 Xiaoxia Sun†
Xiaoxia Sun† Chenchen Guo
Chenchen Guo Khawar Ali
Khawar Ali Qian ZhengQiang WeiYumeng ZhuLi Wang
Qian ZhengQiang WeiYumeng ZhuLi Wang Guishuang Li
Guishuang Li Wenjuan LiBowen ZhengQunwei Bai
Wenjuan LiBowen ZhengQunwei Bai Guang Wu*
Guang Wu*
- College of Life Sciences, Shaanxi Normal University, Xi’an, China
Endoplasmic Reticulum-Associated Degradation (ERAD) is one of the major processes in maintaining protein homeostasis. Class I α-mannosidases MNS4 and MNS5 are involved in the degradation of misfolded variants of the heavily glycosylated proteins, playing an important role for glycan-dependent ERAD in planta. MNS4 and MNS5 reportedly have functional redundancy, meaning that only the loss of both MNS4 and MNS5 shows phenotypes. However, MNS4 is a membrane-associated protein while MNS5 is a soluble protein, and both can localize to the endoplasmic reticulum (ER). Furthermore, MNS4 and MNS5 differentially demannosylate the glycoprotein substrates. Importantly, we found that their gene expression patterns are complemented rather than overlapped. This raises the question of whether they indeed work redundantly, warranting a further investigation. Here, we conducted an exhaustive genetic screen for a suppressor of the bri1-5, a brassinosteroid (BR) receptor mutant with its receptor downregulated by ERAD, and isolated sbi3, a suppressor of bri1-5 mutant named after sbi1 (suppressor of bri1). After genetic mapping together with whole-genome re-sequencing, we identified a point mutation G343E in AT1G27520 (MNS5) in sbi3. Genetic complementation experiments confirmed that sbi3 was a loss-of-function allele of MNS5. In addition, sbi3 suppressed the dwarf phenotype of bri1-235 in the proteasome-independent ERAD pathway and bri1-9 in the proteasome-dependent ERAD pathway. Importantly, sbi3 could only affect BRI1/bri1 with kinase activities such that it restored BR-sensitivities of bri1-5, bri1-9, and bri1-235 but not null bri1. Furthermore, sbi3 was less tolerant to tunicamycin and salt than the wild-type plants. Thus, our study uncovers a non-redundant function of MNS5 in the regulation of ERAD as well as plant growth and ER stress response, highlighting a need of the traditional forward genetic approach to complement the T-DNA or CRISPR-Cas9 systems on gene functional study.
Introduction
In eukaryotic cells, the endoplasmic reticulum (ER) is an important organelle for newly synthesized polypeptides. Since protein folding is not completely precise and easily affected by factors such as amino acid mutations, alterations in post-transcriptional and translational modifications, or biotic and abiotic stress, the newly proteins translocated into the ER are heterogeneous (Howell, 2013; Balchin et al., 2016). As such, a vast majority of the proteins are subjected to a sophisticated and flexible endoplasmic reticulum quality control (ERQC) system that detects the proteins and ensures their correct folding, posttranslational modifications, assembly, and secretion, otherwise the proteins will be degraded with the help of regulatory mechanisms such as molecular chaperones, sugar-binding lectins, and folding enzymes (Ellgaard and Helenius, 2003; Gidalevitz et al., 2013). As such, only terminally correctly folded proteins can reach their final destination (Ellgaard and Helenius, 2003; Strasser, 2018).
The majority of newly synthesized secretory and membrane proteins are N-glycosylated (Aebi, 2013). A specific sequence Asn-X-Ser/Thr (where X can be any amino acid except proline) is recognized by enzyme oligosaccharyltransferase complex (OST) (Kelleher and Gilmore, 2006), which integrates a three-branched tetradecasaccharide precursor Glc3Man9GlcNAC2 (glucose, mannose, and N-acetylglucosamine) from a dolichylpyrophosphate (Dol-PP) carrier to the selected asparagine residues on the nascent peptides (Helenius and Aebi, 2004; Pattison and Amtmann, 2009; Mohorko et al., 2011). Thus, the structure of N-linked glycan plays an important role in protein folding and quality control.
After the rapid removal of the two glucose residues by α-glucosidase I (GI) and α-glucosidases II (GII), the monoglucosylated N-glycan Glc1Man9GlcNAc2 will interact with the two ER-resident lectins, calnexin and calreticulin (CNX and CRT) (Trombetta, 2003; Helenius and Aebi, 2004; Deprez et al., 2005; Williams, 2006; D’Alessio et al., 2010; Stigliano et al., 2011; D’Alessio and Dahms, 2015). This slows the cleavage of the innermost glucose residue by GII, which liberates maturing Man9GlcNAc2-containing glycoproteins from CNX/CRT, thus terminating its folding process in the ER (Caramelo and Parodi, 2008). If the glycoproteins are not properly folded, they will be recognized and reglucosylated by the luminal enzyme UDP-glucose: glycoprotein glucosyltransferase (UGGT) and subjected to additional rounds of CNX/CRT cycle until the protein is fully mature (Sousa and Parodi, 1995; Parodi, 2000; Taylor et al., 2003; Helenius and Aebi, 2004; Caramelo and Parodi, 2007; Jin et al., 2007). The glycoproteins that fail terminally to acquire their native structure are retained in the ER and eventually are selected for a unique degradative mechanism known as ER-associated degradation (ERAD) (McCracken and Brodsky, 1996; Vembar and Brodsky, 2008; Christianson and Ye, 2014). Most of the previous ERAD studies were based on analysis obtained from yeast or mammals (Thibault and Ng, 2012; Christianson and Ye, 2014). The existence of similar ERAD mechanism has also been reported in plants (Strasser, 2018).
In Arabidopsis, the terminal α1,2 Man residue from C-branches of the misfolded glycoproteins is trimmed by the ER-localized α1,2 mannosidase 4 (MNS4) and α-mannosidase 5(MNS5) (Htm1 in yeast, EDEMs in mammals), generating Glc0–1Man7–8GlcNAc2 with a free α1,6 Mannose residue on the C-branch as an N-glycan ERAD signal (Lederkremer and Glickman, 2005; Quan et al., 2008; Clerc et al., 2009; Liebminger et al., 2009; Huttner et al., 2014b; Ninagawa et al., 2014; Schoberer et al., 2019). This glycan signal is recognized by EBS6/AtOS9 (YOS9 in yeast; OS-9 and XTP3-B in mammals) and Hrd3/Sel1L in plants (Denic et al., 2006; Gauss et al., 2006; Hirsch et al., 2009; Hosokawa et al., 2009; Yoshida and Tanaka, 2010; Liu et al., 2011; Su et al., 2011, 2012; Huttner et al., 2012; Ruggiano et al., 2014; Ohta and Takaiwa, 2015). Thus, terminally misfolded glycoproteins carrying an exposed α1,6-mannose residue may be recruited to a membrane-embedded Hrd1 complex (Carvalho et al., 2010; Baldridge and Rapoport, 2016), ubiquitination, and subsequent dislocation into the cytoplasm for degradation (Smith et al., 2011). The Hrd1 complex contains evolutionarily conserved components: EBS5/HRD3A (Liu et al., 2011; Su et al., 2011), Hrd1a/1b (Su et al., 2011), EBS6/AtOS9 (Huttner et al., 2012; Su et al., 2012), their associated E2 conjugase UBC32 (Cui et al., 2012), and plant-specific components: EBS7 (Liu et al., 2015) and PAWH1/PAWH2 (Lin et al., 2019).
In order to decipher the function of genes in a wide range of organisms, many different techniques have been developed over the years, but different approaches often give rise to different phenotypes. One paradoxical example is that the knockouts (via genetic inactivation) of a gene largely do not cause any obvious phenotypic symptoms, while the knockdowns (the reduction of expression) of the same gene exhibit severe biological defects. These phenomena have been previously observed in a number of model systems, including yeast (Jost and Weiner, 2015), Drosophila (Yamamoto et al., 2014), mouse (Dawlaty et al., 2011; Freudenberg et al., 2012), Zebrafish (Rossi et al., 2015; El-Brolosy et al., 2019; Ma et al., 2019), human cell lines (Lin et al., 2007, 2017; Hebbard et al., 2010; Wang et al., 2014; Speers et al., 2016), and Arabidopsis (Braun et al., 2008; Chen et al., 2014; Gao et al., 2015). These results suggest that genetic compensation in response to a gene knockout might occur (El-Brolosy and Stainier, 2017). A previous study had revealed that the ER-localized MNS4 and MNS5 accelerated the demannosylation of the C-branch to generate a terminal α1, 6-linked Man acting as the glycan signal for ERAD, and found that the null mutant mns4-1 or mns5-1 obtained by transfer DNA (T-DNA) insertion could not separately suppress the phenotypes of bri1-5 and bri1-9 mutants that have become excellent materials to study and understand the ERQC in plants (Jin et al., 2007; Hong et al., 2008, 2012). Yet, their double mutant led to the inhibition of the dwarfism of bri1-5 and bri1-9 (Huttner et al., 2014b). These results suggest that MNS4 and MNS5 are functionally redundant to each other by gene duplication or genetic compensation in response to a gene knockout, yet it has not been investigated.
Forward genetics is an effective molecular approach that has led to the identification of several important ERAD complex components in plants (Jin et al., 2007, 2009; Su et al., 2011, 2012; Hong et al., 2012; Liu et al., 2015). Here, we used the EMS-mutagenized approach to isolate another suppressor of bri1-5 mutant (sbi3, suppressor of bri1 3) that carried a point mutation Gly343Glu in MNS5 (AT1G27520). We found that the dwarf phenotype of bri1-5 and bri1-9 was suppressed by the sbi3 mutant. In addition, we found that sbi3 inhibited the degradation of another recently reported ER-retained BRI1 mutant, bri1-235 (Hou et al., 2019), which has a single amino acid substitution from Ser to Phe at position 156 in the less conserved fourth LRR of BRI1 extracellular domain. Moreover, sbi3 led to ER stress and was less tolerant to salt. Therefore, our finding demonstrates that MNS5 has a non-redundant function in the regulation of plant growth, Interestingly, the reverse transcription PCR (RT-PCR) analysis reveals that the expression levels of MNS4 and MNS5 had no change in mutant sbi3, namely a lack of a genetic compensatory response in sbi3. Surprisingly, mns5-1 produces no transcripts (Huttner et al., 2014b), suggesting that the genetic compensatory response in mns5-1 might not be due to the upregulaton of both MNS4 and MNS5. As a result, the cause of the non-redundant function in MNS5 remains to be uncovered. Nevertheless, our finding provides a new avenue for further investigation of the ERAD in planta and raises awareness of the importance of using both forward and reverse genetic studies for gene functions in planta.
Materials and Methods
Isolation of bri1-5 (Ws-2) and bri1-5 Suppressor Mutants
The bri1-5 (Ws-2) seeds were mutagenized with 0.4% ethyl methanesulfonate (Sigma Aldrich). The M2 seeds, derived from around 10,000 M1 plants, were screened on one-half-strength Murashige and Skoog medium. These seeds were stratified in the dark at 4°C for 4 days, and then grown in the light at 22°C for 1 day, in the dark at 22°C for 4 days, and in the light at 22°C for 1 day (Wu et al., 2011). After germination, the seedlings with long hypocotyls were transferred into the soil for continued growth under a 16 h-light/8 h-dark growth condition for 4 weeks in the greenhouse, and mature seeds were then collected. The potential suppressors were genotyped using a bri1-5-dCAPS marker to eliminate pollen or seed contamination. The derived sbi3 bri1-5 homozygous mutants were back-crossed three times to eliminate any unlinked second-site mutations.
Plant Materials and Growth Conditions
The Arabidopsis thaliana ecotypes Wassilewskija-2 (Ws-2) and Columbia-0 (Col-0) were used as the wild-type (WT) control in this study. The mutants bri1-9, bri1-235, bri1-301, bri1-116, det2-1, cpd, and bin2-1 were used in the Col-0 background, the mutant bri1-119 was in Enkheim-2 (En-2), and the mutant bri1-5 was in Ws-2 background. The sbi3 was discovered in the genetic screen for the extragenic suppressor of bri1-5. The mutant sbi3 bri1-5 was crossed into bri1-9, bri1-235, bri1-301, bri1-116, bri1-119, det2-1, cpd, and bin2-1 respectively, to obtain the different background double mutants for genetic analyses. The seeds were surfaced sterilized by washing for 5 min in 75% (v/v) ethanol containing 0.05% (v/v) Tween 20 and for 1 min in 5% NaClO, followed by three-five times washes with sterilized water. Under sterile conditions, the seeds were sown on ½ Murashige and Skoog (MS) medium and plated at 4°C for 2–3 days to break dormancy and increase uniform germination, the seeds were germinated in Petri dishes at 22°C with 70% humidity under long-day (16/8-h light/dark) photoperiod (∼120 μmol.m–2.s–1) condition. One week after germination, seedlings were transferred into soil and grown under the same controlled conditions.
Pavement Cell Analysis: Microscopy and Image Analysis
To observe the profile of the pavement cells, 7-day-old cotyledons were stained in propidium iodide (PI, Sigma) (10 μg/mL in H2O) for 5–10 min and washed three times (10 min each) in deionized water. Stained cotyledons were fixed firmly in water on slides for microscopy. The pavement cells were imaged using a Leica TCS SP8 laser scanning confocal microscope. Images were obtained with × 40 objective for propidium iodide (PI) staining. Images were captured by the following setting: 1.0 μm z-step size, 561 nm laser excitation, and 590–630 nm emission. ImageJ software was used to measure the lobe length, neck width, perimeter, and area1. Circularity was analyzed as previously described by Zhang et al. (2011). At least 30 cells were used for the analysis. The data was recorded and the significance was analyzed using the Student’s paired t-test.
Map-Based Cloning
Map-based cloning was performed as described previously (Lukowitz et al., 2000). The sbi3 bri1-5 (Ws-2) grown in the greenhouse was crossed with Col-0, and the resulted F1 plants were germinated and allowed for self-pollination. The bri1-5–like seedlings with shorter hypocotyls were selected in the segregating F2 population, and grown in the greenhouse to obtain F3 seeds. To map the mutated locus, we screened for seedlings with long hypocotyls exhibiting the suppressed-bri1-5 phenotype in the F3 generation. Genomic DNA from 50 to 100 individual seedlings of the mapping population was extracted. The mutation site was first mapped to a region close to the SSLP markers by Bulked Segregant Analysis using pooled DNA samples. simple sequence length polymorphisms (SSLP) markers were listed in Supplementary Table 1. Meanwhile, the Whole Genome Resequencing was also performed from the genomic DNA of the mapping population by Beijing Nuohe Zhiyuan Technology Co., Ltd. MutMap method was used to rapidly identify the suppressor gene (Abe et al., 2012), and a graph relating single-nucleotide protein (SNP) positions and SNP-index was generated for all 5 Arabidopsis thaliana chromosomes. The SNP index was defined as the ratio between the number of reads of a mutant SNP and the total number of reads corresponding to the SNP. The causative SNP should be shared by all the mutant plants, therefore, SNP-index = 1 harbored the gene responsible for the mutant phenotype, and 0.5 for the unlinked loci to the mutant phenotype.
Generation of Constructs and Transgenic Plants
The SBI3 and sbi3 (G343E) were amplified by PCR from cDNA of WT and sbi3. Both amplified genes were subsequently cloned into a T-Vector PMD19 (TaKaRa), and the cloned genes were verified by DNA sequencing. Later, the T-vectors carrying the genes were digested with KpnI and BamHI and cloned into the binary vector pCHF3 that carries the 35S promoter and a synthetic gene for the green fluorescent protein (GFP) to obtain p35S:SBI3-GFP and p35S:sbi3-GFP constructs, respectively. These resulting constructs were first transformed individually into the sbi3 bri1-5 mutants via Agrobacterium tumefaciens (GV3101)-mediated transformation using the floral-dipping method (Clough and Bent, 1998). The transformants were germinated and screened on 1/2 MS medium with kanamycin (50 mg/L). To confirm the transformants, the transgene from each transgenic line was sequence-verified.
Transcript Analysis by RT-PCR
The two-week-old Arabidopsis seedlings were collected and ground in liquid nitrogen into a fine powder, and their total RNAs were extracted using the TRIzol reagent (Invitrogen) following the manufacturer’s instructions. First-strand complementary DNA (cDNA) was synthesized from 1 μg/2 mg of the total RNA using an M-MLV First Strand cDNA Synthesis Kit (Omega, TQ2501-02, Norcross, GA, United States). The cDNA was then amplified by a Semi-quantitative RT-PCR system with gene-specific primers for CPD, DWF4, BAS1, BRI1, and ACTIN2 to study the expression levels. The PCR amplified cDNA fragments were separated by agarose gel electrophoresis, the ACTIN2 was used as an internal control, RT-PCR experiment was repeated three times. All primers used for RT-PCR were given in Supplementary Table 1.
Western Blot
Arabidopsis seedlings treated with or without kifunensine (Kif), cycloheximide (CHX), MG132 (Abcam), or 24-epibrassinolide (24-eBL, Sigma) were harvested and ground into fine powder in liquid nitrogen. The total protein was suspended in 2x SDS sample buffer [100 mM Tris, pH 6.8, 4% (w/v) SDS, 20% (v/v) glycerol, 0.2% (w/v) bromophenol blue, 2% (v/v) β-Mercaptoethanol] and denatured at 100°C for 10 min. The protein was then centrifuged for 5 min at the top speed, and the resulting supernatants were then resolved on 8% SDS-PAGE gel and transferred into nitrocellulose membrane (Pall Gelman). Anti-GFP antibody (1:1,000 dilution, TransGen, HT801) was used to detect GFP fusion proteins, Anti-BRI1 antibody (1:1,000 dilution, Agrisera) was used to detect the protein expression levels of BRI1/bri1, and BES1 antibody (1:3,000 dilution, kindly provided by J. Li, Lanzhou University, China) was used to detect the phosphorylation status of BES1.
Endo H Treatment
Total proteins were extracted from the 2-week-old grown seedlings with 2×SDS sample buffer. The extracted proteins were denatured at 100°C for 10 min in 1×glycoprotein denaturing buffer and centrifuged for 10 min at 10,000 × g, the resulted supernatant was transferred into a new 1.5 mL tube and incubated with or without Endo H (NEB, New England Biolads) digestion in the 1 × G5 buffer for 1 h at 37°C following the manufacturer’s protocol (New England Biolabs). Both control and Endo H–treated samples were separated by SDS-PAGE and transferred into nitrocellulose membrane (Pall Gelman), and the proteins were quantified by Anti-BRI1 antibody.
Results
The sbi3 Mutation Suppresses Dwarf Phenotypes of bri1-5, bri1-235, and bri1-9
To identify the additional regulators of the ERAD, we performed a genetic screen for a sensitized extragenic suppressor in the bri1-5 mutant that encodes a functionally competent receptor with a mutation in one of the cysteine pairs (C69Y) in the extracellular domain, serving a substrate of ERAD as a misfolded receptor kinase (Noguchi et al., 1999). We identified a putative EMS–mutagenized suppressor, sbi3 that weakly suppressed the dwarf phenotype of bri1-5 mutant. sbi3 bri1-5 exhibited expanded rosette leaves with noticeable petioles at rosette stages (Figure 1A), long siliques, and long floral stems in the soil (Figures 1B,C), longer hypocotyls when grown in the dark (Figure 1D). In mutant sbi3, which was obtained by the hybridization of sbi3 bri1-5 and Ws-2, the bolting time was 5–6 days earlier than that of the wild type Ws-2, and the number of rosette leaves was fewer in sbi3 (Supplementary Figure 1). When the pavement cell shape in Ws-2, bri1-5, sbi3 bri1-5, and sbi3 mutants was examined to understand the cellular mechanism underlying these phenotypes, we found that the mutant bri1-5 exhibited a smaller area and perimeter of pavement cells and defective lobe structure. By contrast, the quantitative analysis results confirmed that pavement cell area and perimeter value were bigger, with smaller circularity value and narrower necks, in the double mutant sbi3 bri1-5 than that of bri1-5 (Figure 1E and Supplementary Figure 2). The morphological analysis of these phenotypes revealed that sbi3 partially inhibited the dwarfing phenotypes of bri1-5. To determine whether BR responses were altered in the sbi3 bri1-5 lines, we performed an exogenous BR (24-eBL) sensitivity assay on root growth to various concentrations of 0–1,000 nM. We found that the increasing concentrations of 24-eBL had little effect on the root elongation of the bri1-5 seedlings, but greatly inhibited the root growth of the Ws-2, sbi3, and the sbi3 bri1-5 seedlings in a dose-dependent manner (Figure 1F). Furthermore, we also examined the effect of exogenously applied Propiconazole (PCZ, Solarbio), a BR biosynthesis inhibitor that blocks the production of BRs, found that the hypocotyl growth of Ws-2, sbi3 bri1-5, and sbi3 was more reduced by PCZ when compared to bri1-5 (Figure 1G). These results confirmed that the sensitivity of double mutant sbi3 bri1-5 to 24-eBL and PCZ had changed in comparison to bri1-5 due to the point mutation in the SBI3 gene.
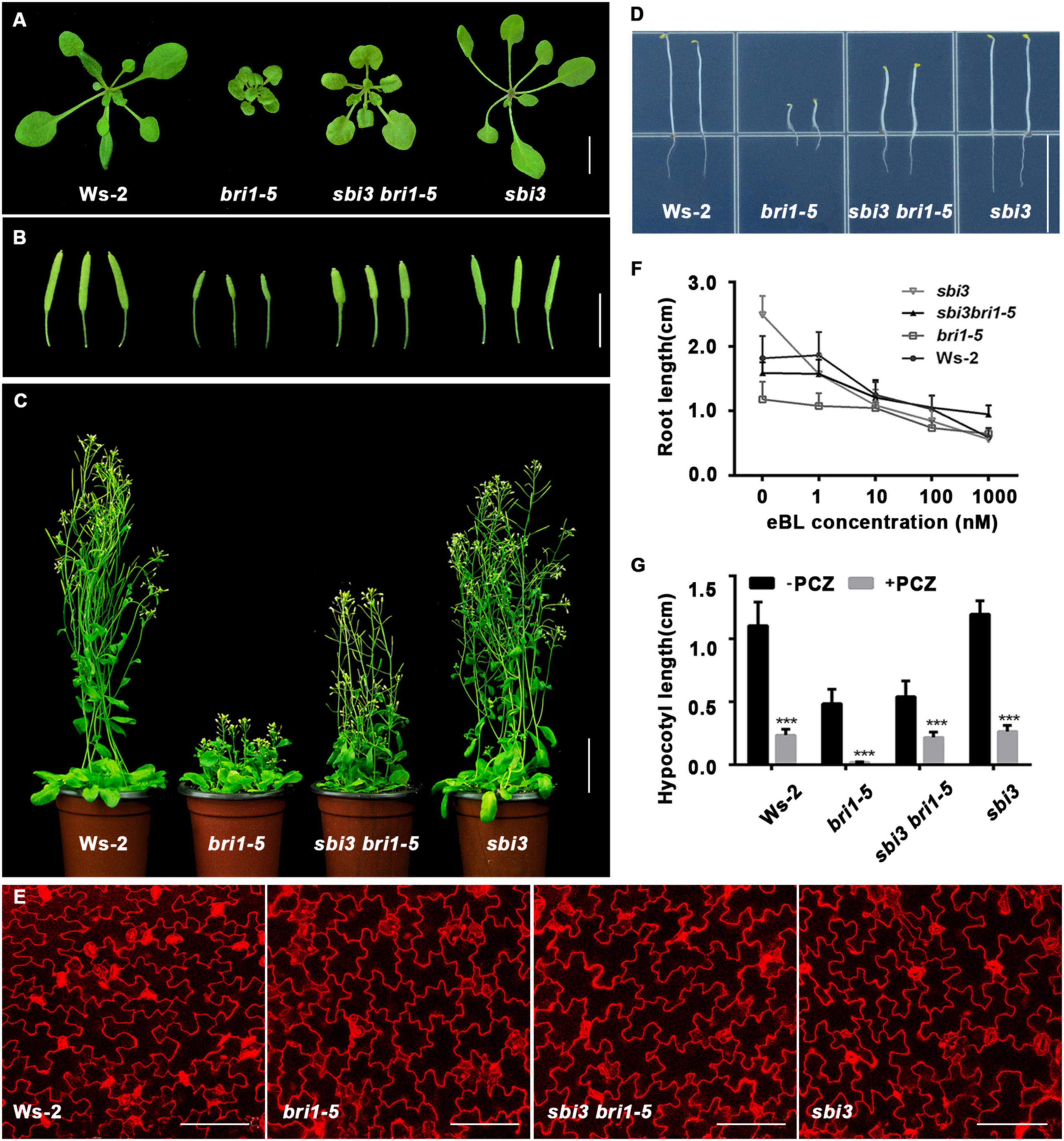
Figure 1. sbi3 mutation partly suppresses bri1-5 dwarfism. (A) Phenotypes of 2-week-old soil-grown seedlings of Ws-2, bri1-5, sbi3 bri1-5, and sbi3. Scale bar, 1 cm. (B) 2-month-old siliques of Ws-2, bri1-5, sbi3 bri1-5, and sbi3. Scale bar, 1 cm. (C) 2-month-old mature plants of Ws-2, bri1-5, sbi3 bri1-5, and sbi3. Scale bar, 3 cm. (D) Hypocotyl comparison of 5-day-old dark-grown seedlings of Ws-2, bri1-5, sbi3 bri1-5, and sbi3. Scale bar, 1.5 cm. (E) The morphology of cotyledon pavement cells of 7-day-old seedlings from Ws-2, bri1-5, sbi3 bri1-5, and sbi3. Cotyledons were stained by propidium iodide (PI). Scale bar, 100 μm. (F) Root growth phenotypes of 8-day-old Ws-2, bri1-5, sbi3 bri1-5, and sbi3 seedlings grown in 1/2 MS medium with different 24-epibrassinolide (24-eBL) concentrations at 22°C under long-day (16/8-h light/dark) condition. Quantitative analysis of root length plotted as the line graph and displayed in panel (F), n ≥ 30 seedlings. Error bar represents ± SD, three independent assays. (G) Hypocotyl growth phenotype of 5-day-old Ws-2, bri1-5, sbi3 bri1-5, and sbi3 seedlings grown in 1/2 MS medium treated with or without 5 μM PCZ in the dark. Quantitative analysis of hypocotyl length of 5-day-old dark-grown seedlings plotted as histograms displayed in panel (G), n ≥ 30 seedlings. ***P < 0.001 as two-way ANOVA with Sidak’s multiple comparisons test.
Consistently, we found that the sbi3 bri1-235 double mutant was also a larger and less compact rosette with taller stature at maturity compared to the bri1-235 mutant (Figures 2A–D). As shown in Figure 2E, the treatment of bri1–235 with increasing concentrations of brassinolide (24-eBL) had a less effect on root growth, compared to Col-0 and sbi3 bri1–235 (Figure 2E). As expected, the hypocotyl of the Col-0, bri1-235, and sbi3 bri1-235 seedlings exhibited hypersensitivity to PCZ (Figure 2F). We, therefore, speculated that sbi3 mutant might broadly block the ERAD in the bri1 mutants with ER-localization.
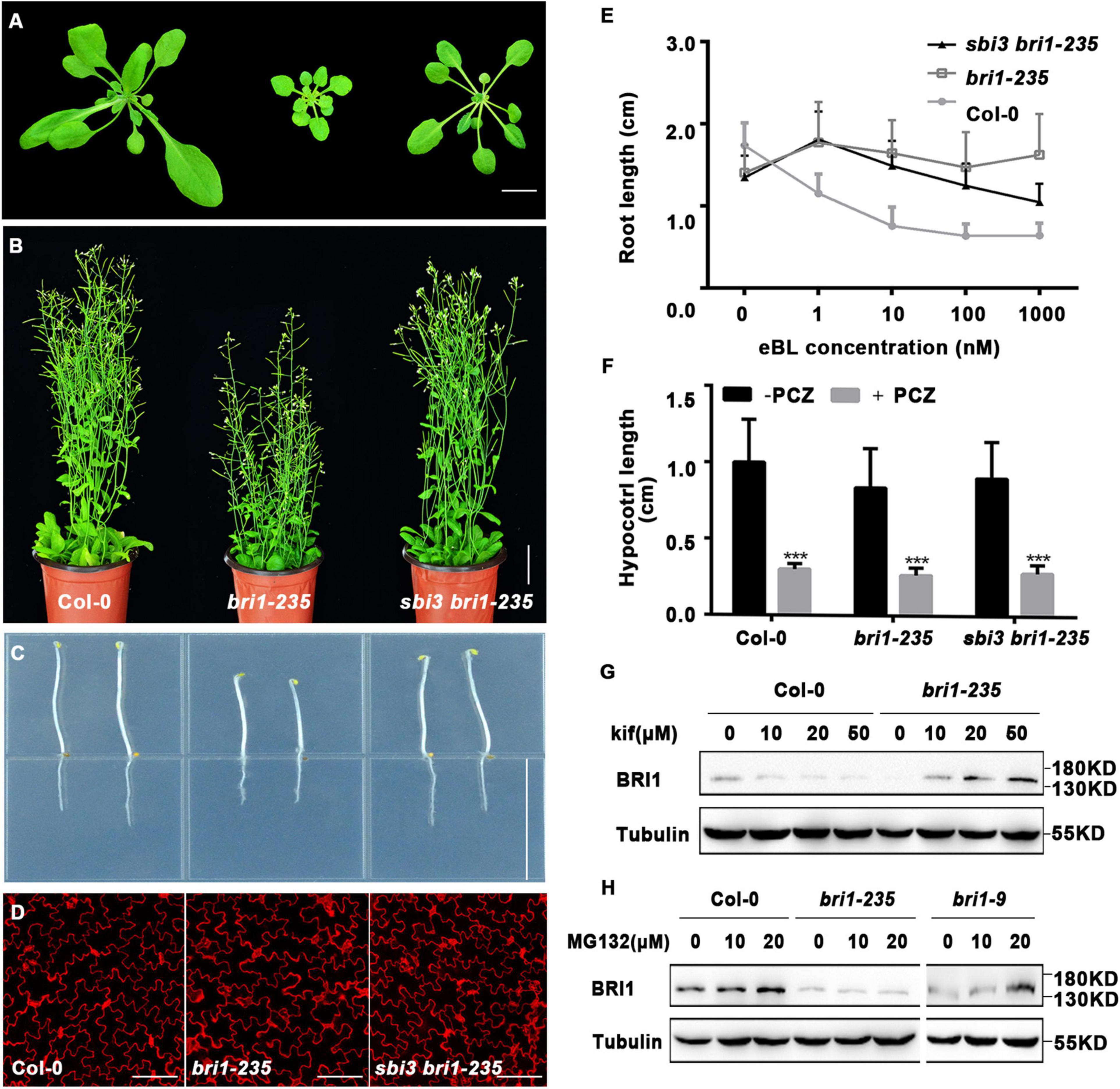
Figure 2. A mutant sbi3 weakly suppresses bri1-235 phenotypes. (A) Phenotypes of two-week-old soil-grown seedlings of Col-0, bri1-235, and sbi3 bri1-235. Scale bar, 1 cm. (B) Phenotypes of 2-month-old mature plants of Col-0, bri1-235, and sbi3 bri1-235. Scale bar, 3 cm. (C) Hypocotyl comparison of 5-day-old dark-grown seedlings of Col-0, bri1-235, and sbi3 bri1-235. Scale bar, 1.5 cm. (D) Morphology of cotyledon pavement cells of 7-day-old seedlings from Col-0, bri1-235, sbi3 bri1-235. Scale bar, 100 μm. (E) The 24-eBL-induced root inhibition assay. Quantitative measurements analysis of root length of 8-day-old Col-0, bri1-235, and sbi3 bri1-235 seedlings were plotted as the line graph and displayed in panel (E), n ≥ 30 seedlings. Error bar represents ± standard deviation (SD), three independent assays were performed with similar results. (F) Measurements of hypocotyl length of 5-day-old dark-grown in 1/2 MS medium treated with or without 5 μM PCZ seedlings were plotted as a histogram. ***P < 0.001. (G) Immunoblot analysis of BRI1 abundance in 2-week-old seedlings incubated with or without Kif for 24 h in liquid half-strength MS medium. The protein abundance of BRI1-235 was increased by treatment with Kif. (H) Immunoblot analysis of BRI1 proteins in 2-week-old seedlings of Col-0, bri1-235, and bri1-9 supplemented with or without MG132 for 24 h in liquid half-strength MS medium.
Given the fact that bri1-235 is retained in the ER (Hou et al., 2019), one-week-old seedlings were transferred to a half-strength MS medium supplemented with or without 10 μM kifunensine (Kif), a widely used inhibitor of α1,2-mannosidases (Tokunaga et al., 2000), for continued growth for 9 days. We found that kif treatment exhibited less compact rosette, with short and radially swollen roots in bri1-235 (Supplementary Figure 3A), which was consistent with findings on bri1-5 and bri1-9 mutants (Hong et al., 2008, 2009). The quantitative analysis of root length revealed similar responses to kif treatment for both bri1-235 and wild-type seedlings (Supplementary Figure 3B).
The above findings suggest that the low BRI1-235 protein abundance in bri1-235 seedlings could have been caused by ERAD. To test this hypothesis, we treated bri1-235 and the wild-type seedlings with Kif. The result showed that Kif treatment significantly increased the abundance level of BRI1-235 in a dose-dependent manner, but had little effect on the BRI1 stability in Col-0 (Figure 2G), suggesting that ER-retained BRI1-235 undergoes ERAD, similar to the findings on bri1-5 and bri1-9 (Hong et al., 2008, 2009). It has been shown previously that ER-retained BRI1 mutant, bri1-5, was degraded by a proteasome-independent ERAD process (Hong et al., 2008), but ERAD of ectopically expressed BRI1-9:GFP involved proteasomes (Hong et al., 2009). To examine the ERAD mechanism of BRI1-235, we treated 2-week-old bri1-235 seedlings with MG132, a widely used proteasome inhibitor that can prevent degradation of proteasome-dependent ERAD substrates (Schmitz and Herzog, 2004). The treatment analysis showed that the BRI1 protein abundance was increased drastically in MG132 treated Col-0 and bri1-9 but not bri1-235 (Figure 2H), indicating that BRI1 protein is degraded by a proteasome-independent ERAD process in bri1-235.
We then asked whether the sbi3 could inhibit the proteasome-dependent ERAD in bri1-9. Indeed, we found that sbi3 mildly rescued the dwarf phenotype of bri1-9 mutant that had a small rosette, a short hypocotyl in the dark, small perimeter, and area, and short inflorescence stems of mature plants (Supplementary Figures 4A–D). In addition, the sbi3 bri1-9 also showed increased sensitivity to exogenous BRs as compared to bri1-9 (Supplementary Figure 4E).
sbi3 Is a New Mutant Allele of MNS5
To understand how sbi3 mutation inhibited the dwarf phenotypes of bri1-5, bri1-235 and bri1-9, we tried to clone the SBI3 gene. We first backcrossed the sbi3 bri1-5 double mutant to bri1-5 to obtain the resulting F1 plants showing the bri1-5 phenotype. The F2 plants showed an approximately 3:1 dwarf-to-normal phenotypic segregation, indicating that sbi3 was a recessive mutant in a single gene. In order to isolate the sbi3 gene, we employed the PCR-based positional cloning approach with pooled genomic DNA of 50-100 F3 that had sbi3 bri1-5-like seedlings derived from the mapping population and located the SBI3 locus to a genomic region close to the SSLP marker chr1-9621kb on the top of chromosome 1 by Bulked segregant analysis (Supplementary Figure 5).
Subsequently, whole-genome re-sequencing (Supplementary Figure 6) revealed higher SNP-index values in the region between 8 and 10 Mb on chromosome 1. Two genes with SNP-index = 1 were identified in the candidate region by comparing with the published reference sequences of Col-0 and Ws-2. An SNP at position chr1-9,576,968 was located in a coding region of AT1G27570 (CDS: G499A, Protein: V167I). But according to the sequence alignment, it was not predicted to be the mutant position because it is not conservative. Another SNP (G343E) was at position chr1-9,560,890 bp, which was a single-nucleotide polymorphism substitution of G to A corresponded to the 11th exon in AT1G27520 (MNS5) between sbi3 bri1-5 and bri1-5. MNS5 encodes a glycosyl hydrolase family 47 known to be a critical ERAD component, suggesting that the non-synonymous mutation identified in G343A (for protein) accounts for suppressor bri1-5 sbi3 phenotype. SBI3/MNS5 encodes a polypeptide of 574-aa, which consists of 15 exons plus 14 introns (Figure 3A). Sequence alignment showed that this mutated G343 residue was absolutely conserved in SBI3/MNS5 among selected species shown in Supplementary Table 2 and Figure 3B.
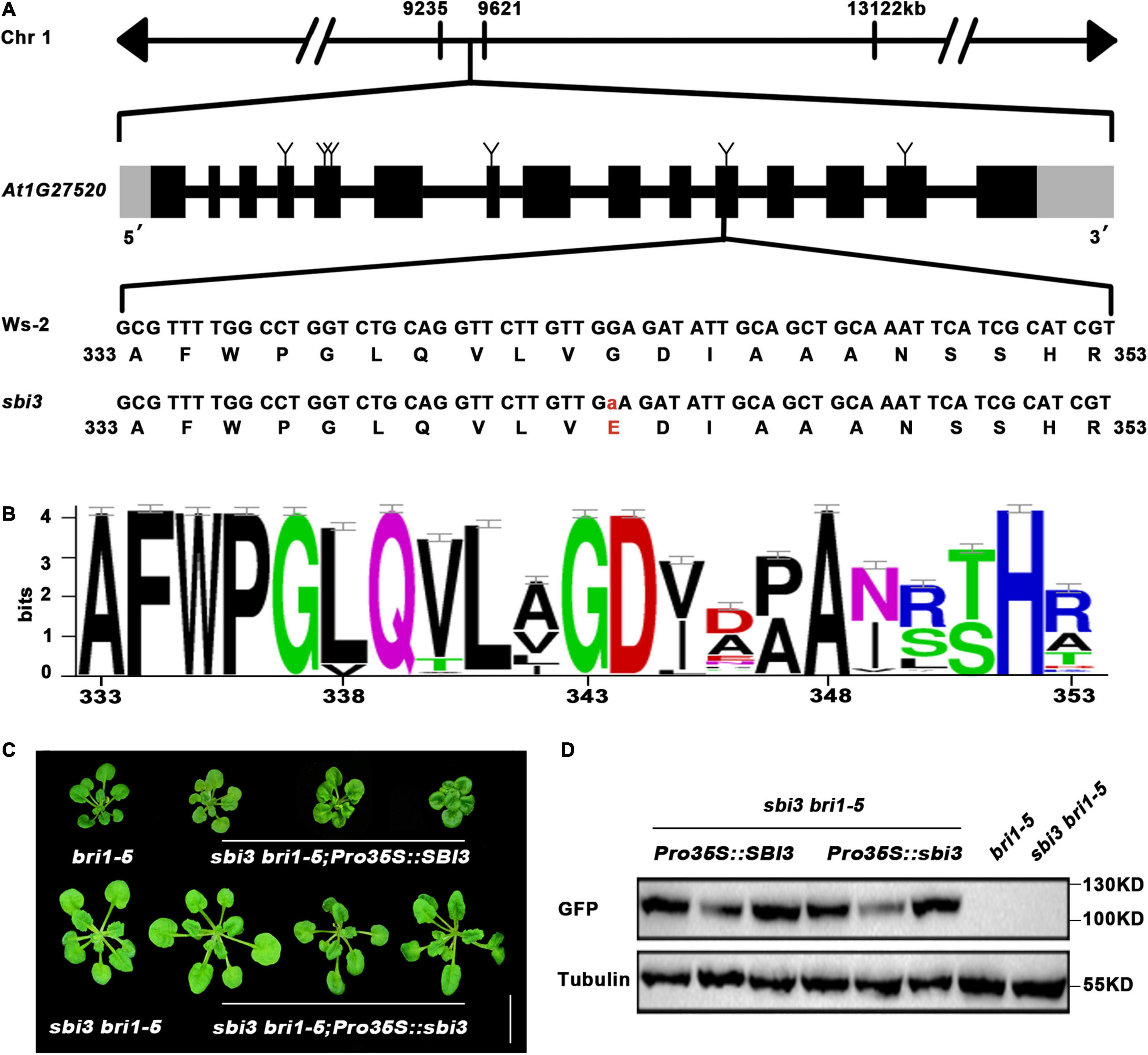
Figure 3. The molecular cloning of sbi3. (A) Schematic presentation of the mutation site in sbi3. (B) Sequence alignment of a small part of the SBI3 protein among different species. G residue at the 343rd position was highly conserved. (C) Three weeks-soil-grown plants of bri1-5, sbi3 bri1-5, three SBI3-complemented sbi3 bri1-9 transgenic lines carrying an SBI3 transgene driven by the 35S promoter, and three independent sbi3 overexpression transgenic lines on sbi3bri1-5 mutants served as control. Scale bar, 1 cm. (D) Protein expression levels of bri1-5, sbi3 bri1-5, and the corresponding transgenic plants with GFP tag shown in panel (C) were detected with anti-GFP antibody. Tubulin served as the loading control.
That MNS5 and SBI3 are the same genes and were further confirmed by two additional experiments. First, this detected SNP was converted to a derived cleaved amplified polymorphic sequence (dCAPS) marker (Neff et al., 1998), which was used to confirm a tight genetic linkage between the G–A mutation and the sbi3 bri1-5 phenotype in several F3 mapping populations. Second, we performed a genetic complementation experiment with SBI3/MNS5–GFP and sbi3-GFP constructs to rescue the sbi3 bri1-5 mutant phenotypes, such that the phenotypes of three independent transgenic lines expressing SBI3/MNS5–GFP but not sbi3-GFP had similar phenotypes to bri1-5, confirming that At1g27520 (MNS5) was indeed SBI3 (Figures 3C,D).
The Molecular Mechanism of sbi3
To understand the underlying biochemical mechanism of which sbi3 restores the BR receptor function of bri1-5, bri1-235, and bri1-9, we checked the expression level of these BR responsive genes (DWF4, CPD, and BAS1) in Ws-2, bri1-5, sbi3 bir1-5, and sbi3 mutants by RT-PCR. It is known that DWF4, CPD (BR biosynthesis genes), and BAS1 (BR inactivation genes) are sensitive feedback regulators for BR signaling (Tanaka et al., 2005). The results showed that the expression level of DWF4 and CPD was significantly downregulated in wild-type, sbi3, and sbi3 bri1-5 compared to the expression level of DWF4 and CPD in bri1-5 plants. On the contrary, the expression level of BAS1 was upregulated in the wild-type, sbi3 and sbi3 bri1-5 compared to bri1-5 (Figure 4A). However, the expression level of BRI1/bri1-5 in SBI3 and sbi3 backgrounds was similar (Figure 4A). However, the immunoblot analysis revealed that the sbi3 mutation greatly elevated the BRI1-5, BRI1-235, and BRI1-9 protein abundance level in double mutants (Figures 4B,C and Supplementary Figure 4F). Together, these data suggested that SBI3 could mediate the BRI1 abundance through a posttranscriptional mechanism.
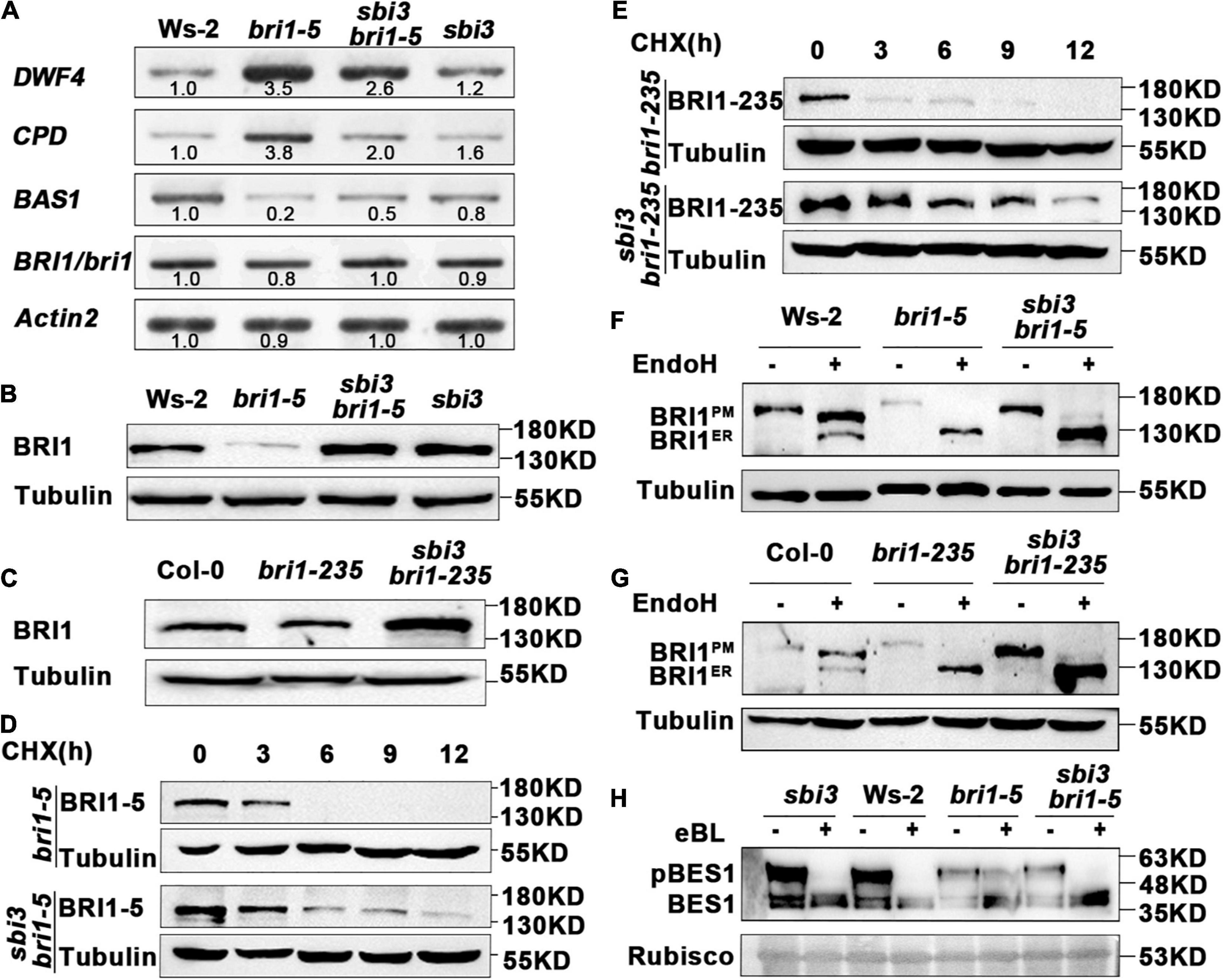
Figure 4. sbi3 mutation inhibits the Endoplasmic Reticulum-Associated Degradation (ERAD) of bri1-5 and bri1-235 through a posttranscriptional mechanism. (A) The expression abundance of transcripts for BR receptor BRI1 in Ws-2, bri1-5, sbi3 bri1-5, and sbi3 seedlings was detected by semi-quantitative RT-PCR. The transcripts of BRI1/bri1 in the wildtype (WT) or the mutant were similar. Actin2 was used as an internal control. N = 3 biological replicates. (B) Western blot analysis of BRI1 protein abundance in Ws-2, bri1-5, sbi3 bri1-5, and sbi3. Extracts were prepared from 14-day-old seedlings grown in 1/2 MS medium. Specific antibodies: Anti-BRI1, Anti-Tubulin (control). (C) Immunoblot analysis of BRI1 protein abundance in Col-0, bri1-235, and sbi3 bri1-235. Specific antibodies: Anti-BRI1; Anti-Tubulin (control). (D) Immunoblot analysis of BRI-5 stability in sbi3 bri1-5 with the anti-BRI1 antibody. Two-week-old seedlings were treated with 180 μM CHX for indicated incubation times. (E) Immunoblot analysis of BRI1-235 stability in sbi3 bri1-235 with the anti-BRI1 antibody. Two-week-old seedlings were treated with 180 μM CHX for indicated incubation times. (F) Endoglycosidase H (EndoH) analysis of Ws-2, bri1-5, sbi3 bri1-5, sbi3. BRI1ER is the ER-localized proteins form, while BRI1PM denotes the localization of BRI1 proteins in the plasma membrane. (G) EndoH analysis of Col-0, bri1-235, and sbi3 bri1-235. (H) Immunoblotting of eBL induced dephosphorylation of sbi3, Ws-2, bri1-5, and sbi3 bri1-5. Rubisco served as a loading control.
To confirm that the increased BRI1-5 abundance was caused by increased synthesis or reduced degradation in sbi3 bri1-5, we treated 2-week-old seedlings of bri1-5 and sbi3 bri1-5 mutants with 180 uM CHX, a widely used protein biosynthesis inhibitor, and then analyzed the BRI1-5 abundance by immunoblot assay. Our findings revealed that CHX caused a rapid disappearance of the mutant BR receptor in bri1-5 lines, but had a much weaker effect on the BRI1-5 abundance in sbi3 bri1-5 lines. Similar to observations in sbi3 bri1-235 and sbi3 bri1-9 mutants, BRI1 became non-detectable after 9 h of CHX treatment in bri1-235 and bri1-9, but BRI1 abundance level in sbi3 bri1-235 and sbi3 bri1-9 was stable (Figures 4D,E and Supplementary Figure 4G). We thus concluded that the observed increased BRI1 abundance in the sbi3 bri1 mutant is largely caused by attenuated degradation rather than by increased protein biosynthesis, supporting a functional role of SBI3 in the ERAD of the mutant BR receptor.
Endoglycosidase H (Endo H) is capable of cleaving N-glycan of ER-retained proteins but not Golgi-processed complex-type N-glycan. Endo H sensitivity assay using an anti-BRI1 antibody provided an accessible biochemical way to examine the subcellular distribution of BRI1, As shown in Figures 4F,G, a small amount of BRI1-5 and BRI1-235 carrying complex-type N-glycan in sbi3 bri1-5 and sbi3 bri1-235, respectively. This effect was more obvious when the sample volume increased or the exposure time was extended, whereas we failed to detect the complex-type N-glycan in bri1-5 or bri1-235 (Figures 4F,G). Consistent with these, BRI1-9 was sensitive, and a minor fraction that was insensitive to Endo H in sbi3 bri1-9 (Supplementary Figure 4H), likely due to the escape of BRI1-5, BRI1-235, and BRI1-9 from the ER, suggesting that sbi3 reduces the stringency of quality control of BRI1-5, BRI1-235, and BRI1-9. We also examined eBL-induced changes in the phosphorylation status of BES1, a marker of BR signaling, BES1 was rapidly dephosphorylated in sbi3 bri1-9, resembling sbi3 and Ws-2. However, little change was found in bri1-5 mutants (Figure 4H).
To investigate whether sbi3 also restored other bri1 mutants, we crossed sbi3 into several other bri1 alleles including bri1–301, bri1–119, and bri1–116. The bri1–301 possesses a missense mutation in the kinase domain of BRI1 (Xu et al., 2008), whereas bri1–119 mutant contains a mutation in the ID-LRR22 domain (Noguchi et al., 1999) while bri1-116 is a null allele (Li and Chory, 1997; Friedrichsen et al., 2000). None of these bri1 mutants tested was suppressed by sbi3 (Figures 5A–C). Our results suggest that sbi3 regulates the abundance of kinase-active and misfolded ER-retained BRI1. Meanwhile, we studied the genetic interaction between sbi3 and BR-deficient mutants det2-1 (de-etiolated 2, a weak BR biosynthetic mutant) (Li et al., 1996) and cpd (a strong BR biosynthetic mutant) (Szekeres et al., 1996). sbi3 slightly rescued the growth retardation phenotype of det2-1, but it had no effect on cpd, probably attributable to a small amount of endogenous BRs in plants (Figures 5D,E). In addition, sbi3 failed to rescue the growth retardation phenotypes of bin2-1 (Li and Nam, 2002), indicating that SBI3 did not act in the BR signaling downstream of BIN2.
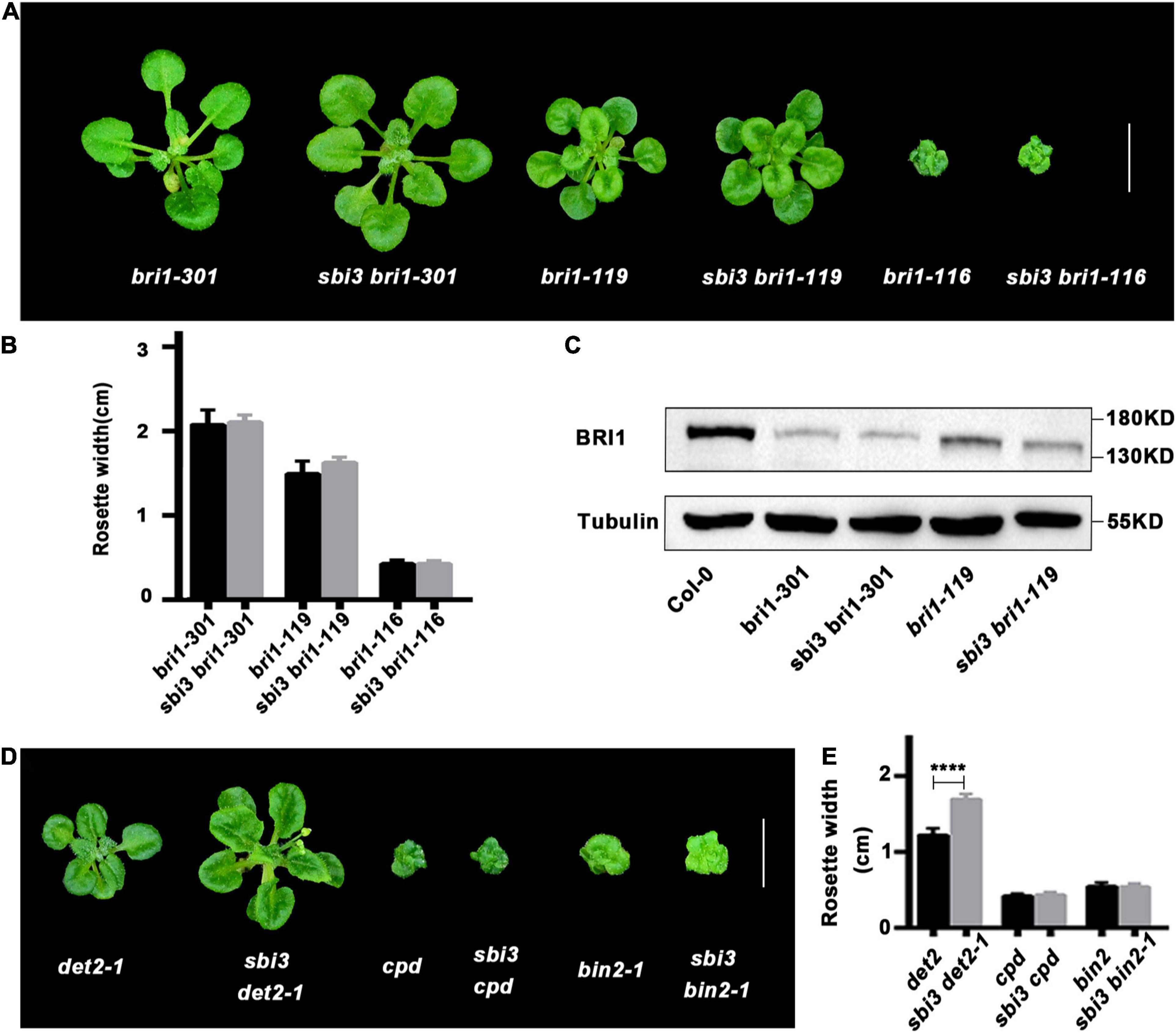
Figure 5. The double mutants of other backgrounds. (A) Phenotypes of bri1-301, bri1-119, bri1-116 and their corresponding double mutants with sbi3 grown in soil for 3 weeks. Scale bar, 1 cm. (B) Comparison of rosette width of 3-week-old plants measured using the ImageJ software. Error bar represents ± standard deviation (SD), n ≥ 10. ***P < 0.001 as two-way ANOVA with Tukey’s multiple comparisons test. (C) Western blotting analysis of BRI1 protein abundance in Col-0, bri1-301, sbi3 bri1-301, bri1-119, and sbi3 bri1-119. (D) Phenotypic comparison of det2-1, cpd, bin2-1 and their corresponding double mutants with sbi3 grown in soil for 3 weeks. Scale bar, 1 cm. (E) The width of rosette leaves of 3-week-old plants were measured using the ImageJ software. Error bar represents ± standard deviation (SD), n ≥ 10. ****P < 0.0001 as one-way ANOVA with Tukey’s multiple comparisons test.
The ER Stress Induced by sbi3
Previous studies have shown that defective mutations of ERAD often cause the accumulation of aberrant proteins, resulting in activation of the unfolded protein response (UPR) pathway, a highly conserved ER stress response pathway. In this pathway, ER chaperones and ERAD components are upregulated in response to agents tunicamycin (tunicamycin, TM, an ER stress-inducing agent that inhibits protein glycosylation) and dithiothreitol (dithiothreitol, DTT, another widely ER stress-inducer that reduces protein disulfide bonds) to maintain proteostasis, such as BIPs, protein disulfide isomerases (PDIs), calreticulins/calnexin (CRT/CNX) (Su et al., 2011; Huttner et al., 2014b; Lin et al., 2019), OS9/EBS6 (Huttner et al., 2012; Su et al., 2012; Lin et al., 2019), HRD3/SEL1L//EBS5, HRD1 (Su et al., 2011; Lin et al., 2019), and EBS7 (Liu et al., 2015). Furthermore, in mutants lacking ERAD components, such as hrd3 (sel1l), os9, mns4 mns5, and pawh1 pawh2 (Liu et al., 2011; Huttner et al., 2012, 2014b; Lin et al., 2019), salt sensitivity was increased.
We found that the expression abundance of PDI5 was increased in sbi3 bri1-235, indicating that the sbi3 mutation activates the UPR pathway (Figure 6A). To test whether the sbi3 mutation affects the plant ER stress tolerance, the seedlings of the Arabidopsis wild type and sbi3 were grown on ½ MS medium containing 0.3 μg/mL TM, and we found that sbi3 is less tolerant to TM (Figure 6B). Consistently, RT-PCR analysis showed that the expression of BIP3 and PDI5 was clearly higher in sbi3 seedlings treated with 5 μg/mL TM for 6 h compared to their controls. However, we could not detect expression differences in BIP3 and PDI5 in their responses to TM treatment in between Ws-2 and sbi3 (Figure 6C), in agreement with the response of mns4-1 mns5-1 to TM treatment (Huttner et al., 2014b). As expected, the expression levels of MNS4 and MNS5 were not upregulated with or without TM treatment in Ws-2 and sbi3 (Figure 6C). In addition, we also found that sbi3 is less tolerant to salt (Figures 6D,E).
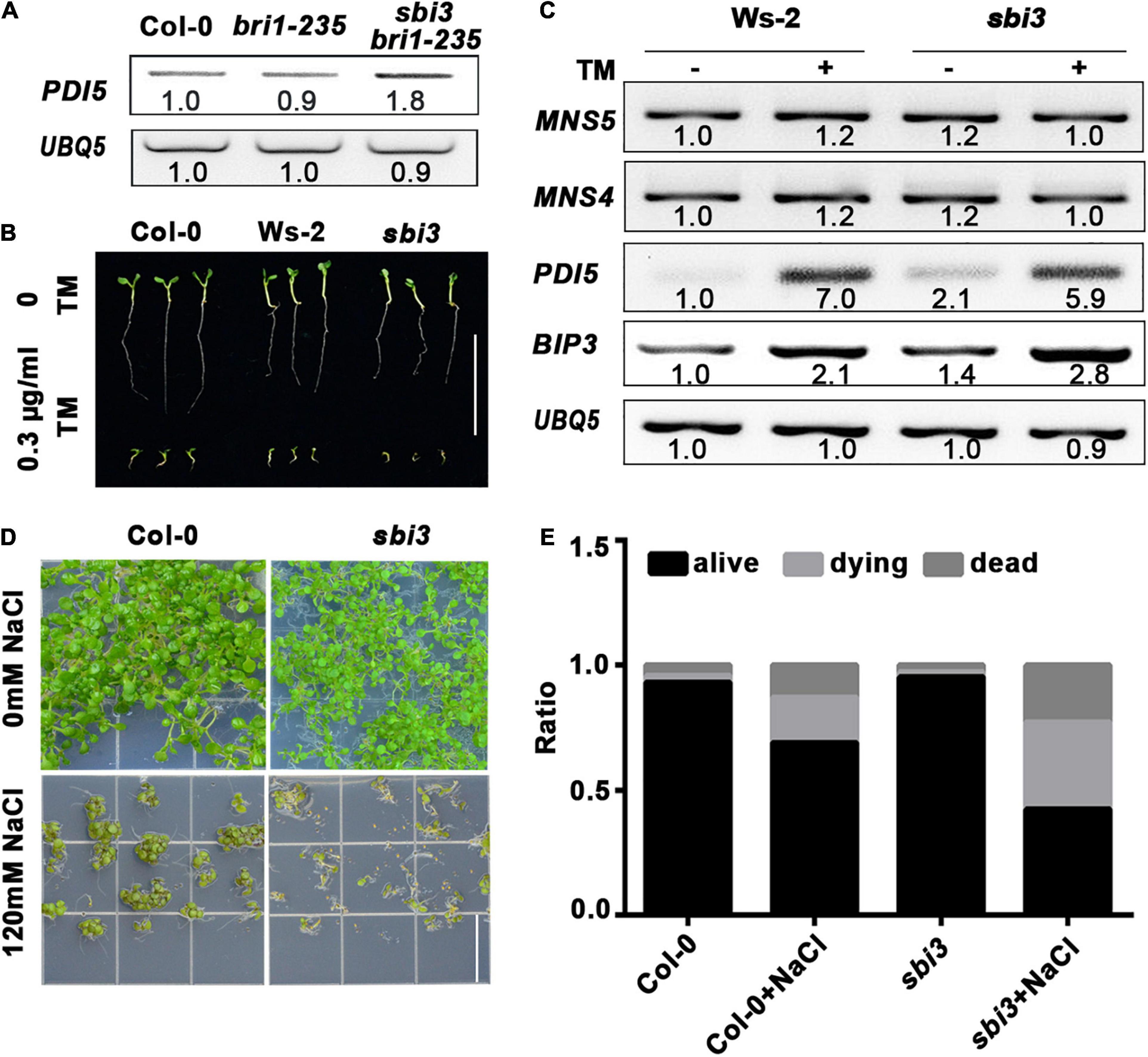
Figure 6. sbi3 exists UPR and is less tolerant to TM and salt. (A) Reverse transcription PCR (RT-PCR) analysis of PDI5 in Col-0, bri1-235, and sbi3 bri1-235, UBQ5 served as a control. (B) Comparison of 7-day-old seedlings of wild type and sbi3 mutant grown in ½ MS with or without 0.3 μg/mL TM. Scale bar = 1 cm. (C) Expression levels of MNS5, MNS4, PDI5, and BIP3 in Ws-2 and sbi3 with or without 5 μg/mL TM for 6 h. UBQ5 served as a control. (D) The photograph of 12-day-old seedlings grown in ½ MS with or without 120 mM NaCl. Scale bar = 1.5 cm. (E) The ratio of the seedlings was shown in the bar graphs, alive (black), dying (light gray), dead (dark gray). These experiments were repeated three times.
Different Expression Patterns of MNS4 and MNS5 in Arabidopsis
The previous study has revealed that MNS4 is a membrane-bound form while MNS5 is a soluble protein. Furthermore, MNS4 and MNS5 differentially demannosylated the glycoprotein reporters (Huttner et al., 2014b). Here, we showed transcriptional divergence in MNS4 and MNS5 across different developmental stages (Figure 7). MNS4 and MNS5 transcripts were widely expressed in different developmental stages, and the MNS5 expression level was higher in the WT seedlings and rosette leaves (Figure 7A). The expression patterns of MNS1 and MNS2 transcripts were similar in ten Arabidopsis developmental stages from dataset AT_AFFY_ATH1-0. Conversely, in most cases, the expression level of MNS5 was higher than that of MNS4 (Figure 7B). Moreover, we performed the phylogenetic analysis and sequence alignments of the Arabidopsis GH47 family that has three branches, MNS1/MNS2, MNS3 and MNS4/MNS5 (Supplementary Figures 7, 8). The homology analysis revealed that the overall sequence identity between MNS1 and MNS2 was 83.54%, while the identity of MNS4 and MNS5 was 44.10% (Supplementary Figure 7B), suggesting that MNS1 and MNS2 are recently divergent while MNS4 and MNS5 are anciently divergent. In fact, both MNS4 and MNS5 presented in the whole kingdom of plants while both MNS1 and MNS2 presented no earlier than vascular plants (Supplementary Table 2). Taken together, MNS4 and MNS5 likely have a significant functional divergence and our finding is consistent with this assessment, which is a non-redundant function in MNS5 in the regulation of ERAD and ER-stress response.
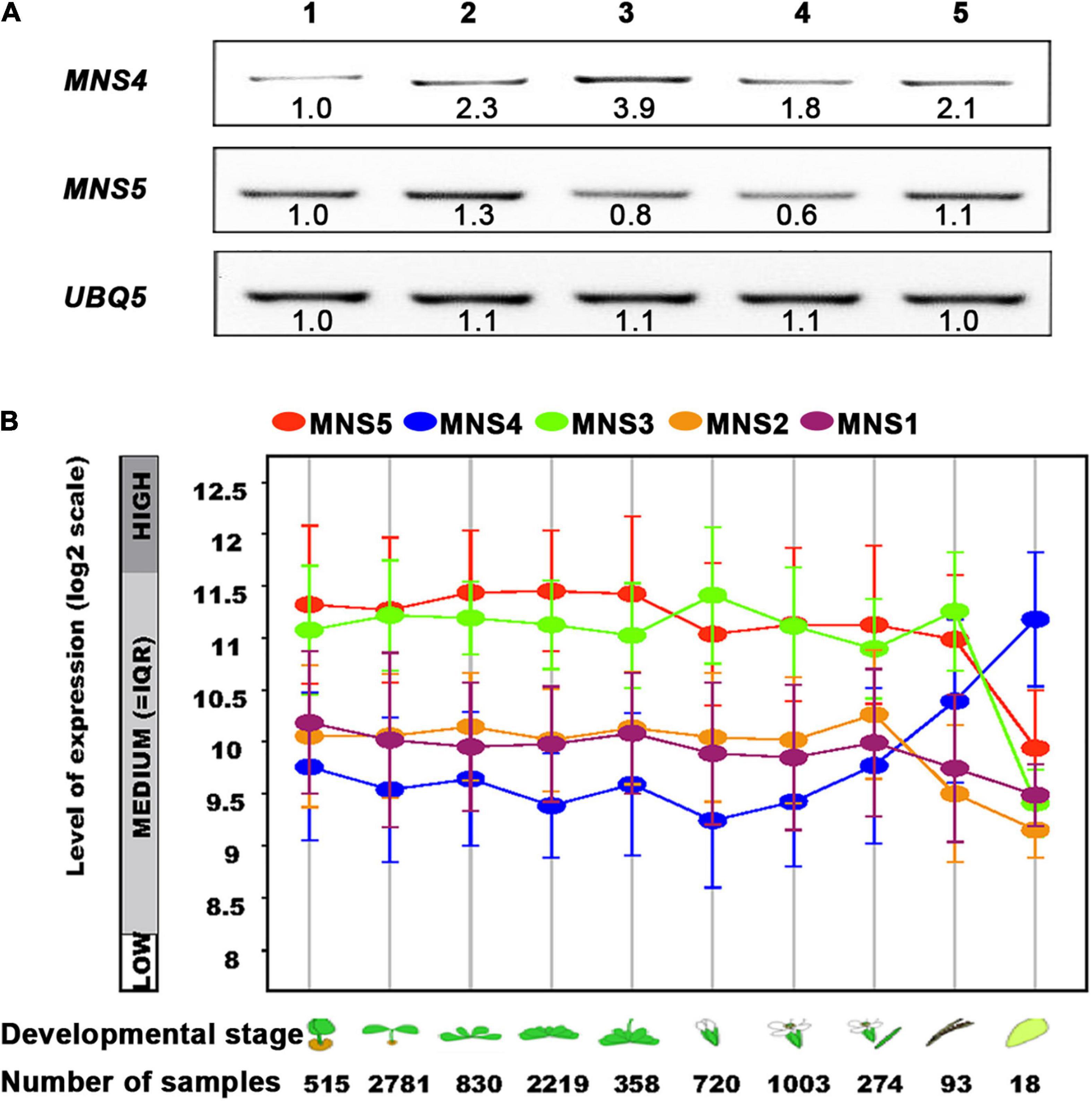
Figure 7. The expression levels of MNS5 and MNS4 in different developmental stages. (A) The transcripts of MNS4 and MNS5 were performed using RT-PCR analysis in WT Ws-2. UBQ5 was a control. 1: 2-weeks-old seedlings, 2: 50-day-old rosette leaves, 3: 50-day-old flowers, 4: 50-day-old siliques, 5: 68-day-old rosette leaves. (B) The transcripts comparison of MNS1-MNS5 was plotted as a line graph during the 10 developmental stages from dataset AT_AFFY_ATH1-0 (https://www.genevestigator.com/gv/plant.jsp). The 10 developmental stages: Germinated, young seedling, rosette, developed rosette, bolting and young, flower, developed flower, flowers, and siliques, mature siliques, senescence.
Discussion
Endoplasmic Reticulum-Associated Degradation is one of the major processes in maintaining proteostasis. Using misfolded transmembrane receptor kinases, several regulatory components of ERAD have been identified. Previous studies have found that BRI1-9 (an ER-retained Ser662Phe mutation in the ligand-binding domain of BRI1) is ubiquitinated and degraded via a classic glycan-dependent, 26S proteasome, and HRD1 complex-mediated pathway after retrotranslocation from the ER into the cytosol (Hong et al., 2009; Liu et al., 2015). On the other hand, the ER-trapped BRI1-5 mutant that carries a Cys69Tyr mutation in the extracellular domain, is regulated by a monoglucosylation (Glc1Man7GlcNA2)-dependent, proteasome-independent ERAD process, but no ubiquitination has been reported so far (Hong et al., 2008; Huttner et al., 2014b). Similar to BRI1-5, SUBEX-C57Y (a misfolded variant of the receptor-like kinase STRUBBELIG’s extracellular domain) is a novel glycoprotein ERAD substrate disposed of by glycan-dependent and non-proteasome dependent route (Huttner et al., 2014a). Unlike the yeast ERAD substrate carboxypeptidase Y, CPY (Jakob et al., 1998; Kostova and Wolf, 2005; Spear and Ng, 2005), none of the three N-glycans on SUBEX-C57Y displays a specific glycan signal for degradation (Huttner et al., 2014a). Furthermore, topologically different folding-defective ERAD substrates do not interfere with the glycan-dependent HRD1 machinery in plants (Shin et al., 2018). Here, we add that the mutant bri1-235 that harbors a Ser156-to-Phe mutation in the less conserved fourth LRR of BRI1 in Arabidopsis is degraded in a proteasome-independent ERAD way (Figures 2G,H). The mutation of the 156th amino acid in bri1-235 resulted in the absence of an N-glycosylation site in position 154 (Supplementary Figure 9), which may affect protein folding and degradation (Hou et al., 2019). It would be of great interest to see if ubiquitination is required in the ERAD substrate BRI1-235. Although a new model of the Arabidopsis Hrd1 complex has been depicted by Lin et al. (2019), it’s not clear whether all ERAD substrates are dependent on the Hrd1 complex -containing ERAD machinery in plants.
The N-glycan analysis of the different mns mutants confirms that two functionally redundant Golgi-α-mannosidases MNS1 and MNS2 act downstream of MNS3 in the Arabidopsis regular N-glycan processing pathway, which readily cleaves off three-terminal α1,2-linked Man residues from the A- and C-branches of Man8GlcNAc2 substrate, resulting in the formation of Man5GlcNAc2 (Liebminger et al., 2009). The ER-type α-mannosidase MNS3 is required for the efficient biosynthetic role by trimming a terminal α1,2-mannose residue from the middle branch (B-branch) of the Man9GlcNAc2 oligosaccharide in the misfolded proteins, forming a Man8GlcNAc2 isomer (Liebminger et al., 2009). MNS3 contains an amino acid tetrapeptide signal motif (LPYS: leucine, proline, tyrosine, serine) in the cytoplasmic tail, acting as a specific Golgi-localization signal (Schoberer et al., 2019).
The other two Arabidopsis class I α-mannosidases MNS4 and MNS5 are not part of the regular N-glycan processing pathway of properly folded secretory glycoproteins. In the ER, MNS4 and MNS5 with a largely redundant function accelerate the demannosylation of the C-branch to generate a terminal α1,6-linked Man, that acts as the glycan signal for ERAD of misfolded variants of BRI1. MNS4 has a transmembrane segment, while MNS5 is soluble (Huttner et al., 2014b). Consistently, they differentially demannosylate glycoprotein substrates, meaning that they share some but not the other substrates (Huttner et al., 2014b). The expression patterns of MNS4 and MNS5 were divergent across 10 Arabidopsis developmental stages from dataset AT_AFFY_ATH1-0. Significantly, in most cases, the expression level of MNS5 was higher than the corresponding MNS4 (Figure 7B). Furthermore, MNS4 and MNS5 presented across the whole plant kingdom, meaning that they have been duplicated and diverged since single-cell plants (Supplementary Table 2). This could mean that MNS4 and MNS5 have a distinct function (significantly functional divergence). Yet, a previous study suggested that MNS4 and MNS5 have a redundant function (Huttner et al., 2014b). Our discovery of a non-redundant function in MNS5 offers new insight into the distinct functions of MNS4 and MNS5. Still, forward genetic mutants of MNS4 are highly sought and screening for additional ERAD substrates that can distinguish the functions of MNS4 and MNS5 are also urgently required. In addition, it is worth examining whether sbi3 protein still has the enzyme activity of MNS5 or has an additional activity.
It is worth noting that mns4-1 or mns5-1 obtained by T-DNA insertion could not suppress the dwarf of bri1-5 and bri1-9, only the deficiency of both MNS4 and MNS5 could (Huttner et al., 2014b). However, our new mutant sbi3 could directly inhibit the phenotype of bri1-5, bri1-9, and bri1-235. In zebrafish, genetic compensation by the transcriptional upregulation of genes related to a mutated gene had recently been proposed as the possible cause for the observed phenotypical discrepancies in different mutants of the same gene (Rossi et al., 2015). Moreover, El-Brolosy et al. (2019), Ma et al. (2019) and Wilkinson (2019) reported an underlying molecular mechanism of the genetic compensation response, which was specifically triggered by PTC (premature termination codons)-bearing mRNA in mutations.
The RT-PCR analysis had revealed that the MNS4 transcripts were upregulated in mns4-1. However, there was no transcript of MNS5 in mns5-1 (Huttner et al., 2014b). We show that transcripts of MNS4 and MNS5 were not upregulated in mutant sbi3. Yet, sbi3 has a clear phenotype but mns5-1 has no detectable phenotype (Figures 1, 2 and Supplementary Figure 4; Huttner et al., 2014b). We thus deduce that the phenotypical discrepancy of sbi3 and mns5-1 are attributed to genetic compensation in mns5-1. Yet, the genetic compensation in mns5-1 might not be caused by the transcription upregulation of the related gene, namely no upregulation of the transcripts of MNS5 (mns5-1) and MNS4 in mns5-1. Therefore, the exact cause of the phenotypical discrepancy of sbi3 and mns5-1 is currently unknown, which is certainly a major part of our future endurance. Nevertheless, our finding raises awareness that the traditional forward genetic approach might still be necessary to complement the T-DNA or CRISPR-Cas9 systems on the study of gene functions in planta.
Data Availability Statement
All datasets generated for these findings are available in the main text and the Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
GW and XS conceived, designed, and coordinated the project. XS and CG performed the genetic mapping and the whole genome re-sequencing. XS and QZ performed protein expression and purification. QW and YZ performed the bioinformatics analysis. GW, XS, and KA wrote the original draft and other authors read and edited the manuscript. All authors interpreted the results.
Funding
This work was supported by the Chinese National Foundation of Science (32070325, 31270324, and 31741014) to GW, the Fundamental Research Funds for Central Universities (GK201101005 and GK202001010) to GW and (2017TS029) to XS, grants from the Chinese Ministry of Education (313034) and (20130202110007) to GW, and the Natural Science Foundation of Shaanxi Province of China (2020JM-268) to GL.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank J. Li (Lanzhou University) for BES1 antibodies.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.873688/full#supplementary-material
Footnotes
References
Abe, A., Kosugi, S., Yoshida, K., Natsume, S., Takagi, H., Kanzaki, H., et al. (2012). Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 30, 174–178. doi: 10.1038/nbt.2095
Aebi, M. (2013). N-linked protein glycosylation in the ER. Biochim. Biophys. Acta 1833, 2430–2437. doi: 10.1016/j.bbamcr.2013.04.001
Balchin, D., Hayer-Hartl, M., and Hartl, F. U. (2016). In vivo aspects of protein folding and quality control. Science 353:aac4354. doi: 10.1126/science.aac4354
Baldridge, R. D., and Rapoport, T. A. (2016). Autoubiquitination of the Hrd1 Ligase Triggers Protein Retrotranslocation in ERAD. Cell 166, 394–407. doi: 10.1016/j.cell.2016.05.048
Braun, N., Wyrzykowska, J., Muller, P., David, K., Couch, D., Perrot-Rechenmann, C., et al. (2008). Conditional repression of AUXIN BINDING PROTEIN1 reveals that it coordinates cell division and cell expansion during postembryonic shoot development in Arabidopsis and tobacco. Plant Cell 20, 2746–2762. doi: 10.1105/tpc.108.059048
Caramelo, J. J., and Parodi, A. J. (2007). How sugars convey information on protein conformation in the endoplasmic reticulum. Semin. Cell Dev. Biol. 18, 732–742. doi: 10.1016/j.semcdb.2007.09.006
Caramelo, J. J., and Parodi, A. J. (2008). Getting in and out from calnexin/calreticulin cycles. J. Biol. Chem. 283, 10221–10225. doi: 10.1074/jbc.R700048200
Carvalho, P., Stanley, A. M., and Rapoport, T. A. (2010). Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell 143, 579–591. doi: 10.1016/j.cell.2010.10.028
Chen, X., Grandont, L., Li, H., Hauschild, R., Paque, S., Abuzeineh, A., et al. (2014). Inhibition of cell expansion by rapid ABP1-mediated auxin effect on microtubules. Nature 516, 90–93. doi: 10.1038/nature13889
Christianson, J. C., and Ye, Y. (2014). Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat. Struct. Mol. Biol. 21, 325–335. doi: 10.1038/nsmb.2793
Clerc, S., Hirsch, C., Oggier, D. M., Deprez, P., Jakob, C., Sommer, T., et al. (2009). Htm1 protein generates the N-glycan signal for glycoprotein degradation in the endoplasmic reticulum. J. Cell Biol. 184, 159–172. doi: 10.1083/jcb.200809198
Clough, S. J., and Bent, A. F. (1998). Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 16, 735–743. doi: 10.1046/j.1365-313x.1998.00343.x
Cui, F., Liu, L., Zhao, Q., Zhang, Z., Li, Q., Lin, B., et al. (2012). Arabidopsis ubiquitin conjugase UBC32 is an ERAD component that functions in brassinosteroid-mediated salt stress tolerance. Plant Cell 24, 233–244. doi: 10.1105/tpc.111.093062
D’Alessio, C., Caramelo, J. J., and Parodi, A. J. (2010). UDP-GlC:glycoprotein glucosyltransferase-glucosidase II, the ying-yang of the ER quality control. Semin. Cell Dev. Biol. 21, 491–499. doi: 10.1016/j.semcdb.2009.12.014
D’Alessio, C., and Dahms, N. M. (2015). Glucosidase II and MRH-domain containing proteins in the secretory pathway. Curr. Protein Pept. Sci. 16, 31–48. doi: 10.2174/1389203716666150213160438
Dawlaty, M. M., Ganz, K., Powell, B. E., Hu, Y. C., Markoulaki, S., Cheng, A. W., et al. (2011). Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell 9, 166–175. doi: 10.1016/j.stem.2011.07.010
Denic, V., Quan, E. M., and Weissman, J. S. (2006). A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell 126, 349–359. doi: 10.1016/j.cell.2006.05.045
Deprez, P., Gautschi, M., and Helenius, A. (2005). More than one glycan is needed for ER glucosidase II to allow entry of glycoproteins into the calnexin/calreticulin cycle. Mol. Cell 19, 183–195. doi: 10.1016/j.molcel.2005.05.029
El-Brolosy, M. A., Kontarakis, Z., Rossi, A., Kuenne, C., Gunther, S., Fukuda, N., et al. (2019). Genetic compensation triggered by mutant mRNA degradation. Nature 568, 193–197. doi: 10.1038/s41586-019-1064-z
El-Brolosy, M. A., and Stainier, D. Y. R. (2017). Genetic compensation: a phenomenon in search of mechanisms. PLoS Genet. 13:e1006780. doi: 10.1371/journal.pgen.1006780
Ellgaard, L., and Helenius, A. (2003). Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 4, 181–191. doi: 10.1038/nrm1052
Freudenberg, J. M., Ghosh, S., Lackford, B. L., Yellaboina, S., Zheng, X., Li, R., et al. (2012). Acute depletion of Tet1-dependent 5-hydroxymethylcytosine levels impairs LIF/Stat3 signaling and results in loss of embryonic stem cell identity. Nucleic Acids Res. 40, 3364–3377. doi: 10.1093/nar/gkr1253
Friedrichsen, D. M., Joazeiro, C. A., Li, J., Hunter, T., and Chory, J. (2000). Brassinosteroid-insensitive-1 is a ubiquitously expressed leucine-rich repeat receptor serine/threonine kinase. Plant Physiol. 123, 1247–1256. doi: 10.1104/pp.123.4.1247
Gao, Y., Zhang, Y., Zhang, D., Dai, X., Estelle, M., and Zhao, Y. (2015). Auxin binding protein 1 (ABP1) is not required for either auxin signaling or Arabidopsis development. Proc. Natl. Acad. Sci. U.S.A. 112, 2275–2280. doi: 10.1073/pnas.1500365112
Gauss, R., Jarosch, E., Sommer, T., and Hirsch, C. (2006). A complex of Yos9p and the HRD ligase integrates endoplasmic reticulum quality control into the degradation machinery. Nat. Cell Biol. 8, 849–854. doi: 10.1038/ncb1445
Gidalevitz, T., Stevens, F., and Argon, Y. (2013). Orchestration of secretory protein folding by ER chaperones. Biochim. Biophys. Acta 1833, 2410–2424. doi: 10.1016/j.bbamcr.2013.03.007
Hebbard, L. W., Maurer, J., Miller, A., Lesperance, J., Hassell, J., Oshima, R. G., et al. (2010). Maternal embryonic leucine zipper kinase is upregulated and required in mammary tumor-initiating cells in vivo. Cancer Res. 70, 8863–8873. doi: 10.1158/0008-5472.CAN-10-1295
Helenius, A., and Aebi, M. (2004). Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73, 1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752
Hirsch, C., Gauss, R., Horn, S. C., Neuber, O., and Sommer, T. (2009). The ubiquitylation machinery of the endoplasmic reticulum. Nature 458, 453–460. doi: 10.1038/nature07962
Hong, Z., Jin, H., Fitchette, A. C., Xia, Y., Monk, A. M., Faye, L., et al. (2009). Mutations of an alpha1,6 mannosyltransferase inhibit endoplasmic reticulum-associated degradation of defective brassinosteroid receptors in Arabidopsis. Plant Cell 21, 3792–3802. doi: 10.1105/tpc.109.070284
Hong, Z., Jin, H., Tzfira, T., and Li, J. (2008). Multiple mechanism-mediated retention of a defective brassinosteroid receptor in the endoplasmic reticulum of Arabidopsis. Plant Cell 20, 3418–3429. doi: 10.1105/tpc.108.061879
Hong, Z., Kajiura, H., Su, W., Jin, H., Kimura, A., Fujiyama, K., et al. (2012). Evolutionarily conserved glycan signal to degrade aberrant brassinosteroid receptors in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 109, 11437–11442. doi: 10.1073/pnas.1119173109
Hosokawa, N., Kamiya, Y., Kamiya, D., Kato, K., and Nagata, K. (2009). Human OS-9, a lectin required for glycoprotein endoplasmic reticulum-associated degradation, recognizes mannose-trimmed N-glycans. J. Biol. Chem. 284, 17061–17068. doi: 10.1074/jbc.M809725200
Hou, Q., Saima, S., Ren, H., Ali, K., Bai, C., Wu, G., et al. (2019). Less conserved LRRs is important for BRI1 folding. Front. Plant Sci. 10:634. doi: 10.3389/fpls.2019.00634
Howell, S. H. (2013). Endoplasmic reticulum stress responses in plants. Annu. Rev. Plant Biol. 64, 477–499. doi: 10.1146/annurev-arplant-050312-120053
Huttner, S., Veit, C., Schoberer, J., Grass, J., and Strasser, R. (2012). Unraveling the function of Arabidopsis thaliana OS9 in the endoplasmic reticulum-associated degradation of glycoproteins. Plant Mol. Biol. 79, 21–33. doi: 10.1007/s11103-012-9891-4
Huttner, S., Veit, C., Vavra, U., Schoberer, J., Liebminger, E., Maresch, D., et al. (2014b). Arabidopsis Class I alpha-mannosidases MNS4 and MNS5 are involved in endoplasmic reticulum-associated degradation of misfolded glycoproteins. Plant Cell 26, 1712–1728. doi: 10.1105/tpc.114.123216
Huttner, S., Veit, C., Vavra, U., Schoberer, J., Dicker, M., Maresch, D., et al. (2014a). A context-independent N-glycan signal targets the misfolded extracellular domain of Arabidopsis STRUBBELIG to endoplasmic-reticulum-associated degradation. Biochem. J. 464, 401–411. doi: 10.1042/BJ20141057
Jakob, C. A., Burda, P., Roth, J., and Aebi, M. (1998). Degradation of misfolded endoplasmic reticulum glycoproteins in Saccharomyces cerevisiae is determined by a specific oligosaccharide structure. J. Cell Biol. 142, 1223–1233. doi: 10.1083/jcb.142.5.1223
Jin, H., Hong, Z., Su, W., and Li, J. (2009). A plant-specific calreticulin is a key retention factor for a defective brassinosteroid receptor in the endoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 106, 13612–13617. doi: 10.1073/pnas.0906144106
Jin, H., Yan, Z., Nam, K. H., and Li, J. (2007). Allele-specific suppression of a defective brassinosteroid receptor reveals a physiological role of UGGT in ER quality control. Mol. Cell 26, 821–830. doi: 10.1016/j.molcel.2007.05.015
Jost, A. P., and Weiner, O. D. (2015). Probing yeast polarity with acute, reversible, optogenetic inhibition of protein function. ACS Synth. Biol. 4, 1077–1085. doi: 10.1021/acssynbio.5b00053
Kelleher, D. J., and Gilmore, R. (2006). An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology 16, 47R–62R. doi: 10.1093/glycob/cwj066
Kostova, Z., and Wolf, D. H. (2005). Importance of carbohydrate positioning in the recognition of mutated CPY for ER-associated degradation. J Cell Sci 118(Pt 7), 1485–1492. doi: 10.1242/jcs.01740
Lederkremer, G. Z., and Glickman, M. H. (2005). A window of opportunity: timing protein degradation by trimming of sugars and ubiquitins. Trends Biochem. Sci. 30, 297–303. doi: 10.1016/j.tibs.2005.04.010
Li, J., and Chory, J. (1997). A putative leucine-rich repeat receptor kinase involved in brassinosteroid signal transduction. Cell 90, 929–938. doi: 10.1016/s0092-8674(00)80357-8
Li, J., Nagpal, P., Vitart, V., McMorris, T. C., and Chory, J. (1996). A role for brassinosteroids in light-dependent development of Arabidopsis. Science 272, 398–401. doi: 10.1126/science.272.5260.398
Li, J., and Nam, K. H. (2002). Regulation of brassinosteroid signaling by a GSK3/SHAGGY-like kinase. Science 295, 1299–1301. doi: 10.1126/science.1065769
Liebminger, E., Huttner, S., Vavra, U., Fischl, R., Schoberer, J., Grass, J., et al. (2009). Class I alpha-mannosidases are required for N-glycan processing and root development in Arabidopsis thaliana. Plant Cell 21, 3850–3867. doi: 10.1105/tpc.109.072363
Lin, A., Giuliano, C. J., Sayles, N. M., and Sheltzer, J. M. (2017). CRISPR/Cas9 mutagenesis invalidates a putative cancer dependency targeted in on-going clinical trials. eLife 6:e24179. doi: 10.7554/eLife.24179
Lin, L., Zhang, C., Chen, Y., Wang, Y., Wang, D., Liu, X., et al. (2019). PAWH1 and PAWH2 are plant-specific components of an Arabidopsis endoplasmic reticulum-associated degradation complex. Nat. Commun. 10:3492. doi: 10.1038/s41467-019-11480-7
Lin, M. L., Park, J. H., Nishidate, T., Nakamura, Y., and Katagiri, T. (2007). Involvement of maternal embryonic leucine zipper kinase (MELK) in mammary carcinogenesis through interaction with Bcl-G, a pro-apoptotic member of the Bcl-2 family. Breast Cancer Res. 9:R17. doi: 10.1186/bcr1650
Liu, L., Cui, F., Li, Q., Yin, B., Zhang, H., Lin, B., et al. (2011). The endoplasmic reticulum-associated degradation is necessary for plant salt tolerance. Cell Res. 21, 957–969. doi: 10.1038/cr.2010.181
Liu, Y., Zhang, C., Wang, D., Su, W., Liu, L., Wang, M., et al. (2015). EBS7 is a plant-specific component of a highly conserved endoplasmic reticulum-associated degradation system in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 112, 12205–12210. doi: 10.1073/pnas.1511724112
Lukowitz, W., Gillmor, C. S., and Scheible, W. R. (2000). Positional cloning in Arabidopsis. Why it feels good to have a genome initiative working for you. Plant Physiol. 123, 795–805. doi: 10.1104/pp.123.3.795
Ma, Z., Zhu, P., Shi, H., Guo, L., Zhang, Q., Chen, Y., et al. (2019). PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components. Nature 568, 259–263. doi: 10.1038/s41586-019-1057-y
McCracken, A. A., and Brodsky, J. L. (1996). Assembly of ER-associated protein degradation in vitro: dependence on cytosol, calnexin, and ATP. J. Cell Biol. 132, 291–298. doi: 10.1083/jcb.132.3.291
Mohorko, E., Glockshuber, R., and Aebi, M. (2011). Oligosaccharyltransferase: the central enzyme of N-linked protein glycosylation. J. Inherit. Metab. Dis. 34, 869–878. doi: 10.1007/s10545-011-9337-1
Neff, M. M., Neff, J. D., Chory, J., and Pepper, A. E. (1998). dCAPS, a simple technique for the genetic analysis of single nucleotide polymorphisms: experimental applications in Arabidopsis thaliana genetics. Plant J. 14, 387–392. doi: 10.1046/j.1365-313x.1998.00124.x
Ninagawa, S., Okada, T., Sumitomo, Y., Kamiya, Y., Kato, K., Horimoto, S., et al. (2014). EDEM2 initiates mammalian glycoprotein ERAD by catalyzing the first mannose trimming step. J. Cell Biol. 206, 347–356. doi: 10.1083/jcb.201404075
Noguchi, T., Fujioka, S., Choe, S., Takatsuto, S., Yoshida, S., Yuan, H., et al. (1999). Brassinosteroid-insensitive dwarf mutants of Arabidopsis accumulate brassinosteroids. Plant Physiol. 121, 743–752. doi: 10.1104/pp.121.3.743
Ohta, M., and Takaiwa, F. (2015). OsHrd3 is necessary for maintaining the quality of endoplasmic reticulum-derived protein bodies in rice endosperm. J. Exp. Bot. 66, 4585–4593. doi: 10.1093/jxb/erv229
Parodi, A. J. (2000). Protein glucosylation and its role in protein folding. Annu. Rev. Biochem. 69, 69–93. doi: 10.1146/annurev.biochem.69.1.69
Pattison, R. J., and Amtmann, A. (2009). N-glycan production in the endoplasmic reticulum of plants. Trends Plant Sci. 14, 92–99. doi: 10.1016/j.tplants.2008.11.008
Quan, E. M., Kamiya, Y., Kamiya, D., Denic, V., Weibezahn, J., Kato, K., et al. (2008). Defining the glycan destruction signal for endoplasmic reticulum-associated degradation. Mol. Cell 32, 870–877. doi: 10.1016/j.molcel.2008.11.017
Rossi, A., Kontarakis, Z., Gerri, C., Nolte, H., Holper, S., Kruger, M., et al. (2015). Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 524, 230–233. doi: 10.1038/nature14580
Ruggiano, A., Foresti, O., and Carvalho, P. (2014). Quality control: ER-associated degradation: protein quality control and beyond. J. Cell Biol. 204, 869–879. doi: 10.1083/jcb.201312042
Schmitz, A., and Herzog, V. (2004). Endoplasmic reticulum-associated degradation: exceptions to the rule. Eur. J. Cell Biol. 83, 501–509. doi: 10.1078/0171-9335-00412
Schoberer, J., Konig, J., Veit, C., Vavra, U., Liebminger, E., Botchway, S. W., et al. (2019). A signal motif retains Arabidopsis ER-alpha-mannosidase I in the cis-Golgi and prevents enhanced glycoprotein ERAD. Nat. Commun. 10:3701. doi: 10.1038/s41467-019-11686-9
Shin, Y. J., Vavra, U., Veit, C., and Strasser, R. (2018). The glycan-dependent ERAD machinery degrades topologically diverse misfolded proteins. Plant J. 94, 246–259. doi: 10.1111/tpj.13851
Smith, M. H., Ploegh, H. L., and Weissman, J. S. (2011). Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science 334, 1086–1090. doi: 10.1126/science.1209235
Sousa, M., and Parodi, A. J. (1995). The molecular basis for the recognition of misfolded glycoproteins by the UDP-Glc:glycoprotein glucosyltransferase. EMBO J. 14, 4196–4203. doi: 10.1002/j.1460-2075.1995.tb00093.x
Spear, E. D., and Ng, D. T. (2005). Single, context-specific glycans can target misfolded glycoproteins for ER-associated degradation. J. Cell Biol. 169, 73–82. doi: 10.1083/jcb.200411136
Speers, C., Zhao, S. G., Kothari, V., Santola, A., Liu, M., Wilder-Romans, K., et al. (2016). Maternal Embryonic Leucine Zipper Kinase (MELK) as a Novel Mediator and Biomarker of Radioresistance in Human Breast Cancer. Clin. Cancer Res. 22, 5864–5875. doi: 10.1158/1078-0432.CCR-15-2711
Stigliano, I. D., Alculumbre, S. G., Labriola, C. A., Parodi, A. J., and D’Alessio, C. (2011). Glucosidase II and N-glycan mannose content regulate the half-lives of monoglucosylated species in vivo. Mol. Biol. Cell 22, 1810–1823. doi: 10.1091/mbc.E11-01-0019
Strasser, R. (2018). Protein quality control in the endoplasmic reticulum of plants. Annu. Rev. Plant Biol. 69, 147–172. doi: 10.1146/annurev-arplant-042817-040331
Su, W., Liu, Y., Xia, Y., Hong, Z., and Li, J. (2011). Conserved endoplasmic reticulum-associated degradation system to eliminate mutated receptor-like kinases in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 108, 870–875. doi: 10.1073/pnas.1013251108
Su, W., Liu, Y., Xia, Y., Hong, Z., and Li, J. (2012). The Arabidopsis homolog of the mammalian OS-9 protein plays a key role in the endoplasmic reticulum-associated degradation of misfolded receptor-like kinases. Mol. Plant 5, 929–940. doi: 10.1093/mp/sss042
Szekeres, M., Nemeth, K., Koncz-Kalman, Z., Mathur, J., Kauschmann, A., Altmann, T., et al. (1996). Brassinosteroids rescue the deficiency of CYP90, a cytochrome P450, controlling cell elongation and de-etiolation in Arabidopsis. Cell 85, 171–182. doi: 10.1016/s0092-8674(00)81094-6
Tanaka, K., Asami, T., Yoshida, S., Nakamura, Y., Matsuo, T., and Okamoto, S. (2005). Brassinosteroid homeostasis in Arabidopsis is ensured by feedback expressions of multiple genes involved in its metabolism. Plant Physiol. 138, 1117–1125. doi: 10.1104/pp.104.058040
Taylor, S. C., Thibault, P., Tessier, D. C., Bergeron, J. J., and Thomas, D. Y. (2003). Glycopeptide specificity of the secretory protein folding sensor UDP-glucose glycoprotein:glucosyltransferase. EMBO Rep. 4, 405–411. doi: 10.1038/sj.embor.embor797
Thibault, G., and Ng, D. T. (2012). The endoplasmic reticulum-associated degradation pathways of budding yeast. Cold Spring Harb. Perspect. Biol. 4:a013193. doi: 10.1101/cshperspect.a013193
Tokunaga, F., Brostrom, C., Koide, T., and Arvan, P. (2000). Endoplasmic reticulum (ER)-associated degradation of misfolded N-linked glycoproteins is suppressed upon inhibition of ER mannosidase I. J. Biol. Chem. 275, 40757–40764. doi: 10.1074/jbc.M001073200
Trombetta, E. S. (2003). The contribution of N-glycans and their processing in the endoplasmic reticulum to glycoprotein biosynthesis. Glycobiology 13, 77R–91R. doi: 10.1093/glycob/cwg075
Vembar, S. S., and Brodsky, J. L. (2008). One step at a time: endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 9, 944–957. doi: 10.1038/nrm2546
Wang, Y., Lee, Y. M., Baitsch, L., Huang, A., Xiang, Y., Tong, H., et al. (2014). MELK is an oncogenic kinase essential for mitotic progression in basal-like breast cancer cells. eLife 3:e01763. doi: 10.7554/eLife.01763
Wilkinson, M. F. (2019). Genetic paradox explained by nonsense. Nature 568, 179–180. doi: 10.1038/d41586-019-00823-5
Williams, D. B. (2006). Beyond lectins: the calnexin/calreticulin chaperone system of the endoplasmic reticulum. J. Cell Sci. 119(Pt 4), 615–623. doi: 10.1242/jcs.02856
Wu, G., Wang, X., Li, X., Kamiya, Y., Otegui, M. S., and Chory, J. (2011). Methylation of a phosphatase specifies dephosphorylation and degradation of activated brassinosteroid receptors. Sci. Signal. 4:ra29. doi: 10.1126/scisignal.2001258
Xu, W., Huang, J., Li, B., Li, J., and Wang, Y. (2008). Is kinase activity essential for biological functions of BRI1? Cell Res. 18, 472–478. doi: 10.1038/cr.2008.36
Yamamoto, S., Jaiswal, M., Charng, W. L., Gambin, T., Karaca, E., Mirzaa, G., et al. (2014). A Drosophila genetic resource of mutants to study mechanisms underlying human genetic diseases. Cell 159, 200–214. doi: 10.1016/j.cell.2014.09.002
Yoshida, Y., and Tanaka, K. (2010). Lectin-like ERAD players in ER and cytosol. Biochim. Biophys. Acta 1800, 172–180. doi: 10.1016/j.bbagen.2009.07.029
Keywords: SBI3, ERAD, BRI1, MNS4, MNS5
Citation: Sun X, Guo C, Ali K, Zheng Q, Wei Q, Zhu Y, Wang L, Li G, Li W, Zheng B, Bai Q and Wu G (2022) A Non-redundant Function of MNS5: A Class I α-1, 2 Mannosidase, in the Regulation of Endoplasmic Reticulum-Associated Degradation of Misfolded Glycoproteins. Front. Plant Sci. 13:873688. doi: 10.3389/fpls.2022.873688
Received: 11 February 2022; Accepted: 14 March 2022;
Published: 19 April 2022.
Edited by:
Els Jm van Damme, Ghent University, BelgiumReviewed by:
Richard Strasser, University of Natural Resources and Life Sciences Vienna, AustriaKazuhito Fujiyama, Osaka University, Japan
Copyright © 2022 Sun, Guo, Ali, Zheng, Wei, Zhu, Wang, Li, Li, Zheng, Bai and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guang Wu, Z3d1M0Bzbm51LmVkdS5jbg==