,000, automatic gain control (AGC) target of 3E6, and maximum injection time (MIT) of 50 ms. The spectra of second-grade MS (MS2) were obtained using the following parameters: resolution of 30,000, MIT of 100 ms, dynamic exclusion duration of 30 s, and AGC target of 1E5. Moreover, the mode of MS2 spectra was high-energy collisional dissociation (HCD), and the collision energy was 28%. For DIA analysis, the same nano-LC system and gradient were used as those employed for data-dependent acquisition analysis. The following were the DIA MS parameters: scan range of 400–1,250 m/z, resolution of 120,000, and MIT of 50 ms. The DIA isolation window was set to 17 m/z with loop count of 50, automatic MIT, resolution of 30,000, stepped collision energy of 22.5%, 25.0%, and 27.5%, and AGC target of 1E6.
Spectronaut was employed to efficiently deconvolute, precisely identify, and quantitatively analyze the data (Bernhardt et al., 2014).
Identification of differentially abundant protein and bioinformatics analysis
The peak areas of an ion pair were extracted using Spectronaut (Bruderer et al., 2015). The error correction and normalization steps were performed using the Msstats software package (Choi et al., 2014). The DAPs fit these two criteria, fold-change ≥ 2 and Q-value ≤ 0.05. The consistency and probability of DAPs were scored and compared with the Swissprot library, and the threshold was set as 1E–5 for protein description. The subcellular localization, protein-protein interaction (PPI) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were performed with these DAPs. A hypergeometric test was used to detect significantly enriched pathways (p < 0.05).
Proteome-transcriptome-associated analysis
We carried out proteome-transcriptome-associated analysis to gain a deeper understanding of the biological functions by combining results of two separate-omics techniques. The samples were analyzed at both mRNA and protein levels. The transcriptome data were retrieved from our previous study (NCBI SRA database under accession no. PRJNA695780) (Wang et al., 2022). Differentially expressed genes (DEGs) were chosen by DESeq software according to the following criteria: fold-change ≥ 2 and Q-value ≤ 0.001. Correlation analyses were performed between DAPs and DEGs of two omics, and the associated DAPs/DEGs were used for expression correlation analysis and metabolic pathway map integration analysis.
Construction of protein-protein interaction network
STRING v11.5 (string-db.org) was applied to analyze the PPIs of DAPs identified in this study and their PPI network was constructed. The minimum required interaction score parameter was set at a high confidence level (0.70).
Quantitative real-time polymerase chain reaction assays
Total RNA was isolated from fruiting bodies using an RNApre Pure Plant Plus Kit (Polyphenolics & Polysaccharides-rich) (Tiangen, Beijing, China). Using FastKing gDNA Dispelling RT SuperMix Kit (Tiangen), the extracted RNA was reverse-transcribed by following the manufacturer’s kit. Specific primers of 16 selected DAPs were designed using Primer 5 (Supplementary Table 1). The 18S gene was used for reference (Zhao et al., 2019). The qRT-PCR amplification was conducted on an ABI StepOne Real-Time polymerase chain reaction (PCR) System (Applied Biosystems, CA, United States). The relative expression levels of target genes were determined using the 2–ΔΔCt method with 3 technical and biological replications (Livak and Schmittgen, 2001).
Statistical analysis
Statistical tests were conducted with SPSS v19.0 (IBM Corp., NY, United States). The differences between means were compared with ANOVA followed by Duncan’s multiple range test. Pearson’s correlation analysis of binary variables was carried out. Level of statistical significance was set at p < 0.05.
Results
Physiological responses to dessication-stress and rehydration
The phenotype of fruiting bodies changed significantly under dessication-stress. In response to dessication, the fruiting bodies shrank and became hard but after rehydration, they began to expand and appeared softened (Figure 1A). Levels of amino acids and biotin in fruiting bodies increased significantly by 40.11 and 22.14%, respectively, under dessication-stress compared with those of the controls (Figures 1B,C). The melanin content increased significantly by > 10-fold compared to the control (Figure 1D). The folate peak was not detected in the control, which might be owing to its extremely low level; but levels increased rapidly under dessication-stress. After rehydration, the decrease in folate was the greatest (21.79%) compared with amino acids, biotin, and melanin (Figure 1E).
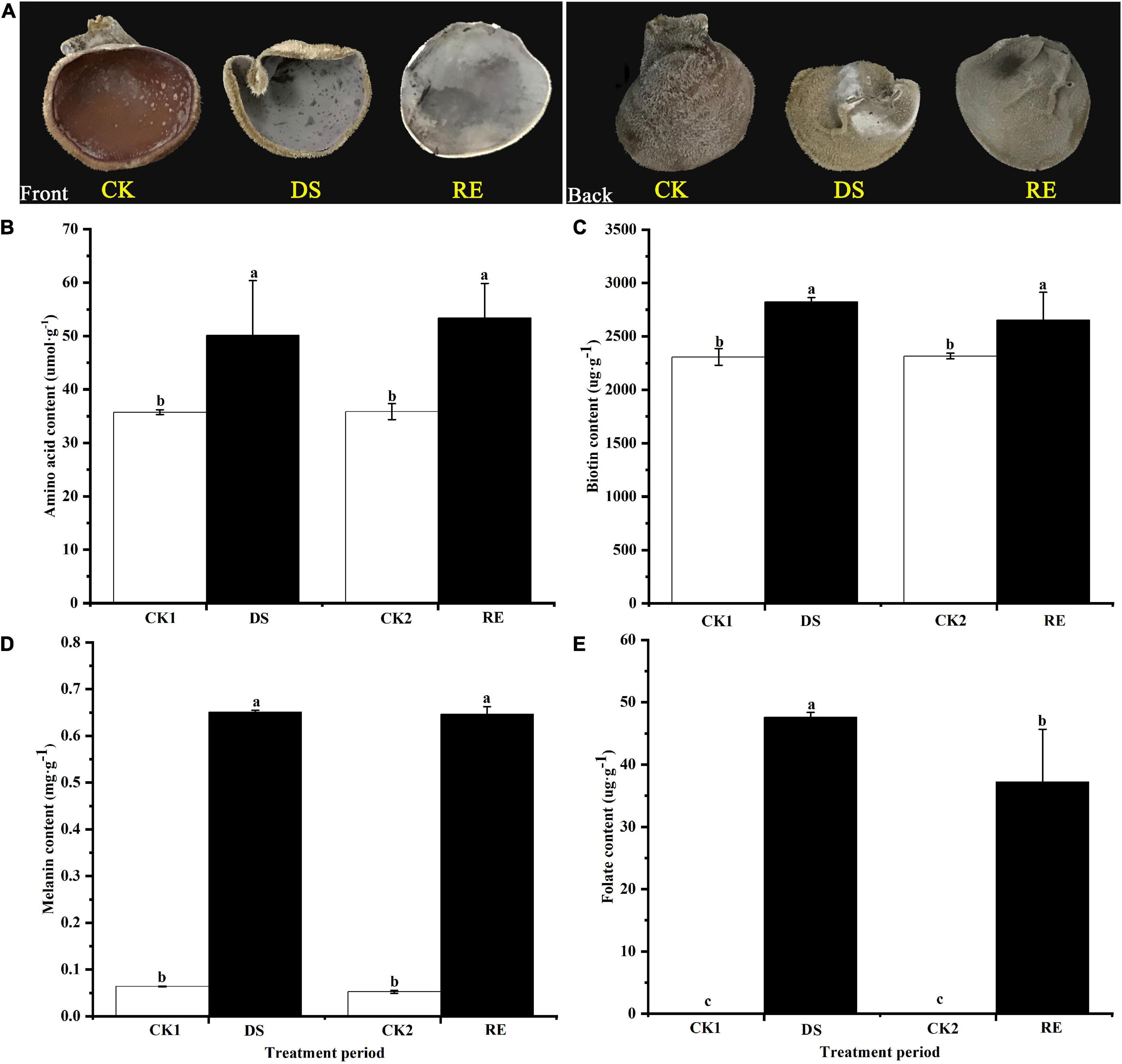
Figure 1. Phenotype and physiological responses to dessication-stress and rehydration in Auricularia fibrillifera. (A) Phenotypes of fruiting bodies. The front is on the left and the back is on the right. (B) Amino acid, (C) biotin, (D) melanin, and (E) folate content. CK1, CK2, DS, and RE represent the parallel control of desiccation-stress, parallel control of rehydration, desiccation-stress, and rehydration process, respectively. Bars indicate mean ± SD (n = 3). Values with different letters are significantly different at p < 0.05.
Differentially abundant proteins under dessication-stress
The proteomic analysis of fruiting bodies was performed during dessication-stress and rehydration stages, and stable results were obtained among the replicates of each treatment (Supplementary Figure 1). A total of 1,572 DAPs (1,005 more-abundant and 567 less-abundant) were observed under dessication-stress, and 10 more-abundant and 11 less-abundant DAPs were found after rehydration (Figure 2A and Supplementary Table 2). Fifteen DAPs appeared under both dessication-stress and rehydration (Figure 2B). The number of DAPs under dessication-stress was 74.86-fold of that after rehydration. Subcellular localization showed that under dessication-stress the DAPs were mainly located in mitochondria, followed by the cytoplasm and extracellular locations (Figure 2C).
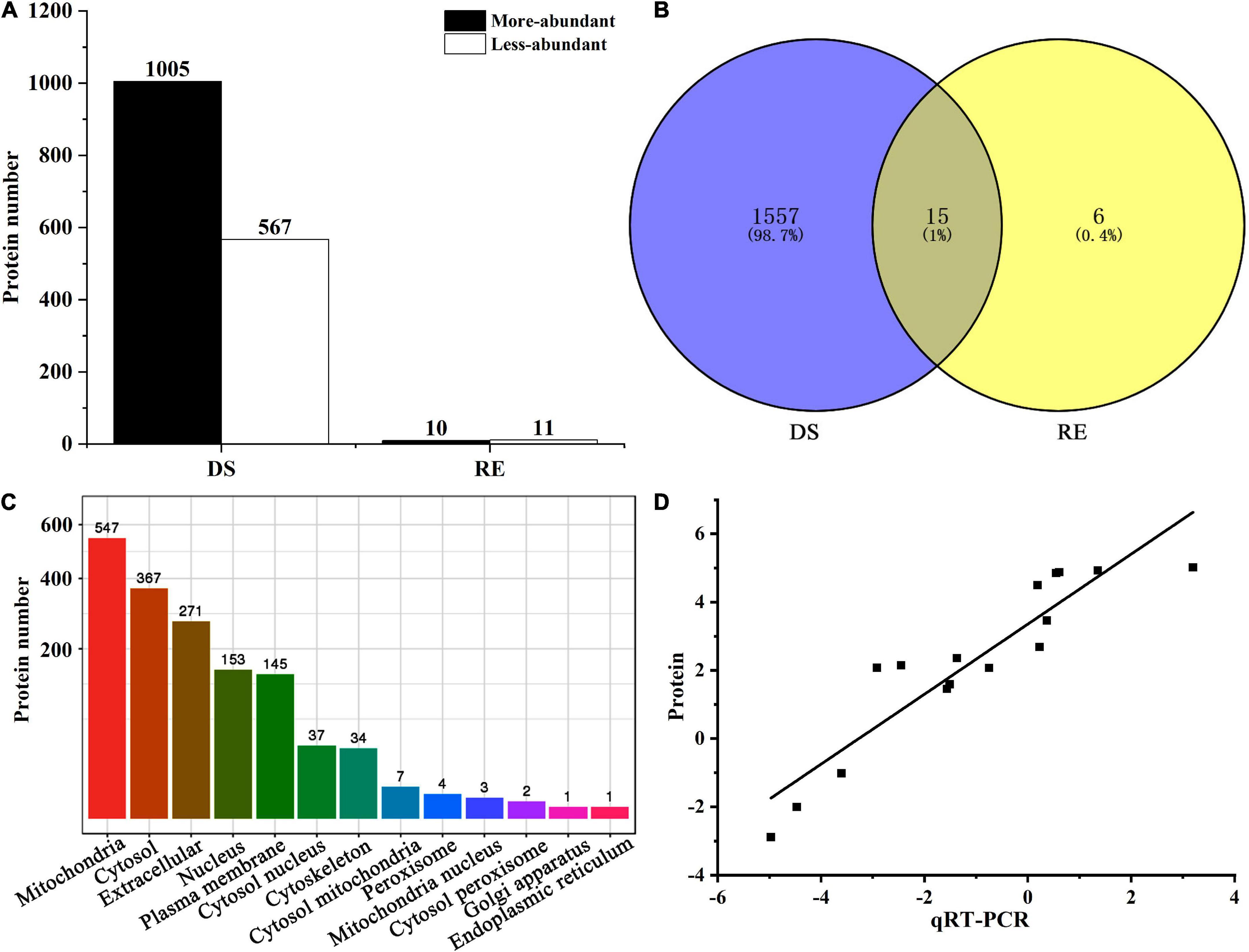
Figure 2. Number (A), Venn diagram (B), subcellular localization (C), and correlation coefficient of differentially abundant proteins between proteome and qRT-PCR (D). DS and RE represent dessication-stress and rehydration, respectively.
To verify the reliability of proteome data, we selected 16 DAPs for qRT-PCR assays (Supplementary Table 3). The correlation coefficient of the fold-change values between proteome and qRT-PCR was statistically significant (r = 0.84, p = 3.80E–7; Figure 2D). The more- or less-abundant of the proteins, as revealed by proteomics analysis, was corroborated by qRT-PCR.
Kyoto encyclopedia of genes and genomes enrichment analysis under dessication-stress
In plants, different proteins coordinate with each other to activate cellular responses, and pathway analysis is helpful in further understanding biological function. Under dessication-stress, there were 874 DAPs annotated to different pathways, among these most DAPs were enriched in “metabolism” pathways, such as “carbohydrate metabolism” (202) and “amino acid metabolism” (133) (Figure 3A and Supplementary Table 4). Many DAPs were also annotated in “translation” (154) and “transport and catabolism” (127) pathways. After rehydration, the DAP number decreased rapidly, and only six DAPs were enriched in KEGG pathways (Figure 3B). Some DAPs were not annotated to any KEGG pathway, while some DAPs were in more than one KEGG pathway at desiccation and rehydration stages. “Glyoxylate and dicarboxylate metabolism,” “folate biosynthesis,” “fructose and mannose metabolism,” “biosynthesis of antibiotics,” “biosynthesis of amino acids,” “phenylalanine, tyrosine and tryptophan biosynthesis,” “biotin metabolism,” and other pathways were significantly enriched during the desiccation stress (Figure 3C). The highest number of DAPs occurred in the “biosynthesis of antibiotics” pathway (Figure 3C). There was no significantly enriched pathway after rehydration.
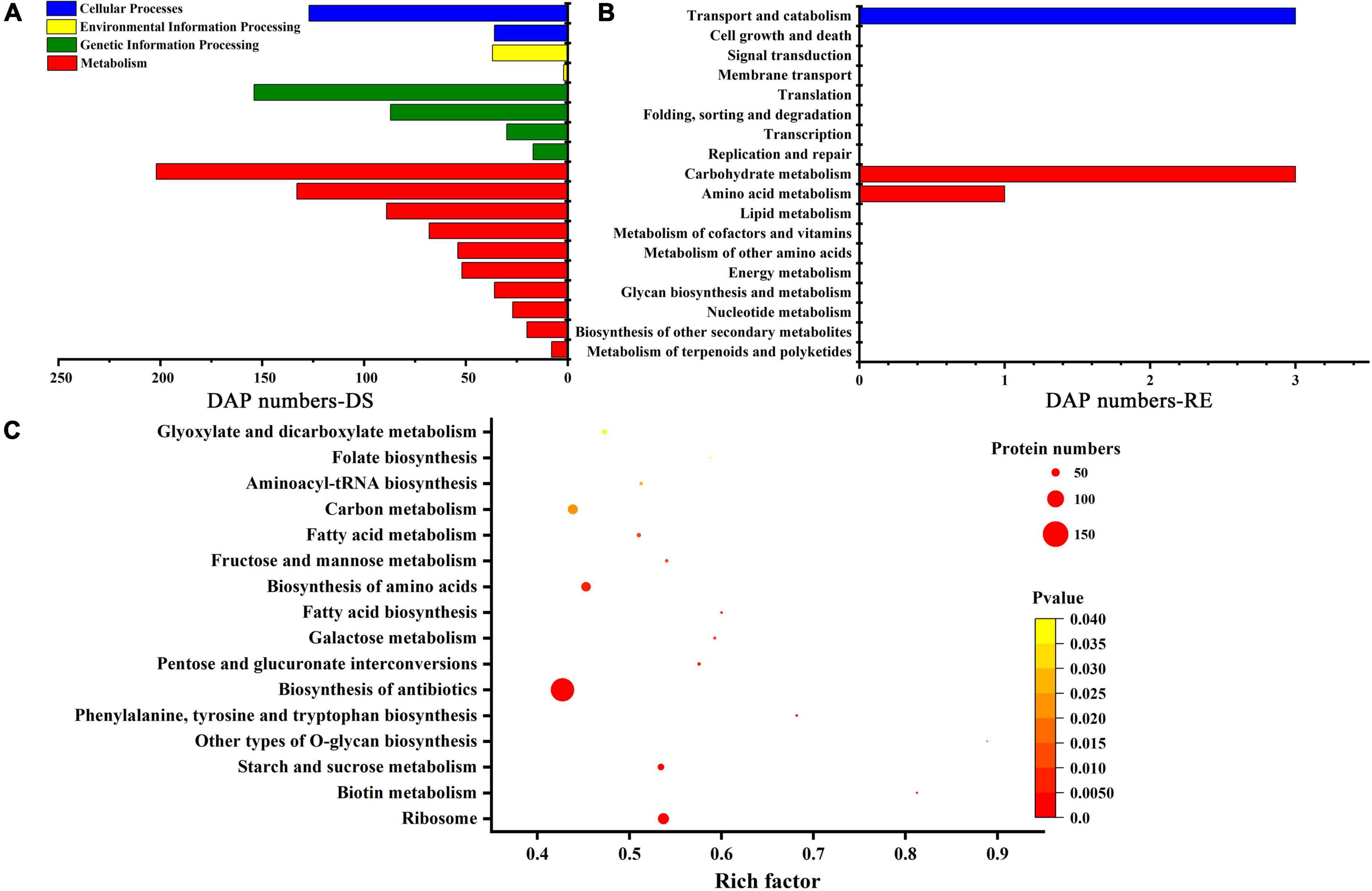
Figure 3. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. (A) KEGG pathways under dessication-stress. (B) KEGG pathways after rehydration. (C) Significantly enriched pathways under dessication-stress. DS, RE, and DAP represent dessication-stress, rehydration, and differentially abundant protein, respectively.
Proteome-transcriptome-associated analysis
The joint proteome and transcriptome analysis was useful in finding the regulation of gene expression (Maier et al., 2009). A total of 391 DAPs were associated with dessication-stress (Supplementary Tables 5, 6). The expression of corresponding DAPs and DEGs were focused mainly on two patterns: (1) both were up-regulated and (2) the DAPs were increased in abundance but the DEGs were down-regulated (Figure 4). The main associated pathways were “starch and sucrose metabolism,” “biosynthesis of antibiotics” and “biosynthesis of amino acids.” Interestingly, in the “biotin metabolism” pathway, all DAPs were associated, and both DAPs and DEGs were up-regulated. In the “folate biosynthesis,” “phenylalanine, tyrosine, and tryptophan biosynthesis” pathways, the associated DAPs were mainly increased in abundance, whereas the corresponding DEGs were expressed in the opposite direction (Figure 4). After rehydration, there were eight associated DAPs (Supplementary Tables 5, 6). Unfortunately, some proteins were not annotated in the KEGG pathway database.
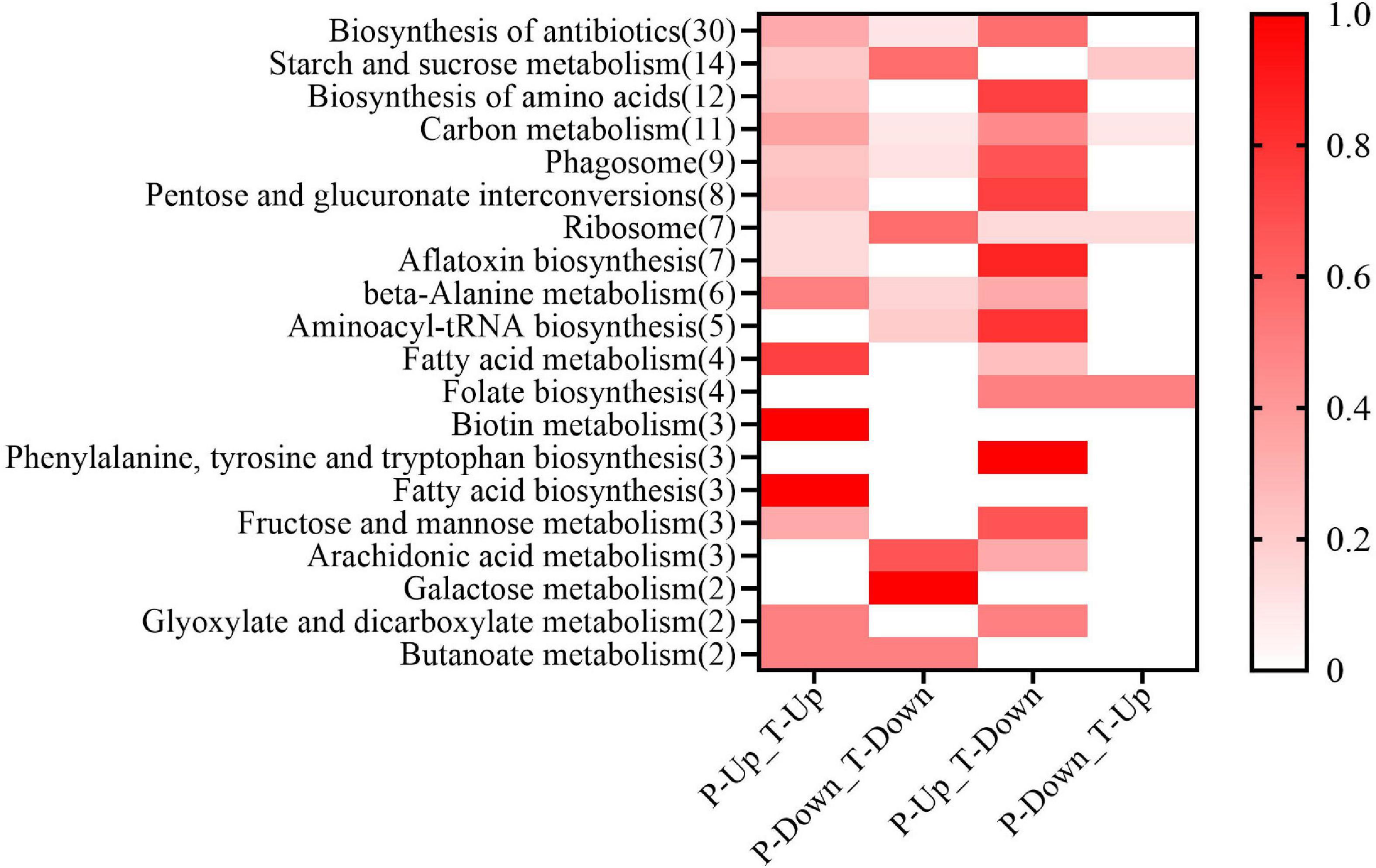
Figure 4. Proteome–transcriptome-associated Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis. In the horizontal axis, T, P, Up, and Down show differentially expressed genes from the transcriptome, differentially abundant proteins from the proteome, up-regulation, and down-regulation, respectively. On the Y axis, the number of differentially abundant proteins in each pathway is shown in parentheses. The color of the heat map represents the proportion of proteins in the pathway.
Protein-protein interaction analysis
Protein-protein interaction is essential for almost every process in cells and may be related to the specific function performed by proteins after binding into complexes through PPI. We selected 133 DAPs related to the dessication-stress response (e.g., stress response, sugar metabolism, and signal transduction) for PPI analysis. A total of 45 nodal DAPs were identified, which were divided into three clusters (Figure 5). Cluster 1 (blue bubbles) included 16 DAPs, which were mainly involved in stress response. CTA1 (catalase A), HOG1 (mitogen-activated protein kinase involved in osmoregulation), and PH1 (glycogen phosphorylase required for the mobilization of glycogen) were increased in abundance, which was conducive to the removal of excess reactive oxygen species, osmotic adjustment, and energy supply under dessication-stress.
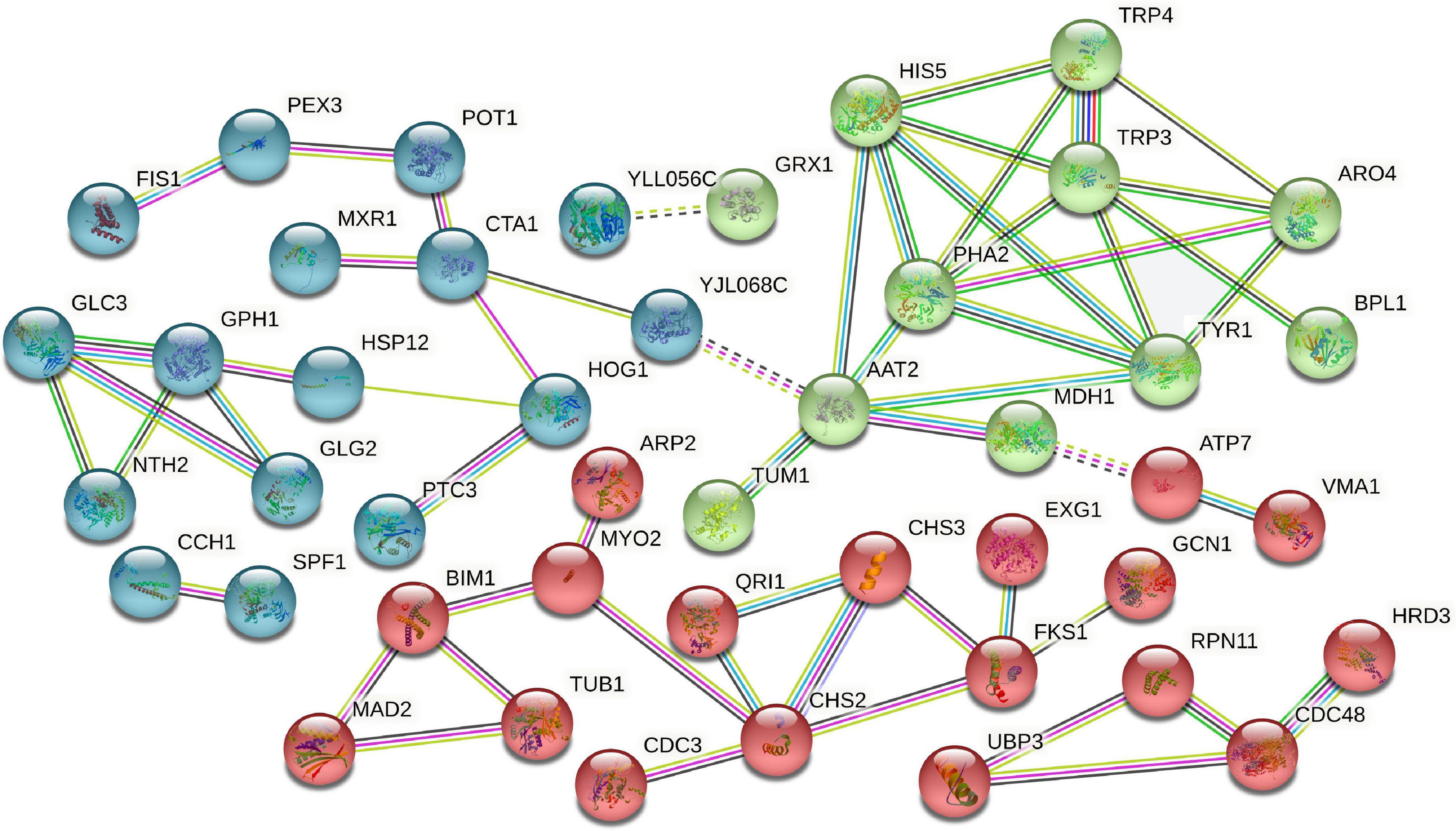
Figure 5. Protein-protein interaction analysis of dessication-responsive differentially abundant proteins. Different line colors represent the types of evidence used in predicting the associations: gene fusion (red), neighborhood (green), co-occurrence across genomes (blue), co-expression (black), experimental (purple), association in curated databases (light blue), or co-mentioned in PubMed abstracts (yellow).
Cluster 2 (green bubbles) included 11 more-abundant DAPs, most of which were related to amino acid metabolism. PHA2 (putative prephenate dehydratase) and TRP3 (multifunctional tryptophan biosynthesis protein) were important hub proteins in this cluster. The most nodal proteins were in the “phenylalanine, tyrosine, and tryptophan biosynthesis” and “tyrosine metabolism” pathways, and might participate in melanin synthesis.
Eighteen DAPs were found in cluster 3 (red bubbles), which were mainly involved in the synthesis and elasticity of the cell wall, such as chitin synthase II (CHS2), chitin synthase III (CHS3), and β-1,3-glucan synthase component (FKS1). These three kinds of proteins were less-abundant under dessication-stress and might contribute to dessication tolerance and the shrinkage of fruiting bodies.
Important proteins associated with the response to dessication-stress
Melanin synthesis
Melanins are derived from tyrosine, and referred to as “fungal armor” due to the ability of the polymer to protect microorganisms against a broad range of toxic insults (Gómez and Nosanchuk, 2003; Glagoleva et al., 2020). Sixteen (12 more-abundant and four less-abundant) DAPs were identified in the “tyrosine metabolism” pathway (Table 1). The fold-change values of more-abundant proteins were higher than those that decreased. Tyrosine synthesis mainly exists in the “phenylalanine, tyrosine, and tryptophan biosynthesis” pathway. Fifteen DAPs were identified in this pathway, they were all increased in abundance. Among them, two proteins that directly promote tyrosine synthesis were histidinol-phosphate aminotransferase (Unigene7245_All, Log2FC = 2.60) and aspartate aminotransferase (CL2639.Contig2_All, Log2FC = 2.78) (Table 1). They provided a reactive substrate for melanin synthesis.
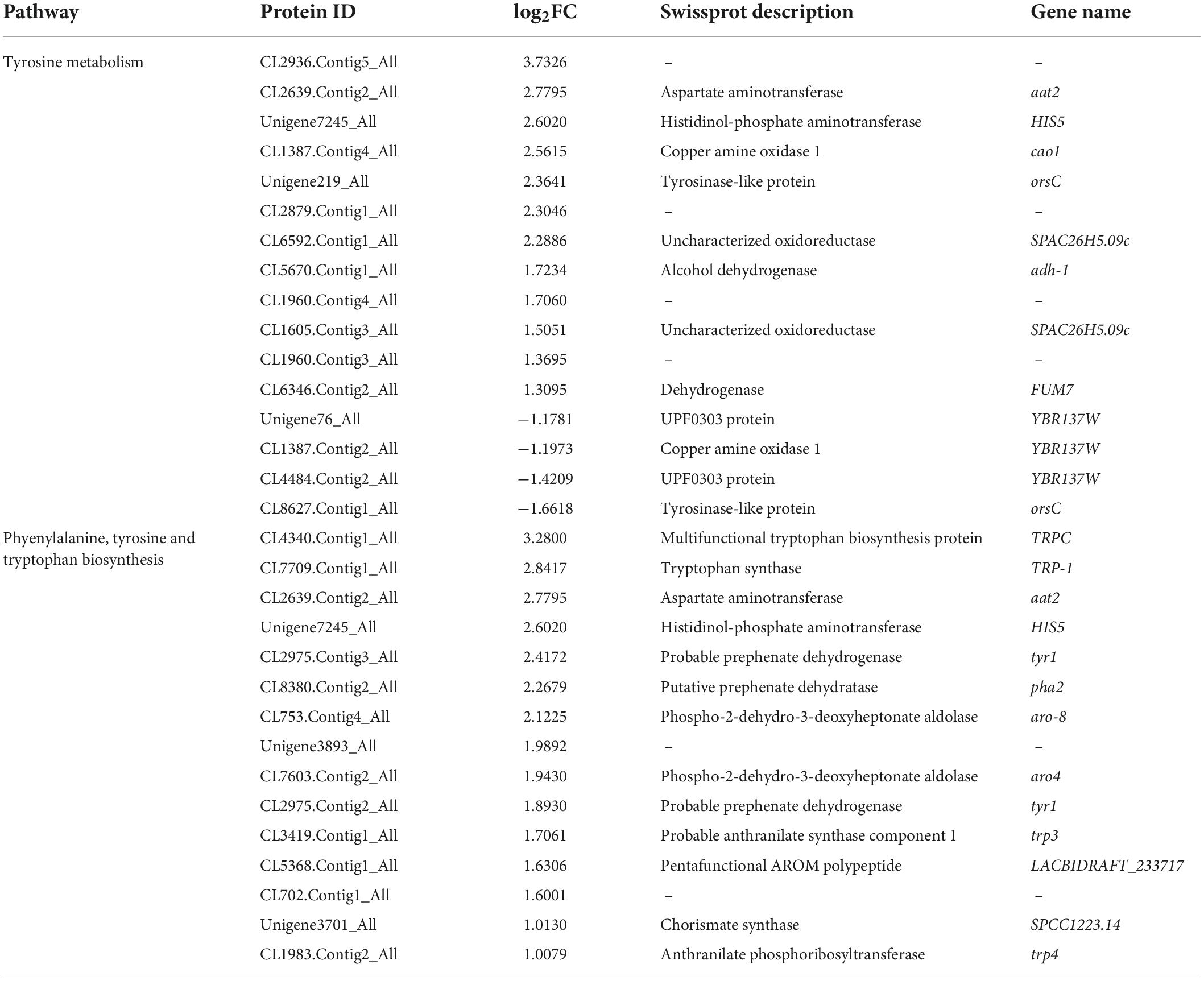
Table 1. Differentially abundant proteins in the “tyrosine metabolism” and “phenylalanine, tyrosine, and tryptophan biosynthesis” pathways.
Vitamin synthesis
Tetrahydrofolic acid (THF) and its derivatives are known as folate or B9 vitamins (Basset et al., 2005). Biotin is also known as vitamin H or B7 (Lazar et al., 2017). The pathways of “folate biosynthesis” and “biotin metabolism” were remarkably enriched under dessication-stress (Figure 3C). In the “folate biosynthesis” pathway, 10 DAPs were identified, including six more-abundant and four less-abundant. Folylpolyglutamate synthase (CL112.Contig1_All) and probable dihydrofolate synthetase (CL8178.Contig1_All) were increased in abundance to promote folate synthesis and were consistent with the folate content (Figure 1D). Thirteen more-abundant DAPs were identified in the “biotin metabolism” pathway (Table 2). Among them, the fold-change value of CL4671.Contig1_All was the highest (4.51) (Table 2). This was consistent with the biotin content (Figure 1B).
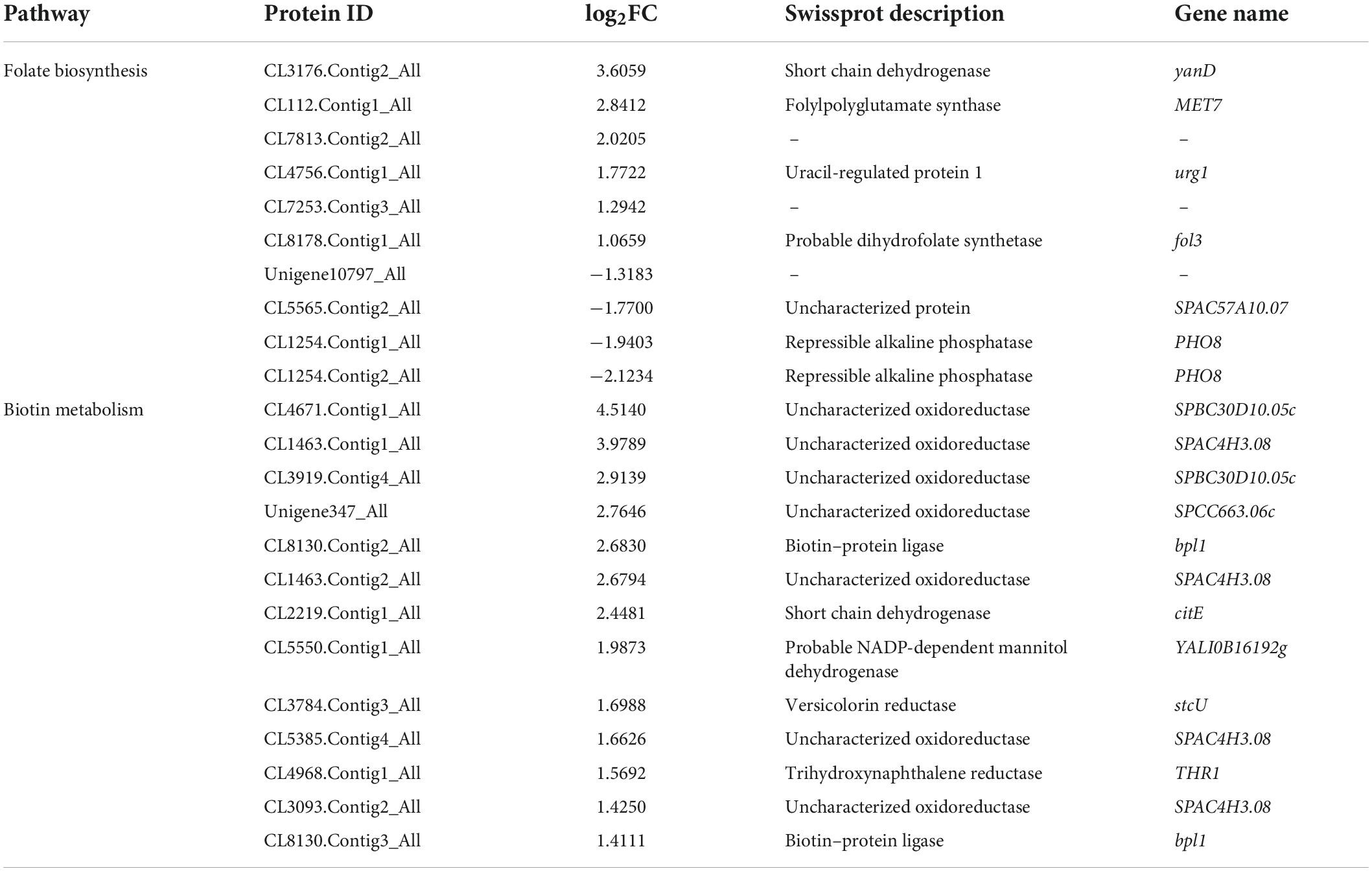
Table 2. Differentially abundant proteins in “folate biosynthesis” and “biotin metabolism” pathways.
Antibiotic synthesis
The associated number of DAPs/DEGs in the “biosynthesis of antibiotics” pathway was the greatest under dessication-stress by proteome–transcriptome-associated analysis. There were 135 DAPs enriched in this pathway, of which 117 DAPs were increased in abundance, and the fold-change value of more-abundant DAPs was much higher than that of the less-abundant value. The top 12 DAPs with | log2FC| ≥ 3 were increased in abundance (Table 3). Two DAPs (CL711.Contig2_All and CL6373.Contig2_All) were involved in isopenicillin-N synthase and glutamate-5-semi aldehyde dehydrogenase, respectively, and could enhance the synthesis of penicillin, cephalosporins, or carbapenem (Figure 6). Other 132 DAPs could promote intermediate synthesis of penicillin, carbapenem, or cephalosporin. For example, fumarase (CL6387.Contig1_All) could catalyze the conversion of fumarate to malate (Nunes-Nesi et al., 2007), then malate is oxidized to oxoloacetate by NAD+-dependent malate dehydrogenase (Nunes-Nesi, 2005). Oxoloacetate facilitates the synthesis of cysteine, and L-cysteine is the precursor for penicillin and cephalosporin synthesis in the “penicillin and cephalosporin biosynthesis” pathway.
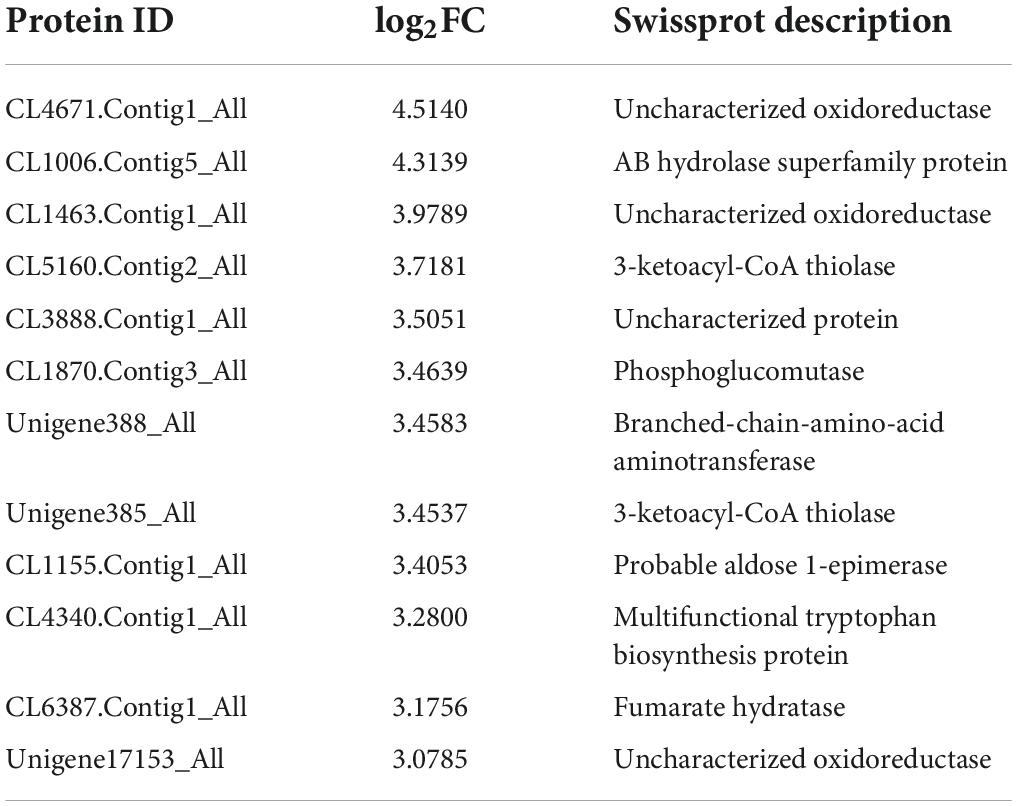
Table 3. Highly differentially abundant proteins in the “biosynthesis of antibiotic” pathway.
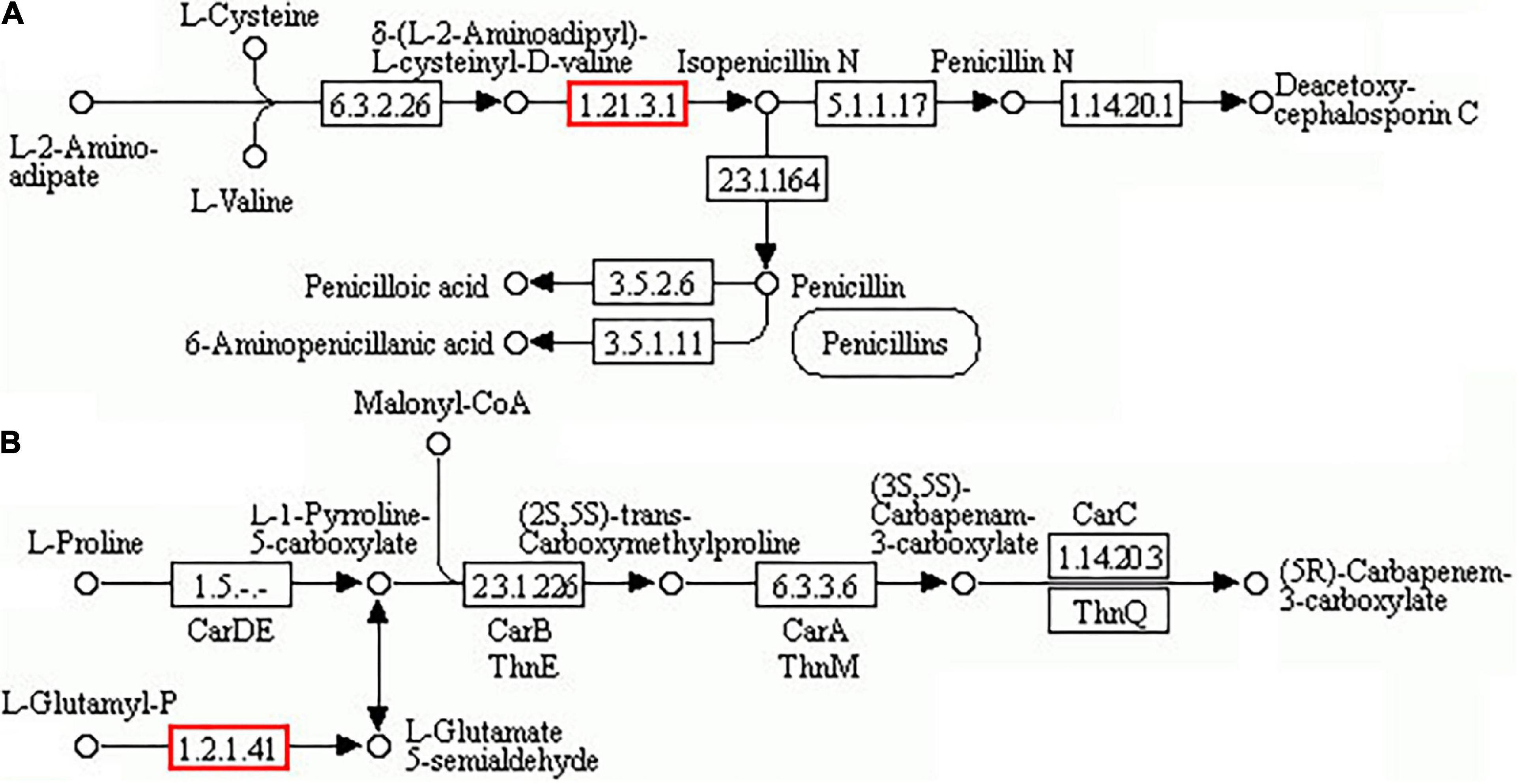
Figure 6. “Biosynthesis of antibiotics” pathway. (A) Biosynthesis of penicillin and cephalosporin. (B) Carbapenem biosynthesis.
Cytoskeleton
The fruiting bodies of A. fibrillifera shrank and became hard under dessication-stress, whereas they recovered rapidly after rehydration (Figure 1A). Long-lived cytoskeleton structure may be an epigenetic regulator of cellular function and fate (Fletcher and Mullins, 2010). A total of 28 (27 more-abundant and one less-abundant) DAPs were associated with cytoskeleton under dessication and rehydration conditions, and the fold-change of more-abundant proteins was much higher than that of less-abundant proteins (Figure 7). These DAPs usually had high fold-change, among which the | log2FC| value of 16 proteins was ≥ 2. CL4846.Contig5_All (Log2FC = 3.87) and CL8973.Contig5_All (Log2FC = 3.55) had the highest fold differences, which play roles in microtubule-related proteins and tubulin alpha chain, respectively (Figure 7). The functions of other significantly more-abundant DAPs were mainly concentrated in actin, tubulin, fimbrin, and cofilin. Fimbrin and cofilin bind to actin to function. The number of DAPs decreased rapidly after rehydration. The DAP (Unigene22219_All) with the highest fold-change (Log2FC = 2.02) was also related to actin (Figure 7).
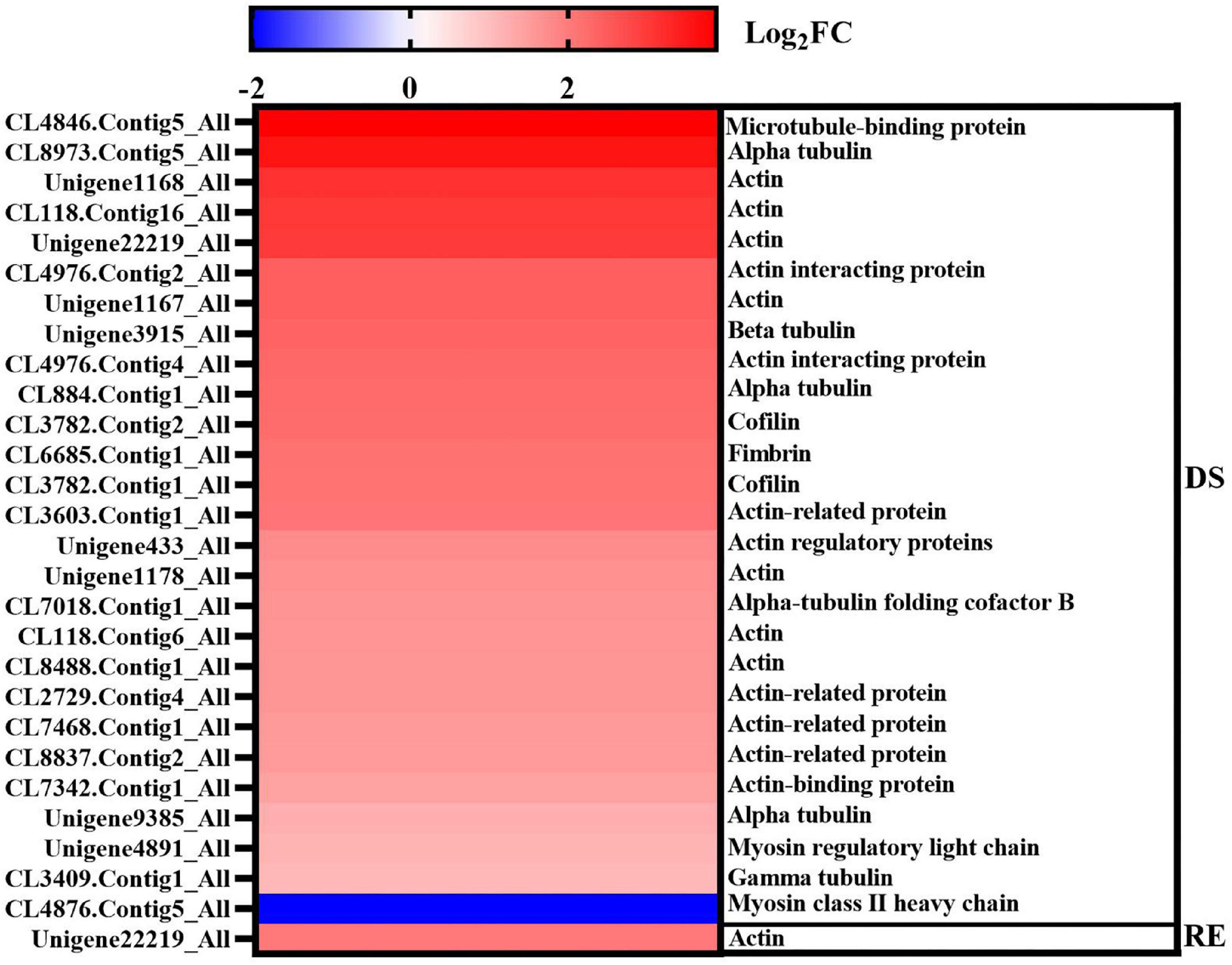
Figure 7. Differentially abundant proteins related to the cytoskeleton under dessication and rehydration. The left and right Y axes represent the protein ID and functional annotation, and DS and RE represent dessication-stress and rehydration, respectively.
Discussion
Proteomics technology is a tool for the comprehensive identification of plant proteins related to drought resistance (Gupta et al., 2020; Qiu et al., 2021). Proteome research has been successfully applied in drought-resistant crops, such as cucumber (Du et al., 2019), corn (Liu et al., 2019), and cassava (Zhao et al., 2015). The strategies of plants to deal with drought stress usually include osmotic regulation, antioxidant capacity, and dehydration tolerance (Zhang, 2007). Similar results were obtained in this experiment. In PPI cluster 1, the DAPs with stronger interactions were concentrated mainly in the above functions. In addition, the DAPs with high fold-change, such as CL4047.Contig1_All (Log2FC = 5.02), CL1516.Contig1_All (Log2FC = 4.93), CL7279.Contig1_All (Log2FC = 4.57), and Unigene10667_All (Log2FC = 4.50) were mainly related to antioxidant mechanisms and heat shock proteins in response to dessication-stress. Meanwhile, the excellent dessication tolerance of A. fibrillifera might be caused by secondary metabolites (melanin, antibiotics, and vitamins).
Melanin response to dessication-stress
Melanin protects dark-pigmented fungi from environmental stresses (Brunskole et al., 2009; Li et al., 2019). Plants highly pigmented are more resistant to biotic and abiotic stresses (Carletti et al., 2014). Melanin has a variety of functions, structures, and forms, which can resist a variety of abiotic factors (Cordero and Casadevall, 2020). Melanin compounds are endowed with excellent photoprotective properties and antioxidative activity (Liberti et al., 2020). Melanin has both free radical scavenging and antioxidant activities. The melanin of Aureobasidium melanogenum XJ5-1 in the Taklimakan Desert plays a vital role in the adaptation of yeasts to drought stress (Jiang et al., 2018). Here, the “phenylalanine, tyrosine and tryptophan biosynthesis” pathway was significantly enriched. TRP3 in this pathway played a central role and had the strongest interaction with other proteins in the PPI analysis. Physiological data showed that the level of melanin increased more than 10-fold under dessication-stress. Tyrosine is the precursor of melanin formation (Gómez and Nosanchuk, 2003; Micillo et al., 2016). In the “tyrosine metabolism” pathway, the melanin-related protein Unigene219_All (Log2FC = 2.36) was significantly increased in abundance too. Under dessication-stress, the DAPs involved in melanin synthesis were significantly increased in abundance, and the melanin content was significantly increased. Proteome data were consistent with the above melanin content. Therefore, specific melanin should contribute to high desiccation tolerance because of its antioxidant and free-radical-scavenging activities in A. fibrillifera.
Vitamin response to dessication-stress
Folate plays a crucial role in overcoming drought-stress during plant development. The foliar application of folate was found to be suitable for drought stress alleviation in Coriandrum sativum L. (Khan et al., 2022). In addition, folates are necessary for the biosynthesis of lignin (Hanson and Gregory, 2011). In this experiment, “folate biosynthesis” and “biotin metabolism” pathways were significantly enriched under dessication-stress. The “folate biosynthesis” pathway was enriched by both transcriptomic and proteomic analysis. The DAP CL112.Contig1_All in this pathway played a role in folylpolyglutamate synthase. This enzyme was very important for maintaining folate homeostasis (Strobbe and Van Der Straeten, 2017). The folate content increased significantly under dessication-stress, whereas it decreased significantly after rehydration. Therefore, folate was the most sensitive to both drought stress and rehydration, and may have a close relation to drought tolerance in A. fibrillifera.
Biotin not only plays a key role in immune regulation in animals but also in the response of plants to various abiotic stresses. Biotin enhances the resistance of Arabidopsis to carbonate stress (Wang et al., 2020). Biotin synthetases and biotin attachment domain containing protein were identified in the root tip of soybean, indicating that biotin and biotinylation were involved in glucose metabolism under drought stress (Wang et al., 2016). In this work, the DAPs and their associated DEGs were up-regulated in the “biotin metabolic” pathway. The significant abundance increase of biotin-related DAPs might supply energy sources to A. fibrillifera under dessication-stress.
Antibiotics involved in dessication tolerance
Antibiotics are a group of secondary metabolites generated by microorganisms or higher organisms in life processes (Mohr, 2016). “Biosynthesis of antibiotics” pathways were significantly enriched in six wheat genotypes under drought stress (Rasool et al., 2022). The enhancement of pyrimidine nucleoside antibiotics can alleviate abiotic stress in Nicotiana tabacum (Song et al., 2022). Penicillins and cephalosporins are the most important classes of β-lactam antibiotics. In this experiment, antibiotic-related DAPs were highly increased in abundance, including fumarate hydratase (FH) (CL6387.Contig1_All), isopenicillin-N synthase (IPNS) (CL711.Contig2_All), and glutamate-5-semialdehyde dehydrogenase (CL6373.Contig2_All), which are beneficial to the synthesis of β-lactam antibiotics. FH facilitates the synthesis of cysteine, which is the precursor for penicillin and cephalosporin synthesis. IPNS can catalyze the specific reaction of L-δ-(α-aminoadipoyl)-L-cysteinyl-D-valine with dioxygen giving isopenicillin-N, a precursor of cephalosporins and penicillins (Rabe et al., 2021). The significant abundance increase of these DAPs under dessication-stress is beneficial to the accumulation of penicillin and cephalosporin in A. fibrillifera. In the “carbapenem biosynthesis” pathway, glutamate-5-semialdehyde dehydrogenase-related DAP was significantly increased in abundance and might be beneficial to the synthesis of carbapenem. In transcriptome-proteome-association analysis, many DEGs/DAPs were enriched in the “biosynthesis of antibiotics” pathway, which could be beneficial to dessication tolerance in A. fibrillifera.
Cytoskeleton response to dessication-stress
The cytoskeleton is the main mechanical structure of cells; it is a complex and dynamic biopolymer network composed of microtubules, actin, and intermediate filaments (Pegoraro et al., 2017). The plant cytoskeleton is associated with plant stress responses, such as drought, salt, and cold (Chun et al., 2021). A balance in the metabolism of cell wall component biosynthesis and cytoskeleton homeostasis can affect the response of cotton fibers to drought stress (Zheng et al., 2014). The microtubule cytoskeleton functions as a sensor for stress response signaling in plants and maintains mechanical stability by forming bundles (Ma and Liu, 2019). Furthermore, actin filaments may control drought-induced signal perception and are involved in regulating the accumulation of HVA1 (a dehydrin-encoding gene) transcripts in barley leaves exposed to drought stress (Śniegowska-Świerk et al., 2016). In this experiment, the fruiting bodies of A. fibrillifera shrink under dessication-stress. A total of 28 DAPs were associated with the cytoskeleton under dessication-stress and rehydration conditions, and the fold-change of more-abundant proteins was much higher than that of less-abundant proteins (Figure 7). The functions of significantly more-abundant DAPs were mainly concentrated in actin, tubulin, fimbrin, and cofilin. Fimbrin and cofilin bind to actin to function. The number of DAPs decreased rapidly after rehydration. The DAP (Unigene22219_All) with the highest fold-change was also related to actin. After rehydration, the fruiting bodies could quickly absorb water and return to the control level, which might be related to the rapid assembly or disassembly of actin. Therefore, the DAPs related to the cytoskeleton might be helpful in adaptation to dessication tolerance and the shape changes of fruiting bodies.
Conclusion
The levels of melanin, amino acids, folate, and biotin in fruiting bodies increased significantly under dessication-stress compared with those of the controls. Folate showed a sensitive response to both dessication-stress and rehydration. In total, 1,572 DAPs were identified under dessication-stress. The number of DAPs decreased rapidly after rehydration. Under dessication-stress, many DAPs were annotated in “amino acid metabolism,” “carbohydrate metabolism,” “folate biosynthesis,” “biotin metabolism,” “biosynthesis of antibiotics,” and “transport and catabolism” pathways. Proteome–transcriptome association analysis revealed that “biosynthesis of antibiotics,” “folate biosynthesis,” “biotin metabolism,” “phenylalanine, tyrosine, and tryptophan biosynthesis” pathways were significantly enriched in response to dessication-stress. The DAPs in these pathways were beneficial to the synthesis of antibiotics, folate, biotin, and melanin, and played important roles in enhancing the dessication tolerance of A. fibrillifera. The findings of transcriptome and physiological analyses were in good agreement with the proteomic data. Some molecular pathways and mechanisms of dessication response are similar between A. fibrillifera and plant species. This work may shed light on the mechanism of dessication tolerance and provide a novel framework for the breeding and cultivation of Auricularia and crops.
Data availability statement
The data presented in this study are deposited in the ProteomeXchange repository, accession number PXD033449.
Author contributions
SZ and GG designed the experiments. HG, XX, YW, and HT were performed material preparation, data collection, and analysis. HG, XX, YW, HT, SZ, and GG wrote the manuscript. All authors have read and approved the final manuscript.
Funding
This study was financially supported by the Science and Technology Program of Guizhou Province [(2022)050].
Acknowledgments
We thank International Science Editing (http//www.internationalscienceediting.com) for editing this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.995810/full#supplementary-material
Supplementary Figure 1 | Heatmap of sample correlation analysis. Both X and Y axes represent samples. The color denotes the correlation coefficient. CK1, CK2, DS, and RE represent the parallel control of desiccation-stress, parallel control of rehydration, desiccation-stress, and rehydration process, respectively. “−1,” “−2,” and “−3” indicate three biological replicates.
References
Ahuja, I., de-Vos, R. C., Bones, A. M., and Hall, R. D. (2010). Plant molecular stress responses face climate change. Trends Plant Sci. 15, 664–674.
Ariyarathne, M. A., and Wone, B. W. M. (2022). Overexpression of the Selaginella lepidophylla bHLH transcription factor enhances water-use efficiency, growth, and development in Arabidopsis. Plant Sci. 315:111129. doi: 10.1016/j.plantsci.2021.111129
Basset, G. J. C., Quinlivan, E. P., Gregory, J. F., and Hanson, A. D. (2005). Folate synthesis and metabolism in plants and prospects for biofortification. Crop Sci. 45, 449–453. doi: 10.2135/cropsci2005.0449
Bernhardt, O., Selevsek, N., Gillet, L., Rinner, O., and Reiter, L. (2014). “Spectronaut: A fast and efficient algorithm for MRM-like processing of data independent acquisition (SWATH-MS) data,” in Proceedings of the 60th American Society for Mass Spectrometry Conference, Vancouver.
Bruderer, R., Bernhardt, O. M., Gandhi, T., Miladinović, S. M., Cheng, L. Y., Messner, S., et al. (2015). Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Mol. Cell. Proteomics 14, 1400–1410. doi: 10.1074/mcp.m114.044305
Brunskole, M., Zorko, K., Kerbler, V., Martens, S., Stojan, J., Gobec, S., et al. (2009). Trihydroxynaphthalene reductase of Curvularia lunata—a target for flavonoid action? Chem. Biol. Interact. 178, 259–267. doi: 10.1016/j.cbi.2008.10.023
Burmasova, M. A., Utebaeva, A. A., Sysoeva, E. V., and Sysoeva, M. A. (2019). Melanins of Inonotus obliquus: Bifidogenic and antioxidant properties. Biomolecules 9:248. doi: 10.3390/biom9060248
Cao, W., Zhou, X., McCallum, N. C., Hu, Z., Ni, Q. Z., Kapoor, U., et al. (2021). Unraveling the structure and function of melanin through synthesis. J. Am. Chem. Soc. 143, 2622–2637. doi: 10.1021/jacs.0c12322
Carletti, G., Nervo, G., and Cattivelli, L. (2014). Flavonoids and melanins: A common strategy across two kingdoms. Int. J. Biol. Sci. 10, 1159–1170. doi: 10.7150/ijbs.9672
Caruso, G., Cavaliere, C., Foglia, P., Gubbiotti, R., Samperi, R., and Laganà, A. (2009). Analysis of drought responsive proteins in wheat (Triticum durum) by 2D-PAGE and MALDI-TOF mass spectrometry. Plant Sci. 177, 570–576. doi: 10.1016/j.plantsci.2009.08.007
Choi, M., Chang, C. Y., Clough, T., Broudy, D., Killeen, T., MacLean, B., et al. (2014). MSstats: An R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 30, 2524–2526. doi: 10.1093/bioinformatics/btu305
Chun, H. J., Baek, D., Jin, B. J., Cho, H. M., Park, M. S., Lee, S. H., et al. (2021). Microtubule dynamics plays a vital role in plant adaptation and tolerance to salt stress. Int. J. Mol. Sci. 22:5957. doi: 10.3390/ijms22115957
Cordero, R. J. B., and Casadevall, A. (2020). Melanin. Curr. Biol. 30, R142–R143. doi: 10.1016/j.cub.2019.12.042
Du, C., Chai, L., Wang, Z., and Fan, H. (2019). Response of proteome and morphological structure to short-term drought and subsequent recovery in Cucumis sativus leaves. Physiol. Plant. 167, 676–689. doi: 10.1111/ppl.12926
Fletcher, D. A., and Mullins, R. D. (2010). Cell mechanics and the cytoskeleton. Nature 463, 485–492. doi: 10.1038/nature08908
Glagoleva, A. Y., Shoeva, O. Y., and Khlestkina, E. K. (2020). Melanin pigment in plants: Current knowledge and future perspectives. Front. Plant Sci. 11:770. doi: 10.3389/fpls.2020.00770
Gómez, B. L., and Nosanchuk, J. D. (2003). Melanin and fungi. Curr. Opin. Infect. Dis. 16, 91–96. doi: 10.1097/01.aco.0000065076.06965.04
Gupta, S., Mishra, S. K., Misra, S., Pandey, V., Agrawal, L., Nautiyal, C. S., et al. (2020). Revealing the complexity of protein abundance in chickpea root under drought-stress using a comparative proteomics approach. Plant Physiol. Biochem. 151, 88–102. doi: 10.1016/j.plaphy.2020.03.005
Hanson, A. D., and Gregory, J. F. (2011). Folate biosynthesis, turnover, and transport in plants. Annu. Rev. Plant Biol. 62, 105–125. doi: 10.1146/annurev-arplant-042110-103819
Hsiao, T. C. (1973). Plant responses to water stress. Annu. Rev. Plant Physiol. 24, 519–570. doi: 10.1146/annurev.pp.24.060173.002511
Isaacson, T., Damasceno, C. M. B., Saravanan, R. S., He, Y. H., Catalá, C., Saladié, M., et al. (2006). Sample extraction techniques for enhanced proteomic analysis of plant tissues. Nat. Protoc. 1, 769–774. doi: 10.1038/nprot.2006.102
Jiang, H., Liu, G. L., Chi, Z., Hu, Z., and Chi, Z. M. (2018). Genetics of trehalose biosynthesis in desert-derived Aureobasidium melanogenum and role of trehalose in the adaptation of the yeast to extreme environments. Curr. Genet. 64, 479–491. doi: 10.1007/s00294-017-0762-z
Khan, M. N., and Komatsu, S. (2016). Proteomic analysis of soybean root including hypocotyl during recovery from drought stress. J. Proteom. 144, 39–50. doi: 10.1016/j.jprot.2016.06.006
Khan, M. T., Ahmed, S., and Shah, A. A. (2022). Regulatory role of folic acid in biomass production and physiological activities of Coriandrum sativum L. under irrigation regimes. Int. J. Phytoremediation 24, 1025–1038. doi: 10.1080/15226514.2021.1993785
Koh, J., Chen, G., Yoo, M. J., Zhu, N., Dufresne, D., Erickson, J. E., et al. (2015). Comparative proteomic analysis of Brassica napus in response to drought stress. J. Proteome Res. 14, 3068–3081. doi: 10.1021/pr501323d
Lazar, N., Fay, A., Nandakumar, M., Boyle, K. E., Xavier, J., Rhee, K., et al. (2017). Control of biotin biosynthesis in mycobacteria by a pyruvate carboxylase dependent metabolic signal. Mol. Microbiol. 106, 1018–1031. doi: 10.1111/mmi.13865
Lesk, C., Rowhani, P., and Ramankutty, N. (2016). Influence of extreme weather disasters on global crop production. Nature 529, 84–87. doi: 10.1038/nature16467
Li, J. J., Zhou, L., Yin, C. M., Zhang, D. D., Klosterman, S. J., Wang, B. L., et al. (2019). The Verticillium dahliae Sho1-MAPK pathway regulates melanin biosynthesis and is required for cotton infection. Environ. Microbiol. 21, 4852–4874. doi: 10.1111/1462-2920.14846
Liberti, D., Alfieri, M. L., Monti, D. M., Panzella, L., and Napolitano, A. (2020). A melanin-related phenolic polymer with potent photoprotective and antioxidant activities for dermo-cosmetic applications. Antioxidants 9:270. doi: 10.3390/antiox9040270
Liu, S., Zenda, T., Dong, A., Yang, Y., Liu, X., Wang, Y., et al. (2019). Comparative proteomic and morpho-physiological analyses of maize wild-type vp16 and mutant vp16 germinating seed responses to peg-induced drought stress. Int. J. Mol. Sci. 20, 1–24. doi: 10.3390/ijms20225586
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Ma, H., and Liu, M. (2019). The microtubule cytoskeleton acts as a sensor for stress response signaling in plants. Mol. Biol. Rep. 46, 5603–5608. doi: 10.1007/s11033-019-04872-x
Ma, H., Xu, X., and Feng, L. (2014). Responses of antioxidant defenses and membrane damage to drought stress in fruit bodies of Auricularia auricula-judae. World J. Microbiol. Biotechnol. 30, 119–124. doi: 10.1007/s11274-013-1416-z
Mahmood, T., Khalid, S., Abdullah, M., Ahmed, Z., Shah, M. K. N., Ghafoor, A., et al. (2019). Insights into drought stress signaling in plants and the molecular genetic basis of cotton drought tolerance. Cells 9:105. doi: 10.3390/cells9010105
Maier, T., Güell, M., and Serrano, L. (2009). Correlation of mRNA and protein in complex biological samples. FEBS Lett. 583, 3966–3973. doi: 10.1016/j.febslet.2009.10.036
Micillo, R., Panzella, L., Koike, K., Monfrecola, G., Napolitano, A., and d’Ischia, M. (2016). “Fifty shades” of black and red or how carboxyl groups fine tune eumelanin and pheomelanin properties. Int. J. Mol. Sci. 17, 1–13. doi: 10.3390/ijms17050746
Mohr, K. I. (2016). History of antibiotics research. Curr. Top. Microbiol. Immunol. 398, 237–272. doi: 10.1007/82_2016_499
Nunes-Nesi, A. (2005). Enhanced photosynthetic performance and growth as a consequence of decreasing mitochondrial malate dehydrogenase activity in transgenic tomato plants. Plant Physiol. 137, 611–622. doi: 10.1104/pp.104.055566
Nunes-Nesi, A., Carrari, F., Gibon, Y., Sulpice, R., Lytovchenko, A., Fisahn, J., et al. (2007). Deficiency of mitochondrial fumarase activity in tomato plants impairs photosynthesis via an effect on stomatal function. Plant J. 50, 1093–1106. doi: 10.1111/j.1365-313x.2007.03115.x
Pampurova, S., and Van Dijck, P. (2014). The desiccation tolerant secrets of Selaginella lepidophylla: What we have learned so far? Plant Physiol. Biochem. 80, 285–290. doi: 10.1016/j.plaphy.2014.04.015
Pegoraro, A. F., Janmey, P., and Weitz, D. A. (2017). Mechanical properties of the cytoskeleton and cells. Cold Spring Harb. Perspect. Biol. 9:a022038. doi: 10.1101/cshperspect.a022038
Qiu, C., Sun, J. H., Shen, J. Z., Zhang, S. N., Ding, Y. Q., Gai, Z. S., et al. (2021). Fulvic acid enhances drought resistance in tea plants by regulating the starch and sucrose metabolism and certain secondary metabolism. J. Proteom. 247:104337. doi: 10.1016/j.jprot.2021.104337
Rabe, P., Kamps, J. J. A. G., Sutherlin, K. D., Linyard, J. D. S., Aller, P., Pham, C. C., et al. (2021). X-ray free-electron laser studies reveal correlated motion during isopenicillin N synthase catalysis. Sci. Adv. 7:eabh0250. doi: 10.1126/sciadv.abh0250
Rasool, F., Khan, M. R., Schneider, M., Uzair, M., Aqeel, M., Ajmal, W., et al. (2022). Transcriptome unveiled the gene expression patterns of root architecture in drought-tolerant and sensitive wheat genotypes. Plant Physiol. Biochem. 178, 20–30. doi: 10.1016/j.plaphy.2022.02.025
Salekdeh, G. H., Siopongco, J., Wade, L. J., Ghareyazie, B., and Bennett, J. (2002). Proteomic analysis of rice leaves during drought stress and recovery. Proteomics 2, 1131–1145.
Sekara, A., Kalisz, A., Grabowska, A., and Siwulski, M. (2015). Auricularia spp. - mushrooms as novel food and therapeutic agents - a review. Sydowia 67, 1–10. doi: 10.12905/0380.sydowia67-2015-001
Śniegowska-Świerk, K., Dubas, E., and Rapacz, M. (2016). Actin microfilaments are involved in the regulation of HVA1 transcript accumulation in drought-treated barley leaves. J. Plant Physiol. 193, 22–25. doi: 10.1016/j.jplph.2016.02.006
Song, Z., Wang, P., Chen, X., Peng, Y., Cai, B., Song, J., et al. (2022). Melatonin alleviates cadmium toxicity and abiotic stress by promoting glandular trichome development and antioxidant capacity in Nicotiana tabacum. Ecotoxicol. Environ. Saf. 236:113437. doi: 10.1016/j.ecoenv.2022.113437
Strobbe, S., and Van Der Straeten, D. (2017). Folate biofortification in food crops. Curr. Opin. Biotechnol. 44, 202–211. doi: 10.1016/j.copbio.2016.12.003
Tsou, C. C., Avtonomov, D., Larsen, B., Tucholska, M., Choi, H., Gingras, A. C., et al. (2015). DIA-Umpire: Comprehensive computational framework for data-independent acquisition proteomics. Nat. Methods 12, 258–264. doi: 10.1038/nmeth.3255
Wang, N., Zhao, J., He, X., Sun, H., Zhang, G., and Wu, F. (2015). Comparative proteomic analysis of drought tolerance in the two contrasting Tibetan wild genotypes and cultivated genotype. BMC Genomics 16:432. doi: 10.1186/s12864-015-1657-3
Wang, X., Oh, M., Sakata, K., and Komatsu, S. (2016). Gel-free/label-free proteomic analysis of root tip of soybean over time under flooding and drought stresses. J. Proteomics 130, 42–55. doi: 10.1016/j.jprot.2015.09.007
Wang, Y., Wang, M., Ye, X., Liu, H., Takano, T., Tsugama, D., et al. (2020). Biotin plays an important role in Arabidopsis thaliana seedlings under carbonate stress. Plant Sci. 300:110639. doi: 10.1016/j.plantsci.2020.110639
Wang, Y., Yang, Z., Shi, L., Yang, R., Guo, H., Zhang, S., et al. (2022). Transcriptome analysis of Auricularia fibrillifera fruit-body responses to drought stress and rehydration. BMC Genomics 23:58. doi: 10.1186/s12864-021-08284-9
Yuan, Y., Wu, F., Si, J., Zhao, Y. F., and Dai, Y. C. (2019). Whole genome sequence of Auricularia heimuer (Basidiomycota, Fungi), the third most important cultivated mushroom worldwide. Genomics 111, 50–58. doi: 10.1016/j.ygeno.2017.12.013
Zandalinas, S. I., Mittler, R., Balfagón, D., Arbona, V., and Gómez-Cadenas, A. (2018). Plant adaptations to the combination of drought and high temperatures. Physiol. Plant 162, 2–12. doi: 10.1111/ppl.12540
Zhang, Q. (2007). Strategies for developing green super rice. Proc. Natl. Acad. Sci. U.S.A. 104, 16402–16409. doi: 10.1073/pnas.0708013104
Zhao, P., Liu, P., Shao, J., Li, C., Wang, B., Guo, X., et al. (2015). Analysis of different strategies adapted by two cassava cultivars in response to drought stress: Ensuring survival or continuing growth. J. Exp. Bot. 66, 1477–1488. doi: 10.1093/jxb/eru507
Zhao, Y., Wang, L., Zhang, D., Li, R., Cheng, T., Zhang, Y., et al. (2019). Comparative transcriptome analysis reveals relationship of three major domesticated varieties of Auricularia auricula-judae. Sci. Rep. 9:78. doi: 10.1038/s41598-018-36984-y
Keywords: Auricularia fibrillifera, proteome, dessication stress, melanin, antibiotics, folate, biotin, cytoskeleton
Citation: Guo H, Xiong X, Wang Y, Tian H, Zhang S and Geng G (2022) Integrative proteomic and physiological analyses of the molecular response to dessication-stress in Auricularia fibrillifera. Front. Plant Sci. 13:995810. doi: 10.3389/fpls.2022.995810
Received: 16 July 2022; Accepted: 26 August 2022;
Published: 21 September 2022.
Edited by:
PingWu Liu, Hainan University, ChinaReviewed by:
Lisa David, North Carolina State University, United StatesSayed Mohammad Mohsin, Sher-e-Bangla Agricultural University, Bangladesh
Copyright © 2022 Guo, Xiong, Wang, Tian, Zhang and Geng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Suqin Zhang, enNxaW4yMDAyQDE2My5jb20=; Guangdong Geng, Z2RnZW5nMjEzQDE2My5jb20=
†These authors have contributed equally to this work