
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 07 April 2022
Sec. Plant Bioinformatics
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.860791
 Haowu Chang1,2
Haowu Chang1,2 Hao Zhang1,2*
Hao Zhang1,2* Tianyue Zhang1
Tianyue Zhang1 Lingtao Su2,3
Lingtao Su2,3 Qing-Ming Qin4
Qing-Ming Qin4 Guihua Li4
Guihua Li4 Xueqing Li1
Xueqing Li1 Li Wang1
Li Wang1 Tianheng Zhao1
Tianheng Zhao1 Enshuang Zhao1
Enshuang Zhao1 Hengyi Zhao1
Hengyi Zhao1 Yuanning Liu1,2
Yuanning Liu1,2 Gary Stacey5
Gary Stacey5 Dong Xu2*
Dong Xu2*
Although growing evidence shows that microRNA (miRNA) regulates plant growth and development, miRNA regulatory networks in plants are not well understood. Current experimental studies cannot characterize miRNA regulatory networks on a large scale. This information gap provides an excellent opportunity to employ computational methods for global analysis and generate valuable models and hypotheses. To address this opportunity, we collected miRNA–target interactions (MTIs) and used MTIs from Arabidopsis thaliana and Medicago truncatula to predict homologous MTIs in soybeans, resulting in 80,235 soybean MTIs in total. A multi-level iterative bi-clustering method was developed to identify 483 soybean miRNA–target regulatory modules (MTRMs). Furthermore, we collected soybean miRNA expression data and corresponding gene expression data in response to abiotic stresses. By clustering these data, 37 MTRMs related to abiotic stresses were identified, including stress-specific MTRMs and shared MTRMs. These MTRMs have gene ontology (GO) enrichment in resistance response, iron transport, positive growth regulation, etc. Our study predicts soybean MTRMs and miRNA-GO networks under different stresses, and provides miRNA targeting hypotheses for experimental analyses. The method can be applied to other biological processes and other plants to elucidate miRNA co-regulation mechanisms.
The growth and development of crops are often restricted due to various environmental stresses, leading to poor harvests and yields below their genetic potential (Ku et al., 2015; Li et al., 2017). In the past decade, microRNAs (miRNAs) have been identified as important gene expression regulatory factors that play an essential role in plant growth and development (Ruiz-Ferrer and Voinnet, 2009). miRNA can target multiple genes, and multiple miRNAs can also target the same gene. miRNAs are involved in the expression of stress-responsive genes and the plant’s ability to adapt to environmental change (Sunkar et al., 2007). Different stresses can induce differential expressions of corresponding miRNAs in plants, while some miRNAs can simultaneously respond to several abiotic stresses (Shukla et al., 2008; Song et al., 2019; Sun et al., 2019). Therefore, studying the cooperative relationship among miRNAs and the interactions with their target genes is essential for understanding the role of miRNAs in controlling plant growth and development.
MicroRNAs may respond to adverse effects on plant growth and development, such as drought, salinity, temperature, and other abiotic environmental factors. It was shown that willow leaves exposed to drought or high temperature induce differential expressions of some miRNAs (Hivrale et al., 2016). For example, miR169c plays a negative regulatory role under drought stress by inhibiting the expression of its target gene nuclear factor Y-A (NF-YA) (Yu Y. et al., 2019). miR172a (Pan et al., 2016) and miR172c (Li et al., 2016) endow plants with a tolerance to salt stress and water deficiency. Meanwhile, miRNAs also indirectly respond to abiotic stress by regulating other biological macromolecules. For example, miR398c can negatively regulate multiple peroxisome-related genes (GmCSD1a/b, GmCSD2a/b/c, and GmCCS) and affect the drought tolerance of the soybean (Zhou et al., 2020). miR166k/o, miR390g, and miR396c/k mediate BX10 (Al-tolerant genotype) root elongation, and miR169r triggers the BD2 (Al-sensitive B genotype) oxidative stress, which in turn triggers a different type of plant aluminum tolerance between BX10 and BD2 (Huang et al., 2018). This indicates that miRNA may regulate plant growth under abiotic stress through a complex network. However, current studies typically explore the role of few miRNA in response to abiotic stresses. From a global view, how miRNAs work together as a co-regulatory mechanism has not been significantly explored.
Several studies have uncovered interesting miRNA interactions. For example, miR160 and miR167 are involved in the adventitious root program of Arabidopsis (Xu et al., 2014c). miR156 and miR172 play a role in the transition of soybean nutrition (Yoshikawa et al., 2013). Transgenic studies of miR482, miR1512, and miR1515 showed that their over-expression may lead to a substantial increase in the number of soybean nodules (Li et al., 2010). Another study verified networks of 365 tissue-specific miRNA–target interactions (MTIs) (Wang et al., 2019). In addition, Ismalia et al. (2019) used SVR to study the interaction between miRNA and lncRNA, constructed a network of miRNA–mRNA, miRNA–lncRNA, and miRNA–mRNA–lncRNA, and recognized their regulatory roles in stress response of Arabidopsis thaliana. Tu et al. (2022) mined the miRNA–lncRNA–TF regulatory network related to leaf and flower development of Liriodendron chinense, and pointed out that lch-lnc7374-miR156h-SPL3 and lch-lnc7374-miR156j-SPL9 are potential regulators of stamen and pistil development, respectively. And the miR157a-SPL and miR160a-ARF modules were validated using RLM-RACE, both of which are involved in leaf and flower development (Tu et al., 2022). The synergistic effects of miRNAs provide a new systematic perspective for the entire microRNome (Xu et al., 2014c), which calls for a global analysis of MTIs. Yang et al. (2021) found that the differential expression of key miRNA–target modules in plants may promote their root growth and development and enhance their tolerance to various stresses. Fu et al. (2019) revealed the response mechanism of potato miRNA–mRNA under alkali stress. It is of great significance to explore the biological mechanism of plants under abiotic stress from the perspective of miRNA–target.
Several methods have been developed and applied to explore this field with the growing miRNA-target data. Shalgi et al. (2007) first constructed a miRNA network from the target genes predicted by PicTar and TargetScan. Xu et al. (2011) constructed a human miRNA–miRNA functional synergy network through co-regulation functional modules. Meanwhile, biclustering was also applied for two different types of objects (gene and miRNA in this case) belonging to the same cluster. Various bi-clustering methods have been developed (Huang and Brutlag, 2001; Yoon and De Micheli, 2005; Caldas and Kaski, 2011; Xie et al., 2019). SAMBA (Tanay et al., 2002), ISA (Bergmann et al., 2003), BIMAX (Prelic et al., 2006), QUBIC (Li et al., 2009), and FABIA (Hochreiter et al., 2010) are some commonly used general algorithms. Contiguous column coherent (CCC) biclustering (Goncalves et al., 2009; Madeira et al., 2010; Medina et al., 2010; Goncalves and Madeira, 2014; Henriques and Madeira, 2014; Henriques et al., 2017) and LateBiccluster (Goncalves and Madeira, 2014) are designed for temporal data analysis. BicPAM (Henriques and Madeira, 2014; Henriques et al., 2017), BicNET (Henriques and Madeira, 2016) and MCbiclust (Bentham et al., 2017) are the latest tools. Pio et al. (2013) applied the biclustering algorithm to predict human miRNA–mRNA modules. The application of biclustering algorithms and miRNA–target regulation module (MTRM) mining is feasible and important for analyzing miRNA regulation mechanisms. Compared with traditional clustering methods, such as Bimax (Prelic et al., 2006) and BiBit (Rodriguez-Baena et al., 2011), CUBiBit (Gonzalez-Dominguez and Exposito, 2019) shortened the computing time and provided an optimized method for finding modules in larger data. However, the result obtained by CUBiBit was mostly a fully-connected bipartite graph, and the relationship between miRNA and the target gene is complex and interactive.
In this study, we proposed a method to obtain the miRNA regulatory modules and analyze their relationship in response to abiotic stresses in the soybean as a means for extending our understanding of soybean resistance mechanisms. Previously, Xu et al. (2014d) provided a soybean miRNA-gene network, SoyFN, based on predicted miRNA targets. However, this work was based only on sequence comparisons, which may result in a high false discovery rate. In contrast, in our work, we collected experimentally proven miRNA–target relationships based on degradome sequencing in the soybean and the stringent homologs of miRNA–target pairs in A. thaliana and M. truncatula. Based on these reliable miRNA–target data, we performed a biclustering analysis. We iteratively fused the overlapping biclusters based on the SoyNet network to obtain the soybean miRNA–target regulatory modules in response to abiotic stresses. We provide soybean MTRMs with high confidence relevant to various stresses, verified by REVIGO analysis to have the concentration of GO functions, and present the miRNA–GO regulatory networks of these modules. Capturing these miRNA–target modules with biological significance expands our understanding of the complex regulatory mechanisms of miRNA. The methods used should be readily applicable to other plant and animal systems where sufficient data exists to perform the analyses.
We collected soybean MTIs from A. thaliana and M. truncatula databases and publications on miRNAs and genes of soybean response to several abiotic stresses. Subsequently, we used homology prediction on the collected MTIs to expand the soybean MTIs. Next, we used the biclustering method to mine the soybean MTRMs to perform overlap analysis to remove the redundancy. Then, based on the soybean gene interaction network, biclustering was applied through multi-level iteration. Finally, based on soybean abiotic stress-related miRNAs and genes, the fusion regulatory module was screened to obtain soybean abiotic stress-related MTRMs. Figure 1 shows a flowchart of our tasks and results.
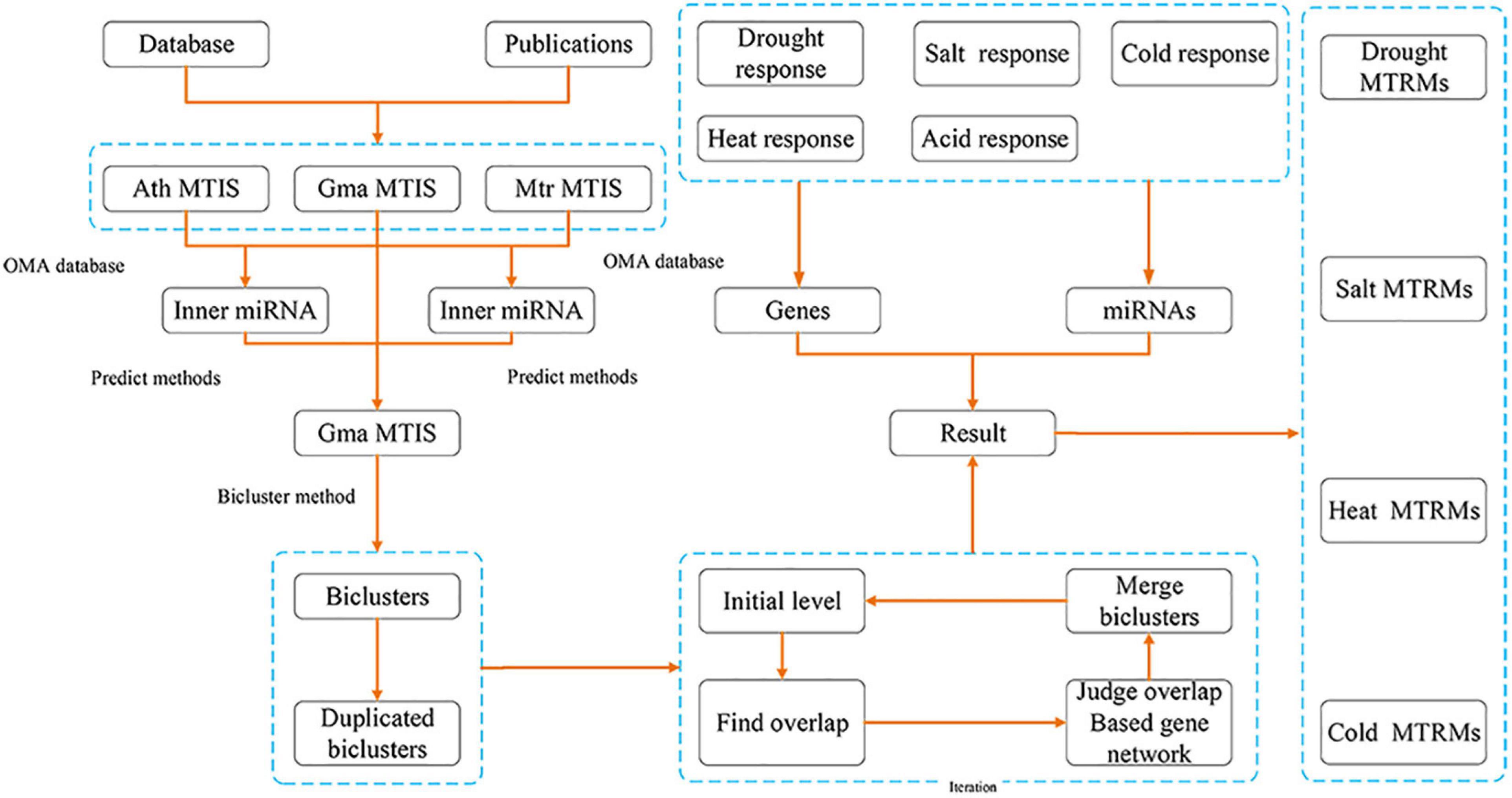
Figure 1. Flowchart of the authors’ research method.
We collected miRNA–target data of A. thaliana, soybean and M. truncatula based on experimentally verified degradome sequencing results from databases [DPMIND, Tarbase, mirTarbase, and Starbase (Sethupathy et al., 2006; Hsu et al., 2011; Yang et al., 2011; Li et al., 2014; Vlachos et al., 2015; Fei et al., 2018)] and publications (Supplementary Table 1). In addition, we collected the miRNA information of the three species from the miRbase (Griffiths-Jones et al., 2008), the gene annotation of the species in the NCBI, EnsemblPlants, and the Phytozome (Goodstein et al., 2012; Howe et al., 2020). We also downloaded the homologous genes of A. thaliana and M. truncatula in Orthologous MAtrix (OMA) (Altenhoff et al., 2011). Besides, we downloaded the soybean cDNA sequence and soybean gene GO annotations from SoyBase (Grant et al., 2010), and obtained soybean gene network data from SoyNet (Kim et al., 2017).
We unified the miRNA and gene formats in the data in various databases and publications, then put the data of the same species together. Next, we annotated the miRNA–target data based on the collected and processed miRNA details and the gene annotations derived from the data of three species, including miRNA target data, related notes, and data sources. Finally, after processing the duplicated data, we obtained the miRNA–target data of the three species.
We chose A. thaliana and M. truncatula to explore the potential targets. A. thaliana as a model plant has rich high-quality data. M. truncatula and soybean are closely related and have many similar biological characteristics. We extracted the miRNA sequence and removed redundant miRNAs with the same sequence in the soybean and A. thaliana. Subsequently, we extracted the target gene corresponding to the miRNA ID. Based on these targeted genes, we obtained soybean genes homologous to these genes from the A. thaliana-soybean homologous genes downloaded by OMA. We assumed that targeting relationships may exist if the sequences coexist and the genes are homologous. Therefore, these homologous genes may be targeted by these miRNAs in soybeans.
Targets obtained only based on homology information may not exist; so, we extracted these miRNA sequences and the cDNA sequence of target genes (SoyBase) and used miRNA-target prediction tools to predict potential relationships. We chose psRNAtarget (Dai and Zhao, 2011), TAPIR (Bonnet et al., 2010), and Targetfinder (Bo and Wang, 2005), whose results were better in non-Arabidopsis plants to predict potential soybean miRNA–target relationships (Srivastava et al., 2014). The three prediction software tools have different scoring methods. We analyzed their respective scores and merged them. The homology extension method for M. truncatula-soybean is the same as above.
The current research on miRNA targeting relationships is mainly based on one-to-one relative targeting. However, the miRNA targeting relationship is a complex interaction. The traditional clustering method is to cluster the same type of data, such as k-means, whose mining results in the miRNA-target regulatory module are poor because the targeting of miRNAs is sparse. The relationship between miRNA and the target gene is a bipartite graph structure; thus, the miRNA–target regulatory group can be found by analyzing the bipartite graph. CUBiBit (Gonzalez-Dominguez and Exposito, 2019) was proposed based on Bimax(Prelic et al., 2006) and BiBit (Rodriguez-Baena et al., 2011), which shortened the computing time and provided an optimized method for finding modules in larger data. We added the miRNA-target data based on the homology expansion predictions from A. thaliana and M. truncatula into the collected soybean miRNA-target data. Then, we extracted the miRNA-Target data with GO annotations and glyma2ID based on the soybean gene annotations of SoyBase. Finally, we used the CUBiBit to perform bi-clustering to obtain the results.
The result obtained by CUBiBit was mostly a fully-connected bipartite graph. However, the relationship between miRNA and target gene is complex and interactive. Therefore, we proposed a method of iterative fusion for MTRM modules based on a gene interaction network (Figure 2).
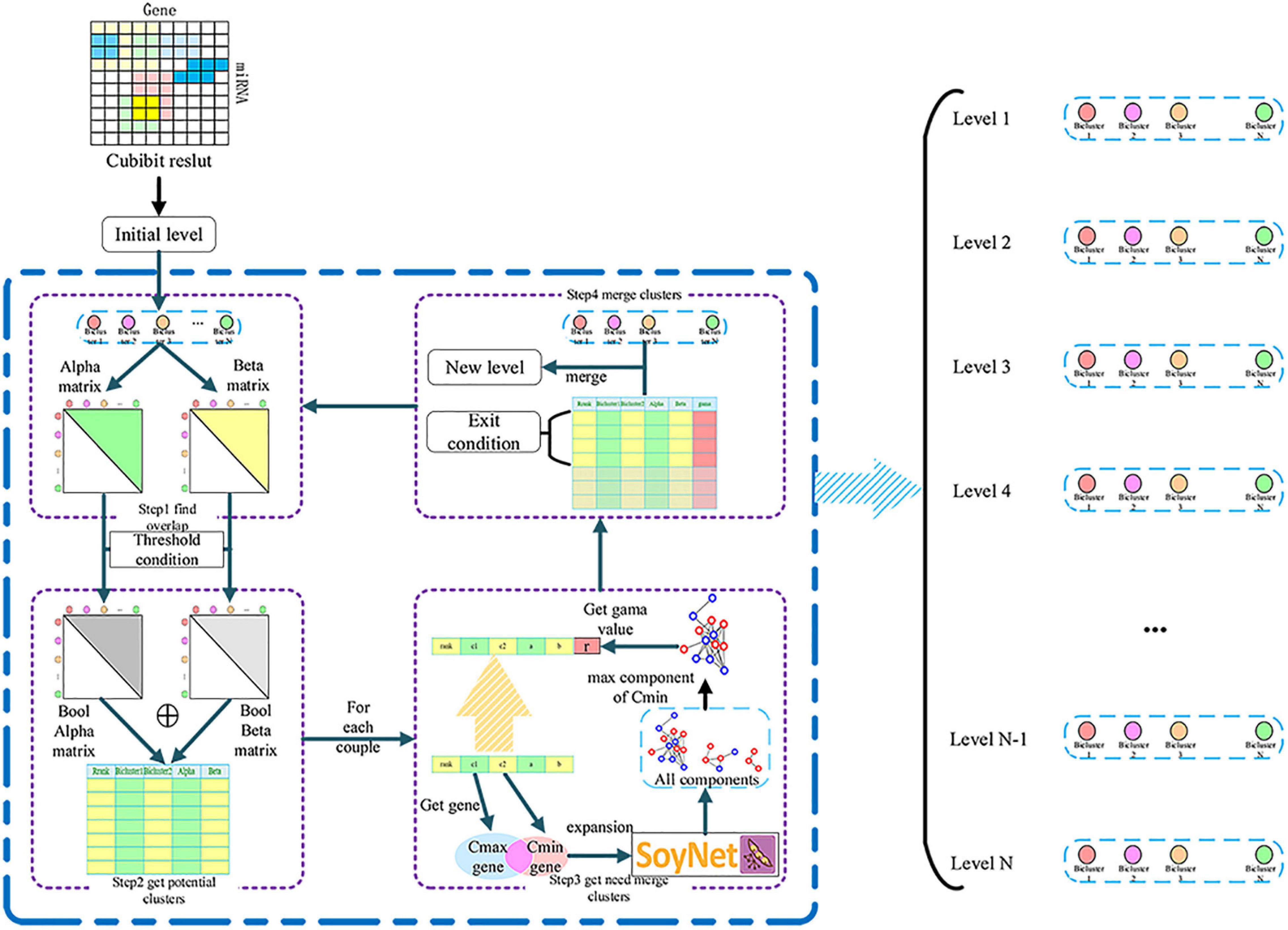
Figure 2. miRNA-target regulatory modules (MTRM) iterative merge algorithm flowchart used to derive a gene interaction network.
We detected the completely included classes in the clustering results and removed the included classes as the initial level result. First, for each class of this level containing miRNAs and genes, we judged the degree of overlap with other classes of miRNA and genes to form alpha and beta matrices, both of which are upper triangular matrices. After that, we set two thresholds of miRNA and genes that can be potentially merged for the two classes. We then recorded the two classes that met the potential fusion class-class table requirements to form a Boolean matrix. The initial alpha threshold was 0.3, and each iteration increased at a pace of 0.05 to conservatively determine the fusionable module and keep this value unchanged after rising to 0.8. It was sufficient if the beta threshold was greater than 0. Next, we extracted the union of two class genes and the network blocks of this pair of genes with a depth of 2 layers based on the SoyNet network for each pair of classes in the potentially merged class-class table. Subsequently, network blocks containing smaller classes were extracted from the obtained block set. We assumed that the network block with the most smaller class genes represented the function of the genes in the smaller class. Therefore, judging the number of genes in the major category of this network block can determine whether the genes of the two categories are similar in function. If the genes of the two classes were concentrated on a network block, which means that their genes interact closely and meet the conditions of potential fusion, the two classes can be merged. We compared the number of genes in the major category with the numbers of genes in all major categories in the sub-category function module to obtain scores and determine the correlation. Finally, we compared the number of genes in the major category with the number in all major categories in the sub-category function module to obtain scores to determine the correlation. The threshold was recorded as gamma. When gamma >0.3 was satisfied, the two classes were merged; otherwise, they would not be merged. For the class pairs that meet the fusion condition, we arranged them in descending order of alpha value and performed top-down non-repetitive fusion. Each class can only merge at most one class in one iteration. A new class set was formed as the new level, and the fusion result was the output. The next iteration would be performed and then another iteration until no fusion class pair could meet the two conditions.
Although this study does not include any experimental validation of our prediction, we assessed the distributions of gene functions indirectly to evaluate whether the results are biologically meaningful. For the results of the above iterative fusion, the enrichment of the classes in each level were separately analyzed. For a bicluster, we extracted its genes, used SoyBase’s GO BP and GO MF for enrichment analysis, and took the corrected GO ID with the smallest p-value as the best enrichment result for this type of cluster. When evaluating each class, the smallest p-value alone was not enough to assess the importance of the class. Instead, we used the cluster score to evaluate the enrichment of all the GO IDs enriched by the class. For all the enriched GO IDs of this class, we screened all the results with a p-value of less than 0.05 and then used Eq. (1) to calculate the cluster score of the class.
Among them, n is the number of gene ontologies enriched in the module, x_i is the number of genes enriched in the i-th GO, and correctPi is the adjusted p-value of the i-th enriched GO.
We collected the miRNAs of soybeans that respond to drought, salt, acid, and low temperature based on our studies of publications (Subramanian et al., 2008; Kulcheski et al., 2011; Li et al., 2011; Sha et al., 2012; Subramanian, 2012; Sunkar et al., 2012; Dong et al., 2013; Zhang et al., 2014, 2018; Balyan et al., 2015; Xu et al., 2016; Zheng et al., 2016; Chen et al., 2018; Gupta et al., 2019; Proust et al., 2019; Yu J.-Y. et al., 2019; Wang et al., 2020). At the same time, we collected the differentially expressed soybean genes under various stresses. We screened these genes with foldchange ≥2 and t-test p-value less than 0.05 as related genes under abiotic stress. Then we marked the genes in the module and calculated the p-value related to abiotic stress based on the hypergeometric distribution. Finally, we screened based on the cluster score calculated by the module, the p-value related to stress, and the proportion of miRNA related to stress. In addition, the screening procedures related to drought and salt stress were consistent with the screening steps of the abiotic stress module.
Based on the results of MTRM mining under stress, we first screened the GO of the enrichment results in the screened module by p-value to remove the GO with a p-value less than 10–5; then, we performed a REVIGO semantic relevance analysis and extraction of concentrated representative GO channels. Based on the MTI data, the miRNA–GO relationship data was constructed through the gene pointed to by the miRNA in the module and the enriched go pathway to which the gene belongs. The relationship between GO is based on the results of REVIGO and the GO similarity calculation. The relationship is presented by setting a threshold to remove some weaker relationships. More detailed parameters are provided here or in the location of the specific figure (Figure 3D).
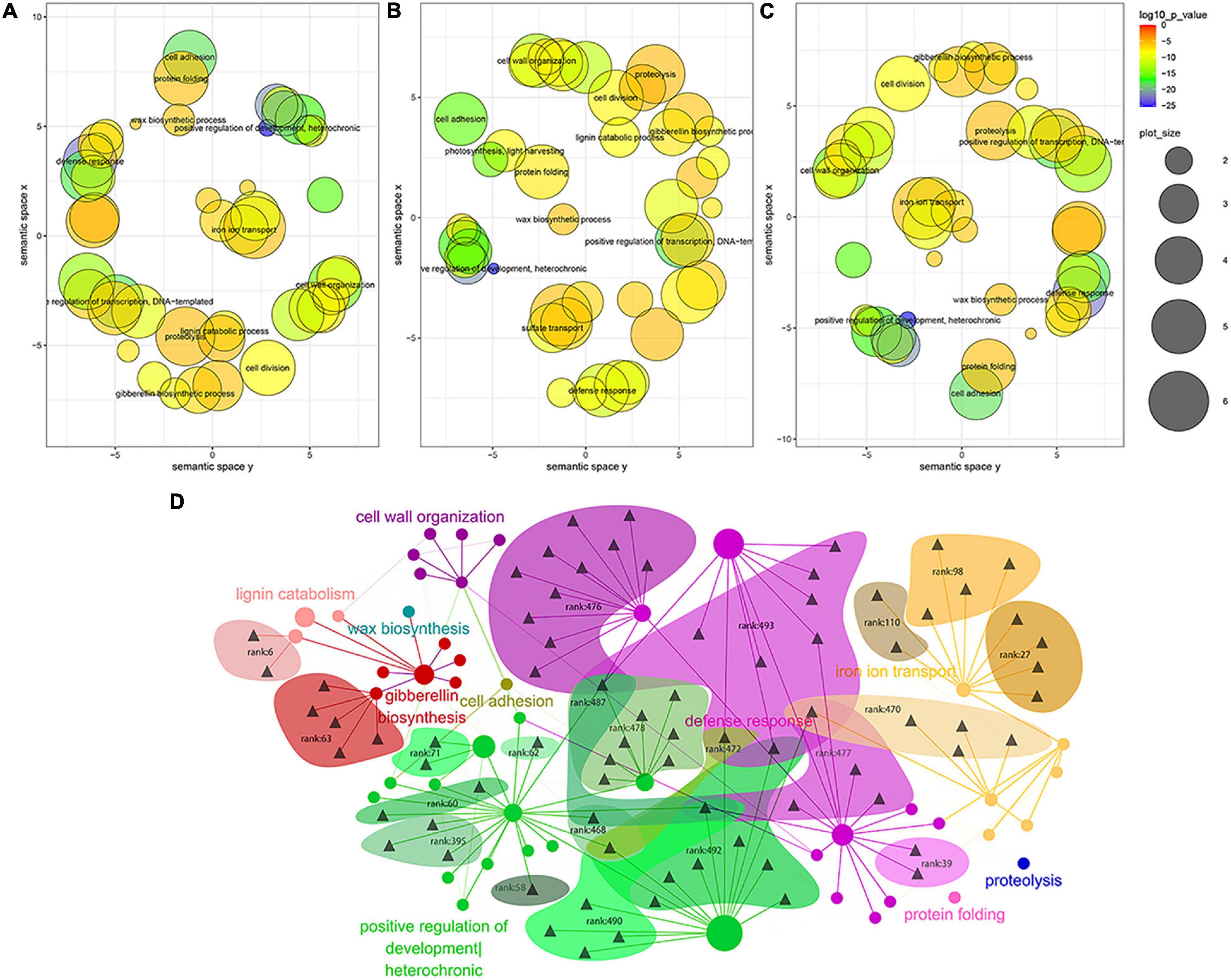
Figure 3. Gene ontology (GO) term analysis of MTRM genes under various abiotic stresses (A–C). (A) GO semantic correlation analysis of abiotic stress, (B) drought stress, and (C) salt stress, and (D) the GO BP regulatory network of cooperative miRNAs under abiotic stresses. Triangles represent different miRNAs and circles represent different GOs. The size of the circle is determined by the number of genes contained in the GO in this article. The color of the circle depends on the representative GO. The areas with different colors show the modules obtained by our method.
We obtained 90,064 confirmed soybean MTIs based on multiple experimental data sources and 1,189 potential soybean MTIs based on homology to experimental data from A. thaliana and M. truncatula. A multi-level iterative bi-clustering analysis resulted in 483 soybean miRNA-target regulatory modules and was evaluated according to GO enrichment function. In addition, we identified 37 abiotic stress-related modules and predicted the underlying miRNA regulatory pathway networks.
We collected soybean miRNA–target data based on databases and related publications. First, we gathered all the soybean MTIs verified by degradome sequencing and biological experiments by mining published data. As a result, we obtained 111,650 pairs of soybean MTIs (Sethupathy et al., 2006; Song et al., 2011; Yang et al., 2011; Shamimuzzaman and Vodkin, 2012; Turner et al., 2012; Fang et al., 2013; Xu et al., 2013, 2014a; Ye et al., 2014; Yan et al., 2015, 2016; Chen et al., 2016, 2017; Ding et al., 2016; Liu et al., 2016; Fei et al., 2018), as shown in Supplementary Table 1. After removing 21,586 redundant pairs of MTIs, 90,064 pairs remained.
To expand MTIs, we predicted the target relationship between potential miRNAs and targeting genes from the MTIs of A. thaliana and M. truncatula based on homology. We obtained 12,094 unique pairs of Arabidopsis MTIs (Addo-Quaye et al., 2008; German et al., 2008; Ding et al., 2012; Xu et al., 2014b; Ma et al., 2018) and 4,394 unique pairs of Medicago MTIs (Devers et al., 2011; Lauressergues et al., 2012; Zhou et al., 2012; Ma et al., 2018) after removing redundant MTIs. Removing any redundant MTIs resulting from identical miRNA sequences, we further validated homology-based MTIs using three miRNA target prediction tools that performed well in general plants, i.e., psRNAtarget, TAPIR, and Targetfinder. In the Arabidopsis MTIs, a total of 961 unique pairs of MTIs were confirmed. In the Medicago MTIs, a total of 986 unique pairs of MTIs were confirmed, as shown in Supplementary Figure 1. There is a high overlap between the two sets of MTIs (Supplementary Table 2). After removing the redundant ones, a total of 1,189 pairs were used to expand soybean MTIs.
We integrated the 90,064 soybean MTIs with the 1,189 MTIs based on homology. We removed MTIs involving genes that do not have the glyma2 ID. A total of 11,018 MTIs were removed, and the remaining 80,235 MTIs were used for analysis in the following tasks.
We applied CUBiBit for bi-clustering analysis, with the smallest scale 2 × 2 or 6 × 2 for miRNA-target modules (i.e., at least two or six target genes and at least two miRNAs in each module), resulting in 15,380 (2 × 2) miRNA-target modules or 2,461 (6 × 2) miRNA-target modules. We contracted the overlapping modules using a multi-level iterative fusion method based on the soybean gene relationship network (see section “Materials and Methods”), yielding 6,577 (2 × 2) and 812 (6 × 2) soybean miRNA-target regulatory modules after removing the modules that were completely included in the preliminary clustering module.
We next merged MTRMs according to the set threshold until the level converged stably (level represents the number of iterations). Each level’s iterative fusion is shown in Figures 4A,B. We compared the iterative results at different scales. Soybean MTRMs at the 2 × 2 scale showed better results at level 10, which contains 2,715 MTRMs. Soybean MTRMs at the 6 × 2 scale showed a better effect at level 7, which contains 483 MTRMs. Comparing the cluster score based on the GO calculation between the two scales of stable convergence (Figure 4C) shows that the cluster score quality at the 6 × 2 scale is higher than that at the 2 × 2 level (Supplementary Table 3). Hence, we used the GO enrichment analysis result on 483 soybean MTRMs obtained at the 6 × 2 level 7.
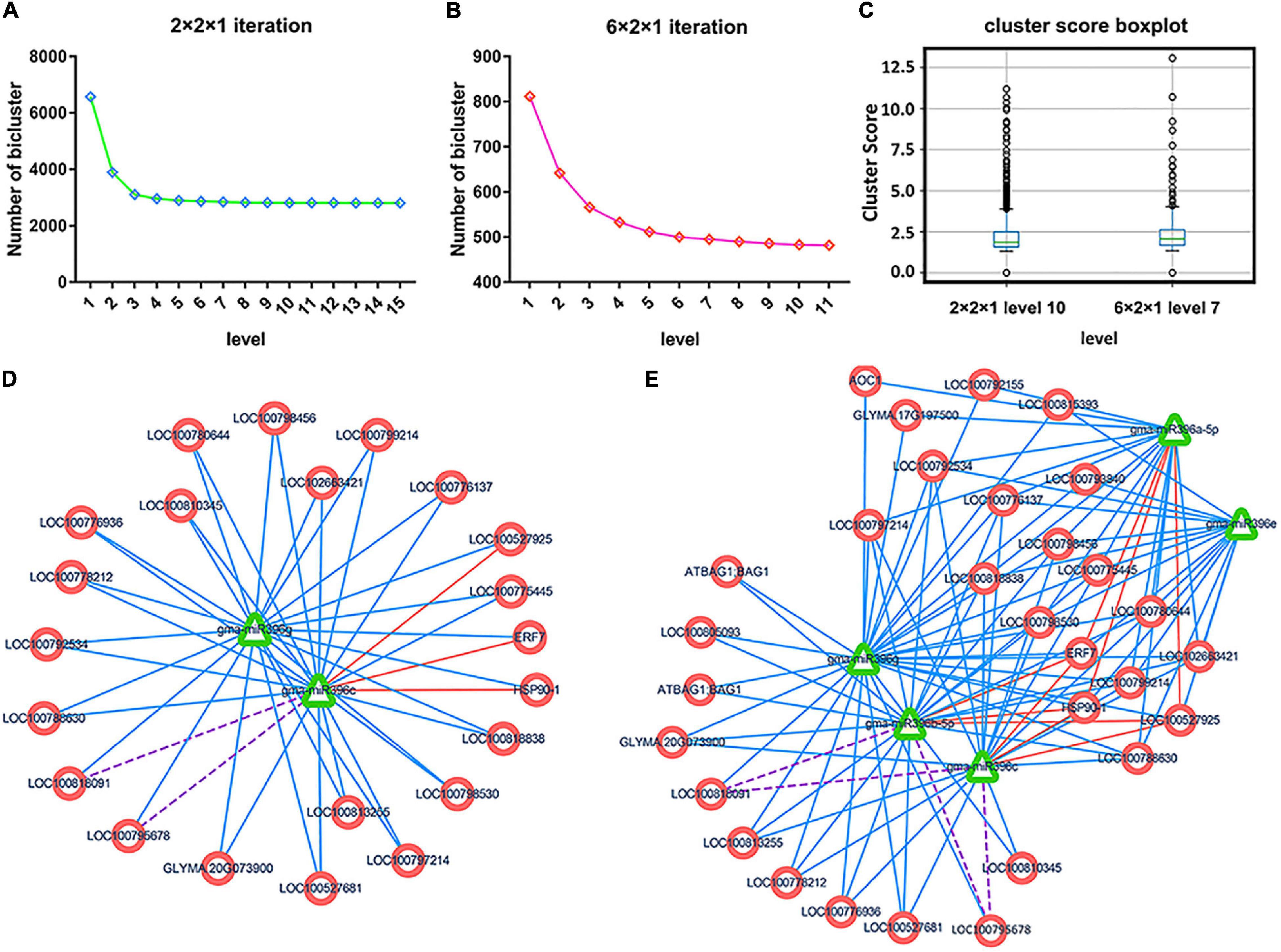
Figure 4. Multi-level iterative biclustering results of soybean MTRMs. (A) Results under different iteration times at the 2 × 2 scale, (B) results under different iteration times at the 6 × 2 scale, and (C) the boxplot of the cluster score is calculated based on the gene ontology (GO) under the two scales when converging to a stable level, where based on the overall distribution, the results at the 6 × 2 scale are better; (D) shows the MTRM bicluster at level 1 before the 6 × 2 scale fusion, and (E) shows the corresponding MTRM bicluster at level 7 after the 6 × 2 scale fusion.
To compare the MTRMs before and after fusion, we extracted an MTRM bicluster, as shown in Figure 4E, from the level 7 clustering results of the 6 × 2 scale and plotted it with the corresponding MTRMs under level 1 before the fusion, as shown in Figure 4E and after Figure 4D, which is a level-7 fusion. The module (1,534) is at level 1 before the fusion has 2 miRNAs and 22 targeted genes. At level 7, the module (1,534) fused an additional three modules, 1,539, 622, and 1,537, and each contains miR396. From the perspective of targeting, the module at level 7 has more miRNA-target interactions than the one at level 1.
We screened 254 GO pathways whose GO biological processes (BP) satisfied the p-value <0.00001 for the GO enrichment from 483 soybean MTRMs obtained at the 6 × 2 scale at level 7. We analyzed the relationship among the enriched GO terms through REVIGO (Supek et al., 2011) with a parameter of 0.5. These GO pathways have a specific aggregation (Figure 5A). MTRMs obtained from a global perspective have several concentrated distributions of GO functions, such as cellular processes, primary metabolism, cell adhesion, hormone response, and negative regulation of biological processes. In addition, there are metachronous positive growth regulations and chalcone biosynthesis. Chalcone plays an important role in soybeans and is involved in the multi-branch pathway of flavonoids and isoflavone biosynthesis (Subramanian et al., 2006). The enrichment results mainly involve positive regulation of development, heterochronic, chalcone biosynthesis, defense response, mitochondrial mRNA modification, sulfate transport, plant-type primary cell wall biogenesis, and cofactor biosynthesis, as shown in Figure 5B.
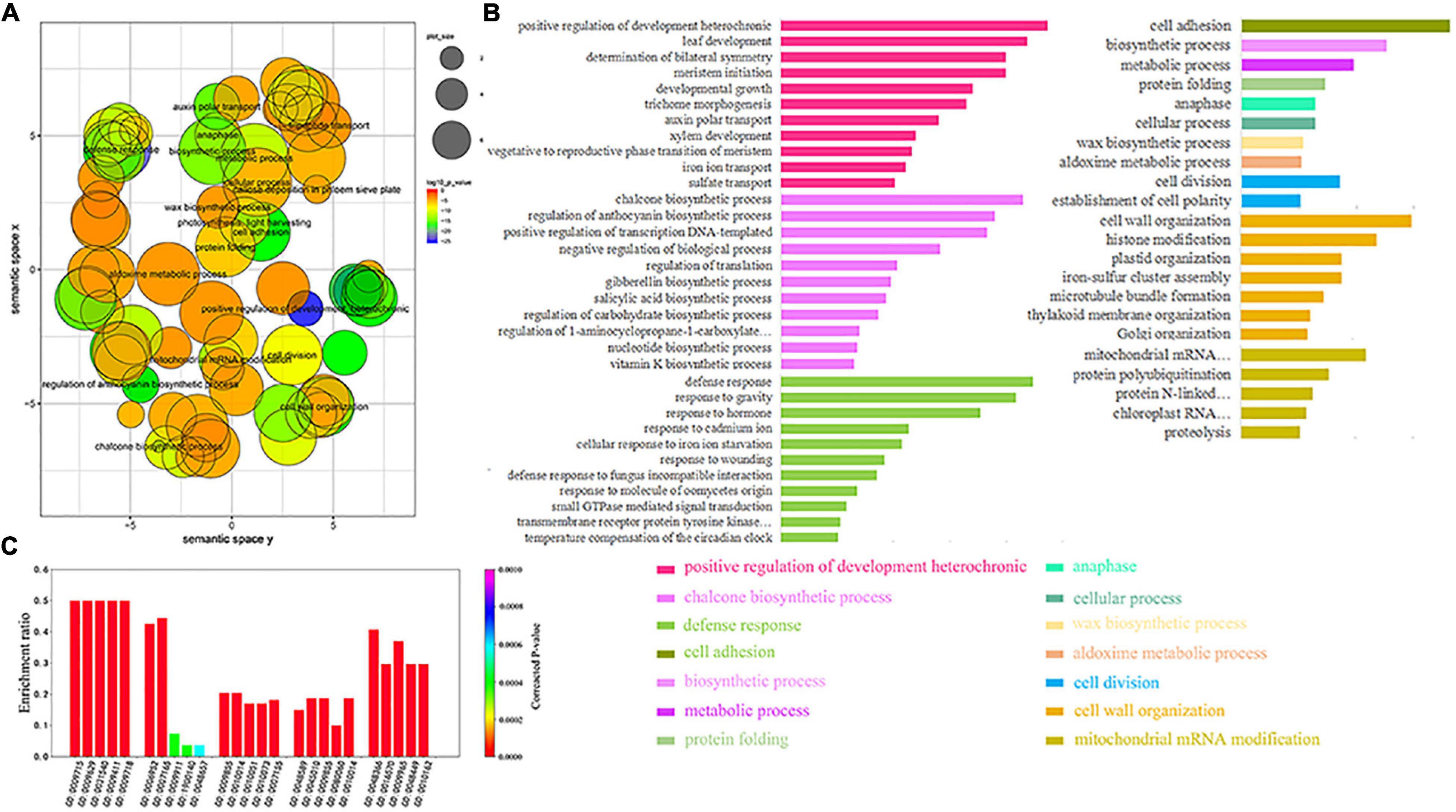
Figure 5. Gene ontology (GO) analysis of soybean MTRMs. (A) Semantic relevance of GO terms wherein the GO pathway has a certain concentration. (B) GO annotation enriched with 483 soybean MTRMs with enrichment results, which mainly involve positive regulation of development, heterochronic, chalcone biosynthesis, defense responses, and mitochondrial mRNA modification. (C) GO enrichment of the top five soybean MTRMs. The listed GO terms were enriched with significant p-values <0.00001.
In addition, we extracted the enrichment results of the top biclusters in terms of cluster score among the 483 MTRMs and selected the top five GO terms of each module, as shown in Figure 5C and Supplementary Table 4.
To explore the biological significance of soybean MTRMs, we collected related soybean miRNAs, which revealed five types of abiotic stresses, involving (1) drought, (2) salt, (3) cold, (4) Pi, (5) phosphorus deficiency based on publications (Subramanian et al., 2008; Kulcheski et al., 2011; Li et al., 2011; Sha et al., 2012; Subramanian, 2012; Sunkar et al., 2012; Dong et al., 2013; Zhang et al., 2014, 2018; Balyan et al., 2015; Xu et al., 2016; Zheng et al., 2016; Chen et al., 2018; Gupta et al., 2019; Proust et al., 2019; Yu J.-Y. et al., 2019; Wang et al., 2020). The function annotations of these miRNAs are shown in Supplementary Table 5. In most processes, soybean miRNA responses are involved in multiple abiotic responses, as shown in Figure 6A. Therefore, mining these miRNAs’ potential cooperative regulatory modules is important to understand their role in modulating soybean stress responses.
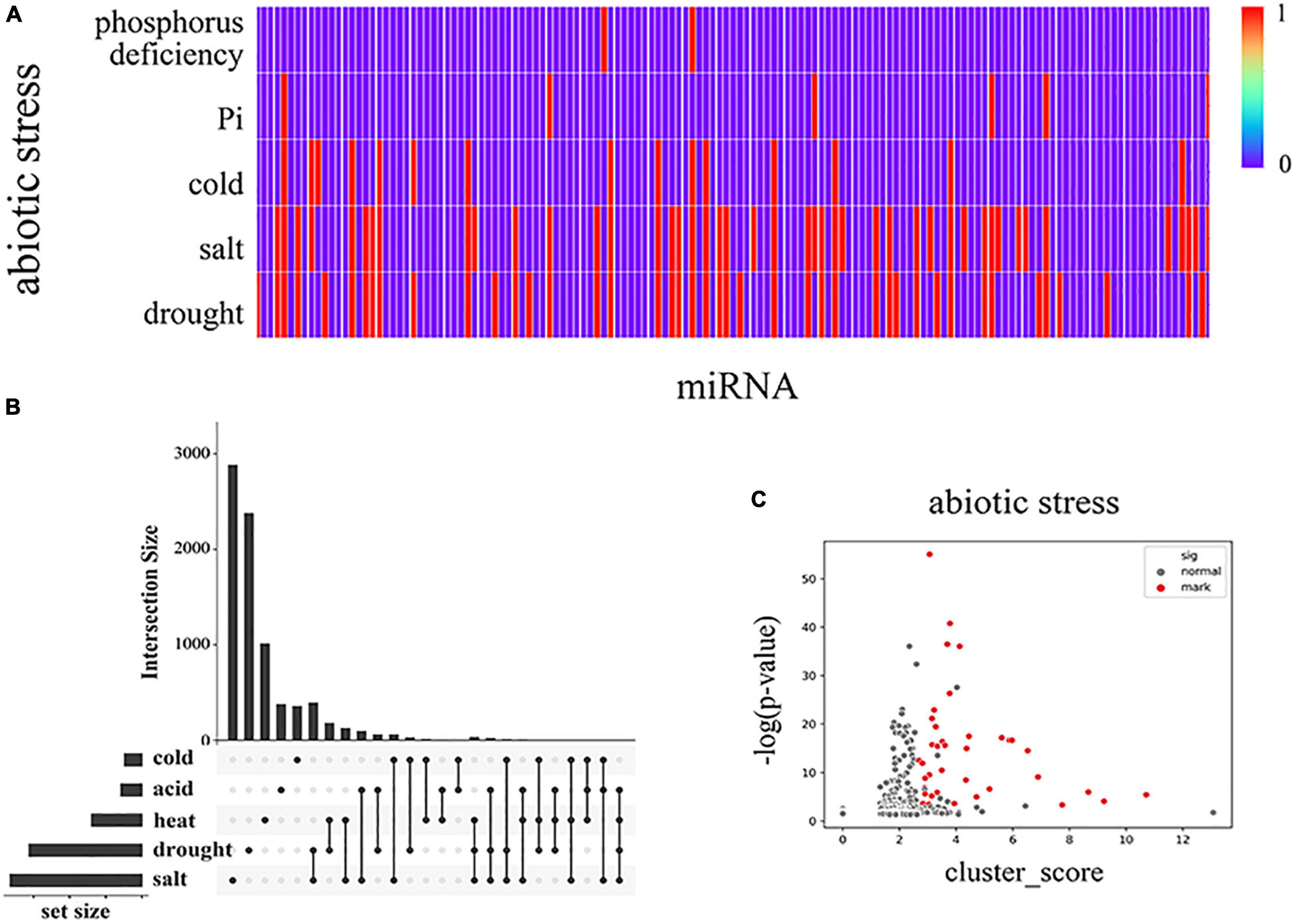
Figure 6. Collected miRNA data on soybeans involved in various abiotic stress responses based on the data statistics from the literature. (A) The distribution of stress types in miRNAs where each vertical line represents one miRNA, and red is marked as relevant, (B) UpSet diagram (Lex et al., 2014) of modular genes under various abiotic stresses within the horizontal correspondence, where dots are used to refer to the corresponding cold stress, acid stress, heat stress, drought stress, and salt stress on the left. The point-to-point connection is realized longitudinally to indicate the intersection between the corresponding data sets, and the upper bar graph shows the number of genes in the intersection. In panel (C), the differentially expressed genes in each MTRM under abiotic stress are shown after screening. We used three indicators to filter the candidate clusters. According to the p-value, the related miRNA purity and the cluster score of each MTRM gene are placed under the corresponding stress. We selected the corresponding threshold, obtained the stress-related MTRMs with higher reliability, and marked them as red dots in Panel (C). Supplementary Figure 2 shows MTRMs under other types of stress.
We correlated the 483 soybean MTRMs obtained by clustering with the functional annotations. We selected miRNAs that responded to drought resistance, salt resistance, heat stress, cold stress, and acid stress. And we performed a statistical analysis on the miRNAs in each of the 483 biclusters. We collected data on the differential expression of soybean genes in the MTRMs under drought, salt, low temperature, cold, and acid stress (Supplementary Table 6). The conditions for screening differentially expressed genes are log2FC > 1, p < 0.05. We obtained 2,145 differentially expressed genes under soybean drought and 1,752 differentially expressed genes under salt treatment. Figure 6B shows the genes in the module together with an abiotic stress diagram. At the same time, we calculated the p-values and FDR. We used the Benjamin Graham formula to correct the p-value of the genes in each MTRM for the differentially expressed genes under abiotic stress scenarios through the hypergeometric distribution, as shown in Figure 6C.
Subsequently, we screened MTRMs related to abiotic stress, drought, and salt stress according to the p-value of differentially expressed genes corresponding to the stress in the MTRMs (p < 0.001, single adversity 0.01), the proportion of the corresponding miRNA family function (miR function ratio), and the cluster score (cluster score > median). The screening results are shown in Supplementary Table 6. We obtained 37 MTRMs related to abiotic stress, including 34 MTRMs related to drought stress, 27 MTRMs related to salt stress, 3 MTRMs related to cold stress, and 21 MTRMs related to heat stress. Figure 7A shows the set relationship of MTRMs involved in a variety of stresses. The data suggest that soybean miRNAs have basic and universal functional modules in their response mechanisms to drought, high salt, high temperature, low temperature, and other abiotic stresses. There are two shared modules (M31 and M493), involving 6 miRNAs and 11 miRNAs (Figure 7B), respectively. The six miRNAs of module M31 belong to the miR156 family. The regulated gene-enriched GO pathway is a transcription regulation, DNA-dependent (p-value 4.24 e-10), and a vegetative phase change regulation with a p-value of 9.24 e-07. The 11 miRNAs of module M493 are mainly in the miR172 family, in addition to miR156, miR1533, miR4374, miR5782, miR3939. The regulated gene-enriched GO pathway involves an oxidation-reduction process (p-value = 4.66 e-12) and a root hair elongation (p-value = 1.63e-08). Among them, miR156 is up-regulated in response to stress under drought conditions, in addition miR156d and miR156c play an important role in the heat tolerance of Arabidopsis (Zheng et al., 2016). miR172b, miR172h, miR172j-5p are down-regulated under drought stress to cope with water stress. miR156 is involved in the regulation of gene expression and signal transduction in response to soybean stimulation in a cold stress environment (Xu et al., 2016). miR156 and miR172 have been confirmed to respond to salt stress in a variety of plants (Sun et al., 2016). Moreover, we also found stress-specific regulatory modules in our results, including 14 drought-specific MTRMs, seven salt-specific MTRMs, and two heat-specific MTRMs (Supplementary Table 7).
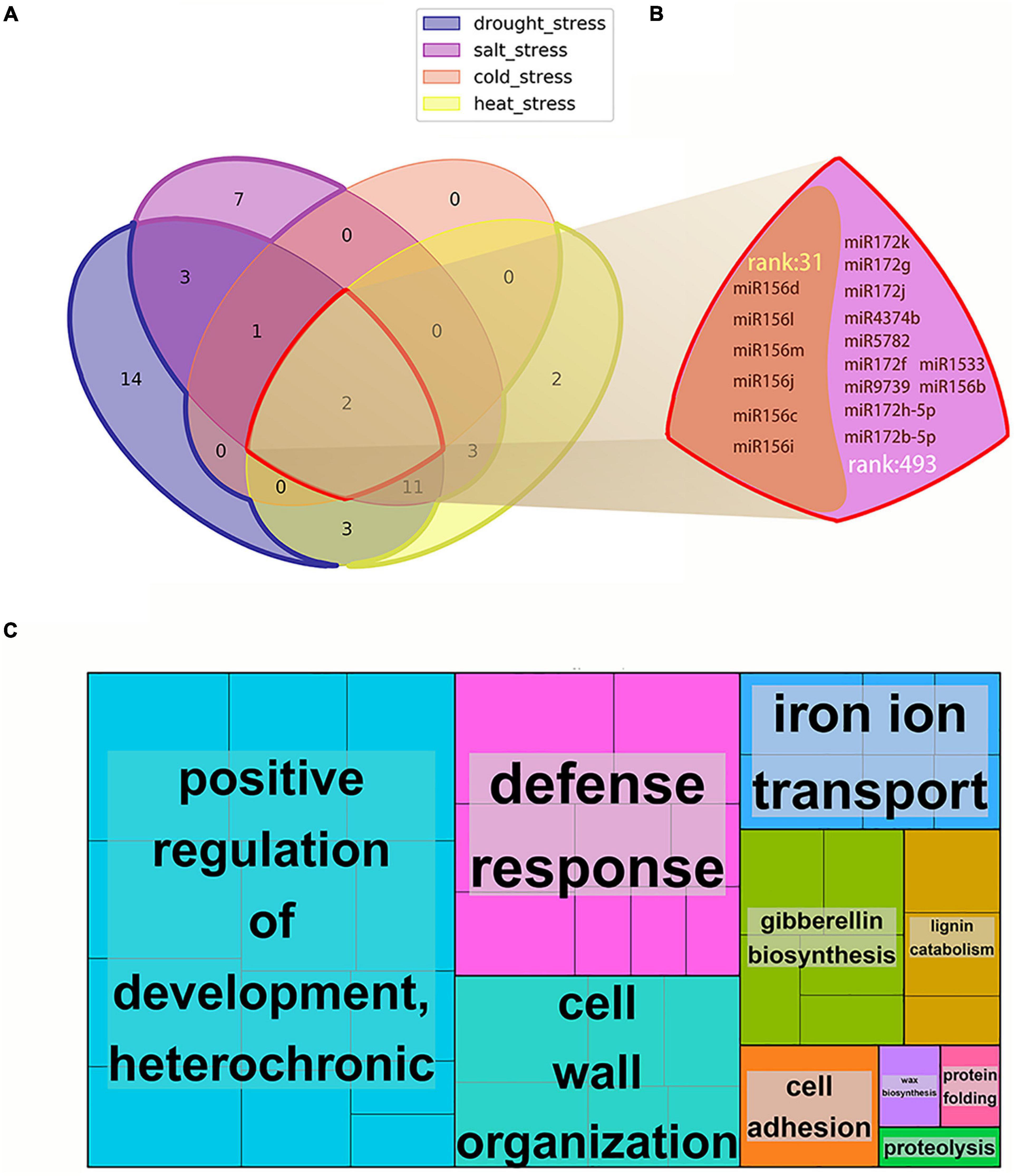
Figure 7. Soybean MTRMs under various abiotic stresses. (A) Venn diagram of 37 kinds of soybean MTRMs under various abiotic stresses, including 14 drought-specific MTRMs, seven salt-specific MTRMs, two heat-specific MTRMs, and two shared MTRMs, (B) the miRNAs in the two shared MTRMs 31 and 493, and (C) a GO Treemap of 37 MTRMs under abiotic stress.
The functions of related miRNA regulatory modules under abiotic stress are mainly concentrated in positive regulation of developmental heterochrony, defense responses, cell wall organization, and other biological processes, as shown in Figure 7C. In addition to plant positive regulation of development and defense response, GO functions such as cell wall organization also produce different response mechanisms under abiotic stresses. For example, salt stress disrupts cell walls integrity (Liu et al., 2021), and cell walls are adaptively regulate under drought stress (Moore et al., 2008). Moreover, plants reduce gibberellin production to reduce growth in order to concentrate energy against stress (Colebrook et al., 2014), sweet briar rose (Rosa rubiginosa L.) adapts to drought conditions by regulating gibberellin (Gadzinowska et al., 2020), and pea seeds adapt to heat stress by reducing gibberellin production (Leitão and Enguita, 2016), Gibberellin in A. thaliana is activated in a low-salt environment (Liu et al., 2013). Thus, GOs enriched in MTRMs play an important role in various stress responses. The data of the top five modules are shown in Supplementary Table 8.
We explored the regulatory pathway network corresponding to the miRNA of the miRNA–target regulatory module in soybean abiotic stress and analyzed the GO terms of MTRM genes under various abiotic stresses. Stringent screening conditions were used, i.e., the p-value of MTRM stress is 0.001, and the GO BP pathway was selected with a p-value of less than 10–5. The REVIGO-based GO language correlation analysis is shown in Figure 3. GO channels with similar functions are closer in the distance in the figure. This is because the small RNAs targeting a certain cluster of GO are functionally close, and the 37 MTRMs of soybeans in abiotic stresses identified in this study mainly focus on resistance response, iron transport, positive growth regulation, and cell wall organization. Under abiotic stress, the cooperating miRNA regulatory modules of the soybean mainly regulate these pathways to respond to the stress environment. Figures 3B,C show the correlation analysis between drought stress and salt stress with specificity.
Subsequently, we constructed the GO BP regulatory network of cooperative miRNAs under soybean abiotic stress for the above main regulatory GO categories and miRNAs, as shown in Figure 3D. Multi-component miRNA families mainly regulate gene expressions related to abiotic stress responses. For example, the miR167 family regulates the resistance response pathway; the miR171 family regulates the gibberellin biosynthesis pathway, while the miR395 family participates in regulating the iron uptake. Moreover, some miRNAs have multiple GO functional partitions, such as miR156b, which regulates developmental growth, the timing of developmental events, the response to hormones, and the response to heavy metal cadmium. The miRNA families and regulatory pathways involved in MTRM are detailed in Supplementary Table 9.
miRNAs are major regulators of plant growth and development. They can also regulate environmental responses (Aukerman and Sakai, 2003; Chen, 2004; Zhu and Helliwell, 2011; Khraiwesh et al., 2012; Mao et al., 2013; Turner et al., 2013; Yan et al., 2013; Wong et al., 2014; Wang et al., 2015; Kulcheski et al., 2016). Hence, the study of the role of miRNAs is crucial—not only to understand the basic events of plant biology but to improve breeding for higher yields and more resilient crop plants. While various papers have noted the role of one or a few miRNAs in regulating plant stress responses, a global analysis of the cooperative interactions is lacking. To study miRNA regulation in response to abiotic response in the soybean, we collected a large number of soybean MTIs. In addition, we proposed a multi-level iterative fusion method of soybean MTRMs based on soybean gene networks.
We mined 483 soybean MTRMs, which provide a data reference for analyzing the cooperative miRNA mechanism of the soybean. Some MTRMs are involved in the biosynthesis of chalcone, which is derived from the general phenylpropanoid pathway that plays a wide variety of roles in soybeans and other plants. In most cases, gene regulation in each MTRMs involved a multi-component miRNA gene family. In some cases, these families were predicted to act cooperatively, which is consistent with the conclusion of Wang et al. (2019). And in the MTRMs we found under abiotic stress in soybean, such as regulatory module M477, which contains miR396, miR172, miR1507 and so on. Among them, soybean miRNA396 and miRNA172 are expressed in soybean drought (Zheng et al., 2016), and miR396s interact with growth-regulating factors (GRFs) to regulate plant growth, development and stress resistance. Liu et al. showed that 7 gma-miR396 (gma-miR396a/b/c/h/e/i/k) and 20 GmGRFs (GmGRF1/2/6-11/13-24) in soybean represent developed a many-to-many network interaction (Liu et al., 2017). Sahito et al. (2017) found that the expression level of soybean NNC1 (Nodule Number Control 1) affects its response to salt stress, while miR172 targets NNC1 and is induced by salt stress. In other plants, the expression of miR396 in rice and Arabidopsis affected the tolerance of plants under the saline-alkali stress (Ning et al., 2019), while another the expression of miR396 in rice was up-regulated under cold conditions (Sun et al., 2019). Sunflower HaWRKY6 (Helianthus annuus) gene expression is related to high-temperature stress, and miR396 has a regulatory effect on this gene (Giacomelli et al., 2012). It can be seen that miR396 has an important regulatory function under abiotic stress such as drought, cold, heat, and salt. Moreover, in the target proteins of the regulatory module M477, in addition to enzymes, transcription factors, etc., we also found some disease resistance-related proteins such as RPM1, RGA2, RGA, and some heat response-related proteins, DnaJ, heat shock 70 kDa protein 14, etc. Jiang et al. (2017) found that PWY-6842 was up-regulated in Arabidopsis under both biotic and abiotic stress. This also indicates that the regulatory mechanism of plants under abiotic stress may have commonalities between the underlying and biotic stress mechanisms. Recent studies have also shown that under biotic and abiotic stress, plants will have a series of signal regulatory networks, such as those mediated by Ca2+, ABA, and G proteins (Ku et al., 2018). The same miRNAs are differentially expressed in adversity (Kar and Raichaudhuri, 2021). It means that the MTRMs of soybean under abiotic stress we excavated have important significance for the regulatory mechanism of soybean under abiotic stress and the coordinated regulation of miRNAs.
Interestingly, we found that miRNAs from different families are also involved in the same regulatory gene clusters, which indicates that different miRNA families may have cross-family cooperative regulatory mechanisms in regulating certain functions. In contrast, miRNAs in the same family can be in different MTRMs; for example, the miR171 family (miR172b-5p, miR172h-5p, miR172f, miR172g, miR172j, and miR172k) are in multiple regulatory modules during drought and salt stress. Such hub miRNAs may be useful research targets for exploring soybean resistance mechanisms and resistance to breeding research under different stresses. After further combining the analysis of differentially expressed genes in soybeans under various stresses, we obtained the miRNA-GO regulatory network under abiotic stress. The GO BP contains a variety of important related pathways for understanding the common mechanisms in stress response. The research covering the plant miRNA regulation module can analyze the coordination mechanism of miRNA from a global perspective and determine the regulation relationship between modules, which may help explore the regulation mechanism of soybean miRNAs.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
HZ and DX conceived and designed the study. LS, Q-MQ, GL, XL, THZ, EZ, HYZ, and LW assembled the data. HC and TYZ performed the analyses and wrote the manuscript. HC wrote the modeling code. YL, HC, and GS assisted with interpreting the results. HZ, DX, and GS reviewed and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (grant no. 62072210) and the US National Science Foundation Plant Genome Program (grant no #IOS-1734145).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank Carla Roberts for thoroughly proofreading this manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.860791/full#supplementary-material
Supplementary Figure 1 | Potential soybean MTIs predicted based on three prediction tools. (A) About 961 pairs of MTIs obtained based on the Arabidopsis data, (B) 986 pairs of MTIs obtained based on the Medicago data, and (C) 1,189 soybean MTIs from the union between panels (A) and (B).
Supplementary Figure 2 | The differentially expressed genes in each MTRM under other stress after the screening.
Supplementary Table 1 | Summary of ATH, GMA, and MTR MTIs sources.
Supplementary Table 2 | Potential targets found by homology expansion.
Supplementary Table 3 | Comparison of the biclustering results of soybean MTRMs.
Supplementary Table 4 | The top 5 GO enrichment of each top 5 soybean MTRMs.
Supplementary Table 5 | miRNA function annotation.
Supplementary Table 6 | Screening results of soybean MTRMs related to abiotic stress, drought and salt stress.
Supplementary Table 7 | Stress-specific MTRMs and shared MTRMs.
Supplementary Table 8 | Top 5 soybean abiotic response MTRMs.
Supplementary Table 9 | GO terms of miRNAs in the soybean abiotic response MTRMs.
Addo-Quaye, C., Eshoo, T. W., Bartel, D. P., and Axtell, M. J. (2008). Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr. Biol. 18, 758–762. doi: 10.1016/j.cub.2008.04.042
Altenhoff, A. M., Schneider, A., Gonnet, G. H., and Dessimoz, C. (2011). OMA 2011: orthology inference among 1000 complete genomes. Nucleic Acids Res. 39, D289–D294. doi: 10.1093/nar/gkq1238
Aukerman, M. J., and Sakai, H. (2003). Regulation of flowering time and floral organ identity by a microRNA and its APETALA2-like target genes. Plant Cell 15, 2730–2741. doi: 10.1105/tpc.016238
Balyan, S. C., Mutum, R. D., Kansal, S., Kumar, S., Mathur, S., and Raghuvanshi, S. (2015). “Insights into the small RNA-mediated networks in response to abiotic stress in plants,” in Elucidation of Abiotic Stress Signaling in Plants: Functional Genomics Gerspectives, Vol. 2, ed. G. K. Pandey (New York, NY: Springer), 45–92. doi: 10.1007/978-1-4939-2540-7_3
Bentham, R. B., Bryson, K., and Szabadkai, G. (2017). MCbiclust: a novel algorithm to discover large-scale functionally related gene sets from massive transcriptomics data collections. Nucleic Acids Res. 45, 8712–8730. doi: 10.1093/nar/gkx590
Bergmann, S., Ihmels, J., and Barkai, N. (2003). Iterative signature algorithm for the analysis of large-scale gene expression data. Phys. Rev. E 67(3 Pt 1):031902. doi: 10.1103/PhysRevE.67.031902
Bo, X. C., and Wang, S. Q. (2005). TargetFinder: a software for antisense oligonucleotide target site selection based on MAST and secondary structures of target mRNA. Bioinformatics 21, 1401–1402. doi: 10.1093/bioinformatics/bti211
Bonnet, E., He, Y., Billiau, K., and Van de Peer, Y. (2010). TAPIR, a web server for the prediction of plant microRNA targets, including target mimics. Bioinformatics 26, 1566–1568. doi: 10.1093/bioinformatics/btq233
Caldas, J., and Kaski, S. (2011). Hierarchical generative biclustering for microRNA expression analysis. J. Comput. Biol. 18, 251–261. doi: 10.1089/cmb.2010.0256
Chen, H., Arsovski, A. A., Yu, K., and Wang, A. (2016). Genome-wide investigation using sRNA-Seq, degradome-seq and transcriptome-seq reveals regulatory networks of microRNAs and their target genes in soybean during soybean mosaic virus infection. PLoS One 11:e0150582. doi: 10.1371/journal.pone.0150582
Chen, H., Arsovski, A. A., Yu, K., and Wang, A. (2017). Deep sequencing leads to the identification of eukaryotic translation initiation factor 5A as a key element in Rsv1-mediated lethal systemic hypersensitive response to soybean mosaic virus infection in soybean. Mol. Plant Pathol. 18, 391–404. doi: 10.1111/mpp.12407
Chen, L., Ding, X., Zhang, H., He, T., Li, Y., Wang, T., et al. (2018). Comparative analysis of circular RNAs between soybean cytoplasmic male-sterile line NJCMS1A and its maintainer NJCMS1B by high-throughput sequencing. BMC Genomics 19:663. doi: 10.1186/s12864-018-5054-6
Chen, X. M. (2004). A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science 303, 2022–2025. doi: 10.1126/science.1088060
Colebrook, E. H., Thomas, S. G., Phillips, A. L., and Hedden, P. (2014). The role of gibberellin signalling in plant responses to abiotic stress (Review). J. Exp. Biol. 217, 67–75. doi: 10.1242/jeb.089938
Dai, X., and Zhao, P. X. (2011). psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 39, W155–W159. doi: 10.1093/nar/gkr319
Devers, E. A., Branscheid, A., May, P., and Krajinski, F. (2011). Stars and symbiosis: microRNA- and microRNA*-mediated transcript cleavage involved in arbuscular mycorrhizal symbiosis. Plant Physiol. 156, 1990–2010. doi: 10.1104/pp.111.172627
Ding, J., Li, D., Ohler, U., Guan, J., and Zhou, S. (2012). Genome-wide search for miRNA-target interactions in Arabidopsis thaliana with an integrated approach. BMC Genomics 13:S3. doi: 10.1186/1471-2164-13-s3-s3
Ding, X., Li, J., Zhang, H., He, T., Han, S., Li, Y., et al. (2016). Identification of miRNAs and their targets by high-throughput sequencing and degradome analysis in cytoplasmic male-sterile line NJCMS1A and its maintainer NJCMS1B of soybean. BMC Genomics 17:24. doi: 10.1186/s12864-015-2352-0
Dong, Z., Shi, L., Wang, Y., Chen, L., Cai, Z., Wang, Y., et al. (2013). Identification and dynamic regulation of microRNAs involved in salt stress responses in functional soybean nodules by high-throughput sequencing. Int. J. Mol. Sci. 14, 2717–2738. doi: 10.3390/ijms14022717
Fang, X., Zhao, Y., Ma, Q., Huang, Y., Wang, P., Zhang, J., et al. (2013). Identification and comparative analysis of cadmium tolerance-associated miRNAs and their targets in two soybean genotypes. PLoS One 8:e81471. doi: 10.1371/journal.pone.0081471
Fei, Y., Wang, R., Li, H., Liu, S., Zhang, H., and Huang, J. (2018). DPMIND: degradome-based plant miRNA-target interaction and network database. Bioinformatics 34, 1618–1620. doi: 10.1093/bioinformatics/btx824
Fu, Y., Mason, A. S., Zhang, Y., Lin, B., Xiao, M., Fu, D., et al. (2019). MicroRNA-mRNA expression profiles and their potential role in cadmium stress response in Brassica napus. BMC Plant Biol. 19:570. doi: 10.1186/s12870-019-2189-9
Gadzinowska, J., Dziurka, M., Ostrowska, A., Hura, K., and Hura, T. (2020). Phytohormone synthesis pathways in sweet briar rose (Rosa rubiginosa L.) seedlings with high adaptation potential to soil drought. Plant Physiol. Biochem. 154, 745–750. doi: 10.1016/j.plaphy.2020.06.018
German, M. A., Pillay, M., Jeong, D.-H., Hetawal, A., Luo, S., Janardhanan, P., et al. (2008). Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat. Biotechnol. 26, 941–946. doi: 10.1038/nbt1417
Giacomelli, J. I., Weigel, D., Chan, R. L., and Manavella, P. A. (2012). Role of recently evolved miRNA regulation of sunflower HaWRKY6 in response to temperature damage. New Phytol. 195, 766–773. doi: 10.1111/j.1469-8137.2012.04259.x
Goncalves, J. P., and Madeira, S. C. (2014). LateBiclustering: efficient heuristic algorithm for time-lagged bicluster identification. IEEE ACM Trans. Comput. Biol. Bioinformatics 11, 801–813. doi: 10.1109/tcbb.2014.2312007
Goncalves, J. P., Madeira, S. C., and Oliveira, A. L. (2009). BiGGEsTS: integrated environment for biclustering analysis of time series gene expression data. BMC Res. Notes 2:124. doi: 10.1186/1756-0500-2-124
Gonzalez-Dominguez, J., and Exposito, R. R. (2019). Accelerating binary biclustering on platforms with CUDA-enabled GPUs. Inf. Sci. 496, 317–325. doi: 10.1016/j.ins.2018.05.025
Goodstein, D. M., Shu, S., Howson, R., Neupane, R., Hayes, R. D., Fazo, J., et al. (2012). Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186. doi: 10.1093/nar/gkr944
Grant, D., Nelson, R. T., Cannon, S. B., and Shoemaker, R. C. (2010). SoyBase, the USDA-ARS soybean genetics and genomics database. Nucleic Acids Res. 38, D843–D846. doi: 10.1093/nar/gkp798
Griffiths-Jones, S., Saini, H. K., van Dongen, S., and Enright, A. J. (2008). miRBase: tools for microRNA genomics. Nucleic Acids Res. 36, D154–D158. doi: 10.1093/nar/gkm952
Gupta, O. P., Dahuja, A., Sachdev, A., Kumari, S., Jain, P. K., Vinutha, T., et al. (2019). Conserved miRNAs modulate the expression of potential transcription factors of isoflavonoid biosynthetic pathway in soybean seeds. Mol. Biol. Rep. 46, 3713–3730. doi: 10.1007/s11033-019-04814-7
Henriques, R., Ferreira, F. L., and Madeira, S. C. (2017). BicPAMS: software for biological data analysis with pattern-based biclustering. BMC Bioinformatics 18:82. doi: 10.1186/s12859-017-1493-3
Henriques, R., and Madeira, S. C. (2014). BicPAM: pattern-based biclustering for biomedical data analysis. Algorithms Mol. Biol. 9:27. doi: 10.1186/s13015-014-0027-z
Henriques, R., and Madeira, S. C. (2016). BicNET: flexible module discovery in large-scale biological networks using biclustering. Algorithms Mol. Biol. 11:14. doi: 10.1186/s13015-016-0074-8
Hivrale, V., Zheng, Y., Puli, C. O. R., Jagadeeswaran, G., Gowdu, K., Kakani, V. G., et al. (2016). Characterization of drought- and heat-responsive microRNAs in switchgrass. Plant Sci. 242, 214–223. doi: 10.1016/j.plantsci.2015.07.018
Hochreiter, S., Bodenhofer, U., Heusel, M., Mayr, A., Mitterecker, A., Kasim, A., et al. (2010). FABIA: factor analysis for bicluster acquisition. Bioinformatics 26, 1520–1527. doi: 10.1093/bioinformatics/btq227
Howe, K. L., Contreras-Moreira, B., De Silva, N., Maslen, G., Akanni, W., Allen, J., et al. (2020). Ensembl genomes 2020-enabling non-vertebrate genomic research. Nucleic Acids Res. 48, D689–D695. doi: 10.1093/nar/gkz890
Hsu, S.-D., Lin, F.-M., Wu, W.-Y., Liang, C., Huang, W.-C., Chan, W.-L., et al. (2011). miRTarBase: a database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. 39, D163–D169. doi: 10.1093/nar/gkq1107
Huang, J. Y., and Brutlag, D. L. (2001). The EMOTIF database. Nucleic Acids Res. 29, 202–204. doi: 10.1093/nar/29.1.202
Huang, S. C., Lu, G. H., Tang, C. Y., Ji, Y. J., Tan, G. S., Hu, D. Q., et al. (2018). Identification and comparative analysis of aluminum-induced microRNAs conferring plant tolerance to aluminum stress in soybean. Biol. Plant. 62, 97–108. doi: 10.1007/s10535-017-0752-5
Ismalia, B., Qiang, K., Luan, Y.-S., and Jun, M. (2019). Predicting miRNA-lncRNA interactions and recognizing their regulatory roles in stress response of plants. Math. Biosci. 312, 67–76. doi: 10.1016/j.mbs.2019.04.006
Jiang, Z., He, F., and Zhang, Z. (2017). Large-scale transcriptome analysis reveals Arabidopsis metabolic pathways are frequently influenced by different pathogens. Plant Mol. Biol. 94, 453–467. doi: 10.1007/s11103-017-0617-5
Kar, M. M., and Raichaudhuri, A. (2021). Role of microRNAs in mediating biotic and abiotic stress in plants. Plant Gene 26:100277. doi: 10.1016/j.plgene.2021.100277
Khraiwesh, B., Zhu, J.-K., and Zhu, J. (2012). Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochim. Biophys. Acta Gene Regul. Mech. 1819, 137–148. doi: 10.1016/j.bbagrm.2011.05.001
Kim, E., Hwang, S., and Lee, I. (2017). SoyNet: a database of co-functional networks for soybean Glycine max. Nucleic Acids Res. 45, D1082–D1089. doi: 10.1093/nar/gkw704
Ku, Y. S., Sintaha, M., Cheung, M. Y., and Lam, H. M. (2018). Plant hormone signaling crosstalks between biotic and abiotic stress responses. Int. J. Mol. Sci. 19:3206. doi: 10.3390/ijms19103206
Ku, Y.-S., Wong, J. W.-H., Mui, Z., Liu, X., Hui, J. H.-L., Chan, T.-F., et al. (2015). Small RNAs in plant responses to abiotic stresses: regulatory roles and study methods. Int. J. Mol. Sci. 16, 24532–24554. doi: 10.3390/ijms161024532
Kulcheski, F. R., de Oliveira, L. F. V., Molina, L. G., Almerao, M. P., Rodrigues, F. A., Marcolino, J., et al. (2011). Identification of novel soybean microRNAs involved in abiotic and biotic stresses. BMC Genomics 12:307. doi: 10.1186/1471-2164-12-307
Kulcheski, F. R., Molina, L. G., da Fonseca, G. C., de Morais, G. L., de Oliveira, L. F. V., and Margis, R. (2016). Novel and conserved microRNAs in soybean floral whorls. Gene 575, 213–223. doi: 10.1016/j.gene.2015.08.061
Lauressergues, D., Delaux, P.-M., Formey, D., Lelandais-Briere, C., Fort, S., Cottaz, S., et al. (2012). The microRNA miR171h modulates arbuscular mycorrhizal colonization of Medicago truncatula by targeting NSP2. Plant J. 72, 512–522. doi: 10.1111/j.1365-313X.2012.05099.x
Leitão, A. L., and Enguita, F. J. (2016). Gibberellins in Penicillium strains: challenges for endophyte-plant host interactions under salinity stress. Microbiol. Res. 183, 8–18. doi: 10.1016/j.micres.2015.11.004
Lex, A., Gehlenborg, N., Strobelt, H., Vuillemot, R., and Pfister, H. (2014). UpSet: visualization of Intersecting Sets. Ieee Trans. Vis. Comput. Graph. 20, 1983–1992. doi: 10.1109/tvcg.2014.2346248
Li, G., Ma, Q., Tang, H., Paterson, A. H., and Xu, Y. (2009). QUBIC: a qualitative biclustering algorithm for analyses of gene expression data. Nucleic Acids Res. 37:e101. doi: 10.1093/nar/gkp491
Li, H., Deng, Y., Wu, T., Subramanian, S., and Yu, O. (2010). Misexpression of miR482, miR1512, and miR1515 increases soybean nodulation. Plant Physiol. 153, 1759–1770. doi: 10.1104/pp.110.156950
Li, H., Dong, Y., Yin, H., Wang, N., Yang, J., Liu, X., et al. (2011). Characterization of the stress associated microRNAs in Glycine max by deep sequencing. BMC Plant Biol. 11:170. doi: 10.1186/1471-2229-11-170
Li, J.-H., Liu, S., Zhou, H., Qu, L.-H., and Yang, J.-H. (2014). starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 42, D92–D97. doi: 10.1093/nar/gkt1248
Li, S., Castillo-Gonzalez, C., Yu, B., and Zhang, X. (2017). The functions of plant small RNAs in development and in stress responses. Plant J. 90, 654–670. doi: 10.1111/tpj.13444
Li, W., Wang, T., Zhang, Y., and Li, Y. (2016). Overexpression of soybean miR172c confers tolerance to water deficit and salt stress, but increases ABA sensitivity in transgenic Arabidopsis thaliana. J. Exp. Bot. 67, 175–194. doi: 10.1093/jxb/erv450
Liu, J., Zhang, W., Long, S., and Zhao, C. (2021). Maintenance of cell wall integrity under high salinity. Int. J. Mol. Sci. 22:3260. doi: 10.3390/ijms22063260
Liu, T., Fang, C., Ma, Y., Shen, Y., Li, C., Li, Q., et al. (2016). Global investigation of the co-evolution of miRNA genes and microRNA targets during soybean domestication. Plant J. 85, 396–409. doi: 10.1111/tpj.13113
Liu, T., Li, Y., Ren, J., Qian, Y., Yang, X., Duan, W., et al. (2013). Nitrate or NaCl regulates floral induction in Arabidopsis thaliana. Biologia 68, 215–222. doi: 10.2478/s11756-013-0004-x
Liu, W. C., Zhou, Y. G., Li, X. W., Wang, X. C., Dong, Y. Y., Wang, N., et al. (2017). Tissue-specific regulation of Gma-miR396 family on coordinating development and low water availability responses. Front. Plant Sci. 8:1112. doi: 10.3389/fpls.2017.01112
Ma, X., Yu, D., Shao, W., Xu, M., Zuo, Z., Wang, H., et al. (2018). Transcriptome-wide identification and characterization of the copper and cadmium stress-responsive small RNAs and their targets in Arabidopsis thaliana. Plant Soil 429, 391–405. doi: 10.1007/s11104-018-3697-3
Madeira, S. C., Teixeira, M. C., Sa-Correia, I., and Oliveira, A. L. (2010). Identification of regulatory modules in time series gene expression data using a linear time biclustering algorithm. IEEE ACM Trans. Comput. Biol. Bioinformatics 7, 153–165. doi: 10.1109/tcbb.2008.34
Mao, G., Turner, M., Yu, O., and Subramanian, S. (2013). miR393 and miR164 influence indeterminate but not determinate nodule development. Plant Signal. Behav. 8, 1559–2316. doi: 10.4161/psb.26753
Medina, I., Carbonell, J., Pulido, L., Madeira, S. C., Goetz, S., Conesa, A., et al. (2010). Babelomics: an integrative platform for the analysis of transcriptomics, proteomics and genomic data with advanced functional profiling. Nucleic Acids Res. 38, W210–W213. doi: 10.1093/nar/gkq388
Moore, J. P., Vicré-Gibouin, M., Farrant, J. M., and Driouich, A. (2008). Adaptations of higher plant cell walls to water loss: drought vs desiccation. Physiol. Plant. 134, 237–245. doi: 10.1111/j.1399-3054.2008.01134.x
Ning, L. H., Du, W. K., Song, H. N., Shao, H. B., Qia, W. C., Amr Sheteiwy, M. S., et al. (2019). Identification of responsive miRNAs involved in combination stresses of phosphate starvation and salt stress in soybean root. Environ. Exp. Bot. 167:103823. doi: 10.1016/j.envexpbot.2019.103823
Pan, W.-J., Tao, J.-J., Cheng, T., Bian, X.-H., Wei, W., Zhang, W.-K., et al. (2016). Soybean miR172a improves salt tolerance and can function as a long-distance signal. Mol. Plant 9, 1337–1340. doi: 10.1016/j.molp.2016.05.010
Pio, G., Ceci, M., D’Elia, D., Loglisci, C., and Malerba, D. (2013). A novel biclustering algorithm for the discovery of meaningful biological correlations between microRNAs and their target genes. BMC Bioinformatics 14:S8. doi: 10.1186/1471-2105-14-s7-s8
Prelic, A., Bleuler, S., Zimmermann, P., Wille, A., Buhlmann, P., Gruissem, W., et al. (2006). A systematic comparison and evaluation of biclustering mexthods for gene expression data. Bioinformatics 22, 1122–1129. doi: 10.1093/bioinformatics/btl060
Proust, H., Moreau, J., Crespi, M., Hartmann, C., and Lelandais-Brière, C. (2019). “Small RNA diversity and roles in model legumes,” in The Model Legume Medicago truncatula, ed. F. de Bruijn (Hoboken, NJ: John Wiley & Sons, Inc.), 948–962. doi: 10.1002/9781119409144.ch123
Rodriguez-Baena, D. S., Perez-Pulido, A. J., and Aguilar-Ruiz, J. S. (2011). A biclustering algorithm for extracting bit-patterns from binary datasets. Bioinformatics 27, 2738–2745. doi: 10.1093/bioinformatics/btr464
Ruiz-Ferrer, V., and Voinnet, O. (2009). Roles of plant small RNAs in biotic stress responses. Annu. Rev. Plant Biol. 60, 485–510. doi: 10.1146/annurev.arplant.043008.092111
Sahito, Z. A., Wang, L. X., Sun, Z. X., Yan, Q. Q., Zhang, X. K., Jiang, Q., et al. (2017). The miR172c-NNC1 module modulates root plastic development in response to salt in soybean. BMC Plant Biol. 17:229. doi: 10.1186/s12870-017-1161-9
Sethupathy, P., Corda, B., and Hatzigeorgiou, A. G. (2006). TarBase: a comprehensive database of experimentally supported animal microRNA targets. RNA 12, 192–197. doi: 10.1261/rna.2239606
Sha, A., Chen, Y., Ba, H., Shan, Z., Zhang, X., Wu, X., et al. (2012). Identification of Glycine max microRNAs in response to phosphorus deficiency. J. Plant Biol. 55, 268–280. doi: 10.1007/s12374-011-0255-4
Shalgi, R., Lieber, D., Oren, M., and Pilpel, Y. (2007). Global and local architecture of the mammalian microRNA-transcription factor regulatory network. PLoS Comput. Biol. 3:e131. doi: 10.1371/journal.pcbi.0030131
Shamimuzzaman, M., and Vodkin, L. (2012). Identification of soybean seed developmental stage-specific and tissue-specific miRNA targets by degradome sequencing. BMC Genomics 13:310. doi: 10.1186/1471-2164-13-310
Shukla, L. I., Chinnusamy, V., and Sunkar, R. (2008). The role of microRNAs and other endogenous small RNAs in plant stress responses. Biochim. Biophys. Acta Gene Regul. Mech. 1779, 743–748. doi: 10.1016/j.bbagrm.2008.04.004
Song, Q.-X., Liu, Y.-F., Hu, X.-Y., Zhang, W.-K., Ma, B., Chen, S.-Y., et al. (2011). Identification of miRNAs and their target genes in developing soybean seeds by deep sequencing. BMC Plant Biol. 11:5. doi: 10.1186/1471-2229-11-5
Song, X., Li, Y., Cao, X., and Qi, Y. (2019). MicroRNAs and their regulatory roles in plant-environment interactions. Annu. Rev. Plant Biol. 70, 489–525. doi: 10.1146/annurev-arplant-050718-100334
Srivastava, P. K., Moturu, T. R., Pandey, P., Baldwin, I. T., and Pandey, S. P. (2014). A comparison of performance of plant miRNA target prediction tools and the characterization of features for genome-wide target prediction. BMC Genomics 15:348. doi: 10.1186/1471-2164-15-348
Subramanian, S. (2012). “microRNA regulation of symbiotic nodule development in legumes,” in MicroRNAs in Plant Development and Stress Responses, Vol. 15, ed. R. Sunkar (Berlin: Springer), 177–195. doi: 10.1007/978-3-642-27384-1_9
Subramanian, S., Fu, Y., Sunkar, R., Barbazuk, W. B., Zhu, J.-K., and Yu, O. (2008). Novel and nodulation-regulated microRNAs in soybean roots. BMC Genomics 9:160. doi: 10.1186/1471-2164-9-160
Subramanian, S., Stacey, G., and Yu, O. (2006). Endogenous isoflavones are essential for the establishment of symbiosis between soybean and Bradyrhizobium japonicum. Plant J. 48, 261–273. doi: 10.1111/j.1365-313X.2006.02874.x
Sun, X., Lin, L., and Sui, N. (2019). Regulation mechanism of microRNA in plant response to abiotic stress and breeding. Mol. Biol. Rep. 46, 1447–1457. doi: 10.1007/s11033-018-4511-2
Sun, Z. X., Wang, Y. N., Mou, F. P., Tian, Y. P., Chen, L., Zhang, S. L., et al. (2016). Genome-wide small RNA analysis of soybean reveals auxin-responsive microRNAs that are differentially expressed in response to salt stress in root apex. Front. Plant Sci. 6:1273. doi: 10.3389/fpls.2015.01273
Sunkar, R., Chinnusamy, V., Zhu, J., and Zhu, J.-K. (2007). Small RNAs as big players in plant abiotic stress responses and nutrient deprivation. Trends Plant Sci. 12, 301–309. doi: 10.1016/j.tplants.2007.05.001
Sunkar, R., Li, Y.-F., and Jagadeeswaran, G. (2012). Functions of microRNAs in plant stress responses. Trends Plant Sci. 17, 196–203. doi: 10.1016/j.tplants.2012.01.010
Supek, F., Bosnjak, M., Skunca, N., and Smuc, T. (2011). REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6:e21800. doi: 10.1371/journal.pone.0021800
Tanay, A., Sharan, R., and Shamir, R. (2002). Discovering statistically significant biclusters in gene expression data. Bioinformatics 18(Suppl. 1) S136–S144. doi: 10.1093/bioinformatics/18.suppl_1.s136
Tu, Z., Xia, H., Yang, L., Zhai, X., Shen, Y., and Li, H. (2022). The roles of microRNA-long non-coding RNA-mRNA networks in the regulation of leaf and flower development in Liriodendron chinense. Front. Plant Sci. 13:816875. doi: 10.3389/fpls.2022.816875
Turner, M., Nizampatnam, N. R., Baron, M., Coppin, S., Damodaran, S., Adhikari, S., et al. (2013). Ectopic expression of miR160 results in auxin hypersensitivity, cytokinin hyposensitivity, and inhibition of symbiotic nodule development in soybean. Plant Physiol. 162, 2042–2055. doi: 10.1104/pp.113.220699
Turner, M., Yu, O., and Subramanian, S. (2012). Genome organization and characteristics of soybean microRNAs. BMC Genomics 13:169. doi: 10.1186/1471-2164-13-169
Vlachos, I. S., Paraskevopoulou, M. D., Karagkouni, D., Georgakilas, G., Vergoulis, T., Kanellos, I., et al. (2015). DIANA-TarBase v7.0: indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res. 43, D153–D159. doi: 10.1093/nar/gku1215
Wang, D., Hao, C., Zhang, L., Zhang, J., Liu, S., Li, Y., et al. (2020). Exosomal miR-125a-5p derived from silica-exposed macrophages induces fibroblast transdifferentiation. Ecotoxicol. Environ. Saf. 192:110253. doi: 10.1016/j.ecoenv.2020.110253
Wang, R., Yang, Z., Fei, Y., Feng, J., Zhu, H., Huang, F., et al. (2019). Construction and analysis of degradome-dependent microRNA regulatory networks in soybean. BMC Genomics 20:534. doi: 10.1186/s12864-019-5879-7
Wang, Y., Li, K., Chen, L., Zou, Y., Liu, H., Tian, Y., et al. (2015). MicroRNA167-directed regulation of the auxin response factors GmARF8a and GmARF8b is required for soybean nodulation and lateral root development. Plant Physiol. 168, 984–999. doi: 10.1104/pp.15.00265
Wong, J., Gao, L., Yang, Y., Zhai, J., Arikit, S., Yu, Y., et al. (2014). Roles of small RNAs in soybean defense against Phytophthora sojae infection. Plant J. 79, 928–940. doi: 10.1111/tpj.12590
Xie, J., Ma, A., Fennell, A., Ma, Q., and Zhao, J. (2019). It is time to apply biclustering: a comprehensive review of biclustering applications in biological and biomedical data. Brief. Bioinformatics 20, 1449–1464. doi: 10.1093/bib/bby014
Xu, F., Liu, Q., Chen, L., Kuang, J., Walk, T., Wang, J., et al. (2013). Genome-wide identification of soybean microRNAs and their targets reveals their organ-specificity and responses to phosphate starvation. BMC Genomics 14:66. doi: 10.1186/1471-2164-14-66
Xu, J., Li, C.-X., Li, Y.-S., Lv, J.-Y., Ma, Y., Shao, T.-T., et al. (2011). MiRNA-miRNA synergistic network: construction via co-regulating functional modules and disease miRNA topological features. Nucleic Acids Res. 39, 825–836. doi: 10.1093/nar/gkq832
Xu, S., Liu, N., Mao, W., Hu, Q., Wang, G., and Gong, Y. (2016). Identification of chilling-responsive microRNAs and their targets in vegetable soybean (Glycine max L.). Sci. Rep. 6:26619. doi: 10.1038/srep26619
Xu, Y., Guo, M., Liu, X., Wang, C., and Liu, Y. (2014c). Inferring the soybean (Glycine max) microRNA functional network based on target gene network. Bioinformatics 30, 94–103. doi: 10.1093/bioinformatics/btt605
Xu, Y., Guo, M., Liu, X., Wang, C., and Liu, Y. (2014d). SoyFN: a knowledge database of soybean functional networks. Database 2014:bau019. doi: 10.1093/database/bau019
Xu, M., Li, Y., Zhang, Q., Xu, T., Qiu, L., Fan, Y., et al. (2014a). Novel miRNA and PhasiRNA biogenesis networks in soybean roots from two sister lines that are resistant and susceptible to SCN race 4. PLoS One 9:e110051. doi: 10.1371/journal.pone.0110051
Xu, M. Y., Zhang, L., Li, W. W., Hu, X. L., Wang, M.-B., Fan, Y. L., et al. (2014b). Stress-induced early flowering is mediated by miR169 in Arabidopsis thaliana. J. Exp. Bot. 65, 89–101. doi: 10.1093/jxb/ert353
Yan, Z., Hossain, M. S., Arikit, S., Valdes-Lopez, O., Zhai, J., Wang, J., et al. (2015). Identification of microRNAs and their mRNA targets during soybean nodule development: functional analysis of the role of miR393j-3p in soybean nodulation. New Phytol. 207, 748–759. doi: 10.1111/nph.13365
Yan, Z., Hossain, M. S., Valdes Lopez, O., Hoang, N. T., Zhai, J., Wang, J., et al. (2016). Identification and functional characterization of soybean root hair microRNAs expressed in response to Bradyrhizobium japonicum infection. Plant Biotechnol. J. 14, 332–341. doi: 10.1111/pbi.12387
Yan, Z., Hossein, M. S., Wang, J., Valdes-Lopez, O., Liang, Y., Libault, M., et al. (2013). miR172 regulates soybean nodulation. Mol. Plant Microbe Interact. 26, 1371–1377. doi: 10.1094/mpmi-04-13-0111-r
Yang, J.-H., Li, J.-H., Shao, P., Zhou, H., Chen, Y.-Q., and Qu, L.-H. (2011). starBase: a database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and degradome-Seq data. Nucleic Acids Res. 39, D202–D209. doi: 10.1093/nar/gkq1056
Yang, Y. H., Li, M. J., Yi, Y. J., Li, R. F., Li, C. X., Yang, H., et al. (2021). Integrated miRNA-mRNA analysis reveals the roles of miRNAs in the replanting benefit of Achyranthes bidentata roots. Sci. Rep. 11:1628. doi: 10.1038/s41598-021-81277-6
Ye, C.-Y., Xu, H., Shen, E., Liu, Y., Wang, Y., Shen, Y., et al. (2014). Genome-wide identification of non-coding RNAs interacted with microRNAs in soybean. Front. Plant Sci. 5:743. doi: 10.3389/fpls.2014.00743
Yoon, S. R., and De Micheli, G. (2005). Prediction of regulatory modules comprising microRNAs and target genes. Bioinformatics 21, 93–100. doi: 10.1093/bioinformatics/bti1116
Yoshikawa, T., Ozawa, S., Sentoku, N., Itoh, J.-I., Nagato, Y., and Yokoi, S. (2013). Change of shoot architecture during juvenile-to-adult phase transition in soybean. Planta 238, 229–237. doi: 10.1007/s00425-013-1895-z
Yu, J.-Y., Zhang, Z.-G., Huang, S.-Y., Han, X., Wang, X.-Y., Pan, W.-J., et al. (2019). Analysis of miRNAs targeted storage regulatory genes during soybean seed development based on transcriptome sequencing. Genes 10:408. doi: 10.3390/genes10060408
Yu, Y., Ni, Z., Wang, Y., Wan, H., Hu, Z., Jiang, Q., et al. (2019). Overexpression of soybean miR169c confers increased drought stress sensitivity in transgenic Arabidopsis thaliana. Plant Sci. 285, 68–78. doi: 10.1016/j.plantsci.2019.05.003
Zhang, S., Wang, Y., Li, K., Zou, Y., Chen, L., and Li, X. (2014). Identification of cold-responsive miRNAs and their target genes in nitrogen-fixing nodules of soybean. Int. J. Mol. Sci. 15, 13596–13614. doi: 10.3390/ijms150813596
Zhang, Z., Dunwell, J. M., and Zhang, Y.-M. (2018). An integrated omics analysis reveals molecular mechanisms that are associated with differences in seed oil content between Glycine max and Brassica napus. BMC Plant Biol. 18:328. doi: 10.1186/s12870-018-1542-8
Zheng, Y., Hivrale, V., Zhang, X., Valliyodan, B., Lelandais-Briere, C., Farmer, A. D., et al. (2016). Small RNA profiles in soybean primary root tips under water deficit. BMC Syst. Biol. 10:126. doi: 10.1186/s12918-016-0374-0
Zhou, Y., Liu, W., Li, X., Sun, D., Xu, K., Feng, C., et al. (2020). Integration of sRNA, degradome, transcriptome analysis and functional investigation reveals gma-miR398c negatively regulates drought tolerance via GmCSDs and GmCCS in transgenic Arabidopsis and soybean. BMC Plant Biol. 20:190. doi: 10.1186/s12870-020-02370-y
Zhou, Z. S., Zeng, H. Q., Liu, Z. P., and Yang, Z. M. (2012). Genome-wide identification of Medicago truncatula microRNAs and their targets reveals their differential regulation by heavy metal. Plant Cell Environ. 35, 86–99. doi: 10.1111/j.1365-3040.2011.02418.x
Keywords: miRNA co-regulation, abiotic stress tolerance in soybeans, homology expansion, bi-clustering, miRNA–target
Citation: Chang H, Zhang H, Zhang T, Su L, Qin Q-M, Li G, Li X, Wang L, Zhao T, Zhao E, Zhao H, Liu Y, Stacey G and Xu D (2022) A Multi-Level Iterative Bi-Clustering Method for Discovering miRNA Co-regulation Network of Abiotic Stress Tolerance in Soybeans. Front. Plant Sci. 13:860791. doi: 10.3389/fpls.2022.860791
Received: 23 January 2022; Accepted: 24 February 2022;
Published: 07 April 2022.
Edited by:
Ziding Zhang, China Agricultural University, ChinaReviewed by:
Ji Huang, Nanjing Agricultural University, ChinaCopyright © 2022 Chang, Zhang, Zhang, Su, Qin, Li, Li, Wang, Zhao, Zhao, Zhao, Liu, Stacey and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hao Zhang, emhhbmdoQGpsdS5lZHUuY24=; Dong Xu, eHVkb25nQG1pc3NvdXJpLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.