 Wuxian Zhou
Wuxian Zhou Xiaogang Jiang
Xiaogang Jiang Xuhui Tan
Xuhui Tan Meide Zhang
Meide Zhang- Key Laboratory of Biology and Cultivation of Herb Medicine, Ministry of Agriculture and Rural Affairs, Institute of Chinese Herbal Medicines, Hubei Academy of Agricultural Sciences, Enshi, China
Codonopsis tangshen Oliv (C. tangshen) is an important Chinese traditional medicinal plant with various health benefits. However, the growth of C. tangshen are seriously affected by continuous cropping, which led to the decrease of the yield and quality. A field experiment was conducted to learn the effects of soil amendments on the growth of C. tangshen under continuous cropping condition, and the biological events which occurred at molecular level were investigated. The results indicated that the content of chlorophyll a (Chl a), chlorophyll b (Chl b), and carotenoid (Car) was significantly higher in SCPM (silicon-calcium-potassium-magnesium fertilizer), SCPMA (SCPM combined with azoxystrobin) and SCPMAOM (SCPM combined with azoxystrobin and organic manure) treatments. Moreover, the yield and the levels of alkaloid, polysaccharide, flavone and total protein in the treatments of SCPM, SCPMA and SCPMAOM were significantly higher than those in the control, and these indexes were all highest in the SCPMAOM treatment. RNA-sequencing (RNA-Seq) is an economical and efficient method to obtain genetic information for species with or without available genome data. In this study, RNA-Seq was performed to understand how continuously cropped C. tangshen responded to the soil amendments at the transcriptome level. The number of differentially expressed genes (DEGs) were as follows: CK vs. SCPM (719 up- and 1456 down-), CK vs. SCPMA (1302 up- and 1748 down-), CK vs. SCPMAOM (1274 up- and 1678 down-). The soil amendments affected the growth of C. tangshen mainly by regulating the genes involved in pathways of ‘photosynthesis,’ ‘plant hormone signal transduction,’ ‘biosynthesis of unsaturated fatty acids,’ ‘phenylpropanoid biosynthesis,’ and ‘starch and sucrose metabolism,’ etc. qRT-PCR was performed to validate the expressions of 10 target genes such as CP26, PsaF, and POX, etc., which verified the reliability of RNA-Seq results. Overall, this study revealed the roles and underlying mechanisms of the soil amendments in regulating the growth of continuously cropped C. tangshen at transcriptome level. These findings are beneficial for improving the continuous cropping tolerance and may be valuable for future genetic improvement of C. tangshen.
Introduction
Codonopsis tangshen Oliv., a perennial herbaceous species, is an important medicinal herb and was highly valued in traditional Chinese medicine (Xu et al., 2021). The dried root of C. tangshen mainly contains lobetyolin, polysaccharides and saponin, and is usually used for curing spleen deficiency, hypertensive disease and hypoglycemic disease (Wu et al., 2020; Xu et al., 2021). Moreover, C. tangshen is not only a medicinal herb, but also a food material, and sometimes is used as a succedaneum for Panax ginseng due to its similar medicinal value and low price (Tsai and Lin, 2008). However, our previous researches revealed that continuous cropping obstacle (also known as replant disease) was a significant limiting factor for the growth and development of C. tangshen (He et al., 2019; Zhang et al., 2021), and thus seriously affected the production and led to the shortage of C. tangshen supply. Consequently, C. tangshen grown in the continuously cropped land showed smaller leaves, thinner stems, lower biomass, and became more rigid than those grown in the non-continuously cropped land (He et al., 2019). In addition, continuous cropping led to a decrease in rhizospheric soil bacterial abundance, and altered the microbial community structure, thereby resulting in a poor growth of C. tangshen under continuous cropping system (Zhang et al., 2021). Therefore, the studies on alleviating negative effects on C. tangshen growth induced by continuous cropping were of great significance in agricultural production.
There are lots of methods to eliminate the continuous cropping obstacle of crops, and application of soil amendments is the most widely used measure in agricultural practice. Silicon–calcium–potassium–magnesium fertilizer (SCPM) is an alkaline fertilizer formed by calcination of phosphogypsum and potassium feldspar at high temperature, and is widely used in the remediation of acidified paddy soil (Ji et al., 2019). Organic manure (OM) has been widely used in agriculture to improve the soil environment. OM combined with biochar amendments could promote the growth of Xinjiang cotton by ameliorating the soil in continuous cropping system (Zhang et al., 2020). Azoxystrobin (AZO) is a fungicide which inhibits fungi by disrupting ATP production in fungal mitochondria (Sun et al., 2018). Wang et al. (2022) found that application of appropriate AZO combined with chloropicrin fumigation could promote the growth and phosphorus amount of ginger rhizomes. RNA-Seq has become an indispensable tool for transcriptome-wide analysis of differential gene expression and differential splicing of mRNAs in the past decade (Stark et al., 2019). Chen et al. (2020) reported the transcriptome change of continuously cropped strawberry in response to a soil amendment and indicated that strawberry plants reallocated defense resources to development when soil amendments alleviated the stress caused by continuous cropping. Lu et al. (2020) characterized the transcriptional response of continuously cropped soybeans to Funneliformis mosseae and found that the increased activity of some disease-resistant genes may partly account for the ability of the plants to resist diseases. Our previous study indicated that the soil amendments including OM and SCPM could effectively relieve the continuous cropping obstacles to C. tangshen cultivation (Zhou et al., 2021), but the recovery mechanisms underlying this phenomenon is still remain unclear. Based on these limited reports, here we hypothesized that soil amendments might directly affect the transcriptome profiles of continuously cropped plants, and therefore induce physiological and phenotypical variations.
In this study, we performed a transcriptome analysis to learn the soil amendments induced response in continuously cropped C. tangshen and explore the biological pathways involved in the soil remediation process. Moreover, we addressed the question of whether and how the increased tolerance of C. tangshen was associated with the maintenance of photosynthesis, plant hormone signal transduction, biosynthesis of unsaturated fatty acids, phenylpropanoid biosynthesis, and starch and sucrose metabolism, etc. The present study provided a technical support for improving the tolerance of C. tangshen to continuous cropping and laid a foundation for unraveling the molecular mechanisms of soil amendments in regulating the growth of continuously cropped C. tangshen at transcriptome level.
Materials and methods
Plant materials and experimental design
The seeds of C. tangshen were used as experimental materials. The field experiment was conducted in Xintian Village (30°32’16”N, 109°12’45”E, 1738 m above sea level), Banqiao Township, Enshi City, Hubei Province, China. A field previously cultivated with C. tangshen for 3 years was selected as experiment plot. This study consisted of four treatments, i.e., (1) CK, conventional fertilization; (2) SCPM, application of 3000 kg/hm2 silicon-calcium-potassium-magnesium fertilizer; (3) SCPMA, application of 3000 kg/hm2 SCPM combined with azoxystrobin (A) 500 times solution; (4) SCPMAOM, application of 3000 kg/hm2 SCPM and 4500 kg/hm2 organic manure (OM), combined with azoxystrobin 500 times solution. All the amendments were used following the manufacturer’s recommendations. There were three replicates (plots) for each treatment, and each plot size was 20 m2. Ditches were set up between the plots (with an interval of 30 cm) to prevent mutual infiltration of soil amendments. The nitrogen application rate was 346.5 kg.hm–2, and m (N): m (P2O5): m (K2O) = 1:0.72:0.65. Urea, calcium magnesium phosphate fertilizer and potassium sulfate were used as nitrogen, phosphorus and potassium fertilizer, respectively. All the treatments maintained the same application rate of nitrogen, phosphorus and potassium by adjusting the ratio of base fertilizer.
On March 20, 2019, all the amendments were applied to the soil, and the fertilizer of 50% nitrogen, 100% phosphorus and 100% potassium were also applied at the same time, then the plots were fully plowed and the C. tangshen seeds were sown on April 11, 2019. In addition, the remaining 50% nitrogen fertilizer was applied at the seedling stage (20%) and flowering stage (30%) of C. tangshen. Moreover, the leaves of the same part were collected on June 6, 2020, and were separated into two parts. One part was used to measure the physiological indexes such as chlorophyll (Chl), superoxide dismutase (SOD), peroxidase (POD), catalase (CAT), soluble sugar (SS), soluble protein (SP), superoxide anion radical (SAR), and malondialdehyde (MDA). The other part was immediately stored in liquid nitrogen and brought back to the laboratory for transcriptome sequencing. The fresh roots of C. tangshen were harvested on September 8, 2020, then washed and dried at 60°C. The quality indexes, such as lobetyolin, polysaccharides and alkaloids were measured.
Measurement of physiological and biochemical traits
The contents of Chl, MDA and SP, and the activities of antioxidant enzymes such as POD, SOD and CAT, etc, were measured according to the previous method (He et al., 2019). Yield, lobetyolin, polysaccharide, etc., were measured according to our previous study (Zhang et al., 2021).
RNA extraction and cDNA library construction
Total RNA was extracted from the leaf samples using TRIzol reagent (Invitrogen, New York, CA, United States). RNA samples’ concentration, purity and integrity were detected using the Agilent 2100 Bioanalyzer (Agilent, Palo Alto, CA, United States) to ensure high-quality samples for transcriptome sequencing. About 1 μg RNA per sample was used to construct sequencing library. In brief, mRNA was purified from the total RNA with poly-T oligo-attached magnetic beads. Then, mRNA was interrupted to 200–300 bp by ion interruption. First-strand cDNA was generated using random hexamer primer and M-MuLV Reverse Transcriptase, followed by the synthesis of second-strand cDNA using DNA Polymerase I and RNase H. In total, 12 libraries were generated using NEBNext®Ultra™ RNA Library Prep Kit for Illumina® (NEB, Lincoln, NE, United States), following the manufacturer’s recommendations. Then PCR enrichment was carried out using Phusion High-Fidelity DNA polymerase, Universal PCR primers and Index Primer. Subsequently, the fragments about 450 bp in length were selected. Afterward, cDNA libraries were purified and quantified by the Agilent 2100 Bioanalyzer. In the end, the libraries were sequenced using an Illumina Hiseq 6000 platform in Shanghai Personal Biotechnology Co., Ltd (Shanghai, China).
Bioinformatics analysis
Processing and assembling of illumina reads
Firstly, the reads numbers, base numbers, Q20, Q30, GC-content, and N (%) were counted based on raw data. Then, clean reads were obtained by removing low-quality reads and reads containing adapters. Subsequently, based on high quality clean data, transcriptome assembly was performed using the Trinity (Version 2.10.0; Grabherr et al., 2011), then the transcript and unigene were identified and used for further analysis.
Gene expression analysis
The read count value of each gene was quantified using RSEM (RNA-Seq by Expectation Maximization) (Li and Dewey, 2011), and FPKM (Fragments Per Kilobase of exon per Million mapped fragments) was used for estimating the gene expression level. Then, differential gene expression analysis of two groups was performed using DESeq2 R package (Version 1.10.1; Love et al., 2014) based on the model of negative binomial distribution. The P-values were adjusted to control the false discovery rate with the Benjamini and Hochberg’s method. Genes with an adjusted p value < 0.05, | log2 Fold Change| > 1 were taken as differentially expressed gene (DEG).
Hierarchical cluster, gene ontology, and kyoto encyclopedia of genes and genomes pathway enrichment analysis
Hierarchical clustering was carried out for DEGs. The FPKM counts for each unigene were two-way clustered using gplots in the R package (R Core Team, 2012). Gene Ontology (GO) enrichment of the differentially expressed genes (DEGs) was analyzed by the top GO R packages (Young et al., 2010). Kyoto encyclopedia of genes and genomes (KEGG) enrichment of DEGs between two groups was analyzed by the R package pathfinder (Ülgen et al., 2019).
Validation of RNA-seq data by real-time quantitative PCR
Real-time quantitative RT-PCR was performed to validate the RNA-Seq results according to the previous method (Li et al., 2017a). The GAPDH gene was used as a reference gene. The qRT-PCR primers in this study are listed in Supplementary Table 1. The 2–ΔΔCT method was used to determine the relative expression of target genes (Kenneth and Thomas, 2002).
Statistical analysis
The plant sampling in this study followed the principle of random sampling. The data are presented as the mean of three replications and corresponding standard deviation. The differences between the samples were determined by Duncan’s multiple range test at p < 0.05. Pearson correlation analysis was performed to analyze the correlations among samples in different treatments. Statistics and correlation analysis were performed using SPSS v20.0 (SPSS Inc., United States). Principal component analysis (PCA) was performed in Canoco 5.0. Bar charts were plotted using OriginPro 2021. Heatmaps and hierarchical clustering were performed using R package models or TBtools software (Version 1.068).
Results
Physiological and biochemical properties
The morphological observation showed that the leaves of C. tangshen in SCPM, SCPMA and SCPMAOM treatments were larger and greener than those in the control (Supplementary Figure 1). Physiological and biochemical properties were compared among remediation groups and control group. Compared with the control (CK), the Chl a, Chl b, and Car levels were significantly higher in SCPM, SCPMA and SCPMAOM, and the highest levels of these indexes were occurred in SCPMAOM (Table 1). Specifically, the SCPMAOM treatment increased the Chl a, Chl b and Car levels by 24.5, 23.1, and 23.6%, respectively, compared with the control. The SOD activity and MDA content were significantly down-regulated in the three treatments, compared with the control, and with lowest levels in SCPMAOM. Moreover, the SAR and SS levels were significantly decreased in SCPM, SCPMA and SCPMAOM, while the SP level was significantly increased in these three treatments, compared with the control (Table 2).
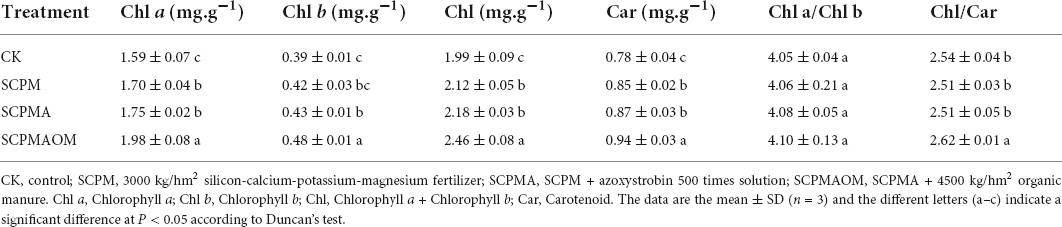
Table 1. Content of chlorophyll and carotenoid in C. tangshen leaves in different treatments.
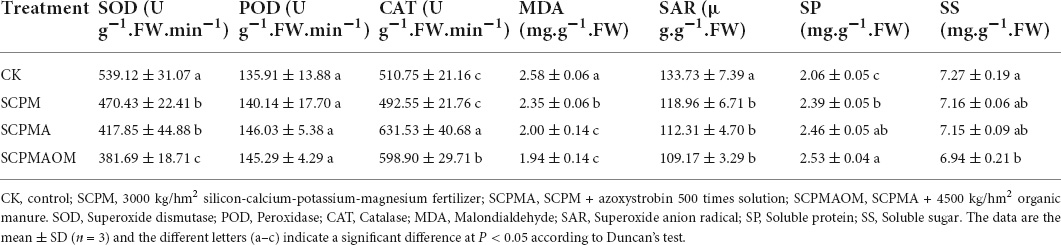
Table 2. Superoxide dismutase (SOD), POD, CAT activities and MDA, SAR, SP, and SS levels in C. tangshen leaves in different treatments.
In this study, important indexes such as yield, lobetyolin, polysaccharide, flavone, alkaloid and total protein were measured to learn the effects of soil amendments on yield and quality of C. tangshen under continuous cropping condition (Table 3). The results showed that the yield and alkaloid levels of SCPM, SCPMA, and SCPMAOM treatments were significantly higher than those of the control, and the yield of SCPMAOM treatment (7179.5 kg/hm2) was 10 times higher than that of the control (683.8 kg/hm2). Meanwhile, the SCPMAOM treatment increased the polysaccharide, flavone and total protein levels by 8.5, 22.8, and 43.0%, compared with the control.
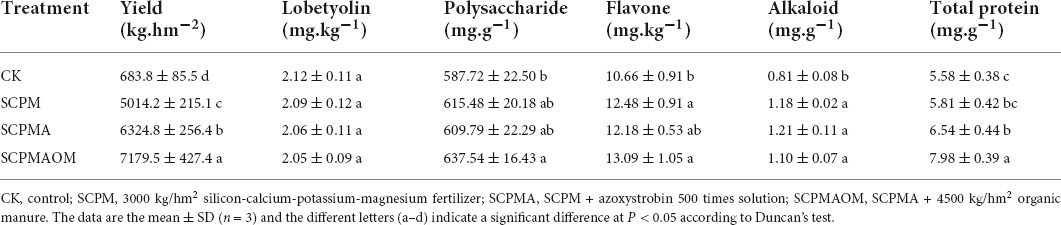
Table 3. Yield and quality of C. tangshen in different treatments.
Transcriptome sequencing and assembly
To comprehensively explore the molecular response of continuously cropped C. tangshen to soil remediation. 12 cDNA libraries were constructed from the leaves of C. tangshen in different treatments including CK, SCPM, SCPMA, and SCPMAOM. All the libraries were sequenced using the Illumina HiSe 6000 platform. The results of sequence assembly were presented in Table 4. The clean reads of CK, SCPM, SCPMA and SCPMAOM treatments were 38715892, 39901087, 40037621, and 40499491, respectively. In addition, the Q20 values were 97.73, 97.64, 97.61, and 97.74%, and the Q30 values were 93.91, 93.75, 93.68, and 93.98%, for CK, SCPM, SCPMA, and SCPMAOM treatments, respectively, which indicated the high quality of the transcriptome sequencing.
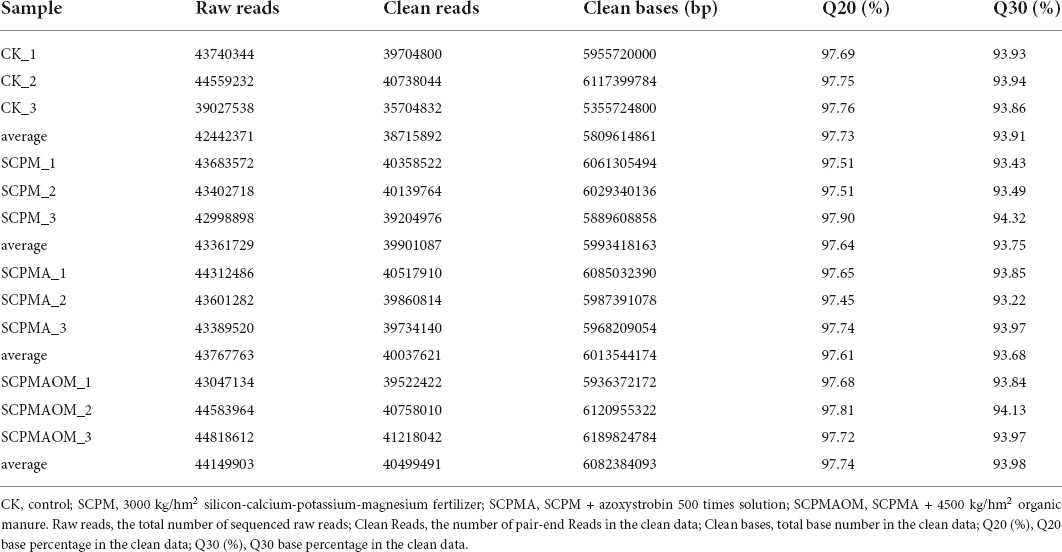
Table 4. Overview of the sequencing data.
Differential expression genes analysis
Large number of DEGs were identified in the comparisons including R1 (CK vs. SCPM), R2 (CK vs. SCPMA), R3 (CK vs. SCPMAOM), R4 (SCPM vs. SCPMA), R5 (SCPM vs. SCPMAOM), R6 (SCPMA vs. SCPMAOM). As shown in Figure 1A, the number of identified DEGs were as follows: R1 (719 up- and 1456 down-), R2 (1302 up- and 1748 down-), R3 (1274 up- and 1678 down-), R4 (291 up- and 287 down-), R5 (682 up- and 355 down-), R6 (646 up- and 312 down-). Venn diagram was performed to analyze the DEGs distribution in comparison group 1 (R1, R2, R3) (Figure 1B) and group 2 (R4, R5, R6) (Figure 1C). The DEGs of R2 (CK vs. SCPMA) and R6 (SCPM vs. SCPMAOM) occupied the largest proportion of total DEGs in comparison group 1 and group 2, respectively. Moreover, the shared DEGs were 979 and 4 in comparison group 1 and group 2, respectively (Figures 1B,C). To evaluate the repeatability of biological repeats, the correlations among different treatments were analyzed. The results showed that the correlation coefficients among three biological repeats in the same treatments were all more than 0.96, which were significantly higher than those among samples from different treatments (Supplementary Figure 2). Furthermore, principal component analysis showed good intragroup correlation and high repeatability (Supplementary Figure 3), suggesting the samples were reliable for use in subsequent data analysis.
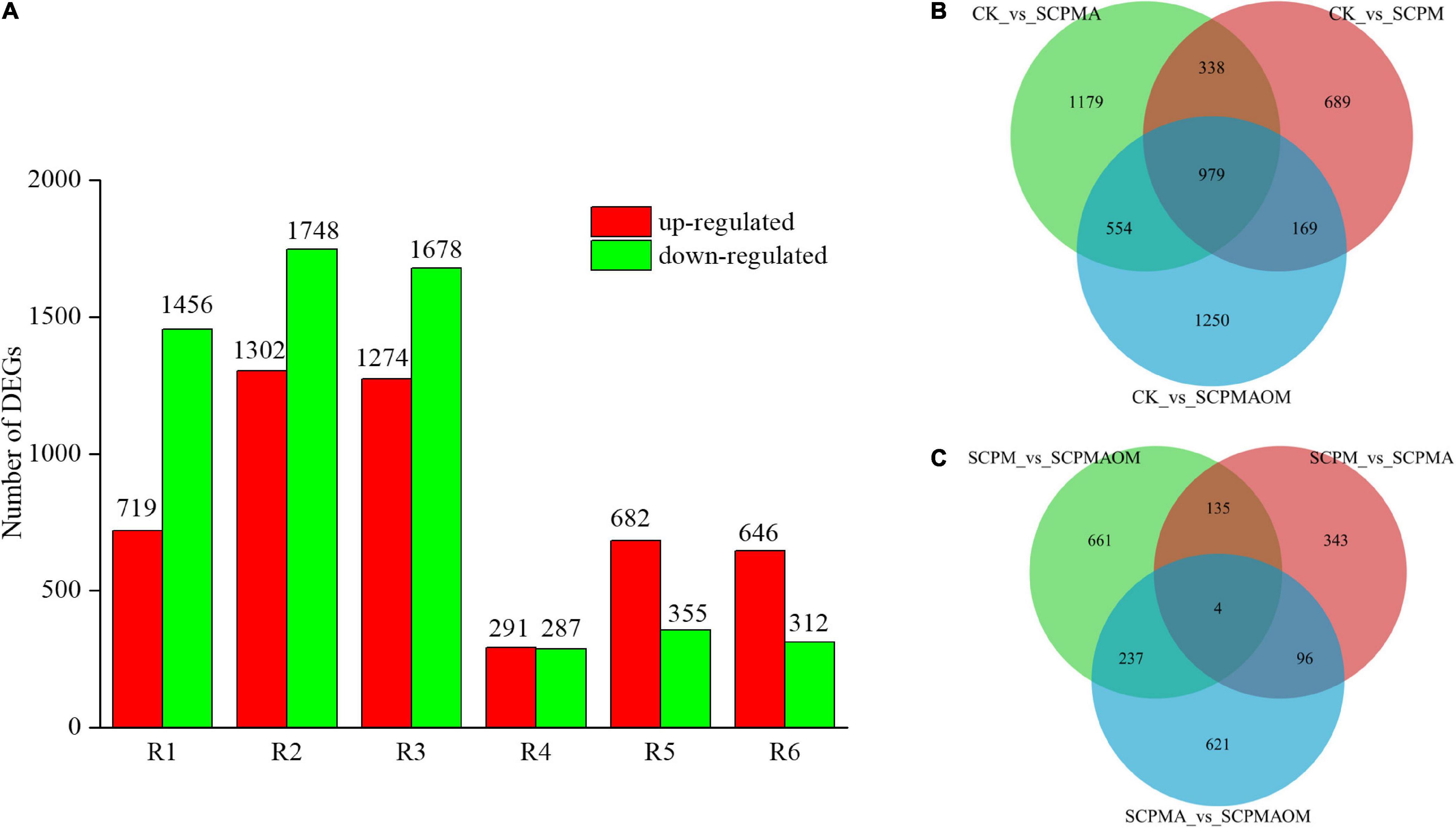
Figure 1. Statistical analysis of the DEGs in different comparisons. (A) The number of up/down-regulated genes in different treatments. (B) Venn diagram of the DEGs from R1, R2, and R3. (C) Venn diagram of the DEGs from R4, R5, and R6. R1, CK vs. SCPM; R2, CK vs. SCPMA; R3, CK vs. SCPMAOM; R4, SCPM vs. SCPMA; R5, SCPM vs. SCPMAOM; R6, SCPMA vs. SCPMAOM.
Gene ontology and kyoto encyclopedia of genes and genomes annotation of differentially expressed genes
Gene ontology classification was performed to annotate DEGs in different comparisons (R1, R2, R3) (Figure 2). For each comparison, top 10 enriched terms in the categories of cell component (CC), molecular function (MF), and biologic process (BP) were selected for further analysis. In comparison R1, the significantly enriched terms in “CC” were, ‘photosystem’ (20 DEGs), ‘extracellular region’ (39 DEGs), ‘thylakoid part’ (32 DEGs), ‘photosystem I’ (11 DEGs). The major terms of “MF” included ‘oxidoreductase activity’ (171 DEGs), ‘oxidoreductase activity, acting on paired donors’ (52 DEGs), ‘tetrapyrrole binding’ (48 DEGs). For “BP,” the DEGs were significantly classified into terms including ‘oxidation-reduction process’ (144 DEGs), ‘photosynthesis’ (31 DEGs), and ‘protein-chromophore linkage’ (12 DEGs).
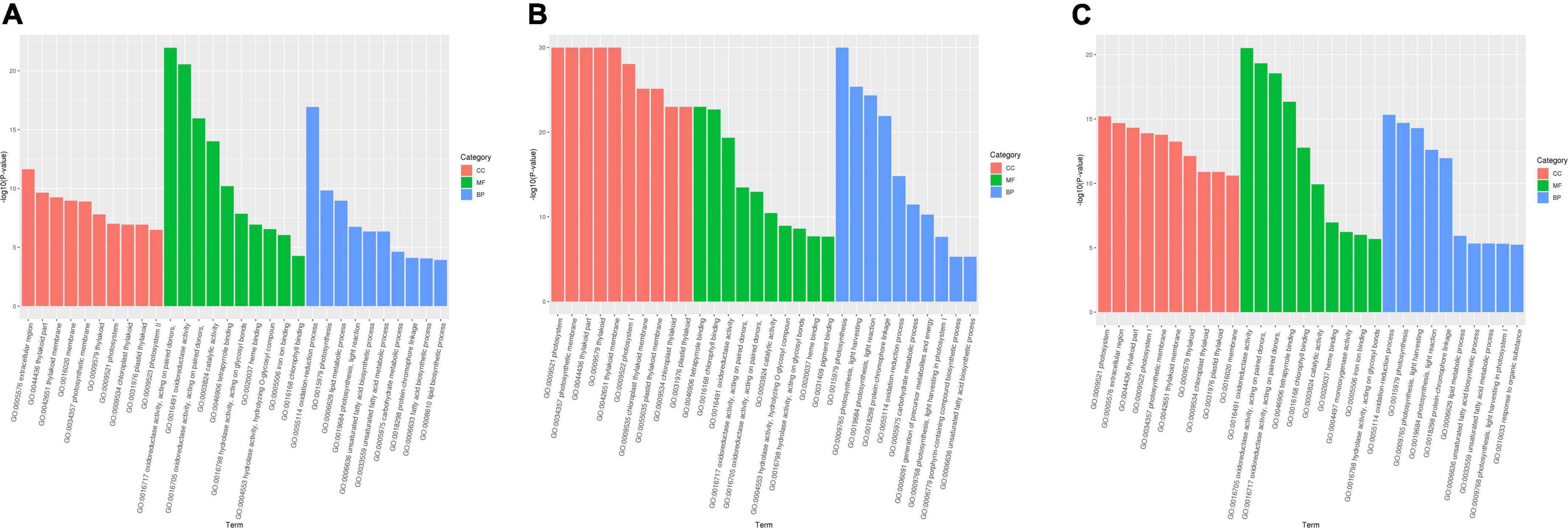
Figure 2. Histogram of GO classification. (A) DEGs in R1 comparison, (B) DEGs in R2 comparison, (C) DEGs in R3 comparison. The DEGs were annotated into three main categories: biological process, cellular component and molecular function. R1, CK vs. SCPM; R2, CK vs. SCPMA; R3, CK vs. SCPMAOM.
KEGG enrichment analysis was conducted to classify the DEGs from comparisons R1, R2 and R3 into 20 significant pathways (P < 0.05). In R1, the main enriched pathways were ‘biosynthesis of unsaturated fatty acids’ (29 DEGs), ‘photosynthesis’ (17 DEGs), ‘phenylpropanoid biosynthesis’ (29 DEGs), ‘photosynthesis-antenna proteins’ (12 DEGs), ‘starch and sucrose metabolism’ (25 DEGs), and ‘plant hormone signal transduction’ (Figure 3A). For R2, the remarkably enriched pathways were ‘photosynthesis-antenna proteins’ (38 DEGs), ‘photosynthesis’ (41 DEGs), ‘biosynthesis of unsaturated fatty acids’ (25 DEGs), and ‘carbon fixation in photosynthetic organisms’ (37 DEGs) (Figure 3B). The DEGs of R3 were mainly involved in ‘biosynthesis of unsaturated fatty acids’ (31 DEGs), ‘photosynthesis-antenna proteins’ (27 DEGs), ‘α-linolenic acid metabolism’ (17 DEGs), and ‘phenylpropanoid biosynthesis’ (DEGs) (Figure 3C). In the comparisons of R1, R2, and R3, the up-regulated DEGs were mainly enriched in the pathways like ‘photosynthesis,’ ‘photosynthesis-antenna proteins,’ and ‘plant hormone signal transduction,’ while the down-regulated DEGs were mainly associated with the pathways including ‘biosynthesis of unsaturated fatty acids,’ ‘phenylpropanoid biosynthesis,’ and ‘starch and sucrose metabolism.’ The details of GO and KEGG annotation in these comparisons were shown in Supplementary Table 2.
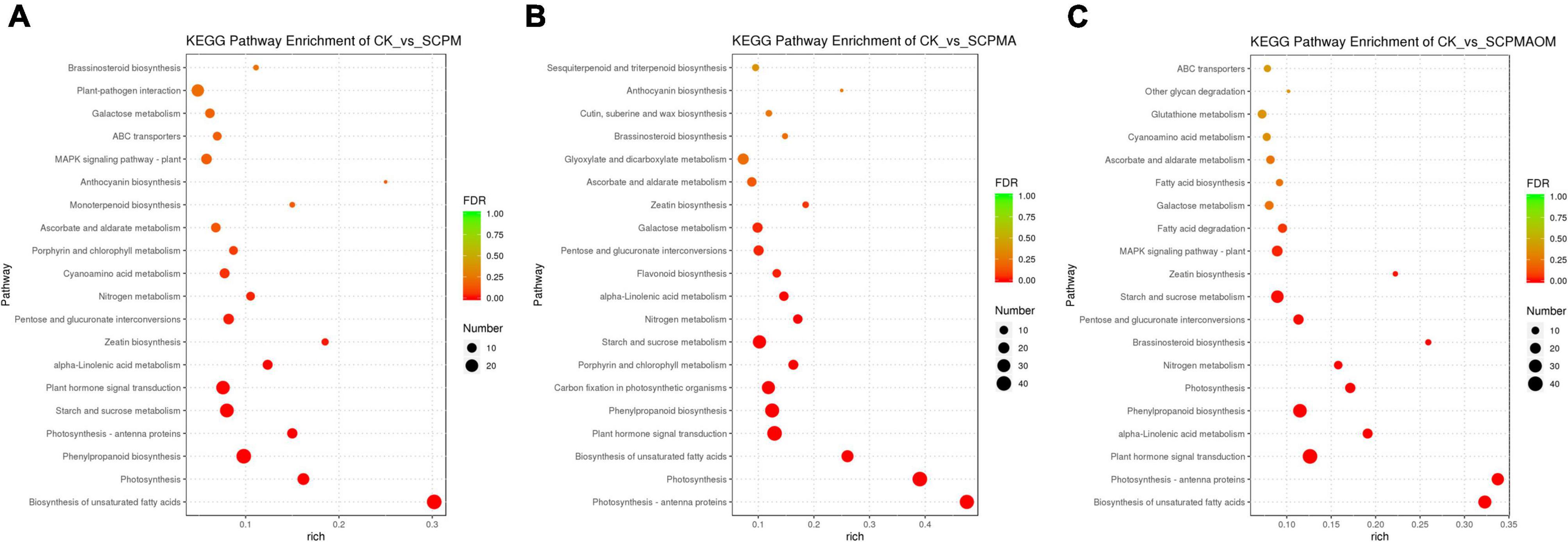
Figure 3. The KEGG pathways enrichment analysis of DEGs from different comparisons. (A) KEGG pathways enriched in R1 comparison. (B) KEGG pathways enriched in R2 comparison. (C) KEGG pathways enriched in R3 comparison. R1, CK vs. SCPM; R2, CK vs. SCPMA; R3, CK vs. SCPMAOM.
Differentially expressed genes related to photosynthesis pathway
A majority of DEGs involved in photosynthesis were identified (Figure 4). In the R1 comparison, 16 up-regulated DEGs were involved in the biological process including photosystem II (6 DEGs), cytochrome b6/f complex (2 DEGs), photosynthetic electron transport (3 DEGs), photosystem I (3 DEGs) and F-type ATPase (2 DEGs). The equivalent distribution for other comparisons were as follows: R2 comparison, 9, 1, 3, 8, and 4 DEGs, respectively; R3 comparison, 5, 4, 2, 5, and 1 DEGs, respectively. The genes encoding PsbO, PsbQ, and Psb27 in photosystem II, PsaF and PsaN in photosystem I, as well as PetG in cytochrome b6/f complex, were all up-regulated in the three treatments (SCPM, SCPMA, and SCPMAOA). For photosynthetic electron transport, the genes including PetE, PetF were significantly up-expressed in these three treatments. In addition, the delta gene associated with the critical element F-type ATPase was up-regulated in all the remediation treatments (Figure 4). The results showed that all the DEGs involved in photosynthesis pathway were up-regulated in SCPM, SCPMA and SCPMAOM treatments, compared with the control, thus indicating the significantly up-expressed genes especially the crucial DEGs related to photoreaction and cal-vin cycle might greatly promote the photosynthesis efficiency.
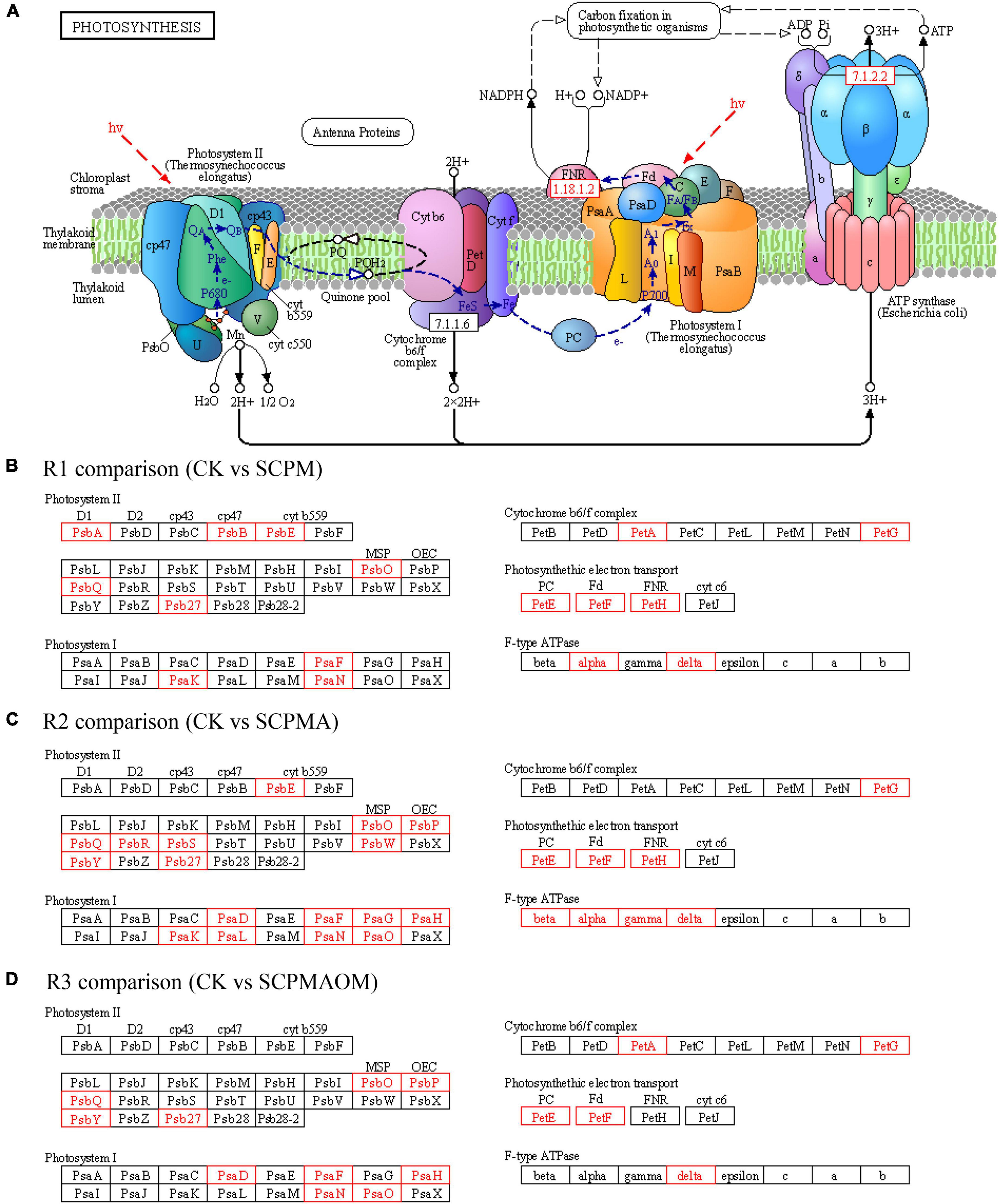
Figure 4. Differentially expressed genes (DEGs) associated with photosynthesis pathway (A) in R1 (B), R2 (C), and R3 (D) comparisons. Red frames denoted up-regulated genes, while black frames denoted genes showing no differential expression. R1, CK vs. SCPM; R2, CK vs. SCPMA; R3, CK vs. SCPMAOM.
Differentially expressed genes associated with hormone signal transduction pathway
Phytohormones play important roles in plant growth and regulation. Multiple genes related to hormone signal transduction pathway were modulated in all the remediation treatments (SCPM, SCPMA, and SCPMAOM) (Figure 5). The DEGs involved in the pathway of auxin signal transduction, such as SAUR, GH3, and AUX/IAA, were significantly up-regulated, compared with the control. In brassinosteroid signal transduction pathway, the CYCD3 gene was significantly up-regulated. In addition, the A-ARR genes associated with cytokinin signal transduction pathway were greatly stimulated in all the treatments. Contrarily, the pathways of ethylene and jasmonic acid signal transductions, were remarkably inhibited in all the remediation treatments. The ERF1/2 and JAZ genes involved in these pathways were significantly down-regulated, thereby slowing down the senescence of C. tangshen in the remediation treatments.
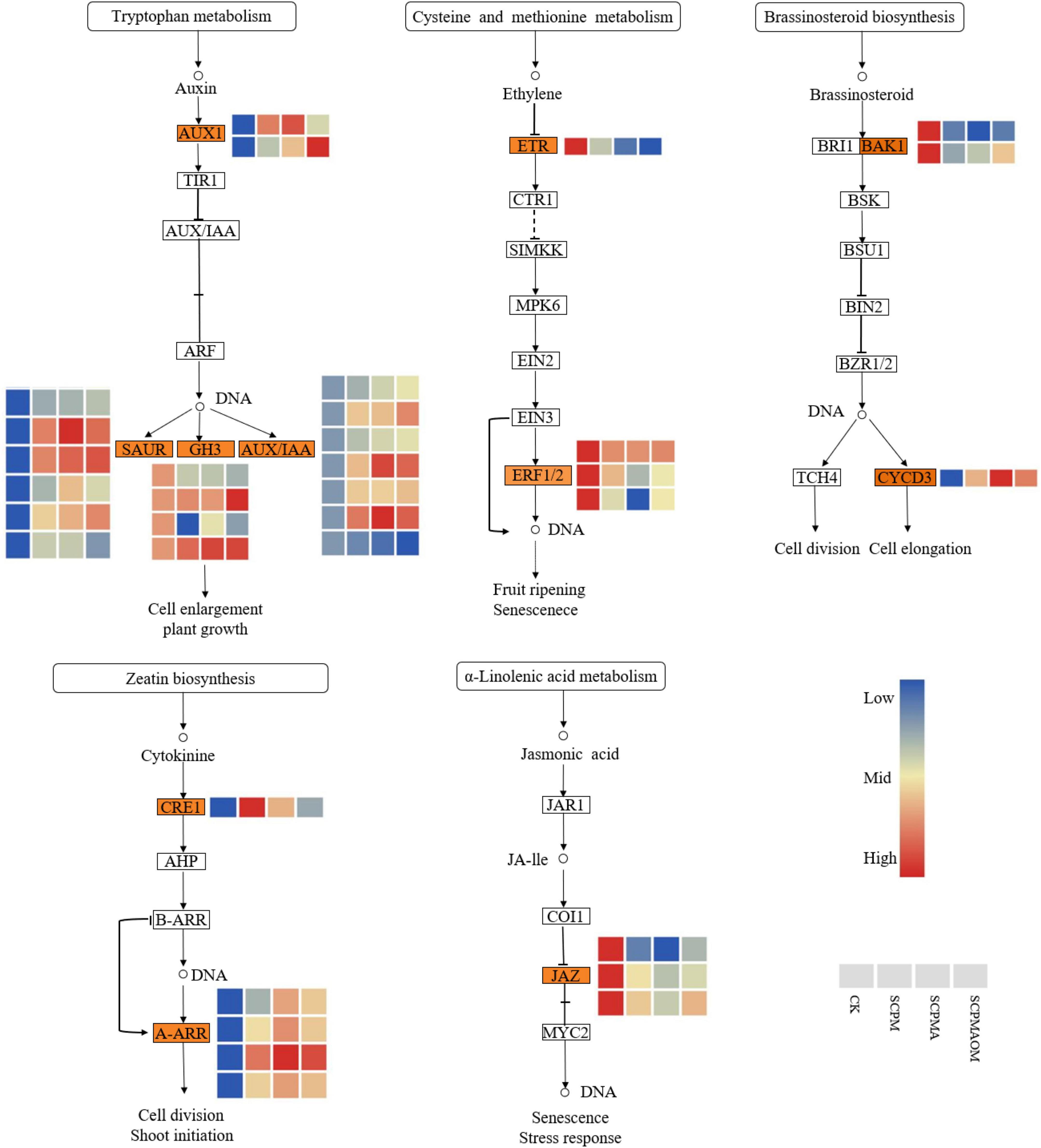
Figure 5. Heat map of DEGs involved in plant hormone signal transduction pathways. Red color denoted genes with high expression level, while blue color denoted genes with low expression level. The relative expression levels of DEGs were calculated using the log2 ratio.
Differentially expressed genes involved in phenylpropanoid biosynthesis pathway
A total of 35 DEGs were annotated into phenylpropanoid biosynthesis pathway. These DEGs were involved in biosynthesis of coumarin, lignin monomers and monoterpenoid (Figure 6). Out of these DEGs, eight genes encoding β-glucosidase (BGL) were remarkably down-regulated in the C. tangshen treated with soil amendments. Moreover, 10 genes encoding peroxidase (POD) were down-regulated in the three remediation treatments. Two genes encoding cinnamyl alcohol dehydrogenase (CAD) were significantly down-regulated in all the remediation treatments. Additionally, two genes encoding 4-coumarate–CoA ligase (4CL) were significantly down-regulated in the three remediation treatments, which might indicate that the biosynthesis of lignin was inhibited in the remediation process.
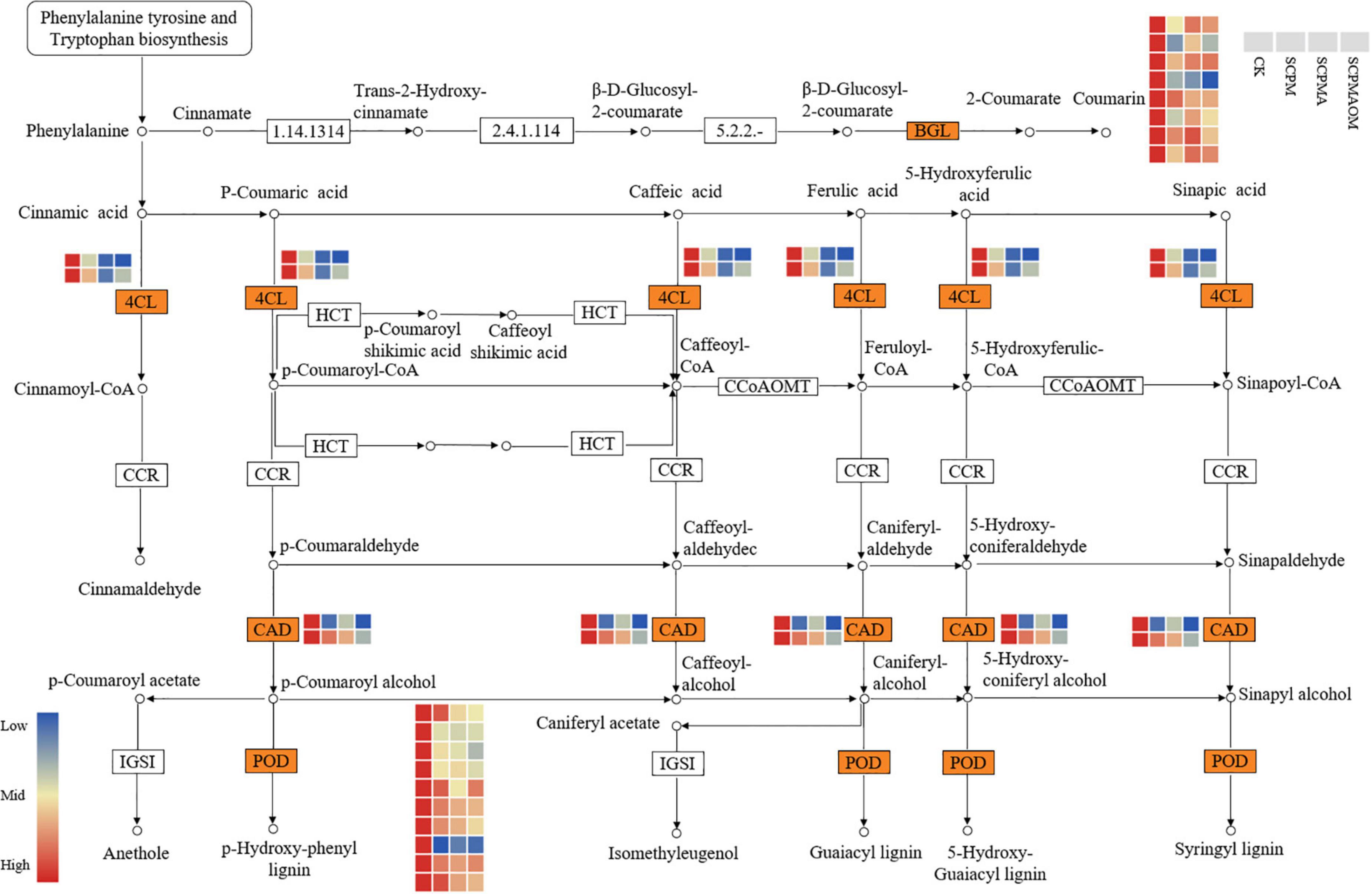
Figure 6. Heat map of DEGs related to phenylpropanoid biosynthesis. Red color denoted genes with high expression level, while blue color denoted genes with low expression level. The relative expression levels of DEGs were calculated using the log2 ratio.
Differentially expressed genes related to biosynthesis of unsaturated fatty acids
Twenty seven of 29, 23 of 25, and 28 of 31 DEGs involved in unsaturated fatty acids biosynthesis were down-regulated in R1, R2, and R3 comparisons, respectively. The results showed that all the genes encoding fatty acid desaturase (FAD) were significantly down-regulated in all the remediation treatments, compared with the control (Figure 7A). Three genes encoding 3-ketoacyl-CoA thiolase (3-KAT), were significantly down-regulated in the three remediation treatments, and the expression levels were lowest in the SCPMAOM treatment. In addition, one gene encoding acyl-coenzyme A oxidase 3 (ACOX3), which was involved in the first step of fatty acids β-oxidation, was significantly down-regulated in all the treatments. Moreover, the ACOX3 gene also participated in the pathway of α-linolenic acid metabolism. Additionally, in the jasmonic acid biosynthesis pathway, one gene encoding allene oxide synthase (AOS) was significantly down-regulated in the three remediation treatments, compared with the control. Interestingly, three 3-KAT genes involved in unsaturated fatty acids biosynthesis pathway were also showed in α-linolenic acid metabolism (Figure 7B). Moreover, the expression levels of three genes encoding linoleate 13S-lipoxygenase (LOX) were significantly lower in the three remediation treatments than those in the control.
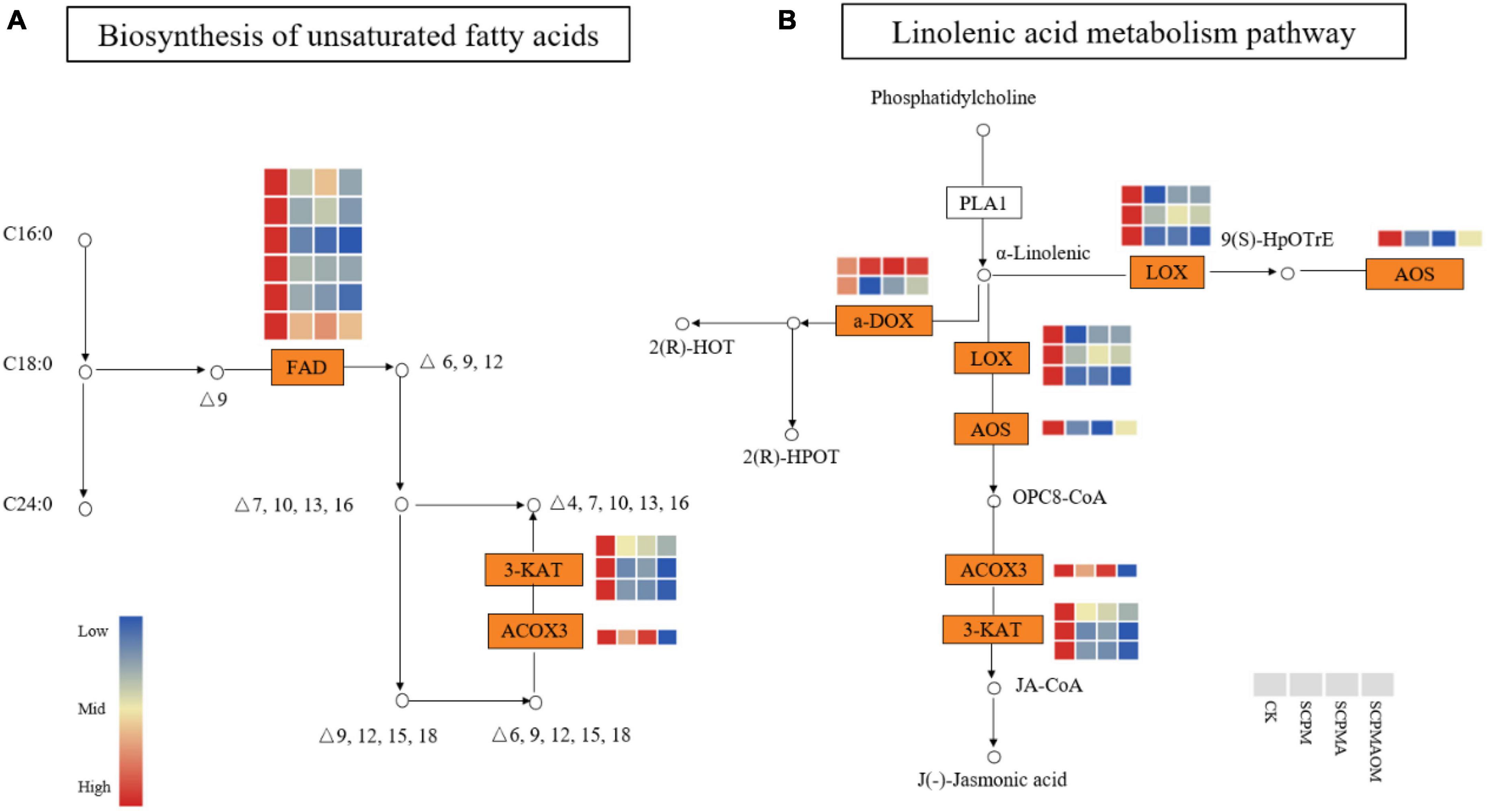
Figure 7. The expression patterns of DEGs in the biosynthetic pathways of unsaturated fatty acid (A) and α-linolenic acid (B). Red color denoted genes with high expression level, while blue color denoted genes with low expression level. The relative expression levels of DEGs were calculated using the log2 ratio.
Differentially expressed genes associated with starch and sucrose metabolism
Multiple genes were significantly enriched in the pathway of starch and sucrose metabolism in C. tangshen treated with soil amendments (Figure 8). The results showed that 25, 32, and 28 DEGs were involved in starch and sucrose metabolism in R1, R2, and R3 comparisons, respectively. Generally, α-amylase (AMY) and isoamylase (ISA) participate in the conversion of starch to maltose. AMY gene was significantly down-regulated, while ISA gene was significantly up-regulated in the SCPMAOM treatment. In the biological process of sucrose metabolism, six genes encoding β-fructofuranosidase (INV) were significantly down-regulated, while two genes encoding sucrose-phosphate synthase (SPS) were significantly up-regulated in the three remediation treatments. Moreover, in the initial stage of UDP-glucose metabolism, four genes encoding β-glucosidase (BGL) and one gene encoding α, α-trehalose-phosphate synthase (TPS) were all down-regulated in the three treatments.
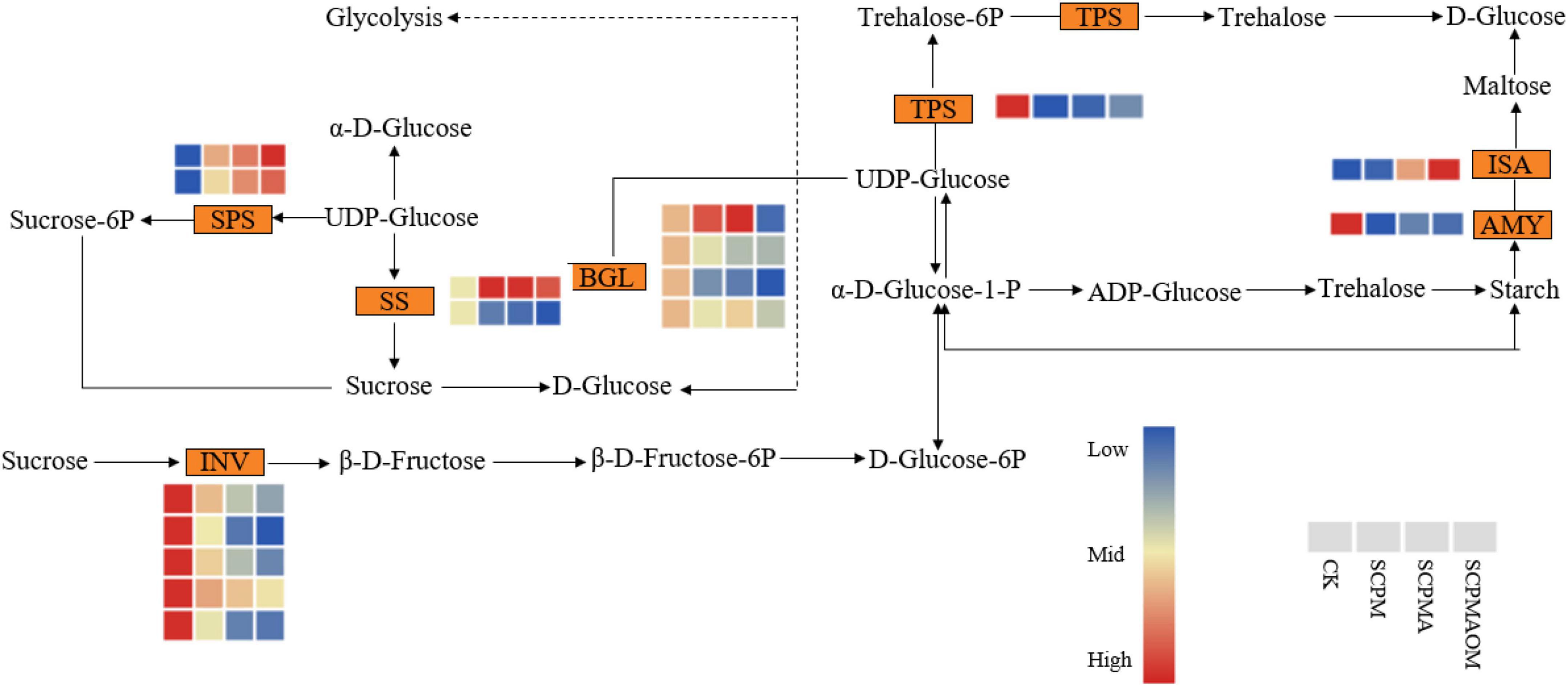
Figure 8. Differentially expressed genes related to the starch and sucrose metabolism pathways in the different treatments. Red color denoted genes with high expression level, while blue color denoted genes with low expression level. The relative expression levels of DEGs were calculated using the log2 ratio.
Validation of the differentially expressed genes by qRT-PCR
Ten unigenes responsive to soil remediation were chosen to validate the RNA-Seq results by qRT-PCR (Figure 9). The expression levels of five photosynthesis-related unigenes (CP26, PsaF, TL19, PsbQ, and Psb27) were significantly increased in the three remediation treatments, while five genes (POX, PER, RBOHD, 2-ODD, and AOO) involved in antioxidant system and redox reaction were remarkably down-regulated in all the remediation treatments. In summary, the expression profiles of these unigenes based on the qRT-PCR were consistent with the RNA-Seq data, indicating the RNA-Seq results were reliable and accurate.
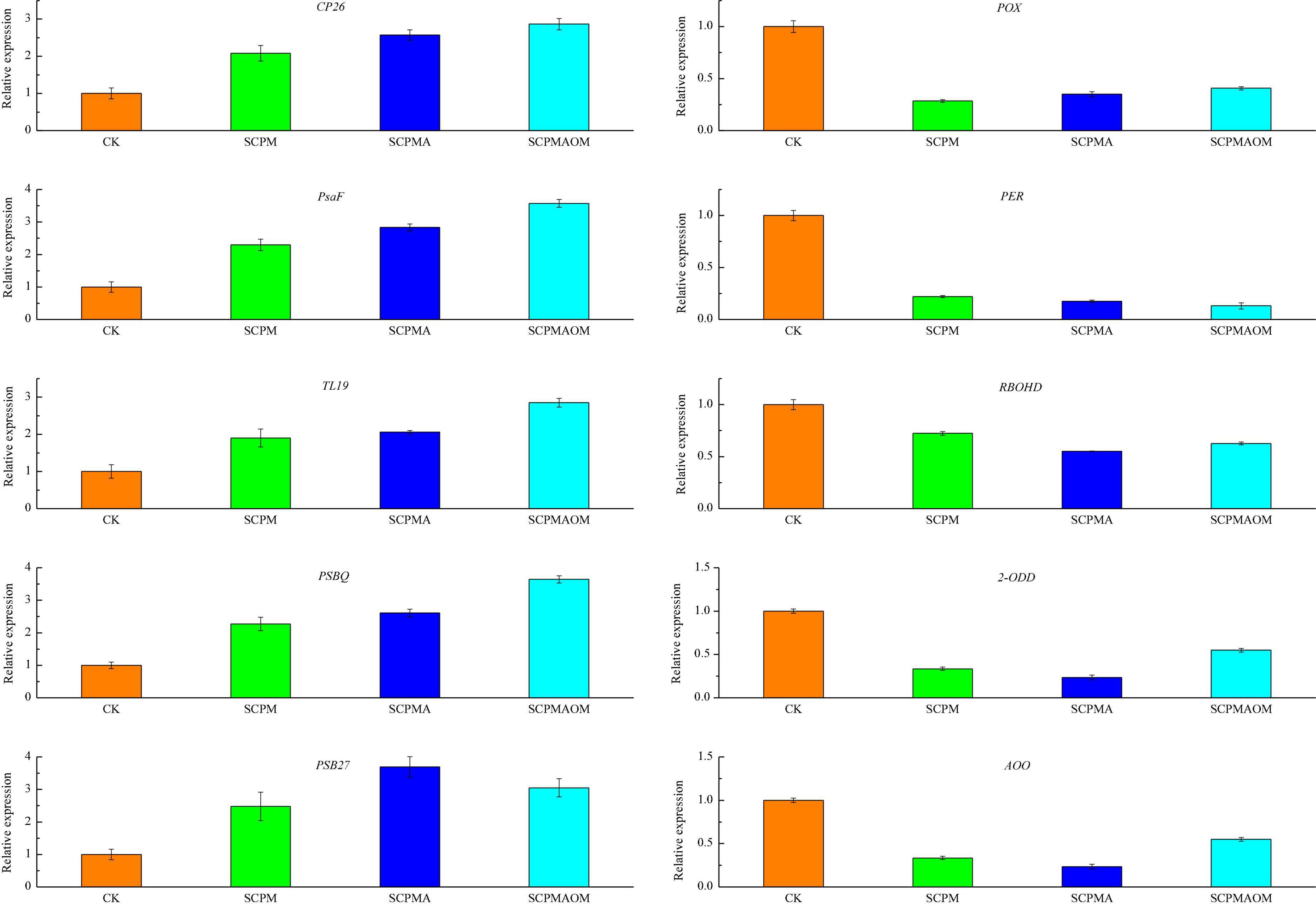
Figure 9. The qRT-PCR analysis of 10 DEGs in the leaves of C. tangshen in different treatments. GAPDH gene was used as the reference gene for normalization of gene expression level. Three technical replicates were performed.
Discussion
Physiological and biochemical responses
Continuous cropping obstacle is a serious problem for agricultural production of C. tangshen (He et al., 2019; Zhang et al., 2021). In this study, the treatments such as SCPM, SCPMA, and SCPMAOM greatly alleviated the adverse effects of continuous cropping on C. tangshen. Photosynthetic pigments, such as chlorophyll a, chlorophyll b and carotenoids are essential for plant growth and development (Kamble et al., 2015). In the present work, the chlorophyll (a, b), total chlorophyll and carotenoid levels were higher in the remediation treatments than those in the control. This was consistent with the results of Zhou et al. (2021), which indicated that organic manure (OM) and silicon–calcium–potassium–magnesium fertilizer (SCPM) could remarkably increase the chlorophyll and carotenoid content in continuously cropped C. tangshen. Moreover, the chlorophyll and carotenoid levels were significantly higher in SCPMAOM than those in CK, SCPM, and SCPMA, indicating that silicon-calcium-potassium-magnesium fertilizer, azoxystroxbin and organic manure have synergistic effects on photosynthetic metabolism in C. tangshen.
Antioxidant enzymes, e.g., SOD, POD and CAT, are important proteins related to the protection of the plasma membrane through prevention of peroxidation in plants (Gill and Tuteja, 2010; Kapoor et al., 2019). In this study, the significantly decreased SOD and slightly increased POD activities were occurred in the three remediation treatments (SCPM, SCPMA, and SCPMAOM), and the CAT activity was significantly increased in SCPMA and SCPMAOM treatments, which was basically consistent with previous study (Zhou et al., 2021). These results suggested the increased POD and CAT activities might be involved in the prevention of peroxidation in C. tangshen. Comprehensively, the levels of yield and alkaloid were significantly increased in the three remediation treatments, and the yield of SCPMAOM treatment was 10 times higher than that of the control. Moreover, the levels of polysaccharide, flavone, alkaloid and total protein were all increased in the three remediation treatments and these indexes reached significant level (p < 0.05) in SCPMAOM treatment. This indicating that the comprehensive stimulative effects on yield and quality (polysaccharide, flavone, alkaloid, and total protein) followed the order of SCPMAOM > SCPMA > SCPM > CK, which further certificated that silicon-calcium-potassium-magnesium fertilizer, azoxystroxbin and organic manure had cumulative effects on the growth and development of C. tangshen under continuous cropping system.
Differentially expressed genes related to photosynthesis
Photosynthetic apparatuses represented by photosystem II, cytochrome b6/f complex, photosystem I and F-type ATPase are easily influenced by environmental changes (Dey et al., 2022). Numerous studies in the past decades have demonstrated that the extrinsic PsbO, PsbP, and PsbQ proteins play important roles in maintaining optimal manganese, calcium and chloride concentrations at the active site of photosystem II, and chemical or genetic removal of these components will induce multiple and profound defects in photosystem II function and oxygen-evolving complex stability (Bricker and Frankel, 2011; Ifuku and Nagao, 2021). In this study, the PsbO, PsbP, and PsbQ genes in C. tangshen were significantly up-regulated in all the remediation treatments, indicating that the soil amendments might promote the photosynthetic metabolism of C. tangshen by enhancing the functions of photosystem II. Similarly, the significantly up-regulated genes of PsaF and PsaN in photosystem I might also play a pivotal role in stimulating the photosynthesis of C. tangshen under remediation system.
Cytochrome b6f complex is an enzyme found in plants, cyanobacteria, and green algae which catalyzes the electron transport in the rate-limiting step of oxygenic photosynthesis (Mehling, 2020). The results showed that the PetA and PetG genes were greatly up-regulated in both SCPM and SCPMAOM treatments, demonstrating that PetA and PetG might play a key role in the electron transfer involved in the photosynthesis of C. tangshen under remediation system. F-type ATPase, which resides in the chloroplast thylakoid membranes, can transform ADP into ATP and provide energy for carbon fixation in photosynthetic organism (Kühlbrandt, 2019). The results showed that ATPase components including beta, alpha, gamma, and delta subunits were integrated and activated in the ATP synthesis pathway, indicating plenty of ATP was synthesized, which contributed to the carbon fixation in photosynthetic organism in C. tangshen during the soil remediation. Interestingly, the enhanced expressions of genes involved in photosynthetic metabolism were consistent with the increased chlorophyll and carotenoid levels in soil remediation treatments, indicating that the soil amendments might promote the growth of continuously cropped C. tangshen by improvement of photosynthesis.
Differentially expressed genes associated with plant hormone signal transduction
Plant hormones are always associated with the regulation of growth and environmental adaptability in higher plants (Kou et al., 2021). Auxin (IAA) plays a key role in regulating plant growth, development and response to environmental stresses (Liu et al., 2014; Singh et al., 2021). In this study, most of the genes involved in IAA signal transduction pathway, such as SAUR, GH3, and AUX1, were significantly up-regulated in the soil remediation treatments, thereby stimulating the cell enlargement and plant growth. This result was in accordance with the higher yield in the soil remediation treatments (Table 3). Brassinosteroids (BRs) are steroid hormones which are essential for plant growth and development. These hormones control the division, elongation and differentiation of various cell types throughout the entire plant life cycle (Planas-Riverola et al., 2019). The D-type cyclin (CYCD) gene family plays an important role in promoting cell division in BR signal transduction (Dewitte et al., 2007). In our study, the expression levels of CYCD3 genes were significantly higher in the soil remediation treatments than those in the control, which might improve the mitotic activity in cambium. Hence, it was comprehensible that the stronger roots and higher yields of C. tangshen were observed in SCPM, SCPMA, and SCPMAOM treatments, compared with the control (Table 3).
The phytohormone cytokinin (CK) plays diverse roles in plant development, cell division and shoot initiation (Kieber and Schaller, 2018). In our study, the expression levels of CRE1 and A-ARR genes involved in the CK signal transduction were significantly increased by all the soil remediation treatments, which might finally promote the cell division and shoot meristem initiation in C. tangshen. Ethylene-responsive transcription factors (ERFs) play important roles in tolerance to biotic and abiotic stresses by regulating the expression of stress-responsive genes (Wu et al., 2014). The expressions of ERF1/2 genes were greatly decreased by all the remediation treatments, indicating that the defensive response was not completely activated due to the slighter stress in the soil remediation treatments. Jasmonic acid (JA) plays important roles in plant growth and development, especially plant responses to biotic and abiotic stresses (Ruan et al., 2019). In the present work, the JAZ genes involved in the JA signal transduction pathway were significantly down-regulated in the soil remediation treatments, indicating that the soil remediation measures might alleviate the continuous cropping induced stress in C. tangshen, which resulting in the down-regulation of JAZ genes in remediation system.
Differentially expressed genes involved in phenylpropanoid biosynthesis
Reportedly, the phenylpropanoid biosynthesis is usually involved in defense metabolisms (Yadav et al., 2020). For instance, some phenylpropanoids are important substrates for the synthesis of lignins and play key roles as protectants, phytoalexins and antioxidants in most of the higher plants (Deng and Lu, 2017). The genes associated with lignin, coumarin, and monoterpenoid biosynthesis, such as BGL, 4CL, and CAD, were involved in the biological responses to various stresses (Krithika et al., 2015; Baba et al., 2017; Qiu et al., 2018). In this study, most of the genes encoding BGL, 4CL, and CAD were significantly down-regulated in the soil remediation treatments, indicating that the C. tangshen in remediation treatments were grown in a better environmental condition than those in the control. Peroxidase (POD) is involved in antioxidant activity and cell wall lignification, which protect plants from damages induced by various stress (Lee et al., 2021). Previous studies showed that greatly expressed POD genes were found involved in antioxidant activity and lignin synthesis in chestnut and tea plants (Cao et al., 2020; Wu et al., 2022). In this study, most of the POD genes were significantly down-regulated in the three soil remediation treatments (SCPM, SCPMA, and SCPMAOM), which again validated the functions of soil amendments in alleviating the continuous cropping induced stress.
Differentially expressed genes related to unsaturated fatty acid metabolism
Some fatty acids play critical roles in plant defense against abiotic stress conditions (Begara-Morales et al., 2021). Previous studies showed that unsaturated fatty acids were closely related to membrane stability, and positively correlated with the degree of environmental stress (Liu et al., 2011; Chen et al., 2022). Fatty acid desaturase 2 (FAD2) catalyses oleic acid (OA) to linoleic acid (LA) in the second step of fatty acid desaturation pathway (Xue et al., 2017; Fan et al., 2019). In this study, all the FAD2 genes were down-regulated in the three remediation treatments, which might lead to the decreased biosynthesis of unsaturated fatty acids, indicating that the abiotic stress in C. tangshen were mitigated by soil remediation treatments. In α-linolenic acid metabolism, acyl-coenzyme A oxidase 3 (ACOX3) and 3-ketoacyl-CoA thiolase (3-KAT) were involved in the first and second step of fatty acid β-oxidative cracking reactions, respectively (Froman et al., 2000; Sundaramoorthy et al., 2006). Our study showed that all the ACOX3 and 3-KAT genes were significantly down-regulated in the three remediation treatments, and the lowest expressions were observed in SCPMAOM treatment, indicating less α-linolenic acids were accumulated in SCPMAOM treatment. Thus, we speculated that SCPMAOM showed the best remediation effects on the continuous cropping soil.
Differentially expressed genes engaged in starch and sucrose metabolism
α-amylase (AMY) was involved in starch conversion into dextrins and fermentable sugars in crops like corn, barley, potato, or rice (Li et al., 2022), and isoamylase (ISA) could hydrolyze α-1,6-glucosidic linkages in amylopectin to yield amylose and oligosaccharides (Li et al., 2017b). In this study, the AMY gene was significantly down-regulated in the three remediation treatments, which suggested the degradation of starch might be reduced and the accumulation of starch promoted the growth and development of C. tangshen in remediation treatments. However, the ISA expression change was inconsistent with that of AMY, suggesting the complexity of starch metabolism in C. tangshen and further verification was required. In sucrose metabolism, β-fructofuranosidase (INV) possesses the transfructosylation activity of transferring sucrose into fructose and glucose, while sucrose-phosphate synthase (SPS) is related to sucrose synthesis (Anur et al., 2020). The results showed that the INV genes were significantly down-regulated, while the SPS genes were significantly up-regulated in the SCPM, SCPMA, and SCPMAOM treatments, indicating the degradation of sucrose was inhibited and the sucrose biosynthesis was stimulated, which might gradually promote the growth and development of C. tangshen in remediation treatments. Additionally, the highest expression of SPS and the lowest expression of INV were observed in the SCPMAOM treatment, which again validated the best remediation effects of SCPMAOM.
Conclusion
This study firstly demonstrated how continuously cropped C. tangshen responded to soil amendments at the transcriptional level. The yield and quality was improved by the remediation treatments, and the SCPMAOM treatment (3000 kg/hm2 silicon-calcium-potassium-magnesium fertilizer combined with 500 times azoxystrobin solution and 4500 kg/hm2 organic manure) showed the best remediation effects on the continuous cropping soil. The results revealed that the soil amendments improved the tolerance of C. tangshen to continuous cropping by regulating the genes involved in pathways like ‘photosynthesis,’ ‘photosynthesis-antenna proteins,’ ‘plant hormone signal transduction,’ ‘phenylpropanoid biosynthesis,’ ‘biosynthesis of unsaturated fatty acids,’ and ‘starch and sucrose metabolism’ (Figure 10). The synergies of these pathways improved the growth and development of continuously cropped C. tangshen in soil remediation treatments. This study was beneficial for unraveling the molecular mechanisms of soil amendments in regulating the growth of continuously cropped C. tangshen at transcriptome level and provided a technical support for improving the tolerance of C. tangshen to continuous cropping.
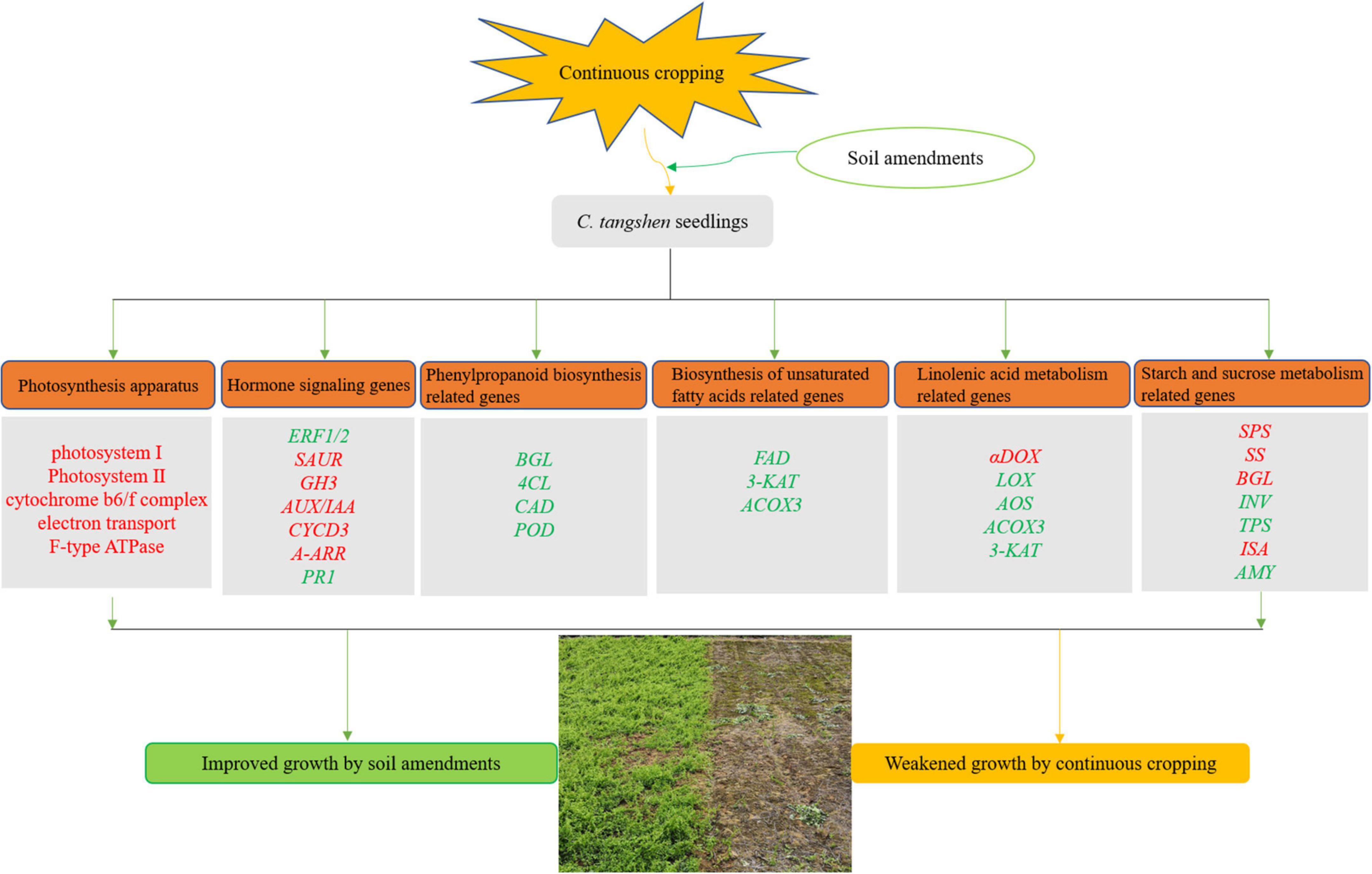
Figure 10. A hypothetical model of soil amendments induced response in continuously cropped C. tangshen. The apparatus or genes labeled with red were up regulated, while labeled with green were down regulated.
Data availability statement
The original contributions presented in this study are publicly available. This data can be found here: https://ngdc.cncb.ac.cn/, CRA006788.
Author contributions
WZ and XJ designed the experiment and completed the manuscript writing. XT and JY participated in the experimental design. WZ, XJ, DL, HW, and XL carried out the experiment and processed the experimental data. MZ reviewed and finalized the manuscript. All authors reviewed and approved the manuscript.
Funding
This work was supported by The Special Fund for the Construction of Modern Agricultural Industrial Technology System (CARS-21) and Hubei Technology Innovation Center for Agricultural Sciences-2020 key technology research and demonstration project of safe and efficient production of genuine medicinal materials (2020-620-000-002-04).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.972804/full#supplementary-material
Supplementary Figure 1. Leaf morphology of Codonopsis tangshen.
Supplementary Figure 2. Correlation analysis of gene expression profiles.
Supplementary Figure 3. The principal component analysis (PCA) of gene expression profiles.
Supplementary Table 1. The information of primers used for qRT-PCR.
Supplementary Table 2. Go and KEGG annotation of CK vs SCPM, CK vs SCPMA, and CK vs SCPMAOM comparisons.
References
Anur, R. M., Mufithah, N., Sawitri, W. D., Sakakibara, H., and Sugiharto, B. (2020). Overexpression of sucrose phosphate synthase enhanced sucrose content and biomass production in transgenic sugarcane. Plants-Basel 9:200. doi: 10.3390/plants9020200
Baba, S. A., Vishwakarma, R. A., and Ashraf, N. (2017). Functional characterization of CsBGlu12, a β-glucosidase from Crocus sativus, provides insights into its role in abiotic stress through accumulation of antioxidant flavonols*. J. Biol. Chem. 292, 4700–4713. doi: 10.1074/jbc.M116.762161
Begara-Morales, J. C., Mata-Pérez, C., Padilla, M. N., Chaki, M., Valderrama, R., Aranda-Caño, L., et al. (2021). Role of electrophilic nitrated fatty acids during development and response to abiotic stress processes in plants. J. Exp. Bot. 72, 917–927. doi: 10.1093/jxb/eraa517
Bricker, T. M., and Frankel, L. K. (2011). Auxiliary functions of the psbO, psbP and psbQ proteins of higher plant photosystem II: a critical analysis. J. Photochem. Photobiol. B-Biol. 104, 165–178. doi: 10.1016/j.jphotobiol.2011.01.025
Cao, H., Li, J., Ye, Y., Lin, H., Hao, Z., Ye, N., et al. (2020). Integrative transcriptomic and metabolic analyses provide insights into the role of trichomes in tea plant (Camellia sinensis). Biomolecules 10:311. doi: 10.3390/biom10020311
Chen, G., Hou, Y., Zheng, Y., and Jin, P. (2022). 2,4-epibrassinolide enhance chilling tolerance of loquat fruit by regulating cell wall and membrane fatty acid metabolism. Sci. Hort 295:110813. doi: 10.1016/j.scienta.2021.110813
Chen, P., Wang, Y. Z., Liu, Q. Z., Li, W. H., Li, H. Q., Li, X. Y., et al. (2020). Transcriptomic analysis reveals recovery strategies in strawberry roots after using a soil amendment in continuous cropping soil. BMC Plant Biol. 20:5. doi: 10.1186/s12870-019-2216-x
Deng, Y., and Lu, S. (2017). Biosynthesis and regulation of phenylpropanoids in plants. Crit. Rev. Plant Sci. 36, 257–290. doi: 10.1080/07352689.2017.1402852
Dewitte, W., Scofield, S., Alcasabas, A. A., Maughan, S. C., Menges, M., Braun, N., et al. (2007). Arabidopsis CYCD3 D-type cyclins link cell proliferation and endocycles and are rate-limiting for cytokinin responses. Proc. Natl. Acad. Sci. U S A. 104, 14537–14542. doi: 10.1073/pnas.0704166104
Dey, P., Datta, D., Pattnaik, D., Dash, D., Saha, D., Panda, D., et al. (2022). "Physiological, biochemical, and molecular adaptation mechanisms of photosynthesis and respiration under challenging environments. Plant Perspect. Global Climate Changes 2022, 79–100.
Fan, R., Li, L., Cai, G., Ye, J., Liu, M., Wang, S., et al. (2019). Molecular cloning and function analysis of FAD2 gene in Idesia Polycarpa. Phytochemistry 168:112114. doi: 10.1016/j.phytochem.2019.112114
Froman, B., Edwards, P., Bursch, A., and Dehesh, K. (2000). ACX3, a novel medium-chain acyl-coenzyme a oxidase from Arabidopsis. Plant Physiol. 123, 733–742. doi: 10.1104/pp.123.2.733
Gill, S., and Tuteja, N. (2010). Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol. Biochem. 48, 909–930. doi: 10.1016/j.plaphy.2010.08.016
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
He, Y., Zhang, M., Zhou, W., Ai, L., et al. (2019). Transcriptome analysis reveals novel insights into the continuous cropping induced response in Codonopsis Tangshen, a medicinal herb. Plant Physiol. Bioch. 141, 279–290. doi: 10.1016/j.plaphy.2019.06.001
Ifuku, K., and Nagao, R. (2021). Evolution and Function of the Extrinsic Subunits of Photosystem II, Photosynthesis: Molecular Approaches to Solar Energy Conversion. Berlin: Spring.
Ji, J. H., Li, X. H., Liu, X. M., Hou, H. Q., Liu, Y. R., Lyu, Z. Z., et al. (2019). Effects of fertilizer of calcium silicon magnesium potassium on the dynamics of soil acidity and exchangeable base cation in paddy field of southern China. J. Appl. Ecol. 30, 583–592. doi: 10.13287/j.1001-9332.201902.024
Kamble, P., Giri, S., Mane, R., and Tiwana, A. (2015). Estimation of chlorophyll content in young and adult leaves of some selected plants. Univers. J. Environ. Res. Tech. 5, 306–310. doi: 10.1016/j.agrformet.2009.01.005
Kapoor, D., Singh, S., Kumar, V., Romero, R., Prasad, R., and Singh, J. (2019). Antioxidant enzymes regulation in plants in reference to reactive oxygen species (ROS) and reactive nitrogen species (RNS). Plant Gene. 19:100182. doi: 10.1016/j.plgene.2019.100182
Kenneth, J. L., and Thomas, D. S. (2002). Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Meth 25, 402–408. doi: 10.1006/meth.2001.1262
Kieber, J. J., and Schaller, G. E. (2018). Cytokinin signaling in plant development. Dev 145:dev149344. doi: 10.1242/dev.149344
Kou, X., Feng, Y., Yuan, S., Zhao, X., Wu, C., Wang, C., et al. (2021). Different regulatory mechanisms of plant hormones in the ripening of climacteric and non-climacteric fruits: a review. Plant Mol. Biol. 2021, 477–497. doi: 10.1007/s11103-021-01199-9
Krithika, R., Srivastava, P., Bajaj, R., Kolet, S., Chopade, M., Soniya, M., et al. (2015). Characterization of 10-hydroxygeraniol dehydrogenase from Catharanthus roseus reveals cascaded enzymatic activity in iridoid biosynthesis. Sci. Rep. 5:8258. doi: 10.1038/srep08258
Kühlbrandt, W. (2019). Structure and mechanisms of F-type ATP synthases. Annu. Rev. Biochem. 88, 515–549.
Lee, C. J., Park, S. U., Kim, S. E., Lim, Y. H., Ji, C. Y., Kim, Y. H., et al. (2021). Overexpression of IbLfp in sweetpotato enhances the low-temperature storage ability of tuberous roots. Plant Physiol. Bioch. 167, 577–585. doi: 10.1016/j.plaphy.2021.08.041
Li, B., and Dewey, C. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 12:323. doi: 10.1186/1471-2105-12-323
Li, D., Chen, C., Tao, Y., Huang, Y., Wang, P., and Han, Y. (2022). Changes in the structural and catalytic characteristics of α-amylase under moderate electric field. Food Hydrocolloid 130:107717. doi: 10.1016/j.foodhyd.2022.107717
Li, H., Hu, T., Amombo, E., and Fu, J. (2017a). Transcriptome profilings of two tall fescue (Festuca arundinacea) cultivars in response to lead (Pb) stress. BMC Genomics 18:145. doi: 10.1186/s12864-016-3479-3
Li, Y., Xu, J., Zhang, L., Ding, Z., Gu, Z., and Shi, G. (2017b). Investigation of debranching pattern of a thermostable isoamylase and its application for the production of resistant starch. Carbohyd. Res. 44, 93–100. doi: 10.1016/j.carres.2017.05.016
Liu, H., Song, L., You, Y., Li, Y., Duan, X., Jiang, Y., et al. (2011). Cold storage duration affects litchi fruit quality, membrane permeability, enzyme activities and energy charge during shelf time at ambient temperature. Postharvest. Biol. Tec. 60, 24–30. doi: 10.1016/j.postharvbio.2010.11.008
Liu, L., Guo, G., Wang, Z., Ji, H., and Xia, L. (2014). Auxin in Plant Growth and Stress Responses. New York, NY: Springer Science:, doi: 10.1007/978-1-4939-0491-41
Love, M., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Lu, C. C., Guo, N., Yang, C., Sun, H. B., and Cai, B. Y. (2020). Transcriptome and metabolite profiling reveals the effects of Funneliformis mosseae on the roots of continuously cropped soybeans. BMC Plant Biol. 20:479. doi: 10.21203/rs.3.rs-31120/v1
Maiorano, A., Piccoli, R., Sabino, E., and Rodrigues, M. (2008). Microbial production of fructosyltransferases for synthesis of pre-biotics. Biotechnol. Lett. 30, 1867–1877. doi: 10.1007/s10529-008-9793-3/qr {Citethefollowingreferencesinsidethetext.} Maioranoetal., 2008; Santneretal., 2009; Yoshidaetal., 2001.
Mehling, M. (2020). Investigating photosynthetic stability: relation between thylakoid lipid content and the stability of the cytochrome b6f complex. J. Purdue Undergrad. Res. 10:15. doi: 10.7771/2158-4052.1463
Planas-Riverola, A., Gupta, A., Betegón-Putze, I., Bosch, N., Ibañes, M., and Caño-Delgado, A. I. (2019). Brassinosteroid signaling in plant development and adaptation to stress. Development 146:dev151894. doi: 10.1242/dev.151894
Qiu, W., Song, X., Han, X., Liu, M., Qiao, G., and Zhuo, R. (2018). Overexpression of sedum alfredii cinnamyl alcohol dehydrogenase increases the tolerance and accumulation of cadmium in arabidopsis. Environ. Exp. Bot. 155, 566–577. doi: 10.1016/j.envexpbot.2018.08.003
R Core Team (2012). R: a Language and Environment for Statistical Computing. Vienna: R foundation for statistical computing.
Ruan, J., Zhou, Y., Zhou, M., Yan, J., Khurshid, M., Weng, W., et al. (2019). Jasmonic acid signaling pathway in plants. Int. J. Mol. Sci. 20:2479. doi: 10.3390/ijms20102479
Santner, A., Calderon-Villalobos, L., and Estelle, M. (2009). Plant hormones are versatile chemical regulators of plant growth. Nat. Chem. Biol. 5, 301–307. doi: 10.1038/nchembio.165
Singh, H., Bhat, J. A., Singh, V. P., Corpas, F. J., and Yadav, S. R. (2021). Auxin metabolic network regulates the plant response to metalloids stress. J. Hazard. Mater. 405:124250. doi: 10.1016/j.jhazmat.2020.124250
Stark, R., Grzelak, M., and Hadfield, J. (2019). RNA sequencing: the teenage years. Nat. Rev. Genet. 20, 631–656. doi: 10.1038/s41576-019-0150-2
Sun, Z., Zhang, C., Li, G., Lin, Q., and Zhao, X. (2018). Does soil amendment alter reactive soil N dynamics following chloropicrin fumigation? Chemosphere 212, 563–571. doi: 10.1016/j.chemosphere.2018.08.084
Sundaramoorthy, R., Micossi, E., Alphey, M. S., Germain, V., Bryce, J. H., Smith, S. M., et al. (2006). The crystal structure of a plant 3-ketoacyl-coA thiolase reveals the potential for redox control of peroxisomal fatty acid β-oxidation. J. Mol. Biol. 359, 347–357. doi: 10.1016/j.jmb.2006.03.032
Tsai, T. H., and Lin, L. C. (2008). Phenolic glycosides and pyrrolidine alkaloids from Codonopsis tangshen. Chem. Pharm. Bull. 56, 1546–1550. doi: 10.1248/cpb.56.1546
Ülgen, E., Ozisik, O., and Sezerman, O. (2019). PathfindR: an R package for comprehensive identification of enriched pathways in omics data through active subnetworks. Front. Genet. 10:858. doi: 10.3389/fgene.2019.00858
Wang, Y., Yang, X., Xu, M., and Violette, G. (2022). Effects of chloropicrin fumigation and azoxystrobin application on ginger growth and phosphorus uptake. Ecotox. Environ. Safe 232:113246. doi: 10.1016/j.ecoenv.2022.113246
Wu, D., Ji, J., Wang, G., Guan, C., and Jin, C. (2014). LchERF, a novel ethylene-responsive transcription factor from Lycium chinense, confers salt tolerance in transgenic tobacco. Plant Cell Rep. 33, 2033–2045. doi: 10.1007/s00299-014-1678-4
Wu, Q., Luo, M., Yao, X., and Yu, L. (2020). Purification, structural characterization, and antioxidant activity of the COP-W1 polysaccharide from codonopsis tangshen oliv. Carbohyd. Polym. 236:116020. doi: 10.1016/j.carbpol.2020.116020
Wu, T., Jiang, Y., Li, M., Pu, D., Shi, M., and Lan, Z. (2022). RNA-seq analysis reveals the potential mechanism of improved viability and product quality of tea plants through intercropping with Chinese chestnut. Plant Growth Regul. 96, 177–193. doi: 10.1007/s10725-021-00768-8
Xu, X., Xu, H., Shang, Y., Zhu, R., Hong, X., Song, Z., et al. (2021). Development of the general chapters of the Chinese Pharmacopoeia 2020 edition: a review. J. Pharm. Anal. 11, 398–404. doi: 10.1016/j.jpha.2021.05.001
Xue, Y., Yin, N., Chen, B., Liao, F., Win, A., Wang, R., et al. (2017). Molecular cloning and expression analysis of two FAD2 genes from chia (Salvia hispanica). Acta Physiol. Plant 39:95. doi: 10.1007/s11738-017-2390-0
Yadav, V., Wang, Z., Wei, C., Amo, A., Ahmed, B., Yang, X., et al. (2020). Phenylpropanoid pathway engineering: an emerging approach towards plant defense. Pathogens 9:312. doi: 10.3390/pathogens9040312
Yoshida, M., Muneyuki, E., and Hisabori, T. (2001). ATP synthase - a marvellous rotary engine of the cell. Nat. Rev. Mol. Cell Bio. 2, 669–677. doi: 10.1038/35089509
Young, M., Wakefield, M., Smyth, G., and Oshlack, A. (2010). Gene ontology analysis for RNA-SEQ: accounting for selection bias. Genome Biol. 11:R14.
Zhang, M., He, Y., Zhou, W., Ai, L., Liu, H., Chen, L., et al. (2021). Effects of continuous cropping of Codonopsis Tangshen on rhizospheric soil bacterial community as determined by pyrosequencing. Diversity 13:317. doi: 10.3390/d13070317
Zhang, Z., Dong, X., Wang, S., and Pu, X. (2020). Benefits of organic manure combined with biochar amendments to cotton root growth and yield under continuous cropping systems in Xinjiang. China. Sci. Rep. 10:4718. doi: 10.1038/s41598-020-61118-8
Keywords: Codonopsis tangshen, continuous cropping, soil remediation, RNA-Seq, metabolic pathway, qRT-PCR
Citation: Zhou W, Jiang X, Tan X, Li D, Wang H, You J, Li X and Zhang M (2022) Transcriptome analysis provides novel insights into the soil amendments induced response in continuously cropped Codonopsis tangshen. Front. Plant Sci. 13:972804. doi: 10.3389/fpls.2022.972804
Received: 19 June 2022; Accepted: 29 July 2022;
Published: 12 August 2022.
Edited by:
Shah Fahad, The University of Haripur, PakistanReviewed by:
Shah Saud, Linyi University, ChinaSubhan Danish, Bahauddin Zakariya University, Pakistan
Copyright © 2022 Zhou, Jiang, Tan, Li, Wang, You, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meide Zhang, ZW1haWx0b2VjaG9AMTYzLmNvbQ==
†These authors have contributed equally to this work