 Man Zhang
Man Zhang Wenhui Cheng
Wenhui Cheng Xi Yuan
Xi Yuan Tangren Cheng
Tangren Cheng Qixiang Zhang
Qixiang Zhang- Beijing Key Laboratory of Ornamental Plants Germplasm Innovation and Molecular Breeding, Beijing Laboratory of Urban and Rural Ecological Environment, Key Laboratory of Genetics and Breeding in Forest Trees and Ornamental Plants of Ministry of Education, National Engineering Research Center for Floriculture, School of Landscape Architecture, Beijing Forestry University, Beijing, China
MicroRNAs is one class of small non-coding RNAs that play important roles in plant growth and development. Though miRNAs and their target genes have been widely studied in many plant species, their functional roles in floral bud break and dormancy release in woody perennials is still unclear. In this study, we applied transcriptome and small RNA sequencing together to systematically explore the transcriptional and post-transcriptional regulation of floral bud break in P. mume. Through expression profiling, we identified a few candidate genes and miRNAs during different developmental stage transitions. In total, we characterized 1,553 DEGs associated with endodormancy release and 2,084 DEGs associated with bud flush. Additionally, we identified 48 known miRNAs and 53 novel miRNAs targeting genes enriched in biological processes such as floral organ morphogenesis and hormone signaling transudation. We further validated the regulatory relationship between differentially expressed miRNAs and their target genes combining computational prediction, degradome sequencing, and expression pattern analysis. Finally, we integrated weighted gene co-expression analysis and constructed miRNA-mRNA regulatory networks mediating floral bud flushing competency. In general, our study revealed the miRNA-mediated networks in modulating floral bud break in P. mume. The findings will contribute to the comprehensive understanding of miRNA-mediated regulatory mechanism governing floral bud break and dormancy cycling in wood perennials.
Introduction
Bud dormancy is an important adaptive strategy for perennial plant species to survive harsh environmental conditions (Rohde and Bhalerao, 2007). Perennial trees in temperate regions went through seasonal cycling of growth and dormancy to preserve vegetative or reproductive primordium during harsh winters (Yang et al., 2021). Bud dormancy can be classified into three categories: paradormancy, endodormancy, and ecodormancy (Lang et al., 1987). Paradormancy is known as growth inhibition controlled by internal hormones and organ competition. Endodormancy is defined as inhibited bud growth that can only be overcome by chilling temperatures. Bud endodormancy is usually triggered by shortening photoperiod and/or low temperature (Cooke et al., 2012; van der Schoot et al., 2013). After accumulating sufficient chilling, endodormant buds enter ecodormancy state and acquire competence to resume vegetative growth or flowering in suitable conditions (Rinne et al., 2011; van der Schoot et al., 2013). Unlike annual or biennial plants, floral initiation and blooming is interposed by bud dormancy (Ramos et al., 2018; Hsiang et al., 2021). In Prunus species, floral induction started in late summer with reproductive organs differentiating and developing before dormancy period (Julian et al., 2011). When the chilling requirement is fulfilled, the arrested floral tissues continue to grow and mature, followed by the subsequent production of gametes and blooming in the following spring (Lloret et al., 2018). In the background of global climate change, warm winters and irregular weather changes have been disrupting important phenological events, such as timing of bud break and flowering (Hatfield and Prueger, 2015). Insufficient chilling in floral buds can lead to erratical flowering, deformed flowers, and reduced fruit set (Luedeling, 2012; Ladwig et al., 2019). Therefore, it is important to understand the regulation mechanism underlying floral bud break and flowering in perennial trees.
Many recent studies have been conducted in elucidating the molecular control of floral bud dormancy cycling in deciduous fruit trees, such as pear, peach, apple (Yang et al., 2021). Till now, a large number of transcription factors were characterized in regulating floral bud dormancy and development. For example, DAM (DORMANCY ASSOCIATED MADS-BOX) transcription factors were firstly identified in the evergrowing peach mutants and were found essential for dormancy establishment and maintenance (Yamane et al., 2011). The over-expression of DAM genes led to delayed bud break in apple (Rongmei et al., 2017). CBFs (C-repeat binding factors) is a group of transcription factors that regulate DAM genes by binding to their promoter regions upon cold induction (Niu et al., 2016). In peach, the TCP (TEOSINTE BRANCHED1/CYCLOIDEA/PROLIFERATING CELL FACTOR) transcription factor, TCP20 can interact with ABF2 (ABSCISIC ACID RESPONSIVE ELEMENTS-BINDING FACTOR 2) and promote dormancy release by repressing DAM5 and DAM6 expression (Leubner et al., 2020). Additionally, HD-ZIP family transcription factors are also involved in regulating bud dormancy. For example, HB22 (HOMEOBOX PROTEIN 22) regulates DAM6 expression by binding the promoter of DAM1 in pear (Yang et al., 2018). Epigenetic modifications also participate in regulating bud dormancy transition. It is reported that the modified level of H3K27 trimethylation and H3K4 trimethylation were associated with DAM gene transcription during bud break (Zhu et al., 2020). Despite the advancements in understanding transcriptional regulation, how post-transcriptional mechanism is involved in regulating floral bud break in temperate perennials still remains elusive.
MicroRNAs (miRNAs) is a class of endogenous, small non-coding RNA molecules of 18–24 nucleotides that can modulate gene expression post-transcriptionally in both plants and animals (Rubio-Somoza and Weigel, 2011; Spanudakis and Jackson, 2014). MiRNAs bind to the coding region or UTR region of target genes through near-perfect base complementation and induced target gene silencing by endonucleolytic cleavage in plants or translational inhibition in animals (Yu et al., 2017). MiRNAs was first transcribed by RNA polymerase II into primary miRNAs (pri-miRNA), which were cropped into the stem-loop structured precursors (pre-miRNAs), exported to cytoplasm by exportin, and subsequently cleaved by cytoplasmic RNase III Dicer, generating miRNA duplex (Kim, 2004; Lee et al., 2004; Strzyz, 2021). The unwinding duplex was further incorporated into AGO proteins to assemble RISC (RNA-induced silencing complex) that targets mRNA for cleavage or translation repression (Betancur et al., 2012; Strzyz, 2021). In plants, miRNAs play a central role in many developmental processes including patterning of leaf and flower organs, timing of phase transition, seed development, biotic and abiotic responses through targeting transcriptional factor encoding genes (D’Ario et al., 2017; Fang and Wang, 2021). The role of miRNAs in chilling-induced dormancy release has been investigated in a few woody perennials (Yu et al., 2021). In Pyrus pyrifolia, miRNA6390 was found to target DAM gene transcripts for degradation and promote dormancy release (Niu et al., 2016). MiR160, involved in auxin signaling pathway, was found significantly induced after chilling treatment in dormant cambium tissues in poplar (Ding et al., 2014). In tea tree, miR169, miR408, miR414, and miR782 were found up-regulated and miR828, miR1864, miR852, and miR1425 were down-regulated as vegetative buds exit dormancy (Jeyaraj et al., 2014). A recent study of ncRNAs (non-coding RNAs) during dormancy transition in peach identified two essential miRNAs, miR6286 targeting ASPARAGINE-RICH PROTEIN encoding gene involved in abscisic acid signaling and miR2275 that regulates anther microsporogenesis at the stage of dormancy release (Yu et al., 2021). So far, the molecular function of miRNAs during floral bud dormancy transition has not been fully examined yet.
Prunus mume Sieb. et Zucc., also known as Japanese apricot, is one of the world’s important perennial fruit crops (Wu et al., 2019). P. mume was first originated from southern China and has a domestication history over 1,000 years (Gao et al., 2012). The fruits of P. mume have many health-beneficial physiochemicals and can be processed into beverage, jams, or used for culinary purposes (Kim et al., 2018; Bailly, 2020). Moreover, being one of the earliest flowering woody perennial, P. mume encompass a great number of varieties and has been widely used for ornamental and landscape purposes (Zhang Q. et al., 2018). Despite extensive efforts in understanding the genetic control of bud break and blooming in P. mume, the molecular network is far from complete (Zhao et al., 2018a,b; Zhang et al., 2021b). The knowledge of dormancy release regulation in P. mume will facilitate our understanding into the post-transcriptional regulation of bud dormancy cycling in temperate tree species (Zhang et al., 2021b). In this study, we applied transcriptome and small RNA sequencing analysis to analyze the global expression change of mRNAs and miRNAs during floral bud break in P. mume. By combining computational prediction and degradome sequencing, we validated the regulatory relationship between miRNAs and their targeting genes. With weighted co-expression analysis, we screened out a number of miRNA-mRNA regulatory pairs and constructed the miRNA mediated regulatory networks that associated with chilling-induced floral bud break in P. mume. In general, our study provided novel candidate microRNAs and regulatory genes for future functional investigations.
Materials and methods
Dormancy status evaluation
Floral bud phenology was surveyed for P. mume cultivar “Subaitaige” grown in Jiufeng International Germplasm Garden (Beijing, China) from October until full bloom during 2020–2021. The air temperature was recorded hourly since October in 2021. To assess the dormancy status of floral bud, we performed bud break competency tests every 2 weeks following the method described by Ionescu et al. (2017). One-year old shoots with at least 10 lateral floral buds were collected every 2 weeks, immersed in water, and incubated in the phytotron under forcing conditions (16 h day/8 h night, 22°C/20°C, 70% relative humidity). The basal part of all branches was cut every week to avoid xylem clogging and the water was changed every 2 days. Bud break rate (BBR) was measured as the percentage of floral buds that flushed after 2 weeks incubation. To evaluate the chilling requirement, we determined the time point of endodormancy release as the date when 50% of all floral buds become competent to flush under forcing conditions (Hsiang et al., 2021). Chilling hours was calculated based on the chilling hour model (0∼7.2°C) to measure the progression of chilling accumulation.
Sample collection
Floral buds were collected at four developmental stages (endodormancy I, endodormancy II, ecodormancy, and bud flush) with three biological replicates during bud break process. Samples were instantly frozen in liquid nitrogen and total RNA was extracted from the flower buds using Trizol reagent kit (Invitrogen, Carlsbad, CA, United States) according to the manufacturer’s protocol. The quantity and purity of total RNA was assessed with Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, United States) and verified on 1% RNase free agarose gel.
Transcriptome sequencing and analysis
The sample mRNA was enriched with Oligo (dT) beads, reverse transcribed, and prepared to construct cDNA libraries. Twelve sequencing libraries were sequenced on the Illumina HiSeq™ 2500 platform in paired-end mode (Gene Denovo Biotechnology Co., Guangzhou, China). Raw reads were obtained and low quality reads were filtered with software fastp (version 0.18.0) (Chen et al., 2018). The raw data was uploaded into the NCBI Sequence Read Archive (accession number: PRJNA833165). Clean paired-end reads were aligned to the genome of P. mume (Zheng et al., 2021) with HISAT2 (Kim et al., 2015). Based on the alignment, transcript abundance was estimated using StringTie v1.3.1 (Pertea et al., 2015, 2016). Read counts were normalized to FPKM value (fragment per kilobase of transcript per million mapped reads) using RSEM software (Li and Dewey, 2011). Differentially expressed genes (DEGs) were defined as genes with | log2FC| > 1.5 and adjusted P < 0.05 in each two sample stage comparison using DESeq R package (Love et al., 2014). The expression patterns of the DEGs were visualized with “pheatmap” R package1.
Small RNA sequencing and analysis
Small RNAs of 18–30 nucleotides were isolated from total RNA, reverse-transcribed, and amplified by PCR. Twelve small-RNA sequencing libraries were constructed following recommended protocol and were sequenced on Illumina HiSeq™ 2500 platform by Gene denovo Biotechnology Co., Ltd. (Guangzhou, China). Finally, the sequencing data was deposited in the NCBI Sequence Read Archive (accession number: PRJNA832606).
Raw reads were filtered to remove low quality reads (reads containing bases with Q < 20, reads with more than 5% N, reads with contaminated 5′ adaptor, reads without 3′ adaptor, or reads with polyA tail) and adaptors were trimmed off using software Fastx-toolkit (Gordon and Hannon, 2010). Clean reads were compared with small RNAs in GeneBank database v236.0 (Benson et al., 2007) and Rfam database v14.0 (Griffiths-Jones, 2004) to remove sequence tags annotated as ncRNAs (rRNA, scRNA, snoRNA, snRNA, and tRNA). Additionally, we aligned the clean reads to the genome of P. mume to remove fragments that mapped to exons, introns, or repeated sequences using SOAP v2.20 (Li R. et al., 2009). The remaining tags were searched against miRBase v22.1 database to identify known miRNAs (Griffiths-Jones, 2006). The miRNAs with less than two mismatches or those with at least 16 bp overlap with known miRNAs were assigned to the same miRNA family (Goldstien et al., 2010). Unannotated tags were used to identify novel miRNAs based on their hairpin structures with software miRDeep2 (Friedländer et al., 2012) following standard protocols (Min and Yoon, 2010). The newly identified miRNAs and their precursor sequences were folded into secondary structure and were visualized with RNAfold2.
The transcription level of miRNA was estimated and normalized to TPM (transcripts per million) value following the formula:
The expression levels of known and novel miRNAs were compared between adjacent developmental stages using DESeq2 software. Differentially expressed miRNAs (DEmiRs) were identified as miRNAs with log2(FC) ≥ 1 and FDR corrected p-value < 0.05. To examine the expression patterns of DEmiRs, we visualized the TPM values of DEmiRs using “pheatmap” R package.
MicroRNA-target gene identification
The target genes of miRNAs were predicted by searching against P. mume reference genome using software Patmatch (Yan et al., 2005) based on the following criteria: (1) MFE (minimum free energy) of miRNA-target duplex ≥ 74%; (2) mismatches between the sRNA and their targets (G-U as 0.5 mismatch) ≤ 4; (3) adjacent mismatches allowed in miRNA-target duplex ≤ 2; (4) no adjacent mismatches allowed in position 2–12 of miRNA-target duplex; (5) no mismatches allowed in position 10–11 of miRNA-target duplex; (6) mismatches allowed in position 2–12 of miRNA-target duplex ≤ 2.5. To validate the miRNA-mRNA relationship, equal amounts of RNA from all samples were pooled to construct degradome sequencing library. The cleaved RNA products were ligated with adaptors, reverse transcribed and sequenced on Illumina HiSeq™ 2500 platform. Raw reads from degradome library were processed in a similar fashion as described above and the clean reads annotated as ncRNAs were removed. The remaining clean tags were analyzed with CleaveLand pipeline v3.0 to predict miRNA cleavage sites. Potential target genes were identified as those with alignment score ≤ 4 and no mismatches between 10th and 11th nucleotides of miRNAs. The target genes were classified into five categories based on raw tag sequence abundance within cleavage sites and those within category 0–2 were kept as true targets (Yu et al., 2021). The raw degradome sequencing data was available from NCBI Sequence Read Archive (accession number: PRJNA832060).
Co-expression network analysis of mRNA and miRNAs
To identify co-regulatory genes or miRNAs that associated with the progression of bud dormancy release, we performed weighted gene co-expression network analysis (WGCNA) on all genes and miRNAs separately with WGCNA (v1.47) package in R. WGCNA is a scale-free network construction approach that finds clusters of genes with highly correlated expression profiles (Langfelder and Horvath, 2008). For the gene network construction, we first removed lowly expressed genes and kept only genes with FPKM ≥ 2 across 70% samples and SD > 0.25. Then we estimated the Pearson’s correlation coefficients among genes based on the FPKM values. The correlation matrix was converted into adjacency matrix (soft threshold power = 9). Subsequently, we transformed the adjacency matrix into topological overlap measures (TOM) and performed hierarchical clustering to group all genes into clusters using Dynamic Tree Cut algorithm. Similarly, the weighted co-expression network of miRNAs was constructed to group all miRNAs (TPM ≥ 2 across 70% samples; SD > 0.25) into associated modules following standard WGCNA protocols (soft-threshold power = 8). All genes or miRNAs were assigned to co-expressed modules that were named with colors. The minimum module size was set as 50 and the threshold of the module similarity was set at 0.8. Using bud break rate as phenotype, we identified most relevant modules of genes or miRNAs as candidate regulators determining dormancy status during floral bud break. The hub genes or miRNAs were selected as those with top 0.1% module membership for each relevant module.
MiRNA-mRNA regulatory network construction
To investigate the miRNA-mRNA regulatory mechanism during floral bud break, we performed an integrative analysis on the miRNA and mRNA co-expression networks. We mapped all DEGs and the target genes of DEmiRs identified in previous sections to the co-expressed gene network with their correlations integrated. Differentially expressed transcription factors (TFs) and hub genes were identified from the trait-related gene modules and the miRNA-TF gene pairs were added to the miRNA-mRNA network. Finally, the network was visualized by Cytoscape software (version 3.7.2) (Shannon et al., 2003).
Functional enrichment analysis
To detect the biological function of candidate genes, we performed functional enrichment analysis on the DEG sets and target gene sets of DEmiRs. Genes were mapped to Gene Ontology (GO) terms of biological progress (BP), molecular function (MF), and cell component (CC) in the GO database (Gene Ontology Consortium, 2004). GO terms with FDR corrected p-values < 0.05 were considered as significantly enriched in hypergeometric tests (Yan et al., 2020). P-values were calculated as follows:
Where N is the total number of background genes, n is the number of candidate genes in N, M is the total number of genes annotated to the GO term, and m is the number of candidate genes in M. The p-values were adjusted with FDR (false discovery rate) and the GO terms with adjusted p-values ≤ 0.05 were considered as significantly enriched among the candidate genes. To understand the metabolic or signal transduction pathways, we also mapped candidate gene sets to the KEGG (Kyoto Encyclopedia of Genes and Genomes) database and considered pathways with FDR ≤ 0.05 as significantly enriched following the same hypergeometric tests.
Expression pattern analysis with quantitative real-time PCR
To validate the expression pattern of miRNAs and their target genes, we performed quantitative real-time PCR (qRT-PCR) assays. We first extracted RNA from the same samples with Plant RNA Kit (Omega Bio-Tek, United States) and reverse transcribed using PrimeScript RT Kit (Takara Bio, Japan). The qRT-PCR assays of candidate genes were conducted using TB Green® Premix Ex Taq™ II (Takara Bio, Japan) on CFX96 Real-Time PCR Detection System (Bio-Rad Laboratories, Inc., United States) following protocol: 95°C for 5 min, followed by 40 cycles at 95°C for 5 s, 60°C for 30 s and 72°C for 30 s. The relative gene expression level was computed with 2–ΔΔCt method using PP2A as the reference gene for candidate genes. Small RNA was extracted with Plant miRNA Kit (Omega, China) following the manufacturer’s protocols. Mature miRNAs were reverse transcribed into cDNA using Mir-X™ miRNA First-Strand Synthesis Kit (Code No. 638315, Takara, China). The qPCR was carried out using miRNA SYBR Green RT-qPCR Kit (Takara, China) following similar temperature settings. The relative expression level of miRNA was estimated using 5S rRNA as internal reference. Primer pairs of candidate genes and miRNAs were designed with NCBI primer-blast and were provided (Supplementary Table 1).
Results
Physiological changes during floral bud break
During the progression of floral bud break, we tracked the morphological changes of P. mume “Subaitaige” and evaluated the bud break rate as an indicator of dormancy depth from dormancy induction to bud flush during 2020–2021 (Figure 1A). Based on bud dormancy competency test, we estimated the date of endodormancy break to be between November 29th and October 12th, 2020 and we observed 5% blooming on February 10th, 2021 (Figure 1A). Using the chilling hour model, we calculated total chilling hours (CH) required for cultivar “Subaitage” as approximately 320 CHs, which started to accumulate since October 12th, 2020 (Figure 1B). In P. mume, floral organ initiation was induced in late summer with floral primordium continuing to develop throughout winter (Zhang et al., 2021a). In early November, floral buds were maintained in endodormant state with floral organ primordiums, including sepal, petal, stamen, and pistil fully differentiated. During the chilling accumulation process, endodormant flower buds continued to develop. From late December to January, green sepals were revealed, floral bud enlarged rapidly with floral organs, such as stamen and ovule matured before natural bud flush, suggesting the complete flower organ differentiation after dormancy release (Figure 1A). To understand the molecular dynamics during dormancy transition, we selected samples of four time points corresponding to floral buds of average bud break percentage at 9.1%, 42.1%, 59.02%, and 88.3% for the following analysis (Figure 1B). We termed the four sampling stages as endodormancy I (Endodor I), endodormancy II (Endodor II), ecodormancy (Ecodor), and bud flush (Bflush) on the basis of bud morphology and chilling accumulation.
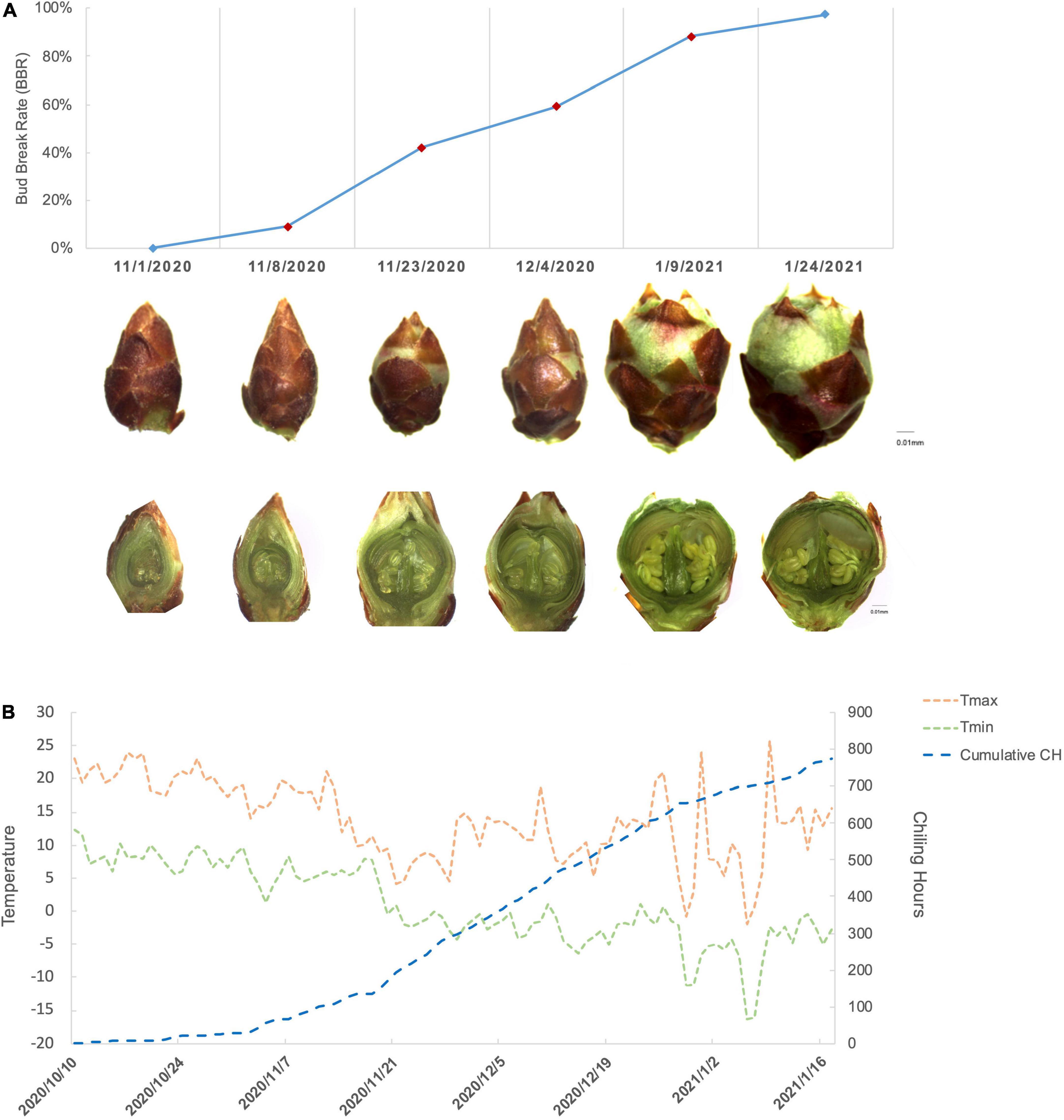
Figure 1. The characterization of floral bud development for P. mume cultivar “Subaitaige” during dormancy release in 2020–2021. (A) The floral bud dormancy status and inner structure changes of floral bud sampled during dormancy release and bud flush. The longitudinal sections of flower buds were compared between sampling time points. (B) The daily maximum temperature, minimum temperature, and cumulative chilling hours were evaluated since October 10th, 2020 till January 24th, 2021 using chilling hour model.
Transcriptome analysis and differentially expressed gene identification
To identify genes that participate in regulating bud dormancy release, we performed RNA-seq analysis on floral bud tissues of four developmental stages. We constructed 12 sequencing libraries and obtained a total of 80.3 Gb raw reads. After quality control, we generated 79.5 Gb clean reads with total mapping rate between 93 and 94% across samples (Table 1). Principal component analysis (PCA) revealed that the first two principal components explained 47.8 and 33.2% of total variation (Supplementary Figure 1A). Pearson correlation among samples indicated that samples of similar physiological state are more closely related (Supplementary Figure 1B). These results confirmed that the transcriptional state is consistent with physiological differences among flower bud samples.
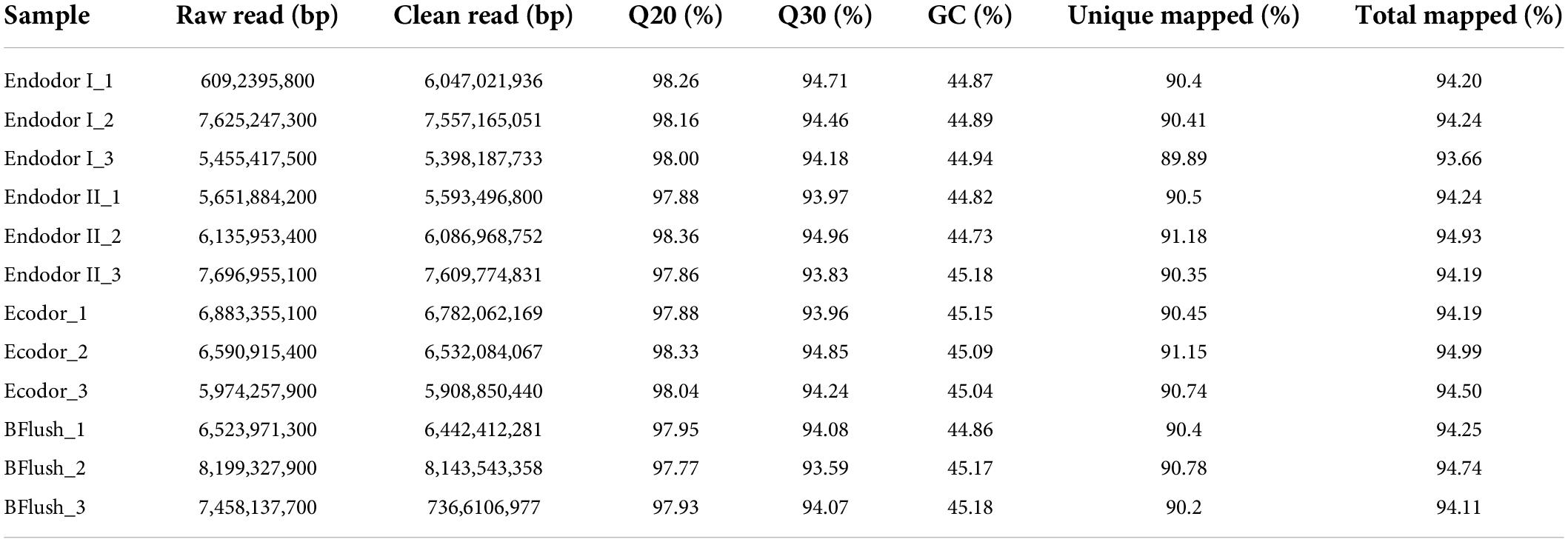
Table 1. Summary table of the transcriptome sequencing data of four developmental stages.
Differential expression analyses were performed by comparing gene expression between adjacent developmental stages. Among 30,082 genes investigated, we detected 2,410 genes differentially expressed between endodormancy stage I and II (Figure 2A). These include 920 up-regulated and 1,490 down-regulated DEGs (Figure 2A). Between endodormancy and ecodormancy, a total of 1,553 DEGs were detected including 896 up-regulated and 657 down-regulated genes (Figure 2A). Additionally, 2,084 DEGs were identified in the comparison between ecodormancy and bud flush stage (Figure 2A). By comparing the DEGs between stage comparisons, we identified 1,328, 503, and 1,161 DEGs that are specific to the comparison endodor I vs. endodor II, endodor II vs. ecodor, and ecodor-vs.-bud flush, respectively (Figure 2B). A total of 287 DEGs were shared by all three stage comparisons (Figure 2B).
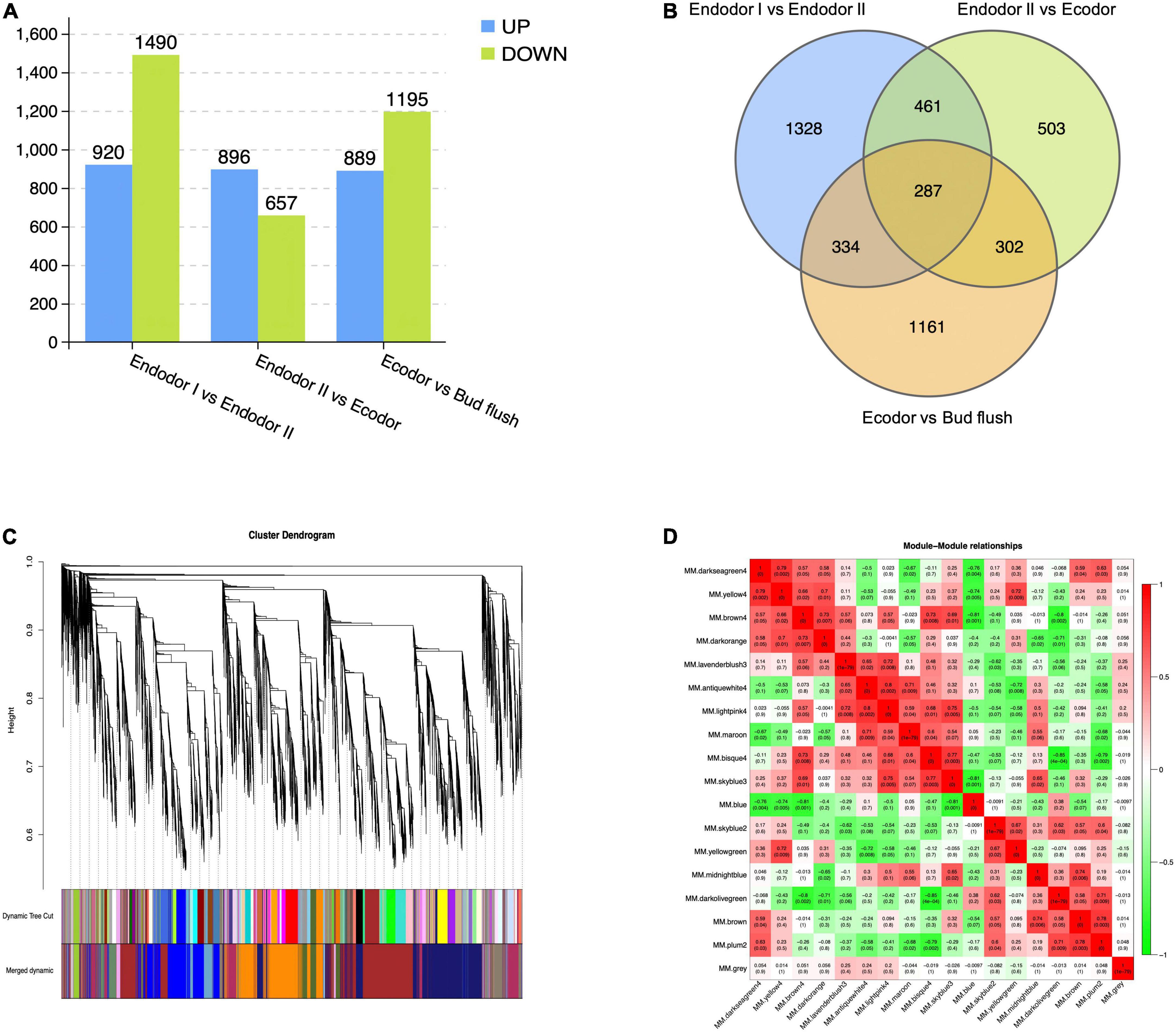
Figure 2. The identification of differentially expressed genes and co-expressed gene modules from WGCNA analysis. (A) The number of up-regulated or down-regulated DEGs among stage comparisons (Endodor I vs. Endodor II, Endodor II vs. Ecodor, and Ecodor-vs.-Bud flush). (B) Venn diagram of DEGs identified across stage comparisons (Endodor I vs. Endodor II, Endodor II vs. Ecodor, and Ecodor-vs.-Bud flush). (C) Hierarchical clustering dendrogram showing 17 co-expressed gene modules in designated colors from WGCNA analysis. (D) Heatmaps showing Pearson correlation among eigengenes of co-expressed gene modules. The Pearson correlation coefficients was colored according to the score.
Functional enrichment analysis of differentially expressed genes
To further investigate the function of DEGs, we performed GO and KEGG pathway enrichment analysis on the DEG sets. The DEGs between endodormancy stage I and II are significantly enriched in biological processes including response to carbohydrate stimulus, response to ethylene stimulus, response to abscisic acid stimulus, response to cold, and response to gibberellin stimulus (Supplementary Table 2). KEGG enrichment analysis confirmed that biosynthesis of secondary metabolites, carbohydrate metabolism, and DNA replication are three most enriched pathways (Supplementary Figure 2A). The DEGs during the transition from endodormancy to ecodormancy were mainly participated in reproductive developmental process, response to temperature stimulus, and anatomical structure development (Supplementary Table 2). The KEGG pathways including photosynthesis, DNA replication, and carbon fixation were significantly enriched (Supplementary Figure 2B). Finally, DEGs between ecodormancy and bud flush were mostly involved in biological processes including DNA replication, flower development, gametophyte development, and response to hormone stimulus (Supplementary Table 2). Pathways such as DNA replication, phenylpropanoid biosynthesis, flavonoid biosynthesis, and plant hormone signal transduction are mostly over-represented in this stage comparison (Supplementary Figure 2C). These results illustrated the important biological processes and metabolic pathways of these DEGs during dormancy transition process in P. mume.
Co-expression network analysis of mRNA
To identify the co-regulatory factors during floral bud break in P. mume, we performed weighted gene co-expression network analysis (WGCNA) and grouped 15,381 genes into 17 co-expressed modules with their pairwise correlation evaluated (Figures 2C,D). Among all modules, brown module contained the maximum number of genes (3,259 genes), while the yellow module contained 64 genes (Figures 2C,D). To further characterize the gene modules essential to the progression of floral bud development, we estimated the correlation between module eigengenes and bud break rate. We identified three gene modules, including module blue, module brown, and module darkseagreen4 that were significantly associated with bud flush rate (Supplementary Table 3 and Figure 3). The eigengene of module blue was positively correlated with BBR (r = 0.81, p-value = 0.002) (Supplementary Table 3 and Figure 3). On the other hand, eigengenes of module brown (r = −0.91, p-value = 4e–5) and darkseagreen4 (r = −0.84, p-value = 7e–4) were negatively correlated with BBR (Supplementary Table 3 and Figure 3).
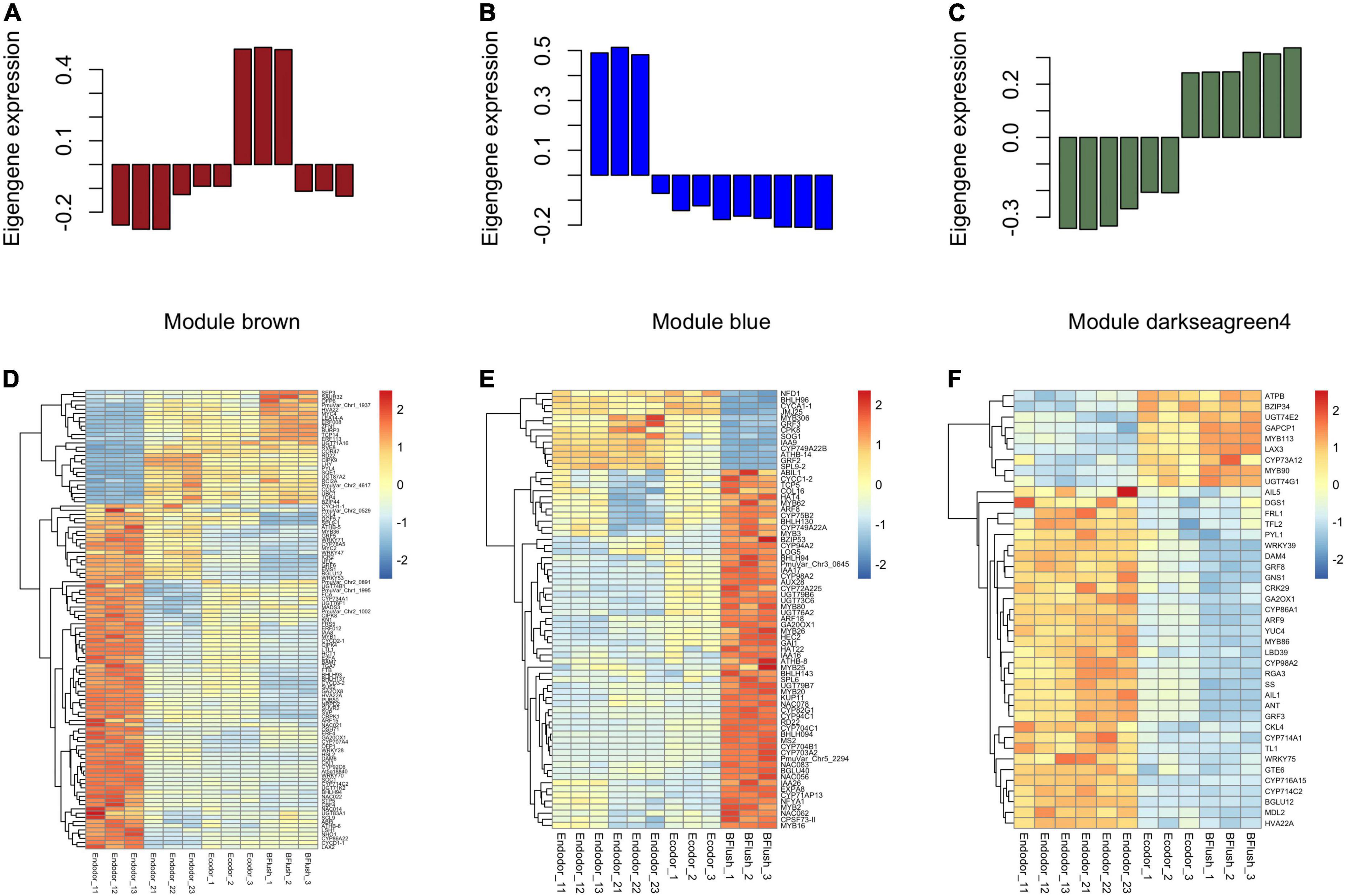
Figure 3. The co-expressed gene modules associated with dormancy transition in the WGCNA analysis. (A–C) The barplot displaying the normalized eigengene expression for module brown (A), module blue (B), and module darkseagreen4 (C). (D–F) The heatmaps showing the expression pattern of differentially expressed genes within module brown (D), module blue (E), and module darkseagreen4 (F).
The eigengene of module brown increased gradually as dormancy depth decreased during chilling accumulation but significantly dropped during bud flush (Figure 3A). Genes within module brown were mostly involved in biological processes such as reproductive organ development, meristem development, and response to dsRNA. The brown module genes annotated to floral organ development included EMS1 (EXCESS MICROSPOROCYTES1) that encodes a basic leucine zipper (bZIP) transcription factor regulating microsporogenesis (Zhao et al., 2002), E-class MADS-box transcription factor SEP3 (SEPALLATA3) (Immink et al., 2009), and LUG (LEUNIG), a transcriptional repressor regulating gynoecium and ovule development in Arabidopsis (Liu et al., 2000; Supplementary Table 4). Among the DEGs, we detected a few transcription factors that are known to be associated with bud dormancy control (Supplementary Table 4). For example, the transcript levels of DAM6 and SVP significantly decreased as chilling accumulates (Figure 3D). SVP (SHORT VEGETATIVE PHASE), a MADS-box transcription factor, acts as a floral repressor in Arabidopsis (Wang et al., 2018). In poplar, SVP-like genes can integrate the short-photoperiod and low-temperature induced endodormancy pathways to promote ABA signaling and bud dormancy establishment (Singh et al., 2018). SOC1 displayed decreasing expression pattern in the early stage of endodormancy release (Figure 3D). SOC1 is another MADS-box transcription factor acting downstream of FT (FLOWERING LOCUS T) in promoting flowering in Arabidopsis (Lee and Lee, 2010). In Prunus avium, SOC1 can interact with DAM5 in vivo, suggesting their interaction is required for floral bud dormancy regulation in trees (Wang et al., 2020). We also identified a few chilling inducible genes, such as the CBF4 (C-REPEAT/DRE BINDING FACTOR 4), dehydrin protein COR47 (COLD-REGULATED 47), and RCI2A (RARE-COLD-INDUCIBLE 2A). CBF4 is a member of ERF/AP2 transcription factor induced by cold temperatures and can activate the downstream dehydrin COR family proteins, such as COR47, through binding to the C-repeat core regulatory motif (Welling and Palva, 2008). The over-expression of CBF4 in Arabidopsis leads to higher tolerance to cold and drought (Haake et al., 2002). In perennial trees, CBFs, with their expression triggered by cold, can induce the transcription of DAM5 and DAM6 to establish dormancy (Niu et al., 2016; Zhao et al., 2018a).
Module blue is consisted of 2,064 genes with its eigengene significantly decreased during the dormancy transition period (Figure 3B). The GO enrichment analysis showed that genes within module blue are mostly involved in biological processes including anatomical structure development, gametophyte development, and pollen development. DEGs related to female gametophyte development within blue module include NFD1 (NUCLEAR FUSION DEFECTIVE 1) and CPSF73-II (CLEAVAGE AND POLYADENYLATION SPECIFICITY FACTOR 73 KDA SUBUNIT-II) (Supplementary Table 5). The pollen development associated DEGs include CYP703A2 (CYTOCHROME P450, FAMILY 703, SUBFAMILY A, POLYPEPTIDE 2), MS2 (MALE STERILITY 2), and KUP11 (K+ UPTAKE PERMEASE 11) (Supplementary Table 5). These gametophyte development related genes were significantly up-regulated after endodormancy release and maintained high expression level during bud flush (Figure 3E). A few differentially expressed transcription factors from module blue were detected. For example, ATHB-8 (ARABIDOPSIS THALIANA HOMEOBOX 8) and ATHB-14 encode HD-ZIP (homeodomain leucine zipper) transcription factors that are known to regulate SAM (shoot apical meristem) and vascular patterning in Arabidopsis (Ramachandran et al., 2017). Two MYB transcription factors, MYB5 (MYB DOMAIN PROTEIN 5) that possibly involved in the formation of endosperm layers (Li S. F. et al., 2009) and MYB26 regulating anther dehiscence were found differentially expressed (Yang et al., 2017; Supplementary Table 5).
Genes within module darkseagreen4 are mostly annotated to biological processes including anatomical structure development, response to stress, and regulation of cell size. The eigengene of module darkseagreen4 dramatically increased during the progression of bud dormancy break (Figure 3C). Among the darkseagreen4 module genes, we identified a few AP2 family transcription factors that may putatively regulate floral bud break, such as ANT (AINTEGUMENTA) required for cell proliferation control during bud break in poplar, AIL1 (AINTEGUMENTA-LIKE 1) and AIL5 with partially overlapping functions with ANT in flower development functions (Krizek, 2015; Supplementary Table 6). These genes were highly expressed during endodormancy release but were decreased slightly afterward, suggesting their role in floral organ development (Figure 3F). Two GRFs, GRF5 (GROWTH-REGULATING FACTOR 5) and GRF8, both encode transcriptional activators of pistil development were also identified in module darkseagreen4 (Liang et al., 2013; Supplementary Table 6). These two genes were highly expressed in dormant floral buds but then dropped after complete floral organ development (Figure 3F).
Small RNA sequencing of floral bud
To study the functional role of miRNAs in bud break process, we constructed 12 small RNA sequencing libraries using the same samples representing four different dormancy stages and obtained approximately 15.6 million raw reads for each library (Table 2). An average of 14.9 million clean tags (95.24%) were retained after removing low quality reads and the unique tags were used for downstream analysis (Table 2). Among all libraries, the length of small RNAs ranged from 18 to 30 nt with two abundance peaks at 21 and 24 nt (Figure 4A). Small RNAs of 21 and 24 nt were mostly enriched accounting for 36.7 and 35.1% on average among total clean tags, respectively (Figure 4A). The small RNAs were first annotated by blasting against NCBI GenBank and Rfam databases to annotate non-coding RNAs. Among all annotated tags, 19.34–23.75, 20.12–20.43%, 11.97–12.67%, and 16.73–22.92% were annotated as non-coding RNAs for endodormancy I, endodormancy II, ecodormancy, and bud-flush samples, respectively (Figure 4B and Supplementary Table 7). All clean tags were also mapped to the reference genome of P. mume (Zheng et al., 2021) with mapping rate ranging from 72.1 to 75.9% across 12 sequencing libraries (Supplementary Table 7).

Table 2. Summary statistics of sequence reads generated from 12 small RNA sequencing libraries.
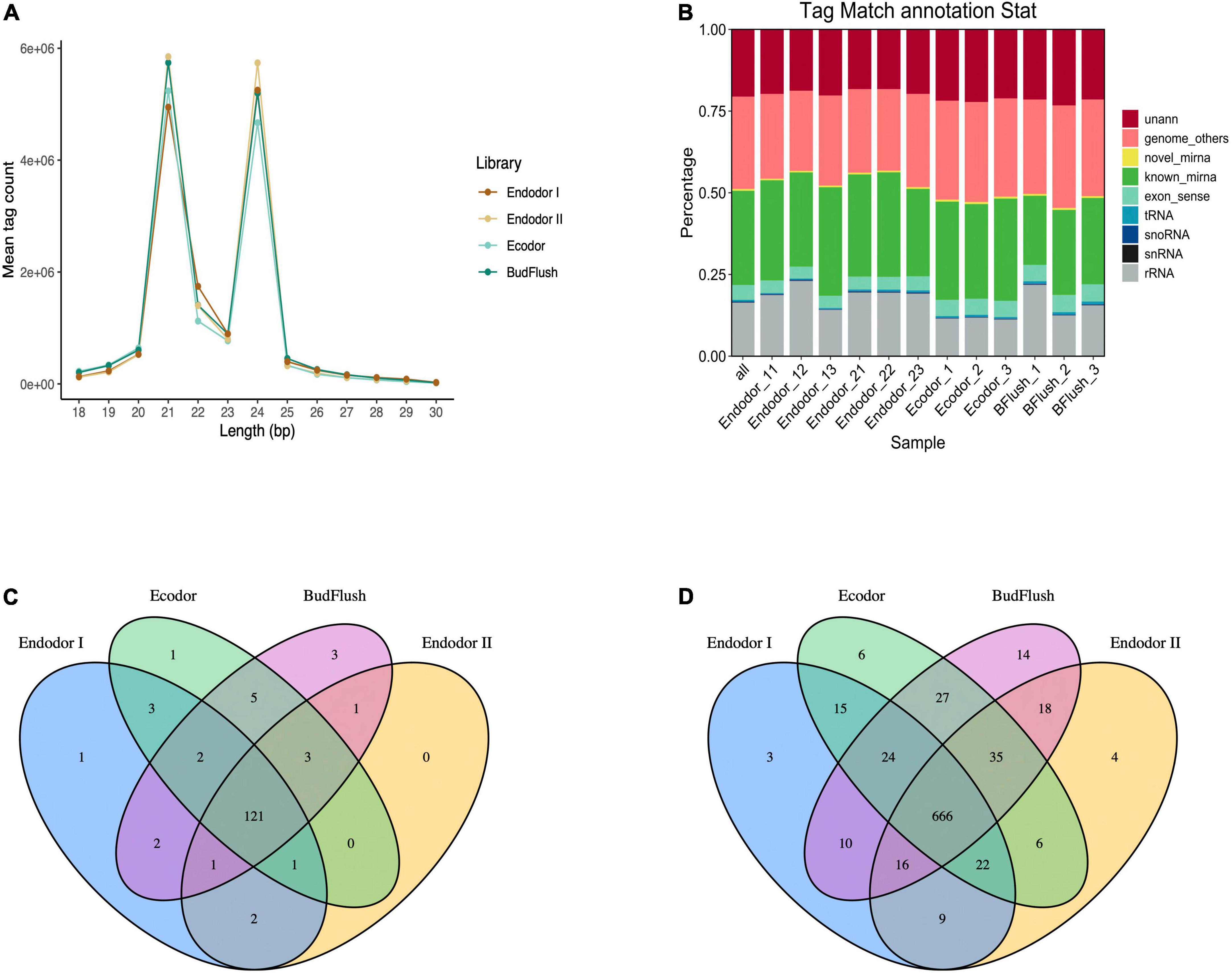
Figure 4. Overview of small RNA sequencing analysis. (A) The size distribution of small RNAs generated across four developmental stages. (B) The annotation of clean sequence tags across 12 small RNA sequencing libraries of four developmental stages. (C,D) The Venn diagram of known miRNAs (C) and novel miRNAs (D) identified across four developmental stages.
Known and novel mRNA identification
After excluding ncRNAs and reads mapped to exons, introns or repeat sequences, we annotated the remaining small RNAs by blasting against miRBase 22.0 database (Kozomara et al., 2019). In total, we identified 146 known miRNAs, among which 121 miRNAs were present in floral bud tissues of all developmental stages (Figure 4C and Supplementary Table 8). Mostly of the known miRNAs were present among 12 floral bud samples with only few miRNAs specific to certain developmental stages. For example, miR11544-z was specific to endodormancy I stage, miR7782-y specific to ecodormancy stage, and miR6294-z, miR5083-z, miR172-x specific for samples of bud-flush stage, respectively (Figure 4C). The known miRNAs were mostly 21 nt in length with hairpin structures ranging from 80 to 366 nt (Supplementary Table 8). Additionally, 875 novel miRNAs were identified using Mireap software with secondary structures predicted with mirDeep2 (Figure 4D). Of all novel miRNAs, 666 miRNAs were shared across all sequencing libraries (Figure 4D).
Differential expression analysis of microRNAs
In order to identify differentially expressed miRNAs (DEmiRs) during floral bud break in P. mume, we compared the normalized expression of miRNAs between every two adjacent stages. By summing up all DEmiRs, we identified 41 known miRNAs and 53 novel miRNAs that were significantly differentially expressed (Figures 5A,B and Supplementary Figures 3A,B). Among them, 63, 83, and 93 DEmiRs were detected in the comparison of endodormancy I-vs.-endodormancy II, endodormancy II-vs.-ecodormancy, and ecodormancy-vs.-bud flush, respectively (Figure 5A and Supplementary Figure 3A). Among the DEmiRs, four known miRNAs (miR156-x, miR157-x, miR2275-x, and miR408-z) and eight novel miRNAs were common to all three comparisons (Figure 5A and Supplementary Figure 3A). We performed hierarchical clustering analysis and grouped the 41 known DEmiRs into five major clusters based on their expression profiles (Figure 5B). Cluster I contained 18 known DEmiRs, including miR157-x/y, miR156-x/y, miR396-z, and miR828-y that were highly expressed in flushed floral buds (Figure 5B). In cluster II, DEmiRs such as miR167-y, miR398-x, miR477-x were significantly induced in floral buds after endo-dormancy release (Figure 5B). Cluster III is consisted of a few miRNAs (such as miR162-x, miR2275-x/y, miR3630-y) that were highly expressed in stage endodormancy II but their expression decreased afterward (Figure 5B). The fourth group included miR3627-x/y, miR4414-x, and miR5225-x, whose expression remained high in endo-dormant floral buds but decreased as floral bud exit dormancy. Known DEmiRs within cluster V showed mixed expression pattern during the process of dormancy break (Figure 5B).
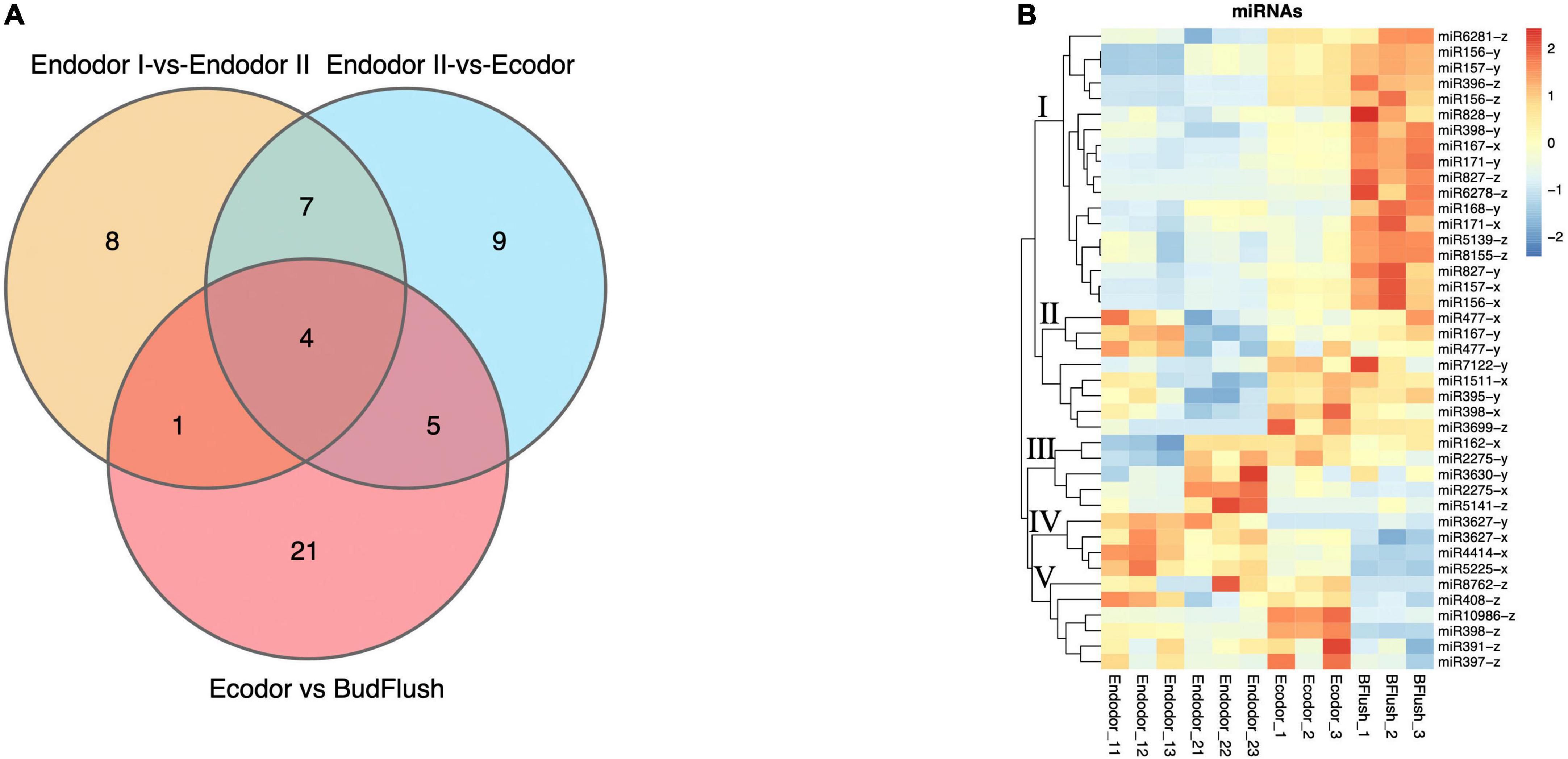
Figure 5. The differential expression analysis identified known miRNAs differentially expressed across stage comparisons. (A) Venn diagram of known DEmiRNAs identified across four stage comparisons. (B) Expression heatmap of known miRNAs displaying their expression pattern across four developmental stages.
Target gene identification of differentially expressed miRNAs
To understand the functions of differentially expressed miRNAs, we performed degradome sequencing on sample pools and obtained 22,942,638 clean reads for subsequent analysis (Supplementary Table 9). By intersecting the target genes predicted computationally and those supported with degradome data, we identified 80 valid target genes for 33 known miRNAs, most of which are transcription factors (Table 3). For example, miR164 family, which targets NAC domain transcription factors, was found to target NAC021 (PmuVar_Chr3_1567) and NAC098 (PmuVar_Chr3_1908) for degradation (Table 3). Similarly, miR166 target three HD-ZIP transcription factors including REV, ATHB-8, and ATHB-14 (Table 3).
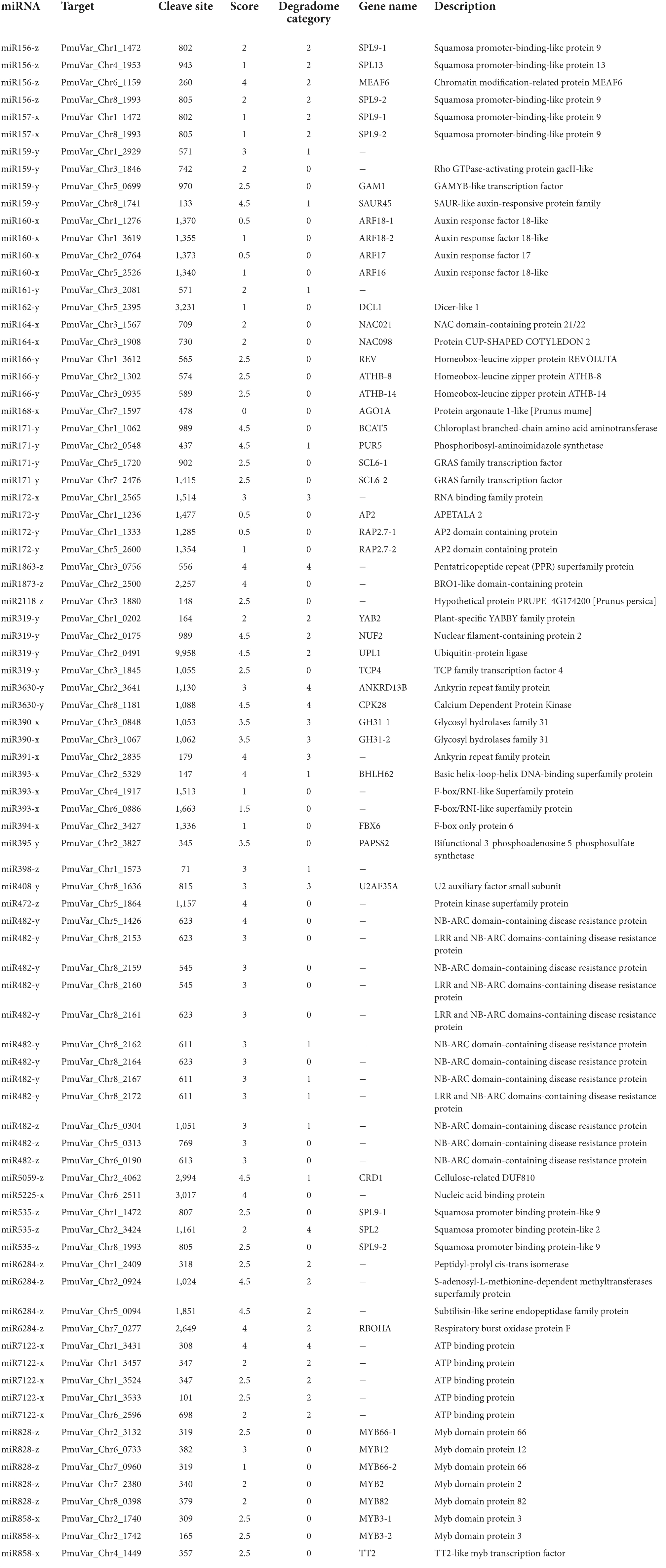
Table 3. Target genes of known miRNAs validated with degradome sequencing.
With functional enrichment analysis, we detected a number of biological pathways enriched among the target genes of DEmiRs. Between two endo-dormancy stages, regulation of organ morphogenesis, single-organism transport, and fatty acid metabolic process were mostly over-represented GO terms (Supplementary Table 10). Target genes of DEmiRs between endodormancy and ecodormancy were mostly involved in lignin metabolic process, phenylpropanoid metabolic process, and regulation of organ formation (Supplementary Table 10). Finally, the DEmiRs detected between ecodormancy and bud flush stages are targeting genes related to carbohydrate transport and secondary metabolic process (Supplementary Table 10). With KEGG pathway enrichment analysis, we observed genes of metabolic pathways such as biosynthesis secondary metabolites, plant hormone signal transduction, and starch and sucrose metabolism were enriched for DEmiRs across all three stage comparisons (Supplementary Table 11).
Relationship analysis of the expression pattern of differentially expressed miRNAs and their target genes
To explore the regulatory mechanism of miRNAs, we analyzed the expression pattern of DEmiRs and their target genes. The Pearson correlation coefficients between the transcript level of DEmiRs and their target genes across four developmental stages ranged from −0.997 to 0.994. Among them, we observed antagonized expression patterns among many known miRNAs-TF pairs. For example, miR156 and miR157 are two miRNA families that were previously reported to regulate reproductive meristem transition and flowering time by repressing the SPL (SQUAMOSA PROMOTER BINDING PROTEIN-LIKE) transcription factors in Arabidopsis (Sharma et al., 2016; Millar et al., 2018). During dormancy release, the expression of miR156 and miR157 family miRNAs increased as floral bud exit dormancy, whereas their target genes including SPL13, SPL9-1, and SPL9-2 showed decreasing expression patterns (Figures 6A,B). Among different members of the miR156-157 family, miR156-y and miR157-y were relatively low-expressed throughout the developmental process (Figure 6A). With degradome sequencing, the cleavage sites of miR156-157 targeted genes were detected within SPL9 and SPL13 (Supplementary Figures 4A,B). The miRNAs of miR171 family were found constantly increasing during the transition from endodormancy to bud flush. Their target genes, SCL6-1 (SCARECROW-LIKE PROTEIN 6) and SCL6-2, two GARS family transcription factors were induced during endodormancy release, but their expression levels decreased slightly during bud break (Figures 6C,D).
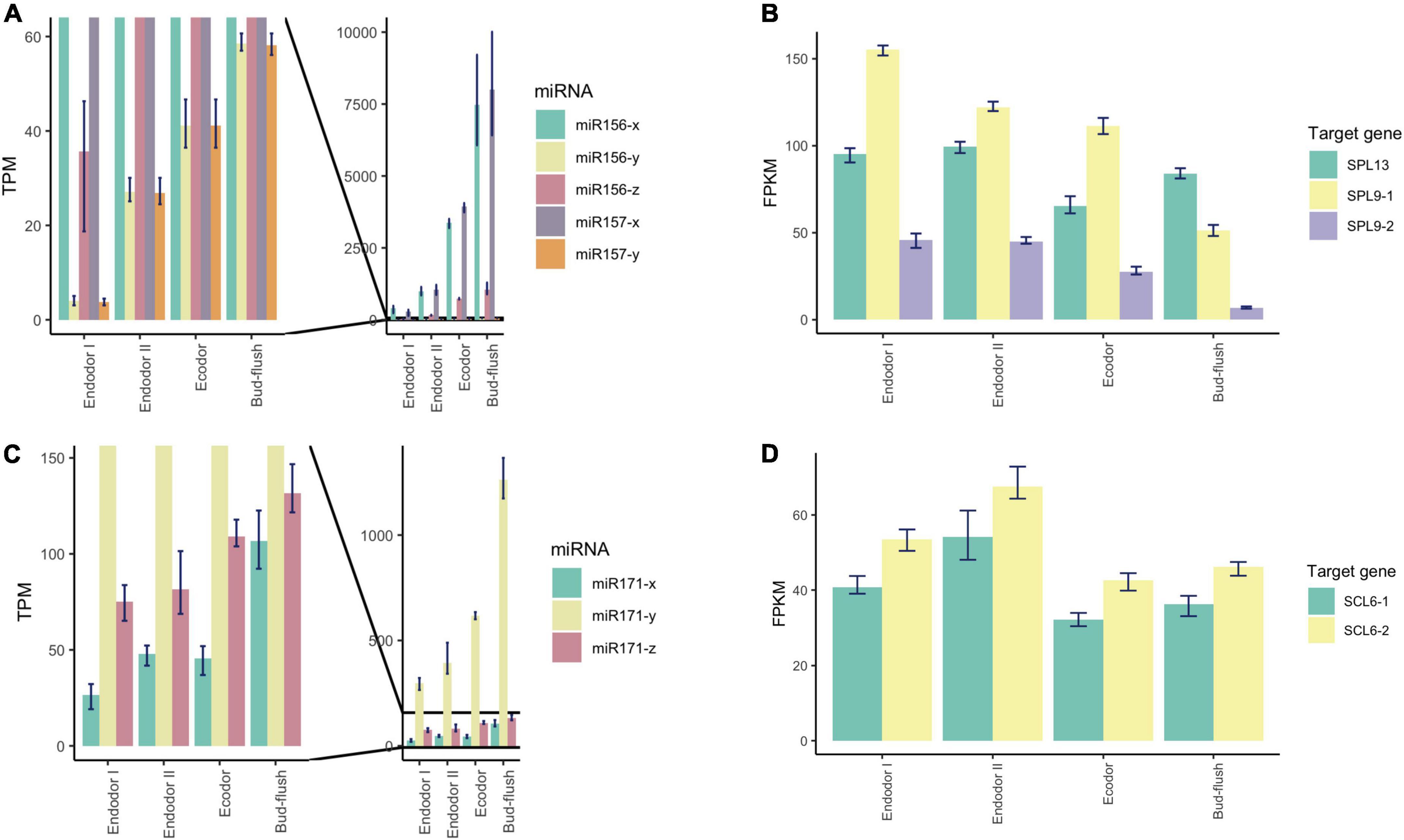
Figure 6. Expression pattern analysis of miR156, miR157, miR171, and their target genes during floral bud break in P. mume. (A,B) The barplots revealing the expression levels of miR156/157-x/y/z (A) and their target genes (B) during floral bud break process. (C,D) The barplots revealing the expression levels of miR171-x/y/z (C) and their target genes (D) during floral bud break.
Among the conserved miRNA families, miR172 is another key miRNA group that regulate age-dependent flowering timing by targeting AP2/ERF transcription factors (Wu et al., 2009). In P. mume, the level of miR172-y first decreased during the endodormancy transition to ecodormancy, however, was significantly increased during bud flush (Figure 7A). On the other hand, miR172-x was slightly up-regulated during bud break and was relatively low-expressed comparing to miR172-y (Figure 7A). Their target genes displayed the opposite expression patterns, where AP2 and RAP2.7 first increased during endodormancy release but then remained at lower level during bud flushing (Figure 7B). MiR858 was previously reported to regulate plant growth and flowering timing. The loss function of miR858 can cause early flowering, while its over-expression induces retarded plant growth and delayed flowering time (Sharma et al., 2016). In our study, miR858-x was significantly down-regulated during the floral bud break, while miR858-y slightly increased despite low expression level (Figure 7C). Among their target genes, MYB3-1 was slightly increased, whereas the transcription level of its paralog MYB3-2 slightly dropped during bud flushing (Figure 7D). The contrasting expression patterns suggest that different members from the same miRNA family may have divergent roles by targeting different target genes. The cleavage events of these miRNAs were verified within their target genes in the degradome analysis (Supplementary Figure 4).
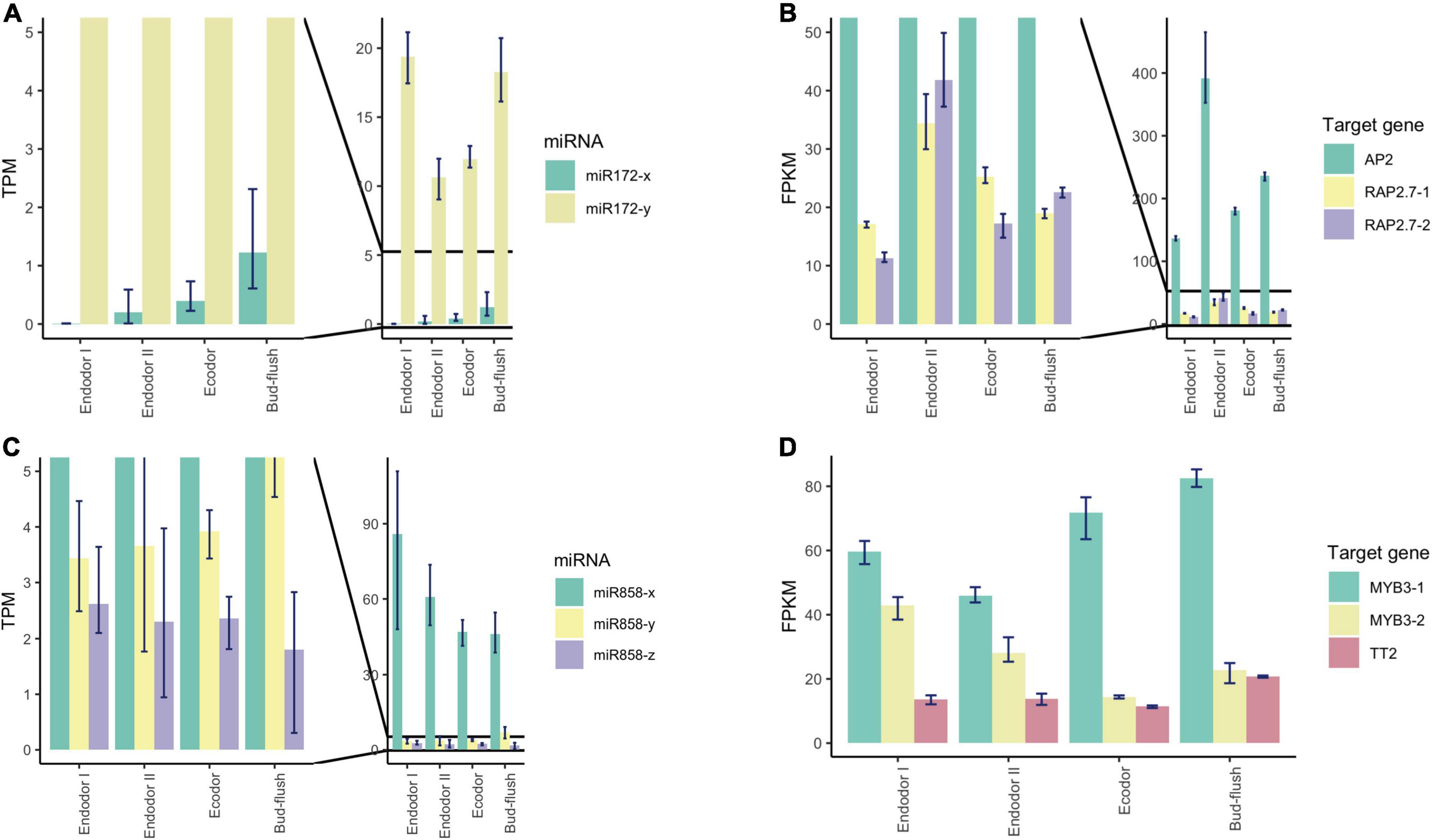
Figure 7. Expression pattern analysis of miR172, miR858, and their target genes during floral bud break in P. mume. (A,B) The barplots revealing the expression levels of miR172-x/y (A) and their target genes (B) during floral bud break process. (C,D) The barplots revealing the expression levels of miR858-x/y/z (C) and their target genes (D) during floral bud break.
The miR319 family is known to regulate TCP transcription factors, which is a class of plant-specific transcription factors that regulate cell proliferation and differentiation (Li et al., 2021). In peach, TCP20 regulate DAM5 and DAM6 by binding to the cis-acting element of their promoters (Wang et al., 2019). TCP18, acting downstream of SVL (SHORT VEGETATIVE PHASE-LIKE), is a negative regulator of vegetative bud break in poplar (Singh et al., 2018). In our study, we detected significantly decreasing transcript level of miR319-y, while miR319-x and miR319-z remained lowly expressed (Supplementary Figure 5A). Their target genes, TCP2 and TCP4, on the other hand, were up-regulated first and then were down-regulated after endodormancy release (Supplementary Figure 5B). Significantly increasing transcription of miR164-x was observed during floral bud break (Supplementary Figure 5C). MiR164 was characterized to regulate root formation and drought resistance in maize and rice (Li et al., 2012; Fang et al., 2014). Their target genes NAC021, NAC098, and NAC100 were slightly decreased during dormancy release (Supplementary Figure 5D). We also detected a few DEmiRs with no sliced target genes found in degradome analysis. For example, we observed that the expression of miR2275-x and miR2275-y were up-regulated before dormancy release, but were significantly dropped after dormancy release (Supplementary Figure 5E). While their predicted target genes (PmuVar_Chr2_1878 and PmuVar_Chr3_0920) decreased slightly and then increased significantly during bud break (Supplementary Figure 5F). It is reported that miR2275 was present in eudicot plants and are especially enriched in meiotic stages (Xia et al., 2019). In flowering plants, miR2275 can trigger the production of 24-nt phasiRNAs that regulate pollen development (Xia et al., 2019). It is likely that transcriptional change of miR2275 is associated with anther microsporogenesis in P. mume during floral bud break.
Co-expression network analysis of miRNA
Weighted co-expression network analysis approach was also applied on miRNAs to investigate the role of regulatory miRNA networks related to dormancy release. The co-expression analysis grouped 759 microRNAs into two modules (MEs), module blue and module turquoise (Supplementary Figures 6A,B). Module turquoise contained 499 miRNAs (103 known miRNAs and 396 novel miRNAs) with its eigengene remained low-expressed at endodormancy stages but was strongly up-regulated after bud-burst (Supplementary Figure 6C). The rest 260 miRNAs (18 known miRNAs and 242 novel miRNAs) were clustered into module blue with the expression pattern specific to certain samples (Supplementary Figure 6D). We also carried out module-trait association analysis to identify miRNA modules correlated with bud break rate (Supplementary Figure 6B). We found module turquoise is highly correlated with BBR (r = 0.93, p-value = 1.0e–5). A number of known miRNAs were detected in module turquoise, such as miR156/157-x/y, miR171, miR172-y, and miR319-x/y that were differentially expressed during the process of floral bud bursting.
Integrated miRNA-transcription factor regulatory network during dormancy release
MiRNAs function post-transcriptionally to regulate protein-coding genes through initiating cleavage or degradation (Rubio-Somoza and Weigel, 2011). Most miRNA targeted genes are transcription factors that mediate plant growth and development through regulating downstream genes (Samad et al., 2017). To identify the potential miRNA-TF-mRNA regulatory network, we first mapped all DEGs to the BBR-associated gene modules and detected 650 DEGs belonging to module blue, 894 DEGs belonging to module brown, and 164 DEGs belonging to module darkseagreen4. The differentially expressed transcription factors were extracted from these three modules along with their potential gene targets selected as the most connected genes based on the correlation matrix. Furthermore, we mapped the transcription factors targeted by DEmiRs to the co-expression network and integrated the miRNA-TF regulatory pairs (Figure 8).
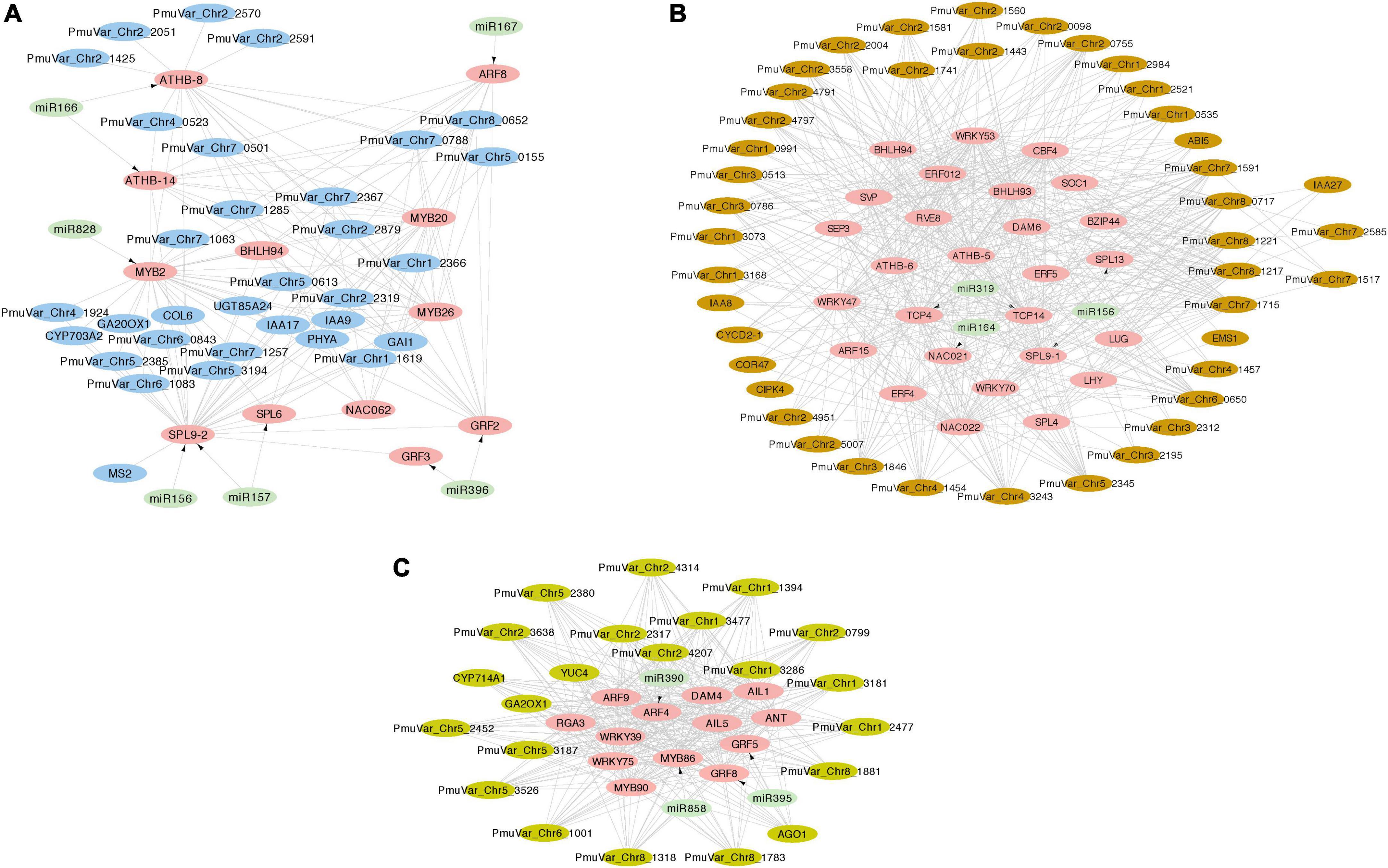
Figure 8. Integrated miRNA-TF regulatory networks of three co-expression gene modules. The transcription factors and target genes of DEmiRs were identified from module blue (A), module brown (B), and module darkseagreen4 (C). The pink nodes represent hub transcription factors, the light green nodes represent miRNAs, and the other putative regulated genes were colored with their corresponding module color. The arrows represent the regulation relationship between miRNAs and their target TFs. The edges connecting TFs and other genes represent co-expression relationship.
In general, the co-expression analysis revealed complex miRNA-TF-mRNA networks modulating floral bud break in P. mume. Module blue is consisted of a few hub transcription factors, including ATHB-8 and ATHB-14 targeted by miR166, GRF2 (GROWTH REGULATOR FACTOR 2) and GRF3 targeted by miR396, ARF8 (AUXIN RESPONSE FACTOR 8) mediated by miR167, MYB2 targeted by miR828, and SBP transcription factors SPL9-2 (SQUAMOSA PROMOTER-BINDING-LIKE PROTEIN 9) and SPL6 targeted by miR156/157 (Figure 8A). Moreover, a few transcription factors related to hormonal signaling were co-expressed in module blue, such as IAA9 (INDOLEACETIC ACID-INDUCED PROTEIN 9) and IAA17 from AUX/IAA transcriptional regulator family, and GARS transcription factor GAI (GIBBERELLIC ACID INSENSITIVE) (Figure 8A). In module brown, we detected CBF4 (C-REPEAT-BINDING FACTOR 4) and DAM6 (DORMANCY ASSOCIATED MADS-BOX 6), which form a regulatory module in regulating flower bud dormancy in P. mume (Zhao et al., 2018b; Figure 8B). Among the co-expressed transcription factors, NAC021 targeted by miR164, TCP4, and TCP14 targeted by miR319, SPL9-1 targeted by miR156, IAA8 and IAA27, and ARF15 are putative transcription factors essential in regulating bud dormancy process in P. mume. The network analysis also identified a few connected genes putatively acting downstream of these transcription factors, such as UDP-Glycosyltransferase superfamily proteins (UGT71K2 and UGT87A2), and Cyclin-D2, and CIPK4 (CBL-INTERACTING PROTEIN KINASE 4) (Figure 8B). Module darkseagreen4 also contained a few characterized bud break regulators in perennial tree species, such as DAM4 (DORMANCY ASSOCIATED MADS-BOX 4), ANT (AINTEGUMENTA), and GA2OX1 (GIBBERELLIN 2-OXIDASE 1) (Yang et al., 2021). We also identified a few miRNA-TF pairs, including miRNA390-ARF4, miR396-GRF5/GRF8, and miR585-MYB86 (Figure 8C). The co-expression network analysis provided a list of candidate genes with functions uncharacterized in floral bud break or dormancy cycling. These co-expressed genes are putative regulators or miRNA/TF target genes worthy of future examinations.
Expression validation of differentially expressed miRNAs and their targets with quantitative real-time PCR
To validate the sequencing results, we performed qRT-PCR analysis on a few differentially expressed miRNAs and their target genes. We examined the relative expression level of seven conserved miRNAs (miR156-x, miR157-x, miR160-x, miR172-x, miR172-y, miR2275-x, and miR2275-y) and their target genes across four developmental stages during floral bud dormancy release (Figure 9). As shown, the qRT-PCR results were consistent with the high-throughput sequencing data (Figure 9). miR156-x and miR157-x were gradually increased as floral bud exit dormancy, whereas their target SPL transcription factors (SPL9-1, SPL9-2, SPL13) were first up-regulated but then were slightly down-regulated (Figure 9A). Similarly, the expression of miR172-x constantly increased with their target genes AP2 and RAP2.7 transcripts displaying contrasting expression pattern as floral bud exit dormancy (Figure 9B). The expression of miR160-x was repressed during early stages of bud dormancy release, however, was significantly increased in the bud flush process. On the other hand, the ARF transcription factors including ARF16, ARF17, and ARF18 were up-regulated during dormancy release (Figure 9C). miR2275-x and miR2275-y were first up-regulated and then down-regulated after exit endodormancy. Their predicted target gene PmuVar_Chr3_0920 was found up-regulated during bud break (Figure 9D). In general, the negative correlation between expression profiles of miRNAs and their target genes were confirmed.
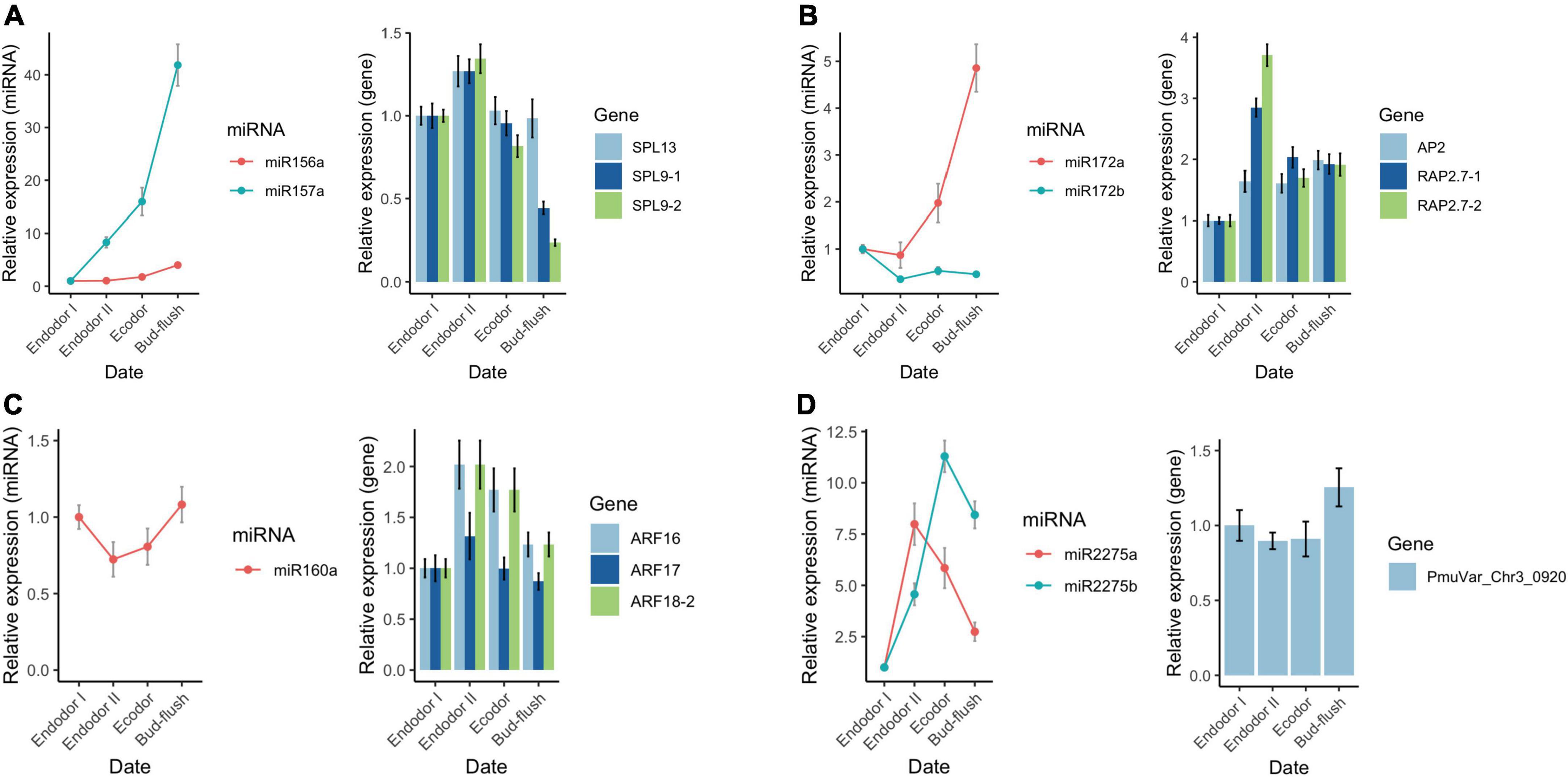
Figure 9. qRT-PCR analysis of the expression pattern of DEGs and DEmiRs during floral bud break in P. mume. The relative expression level of miRNAs within miR156/157 family (A), miR172 family (B), miR160 (C), and miR2275 (D) were analyzed across four developmental stages using 5S rRNA as internal reference. Similarly, the relative expression level of their corresponding target genes were analyzed with PP2A as reference gene in the qRT-PCR assays.
Discussion
Floral bud flush in deciduous trees is a multifaceted physiological process that involves floral bud initiation, dormancy cycling, and floral organ development (Cooke et al., 2012). In temperate fruit trees, the onset of floral bud initiation occurred in late summer with floral primordia differentiated sequentially in the order of sepals, petals, stamens, and carpels (Koutinas et al., 2014). All flower primordia continue to develop for several months before entering dormancy period in winter. In Prunus fruit trees, the internal floral structures of endodormant buds continued to develop during chilling accumulation (Fadón et al., 2018; Hsiang et al., 2021). After the fulfillment of chilling requirement, floral organs continue to expand and mature and finally bloom under warm conditions (Ito et al., 2016). The timing of reproductive bud break is one of the most important adaptive trait that ensures plant survival and synchronized the reproductive activities with seasonal environmental changes (Zhang et al., 2019; Fadón et al., 2020). In the context of global warming, it is crucial to understand the control of flowering phenology in deciduous tree species (Zhang et al., 2021b). Till now, the molecular mechanism underlying flower bud development and dormancy cycling is extensively explored among woody perennial species. However, the knowledge regarding to the microRNA-mediated gene regulatory networks during this process is still limited.
Candidate genes related to floral bud dormancy transition in Prunus mume
In this study, we examined the morphological change of floral bud and observed floral bud enlargement accompanied by organ development and maturation. Based on the bud break competence tests, we proposed four sampling time points corresponding to key developmental stages during floral bud break. Our transcriptome analysis revealed 1,553 DEGs in endodormancy-vs.-ecodormancy comparison and 2,084 DEGs in ecodormancy-vs.-bud-burst comparison. Genes associated with endodormancy release were over-represented in biological processes, such as response to hormones (ethylene, abscisic acid, gibberellin, etc.), response to cold, and flower organ development, suggesting that the transition process from endodrmancy to ecodormancy requires complex regulatory networks coordinating together (Fan et al., 2015). Ecodormancy is a short-term period of inhibited growth and can proceed to bud break in warmer climates (Ueno et al., 2013). Genes differentially expressed between ecodormancy and bud flush stage are mostly involved in floral whorl development and gametophyte production. These results are consistent with the activated reproductive organ development at ecodormancy in apricot and peach (Yu et al., 2020).
With co-expression analysis, we identified three clusters of co-expressed genes with distinctive expression profiles highly correlated with the level of dormancy depth during floral bud break. Among the candidate genes, DAM4-6 are well-known transcription factors that are required for endodormancy maintenance in perennial trees. Previous studies have reported that the expression of DAM4-6 is suppressed by chilling temperatures to ensure dormancy release and the expression of DAM5 and DAM6 was found inversely correlated with bud break rate in peach and Japanese apricot (Jiménez et al., 2010; Zhu et al., 2020). The deletion of DAMs in peach lead to dormancy-incapable mutants (Bielenberg et al., 2008). The silencing of DAMs in apple using RNA interference lead to ever-growing apple that fail to enter bud dormancy in winter (Wu et al., 2021). Moreover, we identified a few transcription factors that putatively interact with DAMs, such as CBF4 and SOC1. In P. mume, CBFs can regulate DAM6 through binding its promoter or forming protein dimers with DAM6 (Zhao et al., 2018a). SOC1 protein can also interact with DAM1 and DAM5 in vivo and in vitro in P. avium (Wang et al., 2020). The regulatory module of DAMs and their putative interacting genes suggested the conserved molecular mechanism underlying dormancy control across Prunus tree species. In addition to DAMs, we identified SVP, another MADS-box transcription factor that is crucial to promoting bud endodormancy in poplar (Singh et al., 2018). SVL can also inhibit gibberellin biosynthetic genes, GA20OX1 and GA20OX2 to deepen the endodormancy state (Singh et al., 2018). In our study, we identified ANT (AINTEGUMENTA) along with its paralogs AIL1 (AINTEGUMENTA-LIKE 1) and AIL5 (AINTEGUMENTA-LIKE 5), and CYCDs, which are likely required for cell proliferation during floral organ development and expansion during bud break. During dormancy induction in poplar, the transcription of ANT is strongly inhibited, resulting in growth cessation and bud formation (Azeez et al., 2014). ANT is also critical for ovule initiation and formation in Arabidopsis (Omidbakhshfard et al., 2015). CYCD3.1 is a D-type cyclin required to promote cell division during bud break in poplar (Azeez et al., 2021).
Hormonal signaling is also essential for floral development and dormancy transition (Liu and Sherif, 2019). ABA and gibberellin are two plant hormones that are known to regulate dormancy and seed germination (Yu et al., 2013). ABI3 (ABSCISIC ACID-INSENSISTIVE 3) is a key factor acting downstream of ABA signaling. In previous studies, overexpressing ABI3 caused early bud set in poplar (Rohde et al., 2002). In our study, the expression of ABI5 was significantly repressed, indicating that ABA deactivation is also required for dormancy release in floral buds. In early stage of chilling accumulation, the expression level of GA biosynthetic genes, such as GA20OX1, was relatively low in endodormant buds and then strongly up-regulated after exit endodormancy. GA2OX1 and GA2OX8, involved in GA catabolic pathway, were strongly down-regulated from dormancy release to floral bud break (Figure 3). The balance between ABA and GA signaling is crucial in regulating dormancy cycling (Zhang Z. et al., 2018). Auxin is also an important plant hormone pivotal for dormancy transition in perennial tree species (Liu and Sherif, 2019). In the auxin-signaling pathway, IAA/AUX transcriptional factors can inhibit the ARFs activities through protein interactions (Leyser, 2017). We observed constantly decreasing level of ARF9, ARF15, and IAA9, and increasing level of ARF8, ARF18, IAA16, IAA26, and IAA17 during the process of floral bud break. These auxin-mediated genes are likely implicated in regulating floral bud break in P. mume. Additionally, we detected the differential expression of a few ERF (ETHYLENE RESPONSE FACTOR) transcription factors, such as ERF4, ERF5, and ERF113. These ERFs are likely required in regulating abiotic stresses responses relevant to the process of floral bud break in P. mume.
MicroRNAs associated with floral bud dormancy release in Prunus mume
Small RNAs are non-coding RNAs ranging from 20 to 24 nt and can be categorized into two major categories: small interfering RNAs (siRNA) and microRNAs (miRNA). miRNAs are usually generated from single-stranded RNA transcripts forming hairpin structure, which can be processed into mature miRNAs and fine-tune gene expression post-transcriptionally (Liu et al., 2020). Unlike miRNAs in animals, plant miRNAs mostly bind to RNA sequences within coding region of their target genes through near-perfect complementarity and direct mRNA cleavage for degradation (Millar and Waterhouse, 2005; Axtell, 2008). It has been demonstrated that the miRNA-mediated gene silencing is essential to many important biological processes including timing of development, pattern formation, response to environmental stimulus (Cui et al., 2017). With the emergence of high-throughput sequencing technology and bioinformatics tools, miRNAs and their functional roles were investigated in diverse developmental processes among a large number of plant species (Qin et al., 2014).
To identify key miRNAs involved in dormancy release in P. mume, we performed small RNA sequencing on floral buds and identified 41 known miRNAs and 53 novel miRNAs differentially expressed across different developmental stage comparisons. Their target genes were enriched in biological pathways, such as floral organ morphogenesis, hormonal signaling, secondary metabolism biosynthesis, and carbohydrate metabolism. Previous studies reported a few miRNAs related to dormancy cycling in perennial tree species (Ding et al., 2016). In poplar, miR169 was found to repress HAP2 (HEME ACTIVATOR PROTEIN 2) in dormant cambium, vegetative bud and floral bud tissues (Ding et al., 2016). The transcription of HAP2 rapidly declined as dormant bud resumes growth in the next spring (Potkar et al., 2013). In our study, miR169 maintained a constant level during dormancy release but slightly decreased after bud flush in our study. Another important miRNA identified as dormancy regulator is miR6390, which target DAM (DORMANCY-ASSOCIATED MADS-BOX) transcripts during floral bud dormancy release in pear (Niu et al., 2016). However, we fail to recognize miR6390 in our small RNA sequencing analysis. It is likely that there exist species-specific miRNAs that regulate dormancy release across tree species. One recent study identified a Prunus specific microRNA miR6285, which is differentially expressed between endodormancy and ecodormancy (Yu et al., 2021). In peach, miR6285 is a cold-responsive microRNA targeting gene NRP (ASPARAGINE-RICH PROTEIN) (Yu et al., 2021). We observed significantly increased level of miR6285 during dormancy release, which is consistent with its expression pattern in peach. However, we fail to validate the target gene for miR6285 with degradome sequencing, which is likely due to the low expression level of NRP during dormancy exit. Yu et al. also identified miR2275 that may be involved in pollen development at ecodormancy (Yu et al., 2021). We observed constantly increasing expression of miR2275 during chilling-induced dormancy release, however, miR2275 transcript level significantly dropped during the transition from ecodormancy to bud flush. In peach, miR2275 is predicted to target lncRNAs to generate phasiRNAs that is proposed to regulate microsporogenesis and pollen meiosis (Yu et al., 2021). It is likely that the functional role of miR2275 is conserved across Prunus species.
In addition to miRNAs characterized in previous studies, we also identified a few candidate miRNAs with expression profiles correlated with floral bud flush rate during dormancy transition. Among them, miR156/157-x/y, miR171, miR172-y, and miR319-x were identified. Previous studies have revealed that SPL transcription factors, which are target genes of miR156/157, play an important role in regulating vegetative-to-adult phase transition, pollen production, and floral organ elongation (including petals, sepals, and siliques) in Arabidopsis (Tsiantis et al., 2016). As plant age increases, the reduced expression of miR156 leads to increased transcript level of their target SPL transcription factors, which directly activate downstream MADS-box genes to promote flowering (Wang et al., 2009; Teotia and Tang, 2015). In the present study, miRNAs from miR156/157 family were found significantly induced with their target genes displaying contrasting expression pattern during floral bud break. The functional role of miR156/157 may be associated with floral organ expansion after the dormancy release. MiR172 is another miRNA family that controls vegetative-to-adult phase transition by targeting AP2-like transcription factors, such as AP2 in Arabidopsis (Aukerman and Sakai, 2003). The overexpression of miR172 triggered early flowering and disrupted floral organ by antagonizing AG (AGAMOUS) activity in Arabidopsis (Aukerman and Sakai, 2003). MiR172-x was found highly induced in the flushed floral bud, while AP2 and RAP2.7 transcripts declined after floral bud exit endodormancy. The temporal miR172-mediated down-regulation of AP2 indicated the reduced AP2 activity in specifying floral organ identity in matured flowers (Wollmann et al., 2010). Scarecrow-like transcription factors are known targets of miR171 and were reported to regulate meristem cell maintenance, polar organization and chlorophyll synthesis (Qu et al., 2014). The overexpression of miR171 in Arabidopsis leads to plants with reduced shoot branching, increased chlorophyll accumulation, abnormal leaf and flower patterning (Curaba et al., 2013; Qu et al., 2014). During the transition from ecodormancy to bud flush, SCL6 transcripts decreased as the level of miR171 dramatically increases, which may be associated with the gibberellin level change and inflorescence branching (Curaba et al., 2013). In general, we confirmed the contrasting expression pattern between many DEmiRNAs and their target genes with qRT-PCR assays. However, functional experiments are still required to clarify the regulatory mechanism of these microRNAs and their target transcription factors in controlling floral bud break in P. mume.
Putative microRNA-transcription factor regulatory network controlling floral bud break
Plant microRNAs are known to target genes that are often transcription factors or genes mediating protein ubiquitination such as F-box genes (Samad et al., 2017). Ancient miRNAs play relevant functions across different plant species by targeting conserved miRNA-binding sites (Axtell, 2008). Based on sequence conservation, many bioinformatics tools were developed to search for target genes of miRNAs in new species (Chorostecki et al., 2012). In two previous small RNA studies of P. mume, bioinformatics approach was employed to reveal miRNA-mRNA pairs (Gao et al., 2012; Wang et al., 2013). One possible problem with pure computational prediction is high false positive rate (Naoumkina et al., 2016). Degradome sequencing is a modified 5′-RACE (Rapid Amplification of cDNA Ends) approach combined with high-throughput sequencing and has recently been used to find accurate miRNA-target relationships in many plant species (Candar-Cakir et al., 2016). In our study, we combined the computational prediction with degradome sequencing to identify miRNA-mRNA regulation relationship. We further compared the expression pattern of miRNAs and their target genes to characterize miRNA-mRNA pairs that possibly regulate dormancy release and flower opening in P. mume. Many target genes displayed contrasting expression pattern with their miRNAs, confirming the negative regulation between miRNAs and their targets. However, we validated only limited number of cleavage events for miRNAs, leaving out a large number of miRNAs with no target gene validated in the degradome analysis. One possible explanation could be the expression of those miRNAs was too low to cleave their target, or the sliced targets were below the level of detection in degradome sequencing (Liu et al., 2014a).
Transcription factors often activate or repress target gene expression through binding to cis-regulatory elements within target gene promoters (Maston et al., 2006). On the other hand, their expression can be regulated post-transcriptionally by miRNAs (Yu et al., 2017). To further understand the cross-talk between TFs and miRNAs, we investigated the regulatory relationship among miRNA-TFs and mRNA-mRNAs. By applying weighted co-expression network analysis (WGCNA) approach, we identified co-expressed gene modules and miRNA modules sharing similar expression patterns during dormancy release. With trait-module association analysis, we identified three gene modules (module blue, module brown, and module darkseagreen4) and one miRNA module (module turquoise) that were related to the progression of dormancy release. Co-expression network is constructed based on the pairwise correlation among genes sharing similar spatial or temporal expression profiles. The connected genes are assumed to be either directly or indirectly regulated by co-regulators (Stuart et al., 2003). Therefore, we screened the differentially expressed transcription factors as well as highly connected neighboring genes from the trait-associated modules and integrated their co-expression network with miRNA-TF regulatory pairs.
In the integrated miRNA-mRNA network, we identified a few ARFs, including ARF8, ARF4, and ARF15 across three co-expression modules. Previous studies have reported that ARF2-4 are targets of trans-acting siRNAs generated from miR390-targeted TAS3 gene, ARF6 and ARF8 are targets of miR167, and ARF10, ARF16-17 are targets of miR160 in Arabidopsis (Axtell et al., 2006; Wu et al., 2006; Liu et al., 2010). miR167 is known to mediate stem elongation and flower organ outgrowth in Arabidopsis (Nagpal et al., 2005). The over-expression of miR167 lead to down-regulation of ARF6 and ARF8, which further results in shortened petals, stamens, styles, and female sterility in tomato (Liu et al., 2014b). In our study, we found the expression of ARF8 first decreased as endodormant floral buds accumulate chilling, but then strongly increased during bud flush. miR167, on the other hand, was significantly repressed during endodormancy release. The dramatic expression changes of miR167-ARF8 module suggested their possible role in floral organ growth and female fertility in P. mume. miR390 is implicated in regulating leaf patterning and developmental timing by repressing ARF2, ARF3, and ARF4 (Fahlgren et al., 2006). We observed constantly increasing level of ARF4 during flower bud flush, which indicate its activity may be required in the female and male gametophyte development in P. mume (Liu et al., 2018). miR396 with four targeting GRFs (GRF2, GRF3, GRF5, GRF8) were identified in module blue and module darkseagreen4. Recent findings have shown that miR396, targeting GRF1-4 and GRF7-9, is essential to regulate leaf morphogenesis, root and flower development (Samad et al., 2017). In Arabidopsis, miR396 is important in control sepal-petal identity and pistil development by regulating GRF transcript levels (Liang et al., 2013). During endodormancy release, miR396 was found decreased with its targeting GRFs increasing, indicating that miR396-GRFs possibly involved in reproductive organ development in P. mume.
Furthermore, miR319-TCP was previously reported to function in leaf development, branching, flower development and gamete production (Fang et al., 2021). TCP transcription factors can be grouped into two classes, where class I members are mainly cell cycling activators and class II are cell cycling inhibitors. The class I transcription factors, such as TCP14 was found involved in cell proliferation during leaf and flower development (Kieffer et al., 2011). In class II, TCP2, TCP3, and TCP4 are important in regulating leaf development (Manassero et al., 2013). In our study, we observed constantly increasing level of TCP4 and TCP14 along with decreasing level of miR319-y. Another miRNA-TF pair, miR166-ATHB-8/ATHB-14 was detected in module blue. miR165/166 downregulate HD-ZIP transcription factors in regulating shoot apical meristem activity and flower development through WUSCHEL pathway (Jung and Park, 2007). It is likely these miRNA-TF pairs are involved in cell proliferation, organ polarity and vascular patterning during flower development in P. mume. The co-expression network analysis also detected co-expressed genes that may be regulated directly or indirectly by transcription factors. For example, we detected ANT, AIL1, and AIL5 co-expressed with ARFs and GRFs within module darkseagreen4. In Arabidopsis, ARF5 upregulates the gene expression of ANT, AIL1, and AIL6 to specify and promote flower primordia (Krizek et al., 2020). On the other hand, ANT/AIL, GRF, and ANGUSTIFOLIA3/GRF-INTERACTING FACTOR (AN3/GIF) family genes form regulatory modules to regulate cell proliferation and organ size in leaf or flower primordia (Krizek et al., 2021). Therefore, the proposed miRNA-TF mediated regulatory modules revealed potential players essential to different developmental processes occurred during endodormancy release and floral bud flush in P. mume.
Conclusion
In this study, we performed comprehensive analysis using transcriptome and small RNA sequencing to reveal candidate genes and miRNAs functioning in floral bud break in P. mume. With differential expression analysis, we identified a number of DEGs that participated in hormonal responses, flower organ development and gametophyte production during dormancy transition. The genome-wide characterization of miRNAs identified 41 known miRNAs and 53 novel miRNAs differentially expressed during this period. Combining computational prediction tool and degradome sequencing, we identified target genes for miRNAs and validated the expression correlation between target genes and their corresponding miRNAs. With weighted co-expression analysis, we characterized the modules of genes and miRNAs displaying expression profiles that highly associated with floral bud flush competency. Based on co-expressed gene candidates, we further integrated the miRNA-TF regulatory pairs to construct the miRNA-mRNA regulatory network that possibly involved in regulating different aspects of dormancy release and floral bud break in P. mume. To conclude, these findings provide valuable information for understanding microRNA-mediated regulatory networks in floral bud development in P. mume and other woody perennials.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA833165; https://www.ncbi.nlm.nih.gov/, PRJNA832606; and https://www.ncbi.nlm.nih.gov/, PRJNA832060.
Author contributions
MZ and QZ conceived and designed the study. MZ performed most of the experiment, analyzed the data, and wrote the manuscript. WC and XY contributed to the sample collection and qRT-PCR assays. TC and JW provided help with the data analysis. All authors contributed to the article and approved the submitted version.
Funding
This work was financially supported by the National Natural Science Foundation of China (31902045), the Fundamental Research Funds for the Central Universities (2021ZY44), and the Special Fund for Beijing Common Construction Project.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.931454/full#supplementary-material
Footnotes
- ^ https://CRAN.R-project.org/package=pheatmap
- ^ http://rna.tbi.univie.ac.at//cgi-bin/RNAWebSuite/RNAfold.cgi
References
Aukerman, M. J., and Sakai, H. (2003). Regulation of flowering time and floral organ identity by a MicroRNA and its APETALA2-like target genes. Plant Cell 15, 2730–2741. doi: 10.1105/tpc.016238
Axtell, M. (2008). Evolution of microRNAs and their targets: are all microRNAs biologically relevant? Biochim. Biophys. Acta 1779, 725–734. doi: 10.1016/j.bbagrm.2008.02.007
Axtell, M. J., Jan, C., Rajagopalan, R., and Bartel, D. P. (2006). A two-hit trigger for siRNA biogenesis in plants. Cell 127, 565–577. doi: 10.1016/j.cell.2006.09.032
Azeez, A., Miskolczi, P., Tylewicz, S., and Bhalerao, R. P. (2014). A tree ortholog of APETALA1 mediates photoperiodic control of seasonal growth. Curr. Biol. 24, 717–724. doi: 10.1016/j.cub.2014.02.037
Azeez, A., Zhao, Y. C., Singh, R. K., Yordanov, Y. S., Dash, M., Miskolczi, P., et al. (2021). EARLY BUD-BREAK 1 and EARLY BUD-BREAK 3 control resumption of poplar growth after winter dormancy. Nat. Commun. 12:1123. doi: 10.1038/s41467-021-21449-0
Bailly, C. (2020). Anticancer properties of Prunus mume extracts (Chinese plum, Japanese apricot). J. Ethnopharmacol. 246, 112215. doi: 10.1016/j.jep.2019.112215
Benson, D. A., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J., and Wheeler, D. L. (2007). GenBank. Nucleic Acids Res. 36, D25–D30. doi: 10.1093/nar/gkm929
Betancur, J. G., Yoda, M., and Tomari, Y. (2012). miRNA-like duplexes as RNAi triggers with improved specificity. Front. Genet. 3:127. doi: 10.3389/fgene.2012.00127
Bielenberg, D. G., Wang, Y., Li, Z., Zhebentyayeva, T., Fan, S., Reighard, G. L., et al. (2008). Sequencing and annotation of the evergrowing locus in peach [Prunus persica (L.) Batsch] reveals a cluster of six MADS-box transcription factors as candidate genes for regulation of terminal bud formation. Tree Genet. Genomes 4, 495–507. doi: 10.1007/s11295-007-0126-9
Candar-Cakir, B., Arican, E., and Zhang, B. (2016). Small RNA and degradome deep sequencing reveals drought-and tissue-specific micrornas and their important roles in drought-sensitive and drought-tolerant tomato genotypes. Plant Biotechnol. J. 14, 1727–1746. doi: 10.1111/pbi.12533
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Chorostecki, U., Crosa, V. A., Lodeyro, A. F., Bologna, N. G., Martin, A. P., Carrillo, N., et al. (2012). Identification of new microRNA-regulated genes by conserved targeting in plant species. Nucleic Acids Res. 40, 8893–8904. doi: 10.1093/nar/gks625
Cooke, J. E. K., Eriksson, M. E., and Junttila, O. (2012). The dynamic nature of bud dormancy in trees: environmental control and molecular mechanisms. Plant Cell Environ. 35, 1707–1728. doi: 10.1111/j.1365-3040.2012.02552.x
Cui, J., You, C., and Chen, X. (2017). The evolution of microRNAs in plants. Curr. Opin. Plant Biol. 35, 61–67. doi: 10.1016/j.pbi.2016.11.006
Curaba, J., Talbot, M., Li, Z., and Helliwell, C. (2013). Over-expression of microRNA171 affects phase transitions and floral meristem determinancy in barley. BMC Plant Biol. 13:6. doi: 10.1186/1471-2229-13-6
D’Ario, M., Griffiths-Jones, S., and Kim, M. (2017). Small RNAs: big impact on plant development. Trends Plant Sci. 22, 1056–1068. doi: 10.1016/j.tplants.2017.09.009
Ding, Q., Zeng, J., and He, X. Q. (2014). Deep sequencing on a genome-wide scale reveals diverse stage-specific microRNAs in cambium during dormancy-release induced by chilling in poplar. BMC Plant Biol. 14:267. doi: 10.1186/s12870-014-0267-6
Ding, Q., Zeng, J., and He, X.-Q. (2016). MiR169 and its target PagHAP2-6 regulated by ABA are involved in poplar cambium dormancy. J. Plant Physiol. 198, 1–9. doi: 10.1016/j.jplph.2016.03.017
Fadón, E., Fernandez, E., Behn, H., and Luedeling, E. (2020). A conceptual framework for winter dormancy in deciduous trees. Agronomy 10:241. doi: 10.3390/agronomy10020241
Fadón, E., Herrero, M., and Rodrigo, J. (2018). Dormant flower buds actively accumulate starch over winter in sweet cherry. Front. Plant Sci. 9:171. doi: 10.3389/fpls.2018.00171
Fahlgren, N., Montgomery, T. A., Howell, M. D., Allen, E., Dvorak, S. K., Alexander, A. L., et al. (2006). Regulation of AUXIN RESPONSE FACTOR3 by TAS3 ta-siRNA affects developmental timing and patterning in Arabidopsis. Curr. Biol. 16, 939–944. doi: 10.1016/j.cub.2006.03.065
Fan, Z., Li, J., Li, X., Wu, B., Wang, J., Liu, Z., et al. (2015). Genome-wide transcriptome profiling provides insights into floral bud development of summer-flowering Camellia azalea. Sci. Rep. 5:9729. doi: 10.1038/srep09729
Fang, L., and Wang, Y. (2021). MicroRNAs in woody plants. Front. Plant Sci. 12:686831. doi: 10.3389/fpls.2021.686831
Fang, Y., Xie, K., and Xiong, L. (2014). Conserved miR164-targeted NAC genes negatively regulate drought resistance in rice. J. Exp. Bot. 65, 2119–2135. doi: 10.1093/jxb/eru072
Fang, Y., Zheng, Y., Lu, W., Li, J., Duan, Y., Zhang, S., et al. (2021). Roles of miR319-regulated TCPs in plant development and response to abiotic stress. Crop J. 9, 17–28.
Friedländer, M. R., Mackowiak, S. D., Li, N., Chen, W., and Rajewsky, N. (2012). miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 40, 37–52. doi: 10.1093/nar/gkr688
Gao, Z., Shi, T., Luo, X., Zhang, Z., Zhuang, W., and Wang, L. (2012). High-throughput sequencing of small RNAs and analysis of differentially expressed microRNAs associated with pistil development in Japanese apricot. BMC Genomics 13:371. doi: 10.1186/1471-2164-13-371
Gene Ontology Consortium (2004). The gene ontology (GO) database and informatics resource. Nucleic Acids Res. 32, 258D–261D. doi: 10.1093/nar/gkh036
Goldstien, S. J., Liang, C., Zhang, X., Zou, J., Xu, D., Su, F., et al. (2010). Identification of miRNA from Porphyra yezoensis by high-throughput sequencing and bioinformatics analysis. PLoS One 5:e10698. doi: 10.1371/journal.pone.0010698
Gordon, A., and Hannon, G. J. (2010). FASTX-Toolkit: FASTQ/a Short-Reads Pre-Processing Tools. Available Online at: http://hannonlab.cshl.edu/fastx_toolkit (accessed July 6, 2021).
Griffiths-Jones, S. (2004). Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, D121–D124. doi: 10.1093/nar/gki081
Griffiths-Jones, S. (2006). miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 34, D140–D144. doi: 10.1093/nar/gkj112
Haake, V., Cook, D., Riechmann, J. L., Pineda, O., Thomashow, M. F., and Zhang, J. Z. (2002). Transcription factor CBF4 is a regulator of drought adaptation in Arabidopsis. Plant Physiol. 130, 639–648. doi: 10.1104/pp.006478
Hatfield, J. L., and Prueger, J. H. (2015). Temperature extremes: effect on plant growth and development. Weather Clim. Extrem. 10, 4–10. doi: 10.1016/j.wace.2015.08.001
Hsiang, T.-F., Lin, Y.-J., Yamane, H., and Tao, R. (2021). Characterization of Japanese apricot (Prunus mume) floral bud development using a modified BBCH scale and analysis of the relationship between BBCH stages and floral primordium development and the dormancy phase transition. Horticulturae 7:142. doi: 10.3390/horticulturae7060142
Immink, R. G. H., Tonaco, I. A. N., de Folter, S., Shchennikova, A., van Dijk, A. D. J., Busscher-Lange, J., et al. (2009). SEPALLATA3: the ‘glue’ for MADS box transcription factor complex formation. Genome Biol. 10:R24. doi: 10.1186/gb-2009-10-2-r24
Ionescu, I. A., López-Ortega, G., Burow, M., Bayo-Canha, A., Junge, A., Gericke, O., et al. (2017). Transcriptome and metabolite changes during hydrogen cyanamide-induced floral bud break in sweet cherry. Front. Plant Sci. 8:1233. doi: 10.3389/fpls.2017.01233
Ito, A., Saito, T., Sakamoto, D., Sugiura, T., Bai, S., Moriguchi, T., et al. (2016). Physiological differences between bud breaking and flowering after dormancy completion revealed by DAM and FT/TFL1 expression in Japanese pear (Pyrus pyrifolia). Tree Physiol. 36, 109–120. doi: 10.1093/treephys/tpv115
Jeyaraj, A., Chandran, V., and Gajjeraman, P. (2014). Differential expression of microRNAs in dormant bud of tea [Camellia sinensis (L.) O. Kuntze]. Plant Cell Rep. 33, 1053–1069. doi: 10.1007/s00299-014-1589-4
Jiménez, S., Reighard, G. L., and Bielenberg, D. G. (2010). Gene expression of DAM5 and DAM6 is suppressed by chilling temperatures and inversely correlated with bud break rate. Plant Mol. Biol. 73, 157–167. doi: 10.1007/s11103-010-9608-5
Julian, C., Rodrigo, J., and Herrero, M. (2011). Stamen development and winter dormancy in apricot (Prunus armeniaca). Ann. Bot. 108, 617–625. doi: 10.1093/aob/mcr056
Jung, J.-H., and Park, C.-M. (2007). MIR166/165 genes exhibit dynamic expression patterns in regulating shoot apical meristem and floral development in Arabidopsis. Planta 225, 1327–1338. doi: 10.1007/s00425-006-0439-1
Kieffer, M., Master, V., Waites, R., and Davies, B. (2011). TCP14 and TCP15 affect internode length and leaf shape in Arabidopsis. Plant J. 68, 147–158. doi: 10.1111/j.1365-313X.2011.04674.x
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Kim, H.-W., Han, S. H., Lee, S.-W., and Suh, H. J. (2018). Effect of isomaltulose used for osmotic extraction of Prunus mume fruit juice substituting sucrose. Food Sci. Biotechnol. 27, 1599–1605. doi: 10.1007/s10068-018-0408-z
Kim, V. N. (2004). MicroRNA precursors in motion: exportin-5 mediates their nuclear export. Trends Cell Biol. 14, 156–159. doi: 10.1016/j.tcb.2004.02.006
Koutinas, N., Pepelyankov, G., and Lichev, V. (2014). Flower induction and flower bud development in apple and sweet cherry. Biotechnol. Biotechnol. Equip. 24, 1549–1558. doi: 10.2478/v10133-010-0003-9
Kozomara, A., Birgaoanu, M., and Griffiths-Jones, S. (2019). miRBase: from microRNA sequences to function. Nucleic Acids Res. 47, D155–D162. doi: 10.1093/nar/gky1141
Krizek, B. A. (2015). AINTEGUMENTA-LIKE genes have partly overlapping functions with AINTEGUMENTA but make distinct contributions to Arabidopsis thaliana flower development. J. Exp. Bot. 66, 4537–4549. doi: 10.1093/jxb/erv224
Krizek, B. A., Bantle, A. T., Heflin, J. M., Han, H., Freese, N. H., and Loraine, A. E. (2021). AINTEGUMENTA and AINTEGUMENTA-LIKE6 directly regulate floral homeotic, growth, and vascular development genes in young Arabidopsis flowers. J. Exp. Bot. 72, 5478–5493. doi: 10.1093/jxb/erab223
Krizek, B. A., Blakley, I. C., Ho, Y. Y., Freese, N., and Loraine, A. E. (2020). The Arabidopsis transcription factor AINTEGUMENTA orchestrates patterning genes and auxin signaling in the establishment of floral growth and form. Plant J. 103, 752–768. doi: 10.1111/tpj.14769
Ladwig, L. M., Chandler, J. L., Guiden, P. W., and Henn, J. J. (2019). Extreme winter warm event causes exceptionally early bud break for many woody species. Ecosphere 10:e02542. doi: 10.1002/ecs2.2542
Lang, G. A., Early, J. D., Martin, G. C., and Darnell, R. L. (1987). Endo-, para-,and ecodormancy : physiological terminology and classification for dormancy research. HortScience 22, 371–377.
Langfelder, P., and Horvath, S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9:559. doi: 10.1186/1471-2105-9-559
Lee, J., and Lee, I. (2010). Regulation and function of SOC1, a flowering pathway integrator. J. Exp. Bot. 61, 2247–2254. doi: 10.1093/jxb/erq098
Lee, Y., Kim, M., Han, J., Yeom, K. H., Lee, S., Baek, S. H., et al. (2004). MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 23, 4051–4060. doi: 10.1038/sj.emboj.7600385
Leubner, G., Li, L., Li, D., Gao, D., Chen, X., Fu, X., et al. (2020). Transcription factor TCP20 regulates peach bud endodormancy by inhibiting DAM5/DAM6 and interacting with ABF2. J. Exp. Bot. 71, 1585–1597.
Li, B., and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323
Li, J., Guo, G., Guo, W., Guo, G., Tong, D., Ni, Z., et al. (2012). miRNA164-directed cleavage of ZmNAC1 confers lateral root development in maize (Zea mays L.). BMC Plant Biol. 12:220. doi: 10.1186/1471-2229-12-220
Li, R., Yu, C., Li, Y., Lam, T. W., Yiu, S. M., Kristiansen, K., et al. (2009). SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25, 1966–1967. doi: 10.1093/bioinformatics/btp336
Li, S. F., Milliken, O. N., Pham, H., Seyit, R., Napoli, R., Preston, J., et al. (2009). The Arabidopsis MYB5 transcription factor regulates mucilage synthesis, seed coat development, and trichome morphogenesis. Plant Cell 21, 72–89. doi: 10.1105/tpc.108.063503
Li, Y., An, S., Cheng, Q., Zong, Y., Chen, W., Guo, W., et al. (2021). Analysis of evolution, expression and genetic transformation of TCP transcription factors in blueberry reveal that VcTCP18 negatively regulates the release of flower bud dormancy. Front. Plant Sci. 12:697609. doi: 10.3389/fpls.2021.697609
Liang, G., He, H., Li, Y., Wang, F., and Yu, D. (2013). Molecular mechanism of microRNA396 mediating pistil development in Arabidopsis. Plant Physiol. 164, 249–258. doi: 10.1104/pp.113.225144
Liu, F., Dong, H., Mei, Z., and Huang, T. (2020). Investigation of miRNA and mRNA Co-expression network in ependymoma. Front. Bioeng. Biotechnol. 8:177. doi: 10.3389/fbioe.2020.00177
Liu, J., and Sherif, S. M. (2019). Hormonal orchestration of bud dormancy cycle in deciduous woody perennials. Front. Plant Sci. 10:1136. doi: 10.3389/fpls.2019.01136
Liu, J. Z., McRae, A. F., Nyholt, D. R., Medland, S. E., Wray, N. R., Brown, K. M., et al. (2010). A versatile gene-based test for genome-wide association studies. Am. J. Hum. Genet. 87, 139–145. doi: 10.1016/j.ajhg.2010.06.009
Liu, N., Tu, L., Tang, W., Gao, W., Lindsey, K., and Zhang, X. (2014a). Small RNA and degradome profiling reveals a role for miRNAs and their targets in the developing fibers of Gossypium barbadense. Plant J. 80, 331–344. doi: 10.1111/tpj.12636
Liu, N., Wu, S., Van Houten, J., Wang, Y., Ding, B., Fei, Z., et al. (2014b). Down-regulation of AUXIN RESPONSE FACTORS 6 and 8 by microRNA 167 leads to floral development defects and female sterility in tomato. J. Exp. Bot. 65, 2507–2520. doi: 10.1093/jxb/eru141
Liu, Z., Franks, R. G., and Klink, V. P. (2000). Regulation of gynoecium marginal tissue formation by LEUNIG and AINTEGUMENTA. Plant Cell 12, 1879–1891. doi: 10.2307/3871199
Liu, Z., Miao, L., Huo, R., Song, X., Johnson, C., Kong, L., et al. (2018). ARF2–ARF4 and ARF5 are essential for female and male gametophyte development in Arabidopsis. Plant Cell Physiol. 59, 179–189. doi: 10.1093/pcp/pcx174
Lloret, A., Badenes, M. L., and Ríos, G. (2018). Modulation of dormancy and growth responses in reproductive buds of temperate trees. Front. Plant Sci. 9:1368. doi: 10.3389/fpls.2018.01368
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Luedeling, E. (2012). Climate change impacts on winter chill for temperate fruit and nut production: a review. Sci. Hortic. 144, 218–229. doi: 10.1016/j.scienta.2012.07.011
Manassero, N. G. U., Viola, I. L., Welchen, E., and Gonzalez, D. H. (2013). TCP transcription factors: architectures of plant form. Biomol. Concepts 4, 111–127. doi: 10.1515/bmc-2012-0051
Maston, G. A., Evans, S. K., and Green, M. R. (2006). Transcriptional regulatory elements in the human genome. Annu. Rev. Genomics Hum. Genet. 7, 29–59. doi: 10.1146/annurev.genom.7.080505.115623
Millar, A., He, J., Xu, M., Willmann, M. R., McCormick, K., Hu, T., et al. (2018). Threshold-dependent repression of SPL gene expression by miR156/miR157 controls vegetative phase change in Arabidopsis thaliana. PLoS Genet. 14:e1007337. doi: 10.1371/journal.pgen.1007337
Millar, A. A., and Waterhouse, P. M. (2005). Plant and animal microRNAs: similarities and differences. Funct. Integr. Genomics 5, 129–135. doi: 10.1007/s10142-005-0145-2
Min, H., and Yoon, S. (2010). Got target : computational methods for microRNA target prediction and their extension. Exp. Mol. Med. 42, 233–244. doi: 10.3858/emm.2010.42.4.032
Nagpal, P., Ellis, C. M., Weber, H., Ploense, S. E., Barkawi, L. S., Guilfoyle, T. J., et al. (2005). Auxin response factors ARF6 and ARF8 promote jasmonic acid production and flower maturation. Development 132, 4107–4118. doi: 10.1242/dev.01955
Naoumkina, M., Thyssen, G. N., Fang, D. D., Hinchliffe, D. J., Florane, C. B., and Jenkins, J. N. (2016). Small RNA sequencing and degradome analysis of developing fibers of short fiber mutants Ligon-lintles-1 (Li 1) and -2 (Li 2) revealed a role for miRNAs and their targets in cotton fiber elongation. BMC Genomics 17:360. doi: 10.1186/s12864-016-2715-1
Niu, Q., Li, J., Cai, D., Qian, M., Jia, H., Bai, S., et al. (2016). Dormancy-associated MADS-box genes and microRNAs jointly control dormancy transition in pear (Pyrus pyrifolia white pear group) flower bud. J. Exp. Bot. 67, 239–257. doi: 10.1093/jxb/erv454
Omidbakhshfard, M. A., Proost, S., Fujikura, U., and Mueller-Roeber, B. (2015). Growth-regulating factors (GRFs): a small transcription factor family with important functions in plant biology. Mol. Plant 8, 998–1010. doi: 10.1016/j.molp.2015.01.013
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T., and Salzberg, S. L. (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667. doi: 10.1038/nprot.2016.095
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., and Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Potkar, R., Recla, J., and Busov, V. (2013). ptr-MIR169 is a posttranscriptional repressor of PtrHAP2 during vegetative bud dormancy period of aspen (Populus tremuloides) trees. Biochem. Biophys. Res. Commun. 431, 512–518. doi: 10.1016/j.bbrc.2013.01.027
Qin, Z., Li, C., Mao, L., and Wu, L. (2014). Novel insights from non-conserved microRNAs in plants. Front. Plant Sci. 5:586. doi: 10.3389/fpls.2014.00586
Qu, L.-J., Ma, Z., Hu, X., Cai, W., Huang, W., Zhou, X., et al. (2014). Arabidopsis miR171-targeted scarecrow-like proteins bind to GT cis-elements and mediate gibberellin-regulated chlorophyll biosynthesis under light conditions. PLoS Genet. 10:e1004519. doi: 10.1371/journal.pgen.1004519
Ramachandran, P., Carlsbecker, A., and Etchells, J. P. (2017). Class III HD-ZIPs govern vascular cell fate: an HD view on patterning and differentiation. J. Exp. Bot. 68, 55–69. doi: 10.1093/jxb/erw370
Ramos, A., Rapoport, H. F., Cabello, D., and Rallo, L. (2018). Chilling accumulation, dormancy release temperature, and the role of leaves in olive reproductive budburst: evaluation using shoot explants. Sci. Hortic. 231, 241–252. doi: 10.1016/j.scienta.2017.11.003
Rinne, P. L. H., Welling, A., Vahala, J., Ripel, L., Ruonala, R., Kangasjärvi, J., et al. (2011). Chilling of dormant buds hyperinduces FLOWERING LOCUS T and recruits GA-inducible 1,3-β-glucanases to reopen signal conduits and release dormancy in Populus. Plant Cell 23, 130–146. doi: 10.1105/tpc.110.081307
Rohde, A., and Bhalerao, R. P. (2007). Plant dormancy in the perennial context. Trends Plant Sci. 12, 217–223. doi: 10.1016/j.tplants.2007.03.012
Rohde, A., Prinsen, E., De Rycke, R., Engler, G., Van Montagu, M., and Boerjan, W. (2002). PtABI3 impinges on the growth and differentiation of embryonic leaves during bud set in poplar. Plant Cell 14, 1885–1901. doi: 10.1105/tpc.003186
Rongmei, W., Sumathi, T., Sakuntala, K., Tustin, S. D., Hellens, R. P., Allan, A. C., et al. (2017). SVP-like MADS box genes control dormancy and budbreak in apple. Front. Plant Sci. 8:477. doi: 10.3389/fpls.2017.00477
Rubio-Somoza, I., and Weigel, D. (2011). MicroRNA networks and developmental plasticity in plants. Trends Plant Sci. 16, 258–264. doi: 10.1016/j.tplants.2011.03.001
Samad, A. F. A., Sajad, M., Nazaruddin, N., Fauzi, I. A., Murad, A. M. A., Zainal, Z., et al. (2017). MicroRNA and transcription factor: key players in plant regulatory network. Front. Plant Sci. 8:565. doi: 10.3389/fpls.2017.00565
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Sharma, D., Tiwari, M., Pandey, A., Bhatia, C., Sharma, A., and Trivedi, P. K. (2016). MicroRNA858 is a potential regulator of phenylpropanoid pathway and plant development. Plant Physiol. 171, 944–959. doi: 10.1104/pp.15.01831
Singh, R. K., Maurya, J. P., Azeez, A., Miskolczi, P., Tylewicz, S., Stojkoviè, K., et al. (2018). A genetic network mediating the control of bud break in hybrid aspen. Nat. Commun. 9:4173. doi: 10.1038/s41467-018-06696-y
Spanudakis, E., and Jackson, S. (2014). The role of microRNAs in the control of flowering time. J. Exp. Bot. 65, 365–380. doi: 10.1093/jxb/ert453
Strzyz, P. (2021). microRNA communication in plants. Nat. Rev. Mol. Cell Biol. 22, 775–775. doi: 10.1038/s41580-021-00431-0
Stuart, J. M., Segal, E., Koller, D., and Kim, S. K. (2003). A gene-coexpression network for global discovery of conserved genetic modules. Science 302, 249–255. doi: 10.1126/science.1087447
Teotia, S., and Tang, G. (2015). To bloom or not to bloom: role of MicroRNAs in plant flowering. Mol. Plant 8, 359–377. doi: 10.1016/j.molp.2014.12.018
Tsiantis, M., Xu, M., Hu, T., Zhao, J., Park, M.-Y., Earley, K. W., et al. (2016). Developmental functions of miR156-regulated SQUAMOSA PROMOTER BINDING PROTEIN-LIKE (SPL) genes in Arabidopsis thaliana. PLoS Genet. 12:e1006263. doi: 10.1371/journal.pgen.1006263
Ueno, S., Klopp, C., Leplé, J. C., Derory, J., Noirot, C., Léger, V., et al. (2013). Transcriptional profiling of bud dormancy induction and release in oak by next-generation sequencing. BMC Genomics 14:236. doi: 10.1186/1471-2164-14-236
van der Schoot, C., Paul, L. K., and Rinne, P. L. H. (2013). The embryonic shoot: a lifeline through winter. J. Exp. Bot. 65, 1699–1712. doi: 10.1093/jxb/ert413
Wang, J., Gao, Z., Li, H., Jiu, S., Qu, Y., Wang, L., et al. (2020). Dormancy-associated MADS-Box (DAM) genes influence chilling requirement of sweet cherries and co-regulate flower development with SOC1 gene. Int. J. Mol. Sci. 21:921. doi: 10.3390/ijms21030921
Wang, J.-W., Czech, B., and Weigel, D. (2009). miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell 138, 738–749. doi: 10.1016/j.cell.2009.06.014
Wang, Q., Xu, G., Zhao, X., Zhang, Z., Wang, X., Liu, X., et al. (2019). Transcription factor TCP20 regulates peach bud endodormancy by inhibiting DAM5/DAM6 and interacting with ABF2. J. Exp. Bot. 71, 1585–1597. doi: 10.1093/jxb/erz516
Wang, T., Pan, H., Wang, J., Yang, W., Cheng, T., and Zhang, Q. (2013). Identification and profiling of novel and conserved microRNAs during the flower opening process in Prunus mume via deep sequencing. Mol. Genet. Genomics 289, 169–183. doi: 10.1007/s00438-013-0800-6
Wang, Z., Wang, F., Hong, Y., Yao, J., Ren, Z., Shi, H., et al. (2018). The flowering repressor SVP confers drought resistance in Arabidopsis by regulating abscisic acid catabolism. Mol. Plant 11, 1184–1197. doi: 10.1016/j.molp.2018.06.009
Welling, A., and Palva, E. T. (2008). Involvement of CBF transcription factors in winter hardiness in birch. Plant Physiol. 147, 1199–1211. doi: 10.1104/pp.108.117812
Wollmann, H., Mica, E., Todesco, M., Long, J. A., and Weigel, D. (2010). On reconciling the interactions between APETALA2, miR172 and AGAMOUS with the ABC model of flower development. Development 137, 3633–3642. doi: 10.1242/dev.036673
Wu, G., Park, M. Y., Conway, S. R., Wang, J.-W., Weigel, D., and Poethig, R. S. (2009). The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 138, 750–759. doi: 10.1016/j.cell.2009.06.031
Wu, M. F., Tian, Q., and Reed, J. W. (2006). Arabidopsis microRNA167 controls patterns of ARF6 and ARF8 expression, and regulates both female and male reproduction. Development 133, 4211–4218. doi: 10.1242/dev.02602
Wu, R., Cooney, J., Tomes, S., Rebstock, R., Karunairetnam, S., Allan, A. C., et al. (2021). RNAi-mediated repression of dormancy-related genes results in evergrowing apple trees. Tree Physiol. 41, 1510–1523. doi: 10.1093/treephys/tpab007
Wu, X., Shi, T., Iqbal, S., Zhang, Y., Liu, L., and Gao, Z. (2019). Genome-wide discovery and characterization of flower development related long non-coding RNAs in Prunus mume. BMC Plant Biol. 19:64. doi: 10.1186/s12870-019-1672-7
Xia, R., Chen, C., Pokhrel, S., Ma, W., Huang, K., Patel, P., et al. (2019). 24-nt reproductive phasiRNAs are broadly present in angiosperms. Nat. Commun. 10:627. doi: 10.1038/s41467-019-08543-0
Yamane, H., Ooka, T., Jotatsu, H., Hosaka, Y., Sasaki, R., and Tao, R. (2011). Expressional regulation of PpDAM5 and PpDAM6, peach (Prunus persica) dormancy-associated MADS-box genes, by low temperature and dormancy-breaking reagent treatment. J. Exp. Bot. 62, 3481–3488. doi: 10.1093/jxb/err028
Yan, G., Zhang, J., Jiang, M., Gao, X., Yang, H., and Li, L. (2020). Identification of known and novel MicroRNAs in raspberry organs through high-throughput sequencing. Front. Plant Sci. 11:728. doi: 10.3389/fpls.2020.00728
Yan, T., Yoo, D., Berardini, T. Z., Mueller, L. A., Weems, D. C., Weng, S., et al. (2005). PatMatch: a program for finding patterns in peptide and nucleotide sequences. Nucleic Acids Res. 33, W262–W266. doi: 10.1093/nar/gki368
Yang, C., Song, J., Ferguson, A. C., Klisch, D., Simpson, K., Mo, R., et al. (2017). Transcription factor MYB26 is key to spatial specificity in anther secondary thickening formation. Plant Physiol. 175, 333–350. doi: 10.1104/pp.17.00719
Yang, Q., Gao, Y., Wu, X., Moriguchi, T., Bai, S., and Teng, Y. (2021). Bud endodormancy in deciduous fruit trees: advances and prospects. Hortic. Res. 8:139. doi: 10.1038/s41438-021-00575-2
Yang, Q., Niu, Q., Li, J., Zheng, X., Ma, Y., Bai, S., et al. (2018). PpHB22, a member of HD-Zip proteins, activates PpDAM1 to regulate bud dormancy transition in ‘Suli’ pear (Pyrus pyrifolia White Pear Group). Plant Physiol. Biochem. 127, 355–365. doi: 10.1016/j.plaphy.2018.04.002
Yu, H., Shu, K., Zhang, H., Wang, S., Chen, M., Wu, Y., et al. (2013). ABI4 regulates primary seed dormancy by regulating the biogenesis of abscisic acid and gibberellins in Arabidopsis. PLoS Genet. 9:e1003577. doi: 10.1371/journal.pgen.1003577
Yu, J., Bennett, D., Dardick, C., Zhebentyayeva, T., Abbott, A. G., Liu, Z., et al. (2021). Genome-wide changes of regulatory non-coding RNAs reveal pollen development initiated at ecodormancy in peach. Front. Mol. Biosci. 8:612881. doi: 10.3389/fmolb.2021.612881
Yu, J., Conrad, A. O., Decroocq, V., Zhebentyayeva, T., Williams, D. E., Bennett, D., et al. (2020). Distinctive gene expression patterns define endodormancy to ecodormancy transition in apricot and peach. Front. Plant Sci. 11:180. doi: 10.3389/fpls.2020.00180
Yu, Y., Jia, T., and Chen, X. (2017). The ‘how’ and ‘where’ of plant microRNAs. New Phytol. 216, 1002–1017. doi: 10.1111/nph.14834
Zhang, M., Yang, Q., Yuan, X., Yan, X., Wang, J., Cheng, T., et al. (2021b). Integrating genome-wide association analysis with transcriptome sequencing to identify candidate genes related to blooming time in Prunus mume. Front. Plant Sci. 12:690841. doi: 10.3389/fpls.2021.690841
Zhang, M., Li, P., Yan, X., Wang, J., Cheng, T., and Zhang, Q. (2021a). Genome-wide characterization of PEBP family genes in nine Rosaceae tree species and their expression analysis in P. mume. BMC Ecol. Evol. 21:32. doi: 10.1186/s12862-021-01762-4
Zhang, M., Suren, H., and Holliday, J. A. (2019). Phenotypic and genomic local adaptation across latitude and altitude in Populus trichocarpa. Genome Biol. Evol. 11, 2256–2272. doi: 10.1093/gbe/evz151
Zhang, Q., Zhang, H., Sun, L., Fan, G., Ye, M., Jiang, L., et al. (2018). The genetic architecture of floral traits in the woody plant Prunus mume. Nat. Commun. 9:1702. doi: 10.1038/s41467-018-04093-z
Zhang, Z., Zhuo, X., Zhao, K., Zheng, T., Han, Y., Yuan, C., et al. (2018). Transcriptome profiles reveal the crucial roles of hormone and sugar in the bud dormancy of Prunus mume. Sci. Rep. 8:5090. doi: 10.1038/s41598-018-23108-9
Zhao, D.-Z., Wang, G.-F., Speal, B., and Ma, H. (2002). The EXCESS MICROSPOROCYTES1 gene encodes a putative leucine-rich repeat receptor protein kinase that controls somatic and reproductive cell fates in the Arabidopsis anther. Genes Dev. 16, 2021–2031. doi: 10.1101/gad.997902
Zhao, K., Zhou, Y., Ahmad, S., Yong, X., Xie, X., Han, Y., et al. (2018a). PmCBFs synthetically affect PmDAM6 by alternative promoter binding and protein complexes towards the dormancy of bud for Prunus mume. Sci. Rep. 8:4527. doi: 10.1038/s41598-018-22537-w
Zhao, K., Zhou, Y., Li, Y., Zhuo, X., Ahmad, S., Han, Y., et al. (2018b). Crosstalk of PmCBFs and PmDAMs based on the changes of phytohormones under seasonal cold stress in the stem of Prunus mume. Int. J. Mol. Sci. 19:15. doi: 10.3390/ijms19020015
Zheng, T., Li, P., Zhuo, X., Liu, W., Qiu, L., Li, L., et al. (2021). The chromosome−level genome provides insight into the molecular mechanism underlying the tortuous−branch phenotype of Prunus mume. New Phytol. 235, 141–156. doi: 10.1111/nph.17894
Keywords: transcriptome analysis, small RNA sequencing, co-expression network, floral bud break, dormancy release, Prunus mume
Citation: Zhang M, Cheng W, Yuan X, Wang J, Cheng T and Zhang Q (2022) Integrated transcriptome and small RNA sequencing in revealing miRNA-mediated regulatory network of floral bud break in Prunus mume. Front. Plant Sci. 13:931454. doi: 10.3389/fpls.2022.931454
Received: 29 April 2022; Accepted: 30 June 2022;
Published: 22 July 2022.
Edited by:
Xiaowu Wang, Biotechnology Research Institute (CAAS), ChinaReviewed by:
Rui Xia, South China Agricultural University, ChinaZhigang Wei, Chinese Academy of Forestry, China
Copyright © 2022 Zhang, Cheng, Yuan, Wang, Cheng and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qixiang Zhang, enF4YmpmdUAxMjYuY29t