 Ali Raza1
Ali Raza1 Yasir Sharif2
Yasir Sharif2 Kun Chen2
Kun Chen2 Lihui Wang2
Lihui Wang2 Huiwen Fu2
Huiwen Fu2 Yuhui Zhuang3
Yuhui Zhuang3 Annapurna Chitikineni4
Annapurna Chitikineni4 Hua Chen1,2
Hua Chen1,2 Chong Zhang1,2
Chong Zhang1,2 Rajeev K. Varshney1,4*
Rajeev K. Varshney1,4* Weijian Zhuang1,2*
Weijian Zhuang1,2*- 1Key Laboratory of Ministry of Education for Genetics, Breeding and Multiple Utilization of Crops, Center of Legume Plant Genetics and Systems Biology, College of Agriculture, Oil Crops Research Institute, Fujian Agriculture and Forestry University (FAFU), Fuzhou, China
- 2College of Plant Protection, Fujian Agriculture and Forestry University (FAFU), Fuzhou, China
- 3College of Life Science, Fujian Agriculture and Forestry University (FAFU), Fuzhou, China
- 4Murdoch’s Centre for Crop and Food Innovation, State Agricultural Biotechnology Centre, Food Futures Institute, Murdoch University, Murdoch, WA, Australia
Ascorbate peroxidase (APX), an important antioxidant enzyme, plays a significant role in ROS scavenging by catalyzing the decrease of hydrogen peroxide under various environmental stresses. Nevertheless, information about the APX gene family and their evolutionary and functional attributes in peanut (Arachis hypogea L.) was not reported. Therefore, a comprehensive genome-wide study was performed to discover the APX genes in cultivated peanut genome. This study identified 166 AhAPX genes in the peanut genome, classified into 11 main groups. The gene duplication analysis showed that AhAPX genes had experienced segmental duplications and purifying selection pressure. Gene structure and motif investigation indicated that most of the AhAPX genes exhibited a comparatively well-preserved exon-intron pattern and motif configuration contained by the identical group. We discovered five phytohormones-, six abiotic stress-, and five growth and development-related cis-elements in the promoter regions of AhAPX. Fourteen putative ah-miRNAs from 12 families were identified, targeting 33 AhAPX genes. Furthermore, we identified 3,257 transcription factors from 38 families (including AP2, ARF, B3, bHLH, bZIP, ERF, MYB, NAC, WRKY, etc.) in 162 AhAPX genes. Gene ontology and KEGG enrichment analysis confirm the role of AhAPX genes in oxidoreductase activity, catalytic activity, cell junction, cellular response to stimulus and detoxification, biosynthesis of metabolites, and phenylpropanoid metabolism. Based on transcriptome datasets, some genes such as AhAPX4/7/17/77/82/86/130/133 and AhAPX160 showed significantly higher expression in diverse tissues/organs, i.e., flower, leaf, stem, roots, peg, testa, and cotyledon. Likewise, only a few genes, including AhAPX4/17/19/55/59/82/101/102/137 and AhAPX140, were significantly upregulated under abiotic (drought and cold), and phytohormones (ethylene, abscisic acid, paclobutrazol, brassinolide, and salicylic acid) treatments. qRT-PCR-based expression profiling presented the parallel expression trends as generated from transcriptome datasets. Our discoveries gave new visions into the evolution of APX genes and provided a base for further functional examinations of the AhAPX genes in peanut breeding programs.
Introduction
Plants are regularly subjected to various environmental factors (abiotic and biotic), which substantially influence crop productivity and cause challenges to food security (Sabagh et al., 2021; Mir et al., 2022; Raza et al., 2022a,b; Saeed et al., 2022; Sharma et al., 2022). These factors can enhance the generation of reactive oxygen species (ROS), damaging cellular systems and supermolecules consisting of DNA, proteins, and lipids, and ultimately leading to cell death (Fahad et al., 2015, 2017; Mittler, 2017; Hasanuzzaman et al., 2020). ROS are mainly produced in different locations including chloroplast, apoplast, plasma membrane, mitochondrion, endoplasmic reticulum, peroxisomes, and cell walls (Mittler, 2017; Hasanuzzaman et al., 2020). In plants, ROS are formed as chemical by-products due to the imperfect decline of oxygen metabolism. Further, ROS are considered as signaling elements that regulate stress tolerance mechanisms in plant molecular biology (Das and Roychoudhury, 2014; Mittler, 2017; Hasanuzzaman et al., 2020). Current progress has revealed that ROS homeostasis is essential for maintaining typical cellular characteristics (Mittler, 2017; Hasanuzzaman et al., 2020, 2021). Subsequently, for regular ROS signaling, plants have developed defense systems including enzymatic and non-enzymatic antioxidant enzymes to maintain the equilibrium between ROS-scavenging and production under stress conditions (Das and Roychoudhury, 2014; Mittler, 2017; Hasanuzzaman et al., 2020, 2021).
In plants, among diverse antioxidant enzymes entailed in ROS-scavenging mechanisms, ascorbate peroxidase (APX; EC, 1.11.1.11) belongs to the heme peroxidase superfamily (Hodges et al., 1999; Teixeira et al., 2004; Lazzarotto et al., 2011). In higher plants, APXs are one of the main antioxidant enzymes involved in regulating the ascorbate-glutathione cycle and take parts to scavenge hydrogen peroxide (H2O2) from chloroplast and the cytoplasm. Mainly, it utilized the ascorbic acid as an electron giver to scavenge H2O2 generated in plants and thus enhances tolerance to oxidative and other stresses in plants (Cao et al., 2017; Pandey et al., 2017; Hasanuzzaman et al., 2021; Raza et al., 2021a). Additionally, APX enzymes are automated by APX gene family involved in stress tolerance has been thoroughly explored in diverse plant species using various in silico approaches. For example, five APX genes have been discovered in wild watermelon (Citrullus lanatus) (Malambane et al., 2018); six in shrub (Ammopiptanthus nanus) (Wang et al., 2022); eight in rice (Oryza sativa L.) (Teixeira et al., 2004) and Arabidopsis thaliana (Panchuk et al., 2002, Panchuk et al., 2005); nine in sorghum (Sorghum bicolor L.) (Akbudak et al., 2018); 13 in kiwifruit (Actinidia chinensis) (Liao et al., 2020); 16 APX genes in tomato (Solanum lycopersicum L.) (Najami et al., 2008); 21 in wheat (Triticum aestivum L.) (Tyagi et al., 2020); and 26 in cotton (Gossypium hirsutum L.) (Tao et al., 2018). Nevertheless, the APX gene family in peanut (Arachis hypogea L.) has not been systematically reported, and their roles in peanut development and stress tolerance still remain ambiguous.
Cultivated peanut/groundnut (A. hypogaea L.), an allotetraploid crop, is one of the most valuable and economic oilseed food crops globally (Agarwal et al., 2018; Bertioli et al., 2019; Chen X. et al., 2019; Zhuang et al., 2019). This crop is being widely cultivated in the tropical and subtropical regions globally; however, several abiotic and biotic factors significantly affect its growth and production, including many important agronomic traits (Agarwal et al., 2018; Gangurde et al., 2020, 2021; Kumar et al., 2020; Pandey et al., 2020; Shasidhar et al., 2020; Sinha et al., 2020; Jadhav et al., 2021; Soni et al., 2021; Aravind et al., 2022; Bomireddy et al., 2022; Liu et al., 2022; Patel et al., 2022). Therefore, it is vital to identify new potential genes associated with multiple stress tolerance and trait improvement in peanut for better protein-rich food supply, particularly in Asian and African countries. In this regard, the recently sequenced peanut genome and recent advances in genomics-assisted breeding make it easier for us to carry out a comprehensive systematic analysis of new gene families (Varshney et al., 2019, 2020, 2021a,b). To our best knowledge, APX gene family was yet to be comprehensively characterized in peanut. Thus, the current study performed a genome-wide identification and characterized the APX gene family in peanut (AhAPX). Several in silico analysis, such as characterization, genomic evolution, gene structure, conserved motifs, cis-regulatory elements, putative miRNA and transcription factors, functional annotations, etc., were utilized to get insights into the novel roles of AhAPX genes. Furthermore, their expression profiling in diverse tissues/organs, under phytohormones and abiotic stress conditions were also performed using transcriptome and qRT-PCR techniques. In short, this report offered evolutionary and functional roles of AhAPX genes which could open new windows for further functional studies on the novel roles of AhAPX genes in peanut breeding programs under stress conditions.
Materials and Methods
Discovery and Physicochemical Features of APX Genes
As explained earlier (Li et al., 2021; Raza et al., 2021b; Su et al., 2021), two approaches, i.e., BLASTP and the Hidden Markov Model (HMM), were applied to identify APX genes in the peanut (A. hypogea) genome. The peanut genome sequence was taken from peanut Genome Resource (PGR) database1 (Zhuang et al., 2019). In the first approach, the sequences of eight Arabidopsis thaliana APX genes were gained from TAIR Arabidopsis genome database2 (Rhee et al., 2003). Then, these sequences were utilized as a query to perform the BLASTP against peanut genome. In the second approach, HMMER 3.13 (Finn et al., 2015) software was employed to seek out the APX genes with default controls. Later, the HMM file of the ascorbic acid peroxidase domain (PF00141) was retrieved from the Pfam database4 (El-Gebali et al., 2019). Lastly, the sequences comprising the PF00141 domain were chosen as putative APX genes, and finally, 166 AhAPX genes were discovered by uniting the results obtained from both approaches in the peanut genome. Following the same approaches, APX genes were also discovered in diploid parents, i.e., A. duranensis (90 genes; AdAPX1-AdAPX90) and A. ipaensis (102 genes; AiAPX1-AiAPX102). Their genome sequences were downloaded from PeanutBase database5 (Dash et al., 2016). The detailed information (including gene name, gene ID, and protein sequences) of all identified APX genes is given in Supplementary Table 1.
Physicochemical features of AhAPX were assessed utilizing the ProtParam tool6 in the ExPASy server (Gasteiger et al., 2005). Subcellular localization of AhAPX proteins was estimated from CELLO v.2.57 (Yu et al., 2006). Exon-intron configuration of all AhAPX were determined using TBtools software (v1.09867)8 (Chen et al., 2020). The conserved motifs of AhAPX sequences were documented using the MEME website9 (Bailey et al., 2009).
Evaluation of Chromosomal Location, Phylogenetic Relationships, and Synteny Analysis of APX Genes
The data about the chromosomal location of AhAPX was attained from the PGR database, and the TBTools was utilized to map the genes on chromosomes. To discover the evolutionary link of the APX proteins, a phylogenetic tree among A. hypogea (AhAPXs), A. duranensis (AdAPXs), A. ipaensis (AiAPXs), and A. thaliana (AtAPXs) was created. Multiple sequence alignment was implemented using MEGA7 software10 (Kumar et al., 2018). The neighbor-joining (NJ) method was undertaken to design a phylogenetic tree with 1,000 bootstrap replicates and iTOL was used to beautify the tree11 (Letunic and Bork, 2021).
The syntenic associations of APX genes between A. hypogea, A. duranensis, A. ipaensis, and A. thaliana were executed through the MCScanX toolkit and were pictured by the Advance Circos package in the TBTools software (Chen et al., 2020). Additionally, the multiple collinearity analysis of APX genes was completed via multiple synteny Plot packages in TBTools software. The Ka/Ks ratios of all AhAPX were predicted via simple Ka/Ks calculator in TBTools software.
Prediction of cis-Regulatory Elements in the AhAPX Promoters
To predict the putative cis-regulatory elements in the AhAPX promoters, the 2 Kb sequences upstream of start codons were separated from the peanut genome. The promoter sequences of all AhAPX genes were observed with PlantCARE website12 (Lescot et al., 2002), and the picture was illustrated using TBtools software.
Prediction of Putative miRNAs Targeting AhAPX Genes and Functional Annotation Evaluation
The CDS of all AhAPX was used to predict the miRNA target sites with psRNATarget website13 (Dai et al., 2018) with default considerations. The interactive network figure among the putative miRNAs and AhAPX genes was made via Cytoscape software (v3.9)14 (Shannon et al., 2003). Gene ontology (GO) and Kyoto encyclopedia of genes and genomics (KEGG) annotation evaluation was undertaken by submitting all AhAPX protein sequences to the eggNOG v4.015 (Powell et al., 2014). At the same time, GO and KEGG enrichment evaluations were performed with TBtools software.
Prediction of Transcription Factor Regulatory Network of AhAPX Genes
To predict the putative transcription factors (TFs) and regulatory network, the 500 bp nucleotide sequences from upstream regions of AhAPX genes were removed and complied to the PlantRegMap (Transcriptional Regulatory Map)16 with p-value ≤ 1e–6 (Tian et al., 2020). The regulatory network of predicted TFs and AhAPX genes was created with Cytoscape v3.9 software.
Expression Profiling of AhAPX Genes
The expression levels of all AhAPX genes at diverse developmental tissues/organs (embryo, cotyledon, testa, pericarp, peg, root and stem, root nodule, root tip, root, step tip, stem, leaf, and flower), under various hormones (ethylene, abscisic acid, paclobutrazol, brassinolide, and salicylic acid), and abiotic stress (drought and cold) conditions were evaluated using openly available transcriptome dataset of cultivated peanut (cultivar Shitouqi) at PGR database (see text footnote 1; BioProject PRJNA480120) (Zhuang et al., 2019). The detailed procedure for sample harvesting and data analysis is presented in our recent paper (Zhuang et al., 2019). Owing to the great differences in the expression trends, we normalize the log2 of fragments per kilobase of transcript per million (FPKM) values. Finally, the circular heat maps were designed by TBtools software.
Plant Material and Stress Conditions
In this study, a widely cultivated peanut variety in southeast China, “Minhua-6” was used for stress treatments. The same variety was also used for transcriptome analysis in our recent paper (Zhuang et al., 2019). The seeds of the “Minhua-6” cultivar were obtained from the FAFU, Fuzhou, China. The vigor seeds were cultured on small pots having a mix of vermicompost. For stress treatment, germinated seedlings at the four-leaf stage were exposed to cold stress at 4°C and ABA (10 μg mL–1) for 0 (CK), 3, 6, 9, and 12 h with three biological repetitions. All of the samples were instantly frozen in liquid nitrogen and were kept at -80°C until RNA extraction.
RNA Extraction and qRT-PCR-based Expression Analysis
Total RNA was isolated utilizing the CTAB method as described in our recent work (Sharif et al., 2021), and cDNA was prepared with the help of Evo M-MLV RT Kit with gDNA Clean for qPCR II (Code No. AG11711; Hunan Aikerui Biological Engineering Co., Ltd., China) following the developer guidelines. The comprehensive information on qRT-PCR reaction has been described in our recent work (Sharif et al., 2021). The peanut Actin gene was used as a housekeeping gene to stabilize the expression (Chi et al., 2012). The expression data of three biological repeats were normalized using the 2–ΔΔCT method (Livak and Schmittgen, 2001). All the primers used for qRT-PCR are given in Supplementary Table 2. The graphs were made with GraphPad Prism v9.0.0 software17 (Swift, 1997).
Results
Comprehensive Characterization of AhAPX Genes in Peanut Genome
In this study, a total of 166 AhAPX genes were discovered in the peanut genome (Table 1). Henceforward, these genes are labeled as “AhAPX1–AhAPX166.” These genes were irregularly mapped in the cultivated peanut genome. The maximum number (15) of AhAPX genes were mapped on Chr14, followed by Chr04/Chr11 with 11 genes on each chromosome. While, Chr01/Chr06/Chr10/Chr20 were found to have ten genes, followed by Chr05/Chr15/Chr16 with nine genes, Chr07/Chr19 with eight genes, Chr09/Chr13 with seven genes, Chr03/Chr08/Chr17/Chr18 with six genes on each chromosome. The lowest number (1 and 4) of AhAPX genes were mapped on Chr02 and Chr12, respectively. Notably, three AhAPX genes (AhAPX1/2/3) were also mapped on an unassembled region (Chr00) (Figure 1).
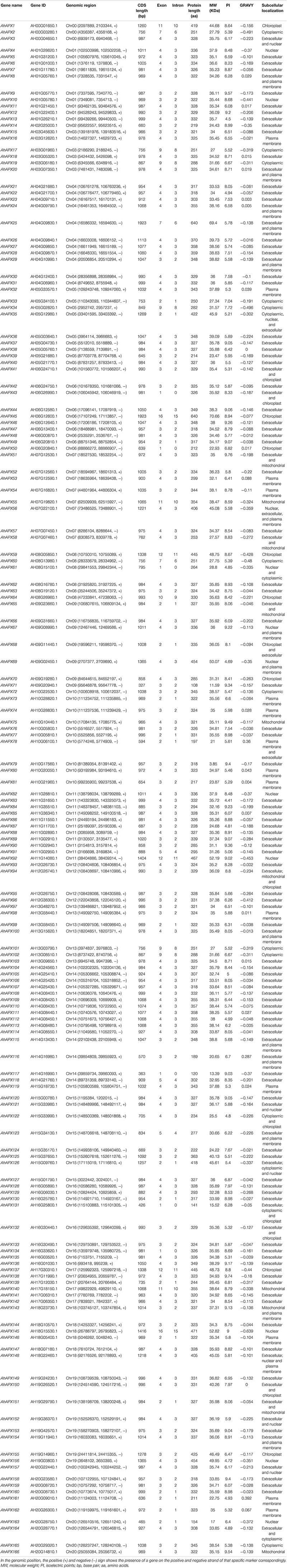
Table 1. The data of 166 AhAPX genes identified in peanut genome.
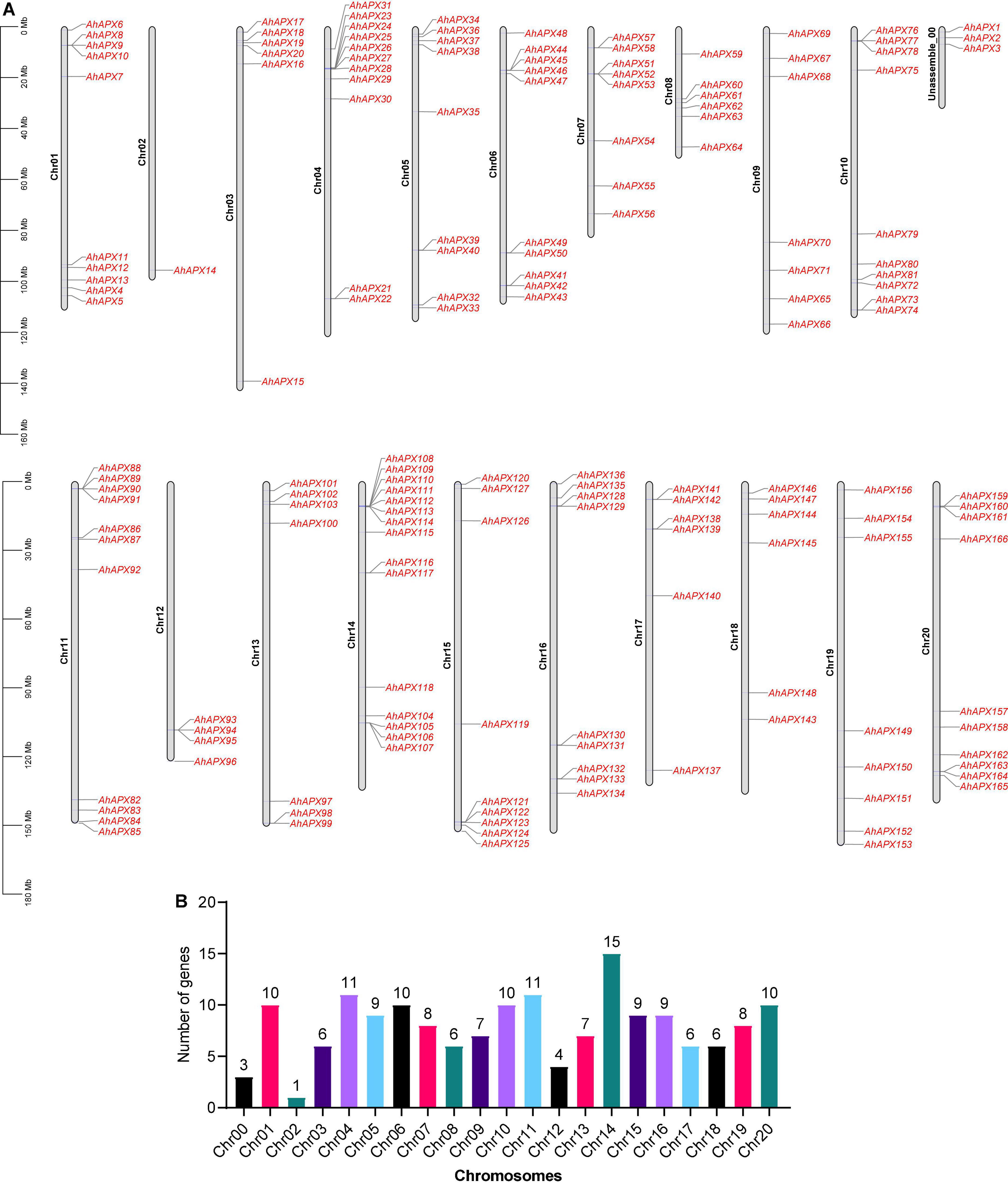
Figure 1. Chromosomal mapping of AhAPX genes on A. hypogea genome. (A) Distribution map of AhAPX genes on 20 chromosomes, including one unassembled region. Representative chromosome numbers are shown on the left side of each bar. The scale on the left side represents the chromosomal distance and is in megabases (Mb). (B) Graph indicates the number of AhAPX genes mapped on each chromosome.
Comprehensive information of all predicted 166 AhAPX genes is presented in Table 1. In short, the CDS length varied from 327 bp (AhAPX71) to 1,923 bp (AhAPX25/45), and the amino acid length assorted from 108 (AhAPX71) to 640 (AhAPX25/45) amino acids. The number of exons varied from one (AhAPX43/50/61/117/131/134) to 16 (AhAPX45/145) (Table 1). Particularly, only two genes (AhAPX45 and AhAPX145) had the uppermost number of introns (i.e., 15), and quite a few genes lack introns (i.e., AhAPX43/50/61/117/131/134) (Table 1). The anticipated molecular weights (MW) of the 166 AhAPX proteins increased from 3.85 kDa (AhAPX79) to 70.66 kDa (AhAPX45), the isoelectric points (PI) extended from 4.41 (AhAPX83) to 9.76 (AhAPX51), and the GRAVY ranged from -0.639 (AhAPX145) to 0.392 (AhAPX161). The transformations in MW and PI are primarily due to the elevated content of necessary amino acids and post-translational alterations. The in silico subcellular localization discovered that 115 AhAPX proteins were situated on the extracellular matrix, 14 AhAPX proteins on plasma membrane, 12 AhAPX proteins on cytoplasm, nine AhAPX proteins on chloroplast, and five AhAPX proteins on mitochondrion (Table 1). Notably, some AhAPX proteins were found to be located in more than one location (Table 1).
On the other hand, eight genes (AtAPXs) from A. thaliana, 90 genes (AdAPX1-AdAPX90) from A. duranensis, and 102 genes (AiAPX1-AiAPX102) from A. ipaensis genomes were also recognized to study the evolution of APX genes between tetraploid and diploid parents (Supplementary Table 1).
Insights From Phylogenetic Relationships of APX Proteins
To determine the in-depth evolutionary and phylogenetic history between the AhAPX (166 members), AdAPX (90 members), AiAPX (102 members), and AtAPX proteins (8 members), an unrooted phylogenetic tree was built by a multiple sequence alignment, which was divided into 11 main groups (group1–group11) (Figure 2). The discoveries exposed that group1 comprised of seven APX members (2 AhAPX, 2 AiAPX, and 3 AdAPX) followed by group4/5 (eight APX members), and group3 (14 APX members). Notably, the maximum number of APX members (66 AhAPX, 39 AiAPX, and 33 AdAPX) were found in group 11 followed by group7 (38 APX members), group2/6 (37 APX members), group8/9 (28 APX members), and group10 (24 APX members) (Figure 2). All AtAPX members were clustered only in one group, i.e., group 2. In general, APXs grouped into the indistinguishable sub-group may retain corresponding functions. It is worth stating that A. hypogea APX (AhAPXs) were distributed in each group with homologs from A. duranensis, A. ipaensis, and A. thaliana., and group11 was detected to have more AhAPX members than the other 10 groups (Figure 2). Furthermore, it was observed that the AhAPXs showed a greater phylogenetic network with the AdAPXs and AiAPXs in each group.
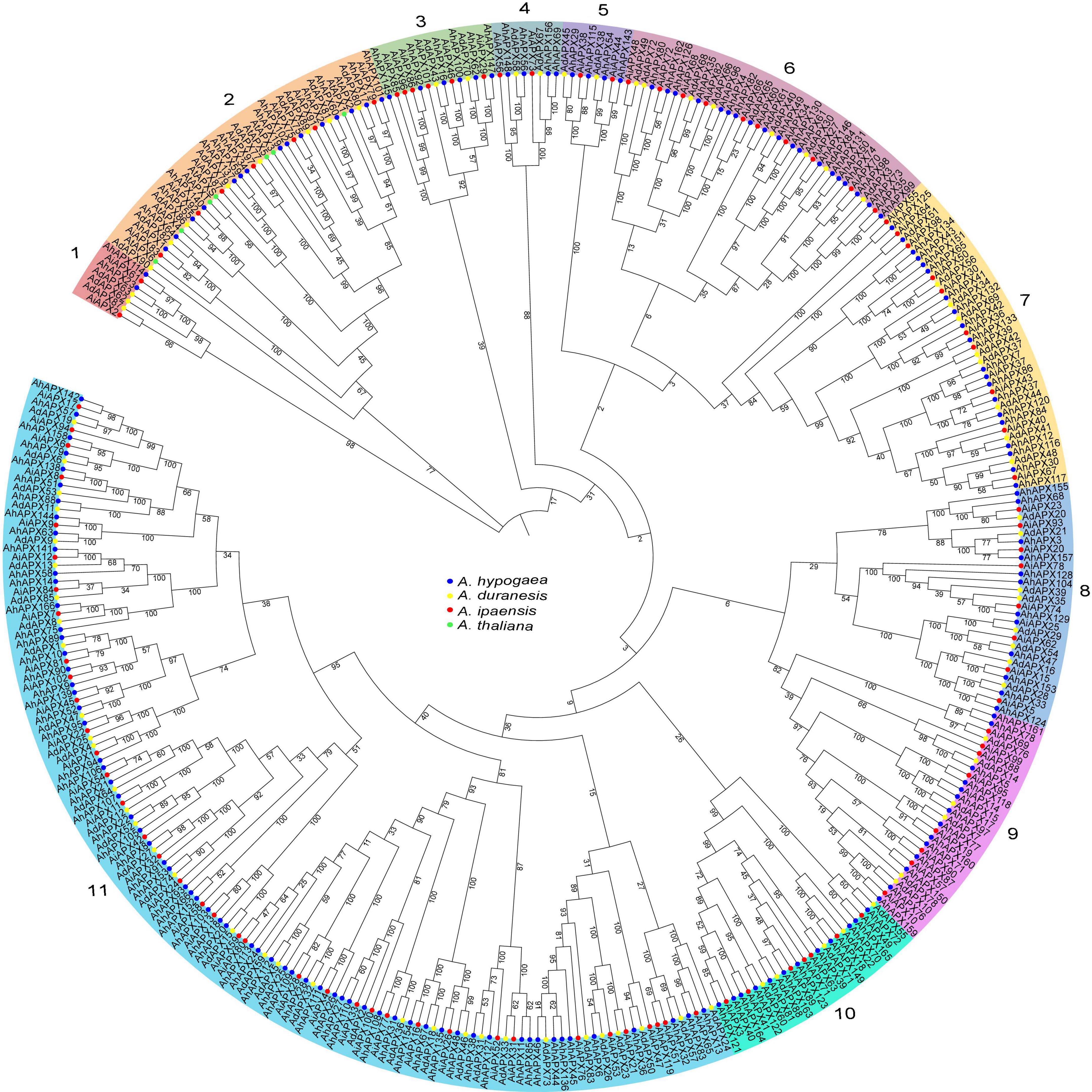
Figure 2. An unrooted neighbor-joining phylogenetic tree of APX proteins from A. hypogea, A. duranensis, A. ipaensis, and A. thaliana. On the whole, 166 AhAPXs from A. hypogea (blue circles), 90 AdAPXs from A. duranensis (yellow circles), 102 AiAPXs from A. ipaensis (red circles), and 8 AtAPXs from Arabidopsis thaliana (green circles) were clustered into 11 groups based on sequence similarities, domain, and 1,000 bootstrap values. The percentage of bootstrap values is shown in the notes.
Insights Into Synteny and Collinearity of APX Genes
Gene duplications (i.e., tandem and segmental) are thought to be the main factors in supporting the expansion and evolution of new gene families in plants (Cannon et al., 2004). Hence, gene duplication procedures were assessed between AhAPXs, AdAPXs, AiAPXs, and AtAPXs (Supplementary Table 3). The results of gene duplication study showed that there were 92 AhAPX gene pairs, and these pairs were unevenly mapped on different chromosomes (Figure 3 and Supplementary Table 3). Mainly, chromosome 13 had a maximum number (i.e., 16) of AhAPX gene pairs, followed by chromosome 5 with 12 AhAPX gene pairs. The least number of gene pairs (i.e., two) was discovered on chromosome 12, and no gene pair was found on chromosome 2 (Figure 3 and Supplementary Table 3). The results reveal that segmental duplications have contributed to the expansion of AhAPX genes in the cultivated peanut genome (Supplementary Table 3). Notably, no tandem duplicated gene pairs were identified.
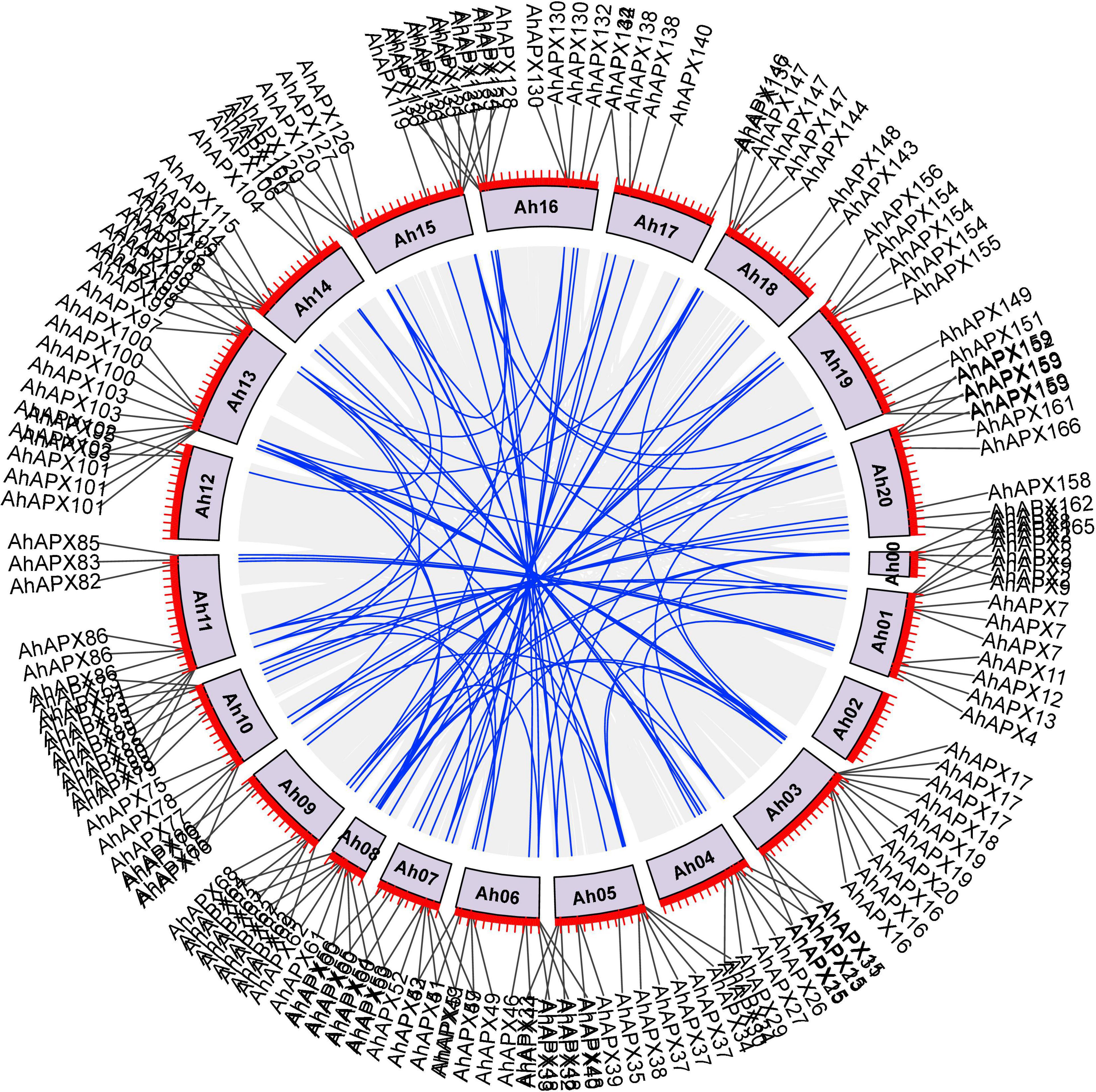
Figure 3. Chromosomal positions and inter-chromosomal groups of duplicated AhAPX gene pairs. Gray lines in the background demonstrate all syntenic blocks in the A. hypogea genome, and the blue lines exhibit the segmental or tandem duplication network zones among AhAPX genes. The near location of AhAPX genes is marked with black lines outside with chromosome names.
Similarly, 10 duplicated gene pairs were detected between AhAPX and AtAPX (Supplementary Figure 1 and Supplementary Table 3); 171 pairs between AhAPX and AiAPX (Supplementary Figure 2 and Supplementary Table 3); and 160 pairs between AhAPX and AdAPX (Supplementary Figure 3 and Supplementary Table 3). All these gene pairs were irregularly mapped on different chromosomes. Taken together, these conclusions explained that the duplication activities played a vital role in enlarging the APX genes between diploid and tetraploid parents. Further, it can also be concluded that A. hypogea might have lost some genes during genome evolution.
Collinearity analysis was carried out to review the evolutionary association of the APX genes between A. hypogea, A. duranensis, A. ipaensis, and A. thaliana (Figure 4 and Supplementary Table 3). The results discovered a strong orthologous of APX genes among these four species (Figure 4). On the whole, several A. hypogea genes presented syntenic networks with different AdAPX, AiAPX, and AtAPX genes. Particularly, only one gene (AhAPX14) at chromosome Ah2 exhibited a syntenic connection with AdAPX85 gene at chromosome Ad02 (Figure 4 and Supplementary Table 3), while other homologous genes present on other A. hypogea chromosomes also showed a syntenic relationship with many AdAPX, AiAPX and AtAPX genes (Figure 4 and Supplementary Table 3). These findings indicate that whole-genome or segmental duplication procedures are considered a main evolutionary force in the evolution of AhAPX genes in the peanut genome (Figure 4 and Supplementary Table 3).
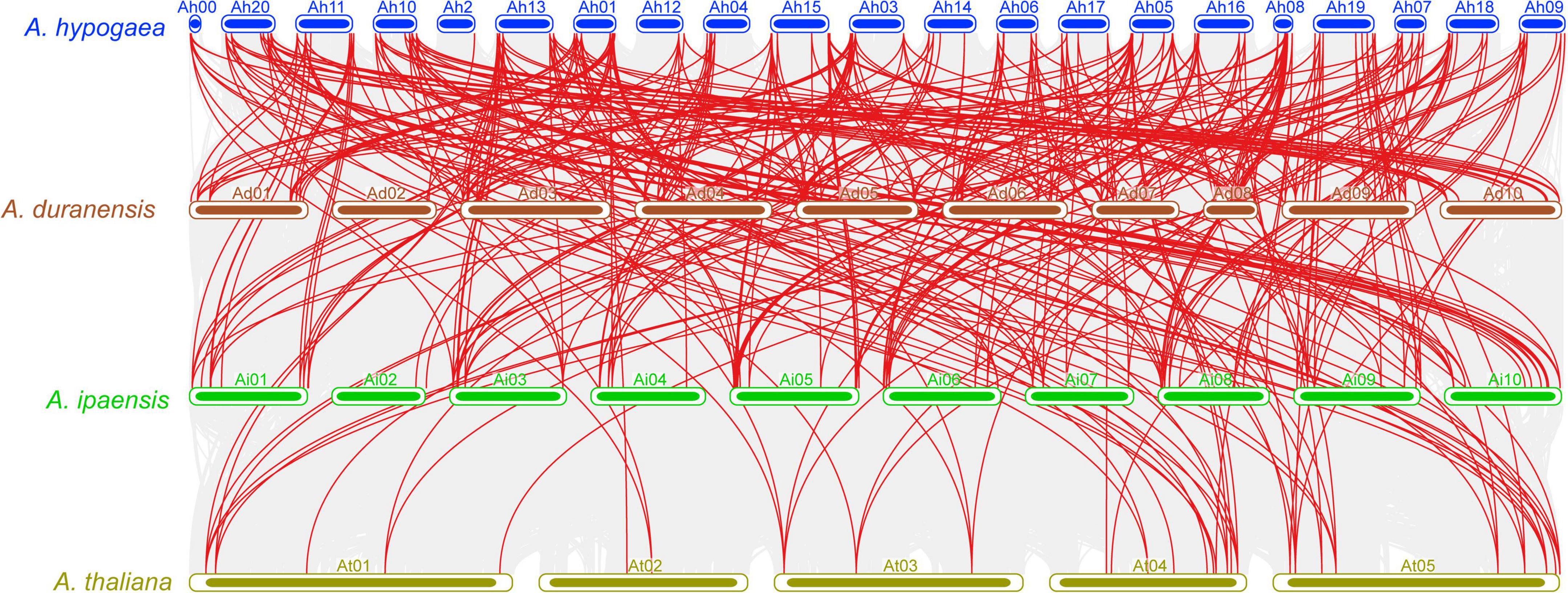
Figure 4. Multiple collinearity analysis of APX genes between A. hypogea, A. duranensis, A. ipaensis, and A. thaliana chromosomes. Gray lines in the background specify the collinear blocks within A. hypogea and other three genomes, while the red lines highlight the syntenic APX orthologous gene pairs.
The Ka/Ks ratio is considered as a huge diagnostic marker in evaluating the sequence evolution in terms of selection pressures and duplication types (Hurst, 2002). Thus, to understand the evolutionary story of the AhAPX, the Ka, Ks, and Ka/Ks ratio was revealed (Supplementary Table 3). The dataset unveiled that all duplicated AhAPX gene pairs had a Ka/Ks ratio of <1 (Supplementary Table 3), demonstrating that the AhAPX genes may have experienced strong purifying selective pressure and segmental duplications throughout the evolution procedure (Supplementary Table 3).
Insights Into Gene Structures and Conserved Motifs of AhAPX Genes
The exon-intron arrangements and conserved motifs of the AhAPX genes were analyzed to get insights into the advancement of the APX family genes in peanut genome (Figure 5 and Supplementary Table 4). The outcomes revealed that the number of exons and introns varied from 16 to 1 and 0 to 15, respectively (Figure 5B and Supplementary Table 10). In short, 6 genes have 1 exon and zero intron; 13 genes have 2 exons and 1 intron; 5 genes have 3 exons and 2 introns; 18 genes have 2 exons and 1 intron; 41 genes have 3 exons and 2 introns; 81 genes have 4 exons and 3 introns; 3 genes have 5/7 exons and 4/6 introns; 5 genes have 9 exons and 8 introns; only 1 gene has 10 exons and 9 introns; 3 genes have 11/12 exons and 10/11 introns; and only 2 genes have a maximum number of exons (16) and introns (15) (Figure 5B and Supplementary Table 10). Above all, genes belonging to the same sub-tree almost had parallel structures apart from a few genes (Figure 5B). Among all genes, AhAPX149 possess the longest structure, and only a few genes have a complex structure, such as AhAPX17, AhAPX19, AhAPX34, AhAPX45, AhAPX55, AhAPX59, AhAPX64, AhAPX92, AhAPX101, AhAPX102, and AhAPX145 (Figure 5B). Exon loss or gain has been found during the evolution of APX family genes. The results recommended that APX genes held a somewhat frequent exon-intron composition throughout the evolution of peanut genome. Furthermore, AhAPX gene participants inside a sub-tree had exceptionally corresponding gene structures, steady with their phylogenetic clusters.
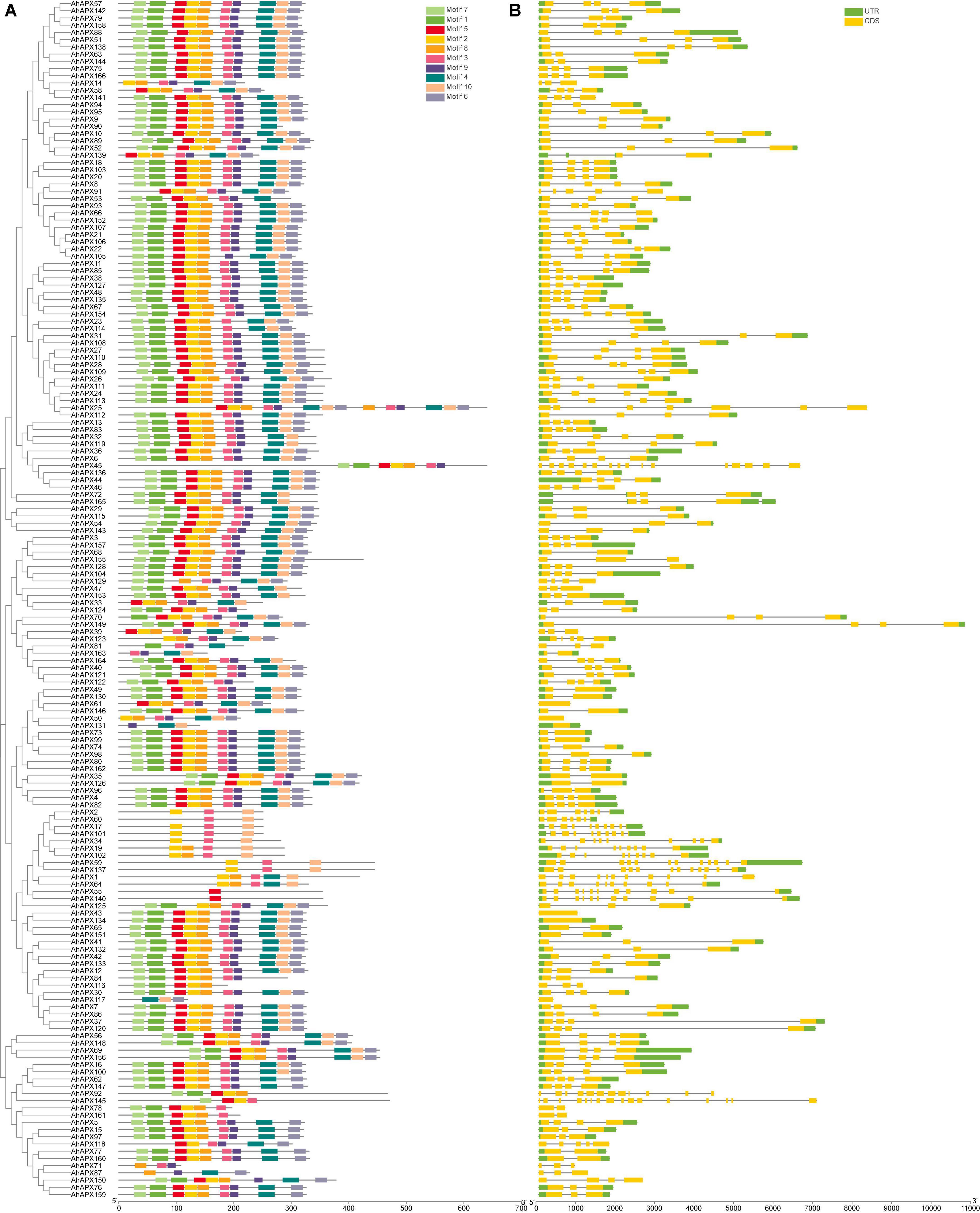
Figure 5. The gene structure and motif analysis of the AhAPX genes. (A) Arrangements of predicted conserved motifs in AhAPXs. Unique color boxes exhibit individual motifs. (B) The gene structure of the AhAPXs. Light green color indicates the UTR regions; yellow color shows the CDS regions, and gray horizontal line displays the introns.
The conserved motif of the AhAPX genes ranged from one (AhAPX55/140) to three (AhAPX2/71/117/131) (Figure 5A). In total, 10 conserved motifs were recognized, and their complete dataset, including motif names, sequences, width, and E-value, is given in Supplementary Table 4. Similar to gene structure, the motif distributions were also similar within the sub-trees (Figure 5A), while some motifs were found to be specific to some genes. For instance, some genes such as AhAPX2/17/101/34/59/137 were limited to motifs 2, 3, and 10. While AhAPX60 gene only contained motifs 3 and 10; AhAPX71 contained motifs 3, 8, and 9; AhAPX117 contained motifs 4, 6, and 10; AhAPX131 contained motifs 4, 9, and 10; and AhAPX71 contained motifs 3, 8, and 9 (Figure 5A). Almost all other motifs were present on all genes except in a few cases (Figure 5A). In summary, the consistency of gene organizations within sub-trees was credibly constant by appraising the conserved motif structures, gene structures, and phylogenetic relations, representing that the APX proteins have enormously well-sustained amino acid deposits and APX members belonging to the same tree may hold corresponding roles.
Cis-Elements: Key Players in the Promoter Regions of AhAPX Genes
To better understand the regulatory role of AhAPX genes toward peanut growth and development, and tolerance to abiotic stress and phytohormones treatment, cis-regulatory elements in the promoter of AhAPX were explored. The complete dataset of cis-elements is presented in Supplementary Table 5. We emphasized and recognized three categories of cis-elements, including abiotic stress-responsive, phytohormones responsive, and growth and development responsive elements (Figures 6, 7 and Supplementary Table 5). Mainly, six abiotic stress-responsive (drought, light, low temperature, wound, defense and stress, and anaerobic) elements were detected. These elements consist of I-box, ATCT-motif, Box 4, GT1-motif, GA-motif, etc. (light-responsive, 77%), ARE (13%), MBS (3%), TC-rich repeats (3%), LTR (3%), and WUN-motif (0.15%) (Figures 7A,B and Supplementary Table 5). Overall, results showed that most of the abiotic stress-related elements were predicted to be specific to some genes and unevenly distributed (Figure 6 and Supplementary Table 5), indicating their defensive role against stress conditions.

Figure 6. Analysis of cis-regulatory elements in the AhAPX promoter regions. Diverse cis-elements with functional resemblance are represented by similar colors.
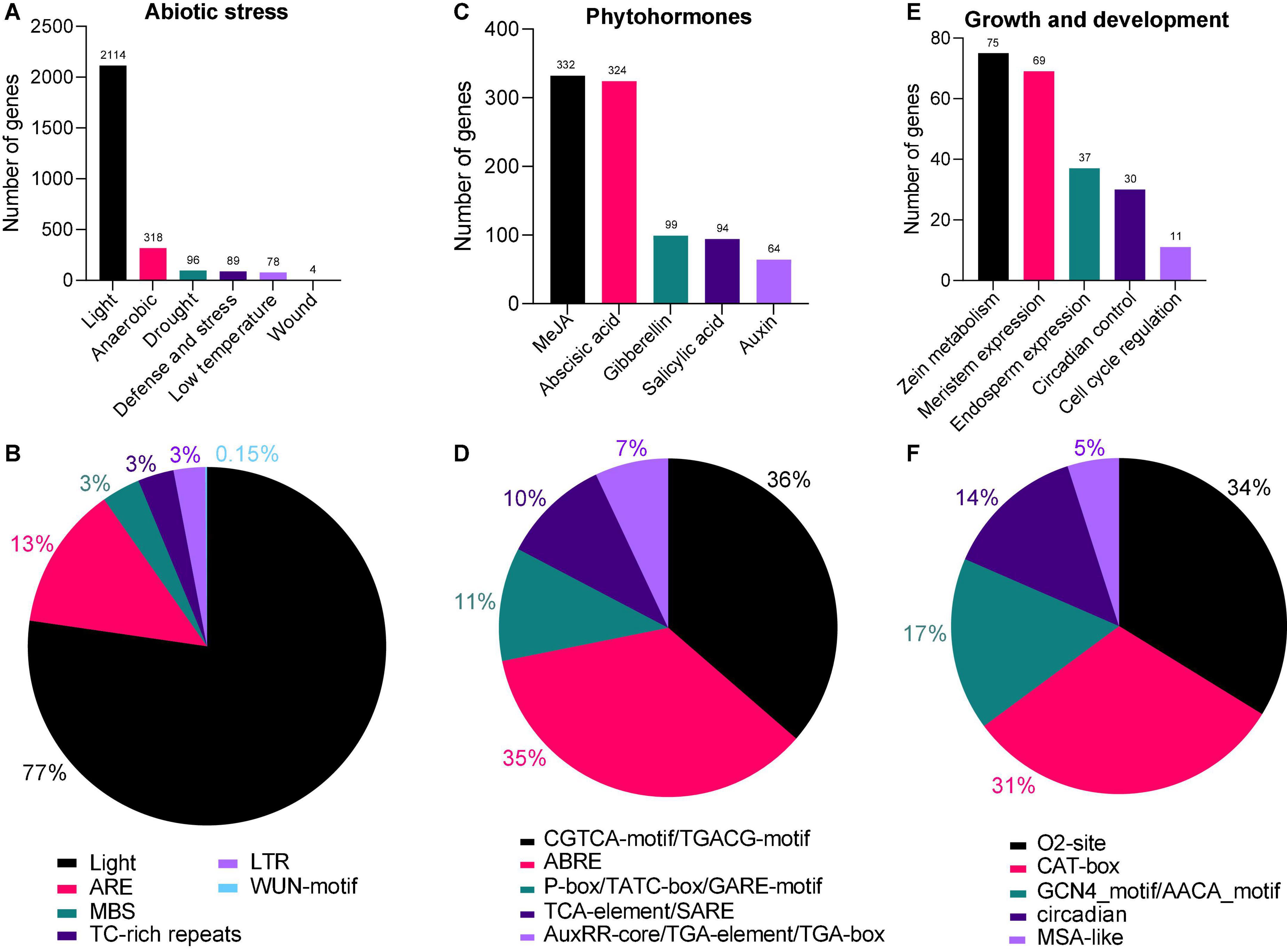
Figure 7. Cis-regulatory elements in the AhAPX promoter regions. (A,C,E) The total number of AhAPX genes involved in abiotic stress, phytohormones, and growth and development categories of cis-elements. (B,D,F) The percentage (%) ratio of the several cis-elements from each category is shown in pie charts, i.e. (B) abiotic stress-responsive, (D) phytohormones responsive, and (F) plant growth and development responsive. Diverse colors signify different cis-acting elements and their ratio present in AhAPX genes.
Likewise, five phytohormone-responsive elements [methyl jasmonate (MeJA), abscisic acid, gibberellin, salicylic acid, and auxin] consist of CGTCA-motif/TGACG-motif (36%), ABRE (35%), P-box/TATC-box/GARE-motif (11%), TCA-element/SARE (11%), and AuxRR-core/TGA-element/TGA-box (7%) (Figures 7C,D and Supplementary Table 5). Some of the elements were found to be specific to some genes and unevenly distributed (Figure 6 and Supplementary Table 5). These outcomes suggest that element-specific genes could be considered as candidate players for further functional studies to reveal their protective role under hormone treatments.
Moreover, five growth and development-related (zein metabolism, meristem expression, endosperm expression, circadian control, and cell cycle regulation) elements were discovered. These key elements include O2-site (34%), CAT-box (31%), GCN4_motif/AACA_motif (17%), circadian (14%), and MSA-like (5%) (Figures 7E,F and Supplementary Table 5), suggesting their dynamic role in different growth and developmental stages of peanut. In a nutshell, these discoveries suggested that some of the key elements are widely and randomly distributed in some genes, while some of the elements are found to be specific to some genes. It can be concluded that the expression profiles of AhAPX genes may fluctuate under different developmental stages, phytohormone and abiotic stress conditions.
Genome-Wide Investigation of miRNAs Targeting AhAPX Genes
To better comprehend the miRNA-arbitrated post-transcriptional alteration of AhAPX genes, we identified 14 miRNAs targeting 33 genes (Figure 8A and Supplementary Table 6). These miRNAs belong to 12 different families. To give an overview, the miRNA-targeted sites of AhAPX29 and AhAPX147 are shown in Figures 8B,C, whereas the complete dataset of all miRNAs targeted sites/genes is provided in Supplementary Table 6. The results showed that ahy-miR159 and ahy-miR3513-3P targeted the most number (5) of genes. Three miRNAs, including ahy-miR3518, ahy-miR3520-3P, and ahy-miR3513-5P targeted four genes, followed by ahy-miR3520-5P that targeted three genes (AhAPX38, AhAPX127, and AhAPX118). While six miRNAs including ahy-miR3512, ahy-miR3510, ahy-miR167-3P, ahy-miR3514-5P, ahy-miR3509-3P, and ahy-miR3508 targeted two different genes individually. Notably, only two miRNAs (ahy-miR156b-5p and ahy-miR3516) targeted one gene, AhAPX155 and AhAPX128, respectively (Figure 8A and Supplementary Table 6). Some common genes like AhAPX29, AhAPX62, AhAPX115, AhAPX147, AhAPX74, and AhAPX98 are found to be targeted by more than one miRNA. Hence, the expression profiling of these predicted miRNAs and their targeted genes necessitates confirmation to oversee their biological roles in the cultivated peanut genome.
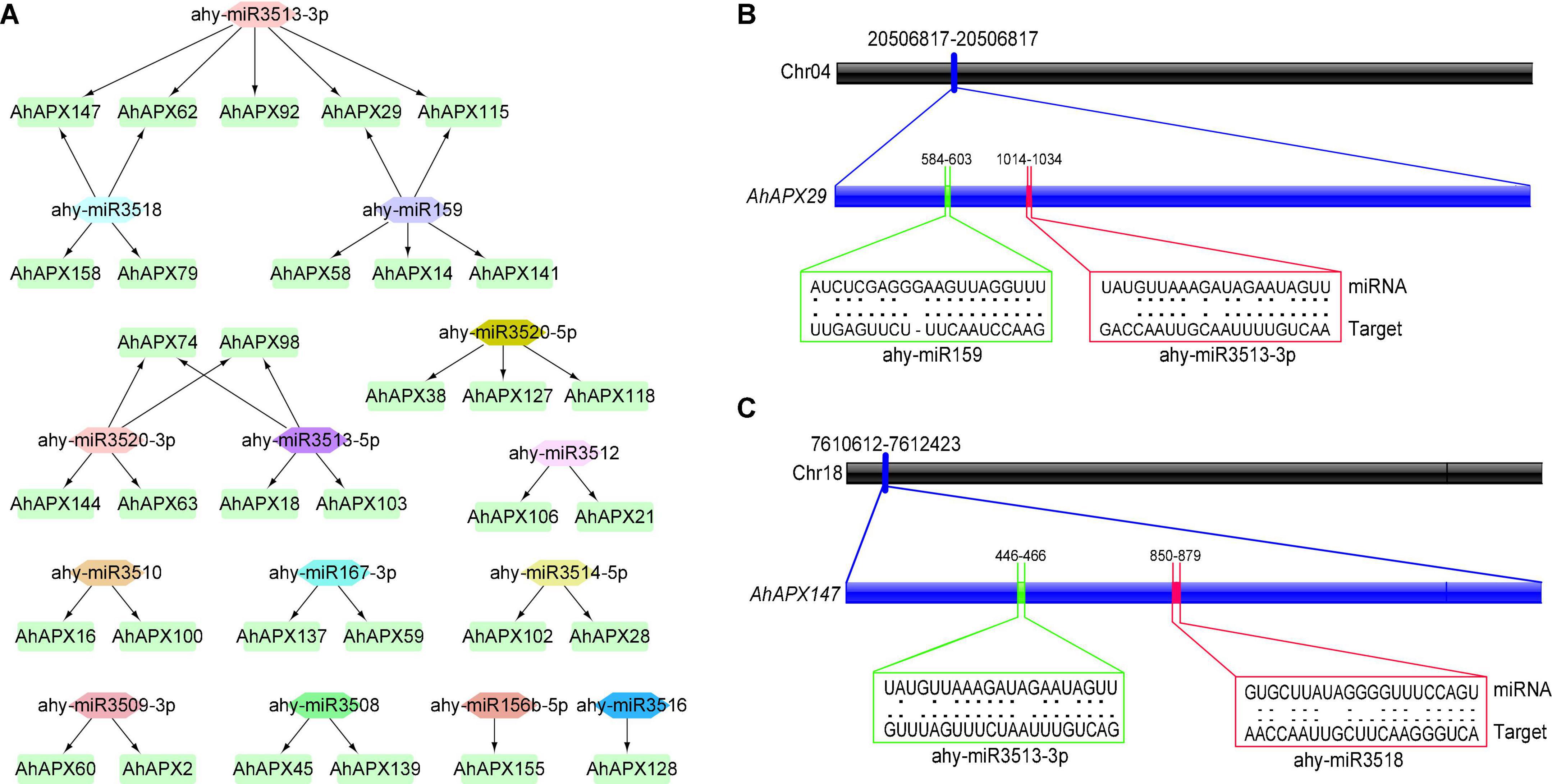
Figure 8. miRNA targeting AhAPX genes. (A) Network map of projected miRNA targeting AhAPX genes. The green boxes correspond to AhAPX genes, and various color shapes indicate predicted miRNAs. (B) The graphic illustration indicates the AhAPX29 gene is targeted by miRNAs (ahy-miR159 and ahy-miR3513-3p). (C) The graphic illustration shows the AhAPX147 gene is targeted by miRNAs (ahy-miR3518 and ahy-miR3513-3p). Black thick bar indicates the chromosomal location of gene. Blue thick bar indicates the location of miRNAs on gene sequence. The RNA sequence of each complementary site from 5′ to 3′ and the predicted miRNA sequence from 3′ to 5′ are shown in the green and red line boxes. The complete dataset of predicted miRNAs is shown in Supplementary Table 6.
Transcription Factor Regulatory Network of AhAPX Genes
To get further insights into the regulatory role of transcription factors (TFs) in regulating the transcription of AhAPX genes, we identified 3,257 TFs in 162 AhAPX genes (Figure 9 and Supplementary Table 7). The results showed that these TFs belong to 38 diverse TFs families, including AP2, ARF, B3, bHLH, bZIP, Dof, ERF, MYB, NAC, WRKY, HSF, GATA, etc. (Figure 9 and Supplementary Table 7). The amplest TFs families were Dof (742 members), ERF (698 members), MYB (545 members), BBR-BPC (344 members), NAC (308 members), WRKY (238 members), GATA (223 members), MIKC_MADS (210 members), C2H2 (177 members), bHLH/bZIP (163 members), B3 (157 members), AP2 (154 members), and HSF (102 members) (Figure 9B and Supplementary Table 7). However, the least ample TFs families were ARR-B/RAV/SRS (2 members), followed by GrBP (4 members), S1Fa-like (6 members), SBP (7 members), C3H (8 members), etc. (Figure 9B and Supplementary Table 7). In contrast, other TFs families contained less than 100 members. Nearly, all 162 AhAPX genes were anticipated to be targeted by various TFs belonging to diverse families. For instance, AhAPX150 gene was abundantly tarted by 314 TFs, followed by AhAPX56 by 172 TFs, AhAPX148 by 145 TFs, AhAPX55 by 107 TFs, AhAPX92 by 93 TFs, AhAPX45 by 96 TFs., etc. (Figure 9 and Supplementary Table 7). Some genes were nominally targeted, e.g., AhAPX5/79/99 by 1 TF, AhAPX83/90/93 by 2 TFs, AhAPX9/15 by 3 TFs, AhAPX7 by 4 TFs, AhAPX8/70/80 by 5 TFs., etc. (Figure 9 and Supplementary Table 7). Overall, these results showed that abiotic and phytohormone-related TFs could be engineered to develop improved peanut cultivars.
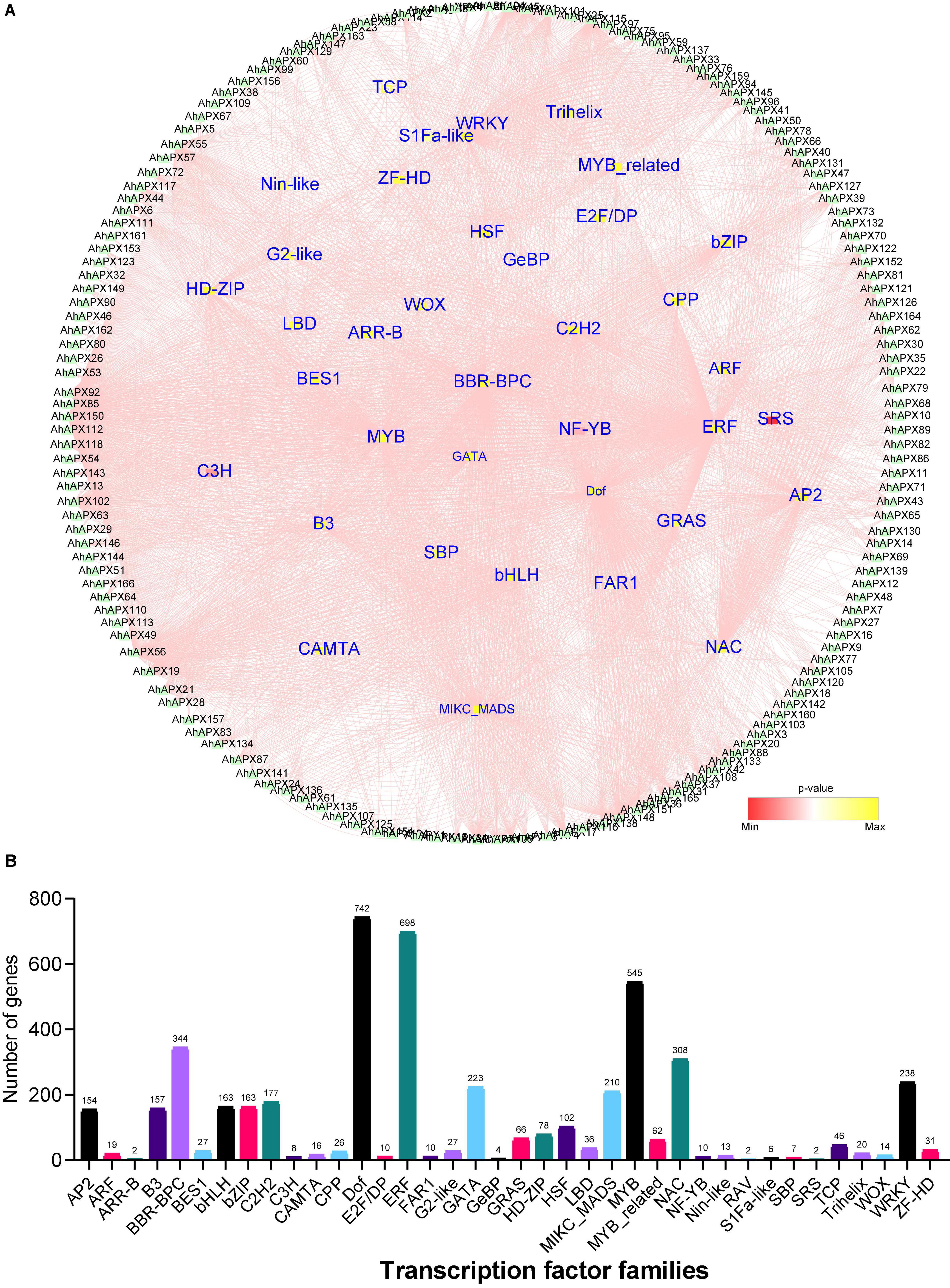
Figure 9. The regulatory network of putative transcription factors of AhAPX genes. (A) Circular network of transcription factors. AhAPX genes are shown around the circle. The small inner circles denote the transcription factors, and their color pink to yellow represents the degree of interaction. (B) Graph indicates the number of AhAPX genes and their representative putative transcription factor families. The complete dataset of putative transcription factors is shown in Supplementary Table 7.
Gene Ontology and Kyoto Encyclopedia of Genes and Genomics Enrichment Analysis of AhAPX Genes
To advance our knowledge about the dynamic roles of AhAPX genes at molecular level, GO and KEGG enrichment analysis were performed (Figure 10 and Supplementary Table 8). The GO annotation outcomes of biological process (BP), molecular function (MF), and cellular component (CC) classes presented quite a few substantially enriched terms (Figure 10A and Supplementary Table 8). For instance, in MF class, the highly enriched terms were cytochrome-c peroxidase activity (GO:0004130), oxidoreductase activity (GO:0016491), catalytic activity (GO:0003824), antioxidant activity (GO:0016209), and peroxidase activity (GO:0004601). In CC class, the most enriched terms were cell wall (GO:0005618), and cell junction (GO:0030054). Whereas in BP class, the highly enriched terms were cellular response to stimulus (GO:0051716), cellular detoxification (GO:1990748), response to chemical (GO:0042221), hydrogen peroxide catabolic process (GO:0042744), response to zinc ion (GO:0010043), modulation by symbiont of host defense response (GO:0052031), obsolete oxidation-reduction process (GO:0055114), detoxification (GO:0098754)., etc. (Figure 10A and Supplementary Table 8).
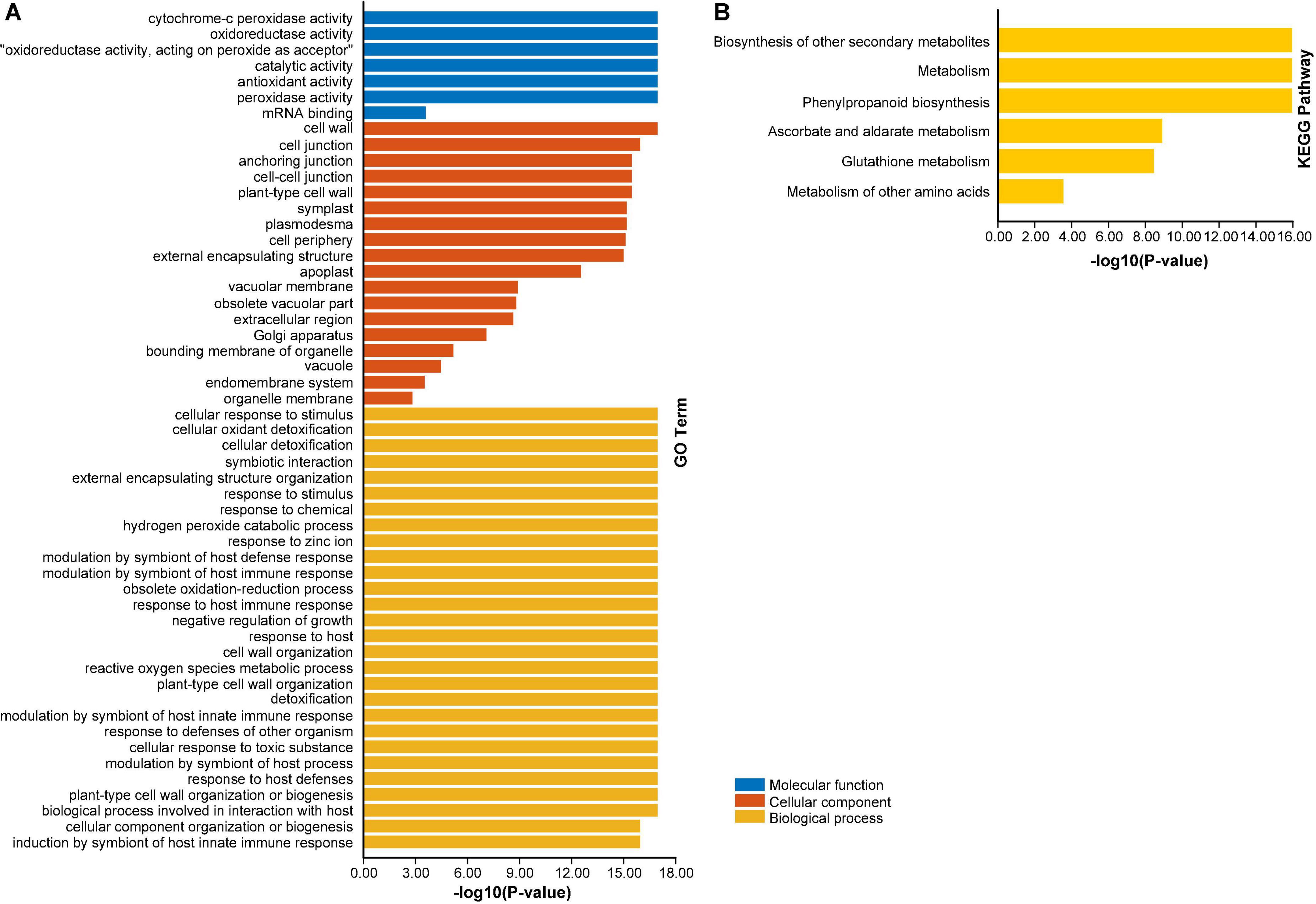
Figure 10. Gene ontology (GO) and KEGG enrichment analysis of AhAPX genes. (A) The highly enriched GO terms from MF, CC, BP classifications in AhAPX genes. (B) The highly enriched KEGG pathways in AhAPX genes.
Furthermore, KEGG pathway enrichment study discovered six pathways participating in diverse functions of AhAPX genes (Figure 10B and Supplementary Table 8). The highly enriched pathways include biosynthesis of other secondary metabolites (B09110), metabolism (A09100), phenylpropanoid biosynthesis (00940), followed by ascorbate and aldarate metabolism (00053), glutathione metabolism (00480), and metabolism of other amino acids (B09106) (Figure 10B and Supplementary Table 8). Briefly, it can be concluded that GO and KEGG enrichment study validates the functional contribution of AhAPX genes in several cellular, molecular, and biological processes, that are associated with antioxidant defense systems, ROS scavenging, response to stresses, and biosynthesis of different metabolites.
Expression Profiling of AhAPX Genes at Diverse Developmental Tissues
The expression profiling of 166 AhAPX genes was observed in various tissues and organs, including embryo, cotyledon, testa, pericarp, peg, root and stem, root nodule, root tip, root, step tip, stem, leaf, and flower using openly available transcriptome dataset (Supplementary Table 9). Overall, the expression heatmap indicated that only a few genes were highly expressed in certain organs/tissues (Figure 11 and Supplementary Table 9). For example, some genes including AhAPX4, AhAPX7, AhAPX17, AhAPX19, AhAPX28, AhAPX42, AhAPX51, AhAPX76, AhAPX77, AhAPX82, AhAPX86, AhAPX101, AhAPX102, AhAPX130, AhAPX133, and AhAPX160 were highly expressed in almost all the organs/tissues (Figure 11). While some genes were found to be specific to some tissues like AhAPX12 showed considerable expression in cotyledon, root and stem, root tip, and stem; AhAPX109, AhAPX111, and AhAPX13 expressed in stem, roots and peg; AhAPX135 expressed in pericarp; and AhAPX138 expressed in cotyledon (Figure 11). Particularly, a few genes also exhibited modest expressions in a variety of tissues. On the whole, expression dataset shows that some particular genes may substantially participate in peanut growth and development. Hence, the functional characterization of these genes may perhaps be carried out in future studies.
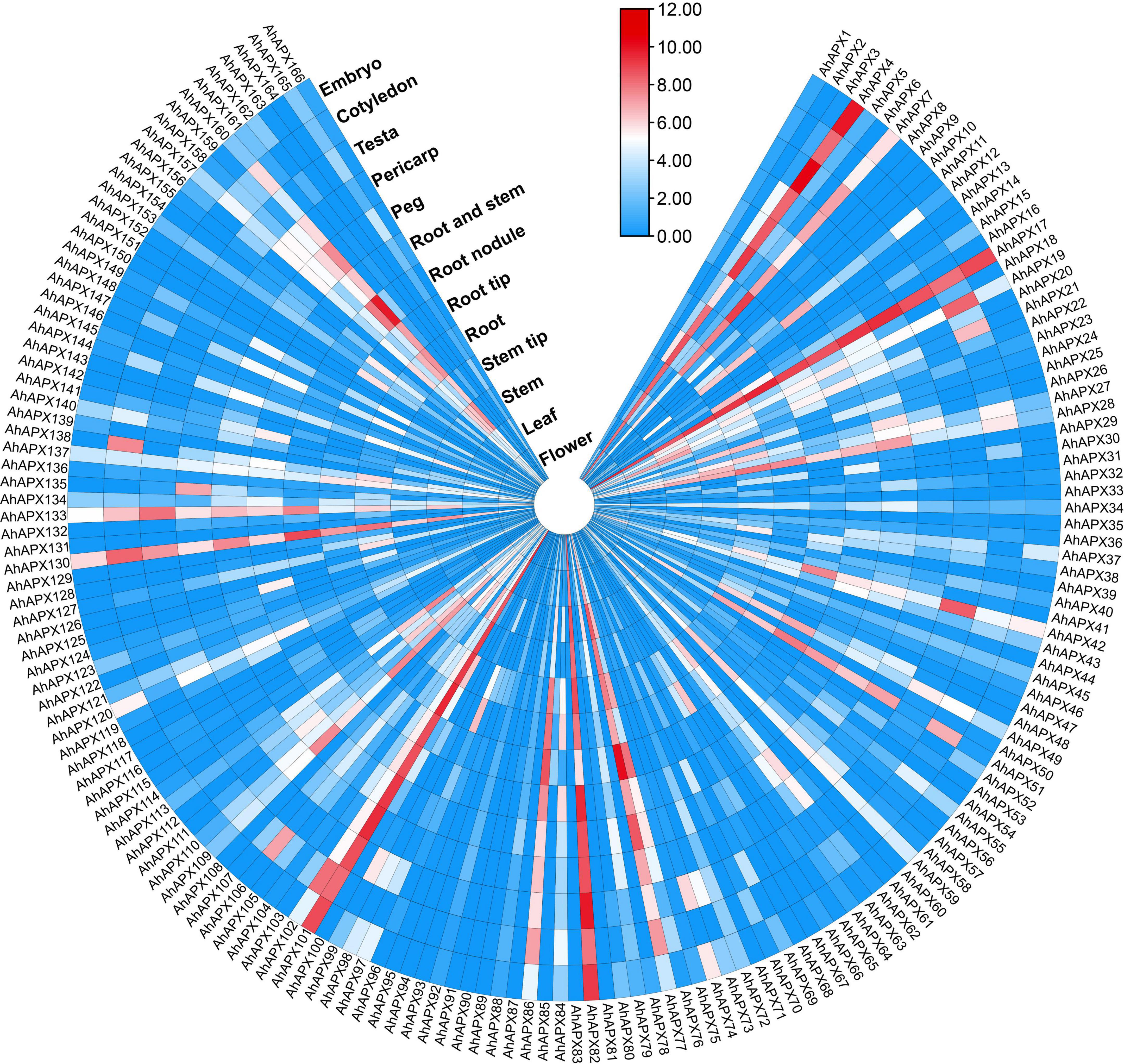
Figure 11. Expression profiling of AhAPX genes in various developmental tissues of peanut. In the expression bar, the red, white and blue colors show high to low expression levels. The circular heat map was designed by taking log2 of fragments per kilobase of transcript per million (FPKM) values.
Expression Profiling of AhAPX Genes Under Abiotic Stress and Hormones Treatments
To further study the contribution of all AhAPX genes toward abiotic and hormones stress tolerance in peanut, an openly available transcriptome dataset was used to evaluate the expression levels (Figure 12 and Supplementary Table 9). Similar to tissue-specific trend, only a few genes showed higher expressions in both cold and drought stresses. For instance, AhAPX4, AhAPX17, AhAPX19, AhAPX82, AhAPX101, and AhAPX102 were highly expressed under stress (cold and drought) and CK conditions. Likewise, some genes also showed moderate expression levels, such as AhAPX27, AhAPX34, AhAPX51, AhAPX55, AhAPX59, AhAPX113, AhAPX137, AhAPX138, AhAPX140, and AhAPX157 under stress (cold and drought) and normal conditions. On the other hand, AhAPX720, AhAPX21, AhAPX51, AhAPX77, AhAPX106, AhAPX130, AhAPX158, and AhAPX160 displayed considerable expression under cold stress compared to CK conditions (Figure 12A).
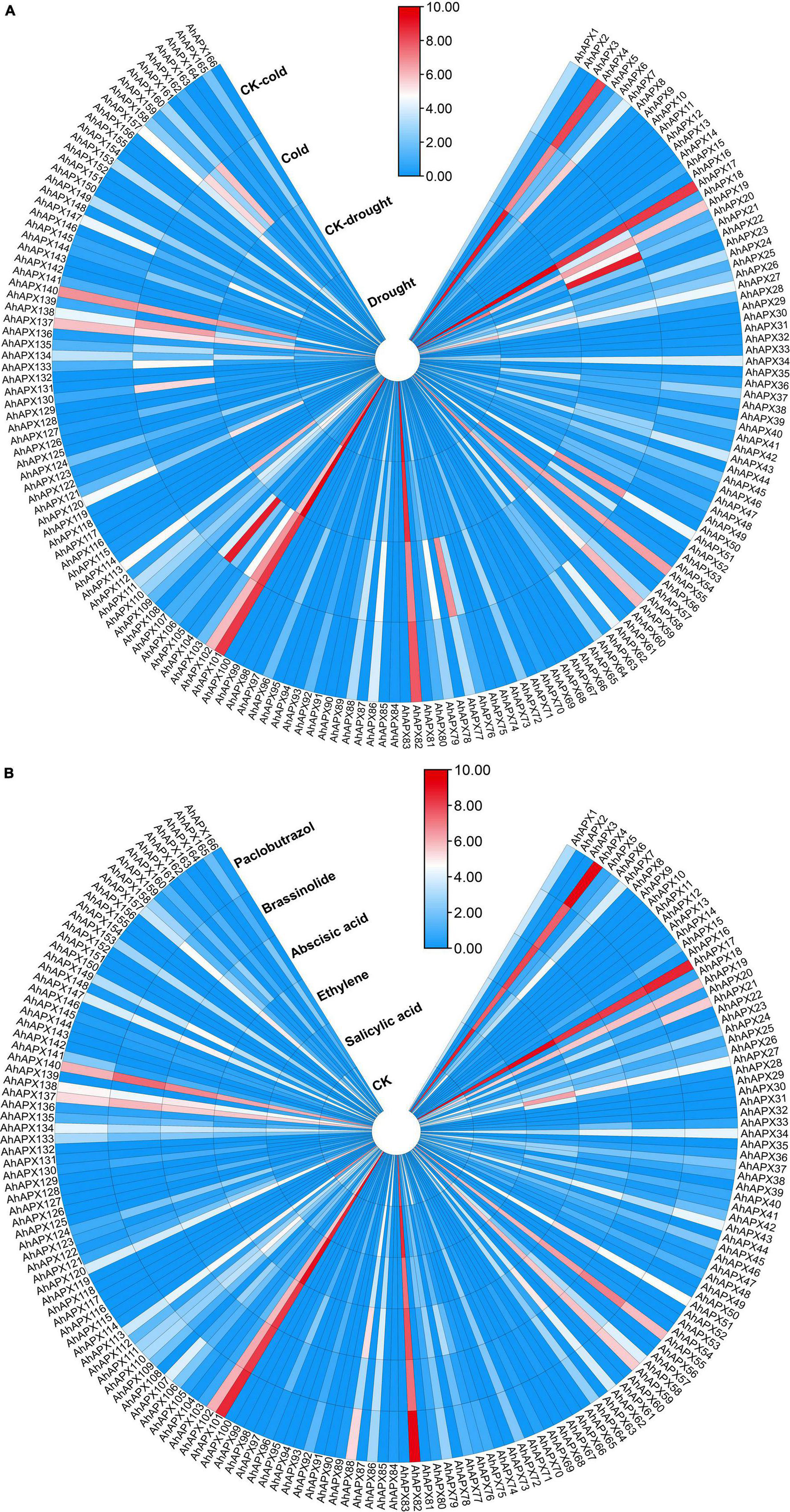
Figure 12. Expression profiling of AhAPX genes under different (A) abiotic stress, including cold and drought stress, and (B) phytohormone treatments such as abscisic acid, salicylic acid, ethylene, brassinolide, and paclobutrazol. In both maps, the label “CK” refers to the control plants in comparison to the stress-treated plants. In the expression bar, the red, white and blue colors show high to low expression levels. The circular heat map was designed by taking log2 of fragments per kilobase of transcript per million (FPKM) values.
Under phytohormones treatments, AhAPX4, AhAPX17, AhAPX55, AhAPX59, AhAPX82, AhAPX101, AhAPX102, AhAPX137, and AhAPX140 displayed significantly higher expression patterns throughout the treatments. In comparison to CK, some genes are specifically expressed under certain hormones, such as AhAPX21 under paclobutrazol, AhAPX27 under ethylene and abscisic acid, AhAPX51 under abscisic acid, and AhAPX88 under abscisic acid and paclobutrazol (Figure 12B). Notably, most of the genes did not show any expression under any type of stress conditions. The candidate genes with higher expression could be genetically engineered to improve the tolerance against multiple hormones and abiotic stress (cold and drought) conditions.
qRT-PCR-Based Expression Profiling of AhAPX Genes Under Cold and ABA Treatment
For qRT-PCR-based expression profiling, 10 highly upregulated AhAPX genes were selected based on transcriptome datasets to validate their transcript levels under ABA and cold treatment at various time points (Figure 13). Under ABA treatment, almost all genes demonstrated higher expression levels at all time points compared to CK, excluding a few cases. Such as, AhAPX55 and AhAPX140 showed relatively low expression at 9 and 12 h compared to CK and other time points (Figure 13A). In response to cold stress, although all the genes were upregulated; nevertheless, some genes showed relatively low expression levels compared to CK, such as AhAPX4, AhAPX19, AhAPX55, AhAPX82, AhAPX102, AhAPX137, and AhAPX140. Whereas AhAPX17 and AhAPX59 showed considerably higher expression than CK (Figure 13B). In short, all the preferred genes display parallel expression trends (i.e., upregulated) to those developed from transcriptome datasets (Supplementary Figure 4), therefore representing the reliability of the transcriptome datasets.
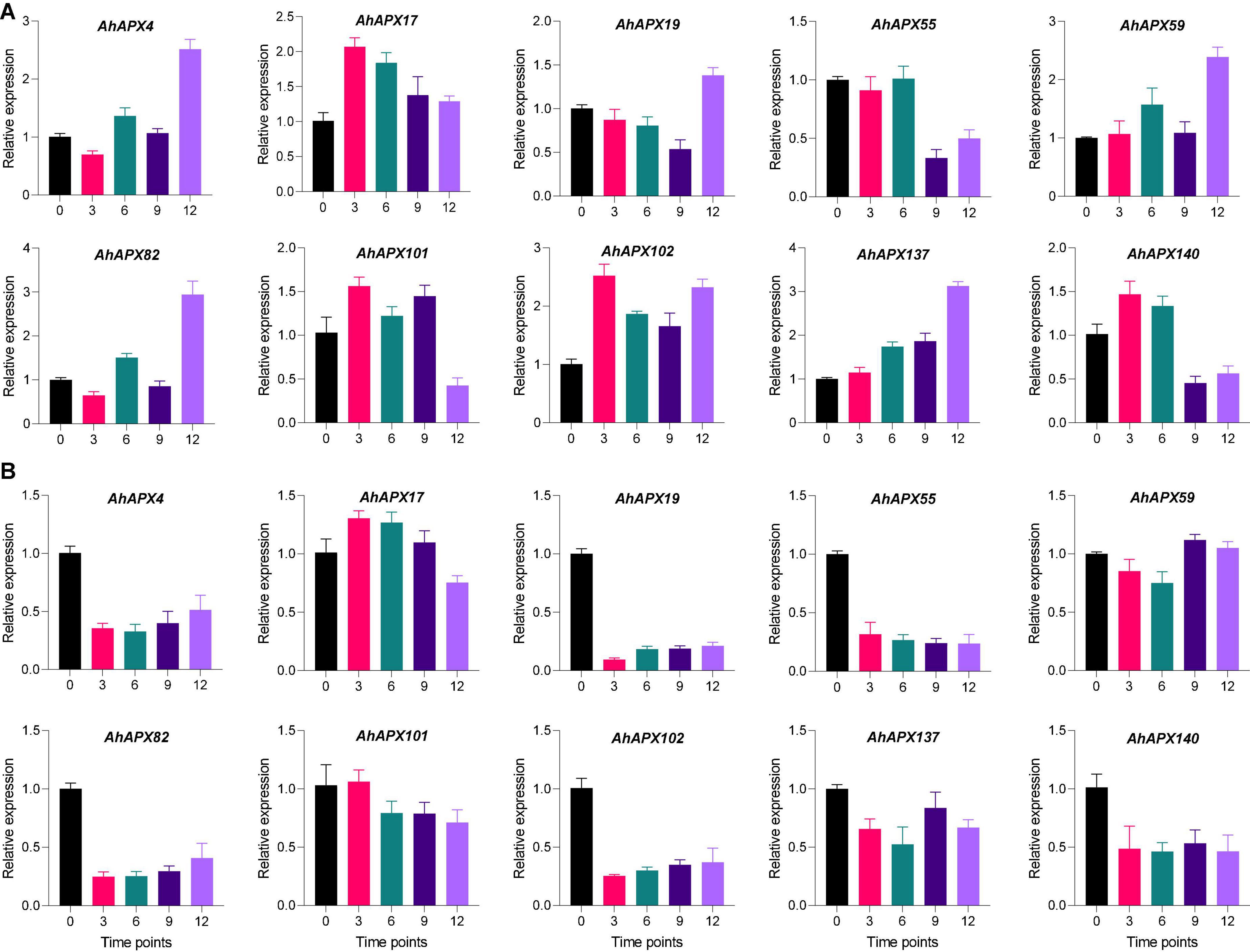
Figure 13. Expression profiling of the AhAPX genes under (A) abscisic acid and (B) cold stress treatments. The time points include 0 (CK), 3, 6, 9, and 12 h when the samples were picked for expression analysis after the stress treatment. Error bars signify the SD of three biological repeats (n = 3).
Discussion
Characterization and Evolution of APX Gene Family in Plants
Cultivated peanut is widely known as an essential oilseed, protein-enrich food crop worldwide and retains important breeding traits during domestication (Zhuang et al., 2019; Bohra et al., 2022). Even so, peanut production is still substantially influenced by numerous biotic and abiotic factors (Agarwal et al., 2018; Gangurde et al., 2020, 2021; Kumar et al., 2020; Shasidhar et al., 2020; Sinha et al., 2020; Jadhav et al., 2021; Pandey et al., 2021; Soni et al., 2021; Aravind et al., 2022; Bomireddy et al., 2022; Liu et al., 2022; Patel et al., 2022). When plants are exposed to diverse abiotic and biotic factors, APX enzyme as a primary marker can quickly eliminate unnecessary H2O2 (i.e., ROS scavenging) from plant cells by adjusting several physiological and biochemical activities to safeguard cells from the noxiousness of overproduction of ROS (Das and Roychoudhury, 2014; Mittler, 2017; Hasanuzzaman et al., 2020, 2021). During the past few years, excessive advancement has been achieved in studying the mode of action of APX genes; however, their vital role still requires more examination. Recent peanut genome sequencing data allow us to comprehensively discover new gene family members and recognize their functional and defensive mechanisms against stress conditions.
Usually, APX gene family of plants comprises a few genes. In this study, 166 AhAPX genes have been discovered in peanut genome (Supplementary Table 10), a larger APX family than previously reported APX gene families in watermelon (Malambane et al., 2018), shrub (Wang et al., 2022), rice (Teixeira et al., 2004), A. thaliana (Panchuk et al., 2002, Panchuk et al., 2005), sorghum (Akbudak et al., 2018), kiwifruit (Liao et al., 2020), tomato (Najami et al., 2008), wheat (Tyagi et al., 2020), and cotton (Tao et al., 2018). Deviations in the APX members amongst diverse plant species may perhaps be attributed to gene duplication events involving tandem and segmental repeats and play a role in expanding APXs for deviation. Repetition of APX genes was also discovered in several plant species (Teixeira et al., 2004; Panchuk et al., 2005; Akbudak et al., 2018; Liao et al., 2020; Wang et al., 2022). Our outcomes confirmed that AhAPXs had suffered segmental duplications (Supplementary Table 3). Subsequently, these reports recommended that AhAPXs duplicate cases may possibly play an essential role in gene evolution.
Previous studies showed that APX family genes are usually clustered into four major groups based on their subcellular localization or tree topologies (Pandey et al., 2011; Malambane et al., 2018; Tyagi et al., 2020). In the present study, all APX genes from four plant species were grouped into 11 main groups based on tree topologies and sequence similarities (Figure 2). This grouping was also recently supported by a new study on brassica crops (B. napus and B. rapa), where all APX genes were grouped into 13 subfamilies (Ma et al., 2021). Further, gene structure analysis also showed that genes belonging to the same subtree possess almost similar exon-intron patterns, ranging from 16 to 1 (exons) and 0 to 15 (introns) (Figure 5B and Supplementary Table 10). A significant difference was observed in gene structures where some genes have many exons-introns while some lack introns. Similar gene structure patterns were also reported in previous reports, such as in wheat number of exons extended from 7 to 12 (Tyagi et al., 2020). In Actinidia chinensis, the gene structure possesses 4–22 introns (Liao et al., 2020), which are higher than our observations. The exon-intron organization disparity was practiced by three important methods (exon/intron gain/loss, exonization/pseudoexonization, and insertion/deletion), and they are clearly supported by structural inconsistency (Xu et al., 2012). Notably, the AhAPX genes in each group almost exhibited comparable exon-intron group and conserved motifs (Figure 5), suggesting that these genes may possibly contribute to the similar tasks associated with several abiotic stressors. These outcomes are in agreement with earlier studies of kiwifruit (Liao et al., 2020), and wheat (Tyagi et al., 2020), where genes inside the same group comprise distinct structures and motifs organizations.
The Contribution of APX Genes Toward Stress Responses and Tolerance Mechanisms
To boost our understanding into the involvement of AhAPX genes contrary to numerous environmental factors, cis-elements were predicted in the promoter of AhAPX genes. The discoveries showed that three types of cis-elements were recognized, i.e., abiotic stress, phytohormones, and growth and development-related elements (Figures 6, 7). Recent studies show that the cis-elements in APX genes contribute to the plant abiotic and phytohormones stress responses. Similar types of abiotic and phytohormone-related cis-regulatory elements have been identified in previous studies (Akbudak et al., 2018; Malambane et al., 2018; Tao et al., 2018; Liao et al., 2020; Tyagi et al., 2020; Wang et al., 2022). Furthermore, AhAPXs gene functions were further predicted by GO enrichment analysis (Figure 10), which also supported the role of these genes in ROS scavenging and stress response mechanisms. To get further insights into the role of AhAPX genes, their expression levels were examined under various hormones and abiotic stress treatments (Figures 12, 13). Our results showed that a few genes significantly contribute to specific stress responses like cold, drought, and ABA. These results are in agreement with the previous reports of Akbudak et al. (2018), where some SbAPX genes were significantly induced by drought stress in the leaves and roots of two genotypes. Similarly, many genes showed higher expression levels in A. nanus under cold and osmotic stress (Wang et al., 2022). Many BrAPX and BnAPX genes showed higher expression trends in cold-tolerant varieties in response to cold stress (Ma et al., 2021). Under drought stress, most of the ClAPX genes were significantly upregulated and displayed elevated expression in watermelon (Malambane et al., 2018). These conclusions can enhance our perception of AhAPX genes under various stress conditions, especially cold and drought.
Recent reports also suggest that manipulating APX genes could contribute to stress tolerance in plants. For instance, a novel ScAPX6 gene from sugarcane was overexpressed in tobacco (Nicotiana benthamiana), and transgenic plants showed improved resistance to the biotic stress (Pseudomonas solanacearum and Fusarium solani) by positively regulating the phytohormones contents (Liu et al., 2018). The overexpression of PcAPX from Chinese white poplar (Populus tomentosa) improves tolerance to multiple stresses, including salinity, drought, and oxidative stress in transgenic tobacco plants by improving biochemical mechanisms (Cao et al., 2017). Likewise, the overexpression of Populus peroxisomal PpAPX gene enhances drought stress tolerance in transgenic tobacco plants (Li et al., 2009). Transgenic tobacco overexpressing cytosolic APX gene alleviated the drought stress tolerance (Faize et al., 2011). Ectopic overexpression of the peroxisomal SbpAPX gene improves salinity tolerance in transgenic peanut (Singh et al., 2014). So far, this is the only APX gene that has been functionally characterized in peanut. These studies recommend that the genetic engineering of APX genes is of great importance in conferring various stress tolerance in crop plants, including peanut.
Among various identified TFs, ERF TFs have been functionally characterized from peanut. The results exhibited that overexpression of AhERF008 impaired the root magnitude of A. thaliana; however, overexpression of AhERF019 improved tolerance to heat, salinity and drought stresses in A. thaliana (Wan et al., 2014). Ectopic overexpression of MYB repressor gene (GmMYB3a) increases drought tolerance and physiological mechanisms in transgenic peanut under drought stress (He et al., 2020). Another NAC TF gene (AhANC4) from peanut enhances drought tolerance in transgenic tobacco plants due to improved stomatal closure and advanced water use efficiency (Tang et al., 2017). A novel WRKY TF gene (AhWRKY75) improved salinity tolerance in transgenic peanut plants by improving antioxidant mechanisms, ROS scavenging, stomatal conductance, and photosynthesis under salinity stress (Zhu et al., 2021). All these studies suggest that the genetic engineering of TF is a promising approach to improve peanut performance under stressed conditions.
The Contribution of APX Genes in Numerous Organs/Tissues
Here, the tissue-specific expression profiling of 166 AhAPX genes was carried out in various organs/tissues using publically available transcriptome datasets. Overall, the results showed that only a few AhAPX genes showed higher expression levels, particularly in roots, stem, leaf, peg, pericarp, testa, and flowers (Figure 11). In A. nanus, the RNA-seq data was used to observe the expression levels in leaves. The results displayed that only one gene showed substantially higher expression in leaf (Wang et al., 2022). In wheat, most of the genes showed higher expression patterns in root, stem, leaf, spike, and grain. Especially, almost half of the APX genes were found to be leaf-specific due to significantly higher expression (Tyagi et al., 2020). In A. chinensis, qRT-PCR-based expression profiling of 13 AhAPX genes was performed in various fruit developmental stages. The outcomes demonstrated that eight AcAPX genes had the utmost expression patterns during the color turn-off phase (Liao et al., 2020). It can be concluded that the tissue-specific APX genes (such as AhAPX4, AhAPX17, AhAPX77, AhAPX82, AhAPX101, and AhAPX130) could be considered as target candidates for further molecular studies to fully reveal their role and mechanisms in peanut growth and development.
MicroRNA: Emerging Players for Crop Improvement and Stress Tolerance
MicroRNAs (miRNAs) are a group of tiny-non-coding RNAs formed from individual-strand hairpin RNA precursors. These miRNAs switch gene expression by attaching to corresponding sequences surrounded by target mRNAs (Jamla et al., 2021; Patil et al., 2021). Extensive progress has been put together in finding the targets of peanut miRNAs that contribute to various stresses and developmental activities (Zhao et al., 2010, 2015; Chi et al., 2011; Zhang et al., 2017; Chen H. et al., 2019; Figueredo et al., 2020; Tong et al., 2021). The present predicted 14 miRNAs belonging to 12 different families targeting 33 AhAPX genes (Figure 8 and Supplementary Table 6). Notably, none of the previous studies predicted the miRNAs that can target APX genes, expect one study. A recent study supports our findings where 51 miRNAs have been identified targeting 29 TaAPX genes in wheat (Tyagi et al., 2020). However, these target genes are yet to be characterized in wheat. In another study, a new miRNA (ath-miR447a-3p) was found to be targeting APX3 gene, and its expression analysis showed that it negatively regulated the expression of APX3, which is directly involved in the APX synthesis under drought stress in Zanthoxylum bungeanum (Fei et al., 2020).
However, some of the identified miRNAs have been reported to take part in stress tolerance and developmental processes. For instance, spatio-temporal expression patterns of miRNA159 family representatives have been found targeting MYB genes in grapevine (Vitis vinifera L.). The results showed that miRNA159c-VvGAMYB module is involved in gibberellin-tempted parthenocarpy in grapevine (Wang et al., 2018). Another study discovered that miR167A is the main member of miR167 family that regulates the A. thaliana reproduction. Further, miR167A acts as a parental gene that works mostly via ARF6 and ARF8 genes in maternal management of embryonic and seed growth (Yao et al., 2019). A member of miRNA156 family has been reported to be involved in the interaction between ABA and miRNA156, which regulates the expression profile of anthocyanin biogenesis genes in drought-stressed plants (González-Villagra et al., 2017). Notably, several miRNA families such as miR3513, miR3518, miR3520, miR3513, miR3516, etc., have not been functionally characterized; therefore, the future work could also be focused on these unique miRNAs to reveal their potential in plant growth and development. Moreover, the expression profiling of prophesied miRNAs and their targeted genes demands validation to direct their biological roles in the peanut breeding programs.
Conclusion
Altogether, we recognized 166 putative AhAPX genes in the cultivated peanut genome, which are mapped on all chromosomes, including unassembled ones. Comprehensive in silico examination of AhAPX genes, i.e., characterization, evolution, gene structure, conserved motifs, cis-elements, putative miRNA and TFs prediction, GO and KEGG enrichment were executed to increase our understanding of AhAPX genes in peanut. Their expression trends were also evaluated in various developmental organs/tissues, phytohormones, and abiotic stress conditions. In brief, this report set the foundation for further functional experiments (such as overexpression, gene editing via CRISPR/Cas system, etc.) of some candidate genes such as AhAPX4/17/19/55/59/82/101/102/137 and AhAPX140, which can advance the peanut breeding programs under undesirable stress conditions.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI BioProject–PRJNA480120.
Author Contributions
AR and WZ conceived the idea. AR analyzed the data and wrote the manuscript. YS and KC helped with qRT-PCR analysis. YS, KC, CZ, LW, HF, AC, and HC helped in literature search and data analysis. WZ and RKV supervised the research, and reviewed and improved the manuscript. All authors have read and approved the final version of the manuscript.
Funding
This work was supported by grants from the National Science Foundation (NSF) of China (U1705233, 32072103, and 31701463), the Science and Technology Foundation of Fujian Province of China (2021N5007), and Fujian Agriculture and Forestry University, Fuzhou, China. RKV is thankful to Food Futures Institute, Murdoch University, Australia, for supporting this study in part.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
RKV is thankful to Food Futures Institute, Murdoch University, Australia, for supporting this study in part.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.962182/full#supplementary-material
Footnotes
- ^ http://peanutgr.fafu.edu.cn/
- ^ http://www.arabidopsis.org/
- ^ http://www.hmmer.org/
- ^ http://pfam.xfam.org/
- ^ https://www.peanutbase.org/
- ^ http://web.expasy.org/protparam/
- ^ http://cello.life.nctu.edu.tw/
- ^ https://github.com/CJ-Chen/TBtools
- ^ https://meme-suite.org/meme/db/motifs
- ^ https://megasoftware.net/home
- ^ https://itol.embl.de/
- ^ http://bioinformatics.psb.ugent.be/webtools/plantcare/html/
- ^ https://www.zhaolab.org/psRNATarget/home
- ^ https://cytoscape.org/download.html
- ^ http://eggnog-mapper.embl.de/
- ^ http://plantregmap.gao-lab.org/binding_site_prediction.php
- ^ https://www.graphpad.com/
References
Agarwal, G., Clevenger, J., Pandey, M. K., Wang, H., Shasidhar, Y., Chu, Y., et al. (2018). High-density genetic map using whole-genome resequencing for fine mapping and candidate gene discovery for disease resistance in peanut. Plant Biotechnol. J. 16, 1954–1967. doi: 10.1111/pbi.12930
Akbudak, M. A., Filiz, E., Vatansever, R., and Kontbay, K. (2018). Genome-wide identification and expression profiling of ascorbate peroxidase (APX) and glutathione peroxidase (GPX) genes under drought stress in sorghum (Sorghum bicolor L.). J. Plant Growth Regulat. 37, 925–936.
Aravind, B., Nayak, S. N., Choudhary, R. S., Gandhadmath, S. S., Prasad, P., Pandey, M. K., et al. (2022). “Integration of genomics approaches in abiotic stress tolerance in groundnut (L.): an overview,” in Genomic Designing for Abiotic Stress Resistant Oilseed Crops, ed. C. Kole (Cham: Springer), 149–197.
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37(Suppl._2), W202–W208.
Bertioli, D. J., Jenkins, J., Clevenger, J., Dudchenko, O., Gao, D., Seijo, G., et al. (2019). The genome sequence of segmental allotetraploid peanut Arachis hypogaea. Nat. Genet. 51, 877–884. doi: 10.1038/s41588-019-0405-z
Bohra, A., Tiwari, A., Kaur, P., Ganie, S. A., Raza, A., Roorkiwal, M., et al. (2022). The key to the future lies in the past: insights from grain legume domestication and improvement should inform future breeding strategies. Plant Cell Physiol. [Online ahead of print]. doi: 10.1093/pcp/pcac086
Bomireddy, D., Gangurde, S. S., Variath, M. T., Janila, P., Manohar, S. S., Sharma, V., et al. (2022). Discovery of major quantitative trait loci and candidate genes for fresh seed dormancy in groundnut. Agronomy 12:404.
Cannon, S. B., Mitra, A., Baumgarten, A., Young, N. D., and May, G. (2004). The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 4:10. doi: 10.1186/1471-2229-4-10
Cao, S., Du, X.-H., Li, L.-H., Liu, Y.-D., Zhang, L., Pan, X., et al. (2017). Overexpression of Populus tomentosa cytosolic ascorbate peroxidase enhances abiotic stress tolerance in tobacco plants. Russ. J. Plant Physiol. 64, 224–234.
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chen, H., Yang, Q., Chen, K., Zhao, S., Zhang, C., Pan, R., et al. (2019). Integrated microRNA and transcriptome profiling reveals a miRNA-mediated regulatory network of embryo abortion under calcium deficiency in peanut (Arachis hypogaea L.). BMC Genomics 20:392. doi: 10.1186/s12864-019-5770-6
Chen, X., Lu, Q., Liu, H., Zhang, J., Hong, Y., Lan, H., et al. (2019). Sequencing of cultivated peanut, Arachis hypogaea, yields insights into genome evolution and oil improvement. Mol. Plant 12, 920–934. doi: 10.1016/j.molp.2019.03.005
Chi, X., Hu, R., Yang, Q., Zhang, X., Pan, L., Chen, N., et al. (2012). Validation of reference genes for gene expression studies in peanut by quantitative real-time RT-PCR. Mol. Genet. Genomics 287, 167–176.
Chi, X., Yang, Q., Chen, X., Wang, J., Pan, L., Chen, M., et al. (2011). Identification and characterization of microRNAs from peanut (Arachis hypogaea L.) by high-throughput sequencing. PLoS One 6:e27530. doi: 10.1371/journal.pone.0027530
Dai, X., Zhuang, Z., and Zhao, P. X. (2018). psRNATarget: a plant small RNA target analysis server (2017 release). Nucleic Acids Res. 46, W49–W54. doi: 10.1093/nar/gky316
Das, K., and Roychoudhury, A. (2014). Reactive oxygen species (ROS) and response of antioxidants as ROS-scavengers during environmental stress in plants. Front. Environ. Sci. 2:53. doi: 10.3389/fenvs.2014.00053
Dash, S., Cannon, E. K., Kalberer, S. R., Farmer, A. D., and Cannon, S. B. (2016). “PeanutBase and other bioinformatic resources for peanut,” in Peanuts: Genetics, Processing, and Utilization, ed. H. Thomas (Amsterdam: Elsevier), 241–252.
El-Gebali, S., Mistry, J., Bateman, A., Eddy, S. R., Luciani, A., Potter, S. C., et al. (2019). The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432.
Fahad, S., Bajwa, A. A., Nazir, U., Anjum, S. A., Farooq, A., Zohaib, A., et al. (2017). Crop production under drought and heat stress: plant responses and management options. Front. Plant Sci. 8:1147. doi: 10.3389/fpls.2017.01147
Fahad, S., Nie, L., Chen, Y., Wu, C., Xiong, D., Saud, S., et al. (2015). “Crop plant hormones and environmental stress,” in Sustainable Agriculture Reviews. Sustainable Agriculture Reviews, Vol. 15, ed. E. Lichtfouse (Cham: Springer), 371–400.
Faize, M., Burgos, L., Faize, L., Piqueras, A., Nicolas, E., Barba-Espin, G., et al. (2011). Involvement of cytosolic ascorbate peroxidase and Cu/Zn-superoxide dismutase for improved tolerance against drought stress. J. Exp. Bot. 62, 2599–2613.
Fei, X., Li, J., Kong, L., Hu, H., Tian, J., Liu, Y., et al. (2020). miRNAs and their target genes regulate the antioxidant system of Zanthoxylum bungeanum under drought stress. Plant Physiol. Biochem. 150, 196–203.
Figueredo, M. S., Formey, D., Rodríguez, J., Ibáñez, F., Hernández, G., and Fabra, A. (2020). Identification of miRNAs linked to peanut nodule functional processes. J. Biosci. 45, 1–7.
Finn, R. D., Clements, J., Arndt, W., Miller, B. L., Wheeler, T. J., Schreiber, F., et al. (2015). HMMER web server: 2015 update. Nucleic Acids Res. 43, W30–W38. doi: 10.1093/nar/gkv397
Gangurde, S. S., Nayak, S. N., Joshi, P., Purohit, S., Sudini, H. K., Chitikineni, A., et al. (2021). Comparative transcriptome analysis identified candidate genes for late leaf spot resistance and cause of defoliation in groundnut. Int. J. Mol. Sci. 22:4491. doi: 10.3390/ijms22094491
Gangurde, S. S., Wang, H., Yaduru, S., Pandey, M. K., Fountain, J. C., Chu, Y., et al. (2020). Nested-association mapping (NAM)-based genetic dissection uncovers candidate genes for seed and pod weights in peanut (Arachis hypogaea). Plant Biotechnol. J. 18, 1457–1471. doi: 10.1111/pbi.13311
Gasteiger, E., Hoogland, C., Gattiker, A., Wilkins, M. R., Appel, R. D., and Bairoch, A. (2005). “Protein identification and analysis tools on the ExPASy server,” in The Proteomics Protocols Handbook, ed. J. M. Walker (Totowa, NJ: Humana Press), 571–607.
González-Villagra, J., Kurepin, L. V., and Reyes-Díaz, M. M. (2017). Evaluating the involvement and interaction of abscisic acid and miRNA156 in the induction of anthocyanin biosynthesis in drought-stressed plants. Planta 246, 299–312.
Hasanuzzaman, M., Bhuyan, M., Zulfiqar, F., Raza, A., Mohsin, S. M., Mahmud, J. A., et al. (2020). Reactive oxygen species and antioxidant defense in plants under abiotic stress: revisiting the crucial role of a universal defense regulator. Antioxidants 9:681. doi: 10.3390/antiox9080681
Hasanuzzaman, M., Raihan, M., Hossain, R., Masud, A. A. C., Rahman, K., Nowroz, F., et al. (2021). Regulation of reactive oxygen species and antioxidant defense in plants under salinity. Int. J. Mol. Sci. 22:9326.
He, Y., Mu, S., He, Z., Wang, B., and Li, Y. (2020). Ectopic expression of MYB repressor GmMYB3a improves drought tolerance and productivity of transgenic peanuts (Arachis hypogaea L.) under conditions of water deficit. Transgen. Res. 29, 563–574. doi: 10.1007/s11248-020-00220-z
Hodges, D. M., DeLong, J. M., Forney, C. F., and Prange, R. K. (1999). Improving the thiobarbituric acid-reactive-substances assay for estimating lipid peroxidation in plant tissues containing anthocyanin and other interfering compounds. Planta 207, 604–611. doi: 10.1007/s00425-017-2699-3
Hurst, L. D. (2002). The Ka/Ks ratio: diagnosing the form of sequence evolution. Trend. Genet. 18, 486–486. doi: 10.1016/s0168-9525(02)02722-1
Jadhav, M. P., Gangurde, S. S., Hake, A. A., Yadawad, A., Mahadevaiah, S. S., Pattanashetti, S. K., et al. (2021). Genotyping-by-sequencing based genetic mapping identified major and consistent genomic regions for productivity and quality traits in peanut. Front. Plant Sci. 12:668020. doi: 10.3389/fpls.2021.668020
Jamla, M., Patil, S., Joshi, S., Khare, T., and Kumar, V. (2021). MicroRNAs and their exploration for developing heavy metal-tolerant plants. J Plant Growth Regulat. 9, 1–17.
Kumar, R., Janila, P., Vishwakarma, M. K., Khan, A. W., Manohar, S. S., Gangurde, S. S., et al. (2020). Whole-genome resequencing-based QTL-seq identified candidate genes and molecular markers for fresh seed dormancy in groundnut. Plant Biotechnol. J. 18, 992–1003. doi: 10.1111/pbi.13266
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lazzarotto, F., Teixeira, F. K., Rosa, S. B., Dunand, C., Fernandes, C. L., de Vasconcelos Fontenele, A., et al. (2011). Ascorbate peroxidase-related (APx-R) is a new heme-containing protein functionally associated with ascorbate peroxidase but evolutionarily divergent. New Phytol. 191, 234–250. doi: 10.1111/j.1469-8137.2011.03659.x
Lescot, M., Déhais, P., Thijs, G., Marchal, K., Moreau, Y., Van de Peer, Y., et al. (2002). PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327. doi: 10.1093/nar/30.1.325
Letunic, I., and Bork, P. (2021). Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Li, W., Huai, X., Li, P., Raza, A., Mubarik, M. S., Habib, M., et al. (2021). Genome-wide characterization of glutathione peroxidase (GPX) gene family in rapeseed (Brassica napus L.) revealed their role in multiple abiotic stress response and hormone signaling. Antioxidants 10:1481. doi: 10.3390/antiox10091481
Li, Y. J., Hai, R. L., Du, X. H., Jiang, X. N., and Lu, H. (2009). Over-expression of a Populus peroxisomal ascorbate peroxidase (PpAPX) gene in tobacco plants enhances stress tolerance. Plant Breed. 128, 404–410.
Liao, G.-L., Liu, Q., Li, Y.-Q., Zhong, M., Huang, C.-H., Jia, D.-F., et al. (2020). Identification and expression profiling analysis of ascorbate peroxidase gene family in Actinidia chinensis (Hongyang). J. Plant Res. 133, 715–726. doi: 10.1007/s10265-020-01206-y
Liu, F., Huang, N., Wang, L., Ling, H., Sun, T., Ahmad, W., et al. (2018). A novel L-ascorbate peroxidase 6 gene, ScAPX6, plays an important role in the regulation of response to biotic and abiotic stresses in sugarcane. Front. Plant Sci. 8:2262. doi: 10.3389/fpls.2017.02262
Liu, Y., Shao, L., Zhou, J., Li, R., Pandey, M. K., Han, Y., et al. (2022). Genomic insights into the genetic signatures of selection and seed trait loci in cultivated peanut. J. Adv. Res. doi: 10.1016/j.jare.2022.01.016
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2- ΔΔCT method. Methods 25, 402–408.
Ma, L., Bai, J., Xu, J., Qi, W., Li, H., Fang, Y., et al. (2021). Identification of Proteins Involved in Response to Cold Stress and Genome-Wide Identification and Analysis of the APX Gene Family in Winter Rapeseed (Brassica rapa L.). Available online at: https://www.researchsquare.com/article/rs-538668/v1.pdf (accessed June 7, 2021)
Malambane, G., Tsujimoto, H., and Akashi, K. (2018). The cDNA structures and expression profile of the ascorbate peroxidase gene family during drought stress in wild watermelon. J. Agric. Sci. 10, 56–71.
Mir, R. A., Bhat, B. A., Yousuf, H., Islam, S. T., Raza, A., Rizvi, M. A., et al. (2022). Multidimensional role of silicon to activate resilient plant growth and to mitigate abiotic stress. Front. Plant Sci. 13:819658. doi: 10.3389/fpls.2022.819658
Najami, N., Janda, T., Barriah, W., Kayam, G., Tal, M., Guy, M., et al. (2008). Ascorbate peroxidase gene family in tomato: its identification and characterization. Mol. Genet. Genomics 279, 171–182. doi: 10.1007/s00438-007-0305-2
Panchuk, I. I, Volkov, R. A., and Schoffl, F. (2002). Heat stress-and heat shock transcription factor-dependent expression and activity of ascorbate peroxidase in Arabidopsis. Plant Physiol. 129, 838–853. doi: 10.1104/pp.001362
Panchuk, I. I., Zentgraf, U., and Volkov, R. A. (2005). Expression of the Apx gene family during leaf senescence of Arabidopsis thaliana. Planta 222, 926–932. doi: 10.1007/s00425-005-0028-8
Pandey, M., Gangurde, S., Sharma, V., Pattanashetti, S., Naidu, G., Faye, I., et al. (2021). Improved genetic map identified major qtls for drought tolerance-and iron deficiency tolerance-related traits in groundnut. Genes 30:37. doi: 10.3390/genes12010037
Pandey, M. K., Gangurde, S. S., Sharma, V., Pattanashetti, S. K., Naidu, G. K., Faye, I., et al. (2020). Improved genetic map identified major QTLs for drought tolerance-and iron deficiency tolerance-related traits in groundnut. Genes 12:37.
Pandey, S., Fartyal, D., Agarwal, A., Shukla, T., James, D., Kaul, T., et al. (2017). Abiotic stress tolerance in plants: myriad roles of ascorbate peroxidase. Front. Plant Sci. 8:581. doi: 10.3389/fpls.2017.00581
Pandey, S., Negi, Y., Marla, S., and Arora, S. (2011). Comparative in silico analysis of ascorbate peroxidase protein sequences from different plant species. J. Bioeng. Biomed. Sci. 1:2. doi: 10.4238/2013.February.27.3
Patel, J., Khandwal, D., Choudhary, B., Ardeshana, D., Jha, R. K., Tanna, B., et al. (2022). Differential physio-biochemical and metabolic responses of peanut (Arachis hypogaea L.) under multiple abiotic stress conditions. Int. J. Mol. Sci. 23:660. doi: 10.3390/ijms23020660
Patil, S., Joshi, S., Jamla, M., Zhou, X., Taherzadeh, M. J., Suprasanna, P., et al. (2021). MicroRNA-mediated bioengineering for climate-resilience in crops. Bioengineered 12, 10430–10456. doi: 10.1080/21655979.2021.1997244
Powell, S., Forslund, K., Szklarczyk, D., Trachana, K., Roth, A., Huerta-Cepas, J., et al. (2014). eggNOG v4.0: nested orthology inference across 3686 organisms. Nucleic Acids Res. 42, D231–D239. doi: 10.1093/nar/gkt1253
Raza, A., Hussain, S., Javed, R., Hafeez, M. B., and Hasanuzzaman, M. (2021a). “Antioxidant defense systems and remediation of metal toxicity in plants,” in Approaches to the Remediation of Inorganic Pollutants, ed. M. Hasanuzzaman (Singapore: Springer), 91–124.
Raza, A., Su, W., Gao, A., Mehmood, S. S., Hussain, M. A., Nie, W., et al. (2021b). Catalase (CAT) gene family in rapeseed (Brassica napus L.): genome-wide analysis, identification, and expression pattern in response to multiple hormones and abiotic stress conditions. Int. J. Mol. Sci. 22:4281. doi: 10.3390/ijms22084281
Raza, A., Tabassum, J., Mubarik, M., Anwar, S., Zahra, N., Sharif, Y., et al. (2022a). Hydrogen sulfide: an emerging component against abiotic stress in plants. Plant Biol. 24, 540–558.
Raza, A., Tabassum, J., Zahid, Z., Charagh, S., Bashir, S., Barmukh, R., et al. (2022b). Advances in “Omics” approaches for improving toxic metals/metalloids tolerance in plants. Front. Plant Sci. 12:794373. doi: 10.3389/fpls.2021.794373
Rhee, S. Y., Beavis, W., Berardini, T. Z., Chen, G., Dixon, D., Doyle, A., et al. (2003). The Arabidopsis Information Resource (TAIR): a model organism database providing a centralized, curated gateway to Arabidopsis biology, research materials and community. Nucleic Acids Res. 31, 224–228. doi: 10.1093/nar/gkg076
Sabagh, A. E., Mbarki, S., Hossain, A., Iqbal, M., Islam, M., Raza, A., et al. (2021). Potential role of plant growth regulators in administering crucial processes against abiotic stresses. Front. Agron. 3:648694. doi: 10.3389/fagro.2021.648694
Saeed, F., Chaudhry, U. K., Bakhsh, A., Raza, A., Saeed, Y., Bohra, A., et al. (2022). Moving beyond DNA sequence to improve plant stress responses. Front. Genet. 13:874648. doi: 10.3389/fgene.2022.874648
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Sharif, Y., Chen, H., Deng, Y., Ali, N., Khan, S. A., Zhang, C., et al. (2021). Cloning and functional characterization of a pericarp abundant expression promoter (AhGLP17-1P) from peanut (Arachis hypogaea L.). Front. Genet. 12:821281. doi: 10.3389/fgene.2021.821281
Sharma, M., Kumar, P., Verma, V., Sharma, R., Bhargava, B., and Irfan, M. (2022). Understanding plant stress memory response for abiotic stress resilience: molecular insights and prospects. Plant Physiol. Biochem. 179, 10–24. doi: 10.1016/j.plaphy.2022.03.004
Shasidhar, Y., Variath, M. T., Vishwakarma, M. K., Manohar, S. S., Gangurde, S. S., Sriswathi, M., et al. (2020). Improvement of three popular Indian groundnut varieties for foliar disease resistance and high oleic acid using SSR markers and SNP array in marker-assisted backcrossing. Crop J. 8, 1–15.
Singh, N., Mishra, A., and Jha, B. (2014). Ectopic over-expression of peroxisomal ascorbate peroxidase (SbpAPX) gene confers salt stress tolerance in transgenic peanut (Arachis hypogaea). Gene 547, 119–125. doi: 10.1016/j.gene.2014.06.037
Sinha, P., Bajaj, P., Pazhamala, L. T., Nayak, S. N., Pandey, M. K., Chitikineni, A., et al. (2020). Arachis hypogaea gene expression atlas for fastigiata subspecies of cultivated groundnut to accelerate functional and translational genomics applications. Plant Biotechnol. J. 18, 2187–2200. doi: 10.1111/pbi.13374
Soni, P., Pandey, A. K., Nayak, S. N., Pandey, M. K., Tolani, P., Pandey, S., et al. (2021). Global transcriptome profiling identified transcription factors, biological process, and associated pathways for pre-harvest aflatoxin contamination in groundnut. J. Fungi 7:413. doi: 10.3390/jof7060413
Su, W., Raza, A., Gao, A., Jia, Z., Zhang, Y., Hussain, M. A., et al. (2021). Genome-wide analysis and expression profile of superoxide dismutase (SOD) gene family in rapeseed (Brassica napus L.) under different hormones and abiotic stress conditions. Antioxidants 10:1182. doi: 10.3390/antiox10081182
Swift, M. L. (1997). GraphPad prism, data analysis, and scientific graphing. J. Chem. Informat. Comput. Sci. 37, 411–412.
Tang, G., Shao, F., Xu, P., Shan, L., and Liu, Z. (2017). Overexpression of a peanut NAC gene, AhNAC4, confers enhanced drought tolerance in tobacco. Russ. J. Plant Physiol. 64, 525–535. doi: 10.3389/fpls.2022.817106
Tao, C., Jin, X., Zhu, L., Xie, Q., Wang, X., and Li, H. (2018). Genome-wide investigation and expression profiling of APX gene family in Gossypium hirsutum provide new insights in redox homeostasis maintenance during different fiber development stages. Mol. Genet. Genomics 293, 685–697. doi: 10.1007/s00438-017-1413-2
Teixeira, F. K., Menezes-Benavente, L., Margis, R., and Margis-Pinheiro, M. (2004). Analysis of the molecular evolutionary history of the ascorbate peroxidase gene family: inferences from the rice genome. J. Mol. Evol. 59, 761–770. doi: 10.1007/s00239-004-2666-z
Tian, F., Yang, D.-C., Meng, Y.-Q., Jin, J., and Gao, G. (2020). PlantRegMap: charting functional regulatory maps in plants. Nucleic Acids Res. 48, D1104–D1113. doi: 10.1093/nar/gkz1020
Tong, B., Shi, Y., Ntambiyukuri, A., Li, X., Zhan, J., Wang, A., et al. (2021). Integration of Small RNA and degradome sequencing reveals the regulatory network of al-induced programmed cell death in peanut. Int. J. Mol. Sci. 23:246. doi: 10.3390/ijms23010246
Tyagi, S., Verma, P. C., Singh, K., and Upadhyay, S. K. (2020). Molecular characterization of ascorbate peroxidase (APX) and APX-related (APX-R) genes in Triticum aestivum L. Genomics 112, 4208–4223. doi: 10.1016/j.ygeno.2020.07.023
Varshney, R. K., Bohra, A., Roorkiwal, M., Barmukh, R., Cowling, W. A., Chitikineni, A., et al. (2021a). Fast-forward breeding for a food-secure world. Trends Genet. 37, 1124–1136. doi: 10.1016/j.tig.2021.08.002
Varshney, R. K., Bohra, A., Yu, J., Graner, A., Zhang, Q., and Sorrells, M. E. (2021b). Designing future crops: genomics-assisted breeding comes of age. Trends Plant Sci. 26, 631–649. doi: 10.1016/j.tplants.2021.03.010
Varshney, R. K., Pandey, M. K., Bohra, A., Singh, V. K., Thudi, M., and Saxena, R. K. (2019). Toward the sequence-based breeding in legumes in the post-genome sequencing era. Theor. Appl. Genet. 132, 797–816. doi: 10.1007/s00122-018-3252-x
Varshney, R. K., Sinha, P., Singh, V. K., Kumar, A., Zhang, Q., and Bennetzen, J. L. (2020). 5Gs for crop genetic improvement. Curr. Opin. Plant Biol. 56, 190–196. doi: 10.1016/j.pbi.2019.12.004
Wan, L., Wu, Y., Huang, J., Dai, X., Lei, Y., Yan, L., et al. (2014). Identification of ERF genes in peanuts and functional analysis of AhERF008 and AhERF019 in abiotic stress response. Funct. Integr. Genomics 14, 467–477. doi: 10.1007/s10142-014-0381-4
Wang, C., Jogaiah, S., Zhang, W., Abdelrahman, M., and Fang, J. G. (2018). Spatio-temporal expression of miRNA159 family members and their GAMYB target gene during the modulation of gibberellin-induced grapevine parthenocarpy. J. Exp. Bot. 69, 3639–3650. doi: 10.1093/jxb/ery172
Wang, Y., Cao, S., Sui, X., Wang, J., Geng, Y., Gao, F., et al. (2022). Genome-wide characterization, evolution, and expression analysis of the ascorbate peroxidase and glutathione peroxidase gene families in response to cold and osmotic stress in Ammopiptanthus nanus. J. Plant Growth Regul. 1–21. doi: 10.1007/s00344-021-10570-5
Xu, G., Guo, C., Shan, H., and Kong, H. (2012). Divergence of duplicate genes in exon–intron structure. Proc. Natl. Acad. Sci. 109, 1187–1192.
Yao, X., Chen, J., Zhou, J., Yu, H., Ge, C., Zhang, M., et al. (2019). An essential role for miRNA167 in maternal control of embryonic and seed development. Plant Physiol. 180, 453–464. doi: 10.1104/pp.19.00127
Yu, C. S., Chen, Y. C., Lu, C. H., and Hwang, J. K. (2006). Prediction of protein subcellular localization. Proteins 64, 643–651.
Zhang, T., Hu, S., Yan, C., Li, C., Zhao, X., Wan, S., et al. (2017). Mining, identification and function analysis of microRNAs and target genes in peanut (Arachis hypogaea L.). Plant Physiol. Biochem. 111, 85–96. doi: 10.1016/j.plaphy.2016.11.018
Zhao, C., Xia, H., Cao, T., Yang, Y., Zhao, S., Hou, L., et al. (2015). Small RNA and degradome deep sequencing reveals peanut microRNA roles in response to pathogen infection. Plant Mol. Biol. Rep. 33, 1013–1029.
Zhao, C.-Z., Xia, H., Frazier, T. P., Yao, Y.-Y., Bi, Y.-P., Li, A.-Q., et al. (2010). Deep sequencing identifies novel and conserved microRNAs in peanuts (Arachis hypogaea L.). BMC Plant Biol. 10:3. doi: 10.1186/1471-2229-10-3
Zhu, H., Jiang, Y., Guo, Y., Huang, J., Zhou, M., Tang, Y., et al. (2021). A novel salt inducible WRKY transcription factor gene, AhWRKY75, confers salt tolerance in transgenic peanut. Plant Physiol. Biochem. 160, 175–183. doi: 10.1016/j.plaphy.2021.01.014
Keywords: abiotic stress, antioxidant, drought, genomics, gene ontology, legume, miRNAs, stress responses
Citation: Raza A, Sharif Y, Chen K, Wang L, Fu H, Zhuang Y, Chitikineni A, Chen H, Zhang C, Varshney RK and Zhuang W (2022) Genome-Wide Characterization of Ascorbate Peroxidase Gene Family in Peanut (Arachis hypogea L.) Revealed Their Crucial Role in Growth and Multiple Stress Tolerance. Front. Plant Sci. 13:962182. doi: 10.3389/fpls.2022.962182
Received: 06 June 2022; Accepted: 23 June 2022;
Published: 09 September 2022.
Edited by:
Shah Fahad, The University of Haripur, PakistanReviewed by:
Engin Yol, Akdeniz University, TurkeyMuhammad Azhar Nadeem, Sivas University of Science and Technology, Turkey
Copyright © 2022 Raza, Sharif, Chen, Wang, Fu, Zhuang, Chitikineni, Chen, Zhang, Varshney and Zhuang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weijian Zhuang, weijianz@fafu.edu.cn; Rajeev K. Varshney, rajeev.varshney@murdoch.edu.au