
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 03 May 2022
Sec. Plant Bioinformatics
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.892642
This article is part of the Research TopicGenetic Improvement of Triticeae Crops Based on High-throughput Phenotyping: Molecular Design for Yield, Resistance and ToleranceView all 14 articles
 Xiaojun Zhang1†
Xiaojun Zhang1† Linyi Qiao1,2†
Linyi Qiao1,2† Xin Li1
Xin Li1 Zujun Yang3
Zujun Yang3 Cheng Liu4Huijuan Guo1Jun Zheng5Shuwei Zhang1Lifang Chang1Fang Chen1
Cheng Liu4Huijuan Guo1Jun Zheng5Shuwei Zhang1Lifang Chang1Fang Chen1 Juqing Jia1
Juqing Jia1 Liuling Yan2*
Liuling Yan2* Zhijian Chang1*
Zhijian Chang1*The number of spikelets per spike is an important trait that directly affects grain yield in wheat. Three quantitative trait loci (QTLs) associated with spikelet nodes per spike (SNS) were mapped in a population of recombinant inbred lines generated from a cross between two advanced breeding lines of winter wheat based on the phenotypic variation evaluated over six locations/years. Two of the three QTLs are QSns.sxau-2A at the WHEATFRIZZY PANICLE (WFZP) loci and QSns.sxau-7A at the WHEAT ORTHOLOG OF APO1 (WAPO1) loci. The WFZP-A1b allele with a 14-bp deletion at QSns.sxau-2A was associated with increased spikelets per spike. WAPO-A1e, as a novel allele at WAPO1, were regulated at the transcript level that was associated with the SNS trait. The third SNS QTL, QSns.sxau-7D on chromosome 7D, was not associated with homoeologous WAPO-D1 or any other genes known to regulate SNS. The favorable alleles for each of WZFP-A1, WAPO-A1, and QSns.sxau-7D are identified and incorporated to increase up to 3.4 spikelets per spike in the RIL lines. Molecular markers for the alleles were developed. This study has advanced our understanding of the genetic basis of natural variation in spikelet development in wheat.
Wheat (Triticum aestivum L., AABBDD, 2n = 6x = 42) is the most widely grown crop in the world, and its grain is a staple food of humans. Wheat grain yield can be divided into three yield components: spikes per unit of area, grain number per spike, and grain weight (Zhang et al., 2018). Grain number per spike can be subdivided into spikelet number per spike and grain number per spikelet, and the number of spikelets per spike is thus an important subcomponent that can be directly used to improve grain yield (Guo et al., 2015).
Common wheat has a normal spike head that looks a square or “Q” (Muramatsu, 1963; Faris et al., 2003). Compared the square spike, a spike that is shorter or compact is called club spike in T. aestivum ssp. Compactum, whereas a spike that is longer or loose is called spelt spike in T. aestivum ssp. spelta (Faris et al., 2003; Simons et al., 2006; Johnson et al., 2008; Cui et al., 2012; Faris et al., 2014). The gene controlling the square shape is “Q” on chromosomes 5A that was cloned in previous studies (Faris et al., 2003; Simons et al., 2006). The gene controlling the club spike is “C,” and the “C” is not known yet but was mapped on chromosomes 2D (Johnson et al., 2008; Fan et al., 2019).
A normal wheat spike has one single spikelet on one node of the spike rachis (McMaster, 2005). However, a spike may have a supernumerary spikelet on the same node in a vertical or horizontal pattern, forming twin spikelets and even triple spikelets (Wang et al., 2016). The supernumerary spikelets are generated on the basal and central rachis nodes but not on the upper rachis nodes on the same spike (Sears, 1954). A spike of a normal wheat cultivar may generate 15–25 spikelets, each of which develops as a terminal meristem on the spike (Rawson, 1970). The developed spikelet may advance to differentiate into the floral meristem consisting of 5–10 florets encompassed by two small bract leaves to form fertile spikelets containing grains or may abort to form sterile spikelets without grains (Pennell and Halloran, 1984; Gonzalez et al., 2011; Guo and Schnurbusch, 2015; Sakuma et al., 2019).
Numerous efforts have been made to identify quantitative trait loci (QTLs) for spikelets per spike due to their relatively higher heritability than other grain yield components/subcomponents (Zhang et al., 2018; Kuzay et al., 2019). Many QTLs have been identified in association analyses of wheat panels with various genetic backgrounds (Boeven et al., 2016; Würschum et al., 2018; Voss-Fels et al., 2019; Ward et al., 2019) and biparental populations in both hexaploid and tetraploid wheat (Quarrie et al., 2006; Faris et al., 2014; Xu et al., 2014; Luo et al., 2016; Zhai et al., 2016). In two recent independent studies (Kuzay et al., 2019; Voss-Fels et al., 2019), the TraesCS7A01G481600 gene on the long arm of chromosome 7A, which is orthologous to ABERRANT PANICLE ORGANIZATION 1 (APO1), known to affect panicle development and spikelet number in rice (Ikeda et al., 2013), was reported to be the candidate gene regulating spikelets per spike in wheat. The TraesCS7A01G481600 locus was found by fine mapping of segregation populations and association analyses of numerous cultivars.
Supernumerary spikelets are genetically controlled by a single recessive gene (Patil, 1958; Martinek and Bednar, 2001; Aliyeva and Aminov, 2011) or two recessive genes (Sharman, 1967; Millet, 1986; Peng et al., 1998; Yang et al., 2005). A major recessive gene on chromosome 2A and numerous minor genes, including one on chromosome 2B, are reported to be involved in the development of supernumerary spikelets (Klindworth et al., 1990; Zhang et al., 2013). Strong inhibitors of supernumerary spikelets may be located on chromosomes 2DS and 2AL in Chinese Spring (Sears, 1954; Zhang et al., 2013). All of these previous studies have pointed to the homoeologous group 2 chromosomes in the control of supernumerary spikelets in wheat. The wheat FRIZZY PANICLE (WFZP) gene, which encodes an APETALA2/ethylene response transcription factor, may be involved in supernumerary spikelets in common wheat (Dobrovolskaya et al., 2015). Non-sense mutations in WFZP-A1 on chromosome arm 2AS and WFZP-D1 on chromosome arm 2DS cause supernumerary spikelets or a so-called multirow spike (Dobrovolskaya et al., 2017). In addition, the TB1 gene, which encodes a transcription factor in a class II TCP (TEOSINTE BRANCHED1/CYCLOIDEA/PCF1), may control paired spikelet development in a dosage-dependent manner (Dobrovolskaya et al., 2015; Poursarebani et al., 2015; Dixon et al., 2018).
Previous studies identified the functional genes for spikelet development, laying a foundation for allelic variation in the candidate genes for QTLs for the same traits that are mapped in different populations. In this study, we mapped three major QTLs for spikelet nodes per spike (SNS) in a biparental population in common wheat: the first is associated with WFZP-A1 on chromosome arm 2AS, the second is associated with WHEAT ORTHOLOG OF APO1 on chromosome arm 7AL (WAPO-A1), and the third is in a previously uncharacterized region on chromosome 7D. These findings have facilitated pyramiding the favorable alleles for the genes/QTLs into a single genotype using molecular markers to increase spikelets per spike in breeding new wheat varieties.
“SY95-71” (with a pedigree of Fan6//Fan6/Eronga83) was developed by the Triticeae Research Institute of Sichuan Agricultural University in the 1990s, and “CH7034” (with a pedigree of Jing411/Xiaoyan7430//Zhong8601) was developed by the College of Agronomy of Shanxi Agricultural University. The two advanced breeding lines have obvious differences in spike morphology. A population of 184 F2:10 recombinant inbred lines (RILs) derived from the cross SY95-71 × CH7034 was developed to map genes for important traits.
The 184 RILs of the SY95-71 × CH7034 population were tested in six locations/years, including Chengdu (104°13′E, 30°47′N) in 2014 and 2015, Yuncheng (110°53′E, 35°00′N) in 2016, and Linfen (111°31′E, 36°54′N) in 2015 and 2016. The field experiments were arranged in a randomized complete block design with three replications in all of the trials. Each line was grown in a single 2-m row with 0.25 m between rows. Twenty seeds were planted in each row with plant spacing of 0.1 m. Nitrogen and superphosphate fertilizers in the six environmental trials were applied at sowing at rates of 120 and 130 kg ha–1, respectively. Field management was carried out with standard agronomic practices according to wheat production throughout the growing season to obtain even crop stands (Ma et al., 2019). The number of spikelet nodes per spike was characterized in the main spikes of 15 randomly selected plants from each line at physiological maturity and was calculated as the mean. In addition, three plants from each RIL were planted in the greenhouse of Oklahoma State University in 2019 to determine whether the supernumerary spikelet phenotype was associated with the QTLs for the SNS trait. To avoid any confusion, the number of spikelet nodes per spike in a regular pattern is referred to as SNS, and the total number of spikelets per spike in a pattern with supernumerary spikelets is referred to as SSS.
The 184 RILs of the SY95-71/CH7034 population and their parents were genotyped using 17K SNP chips with 17,526 markers and 35K DArT SNP chips containing 36,420 markers at Diversity Arrays Technology Pty Ltd. (Canberra, Australia1). The single-nucleotide polymorphism (SNP) marker sequences were used to BLAST the Chinese Spring sequence [International Wheat Genome Consortium (IWGSC) RefSeq v1.1] to determine the physical positions of each marker. The SNP markers that had less than 20% missing values and contained different recombinant information were selected in the final mapping.
The SNP markers were used to construct linkage groups using JoinMap 4.0 (Van-Ooijen, 2006). The genetic distance between markers was estimated using the Kosambi mapping function, and a QTL for SNSs was claimed when the value of the logarithm of the odds (LOD) threshold for significance was above 2.5 in the interval mapping program in MapQTL 6.0 (Van-Ooijen, 2009). A marker with the maximum LOD value was used to determine the chromosomal position of the QTL peak.
Those genes under or close to the peak of the identified QTLs were considered candidates causing the QTLs. Specific primers were designed to isolate and sequence the candidate genes from each of the two parents to determine the allelic variation.
A diagnostic molecular marker for allelic variation in WFZP-A1 was developed. The primers WFZP-Indel-adF2 (5′-GCCAGCCAACCTCACTTCACTTC-3′) and WFZP-Indel-aR2 (5′-TGGCCGAGGACGCGGCGT-3′) were used to amplify the 449-bp CH7034 allele and the 463-bp SY95-71 allele with the following PCR amplification conditions: 94°C for 3 min, 40 cycles of 94°C for 30 s, 64°C for 30 s, and then 72°C for 40 s, with a final extension of 72°C for 10 min. The PCR products was differentiated on a 2.5% agarose gel at 180 V for 1.5 h.
WAPO-A1 was isolated using the forward primer WAPO-A1F2 (5′-AGGAGATCAATGTTTTTAGGACAATAGGA-3′) and the reverse primer WAPO-A1R6 (5′-GAGCAGGGTGTAGCAACTAGT-3′) with the following PCR amplification conditions: 94°C for 3 min, 40 cycles of 94°C for 30 s, 58°C for 30 s, and then 72°C for 2 min, with a final extension of 72°C for 10 min. A 2-bp deletion was found to distinguish between the two alleles after the PCR products were digested with restriction enzyme HypCH4V. The primers WAPOA1-Indel-F1 (5′-CACAAACATATTGAATGAAAGTTCC-3′) and WAPOA1-Indel-R3 (5′-CATTATATCTTAAACTATATGATGTCC-3′) were used to amplify a fragment containing the 2-bp deletion with the following PCR amplification conditions: 94°C for 3 min, 38 cycles of 94°C for 30 s, 58°C for 30 s, and then 72°C for 30 s, with a final extension of 72°C for 10 min. The digested PCR products were 42 and 70 bp for the CH7034 allele but 110 bp for the SY95-71 allele. The PCR products were separated and scored on an 8% non-denaturing polyacrylamide gel.
WAPO-D1 was isolated using the forward primer WAPO-D1F1 (5′-CAGACGCAGAAAGGAAAACCCTAAGGCAACTT-3′) and the reverse primer WAPO-D1R2 (5′-TGCTAAGAACAGAACAACAAATATACTACCC-3′) with the following PCR amplification conditions: 94°C for 3 min, 40 cycles of 94°C for 30 s, 58°C for 30 s, and then 72°C for 3 min, with a final extension of 72°C for 10 min.
Quantitative real-time PCR (qRT–PCR) was used to determine the transcriptional levels of WAPO-A1 and WAPO-D1 in SY95-71 and CH7034 cells with SYBR Green PCR Master Mix, and actin was used as an endogenous control. Total RNA was extracted from the leaves using TRIzol® reagent (Invitrogen, Carlsbad, CA, United States). cDNA was synthesized from 1 μg of RNA treated with deoxyribonuclease I using a SuperScript™ II Reverse Transcriptase kit and with an oligo (dT)20 primer (Invitrogen). Primers for the gene transcripts were UFO-A-RT-F2 (5′-CTCACTCACTCTCACTCCACG-3′) and UFO-A-RT-R2 (5′-GGTGGTGAGGCAGTAGGTTC-3′) (Kuzay et al., 2019) for WAPO-A1 and WAPO1-D1-RT-F (5′-CTCACTCTCCCTCCCACCACA-3′) and WAPO1-D1-RT-R (5′-GGTGGTGAGGCAGTAGGTTC-3′) for WAPO-D1. qRT–PCR was performed on a CFX96™ Real-time PCR System (Bio–Rad Laboratories, Hercules, CA, United States) using iQTM SYBR® Green Supermix (Bio–Rad Laboratories, Hercules, CA, United States), with actin used as an endogenous control. Gene transcript levels were calculated by the 2–ΔΔCT method, where CT is the threshold cycle.
No supernumerary spikelet on a spikelet node was observed in either of the two parental lines SY95-71 and CH7034 when tested in different years/locations (Figure 1A). However, supernumerary spikelets were observed in some RILs derived from the two parental lines (Figure 1B), indicating that the supernumerary spikelets were generated due to recombination of genes from the two parental lines. The SNS trait in the SY95-71/CH7034 RIL population showed clear segregation, as illustrated in Figure 1C.

Figure 1. Morphological traits of spikes in SY95-71 and CH7034. (A) Comparison of spike between SY95-71 and CH7034. The image shows no supernumerary spikelets in either SY95-71 or CH7034. (B) Dissection of supernumerary spikelets. RIL spikes were segregated with supernumerary spikelets (the left side) and without supernumerary spikelets (right). Supernumerary spikelets are shown on spikes within a red square (upper) and dissected spikes (lower). (C) Spikelet nodes on spikes. RIL were segregated to have less SNS (left) and more SNS (right). Bars, 1 cm.
Statistical analysis showed that among the 184 RILs the SNS association under the field conditions was significant (T > | t0.09|, Supplementary Table 1), indicating that major gene(s) might be responsible for SNS segregation in this population, regardless of the difference in changes in climate or environments in the field.
A total of 17,512 SNP markers was eventually generated from the 17K SNP chips, and a total of 36,420 SNP markers was generated from the 35K DArT SNP chips for the 184 RILs of the SY95-71/CH7034 population. According to their sequences, these SNP markers were assembled into 21 chromosomes, forming genetic maps for the RIL population. The total length of the 21 chromosomes containing the 2,347 SNP markers was 4,192 cM, with a marker density of 1.78 cM per marker. Detailed information on the sequence and genetic distances of the SNP markers on the three SNS QTLs mapped in this study is provided in Supplementary Table 2. The SX plus 8-digit SNP codes served as the reference number for each SNP from 17K SNP chips, and the SX-D plus 8-digit SNP codes served as the reference number for each SNP from the 35K DArT chips.
A total of 1,738 SNP markers, including 797 from 17K SNP chips and 941 from 35K DArT SNP chips, were assembled into chromosome 2A, which was confirmed by the IWGSC RefSeq v1.1 sequences. On the basis of whole-chromosome QTL scanning using interval mapping (IM) analysis, a QTL, QSns.sxau-2A, was found on this chromosome (Figure 2A). The LOD value at the peak position of QSns.sxau-2A ranged from 4.89 to 11.13 in six experimental locations/years, and this QTL alone explained 11.65 or 24.20% of the total phenotypic variation. The 48 markers on chromosome 2A were selected as a linkage group to indicate the genetic distance and physical location of the QSns.sxau-2A locus (Figure 2B).
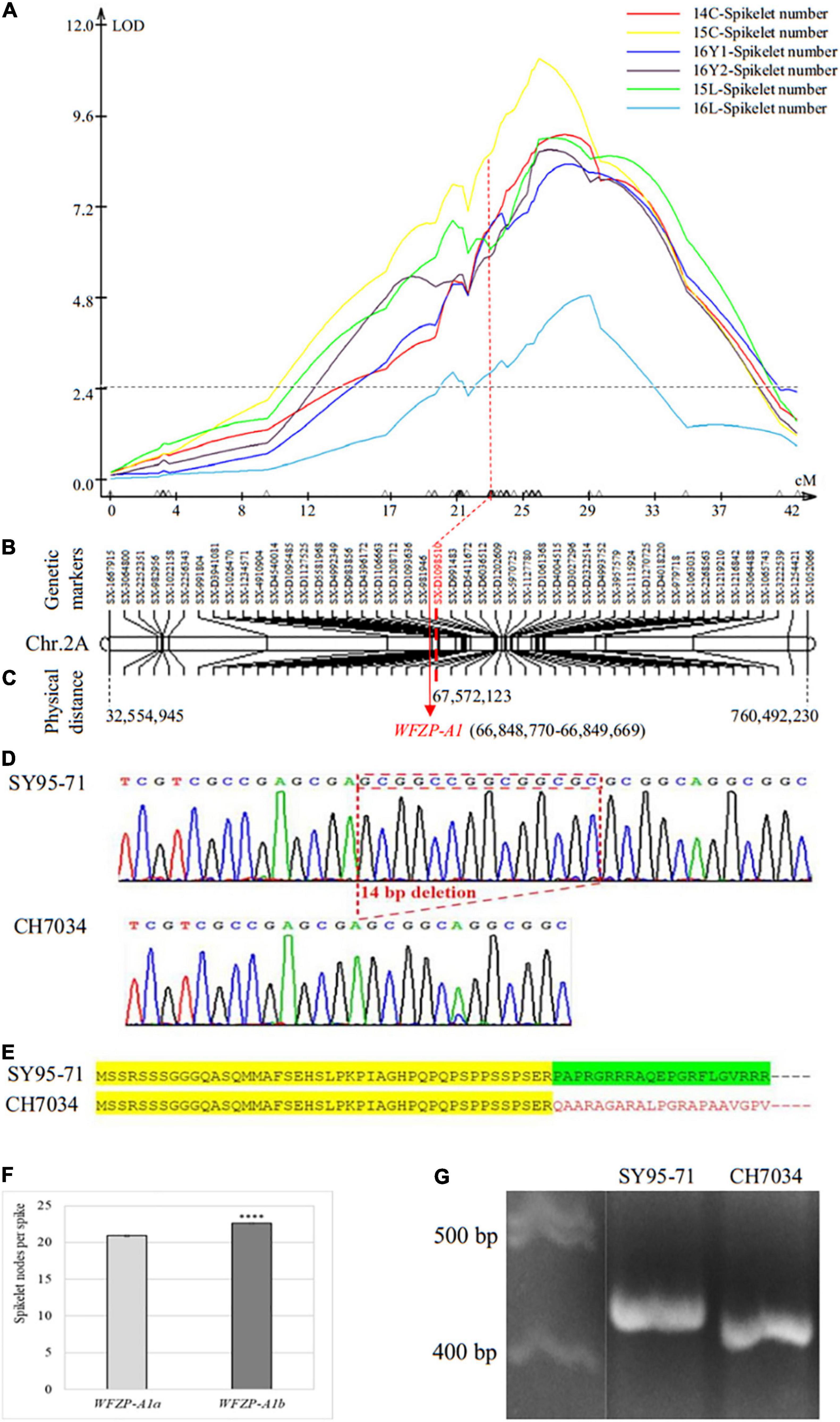
Figure 2. Genetic and physical maps of SNS QTL QSns.sxau-2A and allelic variation in the candidate gene WFZP-A1. (A) Mapping of the QSns.sxau-2A locus. The phenotypic data was collected in Chengdu in 2014 (14C) and 2015 (15C), two experimental locations in Yuncheng in 2016 (16Y1 and 16Y2), and Linfen in 2015 (15L) and 2016 (16L). LOD value is indicated on Y axis, and genetic distance (cM) is indicated on X axis. (B) Genetic map of QSns.sxau-2A. The SX plus 8-digit SNP codes are markers from SNP from 17K SNP chips, and the SX-D plus 8-digit SNP codes are markers from the 35K DArT chips. (C) Physical map of QSns.sxau-2A. The physical locations of markers and candidate gene WFZP-A1 are shown based on the Chinese Spring genome sequence of RefSeq v1.1. (D) Allelic variation in WFZP-A1. The 14-bp sequence that is placed in a red square is present in SY95-71 but absent in CH7034. (E) Deduced WFZP-A1 proteins in SY95-71 and CH7034. Conserved amino acids before the deletion in both SY95-71 and CH7034 are highlighted in yellow, functional amino acids after the deletion in SY95-71 are highlighted in green, and altered amino acids after the deletion in CH7034 are indicated in red. Dots indicate more amino acids toward to C terminus. (F) Comparison of SNS between the SY95-71 allele and the CH7034 allele. A t-test was performed to show significant level in SNS between the two alleles. The bars indicate standard error. “****” indicates significant difference (p < 0.0001). (G) A diagnostic PCR marker for WFZP-A1 between the SY95-71 and CH7034. The marker was developed to distinguish between the SY95-71 allele without the 14-bp deletion and the CH7034 allele with the 14-bp deletion.
The WFZP-A1 gene at position 66,848 kb was close to marker SX-D1098510, which was under the peak of QSns.sxau-2A (Figure 2C); hence, WFZP-A1 was considered a candidate gene causing QSns.sxau-2A. Sequencing results revealed that the SY95-71 allele showed the same sequence as the WFZP-A1 gene of Chinese Spring, which is referred to as the WFZP-A1a allele, whereas the CH7034 allele had a 14-bp deletion in its coding region, which is referred to as the WFZP-A1b allele (Figure 2D). The 14-bp insertion in the WFZP-A1a allele could cause it to gain its function at the protein level, or the 14-bp deletion in the WFZP-A1b allele could cause it to lose its function at the protein level (Figure 2E). No association was found between the WFZP-A1 marker and the SSS trait in this study.
On average, over the six experimental locations/years, the CH7034 WFZP-A1b allele (22.6 ± 0.1, n = 593) set 1.7 more spikelet nodes per spike than the SY95-71 WFZP-A1a allele (20.9 ± 0.1, n = 450) at the QSns.sxau-2A locus (p = 8.68E-27) (Figure 2F). Based on the 14-bp indel, a diagnostic marker for WFZP-A1 was developed (Figure 2G).
A QTL analysis revealed that SNS was associated with 65 markers on chromosome 7A (QSns.sxau-7A). The 1,730 SNP markers including 844 from 17K SNP chips and 886 from 35K DArT SNP chips were assembled into chromosome 7A, but only 43 markers on chromosome arm 7AL were selected as a linkage group to indicate the genetic distance and physical location of the QSns.sxau-7A locus (Figure 3A). The LOD values at the peak position of QSns.sxau-7A ranged from 3.20 to 11.00 in five experimental locations/years, and this QTL alone explained 9.17–28.18% of the total phenotypic variation.
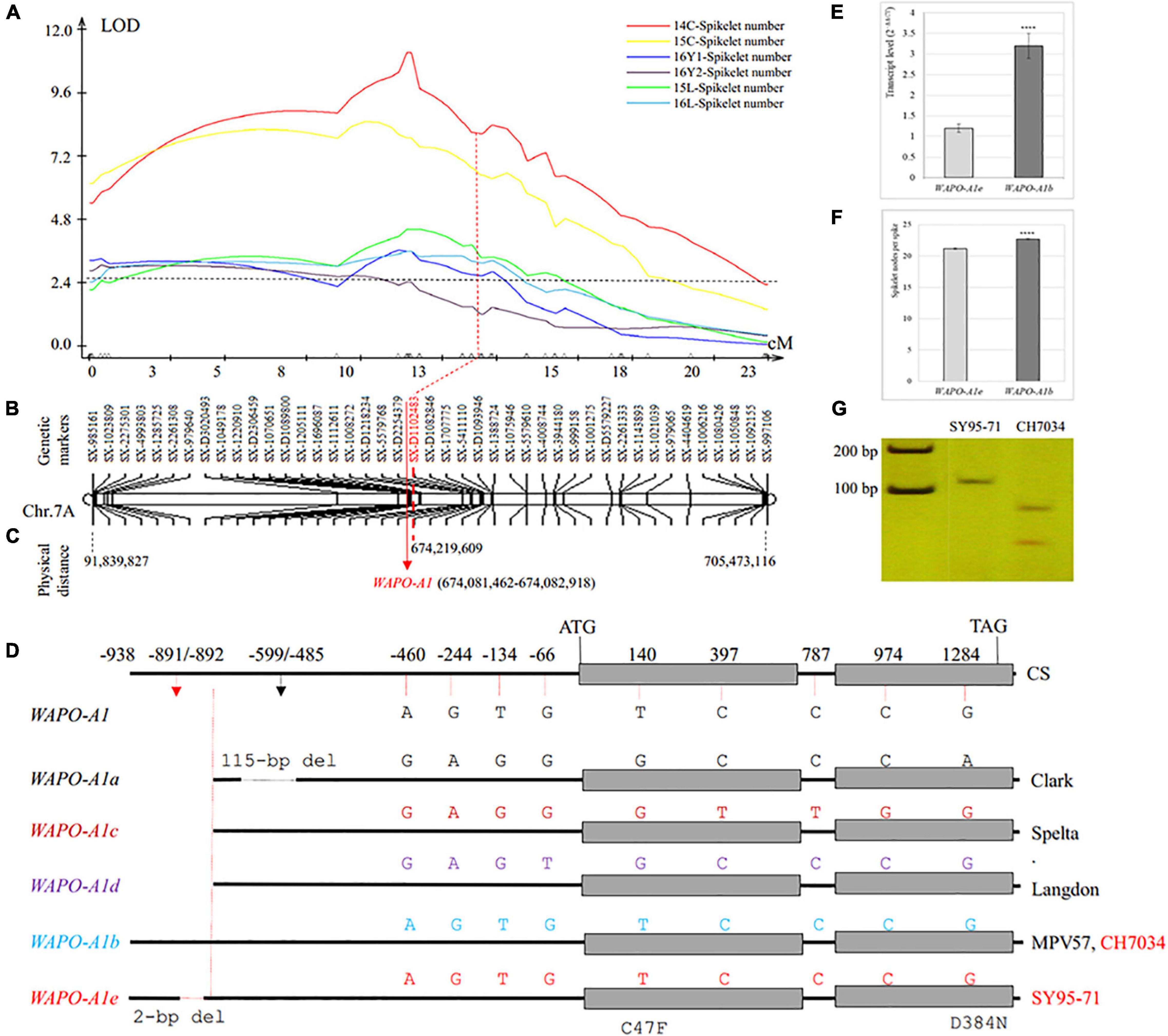
Figure 3. Genetic and physical maps of SNS QTL QSns.sxau-7A and allelic variation in the candidate gene WAPO-A1. (A) Mapping of the QSns.sxau-7A locus. The phenotypic data was collected in Chengdu in 2014 (14C) and 2015 (15C), two experimental locations in Yuncheng in 2016 (16Y1 and 16Y2), and Linfen in 2015 (15L) and 2016 (16L). LOD value is indicated on Y axis, and genetic distance (cM) is indicated on X axis. (B) Genetic map of QSns.sxau-7A. The SX plus 8-digit SNP codes are markers from SNP from 17K SNP chips, and the SX-D plus 8-digit SNP codes are markers from the 35K DArT chips. (C) Physical map of QSns.sxau-7A. The physical locations of markers and candidate gene WAPO-A1 are shown based on the Chinese Spring genome sequence of RefSeq v1.1. (D) A diagram of differences in multiple WAPO-A1 alleles. Allelic variation in WAPO-A1a through WAPO-A1d is cited from the previous study (Kuzay et al., 2019). CH7034 has the same WAPO-A1b allele as MPV57. SY95-71 has WAPO-A1e, which is a novel allele. The only difference between WAPO-A1b and WAPO-A1e is that WAPO-A1e has a 2-bp deletion in its promoter. (E) Comparison of transcript levels between the SY95-71 WAPO-A1e allele and the CH7034 WAPO-A1b allele. (F) Comparison of SNS between the SY95-71 WAPO-A1e allele and the CH7034 WAPO-A1b allele. (E–F) A t-test was performed to show differences in the transcript levels between the two alleles, with “****” indicating a significant difference (p < 0.0001). The bars indicate standard error. (G) A diagnostic PCR marker for WAPO-A1 between the SY95-71 and CH7034. The PCR products were digested with restriction enzyme HypCH4V to distinguish between the SY95-71 WAPO-A1e allele with the 2-bp deletion and the CH7034 WAPO-A1b allele without the 2-bp deletion.
The WAPO-A1 gene (TraesCS7A02G481600) was considered a candidate gene causing QSns.sxau-7A, as it was close to marker SX-D1102483 that was under the peak of QSns.sxau-7A (Figures 3B,C). Primers that were reported to specifically amplify WAPO-A1 (Kuzay et al., 2019) were used to isolate this gene from two parental lines. No difference was found between the CH7034 and WAPO-A1b alleles in the sequenced 2,439-bp region, including the 938-bp promoter region, 1,457 bp from the start codon to the stop codon, and 42 bp at the 3′ end of the gene. Compared with CH7034, SY95-71 had a 2-bp deletion at positions –891 to –892 bp, which was only the difference between the two parental lines (Figure 3D). Compared with the four reported alleles (WAPO-A1a through -A1d), the WAPO-A1 allele in SY95-71 was novel and was thus named WAPO-A1e (Figure 3D).
The WAPO-A1 transcript levels in 1-cm young spikes were compared between the WAPO-A1b and WAPO-A1e alleles. The CH7034 WAPO-A1b allele showed 2.6 times higher transcript levels than the WAPO-A1e allele (p = 0.0001) (Figure 3E). This result suggests that either the WAPOA1b promoter might increase its transcript levels due to the absence of the 2-bp deletion or the WAPO-A1e promoter reduces its transcript levels due to the presence of the 2-bp deletion.
The CH7034 WAPO-A1b allele (22.7 ± 0.1, n = 554) showed 1.5 more spikelet nodes per spike than the SY95-71 WAPO-A1e allele (21.2 ± 0.1, n = 435) at the QSns.sxau-7A locus (p = 1.54E-19), based on the averaged SNS from the six experimental locations/years (Figure 3F). The results showed an association of the SNS phenotype with the 2-bp deletion genotype. A diagnostic marker was developed to distinguish WAPO-A1e that had the 2-bp deletion from WAPO-A1b that did not have the 2-bp deletion (Figure 3G).
The third SNS QTL was located on chromosome 7D (QSns.sxau-7D), where only SNP markers, including 8 from 17K SNP chips and 24 from 35K DArT SNP chips, were assembled into the whole chromosome, spanning 22 cM in genetic distance (Figure 4A). QSns.sxau-7D explained up to 17.92% of the total phenotypic variation in five of the six experimental locations/years, and the effect was not significant in one experimental location/year. The marker SX-D1124962 closest to the peak of QSns.sxau-7D was associated with the SNS trait, the SY95-71 allele (22.1 ± 0.1, n = 561) versus the CH7034 allele (21.8 ± 0.1, n = 536) (p = 0.091) (Figures 4B–D).
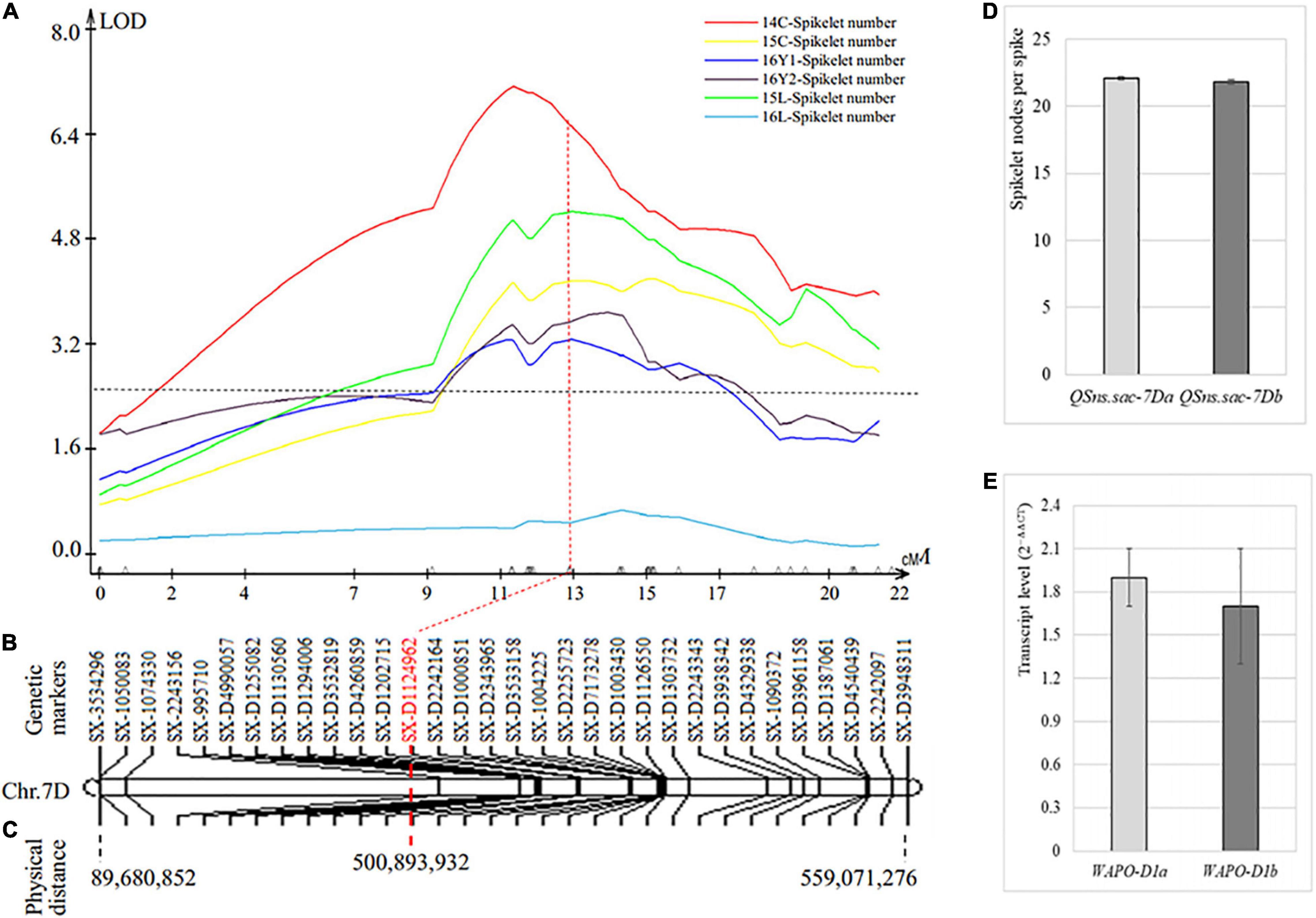
Figure 4. Genetic and physical maps of SNS QTL QSns.sxau-7D. (A) Mapping of the QSns.sxau-7D locus. The phenotypic data was collected in Chengdu in 2014 (14C) and 2015 (15C), two experimental locations in Yuncheng in 2016 (16Y1 and 16Y2), and Linfen in 2015 (15L) and 2016 (16L). LOD value is indicated on Y axis, and genetic distance (cM) is indicated on X axis. (B) Genetic map of QSns.sxau-7D. The SX plus 8-digit SNP codes are markers from SNP from 17K SNP chips, and the SX-D plus 8-digit SNP codes are markers from the 35K DArT chips. (C) Physical map of QSns.sxau-7D. The physical locations of markers are shown based on the Chinese Spring genome sequence of RefSeq v1.1. (D) Comparison of SNS between the SY95-71 QSns.sxau-7D allele and the CH7034 QSns.sxau-7D allele. (E) Comparison of WAPO-D1 transcript levels between the SY95-71 and the CH7034, without significant difference.
The complete gene of homoeologous WAPO-D1 on chromosome 7D (TraesCS7D02G468700) was isolated and sequenced, and the sequenced region, including 564 bp upstream from the start codon, 1,488 bp from the start codon to the stop codon, and 196 bp at the 3′ end of the gene, showed no difference between the SY95-71 and CH7034 alleles. Both SY95-71 and CH7034 had the same WAPO-D1 sequence as Chinese Spring. WAPO-D1 showed no significant difference in its transcript level between the two alleles (Figure 4E). The SNP at SX-D1124962 may be the closest marker to the gene causing QSns.sxau-7D.
Further analyses were performed on each of the three SNS QTLs as independent genetic factors. Eight genotypes generated from these three genes/QTLs, each with two alleles, were analyzed for their SNS phenotypes.
These eight genotypes were classified for the three QTLs in the order of WFZP-A1, WAPO-A1, and QSns.sxau-7D. The SY95-71 allele was designated A, and the CH7034 allele was designated B. The contribution of each genotype to the phenotype was assessed by comparing the SNS mean, given the lack of epistatic interactions. The genotype expected to generate the highest SNS should be the recombinant (B_B_A), containing all the WFZP-A1b and WAPO-A1b alleles from CH7034 and QSns.sxau-7Da from SY95-71, whereas the genotype expected to generate the lowest SNS should be the recombinant (A_A_B), containing all the WFZP-A1a and WAPO-A1e alleles from SY95-71 and QSns.sxau-7Da from CH7034. The incorporated B_B_A genotype generated 23.4 ± 0.2 (n = 149), which was 3.4 more spikelets than the incorporated A_A_B genotype 20 ± 0.2 (n = 124). The remaining six genotypes showed intermediate SNS scores (Figure 5A).

Figure 5. Overall analyses of SNS and fertile spikelets. (A) Comparison of SNS among eight genotypes. These eight genotypes are in the order of WFZP-A1, WAPO-A1, and QSns.sxau-7D, with A for the SY95-71 allele and B for the CH7034 allele. The averaged SNS over the six experimental locations were comparisons. A t-test was performed to show significant levels in difference of SNS between the two alleles, with a though e for a significant difference (P < 0.05). (B) Correlation of SNS and fertile spikelets. The averaged SNS and fertile spikelets over the six experimental locations were analyzed their correlations.
The recently released genome sequences of 10 wheat cultivars provided haplotype information for the frequencies of favorable alleles, WFZP-A1 and WAPO-A1, from CH7034. Only cultivar Norin61 had the same WFZP-A1b allele with the 14-bp deletion as CH7034. All other cultivars showed 100% identity to SY95-71 without the 14-bp deletion, except Spelt, which had a SNP in the WFZP-A gene region.
CH7034 showed 100% identity in the complete 2,439-bp sequence of the WAPO-A1b allele as three wheat cultivars: Mace, Norin61, and Stanley. None of these ten cultivars showed 100% identity to the complete 2,437 bp sequence of the SY95-71 WAPO1-A1e allele, but the 2-bp deletion in WAPO1-A1e was found to exist in six wheat cultivars: Arina, Jaggar, Julius, Lancer, Landmark, and SY_Mattis. These cultivars were found to have the WAPO1-A1a allele, but the sequence of the WAPO1-A1a allele did not include the 2-bp deletion region identified in a previous study (Figure 3D; Kuzay et al., 2019; Voss-Fels et al., 2019). The last cultivar, Spelt, had no 2-bp deletion, but had a WAPO1-A1 that was different from WAPO1-A1a through WAPO1-A1e.
Correlation analysis examined the correlation of the total SNS with fertile spikelets per spike and sterile spikelets per spike over the six experimental locations/years. The total SNS was positively correlated with fertile spikelets per spike (R2 = 0.922, n = 184) (Figure 5B). The results indicated that fertile spikelets were increased through spikelet development in wheat.
Allelic variations in the two advancing lines were identified being associated with three QTLs for SNS. Among the three SNS QTLs identified in this study, QSns.sxau-2A was most stably and consistently observed under six different experimental locations/years, but QSns.sxau-7D was mostly regulated by the environments. The study provides genetic information for grains per spike that can be increased through regular spikelets per spike, in addition to the spikelet fertility rate and grain number per spikelet in wheat (Guo et al., 2015; Zhang et al., 2018).
Previous studies have suggested that mutants in WFZP genes are important resources for increasing supernumerary spikelets to increase grains per spike in bread wheat (Dobrovolskaya et al., 2016; Dobrovolskaya et al., 2017; Zhang et al., 2017; Du et al., 2021; Li et al., 2021). While WFZP-D1 lesions were combined with a frame shift mutation in WFZP-A1 due to the 14-bp deletion, supernumerary spikelets were detectable. In this study, the CH7034 WFZP-A1b allele was not associated with the supernumerary spikelet trait that was observed in the segregation population. No QTL for supernumerary spikelets was observed in the orthologous WFZP-D region or any other genomic region. These results indicated that only the CH7034 WFZP-A1b allele was unable to generate supernumerary spikelets and that the supernumerary spikelet trait may be regulated by other unknown genetic factors in common wheat. Surprisingly, the CH7034 WFZP-A1b allele with the 14-bp deletion was found to positively regulate the SNS number. This observation suggested that wild-type WFZP-A1a might be a repressor of spikelet development, although the possibility that mutated WFZP-A1b may gain new function due to the presence of deduced new amino acids cannot be excluded. Gene editing technology can be used to edit three homoeologous WFZP genes to increase grain number to improve bread wheat yield in future studies. WAPO1 on wheat chromosome arm 7AL was identified as the best candidate gene regulating spikelet number per spike based on a fine genetic map, a genome-wide association analysis, and the known functions of the orthologous APO1 gene in rice (Kuzay et al., 2019; Voss-Fels et al., 2019). Since then, more robust evidence from numerous materials with various genetic backgrounds that were tested under different environments and from mutants and transgenic wheat (Ma et al., 2019; Ding et al., 2021; Jones et al., 2021; Kuzay et al., 2022) has demonstrated that WAPO-A1 is the causal gene for the previously reported QTLs. WAPO-A1b in haplotypes such as Berkut had more than threefold higher transcript levels than WAPO-A1a with the 115-bp deletion and three SNPs in the promoter in haplotypes such as RAC875, suggesting that the reduced transcript level of WAPO-A1a was due to the 115-bp deletion and three SNPs (Kuzay et al., 2019; Voss-Fels et al., 2019). A previous study also suggested that the reduced SNS by WAPO-A1a may be due to the effects of reduced transcript levels or two point mutations in its protein (C47F and D384N) that cannot be separated in the experiment. In this study, WAPO-A1b and WAPO-A1e encoded the same protein sequence but showed differences in their effects on the SNS trait, excluding the possibility that the SNS trait was regulated at the protein level.
WAPO-A1b and WAPO-A1e showed differences at the transcript level, indicating that the SNS trait was regulated at the transcript level. WAPO-A1e in SY95-71 has the same DNA sequence as WAPO-A1b that does not have the previously reported 115-bp deletion or three SNP regions in CH7034. However, further sequencing showed that WAPO-A1e had a 2-bp deletion in upstream of the promoter, facilitating mapping of WAPO-A1 in this study. It is noteworthy that WAPO-A1 in another SY95-71 showed the 115-bp deletion (Ma et al., 2019). It is likely that the two SY95-71 breeding lines might have the same source but different genetic backgrounds that were segregated in progeny plants. WAPO-A1b in CH7034 showed higher transcript levels than WAPO-A1e with the 2-bp deletion in the promoter in SY95-71, suggesting that the transcript level of WAPO-A1e was reduced, as observed in WAPO-A1a. It is likely that the promoter is involved not only in the previously reported 115-bp deletion and three SNP regions ranging from positions –599 to –485 but also in the 2-bp deletion at positions –891 and –892. Further sequencing results at positions up to –2,565 bp revealed seven SNPs further upstream, including six SNPs at positions 2,210, 2,217, 2,243, 2,255, 2,272, and 2,341 bp between the SY95-71 and CH7034 alleles at WAPO-A1. Further work is needed to identify actual binding sites in the WAPO1 promoter.
The tight association between the two SNS QTLs and two genes, WFZP-A1 and WAPO-A1 involved in spikelet development, suggests that each of the spikelet development genes was considered as a candidate gene for one SNS QTL. However, the candidate genes need to be validated in large populations. In the present study the limited size of the RIL population was used for mapping only. Specific RILs can be selected to develop backcross populations, in which only one locus of the three QTLs is heterozygous while the other two are fixed for the same allele; therefore, each of these backcross populations can be used to clone each of the three QTLs in future projects. Diagnostic markers for the WFZP-A1b allele, which should be selected, and the WAPO1e allele, which should be selected against, will provide molecular tools to support the modification of wheat spikes in wheat breeding.
In summary, this study identified allelic variation in three QTLs associated with SNS in the same mapping population, two of which were associated with the genes known to involve in spikelet development in wheat. The findings that WFZP-A1b with the 14-bp deletion increased spikelets per spike and the WFZP-A1e as a novel allele was regulated at the transcript level advanced understanding of these known genes. The molecular markers for these allelic variations can be used to incorporate and pyramid the favorable alleles into single lines for breeding of new varieties. Improved understanding of the genetic basis of natural variation in the spikelet development will be particularly important to wheat production worldwide.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
XZ and LQ performed most of the phenotyping and molecular experiments. XL, CL, HG, SZ, LC, FC, and JJ undertook part of the field and laboratory work. ZY participated in the construction of the RILs. JZ contributed the RILs’ phenotypic data in Linfen. ZC designed and directed the field experiments. LY directed molecular research work. LY, ZC, and LQ wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was financially supported by the Research Program Sponsored by State Key Laboratory of Sustainable Dryland Agriculture (in Preparation) of Shanxi Agricultural University (202002-3), Shanxi Scholarship Council of China (2021-070), the Biological Breeding Engineering of Shanxi Agricultural University (YZGC093), the Service Project from Ministry of Agriculture and Rural Affairs of China (19211089), and the Key Research and Development Program of Shanxi Province (201903D221073).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with one of the authors CL.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Ennian Yang in Sichuan Academy of Agricultural Sciences for providing the experimental field in Sichuan.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.892642/full#supplementary-material
Aliyeva, A. J., and Aminov, N. K. (2011). Inheritance of the branching in hybrid populations among tetraploid wheat species and the new branched spike line 166-schakheli. Genet. Resour. Crop Evol. 58, 621–628. doi: 10.1007/s10722-011-9702-9
Boeven, P. H. G., Longin, C. F. H., Leiser, W. L., Kollers, S., Ebmeyer, E., and Würschum, T. (2016). Genetic architecture of male floral traits required for hybrid wheat breeding. Theor. Appl. Genet. 129, 2343–2357. doi: 10.1007/s00122-016-2771-6
Cui, F., Ding, A., Li, J., Zhao, C., Wang, L., Wang, X., et al. (2012). QTL detection of seven spike-related traits and their genetic correlations in wheat using two related RIL populations. Euphytica 186, 177–192. doi: 10.1007/s10681-011-0550-7
Ding, P. Y., Zhou, J. G., Zhao, C. H., Tang, H. P., Mu, Y., and Tang, L. W. (2021). Dissection of haplotypes, geographical distribution and breeding utilization of WAPO1 associated with spike development in wheat. Acta. Agron. Sin. 11, 1–16.
Dixon, L. E., Greenwood, J. R., Bencivenga, S., Zhang, P., Cockram, J., Mellers, G., et al. (2018). TEOSINTE BRANCHED1 regulates inforescence architecture and development in bread wheat (Triticum aestivum). Plant Cell 30, 563–581. doi: 10.1105/tpc.17.00961
Dobrovolskaya, O. B., Amagai, Y., Popova, K. I., Dresvyannikova, A. E., Martinek, P., Krasnikov, A. A., et al. (2017). Genes wheat frizzy panicle and sham ramification 2 independently regulate differentiation of floral meristems in wheat. BMC Plant Biol. 17:252. doi: 10.1186/s12870-017-1191-3
Dobrovolskaya, O. B., Pont, C., Orlov, Y. L., and Salse, J. (2016). Development of new SSR markers for homoeologous WFZP gene loci based on the study of the structure and location of microsatellites in gene rich regions of chromosomes 2AS, 2BS, and 2DS in bread wheat. Russ. J. Genet. 19, 330–337. doi: 10.1134/S2079059716030023
Dobrovolskaya, O. B., Pont, C., Sibout, R., Martinek, P., Badaeva, E., Murat, F., et al. (2015). FRIZZY PANICLE drives supernumerary spikelets in bread wheat. Plant Physiol. 167, 189–199. doi: 10.1104/pp.114.250043
Du, D. J., Zhang, D. X., Yuan, J., Feng, M., Li, Z. J., Wang, Z. H., et al. (2021). FRIZZY PANICLE defines a regulatory hub for simultaneously controlling spikelet formation and awn elongation in bread wheat. New Phytol. 231, 814–833. doi: 10.1111/nph.17388
Fan, X. L., Cui, F., Ji, J., Zhang, W., Zhao, X. Q., Liu, J. J., et al. (2019). Dissection of pleiotropic QTL regions controlling wheat spike characteristics under different nitrogen treatments using traditional and conditional QTL mapping. Front. Plant Sci. 10:187. doi: 10.3389/fpls.2019.00187
Faris, J. D., Fellers, J. P., Brooks, S. A., and Gill, B. S. (2003). A bacterial artificial chromosome contig spanning the major domestication locus Q in wheat and identification of a candidate gene. Genetics 164, 311–321. doi: 10.1093/genetics/164.1.311
Faris, J. D., Zhang, Q. J., Chao, S. M., Zhang, Z. C., and Xu, S. S. (2014). Analysis of agronomic and domestication traits in a durum × cultivated emmer wheat population using a high-density single nucleotide polymorphism-based linkage map. Theor. Appl. Genet. 127, 2333–2348. doi: 10.1007/s00122-014-2380-1
Gonzalez, F. G., Miralles, D. J., and Slafer, G. A. (2011). Wheat floret survival as related to pre-anthesis spike growth. J. Exp. Bot. 62, 4889–4901. doi: 10.1093/jxb/err182
Guo, J., Zhang, Y., Shi, W. P., Zhang, B. Q., Zhang, J. J., Xu, Y. H., et al. (2015). Association analysis of grain-setting rate at the apical and basal spikelets in bread wheat (Triticum aestivum L.). Front. Plant Sci. 6:1029. doi: 10.3389/fpls.2015.01029
Guo, Z. F., and Schnurbusch, T. (2015). Variation of floret fertility in hexaploid wheat revealed by tiller removal. J. Exp. Bot. 19, 5945–5958. doi: 10.1093/jxb/erv303
Ikeda, M., Miura, K., Aya, K., Kitano, H., and Matsuoka, M. (2013). Genes offering the potential for designing yield-related traits in rice. Curr. Opin. Plant Biol. 16, 213–220. doi: 10.1016/j.pbi.2013.02.002
Johnson, E. B., Nalam, V. J., Zemetra, R. S., and Riera-Lizarazu, O. (2008). Mapping the compactum locus in wheat (Triticum aestivum L.) and its relationship to other spike morphology genes of the triticeae. Euphytica 163, 193–201. doi: 10.1007/s10681-007-9628-7
Jones, B. H., Blake, N. K., Heo, H. Y., Martin, J. M., Torrion, J. A., and Talbert, L. E. (2021). Allelic response of yield component traits to resource availability in spring wheat. Theor. Appl. Genet. 134, 603–620. doi: 10.1007/s00122-020-03717-7
Klindworth, D. L., Williams, N. D., and Joppa, L. R. (1990). Chromosomal location of genes for supernumerary spikelet in tetraploid wheat. Genome 33, 515–520. doi: 10.1139/g90-076
Kuzay, S., Lin, H., Li, C., Chen, S., Woods, D. P., Zhang, J., et al. (2022). WAPO-A1 is the causal gene of the 7AL QTL for spikelet number per spike in wheat. PLoS Genet. 18:e1009747. doi: 10.1371/journal.pgen.1009747
Kuzay, S., Xu, Y. F., Zhang, J. L., Katz, A., Pearce, S., Su, Z. Q., et al. (2019). Identification of a candidate gene for a QTL for spikelet number per spike on wheat chromosome arm 7AL by high-resolution genetic mapping. Theor. Appl. Genet. 132, 2689–2705. doi: 10.1007/s00122-019-03382-5
Li, Y. P., Li, L., Zhao, M. C., Guo, L., Guo, X. X., Zhao, D., et al. (2021). Wheat frizzy panicle activates vernalization1-A and HOMEOBOX4-A to regulate spike development in wheat. Plant Biotechnol. J. 19, 1141–1154. doi: 10.1111/pbi.13535
Luo, W., Ma, J., Zhou, X. H., Sun, M., Kong, X. C., Wei, Y. M., et al. (2016). Identification of quantitative trait loci controlling agronomic traits indicates breeding potential of tibetan semiwild wheat (Triticum aestivum ssp. tibetanum). Crop Sci. 56, 2410–2420. doi: 10.2135/cropsci2015.11.0700
Ma, J., Ding, P. Y., Liu, J. J., Li, T., Zou, Y. Y., Habib, A., et al. (2019). Identification and validation of a major and stably expressed QTL for spikelet number per spike in bread wheat. Theor. Appl. Genet. 132, 3155–3167. doi: 10.1007/s00122-019-03415-z
Martinek, P., and Bednar, J. (2001). “Changes of spike morphology (multirow spike-MRS, long glumes-LG) in wheat (Triticum aestivumL.) and their importance for breeding,” in Proceedings of the International Conference “Genetic Collections, Isogenic and Alloplasmic Lines”, Novosibirsk, 192–194.
McMaster, G. S. (2005). Phytomers, phyllochrons, phenology and temperate cereal development. J. Agr. Sci. 143, 137–150. doi: 10.1017/S0021859605005083
Millet, E. (1986). Genetic control of heading date and spikelet number in common wheat (Triticum aestivum L.) line ‘Noa’. Theor. Appl. Genet. 72, 105–107. doi: 10.1007/BF00261463
Muramatsu, M. (1963). Dosage effect of the spelta gene q of hexaploid wheat. Genetics 48:469–482. doi: 10.1093/genetics/48.4.469
Peng, Z. S., Liu, D. C., Yen, C., and Yang, J. L. (1998). Genetic control of supernumerary spikelet in common wheat line LYB. Wheat Inform. Ser. 86, 6–12.
Pennell, A. L., and Halloran, G. M. (1984). Influence of time of sowing, photoperiod and temperature on supernumerary spikelet expression in wheat (Triticum). Can. J. Bot. 62, 1687–1692. doi: 10.1139/b84-228
Poursarebani, N., Seidensticker, T., Koppolu, R., Trautewig, C., Gawronski, P., Bini, F., et al. (2015). The genetic basis of composite spike form in barley and ‘miracle-wheat’. Genetics 201, 155–165. doi: 10.1534/genetics.115.176628
Quarrie, S. A., Quarrie, S. P., Radosevic, R., Rancic, D., Kaminska, A., Barnes, J. D., et al. (2006). Dissecting A wheat QTL for yield present in a range of environments: from the QTL to candidate genes. J. Exp. Bot. 57, 2627–2637. doi: 10.1007/s00284-007-9035-2
Rawson, H. M. (1970). Spikelet number, its control and relation to yield per ear in wheat. Aust. J. Biol. Sci. 23, 1–15. doi: 10.1071/bi9700001
Sakuma, S., Golan, G., Guo, Z. F., Ogawa, T., Tagiri, A., Sugimoto, K., et al. (2019). Unleashing floret fertility in wheat through the mutation of a homeobox gene. Proc. Natl. Acad. Sci. U.S.A. 116, 5182–5187. doi: 10.1073/pnas.1815465116
Sharman, B. C. (1967). Interpretation of the morphology of various naturally occurring abnormalities of the inflorescence ofwheat (Triticum). Can. J. Bot. 45, 2073–2080. doi: 10.1139/b67-224
Simons, K. J., Fellers, J. P., Trick, H. N., Zhang, Z., Tai, Y. S., Gill, B. S., et al. (2006). Molecular characterization of the major wheat domestication gene Q. Genetics 172, 547–555. doi: 10.1534/genetics.105.044727
Van-Ooijen, J. W. (2006). JoinMap® 4.0: Software for the Calculation of Genetic Linkage Maps In Experimental Populations. Wageningen: Kyazma BV.
Van-Ooijen, J. W. (2009). MapQTL® 6.0: Software for the Mapping Fo Quantitative Trait Loci In Experimental Populations of Diploid Species. Wageningen: Kyazma BV.
Voss-Fels, K. P., Keeble-Gagnère, G., Hickey, L. T., Tibbits, J., Nagornyy, S., Hayden, M. J., et al. (2019). High-resolution mapping of rachis nodes per rachis, a critical determinant of grain yield components in wheat. Theor. Appl. Genet. 132, 2707–2719. doi: 10.1007/s00122-019-03383-4
Wang, Y., Miao, F., and Yan, L. L. (2016). Branching shoots and spikes from lateral meristems in bread wheat. PLoS One 11:e0151656. doi: 10.1371/journal.pone.0151656
Ward, B. P., Brown-Guedira, G., Kolb, F. L., Van Sanford, D. A., Tyagi, P., Sneller, C. H., et al. (2019). Genome-wide association studies for yield-related traits in soft red winter wheat grown in Virginia. PLoS One 14:e0208217. doi: 10.1371/journal.pone.0208217
Würschum, T., Leiser, W. L., Langer, S. M., Tucker, M. R., and Longin, C. F. H. (2018). Phenotypic and genetic analysis of spike and kernel characteristics in wheat reveals long-term genetic trends of grain yield components. Theor. Appl. Genet. 131, 2071–2084. doi: 10.1007/s00122-018-3133-3
Xu, Y. F., Wang, R. F., Tong, Y. P., Zhao, H. T., Xie, Q. G., Liu, D. C., et al. (2014). Mapping QTLs for yield and nitrogen-related traits in wheat: influence of nitrogen and phosphorus fertilization on QTL expression. Theor. Appl. Genet. 127, 59–72. doi: 10.1007/s00122-013-2201-y
Yang, W. Y., Lu, B. R., Hu, X. R., Yu, Y., and Zhang, Y. (2005). Inheritance of the triple-spikelet character in a Tibetan landrace of common. Genet. Resour. Crop Evol. 52, 847–851. doi: 10.1007/s10722-003-6089-2
Zhai, H. J., Feng, Z. Y., Li, J., Liu, X. Y., Xiao, S. H., Ni, Z. F., et al. (2016). QTL analysis of spike morphological traits and plant height in winter wheat (Triticum aestivum L.) using a high-density SNP and SSR-based linkage map. Front. Plant Sci. 7:1617. doi: 10.3389/fpls.2016.01617
Zhang, J. L., Gizaw, S. A., Bossolini, E., Hegarty, J., Howell, T., Carter, A. H., et al. (2018). Identification and validation of QTL for grain yield and plant water status under contrasting water treatments in fall-sown spring wheats. Theor. Appl. Genet. 131, 1741–1759. doi: 10.1007/s00122-018-3111-9
Zhang, R. Q., Hou, F., Chen, J., Chen, S. L., Xing, L. P., Feng, Y. G., et al. (2017). Agronomic characterization and genetic analysis of the supernumerary spikelet in tetraploid wheat (Triticum turgidum L.). J. Integ. Agric. 16, 1304–1311. doi: 10.1016/S2095-3119(16)61469-7
Keywords: spikelet node number per spike, spike development, WFZP gene, WAPO gene, wheat
Citation: Zhang X, Qiao L, Li X, Yang Z, Liu C, Guo H, Zheng J, Zhang S, Chang L, Chen F, Jia J, Yan L and Chang Z (2022) Genetic Incorporation of the Favorable Alleles for Three Genes Associated With Spikelet Development in Wheat. Front. Plant Sci. 13:892642. doi: 10.3389/fpls.2022.892642
Received: 09 March 2022; Accepted: 14 April 2022;
Published: 03 May 2022.
Edited by:
Pengtao Ma, Yantai University, ChinaReviewed by:
Hongjie Li, Institute of Crop Sciences (CAAS), ChinaCopyright © 2022 Zhang, Qiao, Li, Yang, Liu, Guo, Zheng, Zhang, Chang, Chen, Jia, Yan and Chang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liuling Yan, bGl1bGluZy55YW5Ab2tzdGF0ZS5lZHU=; Zhijian Chang, d3JjempAMTI2LmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.