 Jiao Zhang
Jiao Zhang Xiulian Chi
Xiulian Chi Juying Zhong1
Juying Zhong1 Alisdair Fernie
Alisdair Fernie Saleh Alseekh
Saleh Alseekh Luqi Huang
Luqi Huang Dan Qian
Dan Qian- 1Beijing Key Laboratory of Research of Chinese Medicine on Prevention and Treatment for Major Diseases, Experimental Research Center, China Academy of Chinese Medical Sciences, Beijing, China
- 2State Key Laboratory Breeding Base of Dao-di Herbs, National Resource Center for Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing, China
- 3Max Planck Institute of Molecular Plant Physiology, Potsdam, Germany
The internal transcribed spacer (ITS) is one of the most extensively sequenced molecular markers in plant systematics due to its generally concerted evolution. While non-concerted evolution has been found in some plant taxa, such information is missing in Lycium. Molecular studies of six species and two variants of the genus Lycium revealed high levels of intra- and inter-individual polymorphism in the ITS, indicating non-concerted evolution. All genomic DNA ITS paralogues were identified as putative pseudogenes or functional paralogues through a series of comparisons of sequence features, including length and substitution variation, GC content, secondary structure stability, and the presence of conserved motifs in the 5.8S gene, and the rate of evolution. Approximately, 60% of ITS pseudogenes could be easily detected. Based on phylogenetic analysis, all pseudogenes were highly distinct from their corresponding functional copies, tended to evolve neutrally, and clustered randomly together in the evolutionary tree. The results probably suggest that this ITS non-concerted evolution is related to the recent divergence between tandem repeats within the Lycium genome and hybridization between species. Our study complements those of pseudogenes in plant taxa and provides a theoretical basis for the phylogeny and genetic origin of the genus Lycium while having important implications for the use of ITS molecular markers for phylogenetic reconstruction.
Introduction
Concerted evolution is a form of multigene family evolution in which the genes in a gene family or cluster are assumed to evolve in concert as a unit (Elder and Turner, 1995; Liao, 1999). The best-known example of concerted evolution is among multicopy nuclear ribosomal DNA (nrDNA) genes. In higher plants, nrDNA, a multigene family consisting of a number of highly repetitive sequences, is often found in tens of thousands of copies that are repeated in tandem within the genome and distributed on one or more pairs of chromosomes (Eickbush and Eickbush, 2007). Each repeat unit consists of three rRNAs (18S, 5.8S, and 26S), and the genes are separated from each other by internal transcribed spacers (ITS1 and ITS2) (Tian and Li, 2002; Álvarez and Wendel, 2003; Li et al., 2011). The ITS regions (ITS1, 5.8S, and ITS2), which are part of nrDNA, are among the most widely sequenced molecular markers in plant systematics and DNA barcoding studies because of their rapid concerted evolution within and between constituent subunits, fast evolution, and short length and the availability of universal primers (Álvarez and Wendel, 2003; Li et al., 2011; Schoch et al., 2012). The ITS region generally undergoes rapid concerted evolution via unequal crossing over, high-frequency gene conversion, and large deletions (Ganley and Kobayashi, 2007, 2011). Hence, intra-individual polymorphism has generally been considered an exception. However, with the development of sequencing technology, some studies have revealed intra-individual ITS polymorphisms in a range of taxa (Muir et al., 2001; Razafimandimbison et al., 2004; Wei and Wang, 2004; Harpke and Peterson, 2006, 2008a; Zheng et al., 2008; Xiao et al., 2010; Camila De Sousa et al., 2011; Zhou et al., 2013; Xu et al., 2015, 2017; Huang et al., 2016; Asanuma et al., 2019; Li et al., 2019), including non-hybrid diploids. Some ITS copies may even become pseudogenes due to functional degradation.
Pseudogenes are disabled copies of protein-coding genes, often referred to as genomic fossils (Balasubramanian et al., 2009; Sisu et al., 2014). However, in the context of phylogenetic reconstruction, pseudogenes (nrDNA or protein coding) can be defined as sequences whose nucleotide divergence pattern has, irrespective of their expression patterns, not been constrained by function (Bailey, 2003). With the intense interest of scholars in ITS pseudogenes, several methods for identifying ITS pseudogenes have been established, including methods considering 5.8S insertions and variants, lower secondary structure stability and GC content, higher methylation-related mutations throughout the ITS region, higher sequence diversity, and tree-based approaches (Bailey, 2003). On the one hand, pseudogenes are assumed to be free from selection pressure and functional constraints, allowing them to accumulate many mutations, and randomly aggregate in phylogenetic trees because of long-branch attraction (LBA), which prevents an accurate understanding of phylogenetic relationships among taxa and confounds attempts to recover correct phylogenetic species relationships (Kita and Ito, 2000; Mayol and Rosselló, 2001). On the other hand, ITS pseudogenes may be useful for phylogenetic analyses of closely related species when functional copies provide too little variation. ITS pseudogenes have been well obtained and studied in some plant taxa. In a study of Naucleeae (Razafimandimbison et al., 2004), only a small number of functional ITS sequences were identified, but a large number of ITS pseudogenes were found. These ITS pseudogenes have been used for phylogenetic reconstruction owing to their recent origin. Interestingly, the ITS pseudogenes found in Corymbia (Ochieng et al., 2007) recovered a better phylogeny than did functional ITS sequences. Furthermore, in research on Cycas (Xiao et al., 2010), ITS pseudogenes were found to be even more suitable than sister taxa for use as outgroups. It is undoubtedly important to identify ITS pseudogenes and evaluate their utility in phylogenetic reconstruction, but studies on ITS pseudogenes have been limited to only a few species, with considering more attention needed. Consequently, analysis of more species and adequate sampling of ITS copies will shed light on the mechanisms of concerted evolution, at the same time, may provide new genetic evidence for phylogenetic relationships and speciation.
Goji berry (also known as wolfberry) is an important and widely used source of edible and medicinal fruit in China. Recently, Goji berries, a so-called superfood, have become increasingly popular in Europe and the United States because of their health benefits. Goji berry comes from the fruits of the genus Lycium, a genus of both medicinal and economical importance in the Solanaceae family consisting of ~80 species worldwide, including the Mediterranean region and across Asia and Australia. It is widely cultivated in northwestern China as an economic plant. Due to the potential use of Goji berry as a specialty crop, production practices are under development in parts of southern Europe, such as Italy and Greece. There are seven species and three variants of Lycium in China, where wild populations are widely distributed. Compared with wild species, Goji cultivars were mainly derived from Lycium barbarum and are distributed in northwestern China. Several previous studies revealed that ITS sequences can be effectively used for species identification and genetic relationship studies of Lycium (Shi et al., 2008; Li et al., 2014). However, when amplifying the entire ITS, it was hard to obtain amplification and sequencing products (Xin et al., 2013). Subsequently, a study also found inaccessible or nested peaks in PCR product sequencing of Lycium and presumed the existence of ITS polymorphisms and pseudogenes in this genus (Chen et al., 2017). Although the popularity of wolfberry, Lycium, is attracting increasing attention, a gap remains in the systematic study of ITS sequences in the genus.
Based on our previous findings of polymorphisms in cultivated Lycium (Zhang et al., 2022), we performed sufficient sampling and systematically analyzed the nrDNA ITS sequences (ITS1, 5.8S, and ITS2) copied from the genus Lycium in this work. The objectives of this study were to reveal the level and pattern of intra- and inter-species ITS nrDNA variation, to verify the existence of nrDNA ITS pseudogenes and non-concerted evolution in Lycium, and to estimate the pseudogene effects on phylogenetic reconstruction. To the best of our knowledge, this is the first systematic study of incomplete ITS concerted evolution and pseudogenes in the genus Lycium.
Materials and methods
Plant material
In this study, 329 individuals of six species and two variants from Lycium were sampled. All samples were collected from fresh leaves in the field. The samples used for direct sequencing and genomic DNA cloning sequencing (Lycium dasystemum, Lycium truncatum, Lycium ruthenicum, L. barbarum var. auranticarpum, and Lycium chinense) were dried with discolored silica gel and stored in room temperature, and those for cDNA cloning sequencing (L. barbarum, L. chinense var. potaninii and Lycium amarum) were treated with liquid nitrogen and stored at −80°C (Supplementary Table S1).
Nucleic acid isolation, PCR and RT–PCR, cloning, and sequencing
Genomic DNA was extracted from gel-dried leaves following the instruction of the Plant Genomic DNA Kit (TIANGEN, Beijing). RNA was extracted from fresh leaves using the RNA Easy Fast Plant Tissue Kit (TIANGEN, Beijing) and then treated with DNaseI to exclude DNA contaminants before first-strand cDNA synthesis was performed using the Fastking RT Kit (with gDNase). PCR amplification was carried out with two primer pairs, ITS1 (5'-TCCGTAGGTGAACCTGCGG3') and ITS4 (5'-TCCTCCGCTTATTGATAGC-3') (White et al., 1990), and P1 (5'-AACCTGCGGAAGGATCATTGTC-3') and P2 (5'-TGATATGCTTAAACTCAGCGGGTA-3') (Shi et al., 2008). PCR final reactions were performed in a 50 μl volume containing 20–50 ng of DNA template, 25 μl of 2 × SuperMix buffer, and each primer at 0.2 μM. The PCR program settings were as follows: 94°C for 5 min, 40 cycles of 94°C for 30 s, 56°C for 30 s, and 72°C for 45 s, and a final extension step of 72°C for 10 min. PCR and RT–PCR products were detected by electrophoresis in 1.0% agarose gel and then purified with a Thermo Scientific Gene JET Gel Extraction Kit. We cloned PCR products by using a Lethal Based Fast Cloning Kit. Eleven genomic clones per individual were obtained/sequenced, and 11 cDNA clones from L. chinense var. potaninii and L. amarum were sequenced with two primer pairs on an automatic DNA sequencer (ABI 3730XL, Thermo Fisher Scientific, USA) through SinoGenoMax. The PCR products were also directly sequenced.
Data analyses
All the sequences were aligned with SeqMan (Clewley, 1995) (DNAStar, Madison, WI) and corrected manually. The boundaries of the ITS1, 5.8S, and ITS2 regions were determined based on the available ITS sequences of Lycium in GenBank.
To evaluate the presence of ITS pseudogenes in Lycium, the following analyses were carried out: (i) ITS sequence length, GC content, and NJ unrooted evolutionary trees were determined using MEGA 7.0 (Kumar et al., 2016). (ii) Three conserved patterns unique to seed plants were detected for 5.8S (motif 1: CGATGAAGACGTAGC; motif 2: GAATTGCAGAATCC; motif 3: TTTGAACGCA) (Harpke and Peterson, 2008b). (iii) Secondary structure prediction and estimation of the minimum free energy (ΔG at 37°C) were performed by using the web program Mfold (Zuker, 2003).
Genetic diversity information was evaluated based on the mean nucleotide difference number (K) and nucleotide diversity (π) of functional sequences and pseudogenes using DnaSP (Librado and Rozas, 2009), and a neutrality test (Fu and Li's D) was performed to further validate the putative pseudogenes and functional genes that we identified.
Tandem Repeats Finder (Benson, 1999) was used to detect repeats in ITS sequences. Differences in substitution rates can distinguish functional genes from pseudogenes (Buckler and Holtsford, 1996). The distribution and pattern of nucleotide substitutions in all sequences obtained with both primer pairs were studied using HYPERMUT (Rose and Korber, 2000) with cDNA clone CHB1-1 and putative functional copy LC17-1 as reference sequences, respectively.
Results
The evolutionary pattern of nrDNA (including ITS) is usually concerted evolution, with few variations between copies, which provides a basis for direct sequencing of PCR products of ITS sequences. Therefore, we directly sequenced 311 individuals using the universal primers ITS1/ITS4, only 76 sequences succeeded in sequencing, and most samples failed to sequence or showed nested peaks. Although we have tried some approaches (optimizing PCR parameters, adding different concentrations of PCR synergist DMSO), we have still not been able to solve the problem in PCR directly sequenced. To improve the phenomenon, we selected 11 samples for cloning analysis (Supplementary Table S2). To get functional genes for pseudogene-aligned comparative analysis, we performed cDNA cloning and sequencing for samples that met the RNA extraction requirements (L. chinense var. potaninii and L. amarum).
A total of 309 ITS sequences were obtained from 329 samples of Lycium. After removing the repeat sequences, the total number of sequences was reduced to 268. Two pairs of PCR primers (ITS1/ITS4 and P1/P2) were used to amplify the ITS region. A total of 189 sequences were amplified with ITS1/ITS4, including 76 from direct sequencing, 82 from DNA cloning, and 31 from cDNA cloning. A total of 79 sequences were amplified with P1/P2, including 26 from DNA cloning and 53 from cDNA cloning. Five of these sequences (LC25-9-1, LC25-9-4, LC25-9-6, LC25-9-8, and CDN1-2) were excluded from the subsequent data analysis because of more than 100 bp deletion.
ITS pseudogenes were identified by nucleotide variations combined with sequence length, GC content, 5.8S conserved motifs, secondary structure stability and minimum free energy, evolutionary patterns, and phylogeny. We totally identified 154 putative pseudogene sequences out of 268 sequences, of which primer pair ITS1/ITS4 obtained 151 pseudogene sequences, while primer pair P1/P2 obtained only 3 pseudogene sequences. Meanwhile, all 76 sequences directly sequenced by PCR products were identified as putative pseudogene sequences.
Length and repeats of the ITS region in Lycium
All ITS sequences in six species and two variants were obtained by performing direct sequencing, genomic DNA cloning, and cDNA cloning experiments. After amplification with the primer pair ITS1/ITS4, the length of the entire ITS region varied from 532 to 691 bp, with an alignment of 729 bp long. For the whole ITS, the 5.8S gene was relatively conserved, with amplicon lengths ranging from 150 to 169 bp. In contrast, the ITS1 and ITS2 regions displayed length variations from 168 to 260 bp and 185 to 294 bp, respectively. Among them, three samples in the ITS1 region (LBVA1-2, LBVA1-6, and LC17-1-12) were significantly shorter than the others, with a length of only 170 bp. In addition, the length of functional ITS cDNA paralogues showed similar variation and ranged from 119 to 286 bp, 149 to 172 bp, and 183 to 275 bp for ITS1, 5.8S, and ITS2, respectively. The average lengths of the ITS regions varied greatly among the species, with L. amarum being the only exception, showing a relatively uniform length of 540 bp.
The amplicon lengths of ITS, ITS1, 5.8S, and ITS2 ranged from 540 to 698 bp, 190 to 268 bp, 154 to 179 bp, and 250 to 272 bp, respectively, when using the primer pair P1/P2. In contrast to the amplification sequences obtained with the primer pair ITS1/ITS4, the 5.8S gene and ITS2 region were conserved in terms of length, and the length of functional ITS cDNA paralogues showed similar variants, including in L. amarum.
Using Tandem Repeats Finder, 14 and nine putative repeats were detected among the ITS sequences with the primer pairs ITS1/ITS4 and P1/P2, respectively. For the primer pair ITS1/ITS4, most of the repeats were located in the 5.8S region, while the L. amarum repeats were located in the ITS2 region. Compared to those for the primer pair ITS1/ITS4, most of the repeats detected with the primer pair P1/P2 were located in the ITS1 or ITS2 region, and repeats in L. amarum were located in the 5.8S region (Table 1).
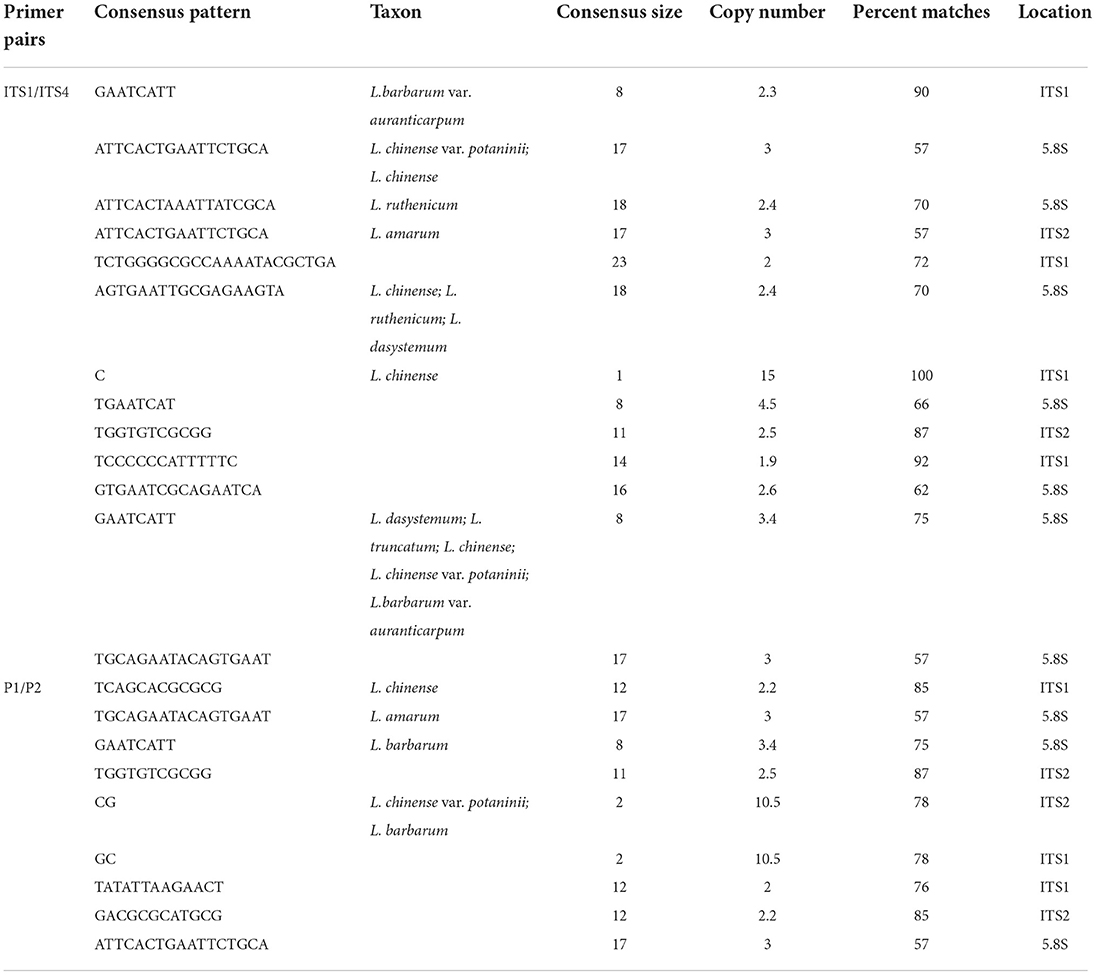
Table 1. Tandem repeats found among ITS sequences in Lycium.
For both primer pairs, the number of repeated sequences was significantly higher in wild samples than in cultivated samples. The percentages of repeated sequences in cultivated and wild samples were 44.4% and 46% for P1/P2 and 82.2% and 92.4% for ITS1/ITS4, respectively. In addition, approximately, 55.6% of the sequences obtained with primer pair P1/P2 contained two repeats, while those obtained with primer pair ITS1/ITS4 mostly had one repeat, approximately 52.3% (Supplementary Table S3).
GC content of the ITS region
Distinct clusters of clones emerged when plotting the GC contents of the ITS sequences of the two primer pairs against each other (Figures 1A,B). In most instances, the GC content correlated well between the two sets of ITS sequences. The putative functional ITS paralogues possessed high GC values in the spacers (59.8 and 59.0% in ITS1, 60.8 and 60.4% in ITS2). The GC values for 5.8S were markedly lower (53.1 and 50.9%). The average GC contents of ITS1, 5.8S, and ITS2 putative pseudogenes were 47.9% and 43.4, 46.1, and 47.1%, and 50.8% and 52.6%, respectively, approximately 2–12% lower than those of putative functional paralogues (Table 2). However, the GC contents of the functional ITS cDNA paralogues were similar to those of the putative pseudogenes, and all were lower than those of the putative functional sequences (54.2 or 54.9% for ITS1, 49.4 or 47.9% for 5.8S, and 55.8 or 56.4% for ITS2).
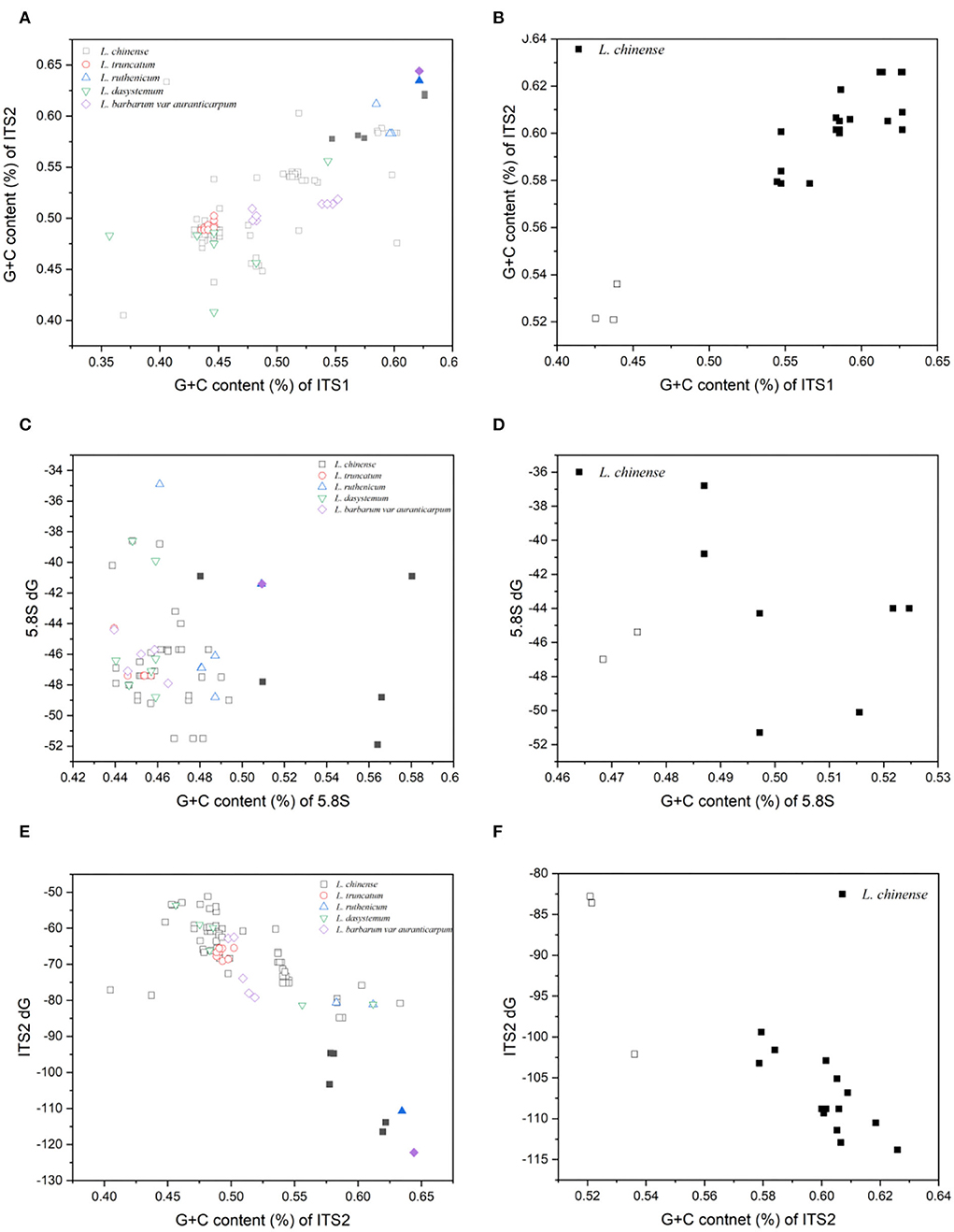
Figure 1. Plot of the G + C content of all paralogues obtained from five Lycium species. (A,B) ITS1 vs. ITS2. (C,D) 5.8S vs. the minimum free energy (ΔG at 37°C) of the secondary structure of 5.8S. (E,F) ITS2 vs. the minimum free energy (ΔG at 37°C) of the secondary structure of ITS2. The left panel shows the results for primer pair ITS1/ITS4, and the right panel shows those for primer pair P1/P2. Shaded symbols indicate putative functional sequences and blank symbols indicate putative pseudogenes.
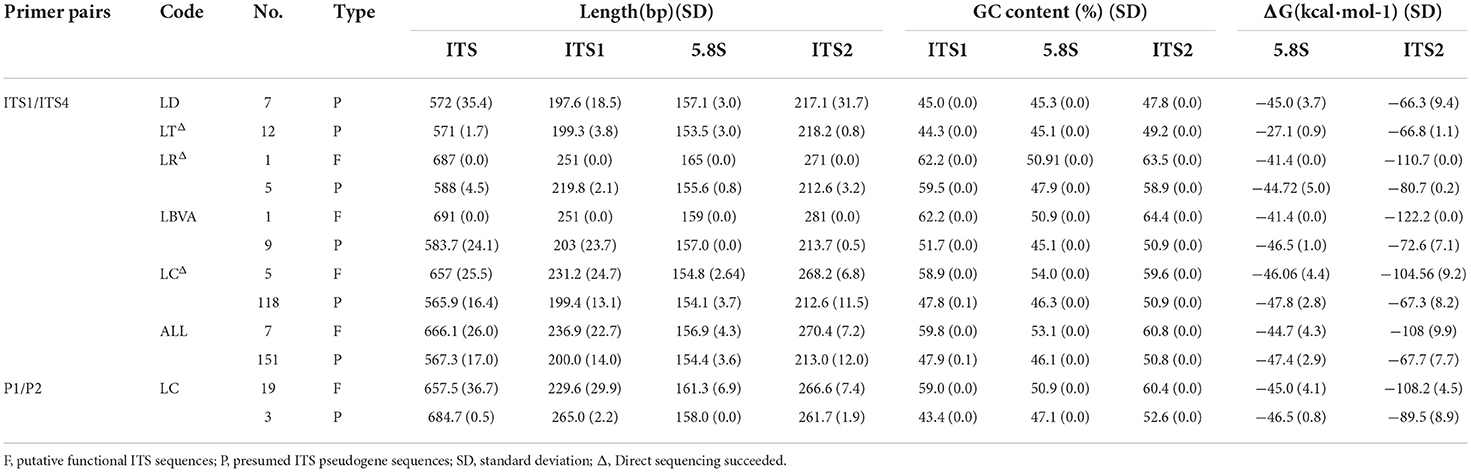
Table 2. Average lengths and GC contents of ITS regions and free energy of 5.8S secondary structures of five species of Lycium.
Secondary structure minimum free energy of 5.8S and ITS2 rDNA
The minimum free energy (ΔG at 37°C) of the 5.8S and ITS2 secondary structures was predicted for all sequences. The minimum free energies for 5.8S and ITS2 of the primer pair ITS1/ITS4 were −54.6 to −34.6 kcal/mol and −122.2 to −35.5 kcal/mol, respectively. The primer pair P1/P2 yielded structures with free energies of −51.3 to −29.2 kcal/mol and −113.8 to −40.7 kcal/mol, respectively (Table 2).
Plotting the minimum free energy values of 5.8S and ITS2 vs. their GC contents resulted in similar distributions between the compared to the GC values in the spacers (Figures 1C–F). In the ITS2 region, the average value for presumed functional paralogues was significantly lower than that for the putative pseudogenes (−108 or −108.2 kcal/mol for functional copies vs. −67.7 or −89.5 kcal/mol for pseudogenes). Consistent with the GC content, the presumed functional copies showed far less variation (Table 2, Figures 1A,B).
Nucleotide diversity and neutrality test
Sequence diversity was assessed by the average number of nucleotide differences (K) and nucleotide diversity (π), revealing that the sequence diversity of all putative pseudogenes was significantly higher than that of the putative functional sequences. In particular, the average number of nucleotide differences and nucleotide diversity of pseudogenes in the ITS1 region were 1.09 and 1.34 times higher, respectively, than those of functional genes, indicating that the ITS1 region accumulated more variants (Table 3).
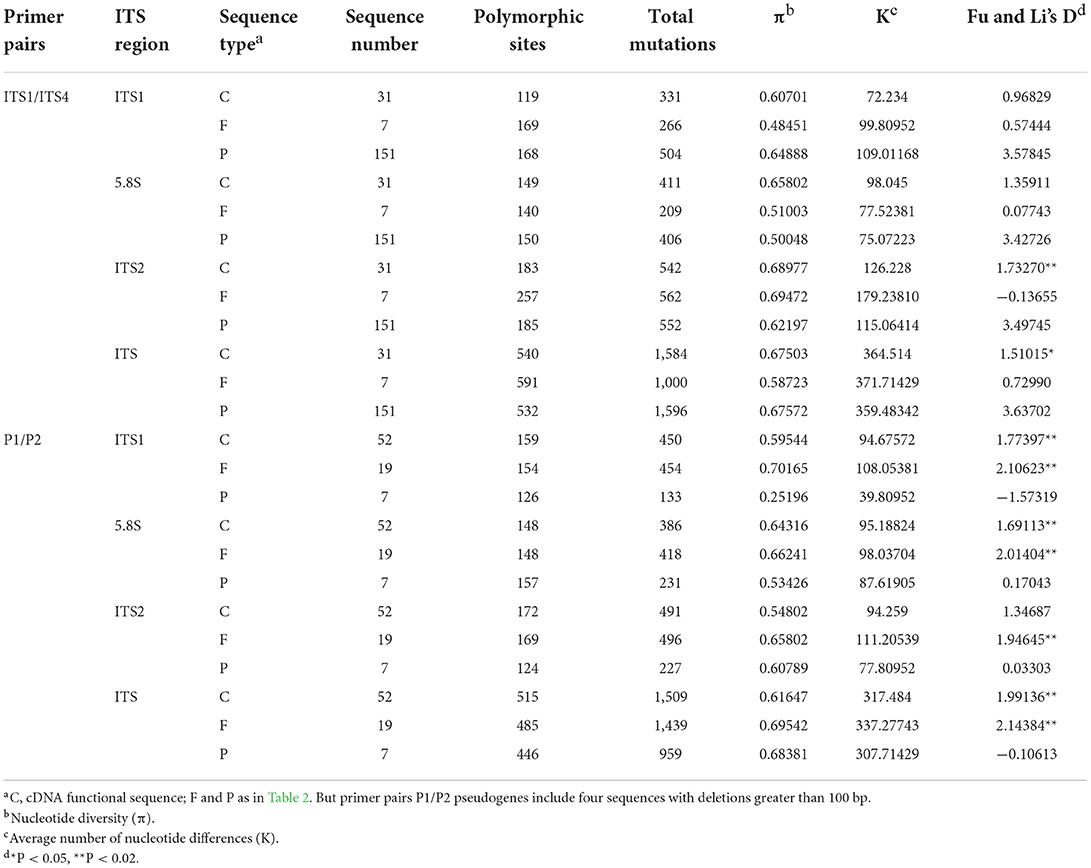
Table 3. Nucleotide diversity of individual parts and the entire ITS region in Lycium.
The nucleotide diversity and an average number of nucleotide differences of pseudogene sequences differed among the three regions. In terms of nucleotide diversity, the three regions ranked as ITS2>ITS1>5.8S, while in terms of an average number of nucleotide differences, they ranked ITS1>ITS2>5.8S.
Interestingly, when using the primer pair P1/P2 to verify the results against a widely distributed species (L. chinense), we found that the orders of the three regions in terms of π and K values (ITS1>5.8S>ITS1 and 5.8S>ITS2>ITS1) were significantly different from those of the primer pair ITS1/ITS4.
Evolutionary neutrality was assessed using Fu and Li's D test on the total number of segregating sites. Although, the values of putative functional sequences obtained with ITS1/ITS4 did not show significant deviations because the presumed functional sequences accounted for only 4.6% of the pseudogene sequences. Both the functional cDNA sequences and the assumed functional sequences from primer pair P1/P2 displayed marked deviations (P < 0.02), indicating a deviation from neutral evolution.
Three conserved motifs in 5.8S rDNA
In this study, motif 3 was the most conserved ITS sequence, with 91% of the sequences containing this motif. Nineteen sequences (six obtained with the ITS1/ITS4 primers and 13 with the P1/P2 primers) had at least one of the three conserved motifs of 5.8S and were therefore identified as putative functional sequences. A total of 161 sequences whose 5.8S did not contain three conserved motifs or contained at least one conserved motif with a base variation were determined to be putative pseudogenes. The base variants of the three conserved motifs of the pseudogenes showed some regularity, with the combination of T → C at position 13 of motif 1 and C → T at position 13 of motif 2 being the major variant, accounting for 83% of the overall variants.
However, only six of the 83 functional cDNA sequences contained three conserved motifs, and most of the sequences (approximately 92.8%) did not contain three conserved motifs or had base mutations, which were mainly G → A at position 12 of motif 1 and A → G at position 3 of motif 2.
Nucleotide substitution patterns
The nucleotide variation profiles display the physical location of nucleotide substitutions in all putative pseudogenes from primer pair ITS1/ITS4 and all functional cDNAs and putative functional genomic paralogues from primer pair P1/P2 (Figures 2A,B).
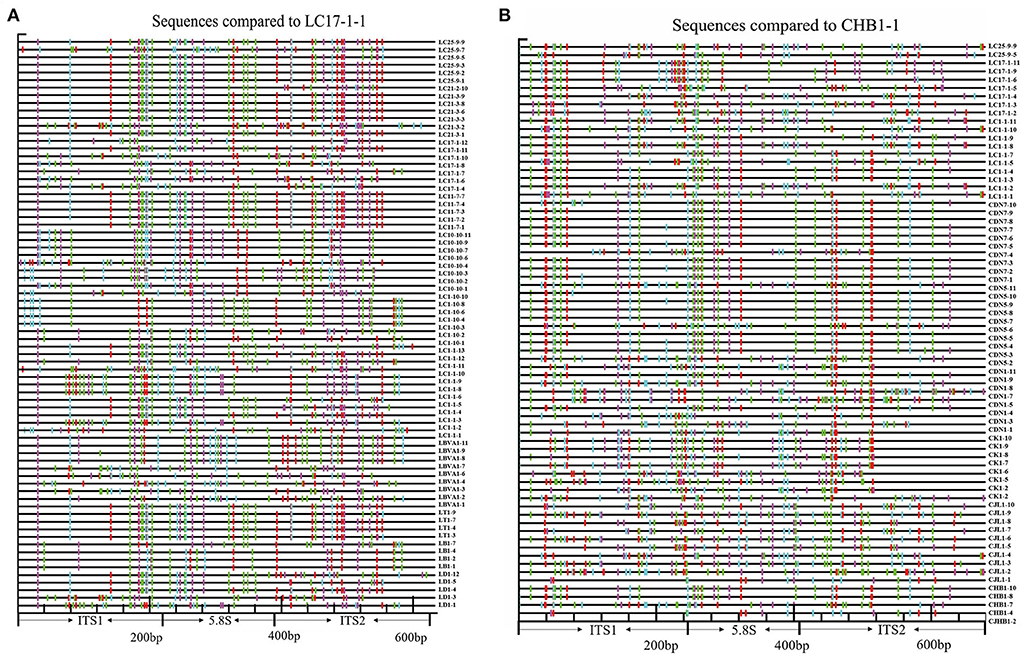
Figure 2. Schematic illustration of the distribution of substitution sites across the entire ITS region. (A) Assumed pseudogenes (excluding sequences obtained via direct sequencing) were yielded by primers ITS1-ITS4 using the putative functional sequence from LC17-1-1 as a reference. (B) Putative functional copies and cDNA paralogues from primers P1-P2 using CHB1-1 as a reference (red = GG > AG, cyan = GA > AA, green = GC > AC, magenta = GT > AT, black = not a G > A transition, yellow = gap) (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article).
Compared to the putative functional gene LC17-1-1, all putative pseudogenes obtained with the primer pair ITS1/ITS4 accumulated more substitutions (Figure 2A), especially the pseudogene sequences obtained by direct sequencing (Supplementary Figure S1), which were randomly distributed throughout the ITS region. In contrast, compared with the functional cDNA ITS paralogue CHB1-1, all functional cDNA and presumed functional genomic sequences obtained with P1/P2 had fewer substitutions, particularly the cDNA of the L. barbarum cultivar (Figure 2B). In addition, ITS sequences from the same population showed similar substitutions.
Phylogenetic tree
In this study, two different phylogenetic trees (A and B) were obtained by phylogenetic analysis of sequences obtained with the two primer pairs (Figures 3A,B). Tree A is the neighbor-joining (NJ) tree constructed for all sequences from primer pair ITS1/ITS4 (Figure 3A). In the NJ tree, all functional cDNA and putative functional genomic ITS copies are grouped together in a monophyletic clade with high bootstrap support (NJ bootstrap support (NJBS)=75%), with the exception of four cDNAs (CHB1-9, CHB1-11, CJL1-10, and CK1-1). The pseudogenes were found to be polyphyletic and randomly clustered into three major clades in the NJ tree. Clade I included all LC7 and LC16 sequences obtained with direct sequencing, as well as LC10 sequences obtained with direct sequencing and ITS genomic DNA copies. Clade II contained all LR, LBVA, LC1, and LC17 ITS sequences obtained with DNA sequencing, but the LC of other regions and LT and LD ITS sequences were mixed together in clade III.
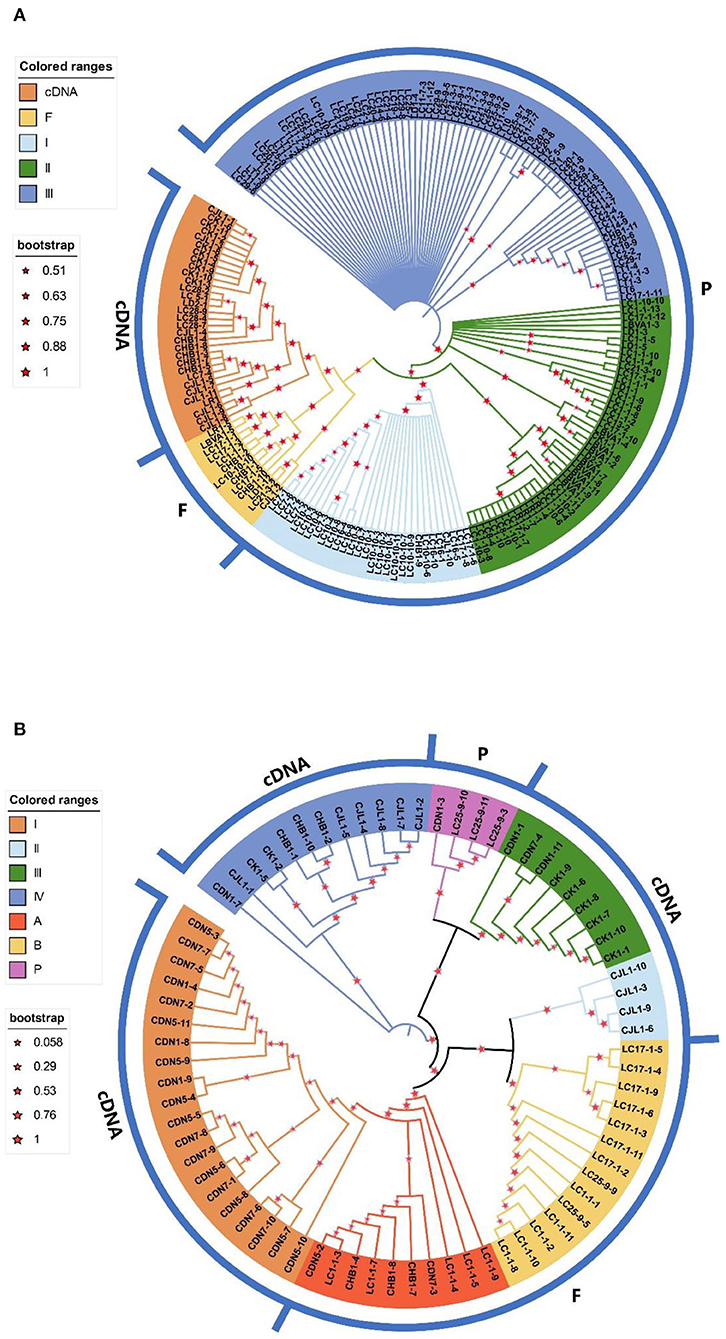
Figure 3. Neighbor-joining trees constructed by using the P-distance matrix from the entire ITS region of all paralogues (1,000 bootstraps). (A) The phylogenetic tree constructed using all sequences from the primer pair ITS1/ITS4. (B) The phylogenetic tree constructed using all sequences from the primer pair P1/P2. F represents presumed functional ITS sequences, P represents putative ITS pseudogene sequences, and cDNA represents ITS cDNA sequence bootstrap values >50%.
Tree B is the NJ tree constructed by all ITS sequences from the primer pair P1/P2 (Figure 3B). The three putative pseudogenes (LC25-9-3, LC25-9-10, and LC25-9-11) are grouped onto one branch with high support. All functional cDNAs and presumed functional genomic ITS sequences were identified as polyphyletic and clustered onto four and two major branches in the NJ tree, respectively.
Clade I (L. barbarum) of functional cDNA and clade A of putative functional sequences grouped together in a monophyletic clade with high bootstrap support (NJBS=100%), and clade II (L. chinense var. potaninii) and clade B also clustered together in a monophyletic clade (NJBS=88%). The difference is that clades III and IV consisted of only functional cDNAs, while clade III consisted mainly of L. amarum and clade IV included L. amarum and L. chinense var. potaninii. The putative functional ITS copies of the two clades differed significantly, with the class B sequences being more numerous (including all genomic ITS sequences of three regions of L. chinense) and containing only conserved motif 2, whereas the A clade was composed of only five sequences of the same region, and all had conserved motifs 2 and 3 (Figure 3B).
The phylogenetic trees obtained for the two primer pairs were generally consistent with those obtained for resequencing data.
Discussion
Identification and characterization of ITS pseudogenes in Lycium
The highly repetitive nrDNA ITS exists in thousands of copies, and its loci are prone to loss-of-function mutations, such as insertions, deletions, or code shifts, during the evolutionary process, resulting in pseudogenes. Although there are some reports about ITS pseudogenes in some plants (Buckler and Holtsford, 1996; Muir et al., 2001; Razafimandimbison et al., 2004; Wei and Wang, 2004; Harpke and Peterson, 2006, 2008a; Zheng et al., 2008; Xiao et al., 2010; Camila De Sousa et al., 2011; Zhou et al., 2013; Xu et al., 2015, 2017; Huang et al., 2016; Asanuma et al., 2019), the severe heterozygosity of ITS sequences can lead to a search for alternative molecular markers. This is also one of the reasons why there have been fewer studies on pseudogenes. Meanwhile, the appearance of nested peaks provides opportunities for us to discover ITS polymorphisms and pseudogenes.
In this study, approximately, 60% of the 268 sequences obtained may have lost function and were identified as putative pseudogenes. Surprisingly, pseudogenes were significantly more common in the wild samples than in the cultivars for both primer pairs. Among the sequences obtained from primer pair ITS1/ITS4, 91% in the cultivar were pseudogenes, while more than 95% of the sequences in the wild samples were identified as pseudogenes. Approximately, 31% of the cultivars and 35% of the wild samples had pseudogenes when using sequences obtained with the primer pair P1/P2 (Supplementary Table S4).
Compared with functional ITS copies, putative pseudogenes had a significantly lower GC content, a lower secondary structure minimum free energy for 5.8S and ITS2, lacked conserved motifs in the 5.8S of seed plants, and increased nucleotide substitutions and sequence diversity, including the average number of nucleotide differences (K) and nucleotide diversity (π). The criteria that we used to identify pseudogenes were simple and effective and even showed that some of the sequences used in previous phylogenetic studies of Lycium were actually pseudogenes (Li et al., 2014; Shi et al., 2016).
The 5.8S gene is generally regarded as highly conserved and therefore often excluded in some studies when phylogenetic analyses are performed (Shi et al., 2016; Chen et al., 2017). It was also reported that the 5.8S gene is the most reliable indicator of functionality within the ITS region, and Yokota et al. (1989) concluded that the GC content of 5.8S functional copies in plants ranged from 50.6 to 59.3%. The average GC contents of 5.8S functional copies obtained with the two primer pairs in Lycium were 53.1% and 50.9%, which were similar to the values reported by Yokota et al. The average values of putative pseudogenes were 46.1 and 45.1%, which were significantly lower, indicating that frequent mutations of bases at their methylation C sites and their low GC content may lead to loss of gene function and may be the origin of pseudogenes (Bailey et al., 2003).
Evolutionary constraints can be detected by inspecting low-energy secondary structure models (Wolf et al., 2005). The minimum free energy of the secondary structure of the 5.8S and ITS2 regions in putative pseudogenes was clearly high, indicative of their lowered stability. In contrast, the secondary structure of ITS2 was more conserved in the functional sequence than that of 5.8S, as shown in Figures 1C–F. Hence, we speculated that ITS2 may be more useful for the identification of ITS pseudogenes in Lycium, as revealed in other taxa (Zheng et al., 2008). This may be related to the conformation of ITS2 and the spacer removal mechanism from primary transcripts (Mai and Coleman, 1997). Therefore, the low GC content and stabilized secondary structure may be reasons for the preferential pseudogene amplification in this study. In addition, all three conserved motifs specific to seed plants in the 5.8S region of the putative pseudogene were found to have base mutations. Furthermore, the pseudogenes had more significant sequence diversity. As indicated by Fu and Li's D test, the pseudogenes were not subject to selection pressure, and nucleotide substitutions are not functionally restricted during evolution and tend to evolve neutrally, allowing them to evolve faster than functional copies. Conversely, putative functional ITS paralogues were found to be evolutionarily restricted. This may be related to the maintenance of specific secondary structures during mature RNA processing (Mai and Coleman, 1997).
ITS polymorphism and incomplete concerted evolution in Lycium
With the increasing development of sequencing technology, many ITS polymorphisms have been discovered among plant taxa (Muir et al., 2001; Wei and Wang, 2004; Zheng et al., 2008; Xiao et al., 2010; Xu et al., 2015). In our study, we found ITS polymorphisms in Lycium, as reflected not only in genomic DNA of different species and individuals but also in functional cDNA, indicating incomplete coevolution. This may be one of the reasons for the low success rate of ITS sequencing in DNA barcoding studies of Lycium (Ochieng et al., 2007; Chen et al., 2017). This finding also indicates the risk of using single molecular markers in plant molecular identification and improves the value of the genetic information of ITS nrDNA sequences lacking concerted evolution in the genus Lycium.
In this study, we found that wild samples had more extensive ITS polymorphisms than cultivars (Zhang et al., 2022). Unexpectedly, the polymorphism of the Lycium-specific primer pair P1/P2 was significantly lower than that of the universal primer pair ITS1/ITS4, with the putative functional sequences obtained with the latter representing no more than 10% of all sequences, while primer pair P1/P2 yielded more than 60% functional sequences (Supplementary Table S4). ITS1/ITS4 is the most popular universal primer pair for ITS fragment application, which is high universality and easy to get non-target amplicons from fungi when amplifying plant ITS fragments. This non-specific amplification phenomenon is unavoidable because fungi are symbiotic with plants in the natural ecosystems (Rodriguez et al., 2009), which may result in low PCR and sequencing success rates in this study. P1/P2 were originally designed for the identification of species in the Lycium genus (Shi et al., 2008). This group specificity primer pair could explain the lower pseudogene identification rate when amplifying with the P1/P2. Therefore, specific primers may be more appropriate than universal primers when performing species identification studies. Although specific primers reduced the amplification of non-targeted templates, the incomplete concerted evolution of multiple copies of ITS still exists.
In addition, we detected paralogous homologs with different ITS sequences in the same individual, including putative pseudogenes and functional paralogues. Several different pseudogenes were even detected in the same individual. For example, three different types of pseudogenes were detected in LC17-1 (Figure 3A), forming multiple lineages in the phylogenetic tree, with LC17-1-6 and LC17-1-8 clustered in clade I, LC17-1-4, LC17-1-7, LC17-1-10 and LC17-1-12 clustered in clade II, and LC17-1-11 in clade III.
The high degree of ITS polymorphism in Lycium was reflected not only in the GC content (40.22–57.27%) but also in the sequence length. The length ranged from 515 bp to 715 bp among all sequences, with a variation of approximately 200 bp, and intra-individual sequences were also found to differ by more than 100 bp (e.g., the shortest ITS sequence in LBVA was LBVA1-4 (540 bp), and the longest ITS sequence was 691 bp) and mainly occurred in the ITS1 region. While previous studies have shown that large ITS length variation occurs mainly among species, the presence of intra-individual variation of more than 100 bp in Cycas (Xiao et al., 2010) and Lespedeza (Xu et al., 2017) better supports our results. As in Cycas, no repeats were detected between ITS sequences in Lycium using Tandem Repeats Finder using the strictest criteria, but different short repeats (2–23 bp) were detected in all species under the most relaxed search options. These findings indicate that the presence of indels and tandem repeats plays an important role in the large amount of ITS length variation observed in Lycium.
The high degree of intra- and inter-individual polymorphism also suggests non-concerted ITS evolution in Lycium. Several biological processes might retard or disrupt concerted evolution, such as polyploidization (Karvonen and Savolainen, 1993; Suh et al., 1993), agamospermy (Campbell et al., 1997), multiple nucleolar organizer regions (NORs) (Karvonen and Savolainen, 1993; Lubaretz et al., 1996; Quijada et al., 1998; Won and Renner, 2005), longer generation times (Sang et al., 1995), and hybridization (Muir et al., 2001; Won and Renner, 2005; Grimm and Denk, 2008; Hribova et al., 2011). Lycium is a diploid plant (2n = 24 chromosomes). Therefore, genomic duplication, and therefore duplication of nrDNA loci, may not be the cause of the within-genome polymorphism observed here. The disadvantageous position of NORs (Komarova et al., 2004) on chromosomes or inactive loci without functional constraints can also lead to a slow rate of concerted evolution. Unfortunately, there is an absence of information about the number of locations of NORs in Lycium, but most diploid angiosperms have only one or two NORs per genome (Wendel et al., 1995). Thus, a large number of NORs may also not be the major reason for the intra-individual polymorphism and large difference in resolution patterns of the ITS clones in Lycium.
Hybridization is a common process that results in different copies and accounts for the high level of intra-individual polymorphism in ITS sequences found in many other plant groups, such as Pyrus (Zheng et al., 2008), Cycas (Xiao et al., 2010), Camellia sinensis (Xu et al., 2015), and Lespedeza (Xu et al., 2017). It is well known that Lycium is an allogamous plant, and interspecific hybridization is widespread. Therefore, it appears plausible that hybridization created a similarly high level of intra-individual ITS polymorphism in the genus. The impact of hybridization on polymorphism depends on the sequence divergence of the parents (Campbell et al., 1997); however, the number and distribution of NORs in Lycium are not clear. To explain the different degrees and sequence patterns of intra-individual ITS polymorphisms in Lycium, further related research is needed, including sequence data from other single-copy and unlinked loci, such as nuclear introns.
Origin and phylogenetic utility of ITS pseudogenes in Lycium
We identified different types of pseudogenes in Lycium, and therefore, it is important to reveal their origin and explore their phylogenetic utility.
Compared to functional copies, pseudogenes are less functionally restricted and evolve independently at different rates, which may contribute to studies of evolutionary genetics (Ochieng et al., 2007). The ITS pseudogenes in Lycium were found to evolve at a high substitution rate under the standard neutral model (Table 3), and the ITS phylogeny (Figure 3) showed that functional paralogues and pseudogenes could be well separated and displayed a different phylogenetic pattern. However, due to the high number of acquired mutations, these pseudogenes clustered together randomly (Figure 3), indicating that they no longer interact genetically with functional ITS copies and could be used as another novel resource to infer phylogenetic relationships. In addition, considering the high sequence divergence between ITS pseudogenes and their corresponding functional copies, we hypothesize that the ITS pseudogenes in Lycium originated earlier.
Some studies have considered the phylogenetic consequences of pseudogenes. In the study of Pyrus (Zheng et al., 2008), it was found that functional ITS copies led to a confusing and poorly resolved phylogeny as a result of low sequence divergence, while certain types of pseudogenes and some relict pseudogenes offered more credible insight into the evolutionary history of Pyrus species. Similar results were obtained in Naucleeae (Razafimandimbison et al., 2004), clearly demonstrating that divergent putative pseudogenes are useful for phylogenetic analysis, especially when sequences of their functional counterparts are not available. Although the above two examples show that pseudogenes can be useful to some extent in resolving phylogenetic relationships in closely related species, in most cases, non-disrupted ITS evolution owing to the presence of pseudogenes poses many difficulties and even leads to misinterpretation of evolutionary relationships among taxa. In this study, divergent pseudogenes in Lycium accumulated large numbers of homoplastic mutations, which may be due to LBA (Anderson and Swofford, 2004), leading to random relationships among species; therefore, if pseudogenes are used together with functional sequences for phylogenetic relationship reconstruction, it will affect the stability of the system topology and preclude an accurate understanding of the phylogenetic relationships among taxa, resulting in the constructed gene trees not reflecting true evolutionary relationships. Similar results were found in Cycas (Xiao et al., 2010) and Lespedeza (Xu et al., 2017). Recombination of divergent sequences following hybridization leads to chimeric DNA sequences, which may also lead to incorrect tree inference and misestimation of evolutionary relationships. Álvarez and Wendel (2003) analyzed the recombination of divergent sequences after hybridization in several different plants and found that this process may obscure the true phylogenetic signal. Thus, when using ITS markers to reconstruct phylogenies and study the evolution process after hybridization, attention must be paid to parental homozygosity and the effects of pseudogenes.
When we validated some previous studies, we were surprised that all the putative pseudogenes and functional sequences had been clustered into monophyletic groups with high support in the evolutionary tree (Li et al., 2014; Shi et al., 2016; Chen et al., 2017) (Supplement Figure S2). In summary, although ITS pseudogenes may obscure the true phylogenetic relationships in Lycium, they can better explain the formation of clustering branches in phylogenetic trees reported in some previous studies.
Conclusion
To the best of our knowledge, this is the first report of non-concerted evolution and pseudogenes of ITS sequences in Lycium. We revealed high polymorphism in both genomic DNA and functional cDNA ITS sequences, indicating incomplete concerted evolution. Approximately, 60% of the sequences degenerated into pseudogenes out of functional constraints and clustered randomly together in the evolutionary tree and thus may not be suitable for phylogenetic investigation. Nevertheless, the nested peaks, low identification rates, and branch formation on the evolutionary tree in previous identification studies of Lycium can be well explained from the perspective of ITS polymorphisms and pseudogenes. In summary, the study of ITS polymorphisms in Lycium not only reveals new taxa with pseudogenes but also provides a reference for germplasm resources and DNA barcoding studies of Lycium.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author/s.
Author contributions
JZha: methodology, data curation, and writing—original draft. XC: resources and investigation. JZho: methodology and analysis. AF: writing—review and editing. SA: formal analysis and validation. LH: conceptualization, supervision, and writing—review and editing. DQ: conceptualization, funding acquisition, project administration, and writing—review and editing. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the Fundamental Research Funds for the Central Public Welfare Research Institutes (JJPY2022008, ZZ13-YQ-079, and XTCX2021001), the National Natural Science Foundation of China (81803671), and the Scientific and technological innovation project of China Academy of Chinese Medical Sciences (CI2021A041 and CI2021B014).
Acknowledgments
We appreciate Ruyu Yao and Jianxiang Wang for their help with the materials collected for this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.984579/full#supplementary-material
References
Álvarez, I., and Wendel, J. F. (2003). Ribosomal ITS sequences and plant phylogenetic inference. Mol. Phylogenet. Evol. 29, 417–434. doi: 10.1016/S1055-7903(03)00208-2
Anderson, F. E., and Swofford, D. L. (2004). Should we be worried about long-branch attraction in real data sets? Investigations using metazoan 18s rDNA. Mol. Phylogenet. Evol. 33, 440–451. doi: 10.1016/j.ympev.2004.06.015
Asanuma, M., Zhu, S., Okura, N., Cai, S., Yoshimatsu, K., et al. (2019). Genetic polymorphism of Japanese cultivated Rheum species in the internal transcribed spacer region of nuclear ribosomal DNA. J. Nat. Med. 73, 541–554. doi: 10.1007/s11418-019-01298-4
Bailey, C. (2003). Characterization of angiosperm nrDNA polymorphism, paralogy, and pseudogenes. Mol. Phylogenet. Evol. 29, 435–455. doi: 10.1016/j.ympev.2003.08.021
Bailey, J. A., Liu, G., and Eichler, E. E. (2003). An Alu transposition model for the origin and expansion of human segmental duplications. Am. J. Hum. Genet. 73, 823–834. doi: 10.1086/378594
Balasubramanian, S., Zheng, D., Liu, Y., Fang, G., Frankish, A., Carriero, N., et al. (2009). Comparative analysis of processed ribosomal protein pseudogenes in four mammalian genomes. Genome Biol. 10, R2. doi: 10.1186/gb-2009-10-1-r2
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Buckler, T. E. S., and Holtsford, T. P. (1996). Zea ribosomal repeat evolution and substitution patterns. Mol. Biol. Evol. 13, 623–632. doi: 10.1093/oxfordjournals.molbev.a025622
Camila De Sousa, Q., Flávia Reis De Carvalho, B., and Luiz Orlando De, O. (2011). Evolution of the 5.8S nrDNA gene and internal transcribed spacers in Carapichea ipecacuanha (Rubiaceae) within a phylogeographic context. Mol. Phylogenet. Evol. 59, 293–302. doi: 10.1016/j.ympev.2011.01.013
Campbell, C. S., Wojciechowski, M. F., Baldwin, B. G., Alice, L. A., and Donoghue, M. J. (1997). Persistent nuclear ribosomal DNA sequence polymorphism in the Amelanchier Agamic complex (Rosaceae). Mol. Biol. Evol. 14, 81–90. doi: 10.1093/oxfordjournals.molbev.a025705
Chen, J., Zhao, M., and Jiang, S. (2017). Identification, phylogenetic relationship analysis of Lycium based on rbcL-a and ITS sequence and the discovery of ITS pseudogene. Biotech Bull 33, 123–139. doi: 10.13560/j.cnki.biotech.bull.1985.2017.05.018
Clewley, J. P. (1995). Macintosh sequence analysis software. DNAStar's lasergene. Mol Biotechnol 3, 221–224. doi: 10.1007/BF02789332
Eickbush, T. H., and Eickbush, D. G. (2007). Finely orchestrated movements: evolution of the ribosomal RNA genes. Genetics 175, 477–485. doi: 10.1534/genetics.107.071399
Elder, J. F., and Turner, B. J. (1995). Concerted evolution of repetitive DNA sequences in eukaryotes. Q. Rev. Biol. 70, 297–320. doi: 10.1086/419073
Ganley, A. R. D., and Kobayashi, T. (2007). Highly efficient concerted evolution in the ribosomal DNA repeats: total rDNA repeat variation revealed by whole-genome shotgun sequence data. Genome Res. 17, 184–191. doi: 10.1101/gr.5457707
Ganley, A. R. D., and Kobayashi, T. (2011). Monitoring the rate and dynamics of concerted evolution in the ribosomal DNA repeats of Saccharomyces cerevisiae using experimental evolution. Mol. Biol. Evol. 28, 2883–2891. doi: 10.1093/molbev/msr117
Grimm, G. W., and Denk, T. (2008). ITS evolution in Platanus (Platanaceae): homoeologues, pseudogenes and ancient hybridization. Ann. Bot. 101, 403–419. doi: 10.1093/aob/mcm305
Harpke, D., and Peterson, A. (2006). Non-concerted ITS evolution in Mammillaria (Cactaceae). Mol. Phylogenet. Evol. 41, 579–593. doi: 10.1016/j.ympev.2006.05.036
Harpke, D., and Peterson, A. (2008a). Extensive 5.8S nrDNA polymorphism in Mammillaria (Cactaceae) with special reference to the identification of pseudogenic internal transcribed spacer regions. J. Plant Res. 121, 261–270. doi: 10.1007/s10265-008-0156-x
Harpke, D., and Peterson, A. (2008b). 5.8s motifs for the identification of pseudogenic its regions. Botany 86, 300–305. doi: 10.1139/B07-134
Hribova, E., Cizkova, J., Christelova, P., Taudien, S., de Langhe, E., et al. (2011). The ITS1-5.8S-ITS2 sequence region in the Musaceae: structure, diversity and use in molecular phylogeny. PLoS ONE. 6, e17863. doi: 10.1371/journal.pone.0017863
Huang, J., Li, L., van der Werff, H., Li, H., Rohwer, J. G., et al. (2016). Origins and evolution of cinnamon and camphor: a phylogenetic and historical biogeographical analysis of the Cinnamomum group (Lauraceae). Mol. Phylogenet. Evol. 96, 33–44. doi: 10.1016/j.ympev.2015.12.007
Karvonen, P., and Savolainen, O. (1993). Variation and inheritance of ribosomal DNA in Pinus sylvestris L. (Scots pine). Heredity 71, 614–622. doi: 10.1038/hdy.1993.186
Kita, Y., and Ito, M. (2000). Nuclear ribosomal ITS sequences and phylogeny in East Asian Aconitum subgenus Aconitum (Ranunculaceae), with special reference to extensive polymorphism in individual plants. Plant Syst. Evolut. 225, 1–13. doi: 10.1007/BF00985455
Komarova, N. Y., Grabe, T., Huigen, D. J., Hemleben, V., and Volkov, R. A. (2004). Organization, differential expression and methylation of rDNA in artificial Solanum allopolyploids. Plant Mol. Biol. 56, 439–463. doi: 10.1007/s11103-004-4678-x
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Li, D., Gao, L., Li, H., Wang, H., Ge, X., Liu, J. Q., et al. (2011). Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants. Proc. Nat. Acad. Sci. 108, 19641–19646. doi: 10.1073/pnas.1104551108
Li, M., Zhao, H., Zhao, F., Jiang, L., Peng, H., Zheng, W., et al. (2019). Alternative analyses of compensatory base changes in an ITS2 phylogeny of Corydalis (Papaveraceae). Ann. Bot. 124, 233–243. doi: 10.1093/aob/mcz062
Li, X., Wang, Y., and Li, J. (2014). Study on 10 species of Lycium L. Genetic relationship based on nrDNA-ITS sequence. Chin. Agri. Sci. Bull. 30, 128–135.
Liao, D. (1999). Concerted evolution: molecular mechanism and biological implications. Am. J. Hum. Genet. 64, 24–30. doi: 10.1086/302221
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Lubaretz, O., Fuchs, J., Ahne, R., Meister, A., and Schubert, I. (1996). Karyotyping of three Pinaceae species via fluorescent in situ hybridization and computer-aided chromosome analysis. Theor. Appl. Genet. 92, 411–416. doi: 10.1007/BF00223687
Mai, J. C., and Coleman, A. W. (1997). The internal transcribed spacer 2 exhibits a common secondary structure in green algae and flowering plants. J. Mol. Evol. 44, 258–271. doi: 10.1007/PL00006143
Mayol, M., and Rosselló, J. A. (2001). Why nuclear ribosomal dna spacers (ITS) tell different stories in Quercus. Mol. Phylogenet. Evol. 19, 167–176. doi: 10.1006/mpev.2001.0934
Muir, G., Fleming, C. C., and Schlötterer, C. (2001). Three divergent rDNA clusters predate the species divergence in Quercus petraea (Matt.) Liebl. and Quercus robur L. Mol. Biol. Evol. 18, 112–119. doi: 10.1093/oxfordjournals.molbev.a003785
Ochieng, J. W., Henry, R. J., Baverstock, P. R., Steane, D. A., and Shepherd, M. (2007). Nuclear ribosomal pseudogenes resolve a corroborated monophyly of the eucalypt genus corymbia despite misleading hypotheses at functional ITS paralogs. Mol. Phylogenet. Evol. 44, 752–764. doi: 10.1016/j.ympev.2007.04.017
Quijada, A., Liston, A., Delgado, P., Vazquez-Lobo, A., and Alvarez-Buylla, E. R. (1998). Variation in the nuclear ribosomal DNA internal transcribed spacer (ITS) region of Pinus rzedowskii revealed by PCR-RFLP. Theor. Appl. Genet. 96, 539–544. doi: 10.1007/s001220050771
Razafimandimbison, S. G., Kellogg, E. A., and Bremer, B. (2004). Recent origin and phylogenetic utility of divergent its putative pseudogenes: a case study from Naucleeae (Rubiaceae). Syst. Biol. 53, 177–192. doi: 10.1080/10635150490423278
Rodriguez, R. J., White, J. J., Arnold, A. E., and Redman, R. S. (2009). Fungal endophytes: diversity and functional roles. New Phytol. 182, 314–330. doi: 10.1111/j.1469-8137.2009.02773.x
Rose, P. P., and Korber, B. T. (2000). Detecting hypermutations in viral sequences with an emphasis on G –> A hypermutation. Bioinformatics 16, 400–401. doi: 10.1093/bioinformatics/16.4.400
Sang, T., Crawford, D., and Stuessy, T. (1995). Documentation of reticulate evolution in peonies (Peonia) using internal transcribed spacer sequences of nuclear ribosomal DNA: implications for biogeography and concerted evolution. Proc. Natl. Acad. Sci 92, 6813–6817. doi: 10.1073/pnas.92.15.6813
Schoch, C. L., Seifert, K. A., Huhndorf, S., Robert, V., Spouge, J. L., Levesque, C. A., et al. (2012). Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for fungi. Proc. Natl. Acad. Sci. USA. 109, 6241–6246. doi: 10.1073/pnas.1117018109
Shi, Z., An, W., and Fan, Y. (2008). Preliminary studies on identification of Lycium Linn. Germplasm resources by nrDNA ITS sequencing. Agri. Sci. Technol. 35–38. doi: 10.16175/j.cnki.1009-4229.2008.01.011
Shi, Z., Wan, R., and Li, Y. (2016). Identification of Lycium L. Germplasm resource based on nrDNA ITS sequence. Hubei Agri. Sci. 55, 5966–5968. doi: 10.14088/j.cnki.issn0439-8114.2016.22.059
Sisu, C., Pei, B., Leng, J., Frankish, A., Zhang, Y., et al. (2014). Comparative analysis of pseudogenes across three phyla. Proc. Nat. Acad. Sci. 111, 13361–13366. doi: 10.1073/pnas.1407293111
Suh, Y., Thien, L., and Reeve, H. (1993). Molecular evolution and phylogenetic implications of internal transcribed spacer sequences of ribosomal DNA in Winteraceae. Am. J. Bot. 80, 1042–1055. doi: 10.1002/j.1537-2197.1993.tb15332.x
Tian, X., and Li, D. (2002). DNA sequences in plant phylogenetic study. Acta Bot Yunnan. 24, 170–184. doi: 10.3969/j.issn.2095-0845.2002.02.004
Wei, X., and Wang, X. (2004). Recolonization and radiation in Larix (Pinaceae): evidence from nuclear ribosomal DNA paralogues. Mol. Ecol. 13, 3115–3123. doi: 10.1111/j.1365-294X.2004.02299.x
Wendel, J. F., Schnabel, A., and Seelanan, T. (1995). Bidirectional interlocus concerted evolution following allopolyploid speciation in cotton (Gossypium). Proc. Nat. Acad. Sci. 92, 280–284. doi: 10.1073/pnas.92.1.280
White, T., Bruns, T., Lee, S., and Taylor, J. (1990). Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. Part Three. Genetics Evolut. 315–322. doi: 10.1016/B978-0-12-372180-8.50042-1
Wolf, M., Achtziger, M., Schultz, J., Dandekar, T., and Muller, T. (2005). Homology modeling revealed more than 20,000 rRNA internal transcribed spacer 2 (ITS2) secondary structures. RNA. 11, 1616–1623. doi: 10.1261/rna.2144205
Won, H., and Renner, S. S. (2005). The internal transcribed spacer of nuclear ribosomal DNA in the gymnosperm Gnetum. Mol. Phylogenet. Evol. 36, 581–597. doi: 10.1016/j.ympev.2005.03.011
Xiao, L., Möller, M., and Zhu, H. (2010). High nrDNA ITS polymorphism in the ancient extant seed plant Cycas: incomplete concerted evolution and the origin of pseudogenes. Mol. Phylogenet. Evol. 55, 168–177. doi: 10.1016/j.ympev.2009.11.020
Xin, T., Yao, H., Gao, H., Zhou, X., Ma, X., et al. (2013). Super food Lycium barbarum (Solanaceae) traceability via an internal transcribed spacer 2 barcode. Food Research International 54, 1699–1704. doi: 10.1016/j.foodres.2013.10.007
Xu, B., Zeng, X., Gao, X., Jin, D., and Zhang, L. (2017). ITS non-concerted evolution and rampant hybridization in the legume genus Lespedeza (Fabaceae). Scientific Rep. 7. doi: 10.1038/srep40057
Xu, J., Xu, Y., Yonezawa, T., Li, L., Hasegawa, M., et al. (2015). Polymorphism and evolution of ribosomal DNA in tea (Camellia sinensis, Theaceae). Mol. Phylogenet. Evol. 89, 63–72. doi: 10.1016/j.ympev.2015.03.020
Yokota, Y., Kawata, T., Iida, Y., Kato, A., and Tanifuji, S. (1989). Nucleotide sequences of the 5.8S rRNA gene and internal transcribed spacer regions in carrot and broad bean ribosomal DNA. J. Mol. Evol. 29, 294–301. doi: 10.1007/BF02103617
Zhang, J., Nie, Y., Guo, W., and Qian, D. (2022). Incomplete concerted evolution of nrDNA ITS in Lycium. Acta Pharm Sin 57, 525–532. doi: 10.16438/j.0513-4870.2021-1303
Zheng, X., Cai, D., Yao, L., and Teng, Y. (2008). Non-concerted ITS evolution, early origin and phylogenetic utility of ITS pseudogenes in Pyrus. Mol. Phylogenet. Evol. 48, 892–903. doi: 10.1016/j.ympev.2008.05.039
Zhou, A., Yun, L., and Li, M. (2013). Intra-genomic polymorphism in the nrDNA ITS sequence of camellia reticulata. Plant Sci. J. 31, 1–10. doi: 10.3724/SP.J.1142.2013.10001
Keywords: Lycium, internal transcribed spacer (ITS), polymorphism, non-concerted evolution, pseudogenes, phylogeny
Citation: Zhang J, Chi X, Zhong J, Fernie A, Alseekh S, Huang L and Qian D (2022) Extensive nrDNA ITS polymorphism in Lycium: Non-concerted evolution and the identification of pseudogenes. Front. Plant Sci. 13:984579. doi: 10.3389/fpls.2022.984579
Received: 02 July 2022; Accepted: 01 August 2022;
Published: 25 August 2022.
Edited by:
Daniel Pinero, National Autonomous University of Mexico, MexicoReviewed by:
Andrea Porceddu, University of Sassari, ItalyCarlos Arbizu, Instituto Nacional de Innovación Agraria (INIA), Peru
Li Gong, Zhejiang Ocean University, China
Copyright © 2022 Zhang, Chi, Zhong, Fernie, Alseekh, Huang and Qian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luqi Huang, aHVhbmdsdXFpMDFAMTI2LmNvbQ==; Dan Qian, cWlhbmRhbkBtZXJjLmFjLmNu