
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 21 June 2022
Sec. Plant Breeding
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.930131
This article is part of the Research Topic Marker-Assisted Selection (MAS) in Crop Plants View all 45 articles
 Juanjuan Feng1,2
Juanjuan Feng1,2 Yongqi Li1,2Jinfa Zhang3
Yongqi Li1,2Jinfa Zhang3 Meng Zhang1
Meng Zhang1 Xuexian Zhang1Kashif Shahzad1Liping Guo1Tingxiang Qi1Huini Tang1Hailin Wang1Xiuqin Qiao1
Xuexian Zhang1Kashif Shahzad1Liping Guo1Tingxiang Qi1Huini Tang1Hailin Wang1Xiuqin Qiao1 Zhongxu Lin2*Chaozhu Xing1*Jianyong Wu1*
Zhongxu Lin2*Chaozhu Xing1*Jianyong Wu1*Hybrid utilization has proficiently increased crop production worldwide. The cytoplasmic male sterility (CMS) system has emerged as an efficient tool for commercial hybrid cotton seed production. The restorer line with dominant Rf2 gene can restore the fertility of the CMS-D8 sterile line. However, the molecular mechanism of fertility restoration remains unclear in CMS-D8 cotton that limits wider utilization of three-line hybrid breeding. In our study, the Pacific Biosciences (PacBio) Iso-Seq technology was applied to understand fertility restoration mechanism of CMS-D8 cotton. In total, 228,106 full-length non-chimeric transcriptome sequences were obtained from anthers of developing flowering buds. The analysis results identified 3,174 novel isoforms, 2,597 novel gene loci, 652 long non-coding RNAs predicted from novel isoforms, 7,234 alternative splicing events, 114 fusion transcripts, and 1,667 genes with alternative polyadenylation. Specially, two novel genes associated with restoration function, Ghir_D05.742.1 and m64033_190821_201011/21103726/ccs were identified and showed significant higher levels of expression in restorer line than sterile and maintainer lines. Our comparative full-length transcriptome analysis provides new insights into the molecular function of Rf2 fertility restorer gene. The results of this study offer a platform for fertility restoration candidate gene discovery in CMS-D8 cotton.
Cotton is prime natural fiber crop widely cultivated worldwide in tropics and subtropics regions. Similar to other crops, the climate change and emergence of new diseases significantly reduce cotton yield and quality. Hybrid breeding is an important measure to reduce recent decline of cotton production (Shahzad et al., 2020). In commercial hybrid seed production, cytoplasmic male sterility (CMS) system is an efficient tool to minimize seed production cost. The CMS is maternally inherited phenomenon, prevents pollen fertility by mitochondrial dysfunction, and facilitates utilization of hybrid vigor (Zhang et al., 2020). The CMS phenomenon exists in more than 150 plants. It has already been exploited in hybrid breeding of many crops such as maize (Laughnan and Gabay-Laughnan, 1983; Jaqueth et al., 2019), rice (Akagi et al., 1994; Chang et al., 2016), pepper (Ma et al., 2013), and sorghum (Tang et al., 1996). In cotton, there are various types of CMS lines, including G. harknessii (D2-2) cytoplasmic male sterile (CMS-D2) lines (Meyer, 1975; Weaver and Weaver, 1977), G. trilobum (D8) cytoplasmic male sterile (CMS-D8) lines (Stewart et al., 1992), 6001A line derived from the wide crossing of G. thurberi and G. hirsutum (Gao et al., 2021), and upland cotton cytoplasmic male sterile lines (104-7A, Xiangyuan A, Jin A; Zhang, 2005). Within plant CMS systems, pollen sterility can be rescued though regulatory mechanism of different nuclear fertility restorer (Rfs) genes (Hanson, 1991; Linke and Börner, 2005; Wu et al., 2011). Different sterile lines have corresponding restorer lines in cotton. The restorer of CMS-D2 contains the Rf1 gene (Weaver and Weaver, 1977), the restorer of CMS-D8 contains Rf2 (Stewart et al., 1992), and fertility of 6001A sterile line can be restored by the D05_PPR-clusters existed in restorer line 7R13 (Gao et al., 2021). Both Rf1 and Rf2 were found to be located on the same chromosome D05 within a genetic distance of 0.9 cM (Zhang and Stewart, 2001b). However, the restorer gene Rf1 of CMS-D2 has the ability to restore fertility of both CMS-D2 and CMS-D8 sterile lines, while fertility of CMS-D2 sterile lines could only be restored by Rf1. Although there have been several reports on the transcriptome of the cotton CMS system (Suzuki et al., 2013; Wu et al., 2017; Yang et al., 2018; Li et al., 2021). However, no study has reported the novel isoforms and novel gene loci of CMS-D8 restorer line compared with G. hirsutum TM-1.
High throughput sequencing technology has recently been made substantial progress to detect complete transcripts, novel genes, isoforms, alternative splicing (AS), open reading frames, and long non-coding RNAs (lncRNAs). Specifically, single-molecule real-time (SMRT) sequencing by PacBio emerged as unique platform to construct full-length transcripts (Roberts et al., 2013). With progress in sequencing technology and bioinformatics, SMRT sequencing was mostly employed in various research projects of corn (Guo et al., 2021), cotton (Li et al., 2021), rice (Schaarschmidt et al., 2020), and clover (Chao et al., 2018). The SMRT sequencing proved more appropriate datasets than short read sequenced technology. It can directly provide all statistics of sequenced RNA without assembly, reads gaps, and high errors (Au et al., 2013; Tilgner et al., 2014; Treutlein et al., 2014; Gordon et al., 2015), this methodology efficiently identified different isoforms of each gene, AS, fusion transcripts, and often improve the accuracy of existing gene models. Furthermore, alignment of different isoforms to the reference genome can effectively identify the modes of alternative splicing of genes. In this way, SMRT improve the accuracy of long isoforms alignment (Kraft and Kurth, 2020). Hence, application of SMRT sequencing can serve as a platform to discover fertility restoration mechanism in CMS-D8 cotton.
For a particular interest, this study performed integrated Iso-seq and RNA-Seq analysis by using mixed anthers of CMS-D8 cotton. Our results characterized full-length transcript differences between restorer, sterile, and the maintainer lines. Further data analysis revealed potential isoforms, alternative splicing events, and fusion transcripts in three lines hybrid cotton. In particular, this study identified 39 novel isoforms that specifically expressed in restorer line. The qRT-PCR analysis stated that Ghir_D05.742.1 and unmapped m64033_190821_201011/21103726/ccs had shown significant higher expression in restorer lines as compared with sterile and the maintainer lines. Our results provide valuable insights into molecular function of fertility restoration in three lines hybrid cotton. These results will offer an important platform to identify the Rf2 restorer genes in CMS-D8 cotton.
The plant material used in this study contained sterile, maintainer and restorer lines of CMS-D8 system. The sterile line (A) has D8 cytoplasm and contained homozygous recessive fertility restorer alleles (rf2rf2), whereas the maintainer line (B) is fertile with upland cotton (AD1) and has homozygous recessive fertility restorer alleles (rf2rf2). The restorer line (R) is fertile with D8 cytoplasm, and homozygous dominant fertility restorer alleles (Rf2Rf2) to recover fertile anther in cotton. The anther from flower bud samples with length of 1–2, 3, and 4 mm were harvested from 100 plants of each line, with three biological replicates. All harvested anther samples were utilized to prepare composite anther sample, snap-frozen in liquid nitrogen, and stored at −80°C before further use. Total RNA was isolated using the Sigma Spectrum Plant Total RNA kit (Sigma-Aldrich, United States) according to the manufacturer’s protocol. RNA concentration, purity, and integrity were monitored by NanoDrop, agarose gel electrophoresis and Agilent 2100, respectively. Poly (A) mRNA was isolated by poly-T oligo attached magnetic beads (Invitrogen). Following fragmentation, the cleaved RNA fragments were reverse-transcribed into a cDNA library following treatment with the TruseqTM RNA sample prep Kit (Illumina, San Diego, USA). After assessing the library quality, PE 2 × 150 sequencing was performed on an Illumina-Hiseq at the Majorbio (Shanghai, China) following the vendor’s recommended protocol.
Equimolar rations of the anthers total RNA from R line of 1–2, 3, and 4-mm buds in length were combined together. The full-length cDNA for library was synthesized from 1 μg of purified polyA (+) RNA with SMARTer™ PCR cDNA Synthesis kit (Clontech). Then, Iso-seq libraries were constructed according to the online available protocol at https://www.pacb.com/wp-content/uploads/Procedure-Checklist-Iso-Seq-Template-Preparation-for-Sequel-Systems.pdf. Once library was prepared and quantified, each SMRT cells were sequenced on the PacBio sequel platform with P6-C4 reagent. PacBio raw data was first handled using the workflow of the PacBio SMRT Analysis software suite.1 Briefly, raw polymerase reads were filtered and trimmed to generate the subreads and read of inserts (ROIs) using the RS_Subreads protocol, requiring a minimum polymerase read length of 50 bp, a minimum polymerase read quality of 0.75, a minimum subread length of 50 bp, and a minimum of one full pass. Full-length and non-chimeric (FLNC) reads were regarded as those containing a 5′ adapter, 3′ adapter, and poly (A) tail in the expected arrangement with no additional copies of the adapter sequence within the ROI. Error correction of FLNC reads was performed with the highly quality Illumina short reads using Proovread version 2.12 with the default parameters. The quality of Illumina short reads was examined using FastQC (v0.11.5).2 Sequencing adaptors and low-quality bases in short reads were trimmed before the error correction of FLNC reads. FLNC reads before and after error correction were, respectively, mapped to the IWGSC RefSeq v1.0 using GMAP (version 2016-09-14).3
According to the read-genome alignments, FLNC reads with the same splicing junctions were collapsed into one isoform. The redundant transcripts were removed through two different ways. First, if all the splicing sites of the same loci transcripts were identical, they were considered one isoform. Secondly, if one isoform was degraded at the 5′ terminal region but the remaining region was consistent with other isoforms, it should be cleaned out. This supporting evidence was examined for the identification of isoforms. The resulted isoforms were retained which was supported with at least two FLNC reads or one FLNC read with percentages of identity higher than 99%, or all junction sites were fully supported by Illumina reads or annotations of the IWGSC RefSeq v1.0. Isoforms that overlapped by at least 20% of their length on the same strand were considered to be from the same gene locus. All detected gene loci and isoforms were matched with the reference annotation to categorize known genes and isoforms as well as novel genes and isoforms. A sequenced gene was regarded to be a novel gene by satisfying any of the following criteria. (i) There is no overlap or an overlap of <20% (ii) The annotated gene has overlap > 20% but the gene direction is inconsistent. In addition, if the sequenced isoform confined one or more new splicing sites, if both sequenced isoform and annotated isoform were not single-exon, the isoform was named as a novel isoform.
The novel isoforms annotations were retrieved from NR, KOG (Karyotic Ortholog Groups), KO, and Swiss-Prot databases (A manually annotated and reviewed protein sequence database) with Diamond software (Gasteiger et al., 2001; Buchfink et al., 2015). KEGG (Kyoto Encyclopedia of Genes and Genome) pathways were searched using KOBAS v2.0 (Xie et al., 2011). GO (Gene Ontology) annotations were obtained by running BLASTX v2.2.26 and BLAST2GO v2.3.5 software (Conesa et al., 2005).
Newly identified isoforms with length ≥ 200 nt were first searched against NCBI’s NR database using BLASTX with default parameters. The isoforms that had BLAST hits with 1E-5 were filtered out, and the remaining isoforms were further evaluated by CPAT v1.2.2.4 The transDecoder software was applied to detect potential coding sequences and to predict ORFs.5 The length of ORFs predicted by TransDecoder.LongOrfs was at least 100 amino acids by default. To increase the sensitivity of ORFs, potential ORF translated proteins were aligned to the Swiss-Prot database with BlastP for homologous protein identification. Protein domain identification was acquired from the Pfam database using Hmmscan (Eddy, 2009; Finn et al., 2014). Subsequently, TransDecoder. Predict was used to filter all predicted ORFs based on the above results, and retained ORFs that had homology to the Swiss-Prot database or with the same domain.
AS events were classified and characterized by comparing different isoforms of the same gene loci using Asprofile (Florea et al., 2013). The alternative polyadenylation (APA) sites for each gene locus were detected using TAPIS (Abdel-Ghany et al., 2016). The number of APA for each gene locus as well as the number of transcripts supporting an APA was provided as data files.
Fusion transcripts were identified by parsing mapped data using the iso-seq fusion transcripts detection software self-developed by Frasergen Inc. (Wuhan, China). A FLNC was considered as a candidate fusion transcript when all of the following criteria were satisfied:
1. A FLNC must be map to 2 or more annotated genes that are at least 10 Kb apart.
2. Alignment to each gene must have >10% FLNC coverage.
3. The total combined FLNC coverage from all alignments must be >99%.
4. Supported by a certain amount of PE reads across the fusion junction.
Illumina RNA-seq data of equally mixed anthers of 1–2-, 3-, and 4-mm flower buds, respectively, A, B, and R lines. The programs TopHat and Cufflinks were used to blast the sequenced reads against the reference genome of G. hirsutum. The analyses of differential genes and transcript expression can evaluate the abundance of gene expression and also reveals new genes that have not been previously annotated in reference genomes. The Fragments Per Kilobase of exon per Million fragments mapped (FPKM) method was used to calculate the abundance of gene expression. DESeq was used to analyze biological duplicate samples obtained from DEG screening, and EBSeq (Leng et al., 2013) was used for non-biological duplicate samples. During the DEG screening, a false discovery rate (FDR) < 0.05 and fold change > 1 were considered standard values. If the DEG fold change was >1, then a FDR < 0.05 was taken to indicate that the DEG was significantly different between the control and test groups.
The transcript levels of DEGs were verified by qRT-PCR. Reverse transcription was accompanied using the PrimeScript™ RT Reagent Kit (TaKaRa, Beijing, China). Trans Start® Mix (Trans gen, Beijing, China) was used according to the manufacturer’s instructions to perform qRT-PCR of the genes. The cotton His3 gene (i.e., histone 3) was used as internal control. The relative gene expression levels were calculated with 2−△△CT method (Schmittgen and Livak, 2008).
To reduce limited capacity of short-read RNA-Seq in CMS, anther-specific full-length transcriptome analysis of R line was performed in this study. High-quality total mRNAs were pooled from 1–2-, 3-, and 4-mm length flower buds to achieve full-length complete transcripts. The SMRT bell library was constructed and sequenced using the PacBio Sequel platform. After filtering, 337,937 polymerase reads were generated in our sequence. These reads represented more than 25.59 G bases with a mean length of 75,716 bp and N50 length of 162,286 bp (Supplementary Table S1; Figure 1A). After eliminating the adapter from polymerase reads approximately 14,877,534 filtered subreads were obtained, with a mean length of 1,649 bp (Supplementary Table S2). A total of 295,042 circular consensus sequences (CCSs) with an average library depth of 46 passes were produced after subreads integration and error correction by multiple sequencing (Supplementary Table S3). The length distribution of CCSs was consistent with the estimated size of the library (Figure 1B). Then, CCSs were calculated as full-length non-chimeric (FLNC) reads. A total of 228,106 reads were considered to be FLNC with lower artificial concatemers accounted for 77.31% of CCSs. The mean length of FLNC reads was accounting 1728 bp (Figure 1C; Supplementary Table S4). Overall, comprehensively full-length transcripts were achieved to accurately construct splice variants. Illumina HiSeq transcripts were employed to further correct the FLNC reads sequenced by the PacBio Sequel platform. Using LoRDEC software (Salmela and Rivals, 2014), the FLNC reads before and after error correction were compared to the reference genome to measure global and local percentage-of-identity (PID; Figure 2). The PID value was up to 98.49% after error correction (Supplementary Table S5). After adjustment, a total of 221,170 high-qualities FLNC reads were obtained for subsequent investigation (Table 1).
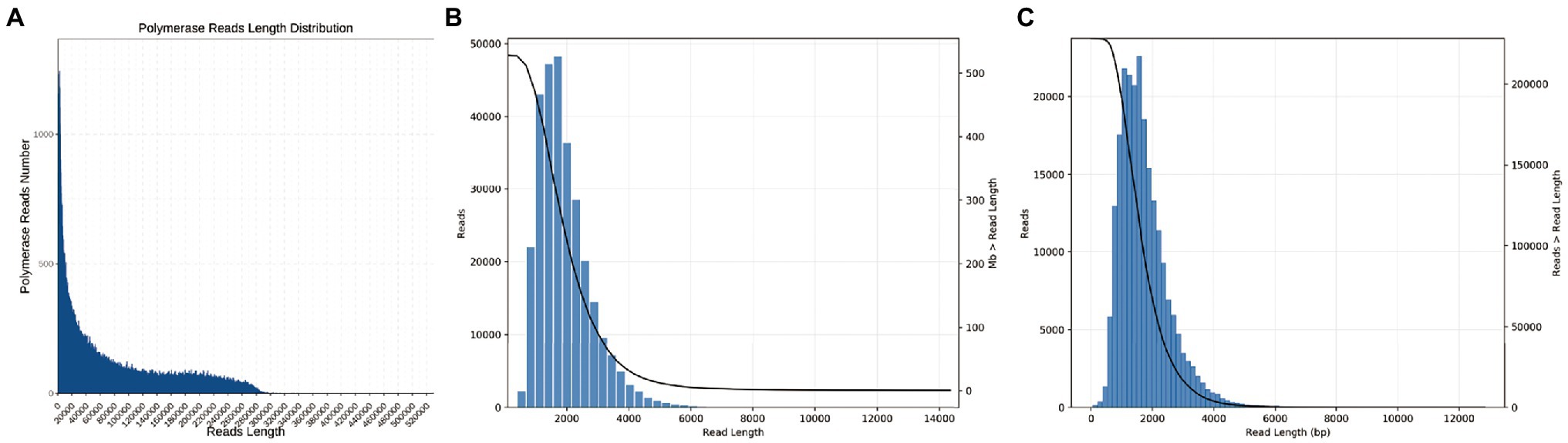
Figure 1. Overall length distribution of the PacBio Sequel data. (A) Number and length distribution of polymerase reads. (B) Number and length distribution of CCSs. (C) Number and length distribution of FLNC reads.
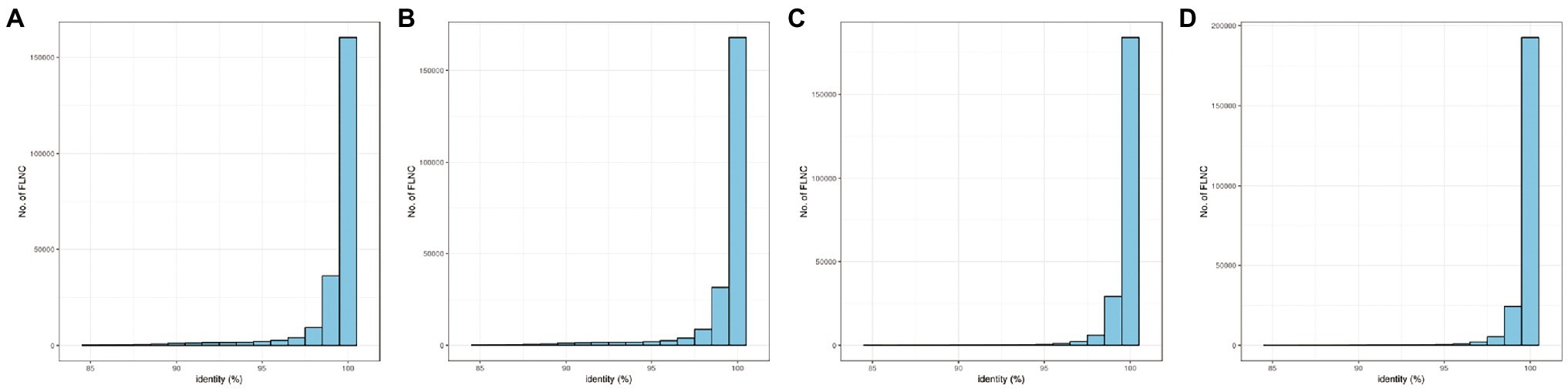
Figure 2. The distribution of PID (percentage-of-identity) before and after error correction. (A) Global PID distribution before error correction. (B) Local PID distribution before error correction. (C) Global PID distribution after error correction. (D) Local PID distribution after error correction.
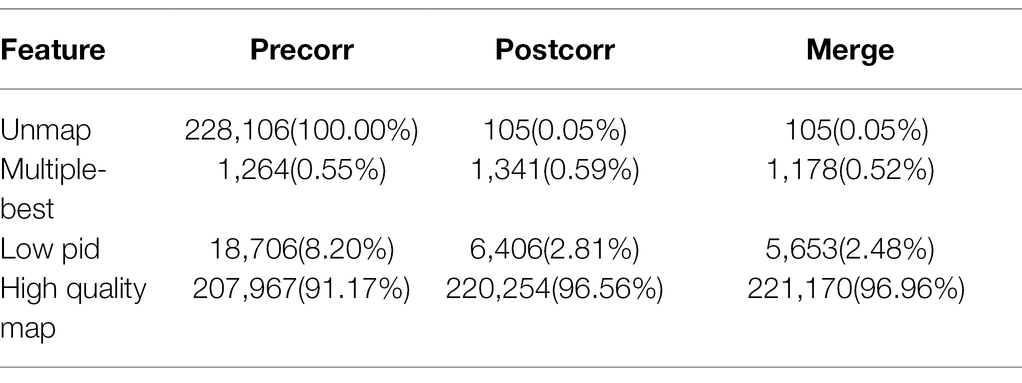
Table 1. Classification of reference genome comparison results.
Error correction accurately mapped FLNC reads to the reference genome including start site, termination site, and splicing site. This was useful to identify gene loci and isoforms. To calculate length density of isoform, we compared the loci coverage of the PacBio dataset with the G. hirsutum_genome_HAU_v1.6 In our data, a total of 221,170 error-corrected FLNC reads covered 38,801 isoforms that were allocated to 27,829 loci. About 3,391 loci were 1–2 kb in length, followed by 2–3 kb (7,413), > 3 kb (3,805), and <1 kb (3,391). In the reference genome, about 115,835 isoforms covered 70,199 loci, most loci distributed at <1 kb (18,751), followed by 1–2 kb (26,033), 2–3 kb (15,038), and >3 kb (10,377; Table 2; Figure 3A). Similarly, loci density of isoforms stated that each locus could produce a unique isoform in the reference genome. However, in our data, ~20,321 (73.02%) loci could produce a unique isoform and more than five isoforms covered about 0.35% of the PacBio annotation loci (Figure 3B). Thus, the PacBio dataset provided higher isoform length diversity and loci density than the reference genome which could help to reveal in-depth fertility restorer function of the R line. Almost 80.57% multi-exon isoform and 86.60% multiple-exon FLNC reads contained the same splice donor site at the 5′ end as the reference annotation. These were regarded as full-length transcript implying a relatively higher integrity in structure variant (Table 3).
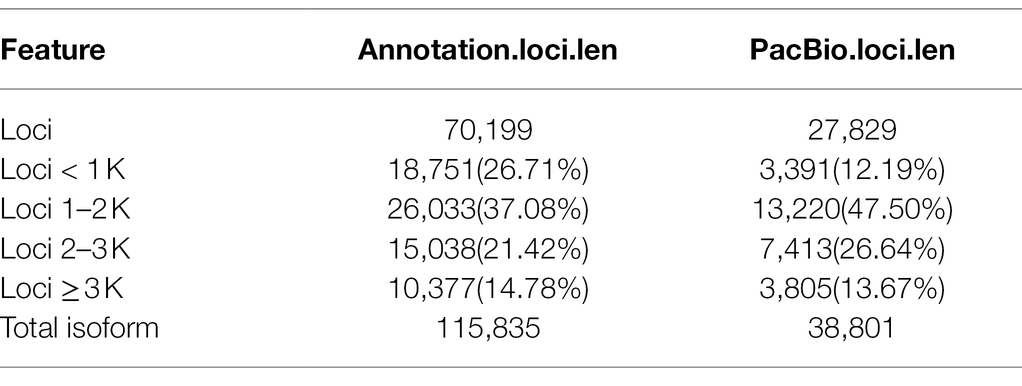
Table 2. Gene structure annotation.
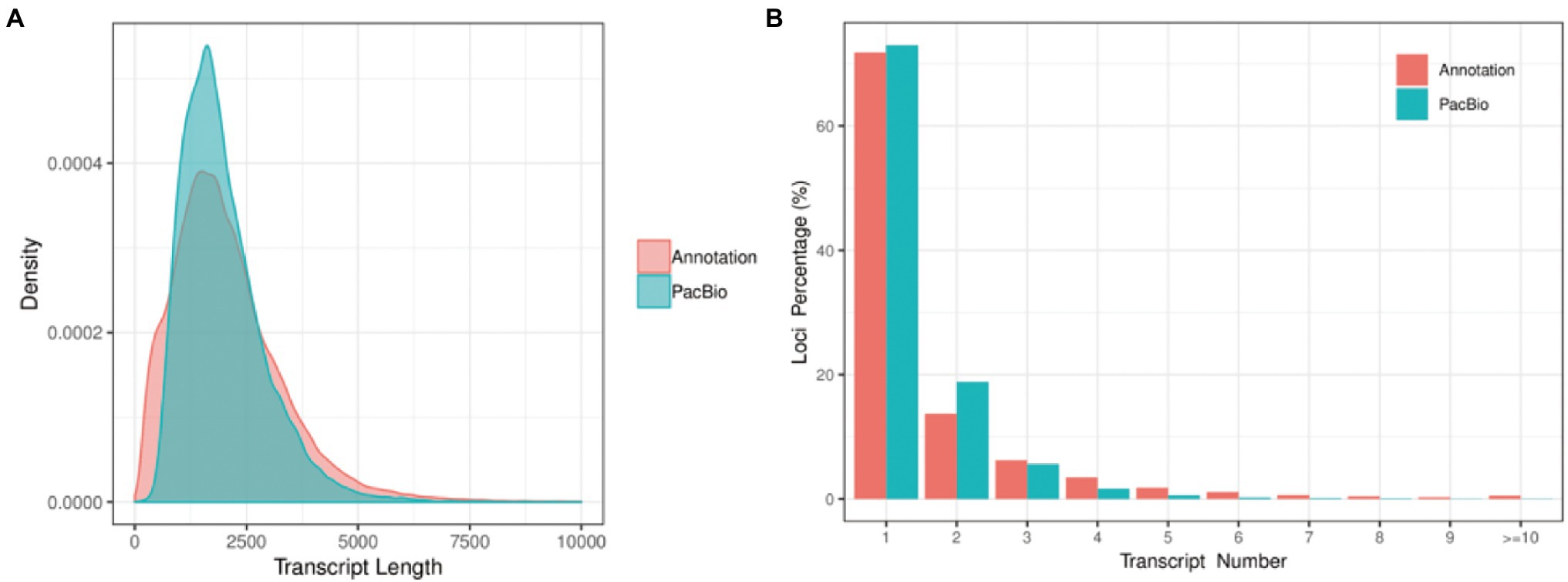
Figure 3. Isoform length density and loci density. (A) The length distribution of all isoforms in the PacBio Sequel platform compared to the reference genome. (B) The number distribution of isoforms from each locus in the PacBio Sequel platform compared to the reference genome.
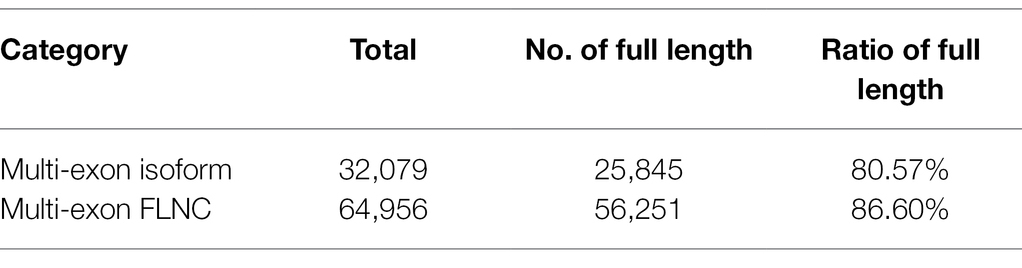
Table 3. Evaluation of full-length transcripts in the PacBio data set.
Moreover, the sequenced gene loci and isoforms were compared with the reference annotation to determine novel gene loci or novel isoforms. The published G. hirsutum TM-1 genome annotation contains 70,199 loci with 115,835 isoforms. In our PacBio dataset, a total of 38,801 isoforms were identified from 27,829 genes. Of which, 18,010 were known isoforms from known genes. In addition, 2,597 transcripts did not overlap with any annotated genes and were considered as novel genes (Supplementary Table S6). Those novel genes were found to produce 3,174 novel isoforms (Figure 4E). In contrast, 17,617 additional novel isoforms were determined from 12,815 known genes. Out of 3,174 novel isoforms, 1,313 (41.37%) were single-exon isoforms, and 1861 (58.63%) were multiple-exon isoforms. Furthermore, among 16,857 known loci novel isoform, there were 1789 (10.61%) single exon isoform and 15,068 (89.39%) multiple exon isoforms. The greater number of identified novel genes and isoforms were useful for reliable genes annotation within the candidate interval of Rf2.
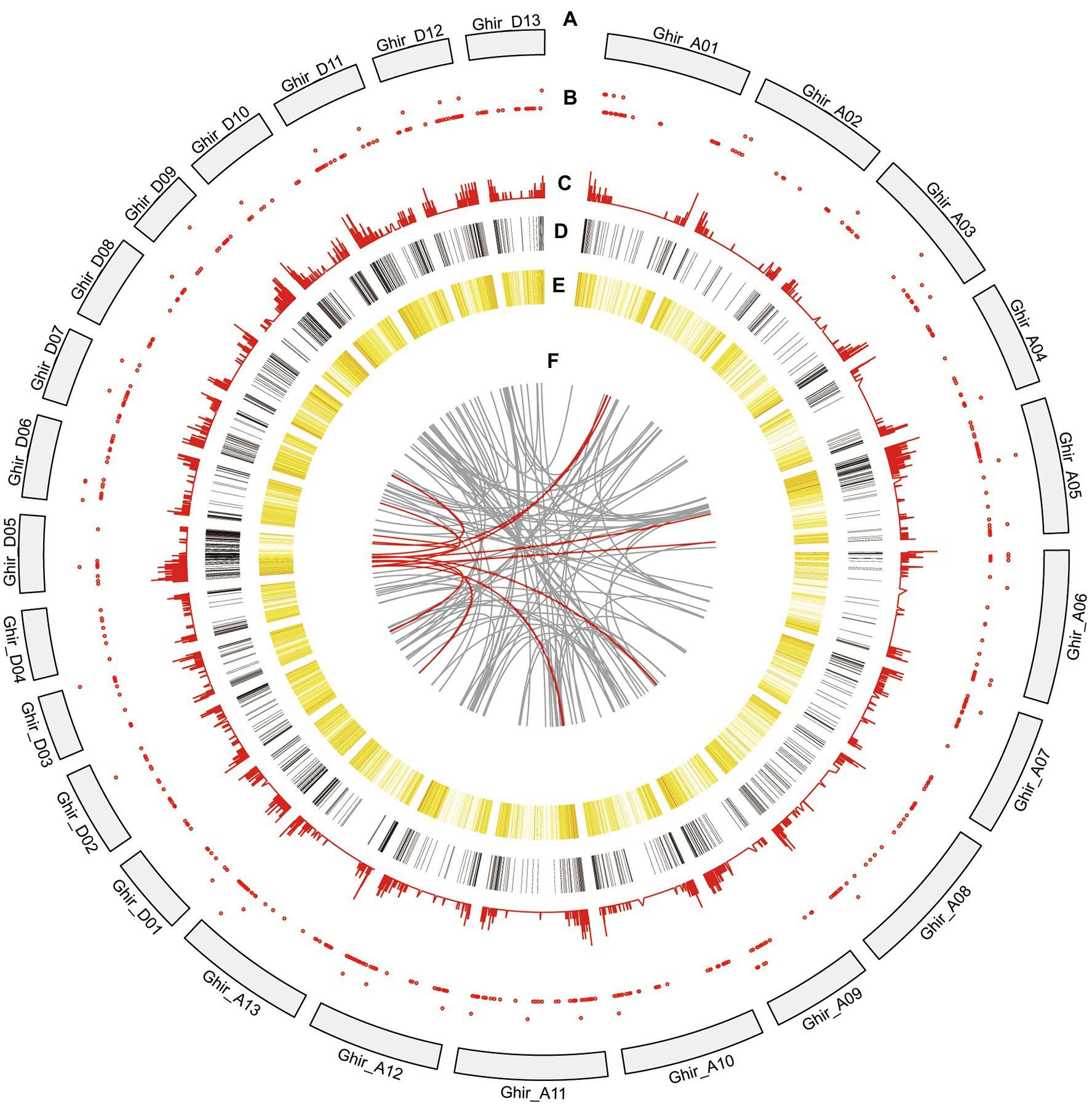
Figure 4. Circos visualization of the PacBio Sequel platform at genome-wide level. (A) Twenty-six chromosomes distribution of G. hirsutum genome. (B) LncRNA density from the PacBio Sequel platform. The closer the red point is to the center, the lower the density. (C) APA sites distribution mapped to G. hirsutum genome. The closer the line is to the center, the lower the density. (D) DEGs of R line compared with A line and B line. The closer the color is to red, the higher the density. Conversely, the closer the color is to black, the lower the density. (E) Novel isoforms density from the PacBio Sequel platform. The closer the color is to red, the higher the density. Conversely, the closer the color is to yellow, the lower the density. (F) Fusion transcripts distribution. Red line represents fusion transcripts which are involved in the D05 chromosome.
In this study, all 3,174 novel isoforms functional annotation were retrieved from different databases. It was observed that 82.04% isoforms were annotated on NCBI non-redundant protein sequences (NR), 26.84 on Gene Ontology (GO), 24.26% on EuKaryotic Orthologous Groups (KO), 16.23% on eukaryotic Ortholog Group (KOG), and 35.07% on Swiss-Prot Protein Sequence (Swiss-Prot) database However, a total of 377 (17.93%) novel isoforms were unannotated (Supplementary Table S7). A total of 175 novel isoforms had significant hits in all five databases (Figure 5A). In the NR database, the largest three groups of novel isoforms were distributed in G. hirsutum (969), G. raimondii (880), and G. arboreum (599; Figure 5B). GO analysis assigned the enrichment of 852 isoforms to various biological process, cellular component, and molecular function. It was found that 863 GO terms were enriched in biological process. Out of which, metabolic (53.40%), cellular (47.18%), and single-organism (23.59%) process had major proportion. In addition, many of the terms in biological process were related with energy metabolism such as ATP metabolic process, ATP synthesis coupled electron transport, oxidation–reduction process, and energy derivation by oxidation of organic compounds. A total of 257 GO terms were detected in category of cellular component. The cell (36.38%), cell part (36.38%), and membrane (34.39%) were the largest three enrichment terms. For a particular interest, six novel isoforms were enriched functions into mitochondrial proton-transporting ATP synthase complex, and catalytic core F(1). Our data showed that 442 GO terms were assigned to molecular function and the most highly abundant terms were catalytic activity (48.94%) and binding (48.63%; Figure 5C). To identify the enrichment pathways, a total of 695 novel isoforms were subjected to 98 KEGG pathways. Novel isoforms in KEGG pathways were consisted of five hierarchies, e.g., cellular processes, environmental information processing, genetic information processing, metabolism, and organismal systems. Among these terms, the most abundant hierarchy was metabolism (366, 52.66%) followed by genetic information processing (236, 33.96%; Figure 5D). KOG analysis had shown that 515 novel isoforms were assigned to 23 categories and the largest three classes were general functional prediction only (87, 16.90%), posttranslational modification, protein turnover, chaperones (68, 13.20%), and Translation, ribosomal structure and biogenesis (63, 12.23%; Figure 5E).
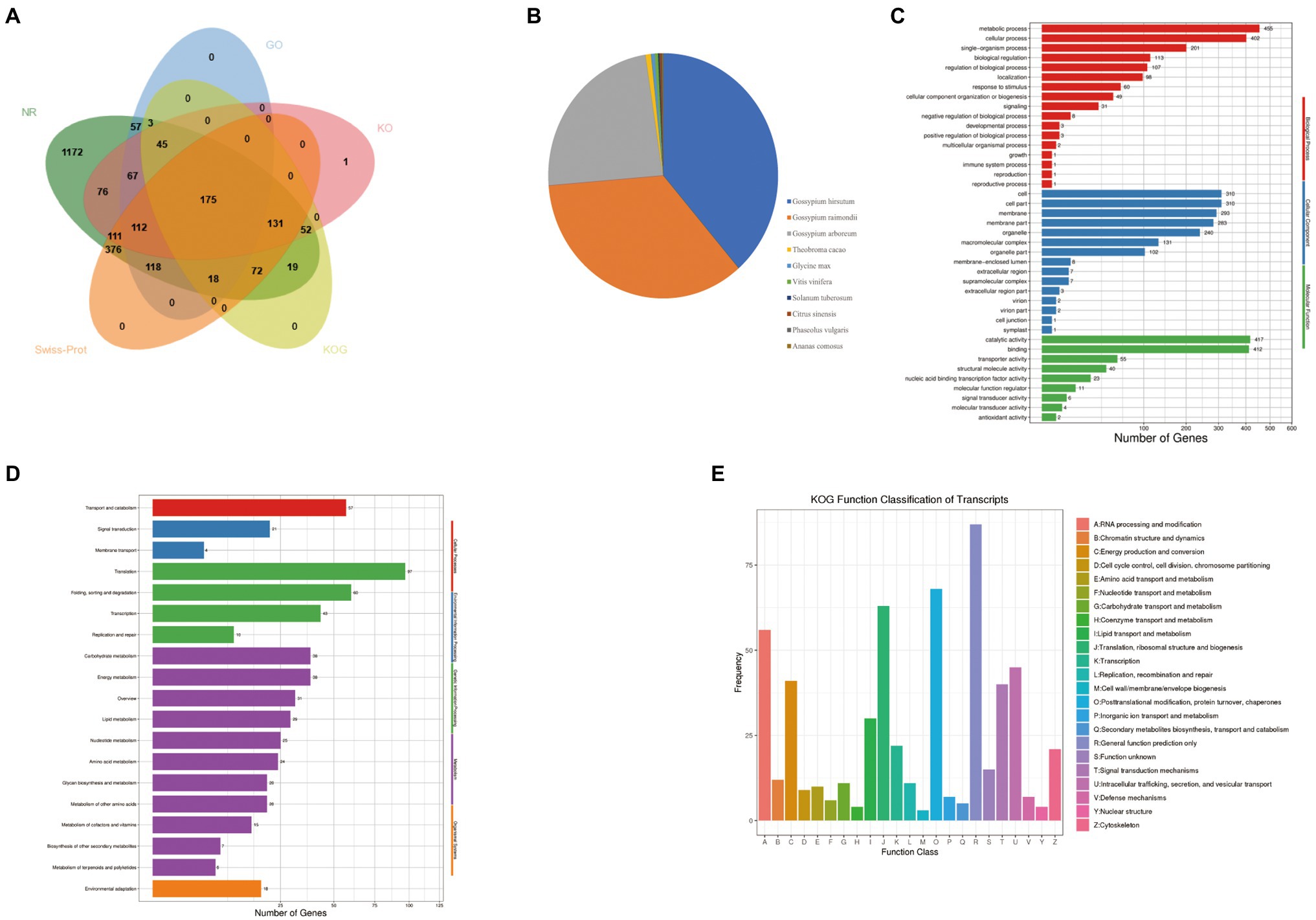
Figure 5. Function annotations of novel isoforms identified by the PacBio Sequel platform. (A) The number statistics of novel isoforms in NR GO, KEGG, KOG databases. (B) Distribution of novel isoforms in Nr homologous top 10 species. (C) Distribution of novel isoforms in GO terms. (D) Distribution of novel isoforms in KEGG pathway. (E) Distribution of novel isoforms in KOG.
LncRNAs perform regulatory functions and play critical role for post-transcription, transcription, and epigenetic changes (Huang et al., 2018). A total of 652 novel isoforms were predicted to be lncRNAs with a mean length of 1,029 bp which counted for 3.25% of all novel isoforms (Supplementary Table S8). About 215 lncRNAs (32.98%) were longer than 1,000 bp and 9 lncRNAs were longer than 3,000 bp. Mapping of the predicted lncRNAs to G. hirsutum 26 chromosomes was presented using Circos visualization software (Figure 4A). It revealed that 652 lncRNAs were randomly distributed (Figure 4B). Moreover, open reading frames (ORFs) were predicted with transDecoder software. It resulted that a total of 16,390 novel isoforms were predicted with ORF. Then, the density and length distributions of coding sequences (CDS) were investigated. The encoded peptide sequences are listed in Supplementary Table S9.
AS increased the complexity of transcriptomes and proteomes in accordance to the diverse splice modes rather than by amplifying the number of genes in cells or tissues (Wang et al., 2008). A total of 27,829 loci corresponding to 38,801 isoforms underwent 7,234 AS events via different spliced modes. It included 616 ES events, 1,670 AA events, 737 AD events, 2,715 IR events, and 1,496 other AS events. These detections stated that the distribution of AS events was much higher in anthers of R line (Figure 6A). Moreover, two or more isoforms were found in 7,234 gene loci in our PacBio Sequel platform analysis. Ten or more splice isoforms were detected in 20 genes (Figure 6B).
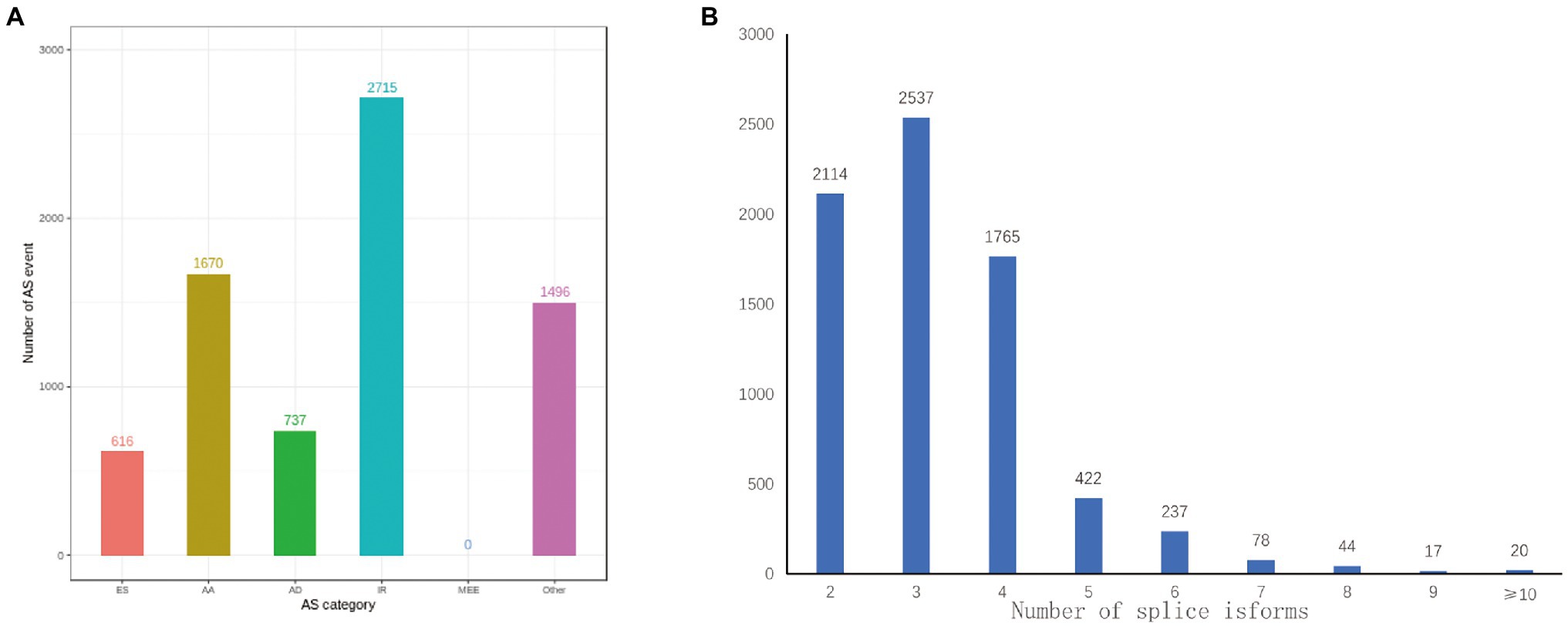
Figure 6. Identification of AS events. (A) The distribution of AS events in loci detected by the PacBio Sequel platform. (B) Distribution of loci that produce two or more splice isoforms detected by the PacBio Sequel platform.
The post-transcriptional modification process of pre mRNA to mature mRNA mainly includes the addition of a 7-methylguanosine cap at the 5′-end, intron splicing, and 3′-end formation by cleavage and polyadenylation (de Almeida et al., 2010). The specific position of the poly-A tail at the 3′-end is variable and this variation may affect the binding of microRNA or RNA-binding protein to mRNA as well as the process of RNA splicing and translation. By investigating the 3′-end of transcripts a total of 10,580 poly-A sites were detected from 8,402 genes. Of which, 1,667 genes showed alternative polyadenylation (APA; Figure 4C). A total of 6,735 genes had at least one poly-A site while 23 genes had more than four poly-A sites (Figure 7A). The largest number of poly-A sites was 10 that were found in Ghir_D06G000070 and Ghir_D11G012090. Then, nucleotide distribution of the 30 nts in upstream and downstream of all poly-A sites were analyzed. Consistent with other species, the poly-A sites from our PacBio dataset showed a nucleotide bias with an enrichment of uracil (U) upstream and adenine (A) downstream (Figure 7B).
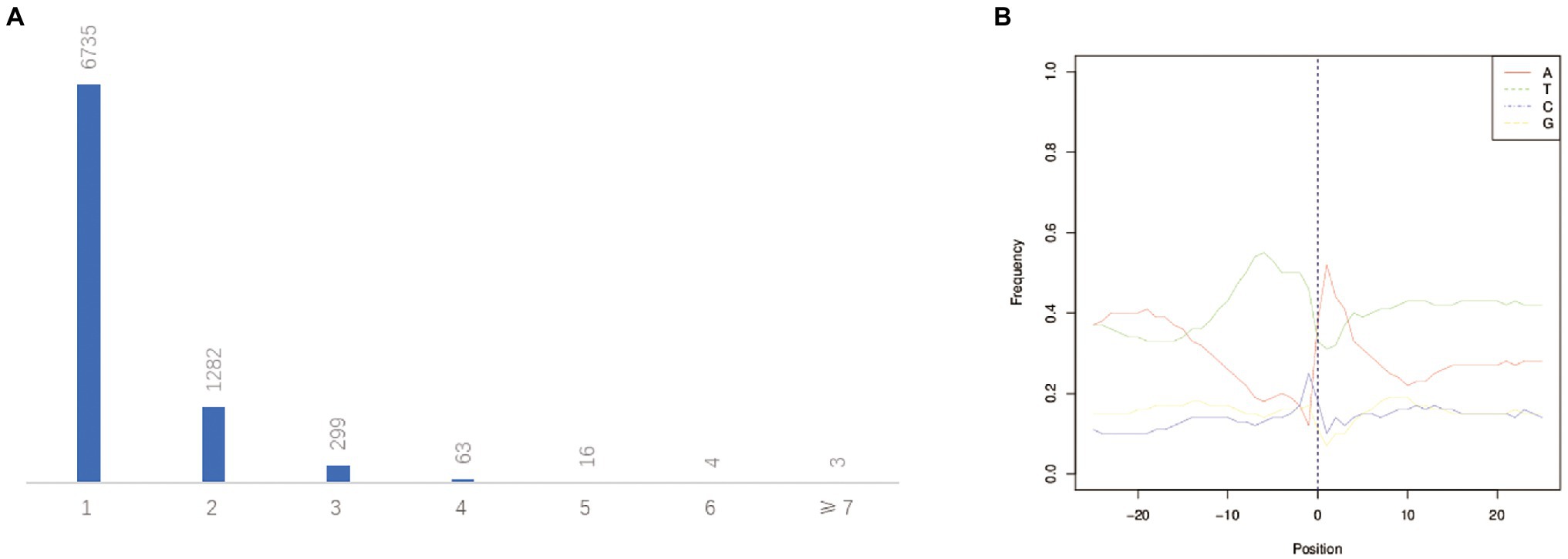
Figure 7. APA analysis predicted by the PacBio Sequel platform. (A) The distribution of poly-A sites per gene. (B) Nucleotide distribution around poly-A cleavage sites.
A fusion transcript refers to a new gene formed by splicing together two or more separate genes which are known as chimeric transcripts. The genomic structural variation, transposition, or trans-splicing after transcription caused generation of fusion transcripts. A total of 114 fusion transcripts related to 225 annotated genes were identified in this study (Supplementary Table S10). According to the chromosomal distribution, 109 inter-chromosome and 5 intra-chromosome fusion transcripts were detected. Previous studies have indicated that most fusion transcripts are composed of two genes (Weirather et al., 2015). Likewise, all the 114 fusion transcripts were composed of two genes in our data. Intriguingly, 11 fusion transcripts were convoluted on the D05 chromosome (Figure 4F). Specifically, m64033_190821_201011/87884027/ccs is composed of Ghir_A01G016800.1 (Niemann-Pick C1 protein) and Ghir_D05G004620.1 (Pollen-specific protein SF21) genes and experimentally validated in anthers of CMS-D8 system using RT-PCR. Thus, the m64033_190821_201011/87884027/ccs is certainly in the anthers transcription (Supplementary Figure S1).
Since, Rf2 is derived from of G. trilobum (Stewart et al., 1992; Zhang and Stewart, 2001a), the sequence of Rf2 might be not available in the reference genome of G. hirsutum. The unmapped reads were further analyzed and 76 isoforms were identified after de-redundancy with ci-hit software, resulting in identification of 10 isoforms which were expressed in the R line with FPKM > 1. As compared with the CMS-D8 A line, the genes that met the default criteria with FDR < 0.05, log FC > 1 or log FC < −1 by DEseq2 software were assigned as DEGs. The 6,084 differentially expressed isoforms from 4,112 DEGs were discovered. Among the differentially expressed isoforms, 3,187 isoforms were up-regulated and 2,897 isoforms were down-regulated (Supplementary Table S11). In DEGs, 2,211 were up-regulated 1,901 were down-regulated. The data also showed that 520 of the novel isoforms were up-regulated and 232 were down-regulated. Comparative analysis showed that 1,456 DEGs (Figure 4D), 161 novel isoforms, and 56 novel genes were upregulated in R line then A line and B line (Figure 8A). Subsequently, novel isoforms specifically expressed in R line with log FC > 3 or FPKM > 1 value and FPKM < 1 in A line and B line were identified. As a result, 39 novel isoforms specifically expressed in R line were screened out. These R line up-regulated genes especially R line-specific genes may co-respond to the expression of restorer gene Rf2 and most likely regulate fertility restoration function in CMS-D8 cotton.
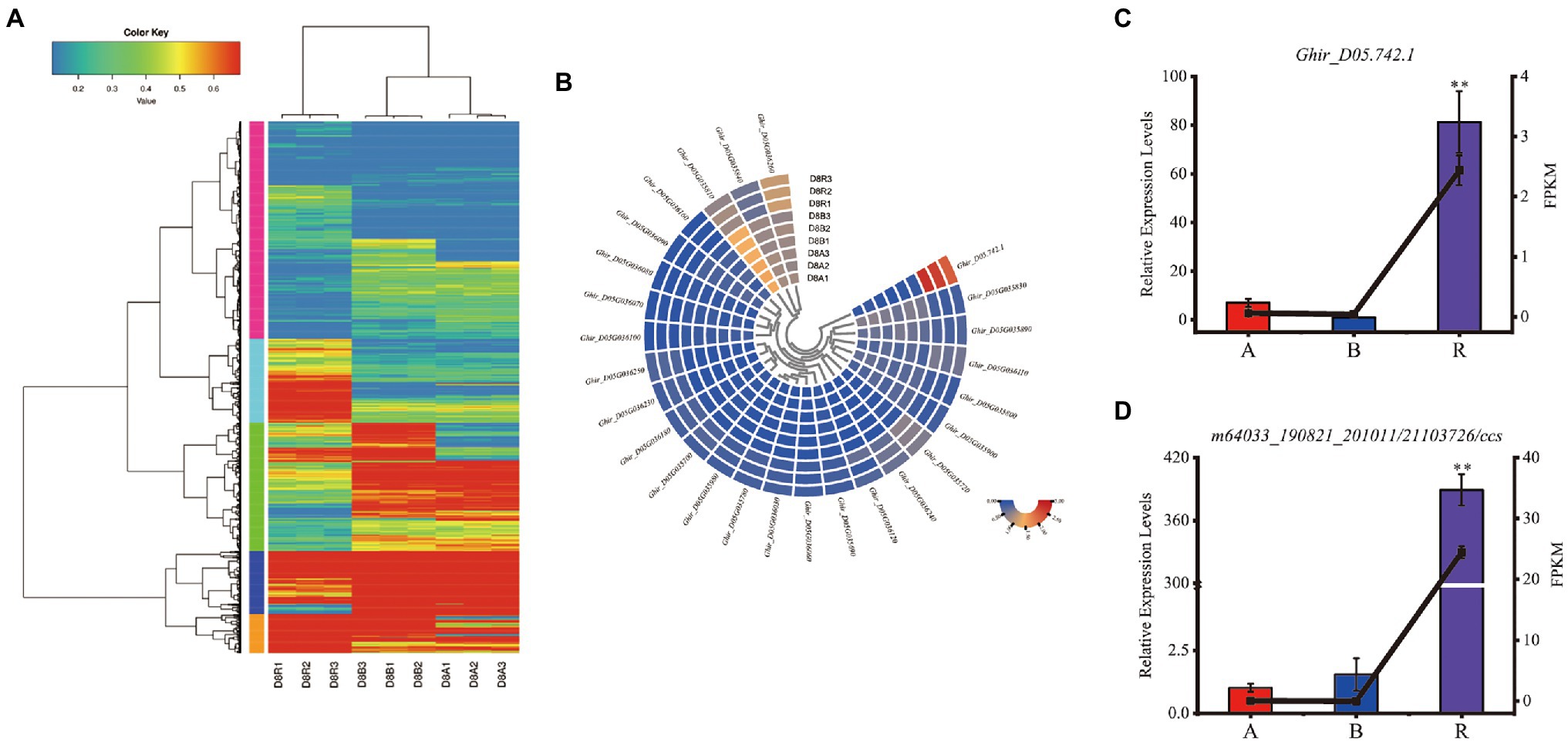
Figure 8. The expression of DEGs. (A) Heat map of DEGs in R line compared with A line and B line. (B) Heat map of FPKM values of differentially expressed genes in candidate interval. A1, A2, A3: sterile line, B1, B2, B3: maintainer line, and R1, R2, R3: restorer line. (C) Expression patterns of Ghir_D05.742.1. (D) Expression patterns of m64033_190821_201011/21103726/ccs. Histograms indicate relative expression levels. Lines indicate FPKM. (**p < 0.01) The asterisks indicate that the difference in gene expression in the A, B, and R lines was highly significant.
According to the previous study, Rf2 is located within the candidate interval of 1.48 Mb on the D05 chromosome (Feng et al., 2021). Hence, the genes in the candidate interval were analyzed in this study. It was determined that 25 of 76 genes were differentially expressed in CMS-D8 system and four novel isoforms were identified including Ghir_D05.742.1 which was specifically expressed (Figure 8B). Considering that the Rf2 gene is a foreign gene introduced into upland cotton, the analysis of unmapped reads shows that one isoform named m64033_190821_201011/21103726/ccs was specifically expressed in R line. The Ghir_D05.742.1 was annotated as disease resistance protein At4g27190-like in NR database of NCBI, but the m64033_190821_201011/21103726/ccs could not be annotated. The qRT-PCR showed that the relative expression levels of Ghir_D05.742.1 and m64033_190821_201011/21103726/ccs in the restorer line was significantly higher than A and B lines (Figures 8C,D). These results suggested that Ghir_D05.742.1 and m64033_190821_201011/21103726/ccs are closely related to Rf2 gene or these two genes are candidate genes of Rf2.
Hybrid breeding has potential to reduce recent the decline in upland cotton production in the world. CMS is a common phenomenon used to produce hybrid seeds in flowering plants. The CMS system restore fertile pollen as a result of the interaction between the mitochondrial and nuclear genomes (Touzet and Meyer, 2014). CMS is required for efficient commercial hybrid breeding of various agronomic crops. Among various techniques of hybrid seed production, the CMS system has been proven to be an ideal and economically effective tool than convention hybrid seed production. The CMS-D8 system in upland cotton with cytoplasm introduced from G. trilobum is one of the main CMS systems in cotton with stable inheritance and complete male abortion. The restorer line of CMS-D8 system contains the restorer Rf2 gene introgressed from G. trilobum. Although, the G. hirsutum genome sequence has been released by cotton researcher community, there are few reports on the full-length transcriptome of cotton, its genome and transcriptome information still need to be further explored. At present, the research of transcript paired-end sequence has extensively been performed in CMS cotton. Wu et al. (2017) discovered 1,464 DEGs by genome-wide comparative transcriptome analysis of CMS-D2 and its maintainer and restorer lines; Yang et al. (2018) discussed the relationship of CMS and its ability to eliminate ROS using the transcriptome of CMS and maintainer line of CMS-D8 cotton. These studies only provided expression changes of transcripts but not reported the novel isoforms and novel genes in cotton CMS systems. Li et al. (2021) recently published the full-length transcripts for restorer line of CMS-D2 system which provided all full-length transcripts with many new isoforms and gens for R line. Since, CMS-D2 aborts in sporophyte and CMS-D8 is gametophyte abortion, the Rf1 of CMS-D2 system was from G. harknessii, and Rf2 of CMS-D8 system source from G. trilobum, and restoration function mechanism may be different. There have already been no reports about the full-length transcriptome of the CMS-D8 restorer line. So, we highly needed the full-length transcriptome to accelerate the research of restoration function of the restorer line in CMS-D8 system. In our study, Iso-seq analysis was performed on the mixed anther, full-length transcripts were identified from 70,199 loci, novel isoform, and fusion transcript, and candidate genes were recognized to facilitate the research on fertility restoration function in R line.
With advance in sequence technology as well as breakthrough in bioinformatics has revealed genetic control of complex trait. Second generation sequencing made vast improvement compared to Sanger sequencing. However, short reads certainly caused assembly gaps and inaccurate detection of isoforms, AS events, and APA sites. In recent years, Pacbio full-length transcriptome sequencing offers an alternative approach to accurately identify AS, APA, and fusion transcripts for target traits (Huang et al., 2022; Tong et al., 2022). Our results identified 7,234 AS events, 1,667 APA, and 114 fusion transcripts that exposed the complexity of transcripts in R line. The functional verification of fusion transcripts has been carried out in some studies in human diseases (Agostini et al., 2018; Siegfried et al., 2019), but rarely reported in plants. This study verified the existence of the fusion transcript, m64033_190821_201011/87884027/cc but unable to study function of this fusion transcript. The m64033_190821_201011/87884027/ccs contains partial sequences of Ghir_A01G016800.1 (Niemann-Pick C1 protein) and Ghir_D05G004620.1 (Pollen-specific protein SF21). It may have the function similar to Ghir_D05G004620.1 which probably regulates anthers development.
Previous studies reported that the restoration of function conditioned by one dominant restorer Rf2 gene in R line of CMS-D8 cotton (Zhang and Stewart, 2001a). At the present, there is no report about the gene expression profiles affected by the Rf2 in R line. Our study analyzed 39 novel genes specifically expressed in R line. The result of qRT-PCR showed that the relative expression levels of Ghir_D05.742.1 and m64033_190821_201011/21103726/ccs in the restorer line were significantly higher than A and B lines. This result revealed specific gene expression of R lines compared with upland cotton (TM-1). Our results provide an important foundation for further studies which aims to explore molecular mechanism of the interactions between the Rf2 and the CMS-D8 cytoplasm.
The Iso-seq developed by Pacific Bio-sciences generates full-length transcripts without assembly. The analyzed full-length transcriptome of R line of CMS-D8 cotton yielded a total of 295,042 CCSs. Of these, 228,106 transcripts were identified as FLNCs. Meanwhile, 3,174 novel isoforms from 2,597 novel gene loci, 652 lncRNAs predicted from novel isoforms, 7,234 AS, 114 fusion transcripts, and 1,667 APA were identified. Further DEGs comparative analysis stated that 161 novel isoforms and 56 novel genes were up-regulated in R line. In particular, 39 novel isoforms specifically showed expression changes in R line. Finally, our study found two key genes named as Ghir_D05.742.1 and m64033_190821_201011/21103726/ccs closely related to restoration function in restorer line of CMS-D8 system. These results provided new insights into novel isoforms, AS events, and candidate gene discovery of R line. Additionally, our datasets will offer a platform to explore in-depth fertility restoration mechanism of R line in CMS-D8 cotton.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: NCBI—PRJNA685585, SAMN28725462, SAMN17088084, and SAMN17088083.
JW, CX, and ZL designed the experiments. LG, TQ, HT, XQ, and HW did the field management. JF, YL, MZ, and XZ performed data analysis and qRT-PCR. JF, JW, JZ, and KS contributed to the preparation of the manuscript. All authors contributed to the article and approved the submitted version.
This work was sponsored by funds from the Zhongyuan Academician Foundation (212101510001), the Agricultural Science, Technology Innovation Program of Chinese Academy of Agricultural Sciences and the Fundamental Research Funds for State Key Laboratory of Cotton Biology (CB2022C05).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors appreciate the field staff for their work from New Mexico Agriculture experiment Station, United States.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.930131/full#supplementary-material
Supplementary Figure S1 | The analysis of m64033_190821_201011/87884027/ccs.
1. ^http://www.pacb.com/products-and-services/analytical-software/smrt-analysis/
2. ^http://www.bioinformatics.babraham.ac.uk/projects/fastqc
3. ^https://github.com/juliangehring/GMAP-GSNAP
4. ^http://lilab.research.bcm.edu/cpat/
5. ^http://transdecoder.sf.net
6. ^https://www.cottongen.org/species/Gossypium_hirsutum/HAU-AD1_genome_v1.0_v1.1
Abdel-Ghany, S. E., Hamilton, M., Jacobi, J. L., Ngam, P., Devitt, N., Schilkey, F., et al. (2016). A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 7:11706. doi: 10.1038/ncomms11706
Agostini, A., Brunetti, M., Davidson, B., Göran Tropé, C., Heim, S., Panagopoulos, I., et al. (2018). Identification of novel cyclin gene fusion transcripts in endometrioid ovarian carcinomas. Int. J. Cancer 143, 1379–1387. doi: 10.1002/ijc.31418
Akagi, H., Sakamoto, M., Shinjyo, C., Shimada, H., and Fujimura, T. (1994). A unique sequence located downstream from the rice mitochondrialatp6 may cause male sterility. Curr. Genet. 25, 52–58. doi: 10.1007/BF00712968
Au, K. F., Sebastiano, V., Afshar, P. T., Durruthy, J. D., Lee, L., Williams, B. A., et al. (2013). Characterization of the human ESC transcriptome by hybrid sequencing. Proc. Natl. Acad. Sci. U. S. A. 110, E4821–E4830. doi: 10.1073/pnas.1320101110
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Chang, Z., Chen, Z., Wang, N., Xie, G., Lu, J., Yan, W., et al. (2016). Construction of a male sterility system for hybrid rice breeding and seed production using a nuclear male sterility gene. Proc. Natl. Acad. Sci. U. S. A. 113, 14145–14150. doi: 10.1073/pnas.1613792113
Chao, Y., Yuan, J., Li, S., Jia, S., Han, L., and Xu, L. (2018). Analysis of transcripts and splice isoforms in red clover (Trifolium pratense L.) by single-molecule long-read sequencing. BMC Plant Biol. 18:300. doi: 10.1186/s12870-018-1534-8
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talón, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
de Almeida, S. F., García-Sacristán, A., Custódio, N., and Carmo-Fonseca, M. (2010). A link between nuclear RNA surveillance, the human exosome and RNA polymerase II transcriptional termination. Nucleic Acids Res. 38, 8015–8026. doi: 10.1093/nar/gkq703
Eddy, S. R. (2009). A new generation of homology search tools based on probabilistic inference. Genome Inform. 23, 205–211. doi: 10.1142/9781848165632_0018
Feng, J., Zhang, X., Zhang, M., Guo, L., Qi, T., Tang, H., et al. (2021). Physical mapping and InDel marker development for the restorer gene Rf(2) in cytoplasmic male sterile CMS-D8 cotton. BMC Genomics 22:24. doi: 10.1186/s12864-020-07342-y
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230. doi: 10.1093/nar/gkt1223
Florea, L., Song, L., and Salzberg, S. L. (2013). Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Res 2:188. doi: 10.12688/f1000research.2-188.v2
Gao, B., Ren, G., Wen, T., Li, H., Zhang, X., and Lin, Z. (2021). A super PPR cluster for restoring fertility revealed by genetic mapping, homocap-seq and de novo assembly in cotton. Theor. Appl. Genet. 135, 637–652. doi: 10.1007/s00122-021-03990-0
Gasteiger, E., Jung, E., and Bairoch, A. (2001). SWISS-PROT: connecting biomolecular knowledge via a protein database. Curr. Issues Mol. Biol. 3, 47–55.
Gordon, S. P., Tseng, E., Salamov, A., Zhang, J., Meng, X., Zhao, Z., et al. (2015). Widespread Polycistronic transcripts in Fungi revealed by single-molecule mRNA sequencing. PLoS One 10:e0132628. doi: 10.1371/journal.pone.0132628
Guo, L., Lu, Y., Li, P., Chen, L., Gou, W., and Zhang, C. (2021). Effects of delayed harvest and additives on fermentation quality and bacterial Community of Corn Stalk Silage. Front. Microbiol. 12:687481. doi: 10.3389/fmicb.2021.687481
Hanson, M. R. (1991). Plant mitochondrial mutations and male sterility. Annu. Rev. Genet. 25, 461–486. doi: 10.1146/annurev.ge.25.120191.002333
Huang, C. K., Lin, W. D., and Wu, S. H. (2022). An improved repertoire of splicing variants and their potential roles in Arabidopsis photomorphogenic development. Genome Biol. 23:50. doi: 10.1186/s13059-022-02620-2
Huang, L., Dong, H., Zhou, D., Li, M., Liu, Y., Zhang, F., et al. (2018). Systematic identification of long non-coding RNAs during pollen development and fertilization in Brassica rapa. Plant J. 96, 203–222. doi: 10.1111/tpj.14016
Jaqueth, J. S., Hou, Z., Zheng, P., Ren, R., Nagel, B. A., Cutter, G., et al. (2019). Fertility restoration of maize CMS-C altered by a single amino acid substitution within the Rf4 bHLH transcription factor. Plant J. 101, 101–111. doi: 10.1111/tpj.14521
Kraft, F., and Kurth, I. (2020). Long-read sequencing to understand genome biology and cell function. Int. J. Biochem. Cell Biol. 126:105799. doi: 10.1016/j.biocel.2020.105799
Laughnan, J. R., and Gabay-Laughnan, S. (1983). Cytoplasmic male sterility in maize. Annu. Rev. Genet. 17, 27–48. doi: 10.1146/annurev.ge.17.120183.000331
Leng, N., Dawson, J. A., Thomson, J. A., Ruotti, V., Rissman, A. I., Smits, B. M., et al. (2013). EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 29, 1035–1043. doi: 10.1093/bioinformatics/btt087
Li, T., Zhang, X., Guo, L., Qi, T., Tang, H., Wang, H., et al. (2021). Single-molecule real-time transcript sequencing of developing cotton anthers facilitates genome annotation and fertility restoration candidate gene discovery. Genomics 113, 4245–4253. doi: 10.1016/j.ygeno.2021.11.014
Linke, B., and Börner, T. J. M. (2005). Mitochondrial effects on flower and pollen development. Mitochodrion 5, 389–402. doi: 10.1016/j.mito.2005.10.001
Ma, Y., Huang, W., Ji, J. J., Gong, Z. H., Yin, C. C., Ahmed, S. S., et al. (2013). Maintaining and restoring cytoplasmic male sterility systems in pepper (Capsicum annuum L.). Genet. Mol. Res. 12, 2320–2331. doi: 10.4238/2013.January.4.8
Meyer, V. G. (1975). Male sterility From Gossypium harknessii. J. Hered. 66, 23–27. doi: 10.1093/oxfordjournals.jhered.a108566
Roberts, R. J., Carneiro, M. O., and Schatz, M. C. (2013). The advantages of SMRT sequencing. Genome Biol. 14:405. doi: 10.1186/gb-2013-14-6-405
Salmela, L., and Rivals, E. (2014). LoRDEC: accurate and efficient long read error correction. Bioinformatics 30, 3506–3514. doi: 10.1093/bioinformatics/btu538
Schaarschmidt, S., Fischer, A., Lawas, L. M. F., Alam, R., Septiningsih, E. M., Bailey-Serres, J., et al. (2020). Utilizing PacBio Iso-Seq for novel transcript and gene discovery of abiotic stress responses in Oryza sativa L. Int. J. Mol. Sci. 21:8148. doi: 10.3390/ijms21218148
Schmittgen, T. D., and Livak, K. J. (2008). Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108. doi: 10.1038/nprot.2008.73
Shahzad, K., Zhang, X., Guo, L., Qi, T., Bao, L., Zhang, M., et al. (2020). Comparative transcriptome analysis between inbred and hybrids reveals molecular insights into yield heterosis of upland cotton. BMC Plant Biol. 20:239. doi: 10.1186/s12870-020-02442-z
Siegfried, A., Rousseau, A., Maurage, C. A., Pericart, S., Nicaise, Y., Escudie, F., et al. (2019). EWSR1-PATZ1 gene fusion may define a new glioneuronal tumor entity. Brain Pathol. 29, 53–62. doi: 10.1111/bpa.12619
Stewart, J., Dugger, C., and Richter, D. (1992). “A new cytoplasmic male sterility and restorer for cotton“, in: Proceedings of Beltwide Cotton Conferences, Nashville, TN); Jan. 6–10.
Suzuki, H., Rodriguez-Uribe, L., Xu, J., and Zhang, J. (2013). Transcriptome analysis of cytoplasmic male sterility and restoration in CMS-D8 cotton. Plant Cell Rep. 32, 1531–1542. doi: 10.1007/s00299-013-1465-7
Tang, H. V., Pring, D. R., Shaw, L. C., Salazar, R. A., Muza, F. R., Yan, B., et al. (1996). Transcript processing internal to a mitochondrial open reading frame is correlated with fertility restoration in male-sterile sorghum. Plant J. 10, 123–133. doi: 10.1046/j.1365-313X.1996.10010123.x
Tilgner, H., Grubert, F., Sharon, D., and Snyder, M. P. (2014). Defining a personal, allele-specific, and single-molecule long-read transcriptome. Proc. Natl. Acad. Sci. U. S. A. 111, 9869–9874. doi: 10.1073/pnas.1400447111
Tong, L., Chen, X., Wang, W., Xiao, Y., Yu, J., Lu, H., et al. (2022). Alternative splicing landscape of small Brown Planthopper and different response of JNK2 isoforms to Rice stripe virus infection. J. Virol. 96, e0171521–e0101721. doi: 10.1128/JVI.01715-21
Touzet, P., and Meyer, E. H. J. M. (2014). Cytoplasmic male sterility and mitochondrial metabolism in plants. Mitochondrion 19, 166–171. doi: 10.1016/j.mito.2014.04.009
Treutlein, B., Gokce, O., Quake, S. R., and Südhof, T. C. (2014). Cartography of neurexin alternative splicing mapped by single-molecule long-read mRNA sequencing. Proc. Natl. Acad. Sci. U. S. A. 111, E1291–E1299. doi: 10.1073/pnas.1403244111
Wang, E. T., Sandberg, R., Luo, S., Khrebtukova, I., Zhang, L., Mayr, C., et al. (2008). Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476. doi: 10.1038/nature07509
Weaver, D. B., and Weaver, J. B. (1977). Inheritance of pollen fertility restoration in cytoplasmic male-sterile upland Cotton1. Crop. Sci. 17, 497–499. doi: 10.2135/cropsci1977.0011183X001700040003x
Weirather, J. L., Afshar, P. T., Clark, T. A., Tseng, E., Powers, L. S., Underwood, J. G., et al. (2015). Characterization of fusion genes and the significantly expressed fusion isoforms in breast cancer by hybrid sequencing. Nucleic Acids Res. 43:e116. doi: 10.1093/nar/gkv562
Wu, J., Gong, Y., Cui, M., Qi, T., Guo, L., Zhang, J., et al. (2011). Molecular characterization of cytoplasmic male sterility conditioned by Gossypium harknessii cytoplasm (CMS-D2) in upland cotton. Euphytica 181, 17–29. doi: 10.1007/s10681-011-0357-6
Wu, J., Zhang, M., Zhang, B., Zhang, X., Guo, L., Qi, T., et al. (2017). Genome-wide comparative transcriptome analysis of CMS-D2 and its maintainer and restorer lines in upland cotton. BMC Genomics 18:454. doi: 10.1186/s12864-017-3841-0
Xie, C., Mao, X., Huang, J., Ding, Y., Wu, J., Dong, S., et al. (2011). KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, W316–W322. doi: 10.1093/nar/gkr483
Yang, L., Wu, Y., Zhang, M., Zhang, J., Stewart, J. M., Xing, C., et al. (2018). Transcriptome, cytological and biochemical analysis of cytoplasmic male sterility and maintainer line in CMS-D8 cotton. Plant Mol. Biol. 97, 537–551. doi: 10.1007/s11103-018-0757-2
Zhang, C. (2005). Preliminary Studies and Breeding on Cytoplasmic male Sterility Three-line of Gossypium hirsutum L. master. Wuhan, China: HuaZhong Agricultural University.
Zhang, J., and Stewart, J. M. (2001a). CMS-D8 restoration in cotton is conditioned by one dominant gene. Crop. Sci. 41, 283–288. doi: 10.2135/cropsci2001.412283x
Zhang, J., and Stewart, J. M. (2001b). Inheritance and genetic relationships of the D8 and D2-2 restorer genes for cotton cytoplasmic male sterility. Crop. Sci. 41, 289–294. doi: 10.2135/cropsci2001.412289x
Keywords: CMS-D8, restorer gene Rf2, full-length transcript, novel isoforms, fusion transcript
Citation: Feng J, Li Y, Zhang J, Zhang M, Zhang X, Shahzad K, Guo L, Qi T, Tang H, Wang H, Qiao X, Lin Z, Xing C and Wu J (2022) Transcript Complexity and New Insights of Restorer Line in CMS-D8 Cotton Through Full-Length Transcriptomic Analysis. Front. Plant Sci. 13:930131. doi: 10.3389/fpls.2022.930131
Edited by:
Baohua Wang, Nantong University, ChinaReviewed by:
Xinlian Shen, Jiangsu Academy of Agricultural Sciences (JAAS), ChinaCopyright © 2022 Feng, Li, Zhang, Zhang, Zhang, Shahzad, Guo, Qi, Tang, Wang, Qiao, Lin, Xing and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhongxu Lin, bGluemhvbmd4dUBtYWlsLmh6YXUuZWR1LmNu; Chaozhu Xing, Y2hhb3podXhpbmdAMTI2LmNvbQ==; Jianyong Wu, ZHIud3VqaWFueW9uZ0BsaXZlLmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.