 Mengdi Li
Mengdi Li Fan Wang
Fan Wang Jiayu Ma
Jiayu Ma Hengzhao Liu
Hengzhao Liu Hang Ye
Hang Ye Peng Zhao
Peng Zhao Jianbo Wang
Jianbo Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 25 April 2022
Sec. Plant Abiotic Stress
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.873071
This article is part of the Research TopicAbiotic Stress: Molecular Genetics and Genomics, Volume IIView all 32 articles
Allopolyploids exist widely in nature and have strong environmental adaptability. The typical allopolyploid Brassica napus L. is a widely cultivated crop, but whether it is superior to its diploid progenitors in abiotic stress resistance and the key genes that may be involved are not fully understood. Cystein-rich polycomb-like protein (CPP) genes encode critical transcription factors involved in the response of abiotic stress, including salt stress. To explore the potential molecular basis of allopolyploid adaptation to salt stress, we comprehensively analyzed the characteristics and salt stress response of the CPP genes in B. napus and its two diploid progenitors in this study. We found some molecular basis that might be associated with the adaptability of B. napus, including the expansion of the CPP gene family, the acquisition of introns by some BnCPPs, and abundant cis-acting elements upstream of BnCPPs. We found two duplication modes (whole genome duplication and transposed duplication) might be the main reasons for the expansion of CPP gene family in B. napus during allopolyploidization. CPP gene expression levels and several physiological indexes were changed in B. napus and its diploid progenitors after salt stress, suggesting that CPP genes might play important roles in the response of salt stress. We found that some BnCPPs might undergo new functionalization or subfunctionalization, and some BnCPPs also show biased expression, which might contribute to the adaptation of B. napus under saline environment. Compared with diploid progenitors, B. napus showed stronger physiological responses, and BnCPP gene expression also showed higher changes after salt stress, indicating that the allopolyploid B. napus had an adaptive advantage under salt stress. This study could provide evidence for the adaptability of polyploid and provide important clues for the study of the molecular mechanism of salt stress resistance in B. napus.
Allopolyploidization is a powerful driving force for plant speciation and species diversity (Abbott et al., 2013; Soltis et al., 2016). This process makes the plants undergo a series of genetic and epigenetic changes, which may lead to the formation of new phenotypes, thus promoting the successful establishment of new allopolyploids and enabling it to adapt to the diverse ecological environment (Adams and Wendel, 2005; Chen, 2007; Jackson and Chen, 2010; Madlung, 2013; Yoo and Wendel, 2014). Natural allotetraploid Brassica napus (2n = 4x = 38, AnAnCnCn) is one of the most widely cultivated oil crops, which was formed by hybridization and genome doubling of diploid B. rapa (2n = 20, Ar) and B. oleracea (2n = 18, Co) about 7,500 years ago, and its domestication and cultivation history were relatively short (Chalhoub et al., 2014). Therefore, natural B. napus and its diploid progenitors, B. rapa and B. oleracea, are a group of ideal material for investigating allopolyploidization event (Prakash et al., 2009). As an allotetraploid, B. napus has advantages than the diploid Brassica species in many aspects such as environmental adaptability (Segraves, 2017). Studies have found that rapeseed can accumulate erucic acid through erucic acid metabolic pathways under salt stress, and high erucic acid genes are related to the stress resistant genes (Ashraf and McNeilly, 2004; Wang et al., 2020), which implied that rapeseed might have the ability to resistant the salt stress. However, the potential molecular basis of the adaptive advantages in allopolyploids was still largely unknown.
Cystein-rich polycomb-like protein (CPP) contains one or two cysteine-rich (CXC) domains and a short sequence R that connects two CXC domains (Riechmann et al., 2000; Andersen et al., 2007; Zhang et al., 2015). Many studies reported CPP genes showed response to a variety of abiotic stresses in plants. For example, the expression of 15 CPP genes was significantly increased after 24 h of drought treatment in soybean (Zhang et al., 2015). CsCPP genes engaged in rapid stress responses under abiotic stresses (such as low temperature, drought, salt and abscisic acid) in cucumber (Zhou et al., 2018). The expression of ZmCPP genes in maize changed under extreme temperature, salt and drought stresses (Song et al., 2016). Studies have also reported the specific function of CPP genes. TSO1, the first CPP gene identified in Arabidopsis thaliana, highly expressed in flowers, developing ovules and microspores of Arabidopsis, and the lack of TSO1 affected the karyokinesis and cytokinesis of cells (Hauser et al., 1998, 2000; Song et al., 2000). Further studies by Wang et al. (2018) found that TSO1 played a key role in regulating cell cycle and cell fate, and was involved in regulating the development of plant stems and roots. In soybean, transcription factor CPP1 was involved in the regulation of the symbiotic nodules development by regulating the expression of GMLBC3 (Cvitanich et al., 2000). In addition, Yang et al. (2008) first identified 8 CPP genes in Arabidopsis and 11 CPP genes in rice, and they found that the CXC domain and the R sequence might have coevolved during evolution. With the release and improvement of genomic information of many species, more CPP gene families in different species have been studied. For example, 20, 13, and 5 CPP genes were identified in soybean, maize, cucumber and wheat, respectively (Zhang et al., 2015; Song et al., 2016; Zhou et al., 2018). In general, the CPP gene family has only been systematically studied in a few species, and most of them are diploid species. Considering that the comparative analysis of CPP gene family between allopolyploid and its diploid parents/progenitors can provide new insights into the molecular basis of environmental adaptation in allopolyploids, it is necessary to identify and analyze CPP gene families in allopolyploid plants.
In this study, we comprehensively analyzed the characteristics and salt stress response (including physiological and gene expression changes) of the CPP gene family in B. napus and its two diploid progenitors, B. rapa and B. oleracea. We also explored the evolutionary changes of the CPP gene family in the allotetraploid B. napus compared with its progenitors, and uncover the potential molecular basis of the advantage of salt stress resistance in allotetraploid B. napus. Our results provide important clues for researchers to explore the roles of CPP genes in salt stress tolerance, and will deepen our understanding of the evolution of the molecular basis for polyploid stress resistance.
All genome data of B. rapa, B. oleracea and B. napus used in this study were obtained from Brassica BRAD database (Cheng et al., 2011). Three methods were used to obtain candidate CPP gene family members. First, 8 AtCPP proteins were used as query sequences (downloaded from TAIR database1) to perform Blastp alignment (E-value < 1e––5) with all protein sequences of three Brassica species. Second, the HMM file of the CXC domain (ID: PF03638) was downloaded from the Pfam database2, and then the software HMMER was used to search this domain in all protein sequences of three species (E-value < 1e––5). Third, the syntenic genes of 8 AtCPP were searched in three Brassica species using the syntenic gene search function of BRAD database. All the members obtained by these three methods (i.e., the union of the members) were candidate members of the CPP gene family. Furthermore, Smart database3, Pfam database and CDD database4 were used to detect the integrity of the CXC domain of all the candidate CPP proteins, and the candidate members with sequence deletion in the domain were removed to obtain the final CPP gene family members. The duplication modes of all CPP genes were analyzed by the online website PlantDGD5. CDS sequences of three Brassica species were downloaded from BRAD database, and the CDS sequences of CPP genes of each species were compared using Blastn program. If the consistency and coverage of two CDS were both greater than 80%, they were considered as segmental duplicated gene pairs (Zhou et al., 2004). The software DNAsp5 (Librado and Rozas, 2009) was used to estimate the non-synonymous (Ka) and synonymous substitution (Ks) values between these gene pairs, and then the selection pressure of CPP genes in B. napus and its progenitors was determined based on Ka/Ks value.
CPP proteins from five species (B. rapa, B. oleracea, B. napus, Arabidopsis and rice) were aligned using the Clustal X program in MEGA software (v7.0; Kumar et al., 2016). According to the alignment results, the MEGA software was used to perform phylogenetic analysis of these CPP proteins utilizing the neighbor-joining (NJ) method with 1000 bootstrap replicates. The phylogenetic tree was decorated by the online website Tree of Life (iTOL6). According to the information provided by Smart database, IBS software (v1.0.3) was used to show the distribution of CXC domain on CPP proteins. The software Clustal W was used to align the CXC domains of all CPP proteins from the three Brassica species. The online website MEME7 was used to predict the characteristic motifs of CPP proteins of three species, and the number and length of the predicted motifs were set as 10 and 6–50 aa, respectively. The structure of the CPP genes was analyzed using the online tool GSDS8.
The location information of all identified CPP genes was obtained from BRAD database, and their positions on chromosomes were visualized using the MapInspector software9, 10. The syntenic CPP genes were searched by the syntenic gene search function of BRAD database, and the synteny relationship between CPP genes was showed by Circos software (Krzywinski et al., 2009).
Online website ExPASy11 was used to predicate the physicochemical properties of CPP proteins, including the number of amino acids, molecular weight (MW), isoelectric point (pI), the grand average of hydropathy (GRAVY) and instability index (II). Subcellular localization of CPP proteins in three Brassica species was predicted using the online tool WoLF PSORT12.
The 2000 bp upstream of the transcription start site of CPP genes were considered to be the promoter region of CPP genes. The cis-acting elements in the promoter region of CPP genes were predicted using website PlantCARE13. Gene expression data from four organs in three Brassica species were derived from our previous studies (Li et al., 2020), and the raw data of which were deposited in the NCBI database (accession number: SRR7816633-SRR7816668). The heatmap was created using the software HemI.
The experimental materials used in this study were B. napus (cv. Darmor), B. rapa (cv. Chiifu) and B. oleracea (cv. Jinzaosheng). Seeds were placed on wet double filter paper in a culture dish for germination in the light incubator (temperature: 22°C, light/dark: 16 h/8 h). When cotyledons of the seedlings were fully unfolded, placed them in black pots in which an equal amount of vermiculite and vegetative soil were mixed. The seedlings then grown in a light incubator under the same conditions, during which 1/2 Hoagland’s nutrient solution were regularly applied. Seedlings growing for 4 weeks were subjected to salt stress. The materials in the experimental group were irrigated with NaCl solution with a concentration of 300 mM (once a day, 30 ml), and the materials in the control group were irrigated with the same amount of distilled water for 4 days. Take the second to fifth leaves of the seedlings and freeze them immediately in liquid nitrogen for further use. Each species/treatment has three biological replicates. The experimental materials used in this study were shown in the Supplementary Figure 1.
Three enzyme activities, including peroxidase (POD), superoxide dismutase (SOD) and catalase (CAT), were quantified according to the method of Cao et al. (2017), with minor modifications as described by Xu et al. (2020). The contents of proline (Pro), soluble sugar, soluble protein, and chlorophyll (Chl a + Chl b) were measured according to Quinet et al. (2012).
Total RNAs from each sample was isolated using the Trizol reagent (Invitrogen, Carlsbad, CA, United States) according to the instructions. The quality of RNA samples was checked on 1% agarose gel. The first strand complementary DNA (cDNA) was synthesized using Moloney Murine Leukemia Virus Reverse Transcriptase (M-MLV RT, Promega, Madison, WI, United States) following the manufacturer’s instructions. Then, cDNA was diluted 10-fold as the templates of qRT-PCR. The qRT-PCR reactions were performed using the ABI Step One Plus Real-Time PCR System (Applied Biosystems, Carlsbad, CA, United States) with the SYBR kit. The program was set to 94°C for 5 min; 38 cycles of 94°C for 30 s, 55°C for 30 s and 72°C for 20 s; and 72°C for 10 min. Three biological and three technical repeats were used for each sample. The ACT2/7 gene was used as internal control to standardize the results and the relative expression level of selected genes was calculated using the delta-delta threshold cycle (2–ΔΔCt) method. Gene-specific primers for qRT-PCR were designed by Primer 5 software and the primer sequences were listed in Supplementary Table 1.
To ensure that CPP gene families in B. napus and its two diploid progenitors are fully identified, we used three methods to obtain candidate members. The three methods include Blastp alignment, HMMER scanning, and syntenic gene searching (see section “Identification and duplication mode of CPP genes” in Materials and Methods section). A total of 18, 19, and 37 candidate CPP genes were initially obtained in B. rapa, B. oleracea and B. napus. The integrity of the CXC domain of all candidate proteins was examined, and which with complete domain were identified as the final CPP gene family members. Finally, 15, 10, and 34 members were identified in B. rapa, B. oleracea, and B. napus (Table 1). The identified members were named according to the previous studies (Li et al., 2019b; Wang et al., 2019; Table 1).
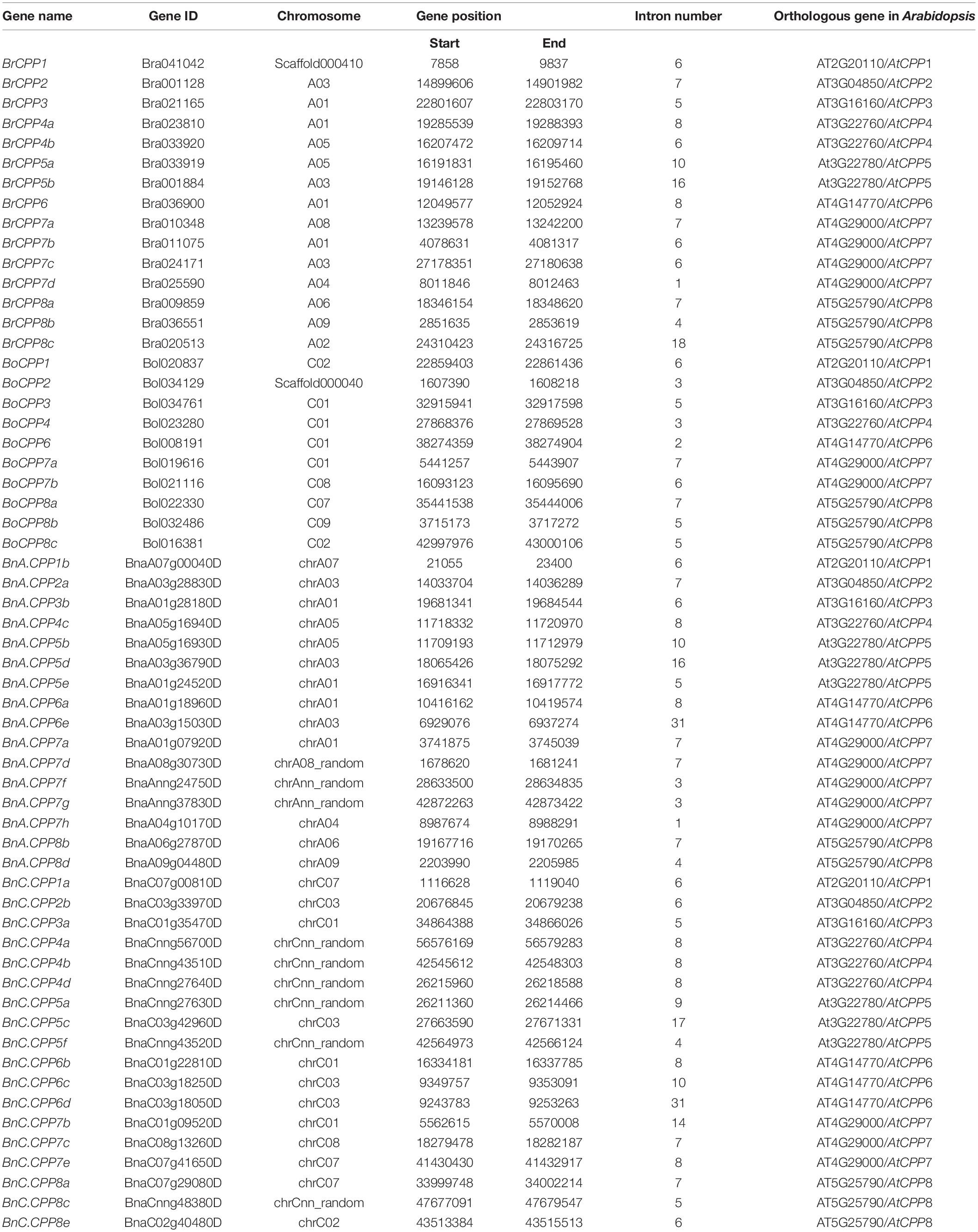
Table 1. The CPP gene family information in B. napus and its diploid progenitors.
As we all know, B. napus is derived from natural hybridization and chromosome doubling of its two diploid progenitors, B. rapa and B. oleracea. Therefore, theoretically, the number of CPP genes in B. napus should be equal to the sum of the number of CPP genes in B. rapa and B. oleracea. In this study, 15 and 10 CPP genes were identified in B. rapa and B. oleracea, respectively, and the sum of CPP genes in the two species was much smaller than the number of CPP genes in B. napus (34). Specifically, the number of each BnCPP genes was greater than or equal to the sum of the corresponding homoeologous genes in the two diploid ancestral species (with one exception, BnCPP8; Supplementary Table 2). Compared with diploid species, the number of CPP1-3 genes in B. napus did not change and the number of CPP4-6 genes in both subgenomes of B. napus increased during allopolyploidization process. Statistically, the number of lost genes (2) was less than the number of increased genes (11) in B. napus (Supplementary Table 2). Therefore, the CPP gene family in B. napus expanded during the allopolyploidization process, although a small number of gene loss events occurred.
To explore the reasons for the expansion of CPP gene family in B. napus, we analyzed the duplication modes of CPP genes (Table 2 and Supplementary Figure 2). Gene duplication modes include whole genome duplication (WGD) and various single gene duplication, and the latter consists of four models, including tandem duplication (TD), proximal duplication (PD), transposed duplication (TRD) and dispersed duplication (DSD; Qiao et al., 2019). We found that CPP genes in B. rapa, B. oleracea and B. napus showed a large number of genome wide duplication, accounting for 60, 50, and 38.2%, respectively, which might be related to the WGD and WGT events of Brassica progenitor. For the four single gene duplication modes, the mechanism of the DSD is unclear (Qiao et al., 2019), so it was excluded from the following comparative analysis. We found that TRD was the most frequent mode (about 50%) in all three species, suggesting that this mode was important for the expansion of the CPP gene family. In addition, although WGD and TRD were the most frequent duplication modes of CPP genes in the three species, the frequency of TRD was higher than that of WGD only in the CPP genes of B. napus. Therefore, we speculated that TRD might greatly promote the expansion of CPP gene family in B. napus during allopolyploidization. Moreover, PD was observed in only one CPP gene in B. rapa, indicating that this duplication mode is not the main source of CPP gene family expansion in B. rapa, B. oleracea and B. napus. In addition, a total of 29 segmental duplicated gene pairs were detected in three Brassica species, and their Ka/Ks values were calculated to explore the selection pressure on CPP genes during evolution (Supplementary Table 3). The results showed that all the values were less than 1, indicating that this family was subjected to purification selection during evolution. The mean value of all Ka/Ks values was 0.42, which might indicate that the purification selection on CPP genes was not strong. One study suggested that CPP genes in plants might be subjected to purification selection only in the conserved domain (Yang et al., 2008). We also visualized these segmental duplication pairs and five duplication modes in Supplementary Figure 3.
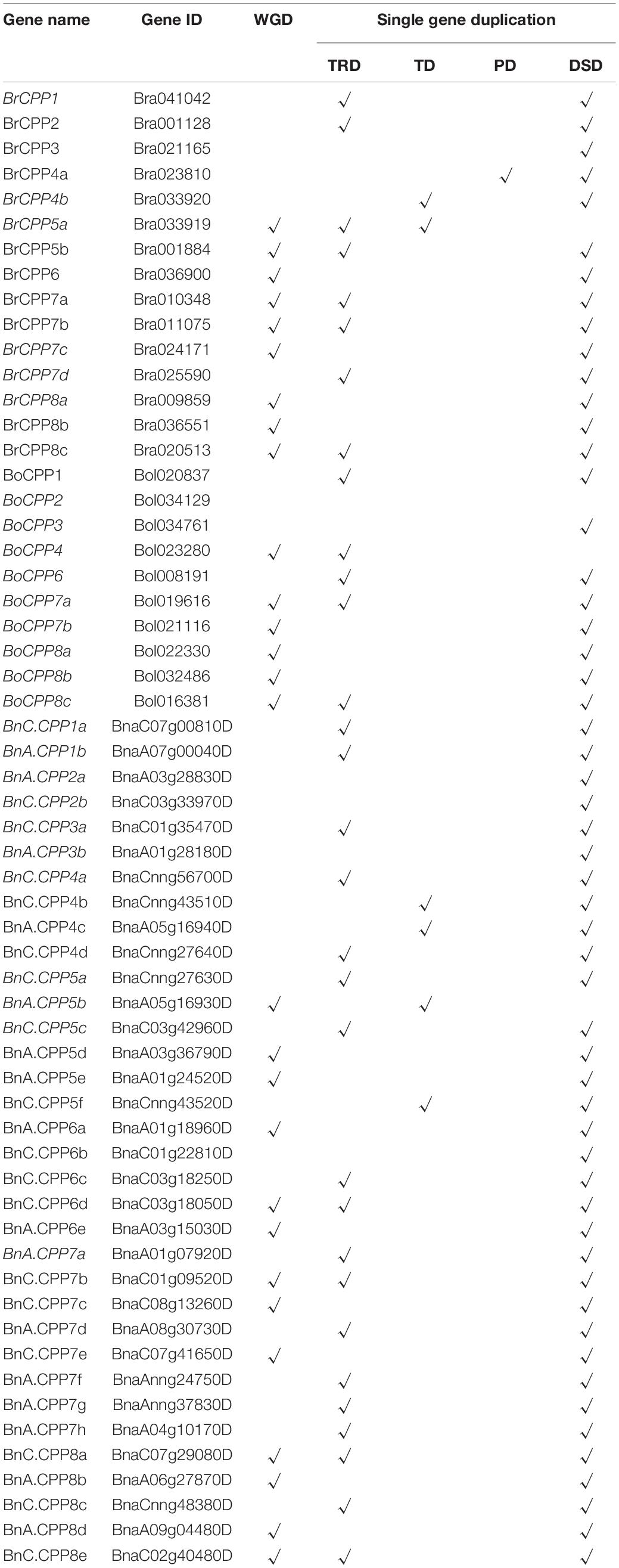
Table 2. Five modes of CPP gene duplication in B. napus and its diploid progenitors.
To better investigate the evolution of CPP genes among three Brassica species, we visualized the syntenic relationship of identified CPP genes. We found that CPP genes in these three Brassica species have a good syntenic relationship, and they were closely related in evolution (Figure 1). There were 8 AtCPP genes, 13 BrCPP genes, 7 BoCPP genes and 13 BnCPP genes, respectively, and these syntenic genes were mainly located in five conserved genome blocks F, H, Q, T, and U (Supplementary Table 4). In the three Brassica species, about 55.9% (33/59) of CPP genes retained some syntenic relationships between species. In addition, we found that the increase or loss of syntenic genes occurred in B. napus after allopolyploidization. For example, the number of AtCPP2 syntenic genes in B. rapa and B. oleracea was 1 and 0 respectively, but the number of AtCPP2 syntenic genes in B. napus increased to two. AtCPP1 had one syntenic gene in B. rapa and B. oleracea respectively, but it was not found in B. napus. AtCPP3 had one syntenic gene in both B. rapa and B. oleracea, which was all located in the LF subgenome (note: LF subgenome stands for less fractioned subgenome; Cheng et al., 2011, 2012), and these two genes were retained in B. napus and were also located in the two LF subgenomes. Therefore, we speculated that AtCPP3 syntenic genes might play an irreplaceable role in the evolution and adaptation of these three Brassica species.
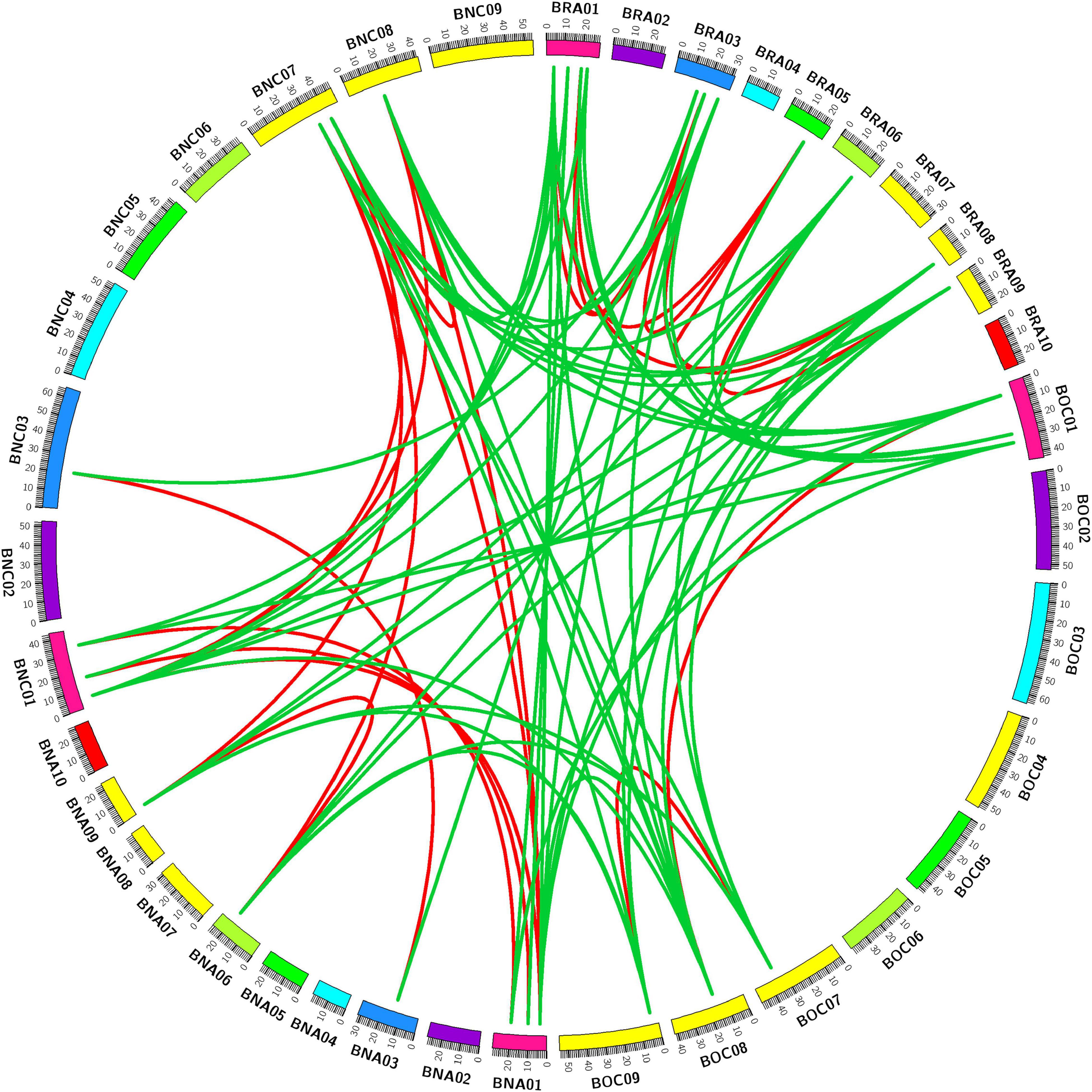
Figure 1. Synteny analysis of CPP genes in genomes of B. rapa, B. oleracea, and B. napus. Red lines and green lines separately linked the syntenic paralogs and orthologs. BRA01–10, ten chromosomes in B. rapa; BOC01–09, nine chromosomes in B. oleracea; BNA01–10, ten chromosomes in An sub-genomes in B. napus; BNC01–09, nine chromosomes in Cn sub-genomes in B. napus.
In addition to focusing on evolutionary changes in the number of CPP genes in B. napus, we also focused on the changes in the relative position of CPP genes on chromosomes. The number of CPP genes in An subgenome (16) was similar to that in the Ar genome (15), but the number of CPP genes in the Cn subgenome (18) was more than that in the Co genome (10). Moreover, we noticed that most of CPP genes in the Cn subgenome (12 out of 18) experienced the TRD duplication event (Table 2), indicating that the number of CPP genes in Cn subgenome might expend after allopolyploidization, which might be related to the fact that more transposable elements were distributed in the Cn subgenome (Chalhoub et al., 2014). In addition, another reason for this phenomenon might be the species-specific loss of CPP genes in Co genome during evolution. We further analyzed the chromosomal location of CPP genes, and a total of 25 BnCPP genes, 14 BrCPP genes and 9 BoCPP genes have been accurately mapped to the specific location of the chromosome (Figure 2). The remaining CPP genes have not been located in the chromosome due to the relatively low degree assembly of the reference genome. CPP genes were not found on 7 chromosomes (An02, An08, An10, Cn04-06, Cn09), 2 chromosomes (Ar07, Ar10) and 4 chromosomes (Co03-06) in B. napus, B. rapa and B. oleracea, respectively. By comparing the distribution of CPP genes on An and Ar subgenome/genome, we found a similar pattern, that is, 4, 3, and 2 CPP genes were distributed on chromosome 1, 3 and 5, respectively, while the remaining CPP genes were distributed on other chromosomes as a single form. However, the distribution of CPP genes on Cn and Co subgenome/genome was significantly different. In addition, about 71.4% (10/14) of CPP genes in Ar genome of B. rapa did not change their relative positions in chromosomes after allopolyploidization, while up to 44.4% (4/9) of CPP genes in Co genome of B. oleracea maintained their original relative positions in chromosomes after allopolyploidization. This phenomenon was consistent with previous studies about the other gene families in B. napus (Li et al., 2019a,b), which implied that there might be more chromosomal rearrangement or homologous exchange events occurring in regions associated with these gene family members in the Cn subgenome than in the An subgenome.
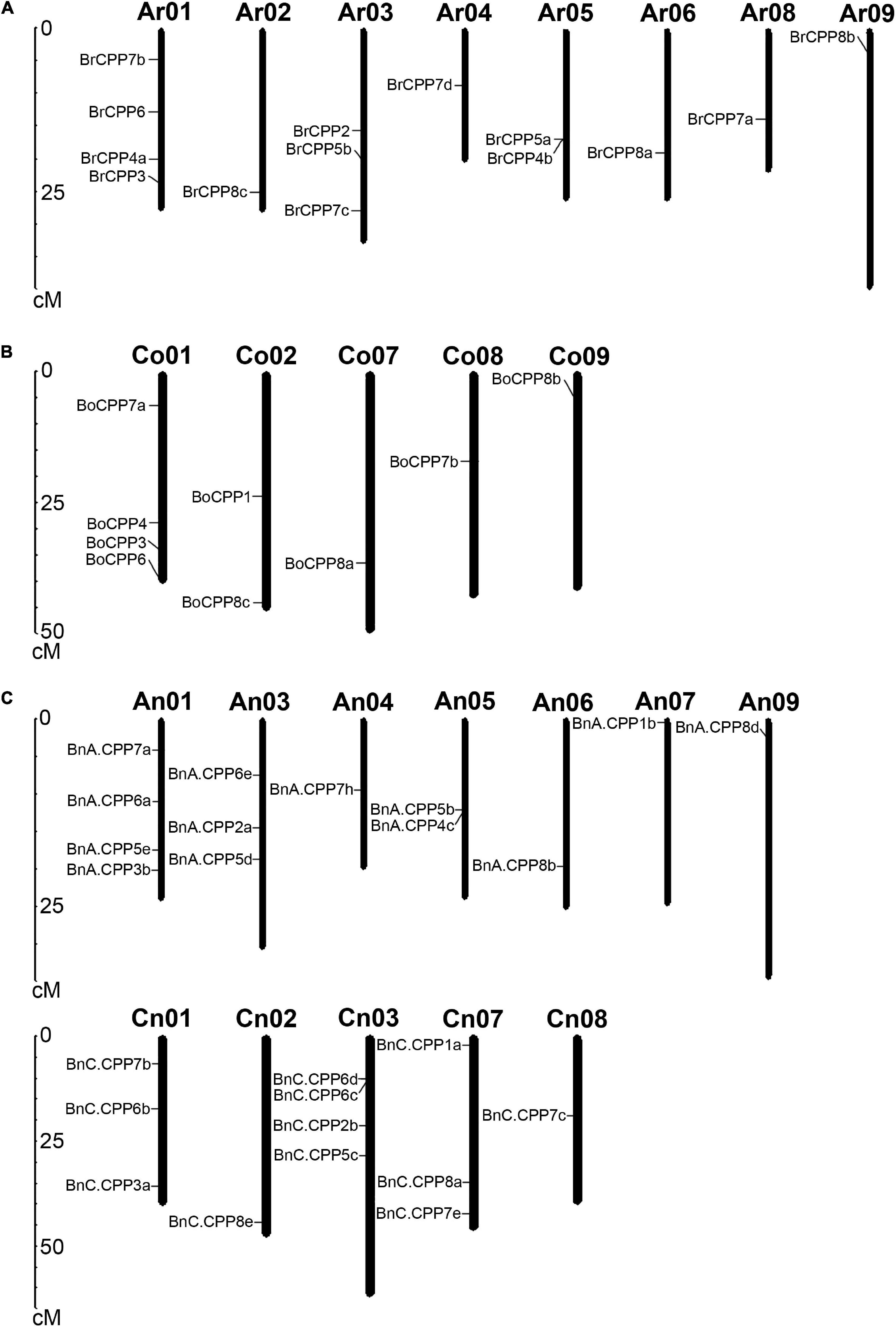
Figure 2. Chromosome distribution of CPP genes in B. rapa (A) and B. oleracea (B) and B. napus (C). All genes located in unassembled scaffolds were not shown in this figure.
We wanted to know what evolutionary changes had taken place in the gene structure of CPP genes after allopolyploidization. To better analyze these changes in terms of the evolutionary relationship, we first conducted phylogenetic analysis of the CPP gene family. CPP proteins of typical monocotyledon rice and dicotyledon Arabidopsis were used as reference proteins, and a phylogenetic tree was constructed based on 78 CPP protein sequences, among which there were 8, 11, 15, 10, and 34 CPP proteins in Arabidopsis, rice, B. rapa, B. oleracea and B. napus, respectively (Figure 3). These CPP proteins can be clearly divided into two clades (I, II) and six subclades (Ia-c, IIa-c), which was consistent with CPP phylogenetic tree in other species (Zhou et al., 2018). We found that the CPP protein of Arabidopsis was present in all six subclades, whereas the CPP protein of rice was found in only three subclades (Ia, Ic, IIb), which might indicate that the evolution of CPP family has gradually diverged after the differentiation of monocotyledon and dicotyledon.
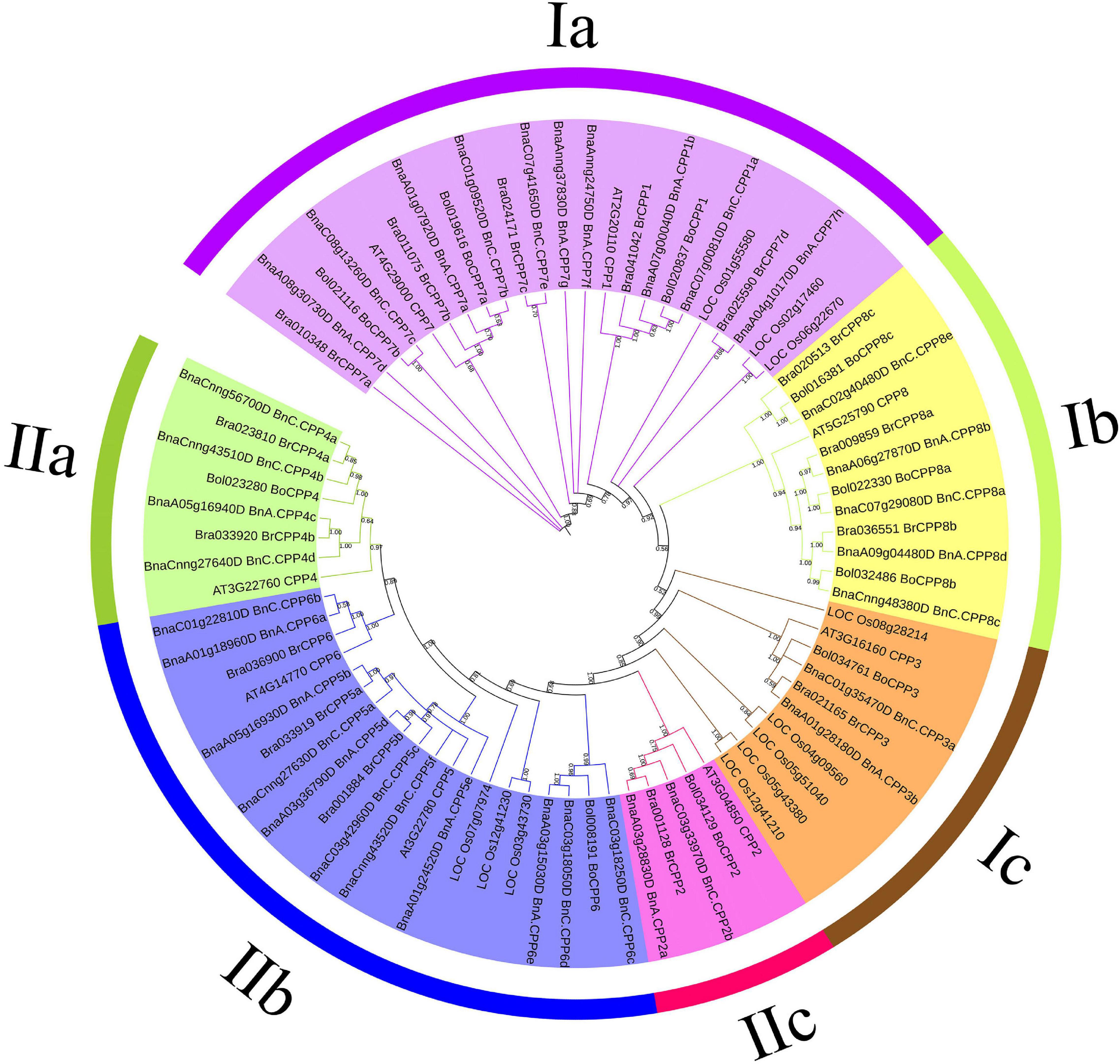
Figure 3. Phylogenetic tree of CPP proteins in B. napus, B. rapa, B. oleracea, Arabidopsis thaliana, and Oryza sativa. The prefixes Bra, Bol, Bna, AT, and LOC stand for B. rapa, B. oleracea, B. napus, A. thaliana and O. sativa, respectively. Only bootstrap values greater than 50% were displayed. All CPP proteins were divided into two clades (I,II) and six subclades (Ia-c,IIa-c).
Next, we analyzed the structure of CPP genes in B. napus and its two diploid progenitors. Compared with many other gene families, members of the CPP gene family have a high diversity of gene structure (Figure 4). We compared the structural diversity of genes in two clades of the phylogenetic tree, and found that both the structural diversity and the mean number of introns of CPP gene in clade II were higher than those in clade I, indicating that the structure of CPP genes in clade I may be more conserved than that in clade II. In addition, we found that BnCPP genes had the highest intron number and structural diversity, while BoCPP genes had the lowest intron number and structural diversity, indicating that CPP genes in B. napus evolved toward increasing intron number and structural diversity after allopolyploidization. To further explore the evolutionary changes in the structure of CPP genes after allopolyploidization, we identified 20 homoeologous gene pairs with potential direct evolutionary relationship and conducted comparative analysis of their gene structure according to Li et al. (2019a; 2019b). Among the 20 homoeologous gene pairs, the intron number of 13 gene pairs (65%) was the same between B. napus and its diploid progenitors, which suggested that CPP genes are conserved to a certain extent at the DNA level after allopolyploidization. Moreover, among the remaining 7 gene pairs, there were 6 gene pairs showed the number of introns in BnCPP gene was greater than that in BrCPP or BoCPP genes. These results indicated that intron increase or loss occurred in some CPP genes after allopolyploidization, and the phenomenon of intron increasing was more common.
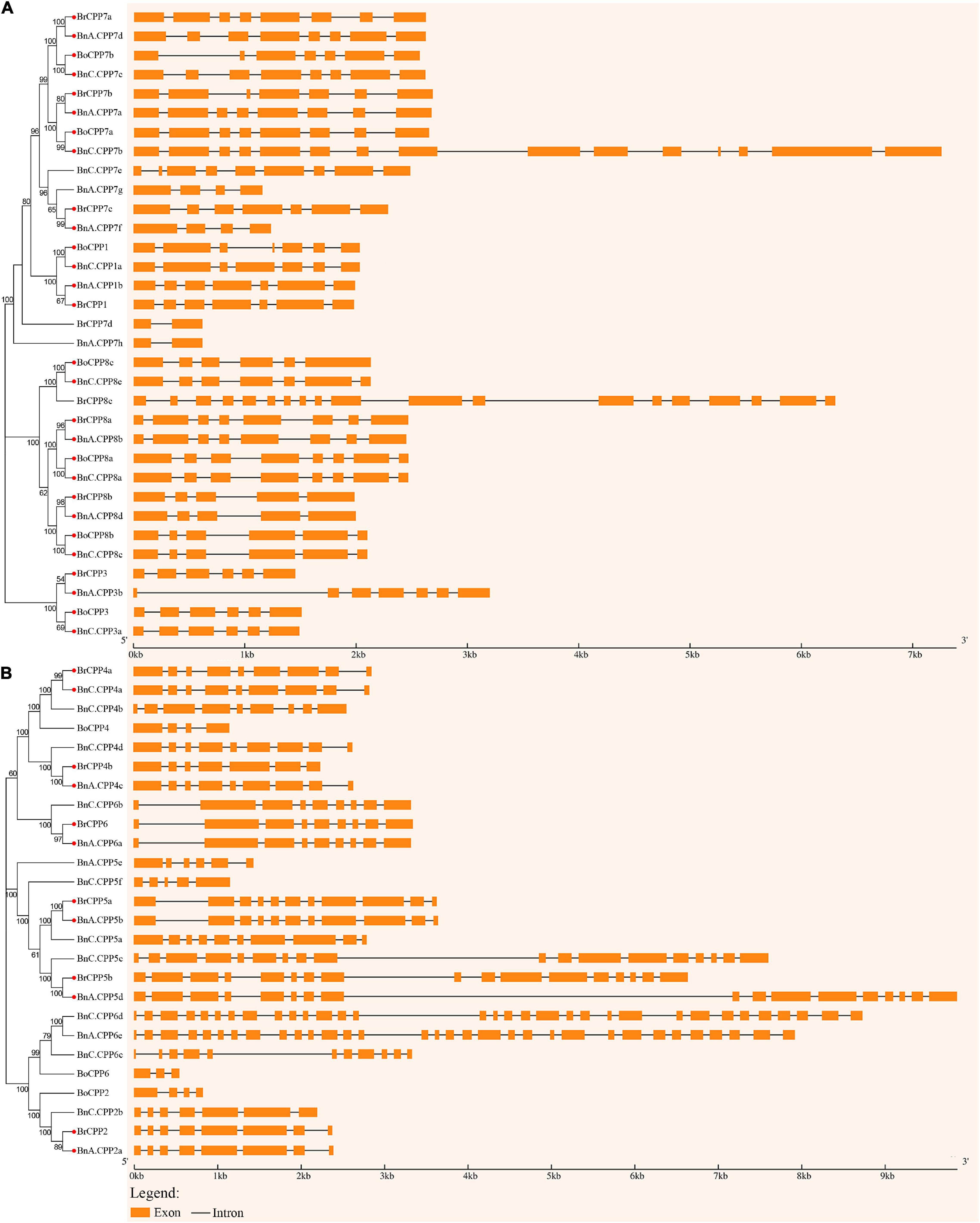
Figure 4. Intron-exon structure of the CPP genes in B. napus and diploid progenitors. The genes on two clades, I (A) and II (B), of the phylogenetic tree were shown separately. The red circle indicates orthologs with potential direct evolutionary relationship.
Analysis of CXC domains in B. napus and its progenitors showed that most CPP proteins in the three Brassica species contained one or two conserved CXC domains, just like CPP proteins in other species (Supplementary Figure 4). However, there were four CPP proteins (BnC.CPP5c, BrCPP5b, BnA.CPP5d, BnC.CPP6c) had four CXC domains, which have not been observed in CPP proteins of other species. This phenomenon might be related to the occurrence of homoeologous exchange events during allopolyploidization (Chalhoub et al., 2014). The alignment results of CXC domains in all identified CPP proteins showed that the two CXC domains and the short-conserved sequence RNPXAFXPK between them were very conserved, and only a few CPP proteins had amino acid deletion or replacement at these locations (Supplementary Figure 5). The motif analysis showed that 6 motifs existed in almost all CPP proteins, which were motifs 1–5 and 9 (Supplementary Figure 6). By analyzing the sequences of these motifs, we found that motifs 2, 4, and 9 were associated with the N-terminal CXC domain of CPP proteins, motif 5 was associated with a short-conserved sequence between two CXC domains, and motifs 1 and 3 were associated with the C-terminal CXC domain of CPP proteins. The motifs of proteins encoded by 20 homoeologous gene pairs with potential direct evolutionary relationship were compared, and we found 15 (75%) BnCPP proteins maintained the similar motif type and arrangement pattern of BrCPP/BoCPP proteins, indicating that the motif of CPP proteins were relatively conservative after allopolyploidization.
We predicted the physicochemical properties of CPP proteins in three Brassica species (Table 3), and the results were as follows. The average lengths of BnCPP, BrCPP and BoCPP proteins were 603, 592, and 381 aa, respectively, and the average molecular weights were 66.2, 65.0, and 41.5 kDa, respectively, which means that the average length and average molecular weight of BnCPP proteins were higher than those of its two progenitors. Furthermore, BnC.CPP7b and BoCPP7a were homoeologous gene pairs with potential direct evolutionary relationship, and we found that the protein sequence length of BnC.CPP7b (1408 aa) is much longer than that of BoCPP7a (578 aa), indicating that the encoding region of CPP gene might have increased after allopolyploidization. It was noteworthy that the length of 6 CPP proteins over 1000 aa were found in three Brassica species, and 5 of them were belonging to clade II in phylogenetic tree (Figure 3). In the three Brassica species, the instability index of all CPP proteins was higher than 40, and the GRAVY of all CPP proteins (except BoCPP2) was lower than 0, indicating that almost all CPP proteins were unstable hydrophilic proteins. The results showed that 44.12% of BnCPP, 53.33% of BrCPP and 30% of BoCPP proteins were acidic proteins (pI < 7), indicating that the ratio of acidic CPP protein in B. napus was between the two diploids. In addition, except for subclade Ic, acidic CPP proteins were found in all the other clades, suggesting that this clade might have diverged from other clades in the process of evolution. Subsequently, we predicted the subcellular localization of CPP proteins in the three Brassica species (Table 3). The results showed that the subcellular localization of CPP protein in three Brassica species was the same as that in other species (Song et al., 2016; Zhou et al., 2018). Most CPP proteins in B. napus and its diploid progenitors were predicted to be located in the nucleus, which may be consistent with the function of CPP proteins as transcription factors.
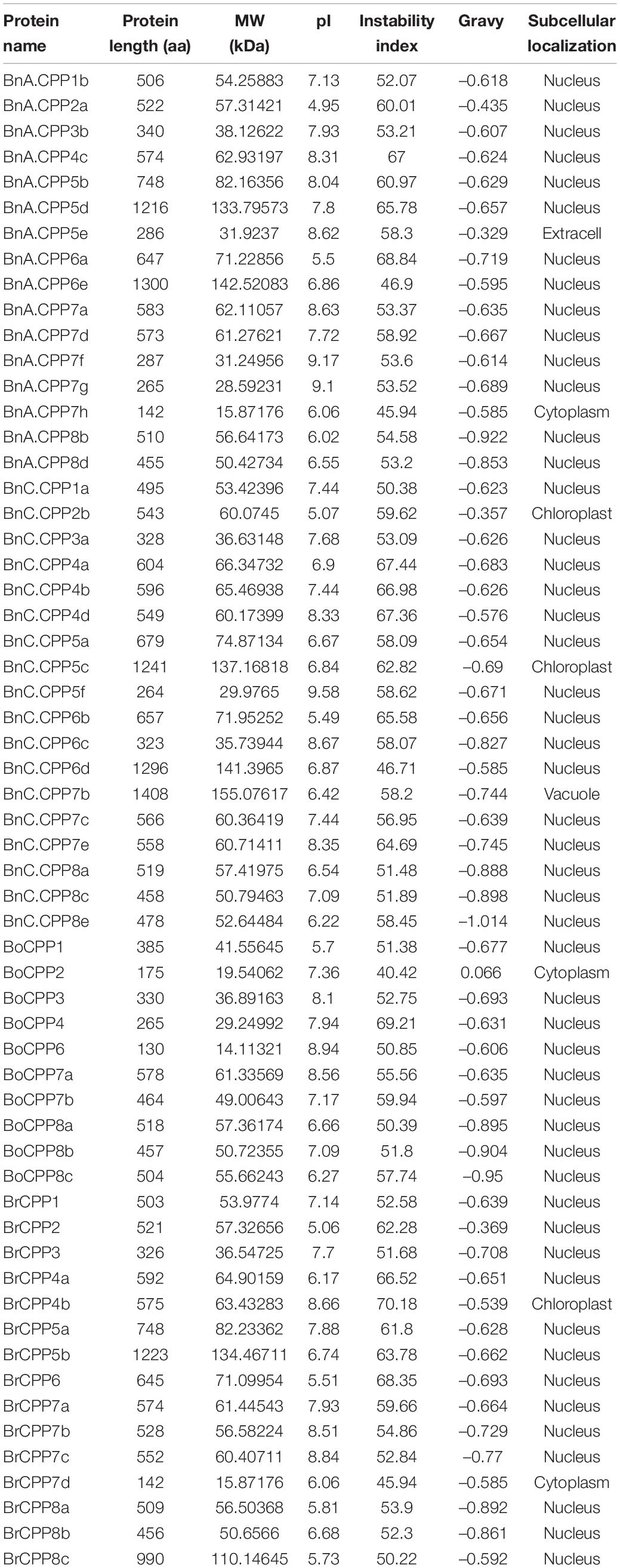
Table 3. The physicochemical parameters and subcellular localization prediction of CPP proteins in B. napus and its diploid progenitors.
We predicted the cis-acting elements in the upstream promoter region of CPP genes in three Brassica species (Figure 5). Fifty-nine cis-acting elements related to plant development and growth, phytohormone responses, stress responses and light responses were identified in the promoter region of CPP genes in B. napus and its progenitors. Among them, the largest number of cis-elements were associated with light responses. Seven identical stress response elements were identified in each of the three Brassica species, and the antioxidant response element (ARE) was found in the promoter region of 52 CPP genes, which was the most common type of cis-acting elements. We hypothesized that these stress-related elements, especially the antioxidant elements, existed in two diploid Brassica progenitors and were entirely preserved during allopolyploidization. We also found some elements related to development and growth that could regulate cell cycle (MSA-like) and affect expression of meristem (CAT-box), and meanwhile, CPP genes have been shown to participate in the coordination of cell cycle and fate (Wang et al., 2018). In addition, a large number of hormone-responsive elements have been found, including methyl jasmonate response elements (CGTCA-motif), abscisic acid response elements (ABRE) and salicylic acid response elements (TCA-element). We further analyzed and compared the cis-acting elements of the promoter regions of 20 homoeologous gene pairs mentioned above, and the results showed that there were four gene pairs (BrCPP7b and BnA.CPP7a, BoCPP1 and BnC.CPP1a, BrCPP8a and BnA.CPP8b, BoCPP8a and BnC.CPP8a) have the same cis-acting element type, indicating that the cis-acting elements of the promoter regions in these CPP genes were conserved during allopolyploidization. Moreover, eight BnCPP genes were found to have more cis-element types or numbers than their diploid progenitors, which might contribute to the regulatory role of CPP gens in the genetics, development and evolution of B. napus.
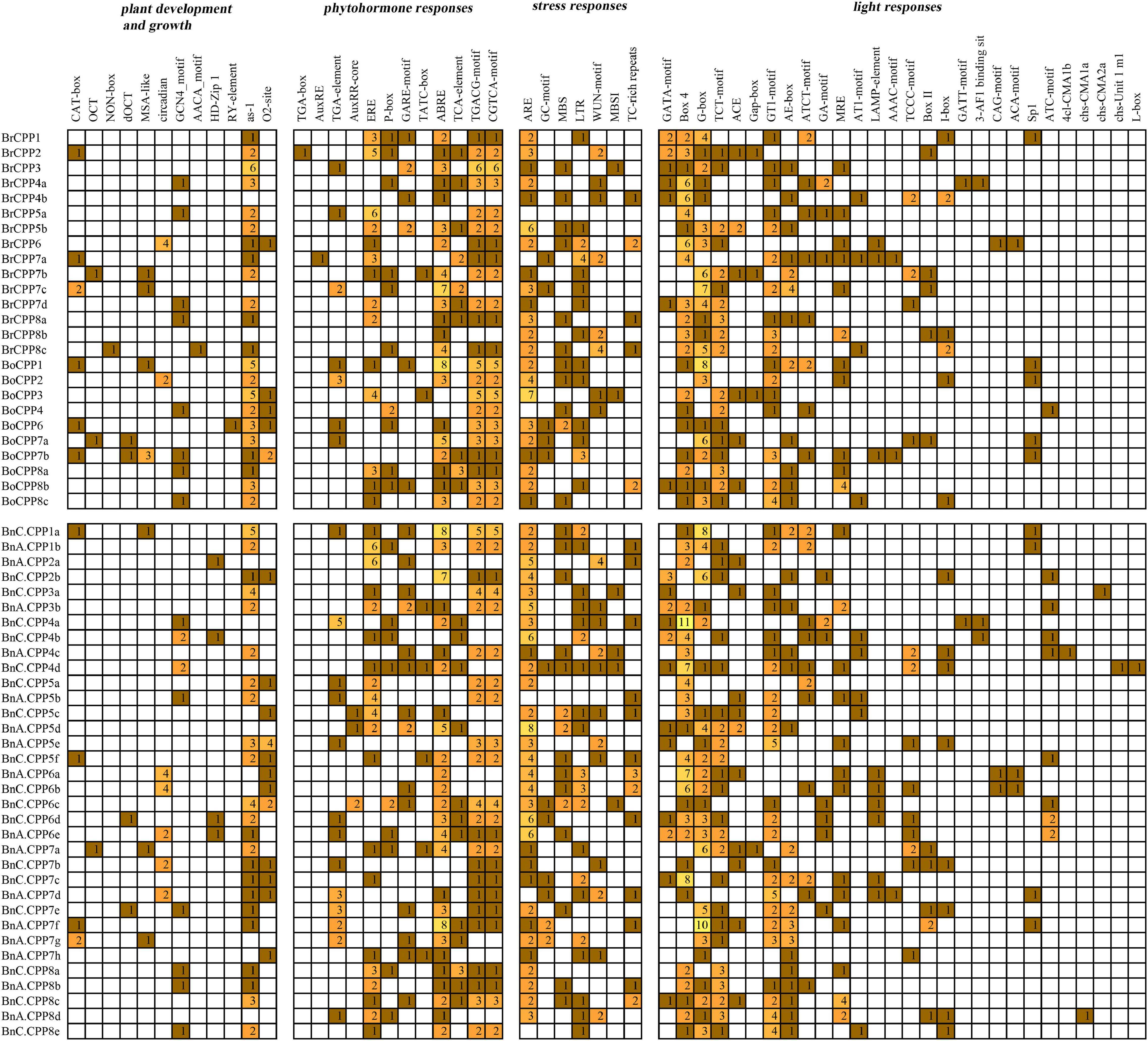
Figure 5. Cis-acting elements on promoters of CPP genes from B. napus and diploid progenitors.
To explore the expression patterns of CPP genes, we analyzed the expression patterns of CPP genes in four organs (stems, leaves, flowers and siliques) in B. napus and its diploid progenitors (Figure 6). CPP genes were widely expressed in the four organs, suggesting that the CPP gene family might have multiple biological functions and might play roles in different organs. Two BnCPP genes (BnA.CPP5e and BnC.CPP5f) were not expressed in the four organs, suggesting that they might not play a role in these organs or might be expressed in specific time and space. The expression patterns of different CPP genes were various in the four organs. For example, the expression level of BrCPP1 was relatively high in flowers, while the expression level of BrCPP4a and BnC.CPP4a was relatively high in siliques. Based on the expression data of 20 homoeologous gene pairs with potential direct evolutionary relationship, we compared the expression patterns between BnCPP and BrCPP/BoCPP genes to explore the evolutionary changes of CPP gene expression during allopolyploidization. We found most of these gene pairs showed different expression patterns in these organs. The expression of some BnCPP genes and their ancestral homoeologous genes in some organs was completely opposite. For example, the expression levels of BrCPP1 and BoCPP8a were the highest in flowers, while the expression levels of BnA.CPP1b and BnC.CPP8a were the lowest in flowers. Similarly, the expression level of BoCPP8c was the lowest in leaves, while the expression level of BnC.CPP8e was the highest in leaves. In addition, some CPP genes were expressed at roughly the same level in different organs, while the expression of their homoeologous genes were significantly different in these organs. For example, the expression level of BoCPP7a in leaves and siliques was identical, while the expression level of BnC.CPP7b in leaves was significantly lower than that in siliques. Moreover, one gene of the homoeologous pairs was expressed and the other was not expressed in some organs. For example, BnA.CPP3b was expressed only in stems, while its homoeologous gene BrCPP3 was expressed in flowers and leaves, but not in stems. These results indicated that the expression patterns of CPP genes might be changed in the process of allopolyploidization. By comparing the expression levels of 13 homoeologous gene pairs with the same gene structure in the four organs, it was found that the majority of them had different expression patterns, that is, the gene expression might change though there was no significant change at the DNA level, which might be related to the regulation of gene expression during allopolyploidization.
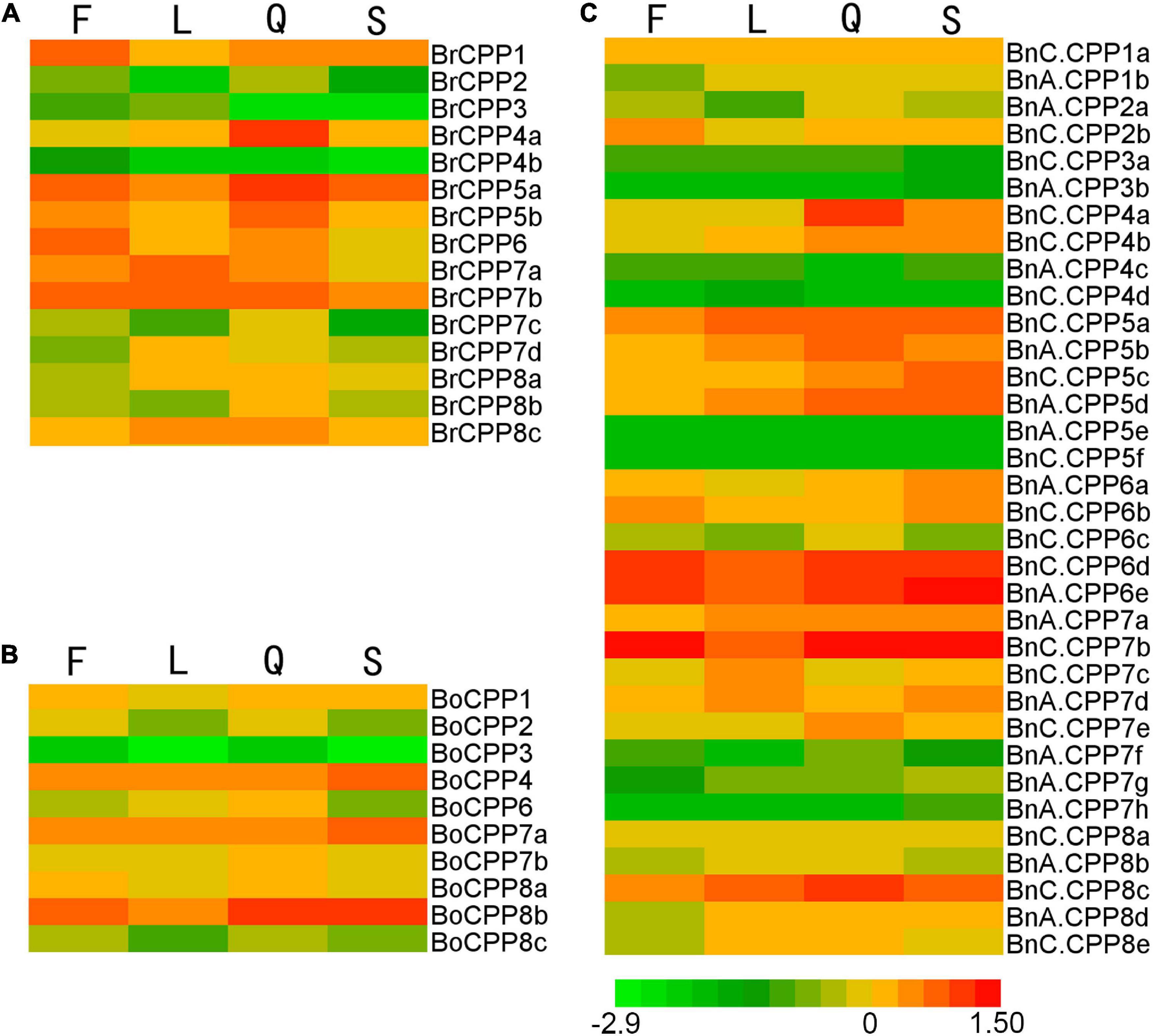
Figure 6. Expression patterns of CPP genes in flowers (F), leaves (L), siliques (Q) and stems (S) from B. rapa (A), B. oleracea (B), and B. napus (C).
To study the expression bias of CPP genes in different organs of allotetraploid B. napus, 20 homoeologous gene pairs were classified into eight groups (CPP1–CPP8) based on their homology. There were four groups (CPP2, CPP4–CPP6) in which the ancestor genes were not present in both B. rapa and B. oleracea, so they could not be analyzed in this way. Finally, four groups of genes (CPP1, CPP3, CPP7, and CPP8) were used for the gene expression bias analysis. The results showed that the expression bias of BnCPP genes was inconsistent in different organs (Supplementary Table 5). Specifically, the expression of BnCPP8 was biased toward B. rapa in all four organs. The expression of BnCPP3 was biased toward B. rapa in flowers, leaves and siliques, while which was biased toward B. oleracea in stems. The expression of BnCPP7 was also biased toward B. rapa in three organs (flowers, stems and siliques), and was biased toward B. oleracea in leaves. The expression of BnCPP1 was biased toward B. rapa only in leaves, and was biased toward B. oleracea in the other three organs (flowers, stems and siliques). We found that 75% of BnCPP gene expression was biased toward B. rapa in flowers, leaves and siliques, while this proportion was only 50% in stems.
Previous studies have shown that CPP gene family can respond to a variety of abiotic stresses, such as low temperature and drought stress. To investigate whether the CPP gene family in three Brassica species responds to salt stress, we analyzed the expression of some CPP genes under salt stress by quantitative real-time PCR (qRT-PCR). CPP genes whose expression was not detected in the experimental material were not analyzed in further study, and we finally selected 12 CPP genes for the qRT-PCR experiment (Supplementary Table 1). The expression levels of 11 CPP genes were significantly changed after salt stress, suggesting that they might be involved in the response to salt stress (Figure 7). Further analysis showed that the expression levels of 8 CPP genes increased significantly after salt stress, while the expression levels of 3 CPP genes decreased significantly. These results suggest that the CPP gene family might adapt to salt stress through two different response mechanisms (positive and negative), and positive regulation might be the main regulatory mode. It was noted that the expression levels of some CPP genes increased sharply after salt stress. For example, the expression levels of BoCPP1 and BoCPP4 were upregulated by 140 and 80 times, respectively, and their expression levels were much higher than those of other up-regulated genes. Therefore, it was speculated that these two BoCPP genes might play dominant roles in salt stress response.
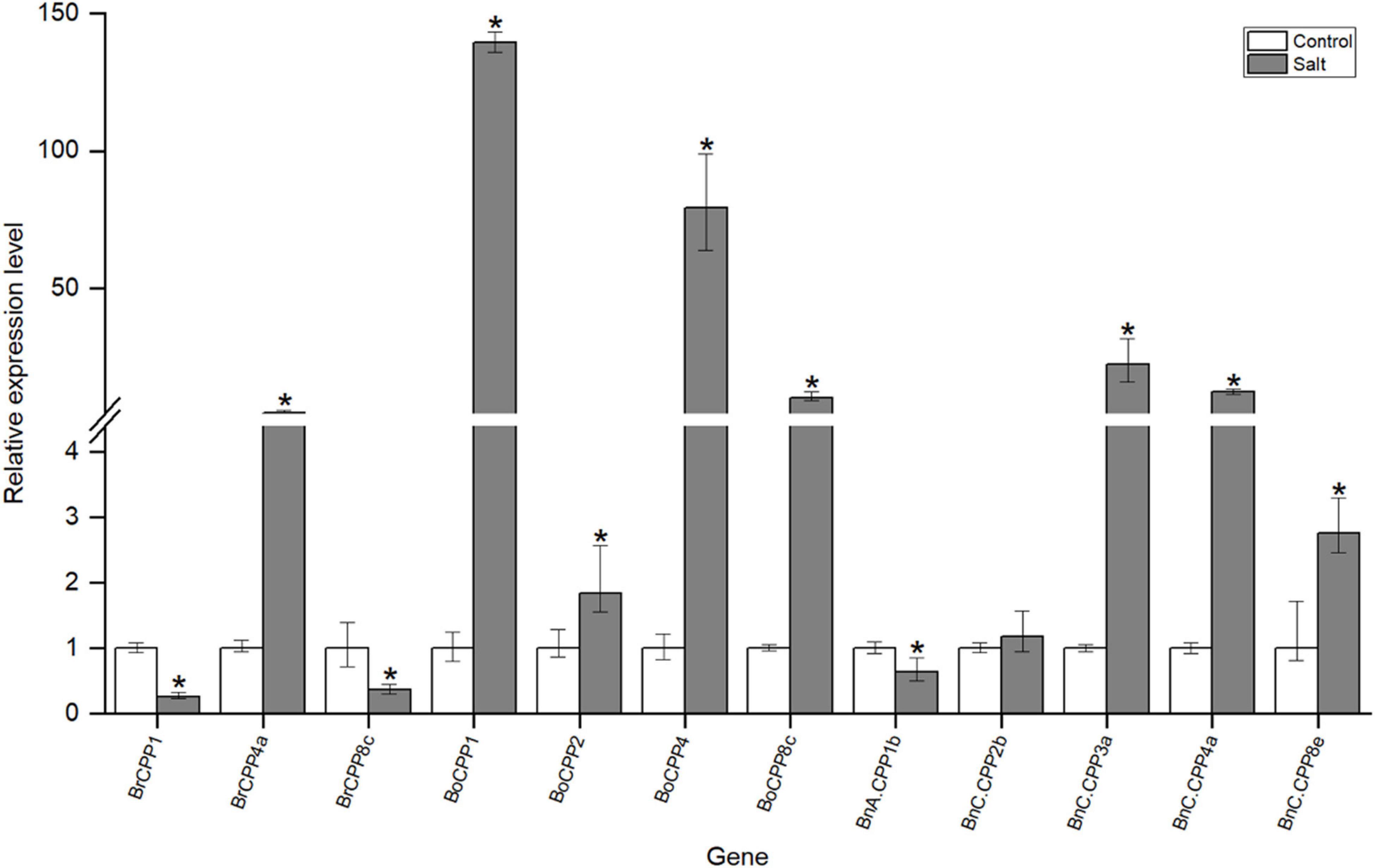
Figure 7. Expression of CPP genes in leaves of B. rapa, B. oleracea, and B. napus. Control: the control group; Salt: salt treatment group; “*” represents a significant difference between the two groups.
In addition, there were three homoeologous gene pairs that have potential direct evolutionary relationship (BrCPP1 and BnA.CPP1b, BrCPP4a and BnC.CPP4a, BoCPP8c and BnC.CPP8e) among the 11 CPP genes whose expression levels changed significantly after salt stress, and the changes of their expression levels after salt stress were compared and analyzed. We found that the expression of the three BnCPP genes and their homoeologous genes in two diploid progenitors showed the same trend after salt stress. In particular, the expression of BrCPP1 and BnA.CPP1b was down-regulated after salt stress, and the expressions of BrCPP4a and BnC.CPP4a and BoCPP8c and BnC.CPP8e were up-regulated after salt stress. However, the expression levels of these BnCPP genes and their corresponding homoeologous genes in two diploid progenitors were different in degree. For example, the expression of BnC.CPP4a was up-regulated about 12.5 times, while the expression of BrCPP4a was up-regulated only about 4.8 times after salt stress, and the expression of BnC.CPP8e increased 2.8 times, while the expression of BoCPP8c increased 10.3 times after salt stress. These results suggested that the response degree of CPP genes to salt stress might change after allopolyploidization.
The superoxide dismutase (SOD) activity of the three Brassica species increased after salt stress, among which the SOD activity of B. rapa increased the most, which was 1.7 times of the control, and the SOD activity of B. oleracea and B. napus was 1.18 times and 1.12 times of the control, respectively (Figure 8). After salt stress, peroxidase (POD) activity of B. oleracea and B. napus increased by 1.62 and 1.43 times, respectively, while POD activity of B. rapa decreased. Similarly, after salt stress, POD activity of B. oleracea and B. napus increased by 1.63 and 1.35 times, respectively, while catalase (CAT) activity of B. rapa decreased. Therefore, the activities of all three enzymes were increased to remove reactive oxygen in B. oleracea and B. napus after salt stress, while only the activity of SOD was increased in B. rapa, and the activities of the other two enzymes were restricted. These results suggested that the response of antioxidant enzyme systems in three Brassica species to salt stress might be different.
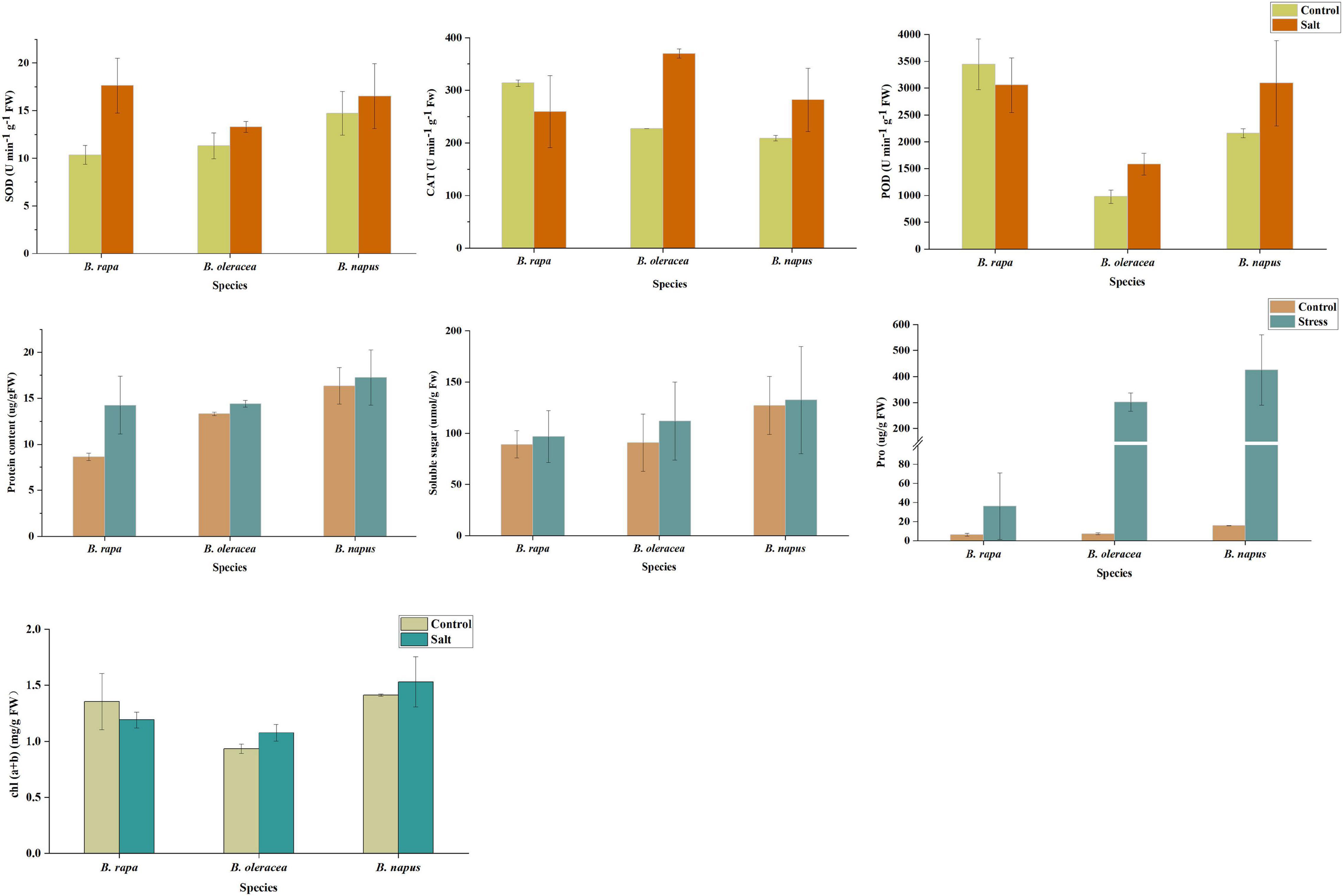
Figure 8. The physiological responses of B. napus and diploid progenitors to salt stress.
Plants can also respond to adverse conditions by accumulating osmotic substances. Soluble protein accumulation occurred in all three Brassica species after salt stress, which was most obvious in B. rapa (Figure 8). For details, after salt stress, the soluble protein content of B. rapa was 1.65 times that of the control, while which of B. oleracea and B. napus was 1.08 and 1.05 times that of the control, respectively. The soluble sugar content of three Brassica species increased after salt stress, and which of B. oleracea increased most. The soluble sugar content of B. oleracea was 1.23 times that of the control, and which of B. rapa and B. napus was 1.09 and 1.04 times that of the control, respectively. In addition, we observed an interesting phenomenon that the content of proline in the three Brassica species increased sharply after salt stress, and the content of proline in B. rapa, B. oleracea, and B. napus were 4.7, 40.9 and 26.3 times higher than that of the control, respectively. These results indicated that these substances played the corresponding osmotic regulation functions to alleviate the salt stress injury of B. napus and its progenitors. Moreover, after salt stress, chlorophyll content in leaves of B. rapa decreased, but which of B. oleracea and B. napus increased, suggesting that the three species might have different salt-stress response mechanisms.
Polyploidization of plants can lead to the enhancement of their environmental adaptability (Segraves, 2017), and the formation and evolution of polyploids are often accompanied by series changes in plant morphology, physiology, genetics and epigenetics (Soltis et al., 2016). A large number of studies have been conducted on the large-scale genetic and epigenetic changes of plants after allopolyploidization, but there were few studies focused on the molecular basis of environmental adaptability on allopolyploids from the perspective of gene family. The allopolyploid B. napus and its two diploid progenitors (B. rapa and B. oleracea) was an appropriate system to study the scientific questions about allopolyploidization. As the transcription factor gene family, CPP gene family played a role in stress response of plants, and this gene family was not reported in Brassica species. In this study, the characteristics and the salt response of CPP gene family in B. napus and its progenitors were compared, which was conducive to exploring the genetic evolution characteristics of CPP gene family and detecting the potential advantageous molecular basis of stress response in allopolyploid B. napus.
The phenomenon of gene family expansion is widespread in the process of plant evolution. For example, the majority of gene families (about 80%) in Arabidopsis thaliana showed expansion in their evolutionary course (Lespinet et al., 2002). In this study, 15, 10, and 34 CPP genes were identified in B. rapa, B. oleracea, and B. napus, and the number of genes in this three Brassica species can be analyzed as follows. Brassica and Arabidopsis diverged from a common ancestor about 14 million years ago (MYA; Song et al., 2020). All 8 CPP genes in Arabidopsis had corresponding homoeologous genes in B. rapa, B. oleracea, and B. napus, indicating that the progenitors of Brassica retained these CPP genes completely after being differentiated from the evolutionary lineage of Arabidopsis. After separating from the evolutionary lineage of Arabidopsis, the progenitors of Brassica underwent a unique whole genome triplication (WGT) event and divided into two diploid species B. rapa and B. oleracea about 3 MYA (Song et al., 2020). Therefore, in theory, if the progenitors of Brassica had not lost genes after the WGT event, there should be three times as many CPP genes in B. rapa and B. oleracea as in Arabidopsis. However, we found that many CPP genes in Arabidopsis have less than three homoeologous genes in diploid B. rapa and B. oleracea (Table 1). Therefore, due to the WGT events, the CPP gene family in Brassica expands compared to Arabidopsis, and at the same time, gene loss events occur in large numbers. In addition, the allotetraploid B. napus was formed by hybridization and genome doubling (i.e., allopolyploidization) of two diploid B. rapa and B. oleracea about 7,500 years ago (Chalhoub et al., 2014). Theoretically, the number of CPP gene family members in B. napus should be equal to the sum of the number of CPP members in the diploid B. rapa and B. oleracea. In this study, we found that the total number of CPP gene family members in B. napus was more than the sum of the two diploid progenitor species (34 > 15 + 10), that is, the CPP gene family also expanded in B. napus during allopolyploidization. Moreover, loss of CPP genes, such as CPP8 homoeologues gene (5 < 3 + 3), was also found during allopolyploidization. From the above analysis, it can be concluded that the CPP gene family expanded in B. napus during evolution, despite the occurrence of gene loss events.
The main duplication modes that cause gene family expansion might be different. Analysis of gene duplication modes in gene families can provide explanations for the expansion of these gene families (Liang et al., 2017). Cannon et al. (2004) studied the duplication modes of 50 gene families in Arabidopsis thaliana, and found that the tandem duplication and segmental duplication were dominant in many gene families. In this study, the duplication modes of the CPP genes were studied, and we found that in addition to a large number of WGD, many TRD were detected in CPP genes of three Brassica species. In addition, unlike the two diploid Brassica species, the proportion of TRD in CPP genes was higher than that of WGD in B. napus. Therefore, it was speculated that TRD may be an important driving force for the expansion of CPP gene family in B. napus during allopolyploidization.
As an important part of structural genes in eukaryotes, the evolution and function of introns are particularly important. In the study of gene families, analyzing the gene structural changes (changes of the number of exon/intron) may help us to understand the evolutionary direction of gene families. In this study, a comparative analysis of the gene structure of 20 homoeologous gene pairs with potential direct evolutionary relationship was conducted. Our results showed that except for 13 homoeologous gene pairs that were highly conserved in intron number after allopolyploidization, the remaining 7 gene pairs showed differences in intron number, and 6 of them showed more introns in BnCPP than in the corresponding ancestral species BrCPP/BoCPP, indicating that most of the CPP genes with structural changes acquired new introns after allopolyploidization. Similar phenomena have been observed in other gene families of B. napus (Li et al., 2019a). Earlier studies have suggested that introns might bring heavy burdens to the organism. For example, on the one hand, intron splicing required a high biological cost (Jiang and Goertzen, 2011). On the other hand, because introns were not subjected to the pressure of natural selection, a large number of mutations were often accumulated in introns, and accumulation of harmful mutations might adversely affect the adaptation of the organism (Lynch, 2006). However, recent studies have consistently suggested that the presence of introns might provide various benefits to the genome and organisms (Mukherjee et al., 2018), of which several have been discovered. First, introns could be used to improve protein diversity through alternative splicing and the fractionation of exons (Lamond, 1999). Second, introns might lead to the production of non-coding RNAs that were involved in some regulatory processes (Ying and Lin, 2005). Third, introns could also play a positive mediating role to enhance/promote the expression of some genes, as researchers have found that genes with higher expression levels have more and longer introns than genes with lower expression levels in rice and Arabidopsis thaliana (Ren et al., 2006). In addition, introns might also play a positive role in transcriptional coupling and mRNA output (Maniatis and Reed, 2002). Therefore, the increase of introns in BnCPP genes after allopolyploidization might be beneficial to the genome evolution of B. napus, but the specific effects still need further studies to prove.
A series of genetic and epigenetic changes, occurring rapidly when polyploids were established, which not only help stabilize the plant genome, but also regulate the expression of genes in the plant (Pikaard, 2001). To understand whether gene expression changes occurred during the formation and evolution of allotetraploid B. napus, the expression pattern of CPP genes in three Brassica species was investigated in this study. The results showed that, on the one hand, except for two BnCPP genes, CPP genes were widely expressed in four organs of the three Brassica species, suggesting that the CPP genes might be indispensable in the genetic, developmental and evolutionary process of three species. On the other hand, we also observed various changes in CPP gene expression during allopolyploidization. For example, most gene pairs, despite having the same gene structure, showed divergence in their expression patterns. Therefore, it was speculated that there might be neofunctionalization or subfunctionalization of CPP genes during allopolyploidization, and these changes might have beneficial effects on the evolution of CPP gene family in B. napus. Moreover, the differentiation of gene function might also reduce the risk of redundant genes being eliminated. In addition, we found that the BnCPP gene showed different bias expression, which was different in four organs. For example, the expression of BnCPP3 and BnCPP7 in three organs were biased toward B. rapa, while the expression of BnCPP1 in three organs was biased toward B. oleracea. It was speculated that such a variety of biased gene expression might help allopolyploid B. napus adapt better to the environment (Samans et al., 2017).
Previous studies have suggested that CPP genes may play a role in response to various abiotic stresses. For example, most CPP genes in the roots of soybean can respond to heat stress, and many CPP genes in the leaves of cucumber show different response patterns and can respond to various stresses such as low temperature, drought, ABA and salt (Zhang et al., 2015; Zhou et al., 2018). B. napus, as an important crop, may be affected by various adverse environment during its growth, among which salting environment is one of the main adverse environments. In this study, we investigated the expression of some CPP genes in B. napus and its progenitors under salt stress. The results showed that CPP gene expression levels were significantly changed in B. napus and its two progenitors after salt stress, suggesting that CPP genes in the three Brassica species might play important roles in the response of salt stress. In addition, CPP genes might be involved in the regulation of salt stress related gene expression through positive or negative regulation, and the former might be dominant. By comparing the expression changes of three homoeologous gene pairs with potential direct evolutionary relationship, we found that the expression trend of CPP genes in B. napus was consistent with that in the progenitors, but the degree of change was different. It was speculated that the function of CPP genes in response to salt stress might be well preserved after allopolyploidization, and the change in response degree might help BnCPP genes to respond better and faster to salt stress. Our study can provide reference for the study of molecular resistance mechanism of B. napus.
Polyploids may have better stress resistance than diploids, and studies have shown that polyploids are more capable of producing beneficial substances and reducing harmful substances than diploids when subjected to abiotic stress. For example, more proline and less malondialdehyde accumulate in tetraploid rice than diploid rice under salt stress, and polyploid beet had lower Na+/K+ ratio than diploid beet in response to salt stress (Tu et al., 2014; Zhu, 2016; Wu et al., 2019). When plants are in an unfavorable environment, reactive oxygen species (ROS; such as O2, H2O2, OH–) accumulate in cells, which gradually damage cells and plants. In order to reduce this damage, plants have built a set of enzymes to maintain the balance of reactive oxygen species in cells. These enzymes include SOD, POD, and CAT. In this study, we systematically studied the physiological responses of B. napus and its progenitors to salt stress, including the activity of SOD, POD, and CAT, and the content of soluble protein, soluble sugar, proline, and chlorophyll. Plants can help them adapt to stress by scavenging ROS accumulated inside cells when they are stressed. Therefore, the increased activities of SOD, POD, and CAT may be related to the response of plants to stress. Proline can be used as an osmotic regulator and hydroxyl radical scavenging agent to help stabilize the subcellular structure of plants, and the accumulation of proline contributes to the resistance of plants (Lv et al., 2011). In addition, plants produce a series of proteins related to stress resistance to avoid injury, and soluble proteins can help plants adapt to adversity as functional proteins or maintainers of osmotic potential (Wu et al., 2004). The increase of soluble sugar content is also a strategy for plants to adapt to adversity, as it not only acts as an osmotic regulator, but also provides carbon framework and energy for the synthesis of other organic solutes (Parankusam et al., 2017). As the basis for material and energy exchange with the outside world, chlorophyll content in plant leaves also changes under stress. The decrease of chlorophyll content was observed in B. rapa after salt stress in this study, which is consistent with most of the previous studies, however, the opposite phenomenon was observed in B. oleracea and B. napus, and the reason may be that the salt stress in our study does not cause rapid decomposition of chlorophyll in the leaves, and this phenomenon may also be related to chloroplast shrinkage (Chaves et al., 2009). In addition, compared with the two diploid progenitors, SOD activity and the accumulation of three osmotic regulatory substances were all the highest in B. napus before salt stress, and SOD activity was still relatively high and the accumulation of three osmotic regulatory substances was still the highest in B. napus after salt stress. Moreover, POD and CAT activities were inhibited and chlorophyll content decreased in B. rapa, while POD and CAT activities and chlorophyll content increased in B. napus under salt stress. These results suggest that not all physiological response pathways in allotetraploid B. napus were superior to their diploid progenitors B. rapa and B. oleracea in salt stress, but overall, B. napus showed a stronger and more stable physiological response in this study.
The CPP gene family in B. napus expanded after allopolyploidization, and the genome doubling and transposon mediated gene replication might be the main reasons for the expansion. All identified CPP members can be clearly divided into two clades (I, II), and the gene structure of CPP in clade I was more conserved than that in clade II. The intron increasing phenomenon was observed in some CPP genes after allopolyploidization. Although the gene structure of some CPP genes were not changed, the expression patterns of them were changed in B. napus after allopolyploidization. Some molecular basis might be associated with the adaptability of B. napus, including the expansion of the CPP gene family, the acquisition of introns by some BnCPPs, and abundant cis-acting elements upstream of BnCPPs. A total of 11 CPP genes potentially involved in the response to salt stress, and the response degree of some CPP genes to salt stress might change in B. napus after allopolyploidization. The physiological response of salt stress was stronger and more stable in B. napus than its two progenitors. Our study provides critical reference for exploring the potential advantageous molecular basis of various stress response in allopolyploids.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
ML conceived and drafted the manuscript, performed the data analysis, software analysis, and correction. FW performed the software analysis, surveyed the literature, validated experiments, and visualized the results. JM conducted software analysis, data analysis, and edited and revised the writing. HL performed the software analysis, surveyed the literature, and visualized the results. HY provided the methodology, performed the software analysis, and edited and revised the writing. PZ provided the methodology, guided the writing and editing, and prodded them. JW conceived the manuscript, did project management and data management, directed writing and editing, and provided funding. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (31970241).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.873071/full#supplementary-material
Supplementary Figure 1 | Phenotype figure of B. rapa, B. napus, and B. oleracea under salt stress.
Supplementary Figure 2 | Bar chart of the number of genes for five duplication modes.
Supplementary Figure 3 | Visual display of segmental duplicated gene pairs and five duplication modes. Blue lines, red lines, and gray lines represented the segmental duplicated gene pairs in B. rapa, B. napus, and B. oleracea, respectively. Red, yellow, green, blue, and pink circular markers represented WGD, TRD, TD, PD, and DSD, respectively.
Supplementary Figure 4 | The CXC domain location of CPP proteins in clade I (A) and clade II (B) from B. napus and its diploid progenitors.
Supplementary Figure 5 | Alignment of the conserved CXC domain sequences of CPP proteins from B. napus and its diploid progenitors.
Supplementary Figure 6 | Conserved motif characterization of CPP proteins in clade I (A) and clade II (B) from B. napus and diploid ancestors. The red circle indicates orthologs with potential direct evolutionary relationship.
Abbott, R., Albach, D., Ansell, S., Arntzen, J. W., Baird, S. J., Bierne, N., et al. (2013). Hybridization and speciation. J. Evol. Biol. 26, 229–246. doi: 10.1111/j.1420-9101.2012.02599.x
Adams, K. L., and Wendel, J. F. (2005). Polyploidy and genome evolution in plants. Curr. Opin. Plant Biol. 8, 135–141. doi: 10.1016/j.pbi.2005.01.001
Andersen, S. U., Algreen-Petersen, R. G., Hoedl, M., Jurkiewicz, A., Cvitanich, C., Braunschweig, U., et al. (2007). The conserved cysteine-rich domain of a tesmin/TSO1-like protein binds zinc in vitro and TSO1 is required for both male and female fertility in Arabidopsis thaliana. J. Exp. Bot. 58, 3657–3670. doi: 10.1093/jxb/erm215
Ashraf, M., and McNeilly, T. (2004). Salinity tolerance in Brassica oilseeds. Crit. Rev. Plant Sci. 23, 157–174. doi: 10.1080/07352680490433286
Cannon, S. B., Mitra, A., Baumgarten, A., Young, N. D., and May, G. (2004). The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 4:10. doi: 10.1186/1471-2229-4-10
Cao, Y., Luo, Q., Tian, Y., and Meng, F. (2017). Physiological and proteomic analyses of the drought stress response in Amygdalus Mira (Koehne) Yü et Lu roots. BMC Plant Biol. 17:53. doi: 10.1186/s12870-017-1000-z
Chalhoub, B., Denoeud, F., Liu, S., Parkin, I. A., Tang, H., Wang, X., et al. (2014). Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345, 950–953. doi: 10.1126/science.1253435
Chaves, M. M., Flexas, J., and Pinheiro, C. (2009). Photosynthesis under drought and salt stress: regulation mechanisms from whole plant to cell. Ann. Bot. 103, 551–560. doi: 10.1093/aob/mcn125
Chen, Z. J. (2007). Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu. Rev. Plant Biol. 58, 377–406. doi: 10.1146/annurev.arplant.58.032806.103835
Cheng, F., Liu, S., Wu, J., Fang, L., Sun, S., Liu, B., et al. (2011). BRAD, the genetics and genomics database for Brassica plants. BMC Plant Biol. 11:136. doi: 10.1186/1471-2229-11-136
Cheng, F., Wu, J., Fang, L., Sun, S., Liu, B., Lin, K., et al. (2012). Biased gene fractionation and dominant gene expression among the subgenomes of Brassica rapa. PLoS One 7:e36442. doi: 10.1371/journal.pone.0036442
Cvitanich, C., Pallisgaard, N., Nielsen, K. A., Hansen, A. C., Larsen, K., Pihakaski-Maunsbach, K., et al. (2000). CPP1, a DNA-binding protein involved in the expression of a soybean leghemoglobin c3 gene. Proc. Natl. Acad. Sci. U.S.A. 97, 8163–8168. doi: 10.1073/pnas.090468497
Hauser, B. A., He, J. Q., Park, S. O., and Gasser, C. S. (2000). TSO1 is a novel protein that modulates cytokinesis and cell expansion in Arabidopsis. Development 127, 2219–2226. doi: 10.1242/dev.127.10.2219
Hauser, B. A., Villanueva, J. M., and Gasser, C. S. (1998). Arabidopsis TSO1 regulates directional processes in cells during floral organogenesis. Genetics 150, 411–423. doi: 10.1093/genetics/150.1.411
Jackson, S., and Chen, Z. J. (2010). Genomic and expression plasticity of polyploidy. Curr. Opin. Plant Biol. 13, 153–159. doi: 10.1016/j.pbi.2009.11.004
Jiang, K., and Goertzen, L. R. (2011). Spliceosomal intron size expansion in domesticated grapevine (Vitis vinifera). BMC Res. Notes 4:52. doi: 10.1186/1756-0500-4-52
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lespinet, O., Wolf, Y. I., Koonin, E. V., and Aravind, L. (2002). The role of lineage-specific gene family expansion in the evolution of eukaryotes. Genome Res. 12, 1048–1059. doi: 10.1101/gr.174302
Li, M., Wang, R., Liang, Z., Wu, X., and Wang, J. (2019a). Genome-wide identification and analysis of the EIN3/EIL gene family in allotetraploid Brassica napus reveal its potential advantages during polyploidization. BMC Plant Biol. 19:110. doi: 10.1186/s12870-019-1716-z
Li, M., Wang, R., Liu, Z., Wu, X., and Wang, J. (2019b). Genome-wide identification and analysis of the WUSCHEL-related homeobox (WOX) gene family in allotetraploid Brassica napus reveals changes in WOX genes during polyploidization. BMC Genomics 20:317. doi: 10.1186/s12864-019-5684-3
Li, M., Wang, R., Wu, X., and Wang, J. (2020). Homoeolog expression bias and expression level dominance (ELD) in four tissues of natural allotetraploid Brassica napus. BMC Genomics 21:330. doi: 10.1186/s12864-020-6747-1
Liang, Y., Wan, N., Cheng, Z., Mo, Y., Liu, B., Liu, H., et al. (2017). Whole-genome identification and expression pattern of the vicinal oxygen chelate family in rapeseed (Brassica napus L.). Front. Plant Sci. 8:745. doi: 10.3389/fpls.2017.00745
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Lv, W. T., Lin, B., Zhang, M., and Hua, X. J. (2011). Proline accumulation is inhibitory to Arabidopsis seedlings during heat stress. Plant Physiol. 156, 1921–1933. doi: 10.1104/pp.111.175810
Lynch, M. (2006). The origins of eukaryotic gene structure. Mol. Biol. Evol. 23, 450–468. doi: 10.1093/molbev/msj050
Madlung, A. (2013). Polyploidy and its effect on evolutionary success: old questions revisited with new tools. Heredity 110, 99–104. doi: 10.1038/hdy.2012.79
Maniatis, T., and Reed, R. (2002). An extensive network of coupling among gene expression machines. Nature 416, 499–506. doi: 10.1038/416499a
Mukherjee, D., Saha, D., Acharya, D., Mukherjee, A., Chakraborty, S., and Ghosh, T. C. (2018). The role of introns in the conservation of the metabolic genes of Arabidopsis thaliana. Genomics 110, 310–317. doi: 10.1016/j.ygeno.2017.12.003
Parankusam, S., Bhatnagar-Mathur, P., and Sharma, K. K. (2017). Heat responsive proteome changes reveal molecular mechanisms underlying heat tolerance in chickpea. Environ. Exp. Bot. 141, 132–144. doi: 10.1016/j.envexpbot.2017.07.007
Pikaard, C. S. (2001). Genomic change and gene silencing in polyploids. Trends Genet. 17, 675–677. doi: 10.1016/s0168-9525(01)02545-8
Prakash, S., Bhat, S., Quiros, C., Kirti, P., and Chopra, V. (2009). Brassica and its close allies: cytogenetics and evolution. Plant Breed Rev. 31:21. doi: 10.1002/9780470593783.ch2
Qiao, X., Li, Q., Yin, H., Qi, K., Li, L., Wang, R., et al. (2019). Gene duplication and evolution in recurring polyploidization-diploidization cycles in plants. Genome Biol. 20:38. doi: 10.1186/s13059-019-1650-2
Quinet, M., Vromman, D., Clippe, A., Bertin, P., Lequeux, H., Dufey, I., et al. (2012). Combined transcriptomic and physiological approaches reveal strong differences between short- and long-term response of rice (Oryza sativa) to iron toxicity. Plant Cell Environ. 35, 1837–1859. doi: 10.1111/j.1365-3040.2012.02521.x
Ren, X. Y., Vorst, O., Fiers, M. W., Stiekema, W. J., and Nap, J. P. (2006). In plants, highly expressed genes are the least compact. Trends Genet. 22, 528–532. doi: 10.1016/j.tig.2006.08.008
Riechmann, J. L., Heard, J., Martin, G., Reuber, L., Jiang, C., Keddie, J., et al. (2000). Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science 290, 2105–2110. doi: 10.1126/science.290.5499.2105
Samans, B., Chalhoub, B., and Snowdon, R. J. (2017). Surviving a genome collision: genomic signatures of allopolyploidization in the recent crop species Brassica napus. Plant Genome 10, 1–15. doi: 10.3835/plantgenome2017.02.0013
Segraves, K. A. (2017). The effects of genome duplications in a community context. New Phytol. 215, 57–69. doi: 10.1111/nph.14564
Soltis, D. E., Visger, C. J., Marchant, D. B., and Soltis, P. S. (2016). Polyploidy: pitfalls and paths to a paradigm. Am. J. Bot. 103, 1146–1166. doi: 10.3732/ajb.1500501
Song, J. M., Guan, Z., Hu, J., Guo, C., Yang, Z., Wang, S., et al. (2020). Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat. Plants 6, 34–45. doi: 10.1038/s41477-019-0577-7
Song, J. Y., Leung, T., Ehler, L. K., Wang, C., and Liu, Z. (2000). Regulation of meristem organization and cell division by TSO1, an Arabidopsis gene with cysteine-rich repeats. Development 127, 2207–2217. doi: 10.1242/dev.127.10.2207
Song, X. Y., Zhang, Y. Y., Wu, F. C., and Zhang, L. (2016). Genome-wide analysis of the maize (Zea may L.) CPP-like gene family and expression profiling under abiotic stress. Genet. Mol. Res. 15:gmr.15038023. doi: 10.4238/gmr.15038023
Tu, Y., Jiang, A., Gan, L., Hossain, M., Zhang, J., Peng, B., et al. (2014). Genome duplication improves rice root resistance to salt stress. Rice 7:15. doi: 10.1186/s12284-014-0015-4
Wang, J., Jin, Z., Zhou, M., Yu, Y., and Liang, M. (2020). Characterization of NF-Y transcription factor families in industrial rapeseed (Brassica napus L.) and identification of BnNF-YA3, which functions in the abiotic stress response. Ind. Crops Prod. 148:112253. doi: 10.1016/j.indcrop.2020.112253
Wang, R., Li, M., Wu, X., and Wang, J. (2019). The gene structure and expression level changes of the GH3 gene family in Brassica napus relative to its diploid ancestors. Genes 10:58. doi: 10.3390/genes10010058
Wang, W., Sijacic, P., Xu, P., Lian, H., and Liu, Z. (2018). Arabidopsis TSO1 and MYB3R1 form a regulatory module to coordinate cell proliferation with differentiation in shoot and root. Proc. Natl. Acad. Sci. U.S.A. 115, e3045–e3054. doi: 10.1073/pnas.1715903115
Wu, G., Lin, L., Jiao, Q., and Li, S. (2019). Tetraploid exhibits more tolerant to salinity than diploid in sugar beet (Beta vulgaris L.). Acta Physiol. Plant. 41:52. doi: 10.1007/s11738-019-2844-7
Wu, Z. H., Zeng, F. H., Ma, S. J., Lin, L. J., Xie, Y. J., and Lu, X. Y. (2004). Effects of exogenous ABA on soluble protein in Cynodon dactylon under PEG stress. Acta Pratac. Sin. 13, 75–78. doi: 10.1088/1009-0630/6/5/011
Xu, C., Li, Z., and Wang, J. (2020). Linking heat and adaptive responses across temporal proteo-transcriptome and physiological traits of Solidago canadensis. Environ. Exp. Bot. 175:104035. doi: 10.1016/j.envexpbot.2020.104035
Yang, Z., Gu, S., Wang, X., Li, W., Tang, Z., and Xu, C. (2008). Molecular evolution of the CPP-like gene family in plants: insights from comparative genomics of Arabidopsis and rice. J. Mol. Evol. 67, 266–277. doi: 10.1007/s00239-008-9143-z
Ying, S. Y., and Lin, S. L. (2005). Intronic microRNAs. Biochem. Biophys. Res. Commun. 326, 515–520. doi: 10.1016/j.bbrc.2004.10.215
Yoo, M. J., and Wendel, J. F. (2014). Comparative evolutionary and developmental dynamics of the cotton (Gossypium hirsutum) fiber transcriptome. PLoS Genet. 10:e1004073. doi: 10.1371/journal.pgen.1004073
Zhang, L., Zhao, H. K., Wang, Y. M., Yuan, C. P., Zhang, Y. Y., Li, H. Y., et al. (2015). Genome-wide identification and expression analysis of the CPP-like gene family in soybean. Genet. Mol. Res. 14, 1260–1268. doi: 10.4238/2015.February.13.4
Zhou, T., Wang, Y., Chen, J. Q., Araki, H., Jing, Z., Jiang, K., et al. (2004). Genome-wide identification of NBS genes in japonica rice reveals significant expansion of divergent non-TIR NBS-LRR genes. Mol. Genet. Genomics 271, 402–415. doi: 10.1007/s00438-004-0990-z
Zhou, Y., Hu, L., Ye, S., Jiang, L., and Liu, S. (2018). Genome-wide identification and characterization of cysteine-rich polycomb-like protein (CPP) family genes in cucumber (Cucumis sativus) and their roles in stress responses. Biologia 73, 425–435. doi: 10.2478/s11756-018-0049-y
Keywords: adaptive advantage, Brassica napus, CPP gene family, gene expression, physiological response, salt stress, allopolyploidization
Citation: Li M, Wang F, Ma J, Liu H, Ye H, Zhao P and Wang J (2022) Comprehensive Evolutionary Analysis of CPP Genes in Brassica napus L. and Its Two Diploid Progenitors Revealing the Potential Molecular Basis of Allopolyploid Adaptive Advantage Under Salt Stress. Front. Plant Sci. 13:873071. doi: 10.3389/fpls.2022.873071
Received: 10 February 2022; Accepted: 29 March 2022;
Published: 25 April 2022.
Edited by:
Rajeev K. Varshney, International Crops Research Institute for the Semi-Arid Tropics (ICRISAT), IndiaReviewed by:
Jinpeng Wang, Institute of Botany (CAS), ChinaCopyright © 2022 Li, Wang, Ma, Liu, Ye, Zhao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianbo Wang, amJ3YW5nQHdodS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.