 Zheng-Ren Zhang
Zheng-Ren Zhang Xue Yang
Xue Yang Wei-Ying Li4
Wei-Ying Li4 Yan-Qiong Peng
Yan-Qiong Peng Jie Gao
Jie Gao- 1CAS Key Laboratory of Tropical Forest Ecology, Xishuangbanna Tropical Botanical Garden, Chinese Academy of Sciences, Mengla, China
- 2University of Chinese Academy of Sciences, Beijing, China
- 3College of Life Sciences, Jilin Agricultural University, Changchun, China
- 4Southwest Research Center for Landscape Architecture Engineering Technology, State Forestry and Grassland Administration, Southwest Forestry University, Kunming, China
As the largest genus in Moraceae, Ficus is widely distributed across tropical and subtropical regions and exhibits a high degree of adaptability to different environments. At present, however, the phylogenetic relationships of this genus are not well resolved, and chloroplast evolution in Ficus remains poorly understood. Here, we sequenced, assembled, and annotated the chloroplast genomes of 10 species of Ficus, downloaded and assembled 13 additional species based on next-generation sequencing data, and compared them to 46 previously published chloroplast genomes. We found a highly conserved genomic structure across the genus, with plastid genome sizes ranging from 159,929 bp (Ficus langkokensis) to 160,657 bp (Ficus religiosa). Most chloroplasts encoded 113 unique genes, including a set of 78 protein-coding genes, 30 transfer RNA (tRNA) genes, four ribosomal RNA (rRNA) genes, and one pseudogene (infA). The number of simple sequence repeats (SSRs) ranged from 67 (Ficus sagittata) to 89 (Ficus microdictya) and generally increased linearly with plastid size. Among the plastomes, comparative analysis revealed eight intergenic spacers that were hotspot regions for divergence. Additionally, the clpP, rbcL, and ccsA genes showed evidence of positive selection. Phylogenetic analysis indicated that none of the six traditionally recognized subgenera of Ficus were monophyletic. Divergence time analysis based on the complete chloroplast genome sequences showed that Ficus species diverged rapidly during the early to middle Miocene. This research provides basic resources for further evolutionary studies of Ficus.
Introduction
Chloroplasts originate from the endosymbiotic relationship between free-living cyanobacteria and eukaryotic cells (Wicke et al., 2011) and are responsible for photosynthesis, which is crucial for plant adaptation and evolution (Yin et al., 2018; Gao et al., 2019b; Thode and Lohmann, 2019). In angiosperms, chloroplast genomes typically exhibit a quadripartite structure, including a pair of inverted repeat (IR) regions, a large single-copy (LSC) region, and a small single-copy (SSC) region (Shinozaki et al., 1986; Abdullah et al., 2021). The number of unique genes generally ranges from 110 to 130 (Green, 2011; Chen et al., 2015; Hu et al., 2016). Furthermore, compared to the nuclear genome, the maternally inherited chloroplast genome is small and haploid, with no recombination and low mutation. Thus, it is highly conserved in structure and gene composition. Based on these characteristics, chloroplast genome sequence data should provide robust solutions for studies on phylogenetics, molecular evolution, population genetics, and phylogeography (Dong et al., 2013; Yang et al., 2020, 2021b; Du et al., 2021).
With the rapid development of DNA barcode identification technology, chloroplast markers have been widely used in inter- and intraspecific analysis of various plant species and populations (Riggins and Seigler, 2012; Li et al., 2015; Wu et al., 2021). For example, the matK and trnH-psbA datasets can identify more than 94% of species in Myristicaceae (Newmaster et al., 2008), and five widely used gene regions (rbcL, matK, rpoC1, trnH-psbA, and atpF-atpH) can correctly identify more than 97% of Canadian temperate flora samples (Burgess et al., 2011). However, none of these Sanger-sequenced chloroplast markers are universal for all plant taxa, and they provide limited information at the subspecies level (Yang et al., 2018; Huang et al., 2021b). In contrast, whole chloroplast genomes provide more extensive and higher resolution information (Wei et al., 2017; Luo et al., 2021).
In plants, chloroplasts not only perform photosynthesis but also play pivotal roles in carbohydrate, protein, and lipid biosynthesis, thus impacting growth and development (Jensen and Leister, 2014). As such, an increasing number of studies have shown that certain chloroplast genes are or have been under positive selection, yielding adaptive changes across taxa (Yang et al., 2005, 2020). For example, to better understand sunlight preferences in Oryza, Gao et al. (2019b) identified 14 chloroplast genes under positive selection in response to diverse habitats. Huang et al. (2021b) and Liu et al. (2021) also identified several positively selected chloroplast genes (e.g., matK, ycf2, accD, clpP, petA, rps13, and rpoC2) involved in adaptation to sunlight intensity in white oak and Bupleurum, respectively. Yao et al. (2019) identified a positively selected gene (rbcL) in Ilex associated with wet and dry habitats, contributing to the adaptation of introgressed individuals to changing environments. Hence, natural selection on the chloroplast genome influences plant adaptation through its evolutionary history (Wu et al., 2020).
Ficus (Moraceae) contains approximately 850 species (Shanahan et al., 2001) divided into six subgenera (Sycomorus, Synoecia, Urostigma, Pharmacosycea, Ficus, and Sycidium). Ficus species are widely distributed across tropical and subtropical regions (Berg and Corner, 2005) and provide essential food resources (infructescences) for frugivorous animals, making them keystone species in tropical forest ecosystems (Dev et al., 2011). Ficus species also exhibit significant coevolutionary relationships with obligate mutualist pollinating wasps (Cruaud et al., 2012). Over the past 25 years, phylogenetic studies on Ficus have been conducted based on nuclear markers [e.g., internal transcribed spacer (ITS), external transcribed spacer (ETS), glyceraldehyde 3-phosphate dehydrogenase (G3pdh), chloroplast expressed glutamine synthetase region (ncpGS), and granule-bound starch synthase (waxy region)] and chloroplast genes (e.g., rbcL and tRNA spacers). However, these studies have been unable to confidently resolve relationships among major groups of figs and have revealed conflicts with traditional morphological classifications (Herre et al., 1996; Weiblen, 2000; Jousselin et al., 2003; Ronsted et al., 2005, 2008; Xu et al., 2011; Cruaud et al., 2012). Bruun-Lund et al. (2017) constructed the first whole chloroplast genome-based phylogenetic tree for figs and detected a high level of cyto-nuclear discordance in several subgenera.
An unambiguously resolved Ficus phylogeny will facilitate key research on diversification, biogeography, and species interactions, and could be achieved using whole chloroplast genomes. Furthermore, comparative analysis of whole chloroplast genomes may provide an accurate and rapid method to discriminate species and subspecies (Li et al., 2015). This contrasts with the use of limited chloroplast regions, which exhibit low resolution in identifying the phylogeographic structure of geographically widespread figs (Huang et al., 2021a) and an inability to differentiate closely related species, such as those within the Ficus auriculata complex, due to high sequence similarity caused by rapid radiation and hybridization (Wu et al., 2003; Berg and Corner, 2005; Zhang et al., 2018, 2019). Whole chloroplast genome sequencing may help reveal divergent hotpot regions to address these and other unanswered questions in Ficus. Furthermore, Ficus species exhibit considerable life-form diversity, including trees, shrubs, stranglers, epiphytes, hemi-epiphytes, climbers, lithophytes, and rheophytes (Harrison et al., 2012; Bruun-Lund et al., 2017; Machado et al., 2018; Clement et al., 2020; Zhang et al., 2020b), and can survive in a variety of ecological niches (Harrison, 2005), indicating that different figs have adapted to diverse habitats. Therefore, chloroplast genomes may exhibit high levels of variation among species due to differing adaptations. Although the availability of sequenced Ficus chloroplast genomes has increased in recent years (Bruun-Lund et al., 2017; Liu et al., 2019; Wang et al., 2021; Lin et al., 2022), large-scale comparative analysis across all six subgenera has not yet been conducted. Therefore, the evolution of Ficus chloroplasts remains poorly understood.
In this study, we sequenced and assembled 13 whole chloroplast genomes from 10 Ficus species. In addition, we assembled 21 plastomes from 13 Ficus species using publicly available next-generation sequencing data. We also downloaded six previously reported Ficus plastomes from the NCBI database. Overall, we obtained the complete chloroplast genomes of 24 Ficus species covering all six subgenera. The aims of this study were to: (1) reveal chloroplast genomic variation across the genus and screen for hypervariable regions among species; (2) identify the adaptive evolution of protein-coding plastid genes within Ficus; and (3) construct phylogenetic trees and estimate divergence times for Ficus based on the whole chloroplast genomes and 40 plastomes of Ficus species obtained from previous research. The results of this study should improve our understanding of chloroplast genome evolution and its contribution to the evolutionary and ecological success of this economically and evolutionarily important genus.
Materials and methods
Genomic DNA extraction, sequencing, assembly, and annotation
We collected 13 individuals representing 10 species of Ficus [F. auriculata, F. oligodon, F. hainanensis, F. beipeiensis, F. religiosa, F. tinctoria, F. ischnopoda, F. langkokensis, F. pumila, and newly identified lineage F. northern (Fn) (Gao et al., unpublished data)] from Xishuangbanna (southern Yunnan), Gengma (northwestern Yunnan), and Chongqing, China. All voucher specimens have been preserved in the Lab of Coevolution Research Group in Xishuangbanna Tropical Botanical Garden, Xishuangbanna, China. We also obtained whole genomic DNA data of 21 samples and two outgroup species (Antiaris toxicaria and Morus alba; both Moraceae) from the BIG Data Center1 under BioProject accession number GSA: PRJCA002187 (Zhang et al., 2020b) and six chloroplast genomes from six species of figs from the NCBI database. In total, we obtained samples from 42 individuals representing 40 accessions and 24 species of Ficus, including at least two species from each of the six subgenera (Supplementary Table 1), and two outgroup taxa.
For the 13 newly sequenced figs, we collected fresh clean leaves and stored them in liquid nitrogen until further processing. From these samples, we extracted genomic DNA using a DNAsecure Plant kit (TIANGEN, Beijing, China). The purified DNA was then used to generate 350-bp fragments for library construction using paired-end sequencing on the Illumina HiSeq 2500 platform (Illumina Inc., San Diego, CA, United States). For the newly sequenced plastomes and downloaded genomes (34 samples in total), we performed de novo assembly using GetOrganelle v1.7.1 (Jin et al., 2020), and checked the generated circular plastid graphs using Bandage v0.8.1 (Wick et al., 2015). We used F. auriculata (accession number: OK078618) as a reference for CPGAVAS2 to automatically annotate the plastome of F. hainanensis (FSP2) and conducted manual adjustments using Geneious Prime 2020.0.5 (Kearse et al., 2012). Thereafter, we manually annotated the remaining 41 plastomes and compared them with the reference sequence FSP2. We deposited the newly generated plastid genome sequences in GenBank (accession numbers ON711000–ON711012) and used OGDRAW2 (Greiner et al., 2019) to visualize the circular genome maps of the Ficus species.
Sequence comparison and simple sequence repeat analyses
We used the mVISTA online tool3 (Frazer et al., 2004) to analyze plastome divergence of all 24 species of Ficus, with F. auriculata (BN-FA10) as a reference. We also aligned the complete chloroplast genome sequences in MAFFT v7.313 using default parameters (Katoh and Standley, 2013) and calculated nucleotide diversity (π) in DnaSP v5.10.01 (Librado and Rozas, 2009) with a window length of 600 bp and step size of 200 bp. We implemented MISA-web4 (Beier et al., 2017) to identify simple sequence repeats (SSRs) in the plastomes of the 24 species. Minimal repeat numbers were set as mononucleotides ≥ 10, dinucleotides ≥ 5, trinucleotides ≥ 4, tetranucleotides ≥ 4, pentanucleotides ≥ 4, and hexanucleotides ≥ 4. Furthermore, we calculated the correlation between chloroplast sequence length and number of SSRs using R (R Core Team, 2013). Spearman rank correlation was used to assess p-values.
Phylogenetic analyses and molecular dating
To gain insight into the phylogeny of figs, we downloaded the chloroplast sequences of individual IR, SSC, and LSC regions of 40 other fig species (Bruun-Lund et al., 2017), and performed phylogenetic analysis of a total of 63 fig species, with two Moraceae species used as the outgroup. All sequences with one IR were aligned with MAFFT v7.313, after which all indels were removed using trimAI (Capella-Gutierrez et al., 2009). Phylogenomic analyses were conducted using maximum-likelihood (ML) and Bayesian inference (BI). The ML tree was reconstructed using IQ-TREE (Nguyen et al., 2015) based on the best-fit nucleotide substitution model selected by ModelFinder (Kalyaanamoorthy et al., 2017). Branch support for the ML tree was evaluated by 10,000 ultrafast bootstrap replicates (Minh et al., 2013). MrBayes 3.2.6 (Ronquist et al., 2012) was used for the BI tree, with 2,000,000 generations under the GTR + F + I + G4 model with three heated chains and one cold chain, where the initial 25% of sampled data were discarded as burn-in. At the end of the run, average standard deviation of split frequencies was < 0.01 and effective sample size (ESS) was larger than 200. All procedures were completed in PhyloSuite v1.2.2 (Zhang et al., 2020a).
We estimated divergence times using BEAST v1.10.4 (Drummond and Rambaut, 2007) with an uncorrelated lognormal relaxed clock. For the tree prior, we applied the Yule and Birth-Death speciation process. The crown divergence time of Ficus [74.9 million years ago (Mya)] and Moraceae (93.1 Mya) was constrained by secondary calibrations using normal distribution based on previous studies (Cruaud et al., 2012; Gardner et al., 2017). We ran Markov Chain Monte Carlo (MCMC) for 800 million generations with sampling every 1,000 cycles. The first 10% of samples were discarded as burn-in. We examined the outputs using Tracer v1.7 (Rambaut et al., 2018) to confirm convergence based on ESS > 200.
To explore the relationship between fig and wasp phylogenies, we directly compared our ML tree of Ficus with a pollinating wasp phylogeny from previous analysis (Cruaud et al., 2012).
Genome-wide scan for positively selected genes
We first calculated the non-synonymous rate (dN), synonymous rate (dS), and substitution ratio (ω = dN/dS) of all 78 chloroplast protein-coding genes using EasyCodeML v.1.31 (Gao et al., 2019a). Purifying selection, neutral selection, and positive selection were interpreted as ω < 1, ω = 1, and ω > 1, respectively.
We extracted each protein-coding gene in Geneious Prime 2020.0.5 and aligned them using MAFFT v7.313. Before calculation, all gaps and stop codons were removed, and multiple aligned genes with similar functions were concatenated into a single matrix, e.g., 11 genes (ndhJ, ndhK, ndhC, ndhB, ndhH, ndhA, ndhI, ndhG, ndhE, ndhD, and ndhF) related to NADH oxidoreductase were concatenated. In total, 17 matrices were obtained, including accD, atp, ccsA, cemA, clpP, matK, ndh, pet, psa, psb, rbcL, rpl, rpo, rps, ycf1, ycf2, and ycf3 plus ycf4. The ML tree was constructed using the above methods (plastome sequences with single IR, SSC, and LSC regions) to detect the selection. Based on the ML tree, we performed selective pressure analysis using branch, site, and branch-site models with the CODEML algorithm in EasyCodeML v.1.31 to estimate ω. Branch models were used to detect different branches in the tree showing significant differences in ω (Yang and Nielsen, 1998, 2002). Two branch models were used: i.e., one-ratio model (M0) assuming the same ω for all branches in the tree, and two-ratio model assuming foreground branches have a specific ω that varies from background branches (Yang, 1998). Site models were used to locate positively selected sites in aligned sequences (Yang and Nielsen, 2002), assuming that ω is the same for all branches in the phylogenetic tree but varies among sites in aligned sequences. Two site models were used: i.e., M1a (nearly neutral) and M2a (positive selection) (Gao et al., 2019a). In the branch-site model, Model Anull assumes that the foreground branch is not under positive selection; conversely, Model A allows positive selection in the foreground branch (Zhang et al., 2005). We performed three pairwise comparisons (i.e., M0/two-ratio model; M1a/M2a; Model Anull/Model A) using likelihood ratio tests to check for significance. In addition, we analyzed the dN, dS, and dN/dS values of all protein-coding gene datasets among the 24 representative fig species with TBtools (Chen et al., 2020).
Results
Structure and features of Ficus plastomes
We obtained the plastomes of 24 species of Ficus. The complete chloroplast genomes ranged in size from 159,929 bp (F. langkokensis) to 160,657 bp (F. religiosa) and displayed a typical quadripartite structure, including a pair of IR regions (IRa and IRb) ranging from 25,830 bp in F. religiosa (W62) to 25,912 bp in F. adhatodifolia and F. langkokensis, a LSC region ranging from 88,215 bp in F. langkokensis to 88,873 bp in F. religiosa (WG62), and a SSC region ranging from 19,871 bp in F. maxima to 20,165 bp in F. erecta (Figure 1 and Supplementary Table 2). GC content ranged from 35.9–36.1%, 33.4–33.7%, 28.9–29.1%, and 42.6–42.7% in the whole chloroplast genomes and LSC, SSC, and IR regions, respectively (Supplementary Table 2).
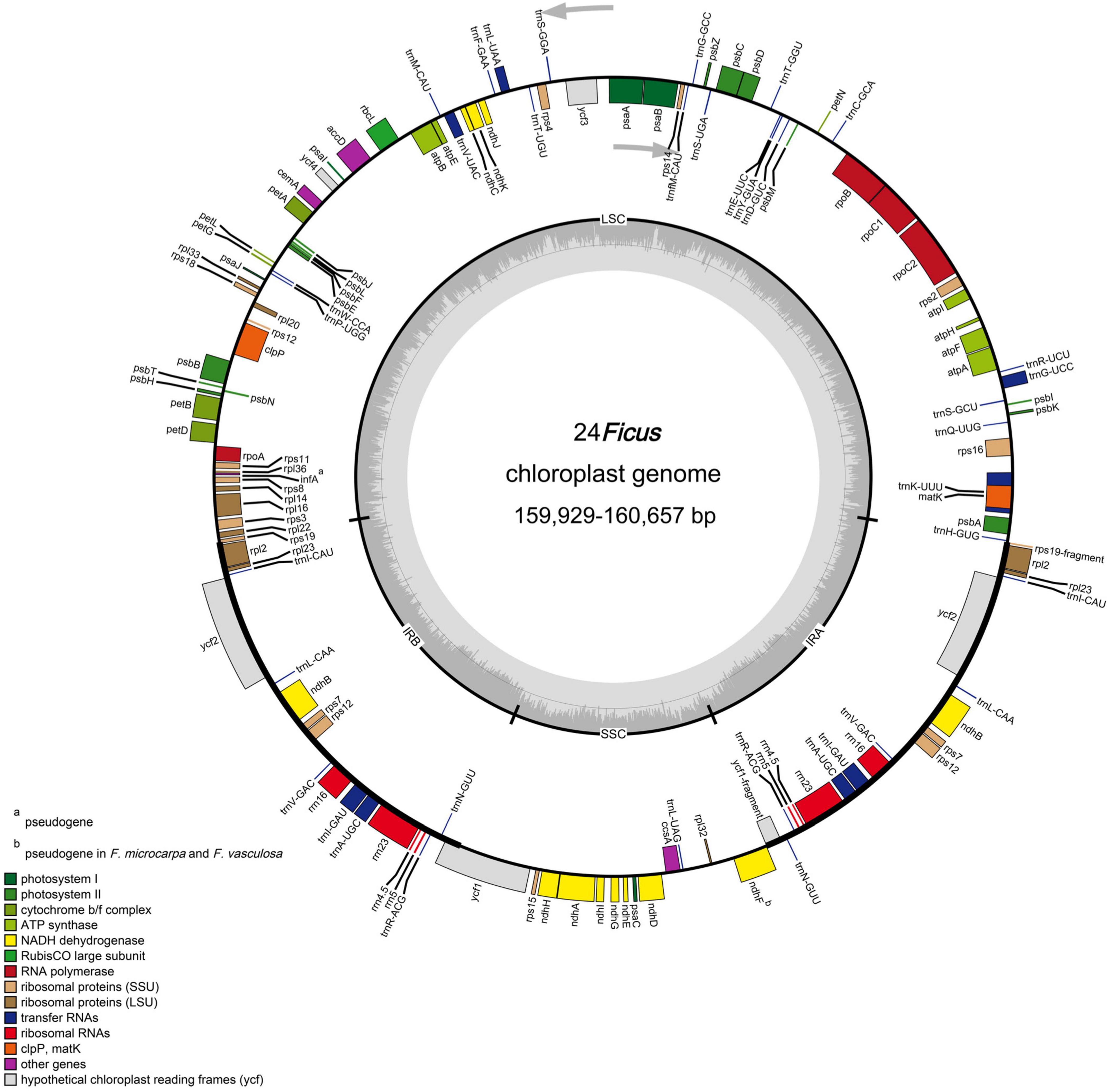
Figure 1. Representative chloroplast genome of Ficus. Genes shown on the outside of the circle are transcribed counter-clockwise, and genes inside are transcribed clockwise. Genes belonging to different functional groups are color-coded. The darker gray in the inner corresponds to GC content, and the lighter gray corresponds to AT content.
The chloroplast genomes each encoded 113 unique genes, consisting of 78 protein-coding genes, 30 transfer RNA (tRNA) genes, four ribosomal RNA (rRNA) genes, and one pseudogene (infA) (Table 1). In addition, ndhF was identified as a pseudogene in F. microcarpa and F. vasculosa. In total, 17 genes were duplicated in IR regions, including six protein-coding genes, seven tRNAs, and all four rRNAs (Table 1). Among them, 15 contained one intron (rpl2, ndhB, trnI-GAU, trnA-UGC, ndhA, rpl16, petD, petB, trnV-UAC, trnL-UAA, rpoC1, atpF, trnG-UCC, rps16, and trnK-UUU) and three contained two introns (ycf3, clpP, and rps12). In addition, rps12 was trans-spliced with two exons, with a 5′ exon in the LSC region and a 3′ exon in the IR region. In all 24 species of Ficus, the rps19 and ycf1 genes crossed the LSC/IRb and SSC/IRb junctions, respectively, and their fragments were present at the IRa/LSC and IRa/SSC borders, respectively. Furthermore, ndhF was traversed at the SSC/IRa junction and rpl2 and trnH-GUG flanked the IRa/LSC junction (Figure 1).
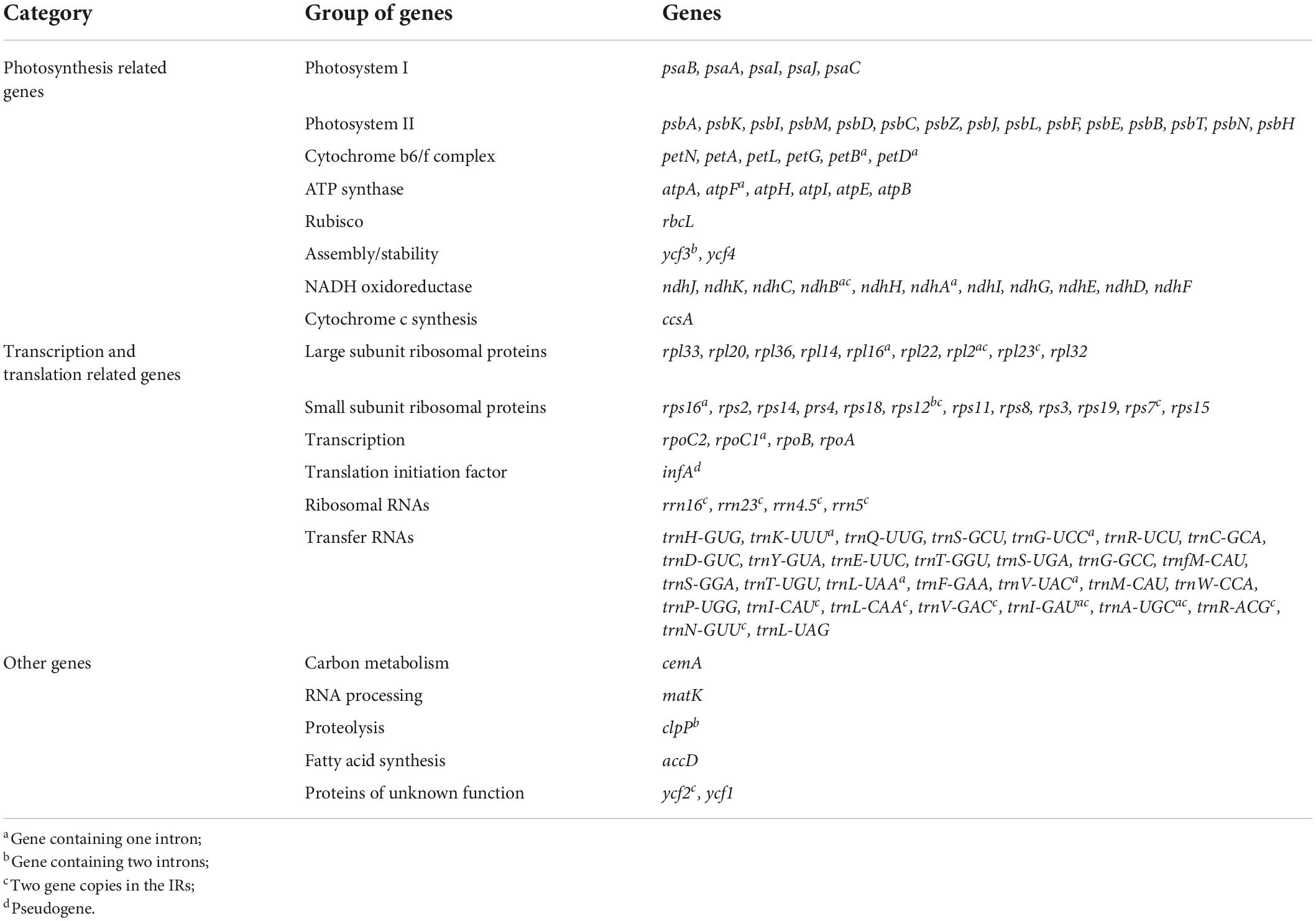
Table 1. Gene composition of Ficus chloroplast genomes.
Simple sequence repeat polymorphism analysis and hypervariable region in Ficus species
We detected five classes of SSRs in the chloroplast genomes of the 24 species of Ficus, including mono-, di-, tri-, tetra-, and pentanucleotide repeats. The total number of SSRs ranged from 67 (F. sagittata) to 89 (F. microdictya) (Supplementary Table 3). The most abundant SSRs were mononucleotides, accounting for 62.69% (F. sagittata) to 74.71% (F. cyathistipula), followed by dinucleotides (19.54% in F. aurea to 28.57% in F. langkokensis), trinucleotides (3.45% in F. cyathistipula to 7.50% in F. cyrtophylla), and tetranucleotides (1.12% in F. microdictya to 2.33% in F. religiosa), with only one pentanucleotide repeat found in F. beipeiensis (1.25%). The mononucleotide SSRs primarily consisted of A or T bases, while most dinucleotide SSRs consisted of AT or TA bases. Based on comparative analysis of SSRs at the subgenus level, mean number of SSRs ranged from 73.00 (subgenus Synoecia) to 86.50 (subgenus Urostigma) (Table 2). Fig species in the Pharmacosycea and Urostigma subgenera contained > 80 SSRs. In contrast, the total number of SSRs in the two Synoecia species was below 80 (Supplementary Table 3). Spearman rank correlation suggested that fig chloroplast sequence length and number of SSRs were strongly correlated (R = 0.48, p < 0.01) (Supplementary Figure 1).
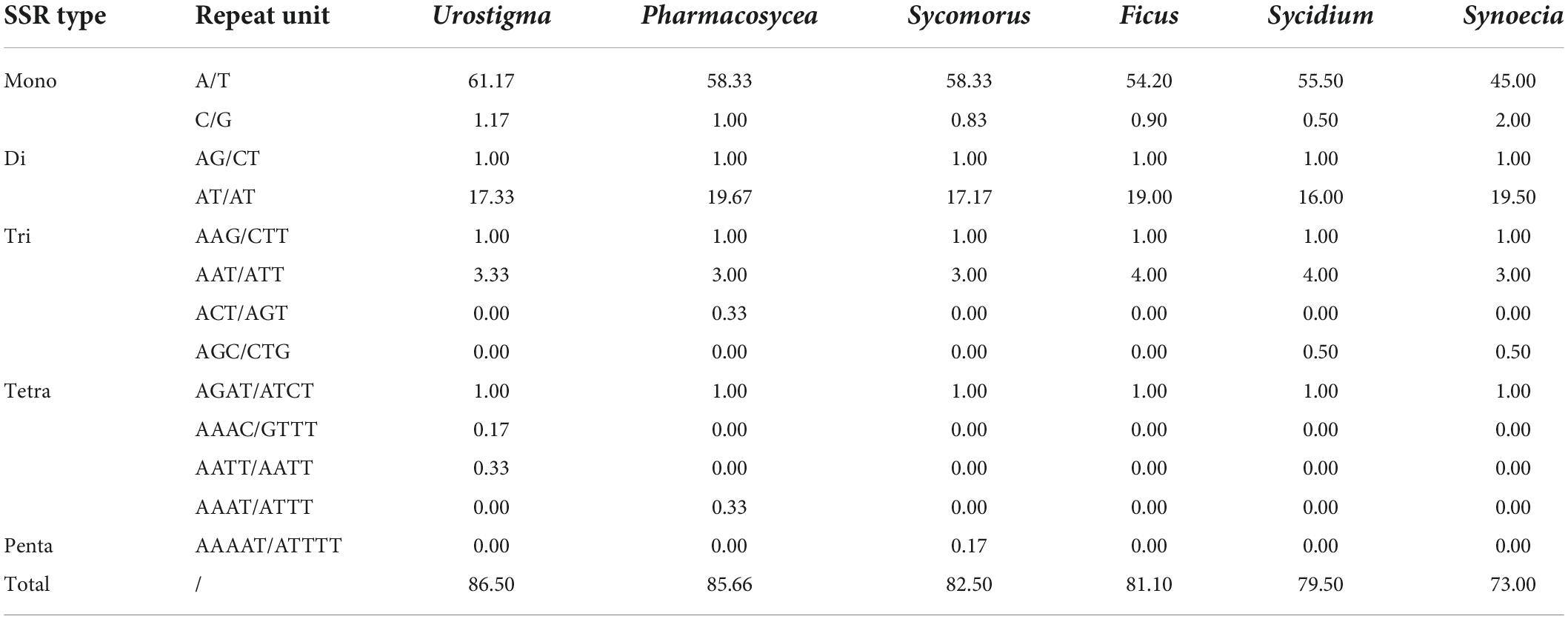
Table 2. Simple sequence repeats (SSRs) types and mean number of chloroplast genomes of six Ficus subgenera.
To reveal hotspot regions for variability among fig chloroplasts, we used the mVISTA online tool to align and compare the plastome sequences, with F. auriculata (BN-FA10) used as a reference (Supplementary Figure 2). Results suggested that the Ficus chloroplast genomes were highly conserved. However, hypervariable regions were detected in some intronic and intergenic regions, including rpl2-trnH(GUG), trnH(GUG)-psbA, trnK(UUU)-rps16, rps16-trnQ(UUG), rpoB-trnC(GCA), petN-psbM, trnE(UUC)-trnT(GGU), trnT(GGU)-psbD, psbZ-trnG(GCC), trnT(UGU)-trnL(UAA), trnL(UAG)-rpl32, and rpl32-ndhF. In contrast, only a few differences were detected in the protein-coding regions, i.e., in ycf2, ycf1, and ndhF. To further assess sequence divergence in the Ficus chloroplast genomes, we calculated the nucleotide diversity (π) in DnaSP, which ranged from 0.0 to 0.01468. We also observed eight highly variable regions (π > 0.010), including six in the LSC region [trnH(GUG)-psbA, trnK(UUU)-rps16, rpoB-trnC(GCA), petN-psbM, trnE(UUC)-psbD, and trnT(UGU)-trnL(UAA)] and two in the SSC region [trnL(UAG)-rpl32 and rpl32-ndhF] (Figure 2). All hotspot regions existed in non-coding regions. The π values in the IR region were all below 0.005.
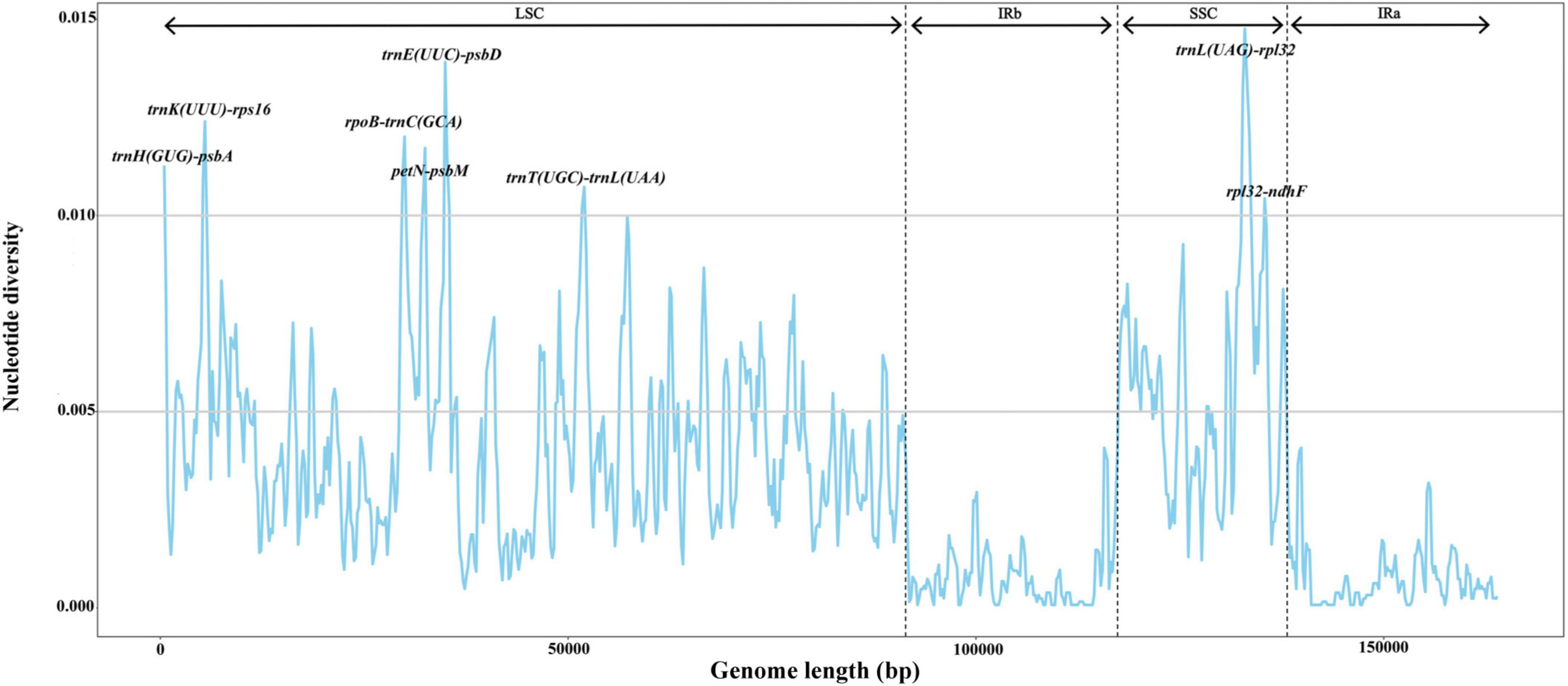
Figure 2. Nucleotide diversity (π) by sliding window analysis in the multiple alignments of 24 Ficus plastomes. Window length: 600 bp, step size: 200 bp. X-axis: the position of the midpoint of a window. Y-axis: the nucleotide diversity of each window.
Phylogenomic analysis and divergence time estimation
We constructed phylogenetic trees of the 63 species of Ficus. The ML and BI trees showed nearly identical topology, with high bootstrap support (BS) or posterior probabilities (Figure 3 and Supplementary Figure 3).
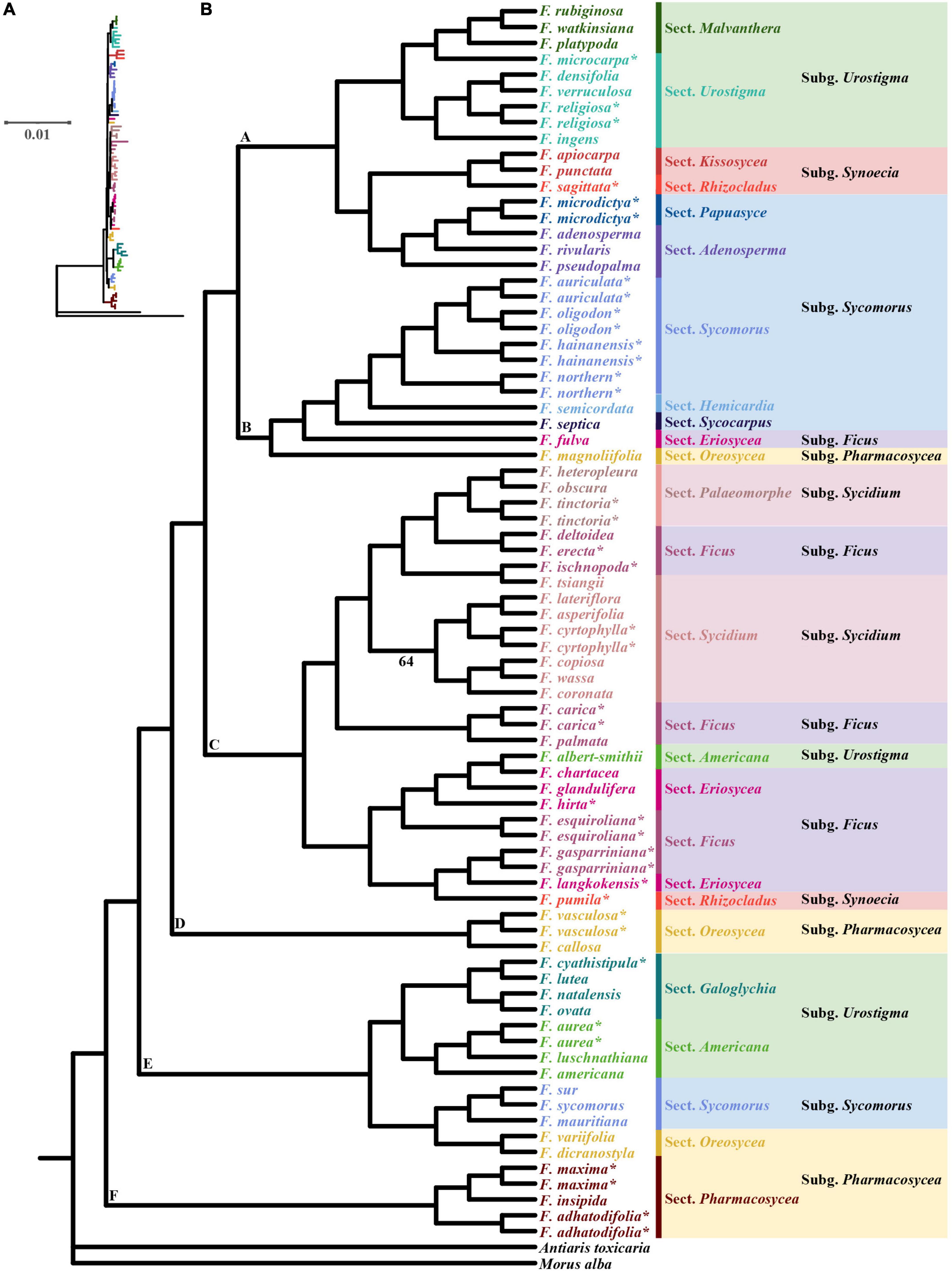
Figure 3. Maximum-likelihood (ML) phylogram (A) and cladogram (B) of 63 Ficus species inferred from whole chloroplast genome sequences. * Indicates the chloroplast was obtained in this study, the other 40 Ficus species were downloaded from Bruun-Lund et al. (2017). Only branches with weak support (bootstrap < 70%) are noted.
The ML tree showed section Pharmacosycea (clade F) as sister to all other figs with high bootstrap support (BS = 100%). After the basal split, a clade of two sections (Galoglychia and Americana) of Urostigma, three species of subgenus Sycomorus section Sycomorus and two species of subgenus Pharmacosycea section Oreosycea diverged (clade E, BS = 100%), and then was a clade comprising two species of subgenus Pharmacosycea sections Oreosycea (clade D, BS = 100%). Thereafter, all Sycidium and Ficus subgenera (except F. fulva, subgenus Ficus section Eriosycea) diverged, containing two species from subgenus Urostigma and Synoecia (clade C, BS = 100%). Clade B (BS = 99%) included the F. auriculata Lour. complex (F. auriculata, F. oligodon, F. hainanensis, and the new lineage, Fn), another two subgenus Sycomorus sections Hemicardia and Sycocarpus, as well as F. fulva and the last species of Pharmacosycea section Oreosycea. Clade A (BS = 97%) included two sections of Urostigma (Malvanthera and Urostigma), Synoecia (Kissosycea and Rhizocladus), and Sycomorus (Papuasyce and Adenosperma), respectively.
We estimated the divergence times of the 63 species of Ficus based on the BI tree. Both the Yule and Birth-Death models suggested that the species shared a common ancestor approximately 74.5 Mya [95% highest posterior density (HPD): 72.58–76.54 Mya] (Figure 4 and Supplementary Figure 4). The Yule model showed that Ficus diverged into all other clades around 40.90 Mya (95% HPD: 23.90–60.11 Mya). Divergence time between the two major clades (clades A and B) was estimated at 33.62 Mya (95% HPD: 19.96–48.75 Mya). Two species (F. vasculosa and F. callosa) in clade C diverged 12.45 Mya (95% HPD: 1.83–27.05 Mya) and species in clade D diverged 37.01 Mya (95% HPD: 21.32–55.58 Mya). For the clade containing F. auriculata Lour. Complex was dated to 7.76 Mya (95% HPD: 1.73–14.55 Mya) (Figure 4). Most divergence times estimated using the Birth-Death model were nearly identical (with slight variation) (Supplementary Figure 4).
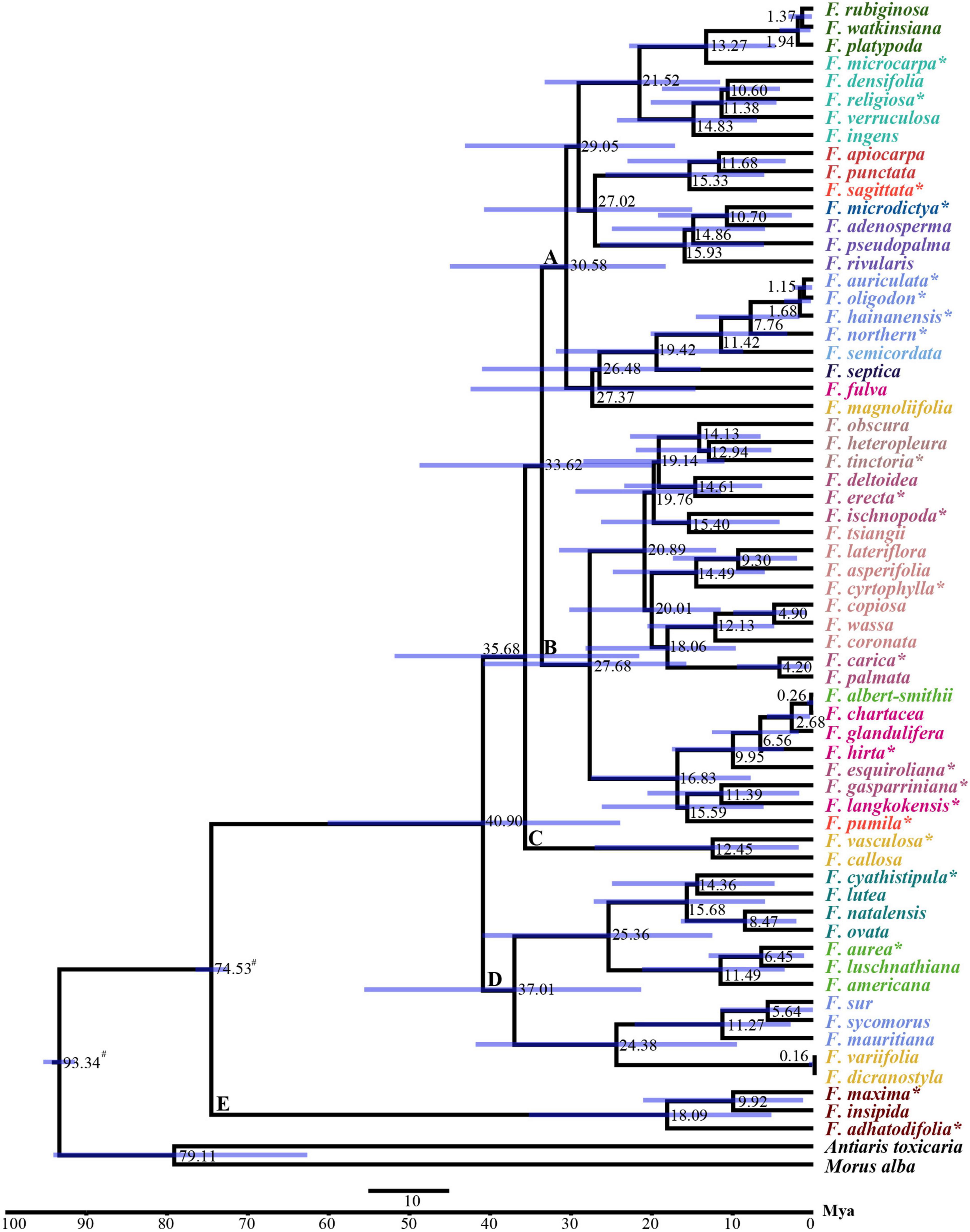
Figure 4. BEAST chronograms of the evolutionary history of Ficus using Yule model. The mean divergence time of the nodes was shown next to the nodes while the blue bars correspond to the 95% highest posterior density (HPD). # Indicates the calibration points, * indicates the chloroplast was obtained in this study, the other 40 Ficus species were downloaded from Bruun-Lund et al. (2017).
Co-phylogenetic comparison of figs and their pollinating wasps showed that the Ficus subgenera were associated with the fig wasp genera. However, we also observed one pollinating wasp genus that was related to more than one Ficus section (Figure 5).
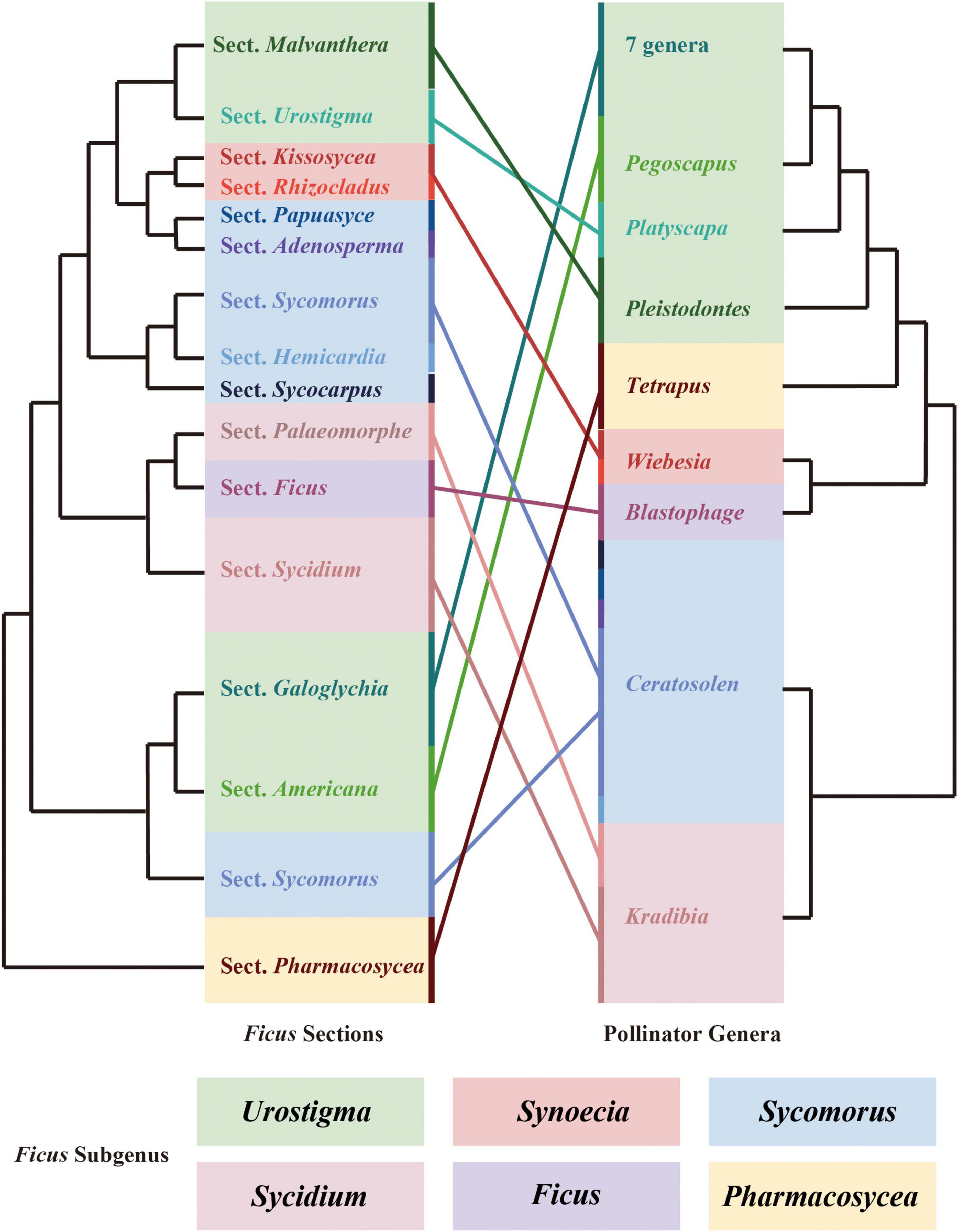
Figure 5. Cophylogenetic comparison of sections of Ficus and genera of pollinating wasps. Both phylogenetic trees were pruned. Colored boxes indicate the subgeneric classification of Ficus. Colored lines connect the sections of Ficus and their related genus of pollinators.
Selective pressure analysis of all plastome protein-coding genes in Ficus
We used branch, site, and branch-site models of the CODEML algorithm in EasyCodeML to analyze the likelihood of positive selection acting on Ficus protein-coding genes. After removing F. microdictya, F. tinctoria, and F. cyrtophylla, the ML tree topology was similar to the above phylogenetic tree (Figure 3 and Supplementary Figure 3) and could be divided into six clades (clades A–F) (Supplementary Figure 5). We selected clades A, B, C, D, E, and F as foreground branches, respectively. In the site model, no positively selected sites were detected (Supplementary Table 4). In the branch-site model, 15 positively selected gene datasets were obtained, but the p-values of the likelihood ratios were > 0.05, except for ccsA (Supplementary Tables 5, 6). In the branch model, M0 revealed that the clpP gene had a ω value > 1 (2.29950) (Supplementary Table 7). We also calculated the pairwise dN/dS ratios for the 17 gene datasets separately (Figure 6). Among the genes, the mean dN/dS ratio of rbcL was the highest (∼1.61), followed by clpP (∼0.91), and positive selection signals were detected in more than half of the pairwise results for both genes (dN/dS ratio > 1). The remaining gene datasets had mean dN/dS ratios varying from 0.09 (cemA) to 0.84 (ycf1). Overall, only three genes (ccsA, clpP, and rbcL) were identified as under positive selection, with all other 75 genes under purifying selection.
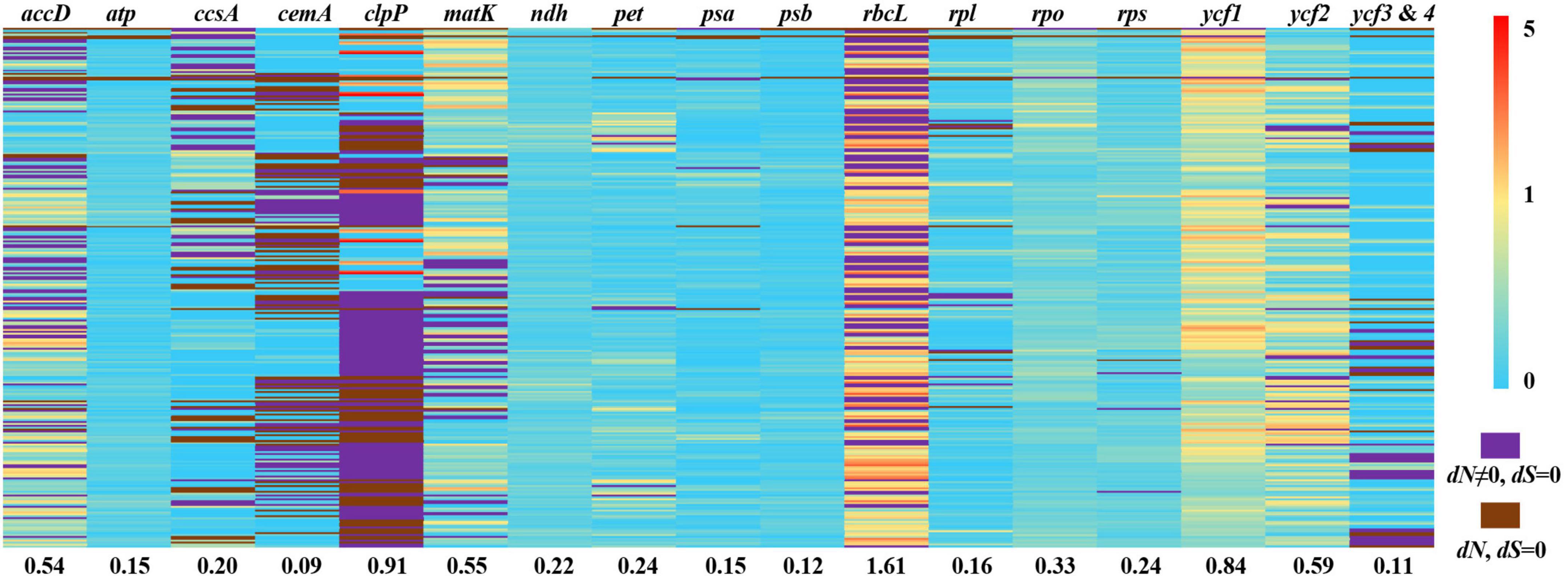
Figure 6. Pairwise non-synonymous rates (dN)/synonymous rates (dS) ratios in Ficus. This heatmap shows pairwise dN/dS ratios of 78 protein-coding genes from 24 Ficus species. The number under each gene dataset represented the mean dN/dS ratio. The order of species pairs showed in Supplementary Table 8.
Discussion
Conserved plastome structure and divergence hotspots in Ficus
Here, we explored the structure and variation in chloroplast genomes covering all six subgenera of the large tropical genus Ficus. Specifically, we showed that the chloroplast genomes of the 24 Ficus species were extremely similar in structure, size, gene content, and order, thus suggesting high conservation. The variation in chloroplast genome size was only ∼0.7 kb. Previous studies have reported that variation in plastome size within a genus is associated with contraction and expansion of IR regions (Ravi et al., 2007). However, gene distribution in all IR boundary regions of the Ficus chloroplast genomes were identical, possibly leading to only minor differences in chloroplast size in the genus. Nevertheless, plastome size in figs may be affected by the number of SSRs. SSRs play essential roles in genome recombination and rearrangement and can be found throughout the genome (Vieira et al., 2016). SSRs exhibit a high rate of polymorphism and significant variation at the species level and are thus valuable markers for studying genetic diversity, population structure, and biogeography within and between fig species (Moe and Weiblen, 2011). We identified 67–89 SSRs in the complete chloroplast genomes of the 24 Ficus species, with varying number among the six subgenera. We also demonstrated that Ficus plastome size increased linearly with increasing SSRs.
Gene number and distribution were conserved among almost all 24 Ficus species, except for ndhF, which was identified as a pseudogene in F. microcarpa and F. vasculosa. Of note, ndhF is a protein-coding gene of the NAD(P)H-dehydrogenase complex involved in the regulation of electron recycling within photosystem I (Krause, 2011; Wicke et al., 2011). The ndhF gene is frequently lost or pseudogenized in heterotrophic plants (Funk et al., 2007; Barrett et al., 2014; Lam et al., 2015), but has also been found pseudogenized or absent in several autotrophic plants, such as Mikania cordata (Asteraceae), Pinus abies (Pinaceae), and Ephedra equisetina (Ephedraceae) (Braukmann et al., 2009; Su et al., 2018; Strand et al., 2019). The pseudogenization of ndhF in F. microcarpa and F. vasculosa is due to a single-base deletion resulting in a premature stop codon. Previous studies have suggested that a functional copy of the gene may have been transferred to the nucleus in this situation (Wakasugi et al., 1994; Sugiura et al., 2003; Su et al., 2018). Further studies on the pseudogenization of ndhF may contribute to our understanding of Ficus evolution and adaptation.
Although the chloroplast genomes were highly conserved among the 24 Ficus species, we identified eight divergence hotspot regions [trnH(GUG)-psbA, trnK(UUU)-rps16, rpoB-trnC(GCA), petN-psbM, trnE(UUC)-psbD, trnT(UGU)-trnL(UAA), trnL(UAG)-rpl32, and rpl32-ndhF] based on mVISTA and sliding window analysis. All regions were found in the single-copy and intergenic regions. As observed in other angiosperms, the IR and coding regions exhibited lower levels of divergence than the single-copy and non-coding regions (Lu et al., 2016; Yin et al., 2018; Wu et al., 2021). Divergence hotpots in chloroplast genomes have been widely utilized for delimitation of closely related species of plants (Bi et al., 2018; Dong et al., 2021). Thus, we propose that the eight highly variable regions identified here may serve as DNA barcodes in Ficus and may be useful for studies on intraspecific phylogeography.
Phylogenetic analysis and divergence time in Ficus
Compared with earlier research on Ficus plastomes sampled from Europe and America (Bruun-Lund et al., 2017), we added 11 new Asian Ficus species and reconstructed the phylogenetic tree based on 63 chloroplasts, which showed highly consistent phylogeny with previous research. The use of additional samples can help resolve the complex Ficus phylogeny with greater confidence (Cruaud et al., 2012). For example, Bruun-Lund et al. (2017) divided species in the subgenus Sycidium section Sycidium into three clades dispersed within the subgenus Ficus and subgenus Sycidium section Palaeomorphe, whereas our ML tree recovered the species as a clade (BS = 64%, except F. tsiangii). However, our chloroplast phylogenetic tree did not support any of the six traditionally recognized subgenera of Ficus as monophyletic. Our placement of section Pharmacosycea (F. maxima and F. adhatodifolia) is consistent with previous study based on a nuclear genomic data, providing strong support that this section is sister to all other figs (Bruun-Lund et al., 2017; Wang et al., 2021). However, a recent study used genome-wide RAD loci and morphological features to infer the phylogenetic relationships in Ficus revealed that long-branch attraction led to section Pharmacosycea was the basal clade (Rasplus et al., 2021). Their results also strongly support the subgenera Sycidium, Sycomorus, and Urostigma as monophyletic clades, and suggest that the polyphyletic nature of these chloroplast genome-based clades may be due to heterogeneity (Rasplus et al., 2021). Thus, more research is needed to determine the reasons for nuclear and plastid discordance in figs, as it is unrealistic to rely on single genomes or small sample sizes to resolve these questions. Our phylogenetic results provided strong support for the monophyly of the F. auriculata Lour. complex. The taxa comprising this complex are sympatric in southeastern and southern China, and subtropical and tropical regions of Southeast Asia (Corner, 1978; Berg, 2007). Among putative species, F. auriculata, F. oligodon, and F. hainanensis are thought to have undergone natural hybridization and introgression, as based on SSR markers (Wei et al., 2014). Additionally, these species share pollinators, which may be the underlying mode of introgression (Wang et al., 2016). In this study, we identified a new genetic lineage (Fn), initially determined using nuclear microsatellite data (Wei et al., 2014). Based on chloroplast genomes, we used two models to estimate divergence times, both of which suggested that figs originated in the Late Cretaceous, approximately 74.5 Mya, then underwent rapid diversification in the early to middle Miocene (21.52–11.68 Mya). This rapid speciation coincided with global warming during the early Miocene and global maximum temperatures during the Miocene Climatic Optimum (MCO; 17–14.5 Mya) (Riera et al., 2021). Rapid speciation in the Miocene detected using the chloroplast genome is similar to that observed using nuclear genomes (Cruaud et al., 2012; Wang et al., 2021).
Co-evolution is defined as an evolutionary process whereby the traits of one species influence the evolution of the traits of another species, and vice versa (Janzen, 1980). The long-term co-evolution of figs and fig wasps has produced several adaptive traits. For example, the head, mandible, antenna, and ovipositor morphology and structure of pollinating wasps are co-adapted to fig traits such as the size of the enclosed inflorescences, aperture of the bracts, length of the styles, morphology of the stigma, and phenology of inflorescences (Ramirez, 1974; Janzen, 1979; Weiblen, 2002). Figs also release specific volatile organic compounds during the receptive phase to attract obligate pollinators and non-pollinating wasps (Hossaert-Mckey et al., 2010; Chen et al., 2016). These co-adaptation traits may have promoted the co-diversification of Ficus and pollinator wasps over the past 75 million years. In this study, we explored the co-phylogeny of Ficus and their pollinators above the species level due to the limited samples and genomic data. Our results supported high co-evolution between Ficus sections and fig wasp genera, as reported in previous morphological (Sonibare et al., 2004) and molecular studies (Machado et al., 2005; Cruaud et al., 2012). In general, closely related fig wasps pollinate closely related fig trees. However, we identified several Ficus sections associated with one pollinating wasp genus. For example, several sections of the subgenus Sycomorus were associated with the Ceratosolen genus in Ficus, and the genus Kradibia pollinated sections Palaeomorphe and Sycidium in the subgenus Sycidium. Thus, at the species level, the general view that one fig wasp species is associated with a particular host fig is challenged by several cases of breakdown of the one-to-one rule in fig-fig wasp mutualism. At present, however, two-thirds of pollinating wasps remain to be identified and a robust fig wasp phylogeny is still lacking, thereby hindering resolution of the incongruencies among fig-fig wasp co-phylogeny. Therefore, further studies with additional Ficus and pollinator wasp samples are needed with advanced genomic markers to construct the fig-fig wasp co-phylogeny (Rasplus et al., 2021).
Adaptive evolution in Ficus
Figs are present in tropical and subtropical regions worldwide and within heterogeneous ecological niches (Harrison, 2005). This suggests the occurrence of adaptive radiation, which may have left a selection footprint on the chloroplast genome. Our results indicated that chloroplast genes clpP, rbcL, and ccsA were subjected to positive selection, indicating that they may have contributed to environmental adaptation in Ficus. The clpP gene, which encodes clpP protease, is also positively selected in several angiosperm lineages, such as Paphiopedilum (Orchidaceae) (Guo et al., 2021), Acacia (Fabaceae) (Dugas et al., 2015), and Bupleurum (Apiaceae) (Huang et al., 2021b), and shows hypervariability in Amaryllidaceae and Papilionoideae (Liu et al., 2022; Moghaddam et al., 2022), suggesting it may have accelerated substitution rates in many angiosperms. Functional studies have also indicated that clpP protease degrades or restores damaged proteins (Wicke et al., 2011), and is important for changes in plant development in response to stress (Erixon and Oxelman, 2008). Hence, positive selection in this gene may help explain the radiation of Ficus into diverse ecological niches. The rbcL gene, which is a photosynthesis-related gene encoding the large subunit of ribulose-1,5-bisphosphate carboxylase/oxygenase (RuBisCO), is commonly under positive selection in terrestrial plants but not in algae (Kapralov and Filatov, 2007; Li et al., 2022), possibly due to more unstable thermal regimes on land (Kapralov and Filatov, 2007). For example, rbcL is under positive selection in shade-tolerant Oryza species, probably in response to adaptive evolution associated with sunlight and thermal conditions (Gao et al., 2019b). In this study, we collected Ficus from habitats with diverse sunlight exposure, and thus rbcL may be involved in adaptation to different light intensities and thermal habitats in this genus. The ccsA (ycf5) gene encodes a protein required for cytochrome biogenesis that mediates the attachment of heme to c-type cytochromes (Xie and Merchant, 1996). Here, this gene was positively selected in clade E (F. aurea and F. cyathistipula). Previous research has reported that ccsA is under positive selection in orchids, including epiphytic species (Dong et al., 2018). In our analysis, F. aurea and F. cyathistipula were epiphytes, and thus ccsA may play an important role in the adaptation of epiphytes to special habitats. Additionally, F. cyathistipula was the only species distributed in the Afrotropics, and positive selection on ccsA may be related to specific environmental conditions, such as light intensity, moisture, and/or temperature. In summary, these three positively selected genes may contribute to the adaptation of different Ficus life-forms to diverse environments and may serve as candidate genes for further research on the mechanisms of adaptive evolution of the genus.
We also found that the remaining 75 genes in the 24 chloroplast genomes of Ficus were under purifying selection, suggesting low synonymous and/or non-synonymous DNA substitution. These results are similar to previous findings showing that most chloroplast genes are under purifying selection in Rosales (Souza et al., 2020; Wang et al., 2020; Yang et al., 2021a) and angiosperm species (Henriquez et al., 2020; Yang et al., 2020; Huang et al., 2021b), with a highly conserved evolutionary history. Purifying selection helps stop mutations from becoming fixed (Wu et al., 2020), thus eliminating deleterious mutations and retaining conserved function in genes (Huang et al., 2021b). In Ficus, purifying selection likely explains the high levels of interspecific conservation among chloroplast genomes.
Conclusion
Our study supported the conserved structure of Ficus chloroplast genomes and revealed eight mutational hotspot regions, which may be utilized as high-resolution DNA markers for figs in future phylogenetic and phylogeographic studies. We detected positive selection in three genes (clpP, rbcL, and ccsA), which may be linked to adaptive evolution in Ficus. Phylogenetic analysis showed that none of the six traditionally recognized subgenera of Ficus were monophyletic. Thus, further research and additional taxonomic sampling are needed to explain the potential discordance with morphology and nuclear genomes. Overall, our study provides a new framework for an improved understanding of species delimitation, genome evolution, and phylogenetic relationships in Ficus.
Data availability statement
The 13 new sequencing data presented in the study are deposited in the NCBI. The link to the repository and accession numbers can be found below: https://www.ncbi.nlm.nih.gov/genbank, ON711000–ON711012.
Author contributions
JG and Y-QP conceived and designed the study. JG and W-YL collected the samples. Z-RZ and XY analyzed the data. JG, Z-RZ, and XY wrote the manuscript. Y-QP gave suggestions during the manuscript writing. All authors contributed to the article and approved the submitted version.
Funding
This study was financially supported by the West Light Foundation of the Chinese Academy of Sciences, Yunnan Fundamental Research Projects (202201AT070217) and National Natural Science Foundation of China (31770701 and 32070487).
Acknowledgments
We thank TopEdit (www.topeditsci.com) for its linguistic assistance during the preparation of this manuscript. We also thank Xin Li for the guide on using the GetOrganelle software. Genomic data processing and analyses were conducted at the High Performance Computing Cluster from the Institutional Center for Shared Technologies and Facilities of Xishuangbanna Tropical Botanical Garden, CAS.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.965335/full#supplementary-material
Footnotes
- ^ https://bigd.big.ac.cn/gsa/
- ^ https://chlorobox.mpimp-golm.mpg.de/OGDraw.html
- ^ https://genome.lbl.gov/vista/mvista/submit.shtml
- ^ https://webblast.ipk-gatersleben.de/misa/
References
Abdullah, Henriquez, C. L., Mehmood, F., Hayat, A., Sammad, A., Waseem, S., et al. (2021). Chloroplast genome evolution in the Dracunculus clade (Aroideae Araceae). Genomics 113, 183–192. doi: 10.1016/j.ygeno.2020.12.016
Barrett, C. F., Freudenstein, J. V., Li, J., Mayfield-Jones, D. R., Perez, L., Pires, J. C., et al. (2014). Investigating the path of plastid genome degradation in an early-transitional clade of heterotrophic orchids, and implications for heterotrophic angiosperms. Mol. Biol. Evol. 31, 3095–3112. doi: 10.1093/molbev/msu252
Beier, S., Thiel, T., Munch, T., Scholz, U., and Mascher, M. (2017). MISA-web: A web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Berg, C. C. (2007). Precursory taxonomic studies on Ficus (Moraceae) for the Flora of Thailand. Thai For. Bull. 35, 4–28
Berg, C. C. and, Corner, E. J. H. (2005). Flora Malesiana Series I - Seed Plants. Leiden: National Herbarium
Bi, Y., Zhang, M. F., Xue, J., Dong, R., Du, Y. P., and Zhang, X. H. (2018). Chloroplast genomic resources for phylogeny and DNA barcoding: A case study on Fritillaria. Sci. Rep. 8:1184. doi: 10.1038/s41598-018-19591-9
Braukmann, T. W., Kuzmina, M., and Stefanovic, S. (2009). Loss of all plastid ndh genes in Gnetales and conifers: Extent and evolutionary significance for the seed plant phylogeny. Curr. Genet. 55, 323–337. doi: 10.1007/s00294-009-0249-7
Bruun-Lund, S., Clement, W. L., Kjellberg, F., and Ronsted, N. (2017). First plastid phylogenomic study reveals potential cyto-nuclear discordance in the evolutionary history of Ficus L. (Moraceae). Mol. Phylogenet. Evol. 109, 93–104. doi: 10.1016/j.ympev.2016.12.031
Burgess, K. S., Fazekas, A. J., Kesanakurti, P. R., Graham, S. W., Husband, B. C., Newmaster, S. G., et al. (2011). Discriminating plant species in a local temperate flora using the rbcL+matK DNA barcode. Methods Ecol. Evol. 2, 333–340. doi: 10.1111/j.2041-210X.2011.00092.x
Capella-Gutierrez, S., Silla-Martinez, J. M., and Gabaldon, T. (2009). TrimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1101/289660
Chen, J., Hao, Z., Xu, H., Yang, L., Liu, G., Sheng, Y., et al. (2015). The complete chloroplast genome sequence of the relict woody plant Metasequoia glyptostroboides Hu et Cheng. Front. Plant Sci. 6:447. doi: 10.3389/fpls.2015.00447
Chen, Y. L., Huang, M. L., Wu, W. S., Wang, A. F., Bao, T., Zheng, C. F., et al. (2016). The floral scent of Ficus pumila var. pumila and its effect on the choosing behavior of pollinating wasps of Wiebesia pumilae. Acta Ecol. Sinica 36, 321–326. doi: 10.1016/j.chnaes.2016.06.008
Clement, W. L., Bruun-Lund, S., Cohen, A., Kjellberg, F., Weiblen, G. D., and Rønsted, N. (2020). Evolution and classification of figs (Ficus, Moraceae) and their close relatives (Castilleae) united by involucral bracts. Bot. J. Linn. Soc. 193, 316–339. doi: 10.1093/botlinnean/boaa022
Corner, E. J. H. (1978). Ficus Dammaropsis and the multibracteate species of Ficus sect. sycocarpus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 281, 373–406. doi: 10.1098/rstb.1978.0003
Cruaud, A., Ronsted, N., Chantarasuwan, B., Chou, L. S., Clement, W. L., Couloux, A., et al. (2012). An extreme case of plant-insect codiversification: Figs and fig-pollinating wasps. Syst. Biol. 61, 1029–1047. doi: 10.1093/sysbio/sys068
Dev, S. A., Kjellberg, F., Hossaert-Mckey, M., and Borges, R. M. (2011). Fine-scale Population Genetic Structure of Two Dioecious Indian Keystone Species. Ficus hispida and Ficus exasperata (Moraceae). Biotropica 43, 309–316. doi: 10.1111/j.1744-7429.2010.00704.x
Dong, S., Ying, Z., Yu, S., Wang, Q., Liao, G., Ge, Y., et al. (2021). Complete chloroplast genome of Stephania tetrandra (Menispermaceae) from Zhejiang Province: Insights into molecular structures, comparative genome analysis, mutational hotspots and phylogenetic relationships. BMC Genomics 22:880. doi: 10.1186/s12864-021-08193-x
Dong, W., Xu, C., Cheng, T., Lin, K., and Zhou, S. (2013). Sequencing angiosperm plastid genomes made easy: A complete set of universal primers and a case study on the phylogeny of saxifragales. Genome Biol. Evol. 5, 989–997. doi: 10.1093/gbe/evt063
Dong, W.-L., Wang, R.-N., Zhang, N.-Y., Fan, W.-B., Fang, M.-F., and Li, Z.-H. (2018). Molecular Evolution of Chloroplast Genomes of Orchid Species: Insights into Phylogenetic Relationship and Adaptive Evolution. Int. J. Mol. Sci. 19:716. doi: 10.3390/ijms19030716
Drummond, A. J., and Rambaut, A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. doi: 10.1186/1471-2148-7-214
Du, Z., Lu, K., Zhang, K., He, Y., Wang, H., Chai, G., et al. (2021). The chloroplast genome of Amygdalus L. (Rosaceae) reveals the phylogenetic relationship and divergence time. BMC Genomics 22:645. doi: 10.1186/s12864-021-07968-6
Dugas, D. V., Hernandez, D., Koenen, E. J., Schwarz, E., Straub, S., Hughes, C. E., et al. (2015). Mimosoid legume plastome evolution: IR expansion, tandem repeat expansions, and accelerated rate of evolution in clpP. Sci. Rep. 5:16958. doi: 10.1038/srep16958
Erixon, P., and Oxelman, B. (2008). Whole-gene positive selection, elevated synonymous substitution rates, duplication, and indel evolution of the chloroplast clpP1 gene. PLoS One 3:e1386. doi: 10.1371/journal.pone.0001386
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Funk, H. T., Berg, S., Krupinska, K., Maier, U. G., and Krause, K. (2007). Complete DNA sequences of the plastid genomes of two parasitic flowering plant species. Cuscuta reflexa and Cuscuta gronovii. BMC Plant Biol. 7:45. doi: 10.1186/1471-2229-7-45
Gao, L. Z., Liu, Y. L., Zhang, D., Li, W., Gao, J., Liu, Y., et al. (2019b). Evolution of Oryza chloroplast genomes promoted adaptation to diverse ecological habitats. Commun. Biol. 2:278. doi: 10.1038/s42003-019-0531-2
Gao, F., Chen, C., Arab, D. A., Du, Z., He, Y., and Ho, S. Y. W. (2019a). EasyCodeML: A visual tool for analysis of selection using CodeML. Ecol. Evol. 9, 3891–3898. doi: 10.1002/ece3.5015
Gardner, E. M., Sarraf, P., Williams, E. W., and Zerega, N. J. C. (2017). Phylogeny and biogeography of Maclura (Moraceae) and the origin of an anachronistic fruit. Mol. Phylogenet. Evol. 117, 49–59. doi: 10.1016/j.ympev.2017.06.021
Green, B. R. (2011). Chloroplast genomes of photosynthetic eukaryotes. Plant J. 66, 34–44. doi: 10.1111/j.1365-313X.2011.04541.x
Greiner, S., Lehwark, P., and Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64. doi: 10.1093/nar/gkz238
Guo, Y. Y., Yang, J. X., Bai, M. Z., Zhang, G. Q., and Liu, Z. J. (2021). The chloroplast genome evolution of Venus slipper (Paphiopedilum): IR expansion. SSC contraction, and highly rearranged SSC regions. BMC Plant Biol. 21:248. doi: 10.1186/s12870-021-03053-y
Harrison, R. D. (2005). Figs and the Diversity of Tropical Rainforests. BioScience 55, 1053–1064. doi: 10.1641/0006-35682005055[1053:FATDOT]2.0.CO;2
Harrison, R. D., Ronsted, N., Xu, L., Rasplus, J. Y., and Cruaud, A. (2012). Evolution of fruit traits in ficus subgenus Sycomorus (Moraceae): To what extent do frugivores determine seed dispersal mode? PLoS One 7:e38432. doi: 10.1371/journal.pone.0038432
Henriquez, C. L., Abdullah, Ahmed, I., Carlsen, M. M., Zuluaga, A., Croat, T. B., et al. (2020). Molecular evolution of chloroplast genomes in Monsteroideae (Araceae). Planta 251:72. doi: 10.1007/s00425-020-03365-7
Herre, E. A., Machado, C. A., Bermingham, E., Nason, J. D., Windsor, D. M., Mccafferty, S. S., et al. (1996). Molecular phylogenies of figs and their pollinator wasps. J. Biogeogr. 23, 521–530. doi: 10.1111/j.1365-2699.1996.tb00014.x
Hossaert-Mckey, M., Soler, C., Schatz, B., and Proffit, M. (2010). Floral scents: Their roles in nursery pollination mutualisms. Chemoecology 20, 75–88. doi: 10.1007/s00049-010-0043-5
Hu, Y., Woeste, K. E., and Zhao, P. (2016). Completion of the chloroplast genomes of five Chinese Juglans and their contribution to chloroplast phylogeny. Front. Plant Sci. 7:1955. doi: 10.3389/fpls.2016.01955
Huang, R., Xie, X., Chen, A., Li, F., Tian, E., and Chao, Z. (2021b). The chloroplast genomes of four Bupleurum (Apiaceae) species endemic to Southwestern China, a diversity center of the genus, as well as their evolutionary implications and phylogenetic inferences. BMC Genomics 22:714. doi: 10.1186/s12864-021-08008-z
Huang, J.-F., Darwell, C. T., and Peng, Y.-Q. (2021a). Homogenized phylogeographic structure across the Indo-Burma ranges of a large monoecious Fig Ficus altissima Blume. Diversity 13:654. doi: 10.3390/d13120654
Janzen, D. H. (1979). How to be a fig. Annu. Rev. Ecol. Evol. Syst. 10, 13–51. doi: 10.1146/annurev.es.10.110179.000305
Janzen, D. H. (1980). When is it coevolution? Evolution 34, 611–612. doi: 10.1111/j.1558-5646.1980.tb04849.x
Jensen, P., and Leister, D. (2014). Chloroplast evolution, structure and functions. F1000prime Rep. 6:40. doi: 10.12703/P6-40
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., Depamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21:241. doi: 10.1186/s13059-020-02154-5
Jousselin, E., Rasplus, J.-Y., and Kjellberg, F. (2003). Convergence and coevolution in a mutualism: Evidence from a molecular phylogeny of Ficus. Evolution 57, 1255–1269. doi: 10.1554/02-445
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., Von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Kapralov, M. V., and Filatov, D. A. (2007). Widespread positive selection in the photosynthetic Rubisco enzyme. BMC Evol. Biol. 7:73. doi: 10.1186/1471-2148-7-73
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Krause, K. (2011). Piecing together the puzzle of parasitic plant plastome evolution. Planta 234, 647–656. doi: 10.1007/s00425-011-1494-9
Lam, V. K., Soto Gomez, M., and Graham, S. W. (2015). The highly reduced plastome of mycoheterotrophic Sciaphila (Triuridaceae) is colinear with its green relatives and is under strong purifying selection. Genome Biol. Evol. 7, 2220–2236. doi: 10.1093/gbe/evv134
Li, C., Liu, Y., Lin, F., Zheng, Y., and Huang, P. (2022). Characterization of the complete chloroplast genome sequences of six Dalbergia species and its comparative analysis in the subfamily of Papilionoideae (Fabaceae). PeerJ. 10:e13570. doi: 10.7717/peerj.13570
Li, X., Yang, Y., Henry, R. J., Rossetto, M., Wang, Y., and Chen, S. (2015). Plant DNA barcoding: From gene to genome. Biol. Rev. Camb. Philos. Soc. 90, 157–166. doi: 10.1111/brv.12104
Librado, P., and Rozas, J. (2009). DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Lin, E., Liao, Z., Xu, X., Zhang, X., and Fang, J. (2022). The complete chloroplast genome of the Chinese banyan tree Ficus microcarpa. Mitochondrial DNA B 7, 423–425. doi: 10.1080/23802359.2021.1993097
Liu, K., Sun, L., Meng, W., Zhu, H., Zhang, D., and Wang, J. (2022). Comparative genomics and phylogenetic perspectives of six fertile Lycoris species endemic to East Asia based on plastome characterization. Nord. J. Bot. 2022:e03412. doi: 10.1111/njb.03412
Liu, X., Chang, E., Liu, J., and Jiang, Z. (2021). Comparative analysis of the complete chloroplast genomes of six white oaks with high ecological amplitude in China. J. For. Res. 32, 2203–2218. doi: 10.1007/s11676-020-01288-3
Liu, Y., Chen, W., Li, F., Li, C., Xie, X., Chao, Z., et al. (2019). The complete chloroplast genome sequence of Ficus hirta (Moraceae). Mitochondrial DNA B 4, 4041–4042. doi: 10.1080/23802359.2019.1689867
Lu, R. S., Li, P., and Qiu, Y. X. (2016). The Complete Chloroplast Genomes of Three Cardiocrinum (Liliaceae) Species: Comparative Genomic and Phylogenetic Analyses. Front. Plant Sci. 7:2054. doi: 10.3389/fpls.2016.02054
Luo, C., Huang, W., Sun, H., Yer, H., Li, X., Li, Y., et al. (2021). Comparative chloroplast genome analysis of Impatiens species (Balsaminaceae) in the karst area of China: Insights into genome evolution and phylogenomic implications. BMC Genomics 22:571. doi: 10.1186/s12864-021-07807-8
Machado, A. F. P., Ronsted, N., Bruun-Lund, S., Pereira, R. A. S., and Paganucci de Queiroz, L. (2018). Atlantic forests to the all Americas: Biogeographical history and divergence times of Neotropical Ficus (Moraceae). Mol. Phylogenet. Evol. 122, 46–58. doi: 10.1016/j.ympev.2018.01.015
Machado, C. A., Robbins, N., Gilbert, M. T. P., and Herre, E. A. (2005). Critical review of host specificity and its coevolutionary implications in the fig/fig-wasp mutualism. PNAS 102, 6558–6565. doi: 10.1073/pnas.0501840102
Minh, B. Q., Nguyen, M. A., and Von Haeseler, A. (2013). Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195. doi: 10.1093/molbev/mst024
Moe, A. M., and Weiblen, G. D. (2011). Development and characterization of microsatellite loci in dioecious figs (Ficus Moraceae). Am. J. Bot. 98, e25–e27. doi: 10.3732/ajb.1000412
Moghaddam, M., Ohta, A., Shimizu, M., Terauchi, R., and Kazempour-Osaloo, S. (2022). The complete chloroplast genome of Onobrychis gaubae (Fabaceae-Papilionoideae): Comparative analysis with related IR-lacking clade species. BMC Plant Biol. 22:75. doi: 10.1186/s12870-022-03465-4
Newmaster, S. G., Fazekas, A. J., Steeves, R. A., and Janovec, J. (2008). Testing candidate plant barcode regions in the Myristicaceae. Mol. Ecol. Resour. 8, 480–490. doi: 10.1111/j.1471-8286.2007.02002.x
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
R Core Team (2013). R: A Language and Environment for Statistical Computing Team. Vienna: R Core Team.
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., and Suchard, M. A. (2018). Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Ramirez, W. (1974). Coevolution of Ficus and Agaonidae. Ann. Mol. Bot. Gard. 61, 770–780. doi: 10.2307/2395028
Rasplus, J.-Y., Rodriguez, L. J., Sauné, L., Peng, Y.-Q., Bain, A., Kjellberg, F., et al. (2021). Exploring systematic biases, rooting methods and morphological evidence to unravel the evolutionary history of the genus Ficus (Moraceae). Cladistics. 37, 402–422. doi: 10.1111/cla.12443
Ravi, V., Khurana, J. P., Tyagi, A. K., and Khurana, P. (2007). An update on chloroplast genomes. Plant Syst. Evol. 271, 101–122. doi: 10.1007/s00606-007-0608-0
Riera, R., Bourget, J., Allan, T., Håkansson, E., and Wilson, M. E. J. (2021). Early Miocene carbonate ramp development in a warm ocean North West Shelf, Australia. Sedimentology 69, 219–253. doi: 10.1111/sed.12917
Riggins, C. W., and Seigler, D. S. (2012). The genus Artemisia (Asteraceae: Anthemideae) at a continental crossroads: Molecular insights into migrations, disjunctions, and reticulations among Old and New World species from a Beringian perspective. Mol. Phylogenet. Evol. 64, 471–490. doi: 10.1016/j.ympev.2012.05.003
Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Ronsted, N., Weiblen, G. D., Clement, W. L., Zerega, N. J. C., and Savolainen, V. (2008). Reconstructing the phylogeny of figs (Ficus. Moraceae) to reveal the history of the fig pollination mutualism. Symbiosis 45, 45–55.
Ronsted, N., Weiblen, G. D., Cook, J. M., Salamin, N., Machado, C. A., and Savolainen, V. (2005). 60 million years of co-divergence in the fig-wasp symbiosis. Proc. Biol. Sci. 272, 2593–2599. doi: 10.1098/rspb.2005.3249
Shanahan, M., So, S., Compton, S., and Corlett, R. (2001). Fig-eating by vertebrate frugivores: A global review. Biol. Rev. Camb. Philos. Soc. 76, 529–572. doi: 10.1017/S1464793101005760
Shinozaki, K., Ohme, M., Tanaka, M., Wakasugi, T., Hayashida, N., Matsubayashi, T., et al. (1986). The complete nucleotide sequence of the tobacco chloroplast genome: Its gene organization and expression. EMBO J 5, 2043–2049. doi: 10.1002/j.1460-2075.1986.tb04464.x
Sonibare, M., Jayeola, A., and Egunyomi, A. (2004). A morphometric analysis of the genus Ficus Linn.(moraceae). Afr. J. Biotechnol. 3, 229–235. doi: 10.5897/AJB2004.000-2043
Souza, U. J. B. D., Vitorino, L. C., Bessa, L. A., and Silva, F. G. (2020). The Complete Plastid Genome of Artocarpus camansi: A High Degree of Conservation of the Plastome Structure in the Family Moraceae. Forests 11:1179. doi: 10.3390/f11111179
Strand, D. D., D’andrea, L., and Bock, R. (2019). The plastid NAD(P)H dehydrogenase-like complex: Structure, function and evolutionary dynamics. Biochem. J. 476, 2743–2756. doi: 10.1042/BCJ20190365
Su, Y., Huang, L., Wang, Z., and Wang, T. (2018). Comparative chloroplast genomics between the invasive weed Mikania micrantha and its indigenous congener Mikania cordata: Structure variation, identification of highly divergent regions, divergence time estimation, and phylogenetic analysis. Mol. Phylogenet. Evol. 126, 181–195. doi: 10.1016/j.ympev.2018.04.015
Sugiura, C., Kobayashi, Y., Aoki, S., Sugita, C., and Sugita, M. (2003). Complete chloroplast DNA sequence of the moss Physcomitrella patens: Evidence for the loss and relocation of rpoA from the chloroplast to the nucleus. Nucleic Acids Res. 31, 5324–5331. doi: 10.1093/nar/gkg726
Thode, V. A., and Lohmann, L. G. (2019). Comparative chloroplast genomics at low taxonomic levels: A case study using Amphilophium (Bignonieae. Bignoniaceae). Front. Plant Sci. 10:796. doi: 10.3389/fpls.2019.00796
Vieira, M. L. C., Santini, L., Diniz, A. L., and Munhoz, C. D. F. (2016). Microsatellite markers: What they mean and why they are so useful. Genet. Mol. Biol. 39, 312–328. doi: 10.1590/1678-4685-GMB-2016-0027
Wakasugi, T., Tsudzuki, J., Ito, S., Nakashima, K., Tsudzuki, T., and Sugiura, M. (1994). Loss of all ndh genes as determined by sequencing the entire chloroplast genome of the black pine Pinus thunbergii. PNAS 91, 9794–9798. doi: 10.1073/pnas.91.21.9794
Wang, G., Cannon, C. H., and Chen, J. (2016). Pollinator sharing and gene flow among closely related sympatric dioecious fig taxa. Proc. Biol. Sci 2016:283. doi: 10.1098/rspb.2015.2963
Wang, G., Zhang, X., Herre, E. A., Mckey, D., Machado, C. A., Yu, W. B., et al. (2021). Genomic evidence of prevalent hybridization throughout the evolutionary history of the fig-wasp pollination mutualism. Nat. Commun. 12:718. doi: 10.1038/s41467-021-20957-3
Wang, L., Wang, J., He, C., Zhang, J., and Zeng, Y. (2020). Characterization and comparison of chloroplast genomes from two sympatric Hippophae species (Elaeagnaceae). J. For. Res. 32, 307–318. doi: 10.1007/s11676-019-01079-5
Wei, R., Yan, Y. H., Harris, A. J., Kang, J. S., Shen, H., Xiang, Q. P., et al. (2017). Plastid phylogenomics resolve deep relationships among eupolypod II ferns with rapid radiation and rate heterogeneity. Genome Biol. Evol. 9, 1646–1657. doi: 10.1093/gbe/evx107
Wei, Z. D., Kobmoo, N., Cruaud, A., and Kjellberg, F. (2014). Genetic structure and hybridization in the species group of Ficus auriculata: Can closely related sympatric Ficus species retain their genetic identity while sharing pollinators? Mol. Ecol. 23, 3538–3550. doi: 10.1111/mec.12825
Weiblen, G. D. (2000). Phylogenetic relationships of functionally dioecious Ficus (Moraceae) based on ribosomal DNA sequences and morphology. A. J. Bot. 87, 1342–1357. doi: 10.2307/2656726
Weiblen, G. D. (2002). How to be a fig wasp. Annu. Rev. Entomol. 47, 299–330. doi: 10.1146/annurev.ento.47.091201.145213
Wick, R. R., Schultz, M. B., Zobel, J., and Holt, K. E. (2015). Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Wicke, S., Schneeweiss, G. M., Depamphilis, C. W., Muller, K. F., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Wu, L., Cui, Y., Wang, Q., Xu, Z., Wang, Y., Lin, Y., et al. (2021). Identification and phylogenetic analysis of five Crataegus species (Rosaceae) based on complete chloroplast genomes. Planta 254:14. doi: 10.1007/s00425-021-03667-4
Wu, Z., Liao, R., Yang, T., Dong, X., Lan, D., Qin, R., et al. (2020). Analysis of six chloroplast genomes provides insight into the evolution of Chrysosplenium (Saxifragaceae). BMC Genomics 21:621. doi: 10.1186/s12864-020-07045-4
Wu, Z. Y., Raven, P. H., Hong, D., Zhou, Z., and Gilbert, M. G. (2003). Flora of China. Beijing: Science Press.
Xie, Z., and Merchant, S. (1996). The plastid-encoded ccsA gene is required for heme attachment to chloroplast c-type cytochromes. J. Biol. Chem. 271, 4632–4639. doi: 10.1074/JBC.271.9.4632
Xu, L., Harrison, R. D., Yang, P., and Yang, D.-R. (2011). New insight into the phylogenetic and biogeographic history of genus Ficus: Vicariance played a relatively minor role compared with ecological opportunity and dispersal. J. Syst. Evol. 49, 546–557. doi: 10.1111/j.1759-6831.2011.00155.x
Yang, Q., Fu, G. F., Wu, Z. Q., Li, L., Zhao, J. L., and Li, Q. J. (2021b). Chloroplast Genome Evolution in Four Montane Zingiberaceae Taxa in China. Front. Plant Sci. 12:774482. doi: 10.3389/fpls.2021.774482
Yang, J., Chiang, Y.-C., Hsu, T.-W., Kim, S.-H., Pak, J.-H., and Kim, S.-C. (2021a). Characterization and comparative analysis among plastome sequences of eight endemic Rubus (Rosaceae) species in Taiwan. Sci. Rep. 11:1152. doi: 10.1038/s41598-020-80143-1
Yang, X., Xie, D. F., Chen, J. P., Zhou, S. D., Yu, Y., and He, X. J. (2020). Comparative analysis of the complete chloroplast genomes in Allium Subgenus Cyathophora (Amaryllidaceae): Phylogenetic relationship and adaptive evolution. Biomed. Res. Int. 2020:1732586. doi: 10.1155/2020/1732586
Yang, Z. (1998). Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol. Biol. Evol. 15, 568–573. doi: 10.1093/oxfordjournals.molbev.a025957
Yang, Z., and Nielsen, R. (1998). Synonymous and nonsynonymous rate variation in nuclear genes of mammals. J. Mol. Evol. 46, 409–418. doi: 10.1007/pl00006320
Yang, Z., and Nielsen, R. (2002). Codon-Substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol. Biol. Evol. 19, 908–917. doi: 10.1093/oxfordjournals.molbev.a004148
Yang, Z., Wong, W. S., and Nielsen, R. (2005). Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 22, 1107–1118. doi: 10.1093/molbev/msi097
Yang, Z., Zhao, T., Ma, Q., Liang, L., and Wang, G. (2018). Comparative genomics and phylogenetic analysis revealed the chloroplast genome variation and interspecific relationships of Corylus (Betulaceae) species. Front. Plant Sci. 9:927. doi: 10.3389/fpls.2018.00927
Yao, X., Tan, Y. H., Yang, J. B., Wang, Y., Corlett, R. T., and Manen, J. F. (2019). Exceptionally high rates of positive selection on the rbcL gene in the genus Ilex (Aquifoliaceae). BMC Evol. Biol. 19:192. doi: 10.1186/s12862-019-1521-1
Yin, K., Zhang, Y., Li, Y., and Du, F. K. (2018). Different natural selection pressures on the atpF gene in evergreen sclerophyllous and deciduous oak species: Evidence from comparative analysis of the complete chloroplast genome of Quercus aquifolioides with other oak species. Int. J. Mol. Sci. 19:1042. doi: 10.3390/ijms19041042
Zhang, X., Wang, G., Zhang, S., Chen, S., Wang, Y., Wen, P., et al. (2020b). Genomes of the Banyan Tree and Pollinator Wasp Provide Insights into Fig-Wasp Coevolution. Cell 183:e17. doi: 10.1016/j.cell.2020.09.043
Zhang, D., Gao, F., Jakovlic, I., Zou, H., Zhang, J., Li, W. X., et al. (2020a). PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi: 10.1111/1755-0998.13096
Zhang, J., Nielsen, R., and Yang, Z. (2005). Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 22, 2472–2479. doi: 10.1093/molbev/msi237
Zhang, L.-F., Zhang, Z., Wang, X., Gao, H.-Y., Tian, H., and Li, H.-Q. (2018). Molecular phylogeny of the Ficus auriculata Complex (Moraceae). Phytotaxa 362:39. doi: 10.11646/phytotaxa.362.1.3
Keywords: Ficus, chloroplast genome, nucleotide diversity hotspots, phylogenetic relationship, adaptive evolution, divergence time
Citation: Zhang Z-R, Yang X, Li W-Y, Peng Y-Q and Gao J (2022) Comparative chloroplast genome analysis of Ficus (Moraceae): Insight into adaptive evolution and mutational hotspot regions. Front. Plant Sci. 13:965335. doi: 10.3389/fpls.2022.965335
Received: 09 June 2022; Accepted: 22 August 2022;
Published: 15 September 2022.
Edited by:
Xiaohua Jin, Institute of Botany (CAS), ChinaReviewed by:
Gang Yao, South China Agricultural University, ChinaWenpan Dong, Beijing Forestry University, China
Copyright © 2022 Zhang, Yang, Li, Peng and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan-Qiong Peng, cGVuZ3lxQHh0YmcuYWMuY24=; Jie Gao, Z2FvamllQHh0Ymcub3JnLmNu
†These authors have contributed equally to this work