 Xiaoyan Liu
Xiaoyan Liu Xun Gong2
Xun Gong2 Min Tang
Min Tang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Plant Sci., 14 July 2022
Sec. Plant Metabolism and Chemodiversity
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.900035
This article is part of the Research TopicInsights in Plant Metabolism and Chemodiversity: 2021View all 14 articles
The Chinese Herbal Medicine (CHM) has been used worldwide in clinic to treat the vast majority of human diseases, and the healing effect is remarkable. However, the functional components and the corresponding pharmacological mechanism of the herbs are unclear. As one of the main means, the high-throughput sequencing (HTS) technologies have been employed to discover and parse the active ingredients of CHM. Moreover, a tremendous amount of effort is made to uncover the pharmacodynamic genes associated with the synthesis of active substances. Here, based on the genome-assembly and the downstream bioinformatics analysis, we present a comprehensive summary of the application of HTS on CHM for the synthesis pathways of active ingredients from two aspects: active ingredient properties and disease classification, which are important for pharmacological, herb molecular breeding, and synthetic biology studies.
The Chinese herbal medicine (CHM) makes a great contribution to the human healthcare and clinical therapy due to its remarkable efficacy and fewer side effects (Flower et al., 2012; Lu et al., 2019; Xiang et al., 2019; Zheng et al., 2020). Since the Qin and Han dynasties, the Chinese ancestors had made the natural plants to cure patients without the knowledge of the chemical constituents, which gradually formed the later mature system of CHM with clarified the properties (He et al., 2015). Artemisia annua, an ancient medicine, grows broadly in China, the province such as, Jiangsu, Shanxi, Guangdong. Artemisinin, the main medicinal ingredient of A. annua, is world-famous for its treatment of malaria. Moreover, both traditional and modern pharmaceutical research imply it has anti-inflammatory, anti-viral, and anti-cancer effects (Nair et al., 2019; Feng et al., 2020; Liu H. et al., 2021). Accumulated evidence suggests that the bioactive components originated from CHM play a non-negligible role in the treatment of diseases (Li et al., 2016; Ji et al., 2019). However, the low-abundance is insufficient to meet the clinical requirements, such as paclitaxel, a well-known natural anti-cancer drug (Sabzehzari et al., 2020; Xiong et al., 2021). Nowadays, with the continuous advances in sequencing technologies for the fine genome assembly, the synthesis of bioactive components, or called cell-Bio-fluid sync, can be well elucidated, which provides abundant genetic resources for life and pharmaceutical sciences.
Direct application of the conventional Sanger sequencing (called first-generation sequencing) on the herbs with large and complicated genomes is grudging because of the low throughput and accuracy (Lo and Shaw, 2019). Instead of it, the next-generation sequencing (NGS, also called second-generation sequencing) was gradually applied since 2010 (Cheng Q. et al., 2021). NGS can perform millions to billions of independent sequencing processes, dramatically increasing sequence output, which includes Illumina Solexa, Roche454, and ABI SOLiD platform. Based on the principle of reversible termination and fluorescently labeled dNTP, Illumina Solexa is sequencing while synthesizing (Guo et al., 2021a). It also has certain drawbacks, such as short read length (usually 200-800 bp), base mismatches, GC preference, and template migration during PCR amplification, which might influence the accuracy and integrity of sequencing data (Cheng Q. et al., 2021; Guo et al., 2021a). Subsequently, the third-generation sequencing (TGS) stands out for the high-throughput sequencing (HTS) technologies as a routine method. Oxford Nanopore and PacBio single-molecule real-time (SMRT) sequencing technology are now the main TGS platforms. Although SMRT can achieve read lengths of 100 kb, additional factors like template breakage, enzyme denaturation, and short library sequences can affect read lengths and accuracy. Unlike SMART sequencing, the Nanopore read length is determined by the length of the DNA molecules to be sequenced rather than the sequencing technique (van Dijk et al., 2018). Here, we mainly reviewed relevant articles from the recent decades and presented a comprehensive summary of the application of HTS on CHM for the synthesis pathways of active ingredients from two aspects: active ingredient properties and disease classification.
The last 5 years have been an era of expansion in medicinal plants genome sequencing, with 2nd and 3rd generation technologies combining to assure long read length, high throughput, and reasonable sequencing price for medicinal plant genomes assembled to the chromosome level. The employment of HTS technologies on data-mining bioactive compounds is shown in Figure 1. This strategy integrates genomics, transcriptomics, and metabolomics data to analyze genomic properties, synthetic and metabolic pathways of bioactive constituents, the overall transcriptional activity of organisms, and pathway regulatory mechanisms that will be revealed to uncover functional genes (see Table 1). The Panax notoginseng genome, for example, has been assembled in five versions. Chen et al. (2017) and Zhang D. et al. (2017) used Illumina technology to assemble a sketch of the P. notoginseng genome, but the assembly was highly fragmented. Fan G. et al. (2020) employed Pacbio and Oxford Nanopore technologies to assemble the genome to the chromosome level in 2020, with significantly improved assembly continuity. The last two versions were assembled more accurately by using a combination of 2nd and 3rd generation sequencing (Jiang Z. et al., 2021; Yang Z. et al., 2021). In contrast to the study by Jiang Z. et al. (2021) and Yang Z. et al. (2021) illustrated dencichine biosynthesis, the other major bioactive compounds derived from P. notoginseng. Vaccinium darrowi, the diploid blueberry, benefits cardiovascular, neural, and retinal. Cui et al. (2022) obtained a de novo genome assembly for V. darrowi according to Oxford Nanopore, Illumina short reads, and Hi-C data. Sapindus mukorossi, an environmentally herb, has been used for treating inflammatory conditions owing to its abundant active compounds. Xue et al. (2022) revealed the first reference genome sequence of S. mukorossi. Li et al. (2022) assembled the high-quality reference genome of Gentiana dahurica using Nanopore long reads, Illumina short reads, and Hi-C technologies, which is the first chromosome-level genome of Gentianaceae. Based on comparative genomic and transcriptome analyses, cytochrome P450 candidate genes related to gentiopicroside biosynthesis were identified. Noteworthy, Wang et al. (2022) employed genome-wide association studies (GWAS) and identified six quantitative trait loci (QTLs) related to fruit traits according to the latest version genome of Dimocarpus longan at the chromosome level with 455.5 Mb assembled into 15 chromosomes. Based on the genome-assembly and the downstream bioinformatics analysis, these articles weremainly focusing on providing new insight for the discovery of novel drug candidates in CHM.
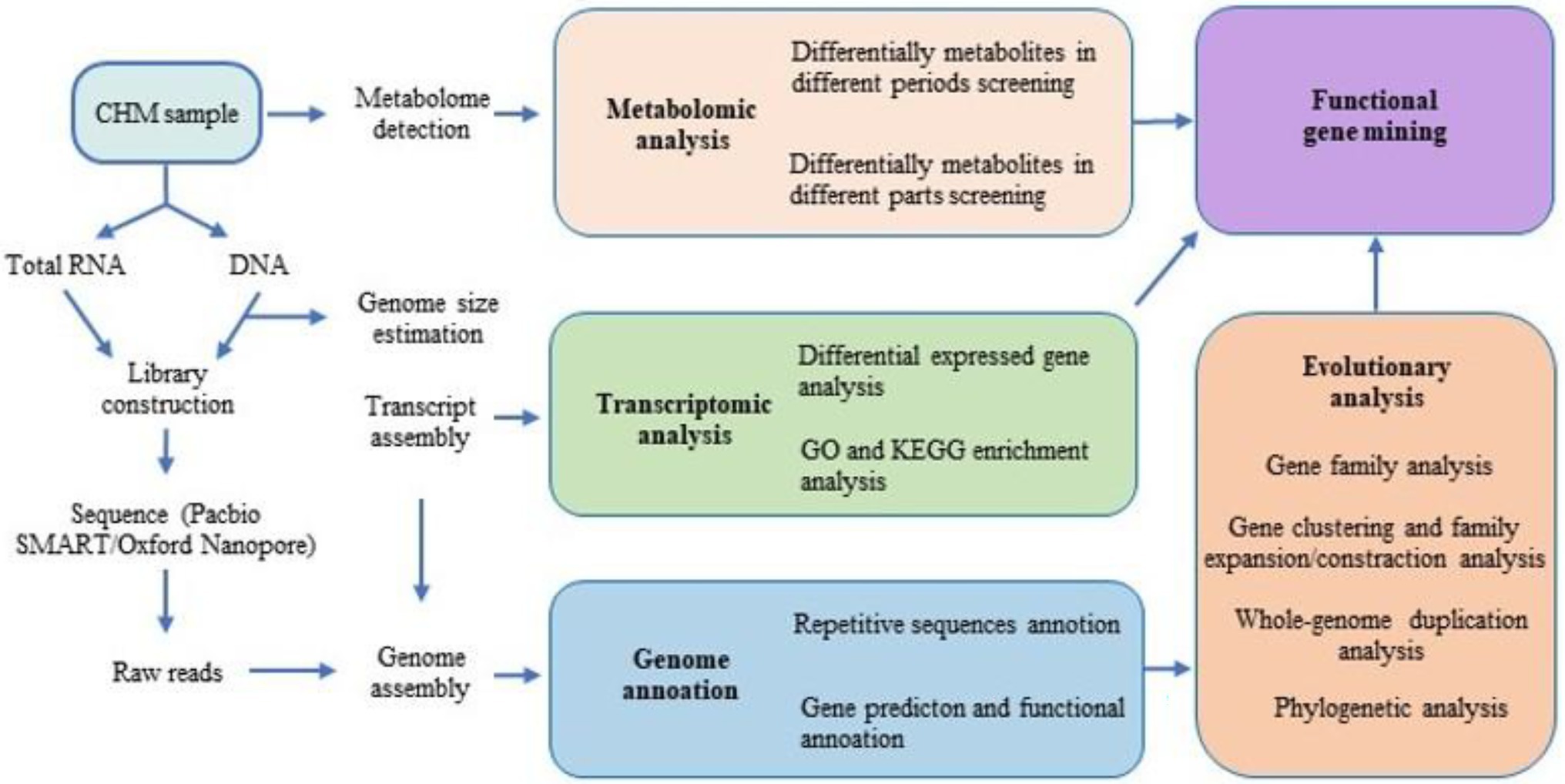
Figure 1. Flow chart of bioactive compound related functional genes discovering based on high-throughput sequencing (HTS).
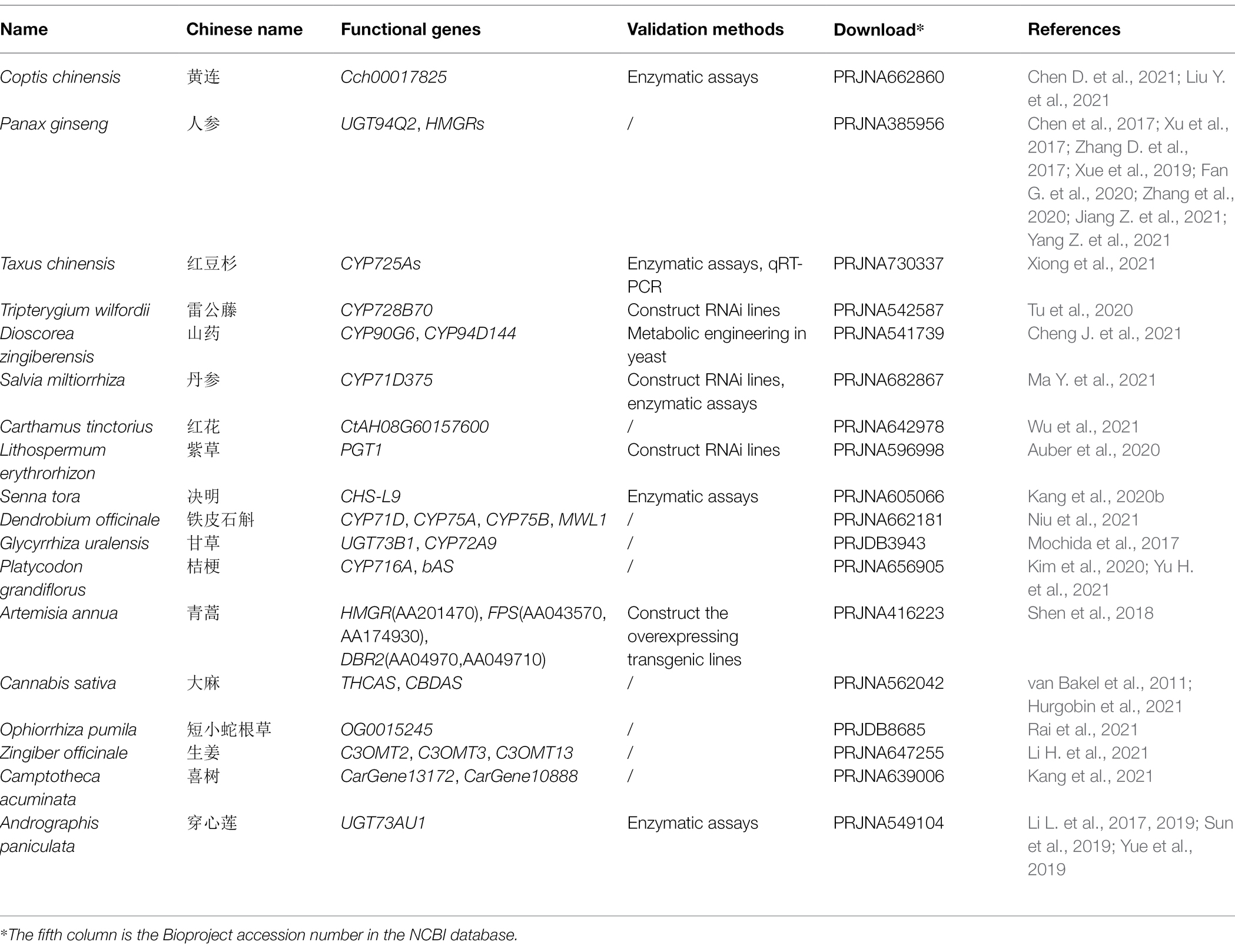
Table 1. Summary of the bioactive compounds related functional genes from the assembly Chinese herbal medicines (CHM) genome.
Based on the accumulation in the sequencing field, many useful bioactive compounds and their varieties have been screened out from the complex mixtures and the clinic effects have been validated (see Figure 2; Supplementary Figure S1). Specially, the secondary metabolites constitute the backbone of many drugs as the active ingredients of the medicinal plants and are widely used in pharmaceutical products. In recent years, due to the innovation of sequencing technology, the HTS accelerates the study of secondary metabolites biosynthetic in the medicinal plants, which indirectly expands the global commercial market of the herb products (Barbosa et al., 2019). In general, the secondary metabolites are divided into seven major groups, namely flavonoid, terpenoid, alkaloid, phenylpropanoid, quinone, tannin, and steroid (Lo and Shaw, 2019; Erb and Kliebenstein, 2020).
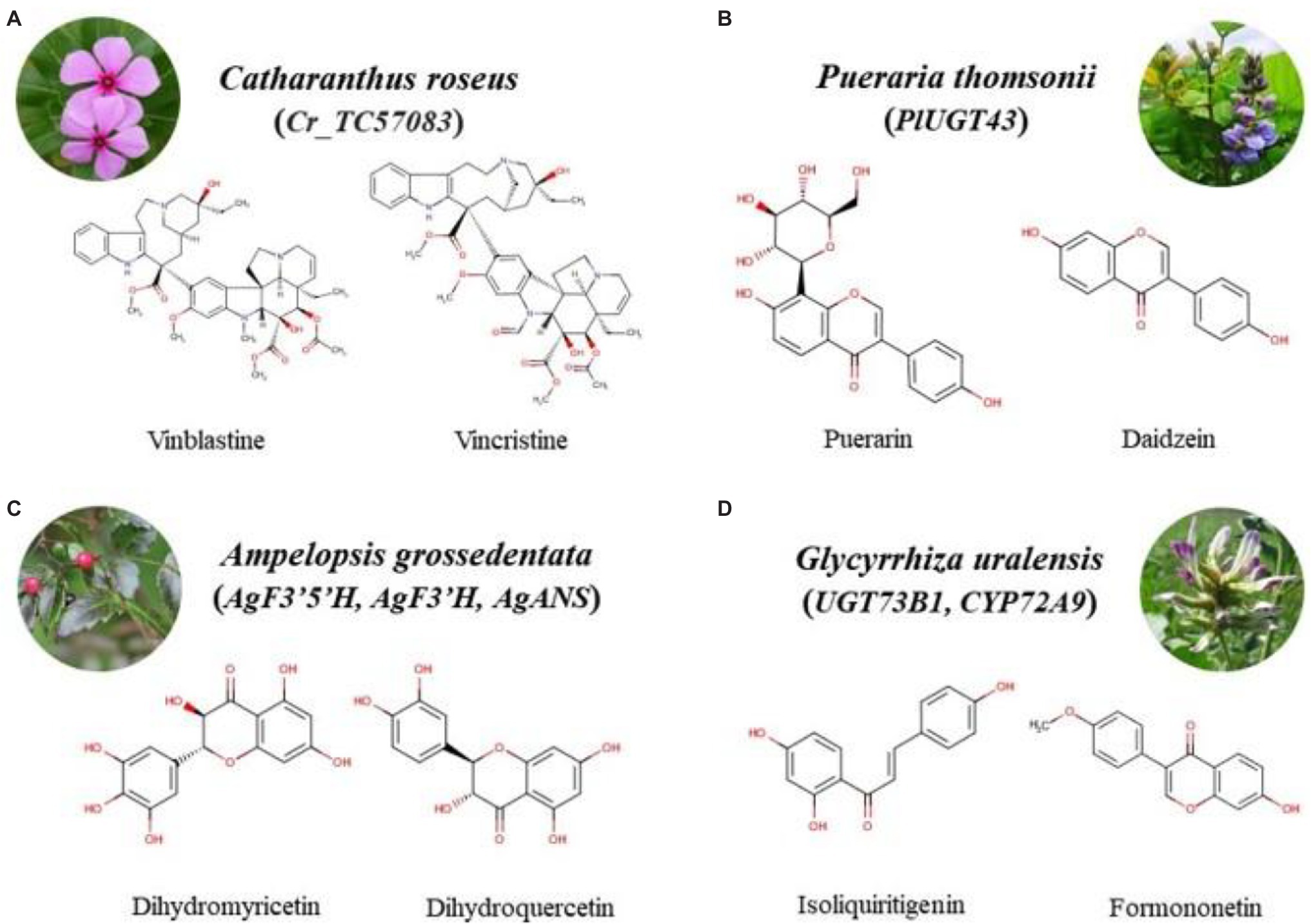
Figure 2. Chemical structure formula of the bioactive compounds from four species. *Bioactive compound-related functional genes are shown in the brackets. (A) Cr_TC57083, the functional gene related to synthesis of vinblastine and vincristine in Catharanthus roseus; (B) PlUGT43, the functional gene involved in synthesis of puerarin and daidzein in Pueraria thomsonii; (C) AgF3’5’H, AgF3’H, AgANS, the functional genes associated with biosynthesis of dihydromyricetin and dihydroquercetin in Ampelopsis grossdentata; (D) UGT73B1, CYP72A9, the functional genes correlation with synthesis of isoliquiritigrnin and formononetin in Glycyrrhiza uralensis.
Terpenoids are hydrocarbon compounds consisting of isoprenoid as structural units (Chen et al., 2011). There are two synthetic pathways for terpenoid in medicinal plants: MVA pathway and DOXP/MEP pathway. In the MVA pathway, IPP is condensed to generate DAMP by IDI, and FDPS converts IPP and DMAPP to FPP, which are the common precursors of sesquiterpenes, triterpene, and sterol (Lichtenthaler, 1999). Monoterpene, diterpene, and tetraterpene produce in the MEP pathway (Rohmer, 1999). The first rate-limiting enzyme in the MVA pathway, 3-hydroxy-3-methylglutaryl-CoA reductase (HMGR), is critical for its regulation (Korth et al., 1997). Cytochrome P450 hydroxylases (CYP450) are used to structurally modify terpene end-products to eventually become a large number of natural products. Tu et al. (2020) reported a reference genome of Tripterygium wilfordii using PacBio long reads, 10X Genomics, and high throughput chromosome conformation capture (Hi-C) data and annotated 28,321 protein-coding genes. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses showed that 951 T. wilfordii-specific genes were especially enriched in terpene synthases. According to the gene-to-metabolite network, 57 CYP genes may be involved in the triptolide biosynthesis pathway were identified, of which 10 candidate genes with tissue-specific were selected for further functional validation. RNAi studies found that the transcript levels of four candidate genes (CYP728B70, TW011445.1, TW012149.1, and TW006625.1) were decreased and triptolide accumulation showed the same results. Additionally, based on substrate feeding and overexpression studies, the findings showed that CYP728B70 was involved in triptolide biosynthesis. This finding provides new insight into triptolide biosynthesis and the theoretical foundation for heterologous bioproduction (Tu et al., 2020). Xu et al. (2017) used the Illumina Hiseq X-Ten platform to sequence the genome and the transcriptome of Panax ginseng and predicted 42,006 protein-coding genes. Weighted gene coexpression network analysis (WGCNA) obtained 15,762 genes positively associated with ginsenosides, which are produced by the precursor IPP via the MVA pathway. BLAST search discovered 31 genes were linked to 10 upstream enzymes. Finally, eight genes encoding HMGRs found that they may perform different roles during ginseng development. This is of great significance for future studies on ginseng breeding and synthetic biology. Xia et al. (2018) reported a high-quality draft genome of Siraitia grosvenorii using SMRT sequencing via PacBio platform and Illumina paired-end reads. According to the genomic information, 127 candidate genes were found in the mogrosides biosynthesis pathway, including SQEs, EPHs, CYP450s, and UGTs. In addition, mogrosides are accumulated during the development of S. grosvenorii fruit. Up-regulated genes in fruit development were found significantly enriched in the sesquiterpenoid and triterpenoid biosynthesis pathways through RNA-seq data and KEGG analysis. This study for S. grosvenorii genome assembly and annotation will contribute to the discovery of new functional genes. Chen et al. performed PacBio sequencing technologies to construct the first full-length transcriptome of Pogostemon cablin and annotated 102 transcripts related to patchoulol biosynthesis. Patchoulo, the main bioactive compounds, among the 39 chemical compositions in P. cablin were detected by GS-MS analysis. Patchoulol synthase (PatPTS) converts famesy1-pp (FPP) to patchouli alcohol (PA). Furthermore, based on the P. cablin full-length transcriptome and transcriptome profiling under MeJA treatment, 427 DEGs were up-regulated in leaves after MeJA treatment, of which HMGR, DXS, HDR, IDI, FDPS, PatPTS genes related to patchouli biosynthesis were up-regulated under MeJA treatment and positively correlated with patchouli content. Although this study did not validate the identified genes using qRT-PCR, it provides a valuable genetic resource for further research in patchouli. Dong et al. (2021) constructed the genome of Magnolia biondii using SMRT via PacBio long read, 10X Genomics and Hi-C data. The chromosome-level reference genome of M. biondii is approximately 2.22 Gb long and predicted 47,547 protein-coding genes. The volatile oil extracted from the flower buds of M. biondii has many pharmacological properties such as anti-inflammatory and is rich in terpenoids, which are catalyzed by terpene synthase (TPS). Based on genomic information and RNA-seq data, 102 TPS genes were identified and the expression profiles showed 33 TPS genes were higher expressed in flowers than in leaves. These findings will improve the understanding of the molecular breeding of M. biondii. Kim et al. (2020) assembled a draft genome of Platycodon grandiflorus through PacBio platform and contained 40,017 protein-coding genes. Gene family expansion and contraction analysis found that CYP76C, CYP72, and CYP716 families in P. grandiflorus underwent expansion. Based on orthologous gene annotation, β-amyrin synthases (bASs) were found that underwent expansion in P. grandifloras. Previous research has revealed that the CYP716 gene family was involved in the platycodon saponins biosynthesis. Additionally, among the expanded gene families, CYP716 and bAS genes were highest expression in roots than other tissues. To investigate the terpenoid biosynthesis pathway in Artemisia argyi, gene expression analysis was performed and found 36,820 non-redundant transcripts, of which 187 transcripts relevant to terpenoid biosynthesis were discovered via KEGG analysis. Among them, eight diterpenoid biosynthesis genes were identified and highly expressed compared to other tissues. Finally, qRT-PCR verified 12 genes that were highest expressed in leaves were consistent with RNA-seq data (Kim et al., 2020). Shen et al. (2018) reported a draft A. annua genome sequence of 1.74 Gb that is assembled by Illumina and PacBio sequencing platform. They annotated 63, 226 protein-coding genes based on expression evidence. Gene expansion and contraction analysis revealed that 7,286 expanded and 3,950 contracted gene families in A. annua. Among the A. annua expanded gene families, TPS families expanded significantly and 122 TPS genes were identified. Subsequently, according to genomic and transcriptomic analyses, sesquiterpenoid synthesis-related genes were found and expressed in specific tissues. Previous studies were conducted to increase the yield of artemisinin by overexpressing the upstream or downstream enzymes, such as FPS and HMGR, but the artemisinin content did not increase significantly. Therefore, this study overexpressed the upstream, midstream and downstream enzymes [HMGR (AA201470), FPS, and DBR2] simultaneously to examine the content of artemisinin, and the revealed that the transgenic plants produced more artemisinin. The present study provides a large number of candidate genes for further enhancement of artemisinin content. Xiong et al. (2021) revealed the Taxus genome with 10.23 Gb assembled into 12 chromosomes using Illumina HiSeq 2500, PacBio Sequel II, and Hi-C data. Based on genomic information and previous literature, 649 CYP450 genes were identified, of which CYP750 and CYP725 families in Taxus underwent significant expansion. Moreover, expression levels of four gene clusters on chromosome 9 where most CYP725A genes located on were significantly up-regulated by jasmonic acid induction. These results suggested that the gene cluster probably contains the majority of the paclitaxel synthesis genes that originated from the evolution of T. chinensis. PlantiSMASH analysis further indicated a gene cluster in group 9.2 that may be associated with terpenoid biosynthesis, including two TS genes, two T5αH genes, and two unknown CYP725As. Biochemical assays demonstrated that TS and T5αH were mainly responsible for catalyzing the first two steps of paclitaxel biosynthesis. Finally, 17 CYP725A genes which were closely related to known paclitaxel biosynthesis genes were identified through a gene-to-gene coregulation network. This article helps to accelerate paclitaxel biosynthesis and Taxus biotechnology applications.
Flavonoids are natural products with a C6-C3-C6 carbon skeleton structure, usually combined with sugars to form glycosides present in medicinal plants, which classified into flavonols, flavones, isoflavones, and anthocyanidins (Ciumarnean et al., 2020). Phenylalanine, the biosynthetic precursor of flavonoids, which is then converted to rutin through 10 enzymes, including phenylalanine deaminase (PAL), cinnamic acid-4-hydroxylase (C4H), 4-coumarate coenzyme A ligase (4CL), chalcone synthase (CHS), chalcone isomerase (CHI), flavanones-3′-hydroxylase (F3’H), flavanones-3′5’-hydroxylase (F3’5’H), flavonol synthase (FLS), glucose/witch hazel transferases (UGT/GT; Zhang L. et al., 2017). CHS is the first key enzyme in the flavonoid synthesis pathway and has been studied more in medicinal plants. In addition, O-methyltransferase (OMT) catalyzes the conversion of root-specific norbornin to other flavones, such as wogonin, isowogonin, and moslosooflavone (Liu W. et al., 2021). Zhao et al. (2019) reported the high-quality reference genome sequence of Scutellaria baicalensis at the chromosome level with 408.14 Mb (93%) assembled into 9 pseudochromosomes using Illumina, PacBio, and Hi-C technology. The article further elucidated the biosynthesis of norwogonin to wogonin catalyzed by 8-O-methyltransferase. Hence, 28,930 genes were annotated by bioinformatics tools based on de novo predictions, homology-based prediction, and RNA-seq data. On the basis of the genomic and transcriptome information, six genes encoding OMTs were found in the tandem repeat region unique to S. baicalensis. Moreover, enzyme activation and RNAi experiments confirmed that SbPFOMTs were involved in the synthesis of Wogonin, and the Wogonin synthesis pathway was resolved. It provides a basis for synthetic biology to obtain baicalein and a reference for genetic analysis of other Labiatae plants (Zhao et al., 2019). Wu et al. (2021) reported the first chromosome-scale reference genome of Carthamus tinctorius through combined PacBio platform and Hi-C mapping and predicted 33,343 protein-coding genes. Among them, seven CHS genes were identified based on the modified flavonoid biosynthetic pathway combined with the KEGG database and related literature. CarCHS5 and CarCHS6 which are unique to C. tinctorius were revealed by Collinearity analysis. These results provide evolutionary insights into the flavonoid biosynthesis in C. tinctorius. Qing et al. (2021) generated the Hemerocallis citrina genome of 3.77 Gb that was assembled by PacBio long reads and Hi-C data. Gene family expansion and contraction analysis revealed that 10,375 gene families in H. citrina underwent expansion, whereas a significant number of gene families (6707) underwent contraction. In addition, the expanded gene families were mainly enriched in flavonoids biosynthesis, which may have contributed to the content of rutin in H. citrina. Among them, 108 genes were identified by homology search. In addition, High-performance liquid chromatography/quadrupole time-of-flight (HPLC-Q-TOF) data revealed that rutin was mainly accumulated in flower buds, 20 candidate genes which mainly expressed in flower buds were identified in combination with transcriptome data. Zhang L. et al. (2017) reported a high-quality assembly of the 489.3 Mb genome of Fagopyrum tataricum at chromosome-scale, and contained 33, 366 protein-coding genes. In F. tataricum genome, nine rutin biosynthesis enzymes were identified, such as CHI (FtPinG0002790600) and F3’H (FtPinG0002353900). Although this study did not further validate the candidate genes, it provided some insights into the biosynthetic pathway of rutin.
Alkaloids are basic nitrogen-containing organic compounds with complex ring structures. It is mainly synthesized with precursors such as tryptophan, tyrosine, phenylalanine, lysine, and arginine. According to the structures, it can be classified into pyridines, indoles, terpenoids, isoquinolines, steroids, etc. (Liu et al., 2019). The previous report showed that CYP80 and CYP719 families catalyze many different reactions in the biosynthesis of BIAs, including hydroxylation, C-C, or C-O coupling, and formation of methylenedioxy bridges (Deng X. et al., 2018). Hu et al. (2021) presented the draft genome sequence of Strobilanthes cusia, constructed using PacBio long reads and Hi-C sequencing data. The draft genome assembly had a size of 913.74 Mb and contained 2,974 coding gene sequences, of which 2,975 DEGs were identified by transcriptome analysis and enriched in phenylpropanoid, flavonoid, and triterpenoid biosynthesis. Based on gene family expansion and contraction analysis, 60 gene families expanded in S. cusia, while 16 gene families contracted. In addition, these expanded gene families were mainly enriched in secondary metabolism pathway according to GO and KEGG analysis. On the basic of homology searching, 18 genes coding IA-related key enzymes were identified as DEGs in the S. cusia genome, such as UGT, CYP450, ASA, TSB, BGL, CS, and EPSPS genes. This study reveals the molecular basis of the accumulated indole alkaloids of S. cusia. Yang et al. (2021a) presented a reference chromosome level genome of Areca catechu by Illumina and PacBio data. The assembled genome was 2.51 Gb with a N50 scaffold size of 1.7 Mb and predicted 31,571 protein-coding genes. Among them, 904 expanded gene families were enriched in secondary metabolites pathways, including flavonoid, terpenoid, and isoquinoline alkaloid biosynthesis. Although this article was not mining deeper into functional genes related to secondary metabolite synthesis, it provides basic insights into areca alkaloid biosynthesis. Liu Y. et al. (2021) assembled a high-quality genome of Coptis chinensis at the chromosome level through integrating Nanopore sequencing, Illumina short reads, and Hi-C technology. Forty-one thousand four protein-coding genes were annotated. In addition, 1,083 gene families underwent expansion were revealed by gene evolution analysis. Among them, two new P450 families are found in early-diverging eudicots: CYP719 and CYP749. According to a comparison of these CYP719 genes, Cch00017825 is clustered on chromosome 3 and expressed significantly in the C. chinensis rhizomes. Liu X. et al. (2017) reported the Macleaya cordata genome with 378 Mb using Illumina HiSeq 2000. Based on the previous report, 39 candidate genes related to SAN and CHE biosynthesis were identified. They performed liquid chromatography coupled with mass spectrometry (LC/MS) to detect the metabolites in M. cordata and obtained the SAN and CHE biosynthesis. Based on the reference genome of M. cordata, 39 genes were identified involved in SAN and CHE biosynthesis pathway. Additionally, SAN and CHE were not accumulated in the stem, 16 candidate genes were selected in combination with RNA-seq data. Finally, Metabolic engineering verified 14 candidate genes involved in catalytic reactions of SAN and CHE biosynthesis. This research could help medicinal plants produce more SAN and CHE. Rai et al. (2021) used PacBio long reads, Illumina short reads, Bionano optical mapping and Hi-C sequencing to assemble the Ophiorrhiza pumila genome. Based on metabolome annotation datasets, 273 nitrogen containing metabolites were annotated, most of them were indole alkaloids (IAs). According to a previous study and comparative genomic analysis, they found that monoterpene indole alkaloids (MIAs) biosynthesis are originated from strictosidine biosynthesis. In the late secoiridoid pathway, OG0014621 (LAMT) was found specifically expanded in O. pumila. Additionally, genes encoding MIA-related enzymes were replicated and obtained, such as STR, SLS, 7-DLH, and 7-DLGT. These suggested that gene clusters include several functional signature genes. The study presents a pangenome model of MIA biosynthesis that will help establish a sustainable supply of camptothecin. Kang et al. (2021) presented a high-quality genome sequence of Camptotheca acuminata, constructed using SMRT sequencing technology from PacBio, Illumina platform, and Hi-C techniques. In C. acuminata, gene expansion and contraction analysis indicated that 2,951 gene families expanded, while 1,733 gene families contracted. Previous research on C. roseus illustrated that CYP72A219 (SLS) and CYP72A224 (7-DLH) play a vital role in indole alkaloids biosynthesis in periwinkle. Hence, on the basic of homology searching and phylogenetic tress, two SLS genes (CacGene13172, CacGene10833) and two 7-DLH genes (CacGene13171, CacGene10832) were identified in C. acuminata genome. This study reveals candidate genes that may play a role in the camptothecin biosynthesis in C. acuminata, providing a basis for future high-yield artificial biosynthesis.
Kang et al. (2020b) presented the chromosome-level genome of Senna tora with 526.4 Mb by PacBio long read sequencing and Illumina data and were assembled into 13 chromosomes using Hi-C data. Metabolite profile analyzed the content of 10 anthraquinone in different developmental stages of seeds and found that aurantio-obtusin was the major bioactive compounds in mature seeds. Furthermore, they predicted 45,268 protein-coding genes. Comparative genomics revealed that 2,874 gene families in S. tora underwent expansion, while 3,371 gene families underwent contraction. Interestingly, the expanded gene families were mainly enriched in phenylpropanoid, isoflavonoid, and terpene biosynthesis. Among the expanded gene families, 16 CHS-L genes were identified due to its rapidly expanded in in S. tora. At stage 4 when anthraquinones began to accumulate, two CHS-L genes (STO07G228250 and STO07G228220) presented high expression levels. Further phylogenetic tree analysis showed that STO07G228250 was more similar to HpPKS and ArOKS, octaketide synthases. Finally, according to ESI-MS spectrum and enzyme activity experiment, STO07G228250 was demonstrated to be involved in the first step of anthraquinone biosynthesis. The study provides a platform for medicinal plant S. tora with high bioactive molecular content.
Yang et al. (2021b) constructed the assembly of 1.79 Gb genome sequence of Arctium lappa and obtained 32,771 protein-coding genes. Based on the genomic information, 616 positively selected candidate genes were discovered in A. lappa. Transcriptome analysis revealed that genes related to lignan biosynthesis in five different stages of A. annua (4CL), dirigent protein (DIR), and hydroxycinnamoyl transferase (HCT) were highly related to arctiin biosynthesis.
Herbs have been demonstrated in studies to help with the treatment of rheumatism, diabetes, cancer, Alzheimer’s disease, and cardiovascular disease (Li X. et al., 2019). Take rheumatism as an example, they treat rheumatism by the following effects: dispelling wind, eliminating the body moisture, removing coldness, clearing heart, dredging the channel, expectorant and diffusing impediment, benefiting Qi and nourishing the blood, and invigorating the kidney and strengthening the spleen.
Callerya speciosa made contributions to treat rheumatism via dredging the channel effects, due to its main medicinal ingredients-isoflavonoids, such as maackiain and formononetin. Yao et al. (2021) used NGS technology to sequence the transcriptome of C. speciosa and identified 4,337 DEGs during the tuberous root development. Among them, 15 genes related to isoflavonoids biosynthesis were found. These results indicated that these genes may be promoted the accumulation of isoflavonoids in the tuberous toot. In addition, qRT-PCR validated the expression pattern of candidate DEGs were consistent with the RNA-seq data. The study provides new insights into the potential mechanisms of isoflavonoid biosynthesis in C. speciosa. Flavonoid, a bioactive compound derived from Fritillaria hupehensis, has often been used as an expectorant to treat rheumatic diseases. Guo K. et al. (2021) performed SMART analysis from the PacBio platform to sequence the full-length cDNA of F. hupehensis. Thirty-four flavonoid biosynthesis unigenes were found using the KEGG pathway, and divided into five branches by blast against model plants. The study provides a valued resource for herb breeding and bioactive compounds for pharmacological application. Asarum sieboldii has abundant medicinal properties, such as anti-inflammatory, antiallergic, and removing coldness. Asarinin and aristolochic acid are bioactive compounds that originated from A. sieboldii. Chen C. et al. (2021) used full-length transcriptome analysis to uncover genes involved in asarinin and aristolochic acid biosynthesis. The result found 63, 023 transcriptional sequences, of which 41 asarinin biosynthesis candidate genes and 56 aristolochic acid biosynthesis candidate genes were identified, including AsCOMT, AsEPI, AsCYP81Q2, AsCYP81Q4, AsCYP81Q7, AsCYP81Q29, AsTYR, AsTYDC, AsNCS, AsNOMT, AsCNMT, and AsCYP80B1. Finally, qRT-PCR data verified 4 genes were significantly expressed in the root, including AsCCR, AsPAL, AsCOMT, and AsCYP81Q. The study will provide a good basis for the production of a valuable, low toxicity active ingredient. Gynostemma pentaphyllum has the pharmacological effect of eliminating the body moisture and clearing the heart. Gypenosides, triterpene saponins, are the main active compounds in G. pentaphyllum. Liang et al. (2019) obtained 140,157 unigenes using PacBio standard analysis pipeline and Illumina data. Among them, 404 gypenoside biosynthetic genes were detected and annotated. GpOSC1, GpCYP89, and GpUGT35 were demonstrated the leading candidate genes for gypenoside biosynthesis by qRT-PCR technology. These findings will lay a new foundation for gypenosides biosynthesis. Akebia trifoliata possesses the properties of strengthening the spleen and relieving pain. Bioactive compounds contribute medicinal effects to A. trifoliata, including triterpenoid saponins, triterpenes, and flavonoids. Huang H. et al. (2021) presented the chromosome level genome sequence of A. trifoliata using Illumina HiSeq X-Ten sequencing technology, SMRT platform from PacBio and Hi-C technique. The genome assembly had a size of 682.14 Mb and predicted 25,598 protein-coding genes. Two hundred forty-six expanded and 473 contracted gene families in A. trifoliata were discovered, according to the phylogenetic tree. Interestingly, the expanded gene families were mainly enriched in terpenoid biosynthesis via KEGG enrichment analysis. Among the expanded gene families, 24 Atrβ-AS genes, 12 UDP-glucoronosyl, three UDP-glucosyltransferase and seven cytochromes P450 gene families were involved in sesquiterpenoid and triterpenoid biosynthesis pathways. In addition, three UDP-glucosyltransferase, 14 cytochrome P450, and two TPS gene families were constructed in the A. trifoliata genome. The findings suggested that these gene families were quickly changing to synthesis varies of triterpenes in A. trifoliata. The study provides a useful genetic resource for pharmacological applications of A. trifoliata. Trillium govanianum contributes to the treatment of rheumatism disease, due to its ability to benefit Qi and nourish the blood. Diosgenin, as steroidal saponins, is considered to be the main bioactive component of T. govanianum. Singh et al. (2017) performed spatial transcriptome analysis of the leaf, fruit, stem, and rhizome tissues of T. govanianum and obtained 69,174 transcripts. As a result, 108 CYP genes and 58 UGTs were identified, of which 87 CYP genes and 49 UGTs were differentially expressed in four tissues. Based on KEGG classification, genes involved in steroidal saponin biosynthesis were divided into three pathways: terpenoid backbone (16 genes), sesquiterpenoid and triterpenoid (two genes), and steroid biosynthesis (14 genes). In addition, qRT-PCR was employed to confirm the expression pattern of 29 genes, which was consistent with the RNA-seq results. Thus, compared with transcriptome sequencing, de novo genome assembly of herbs mines more functional genes involved in the biosynthesis pathways of active ingredients, which is more beneficial to the development of herb molecular breeding.
For many years in India, Gymnema sylvestre, a well-known and valuable medicinal plant, has been used to treat diabetes (Hossain et al., 2016; Tiwari et al., 2017; Pham et al., 2018). Ayachit et al. (2019) performed RNA-seq data to uncover terpenoid biosynthesis genes and identified 111 transcripts involved in the terpenoid biosynthetic pathway, such as mono-terpenes, di-terpenes, tri-terpenes, and ubiquinones. Finally, qRT-PCR verified six transcripts involved in the MEP pathway were a positive correlation to terpenoid biosynthesis. This study provides insights for future functional genomics studies of G. sylvestre. Eriobotrya japonica, a traditional medicine, is beneficial in the treatment of diabetes, due to its variety of active compounds, such as flavonoids and terpenoids (Mogole et al., 2020). Wang (2021) constructed a draft genome of E. japonica to discover medicinal bioactive compounds using HiSeq 4,000 sequencing platform, PacBio long-read sequencing technology, and Hi-C data and obtained 45,492 protein-coding genes. According to gene family expansion and contraction analysis, 483 gene families in E. japonica expanded significantly and were mainly enriched for metabolic pathways in combination with KEGG analysis. Metabolite profiles showed that phenolic acids, flavonoids, and terpenoids were detected abundantly in the E. japonica. Based on genomic information, 71 flavonoid biosynthesis genes were annotated, of which 3 genes encoding key enzyme were identified in the quercetin biosynthesis pathway. In addition, 286 predicted protein-coding genes in phenylpropanoid biosynthesis were identified, only five genes underwent in the expansion family. According to KEGG analysis, 92, 32, 56, and 37 candidate genes were identified involved in terpenoid backbones, monoterpenoids, diterpenoids, and sesquiterpenoid-triterpenoids biosynthesis pathways, respectively. The study provides a valuable introduction for further molecular pharmacological studies of E. japonica. Thousands of years ago, Pueraria thomsonii was used to treat diabetes in the East. Puerarin as the bioactive isoflavones is mainly accumulated in the root of P. thomsoni and has antioxidant and anti-inflammatory properties (Chen et al., 2018; Yang et al., 2019; Luo et al., 2021). He et al. (2019) performed PacBio and Illumina sequencing technology to sequence the P. thomsoni transcriptome and acquired 44,339 transcripts. They discovered 9,225 differentially expressed transcripts (DETs). Among them, 32 genes might be involved in isoflavone production, of which the expression profile of eight genes were confirmed by qRT-PCR which consistent with RNA-Seq data. Glycyrrhiza uralensis, an important medicinal plant of the genus Glycyrrhiza, has been used as TCM. Flavonoids and Glycyrrhizin originated from liquorice possess antioxidative, antidiabetic, and anti-inflammatory effects (Lee et al., 2010; Feng et al., 2013; Lin et al., 2022). Mochida et al. (2017) reported a draft genome of Glycyrrhiza uralensis, based on Illumina short reads and PacBio long reads. The assembled genome is 379 Mb with scaffold N50 of 109 kb, encoding 34,445 predicted genes. On the basis of genomic information, CYP93C, HI4OMT, and 7-IOMT some of which are involved in isoflavonoid biosynthesis were observed and generated a cluster. Based on homolog searching and functional annotation, P450 and UDP-dependent glycosyltransferase (UGT) families involved in triterpenoid saponin biosynthesis were predicted and the expression of bAS, CYP88D6 and CYP72A154 were consistent with the glycyrrhizin yield of the G. uralensis samples. In addition, RNA-seq data revealed that Glyur002597s00038051 and Glyur002597s00038050 which closely homologous to CYP72A9 and UGT73B1 in Arabidopsis thaliana were high correlation of expression patterns. Hence, P450 and UGT genes might be involved in triterpene saponin biosynthesis in G. uralensis. These findings help researchers use genomic resources combined with biosynthetic approaches to create a library of rare natural or novel bioactive compounds to facilitate drug discovery. Sophora flavescens are important traditional medicinal plants with pharmacological properties effective in the treatment of inflammatory disorders including diabetes complications (Guo et al., 2021b). Alkaloids and flavonoids are the major bioactive compounds in root tissues. Wei et al. (2021) performed transcriptome analysis of the periderm, phloem, and xylem tissues of S. flavescens and obtained 58,327 unigenes. High-performance liquid chromatography (HPLC) detected metabolite contents in the root tissues and the results showed that alkaloids contents were highest in the phloem, while flavonoids contents were highest in the xylem. Fifty-two and one hundred thirty-seven CYP transcripts involved in alkaloid biosynthesis were identified and expressed highest in the xylem. Additionally, 37 transcripts were found in flavonoid biosynthesis and expressed highest in the xylem. Correlation analysis found LYSA, AO, PMT transcripts were markedly and positively correlated with alkaloids contents and 4CL, 2’OH, CHI5, CHRI transcripts were markedly and positively correlated with flavonoids contents. These results provide a basis for the molecular breeding of S. flavescens.
Curcumin, the bioactive compound of Curcuma longa, may prevent or reverse Alzheimer’s disease due to its anti-inflammatory and antioxidant activity (Kim et al., 2022). Chakraborty et al. (2021) assembled the draft genome sequence of C. longa using Oxford Nanopore long reads and obtained 50,401 coding gene sequences. Among 10 enzymes involved in the curcuminoid biosynthesis pathway, gene family evolution analysis revealed that two gene families (HCT and OMT genes) appear to be undergoing contraction, while a significant number of gene families (8) appear to be expanding in C. longa. The result suggests that genes related to the curcumin biosynthesis pathway have evolved, providing a genetic basis for its pharmacological properties. Corydalis yanhusuo have been used to relieve neuropathic pain, due to its bioactive compounds-tetrahydropalmatine, a member of BIAs. Xu D. et al. (2021) performed SMRT sequencing from the PacBio platform to sequence the cDNA library from leaves and tubers of C. yanhusuo. Based on the tblastn results, 101 unigenes involved in BIA biosynthetic pathway were founded, of which 36 unigenes were identified as DEGs via expression analysis, owing to the majority of them expressed at a higher degree in tubers which consistent with the metabolome data. In addition, phylogenetic analysis showed that 10 OMT unigenes involved in the final step for tetrahydropalmatine synthesis were identified. This study provides the basis for the subsequent molecular cloning and activity validation of the enzyme. Diosgenin is an anti-inflammatory and antioxidant compound found in the rhizome of Dioscorea zingiberensis. Cheng J. et al. (2021) used Illumina HiSeq and PacBio SMART technologies to sequence the D. zingiberensis genome and annotated 26,022 protein-coding genes. Based on previous study and genomic information, they found DzinCYP90G6 and DzinCYP94D144, two P450 genes, were related to diosgenin biosynthesis using blastp queries. Finally, metabolic engineering verified that coexpression of DzinCYP90G6 and DzinCYP94D144 in yeast was able to produce diosgenin. This study helps to decode the evolutionary trajectory of the biosynthetic pathway of diosgenin, but also provides insights into the enhancement of diosgenin production through biochemical synthesis. Chlorogenic acid (CGA) accumulates in the leaves and bark of Eucommia ulmoides, and it can reduce the concentration of glucose in the blood after the meal and lower blood pressure. Li et al. (2020) used PacBio Sequel platform, Illumina NovaSeq platform, and Hi-C data to assemble a high-quality E. ulmoides genome. Twenty-six thousand one protein-coding genes were predicted. Based on homologous gene comparison in the E. ulmoides genome, 23 candidate genes encoding six key enzymes involved in the CGA biosynthesis pathway were identified, including PAL, 4CL, C4H, C3′H, HCT, and HQT genes. In addition, gene expression profile analysis showed that the expression levels of these genes were higher than other genes. This work will accelerate the understanding of the molecular mechanisms of biosynthesis of other valuable secondary metabolites, such as rutin and quercetin. Dihydroquercetin (DHQ) is a pharmacologically active, which can be converted to dihydromyricetin (DHM). Yu Z. et al. (2021) used HPLC to detect the DHM content in Ampelopsis grossedentata from different geographical locations and divided into two groups: B group (low DHM) and D group (high DHM). Then, Illumina HiSeq 2000 sequencing platform was performed to sequence the transcriptome of A. grossedentata from B and D groups and annotated 57,016 unigenes in D group using seven public protein databases. The differentially expressed gene analysis revealed 926 DEGs in B vs. D, of which 446 up-regulated genes and 480 down-regulated genes. DEGs of 10 structural enzyme genes associated with flavonoids biosynthesis were identified, including PALs, CLs, CHSs, F3’H, F3’5’H, ANS. In addition, qRT-PCR verified the expression level of selected genes which was matched the transcriptome data, including CHSs, F3’H, and F3’5’H. This work will stimulate further genetic research on A. grossedentata and may ultimately lead to genetic improvements in the plant’s DHQ content.
Catharanthus roseus accumulates vinblastine and vincristine, which have long been known to be an anti-cancer drug to cure glioma disease (Skubnik et al., 2021). Verma et al. (2014) performed Illumina platform to sequence the C. roseus transcriptome from leaf, flower, and root and obtained 59,220 unique transcripts. Next, using the CathaCyc database analysis, 30 well-known genes involved in TIA biosynthetic pathways were identified and they are conserved in the sequenced reference genome of tomato, potato, and Arabidopsis at the protein level. Based on the RNA-seq data, most of TIA biosynthesis genes were up-regulated in leaf and root tissues, while Cr_TC35206 and Cr_TC35622 were highly down-regulated in roots, Cr_TC04217 were highly down-regulated in leaves. Finally, they performed qRT-PCR to validate 10 TIA biosynthetic pathway genes and their expression pattern consistent with RNA-seq data (see Figure 2). This research adds to our knowledge of the regulatory systems that control alkaloid production. Andrographis paniculata produces a large number of diterpenoid lactones with antitumor and immunomodulatory effects which are usually used to treat esophageal cancer, including andrographolide and neoandrographolide (Li L. et al., 2017, 2019; Sun et al., 2019; Yue et al., 2019). Sun et al. (2019) assembled A. paniculata genome sequence of 269 Mb at chromosome-scale using Illumina short reads, PacBio long reads, and Hi-C data and predicted 25,428 protein-coding genes. Based on the phylogenetic tree, 1,290 expanded and 5,383 contracted gene families in A. paniculate were discovered. Interestingly, the expand gene families were mainly enriched in the secondary metabolism pathway, such as TPS and CYP gene families, which may have contributed to the synthesis of andrographolide and neoandrographolide. Previous reports suggested that CYPs and 2OGDs may be involved in turing the diterpene backbone into diterpenes. Therefore, 278 CYP genes and 112 putative 2OGDs in the A. paniculata genome. Moreover, diterpene lactones need to be glycosylated to become neandrographolide, and 120 putative UGT genes were identified. Transcriptome data revealed that 18 CYP71and 6 CYP76 family members, 17 2OGDs, and 29 ApUGTs were significantly elevated after MeJA treatment. Finally, UGT73AU1 was confirmed that it converted the andrograpanin to neandrographolide via enzymatic assays. The study provides further understanding of the production of bioactive diterpene lactone components. The antitumor and anticancer medicinal properties of Lantana camara are attributed to its multiple bioactive substances, such as triterpenod which can be used to treat papilloma (Sharma et al., 2007). Shah et al. (2020) performed Illumina sequencing platform to analyze L. camara transcriptome in leaf and root and found 72,877 and 513,985 unigenes from leaf and root tissues, respectively. Moreover, 229 and 943 genes involved in the phenylpropanoid biosynthesis in leaf and root tissues, respectively were identified by pathway analysis. Twenty thousand forty-four DEGs were identified. Among them, 11,496 genes were up-regulated and 8,548 genes were down-regulated. The transcriptome analysis provides the basis for further molecular studies of L. camara. Salvia officinalis is used in medication for treating liver cancer, leukemia, colon cancer through inhibiting cell proliferation and apoptosis (Jantová et al., 2014; Pedro et al., 2016; Jiang et al., 2017). Ali et al. (2017) used Illumina HiSeq 2000 to sequence the transcriptome of S. officinalis leaves and assembled 48,671 unigenes. Among them, 65 unigenes involved in terpene synthase were identified, according to the TPS sequence in the reference database. Furthermore, 11 DEGs were selected to be validated by qRT-PCR. The results showed that eight candidate genes were most highly expressed in young leaves, except for SoGGPS, SoLINS, and SoHUMS genes were most highly expressed in stems. qRT-PCR data are well in agreement with GC–MS analysis data, major terpene groups present in young leaves. In addition, five terpene synthase genes (SoNEOD, SoCINS, SoSABS, SoLINS, and SoTPS6) were selected to overexpress in tobacco, respectively. The results indicate that more terpenoids were produced in transgenic tobacco. 2-methoxy-1,4-naphthoquinone (MNQ) is an active component of Impatiens balsamina with anticancer pharmacological properties. Shikimate and 1,4-dihydroxy-2-naphthoate (DHNA) pathways are responsible for the synthesis of MNQ, which lawsone is first synthesized by oxidative decarboxylation of DHNA and then possibly catalyzed by S-adenosylmethionine-dependent O-methyltransferase (SAM-dependent O-MT), NADH-quinone oxidoreductase, and UDP glycosyltransferases. Foong et al. (2020) reported the transcriptome sequencing of I. balsamina through Illumina HiSeq4000 paired-end sequencing technology and obtained 50,786 unigenes, of which 27, 104, 82, and 122 unigenes related to DHNA pathway, SAM-dependent O-MT activity, NADH-quinone oxidoreductase, and UDP glycosyltransferases, respectively. Among them, five unigenes related to DHNA pathway were highest expressed in early-stage capsule, gene expression of six unigenes were substantially and positively connected with MNQ content, 3 NADH-quinone oxidoreductase and 5 UDP glycosyltransferases were connected with lawsone content significantly. In addition, HPLC analysis indicated that lawsone was highest expressed in mature leaves, whereas MNQ was expressed only in capsules pericarps. Finally, qRT-PCR verified 20 candidate genes were consistent with transcriptome data. Dendrobium officinale is a widely used medicinal plant that creates a variety of bioactive compounds which resist cancer by inhibiting cell proliferation (Luo et al., 2019; Tao et al., 2021). Niu et al. (2021) reported its 1.23 Gb genome, encoding 27,631 predicted genes using PacBio long-reads, Illumina short-reads, and Hi-C data. Additionally, gene family evolution analysis found that 820 gene families expanded in D. officinale, while 975 gene families contracted. Interestingly, the expanded gene families were mainly enriched in polysaccharides, alkaloids and flavonoids biosynthesis according to KEGG enrichment analysis. Based on homolog searching and functional annotation, 268, 98, and 52 genes related to three main bioactive ingredients biosynthesis were identified, including polysaccharides, alkaloids, and flavonoids biosynthesis, respectively. Differential expression profiling revealed 1,677 DEGs, of which 51 DEGs related to polysaccharides, alkaloids, and flavonoids biosynthesis were identified. Additionally, 218 CYP450 genes were found in the D. officinale genome according to homologous search from A. thaliana, of which 29 DEGs were identified using comparative transcriptome analysis and most of them are CYP71 family members which were up-regulated. The results suggested that CYP71 groups make a great contribution in regulating the synthesis pathway of active ingredients. Therefore, the high-quality reference genome reported in this study can contribute to the functional genomics study and molecular breeding of D. officinale. Chen et al. (2012) reported a reference genome assembly of Ganoderma lucidum using Illumina next-generation sequencing and predicted 16,113 genes. Terpenoids and polysaccharides are the main active components of G. lucidum. One hundred ninety-seven CYP functional genes were identified, of which 78 CYP genes expression levels were consistent with terpenoids content in G. lucidum. According to previous literatures, 15 CYP512 and one CYP5144 genes were discovered in the G. lucidum genome, which may be related to triterpenoid biosynthesis. In addition, two polysaccharide genes, two LZ-8 genes, 12 TPS genes were identified. This study will pave the way for the role of G. lucidum in pharmacological and industrial applications (see Figure 3). Zingiber officinale, a gingerol-producing medicinal plant, possesses anti-cancer properties and treats breast cancer by inhibiting apoptosis (Huang P. et al., 2021). Li H. et al. (2021) reported chromosome-level reference genome assembly of Z. officinale using PacBio long reads, Illumina short reads, and Hi-C reads. Based on the assembly Z. officinale genome, they investigated the gene family expansion and contraction and revealed that 1,098 gene families underwent expansion, while 20 gene families underwent contraction. In addition, two expanded gene families (PKS and AOR) were mainly enriched in the biosynthesis of secondary metabolites. UHPLC–MS/MS detected rhizomes of Z. officinale at different stages and revealed that the concentration of active compounds were the lowest in R1, whereas were the highest in R5, such as gingerol analogs-6-gingerol and tetrahydrocurcumin. According to the metabolomic data and previous study, 12 gene families involved in the gingerol biosynthesis pathway were identified, and the expression level of 10 gene families were compatible with the concentration of gingerol. Additionally, C3OMT2, C3OMT3, and C3OMT13 formed a unique clade in the Z. officinale genome which suggested that C3OMTs were likely related to feruloyl-CoA biosynthesis.
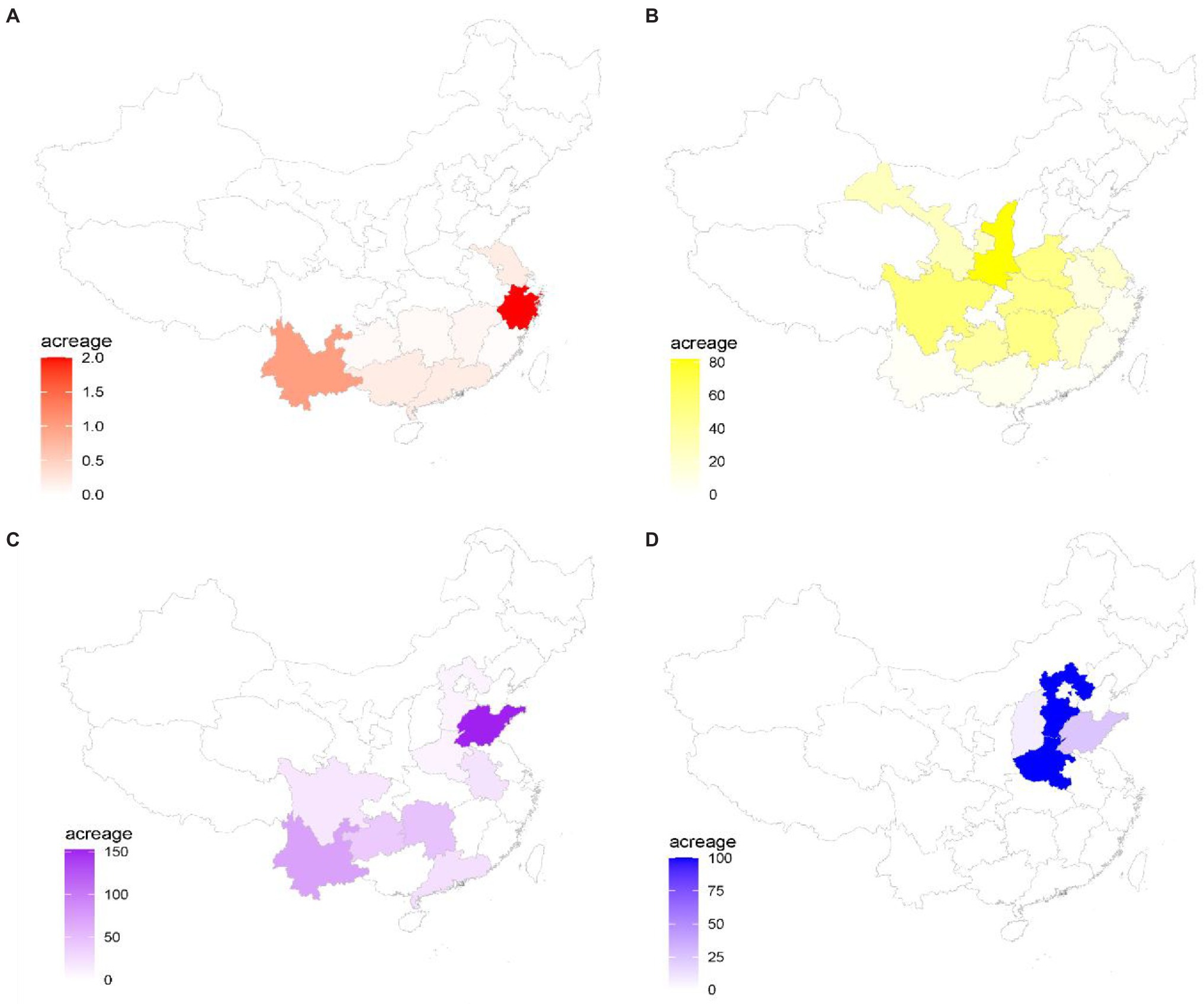
Figure 3. Industry distribution and covered areas of four representative species in China. (A) Dendrobium officinale; (B) Eucommia ulmoides; (C) Zingiber officinale; and (D) Dioscorea zingiberensis. A color scale bar is shown the acreage size of species in each province of China.
The tubers of Ophiopogon japonicus are accumulated in many active substances which contribute significantly to the treatment of cardiovascular diseases through their antioxidant, anti-inflammatory, and cardioprotective pharmacological properties, such as flavonoids, saponins, and polysaccharides (He et al., 2016; Wu et al., 2019; Fan S. et al., 2020). Liu H. et al. (2017) employed the Illumina platform to sequence the transcriptome of different ages tubers (Y1, Y2, Y3) from O. japonicus and generated 96,738 unigenes with de novo assembly. According to searching against the five databases, 77,409 unigenes were annotated among the 96,738 unigenes. Based on the result of gene annotation, 245, 135, and 236 unigenes encoding key enzymes in flavonoid, saponin, polysaccharide biosynthesis were identified, respectively. Most genes related to polysaccharide biosynthesis have the highest expression in Y2, while genes involved in flavonoid and saponin biosynthesis have the highest expression in Y1. qRT-PCR verified the expression level of 17 unigenes were consistent with RNA-Seq data, which were selected from flavonoid, saponin, polysaccharide biosynthesis, respectively. This study can accelerate the understanding of the physiological process of active ingredient synthesis at the molecular level and promote the development of natural drugs. Chrysanthemums have significant protective functions for cardiovascular (Song et al., 2018). Song et al. (2018) performed Oxford Nanopore long reads and Illumina short reads to assemble the genome of Chrysanthemum nankingense. In C. nankingense, gene evolution analysis revealed that 1965 gene families underwent expansion, while a significant number of gene families (1777) underwent contraction. Surprisingly, the expanded gene families were mainly enriched in terpene synthase activity. Among them, 219 TS genes and 708 CYP genes were identified, including seven SQSs, 158 TPSs, and 54 TCCs. Due to, the genes that make up the terpene synthesis pathway are organized into gene clusters, TPS-a/CYP99 and TPS-g/CYP79/CYP76, a new combination, were found in the C. nankingense genome. Further analysis revealed CHR00048430 and CHR00048432 genes from the TPS-a/CYP99 cluster were mainly expressed in roots, whereas CHR00011805 and CHR0001181 from TPS-g/CYP79/CYP76 cluster were mainly expressed in flowers. Additionally, two key flavonoid biosynthesis genes (CHS and CHI) from expansion families were significantly highly expressed in flowers. The study gives genomic data to help researchers better understand the evolutionary history of chrysanthemums. Rehmannia glutinosa possesses a significant effect on treating cardiovascular disease accumulation of iridoids in root tissues is thought to be responsible for its health advantages (Ma L. et al., 2021). Ma L. et al. (2021) reported a chromosome level of 2.49 Gb genome sequence of R. glutinosa and were assembled into 14 chromosomes using Illumina NovaSeq reads, Oxford Nanopore technology, and Hi-C data. Most iridoids exist mainly as glycosides, due to glycosylation being the final step in terpene biosynthesis. In addition, gene family evolution analysis revealed that 6,237 gene families in R. glutinosa underwent expansion, whereas 848 gene families underwent contraction. Eighty-seven TPS and 333 UGT genes were discovered in the expanded gene families, with the majority of them being significantly expressed in roots. The genomic resources provided by this study are essential for the molecular breeding of medicinal plants. Previous research reported that flavonoids derived from Ziziphora bungeana possess the ability to treat cardiovascular disease (Li et al., 2018). He et al. (2020) performed Illumina technology to sequence the transcriptome of Z. bungeana from different tissues and assembled 397,182 unigenes. Firstly, LC/MS detected 12 flavonoid components and linarin was one of the main bioactive compounds which highly accumulated in the inflorescence. Secondly, based on the transcriptome data, 18 candidate genes were identified from assembled unigenes that encode four key enzymes, including PAL, C4H, 4CL, and FNSII. Finally, qRT-PCR data revealed that ZbPAL3, Zb4CL3, ZbCHS1, ZbFNSII, and ZbANS had the highest expression levels in inflorescence, which suggested that they were likely related to the lignin biosynthesis.
Research on medicinal plants has focused on the genetic mechanism of active ingredients synthesis and the subsequent pharmacological effects on diseases treatment. Thus, when researchers collect samples, they need to pay attention to the geographical distribution, different tissues and different developmental stages of the herb, due to these aspects play an important role in the efficacy of the pharmacological components (Duan et al., 2014; Zhang X. et al., 2017). The characteristics summary of the CHM is shown in Table 2. NGS had been widely applied in transcriptome sequencing of medicinal plants to get the expression profile and mined the genes related to bioactive compounds before the advent of TGS. However, due to the short read length, it was not discovering more candidate genes involved in the synthesis of active ingredients. With the aid of matured sequencing technologies, TGS is used to sequence the full-length transcriptome of medicinal plants, making a name for itself in herbal medicine research. Salvia miltiorrhiza, a well-known herbal medicine, is commonly used to treat diabetes. It has various biological functions, such as anti-oxidative stress and anti-inflammatory (Jia et al., 2019; Guo Y. et al., 2021; Yin et al., 2021). For instance, Xu et al. (2015) used SMRT via PacBio long reads to sequence RNA mixes from three root tissues of S. miltiorrhiza and obtained high-quality full-length transcriptome sequences. Furthermore, using Illumina short reads to quantify gene expression in three different tissues and obtained DEGs which elucidated the molecular mechanism of salvinone accumulation in the periderm. What’s more, alternative splicing analysis on the full-length transcripts revealed that many terpenoids and isoprene biosynthesis genes underwent alternative splicing. Based on the above findings, the results suggest that these genes may regulate the diterpene metabolic pathway. Nevertheless, comprehensive analysis of multi-omics data is required for mining pharmacodynamic genes more accurately, such as genomics, transcriptomics, metabolomics, and so on (Ma et al., 2020). Ma Y. et al. (2021) revealed the S. miltiorrhiza genome with 622 Mb using Illumina Hiseq2000 and PacBio RS platform and obtained 33,760 protein-coding genes. CYP450 genes were discovered to be present as a gene cluster after a thorough study of genomic data. Based on this, a CYP71D subfamily, which is significantly expanded in the S. miltiorrhiza genome, was identified using gene expansion and contraction analysis, and four candidate genes were targeted by co-expression analysis for enzyme activity and RNAi studies. The results showed that three of these genes play important roles in the tanshinone biosynthetic pathway, two of which can catalyze the generation of tanshinone characteristic furan rings and one is associated with the hydroxylation process at the C20 position of tanshinones. Based on previous literature, the researchers used the genome-assembly and the downstream bioinformatics analysis to uncover key enzymes genes involved in the biosynthesis pathway of active ingredient. Furthermore, the validation methods for functional genes are shown in Table 1.
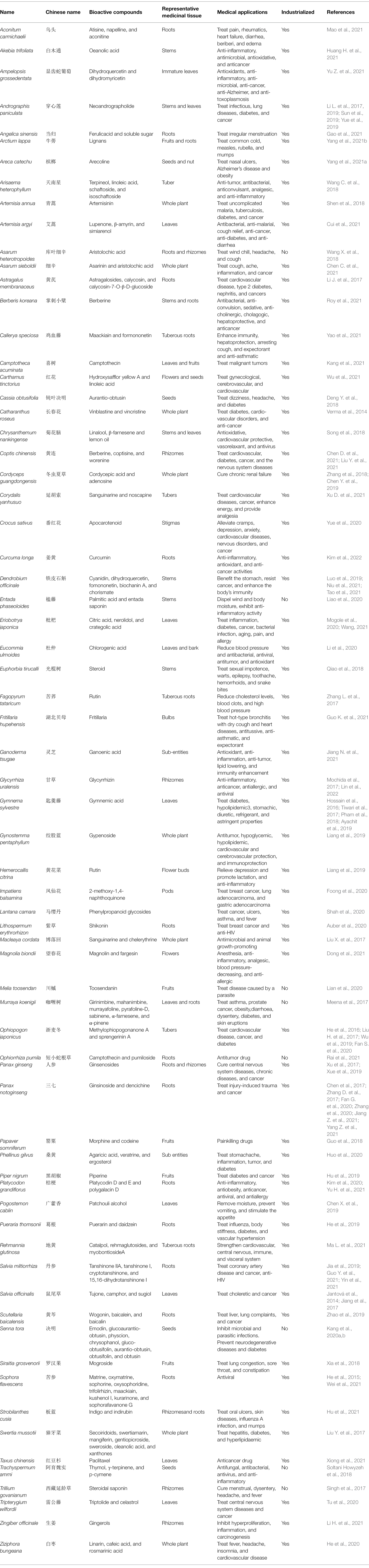
Table 2. Characteristics summary of the CHMs.
Regarding future practical applications, on one hand, combining 2nd and 3rd generation sequencing technologies makes full use of the strengths of each, such as long read length, high throughput, and acceptable sequencing costs. On the other hand, single-cell sequencing technology has great application prospects in the mining of active ingredients of Chinese traditional medicinal herbs. Traditional RNA-Seq technologies sequence RNA extracted from a variety of tissues and cells, ignoring intercellular differences. Single-cell RNA sequencing isolates the target cells from the sample and then sequences them, allowing for the unique characteristics of individual cells. Additionally, scRNA-seq has yielded rich results in the fields of tumors, microorganisms, and so on (Li S. et al., 2021). Xu X. et al. (2021) used scRNA-seq to mine trait genes during maize development. It is more accurate and efficient than traditional RNA-seq and uncovers more information.
This study was designed by MT and GL. Data analysis was performed by JLL. The figures were organized by YL and HTZ. The manuscript was written by XYL and XG. The manuscript was revised by SQ. All authors made a direct and intellectual contribution to this topic and approved the article for publication.
This work was financially supported by grants from the National Natural Science Foundation of China (32002235).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank all the individuals who have helped us in this study. We acknowledge the valuable work of the many investigators whose published articles we were unable to cite owing to space limitations.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.900035/full#supplementary-material
Ali, M., Li, P., She, G., Chen, D., Wan, X., and Zhao, J. (2017). Transcriptome and metabolite analyses reveal the complex metabolic genes involved in volatile terpenoid biosynthesis in garden sage (Salvia officinalis). Sci. Rep. 7:16074. doi: 10.1038/s41598-017-15478-3
Auber, R. P., Suttiyut, T., McCoy, R. M., Ghaste, M., Crook, J. W., Pendleton, A. L., et al. (2020). Hybrid de novo genome assembly of red gromwell (Lithospermum erythrorhizon) reveals evolutionary insight into shikonin biosynthesis. Hortic. Res. 7:82. doi: 10.1038/s41438-020-0301-9
Ayachit, G., Shaikh, I., Sharma, P., Jani, B., Shukla, L., Sharma, P., et al. (2019). De novo transcriptome of Gymnema sylvestre identified putative lncRNA and genes regulating terpenoid biosynthesis pathway. Sci. Rep. 9:14876. doi: 10.1038/s41598-019-51355-x
Barbosa, C., Nogueira, S., Gadanho, M., and Chaves, S. (2019). Study on commercial spice and herb products using next-generation sequencing (NGS). J. AOAC Int. 102, 369–375. doi: 10.5740/jaoacint.18-0407
Chakraborty, A., Mahajan, S., Jaiswal, S. K., and Sharma, V. K. (2021). Genome sequencing of turmeric provides evolutionary insights into its medicinal properties. Commun. Biol. 4:1193. doi: 10.1038/s42003-021-02720-y
Chen, W., Kui, L., Zhang, G., Zhu, S., Zhang, J., Wang, X., et al. (2017). Whole-genome sequencing and analysis of the Chinese herbal plant Panax notoginseng. Mol. Plant 10, 899–902. doi: 10.1016/j.molp.2017.02.010
Chen, X., Li, J., Wang, X., Zhong, L., Tang, Y., Zhou, X., et al. (2019). Full-length transcriptome sequencing and methyl jasmonate-induced expression profile analysis of genes related to patchoulol biosynthesis and regulation in Pogostemon cablin. BMC Plant Biol. 19:266. doi: 10.1186/s12870-019-1884-x
Chen, D. X., Pan, Y., Wang, Y., Cui, Y. Z., Zhang, Y. J., Mo, R. Y., et al. (2021). The chromosome-level reference genome of Coptis chinensis provides insights into genomic evolution and berberine biosynthesis. Hortic. Res. 8:121. doi: 10.1038/s41438-021-00559-2
Chen, C., Shi, X., Zhou, T., Li, W., Li, S., and Bai, G. (2021). Full-length transcriptome analysis and identification of genes involved in asarinin and aristolochic acid biosynthesis in medicinal plant Asarum sieboldii. Genome 64, 639–653. doi: 10.1139/gen-2020-0095
Chen, F., Tholl, D., Bohlmann, J., and Pichersky, E. (2011). The family of terpene synthases in plants: a mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 66, 212–229. doi: 10.1111/j.1365-313X.2011.04520.x
Chen, Y., Wu, Y., Liu, L., Feng, J., Zhang, T., Qin, S., et al. (2019). Study of the whole genome, methylome and transcriptome of Cordyceps militaris. Sci. Rep. 9:898. doi: 10.1038/s41598-018-38021-4
Chen, S., Xu, J., Liu, C., Zhu, Y., Nelson, D. R., Zhou, S., et al. (2012). Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat. Commun. 3:913. doi: 10.1038/ncomms1923
Chen, X., Yu, J., and Shi, J. (2018). Management of diabetes mellitus with puerarin, a natural isoflavone from Pueraria lobata. Am. J. Chin. Med. 46, 1771–1789. doi: 10.1142/S0192415X18500891
Cheng, J., Chen, J., Liu, X., Li, X., Zhang, W., Dai, Z., et al. (2021). The origin and evolution of the diosgenin biosynthetic pathway in yam. Plant Commun. 2:100079. doi: 10.1016/j.xplc.2020.100079
Cheng, Q. Q., Ouyang, Y., Tang, Z. Y., Lao, C. C., Zhang, Y. Y., Cheng, C. S., et al. (2021). Review on the development and applications of medicinal plant genomes. Front. Plant Sci. 12:791219. doi: 10.3389/fpls.2021.791219
Ciumarnean, L., Milaciu, M. V., Runcan, O., Vesa, S. C., Rachisan, A. L., Negrean, V., et al. (2020). The effects of flavonoids in cardiovascular diseases. Molecules 25:4320. doi: 10.3390/molecules25184320
Cui, Y., Gao, X., Wang, J., Shang, Z., Zhang, Z., Zhou, Z., et al. (2021). Full-length transcriptome analysis reveals candidate genes involved in terpenoid biosynthesis in Artemisia argyi. Front. Genet. 12:659962. doi: 10.3389/fgene.2021.659962
Cui, F., Ye, X., Li, X., Yang, Y., Hu, Z., Overmyer, K., et al. (2022). Chromosome-level genome assembly of the diploid blueberry Vaccinium darrowii provides insights into its subtropical adaptation and cuticle synthesis. Plant Commun. :100307. doi: 10.1016/j.xplc.2022.100307
Deng, X., Zhao, L., Fang, T., Xiong, Y., Ogutu, C., Yang, D., et al. (2018). Investigation of benzylisoquinoline alkaloid biosynthetic pathway and its transcriptional regulation in lotus. Hortic. Res. 5:29. doi: 10.1038/s41438-018-0035-0
Deng, Y., Zheng, H., Yan, Z., Liao, D., Li, C., Zhou, J., et al. (2018). Full-length transcriptome survey and expression analysis of Cassia obtusifolia to discover putative genes related to aurantio-obtusin biosynthesis, seed formation and development, and stress response. Int. J. Mol. Sci. 19:2476. doi: 10.3390/ijms19092476
Dong, S., Liu, M., Liu, Y., Chen, F., Yang, T., Chen, L., et al. (2021). The genome of Magnolia biondii Pamp. Provides insights into the evolution of magnoliales and biosynthesis of terpenoids. Hortic. Res. 8:38. doi: 10.1038/s41438-021-00471-9
Duan, X., Zhang, D., Nie, L., and Zang, H. (2014). Rapid discrimination of geographical origin and evaluation of antioxidant activity of Salvia miltiorrhiza var. alba by Fourier transform near infrared spectroscopy. Spectrochim. Acta A Mol. Biomol. Spectrosc. 122, 751–757. doi: 10.1016/j.saa.2013.12.003
Erb, M., and Kliebenstein, D. J. (2020). Plant secondary metabolites as defenses, regulators, and primary metabolites: the blurred functional Trichotomy. Plant Physiol. 184, 39–52. doi: 10.1104/pp.20.00433
Fan, G., Liu, X., Sun, S., Shi, C., Du, X., Han, K., et al. (2020). The chromosome level genome and genome-wide association study for the agronomic traits of Panax notoginseng. iScience 23:101538. doi: 10.1016/j.isci.2020.101538
Fan, S., Zhang, J., Xiao, Q., Liu, P., Zhang, Y., Yao, E., et al. (2020). Cardioprotective effect of the polysaccharide from Ophiopogon japonicus on isoproterenol-induced myocardial ischemia in rats. Int. J. Biol. Macromol. 147, 233–240. doi: 10.1016/j.ijbiomac.2020.01.068
Feng, X., Cao, S., Qiu, F., and Zhang, B. (2020). Traditional application and modern pharmacological research of Artemisia annua L. Pharmacol. Ther. 216:107650. doi: 10.1016/j.pharmthera.2020.107650
Feng, L., Zhu, M. M., Zhang, M. H., Wang, R. S., Tan, X. B., Song, J., et al. (2013). Protection of glycyrrhizic acid against AGEs-induced endothelial dysfunction through inhibiting RAGE/NF-κB pathway activation in human umbilical vein endothelial cells. J. Ethnopharmacol. 148, 27–36. doi: 10.1016/j.jep.2013.03.035
Flower, A., Liu, J. P., Lewith, G., Little, P., and Li, Q. (2012). Chinese herbal medicine for endometriosis. Cochrane Database Syst. Rev. :CD006568. doi: 10.1002/14651858.CD006568.pub3
Foong, L. C., Chai, J. Y., Ho, A. S. H., Yeo, B. P. H., Lim, Y. M., and Tam, S. M. (2020). Comparative transcriptome analysis to identify candidate genes involved in 2-methoxy-1,4-naphthoquinone (MNQ) biosynthesis in Impatiens balsamina L. Sci. Rep. 10:16123. doi: 10.1038/s41598-020-72997-2
Gao, X., Guo, F., Chen, Y., Bai, G., Liu, Y., Jin, J., et al. (2021). Full-length transcriptome analysis provides new insights into the early bolting occurrence in medicinal Angelica sinensis. Sci. Rep. 11:13000. doi: 10.1038/s41598-021-92494-4
Guo, K., Chen, J., Niu, Y., and Lin, X. (2021). Full-length transcriptome sequencing provides insights into flavonoid biosynthesis in Fritillaria hupehensis. Life 11:287. doi: 10.3390/life11040287
Guo, J., Huang, Z., Sun, J., Cui, X., and Liu, Y. (2021a). Research Progress and future development trends in medicinal plant Transcriptomics. Front. Plant Sci. 12:691838. doi: 10.3389/fpls.2021.691838
Guo, J., Li, J., Wei, H., and Liang, Z. (2021b). Maackiain protects the kidneys of type 2 diabetic rats via modulating the Nrf2/HO-1 and TLR4/NF-κB/Caspase-3 pathways. Drug Des. Devel. Ther. 15, 4339–4358. doi: 10.2147/DDDT.S326975
Guo, Y., Sun, J., Zhang, R., Yang, P., Zhang, S., and Wu, Z. (2021). Salvia miltiorrhiza improves type 2 diabetes: a protocol for systematic review and meta-analysis. Medicine 100:e23843. doi: 10.1097/MD.0000000000023843
Guo, L., Winzer, T., Yang, X., Li, Y., Ning, Z., He, Z., et al. (2018). The opium poppy genome and morphinan production. Science 362, 343–347. doi: 10.1126/science.aat4096
He, X., Fang, J., Huang, L., Wang, J., and Huang, X. (2015). Sophora flavescens Ait.: traditional usage, phytochemistry and pharmacology of an important traditional Chinese medicine. J. Ethnopharmacol. 172, 10–29. doi: 10.1016/j.jep.2015.06.010
He, F., Xu, B. L., Chen, C., Jia, H. J., Wu, J. X., Wang, X. C., et al. (2016). Methylophiopogonanone A suppresses ischemia/reperfusion-induced myocardial apoptosis in mice via activating PI3K/Akt/eNOS signaling pathway. Acta Pharmacol. Sin. 37, 763–771. doi: 10.1038/aps.2016.14
He, J., Yang, W., Cheng, B., Ma, L., Tursunjiang, D., Ding, Z., et al. (2020). Integrated metabolomic and transcriptomic profiling reveals the tissue-specific flavonoid compositions and their biosynthesis pathways in Ziziphora bungeana. Chin. Med. 15:73. doi: 10.1186/s13020-020-00354-6
He, M., Yao, Y., Li, Y., Yang, M., Li, Y., Wu, B., et al. (2019). Comprehensive transcriptome analysis reveals genes potentially involved in isoflavone biosynthesis in Pueraria thomsonii benth. PLoS One 14:e0217593. doi: 10.1371/journal.pone.0217593
Hossain, M. U., Khan, M. A., Rakib-Uz-Zaman, S. M., Ali, M. T., Islam, M. S., Keya, C. A., et al. (2016). Treating diabetes mellitus: pharmacophore based designing of potential drugs from Gymnema sylvestre against insulin receptor protein. Biomed. Res. Int. 2016:3187647. doi: 10.1155/2016/3187647
Hu, Y., Ma, D., Ning, S., Ye, Q., Zhao, X., Ding, Q., et al. (2021). High-quality genome of the medicinal plant Strobilanthes cusia provides insights Into the biosynthesis of Indole alkaloids. Front. Plant Sci. 12:742420. doi: 10.3389/fpls.2021.742420
Hu, L., Xu, Z., Wang, M., Fan, R., Yuan, D., Wu, B., et al. (2019). The chromosome-scale reference genome of black pepper provides insight into piperine biosynthesis. Nat. Commun. 10:4702. doi: 10.1038/s41467-019-12607-6
Huang, H., Liang, J., Tan, Q., Ou, L., Li, X., Zhong, C., et al. (2021). Insights into triterpene synthesis and unsaturated fatty-acid accumulation provided by chromosomal-level genome analysis of Akebia trifoliata subsp. australis. Hortic. Res. 8:33. doi: 10.1038/s41438-020-00458-y
Huang, P., Zhou, P., Liang, Y., Wu, J., Wu, G., Xu, R., et al. (2021). Exploring the molecular targets and mechanisms of [10]-gingerol for treating triple-negative breast cancer using bioinformatics approaches, molecular docking, and in vivo experiments. Transl. Cancer Res. 10, 4680–4693. doi: 10.21037/tcr-21-1138
Huo, J., Zhong, S., Du, X., Cao, Y., Wang, W., Sun, Y., et al. (2020). Whole-genome sequence of Phellinus gilvus (mulberry Sanghuang) reveals its unique medicinal values. J. Adv. Res. 24, 325–335. doi: 10.1016/j.jare.2020.04.011
Hurgobin, B., Tamiru-Oli, M., Welling, M. T., Doblin, M. S., Bacic, A., Whelan, J., et al. (2021). Recent advances in Cannabis sativa genomics research. New Phytol. 230, 73–89. doi: 10.1111/nph.17140
Jantová, S., Hudec, R., Sekretár, S., Kučerák, J., and Melušová, M. (2014). Salvia officinalis L. extract and its new food antioxidant formulations induce apoptosis through mitochondrial/caspase pathway in leukemia L1210 cells. Interdiscip. Toxicol. 7, 146–153. doi: 10.2478/intox-2014-0020
Ji, X., Shi, S., Liu, B., Shan, M., Tang, D., Zhang, W., et al. (2019). Bioactive compounds from herbal medicines to manage dyslipidemia. Biomed. Pharmacother. 118:109338. doi: 10.1016/j.biopha.2019.109338
Jia, Q., Zhu, R., Tian, Y., Chen, B., Li, R., Li, L., et al. (2019). Salvia miltiorrhiza in diabetes: a review of its pharmacology, phytochemistry, and safety. Phytomedicine 58:152871. doi: 10.1016/j.phymed.2019.152871
Jiang, N., Hu, S., Peng, B., Li, Z., Yuan, X., Xiao, S., et al. (2021). Genome of Ganoderma species provides insights Into the evolution, conifers substrate utilization, and terpene synthesis for Ganoderma tsugae. Front. Microbiol. 12:724451. doi: 10.3389/fmicb.2021.724451
Jiang, Z., Tu, L., Yang, W., Zhang, Y., Hu, T., Ma, B., et al. (2021). The chromosome-level reference genome assembly for Panax notoginseng and insights into ginsenoside biosynthesis. Plant Commun. 2:100113. doi: 10.1016/j.xplc.2020.100113
Jiang, Y., Zhang, L., and Rupasinghe, H. P. (2017). Antiproliferative effects of extracts from Salvia officinalis L. and saliva miltiorrhiza Bunge on hepatocellular carcinoma cells. Biomed. Pharmacother. 85, 57–67. doi: 10.1016/j.biopha.2016.11.113
Kang, M., Fu, R., Zhang, P., Lou, S., Yang, X., Chen, Y., et al. (2021). A chromosome-level Camptotheca acuminata genome assembly provides insights into the evolutionary origin of camptothecin biosynthesis. Nat. Commun. 12:3531. doi: 10.1038/s41467-021-23872-9
Kang, S. H., Lee, W. H., Lee, C. M., Sim, J. S., Won, S. Y., Han, S. R., et al. (2020a). De novo transcriptome sequence of Senna tora provides insights into anthraquinone biosynthesis. PLoS One 15:e0225564. doi: 10.1371/journal.pone.0225564
Kang, S. H., Pandey, R. P., Lee, C. M., Sim, J. S., Jeong, J. T., Choi, B. S., et al. (2020b). Genome-enabled discovery of anthraquinone biosynthesis in Senna tora. Nat. Commun. 11:5875. doi: 10.1038/s41467-020-19681-1
Kim, J., Kang, S. H., Park, S. G., Yang, T. J., Lee, Y., Kim, O. T., et al. (2020). Whole-genome, transcriptome, and methylome analyses provide insights into the evolution of platycoside biosynthesis in Platycodon grandiflorus, a medicinal plant. Hortic. Res. 7:112. doi: 10.1038/s41438-020-0329-x
Kim, M. J., Park, S. Y., Kim, Y., Jeon, S., Cha, M. S., Kim, Y. J., et al. (2022). Beneficial effects of a combination of Curcuma longa L. and citrus junos against beta-amyloid peptide-induced neurodegeneration in mice. J. Med. Food 25, 12–23. doi: 10.1089/jmf.2021.K.0104
Korth, K. L., Stermer, B. A., Bhattacharyya, M. K., and Dixon, R. A. (1997). HMG-CoA reductase gene families that differentially accumulate transcripts in potato tubers are developmentally expressed in floral tissues. Plant Mol. Biol. 33, 545–551. doi: 10.1023/A:1005743011651
Lee, Y. S., Kim, S. H., Jung, S. H., Kim, J. K., Pan, C. H., and Lim, S. S. (2010). Aldose reductase inhibitory compounds from Glycyrrhiza uralensis. Biol. Pharm. Bull. 33, 917–921. doi: 10.1248/bpb.33.917
Li, J. H., Chen, Z. X., Zhang, X. G., Li, Y., Yang, W. T., Zheng, X. W., et al. (2016). Bioactive components of Chinese herbal medicine enhance endogenous neurogenesis in animal models of ischemic stroke: a systematic analysis. Medicine (Baltimore) 95:e4904. doi: 10.1097/MD.0000000000004904
Li, J., Harata-Lee, Y., Denton, M. D., Feng, Q., Rathjen, J. R., Qu, Z., et al. (2017). Long read reference genome-free reconstruction of a full-length transcriptome from astragalus membranaceus reveals transcript variants involved in bioactive compound biosynthesis. Cell Discov. 3:17031. doi: 10.1038/celldisc.2017.31
Li, X., Liang, Z., Du, J., Wang, Z., Mei, S., Li, Z., et al. (2019). Herbal decoctosome is a novel form of medicine. Sci. China Life Sci. 62, 333–348. doi: 10.1007/s11427-018-9508-0
Li, Q., Tursun, D., Shi, C., Heyrulla, M., Zhang, X., and Yang, W. (2018). Ziziphora clinopodioides flavonoids protect myocardial cell damage from myocardial ischemia-reperfusion injury. Evid. Based Complement. Alternat. Med. 2018:8495010. doi: 10.1155/2018/8495010
Li, Y., Wei, H., Yang, J., Du, K., Li, J., Zhang, Y., et al. (2020). High-quality de novo assembly of the Eucommia ulmoides haploid genome provides new insights into evolution and rubber biosynthesis. Hortic. Res. 7:183. doi: 10.1038/s41438-020-00406-w
Li, H. L., Wu, L., Dong, Z., Jiang, Y., Jiang, S., Xing, H., et al. (2021). Haplotype-resolved genome of diploid ginger (Zingiber officinale) and its unique gingerol biosynthetic pathway. Hortic. Res. 8:189. doi: 10.1038/s41438-021-00627-7
Li, S., Yan, H., and Lee, J. (2021). Identification of gene regulatory networks from single-cell expression data. Methods Mol. Biol. 2328, 153–170. doi: 10.1007/978-1-0716-1534-8_9
Li, T., Yu, X., Ren, Y., Kang, M., Yang, W., Feng, L., et al. (2022). The chromosome-level genome assembly of Gentiana dahurica (Gentianaceae) provides insights into gentiopicroside biosynthesis. DNA Res. 29. doi: 10.1093/dnares/dsac008
Li, L., Yue, G. G. L., Fung, K. P., Yu, J., Lau, C. B. S., and Chiu, P. W. Y. (2019). Combined use of Andrographis paniculata and chemotherapeutics for metastatic oesophageal cancer: a pre-clinical study. Hong Kong Med. J. 25, 43–46.
Li, L., Yue, G. G., Lee, J. K., Wong, E. C., Fung, K. P., Yu, J., et al. (2017). The adjuvant value of Andrographis paniculata in metastatic esophageal cancer treatment - from preclinical perspectives. Sci. Rep. 7:854. doi: 10.1038/s41598-017-00934-x
Lian, X., Zhang, X., Wang, F., Wang, X., Xue, Z., and Qi, X. (2020). Characterization of a 2,3-oxidosqualene cyclase in the toosendanin biosynthetic pathway of Melia toosendan. Physiol. Plant. 170, 528–536. doi: 10.1111/ppl.13189
Liang, T., Zou, L., Sun, S., Kuang, X., Wei, J., Wang, L., et al. (2019). Hybrid sequencing of the Gynostemma pentaphyllum transcriptome provides new insights into gypenoside biosynthesis. BMC Genomics 20:632. doi: 10.1186/s12864-019-6000-y
Liao, W., Mei, Z., Miao, L., Liu, P., and Gao, R. (2020). Comparative transcriptome analysis of root, stem, and leaf tissues of Entada phaseoloides reveals potential genes involved in triterpenoid saponin biosynthesis. BMC Genomics 21:639. doi: 10.1186/s12864-020-07056-1
Lichtenthaler, H. K. (1999). The 1-deoxy-D-xylulose-5-phosphate pathway of isoprenoid biosynthesis in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 50, 47–65. doi: 10.1146/annurev.arplant.50.1.47
Lin, H. C., Paul, C. R., Kuo, C. H., Chang, Y. H., Chen, W. S., Ho, T. J., et al. (2022). Glycyrrhiza uralensis root extract ameliorates high glucose-induced renal proximal tubular fibrosis by attenuating tubular epithelial-myofibroblast transdifferentiation by targeting TGF-β1/Smad/Stat3 pathway. J. Food Biochem. 46:e14041. doi: 10.1111/jfbc.14041
Liu, W., Feng, Y., Yu, S., Fan, Z., Li, X., Li, J., et al. (2021). The flavonoid biosynthesis network in plants. Int. J. Mol. Sci. 22:12824. doi: 10.3390/ijms222312824
Liu, H., Guo, S. S., Lu, L., Li, D., Liang, J., Huang, Z. H., et al. (2021). Essential oil from Artemisia annua aerial parts: composition and repellent activity against two storage pests. Nat. Prod. Res. 35, 822–825. doi: 10.1080/14786419.2019.1599887
Liu, X., Liu, Y., Huang, P., Ma, Y., Qing, Z., Tang, Q., et al. (2017). The genome of medicinal plant Macleaya cordata provides new insights into benzylisoquinoline alkaloids metabolism. Mol. Plant 10, 975–989. doi: 10.1016/j.molp.2017.05.007
Liu, Y., Wang, Y., Guo, F., Zhan, L., Mohr, T., Cheng, P., et al. (2017). Deep sequencing and transcriptome analyses to identify genes involved in secoiridoid biosynthesis in the tibetan medicinal plant Swertia mussotii. Sci. Rep. 7:43108. doi: 10.1038/srep43108
Liu, Y., Wang, B., Shu, S., Li, Z., Song, C., Liu, D., et al. (2021). Analysis of the Coptis chinensis genome reveals the diversification of protoberberine-type alkaloids. Nat. Commun. 12:3276. doi: 10.1038/s41467-021-23611-0
Liu, H., Wang, Y., Wang, T., Ying, X., Wu, R., and Chen, H. (2017). De novo assembly and annotation of the Zhe-Maidong (Ophiopogon japonicus (L.f.) Ker-Gawl) transcriptome in different growth stages. Sci. Rep. 7:3616. doi: 10.1038/s41598-017-03937-w
Liu, C., Yang, S., Wang, K., Bao, X., Liu, Y., Zhou, S., et al. (2019). Alkaloids from traditional Chinese medicine against hepatocellular carcinoma. Biomed. Pharmacother. 120:109543. doi: 10.1016/j.biopha.2019.109543
Lo, Y. T., and Shaw, P. C. (2019). Application of next-generation sequencing for the identification of herbal products. Biotechnol. Adv. 37:107450. doi: 10.1016/j.biotechadv.2019.107450
Lu, Z., Zhong, Y., Liu, W., Xiang, L., and Deng, Y. (2019). The efficacy and mechanism of Chinese herbal medicine on diabetic kidney disease. J. Diabetes Res. 2019:2697672. doi: 10.1155/2019/2697672
Luo, D., Dong, X., Huang, J., Huang, C., Fang, G., and Huang, Y. (2021). Pueraria lobata root polysaccharide alleviates glucose and lipid metabolic dysfunction in diabetic db/db mice. Pharm. Biol. 59, 382–390. doi: 10.1080/13880209.2021.1898648
Luo, Y., Ren, Z., Du, B., Xing, S., Huang, S., Li, Y., et al. (2019). Structure identification of ViceninII extracted from Dendrobium officinale and the reversal of TGF-β1-induced epithelial-Mesenchymal transition in lung adenocarcinoma cells through TGF-β/Smad and PI3K/Akt/mTOR signaling pathways. Molecules 24:144. doi: 10.3390/molecules24010144
Ma, Y., Cui, G., Chen, T., Ma, X., Wang, R., Jin, B., et al. (2021). Expansion within the CYP71D subfamily drives the heterocyclization of tanshinones synthesis in Salvia miltiorrhiza. Nat. Commun. 12:685. doi: 10.1038/s41467-021-20959-1
Ma, L., Dong, C., Song, C., Wang, X., Zheng, X., Niu, Y., et al. (2021). De novo genome assembly of the potent medicinal plant Rehmannia glutinosa using nanopore technology. Comput. Struct. Biotechnol. J. 19, 3954–3963. doi: 10.1016/j.csbj.2021.07.006
Ma, X., Meng, Y., Wang, P., Tang, Z., Wang, H., and Xie, T. (2020). Bioinformatics-assisted, integrated omics studies on medicinal plants. Brief. Bioinform. 21, 1857–1874. doi: 10.1093/bib/bbz132
Mao, L., Jin, B., Chen, L., Tian, M., Ma, R., Yin, B., et al. (2021). Functional identification of the terpene synthase family involved in diterpenoid alkaloids biosynthesis in Aconitum carmichaelii. Acta Pharm. Sin. B 11, 3310–3321. doi: 10.1016/j.apsb.2021.04.008
Meena, S., Rajeev Kumar, S., Dwivedi, V., Kumar Singh, A., Chanotiya, C. S., Akhtar, M. Q., et al. (2017). Transcriptomic insight into terpenoid and carbazole alkaloid biosynthesis, and functional characterization of two terpene synthases in curry tree (Murraya koenigii). Sci. Rep. 7:44126. doi: 10.1038/srep44126
Mochida, K., Sakurai, T., Seki, H., Yoshida, T., Takahagi, K., Sawai, S., et al. (2017). Draft genome assembly and annotation of Glycyrrhiza uralensis, a medicinal legume. Plant J. 89, 181–194. doi: 10.1111/tpj.13385
Mogole, L., Omwoyo, W., and Mtunzi, F. (2020). Phytochemical screening, anti-oxidant activity and α-amylase inhibition study using different extracts of loquat (Eriobotrya japonica) leaves. Heliyon 6:e04736. doi: 10.1016/j.heliyon.2020.e04736
Nair, P., Mall, M., Sharma, P., Khan, F., Nagegowda, D. A., Rout, P. K., et al. (2019). Characterization of a class III peroxidase from Artemisia annua: relevance to artemisinin metabolism and beyond. Plant Mol. Biol. 100, 527–541. doi: 10.1007/s11103-019-00879-x
Niu, Z., Zhu, F., Fan, Y., Li, C., Zhang, B., Zhu, S., et al. (2021). The chromosome-level reference genome assembly for Dendrobium officinale and its utility of functional genomics research and molecular breeding study. Acta Pharm. Sin. B 11, 2080–2092. doi: 10.1016/j.apsb.2021.01.019
Pedro, D. F., Ramos, A. A., Lima, C. F., Baltazar, F., and Pereira-Wilson, C. (2016). Colon Cancer chemoprevention by sage tea drinking: decreased DNA damage and cell proliferation. Phytother. Res. 30, 298–305. doi: 10.1002/ptr.5531
Pham, H. T. T., Hoang, M. C., Ha, T. K. Q., Dang, L. H., Tran, V. O., Nguyen, T. B. T., et al. (2018). Discrimination of different geographic varieties of Gymnema sylvestre, an anti-sweet plant used for the treatment of type 2 diabetes. Phytochemistry 150, 12–22. doi: 10.1016/j.phytochem.2018.02.013
Qiao, W., Li, C., Mosongo, I., Liang, Q., Liu, M., and Wang, X. (2018). Comparative transcriptome analysis identifies putative genes involved in steroid biosynthesis in Euphorbia tirucalli. Gene 9:38. doi: 10.3390/genes9010038
Qing, Z., Liu, J., Yi, X., Liu, X., Hu, G., Lao, J., et al. (2021). The chromosome-level Hemerocallis citrina Borani genome provides new insights into the rutin biosynthesis and the lack of colchicine. Hortic. Res. 8:89. doi: 10.1038/s41438-021-00539-6
Rai, A., Hirakawa, H., Nakabayashi, R., Kikuchi, S., Hayashi, K., Rai, M., et al. (2021). Chromosome-level genome assembly of Ophiorrhiza pumila reveals the evolution of camptothecin biosynthesis. Nat. Commun. 12:405. doi: 10.1038/s41467-020-20508-2
Rohmer, M. (1999). The discovery of a mevalonate-independent pathway for isoprenoid biosynthesis in bacteria, algae and higher plants. Nat. Prod. Rep. 16, 565–574. doi: 10.1039/a709175c
Roy, N. S., Choi, I. Y., Um, T., Jeon, M. J., Kim, B. Y., Kim, Y. D., et al. (2021). Gene expression and isoform identification of PacBio full-length cDNA sequences for berberine biosynthesis in Berberis koreana. Plants 10:1314. doi: 10.3390/plants10071314
Sabzehzari, M., Zeinali, M., and Naghavi, M. R. (2020). Alternative sources and metabolic engineering of Taxol: advances and future perspectives. Biotechnol. Adv. 43:107569. doi: 10.1016/j.biotechadv.2020.107569
Shah, M., Alharby, H. F., Hakeem, K. R., Ali, N., Rahman, I. U., Munawar, M., et al. (2020). De novo transcriptome analysis of Lantana camara L. revealed candidate genes involved in phenylpropanoid biosynthesis pathway. Sci. Rep. 10:13726. doi: 10.1038/s41598-020-70635-5
Sharma, M., Sharma, P. D., Bansal, M. P., and Singh, J. (2007). Synthesis, cytotoxicity, and antitumor activity of lantadene: a congeners. Chem. Biodivers. 4, 932–939. doi: 10.1002/cbdv.200790082
Shen, Q., Zhang, L., Liao, Z., Wang, S., Yan, T., Shi, P., et al. (2018). The genome of Artemisia annua provides insight into the evolution of Asteraceae Family and Artemisinin biosynthesis. Mol. Plant 11, 776–788. doi: 10.1016/j.molp.2018.03.015
Singh, P., Singh, G., Bhandawat, A., Singh, G., Parmar, R., Seth, R., et al. (2017). Spatial transcriptome analysis provides insights of key gene(s) involved in steroidal saponin biosynthesis in medicinally important herb Trillium govanianum. Sci. Rep. 7:45295. doi: 10.1038/srep45295
Skubnik, J., Pavlickova, V. S., Ruml, T., and Rimpelova, S. (2021). Vincristine in combination therapy of cancer: emerging trends in clinics. Biology 10:849. doi: 10.3390/biology10090849
Soltani Howyzeh, M., Sadat Noori, S. A., Shariati, J. V., and Amiripour, M. (2018). Comparative transcriptome analysis to identify putative genes involved in thymol biosynthesis pathway in medicinal plant Trachyspermum ammi L. Sci. Rep. 8:13405. doi: 10.1038/s41598-018-31618-9
Song, C., Liu, Y., Song, A., Dong, G., Zhao, H., Sun, W., et al. (2018). The Chrysanthemum nankingense genome provides insights into the evolution and diversification of Chrysanthemum flowers and medicinal traits. Mol. Plant 11, 1482–1491. doi: 10.1016/j.molp.2018.10.003
Sun, W., Leng, L., Yin, Q., Xu, M., Huang, M., Xu, Z., et al. (2019). The genome of the medicinal plant Andrographis paniculata provides insight into the biosynthesis of the bioactive diterpenoid neoandrographolide. Plant J. 97, 841–857. doi: 10.1111/tpj.14162
Tao, S., Ren, Z., Yang, Z., Duan, S., Wan, Z., Huang, J., et al. (2021). Effects of different molecular weight polysaccharides from Dendrobium officinale Kimura & Migo on human colorectal cancer and transcriptome analysis of differentially expressed genes. Front. Pharmacol. 12:704486. doi: 10.3389/fphar.2021.704486
Tiwari, P., Ahmad, K., and Baig, M. H. (2017). Gymnema sylvestre for diabetes: from traditional herb to future’s therapeutic. Curr. Pharm. Des. 23, 1667–1676. doi: 10.2174/1381612823666161108162048
Tu, L., Su, P., Zhang, Z., Gao, L., Wang, J., Hu, T., et al. (2020). Genome of Tripterygium wilfordii and identification of cytochrome P450 involved in triptolide biosynthesis. Nat. Commun. 11:971. doi: 10.1038/s41467-020-14776-1
van Bakel, H., Stout, J. M., Cote, A. G., Tallon, C. M., Sharpe, A. G., Hughes, T. R., et al. (2011). The draft genome and transcriptome of Cannabis sativa. Genome Biol. 12:R102. doi: 10.1186/gb-2011-12-10-r102
van Dijk, E. L., Jaszczyszyn, Y., Naquin, D., and Thermes, C. (2018). The third revolution in sequencing technology. Trends Genet. 34, 666–681. doi: 10.1016/j.tig.2018.05.008
Verma, M., Ghangal, R., Sharma, R., Sinha, A. K., and Jain, M. (2014). Transcriptome analysis of Catharanthus roseus for gene discovery and expression profiling. PLoS One 9:e103583. doi: 10.1371/journal.pone.0103583
Wang, Y. (2021). A draft genome, resequencing, and metabolomes reveal the genetic background and molecular basis of the nutritional and medicinal properties of loquat (Eriobotrya japonica (Thunb.) Lindl). Hortic. Res. 8:231. doi: 10.1038/s41438-021-00657-1
Wang, X., Hui, F., Yang, Y., and Yang, S. (2018). Deep sequencing and transcriptome analysis to identify genes related to biosynthesis of aristolochic acid in Asarum heterotropoides. Sci. Rep. 8:17850. doi: 10.1038/s41598-018-36316-0
Wang, J., Li, J., Li, Z., Liu, B., Zhang, L., Guo, D., et al. (2022). Genomic insights into longan evolution from a chromosome-level genome assembly and population genomics of longan accessions. Hortic. Res. 9:uhac021. doi: 10.1093/hr/uhac021
Wang, C., Zhu, J., Liu, M., Yang, Q., Wu, J., and Li, Z. (2018). De novo sequencing and transcriptome assembly of Arisaema heterophyllum Blume and identification of genes involved in isoflavonoid biosynthesis. Sci. Rep. 8:17643. doi: 10.1038/s41598-018-35664-1
Wei, G., Chen, Y., Guo, X., Wei, J., Dong, L., and Chen, S. (2021). Biosyntheses characterization of alkaloids and flavonoids in Sophora flavescens by combining metabolome and transcriptome. Sci. Rep. 11:7388. doi: 10.1038/s41598-021-86970-0
Wu, Z., Liu, H., Zhan, W., Yu, Z., Qin, E., Liu, S., et al. (2021). The chromosome-scale reference genome of safflower (Carthamus tinctorius) provides insights into linoleic acid and flavonoid biosynthesis. Plant Biotechnol. J. 19, 1725–1742. doi: 10.1111/pbi.13586
Wu, Z., Zhao, X., Miyamoto, A., Zhao, S., Liu, C., Zheng, W., et al. (2019). Effects of steroidal saponins extract from Ophiopogon japonicus root ameliorates doxorubicin-induced chronic heart failure by inhibiting oxidative stress and inflammatory response. Pharm. Biol. 57, 176–183. doi: 10.1080/13880209.2019.1577467
Xia, M., Han, X., He, H., Yu, R., Zhen, G., Jia, X., et al. (2018). Improved de novo genome assembly and analysis of the Chinese cucurbit Siraitia grosvenorii, also known as monk fruit or luo-han-guo. Gigascience 7:giy067. doi: 10.1093/gigascience/giy067
Xiang, Y., Guo, Z., Zhu, P., Chen, J., and Huang, Y. (2019). Traditional Chinese medicine as a cancer treatment: modern perspectives of ancient but advanced science. Cancer Med. 8, 1958–1975. doi: 10.1002/cam4.2108
Xiong, X., Gou, J., Liao, Q., Li, Y., Zhou, Q., Bi, G., et al. (2021). The Taxus genome provides insights into paclitaxel biosynthesis. Nat. Plants 7, 1026–1036. doi: 10.1038/s41477-021-00963-5
Xu, J., Chu, Y., Liao, B., Xiao, S., Yin, Q., Bai, R., et al. (2017). Panax ginseng genome examination for ginsenoside biosynthesis. Gigascience 6, 1–15. doi: 10.1093/gigascience/gix093
Xu, X., Crow, M., Rice, B. R., Li, F., Harris, B., Liu, L., et al. (2021). Single-cell RNA sequencing of developing maize ears facilitates functional analysis and trait candidate gene discovery. Dev. Cell 56, 557.e6–568.e6. doi: 10.1016/j.devcel.2020.12.015
Xu, D., Lin, H., Tang, Y., Huang, L., Xu, J., Nian, S., et al. (2021). Integration of full-length transcriptomics and targeted metabolomics to identify benzylisoquinoline alkaloid biosynthetic genes in Corydalis yanhusuo. Hortic. Res. 8:16. doi: 10.1038/s41438-020-00450-6
Xu, Z., Peters, R. J., Weirather, J., Luo, H., Liao, B., Zhang, X., et al. (2015). Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 82, 951–961. doi: 10.1111/tpj.12865
Xue, T., Chen, D., Zhang, T., Chen, Y., Fan, H., Huang, Y., et al. (2022). Chromosome-scale assembly and population diversity analyses provide insights into the evolution of Sapindus mukorossi. Hortic. Res. 9:uhac012. doi: 10.1093/hr/uhac012
Xue, L., He, Z., Bi, X., Xu, W., Wei, T., Wu, S., et al. (2019). Transcriptomic profiling reveals MEP pathway contributing to ginsenoside biosynthesis in Panax ginseng. BMC Genomics 20:383. doi: 10.1186/s12864-019-5718-x
Yang, L., Chen, J., Lu, H., Lai, J., He, Y., Liu, S., et al. (2019). Pueraria lobata for diabetes mellitus: past, present and future. Am. J. Chin. Med. 47, 1419–1444. doi: 10.1142/S0192415X19500733
Yang, Y., Huang, L., Xu, C., Qi, L., Wu, Z., Li, J., et al. (2021a). Chromosome-scale genome assembly of areca palm (Areca catechu). Mol. Ecol. Resour. 21, 2504–2519. doi: 10.1111/1755-0998.13446
Yang, Y., Li, S., Xing, Y., Zhang, Z., Liu, T., Ao, W., et al. (2021b). The first high-quality chromosomal genome assembly of a medicinal and edible plant Arctium lappa. Mol. Ecol. Resour. 22, 1493–1507. doi: 10.1111/1755-0998.13547
Yang, Z., Liu, G., Zhang, G., Yan, J., Dong, Y., Lu, Y., et al. (2021). The chromosome-scale high-quality genome assembly of Panax notoginseng provides insight into dencichine biosynthesis. Plant Biotechnol. J. 19, 869–871. doi: 10.1111/pbi.13558
Yao, S., Lan, Z., Huang, R., Tan, Y., Huang, D., Gu, J., et al. (2021). Hormonal and transcriptional analyses provides new insights into the molecular mechanisms underlying root thickening and isoflavonoid biosynthesis in Callerya speciosa (champ. Ex Benth.) Schot. Sci. Rep. 11:9. doi: 10.1038/s41598-020-76633-x
Yin, Z., Wang, X., Yang, X., Chen, Y., Duan, Y., and Han, J. (2021). Salvia miltiorrhiza in anti-diabetic angiopathy. Curr. Mol. Pharmacol. 14, 960–974. doi: 10.2174/1874467214999210111222918
Yu, H., Liu, M., Yin, M., Shan, T., Peng, H., Wang, J., et al. (2021). Transcriptome analysis identifies putative genes involved in triterpenoid biosynthesis in Platycodon grandiflorus. Planta 254:34. doi: 10.1007/s00425-021-03677-2
Yu, Z. W., Zhang, N., Jiang, C. Y., Wu, S. X., Feng, X. Y., and Feng, X. Y. (2021). Exploring the genes involved in biosynthesis of dihydroquercetin and dihydromyricetin in Ampelopsis grossedentata. Sci. Rep. 11:15596. doi: 10.1038/s41598-021-95071-x
Yue, G. G., Li, L., Lee, J. K., Kwok, H. F., Wong, E. C., Li, M., et al. (2019). Multiple modulatory activities of Andrographis paniculata on immune responses and xenograft growth in esophageal cancer preclinical models. Phytomedicine 60:152886. doi: 10.1016/j.phymed.2019.152886
Yue, J., Wang, R., Ma, X., Liu, J., Lu, X., Balaso Thakar, S., et al. (2020). Full-length transcriptome sequencing provides insights into the evolution of apocarotenoid biosynthesis in Crocus sativus. Comput. Struct. Biotechnol. J. 18, 774–783. doi: 10.1016/j.csbj.2020.03.022
Zhang, C., Deng, W., Yan, W., and Li, T. (2018). Whole genome sequence of an edible and potential medicinal fungus, Cordyceps guangdongensis. G3 8, 1863–1870. doi: 10.1534/g3.118.200287
Zhang, D., Li, W., Chen, Z. J., Wei, F. G., Liu, Y. L., and Gao, L. Z. (2020). SMRT- and Illumina-based RNA-seq analyses unveil the ginsinoside biosynthesis and transcriptomic complexity in Panax notoginseng. Sci. Rep. 10:15310. doi: 10.1038/s41598-020-72291-1
Zhang, L., Li, X., Ma, B., Gao, Q., Du, H., Han, Y., et al. (2017). The Tartary buckwheat genome provides insights into Rutin biosynthesis and abiotic stress tolerance. Mol. Plant 10, 1224–1237. doi: 10.1016/j.molp.2017.08.013
Zhang, D., Li, W., Xia, E. H., Zhang, Q. J., Liu, Y., Zhang, Y., et al. (2017). The medicinal herb Panax notoginseng genome provides insights into Ginsenoside biosynthesis and genome evolution. Mol. Plant 10, 903–907. doi: 10.1016/j.molp.2017.02.011
Zhang, X., Zhao, Y., Guo, L., Qiu, Z., Huang, L., and Qu, X. (2017). Differences in chemical constituents of Artemisia annua L from different geographical regions in China. PLoS One 12:e0183047. doi: 10.1371/journal.pone.0183047
Zhao, Q., Yang, J., Cui, M. Y., Liu, J., Fang, Y., Yan, M., et al. (2019). The reference genome sequence of Scutellaria baicalensis provides insights into the evolution of Wogonin biosynthesis. Mol. Plant 12, 935–950. doi: 10.1016/j.molp.2019.04.002
Keywords: Chinese herbal medicine, high-throughput sequencing technologies, genome assembly, bioactive compounds, functional genes
Citation: Liu X, Gong X, Liu Y, Liu J, Zhang H, Qiao S, Li G and Tang M (2022) Application of High-Throughput Sequencing on the Chinese Herbal Medicine for the Data-Mining of the Bioactive Compounds. Front. Plant Sci. 13:900035. doi: 10.3389/fpls.2022.900035
Edited by:
Jian Li Yang, Zhejiang University, ChinaReviewed by:
Niveshika, Banaras University, IndiaCopyright © 2022 Liu, Gong, Liu, Liu, Zhang, Qiao, Li and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Tang, bXQzMTM4QHVqcy5lZHUuY24=; Gang Li, bGlnYW5nMTExNjY2QDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.