
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 13 October 2022
Sec. Plant Systematics and Evolution
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.990064
This article is part of the Research TopicRise to the Challenges in Plastome PhylogenomicsView all 10 articles
 Jianwei Xi1†
Jianwei Xi1† Saibin Lv1†
Saibin Lv1† Weiping Zhang2†
Weiping Zhang2† Jingbo Zhang3*†
Jingbo Zhang3*† Ketao Wang1Haobing Guo4Jie Hu4Yang Yang2
Ketao Wang1Haobing Guo4Jie Hu4Yang Yang2 Jianhua Wang1Guohua Xia1
Jianhua Wang1Guohua Xia1 Guangyi Fan4
Guangyi Fan4 Xinwang Wang5
Xinwang Wang5 Lihong Xiao1*
Lihong Xiao1*Carya, in the Juglandiodeae subfamily, is to a typical temperate-subtropical forest-tree genus for studying the phylogenetic evolution and intercontinental disjunction between eastern Asia (EA) and North America (NA). Species of the genus have high economic values worldwide for their high-quality wood and the rich healthy factors of their nuts. Although previous efforts based on multiple molecular markers or genome-wide SNPs supported the monophyly of Carya and its two EA and NA major subclades, the maternal phylogeny of Carya still need to be comprehensively evaluated. The variation of Carya plastome has never been thoroughly characterized. Here, we novelly present 19 newly generated plastomes of congeneric Carya species, including the recently rediscovered critically endangered C. poilanei. The overall assessment of plastomes revealed highly conservative in the general structures. Our results indicated that remarkable differences in several plastome features are highly consistent with the EA-NA disjunction and showed the relatively diverse matrilineal sources among EA Carya compared to NA Carya. The maternal phylogenies were conducted with different plastome regions and full-length plastome datasets from 30 plastomes, representing 26 species in six genera of Juglandoideae and Myrica rubra (as root). Six out of seven phylogenetic topologies strongly supported the previously reported relationships among genera of Juglandoideae and the two subclades of EA and NA Carya, but displayed significant incongruencies between species within the EA and NA subclades. The phylogenetic tree generated from full-length plastomes demonstrated the optimal topology and revealed significant geographical maternal relationships among Carya species, especially for EA Carya within overlapping distribution areas. The full-length plastome-based phylogenetic topology also strongly supported the taxonomic status of five controversial species as separate species of Carya. Historical and recent introgressive hybridization and plastid captures might contribute to plastome geographic patterns and inconsistencies between topologies built from different datasets, while incomplete lineage sorting could account for the discordance between maternal topology and the previous nuclear genome data-based phylogeny. Our findings highlight full-length plastomes as an ideal tool for exploring maternal relationships among the subclades of Carya, and potentially in other outcrossing perennial woody plants, for resolving plastome phylogenetic relationships.
The plastid, as a key uniparentally inherited organelle in plant cells, carries out not only photosynthesis but also other metabolic processes that mediate the adaptation of the plant to its environment (Dierckxsens et al., 2016). Despite increased interest in phylogenetic and phylogenomic analyses using nuclear genome datasets, plastome-based analyses have also become a powerful solution that have been widely applied to address recalcitrant relationships across the Tree of Life (Hu et al., 2016; Wei et al., 2017; Yan et al., 2018; Song et al., 2020; Wang et al., 2020; Tu et al., 2021; Dong et al., 2022; Liu et al., 2022; Ogoma et al., 2022). The plastome-based phylogenetic approaches play important roles in plant phylogenetics and evolution, such as revealing ancient and recent introgression or hybridization, tracking seed dispersal in phylogeographic studies, and charactering the structural diversity and evolution of organellar genomes (Zhao et al., 2018; Li et al., 2019; Yao et al., 2019; Li et al., 2020; Yang et al., 2021 and references therein). The most recent studies have produced numerous examples of phylogenetic discordance in plastomes or between plastids and nuclear gene trees at various evolutionary scales, which are interpreted as ancient introgressive hybridization, ancient chloroplast capture, or incomplete lineage sorting (Dong et al., 2022; Yang et al., 2021; Zhou et al., 2022). Some studies also emphasized the application of plastome-based analyses to track patterns of geographically structured interspecific gene flow, including several lineages of Fagales such as oak species with overlapping distribution ranges (Pham et al., 2017; Yang et al., 2021; Zhou et al., 2022).
Genus Carya, as the second largest genus in the Juglandiodeae subfamily of Fagales, contains typical temperate-subtropical forest trees for studying the phylogenetic evolution and intercontinental disjunction between eastern Asia (EA) and North America (NA) (Zhang et al., 2013; Huang et al., 2019). Carya is comprised of up to 20 extant species disjunctively distributed in EA and NA, and most species of the genera are also economically important for their valuable timbers and/or edible nut kernels (e.g., the pecan, Chinese hickory and Dabieshan hickory) (Grauke, 2003; Kozlowski et al., 2018; Huang et al., 2019; Xiao. et al., 2021). Based on their morphological characteristics, nineteen extant Carya species (excluding C. dabieshanensis that disputed as a variant of C. cathayensis) are divided into three sections based on morphological characters: section Carya, the true hickories (nine species); section Apocarya, the pecan hickories (four species); and section Sinocarya, the Asian hickories (six species) (Manning and Wayne, 1978; Grauke, 2003). The phylogeny and evolutionary history of Carya has been proposed by multiple studies based on extensive phylogenetic studies, combined with morphology, anatomy, cytology, and DNA sequences of nuclear and organelles (Manos and Stone, 2001; Manos et al., 2007; Zhang et al., 2013; Huang et al., 2019; Mu et al., 2020; Zhang et al., 2021). Zhang et al. (2013) investigated phylogenetic relationships between Carya species in EA and NA by integrating ten molecular markers from plastid (eight) and nuclear (two) with extensive taxon sampling, and they reconstructed the historical biogeography of Carya by integrating macro-fossil, morphological, and molecular data. However, the phylogeny among species in EA or NA subclades were hard to explain by any morphological traits (Zhang et al., 2013). Although the inference of biogeographical history gave more supports for the NA origin of Carya and the hypothetical migratory from NA to Europe and then to EA, evidence was not sufficient only based on a small set of molecular markers and partial macro-fossil records, in particular the omission of the early fossil records from the Kaliningrad region of former Soviet Union in Eurasia (Mai, 1981; Zhang et al., 2021). The most recent reports based on genome-wide SNPs (Huang et al., 2019), integrated RAD-seq and chloroplast genomes (Mu et al., 2020), and fossil-informed models provided strong support for backbone relationships among taxa of Juglandoideae, the monophyly of Carya genus, and its two major subclades in EA-NA (Zhang et al., 2021). However, significant inconsistencies were found among these studies phylogenetic topologies within EA or NA subclades, especially five taxonomically disputed species, C. poilanei, C. sinensis (the synonym name is Annamocarya sinensis), C. dabieshanensis, C. glabra and C. ovalis, that inferred from different molecular markers or datasets. Furthermore, C. illinoinensis, as the most commercially valuable species, has shown that many cultivars of this species were generated from intraspecies hybridization with other potential admixing species, such as C. cordiformis, C. aquatica and C. myristiciformis (Lovell et al., 2021). Recently, based on the pan-genome assemblies of pecan genotypes and resequencing data of multiple genotypes above potential admixing species, several disease-related interspecific genomic introgression blocks have been identified in the genotypes of C. illinoinensis (Lovell et al., 2021). However, the maternal relationships of these pecan varieties with different economically important traits have never been addressed. Meanwhile, the maternal phylogeny of Carya still need to be comprehensively evaluated, and the variation of Carya plastome has never been thoroughly characterized.
Carya poilanei (Chev.), also known as Poilane’s hickory, had three original collections in Vietnam, Laos, and Thailand (Chevalier, 1941; Leroy, 1950; Manning, 1963; Grauke et al., 1991). This rare species was initially described as Juglans poilanei (Chevalier, 1941). It was suspected to be extinct in the wild since the 1950s when Leroy placed the species under Carya (Leroy, 1950; Leroy, 1955; Grauke et al., 1991; Grauke et al., 2016). Until most recently, three subpopulations were rediscovered in the Ailao Mountain, Yunnan province, China, and C. poilanei was instead suggested to be listed as critically endangered (Zhang et al., 2022). Thus, the phylogenetic relationship of C. poilanei with other Carya species needs to be clarified. Carya sinensis (Dode), with a common name of ‘Hui He Tao’ (i.e. beaked walnut or beaked hickory) in China and ‘Cay Cho Dai’ in Vietnam, is narrowly distributed in southern China and northern Vietnam (Chevalier, 1941; Manning and Hjelmqvist, 1951). The species was first described by Dode (1912) and categorized as a separate genus Annamocarya indochinensis (Chevalier, 1941). It was subsequently ascribed to six different genera: Annamocarya, Rhamphocarya, Juglandicarya, Caryojuglans, Juglans, and Carya (Leroy, 1950; Manning and Hjelmqvist, 1951; Scott, 1953; Leroy, 1955; Lu et al., 1999). Although the previous and recent reports based on the molecular markers evidenced the taxon of the tree as a member of section Sinocarya in EA Carya, discordance remains among the phylogenetic topologies that were built based on different data sets (Zhang et al., 2013; Luo et al., 2021). Carya dabieshanensis M. C. Liu & Z. J. Li., historically considered a variant of C. cathayensis, is now treated as an addition to the nomenclature of Chinese hickory (Liu and Li, 1984). However, collections of germplasm and voucher specimens for more thorough comparison are necessary, and the relationship between C. dabieshanensis and C. cathayensis still needs to be addressed (Manos and Stone, 2001). Distribution maps of the widely distributed NA species, Carya glabra (Mill.) Sweet. with the common name pignut hickory, have included C. ovalis (Wangenh.) Sarg. (common name red hickory) since the reduction of C. ovalis to synonymy with C. glabra (Little, 1969). Although C. glabra readily hybridized with C. ovalis when the two occurred together, with hybrids confusing the distinctions between the species (Manning, 1950), the morphological characteristics and their habitats show that they are separate species in the section Carya in NA (Manning, 1950; Grauke, 2003). Nevertheless, C. glabra and some other species in NA Carya were also wrongly identified as other species partially because of the interspecific hybridization in nature stands (Heiges, 1896). These circumstances, together with the inconsistent results based on molecular inferences (Zhang et al., 2013; Huang et al., 2019), make them imperative for a more comprehensive study on the phylogeny of Carya and the development of molecular markers for species identification. Carya illinoinensis (Wangenheim) K. Koch (common name pecan), as a member of the section Apocarya, is native to the United States and Mexico, with over 8,000 years of history (Hester, 1983; Grauke et al., 2011; Wang et al., 2020). The latest microsatellite profiles revealed ‘87MX3-2.11’ to be homozygous (Wang et al., 2020). Owing to the richness in health factors of its nut kernels, C. illinoinensis became the most commercially valuable species of Carya. Commercial production of C. illinoinensis has persisted for about a century and a half, and it has been widely planted across six continents with more than 400 cultivars released so far, including some varieties promoted and planted in large areas (Grauke et al., 2016; Xiao. et al., 2021). Among the cultivars of C. illinoinensis, many were formed by both natural and artificial hybridization (https://cgru.usda.gov/CARYA/PECANS). For example, the varieties ‘Pawnee’, ‘Lakota’ and ‘Elliott’ mentioned above, belong to intraspecies hybrids with interspecies introgression in history, based on whole genome sequencing data (Thompson et al., 2008; Huang et al., 2019; Lovell et al., 2021; Xiao et al., 2021; https://cgru.usda.gov/CARYA/PECANS). However, the maternal relationships of the varieties are still unclear, the investigation of plastome variations inherited from the female parent may provide important clues for tracking both ancient and recent gene flows among different varieties caused by inter- and intraspecies hybridization.
In this study, we newly assembled and characterized the structures and diversity of complete plastomes for the critically endangered C. poilanei and other 18 congeneric species, based on the sequences, generated by high-throughput sequencing technology. To explore the optimal phylogenetic relationship within Carya, the phylogenetic trees were reconstructed respectively by employing the entire or partial plastome sequences and unique protein-coding sequences of 30 plastomes representing all 20 Carya species (19 newly assembled, plus the recent release of C. pallida), plus six representatives from five other genera of Juglandoideae. The maternal relationship between C. dabieshanensis and C. cathayensis was determined by integrating the phylogenetic relationship with the comparison of morphological features, and confirmed the taxonomic status of C. dabieshanensis, C. cathayensis, C. sinensis, C. glabra, and C.,poilanei as separate species in section Sinocarya. The maternal relationships and discordance of species in EA and NA Carya, and several varieties in C. illinoinensis were also discussed based on the reconstruction of plastome phylogenetic topologies of Carya. The results generated here are of great value for the evolution, wild resource conservation and genetic breeding of Carya in the future. Our methodology of exploring the phylogenetic relationships of Carya species in EA or NA, by using the different molecular markers or the informative fragments of plastomes with different evolutionary rates, will provide a good example for the reconstruction of biogeography under subgenus in future, especially for outcrossing perennial tree species.
In this study, fresh, fully expanded young leaves from an adult tree of C. poilanei (Supplementary Figure 1) were collected in July from the eastern edge of the Ailao Mountains in Jianshui County, in the southern Yunnan Province of China. Fresh, fully expanded young leaves from C. illinoinensis (cv. ‘Sioux’), C. cathayensis, C. dabieshanensis C. hunanensis, and C. tonkinensis were collected from an orchard in Zhejiang A&F University, China. The samples of fresh fully expanded young leaves from C. kweichowensis and C. sinensis were collected from the Qiannan Buyi and Miao Autonomous Prefecture, Guizhou Province, China, respectively. All collected leaf samples were immediately immersed in liquid nitrogen for transportation back to the laboratory and stored at -80°C before DNA extraction. The details of the collected samples are listed in Supplementary Table 1.
The trunk, branches, leaves, flowers and fruits of C. dabieshanensis and C. cathayensis planted in the orchard of Zhejiang A&F University, Hangzhou, China, were photographed and characterized by their morphologic traits. Meanwhile, the nuts of other EA Carya species were also photographed.
The high-quality genomic DNA was isolated from approximately 100 mg samples using the E.Z.N.A.® HP Plant DNA Mini kit (Omega Bio-Tek, USA). DNA quality and concentration were assessed in a Qubit 3.0 Fluorometer (Thermo Fisher Scientific Inc., USA), and DNA integrity was evaluated by a 1.0% (W/V) agarose gel.
Approximately 1μg of high-quality genomic DNA from each sample was used for the whole genome sequencing (WGS). The high-throughput sequencing library of C. poilanei was constructed and sequenced using the Illumina NovaSeq 6000 platform (NovoGene, Beijing, China) following the standard procedure of the manufacturer. The MGIEasy DNA Rapid Library Prep Kit (cat.# 1000006985; MGI-Tech., China) was used to construct the sequencing library of C. illinoinensis (cv. ‘Sioux’), C. cathayensis, C. dabieshanensis, C. hunanensis, C. tonkinensis, C. kweichowensis and C. sinensis, and the paired-end 100 bp reads were generated on the BGISEQ-500 platform (BGI, Shenzhen, China) according to the manufacturers’ procedures, respectively. The raw sequence data was submitted to the Sequence Read Archive (SRA) of NCBI (Supplementary Table 1).
To obtain the high-quality plastome assemblies of 19 Carya species, 2 Gb raw reads were randomly selected for each species from NovaSeq 6000 data (C. poilanei), BGISEQ-500 PE100 data (seven species that were sequenced in this study) or from Illumina HiSeq X-TEN data retrieved from the NCBI database (eleven previously sequenced species), respectively. The low-quality reads from all samples, i.e., reads having over 50% bases with a quality value below 12, adaptor only and more than 10% N bases, were filtered out by SOAPnuke software (Chen et al., 2018), separately. The trimmed clean reads including nuclear and organelle genome data were used to assemble the plastomes for each species. The plastomes of 19 Carya species were assembled via NOVOPlasty software (version 3.7), with ‘Seed_RUBP_cp.fasta’ provided by the software as the seed input and the published Juglans regia plastome (accession no. NC_028617.1) as reference (Dierckxsens et al., 2016; Peng et al., 2017). The default K-mer size (K-mer = 39) was firstly selected and multiple K-mers were used for a synchronous assembling test during the assembling to obtain the ideal assemblies.
To verify the correction of the plastome assemblies, PCR amplification and Sanger sequencing were performed in the six EA species and the pecan variety ‘Sioux’, to further confirm the SC-IR boundaries of the assembled sequences, as well as several special genomic regions among EA species. The sequences of the primers are listed in Supplementary Table 2.
The plastome assemblies were annotated using the online program GeSeq – Annotation of Organellar Genomes (Michael et al., 2017). Initial annotation, putative starts, stops, and intron positions were predicted against the annotation of Juglans regia and C. illinoinensis (GenBank accession numbers: NC_028617.1 and NC_041449.1) plastomes. The final annotations were determined by integrating the GeSeq prediction and manual correction. The circular plastome maps of Carya species were constructed using the online visualization program Organellar Genome DRAW version 1.3.1 (OGDRAW) (Stephan et al., 2019). The newly generated complete plastome sequences were deposited in GeneBank (Accession numbers were listed in Supplementary Table 3).
The online program IRscope (Amiryousefi et al., 2018) was used to visualize the boundaries of LSC-IRb, IRb-SSC, SSC-IRa, and IRa-LSC of plastome among the 19 Carya species, The IR expansion and contraction among these Carya species were also compared afterward. DnaSP v. 6.12.03 (Julio et al., 2017) was employed to analyze the nucleotide diversities, sequence polymorphisms, and relative rates of plastome sequence divergence in the eighteen Carya species. In order to calculate the synonymous (Ks) and non-synonymous (Ka) substitution rates and the nucleotide variance (π and θ), we extracted the same individual functional protein-coding exons and aligned them separately using BioEdit v. 7.0.9.0 (Hall, 1999). To obtain selection patterns in protein-coding genes, we calculated the Ka and Ks values of each protein coding gene between two plastomes in 19 Carya species using DnaSP v. 6.12.03, and divided them to take the average to get the Ka/Ks ratio of each gene.
The online tool MIcroSAtellite (MISA, http://pgrc.ipk-gatersleben.de/misa/) was used to discover simple sequence repeats (SSRs or microsatellites) in the eighteen plastomes with the following parameters: ten for mono-nucleotide motifs, six for di-nucleotide, five for tri-nucleotide, and three for tetra-, penta- and hexa-nucleotides, respectively (Sebastian et al., 2017). The repeat numbers in the regions of LSC, SSC, IRa, and IRb were counted.
Codon usage bias was calculated using MEGA X v. 10.1.8 (Kumar et al., 2018) and EMBOSS v.6.3.1 (Peter et al., 2000; Itaya et al., 2013). In general, many genes less than 300 bp in length are believed have no real functions and the encoded proteins may not be accurate and do not necessarily contain domains. Too many such genes may interfere with the accuracy of the results. Therefore, the protein-coding genes with more than 300 nucleotides in length were extracted according to the annotation file of each plastome.
RNA-editing sites of protein-coding genes in Carya plastomes were predicted using the online program Predictive RNA Editor for Plant cp genes (PREP-Cp) suite with a minimal editing score of 0.7 (Mower, 2009) http://prep.unl.edu/).
To construct the phylogenomic trees, 30 plastome sequences were included, including 23 plastomes from 20 species in genus Carya (19 newly assembled Carya species, 2 C. pallida varieties and 2 published C. illinoinensis varieties), 6 representative species from 5 other genera in Juglandoideae (Juglans regia, J. sigillata, Cyclocarya paliurus, Engelhardia roxburghiana, Platycarya strobiacea, and Pterocarya stenoptera), and the Myrica rubra in Myricaceae (as outgroups). In addition, the plastome sequences of seven other plastome assemblies were downloaded from GenBank (Supplementary Table 3). Supplementary Table 3 lists all taxa used in the phylogenomic analyses, including their sampling locations and GenBank accession numbers.
The phylogenetic relationships were inferred based on seven datasets from all 30 plastomes: the full-length sequences of plastomes; the sequences of LSC, SSC, or IR regions; the 79 concatenated protein-coding sequences (CDSs); and the LSC-SSC or LSC-IR-SSC ZZconcatenated sequences. Multiple sequence matrices of the dataset were generated in MAFFT v7.490 under standard parameters (Kazutaka and Standley, 2013), and manually adjusted, respectively. The phylogenetic trees were reconstructed based on Bayesian inference (BI) using MrBayes v3.2.6 (Fredrik et al., 2012), and the maximum-likelihood (ML) method using PHYLIP version 3.698 (http://evolution.genetics.washington.edu/phylip.html). For BI inference, two independent Markov chain Monte Carlo (MCMC) simulations were run for 2,000,000 generations and sampled every 100 generations. MrBayes settings for the best-fit model (GTR+I+G) were selected by AIC in MrModeltest. The first 25% of calculated trees were discarded as burn-in. A consensus tree and Bayesian posterior probabilities (PPBI) were constructed using the remaining trees. The ML trees were reconstructed using in PHYLIP v.7.490 and the bootstrap value was set to 500. Both BI and ML trees were visualized in FIGTREE v1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/).
To facilitate our analyses, we generated a map of modern distribution for each Carya species. Meanwhile, we also generated a map by combining the distributions and the diversity of IR-SC boundaries with the sampling location of each analyzed Carya species using the ArcGIS v10.2 program (Esri. ArcGIS software on https://www.esri.com/).
Using 2-Gb whole-genome resequencing data from each species, we de-novo assembled the complete plastomes of 19 Carya species with the J. regia chloroplast genome as reference (Peng et al., 2017). Boundaries between the regions of inverted repeats (IRs) and single copies (SCs) of the assemblies were verified by PCR (Supplementary Figures 2, 3; Supplementary Table 2). Sequence alignment of the amplificated sequences with our assemblies displayed a perfect match, indicating our assembly’s accuracy and the inaccuracy of the assembly (additional duplication in SSC region with ca. 15 kb in length) of the published C. kweichowensis plastome sequence (Ye et al., 2018). Like those previously published plastomes in Juglans and Carya (Dong et al., 2017; Hu et al., 2017; Peng et al., 2017; Feng et al., 2018; Ye et al., 2018; Zhai et al., 2019; Wang et al., 2020), all the newly assembled Carya plastomes were circular molecules ca. 160 kb in length on average with the typical quadripartite structure, i.e., a pair of inverted repeat (IR) regions, separated by a large single-copy (LSC) region and a small single-copy (SSC) region (Figure 1; Supplementary Figure 2). Among the 19 plastomes of Carya, C. poilanei has the shortest sequence length (158,036 bp), followed by C. tonkinensis (158,076 bp), while C. cathayensis (160,823 bp) has the longest genome (Table 1). C. cathayensis has the longest LSC region (90,114 bp) but C. hunanensis has both the shortest LSC (89,468 bp) and SSC regions (18,730 bp). C. palmeri has the largest IR region (26,004 bp), whilst C. poilanei has the smallest IR region (23,634 bp) and the largest SSC region (20,861 bp). GC content do not display significant variation among the 19 Carya species (36.13% ~ 36.28%), as well as in the regions of LSC (33.71% ~ 33.87%), SSC (29.66% ~ 32.61%) and IRs (42.57% ~ 42.89%) among the 19 Carya plastomes assemblies (Table 1). However, the paired-IR regions have the highest GC content, because of the presence of four pairs of duplicated rRNAs in the IR regions of each species (Table 1).
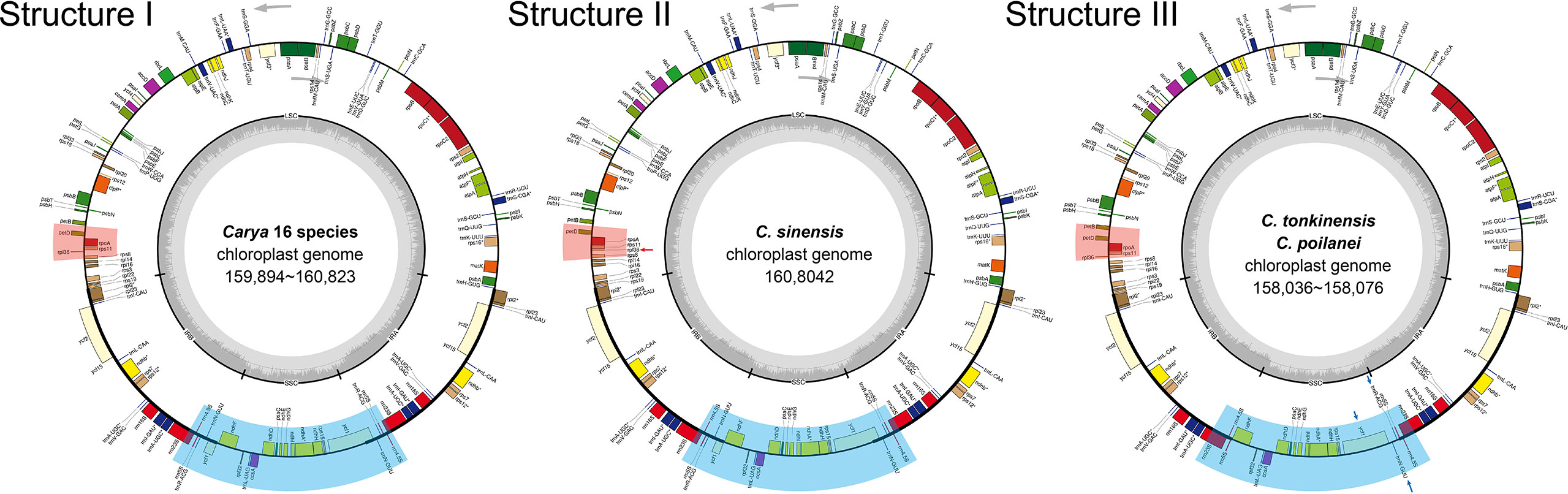
Figure 1 Circular maps of the 19 Carya species plastomes. GC content graphs are included as dark gray bars toward the center of each diagram. Intron-containing genes are marked with (*). Gray arrows indicate the translation direction of protein-coding genes. I - III show the different structures of Carya plastomes. Areas with light red and light blue backgrounds show differences in structure, and detailed differences are indicated by red and blue arrows.
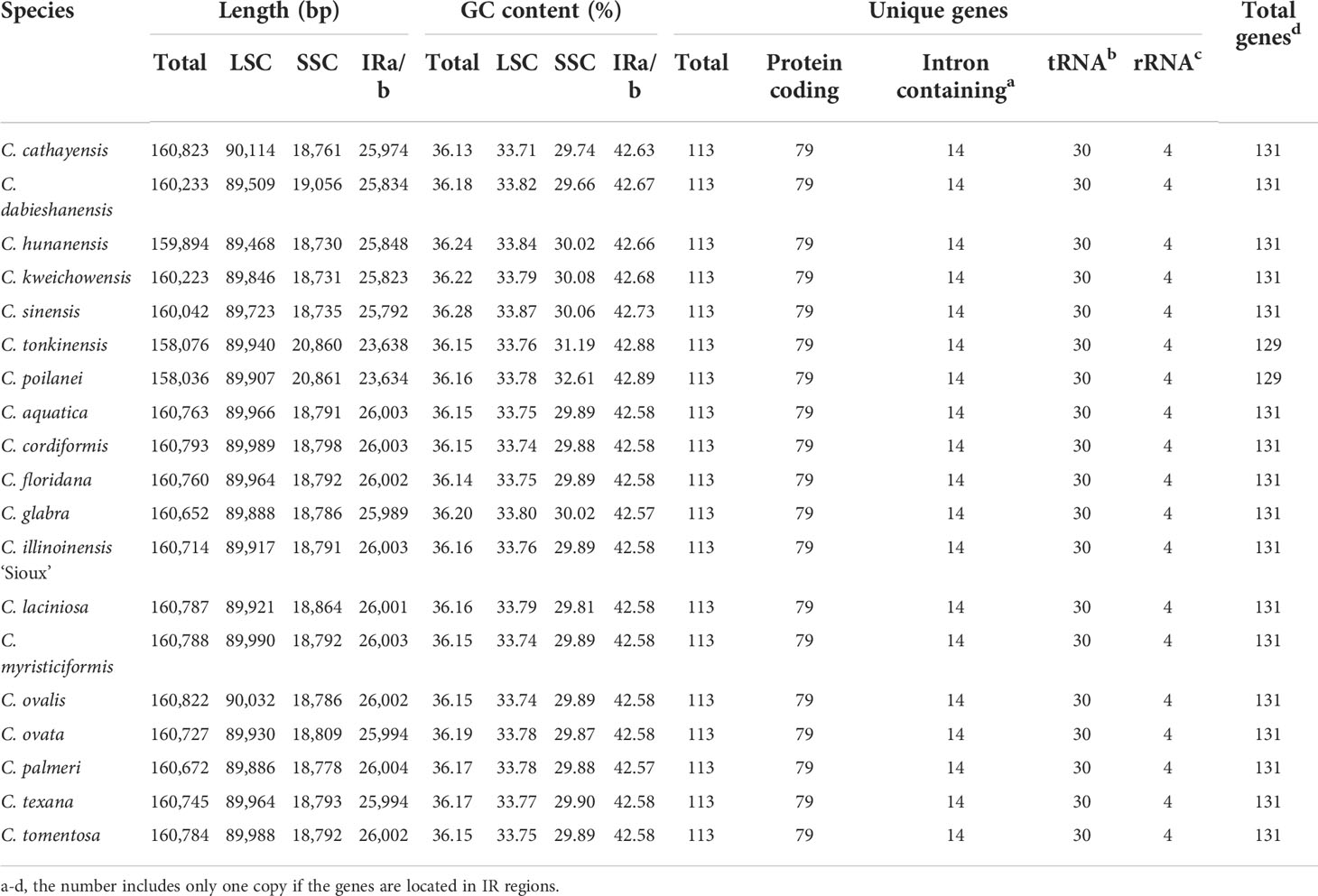
Table 1 Summary of complete plastome features of the 19 newly assembled Carya plastomes.
The 19 Carya complete plastomes have the same amounts of unique genes (a total of 113 unique genes including 79 conserved protein-coding genes, 30 tRNAs, and 4 rRNAs) and introns, which were arranged with a similar gene order (Figure 1; Table 1). Fourteen of the unique genes were intron-containing genes (9 protein-coding genes and 5 tRNA genes), with 1 or 2 introns (Table 2; Supplementary Table 4). Five intron-containing genes (3 protein-coding genes and 4 tRNA genes) are located within the IR regions (Supplementary Table 4). However, the intron size varied among the Carya plastomes, except for ndhB, rps12, trnA-UGC, trnL-UAA and trnV-UAC. The length of introns in the plastomes ranged from 526 bp to 1,229 bp and the longest intron was observed in ndhA. Two protein-coding genes, rpl2 and ycf3, exhibit significant variations in the length of both exons and introns, with considerable differences among species from EA and NA, so as the rpoC1 and trnA-UGC in their intron regions.

Table 2 Genes and contents in the newly assembled Carya plastomes.
Annotation-based circular maps of the 19 Carya plastomes can be categorized into three distinct structures (Figure 1). The major structure (Structure I) is shared by 16 Carya plastomes, with the same number of genes (131 genes, representing 113 unique genes), gene order, and translation direction (Figure 1). Eighteen unique genes were duplicated in the IR regions of these 16 plastomes (Table 2). Structure II is the most different from the major structure (Structure I) with respect to the direction of transcription of rpl36, which uses the complement strand as a transcriptional template in C. sinensis (Figure 1). A sequence loss of ~2.3 kb in length in the IRb region of C. poilanei and C. tonkinensis plastomes leads to distinguished genome structure (Structure III) and decreased gene content (a total of 129 genes). The loss of 2.3 kb sequences results in a contracted IR region and the loss of tRNA genes, trnN-GUU, and trnR-ACG in the IRb region (Figure 1; Table 2).
Despite relative conservation of IR/SC boundaries in plant plastomes, contraction and expansion of the IR-SC border regions are common in the process of plastid evolution, which is the major source of variation in angiosperm-plant plastome length (Raubeson et al., 2007; Wang et al., 2018). We examined the fluctuation of IR-SC borders together with the adjacent genes in the 19 Carya plastomes. Of the IR-SC junctions, IRa/LSC junction has the most conserved border within a gene spacer of rpl2 to trnH-GUG, with contraction sizes of 34 - 80 bp in the IRa region and 15 - 48 bp in the LSC region in the Carya plastomes (Figure 2A). Complete comparisons of these junctions revealed six patterns: Patterns I to VI (Figure 2A). Among them, Patterns I to IV are present in the 7 EA species and Patterns V and VI are present in the 12 NA species (Figure 2).
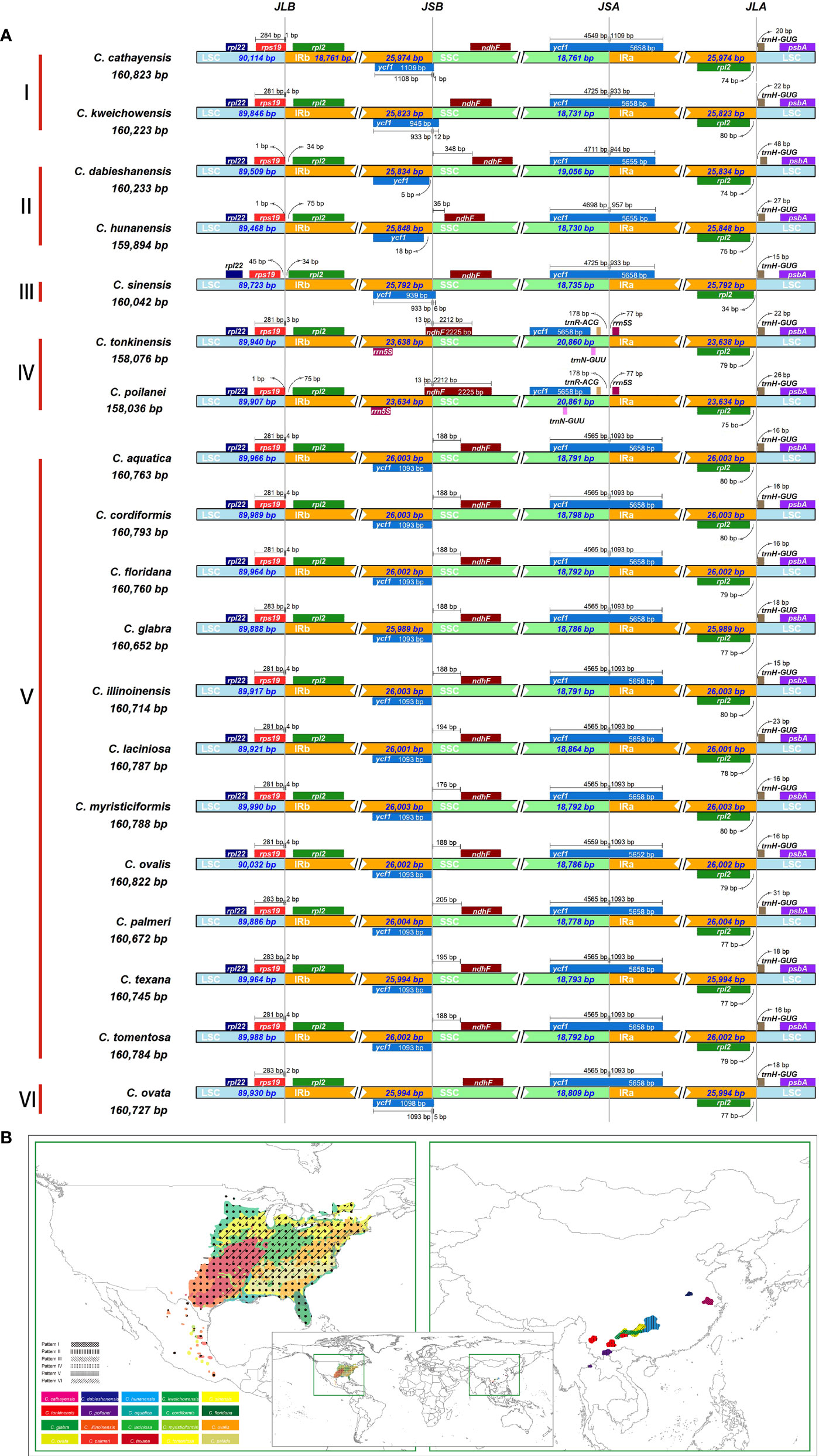
Figure 2 IR junction variation and its diversity among the 19 newly assembled Carya plastomes. (A), Comparison of IRb-LSC, LSC-IRa, IRa-SSC, and SSC-IRb boundaries. (B), The modern distribution and the diversity of IR-SC boundaries.
In details, Pattern I includes two species, C. cathayensis and C. kweichowensis, in which the LSC/IRb border is located within the rps19 gene with the expansion of 1 bp and 4 bp from rps19 of LSC to the IRb region, and the IRb/SSC border is located within the ycf1 fragment with expansion 1 bp and 12 bp from ycf1 fragment of IRb region to the SSC region (Figure 2A). Pattern II consists of C. dabieshanensis and C. hunanensis, in which the LSC/IRb border is within the gene spacer of rps19-rpl2 with 1 bp contraction of rps19 in the LSC region and 34 or 76 bp contraction of rpl2 in the IRb region. The IRb/SSC border within the gene spacer of the ycf1-fragment and ndhF that contracted 5 bp and 14 bp of the ycf1 fragment in the IRb region and 350 and 32 bp in the SSC region, and their SSC/IRa border was similar to Pattern I (Figure 2A). Pattern III includes only C. sinensis, which similarly has borders of LSC/IRb and SSC/IRa with the same plastomes as in Pattern II and IRb/SSC border with the same plastomes as in Pattern I. Pattern IV contains C. poilanei and C. tonkinensis plastomes, with a distinguished IR-SC boundary pattern: IRb/SSC border within the ndhF gene with a 6-bp expansion from the SSC region to the IRb region, and a SSC/IRa border located in the gene spacer between trnR-ACG and rrn5S with contractions of 178 bp and 77 bp for both sides, respectively (Figure 2A). Meanwhile, the LSC/IRb border of the plastomes in Pattern IV is similar to those in Pattern II (Figure 2A). Of the 12 NA Carya species, 11 have the highly conserved SC-IR boundaries and were assigned into Pattern V, which has the LSC/IRb border within rps19 with 2 bp or 4 bp expansion to the IRb region, the IRb/SSC border on the last base of the ycf1-fragment in IRb and SSC/IRa border within the ycf1 gene with 1,093 bp expansion to IRa region (Figure 2A). Pattern VI includes only C. ovata, with the same borders of LSC/IR and SSC/IRa, except for the IRb/SSC border, which was within the ycf1-fragment with 5-bp expansion to the IRa region (Figure 2A). These results indicated more conserved SC-IR boundary patterns among NA Carya plastomes than those in EA that exhibit highly diverse IR-SC boundary patterns (Figure 2B).
Simple-sequence repeats (SSRs), also known as microsatellites, are short tandem repeat DNA sequences that consist of repeating 1-6 nucleotide motifs widely distributed throughout the plastomes, which are important molecular markers for analysis of genetic diversity and relationships between species (Yang et al., 2011; Jiao et al., 2012). We detected 1,652 SSRs in the 19 Carya plastomes and the numbers of SSRs varied among the species, with a range from 76 in C. aquatica and C. texana to 106 in C. poilanei and C. tonkinensis (Figure 3A; Supplementary Table 5). Of these detected SSRs, mononucleotide repeats were the most abundant SSR motifs, which accounted for approximately 85.5% of the total SSRs, and over 99% of the mononucleotide repeats were composed of A/T repeats (Figure 3A; Supplementary Figure 4). While the tri- to hexanucleotide SSRs were occupied 7.75% of the total (Figure 3A; Supplementary Figure 4). In addition, the total number of SSRs in each Carya plastome also revealed significantly different patterns among species in EA (93 - 106) and NA (76 - 82) Carya species, in which the mononucleotide repeats (especially for A/T repeats) are the major contributor (Figure 3A; Supplementary Table 5). Meanwhile, the numbers of SSRs of the plastomes can also be classified into six patterns, corresponding to those of IR-SC boundary patterns (Figures 2, 3A).
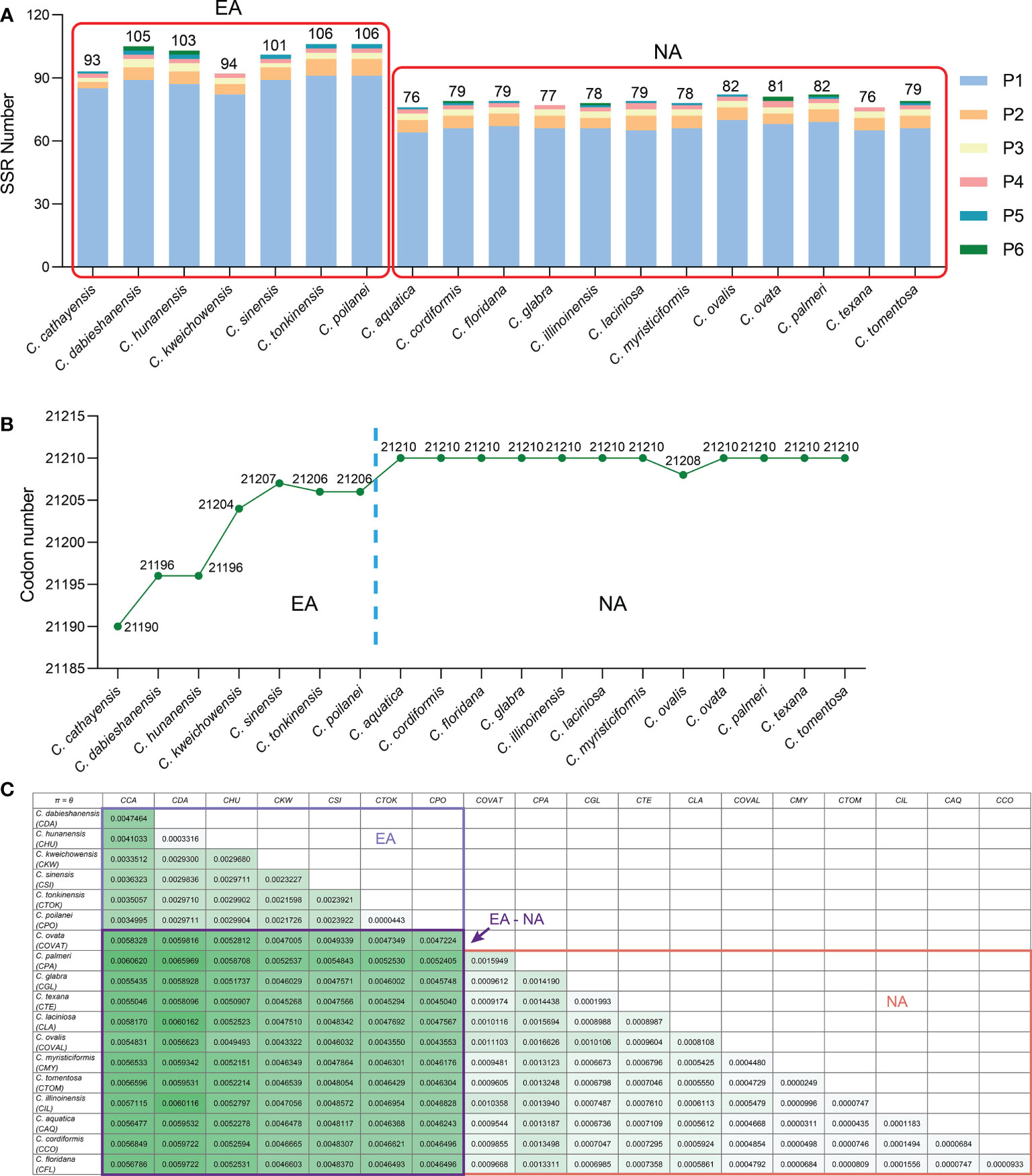
Figure 3 Comparison on SSR numbers, codon number and nucleotide diversities of Carya plastomes. (A), Statistics of SSR number for each Carya plastome. (B), Statistics of codon number for each Carya plastome. (C), The paired nucleotide diversities of Carya plastomes from species in Eastern Asia (EA) or North America (NA), or between two species from Eastern Asia and North America (EA-NA).
Codon distribution analysis of 52 protein-coding genes with a length over 300 bp for each gene sequence in the Carya plastomes revealed a similar codon usage pattern among the plastomes of Carya species and J. regia (Supplementary Table 6). The protein-coding sequences contain a total of 21,190 to 21,210 codons in the Carya plastomes and 21,265 in J. regia, including stop codons. The total number of codons varied among EA Carya, whereas 11 of the NA Carya have the same number of total codons (21,210), with the only exception of NA species in C. ovalis plastome with 21,208 codons (Figure 3B). The total number of codons for each species can also be classified into six patterns as mentioned above.
Other than the codon distribution, we also counted the average effective numbers of codon (ENC) in the plastomes. The results showed that 17 of the 19 assembled Carya plastomes had similar codon usage bias (ENC ranged from 48.883 to 49.001), which were slightly lower than that of J. regia (48.776) (Supplementary Table 6). C. tonkinensis and C. poilanei have ENC = 56.51 and 56.52, which are significantly higher than any of other species. All the Carya plastomes have similar GC content in the codons (37.32% ~ 37.39%) except for C. tonkinensis and C. poilanei plastomes which have lower GC contents for the first (39.59%) and second codons (31.12% and 31.18%) and relatively high GC content (41.26% and 41.20%) for the third position (Supplementary Table 6). Among all codons in the 19 Carya plastomes, leucine is the most abundant amino acid, while cysteine showed the least abundance (Supplementary Figure 6A). The most frequent synonymous codon in the Carya plastomes was AUU, which encodes isoleucine, and the least frequent codon, except for the stop codons, is UGC, which encodes cysteine (Supplementary Figures 5, 6B). Of the 20 amino acids and stop codons, only leucine each has six codon types, while methionine and tryptophan preferred one codon type in the plastomes of 19 Carya species and J. regia (Supplementary Figures 6). In general, AUG encodes not only methionine but also the universal start codon for the nuclear genome of eukaryotes (Thach et al., 1966). We detected that the genes of ndhD, rpl16, ycf15, and rps19 use ACG, ATC, CTG, and GTG encodes (not ATG) to encode the start codon – AUG respectively.
We also assessed the relative synonymous codon usage (RSCU), a good indicator for measuring nonuniform synonymous codon usage bias in coding sequences (Lee et al., 2010). Both methionine and tryptophan have RSCU = 1 (Supplementary Figure 7), indicating that codons AUG and UGG have no bias or preferences. The plastomes of 19 Carya species and J. regia had 30 biased codons with RSCU > 1, and their third position is A/U except for leucine (UUG). UUA has the highest RSCU values (1.94 to 2.03) in leucine in all the plastomes of Carya species and J. regia, and AGC has the lowest RSCU values in leucine (0.38 for Carya species and J. regia). Moreover, leucine showed A or T (U) bias in all synonymous codons: UUA, UUG, CUU, CUC, CUA, and CUG. We observed that the RSCU value for the specific amino acid increased with the number of codons. The following codons have high RSCU frequency (>30%) and fraction: GAU (aspartate), GAA (glutamate), AUU (isoleucine), AAA (lysine), UUA (leucine), AAU (asparagine), UAU (tyrosine), UUU (phenylalanine), and CAA (glutamine); and the bias toward these nine codons was consistent with the low content of GC in the third codon position (Supplementary Figure 7). Our analysis also showed that the RSCU value increased with the quantity of codons coding for a specific amino acid.
We found that 62 post-transcriptional RNAs have editing modifications in 23 protein-coding genes (Supplementary Figure 9; Supplementary Table 7). Genes of ndhB (11 editing sites) and ndhD (10 editing sites) contain the most RNA editing sites, followed by rpoB and rpoC2, with 6 and 5 editing sites respectively. Only one to three RNA editing sites were found in the rest of the 19 protein-coding genes. All of RNA editing sites potentially caused the conversion from cytosine to uracil after transcription, and 43 of them (69.4%) took place at the second nucleotide of codons, with two times the conversion rate compared to the first nucleotide (19, 30.6%) (Supplementary Figure 8). However, no correlation was observed between gene length and gene number (Supplementary Figure 8). Approximately 77% of the RNA modifications resulted in the conversion of hydrophilic to hydrophobic amino acids, mainly serine to leucine or phenylalanine (S to L, 22 editing sites; S to F, 8 editing sites), or proline to leucine (P to L, 8 editing sites; Supplementary Table 7). We also observed the conversion from proline to serine in three editing sites, representing the changes of amino acids from nonpolar to polar. On the species level, the majority of the RNA-editing sites were shared by all the Carya plastomes and only a few were species-specific, for example, CUC to UUU (L to F) was found only in C. poilanei at the amino acid position 71, CGG to UGG (R to W) only in C. sinensis, CCA to UCA (P to S) only in C. floridana, and CUU to UUU (L to F) only in C. laciniosa (Supplementary Table 7).
In order to obtain selection patterns in protein-coding genes, the nonsynonymous (Ka) and synonymous (Ks) substitutions ratios (Ka/Ks = ω) were determined for 79 unique protein-coding genes with the comparison of the 19 Carya plastomes (Supplementary Figure 9; Supplementary Table 8). Among the protein-coding genes, ndhA and petA have Ka/Ks ratios greater than 1.0 (Supplementary Figure 10; Supplementary Table 8), indicating the genes were subjected to positive selection. The rest of the 77 protein-coding genes have Ka/Ks < 1.0 (Supplementary Figure 9; Supplementary Table 8), indicating that purification selection happened only during plastome evolution. Specially, seven genes (atpF, ccsA, matK, rbcL, rpoA, rps15 and ycf1) have the Ka/Ks ratios between 0.5 and 1.0 (Supplementary Figure 9; Supplementary Table 8). These results clearly indicate that protein-coding genes in plastomes of different plant species were subjected to diverse selection pressures.
Two parameters, Pi (π) and theta (θ), were used for measuring the nucleotide variability among protein-coding genes of the 19 Carya plastomes. The value πvaried among the protein-coding genes with a range from 0 to 0.08495 (average π = 0.00326) (Supplementary Table 8). The gene rpl36 (π = 0.08495) was notably variable among the protein-coding genes in the 19 Carya plastomes. The gene ycf1 showed a relatively lowerπ value (0.00493) among the protein-coding genes in the Carya plastomes, although it was commonly used as a representative plant DNA barcoding region (Dong et al., 2015). The variation of the other parameter θ showed the same patterns as the π value among Carya plastomes of the 79 protein-coding genes (Supplementary Table 8). In comparison, plastomes of EA Carya species revealed higher nucleotide diversities (π and θ) than those in NA Carya species (Figure 3C).
The phylogenetic analyses, based on matrices (with indels) from seven datasets containing the full or partial sequence of 30 plastome sequences including 26 Juglandaceae species and one M. rubra (as tree root), resulted in seven different topologies using both Bayesian inference and the ML method (Figure 4; Supplementary Figure 10–15). Among the seven BI trees, one generated from the IR-region dataset displayed a very chaotic phylogenetic relationship among all analyzed species and varieties, and therefore was discarded for further analysis (Supplementary Figure 15). The remaining six BI trees displayed identical topologies (with posterior probability (PP) value = 1) on the intergeneric level in Juglandiodeae. All six topologies supported the two sister subclades of EA and NA in the genus Carya (Figure 4; Supplementary Figures 10–14). When considering the overall supporting rates for the six topologies based on BI, we are confident that the best phylogenetic tree is generated by full-length plastome sequence, followed by LSC, LSC-SSC, SSC, CDS, and LSC-IR-SSC (Figure 4; Supplementary Figures 10–14). Almost all branches of the BI tree inferred from full-length plastomes displayed the highest supports of any of the other 5 phylogenetic trees based on Bayesian inference. The topologies of all ML trees highly supported the backbone relationships among genera of Juglandoideae and the two sister subclades of EA and NA in Carya (Figure 4; Supplementary Figures 10–15). The overall supporting rates were in the same sequence as mentioned above for BI trees (Figure 4; Supplementary Figures 10–15). Excluding the trees based on the IR dataset, the genus Engelhardia formed a sister relationship with five other genera (including Carya) with full supports (PP = 1 or BS = 100) in the remaining partial or full-length plastome sequences-based topologies generated by Bayesian inference and the ML method (Figure 4; Supplementary Figures 10–14). These topologies fully supported the hierarchical clustering of Platycarya, Cyclocarya, Pterocarya, and Juglans, and grouped a sister clade with Carya. Within the Carya monophyletic group, two major subclades, i.e., EA and NA are well-separated (Figure 4; Supplementary Figures 10–14). However, the supporting rates [bootstrap (BS) values] for the branches in all seven ML trees were significantly lower than the PP values in BI trees, and the topologies in the EA and NA subclades varied among the ML trees (Figure 4; Supplementary Figures 10–15). Compared to the corresponding BI tree, similar phylogenetic topologies were obtained using the ML method based on the data set generated by full-length plastome sequences, which also represented the best ML tree although with relatively low supports (BS values) of most branches (Figure 4; Supplementary Figures 10–15). Therefore, the BI trees are considered the primary references in the following analyses of plastome phylogenetic relationships of Carya, and the detailed analyses were mainly based on the BI topology inferred by full-length plastome sequences (Figure 4; Supplementary Figures 10–14).
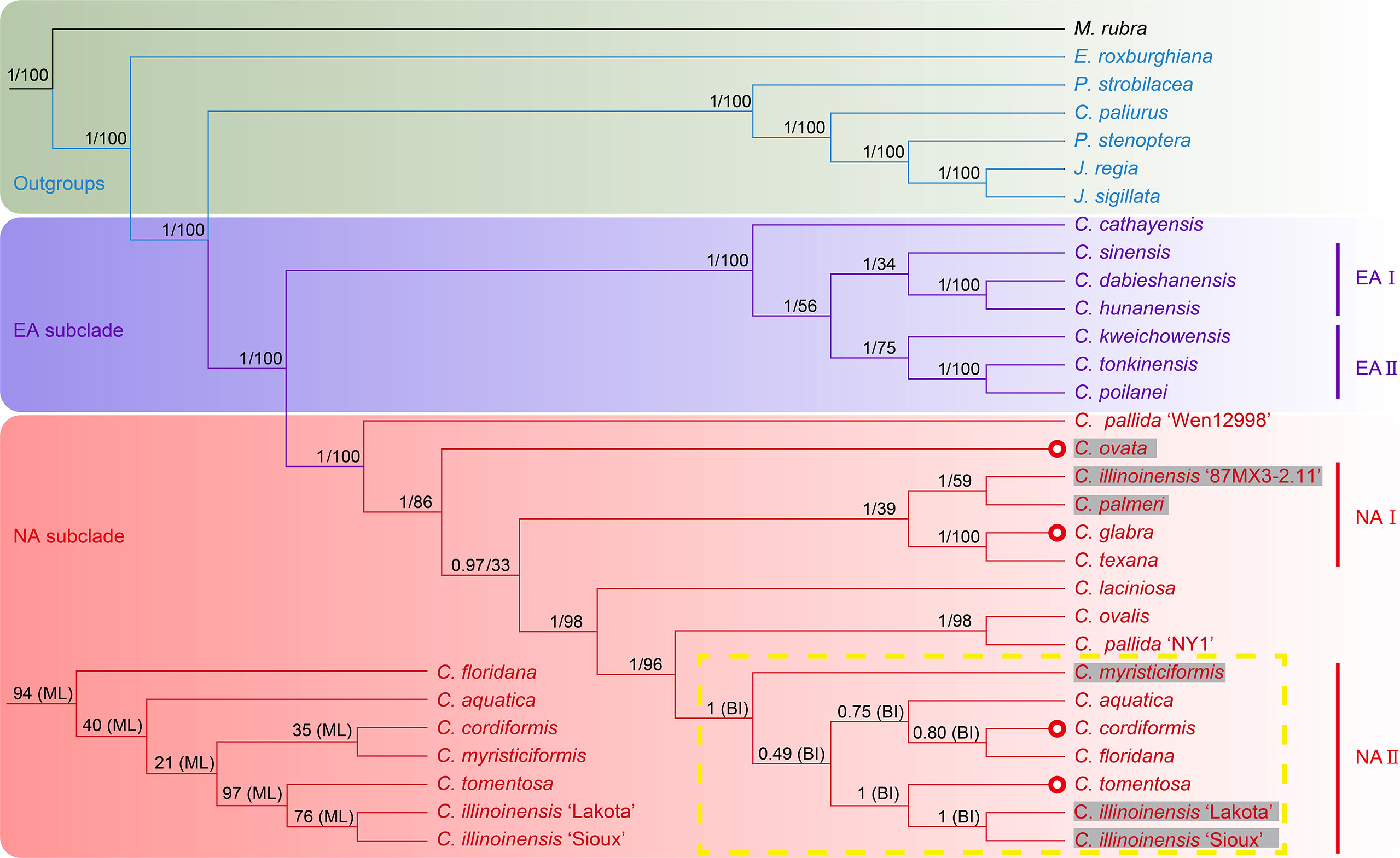
Figure 4 Phylogenetic trees of Carya species and representative species in other genera of Juglandaceae inferred from the datasets of complete plastomes using Bayesian inference and maximum likelihood method. The posterior probability (PP) and bootstrap (BS) values that supported each node are shown above the branches. The colors in green, light purple and pink indicate the outgroups, EA and NA Carya species, respectively. The letter n represents the haplotype, and x represent and the number before it shows the chromosomal set of the haplotype of each species. The numbers after n and x are the chromosomes of haplotype of each species. Red circles show the species in section Apocarya, gray background and the red circles indicate the tropical distributed species/varieties. The lower left quarter and the yellow box respectively show the parts with different topologies in the trees based on ML and BI methods.
The plastome phylogenetic relationships inferred from datasets of full-length, LSC, and LSC-SSC regions of plastomes displayed the same topologies for EA subclade with PP values of 0.997 (LSC BI phylogenetic tree) to 1 (full-length and LSC-SSC BI trees) (Figure 4; Supplementary Figure 10). However, the maternal phylogenetic incongruencies were obtained between species of EA or NA Carya in the topologies inferred from partial or full-length plastome datasets. In contrast, the maternal phylogenetic trees inferred from the datasets of SSC, CDS, and LSC-IR-SSC showed completely different topologies for EA Carya subclades, with relatively low PP values for several internal subclades (Supplementary Figures 11–13). However, all six BI topologies support the sister relationships for two pair of terminal branches, C. dabieshanensis – C. hunanensis and C. tonkinensis – C. poilanei, with supports = 1 (Figure 4; Supplementary Figures 11–13). Within the EA subclade, C. cathayensis was considered the base taxon with PP = 1 in four topologies of full-length, LSC-SSC, SSC and LSC-IR-SSC. Meanwhile, the topologies of full-length and LSC fully supported the sister relationships with the nodes of C. sinensis – C. hunanensis – C. dabieshanensis (designated as EA-I) and C. kweichowensis – C. tonkinensis – C. poilanei (designated as EA-II) (Figure 4; Supplementary Figures 10–13). C. kweichowensis formed a sister node with the terminal branch of C. tonkinensis – C. poilanei in four topologies of full-length, LSC, LSC-SSC, and SSC of plastomes. C. sinensis formed a sister relationship with the terminal branch of C. dabieshanensis – C. hunanensis in the phylogenetic trees of full-length plastomes and LSC-SSC sequences. This relationship was strongly supported in the LSC dataset-based maternal phylogeny but formed a terminal sister relationship with C. kweichowensis in the SSC dataset-based phylogenetic tree. Although C. dabieshanensis and C. cathayensis, have close geographical distribution and similar morphological traits, the former has larger size of fruits and nuts than the latter, and their maternal phylogenetic relationship is relatively far away (Figure 4; Supplementary Figure 17).
In the NA subclade, five out of six BI topologies (except for that inferred from SSC dataset) fully supported the C. pallida var. ‘Wen 12998’ as the basal taxon, while C. pallida variety ‘NY1’ formed a terminal branch with C. ovalis (Figure 4; Supplementary Figures 10–13). All six BI topologies fully supported the terminal branch of C. glabra and C. texana. The variety of C. illinoinensis ‘87MX3-2.11’ displayed a sister relationship with C. palmeri in the full-length and SSC-based phylogenetic trees, and formed a sister relationship with C. laciniosa in the LSC, LSC-SSC, CDS, and LSC-IR-SSC-based phylogenetic trees. The other two varieties of C. illinoinensis – ‘Lakota’ and ‘Sioux’ formed a terminal branch with very strong supports and a sister branch to C. tomentosa in the full-length, LSC, LSC-SSC, and LSC-IR-SSC-based maternal phylogenetic trees. C. aquatica and the terminal branch of C. cordiformis – C. floridana formed a sister note in full-length and LSC plastome sequences-based maternal phylogenetic trees. The phylogenetic relationship of C. ovata varied in five phylogenetic trees (full-length plastomes, LSC, LSC-SSC, SSC, and CDS-based inferences).
In summary, the maternal phylogenetic relationships within the subclades EA or NA were clear based on the topology inference of complete plastomes with high confidence (PP > 0.8) for most nodes and the topology was selected for further discussion on the maternal phylogenetic relationships within EA or NA subclade (Figure 4). The topology of the EA subclade fully supported the earliest divergence of C. cathayensis, which consists of a sister relationship with two sub-sister nodes, C. sinensis – C. hunanensis – C. dabieshanensis (EA-I) and C. kweichowensis – C. tonkinensis – C. poilanei (EA-II), indicating that they came from a common ancestor in the genus. Among the species of the NA subclade, one landrace ‘Wen 12998’ of C. pallida was clustered as a basal taxon and formed a sister relationship with the rest of the NA plastomes. Except for the node in NA-II (PP = 0.502) containing C. tomentosa, two cultivars of C. illinoinensis, and the sister node (PP = 0.755) of C. aquatica, C. cordiformis and C. floridana, the majority nodes of the NA subclade topology displayed very high supporting rates. The topology of NA subclade disrupted the morphological features-based division of the two sections, Carya and Apocarya (Huang et al., 2019), but varieties that are at the same terminal branch or that have a close relationship in the phylogenetic tree indicate that they have close or overlapping distribution regions (Figures 2B, 4; Supplementary Figure 17). Two pairs of terminal branches containing a local collection ‘87MX3-2.11’ of C. illinoinensis (Wang et al., 2020), and C. palmeri (section Carya) section and species C. glabra and C. texana (section Apocarya) consisted of an internal sister node (NA-I) with C. ovata (section Carya) in the NA subclade (Figure 4). In contrast, C. myristiciformis, as the partial sister of the terminal branch of C. ovalis and the C. pallida var. ‘NY1’, showed a relatively distant maternal phylogenetic relationship with the two species (Figure 4). Interestingly, two varieties of C. illinoinensis – ‘Sioux’ and ‘Lakota’ which had different parentages and came from different controlled crosses made in Brownwood, Texas in 1943 and 1964, respectively (Grauke and Thompson, 1996; Wang et al., 2020), also showed distant phylogenetic relationships with the wild seedling ‘87MX3-2.11’ that originated from Oaxaca, Mexico (Wang et al., 2020) (Figure 4).
Being consistent with reports in most flowering plants, Carya plastomes have conservative canonical quadripartite structures including LSC and SSC regions (Figure 1), which are separated by two IR regions that are generally known to play a role in the structural stability of plastomes (Palmer and Thompson, 1982; Bock, 2007). The Carya plastomes are similar in gene content and gene order (113 unique genes) (Figure 1; Tables 1, 2), as reported in many angiosperms (Hu et al., 2016; Xu et al., 2017; Mader et al., 2018; Yang et al., 2018). A strong tendency toward A or U at the third codon position (Clegg et al., 1994; Gao et al., 2017; Mader et al., 2018; Meng et al., 2018) may explain why the A/T content is as high as ~ 64% in Carya plastomes (Table 1). The conservatism of Carya plastomes is also revealed by RNA editing sites and the measurement of selection for each protein-coding gene (Supplementary Table 7).
As the most conserved region in the plastomes, IR expansion or contraction may alter the plastome size and the stability of the genomic structure, which in turn could cause IR-SC boundary shift, repeated sequences (Dugas et al., 2015), and/or duplication or deletion of a certain gene through inversion during evolution (Park et al., 2018). Frequent expansions and contractions at the junctions of IR-SCs have been recognized as evolutionary signals for illustrating the relationships among taxa (Khakhlova and Bock, 2006; Raubeson et al., 2007; Wang et al., 2008; Lu et al., 2018; Park et al., 2018). Moreover, repeated sequences, especially SSRs play key roles in plastid genome rearrangement, divergence, and evolution (Weng et al., 2014). Our analyses reveal the intercontinental disjunctive distribution between and among EA and NA Carya species, reflected by the patterns of IR-SC junctions, the number of SSRs and codons, and the paired nucleotide diversities of Carya plastomes (Figures 2, 3). The diversified features described here for Carya plastomes and the shortened IR regions in C. tonkinensis and C. poilanei could be related to the variation in genome size and the long evolutionary history of Carya species. By comparison, Carya species in EA exhibited higher diversities in plastome features and structure compared to those in NA (Figures 1–3; Table 1), although higher diversities in species and morphology were displayed in NA Carya than those in EA (Huang et al., 2019). The interesting findings combined with geology and vicariance events suggest more diverse matrilineal sources of EA Carya species, in contrast to fewer matrilineal sources of NA Carya species during their evolution. Meanwhile, the changes in climates and habitats especially in the glacial period may speed up the adaptive speciation in NA Carya and lead to higher speciation and morphological diversities in NA than those in EA. It is known that climate cooling is commonly accepted as the main causes of the isolated habitats and the disjunctions of floristic elements between continents (Raven, 1972; Tiffney, 1985; Wen, 1999). One example as addressed by Deng et al. (2017), is the modern Asia-North America disjunct distribution of two taxa from Rubiaceae – Kelloggia and Theligonum formed by the fragmentations of original wide distribution caused by climate cooling. Extinction of Carya in EA and NA might occur commonly during climate change. The rapid uplift of Qinghai-Tibetan Plateau and climate change could be responsible for the modern restricted distribution of Carya in EA (Supplementary Figure 16), while the overlapped distribution area and frequent inter- and intra-species hybridization could account for the relatively low plastome diversity in modern NA Carya species (Supplementary Figure 16).
Our maternal phylogenetic analyses showed that six of the BI-based and all ML-based topologies inferred by different datasets of plastomes support the sister relationship of Carya and the monophyletic Cyclocarya-Juglans-Platycary-Pterocarya clade in Juglandoideae (Figure 4; Supplementary Figures 10–13). Our results also strongly support two monophyletic subclades corresponding to the disjunctive geographical distributions of Carya in EA and NA (Figure 4; Supplementary Figures 10–13). The results are highly consistent with those of previous inferences on the intergeneric phylogenetic relationships in Juglandoideae and the relationships between two subclades in the genus Carya, based on the molecular markers from partial or complete nuclear or/and organelle sequences (Manos and Stone, 2001; Manos et al., 2007; Zhang et al., 2013; Zhang et al., 2019; Mu et al., 2020; Zhang et al., 2021; Zhou et al., 2021). It is known that the plastome is a key organelle in the plant cells, performing photosynthesis and other metabolic processes related to the adaptation of the plant to its environments (Dierckxsens et al., 2016). Although the basic structure of the plastome is highly conserved throughout land plant lineages, it has been proven that differences in the sizes of the complete genome and the protein-coding gene content of the different genome regions related to the difference in selection pressure are informative in phylogeny and evolution for many plant lineages. Although the matriarchal phylogenetic relationships among species and/or varieties within the EA and NA subclades varied among the topologies inferred from different datasets of plastomes, the plastome-based physiologies still provide very important clues to the backbone relationships between EA and NA Carya (Figure 4; Supplementary Figures 10–13). The plastome-based phylogeny of Carya could also provide a good example for exploring the evolution of plastomes as we present in this study.
By comparing the PP and BI values of all nodes among six topologies, almost all the nodes of the BI tree reconstructed from full-length plastomes showed the highest support (PP > 0.8) (Figure 4; Supplementary Figures 10–13). Therefore, the full-length plastome dataset-based BI tree is considered the main reference for further analyses of maternal phylogenetic relationships among Carya species. The maternal phylogenetic relationships among EA Carya species are highly in agreement with their geographical distributions (Figures 2B and 4; Supplementary Figure 17). The maternal phylogenetic positions of the species in NA subclade provide strong support for their overlapping and adjacent distribution patterns (Figures 2B and 4; Supplementary Figure 17), although the topology does not match the morphological features-based division of two sections (Manos and Stone, 2001; Huang et al., 2019). Meanwhile, the topologies among species within EA and NA subclades exhibited significant deviation from the pattern presented by nuclear genome data (Huang et al., 2019). Our results demonstrated that the different contributions of nuclear and organelle genomes, compared to the control, based on the morphological features associated with the different evolutionary rates between organelle and nuclear genomes, showed the difference between topologies based on the different datasets of plastomes in the present study. In general, the nonparental-inherited plastomes are highly conservative in protein-coding gene functions and genome structure, which can also reveal the maternal origin and diversity of a plant (Raubeson and Jansen, 2005). However, the morphological characteristics could be controlled by parental-inherited materials in the nucleus and may be influenced by environment and selection resulting in adaptation, convergent evolution, and species diversity (Givnish, 2016). Taking this into account, it is easy to understand the contradiction between topologies built from plastomes and nuclear data, as both had different evolutionary trajectories.
Carya poilanei, has been regarded as an extinct species in the subfamily Juglandoideae for more than 60 years before its rediscovery in the Ailao Mountain area, Jianshui County, Yunnan Province, China (Zhang et al., 2022). This species has historically been classified as a member of Juglans (Chevalier, 1941), then moved to the genus Carya (Leroy, 1950), and finally botanically characterized (Leroy, 1955). Although it is classified in the section Sinocarya of the genus Carya based on its morphological traits, the phylogenetic and maternal relationships are still unclear. The rediscovery of the species brings this issue back into consideration. Our results indicate that C. poilanei has the highest similarity with C. tonkinensis in plastome structure and features, and it has the closest maternal phylogenetic relationship with C. tonkinensis, which is supported by their closest geographical distribution (Figures 1–4; Tables 1, 2; Supplementary 17). Therefore, we assume that these two species historically shared a common maternal ancestor. In addition, the taxonomic placement of four contentious species C. dabieshanensis, C. (Annamocarya) sinensis, C. glabra, and C. ovalis has been disputed for decades (Chang and Lu, 1979; Thompson and Grauke, 1991; Manos and Stone, 2001; Grauke and Mendoza-Herrera, 2012; Grauke et al., 2016). C. dabieshanensis has been treated as a member of C. cathayensis because of their high similarities in morphological features, except for the larger fruit size of C. dabieshanensis (Liu and Li, 1984). Our previous nuclear genome analyses also supported that C. dabieshanensis is close to C. cathayensis, but not to any members in the section Sinocarya (Huang et al., 2019). This study provided a full comparison of the morphological features between these two species and found no additional differences beyond the genomes (Supplementary Figure 17). However, the plastome features of both species reveal a significant variation in the patterns of IR-SC junction: C. cathayensis belongs to Pattern I with C. kweichowensis, but C. dabieshanensis belongs to Pattern II with C. hunanensis (Figure 2A). The phylogenetic topology built from the complete plastomes also supports that C. dabieshanensis has the close matrilineal relationship to C. hunanensis but not to C. cathayensis (Figure 4). Although taxonomists distinguished C. (Annamocarya) sinensis from the genus Carya by its distinctive taxonomic, botanical, and horticultural characteristics, high throughout genome-wide sequencing technology has provided a credible molecular phylogeny to distinguish plant species (Favre et al., 2020). The plastomes-based phylogenetic tree here also provided solid support that C. (Annamocarya) sinensis is a member of EA Carya (Figure 4). From the plastome phylogenetic tree, C. glabra and C. ovalis were separated in the NA Carya subclade and this is also confirmed by their plastome features, the patterns of IR-SC junction variations (Figures 2A, 4), as well as their morphological differences (Grauke, 2003). As such, the matriarchal-originating plastomes provide an effective tool for taxonomic placement of the outcrossing plant species, at least at genus level.
As mentioned above, maternal phylogenetic analyses based on whole plastomes and partial plastome regions, except for the IR regions, recovered the same topologies and provided substantial support of the six genera of Juglandoideae and two subclades of the genus Carya (Figure 4; Supplementary Figures 10-16). These results were in consistent with our early studies based on nuclear genome data and a few organelle and nuclear gene markers (Zhang et al., 2013; Huang et al., 2019). However, multiple significant inconsistencies between species of EA or NA subclades have been found in the maternal phylogenetic topologies inferred from different plastome regions and the nuclear datasets. These discordances of phylogenetic trees have been reported in numerous studies of plants in the North Hemisphere, including several close relative families of Juglandaceae, such as Betulaceae and Fagaceae in Fagales (Chan and Ragan, 2013; Lemmon and Lemmon, 2013; Zwickl et al., 2014; Stenz et al., 2015; Yang et al., 2019; Yang et al., 2021; Zhou et al., 2022). The inconsistencIES between the phylogenetic relationships based on plastomes and nuclear genes are believed to be based upon hybridization and introgression, incomplete lineage sorting, and/or ancient chloroplast capture, while the gene flow caused by hybridization and introgression, as well as plastid capture between heterocompatible species, is considered to be the source of phylogenetic inconsistencies inferred from different datasets of plastomes (Suh et al., 2015; Yang et al., 2019; Lovell et al., 2021; Yang et al., 2021; Zhou et al., 2022).
Hedrick (2013) found that introgression usually involves a small number of genes or genomic regions but may be of substantial significance. Incomplete lineage sorting largely resulted from ancestral polymorphism spanning multiple speciation events and subsequent random fixation of the polymorphisms in different lineages (Oliver, 2013), and it is common in multi-locus phylogenetic datasets due to rapid diversification (Wang et al., 2022). The entire plastome functions as a single linked locus, with different haplotypes retained through ancient clado-genic events like the alleles of a single nuclear locus (Folk et al., 2017; Lee-Yaw et al., 2019). Natural hybridization and introgression are very common in numerous wind-pollinated plant species, such as Betulaceae (Yang et al., 2019). In such species, plastid DNA is maternally inherited from ovum, whereas nuclear DNA is transmitted parentally through both pollen and ovum. Ancient and recent hybridization and introgression can result in rapid introgression of maternally inherited plastomes and the concerted evolution of the nuclear genes toward the introgressive species, especially for the geographically closely related species, and finally distort the phylogenetic relationships of introgressive species. In Carya, hybridization has been well studied and introgression signatures between species were evident in previous and recent reports (Lovell et al., 2021; Wang et al., 2022). In addition to the incomplete lineage sorting and ancient plastid capture, the obvious incongruencies within subclades of Carya between plastome- and nuclear marker-based topologies in our analysis could partially result from introgressive hybridization. Introgressive hybridization could also account for the phylogenetic incongruencies between varieties of non-monopoly Carya species such as C. illinoinensis and C. pallida.
‘87MX3-2.11’ (C. illinoinensis), a local seedling collection from an autochthonous tree growing near Zaachilla, Oaxaca in Mexico (Wang et al., 2020), was clustered with C. palmeri, which is only distributed in the tropical region in the NA Carya subclade (Figures 2B, 4; Supplementary Figure 16). Meanwhile, two controlled-cross cultivars of C. illinoinensis, ‘Lakota’ and ‘Sioux’, made in Brownwood, Texas, in 1964 and 1943, respectively (Grauke and Thompson, 1996) were grouped together (Figure 4). ‘Schley’ is the maternal parent of ‘Sioux’, while ‘Lakota’ was from a cross of ‘Mahan’ × ‘Major’, and ‘Mahan’ is a progeny of ‘Schley’ (Thompson and Young, 1985; Grauke et al., 2015). Therefore, it is easy to understand the sister relationships between these two cultivars (Figure 4). Meanwhile, the sister group of ‘Lakota’ and ‘Sioux’ demonstrates a close maternal phylogenetic relationship with the widely distributed species C. tomentosa (Figures 2B, 4; Supplementary Figure 17). Our plastome-based phylogenetic analyses also suggested that it is likely that the C. illinoinensis ‘87MXx3-2.11’ share a common matrilineal ancestor with the tropical-originated C. palmeri, and ‘Lakota’ and ‘Sioux’ share a common female ancestor with C. tomentosa because of their overlapping and/or adjacent distribution (Figure 2B; Supplementary Figure 17). In addition, two C. pallida landraces ‘Wen 12998’ and ‘NY1’ collected from different locations of the distribution have close maternal phylogenetic relationships with C. ovata and C. ovalis, respectively. This pattern also supports the model of “the maternal phylogenetic relationship of intra-genus outcrossing species is determined by geographical distribution, and the geographically adjacent species shares a common maternal ancestor adjacent geographical distribution sharing common maternal ancestor” like the relationships among the cultivars in C. illinoinensis (Figure 4). Different varieties within the same species clustered in different phylogenetic positions may be caused by their multiple matrilineal origins, for reasons such as overlapping and/or adjacent distribution regions, the hybridizability of interspecies of Carya species, or diverse plastome structure features, or wind pollination (Wang et al., 2022). These results may partially account for the phylogenetic incongruencies between species in EA or NA subclades inferred from partial or complete plastome datasets or nuclear data. The results generated here indicate that further broader population-wide sampling and their plastome assemblies will be a powerful approach in tracking the matrilineal historical origin of a species, especially for outcrossing species.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, MW410235 MW410236 MW368388 ON568300 MW410227 MW255965 MW368387 MW410228 MW410229 MW410230 MW298527 MW410238 MW410237 MW410231 MW410232 MW440674 MW410233 MW410234 MW421595.
LX: Conceptualization. JX, SL, WZ and JW: Investigation, Resources. GX: Resources. JX and SL: Validation. HG, JH and YY: Data Curation. LX, JX, JZ and KW: Data Curation, Visualization. LX, JX and WZ: Writing Original Draft. LX, JX, WZ and XW: Writing, Review and Editing. LX, JZ: Supervision. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the Chinese Ministry of Science and Technology, China (grant no. 2018YFD1000604), the Natural Science Foundation of Zhejiang Province, China (grant no. Z20C160001), Research and Development Fund of Zhejiang A&F University (grant no. 2018FR002) and the State Key Laboratory of Subtropical Silviculture in Zhejiang A&F University, China (grants no. ZY20180202 and KF201905).
We are grateful to Dr. Zhiduan Chen at Institute of Botany, the Chinese Academy of Sciences, China, for his help and valuable suggestions during the manuscript preparation. We also thank to Drs. Dayong Zhang and Weining Bai at State Key Laboratory of Earth Surface Processes and Resource Ecology and Ministry of Education Key Laboratory for Biodiversity Science and Ecological Engineering, College of Life Sciences, Beijing Normal University, Beijing, China, for the sharing the resequencing data of C. poilanei.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.990064/full#supplementary-material
Amiryousefi, A., Hyvonen, J., Poczai, P. (2018). IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220
Chang, R. H., Lu, A. M. (1979). A study of the genus carya nutt. in china. J. Syst. Evol. 17, 40–44.
Chan, C. X., Ragan, M. A. (2013). Next-generation phylogenomics. Biol. Direct 8, 1–6. doi: 10.1186/1745-6150-8-3.
Chen, Y. X., Chen, Y. S., Shi, C. M., Huang, Z. B., Zhang, Y., Li, S. K., et al. (2018). SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Oxford Open 7, 1–6. doi: 10.1093/gigascience/gix120
Chevalier, A. (1941). Variabilité et Hybridité chez les Noyers. Notes sur des Juglans peu connus, sur l'Annamocarya et un Carya d'Indochine. J. D'agriculture Traditionnelle Et Botanique Appliquée 21, 477–509. doi: 10.3406/jatba.1941.1646
Clegg, M. T., Gaut, B. S., Learn, G. H., Morton, A. B. R. (1994). Rates and patterns of chloroplast DNA evolution. Proc. Natl. Acad. Sci. U. S. A. 91, 6795–6801. doi: 10.1073/pnas.91.15.6795
Dierckxsens, N., Mardulyn, P., Smits, G. (2016). NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45, e18. doi: 10.1093/nar/gkw955
Dong, W., Liu, Y., Li, E., Xu, C., Sun, J., Zhou, S., et al. (2022). Phylogenomics and biogeography of Catalpa (Bignoniaceae) reveal incomplete lineage sorting and three dispersal events. Mol. Phyloget. Evol. 166, 107330. doi: 10.1016/j.ympev.2021.107330
Dong, W., Xu, C., Li, C., Sun, J., Zuo, Y., Shi, S., et al. (2015). Ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 5, 8348. doi: 10.1038/srep08348
Dong, W., Xu, C., Li, W. Q., Xie, X. M., Lu, Y. Z., Liu, Y. L., et al. (2017). Phylogenetic resolution in juglans based on complete chloroplast genomes and nuclear DNA sequences. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.01148
Dugas, D. V., Hernandez, D., Koenen, E. J. M., Schwarz, E., Straub, S., Hughes, C. E., et al. (2015). Mimosoid legume plastome evolution: IR expansion, tandem repeat expansions, and accelerated rate of evolution in clpP. Sci. Rep. 5, 16958. doi: 10.1038/srep16958
Favre, F., Jourda, C., Besse, P., Charron, C. (2020). Genotyping-by-Sequencing technology in plant taxonomy and phylogeny. molecular plant taxonomy: Ile de la réunion. 2222, 167–178. doi: 10.1007/978-1-0716-0997-2_10
Feng, X. J., Yuan, X. Y., Sun, Y. W., Hu, Y. H., Saman, Z., Ouyang, X. H., et al. (2018). Resources for studies of iron walnut (Juglans sigillata) gene expression, genetic diversity, and evolution. Tree Genet. Genomes 14, 1–15. doi: 10.1007/s11295-018-1263-z
Folk, R. A., Mandel, J. R., Freudenstein, J. V. (2017). Ancestral gene flow and parallel organellar genome capture result in extreme phylogenomic discord in a lineage of angiosperms. Syst. Biol. 66, 320–337. doi: 10.1093/sysbio/syw083
Fredrik, R., Maxim, T., Pau, V. D. M., Daniel, L. A., Aaron, D. (2012). MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a Large model space. Systematic Biol. 61, 539–542. doi: 10.2307/41515220
Gao, Q. B., Yan, L., Zhuo-Ma, G., Gornall, R. J., Wang, J. L., Liu, H. R., et al. (2017). Population genetic differentiation and taxonomy of three closely related species of saxifraga (Saxifragaceae) from southern tibet and the hengduan mountains. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.01325
Givnish, T. J. (2016). Convergent evolution, adaptive radiation, and species diversification in plants. Encyclopedia Evolutionary Biol. 1, 362–373. doi: 10.1016/B978-0-12-800049-6.00266-3
Grauke, L. J. (2003). Nut tree culture in north America. Eds.Fulbright, D. (Northern Nut Growers Association, Inc).
Grauke, L. J., Klein, R., Grusak, M. A., Klein, P. (2015). The forest and the trees: Applications for molecular markers in the repository and pecan breeding program. Acta Hortic. 1070, 109–126. doi: 10.17660/ActaHortic.2015.1070.12
Grauke, L. J., Mendoza-Herrera, M. A. (2012). Population structure in the genus carya. Acta Hortic. 948, 143–158. doi: 10.17660/ActaHortic.2012.948.16
Grauke, L. J., Mendoza-Herrera, M. A., Miller, A. J., Wood, B. W. (2011). Geographic patterns of genetic variation in native pecans. Tree Genet. Genomes 7, 917–932. doi: 10.1007/s11295-011-0384-4
Grauke, L. J., Thompson, T. E. (1996). Variability in pecan flowering. Fruit Varieties J. 50, 140–150.
Grauke, L. J., Wood, B. W., Harris, M. K. (2016). Crop vulnerability: Carya. HortScience 51, 653–663. doi: 10.21273/HORTSCI.51.6.653
Grauke, L. J., Wood, B. W., Payne, J. A. (1991). “Genetic resources of carya,” in Vietnam And china. 82nd annual report of the northern nut growers association, vol. 82. , 80–87.
Hall, T. A. (1999). BioEdit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nuclc Acids Symposium Ser. 41, 95–98. doi: 10.1021/bk-1999-0734.ch008
Hedrick, P. W. (2013). Adaptive introgression in animals: examples and comparison to new mutation and standing variation as sources of adaptive variation. Mol. Ecol. 22 (18), 4606–4618. doi: 10.1111/mec.12415
Heiges, S. B. (1896). Nut culture in the united states: Embracing native and introduced species (USDA Pomology Div., Govt. Printing Office: Washington D.C).
Hester, T. R. (1983). Late paleo-Indian occupations at baker cave, southwestern texas. Bull. Tex. Arch. Soc. 53, 101–119.
Huang, Y. J., Xiao, L. H., Zhang, Z. R., Zhang, R., Wang, Z. J., Huang, C. Y., et al. (2019). The genomes of pecan and Chinese hickory provide insights into Carya evolution and nut nutrition. GigaScience 8, 1–17. doi: 10.1093/gigascience/giz036
Hu, H., Hu, Q., Al-Shehbaz, I. A., Luo, X., Zeng, T., Guo, X., et al. (2016 1826). Species delimitation and interspecific relationships of the genus Orychophragmus (Brassicaceae) inferred from whole chloroplast genomes. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.01826
Hu, Y. H., Woeste, K. E., Zhao, P. (2017 1955). Completion of the chloroplast genomes of five chinese juglans and their contribution to chloroplast phylogeny. Front. Plant 7. doi: 10.3389/fpls.2016.01955
Itaya, H., Oshita, K., Arakawa, K., Tomita, M. (2013). GEMBASSY: An EMBOSS associated software package for comprehensive genome analyses. Source Code Biology&Medicine 8, 17. doi: 10.1186/1751-0473-8-17
Jiao, Y., Jia, H. M., Li, X. W., Chai, M. L. (2012). Development of simple sequence repeat (SSR) markers from a genome survey of Chinese bayberry (Myrica rubra). BMC Genomics 13, 201. doi: 10.1186/1471-2164-13-201
Julio, R., Albert, F. M., Carlos, S. J., Sara, G. R., Pablo, L., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of Large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Kazutaka, K., Standley, M. D. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Khakhlova, O., Bock, R. (2006). Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 46, 85–94. doi: 10.1111/j.1365-313X.2006.02673.x
Kozlowski, G., Sébastien, B., Song, Y. G. (2018). Wingnuts (Pterocarya) & walnut family. relict trees: Linking the past, present and future (Natural History Museum Fribourg: Switzerland). Relict trees: linking the past, present and future.Natural History Museum Fribourg, Switzerland.
Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lee, S., Weon, S., Lee, S., Kang, C. (2010). Relative codon adaptation index, a sensitive measure of codon usage bias. Evolutionary Bioinf. Online 6, 47–55. doi: 10.4137/EBO.S4608
Lee-Yaw, J. A., Grassa, C. J., Joly, S., Andrew, R. L., Rieseberg, L. H. (2019). An evaluation of alternative explanations for widespread cytonuclear discordance in annual sunflowers (Helianthus). New Phytol. 221 (1), 515–526. doi: 10.1111/nph.15386
Lemmon, E. M., Lemmon, A. R. (2013). High-throughput genomic data in systematics and phylogenetics. Annu. Rev. Ecol. Evol. Systematics 44, 99–121. doi: 10.1111/nph.15386
Leroy, J. F. (1950). Note sur les noyers (Carya et annamocarya) sauvages d'Indochine. Rev. Internationale Botanique Appliquée Et D'agriculture Tropicale 30, 425–428. doi: 10.3406/jatba.1950.6726
Leroy, J. F. (1955). Etude sur les juglandaceae. Mem. Mus. Hist. Nat. (Paris)II B [Bot.] 6, 246. Available at: https://www.biodiversitylibrary.org/item/266009#page/1/mode/1up.
Li, Y. X., Li, Z. H., Schuitman, A., Chase, M. W., Jin, X. H. (2019). Phylogenomics of orchidaceae based on plastid and mitochondrial genomes. Mol. Phylogenet. Evol. 139, 106540. doi: 10.1016/j.ympev.2019.106540
Li, H., Liu, B., Davis, C. C., Yang, Y. (2020). Plastome phylogenomics, systematics, and divergence time estimation of the Beilschmiedia group (Lauraceae). Mol. Phylogenet. Evol. 151, 106901. doi: 10.1016/j.ympev.2020.106901
Liu, J., Lindstrom, A. J., Gong, X. (2022). Towards the plastome evolution and phylogeny of Cycas l. (Cycadaceae): molecular-morphology discordance and gene tree space analysis. Front. Plant Sci. 22, 116. doi: 10.1186/s12870-022-03491-2
Lovell, J. T., Bentley, N. B., Bhattarai, G., Jenkins, J. W., Sreedasyam, A., Alarcon, Y., et al. (2021) 4125. Four chromosome scale genomes and a pan-genome annotation to accelerate pecan tree breeding. Nat. Commun. 12, 4125. doi: 10.1038/s41467-021-24328-w
Lu, A. M., Stone, D. E., Grauke, L. J. (1999). Juglandaceae, in flora of china. China/Missouri botanical garden: Beijing.
Lu, Q. X., Ye, W. Q., Lu, R. S., Xu, W. Q., Qiu, Y. X. (2018). Phylogenomic and comparative analyses of complete plastomes of croomia and stemona (Stemonaceae). Int. J. Mol. Sci. 19, 2383. doi: 10.3390/ijms19082383
Luo, J., Chen, J., Guo, W., Yang, Z., Lim, K.-J., Wang, Z. (2021). Reassessment of Annamocarya sinesis (Carya sinensis) taxonomy through concatenation and coalescence phylogenetic analysis. Plants 11, 52. doi: 10.3390/plants11010052
Mader, M., Pakull, B., Blanc, J. C., Drewes, M. P., Zoéwindé, B., Bernd, D., et al. (2018). Complete chloroplast genome sequences of four meliaceae species and comparative analyses. Int. J. Mol. Sci. 19, 701. doi: 10.3390/ijms19030701
Mai, D. H. (1981). Der formenkreis der Vietnam-nuß (Carya poilanei (Chev.) Leroy) in Europa. Feddes Repert 92, 339–385. doi: 10.1002/fedr.4910920502
Manning, W. E. (1950). A key to the hickories north of Virginia with notes on the two pignuts Carya glabra and C. ovalis. Rhodora 52, 188–199. doi: 10.2307/23304073
Manning, E. W. (1963). Hickories reported in India and Laos with other notes on Carya in Asia. Brittonia 15, 123–125. doi: 10.2307/2805397
Manning, W. E. (1978). The classification within the juglandaceae. Ann. Missouri Botanical Garden 65, 1058–1087. doi: 10.2307/2398782
Manning, W. E., Hjelmqvist, H. (1951). Annamocarya rhampocarya and Carya sinensis. Bot. Notiser 4, 319–330.
Manos, P. S., Soltis, P. S., Soltis, D. E., Manchester, S. R. (2007). Phylogeny of extant and fossil juglandaceae inferred from the integration of molecular and morphological data sets. Systematic Biol. 56, 412–430. doi: 10.1080/10635150701408523
Manos, P. S., Stone, D. E. (2001). Evolution, phylogeny, and systematics of the juglandaceae. Ann. Missouri Botanical Garden 88, 231–269. doi: 10.2307/2666226
Meng, X. X., Xian, Y. F., Li, X., Dong, Z., Shi, Y. H., Wu, M. L., et al. (2018). Complete chloroplast genomes from sanguisorba: Identity and variation among four species. Molecules 23, 2137. doi: 10.3390/molecules23092137
Michael, T., Pascal, L., Tommaso, P., Ulbricht-Jones, E. S., Axel, F., Ralph, B., et al. (2017). GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. W1. doi: 10.1093/nar/gkx391
Mower, J. P. (2009). The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nuclc Acids Res. 37, W253–W259. doi: 10.1093/nar/gkp337
Mu, X. Y., Tong, L., Sun, M., Zhu, Y. X., Wen, J., Lin, Q. W., et al. (2020). Phylogeny and divergence time estimation of the walnut family (Juglandaceae) based on nuclear RAD-seq and chloroplast genome data. Mol. Phylogenet. Evol. 147, 106802. doi: 10.1016/j.ympev.2020.106802
Ogoma, C. A., Liu, J., Stull, G. W., Wambulwa, M. C., Oyebanji, O., Miline, R. I., et al. (2022). Deep insights into the plastome evolution and phylogenetic relationships of the tribe Urticeae (Family urticaceae). Front. Plant Sci. 13. doi: 10.3389/fpls.2022.870949
Oliver, J. C. (2013). Microevolutionary processes generate phylogenomic discordance at ancient divergences. Evolution 67 (6), 1823–1830. doi: 10.1111/evo.12047
Palmer, J. D., Thompson, W. F. (1982). Chloroplast DNA rearrangements are more frequent when a large inverted repeat sequence is lost. Cell 29, 537–550. doi: 10.1016/0092-8674(82)90170-2
Park, S., An, B., Park, S. J. (2018). Reconfiguration of the plastid genome in Lamprocapnos spectabilis: IR boundary shifting, inversion, and intraspecific variation. Sci. Rep. 8, 13568. doi: 10.1038/s41598-018-31938-w
Peng, S. B., Yang, G. Y., Liu, C. B., Yu, Z. D., Zhai, M. Z. (2017). The complete chloroplast genome of the Juglans regia (Juglandales: Julandaceae). Mitochondrial DNA 28, 407–408. doi: 10.3109/19401736.2015.1127367
Peter, R., Ian, L., Alan, B. (2000). EMBOSS: The european molecular biology open software suite. Trends Genet. 16, 276–277. doi: 10.1016/s0168-9525(00)02024-2
Pham, K. K., Hipp, A. L., Manos, P. S. (2017). And cronn, rA time and a place for everything: phylogentic history and geography as joint predictors of oak plastome phylogeny. C. Genome 60, 720–732. doi: 10.1139/gen-2016-0191
Raubeson, L. A., Jansen, R. K. (2005). “Chloroplast genomes of plants,” in Plant diversity and evolution: Genotypic and phenotypic variation in higher plants.
Raubeson, L. A., Peery, R., Chumley, T. W., Dziubek, C., Fourcade, H. M., Boore, J. L., et al. (2007). Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics 8, 174. doi: 10.1186/1471-2164-8-174
Raven, P. H. (1972). Plant species disjunctions: a summary. Ann. Mo. Bot. Gard. 59, 234–246. doi: 10.2307/2394756
Scott, R. A. (1953). Status of an asiatic member of the juglandaceae regarded as a 'Living fossil'. Am. J. Bot. 40, 666–669. doi: 10.1002/j.1537-2197.1953.tb06538.x
Sebastian, B., Thomas, T., Münch, T., Uwe, S., Martin, M. (2017). MISA-web: A web server for microsatellite prediction. Bioinf. (Oxford England) 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Song, F., Li, T., Burgess, K. S., Feng, Y., Ge, X.-J. (2020). Complete plastome sequencing resolves taxonomic relationships among species of Calligonum l. (Polygonaceae) in China. BMC Plant Biol. 20, 261. doi: 10.1186/s12870-020-02466-5
Stenz, N. W., Larget, B., Baum, D. A., An, C. C. (2015). Exploring tree-like and non-tree-like patterns using genome sequences: An example using the inbreeding plant species Arabidopsis thaliana (L.) heynh. Systematic Biol. 64, 809–823. doi: 10.1093/sysbio/syv039
Stephan, G., Pascal, L., Ralph, B. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nuclc Acids Res. 47, W59–W64. doi: 10.1093/nar/gkz238
Suh, A., Smeds, L., Ellegren, H. (2015). The dynamics of incomplete lineage sorting across the ancient adaptive radiation of neoavian birds. PloS Biol. 13, e1002224. doi: 10.1371/journal.pbio.1002224
Thach, R. E., Dewey, K. F., Brown, J. C., Doty, P. (1966). Formylmethionine codon AUG as an initiator of polypeptide synthesis. Science 153, 416–418. doi: 10.1126/science.153.3734.416
Thompson, T. E., Grauke, L. J., Reid, W. (2008). 'Lakota' pecan. Hortscience Publ. Am. Soc. Hortic. Sci. 43. doi: 10.21273/HORTSCI.43.1.250
Thompson, T. E., Grauke, L. J. (1991). Pecans and other hickories (Carya). Acta Hortic 290, 839–906. doi: 10.17660/ActaHortic.1991.290.19
Tiffney, B. H. (1985). The Eocene north Atlantic land bridge: its importance in tertiary and modern phytogeography of the northern hemisphere. J. Arnold Arboretum 66, 243–273.
Tu, X.-D., Liu, D.-K., Xu, S.-W., Zhou, C.-Y., Gao, X.-Y., Zeng, M.-Y., et al. (2021). Plastid phylogenomics improves resolution of phylogenetic relationship in the cheirostylis and Goodyera clades of Goodyerinae (Orchidoideae, rchidaceae). Mol. Phylogen. Evol. 164, 107269. doi: 10.1016/j.ympev.2021.107269
Wang, R. J., Cheng, C. L., Chang, C. C., Wu, C. L., Su, T. M., Chaw, S. M., et al. (2008). Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evolutionary Biol. 8, 36–51. doi: 10.1186/1471-2148-8-36
Wang, X.-H., Moore, M. J., Barrett, R. L., Landrein, S., Sakaguchi, S., Maki, M., et al. (2020). Plastome phylogenomic insights into the sino-Japanese biogeography of Diabelia (Caprifoliaceae). J. System. Evol. 58 (6), 972–987. doi: 10.1111/jse.12560
Wang, X. W., Rhein, H. S., Jenkins, J., Schmutz, J., Randall, J. J. (2020). Chloroplast genome sequences of Carya illinoinensis from two distinct geographic populations. Tree Genet. Genomes 16, 2–14. doi: 10.1007/s11295-020-01436-0
Wang, Y., Ruhsam, M., Milne, R., Graham, S. W., Li, J., Tao, T., et al. (2022). Incomplete lineage sorting and local extinction shaped the complex evolutionary history of the paleogene relict conifer genus, Chamaecyparis (Cupressaceae). Mol. Phylogen. Evol. 172, 107485. doi: 10.1016/j.ympev.2022.107485
Wang, X. M., Zhou, T., Bai, G. Q., Zhao, Y. M. (2018). Complete chloroplast genome sequence of Fagopyrum dibotrys: Genome features, comparative analysis and phylogenetic relationships. Sci. Rep-Uk 8, 12379. doi: 10.1038/s41598-018-30398-6
Wei, R., Yan, Y.-H., Harris, A. J., Kang, J.-S., Shen, H., Xiang, Q.-P., et al. (2017). Plastid phylogenomics resolve deep relationships among eupolypod II ferns with rapid radiation and rate heterogeneity. Genome Biol. Evol. 9 (6), 1646–1657. doi: 10.1093/gbe/evx1075
Wen, J. (1999). Evolution of eastern Asian and eastern north American disjunct distributions in flowering plants. Annu. Rev. Ecol. Syst. 30, 421–455. doi: 10.1146/annurev.ecolsys.30.1.421
Weng, M. L., Blazier, J. C., Madhumita, G., Jansen, R. K. (2014). Reconstruction of the ancestral plastid genome in geraniaceae reveals a correlation between genome rearrangements, repeats, and nucleotide substitution rates. Mol. Biol. Evol. 31, 645–659. doi: 10.1093/molbev/mst257
Xiao., L., Yu, M., Zhang, Y., Hu, J., Wang, J., Guo, H., et al. (2021). Chromosome-scale assembly reveals the asymmetric paleo-subgenomes evolution and targets for accelerating fungal resistance breeding in nut crop, pecan. Plant Commun. doi: 10.1016/j.xplc.2021.100247
Xu, C., Dong, W., Li, W., Lu, Y., Xie, X., Jin, X., et al (2017). Comparative analysis of six Lagerstroemia complete chloroplast genomes. Front. Plant Sci. 8, 15. doi: 10.3389/fpls.2017.00015
Yan, M., Fritsch, P. W., Moore, M. J., Feng, T., Meng, A., Yang, J., et al. (2018). Plastid phylogenomics resolves infrafamilial relationships of the styracaceae and sheds light on the backbone relationships of the ericales. Mol. Phylogenet. Evol. 121, 198–211.
Yang, Y.-Y., Qu, X.-J., Zhang, R., Stull, G. W., Yi, T.-S. (2021). Plastid phylogenomic analyses of fagales reveal signatures of conflict and ancient chloroplast capture. Mol. Phylogenet. Evol. 163, 107232. doi: 10.1016/j.ympev.2021.107232
Yang, X. Y., Wang, Z. F., Luo, W. C., Guo, X. Y., Zhang, C. H., Liu, J. Q., et al. (2019). Plastomes of betulaceae and phylogenetic implications. J. Systematics Evol. 57, 508–518. doi: 10.1111/jse.12479
Yang, A. H., Zhang, J. J., Yao, X. H., Huang, H. W. (2011). Chloroplast microsatellite markers in Liriodendron tulipifera (Magnoliaceae) and cross-species amplification in L. chinense. Am. J. Bot. 98, e123–e126. doi: 10.3732/ajb.1000532
Yang, Z., Zhao, T. T., Ma, Q. H., Liang, L. S., Wang, G. X. (2018). Comparative genomics and phylogenetic analysis revealed the chloroplast genome variation and interspecific relationships of corylus (Betulaceae) species. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00927
Yao, G., Jin, J. J., Li, H. T., Yang, J. B., Shiva, M. V., Croley, M., et al. (2019). Plastid phylogenomic insights into the evolution of caryophyllales. Mol. Phylogenet. Evol. 134, 74–86. doi: 10.1016/j.ympev.2018.12.023
Ye, L. J., Fu, C. N., Wang, Y. H., Liu, J., Gao, L. M. (2018). Characterization of the complete plastid genome of a Chinese endemic species Carya kweichowensis. Mitochondrial DNA Part B 3, 492–493. doi: 10.1080/23802359.2018.1464414
Zhai, D. C., Yao, Q., Cao, X. F., Hao, Q. Q., Bai, X. H. (2019). Complete chloroplast genome of the wild-type hickory Carya cathayensis. Mitochondrial DNA Part B 4, 1457–1458. doi: 10.1080/23802359.2019.1598815
Zhang, W. P., Bai, W. N., Zhang, D. Y. (2022). The rediscovery of Carya poilanei (Juglandaceae) after 63 years reveals a new record from China. PhytoKeys 188, 73–82. doi: 10.3897/phytokeys.188.77242
Zhang, J. B., Li, R. Q., Xiang, X. G., Manchester, S. R., Li, L., Wang, W., et al. (2013). Integrated fossil and molecular data reveal the biogeographic diversification of the eastern Asian-Eastern north american disjunct hickory genus (Carya nutt.). PloS One 8, e70449. doi: 10.1371/journal.pone.0070449
Zhang, Q. Y., Ree, R. H., Salamin, N., Xing, Y. W., Silvestro, D. (2021). Fossil-informed models reveal a boreotropical origin and divergent evolutionary trajectories in the walnut family (Juglandaceae). Systematic Biol. 0, 1–17. doi: 10.1093/sysbio/syab030
Zhang, B. W., Xu, L. L., Li, N., Yan, P. C., Jiang, X. H., Woeste, K. E., et al. (2019). Phylogenomics reveals an ancient hybrid origin of the persian walnut. Mol. Biol. Evol. doi: 10.1093/molbev/msz112
Zhao, P., Zhou, H. J., Potter, D., Hu, Y. H., Feng, X. J., Dang, M., et al. (2018). Population genetics, phylogenomics and hybrid speciation of juglans in China determined from whole chloroplast genomes, transcriptomes, and genotyping-by-sequencing (GBS). Mol. Phylogenet. Evol. 126, 250–265. doi: 10.1016/j.ympev.2018.04.014
Zhou, H., Hu, Y., Ebrahimi, A., Liu, P., Zhao, P. (2021). Whole genome based insights into the phylogeny and evolution of the juglandaceae. BMC Ecol. Evol. 21, 191. doi: 10.21203/rs.3.rs-495294/v1
Zhou, B.-F., Yuan, S., Crowl, A. A., Liang, Y.-Y., Shi, Y.-Y., Chen, X.-Y., et al. (2022 1320). Phylogenomic analyses highlight innovation and introgression in the continental radiations of fagaceae across the northern hemisphere. Nat. Commun. 13, 1320. doi: 10.1038/s41467-022-28917-1
Keywords: hickory, pecan, nut crop, EA-NA disjunction, structure diversity, plastome phylogeny
Citation: Xi J, Lv S, Zhang W, Zhang J, Wang K, Guo H, Hu J, Yang Y, Wang J, Xia G, Fan G, Wang X and Xiao L (2022) Comparative plastomes of Carya species provide new insights into the plastomes evolution and maternal phylogeny of the genus. Front. Plant Sci. 13:990064. doi: 10.3389/fpls.2022.990064
Received: 09 July 2022; Accepted: 21 September 2022;
Published: 13 October 2022.
Edited by:
Wenpan Dong, Beijing Forestry University, ChinaReviewed by:
Jianqiang Zhang, Shaanxi Normal University, ChinaCopyright © 2022 Xi, Lv, Zhang, Zhang, Wang, Guo, Hu, Yang, Wang, Xia, Fan, Wang and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lihong Xiao, eGlhb2xoQHphZnUuZWR1LmNu; Jingbo Zhang, anpoYW5nNUB2Y3UuZWR1
†These authors have contributed equally to this work and share the first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.