 Yu Sugo1
Yu Sugo1 Hiroshi Ishikita1,2*
Hiroshi Ishikita1,2*- 1Department of Applied Chemistry, The University of Tokyo, Tokyo, Japan
- 2Research Center for Advanced Science and Technology, The University of Tokyo, Tokyo, Japan
Photo-induced charge separation, which is terminated by electron transfer from the primary quinone QA to the secondary quinone QB, provides the driving force for O2 evolution in photosystem II (PSII). However, the backward charge recombination using the same electron-transfer pathway leads to the triplet chlorophyll formation, generating harmful singlet-oxygen species. Here, we investigated the molecular mechanism of proton-mediated QA⋅– stabilization. Quantum mechanical/molecular mechanical (QM/MM) calculations show that in response to the loss of the bicarbonate ligand, a low-barrier H-bond forms between D2-His214 and QA⋅–. The migration of the proton from D2-His214 toward QA⋅– stabilizes QA⋅–. The release of the bicarbonate ligand from the binding Fe2+ site is an energetically uphill process, whereas the bidentate-to-monodentate reorientation is almost isoenergetic. These suggest that the bicarbonate protonation and decomposition may be a basis of the mechanism of photoprotection via QA⋅–/QAH⋅ stabilization, increasing the QA redox potential and activating a charge-recombination pathway that does not generate the harmful singlet oxygen.
Introduction
The driving force for photosynthetic O2 evolution is provided by the photo-induced charge separation at the reaction center of photosystem II (PSII). The electronic excitation of the chlorophyll leads to electron transfer via pheophytin in the D1 protein, PheoD1, to two plastoquinone molecules, QA and QB (Shen, 2015). The terminal electron acceptor QB accepts two electrons via the primary quinone QA and two protons. The non-heme Fe2+ complex, which comprises D1-His215, D2-His214, D1-His272, D2-His268, and bicarbonate (HCO3–), is equidistant from QA and QB (Shevela et al., 2012; Figure 1A). The bidentate bicarbonate is modeled in the majority of the reported PSII structures (e.g., Umena et al., 2011; Gisriel et al., 2022; Figure 1A), whereas not only the bidentate but also monodentate bicarbonate is modeled in the PsbM-deleted PSII
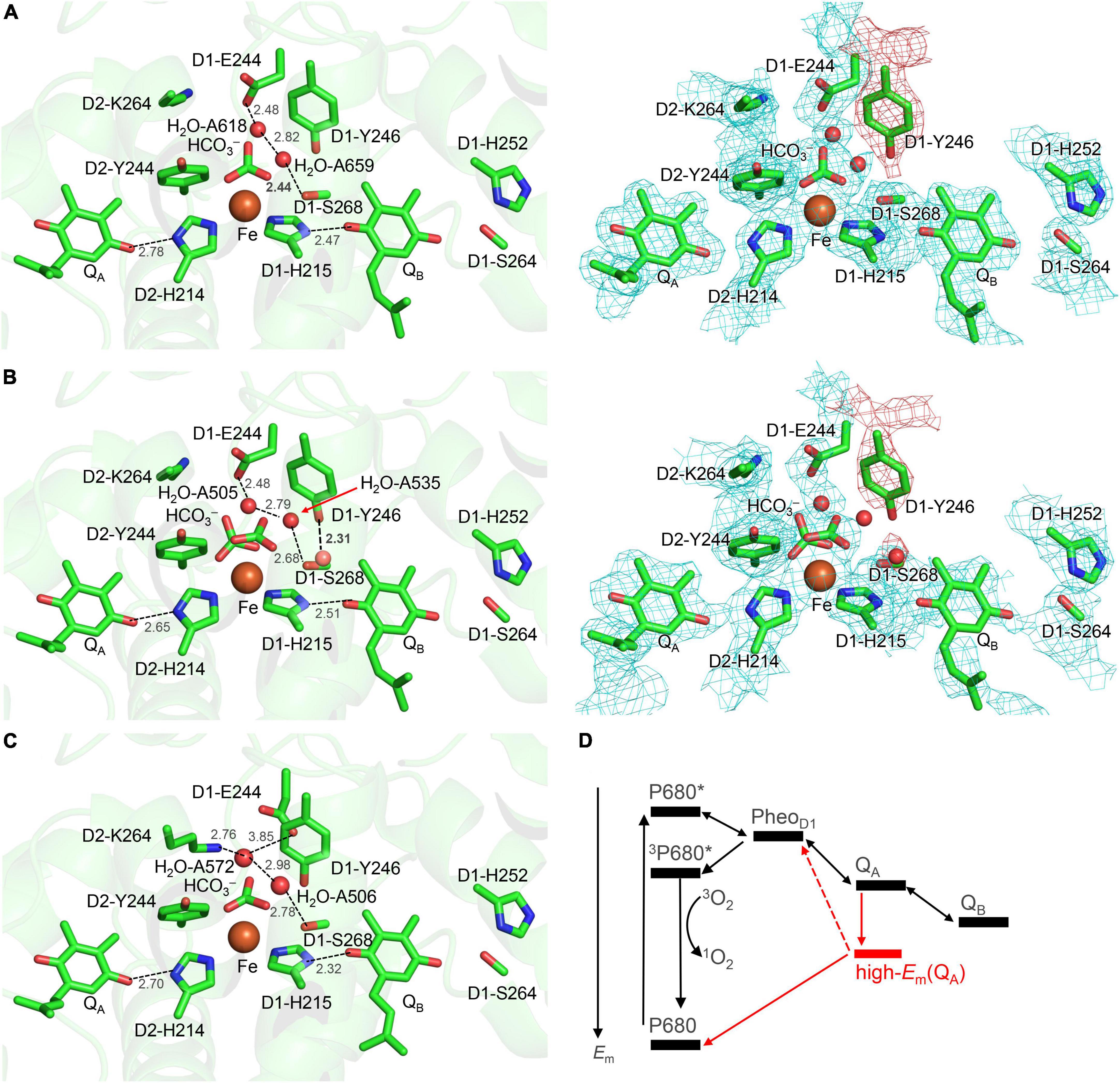
Figure 1. Overview of the quinone binding sites. (A) X-ray crystal structure of PSII from Thermosynechococcus vulcanus (PDB code, 3WU2) (Umena et al., 2011) and electron density map. (B) X-ray crystal structure of the PsbM-deleted PSII from Thermosynechococcus vestitus BP-1 (PDB code, 5H2F) (Uto et al., 2017) and electron density map. (C) Cryo-electron microscopy structure of PSII from Synechocystis sp. PCC 6803 (PDB code, 7N8O) (Ferreira et al., 2004). Dotted lines indicate H-bonds. The density of the D1-Tyr246 moiety, including the adjacent water molecule in the PsbM-deleted structure, is indicated by the red mesh. (D) Energetics of forward and backward electron-transfer processes. Processes occurring via the high-Em QA form are shown in red. P680* denotes the electronically excited P680.
(Figure 1B; Uto et al., 2017). D1-Tyr246 and D2-Tyr244 are located at the bicarbonate binding site (Forsman et al., 2019). The characteristic of D1-Tyr246 is the electron density near the hydroxyl group (Saito et al., 2013), which is clearly observed in the 1.9-Å X-ray diffraction (XRD) (Umena et al., 2011) and X-ray free electron laser (XFEL)-S1 (Suga et al., 2015) structures (Figure 1A). The experimentally observed electron density was not interpreted in 2011 and 2015 (Figure 1A). In 2017, Uto et al. (2017) modeled the corresponding density as a water molecule (B-factor: 40 Å2) forming a significantly short H-bond with the hydroxyl group of D1-Tyr246 (2.31 Å) (Figure 1B). Remarkably, the OD1–Tyr246…Owater distance of 2.3 Å is closer to the O…O distance for a typical Zundel (H2O…H+…H2O) cation (≈2.3 Å). The absence of the corresponding density at the D2-Tyr244 moiety may indicate a functional asymmetry between the QA and QB sides (Saito et al., 2013).
The redox potential (Em) values of –100 to –140 mV for one-electron reduction of QA (QA/QA⋅–) (Krieger and Weis, 1992; Johnson et al., 1995; Krieger et al., 1995; Ishikita and Knapp, 2005a; Shibamoto et al., 2009) and 90 mV for QB/QB⋅– (Kato et al., 2016; De Causmaecker et al., 2019) indicate that exergonic electron transfer occurs under normal functional conditions. Reduced QB accepts the first proton via D1-Ser264 and D1-His252 at the distal carbonyl O site with respect to the non-heme Fe2+ complex (Ishikita and Knapp, 2005a; Saito et al., 2013; Ashizawa and Noguchi, 2014) and the second proton via D1-His215 at the proximal carbonyl O site (Saito et al., 2013, 2020; Kimura et al., 2020); thus, reduced QB forms QBH2 and moves from PSII toward the quinone pool. The depletion of the bicarbonate from the non-heme Fe2+ reduces the rate of the electron transfer from QA to QB (Jursinic et al., 1976; Eaton-Rye and Govindjee, 1988) or the exchange of QBH2 (Siggel et al., 1977; Sedoud et al., 2011).
Under strong light, the plastoquinone pool is fully reduced, and the QB binding site is unoccupied, which inhibits electron transfer and causes QA⋅– to accumulate in PSII (photoinhibition) (Vass et al., 1992; Noguchi, 2002). If the Em gap between QA and PheoD1 is small, backward electron transfer occurs in the form of charge recombination from QA⋅– via PheoD1 to the cationic chlorophyll in the reaction center, forming triplet chlorophyll and generating harmful singlet-oxygen species (Rutherford and Krieger-Liszkay, 2001; Rutherford et al., 2012). QA has been reported to exhibit two forms, a low- and a high-Em(QA) conformation (Figure 1D; Krieger and Weis, 1992; Johnson et al., 1995; Krieger et al., 1995). QA exists in the low-potential form under normal functional conditions. The high-Em(QA) conformation can increase the Em gap between QA and PheoD1, preventing charge recombination via PheoD1 and singlet-oxygen generation under strong light [photoprotection (Rutherford and Krieger-Liszkay, 2001; Rutherford et al., 2012)].
The molecular origin of the high-Em(QA) form had long remained unsolved. Vass et al. (1992) proposed that the QA⋅– stabilization is mediated by protonation. In the 3.5-Å crystal structure of PSII (Ferreira et al., 2004), the OH group of D2-Thr217 forms an H-bond with QA⋅– but not with unprotonated neutral QA (Ishikita and Knapp, 2005a). Thus, H-bond donation of D2-Thr217 to QA⋅– could be a possible mechanism for the formation of the high-Em conformation. However, the corresponding H-bond is unlikely to form in the crystal structure of PSII from Thermosynechococcus vulcanus (Umena et al., 2011) and the recent cryo-electron microscopy structure of PSII from Synechocystis sp. PCC 6803 (Gisriel et al., 2022). The aforementioned crystal structure shows that QA has D2-His214 at the proximal carbonyl O site and the backbone amide group of D2-Phe261 at the distal carbonyl O site as H-bond partners.
The non-heme Fe2+ complex might be involved in the photoprotection mechanism (Diner and Petrouleas, 1987; Muh et al., 2012). Notably, Brinkert et al. (2016) reported that the loss of the bicarbonate ligand from the non-heme Fe2+ complex leads to an increase of 75 mV in Em(QA) in the presence of QB; this suggests that the loss of the bicarbonate ligand is responsible for the formation of the high-Em(QA) conformation. Recent studies have shown that photo-induced CO2 conversion is likely to occur from the bicarbonate ligand at Fe2+ (Shevela et al., 2020). Bicarbonate loss and photo-induced CO2 conversion likely constitute the basis of the photoprotection mechanism. Although the loss of the negatively charged bicarbonate (HCO3–) certainly increases Em(QA), the distance between the bicarbonate ligand and QA is more than 6 Å (Umena et al., 2011). Moreover, whether the proton-mediated QA⋅– stabilization mechanism (Vass et al., 1992) is still relevant to the QA⋅– stabilization remains unclear.
To understand the mechanism of how the loss of the bicarbonate increases Em(QA), we investigated the bicarbonate and QA binding sites in PSII using a quantum mechanical/molecular mechanical (QM/MM) approach based on the PSII crystal structure (Umena et al., 2011).
Materials and methods
Coordinates and atomic partial charges
The atomic coordinates were obtained from the X-ray crystal structure of PSII (PDB code, 3WU2) (Umena et al., 2011). The heavy-atom positions were fixed while the H-atom positions were optimized with CHARMM (Brooks et al., 1983). All titratable groups were ionized if not otherwise specified. D1-His337 was considered to be protonated (Nakamura and Noguchi, 2017). Atomic partial charges were obtained from the CHARMM22 (MacKerell et al., 1998) parameter set for amino acids and previous studies for cofactors (Saito et al., 2015), respectively.
Quantum mechanical/molecular mechanical calculations
The unrestricted density functional theory method was employed with the B3LYP functional and LACVP* basis sets (LANL2DZ (double ζ quality basis set with the Los Alamos effective core potential) for Mn and Ca atoms and 6-31G* for other atoms) (Hay and Wadt, 1985) using the QSite (QSite, 2012) program. Counter ions were added to neutralize the system. In the QM region, all atomic coordinates were relaxed. In the MM region, the H-atom positions were energetically optimized, and the heavy-atom positions were fixed using the OPLS2005 force field because the MM region is used mainly to reproduce (long-distance) electrostatic interactions with the QM region and the heavy-atom positions in the MM region should remain unchanged with respect to those in the original crystal structure. The initial-guess wave functions were obtained using the ligand field theory (Vacek et al., 1999) implemented in the QSite program. Three QM regions were used: (i) [QA, QB, the non-heme Fe complex (bicarbonate if applicable, Fe, and side chains of D1-His215, D1-His272, D2-His214, and D2-His268), side chains of D1-His252, D1-Ser264, and D2-Phe261 (including backbone)] for the analysis of the H-bond between QA and D2-His214; (ii) [QA, QB, the non-heme Fe complex (bicarbonate, Fe, and side chains of D1-His215, D1-His272, D2-His214, and D2-His268), side chains of D1-Tyr246, D1-Ser264, and D1-His252, and the modeled water molecule adjacent to D1-Tyr246] for the analysis of the proton transfer toward the bicarbonate ligand; and (iii) [QA, QB, the non-heme Fe complex (bicarbonate, Fe, and side chains of D1-His215, D1-His272, D2-His214, and D2-His268), side chains of D1-Tyr246, D1-Ser264, D1-His252, and protonated D1-Glu244, and water molecules in the H-bond network (water molecules A618 and A659 and the modeled water molecule adjacent to D1-Tyr246] for the analysis of the proton transfer from D1-Ser268 to the bicarbonate ligand.
All other protein units and cofactors were approximated by the MM force field (i.e., electrostatic influences are sufficiently considered in the MM region). Note that the residues in the proton transfer pathways must be included in the QM region to consider the formation/breakage of the covalent (H-)bonds during proton transfer. See Supplementary material for the atomic coordinates of the resulting QM region. As in a previous study (Chernev et al., 2011), the non-heme Fe complex was in a high-spin state of Fe2+, and the spin multiplicity of the system was set to S = 2 in calculations for neutral QA and S = 5/2 for QA⋅–.
The QM/MM-optimized geometry was used as the initial geometry to analyze the potential energy profiles. For the analysis of the ND2–His214…H+…OQA H-bond, the focusing H atom was moved along the N…O H-bond from 0.02 to 0.10 Å (e.g., 0.02 Å around the local energy minimum in some cases), after which the geometry was optimized by constraining the N…H+ and H+…O distances and energy was calculated. For the analysis of the bidentate-to-monodentate reorientation of the bicarbonate ligand, the focusing OHCO3–…Fe distance was increased by 0.10 Å, after which the geometry was optimized by constraining the OHCO3–…Fe distance and energy was calculated. For the analysis of the release of the bicarbonate ligand from the Fe site, the CHCO3–…Fe distance was increased by 0.10 Å, after which the geometry was optimized by constraining the CHCO3–…Fe distance and energy was calculated. For the analysis of the proton transfer from H3O+ via D1-Tyr246 and the bicarbonate ligand, the focusing H atom was moved away from the electron donor O site from 0.02 to 0.10 Å, after which the geometry was optimized by constraining the Odonor…H distance and energy was calculated. For the analysis of the proton transfer from H3O+ adjacent to D1-Ser268 to the bicarbonate ligand, the focusing H atom was moved to the electron acceptor O site by 0.10 Å, after which the geometry was optimized by constraining the Oacceptor…H distance and energy was calculated. For the analysis of the protonated bicarbonate decomposition to H2O + CO2, the H2O…CO2 distance was increased by 0.10 Å, after which the geometry was optimized by constraining the H2O…CO2 distance and the energy was calculated.
Results
Formation of a low-barrier H-bond between D2-His214 and QA upon bicarbonate loss
The H-bond distance of 2.78 Å between D2-His214 and QA in the PSII crystal structure (Umena et al., 2011; Figure 1A) is closest to 2.76 Å for neutral unprotonated QA in the QM/MM-optimized geometry (Figure 2A). This suggests that QA is neutral QA in the PSII crystal structure. Although the H-bond is shortened to 2.65 Å when QA is reduced to QA⋅–, the potential-energy profile indicates that the H+ is localized at the D2-His214 moiety (Figure 2B).
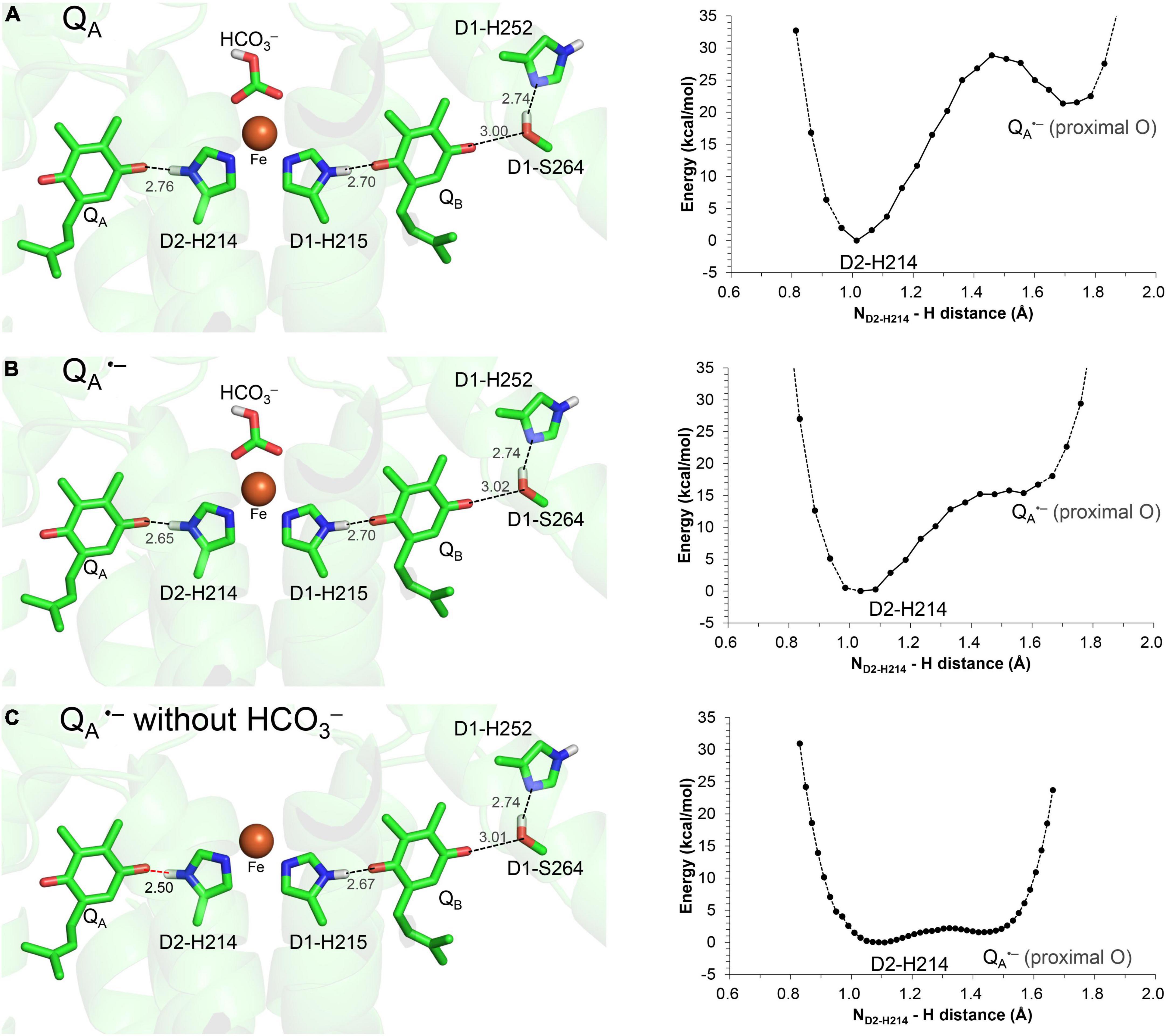
Figure 2. QM/MM-optimized geometry and potential energy profile for the H-bond between QA and D2-His214 prior to electron transfer from QA to QB. (A) Neutral QA. (B) QA⋅–. (C) QA⋅– in the absence of the bicarbonate ligand. D1-Ser264 forms an H-bond with unprotonated D1-His252, as the electron is not transferred to QB (Ishikita and Knapp, 2005a). Note that the geometry of each intermediate conformation is fully QM/MM optimized.
Remarkably, the D2-His214…QA⋅– H-bond transforms into a low-barrier H-bond (2.50 Å) in response to the loss of the bicarbonate ligand (Figure 2C). In addition to the electrostatic contribution of the loss of the negative charge to an increase in Em(QA), the migration of the D2-His214 proton toward QA⋅– along the low-barrier H-bond (0.23 Å) stabilizes QA⋅– significantly. In addition to the loss of a negative charge, this is likely a substantial reason for the 75 mV upshift in Em(QA) upon the loss of the bicarbonate (Brinkert et al., 2016) because QA…HCO3– is not short (6.8 Å).
The formation of the low-barrier H-bond between QA⋅– and D2-His214 suggests that pKa(QA⋅–/QAH) ≈ pKa(D2-His214-NH/N–) in the absence of the bicarbonate. The low-barrier H-bond formation is also observed between D1-His215 and QBH– in PSII (Saito et al., 2013) [and His-L190 and QBH– in photosynthetic reaction centers from purple bacteria (PbRC) (Sugo et al., 2021)] during the QBH2 formation; this suggests that pKa(QBH–/QBH2) ≈ pKa(D1-His215-NH/N–) in the presence of the bicarbonate. pKa(D1-His215-NH/N–) and pKa(D2-His214-NH/N–) for deprotonation of singly protonated to anionic histidine are likely higher [e.g., 13 in the Rieske cluster (Zu et al., 2003; Hsueh et al., 2010)] than pKa for deprotonation of doubly protonated to singly protonated histidine [e.g., 2–9 in protein environments (Grimsley et al., 2009)]. As pKa(QH–/QH2) = 11 for plastoquinone (Q) in water (Hasegawa et al., 2017), a slight decrease in pKa(D2-His214-NH/N–) due to Fe2+ can lead to pKa(Qa⋅–/QAH) ≈ pKa(D2-His214-NH/N–).
In the low-barrier H-bond, the proton can move easily between the two H-bond moieties (Ishikita and Saito, 2014). In the low-barrier H-bond between TyrZ and D1-His190, the 0.35 Å migration of the proton toward TyrZ from D1-His190 can increase Em(TyrZ) by 130 mV (Saito et al., 2020). From the analogy, the > 0.2 Å migration of the proton toward QA⋅– along the low-barrier H-bond (Figure 2C) is the most likely origin of the observed increase of 75 mV in Em(QA) (Brinkert et al., 2016). Thus, QA⋅– can be stabilized irrespective of QA…HCO3– = 6.8 Å.
Energetics of the bicarbonate displacement from the non-heme Fe complex
In QM/MM calculations, the release of the bicarbonate ligand from the binding Fe2+ site is an energetically uphill process (Figure 3A), which suggests that the electrostatic interaction between Fe2+ and HCO3– is not negligibly small as long as the negatively charged HCO3– state is stable at Fe2+.
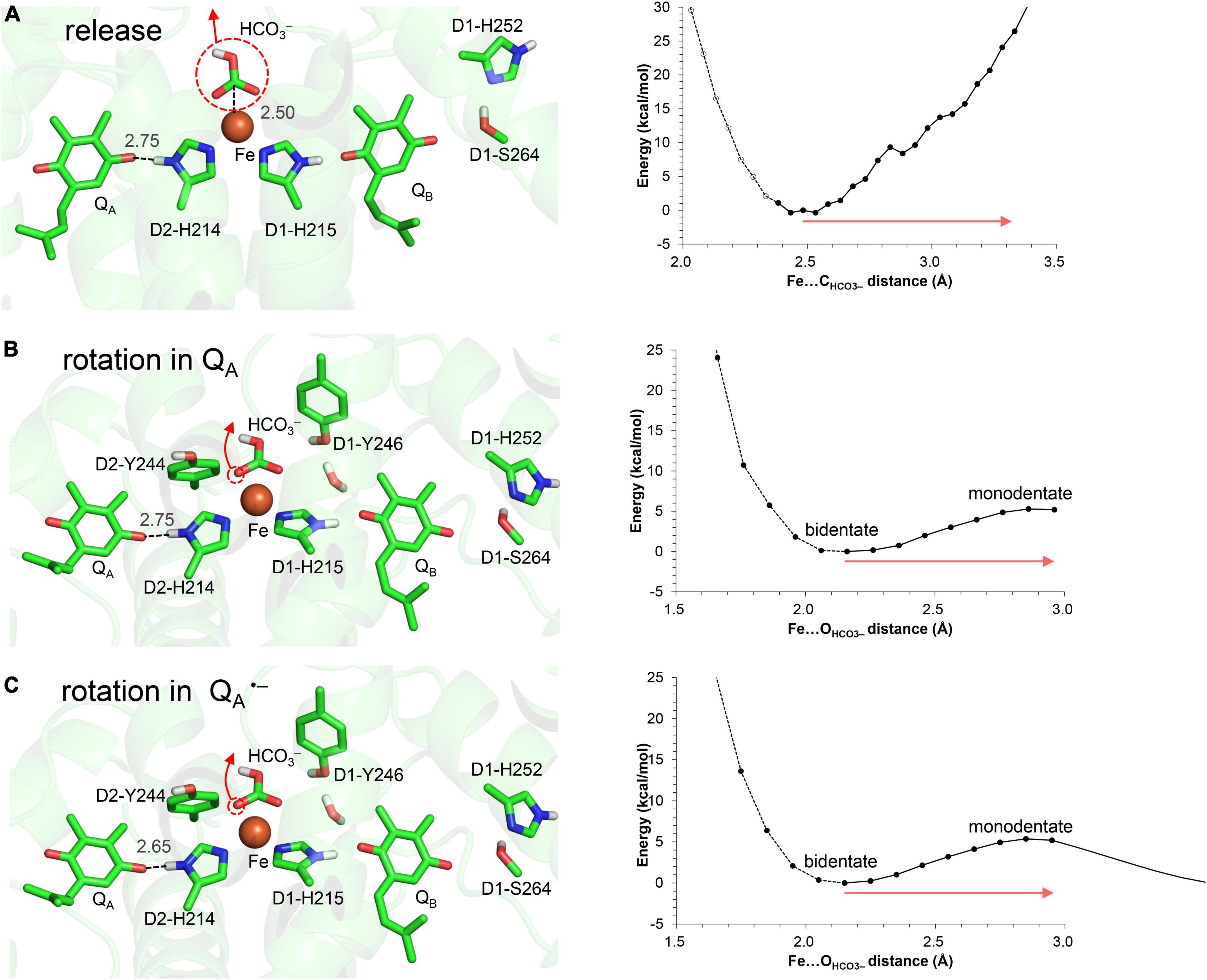
Figure 3. Energy profile for the bicarbonate displacement. (A) Release of the bicarbonate ligand from the Fe2+ site. (B) Bidentate-to-monodentate reorientation of the bicarbonate ligand in neutral QA. (C) Bidentate-to-monodentate reorientation of the bicarbonate ligand in QA⋅–. Red arrows indicate the orientations of the bicarbonate displacement. Note that the geometry of each intermediate conformation is fully QM/MM optimized.
In contrast, the bidentate-to-monodentate reorientation is slightly uphill in QA/QA⋅–, as the monodentate bicarbonate can accept an H-bond from D1-Tyr246 (Figures 3B,C). Note that QA⋅– is stabilized when the hydroxyl group of D1-Tyr246 is oriented toward QA⋅– (Saito et al., 2013). It has been proposed that bicarbonate serves as not only a bidentate but also a monodentate ligand (Hienerwadel and Berthomieu, 1995; Chernev et al., 2011; Uto et al., 2017). In vacuum, the monodentate ligation may become pronounced specifically upon the formation of QA⋅– (Chernev et al., 2011), because the reorientation of the free bicarbonate ligand occurs easily and is the only way to compensate for the electrostatic repulsion against QA⋅–. The corresponding electrostatic repulsion is smaller (bicarbonate…QA⋅– = 6.8 Å) than the interactions with D1-Glu244 (3.4 Å from bicarbonate), D1-Tyr246 (3.2 Å), and D2-Tyr244 (3.1 Å) in the PSII protein environment. The shape of the potential-energy profile remains essentially unaffected in response to changes in the QA redox state (Figures 3B,C). Furthermore, the bidentate ligation is more stable than the monodentate ligation in the PSII protein environment (Figures 3B,C). Thus, the bidentate-to-monodentate reorientation is unlikely to synchronize with unimpaired electron transfer from QA to QB: the reorientation may occur only when the electron transfer is sufficiently slow to compete with the energetically uphill movement of the bicarbonate ligand.
These results suggest that the release of the bicarbonate ligand is energetically more uphill than the reorientation, as long as HCO3– is ligated to the Fe2+ site.
Discussion
Possible mechanisms of bicarbonate loss
The bicarbonate protonation and decomposition can also lead to the bicarbonate loss (e.g., Loerting et al., 2000). This requires the proton transfer pathway toward the bicarbonate binding site. Below we have discussed candidate proton-donor residues near the bicarbonate binding site.
Notably, the bicarbonate binding site is linked with the protein bulk surface via an H-bond network that is formed by D1-Glu244, D1-Tyr246, and D1-Ser264 (Figure 4). D1-Glu244 in the highly charged de-loop region was responsible for the pH dependence of Em for the non-heme Fe complex (Ishikita and Knapp, 2005b) and may be involved in the bicarbonate protonation at lower pH in the Fe2+ state (Kato et al., 2021). In addition, the Em(QA) shift was observed upon the D1-E244A mutation (Forsman et al., 2019). Indeed, D1-Glu244 is close to the bicarbonate (3.37 Å, Figure 1) (Umena et al., 2011). However, according to the geometry of the PSII crystal structure, ionized D1-Glu244 is stabilized by a salt bridge with D2-Lys264 (3.31 Å, Umena et al., 2011) and is unlikely to serve as a proton donor to the bicarbonate. Unless structural changes occur at higher pH, the geometry does not directly support the involvement of D1-Glu244 in the bicarbonate protonation.
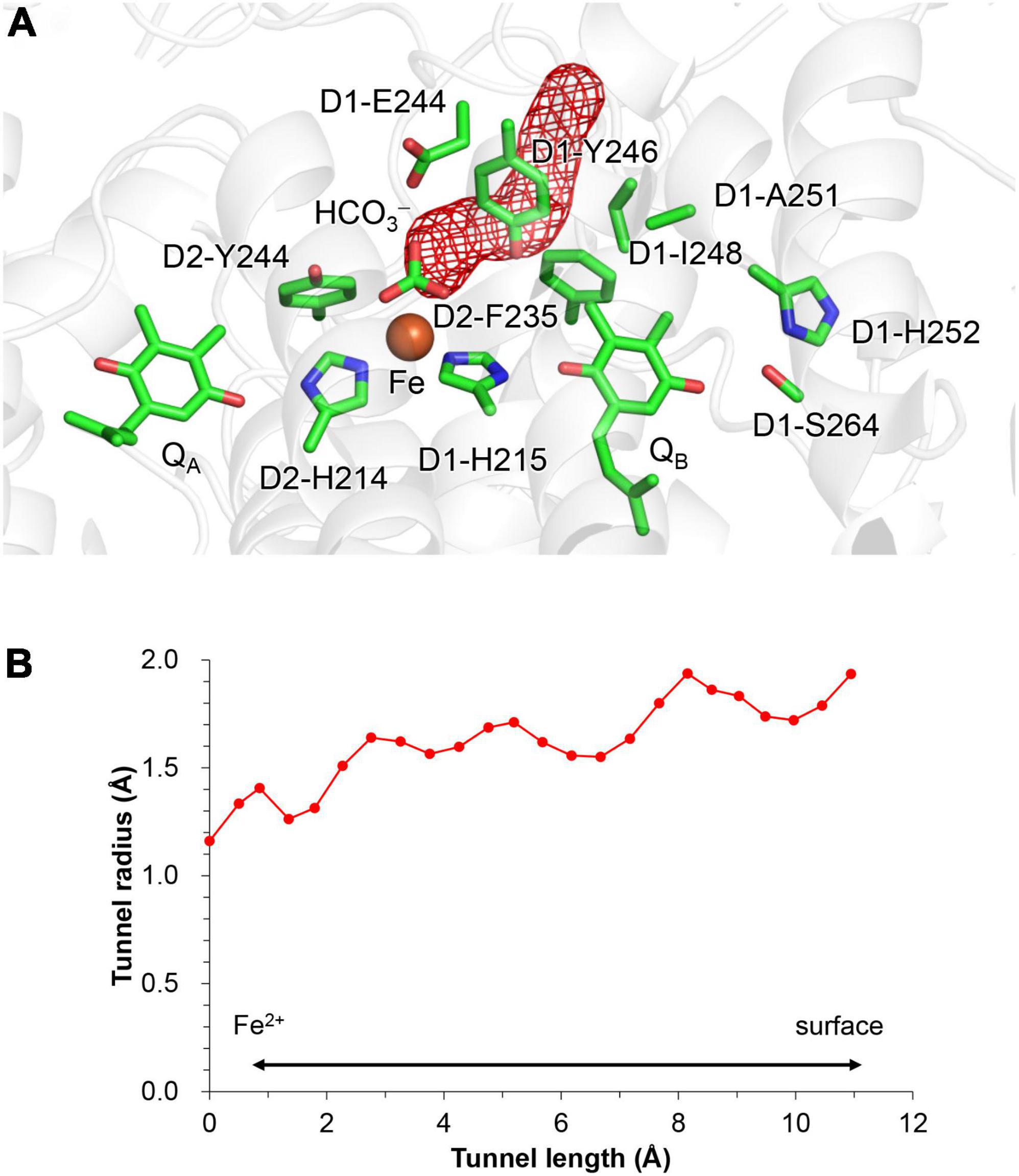
Figure 4. Channel that proceeds from the bicarbonate binding site toward the stromal protein surface. (A) Overview of the hydrophobic channel (red mesh). (B) Cavity radius along the hydrophobic channel. The channel space was analyzed using CAVER (Chovancova et al., 2012).
Although D1-Ser268 does not form an H-bond with the bicarbonate ligand (4.57 Å, Umena et al., 2011), the existence of a water molecule, which is only 2.44 Å away from the D1-Ser268 side chain (2.44 Å, Umena et al., 2011), is remarkable. The short distance might be due to the water molecule being H3O+ [e.g., O…O = 2.4 Å for typical H2O…H3O+ (Mikenda, 1986; Limbach et al., 2009; Figure 1A)]. However, assuming that the water molecule is H3O+ in the QM/MM calculation, H3O+ forms H-bonds with D1-Tyr246 and a water molecule adjacent to D1-Glu244, but not with D1-Ser268; this increases the OD1–Ser268…OH3O+ distance to 2.98 Å (Figure 5). Furthermore, H3O+ is unstable at this site, releasing the proton to ionized D1-Glu244. H3O+ can exist at the D1-Ser268 moiety only when D1-Glu244 is protonated. However, proton transfer from H3O+ at the D1-Ser268 to the bicarbonate ligand is energetically uphill (Figure 5), which suggests that D1-Ser268 is unlikely the proton donor to the bicarbonate. D1-Ser268 and the adjacent water molecule may be more likely to be involved in the proton transfer associated with the QBH2 release, as suggested for mutant PSII proteins (Forsman and Eaton-Rye, 2020).
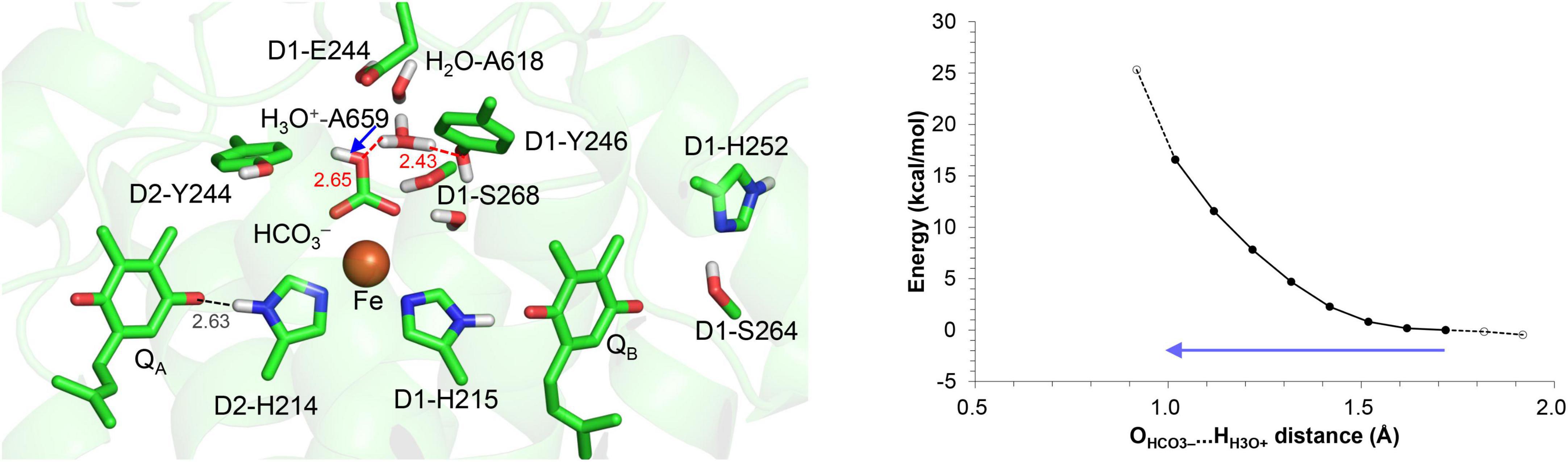
Figure 5. Potential energy profile for proton transfer from H3O+ at D1-Ser268 to the bicarbonate ligand. The blue arrow indicates the orientation of the proton transfer. Note that the geometry of each intermediate conformation is fully QM/MM optimized.
D1-Tyr246 does not form an H-bond with the bicarbonate –OH group (3.80 Å) in the PSII crystal structure (Umena et al., 2011), whereas D1-Tyr246 forms an H-bond with the bicarbonate –OH group (≈2.7 Å) in response to the bidentate-to-monodentate reorientation according to QM/MM calculations (Figure 3). D1-Tyr246 is located at the unique position, the interface between the channel that proceeds from the bicarbonate binding site (Figure 4) and the inner cavity, the QB pocket.
The elongation electron density near the hydroxyl group of D1-Tyr246 (Saito et al., 2013) may be interpreted as a peroxide O with the O–O distance of 1.48 Å, which can be fitted to the density (Figure 1A; Saito et al., 2013). Although the peroxide O is oriented toward the QB binding pocket, the link between the bicarbonate binding site and the QB binding pocket via D1-Tyr246 is weak due to the less polar O site.
Alternatively, the elongation of the density may be interpreted as H3O+. In the crystal structure reported by Uto et al. (2017), the OD1–Tyr246…Owater distance of 2.3 Å is closer to the O…O distance for a typical Zundel (H2O…H+…H2O) cation (≈2.3 Å), which suggests that the density may originate from H3O+ (Figure 1B). In this case, the H+ needs to enter via the QB-exchange channel, possibly during the QB exchange. To investigate whether the uptake and existence of the H+ are energetically possible in the QB binding pocket, QM/MM calculations were performed, removing QB and filling the pocket with water molecules. The potential-energy profile indicates that the proton is energetically stable and populated among the two water molecules that bridge between D1-His215 and D1-Tyr246 (i.e., H2O-4* and H2O-5*), as H3O+ is stabilized by three H-bond acceptor groups (Figure 6). After QB re-enters the binding pocket, the H+ is allowed to exist only at the non-excluded water molecules, e.g., the one adjacent to D1-Tyr246. Indeed, the bulky methoxy groups of ubiquinone in PbRC are replaced with the methyl groups of plastoquinone in PSII, allowing one water molecules to remain at the D1-Tyr246 moiety. Thus, a water molecule can remain at the D1-Tyr246 moiety, the only polar site in the bottleneck of the hydrophobic QB pocket. This is likely the cause of the elongation of the electron density adjacent to the –OH region of D1-Tyr246.
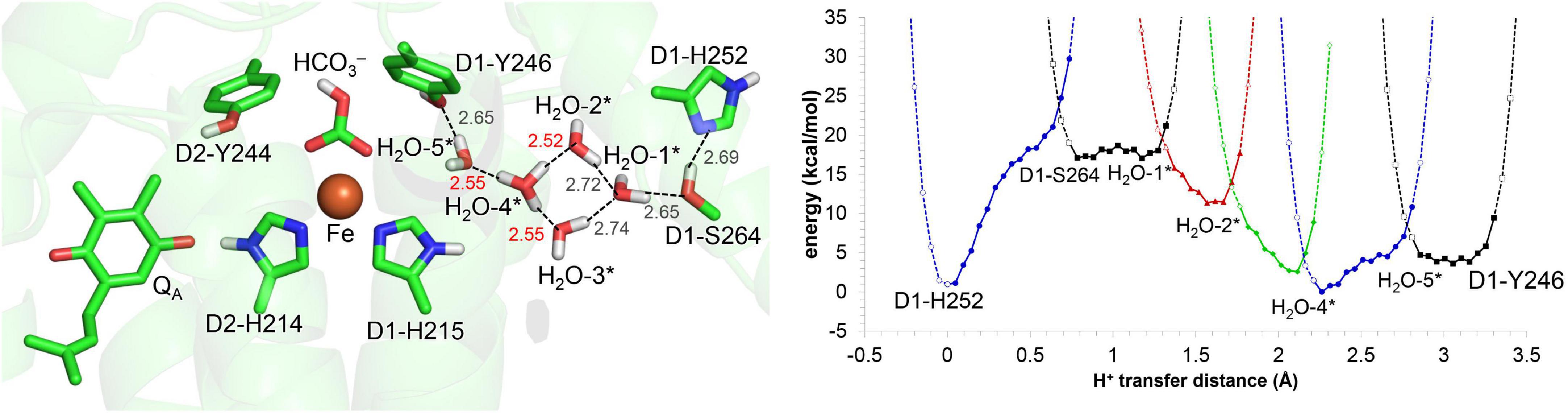
Figure 6. Potential energy profile for proton transfer in the QB binding pocket, adding water molecules (*: H2O-1 to 5). Note that the geometry of each intermediate conformation is fully QM/MM optimized.
When H3O+ is adjacent to D1-Tyr246, the bidentate-to-monodentate reorientation leads to the formation of an H-bond network that proceeds from H3O+ via D1-Tyr246 toward the monodentate bicarbonate (Figure 7A). The potential energy profile for the H-bonds suggests that the H-bond network can serve as a proton-transfer pathway (Figure 7B).
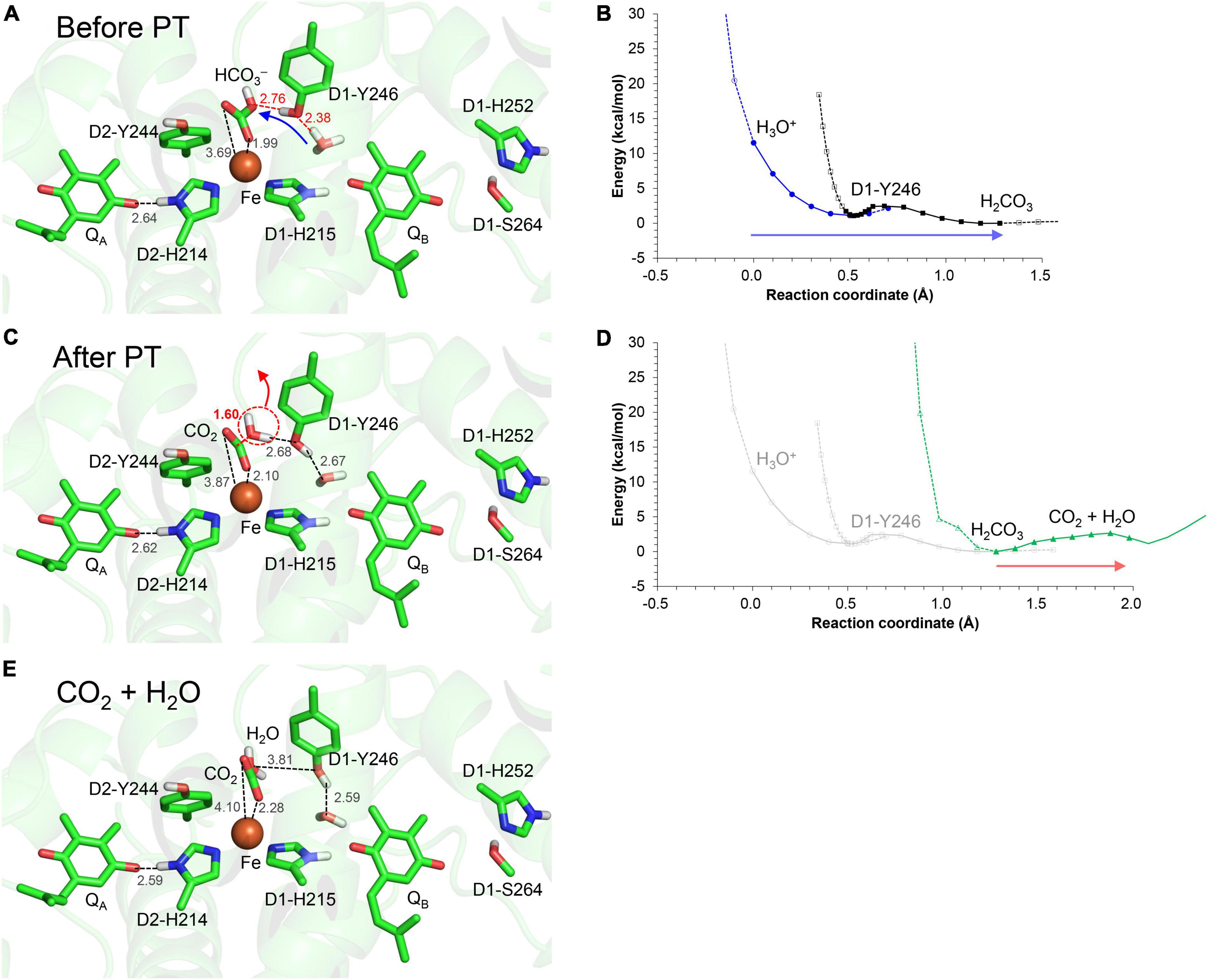
Figure 7. H-bond network via D1-Tyr246 to the monodentate bicarbonate ligand. (A) QM/MM-optimized geometry before proton transfer. (B) Potential energy profile for proton transfer along the H3O+…D1-Tyr246…bicarbonate H-bond network. (C) QM/MM-optimized geometry after proton transfer. (D) Potential energy profile for CO2 release from the monodentate binding site. (E) QM/MM-optimized geometry after decomposition to H2O and CO2. Blue arrows indicate the orientations of the proton transfer. Red arrows indicate the orientations of the structural changes. Note that the geometry of each intermediate conformation is fully QM/MM optimized.
Intriguingly, when the proton arrives at the bicarbonate moiety along the Grotthuss-like proton conduit, the bicarbonate undergoes protonation (Figure 7C) followed by conversion to H2O and CO2 (Figure 7D). As the products move away from the Fe2+ site, the shape of O…C…O becomes linear, i.e., O = C = O (Figure 7E). Thus, the bicarbonate loss can occur through the conversion of the protonated bicarbonate to CO2 and H2O at the Fe2+ moiety (Figure 7D), which is in line with the CO2 release observed on the electron-acceptor side (Shevela et al., 2020).
The bidentate-to-monodentate reorientation of the bicarbonate ligand is a low-barrier process (Figure 3). In the monodentate bicarbonate ligand, the H3O+…D1-Tyr246…HCO3–/QA⋅– H-bond network is formed. The hydroxyl group forms a Grotthuss-like proton transfer pathway, accepting the proton from H3O+ and donating it to the bicarbonate. That is, D1-Tyr246 remains protonated during proton transfer, as suggested using Fourier transform infrared (FTIR) spectroscopy (Takahashi et al., 2009). The absence of the proton donor (H3O+), proton-conducting side chain (D1-Tyr246), and decomposable carboxylic ligand (bicarbonate) in PbRC implies that the proton-mediated QA⋅– stabilization occurs specifically in O2-evolving PSII for photoprotection. The involvement of D1-Tyr246 in the photoprotective role (proton-mediated QA⋅– stabilization) is in line with the findings of mutational studies; the photoautotrophic growth was slower in the D1-Y246F mutant PSII than in wild-type PSII (Kless et al., 1994); the D1-Y246A mutant PSII was also unable to grow photoautotrophically under strong light (Forsman et al., 2019).
The polar –OH region of D1-Tyr246 on the surface of the QB binding pocket can mediate the conduction of the proton to the bicarbonate binding site. The low-barrier bidentate-to-monodentate reorientation contributes to the formation of the “proton wire” (Stuchebrukhov, 2009) between D1-Tyr246 and the unligated –OH site of the bicarbonate. The corresponding H-bond formation with D1-Tyr246 does not occur upon the replacement of bicarbonate with formate, because formate has no –OH site. The absence of the –OH site may also be an origin of why formate substitution inhibits electron transfer from QA to QB (Eaton-Rye and Govindjee, 1988) or the exchange of QBH2 (Sedoud et al., 2011). Thus, the hydrophobic and pre-organized protein electrostatic environment (Warshel, 1998; Warshel et al., 2006) on the bicarbonate binding site is likely to reduce the energy barrier for the bicarbonate protonation with respect to the bulk region, facilitating the direct protonation of the –OH site of the bicarbonate and the release of the product, gaseous CO2, without the formation of the typical (HO)2C = O intermediate (Loerting et al., 2000).
Molecular dynamics simulations suggested that the polar –OH site of the bicarbonate is linked to water molecules in the QB binding pocket via D1-Tyr246 during QBH2 release (Sugo et al., 2022). These water molecules serve as a proton transfer pathway for the reprotonation of D1-His215 (Sugo et al., 2022), the proton donor during QBH–/QBH2 conversion (Saito et al., 2013, 2020). Thus, the link between the polar –OH site and D1-Tyr246 may provide a key to understand how the bicarbonate is associated with QBH2 release, interacting with water molecules in the reprotonation pathway for D1-His215.
Hydrophobic residues that form a CO2-releasing channel
QM/MM calculations suggest that the product, CO2, enters the channel that proceeds via D1-Glu244 and D1-Tyr246 toward the stromal protein surface (Figure 4). The entrance of the channel is hydrophobic, being formed by the side chains of D1-Tyr246, D2-Phe235, and D1-Ile248.
D1-Tyr246 may play two roles in the protonation of bicarbonate. While the polar –OH region mediates the conduction of the proton to the bicarbonate protonation site, the hydrophobic ring region can isolate the bicarbonate from the water molecules in the QB binding pocket, making the bicarbonate binding moiety hydrophobic. The loss of the solvation also increases pKa(HCO3–/H2CO3) with respect to the bulk water region, facilitating the conversion of bicarbonate to gaseous CO2.
Conclusion
In response to the loss of the bicarbonate ligand, a short low-barrier H-bond forms between D2-His214 and QA⋅–, which facilitates the proton migration toward QA⋅– and increases Em(QA) (Figure 2). The loss of bicarbonate may be induced by the protonation. The D1-Glu244 conformation identified in the PSII crystal structure does not support the role in donating a proton to the bicarbonate ligand, whereas D1-Tyr246 may facilitate bicarbonate protonation and decomposition into H2O and CO2 (Figure 7). The channel, which is formed by D1-Tyr246, D2-Phe235, and D1-Ile248, may serve as a CO2-releasing channel (Figure 4). These constitute the basis for the photoprotection mechanism through the high-Em(QA) conformation, which inhibits the backward electron transfer from QA⋅– via the triplet-generating pheophytin/chlorophyll cofactors (Figure 7). The proton-mediated photoprotection mechanism is pronounced exclusively in PSII, as the bicarbonate ligand is replaced with Glu-M234 in PbRC from Rhodobacter sphaeroides (Figure 8). The mechanism of the low-barrier H-bond formation caused by the loss of the bicarbonate ligand may also be associated with the QAH2 formation under high light in PSII (Vass et al., 1992; Noguchi, 2002), as Glu-M234 exists permanently and QA never leaves in PbRC (Wei et al., 2022). These findings could elucidate how the type-II reaction centers show notable structural differences not only on the lumenal oxygen-evolving side but also on the stromal electron acceptor side.
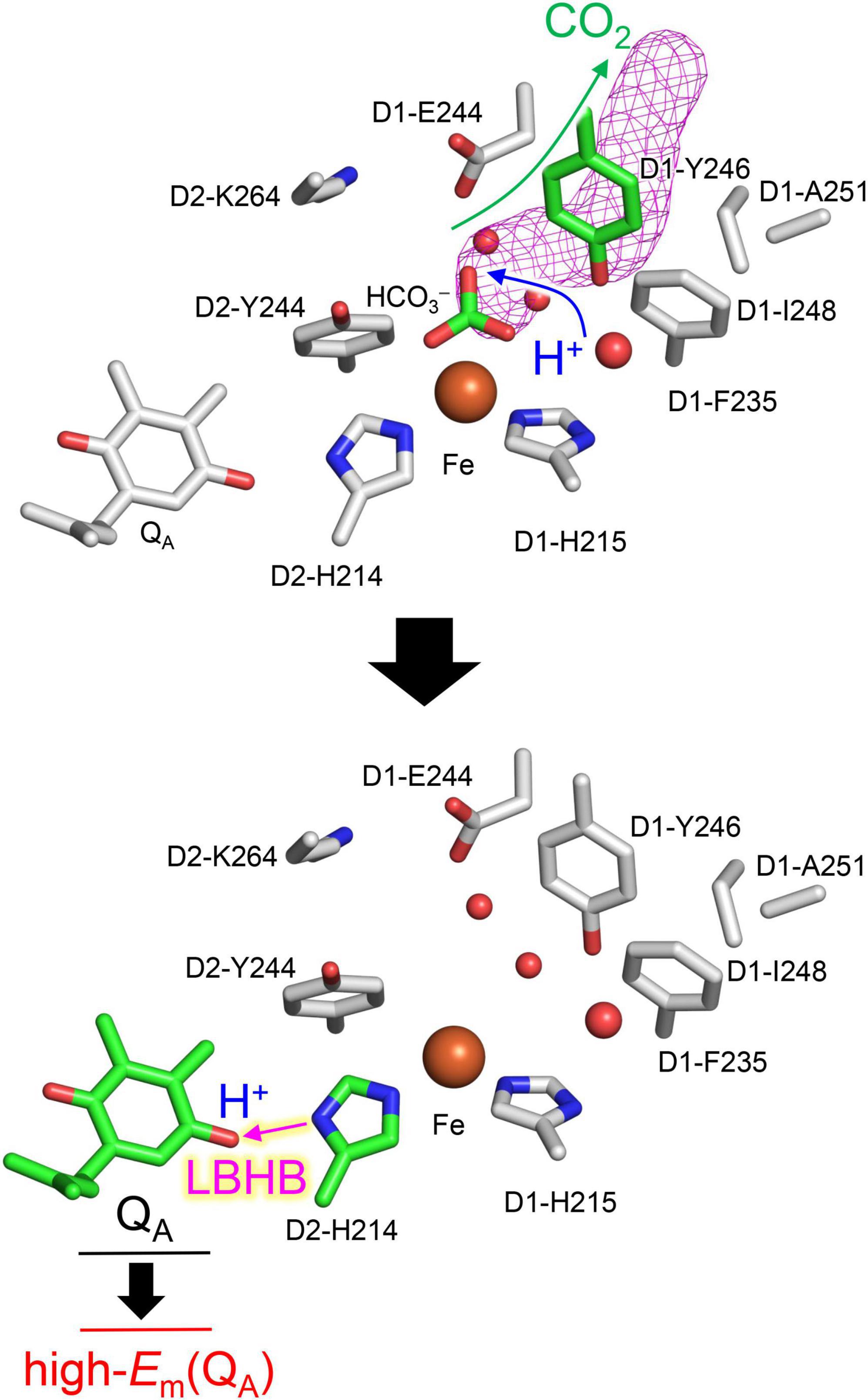
Figure 8. Overview of the proton-mediated photoprotection mechanism in photosystem II.
Data availability statement
The original contributions presented in this study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
HI designed the research and wrote the manuscript. YS and HI performed the research and analyzed the data. Both authors contributed to the article and approved the submitted version.
Funding
This research was supported by JST CREST (JPMJCR1656 to HI), JSPS KAKENHI (JP18H01937, JP18H05155, JP20H03217, and JP20H05090 to HI), and Interdisciplinary Computational Science Program in CCS, University of Tsukuba.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.934736/full#supplementary-material
References
Ashizawa, R., and Noguchi, T. (2014). Effects of hydrogen bonding interactions on the redox potential and molecular vibrations of plastoquinone as studied using density functional theory calculations. Phys. Chem. Chem. Phys. 16, 11864–11876. doi: 10.1039/c3cp54742f
Brinkert, K., De Causmaecker, S., Krieger-Liszkay, A., Fantuzzi, A., and Rutherford, A. W. (2016). Bicarbonate-induced redox tuning in Photosystem II for regulation and protection. Proc. Natl. Acad. Sci. U.S.A. 113, 12144–12149. doi: 10.1073/pnas.1608862113
Brooks, B. R., Bruccoleri, R. E., Olafson, B. D., States, D. J., Swaminathan, S., and Karplus, M. (1983). CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4, 187–217. doi: 10.1002/jcc.540040211
Chernev, P., Zaharieva, I., Dau, H., and Haumann, M. (2011). Carboxylate shifts steer interquinone electron transfer in photosynthesis. J. Biol. Chem. 286, 5368–5374. doi: 10.1074/jbc.M110.202879
Chovancova, E., Pavelka, A., Benes, P., Strnad, O., Brezovsky, J., Kozlikova, B., et al. (2012). CAVER 3.0: a tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput. Biol. 8:e1002708. doi: 10.1371/journal.pcbi.1002708
De Causmaecker, S., Douglass, J. S., Fantuzzi, A., Nitschke, W., and Rutherford, A. W. (2019). Energetics of the exchangeable quinone QB in photosystem II. Proc. Natl. Acad. Sci. U.S.A. 116, 19458–19463. doi: 10.1073/pnas.1910675116
Diner, B. A., and Petrouleas, V. (1987). Q400, the non-heme iron of the photosystem II iron-quinone complex. A spectroscopic probe of quinone and inhibitor binding to the reaction center. Biochim. Biophys. Acta 895, 107–125. doi: 10.1016/S0304-4173(87)80010-1
Eaton-Rye, J. J., and Govindjee. (1988). Electron transfer through the quinone acceptor complex of photosystem II after one or two actinic flashes in bicarbonate-depleted spinach thylakoid membranes. Biochim. Biophys. Acta 935, 248–257. doi: 10.1016/0005-2728(88)90221-6
Ferreira, K. N., Iverson, T. M., Maghlaoui, K., Barber, J., and Iwata, S. (2004). Architecture of the photosynthetic oxygen-evolving center. Science 303, 1831–1838. doi: 10.1126/science.1093087
Forsman, J. A., and Eaton-Rye, J. J. (2020). The D1:Ser268 residue of Photosystem II contributes to an alternative pathway for QB protonation in the absence of bound bicarbonate. FEBS Lett. 594, 2953–2964. doi: 10.1002/1873-3468.13880
Forsman, J. A., Vass, I., and Eaton-Rye, J. J. (2019). D1:Glu244 and D1:Tyr246 of the bicarbonate-binding environment of Photosystem II moderate high light susceptibility and electron transfer through the quinone-Fe-acceptor complex. Biochim. Biophys. Acta 1860:148054. doi: 10.1016/j.bbabio.2019.07.009
Gisriel, C. J., Wang, J., Liu, J., Flesher, D. A., Reiss, K. M., Huang, H.-L., et al. (2022). High-resolution cryo-electron microscopy structure of photosystem II from the mesophilic cyanobacterium, Synechocystis sp. PCC 6803. Proc. Natl. Acad. Sci. U.S.A. 119:e2116765118. doi: 10.1073/pnas.2116765118
Grimsley, G. R., Scholtz, J. M., and Pace, C. N. (2009). A summary of the measured pK values of the ionizable groups in folded proteins. Protein Sci. 18, 247–251. doi: 10.1002/pro.19
Hasegawa, R., Saito, K., Takaoka, T., and Ishikita, H. (2017). pKa of ubiquinone, menaquinone, phylloquinone, plastoquinone, and rhodoquinone in aqueous solution. Photosynth. Res. 133, 297–304. doi: 10.1007/s11120-017-0382-y
Hay, P. J., and Wadt, W. R. (1985). Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 82, 299–310. doi: 10.1063/1.448975
Hienerwadel, R., and Berthomieu, C. (1995). Bicarbonate binding to the non-heme iron of photosystem II investigated by Fourier transform infrared difference spectroscopy and 13C-labeled bicarbonate. Biochemistry 34, 16288–16297. doi: 10.1021/bi00050a008
Hsueh, K. L., Westler, W. M., and Markley, J. L. (2010). NMR investigations of the Rieske protein from Thermus thermophilus support a coupled proton and electron transfer mechanism. J. Am. Chem. Soc. 132, 7908–7918. doi: 10.1021/ja1026387
Ishikita, H., and Knapp, E.-W. (2005a). Control of quinone redox potentials in photosystem II: electron transfer and photoprotection. J. Am. Chem. Soc. 127, 14714–14720. doi: 10.1021/ja052567r
Ishikita, H., and Knapp, E.-W. (2005b). Oxidation of the non-heme iron complex in photosystem II. Biochemistry 44, 14772–14783. doi: 10.1021/bi051099v
Ishikita, H., and Saito, K. (2014). Proton transfer reactions and hydrogen-bond networks in protein environments. J. R. Soc. Interface 11:20130518. doi: 10.1098/rsif.2013.0518
Johnson, G. N., Rutherford, A. W., and Krieger, A. (1995). A change in the midpoint potential of the quinone QA in Photosystem II associated with photoactivation of oxygen evolution. Biochim. Biophys. Acta 1229, 202–207. doi: 10.1016/0005-2728(95)00003-2
Jursinic, P., Warden, J., and Govindjee. (1976). A major site of bicarbonate effect in system II reaction. Evidence from ESR signal IIvf, fast fluorescence yield changes and delayed light emission. Biochim. Biophys. Acta 440, 322–330. doi: 10.1016/0005-2728(76)90066-9
Kato, Y., Nagao, R., and Noguchi, T. (2016). Redox potential of the terminal quinone electron acceptor QB in photosystem II reveals the mechanism of electron transfer regulation. Proc. Natl. Acad. Sci. U.S.A. 113, 620–625. doi: 10.1073/pnas.1520211113
Kato, Y., Watanabe, H., and Noguchi, T. (2021). ATR-FTIR spectroelectrochemical study on the mechanism of the pH dependence of the redox potential of the non-heme iron in photosystem II. Biochemistry 60, 2170–2178. doi: 10.1021/acs.biochem.1c00341
Kimura, M., Kato, Y., and Noguchi, T. (2020). Protonation state of a key histidine ligand in the iron–quinone complex of photosystem II as revealed by light-induced ATR-FTIR spectroscopy. Biochemistry 59, 4336–4343. doi: 10.1021/acs.biochem.0c00810
Kless, H., Oren-Shamir, M., Malkin, S., McIntosh, L., and Edelman, M. (1994). The D-E region of the D1 protein is involved in multiple quinone and herbicide interactions in photosystem II. Biochemistry 33, 10501–10507. doi: 10.1021/bi00200a035
Krieger, A., and Weis, E. (1992). Energy dependent quenching of chlorophyll a fluorescence: the involvement of proton-calcium exchange at photosystem II. Photosynthetica 27, 89–98.
Krieger, A., Rutherford, A. W., and Johnson, G. N. (1995). On the determination of redox midpoint potential of the primary quinone electron transfer acceptor, QA, in photosystem II. Biochim. Biophys. Acta 1229, 193–201. doi: 10.1016/0005-2728(95)00002-Z
Limbach, H.-H., Tolstoy, P. M., Pérez-Hernández, N., Guo, J., Shenderovich, I. G., and Denisov, G. S. (2009). OHO hydrogen bond geometries and NMR chemical shifts: from equilibrium structures to geometric H/D isotope effects, with applications for water, protonated water, and compressed ice. Isr. J. Chem. 49, 199–216. doi: 10.1560/IJC.49.2.199
Loerting, T., Tautermann, C., Kroemer, R. T., Kohl, I., Hallbrucker, A., Mayer, E., et al. (2000). On the surprising kinetic stability of carbonic acid (H2CO3). Angew. Chem. Int. Ed. 39, 891–894. doi: 10.1002/(sici)1521-3773(20000303)39:5<891::aid-anie891>3.0.co;2-e
MacKerell, A. D. Jr., Bashford, D., Bellott, R. L., Dunbrack, R. L. Jr., Evanseck, J. D., Field, M. J., et al. (1998). All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616. doi: 10.1021/jp973084f
Mikenda, W. (1986). Stretching frequency versus bond distance correlation of O-D(H)⋯Y (Y = N, O, S, Se, Cl, Br, I) hydrogen bonds in solid hydrates. J. Mol. Struct. 147, 1–15. doi: 10.1016/0022-2860(86)87054-5
Muh, F., Glockner, C., Hellmich, J., and Zouni, A. (2012). Light-induced quinone reduction in photosystem II. Biochim. Biophys. Acta 1817, 44–65. doi: 10.1016/j.bbabio.2011.05.021
Nakamura, S., and Noguchi, T. (2017). Infrared determination of the protonation state of a key histidine residue in the photosynthetic water oxidizing center. J. Am. Chem. Soc. 139, 9364–9375. doi: 10.1021/jacs.7b04924
Noguchi, T. (2002). Dual role of triplet localization on the accessory chlorophyll in the photosystem II reaction center: photoprotection and photodamage of the D1 protein. Plant Cell Physiol. 43, 1112–1116. doi: 10.1093/pcp/pcf137
Rutherford, A. W., and Krieger-Liszkay, A. (2001). Herbicide-induced oxidative stress in photosystem II. Trends Biochem. Sci. 26, 648–653. doi: 10.1016/s0968-0004(01)01953-3
Rutherford, A. W., Osyczka, A., and Rappaport, F. (2012). Back-reactions, short-circuits, leaks and other energy wasteful reactions in biological electron transfer: redox tuning to survive life in O2. FEBS Lett. 586, 603–616. doi: 10.1016/j.febslet.2011.12.039
Saito, K., Mandal, M., and Ishikita, H. (2020). Redox potentials along the redox-active low-barrier H-bonds in electron transfer pathways. Phys. Chem. Chem. Phys. 22, 25467–25473. doi: 10.1039/d0cp04265j
Saito, K., Rutherford, A. W., and Ishikita, H. (2013). Mechanism of proton-coupled quinone reduction in Photosystem II. Proc. Natl. Acad. Sci. U.S.A. 110, 954–959. doi: 10.1073/pnas.1212957110
Saito, K., Rutherford, A. W., and Ishikita, H. (2015). Energetics of proton release on the first oxidation step in the water-oxidizing enzyme. Nat. Commun. 6:8488. doi: 10.1038/ncomms9488
Sedoud, A., Kastner, L., Cox, N., El-Alaoui, S., Kirilovsky, D., and Rutherford, A. W. (2011). Effects of formate binding on the quinone-iron electron acceptor complex of photosystem II. Biochim. Biophys. Acta 1807, 216–226. doi: 10.1016/j.bbabio.2010.10.019
Shen, J. R. (2015). The structure of photosystem II and the mechanism of water oxidation in photosynthesis. Annu. Rev. Plant. Biol. 66, 23–48. doi: 10.1146/annurev-arplant-050312-120129
Shevela, D., Do, H.-N., Fantuzzi, A., Rutherford, A. W., and Messinger, J. (2020). Bicarbonate-mediated CO2 formation on both sides of photosystem II. Biochemistry 59, 2442–2449. doi: 10.1021/acs.biochem.0c00208
Shevela, D., Eaton-Rye, J. J., Shen, J. R., and Govindjee. (2012). Photosystem II and the unique role of bicarbonate: a historical perspective. Biochim. Biophys. Acta 1817, 1134–1151. doi: 10.1016/j.bbabio.2012.04.003
Shibamoto, T., Kato, Y., Sugiura, M., and Watanabe, T. (2009). Redox potential of the primary plastoquinone electron acceptor QA in photosystem II from Thermosynechococcus elongatus determined by spectroelectrochemistry. Biochemistry 48, 10682–10684. doi: 10.1021/bi901691j
Siggel, U., Khanna, R., Renger, G., and Govindjee. (1977). Investigation of the absorption changes of the plasto-quinone system in broken chloroplasts. The effect of bicarbonate-depletion. Biochim. Biophys. Acta 462, 196–207. doi: 10.1016/0005-2728(77)90202-x
Stuchebrukhov, A. A. (2009). Mechanisms of proton transfer in proteins: localized charge transfer versus delocalized soliton transfer. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 79:031927. doi: 10.1103/PhysRevE.79.031927
Suga, M., Akita, F., Hirata, K., Ueno, G., Murakami, H., Nakajima, Y., et al. (2015). Native structure of photosystem II at 1.95 Å resolution viewed by femtosecond X-ray pulses. Nature 517, 99–103. doi: 10.1038/nature13991
Sugo, Y., Saito, K., and Ishikita, H. (2021). Mechanism of the formation of proton transfer pathways in photosynthetic reaction centers. Proc. Natl. Acad. Sci. U.S.A. 118:e2103203118.
Sugo, Y., Saito, K., and Ishikita, H. (2022). Conformational changes and H-bond rearrangements during quinone release in photosystem II. Biochemistry doi: 10.1021/acs.biochem.1022c00324
Takahashi, R., Boussac, A., Sugiura, M., and Noguchi, T. (2009). Structural coupling of a tyrosine side chain with the non-heme iron center in photosystem II as revealed by light-induced Fourier transform infrared difference spectroscopy. Biochemistry 48, 8994–9001. doi: 10.1021/bi901195e
Umena, Y., Kawakami, K., Shen, J.-R., and Kamiya, N. (2011). Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 473, 55–60.
Uto, S., Kawakami, K., Umena, Y., Iwai, M., Ikeuchi, M., Shen, J. R., et al. (2017). Mutual relationships between structural and functional changes in a PsbM-deletion mutant of photosystem II. Faraday Discuss 198, 107–120. doi: 10.1039/c6fd00213g
Vacek, G., Perry, J. K., and Langlois, J. M. (1999). Advanced initial-guess algorithm for self-consistent-field calculations on organometallic systems. Chem. Phys. Lett. 310, 189–194.
Vass, I., Styring, S., Hundal, T., Koivuniemi, A., Aro, E. M., and Andersson, B. (1992). Reversible and irreversible intermediates during photoinhibition of photosystem II: stable reduced QA species promote chlorophyll triplet formation. Proc. Natl. Acad. Sci. U.S.A. 89, 1408–1412. doi: 10.1073/pnas.89.4.1408
Warshel, A. (1998). Electrostatic origin of the catalytic power of enzymes and the role of preorganized active sites. J. Biol. Chem. 273, 27035–27038. doi: 10.1074/jbc.273.42.27035
Warshel, A., Sharma, P. K., Kato, M., and Parson, W. W. (2006). Modeling electrostatic effects in proteins. Biochim. Biophys. Acta 1764, 1647–1676.
Wei, R. J., Zhang, Y., Mao, J., Kaur, D., Khaniya, U., and Gunner, M. R. (2022). Comparison of proton transfer paths to the QA and QB sites of the Rb. sphaeroides photosynthetic reaction centers. Photosynth. Res. doi: 10.1007/s11120-11022-00906-x
Keywords: photoprotection, photoinhibition, low-barrier hydrogen bond (LBHB), bicarbonate, proton-coupled electron transfer (PCET), formate, D1-Tyr246, QBH2 release/exchange
Citation: Sugo Y and Ishikita H (2022) Proton-mediated photoprotection mechanism in photosystem II. Front. Plant Sci. 13:934736. doi: 10.3389/fpls.2022.934736
Received: 03 May 2022; Accepted: 08 August 2022;
Published: 07 September 2022.
Edited by:
Roland Valcke, University of Hasselt, BelgiumReviewed by:
Juan B. Arellano, Spanish National Research Council (CSIC), SpainJulian Eaton-Rye, University of Otago, New Zealand
Copyright © 2022 Sugo and Ishikita. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hiroshi Ishikita, aGlyb0BhcHBjaGVtLnQudS10b2t5by5hYy5qcA==