 Hua Liu1†
Hua Liu1† Yin Jia2†Yuhong Chai1,3†Sen Wang1Haixia Chen1Xiumei Zhou3
Yin Jia2†Yuhong Chai1,3†Sen Wang1Haixia Chen1Xiumei Zhou3 Conglin Huang1*Shuang Guo4*Dongliang Chen1*
Conglin Huang1*Shuang Guo4*Dongliang Chen1*- 1Institute of Grassland, Flowers and Ecology, Beijing Academy of Agriculture and Forestry Sciences, Beijing, China
- 2College of Landscape Architecture, Sichuan Agricultural University, Chengdu, China
- 3School of Horticulture and Landscape Architecture, Henan Institute of Science and Technology, Xinxiang, China
- 4Chengdu Park City Construction Development Research Institute, Chengdu, China
Chrysanthemum morifolium has ornamental and economic values. However, there has been minimal research on the morphology of the chrysanthemum florets and related genes. In this study, we used the leaves as a control to screen for differentially expressed genes between ray and disc florets in chrysanthemum flowers. A total of 8,359 genes were differentially expressed between the ray and disc florets, of which 3,005 were upregulated and 5,354 were downregulated in the disc florets. Important regulatory genes that control flower development and flowering determination were identified. Among them, we identified a TM6 gene (CmTM6-mu) that belongs to the Class B floral homeotic MADS-box transcription factor family, which was specifically expressed in disc florets. We isolated this gene and found it was highly similar to other typical TM6 lineage genes, but a single-base deletion at the 3′ end of the open reading frame caused a frame shift that generated a protein in which the TM6-specific paleoAP3 motif was missing at the C terminus. The CmTM6-mu gene was ectopically expressed in Arabidopsis thaliana. Petal and stamen developmental processes were unaffected in transgenic A. thaliana lines; however, the flowering time was earlier than in the wild-type control. Thus, the C-terminal of paleoAP3 appears to be necessary for the functional performance in regulating the development of petals or stamens and CmTM6-mu may be involved in the regulation of flowering time in chrysanthemum. The results of this study will be useful for future research on flowering molecular mechanisms and for the breeding of novel flower types.
Introduction
As one of the most important cut flower species, Chrysanthemum morifolium is cultivated worldwide (Teixeira, 2003; Silva et al., 2013). Chrysanthemum flowers have capitula consisting of two types of florets (ray and disc), which are derived from one torus. Capitula are complex structures that include (from the outside to the inside) bracts, a torus, ray florets, and disc florets (Liu et al., 2020). The differentiation and development of ray and disc florets mediate the formation of various flower types in cultivated chrysanthemum varieties (Song et al., 2018). In the same genetic background, the two types of florets vary substantially in their position, fertility, symmetry, organ fusion, pigment composition (Berger et al., 2016; Novaković et al., 2019; Su et al., 2019). However, because of the lack of genome information, the molecular regulation mechanisms involved in the development of the two types of florets in chrysanthemum are still not fully understood.
In a previous study, Liu et al. identified homologs of important regulators of flower development and organ determination, including homologs of Class A, B, C, and E genes as well as two MCMl/AGAMOUS/DEFICIENS/SERUM RESPONSE FACTOR (MADS)-box genes (WUSCL and KNUCL) that were differentially expressed between the ray and disc florets, verifying their important roles in flower development and organ determination (Liu et al., 2016a). Indeed, many previous studies have proved that the expression of homologous genes related to floral organ development in higher plants vary in the flower heads of Asteraceae plants (Ren et al., 2013; Liu et al., 2015, 2016a). Among these genes, the CYCLOIDEA2 (CYC2)-like genes, as well as the Class B and Class C MADS-box genes, may be involved in regulating the distinct development of the two types of florets in Asteraceae (Burke, 2008; Chapman et al., 2012). Currently, among higher plants, floral developmental and regulatory activities have been characterized primarily in A. thaliana, Antirrhinum majus, and other model species (Johannesson et al., 2001; Clark and Coen, 2002; Juntheikki-Palovaara et al., 2014).
Class B genes also help determine petal and stamen development (Mahajan and Yadav, 2014; Sasaki et al., 2014; Prunet et al., 2017). There are two Class B gene lineages, PISTILLATA (PI)/GLOBOSA (GLO) and APETALA3 (AP3)/DEFICIENS (DEF)/Tomato MADS-BOX GENE 6 (TM6; Kramer et al., 1998; Vandenbussche et al., 2003; Zhang et al., 2011). The AP3/DEF/TM6 lineage is further divided into two sublineages (i.e., AP3 and TM6; Vandenbussche et al., 2003). The AP3 and TM6 genes encode proteins that have euAP3 and paleoAP3 motifs, respectively, in the C-terminal regions (Kramer et al., 2006; Ackerman et al., 2008). The AP3 lineage genes mediate petal and stamen development, whereas the TM6 lineage genes mostly influence stamen development (Tsaftaris et al., 2006; Yeoh et al., 2016; Martín-Pizarro et al., 2018). However, the genetic regulatory mechanism underlying the development of chrysanthemum capitula remains unknown. Elucidating the mechanism controlling the development of the two types of chrysanthemum florets in the same genetic background is critical for the breeding of chrysanthemum varieties with enhanced flower types.
Flowering time is an important ornamental trait for flowering plants. Six major floral induction pathways have been identified in A. thaliana, and these pathways converge to regulate a small number of “floral integrator genes,” such as, FLOWERING LOCUS T (FT) and SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 (SOC1), both of which rapidly promote floral development (Fornara et al., 2010). Chrysanthemum is a typical short-day flowering plant and floral induction is controlled mainly through the photoperiod pathway (Oda et al., 2017). As was found in model plants, in chrysanthemum, CONSTANS (CO) is the core gene of the photoperiod pathway, which is induced by circadian clock genes, such as CRYPTOCHROMES (CRYs) and GIGANTEA (GI), and activates FT transcription in leaves (Yang et al., 2017; Sun et al., 2018; Oda et al., 2020). The balance between FT and an ortholog of FT, ANTI-FLORIGENIC FT/TFL1 FAMILY PROTEIN (AFT) determines the progression of floral transition and anthesis (Higuchi et al., 2013; Oda et al., 2020).
Other floral induction pathways, such as the gibberellin (GA), age, and ambient temperature pathways, have been found in chrysanthemum (Sumitomo et al., 2009; Gao et al., 2017; Wei et al., 2017). The ambient temperature pathway converges to regulate the expression of FT by SHORT VEGETATIVE PHASE (SVP). Overexpression of the C. morifolium SVP gene delayed blossoming in transgenic Arabidopsis, indicating that a similar ambient temperature pathway may exist in chrysanthemum (Gao et al., 2017). The GA and age pathways were shown to converge to regulate the expression of SOC1. In chrysanthemum, the nuclear factor Y, subunit B8 gene (CmNFYB8) was shown to influence flowering time by directly regulating the expression of the microRNA cmo-MIR156 in the aging pathway (Wei et al., 2017). GA was found to promote flowering in chrysanthemum by up-regulating FLORICAULA/LEAFY (FL) expression (Sumitomo et al., 2009). Flowering regulation is a complex biochemical process that involves a large number of genes and complex molecular regulation networks. Research on the flowering regulation mechanism has important implications for the accurate regulation of flowering time.
Transcriptome sequencing is important for investigating floral organ development in higher plants because it enables the rapid and efficient screening of many differentially expressed genes (DEGs; Liu et al., 2017; Pei et al., 2017). In floral development research, transcriptome sequencing technology has been widely used for basic screening work (Fu et al., 2018). For example, an earlier transcriptome analysis identified numerous MADS-box genes that are differentially expressed between wild-type (WT) and lip-flap Phalaenopsis mutants, revealing how the orchid labellum differs and why the petal or sepal converts to a labellum in Phalaenopsis (Huang et al., 2015). Another transcriptome analysis, which examined the flowering transformation process in sugar apple, revealed the differential expression of genes related to circadian rhythms and plant signal transduction (Liu et al., 2016b). The transcriptome sequencing of the flower–fruit transformation process of grape trees and other fruit trees identified DEGs related to sugar and hormone signaling pathways (Domingos et al., 2016). Transcriptome sequencing techniques have been used to investigate various stages of floral development in plants such as broccoli, orchid, and grape (Huang et al., 2015; Domingos et al., 2016; Liu et al., 2016a, 2017; Pei et al., 2017; Fu et al., 2018). Therefore, in this study, we applied high-throughput sequencing technology to analyze genes that are differentially expressed between chrysanthemum ray and disc florets.
The transcriptome sequencing and comparative analysis of chrysanthemum ray florets, disc florets, and leaves were performed using Illumina sequencing technology and RNA sequencing (RNA-seq) data. We identified DEGs between ray florets and disc florets to reveal important regulators controlling the differential development of the two floret types. Next, we identified important regulatory genes that may be involved in controlling floral development and floral organ identity. A list was compiled of candidate genes for future functional analyses of flowering regulation in chrysanthemum. We also isolated a TM6 homolog in C. morifolium (CmTM6-mu). This gene shares a high similarity with other TM6 lineage genes (Ackerman et al., 2008; Yeoh et al., 2016; Martín-Pizarro et al., 2018), but a single-base deletion was observed at the 3′ terminal of the open reading frame (ORF), which caused a frameshift and led to the encoding product not having the typical paleoAP3 motif at its C-terminal. The gene function of CmTM6-mu was analyzed by ectopic expression in A. thaliana. Transgenic Arabidopsis showed no petals or stamen developmental affects, as expected; however, an earlier flowering phenotype was observed in almost all the transgenic lines, which indicated that the C-terminus of paleoAP3 is necessary for the functional performance in regulating the development of petals or stamens and that CmTM6-mu may be involved in the regulation of flowering time in chrysanthemum.
Our results will provide researchers with valuable genomic information and candidate genes that are potentially useful for flowering molecular mechanism studies and for the breeding of novel flower types. Furthermore, the data presented herein may be useful for clarifying the molecular mechanisms regulating the differential development of ray and disc florets in chrysanthemum flowers.
Results
Screening, comparison, and analysis of data
Total RNA was extracted from the ray and disc florets of the capitula, as well as the fully extended leaves of chrysanthemum cultivar “Pink Carpet” during the full-bloom stage (Figures 1A–C). The RNA was used to construct nine libraries (i.e., three biological replicates per sample type) for the RNA-seq analysis. Approximately 38–58 million reads were generated per library. After a strict quality control step during which low-quality data were eliminated, ~37–56 million clean reads were retained for each library. Details regarding the sequencing data are provided in Table 1. For each sample, more than 96% of the raw reads were retained as clean reads for all three replicates. Thus, the high-quality sequencing data were appropriate for the subsequent analyses. The HISAT2 program was used to align the clean reads to the Chrysanthemum seticuspe reference genome (Kim et al., 2015). The mapping rates for all samples exceeded 75% (Table 1). Therefore, the sequenced plants were closely related to C. seticuspe.
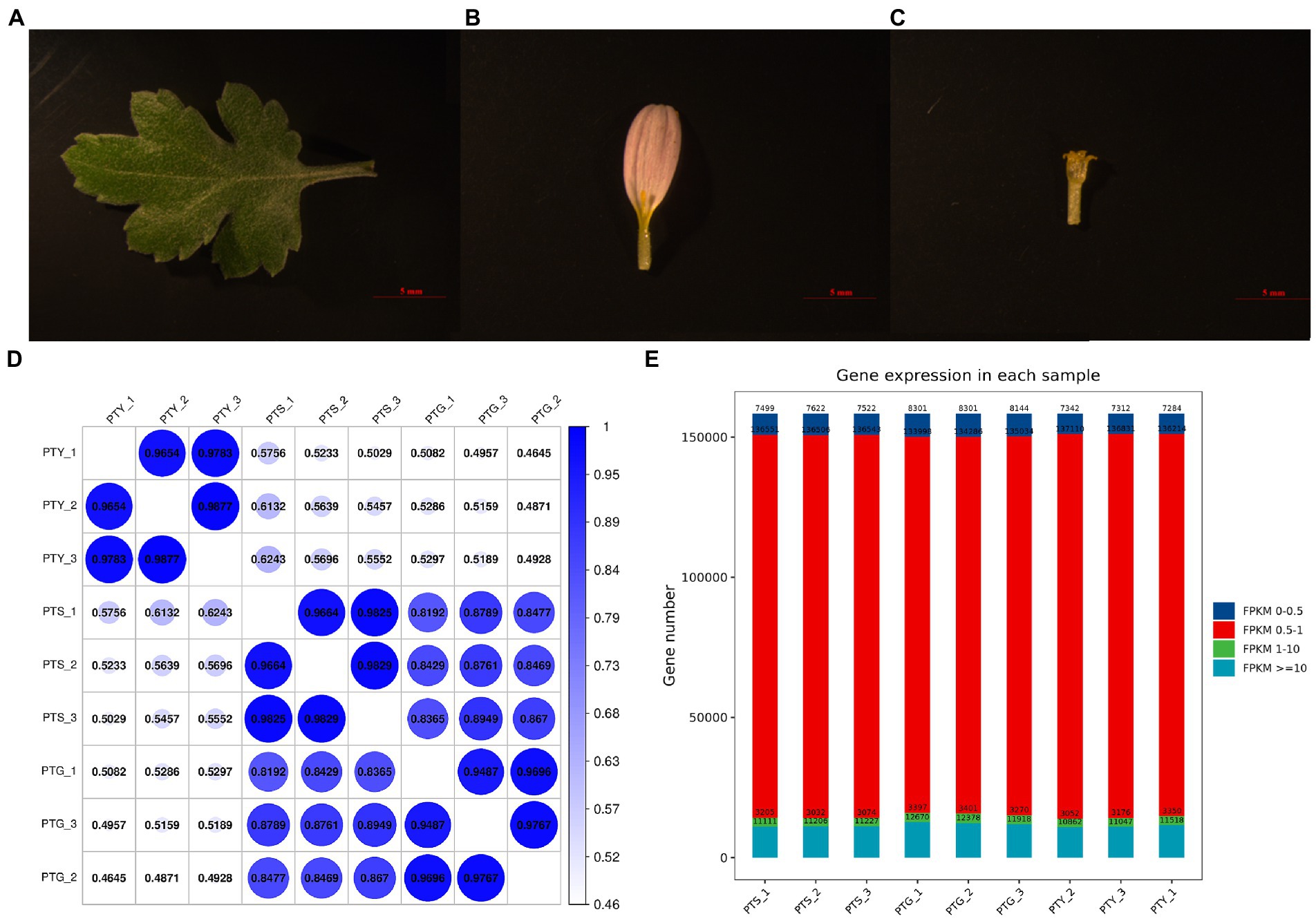
Figure 1. Chrysanthemum samples used for transcriptome sequencing and gene expression levels and correlation analysis results. (A–C) Leaf, ray floret, and disc floret, respectively, of Chrysanthemum morifolium Ramat “Pink Carpet.” (D) Gene expression levels in each sample. (E) Gene expression level statistics of each sample.
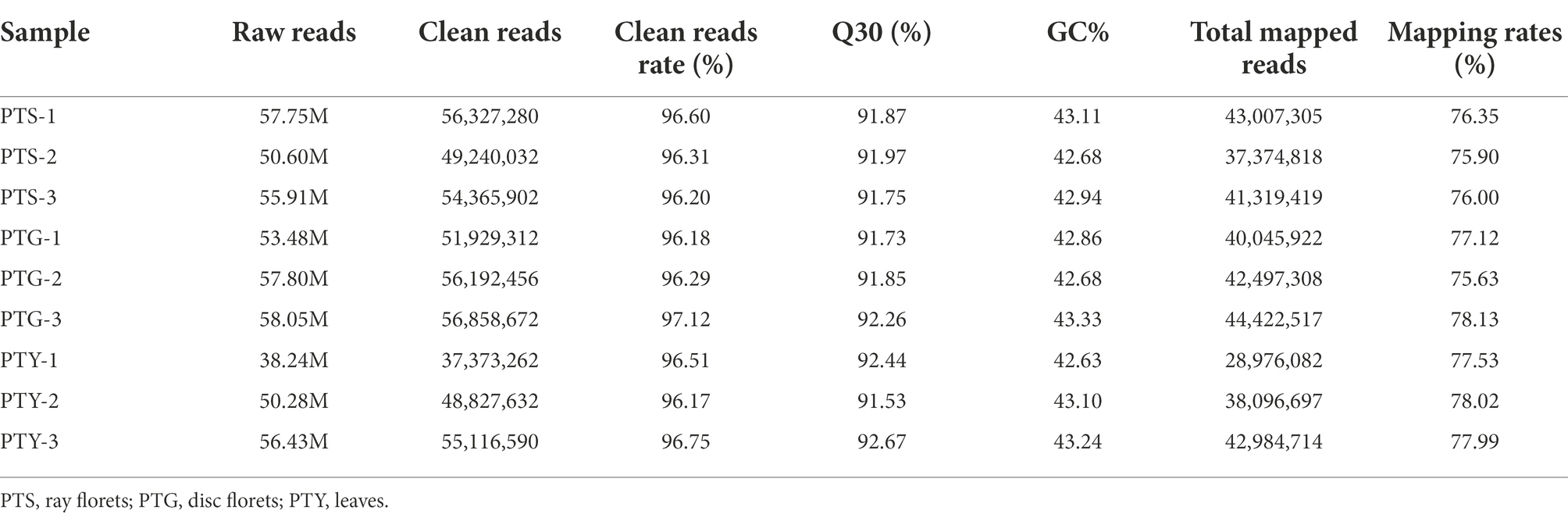
Table 1. General statistics for the Illumina HiSeq 2000 sequencing data.
Using known reference gene sequences and annotation files, sequence similarities were determined, and the expression levels of each protein-coding gene in each sample were determined. The fragments per kilobase of transcript per million mapped reads (FPKM) values calculated using the Cufflinks software were used to represent gene expression levels. The gene expression levels were highly correlated among the three biological replicates for the different sample types (Figure 1D), implying that the sequencing data were reliable. The average FPKM value for the three biological replicates was calculated for each gene in each sample. Most gene expression levels were between 0.5 and 1 (Figure 1E).
Gene annotation and functional classification
A total of 150,346 unigenes were annotated following a BLAST search of seven databases [non-redundant (NR) protein database, Swiss-Prot, Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), EuKaryotic Orthologous Groups (KOG), evolutionary genealogy of genes: Non-supervised Orthologous Groups (eggNOG), and Pfam], leaving 8,021 (5.06%) unannotated unigenes. More specifically, 150,122, 62,321, 55,481, 78,597, 18,304, 93,361, and 87,286 unigenes were annotated on the basis of information in the NR, Swiss-Prot, GO, KOG, KEGG, eggNOG, and Pfam databases, respectively.
The GO database is divided into the following three main categories: biological process, molecular function, and cellular component. A total of 55,481 unigenes were classified into 50 categories: 22 biological process, 13 cellular component, and 15 molecular function categories. The predominant biological process GO terms among the unigenes were “cellular process” and “metabolic process,” whereas “biological adhesion,” “cell killing,” and “locomotion” were relatively uncommon GO terms. The main cellular component GO terms among the unigenes were “cell,” “organelle,” and “cell part,” whereas “nucleoid” was a relatively uncommon GO term. The predominant molecular function GO terms among the unigenes were “binding” and “catalytic activity,” whereas “transcription regulator activity,” “protein tag,” “metallochaperone activity,” “electron carrier activity,” and “receptor regulator activity” were relatively uncommon GO terms (Figure 2A).
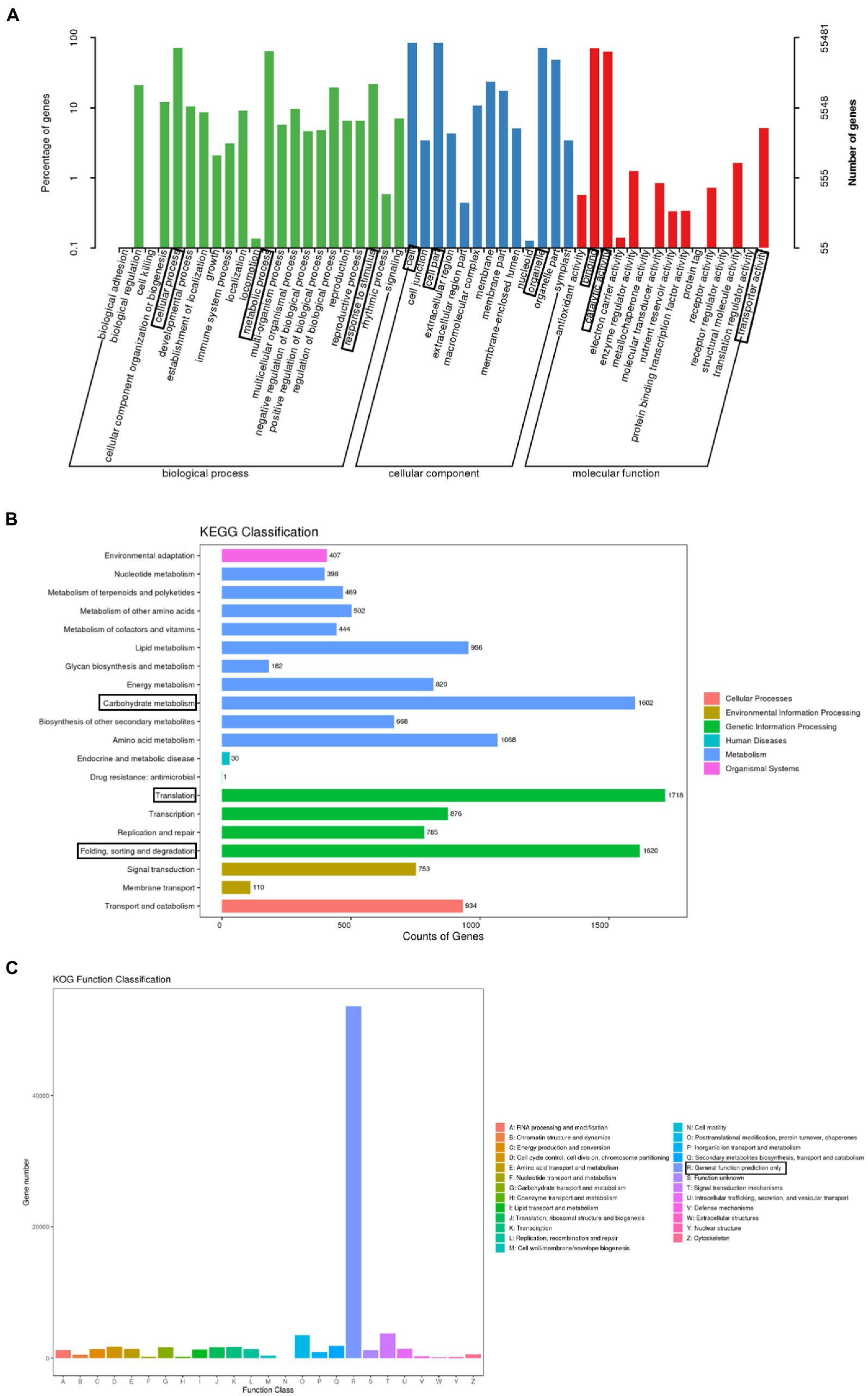
Figure 2. Functional annotations assigned to the unigenes. (A) Enriched gene ontology (GO) terms under the three main categories: biological process, molecular function, and cellular component. The three most significant GO terms in each category are boxed. (B) Enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (level 2) in the six KEGG categories. The three most significant pathways are boxed. (C) Enriched EuKaryotic Orthologous Groups (KOG) functional classifications of identified paralogous proteins. The most significant classification is boxed.
Under the biological process category, a total of 935 unigenes were annotated with at least one of the 8 terms were related to flowering time. They includes “photoperiodism, flowering” (499 genes), “long-day photoperiodism, flowering” (272 genes), “regulation of long-day photoperiodism, flowering” (72 genes), “regulation of photoperiodism, flowering” (42 genes), “short-day photoperiodism, flowering” (22 genes), “negative regulation of long-day photoperiodism, flowering” (17 genes), “negative regulation of short-day photoperiodism, flowering” (eight genes), and “positive regulation of short-day photoperiodism, flowering” (three genes; Table 2).

Table 2. Gene ontology (GO) biological process terms related to flowering time assigned to the unigenes.
The KEGG database divides biological pathways into six categories, each of which is subdivided and displayed in a pathway graph that presents the molecular interaction networks in cells and in particular organisms. Of the 150,346 unigenes, 18,304 were assigned to at least one KEGG pathway, for a total of 127 KEGG pathways. The most represented pathways were “translation” (1,718 unigenes), “folding, sorting and degradation” (1,620 unigenes), and “carbohydrate metabolism” (1,602 unigenes), followed by “amino acid metabolism” (1,068 unigenes), “lipid metabolism” (956 unigenes), and “transport and catabolism” (934 unigenes). Additionally, 876 unigenes were associated with the “transcription” pathway (Figure 2B).
The KOG database is used to identify paralogous proteins. Gene annotation information was used to screen the KOG database. Among the 25 KOG categories, “general function prediction only” (53,642 unigenes) was the largest, followed by “signal transduction mechanisms” (3,087 unigenes) and “posttranslational modification, protein turnover, chaperones” (2,987 unigenes). In contrast, “cell motility” (five unigenes), “extracellular structures” (five unigenes), and “nuclear structure” (seven unigenes) were the smallest categories (Figure 2C).
Differential gene expression analysis
Analyzing the differential expressions of genes between samples provides the basis for subsequent analyses of gene functions. We screened the DEGs among the ray florets, disc florets, and leaves using the following criteria: fold-change >2 and value of p <0.05. Accordingly, 8,359 genes were differentially expressed between ray florets and disc florets, of which the expression levels of 3,005 and 5,354 genes were higher and lower in the disc florets than in the ray florets, respectively (Figure 3A; Supplementary File 1). Moreover, 1,232 genes were differentially expressed specifically between the ray florets and disc florets (Figure 3B).
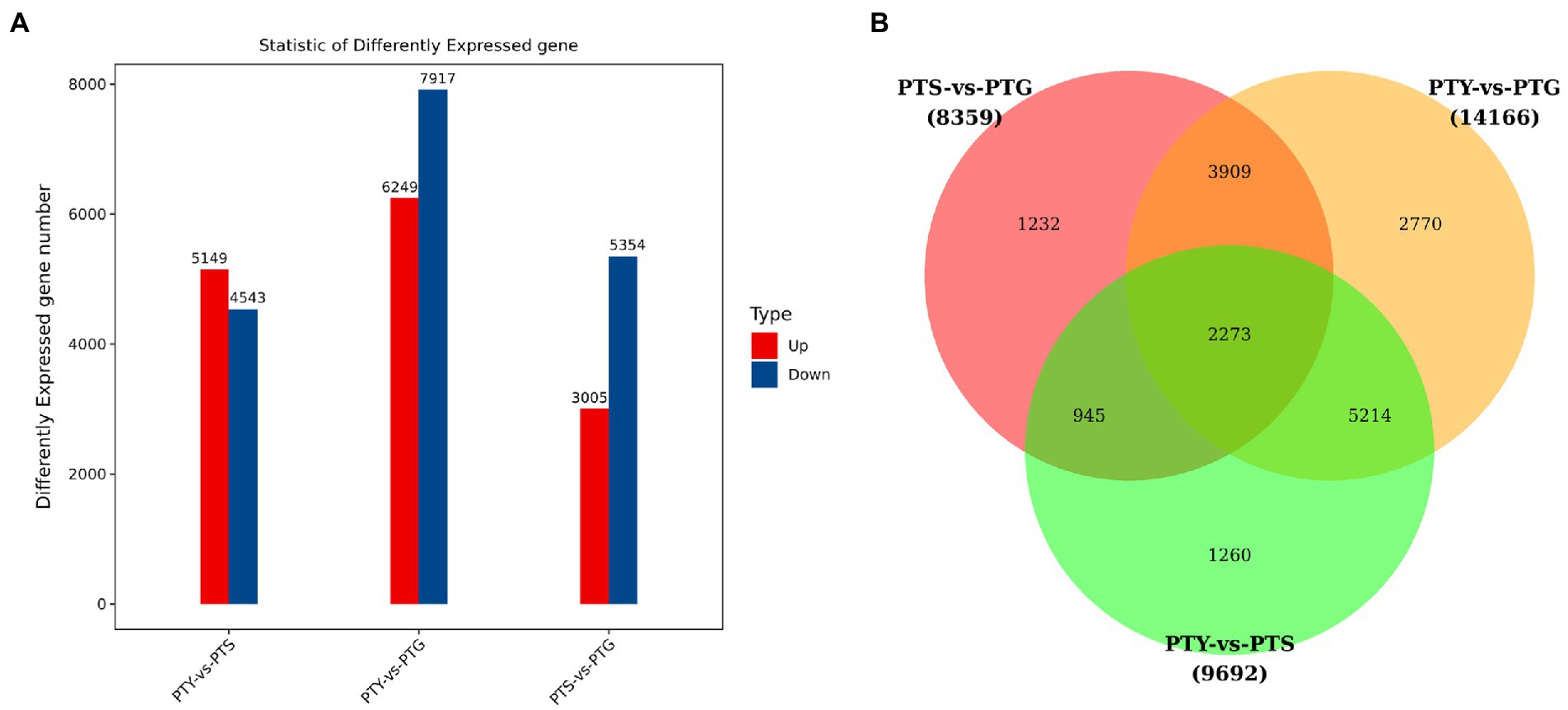
Figure 3. Differential gene expression analysis of leaves, disc florets, and ray florets in chrysanthemum. (A) Differentially expressed genes among the analyzed chrysanthemum samples. (B) Common and unique differentially expressed genes among the analyzed chrysanthemum samples.
Enriched gene ontology terms among the differentially expressed genes
The DEGs were screened for significantly enriched GO functions. Following the annotation of the DEGs with GO terms, the GO classifications were visualized using the WEGO software. The ray florets, disc florets, and leaves were analyzed to reveal the significantly enriched GO terms among the DEGs.
A total of 3,989 GO terms were enriched among the DEGs between the ray florets and disc florets (Supplementary File 2). The most enriched GO terms were mainly related to cellular components (GO:0016021, GO:0005886, and GO:0005634) and molecular functions (GO:0046872 and GO:0005524). In the biological process category, many DEGs were related to “cellular processes,” “metabolic processes,” and “single biological processes.” In the cellular component category, the DEGs were mainly related to “cells,” “cell parts,” “organelles,” and “cell membranes.” In the molecular function category, most of the DEGs were related to “binding function” and “catalytic activity” (Figure 4A).
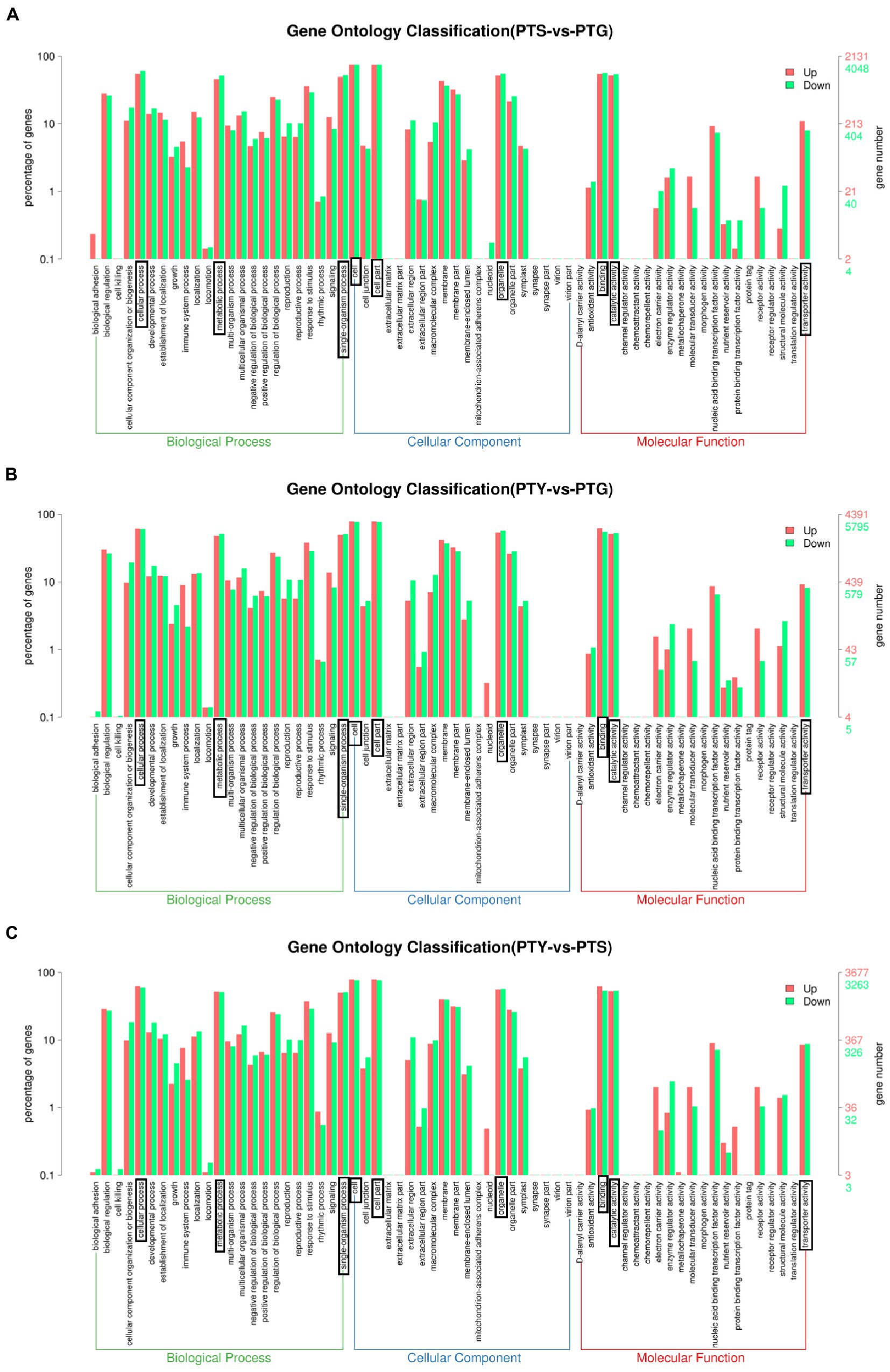
Figure 4. Enriched gene ontology (GO) terms assigned to the differentially expressed genes in comparisons among the leaf, disc floret, and ray floret samples. (A–C) Comparisons between ray florets and disc florets, leaves and disc florets, and leaves and ray florets, respectively. The three most significant GO classifications in each comparison are boxed. PTS, ray florets; PTG, disc florets; PTY, leaves.
An analysis of the significantly enriched GO terms among the DEGs between the disc florets and leaves (Figure 4B) indicated that the distribution of significantly enriched GO terms was similar in the disc florets vs. leaves and disc florets vs. ray florets comparisons. The significantly enriched GO terms and their distribution were also determined for the DEGs between the ray florets and leaves (Figure 4C). The significantly enriched GO terms and the distribution of GO functions were essentially the same for the ray and disc florets.
Significantly enriched pathways among differentially expressed genes
Biological functions in vivo require the coordinated expression of various genes. Pathway analyses are useful for clarifying the biological functions of genes. We used the KEGG database to determine the enriched pathways assigned to the protein-coding genes differentially expressed among the ray florets, disc florets, and leaves (Figure 5). A total of 190 KEGG pathways were enriched among the DEGs between the ray florets and disc florets, including “synthesis” (ko00940), “starch and sucrose metabolism” (ko00500), “plant hormone signal transduction” (ko04075), and “carbon metabolism” (ko01200; Supplementary File 3). The DEGs were mainly related to 20 metabolic pathways (Figure 5A). The most enriched pathway was “phenylpropanoid biosynthesis,” followed by “starch and sucrose metabolism,” “fatty acid metabolism,” and “pentose and glucose.” These genes may be associated with the conversion of uronic acid esters.
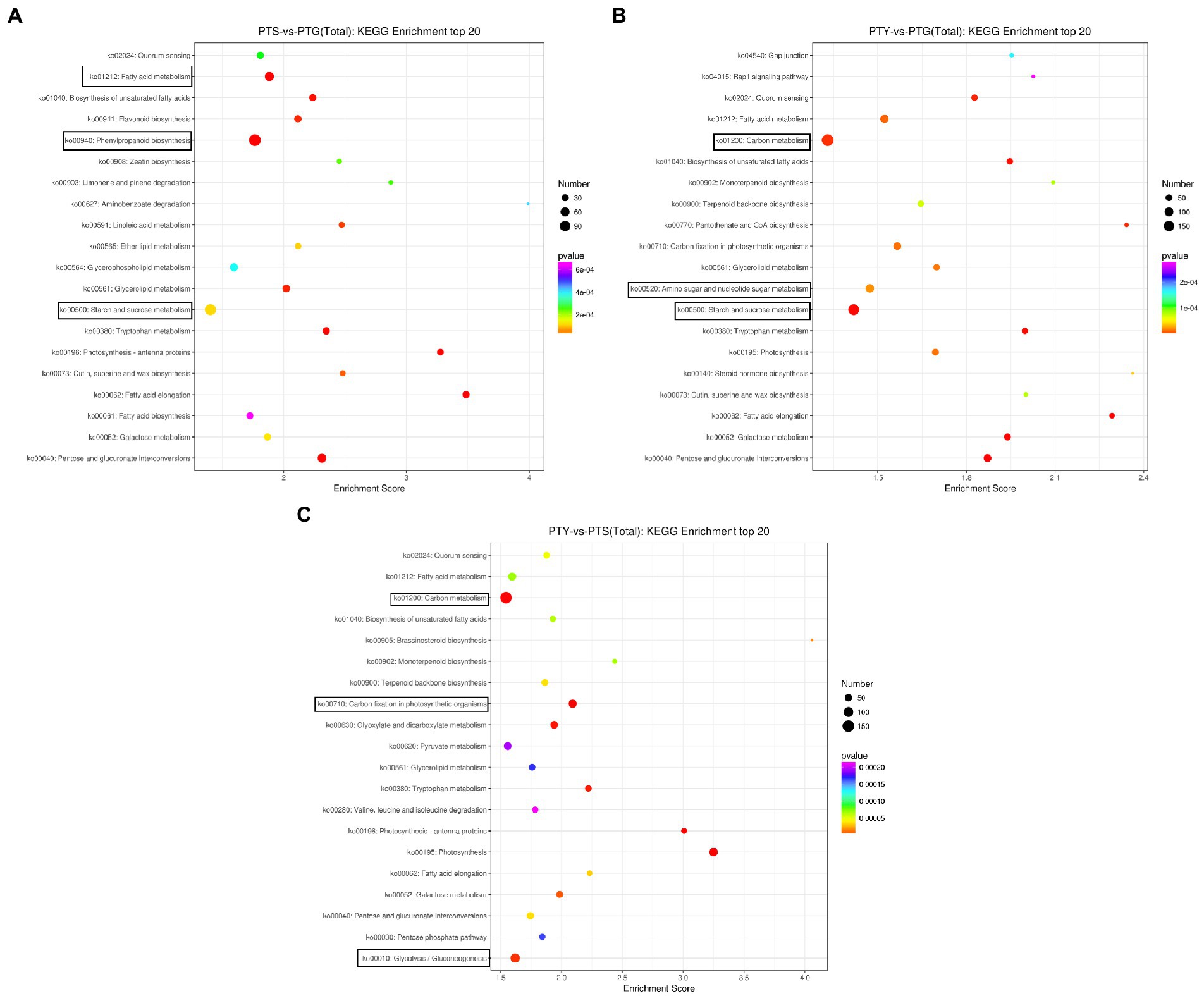
Figure 5. Bubble diagrams of enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways assigned to the differentially expressed genes in comparisons among the leaf, disc floret, and ray floret samples. (A–C) Comparisons between ray florets and disc florets, leaves and disc florets, and leaves and ray florets, respectively. The three most significant KEGG pathways in each comparison are boxed. PTS, ray florets; PTG, disc florets; PTY, leaves.
Of the top 20 enriched KEGG pathways among the DEGs between the disc florets and leaves, “carbon metabolism” and “starch and sucrose metabolism” were the main pathways (Figure 5B). In contrast, “carbon metabolism,” “glycolysis,” “photosynthesis,” and “fatty acid metabolism” were the primary enriched KEGG pathways among the DEGs between the ray florets and leaves (Figure 5C).
Analysis of important differentially expressed genes
Bioinformatics analyses of the significantly enriched GO terms and KEGG pathways may help clarify the functions and metabolic pathways associated with the DEGs. However, to identify the key regulatory genes among the DEGs, we conducted a detailed analysis of the DEGs related to floral development. The results suggested that most of the DEGs are involved in inducing the flowering pathway, activating transcription factors (TFs), and regulating floral organ development and floral symmetry. This study mainly focused on floral organ development and the homologous genes related to the regulation of floral development and floral symmetry. The results of the analysis are listed in Table 3.
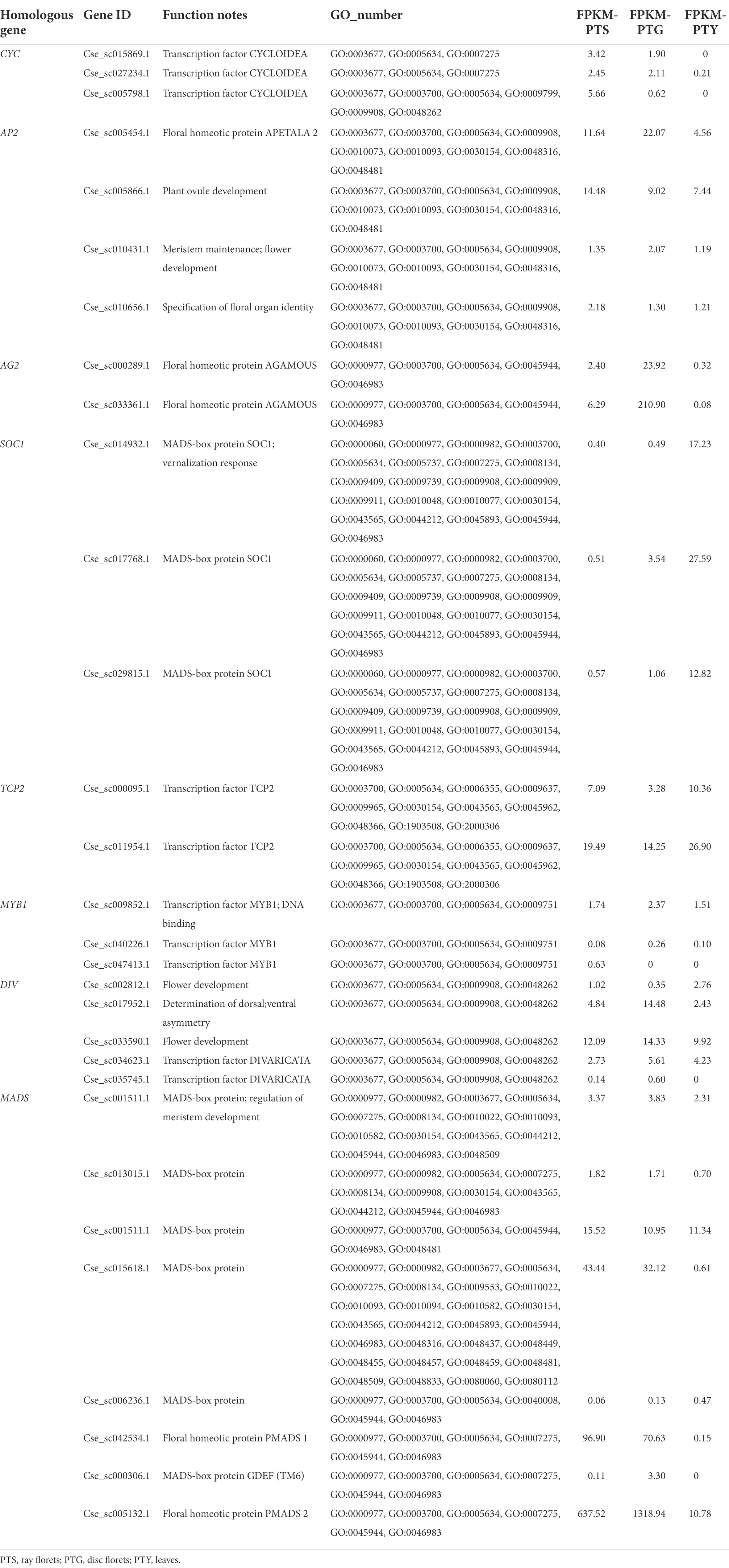
Table 3. Floral development-related differentially expressed genes in comparisons among the leaf, disc floret, and ray floret samples.
On the basis of the annotation information and sequence analyses, the key genes related to the regulation of floral organ development, APETALA2 (AP2) and AGAMOUS2 (AG2), were identified in the transcriptome (Becker and Theissen, 2003; Aida et al., 2008; Wollmann et al., 2010). Moreover, TEOSINTE BRANCHED1/CYCLOIDEA /PROLIFERATING CELL FACTOR (TCP), V-MYB AVIAN MYELOBLASTOSIS VIRAL ONCOGENE HOMOLOG (MYB), and MADS genes, which help regulate flowering, meristem differentiation, and floral symmetry, were also identified, as were CYC, DIVARICATA (DIV), and other homologous genes (Galego and Almeida, 2002; Dornelas et al., 2011; Huang and Irish, 2015; Niwa et al., 2018; Victoria and Minsung, 2017).
TFs are important for plant development at the cellular, tissue, and organ levels (Shin et al., 2002; Hileman, 2014; Lai et al., 2020). Previous studies proved that the TCP, MYB, and MADS TF families, as well as TFs from other families, have key regulatory functions that influence floral development (Kaufmann et al., 2009; Hileman, 2014; Ke et al., 2021; Li et al., 2021). Our RNA-seq analysis revealed that the TCP2, MYB1 (Huang and Irish, 2015; Niwa et al., 2018), and MADS gene expression levels varied significantly among the ray florets, disc florets, and leaves. Additionally, expression-level differences were detected for multiple CYC and DIV genes that regulate floral symmetry. Significant differences in gene expression were also observed between the two floret types. Most TCP family genes and CYC genes were significantly more highly expressed in the ray florets than in the disc florets, whereas the opposite pattern was detected for the AG and SOC family genes (Wollmann et al., 2010; Li et al., 2021). Some AP, MYB, DIV, and MADS family genes were more highly expressed in the ray florets than in the disc florets, but some were more highly expressed in disc florets. These findings imply that changes in the expression levels of floral development-related TFs may cause meristems to differentiate in different directions. These genes should be investigated more thoroughly regarding their effects on chrysanthemum floret development.
Identification of important genes and signaling pathways involved in flowering control in chrysanthemum
Previous studies have identified six major floral induction pathways in A. thaliana, namely the ambient temperature, vernalization, autonomous, photoperiod pathway, GA, and age pathways (Sumitomo et al., 2009; Fornara et al., 2010; Gao et al., 2017; Wei et al., 2017; Oda et al., 2020). In this study, we identified many genes that were homologs of flowering time control genes involved in these floral induction signaling pathways. The expression levels of some of these genes are shown in Figure 6. Chrysanthemum floral induction is controlled mainly through the photoperiod and circadian signaling pathways by important flowering regulator genes, including PHYTOCHROME (PHY), CRY, CONSTITUTIVE PHOTOMORPHOGENIC 1 (COP1), CO, FT, TYPE B-LACTAMASE (TEM), CATION DIFFUSION FACILITATOR (CDF), LATEGIGANTEA (GI), and FLAVIN-BINDING KELCH REPEAT F-box1 (FKF1; Yang et al., 2017; Sun et al., 2018; Oda et al., 2020). We identified homologs of four PHY, three CRY, four COP1, 10 CO, one FT, one TEM, six CDF, four GI, and one FKF1 in the chrysanthemum transcriptome data.
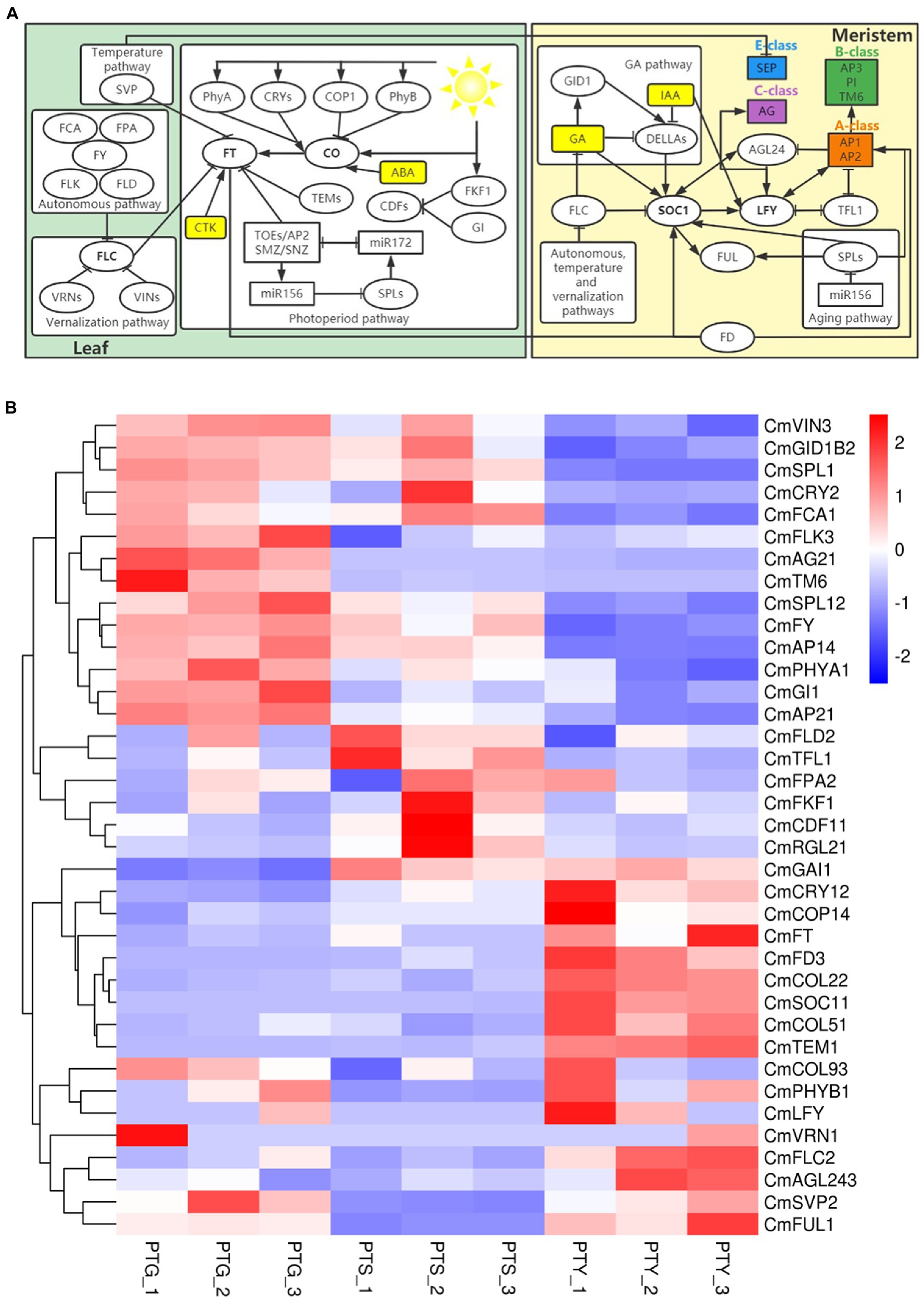
Figure 6. Important genes and signaling pathways involved in flowering control in chrysanthemum. (A) Schematic diagram of the flowering regulatory networks involved in six major floral induction pathways the Chrysanthemum morifolium. Arrows indicate activation. Bars indicate repression. (B) Heat map of the gene expression levels in the six major floral induction pathways. Rows and columns represent genes and samples, respectively. Sample names are provided below the heat map (PTS, ray florets; PTG, disc florets; PTY, leaves). The color scale indicates gene expression fold-changes (red, high expression; blue, low expression). All homologs of the regulators involved in the flowering regulatory networks are listed in Supplementary File 6.
The ambient temperature pathway converges to regulate the expression of FT by SVP, and two homologs of SVP (CmSVP1 and CmSVP2) were identified. Many autonomous pathway genes have been found in Arabidopsis, including FLOWERING LOCUS CA (FCA), FLOWERING LOCUS PA (FPA), FLOWERING LOCUS D (FLD), FLOWERING LOCUS Y (FY), and FLOWERING LOCUS KH DOMAIN (FLK; Eom et al., 2017). These genes promote flowering by inhibiting the expression of FLOWERING LOCUS C (FLC; Yan et al., 2020). We identified homologs of three FCA, four FPA, three FLK, three FLD, and one FY in chrysanthemum. FLC is a MADS-box TF that acts as a potent repressor of flowering in the vernalization pathway of different Arabidopsis varieties (Fornara et al., 2010). We detected homologs of two FLC (CmFLC1 and CmFLC2), one VERNALIZATION INSENSITIVE 3 (VIN3) and one VERNALIZATION 2 (VRN2) in this pathway. In the age pathway, SQUAMOSA PROMOTER BINDING LIKE (SPL) TFs promote flowering by initiating the expression of other TFs, such as LEAFY (LFY), FRUITFULL (FUL), and SOC1 (Fornara et al., 2010). We identified 19 homologs of SPL genes in this pathway.
The GA pathway converges to regulate SOC1, and we identified homologs of three GAINSENSITIVE DWARF1 (GID1) and 15 DELLA in this pathway. We also found homologs of 91 auxin (IAA), 16 cytokinin (CTK), and 11 abscisic acid (ABA), and identified homologs of LFY, FUL, TFL1 (TERMINAL FLOWER 1), AGAMOUS like-24 (AGL24), and bZIP transcription factor FD genes. Details of all the identified homologs of the regulators in the flowering regulatory networks involved in the six major floral induction pathways of C. morifolium are listed in Supplementary File 6. On the basis of the annotations of the transcriptome sequences, homologs of the Class A, B, C, and E genes were identified, including homologs of four AP1, four AP2, three SOC1, two AG2, and one TM6 (Supplementary File 6).
Verification of transcriptome accuracy via a quantitative real-time polymerase chain reaction analysis
The FPKM values indicated that FAMA, DIAMINE OXIDASE (DAO), AINTEGUMENTA (ANT), SMALL AUXIN UP RNA24 (SAUR24), ETHYLENE RESPONSE FACTOR 1B (ERF1B), MYB101, WRKY42, AG2, and AGAMOUS like-11 (AGL11), which are plant growth and development and floral development-related genes (Ohashi-Ito and Bergmann, 2006; Park et al., 2007; Jabbour et al., 2015; Ocarez and Mejía, 2016; Vuosku et al., 2019; Hara-Kitagawa et al., 2020; Niu et al., 2020; Zhang et al., 2020; Juárez-Corona and Stefan, 2021), as well as CYC and TCP2, which are flowering-related TF genes (Huang and Irish, 2015; Victoria and Minsung, 2017), are highly expressed in ray florets. CYC genes include CYC1, CYC2, and CYC3 (Howarth and Donoghue, 2006). CYC and TCP2 are also flower development-related genes (Juntheikki-Palovaara et al., 2014; Li et al., 2021). TM6, a member of MAD-box family, are highly expressed in disc florets, has proved determining the identity of stamen in plants (Tsaftaris et al., 2006; Yeoh et al., 2016; Martín-Pizarro et al., 2018). The expression levels of these genes were verified by qRT-PCR analysis. The results showed that FAMA, ANT, and TCP2 were highly expressed in leaves, DAO, SAUR24, ERF1B, MYB101, WRKY42, AG2, AGL11, and TM6 were highly expressed in disc florets, and CYC1 and CYC2 were highly expressed in ray florets. These results are consistent with the FPKM values obtained from the transcriptome data (Figure 7; Table 3), which confirms the reliability of the transcriptome sequencing results. Relative expression levels were determined using the PP2Ac-encoding gene as a internal control gene (Ren et al., 2013; Liu et al., 2015, 2016a).
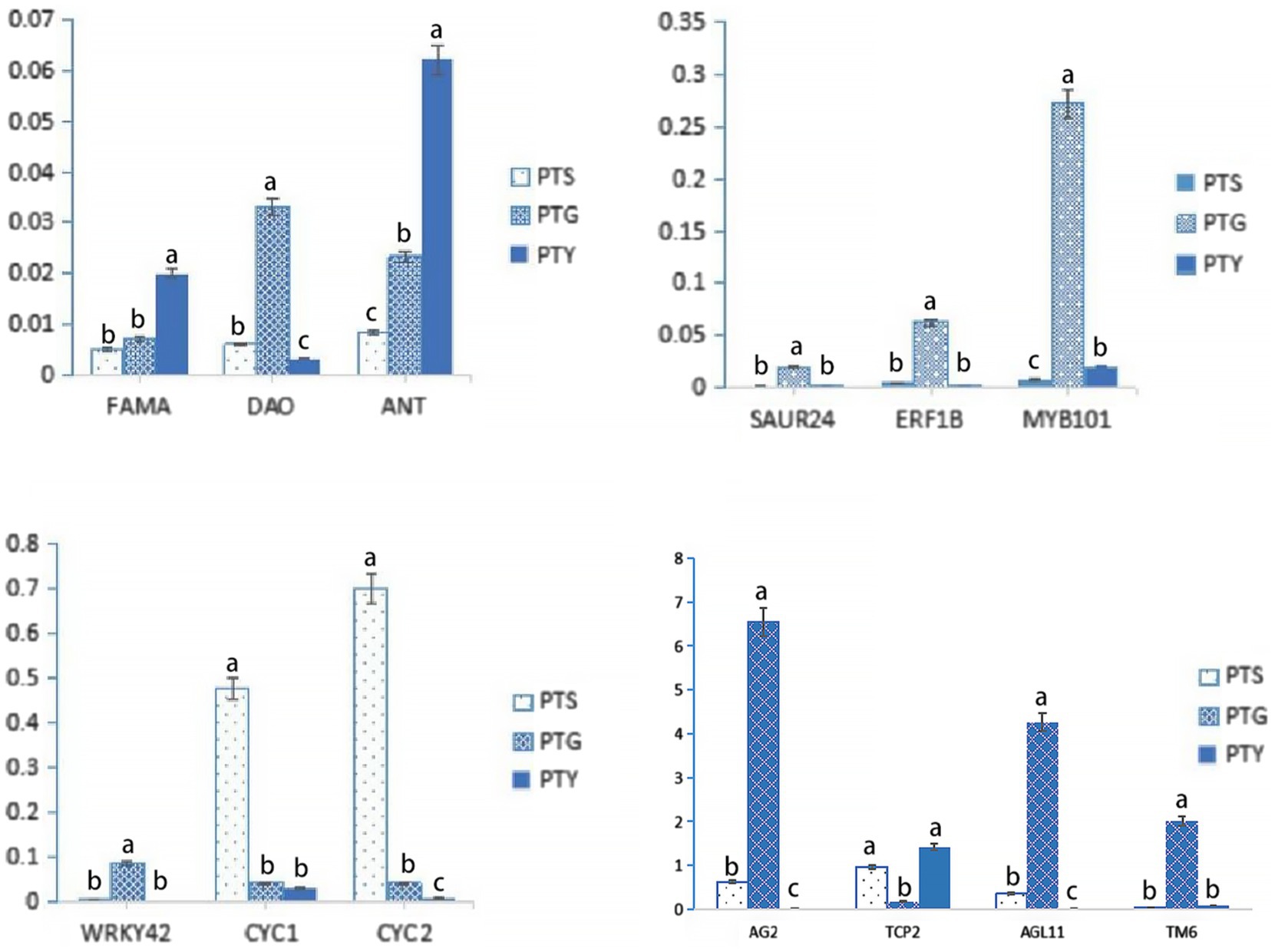
Figure 7. Relative expression levels of 13 floral development-related genes identified in the transcriptome data. Bars indicate standard error. The levels of each gene from the different samples (PTS, ray florets; PTG, disc florets; PTY, leaves) that are marked with the same letter were not significantly different.
Analysis of the CmTM6-mu sequence
In the transcription database, a PTG-specific expression gene, which was annotated as GDEF and belongs to Class B floral homeotic MADS-box TF family, attracted our attention. This may be a key function gene related to disc florets development. This gene was isolated from C. morifolium via RT-PCR using primers designed on the basis of the transcription data. The amplified gene was 690 bp in length and was predicted to encode a protein comprising 229 amino acid residues. The coding product shared a high similarity (96.79%) with Artemisia annua GDEF1 protein (GenBank No. PWA78933) as determined by a BLAST search of the NCBI database. In the phylogenetic tree constructed using AP3 and PI proteins from different plants, the protein encoded by this gene clustered with TM6 lineage proteins (Figure 8; Supplementary File 5). A sequence alignment revealed that the encoded protein was highly similar to AP3-type proteins from other plants (Tsaftaris et al., 2006; Ackerman et al., 2008; Zhang et al., 2011), but the paleoAP3 motif typical of TM6 proteins was not present in the C-terminal region. A single-base deletion (one A base missing) was discovered at the 3′ end (Supplementary File 4), which resulted in a frameshift in the ORF that caused the final 70 amino acid residues at the C-terminal to be replaced with a different 73-amino acid sequence. Hence, this gene was designated as CmTM6-mu. The full CmTM6 gene (i.e., without the deletion) encodes a protein with the expected C-terminal paleoAP3 motif (Figure 9).

Figure 8. Phylogenetic tree with TM6/AP3 lineage and PI/GLO lineage proteins.

Table 5. Specific primers for TM6 gene cloning.

Figure 9. Alignment of the protein sequences encoded by TM6 homologs in different organisms.
Ectopic expression of CmTM6-mu in Arabidopsis thaliana induces early flowering
The 35S:CmTM6-mu construct was inserted into A. thaliana via an Agrobacterium tumefaciens-mediated transformation method (Clough and Bent, 2010). We obtained 34 independent transgenic lines of 35S:CmTM6-mu, and nine transgenic lines were randomly selected for further analysis. Three weeks after sowing, the transgene expression levels in leaves of T3 generation Arabidopsis plants were analyzed by qRT-PCR. In all the tested transgenic lines, the TM6-mu genes were highly expressed; the highest TM6-mu expression level was detected in line #18 (Figures 10A,B). No distinguishable differences were found between transgenic Arabidopsis and wild-type Arabidopsis (Col-0) plants by morphological observation. However, seven of the nine transgenic lines produced earlier flowers than Col-0 Arabidopsis did (Figures 10C,D). The 35S:CmTM6-mu transgenic Arabidopsis lines began to bolt at ~23 days after sowing, whereas the wild-type Col-0 plants started to bolt at ~30 days after sowing, indicating that the transgenic lines flowered 5–7 days earlier than the wild-type plants (Figures 10C,D). The numbers of rosette leaves on the transgenic lines were lower (7 ± 1) than the numbers on the wild-type plants (11 ± 1). These results imply that TM6-mu had no effect on petal and stamen development, but affecting the flowering time in the transgenic Arabidopsis plants.
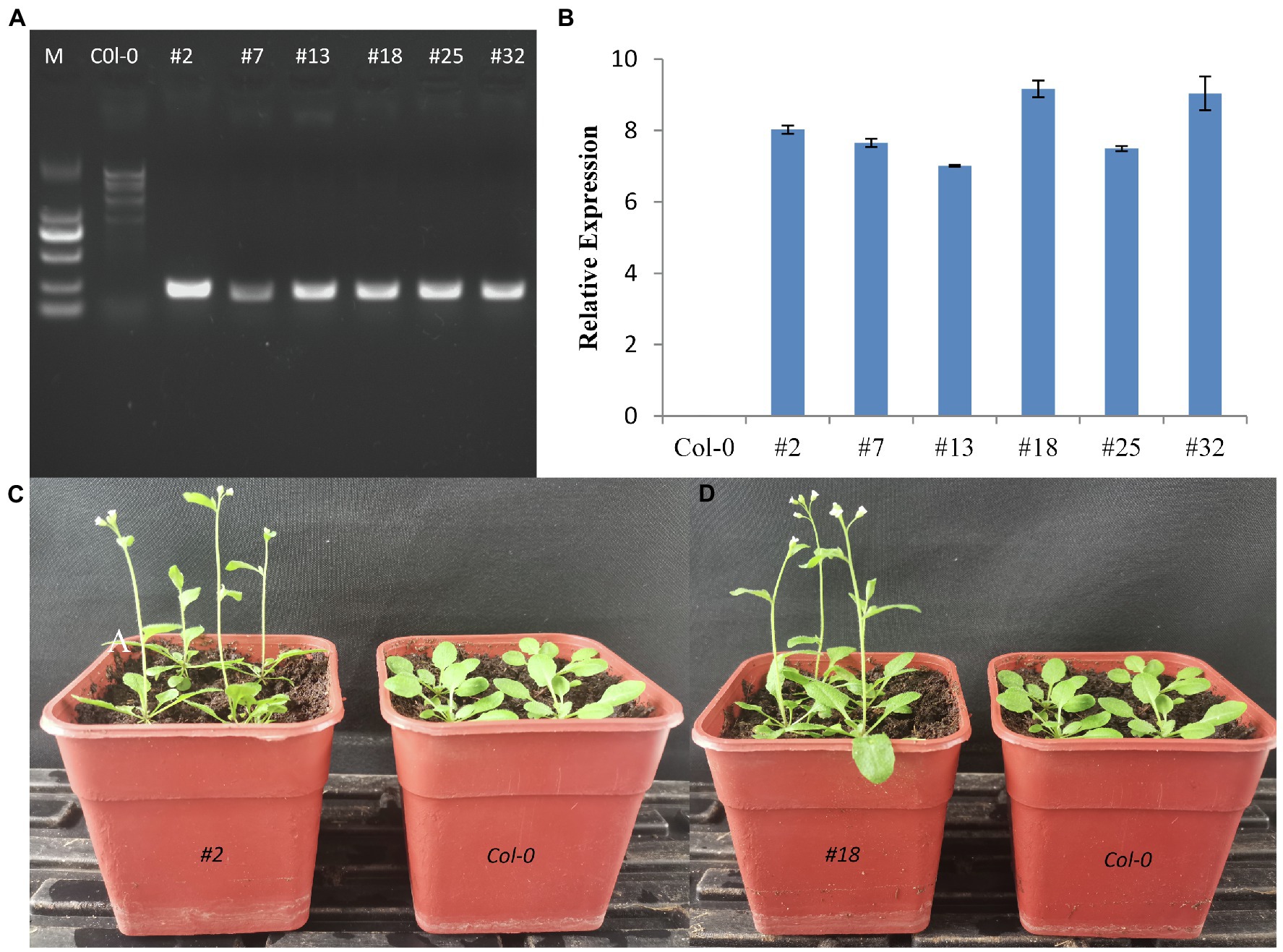
Figure 10. Effect of the ectopic expression of CmTM6-mu in Arabidopsis thaliana. (A) Detection of transgenic A. thaliana by PCR. (B) Relative gene expression levels in the transgenic Arabidopsis lines. (C,D) Phenotypes of wild-type Col-0 plants and Arabidopsis lines #2 and #18 overexpressing CmTM6-mu. respectively.
Discussion
Many important TFs (e.g., TCP, MYB, and MADS) that were differentially expressed between ray and disc florets may be important candidate genes for the floral development regulation mechanism in chrysanthemum
The whole transcriptome analysis of the DEGs between the ray and disc florets of chrysanthemum reflected the complexity of the regulatory machinery underlying the development of the capitula. The CYC and MADS TF families, as well as TFs from other families, have been shown to have key regulatory functions that influence floral development (Kaufmann et al., 2009; Hileman, 2014; Li et al., 2021). In this study, many floral development TF genes from the TCP, MYB, MADS, and other TF families (e.g., AG2, TCP2, CYC1, CYC2, ANT) were differentially expressed between the two floret types.
Earlier research on CYC genes in the model plant snapdragon proved the importance of these genes as regulators of floral symmetry (Clark and Coen, 2002; Galego and Almeida, 2002). The CYC genes also have crucial functions in sunflower, as well as in Gerbera and Senecio species (Burke, 2008; Kim et al., 2008; Chapman et al., 2012; Juntheikki-Palovaara et al., 2014). Moreover, studies on sunflower and Gerbera species, as well as chamomile, proved that CYC genes help regulate the formation of the two floret types in Compositae species (Broholm et al., 2008; Fambrini et al., 2011; Mizzotti et al., 2015; Wen et al., 2019). In our study, the qRT-PCR data were consistent with the FPKM values from the transcriptome data analysis, which confirmed that CYC1 and CYC2 were highly expressed in ray florets and lowly expressed in disc florets (Figure 7; Table 3). Thus, CYC genes are excellent candidates for studying chrysanthemum floral development.
In addition to CYC genes, floral organ-determining genes (e.g., DIV, MYB, and AG2 genes) were differentially expressed in the two floret types. MYB101, AG2, and AGL11 were highly expressed in disc florets, whereas TCP2 was highly expressed in ray florets (Figure 7; Table 3). Most TCP family genes (e.g., TCP2) and CYC genes (CYC1, CYC2) were significantly more highly expressed in the ray florets than they were in the disc florets, whereas the AG family genes (AG2, AGL11) were significantly more highly expressed in disc florets than they were in ray florets. Besides, some AP, MYB, DIV, and MADS family genes were more highly expressed in ray florets than in disc florets, whereas others were more highly expressed in disc florets. These genes likely play important roles in chrysanthemum floret development, and therefore changes in their expression levels may cause meristems to differentiate in different directions. These genes need to be investigated more thoroughly to confirm their effects on chrysanthemum floret development.
Many genes involved in flowering regulatory signaling pathways were identified in chrysanthemum, among them CmTM6 was specifically expressed in disc florets
Flowering is a complex process that is controlled by environmental conditions and developmental regulation. Several flowering regulatory signaling pathways have been identified in A. thaliana, including the ambient temperature, vernalization, autonomous, photoperiod, GA, and age pathways (Sumitomo et al., 2009; Fornara et al., 2010; Gao et al., 2017; Wei et al., 2017; Oda et al., 2020). These different signaling pathways converge on important cross-regulatory genes, such as FLC, SOC1, FT, CO, and LFY. We outlined the gene regulatory networks involved in these pathways (Figure 6A) and identified homologs of the key regulators in chrysanthemum (Supplementary File 6). FLC encodes a MADS-box TF that inhibits flowering, and its expression suppressed flowering in the ambient temperature, autonomous, and vernalization pathways (Fornara et al., 2010; Liu et al., 2016a). We identified two homologs of FLC (CmFLC1 and CmFLC2) in chrysanthemum. FLC inhibits flowering by repressing the expression of SOC1 and FT, which are the early targets of CO, a key regulatory gene in the photoperiod pathway. LFY is a downstream regulatory gene of SOC1 and a key regulator in the specification of floral meristem identity. LFY expression leads to a cascade of transcriptional activities that sustain indeterminate growth of the inflorescence meristem (Irish, 2010). On the basis of the protein annotation of the unigenes, we identified homologs of SOC1 (CmSOC11, CmSOC12, CmSOC13), FT (CmFT), CO (CmCOL91, CmCOL21, CmCOL22, CmCOL1, CmCOL92, CmCOL51, CmCOL93, CmCOL23, CmCOL94, CmCOL52), and LFY (CmLFY).
Plant hormones (e.g., IAA, CTK, GA, ABA) are important endogenous signal participants that play important roles in the flowering process. The mechanisms of hormones in photoperiod and epigenetic regulation have been elucidated (Oda et al., 2017). Synergistic and antagonistic effects of various hormones have been found, and various hormones have been shown to be involved in flower formation regulation pathways mediated by DELLA proteins in the GA pathway (Sumitomo et al., 2009; Gao et al., 2017; Wei et al., 2017). In this study, we identified three homologs of GID1 (CmGID1B1, CmGID1B2, CmGID1A) and 15 homologs of DELLA (e.g., CmGAI1, CmGAI, CmRGL3) in the GA pathway. IAA was the first plant hormone to be identified, and its involvement in the elongation and differentiation of plant cells, seed development, lateral root formation, root and leaf growth, development, and other physiological processes has been widely reported (Dinesh et al., 2016). IAA participates in GA biosynthesis and signal transduction by inhibiting the expression of DELLA (Fu and Harberd, 2003). We identified 91 homologs of IAA (e.g., CmARF3, CmSAUR50, CmIAA1) in flowering regulatory pathways. CTK regulates the division and differentiation of floral meristem cells, down-regulates miR172 expression levels, and promotes AP2 expression (Aukerman and Sakai, 2003; Meijón et al., 2011); however, the mechanism of CTK in floral regulation has not been fully explained. We identified 16 homologs of CTK (e.g., CmLOG5, CmCKX1, CmCKX3) were found in chrysanthemum. Riboni et al. found that ABA activates the expression of FT and TSF to promote Arabidopsis flower bud differentiation (Riboni et al., 2013, 2016). In the ABA signaling network, ABA activates the ABA-response element binding protein (AREB) by promoting CO transcription, ABA insensitive 3 (ABI3) was ubiquitinated by ABA, ABI3-binding CO protein was released to promote flower formation (Zhang et al., 2005; Riboni et al., 2013, 2016). In this study, we identified 11 homologs of ABA (e.g., CmCAR4, CmCAR4, CmAIB) in chrysanthemum.
The Class A, B, C, and E genes specify flower organ identity. Class A genes include AP1 and AP2 (Mahajan and Yadav, 2014; Sasaki et al., 2014; Liu et al., 2016a; Prunet et al., 2017). LFY was shown to directly activate AP1 transcription of Wagner et al. (1999). We identified four homologs of AP1 (CmAP11, CmAP12, CmAP13, CmAP14) and four homologs of AP2 (CmAP21, CmAP22, CmAP23, CmAP24). In most higher plants, Class B genes are divided into three lineages: PI, euAP3, and TM6. (Vandenbussche et al., 2003; Kramer et al., 2006; Ackerman et al., 2008). In this study, several Class B MADS-box genes were identified, and one gene (Cse_sc000306.1) was specifically expressed in disc florets (Table 3; Figure 7). This Class B MADS-box gene was identified as a homolog of the TM6 lineage gene based on the encoded protein sequence and the evolutionary tree that contained all the functional Class B MADS-box genes (Figures 7, 8). In plants, the PI/GLO and AP3/DEF/TM6 genes (i.e., Class B genes) control petal and stamen development (Kramer et al., 1998; Vandenbussche et al., 2003; Zhang et al., 2011). PI/GLO and AP3/DEF/TM6 genes form separate lineages that arose from a duplication event that occurred ~260 million years ago (231–290 million years ago; Kim et al., 2004). Following the divergence of the AP3/DEF/TM6 and PI/GLO lineages, many other duplication events occurred, including one that may be related to the separation of the AP3 lineage from the ancestral TM6 lineage in higher eudicots (Kramer et al., 2006; Broholm et al., 2008). In the C-termini of proteins encoded by the AP3 lineage genes, the paleoAP3 motif has been replaced by the euAP3 motif (Martino et al., 2006; Rijpkema et al., 2006; Cao et al., 2019).
There has been substantial research on the derivation of the euAP3 gene from the paleoAP3 gene, as well as on the mechanism underlying gene functions and expression patterns (Kramer et al., 1998, 2006; Vandenbussche et al., 2003; Zhang et al., 2011). Initially, a new copy of the paleoAP3 gene was produced via a duplication event, after which a frameshift due to a mutation resulted in the paleoAP3 motif at the C-terminus of the encoded protein being replaced by a novel euAP3 motif (Kramer et al., 1998; Zhang et al., 2011). This motif change coincided with the functional differentiation between the proteins (Kramer et al., 1998, 2006). During or after the duplication event that resulted in the euAP3 and TM6 lineages, an important change in the regulatory elements modulated the expression patterns of these genes (Kramer et al., 1998, 2006; Vandenbussche et al., 2003; Zhang et al., 2011). Finally, new gene lineages that varied in terms of functions and expression patterns were created (Kramer et al., 1998, 2006; Vandenbussche et al., 2003; Zhang et al., 2011). Along with gene duplications, gene losses also occurred in some plants, which included the loss of the TM6 lineage genes in A. majus and A. thaliana (Lamb and Irish, 2003; Rijpkema et al., 2006). However, AP3 and TM6 coexist in most higher eudicots (Vandenbussche et al., 2003; Kramer et al., 2006; Tsaftaris et al., 2006; Ackerman et al., 2008). AP3 and TM6 genes have been isolated from plants in the Asteraceae family, such as Gerbera hybrida and sunflower (Helianthus annuus; Shulga et al., 2008; Broholm et al., 2010). Two euAP3 type homologous genes, CMD19 and CDM115, were isolated from chrysanthemum (Shchennikova et al., 2004, 2018), and in this study, we isolated a TM6 type gene from chrysanthemum for the first time.
The euAP3 genes, such as AP3 in A. thaliana and DEF in A. majus, determine petal and stamen development, whereas paleoAP3 lineage genes (TM6) affect only stamen development (Martino et al., 2006; Rijpkema et al., 2006; Cao et al., 2019). In petunia, four Class B genes have been found, including euAP3 lineage PhDEF and paleoAP3 lineage PhTM6 (Rijpkema et al., 2006); PhDEF expression occurred in whorls 2 and 3, whereas PhTM6 expression occurred mainly in whorls 3 and 4 (Rijpkema et al., 2006). Heterologous paleoAP3 genes were able to rescue stamen development in the Arabidopsis ap3 mutant that had both petal and stamen abnormalities (Fang et al., 2014; Jing et al., 2014), which is consistent with the observation that paleoAP3 only affects stamen development. However, overexpression of heterologous paleoAP3 genes in WT model plants seemed to have no phenotypic effects (Zhang et al., 2011; Ai et al., 2017), possibly because there are redundant functional genes in the receiving plants (Fang et al., 2014; Ai et al., 2017). In this study, the TM6 gene in chrysanthemum was specially expressed in disc florets, and its expression was not detected in ray florets and leaves (Table 3; Figure 7). Besides the difference in symmetry, the most significant difference between ray and disc florets is that ray florets have no stamen, whereas disc florets have normal functional stamen. The gene expression patterns that we found in chrysanthemum flowers combined with similar findings reported previously in other plants, support the hypothesis that TM6 acts as the trigger for stamen morphogenesis in chrysanthemum. The absence of stamen in ray florets, which are located in the outer side of the capitulum of chrysanthemum, may be linked to the non-expression of TM6 in ray florets. However, to understand why the expression of the TM6 gene is different between ray and disc florets requires further studies into the complex regulatory network that is involved.
Frameshift in the chrysanthemum TM6 gene regulated flowering time changes in Arabidopsis, indicating that CmTM6-mu, which has a new C-terminal motif, may have novel functions in the regulation of flowering time
We isolated a non-typical TM6 lineage gene from C. morifolium flowers and named it CmTM6-mu. Phylogenetic analysis revealed that the encoded CmTM6-mu protein shared high similarity with TM6 proteins from other plants (Figure 8). The CmTM6-mu protein contained the same conserved MADS (M), intervening (I), and keratin-like (K) domains as other TM6 proteins, but had a completely different C-terminal that lacked the characteristic paleoAP3 motif found in other TM6 proteins (Figure 9). Further analyses detected a single-base deletion that cause a frameshift that changed the C-terminal sequence of the encoded CmTM6-mu protein (Supplementary File 4). Because AP3 and TM6 proteins have the same origin they share high similarity; both of them have conserved M, I, and K domains and variable C-terminals. All the conserved domains are necessary for their function. The M domain is highly conserved among the AP3, TM6, PI, and GLO proteins, and is important for binding to DNA by recognizing CArG sites, as well as acting as a mediator in protein dimerization (Pellegrini et al., 1995; Shore and Sharrocks, 1995; Hassler and Richmond, 2001). The function of the K domain is still unclear. In Arabidopsis, the K domain may mediate the strength and specificity of AP3/PI heterodimerization (Yang et al., 2003). The I domain is located between the M and K domains, and varies in length. The I domain is also necessary for DNA-binding and interactions with other proteins (i.e., heterodimer formation; Riechmann and Meyerowitz, 1997; Yang et al., 2003). The function of the C-terminal of AP3 is also unclear. Analyses of the phenotypes that resulted from ectopic gene expression and rescue experiments involving truncated proteins (i.e., removal of characteristic C-terminal motifs) showed that the C-terminal AtAP3 motif conferred AP3 functionality on the heterologous PI protein (Lamb and Irish, 2003). The C-terminal region is the only significantly different region between AP3 and TM6, which suggests the C-terminus may be responsible for the functional diversity between these proteins (Riechmann and Meyerowitz, 1997; Yang et al., 2003; Tzeng et al., 2004). The conserved M, I, and K domains of CmTM6-mu indicate that it may still have DNA-binding ability and the ability to interact with other proteins. The functions of these domains have not been studied in chrysanthemum, but similarities between the heterologous proteins AP3 (CMD115) and PI (CMD86) have been reported (Shchennikova et al., 2018).
Studies have shown that AP3 functions as a petal and stamen determiner, whereas TM6 only affects stamen development (Fang et al., 2014; Jing et al., 2014). We found that CmTM6 was highly expressed in disc florets (with normal stamen), whereas its expression was almost undetectable in ray florets (stamens abortion) and in leaves, indicating that CmTM6 may be involved in stamen development. However, we do not know if the function of CmTM6-mu, which contains the conserved domains but has a mutation in the C-terminal, has changed. When the 35S:CmTM6-mu construct was transformed into Arabidopsis, an interesting and unexpected floral organ phenotype was observed in the transgenic plants, and earlier flowering occurred in almost all the transgenic lines. Similar results have been reported in other studies. An et al. found that ectopic expression in tobacco of a poplar PtAP3 gene that had a non-conserved paleoAP3 motif caused early flowering (An et al., 2011). Ectopic expression in Arabidopsis of lily LMADS1, a paleoAP3 type gene that shares high homology with AP3 lineage proteins in monocots, was also shown to cause early flowering (Tzeng and Yang, 2001). In chrysanthemum, ectopic expression of a sunflower TM6 lineage gene, HAM91, led to later flowering than in the WT (Shchennikova et al., 2018). Although, the mechanism of florescence change caused by TM6 is not clear, this finding still provides a new insight into the mechanism of flowering regulation in plants that can be clarified in future studies. Given the way that euAP3 originated from paleoAP3, it is possible that CmTM6-mu with the new C-terminal motif may have novel functions related to the regulation of flowering time. Many more studies are needed to confirm these findings.
Conclusion
To screen for key genes that regulate the differentiation of the two types of chrysanthemum florets, we used leaves as a control for a transcriptome sequencing analysis of ray and disc florets. The generated data were analyzed to identify DEGs related to floret differentiation and development. The expression levels and patterns of the DEGs were verified by qRT-PCR analysis. The data presented herein suggest that CYC and MADS genes are involved in regulating the differentiation of the two types of chrysanthemum florets. Important regulatory genes and signaling pathways involved in flowering control were identified. Among them, a TM6 gene (CmTM6-mu) that was specifically expressed in disc florets was found to be highly similar to other typical TM6 lineage genes. Interestingly, a single-base deletion at the 3′ end of the TM6 ORF resulted in a frame shift that caused the paleoAP3 motif to be missing at the C terminus of the encoded protein. Overexpression of CmTM6-mu in Arabidopsis led to earlier flowering of the transgenic lines, which indicated that CmTM6-mu with the new C-terminal motif may have novel functions in the regulation of flowering time. The results of this study provide valuable genomic information and candidate genes that will be useful for studies of flowering molecular mechanisms and for the breeding of novel flower types in chrysanthemums.
Materials and methods
Plant materials and RNA extraction
The ground-cover chrysanthemum variety C. morifolium Ramat “Pink Carpet” grown at the Shunyi Base of the Biotechnology Research Center of the Beijing Academy of Agriculture and Forestry Sciences was used as the study material. Ray and disc florets of the capitula, as well as fully extended leaves of the “Pink Carpet” cultivar were collected during the full-bloom stage (Figures 1A–C), with three biological replicates for each sample type. The collected samples were immediately frozen in liquid nitrogen and transported to the laboratory for storage at −80°C. Total RNA was extracted from the frozen materials using the MiniBEST Plant RNA Extraction Kit (TaKaRa, Dalian, China). The NanoDrop ND2000 spectrophotometer (Thermo Fisher Scientific, Barrington, IL, United States) was used to quantify the extracted RNA and assess RNA quality.
Library construction and transcriptome sequencing
The extracted RNA was treated with DNase to eliminate any residual genomic DNA, after which magnetic beads with oligo-(dT) were used to enrich the eukaryotic mRNA. The mRNA was broken into short fragments. The disrupted mRNA and random hexamer primers were used to synthesize first-strand cDNA. After synthesizing the second cDNA strand, the double-stranded cDNA was purified using a commercial kit (Promega, Madison, WI, United States). The purified cDNA underwent an end-repair step before a poly-A tail and a sequencing adapter were added. Following a size selection step, the sequences were amplified by PCR with 2 x Phanta Max Master Mix (Vazyme, Nanjing, China). The libraries were qualitatively analyzed using the Agilent 2,100 Bioanalyzer (Agilent, Santa Clara, CA, United States) before they were sequenced using the Illumina HiSeq™ 2,500 system (Illumina, San Diego, CA, United States), which generated 125 or 150 bp paired-end reads.
Data analysis
The image data obtained from the Illumina sequencer were used to generate raw data (raw reads) after base calling. The FastQC program1 was used to analyze the base quality of the raw data, and the results were visualized using R statistical software.2 Low-quality sequences were eliminated and Trimmomatic was used to remove the linker sequence (Bolger et al., 2014). The reads in which the initial sequence had a base quality lower than 3 and the terminal sequence had a base quality lower than 20 were removed to obtain high-quality reads (clean reads). The retained sequencing data of all the samples were combined, and the pre-processing quality and base distribution statistics were recorded. The HISAT2 program (Kim et al., 2015) was used to align the clean reads to the C. seticuspe reference genome to obtain information regarding the positions of reads in the reference genome or genes, as well as sequence information unique to the analyzed chrysanthemum materials. Genome comparison results were used to quantify genes. Additionally, the FPKM method (Roberts et al., 2011) was used to analyze the expression of protein-coding genes. The number of reads (Mortazavi et al., 2008), the accuracy of the FPKM calculations, and the correlations between the sequencing results for the different samples, especially the biological replicates, were analyzed to ensure the sequencing data were reliable. The correlations between the protein-coding gene expression levels of the samples were also determined. Correlation coefficients close to 1 were indicative of highly similar expression patterns between samples. Genes differentially expressed between samples were identified using the DESeq software (Anders and Huber, 2010). The baseMean value was used to estimate expression levels. The fold-change in the expression levels was calculated, and the negative binomial distribution test was used to evaluate the significance of the change. When using RNA-seq data to determine whether a gene is differentially expressed in two samples, the following two factors are considered: the expression-level fold-change and the value of p or false discovery rate (FDR; i.e., adjusted value of p). To calculate the FDR, the value of p for the expression in each sample was determined, after which the FDR error control method was used for the multiple hypothesis test correction of the value of ps. The identified DEGs were functionally characterized on the basis of a GO enrichment analysis. Additionally, the KEGG database was used to determine the enriched metabolic pathways associated with the DEGs.
Analysis of differentially expressed genes
The DEGs were screened according to the FPKM values for the unigenes in each sample. The DEGs were identified on the basis of an FPKM ratio between samples that was >2 or <0.5. To control false positives, Benjamini and Hochberg FDR multiple check values were used. The corrected value of p was <0.05. The ray florets and disc florets were compared to screen for DEGs between the two floret types.
Gene expression analysis via qRT-PCR
Total RNA was extracted from the ray florets, disc florets, and leaves as described above. Total RNA was treated with DNase (Promega, Madison, WI, United States) and then reverse-transcribed to cDNA using a commercial reverse transcription system (Tiangen, China). The qRT-PCR analysis was performed using the PikoReal real-time PCR system (Thermo Fisher Scientific, Germany). Each reaction was completed in a total volume of 20 μl, which contained 2 μl first-strand cDNA as the template. The PCR program was as follows: 95°C for 30 s, 40 cycles of 95°C for 5 s and 60°C for 30 s. Details regarding the gene-specific qRT-PCR primers are provided in Table 4. The qRT-PCR experiments were conducted using three biological replicates. Each replicate was analyzed in triplicate. The relative expression levels were calculated according to the 2−ΔΔCt method. The C. morifolium gene encoding protein phosphatase 2A (PP2Ac) was used as the internal control reference gene to determine relative expression levels.
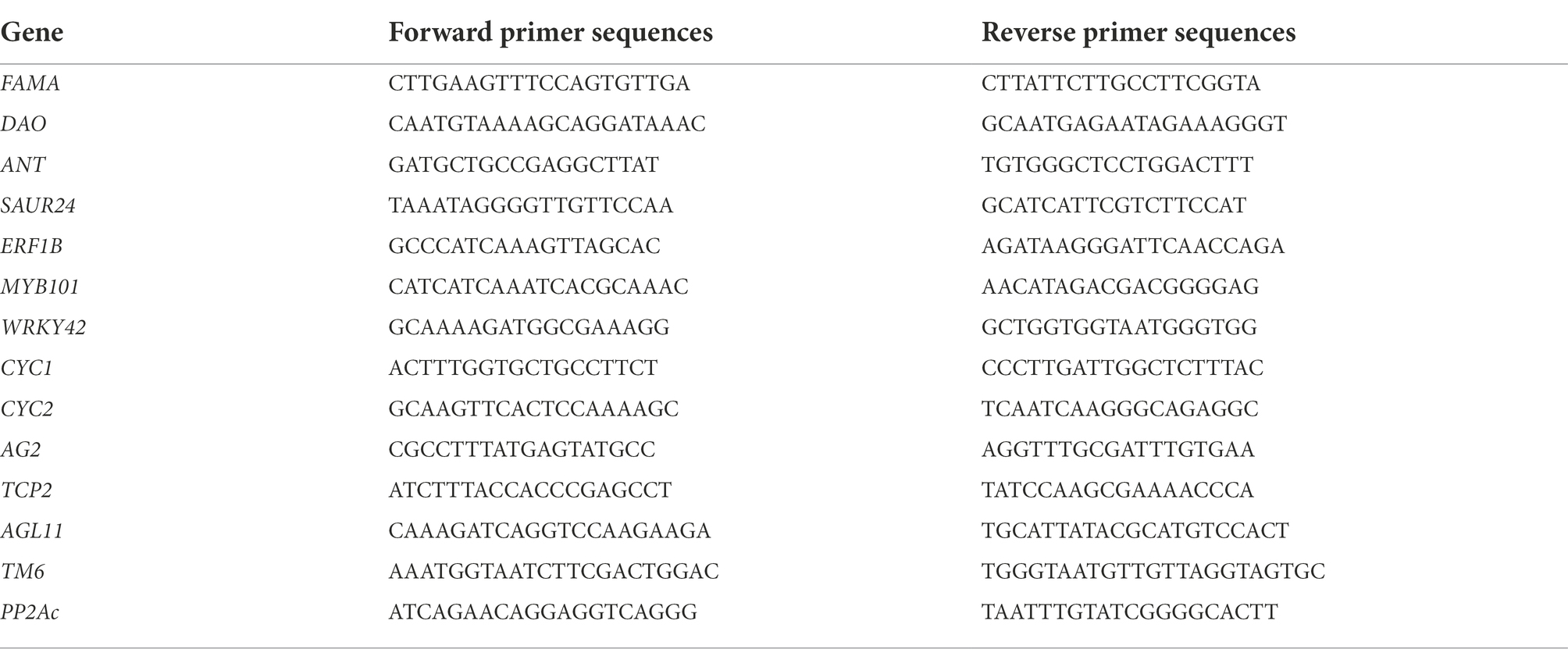
Table 4. Primers for the quantitative real-time PCR analysis.
Molecular cloning of CmTM6-mu
Total RNA was extracted from chrysanthemum flowers using the MiniBEST Plant RNA Extraction Kit (TaKaRa). First-strand cDNA was synthesized using a reverse transcription system (Promega). Primers specific for the TM6 ORF (Table 5) were designed on the basis of a sequence annotated as TM6 in the transcriptome data for chrysanthemum flowers. The PrimeSTAR HS DNA polymerase kit (TaKaRa) was used for amplifying the TM6 ORF. The PCR product was examined by 1% agarose gel electrophoresis. The amplified fragment purified from the agarose gel was inserted into the pGEM-T Easy vector (Promega) and sequenced by the Shanghai Sangon Company.
Sequence alignment and phylogenetic analysis
Sequences were aligned using the DNAStar 5.0 software3 in accordance with the ClustalW method. A phylogenetic tree was constructed using the MEGA program (version 6.0; Tamura et al., 2013). Specifically, the neighbor-joining method was used, with 1,000 bootstrap replicates. The sequences of TM6, PI, and AP3 homologs were obtained from the GenBank database4 (Supplementary File 5).
Construction of the overexpression vector and transformation of Arabidopsis thaliana
The full-length CmTM6-mu cDNA sequence was amplified by PCR using gene-specific primers (Table 5) and then inserted downstream of the 35S promoter in the pCAMBIA1301 vector digested with HindIII and XbaI. The 35S:CmTM6-mu construct in the recombinant plasmid was sequenced to confirm its accuracy before it was introduced into A. tumefaciens strain EHA105 cells via electroporation. The A. tumefaciens-mediated genetic transformation of A. thaliana Col-0 was performed in accordance with a published floral-dip method (Clough and Bent, 2010). The putative transgenic lines were selected on MS medium containing 40 mg/l hygromycin B. The hygromycin B-resistant seedlings (T0 generation) were transferred to soil and grown in a greenhouse at 22°C, with 70% relative humidity and a 16 h light/8 h dark cycle. The presence of the transgene in the transgenic Arabidopsis plants was confirmed by qRT-PCR using the TM6 primers list in Table 4. Phenotypic analyses were performed using the independent T3 transgenic lines. Three biological replicates were used for both the qRT-PCR and phenotypic analyses.
Data availability statement
The data presented in the study are deposited in the repositories of NCBI Sequence Read Archine with BioProject accession number: PRJNA821995 and SRA accession number: SRR18585549–SRR18585557. We confirm the data had been released on 2022-04-11.
Author contributions
YJ, YC, SW, HC, XZ, SG, DC, and HL conducted the experiments. YC and SG analyzed the data and prepared the manuscript. DC and HL reviewed the manuscript. CH provided assistance. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the Youth Fund of Beijing Academy of Agriculture and Forestry Sciences (QNJJ202010), National Natural Science Foundation of China (31901354), the Innovation Foundation of the Beijing Academy of Agriculture and Forestry Sciences (KJCX20200205), and Study on breeding technology and ecological adaptability of Lily for both ornament and food (KM202112448004). These funding bodies did not participate in the design of the study, collection, analysis, and interpretation of data or writing the manuscript.
Acknowledgments
We thank Liwen Bianji (Edanz; www.liwenbianji.cn/) for editing the English text of a draft of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls. 2022.947331/full#supplementary-material
Supplementary File 1 | Differentially expressed genes between disc florets and ray florets of Chrysanthemum morifolium.
Supplementary File 2 | GO categories enriched with expressed genes between disc florets and ray florets of Chrysanthemum morifolium.
Supplementary File 3 | KEGG pathways enriched with expressed genes between disc florets and ray florets of Chrysanthemum morifolium.
Supplementary File 4 | Sequence and mutation information of CmTM6 and CmTM6-mu.
Supplementary File 5 | Details of the proteins used to construct the phylogenetic tree.
Supplementary File 6 | Homologous genes in the flowering regulatory networks involved in the six major floral induction pathways of Chrysanthemum morifolium.
Abbreviations
DEG, Differentially expressed gene; FDR, False discovery rate; FPKM, Fragments per kilobase of transcript per million mapped reads; qRT-PCR, Quantitative real-time PCR; PCR, Polymerase chain reaction; TF, Transcription factor.
Footnotes
1. ^https://www.bioinformatics.babraham.ac.uk/
References
Ackerman, C. M., Yu, Q., Kim, S., Paull, R. E., and Ming, M. R. (2008). B-class MADS-box genes in trioecious papaya: two paleoAP3 paralogs, CpTM6-1 and CpTM6-2, and a PI ortholog CpPI. Planta 227, 741–753. doi: 10.1007/s00425-007-0653-5
Ai, Y., Zhang, C., Sun, Y., Wang, W., and Bao, M. (2017). Characterization and functional analysis of five MADS-box b class genes related to floral organ identification in Tagetes erecta. PLoS One 12:e169777. doi: 10.1371/journal.pone.0169777
Aida, R., Komano, M., Saito, M., Nakase, K., and Murai, K. (2008). Chrysanthemum flower shape modification by suppression of chrysanthemum-AGAMOUS gene. Plant Biotechnol. Nar. 25, 55–59. doi: 10.5511/plantbiotechnology.25.55
An, X., Ye, M., Wang, D., Wang, Z., Cao, G., Zheng, H., et al. (2011). Ectopic expression of a poplar APETALA3-like gene in tobacco causes early flowering and fast growth. Biotechnol. Lett. 33, 1239–1247. doi: 10.1007/s10529-011-0545-4
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genmome Biol. 11:R106. doi: 10.1186/gb-2010-11-10-r106
Aukerman, M. J., and Sakai, H. (2003). Regulation of flowering time and floral organ identity by a microRNA and its APETALA2-like target genes. Plant Cell 15, 2730–2741.
Becker, A., and Theissen, G. (2003). The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol. Phylogenet. Evol. 29, 464–489. doi: 10.1016/S1055-7903(03)00207-0
Berger, B. A., Thompson, V., Lim, A., Ricigliano, V., and Howarth, D. G. (2016). Elaboration of bilateral symmetry across Knautia macedonica capitula related to changes in ventral petal expression of CYCLOIDEA-like genes. EvoDevo 7, 1–10. doi: 10.1186/s13227-016-0045-7
Bolger, A. M., Marc, L., and Bjoern, U. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Broholm, S. K., Pöllänen, E., Ruokolainen, S., Tähtiharju, S., Kotilainen, M., Albert, V. A., et al. (2010). Functional characterization of B class MADS-box transcription factors in Gerbera hybrida. J. Exp. Bot. 61, 75–85. doi: 10.1093/jxb/erp279
Broholm, S. K., Tahtiharju, S., Laitinen, R. A. E., Albert, V. A., Teeri, T. H., and Elomaa, P. (2008). A TCP domain transcription factor controls flower type specification along the radial axis of the Gerbera (Asteraceae) inflorescence. P. Natl. Acad. Sci. U. S. A. 105, 9117–9122. doi: 10.1073/pnas.0801359105
Burke, M. A. C. J. (2008). Positive selection and expression divergence following gene duplication in the sunflower CYCLOIDEA gene family. Mol. Biol. Evol. 25, 1260–1273. doi: 10.1093/molbev/msn001
Cao, X., Liu, X., Wang, X., Yang, M., Giang, T. V., Wang, J., et al. (2019). B-class MADS-box TM6 is a candidate gene for tomato male sterile-1526. Theor. Appl. Genet. 132, 2125–2135. doi: 10.1007/s00122-019-03342-z
Chapman, M. A., Tang, S., Draeger, D., Nambeesan, S., Shaffer, H., Barb, J. G., et al. (2012). Genetic analysis of floral symmetry in van gogh’s sunflowers reveals independent recruitment of CYCLOIDEA genes in the asteraceae. PLoS Genet. 8:e1002628. doi: 10.1371/journal.pgen.1002628
Clark, J. I., and Coen, E. S. (2002). The cycloidea gene can respond to a common dorsoventral prepattern in Antirrhinum. Plant J. 30, 639–648. doi: 10.1046/j.1365-313X.2002.01310.x
Clough, S. J., and Bent, A. F. (2010). Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 16, 735–743. doi: 10.1046/j.1365-313x.1998.00343.x
Dinesh, D. C., Villalobos, L. I. A. C., and Abel, S. (2016). Structural biology of nuclear auxin action. Trends Plant Sci. 21, 302–316. doi: 10.1016/j.tplants.2015.10.019
Domingos, S., Fino, J., Paulo, O. S., Oliveira, C. M., and Goulao, L. F. (2016). Molecular candidates for early-stage flower-to-fruit transition in stenospermocarpic table grape (Vitis vinifera L.) inflorescences ascribed by differential transcriptome and metabolome profiles. Plant Sci. 244, 40–56. doi: 10.1016/j.plantsci.2015.12.009
Dornelas, M. C., Patreze, C. M., Angenent, G. C., and Immink, R. (2011). MADS: the missing link between identity and growth? Trends Plant Sci. 16, 89–97. doi: 10.1016/j.tplants.2010.11.003
Eom, H., Park, S. J., Kim, M. K., Kim, H., and Lee, I. (2017). TAF15b, involved in the autonomous pathway for flowering, represses transcription of FLOWERING LOCUS C. Plant J. 93, 79–91. doi: 10.1111/tpj.13758
Fambrini, M., Salvini, M., and Pugliesi, C. (2011). A transposon-mediate inactivation of a CYCLOIDEA-like gene originates polysymmetric and androgynous ray flowers in Helianthus annuus. Genetica 139, 1521–1529. doi: 10.1007/s10709-012-9652-y
Fang, Z. W., Qi, R., Li, X. F., and Liu, Z. X. (2014). Ectopic expression of FaesAP3, a Fagopyrum esculentum (Polygonaceae) AP3 orthologous gene rescues stamen development in an Arabidopsis ap3 mutant. Gene 550, 200–206. doi: 10.1016/j.gene.2014.08.029
Fornara, F., de Montaigu, A., and Coupland, G. (2010). SnapShot: control of flowering in Arabidopsis. Cell 141:550. doi: 10.1016/j.cell.2010.04.024
Fu, X., and Harberd, N. P. (2003). Auxin promotes Arabidopsis root growth by modulating gibberellin response. Nature 421, 740–743. doi: 10.1038/nature01387
Fu, Q., Niu, L., Chen, M., Tao, Y., Wang, X., He, H., et al. (2018). De novo transcriptome assembly and comparative analysis between male and benzyladenine-induced female inflorescence buds of Plukenetia volubilis. J. Plant Physiol. 221, 107–118. doi: 10.1016/j.jplph.2017.12.006
Galego, L., and Almeida, J. (2002). Role of in the control of dorsoventral asymmetry in flowers. Genes Dev. 16, 880–891. doi: 10.1101/gad.221002
Gao, Y., Fan, M., Yuan, L., Wu, Z., and Zhang, Q. (2017). Overexpression of Chrysanthemum morifolium SVP gene delays blossoming and regulates inflorescence architecture in transgenic Arabidopsis. Can. J. Plant Sci. 97, 1130–1139.
Hara-Kitagawa, M., Unoki, Y., Hihara, S., and Oda, K. (2020). Development of simple PCR-based DNA marker for the red-fleshed trait of a blood peach ‘Tenshin-suimitsuto’. Mol. Breed. 40, 1166–1175. doi: 10.1007/s11032-019-1068-2
Hassler, M., and Richmond, T. J. (2001). The B-box dominates SAP-1-SRF interactions in the structure of the ternary complex. EMBO J. 20, 3018–3028. doi: 10.1093/emboj/20.12.3018
Higuchi, Y., Narumi, T., Oda, A., Nakano, Y., Sumitomo, K., and Fukai, S. (2013). The gated induction system of a systemic floral inhibitor, antiflorigen, determines obligate short-day flowering in chrysanthemums. Proc. Natl. Acad. Sci. 110, 17137–17142. doi: 10.1073/pnas.1307617110
Hileman, L. C. (2014). Bilateral flower symmetry−how, when and why? Curr. Opin. Plant Biol. 17, 146–152. doi: 10.1016/j.pbi.2013.12.002
Howarth, D. G., and Donoghue, M. J. (2006). Phylogenetic analysis of the “ECE” (CYC/TB1) clade reveals duplications predating the core eudicots. Proc. Natl. Acad. Sci. 103, 9101–9106. doi: 10.1073/pnas.0602827103
Huang, T., and Irish, V. (2015). Temporal control of plant organ growth by TCP transcription factors. Curr. Biol. 25, 1765–1770. doi: 10.1016/j.cub.2015.05.024
Huang, J., Lin, C., Cheng, T., Chang, B. C., and Cheng, S. (2015). A de novo floral transcriptome reveals clues into Phalaenopsis orchid flower development. PLoS One 10:e123474. doi: 10.1371/journal.pone.0123474
Irish, V. F. (2010). The flowering of Arabidopsis flower development. Plant J. Cell Mol. Biol. 61, 1014–1028. doi: 10.1111/j.1365-313X.2009.04065.x
Jabbour, F., Udron, M., Le Guilloux, M., Gonalves, B., and Domenica, M. (2015). Flower development schedule and AGAMOUS-like gene expression patterns in two morphs of Nigella damascena (Ranunculaceae) differing in floral architecture. Bot. J. Linn. Soc. 178, 608–619. doi: 10.1111/boj.12297
Jing, D., Liu, Z., Zhang, B., Ma, J., Han, Y., and Chen, F. (2014). Two ancestral APETALA3 homologs from the basal angiosperm Magnolia wufengensis (Magnoliaceae) can affect flower development of Arabidopsis. Gene 537, 100–107. doi: 10.1016/j.gene.2013.11.076
Johannesson, H., Wang, Y., and Engström, P. (2001). DNA-binding and dimerization preferences of Arabidopsis homeodomain-leucine zipper transcription factors invitro. Plant Mol. Biol. 45, 63–73. doi: 10.1023/A:1006423324025
Juárez-Corona, N., and Stefan, D. F. (2021). ANT and AIL6: masters of the master regulators during flower development. J. Exp. Bot. 72, 5263–5266. doi: 10.1093/jxb/erab235
Juntheikki-Palovaara, I., Tahtiharju, S., Lan, T., Broholm, S. K., Rijpkema, A. S., Ruonala, R., et al. (2014). Functional diversification of duplicated CYC2 clade genes in regulation of inflorescence development in Gerbera hybrida (Asteraceae). Plant J. 79, 783–796. doi: 10.1111/tpj.12583
Kaufmann, K., Muiño, J. M., Jauregui, R., Airoldi, C. A., Smaczniak, C., Krajewski, P., et al. (2009). Target genes of the MADS transcription factor SEPALLATA3: integration of developmental and hormonal pathways in the arabidopsis flower. PLoS Biol. 7:e1000090. doi: 10.1371/journal.pbio.1000090
Ke, Y., Zheng, Q., Yao, Y., Ou, Y., Chen, J., Wang, M., et al. (2021). Genome-wide identification of the MYB gene family in cymbidiumensifolium and its expression analysis in different flower colors. Int. J. Mol. Sci. 22:13245. doi: 10.3390/ijms222413245
Kim, M., Cui, M., Cubas, P., Gillies, A., Lee, K., Chapman, M. A., et al. (2008). Regulatory genes control a key morphological and ecological trait transferred between species. Science 322, 1116–1119. doi: 10.1126/science.1164371
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Kim, S., Yoo, M., Albert, V. A., Farris, J. S., Soltis, P. S., and Soltis, D. E. (2004). Phylogeny and diversification of B-function MADS-box genes in angiosperms: evolutionary and functional implications of a 260-million-year-old duplication. Am. J. Bot. 91, 2102–2118. doi: 10.3732/ajb.91.12.2102
Kramer, E. M., Dorit, R. L., and Irish, V. F. (1998). Molecular evolution of genes controlling petal and stamen development: duplication and divergence within the APETALA3 and PISTILLATA MADS-box gene lineages. Genetics 149, 765–783. doi: 10.1093/genetics/149.2.765
Kramer, E. M., Su, H. J., Wu, C. C., and Hu, J. M. (2006). A simplified explanation for the frameshift mutation that created a novel C-terminal motif in the APETALA3 gene lineage. BMC Evol. Biol. 6:30. doi: 10.1186/1471-2148-6-30
Lai, X., Arnaud, S., Jérémy, L., Véronique, H., Franois, P., and Chloe, Z. (2020). Genome-wide binding of SEPALLATA3 and AGAMOUS complexes determined by sequential DNA-affinity purification sequencing. Nucleic Acids Res. 48, 9637–9648. doi: 10.1093/nar/gkaa729
Lamb, R. S., and Irish, V. F. (2003). Functional divergence within the APETALA3/PISTILLATA floral homeotic gene lineages. P. Natl. Acad. Sci. U. S. A. 100, 6558–6563. doi: 10.1073/pnas.0631708100
Li, X., Zhang, G., Liang, Y., Hu, L., Zhu, B., Qi, D., et al. (2021). TCP7 interacts with nuclear factor-Ys to promote flowering by directly regulating SOC1 in Arabidopsis. Plant J. 108, 1493–1506. doi: 10.1111/tpj.15524
Liu, K., Feng, S., Pan, Y., Zhong, J., Chen, Y., Yuan, C., et al. (2016a). Transcriptome analysis and identification of genes associated with floral Transitionand flower development in sugar apple (Annona squamosa L.). Front. Plant Sci. 7:1695. doi: 10.3389/fpls.2016.01695
Liu, H., Luo, C., Chen, D., Wang, Y., and Huang, C. (2020). Whole-transcriptome analysis of differentially expressed genes in the mutant and normal capitula of Chrysanthemum morifolium. BMC Genomic Data. 22:2. doi: 10.21203/rs.3.rs-27505/v2
Liu, H., Sun, M., Du, D., Pan, H., Cheng, T., Wang, J., et al. (2015). Whole-Transcriptome analysis of differentially expressed genes in the vegetative buds, floral buds and buds of chrysanthemum morifolium. PLoS One 10:e128009. doi: 10.1371/journal.pone.0128009
Liu, H., Sun, M., Du, D., Pan, H., Cheng, T., Wang, J., et al. (2016b). Whole-transcriptome analysis of differentially expressed genes in the ray florets and disc florets of Chrysanthemum morifolium. BMC Genomics 17:398. doi: 10.1186/s12864-016-2733-z
Liu, F., Wang, Y., Ding, Z., Zhao, L., Xiao, J., Wang, L., et al. (2017). Transcriptomic analysis of flower development in tea (Camellia sinensis (L.)). Gene 631, 39–51. doi: 10.1016/j.gene.2017.08.013
Mahajan, M., and Yadav, S. K. (2014). Gain of function mutation in tobacco MADS box promoter switch on the expression of flowering class B genes converting sepals to petals. Mol. Biol. Rep. 41, 705–712. doi: 10.1007/s11033-013-2909-4
Martino, G. D., Pan, I., Emmanuel, E., and Irish, L. V. F. (2006). Functional analyses of two tomato APETALA3 genes demonstrate diversification in their roles in regulating floral development. Plant Cell 18, 1833–1845. doi: 10.1105/tpc.106.042978
Martín-Pizarro, C., Triviño, J. C., and Posé, D. (2018). Functional analysis of TM6 MADS-box gene in the Octoploid strawberry by CRISPR/Cas9 directed mutagenesis. J. Exp. Bot. 70, 885–895. doi: 10.1101/351296
Meijón, M., Cañal, M. J., Valledor, L., Rodríguez, R., and Feito, I. (2011). Epigenetic and physiological effects of gibberellin inhibitors and chemical pruners on the floral transition of azalea. Physiol. Plant. 141, 276–288. doi: 10.1111/j.1399-3054.2010.01430.x
Mizzotti, C., Fambrini, M., Caporali, E., Masiero, S., and Pugliesi, C. (2015). A CYCLOIDEA-like gene mutation in sunflower determines an unusual floret type able to produce filled achenes at the periphery of the pseudanthium. Botany 93, 171–181. doi: 10.1139/cjb-2014-0210
Mortazavi, A., Williams, B. A., Mccue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi: 10.1038/nmeth.1226
Niu, F., Cui, X., Zhao, P., Sun, M., and Jiang, Y. (2020). WRKY42 transcription factor positively regulates leaf senescence through modulating SA and ROS synthesis in Arabidopsis thaliana. Plant J. 104, 171–184. doi: 10.1111/tpj.14914
Niwa, T., Suzuki, T., Takebayashi, Y., Ishiguro, R., Higashiyama, T., Sakakibara, H., et al. (2018). Jasmonic acid facilitates flower opening and floral organ development through the upregulated expression of SlMYB21 transcription factor in tomato. Biosci. Biotechnol. Biochem. 82, 292–303. doi: 10.1080/09168451.2017.1422107
Novaković, J., Rajčević, N., Garcia-Jacas, N., Susanna, A., Marin, P. D., and Janaćković, P. (2019). Capitula essential oil composition of seven Centaurea species (sect. Acrocentron, Asteraceae)-taxonomic implication and ecological significance. Biochem. Syst. Ecol. 83, 83–90. doi: 10.1016/j.bse.2019.01.010
Ocarez, N., and Mejía, N. (2016). Suppression of the D-class MADS-box AGL11 gene triggers seedlessness in fleshy fruits. Plant Cell Rep. 35, 239–254. doi: 10.1007/s00299-015-1882-x
Oda, A., Higuchi, Y., and Hisamatsu, T. (2017). Photoperiod-insensitive floral transition in chrysanthemum induced by constitutive expression of chimeric repressor CsLHY-SRDX. Plant Sci. 259, 86–93. doi: 10.1016/j.plantsci.2017.03.007
Oda, A., Higuchi, Y., and Hisamatsu, T. (2020). Constitutive expression of CsGI alters critical night length for flowering by changing the photo-sensitive phase of anti-florigen induction in chrysanthemum. Plant Sci. 293:110417. doi: 10.1016/j.plantsci.2020.110417
Ohashi-Ito, K., and Bergmann, D. C. (2006). Arabidopsis FAMA controls the final proliferation/differentiation switch during Stomatal Developemnt. Plant Cell 18, 2493–2505. doi: 10.1105/tpc.106.046136
Park, J. E., Kim, Y. S., Yoon, H. K., and Park, C. M. (2007). Functional characterization of a small auxin-up RNA gene in apical hook development in Arabidopsis. Plant Sci. 172, 150–157. doi: 10.1016/j.plantsci.2006.08.005
Pei, X., Jing, Z., Tang, Z., and Zhu, Y. (2017). Comparative transcriptome analysis provides insight into differentially expressed genes related to cytoplasmic male sterility in broccoli (Brassica oleracea var. Italica). Sci. Hortic 217, 234–242. doi: 10.1016/j.scienta.2017.01.041
Pellegrini, L., Tan, S., and Richmond, T. J. (1995). Structure of serum response factor core bound to DNA. Nature 376, 490–498. doi: 10.1038/376490a0
Prunet, N., Yang, W., Das, P., Meyerowitz, E. M., and Jack, T. P. (2017). SUPERMAN prevents class B gene expression and promotes stem cell termination in the fourth whorl of Arabidopsis thaliana flowers. P. Natl. Acad. Sci. U. S. A. 114, 7166–7171. doi: 10.1073/pnas.1705977114
Ren, H., Zhu, F., Zheng, C., Sun, X., Wang, W., and Shu, H. (2013). Transcriptome analysis reveals genes related to floral development in chrysanthemum responsive to photoperiods. Biochem. Genet. 51, 20–32. doi: 10.1007/s10528-012-9541-1
Riboni, M., Galbiati, M., Tonelli, C., and Conti, L. (2013). GIGANTEA enables drought escape response via abscisic aciddependent activation of the florigens and suppressor of overexpression of constans1. Plant Physiol. 162, 1706–1719. doi: 10.1104/pp.113.217729
Riboni, M., Robustelli, T. A., Galbiati, M., Tonelli, C., and Conti, L. (2016). ABA-dependent control of GIGANTEA signalling enables drought escape via up-regulation of FLOWERING LOCUS T in Arabidopsis thaliana. J. Exp. Bot. 67, 6309–6322. doi: 10.1093/jxb/erw384
Riechmann, J. L., and Meyerowitz, E. M. (1997). Determination of floral organ identity by Arabidopsis MADS domain homeotic proteins AP1, AP3, PI, and AG is independent of their DNA-binding specificity. Mol. Biol. Cell 8, 1243–1259. doi: 10.1091/mbc.8.7.1243
Rijpkema, A., Royaert, S., Zethof, J., van der Weerden, G., Gerats, T., and Vandenbussche, M. (2006). Analysis of the petunia TM6 MADS box gene reveals functional divergence within the DEF/AP3 lineage. Plant Cell 18, 1819–1832. doi: 10.1105/tpc.106.042937
Roberts, A., Trapnell, C., Donaghey, J., Rinn, J. L., and Pachter, L. (2011). Improving RNA-seq expression estimates by correcting for fragment bias. Genome Biol. 12:R22. doi: 10.1186/gb-2011-12-3-r22
Sasaki, K., Yamaguchi, H., Nakayama, M., Aida, R., and Ohtsubo, N. (2014). Co-modification of class B genes TfDEF and TfGLO in Torenia fournieri Lind. Alters both flower morphology and inflorescence architecture. Plant Mol. Biol. 86, 319–334. doi: 10.1007/s11103-014-0231-8
Shchennikova, A. V., Shulga, O. A., Immink, R., and Angenent, S. G. C. (2004). Identification and characterization of four chrysanthemum MADS-box genes, belonging to the APETALA1/FRUITFULL and SEPALLATA3 subfamilies. Plant Physiol. 134, 1632–1641. doi: 10.1104/pp.103.036665
Shchennikova, A. V., Shulga, O. A., and Skryabin, K. G. (2018). Diversification of the homeotic AP3 clade MADS-box genes in Asteraceae species Chrysanthemum morifolium L. And Helianthus annuus L. Dokl. Biochem. Biophys. 483, 348–354. doi: 10.1134/S1607672918060145
Shin, B., Choi, G., Yi, H., Yang, S., and Choi, G. (2002). AtMYB21, a gene encoding a flower-specific transcription factor, is regulated by COP1. Plant J. 30, 23–32. doi: 10.1046/j.1365-313X.2002.01264.x
Shore, P., and Sharrocks, A. D. (1995). The MADS-box family of transcription factors. FEBS J. 1995, 87–99. doi: 10.1007/978-3-642-85252-7_7
Shulga, O. A., Shchennikova, A. V., Angenent, G. C., and Skryabin, K. G. (2008). MADS-box genes controlling inflorescence morphogenesis in sunflower. Ontogenez 39, 2–5. doi: 10.1134/S1062360408010025
Silva, J. A. T. D., Shinoyama, H., Aida, R., Matsushita, Y., Raj, S. K., and Chen, F. (2013). Chrysanthemum biotechnology: quo vadis? Crit. Rev. Plant Sci. 32, 21–52. doi: 10.1080/07352689.2012.696461
Song, X., Zhao, X., Fan, G., Gao, K., Dai, S., Zhang, M., et al. (2018). Genetic analysis of the corolla tube merged degree and the relative number of ray florets in chrysanthemum (Chrysanthemum×morifolium Ramat.). Sci. Hortic. 242, 214–224. doi: 10.1016/j.scienta.2018.07.010
Su, J., Jiang, J., Zhang, F., Liu, Y., and Chen, F. (2019). Current achievements and future prospects in the genetic breeding of chrysanthemum: a review. Hortic Res. 6:19. doi: 10.1038/s41438-019-0193-8
Sumitomo, K., Li, T., and Hisamatsu, T. (2009). Gibberellin promotes flowering of chrysanthemum by upregulating CmFL, a chrysanthemum FLORICAULA/LEAFY homologous gene. Plant Sci. 176, 643–649. doi: 10.1016/j.plantsci.2009.02.003
Sun, X., Wang, X., Zhang, C., Xing, S., and Shu, H. (2018). Cloning, sequence, and expression analyses of the Chrysanthemum morifolium flowering-related gene CmCOL (CONSTANS-like). Gene Reports. 12, 81–88. doi: 10.1016/j.genrep.2018.05.012
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Teixeira, J. A. (2003). Chrysanthemum: advances in tissue culture, cryopreservation, postharvest technology, genetics and transgenic biotechnology. Biotechnol. Adv. 21, 715–766. doi: 10.1016/S0734-9750(03)00117-4
Tsaftaris, A. S., Polidoros, A. N., Pasentsis, K., and Kalivas, A. (2006). Tepal formation and expression pattern of B-class paleoAP3-like MADS-box genes in crocus (Crocus sativus L.). Plant Sci. 170, 238–246. doi: 10.1016/j.plantsci.2005.08.008
Tzeng, T. Y., and Yang, C. H. (2001). A MADS box gene from Lily (Lilium Longiflorum) is sufficient to generate dominant negative mutation by interacting with PISTILLATA (PI) in Arabidopsis thaliana. Plant Cell Physiol. 42, 1156–1168.
Tzeng, T. Y., Liu, H. C., and Yang, C. H. (2004). The c-terminal sequence of LMADS1 is essential for the formation of homodimers for b function proteins. J. Biol. Chem. 279, 10747–10755. doi: 10.1074/jbc.M311646200
Vandenbussche, M., Theissen, G., Van de Peer, Y., and Gerats, T. (2003). Structural diversification and neo-functionalization during floral MADS-box gene evolution by C-terminal frameshift mutations. Nucleic Acids Res. 31, 4401–4409. doi: 10.1093/nar/gkg642
Victoria, S., and Minsung, K. (2017). Re”CYC”ling molecular regulators in the evolution and development of flower symmetry. Semin. Cell Dev. Biol. 79, 16–26. doi: 10.1016/j.semcdb.2017.08.052
Vuosku, J., Muilu-Mkel, R., Avia, K., Suokas, M., and Sarjala, T. (2019). Thermospermine synthase (ACL5) and diamine oxidase (DAO) expression is needed for zygotic embryogenesis and vascular development in scots pine. Front. Plant Sci. 10:1600. doi: 10.3389/fpls.2019.01600
Wagner, D., Sablowski, R. W., and Meyerowitz, E. M. (1999). Transcriptional activation of APETALA1 by LEAFY. Science 285, 582–584. doi: 10.1126/science.285.5427.582
Wei, Q., Ma, C., Xu, Y., Wang, T., Chen, Y., Lü, J., et al. (2017). Control of chrysanthemum flowering through integration with an aging pathway. Nat. Commun. 8:829. doi: 10.1038/s41467-017-00812-0
Wen, X., Qi, S., Huang, H., Wu, X., and Dai, S. (2019). The expression and interactions of ABCE-class and CYC2-like genes in the capitulum development of Chrysanthemum lavandulifolium and C.×morifolium. Plant Growth Regul. 88, 205–214. doi: 10.1007/s10725-019-00491-5
Wollmann, H., Mica, E., Todesco, M., Long, J. A., and Weigel, D. (2010). On reconciling the interactions between APETALA2, miR172 and AGAMOUS with the ABC model of flower development. Development 137, 3633–3642. doi: 10.1242/dev.036673
Yan, Z., Shi, H., Liu, Y., Jing, M., and Han, Y. (2020). KHZ1 and KHZ2, novel members of the autonomous pathway, repress the splicing efficiency of FLC pre-mRNA in Arabidopsis. J. Exp. Bot. 71, 1375–1386. doi: 10.1093/jxb/erz499
Yang, Y., Fanning, L., and Jack, T. (2003). The K domain mediates heterodimerization of the Arabidopsis floral organ identity proteins, APETALA3 and PISTILLATA. Plant J. 33, 47–59. doi: 10.1046/j.0960-7412.2003.01473.x
Yang, L., Fu, J., Qi, S., Hong, Y., Huang, H., and Dai, S. (2017). Molecular cloning and function analysis of ClCRY1a and ClCRY1b, two genes in Chrysanthemum lavandulifolium that play vital roles in promoting floral transition. Gene 617, 32–43. doi: 10.1016/j.gene.2017.02.020
Yeoh, Y. K., Yuji, S., Parks Donovan, H., and Philip, H. (2016). Comparative genomics of candidate phylum TM6 suggests that parasitism is widespread and ancestral in this lineage. Mol. Biol. Evol. 33, 915–927. doi: 10.1093/molbev/msv281
Zhang, X., Garreton, V., and Chua, N. H. (2005). The AIP2 E3 ligase acts as a novel negative regulator of ABA signaling by promoting ABI3 degradation. Genes Dev. 19, 1532–1543. doi: 10.1101/gad.1318705
Zhang, H., Pan, X., Liu, S., Lin, W., and Zhang, X. (2020). Genome-wide analysis of AP2/ERF transcription factors in pineapple reveals functional divergence during flowering induction mediated by ethylene and floral organ development. Genomics 113, 474–489. doi: 10.1016/j.ygeno.2020.10.040
Keywords: Chrysanthemum morifolium, ray florets, disc florets, transcriptome, RNA-seq, differentially expressed genes
Citation: Liu H, Jia Y, Chai Y, Wang S, Chen H, Zhou X, Huang C, Guo S and Chen D (2022) Whole-transcriptome analysis of differentially expressed genes between ray and disc florets and identification of flowering regulatory genes in Chrysanthemum morifolium. Front. Plant Sci. 13:947331. doi: 10.3389/fpls.2022.947331
Edited by:
Tangchun Zheng, Beijing Forestry University, ChinaReviewed by:
Zongda Xu, Shandong Agricultural University, ChinaYaping Kou, Institute of Vegetables and Flowers (CAAS), China
Sagheer Ahmad, Guangdong Academy of Agricultural Sciences, China
Copyright © 2022 Liu, Jia, Chai, Wang, Chen, Zhou, Huang, Guo and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Conglin Huang, Y29uZ2xpbmhAMTI2LmNvbQ==; Shuang Guo, c2NueWR4Z3NAMTYzLmNvbQ==; Dongliang Chen, ZGxjaGVuMTk4NEAxMjYuY29t
†These authors have contributed equally to this work and share first authorship