
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 14 February 2022
Sec. Plant Bioinformatics
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.818206
This article is part of the Research Topic Approaches and Applications in Plant Genome Assembly and Sequence Analysis View all 13 articles
 Yuanzhao Yang1
Yuanzhao Yang1 Xiaodan Liu1
Xiaodan Liu1 Xiaoqing Shi1
Xiaoqing Shi1 Jiao Ma1Xinmei Zeng1Zhangshun Zhu1Fangwen Li1Mengyan Zhou2Xiaodan Guo2Xiaoli Liu1*
Jiao Ma1Xinmei Zeng1Zhangshun Zhu1Fangwen Li1Mengyan Zhou2Xiaodan Guo2Xiaoli Liu1*Hibiscus mutabilis (cotton rose) is a deciduous shrub or small tree of the Malvaceae family. Here, we report a chromosome-scale assembly of the H. mutabilis genome based on a combination of single-molecule sequencing and Hi-C technology. We obtained an optimized assembly of 2.68 Gb with a scaffold N50 length of 54.7 Mb. An integrated strategy of homology-based, de novo, and transcriptome-based gene predictions identified 118,222 protein-coding genes. Repetitive DNA sequences made up 58.55% of the genome, and LTR retrotransposons were the most common repetitive sequence type, accounting for 53.15% of the genome. Through the use of Hi-C data, we constructed a chromosome-scale assembly in which Nanopore scaffolds were assembled into 46 pseudomolecule sequences. We identified important genes involved in anthocyanin biosynthesis and documented copy number variation in floral regulators. Phylogenetic analysis indicated that H. mutabilis was closely related to H. syriacus, from which it diverged approximately 15.3 million years ago. The availability of cotton rose genome data increases our understanding of the species’ genetic evolution and will support further biological research and breeding in cotton rose, as well as other Malvaceae species.
Hibiscus mutabilis is one of the most popular tree species in the Malvaceae family, which includes species such as Gossypium raimondii and Hibiscus syriacus (Rose of Sharon). Some members of the Malvaceae have relatively high economic value. For example, cotton is the largest source of natural textile fibers in the world, and over 90% of its annual fiber production comes from allotetraploid cotton (G. hirsutum and G. barbadense) (Wang et al., 2019). Additionally, many Malvaceae species are used as ornamentals because of their flowers. H. syriacus is an important horticultural species whose attractive white, pink, red, lavender, or purple flowers are displayed over a long bloom period, although individual flowers last only a day in the landscape (Kim et al., 2016). This study of H. mutabilis (2n = 92) (Li et al., 2015) focuses primarily on its ornamental characters, including its flower colors, long bloom time, and floral development and morphogenesis. In addition to its ornamental value, H. mutabilis is also an ingredient in local herbal remedies. It is thought to cool the blood, relieve toxins, reduce swelling, and alleviate pain, and it has long been used in the treatment of ulcers, swelling, herpes zoster, scalding, bruises, etc. (Liu et al., 2015). The complete genome sequence of an organism provides a large amount of information for subsequent biological studies (Yang et al., 2005). The H. mutabilis genome sequencing project is therefore extremely valuable for breeding, comparative genomics research, and other activities.
H. mutabilis has been cultivated for more than 2,000 years south of the Yangtse River; it is also the city flower of Chengdu and has great significance for the city. Commonly used as an ornamental species, its attractive purple, red, pink, or white flowers are displayed over a long bloom period (3–4 months or more), although its individual flowers last for less than 48 h (Figure 1). During flower development, the floral color of some varieties shows little change, but that of other varieties undergoes a marked change from white to pink within a single day. This interesting dynamic phenomenon can be seen in the cultivars ‘Drunk girl’ and ‘Bairihuacai’ and occurs during the process of individual flower development, unlike the color differences found in distinct cultivars of chrysanthemum (Ohmiya et al., 2006), Narcissus pseudonarcissus (Li et al., 2018), or Brassica napus (Zhang et al., 2015).

Figure 1. Hibiscus mutabilis floral morphology. (A) Dynamic change in the flower color of H. mutabilis f. mutabilis ‘Drunk girl’ from white to pink. (B) A variety of colors found in different color cultivars of H. mutabilis, and from left to right in turn are H. mutabilis ‘Single-Petal Pink,’ H. mutabilis ‘Single-Petal White’ and H. mutabilis ‘Purple silk.’
The development of genomic resources and molecular breeding technologies holds promise for targeted character improvement of H. mutabilis in the near future. Recently, a 1.75 Gb draft genome of H. syriacus was assembled, and a chromosome-scale genome of H. cannabinus was published in 2020 (Zhang et al., 2020). Many breeding systems and novel varieties have been produced using traditional methods to meet horticultural requirements, but a completed genomic sequence will accelerate the breeding of cotton rose.
The many flower colors of cotton rose give it high ornamental value and reflect the complexity of the underlying flavonoid metabolic pathway. One endpoint of flavonoid biosynthesis is the production of anthocyanins, pigments that produce the colors of many flowers, fruits, and other plant tissues (Koes et al., 1994). Chalcone synthase (CHS), chalcone isomerase (CHI), flavanone 3-hydroxylase (F3H), dihydroflavonol reductase (DFR), anthocyanidin synthase (ANS), and flavonoid 3-O-glucosyltransferase (UFGT) all function in the synthesis of anthocyanins and anthocyanidins, their aglycone counterparts. CHS represents the first committed step in the flavonoid pathway (Meer et al., 1993). The second step is performed by CHI, which acts on the yellow naringenin chalcone product of CHS, catalyzing its isomerization to the colorless flavanone naringenin (Moustafa and Wong, 1967). Dihydroflavonols are subsequently reduced to leucoanthocyanidins by DFR (Durbin et al., 2004). ANS catalyzes the formation of cyanidin from leucoanthocyanidin and is the penultimate step in the biosynthesis of the anthocyanin class of flavonoids (Figure 2). Despite recent progress in understanding H. mutabilis anthocyanidin biosynthesis, the lack of a genome sequence has hampered efforts to elucidate the molecular and genetic determinants of this trait, which underlies the dynamic phenomenon of flower color development. Genome and transcriptome sequences are needed in order to fully analyze the molecular mechanisms of anthocyanidin biosynthesis.
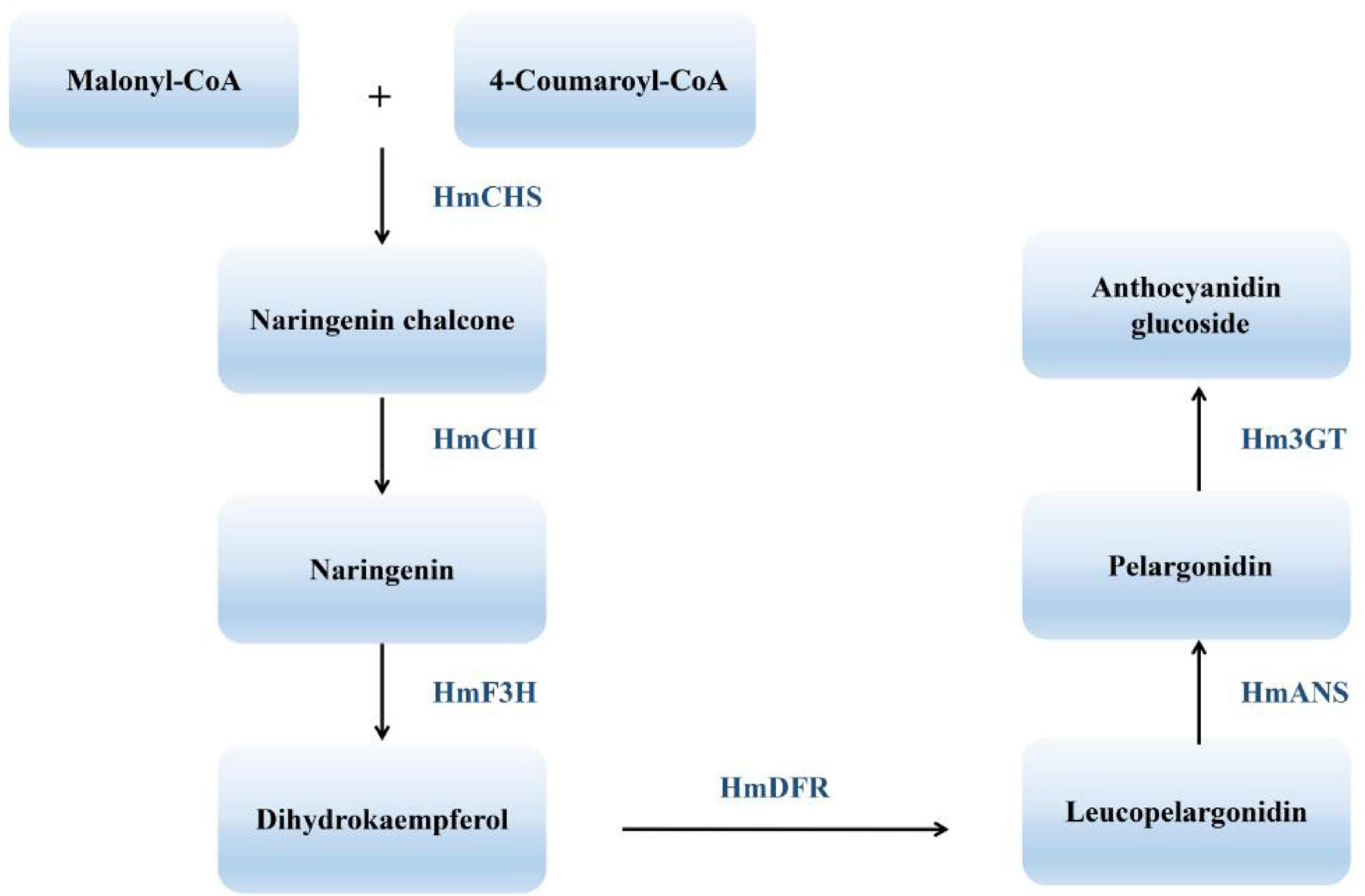
Figure 2. An abbreviated diagram of the flavonoid pathway that produces anthocyanins. CHS, chalcone synthase; CHI, chalcone isomerase; F3H, flavanone 3-hydroxylase; DFR, dihydroflavonol 4-reductase; ANS, anthocyanidin synthase; 3GT, 3-O-glucosyl transferase.
In the present study, we generated a reference genome for H. mutabilis using a combination of single-molecule sequencing and Hi-C technology. We identified functional genes involved in the biosynthesis of anthocyanins based on homology searches and functional annotations. We also investigated copy number variation in floral regulators among multiple species to gain insight into the evolution of flowering phenotypes in H. mutabilis. The genomic resources developed here will be useful for further experimentation, cultivation, and breeding of H. mutabilis and other Malvaceae species.
The H. mutabilis material sequenced in this study was the stably heritable single-petal white color cultivars, which is cultivated in the nursery of the Chengdu Botanical Garden (CDBG), Sichuan, China. The breeding system of H. mutabilis belongs to allogamy. Seeds of ‘single-petal white’ were collected in the laboratory of the CDBG. Young leaves (∼3 cm width) were harvested to extract high-quality DNA for Illumina and Oxford Nanopore Technology (ONT) sequencing. For transcriptome sequencing, petals were manually collected from three color cultivars (‘single-petal white,’ ‘single-petal pink,’ and ‘Purple silk’) and at three stages of color development in ‘Drunk girl’ (white, blended white and pink, and fully pink). Flowers at the same stage from individual H. mutabilis plants were pooled and divided into three samples. These samples were immediately frozen in liquid nitrogen and then used for RNA sequencing.
High-quality H. mutabilis genomic DNA was extracted from young leaves with a DNA secure Plant Kit (TIANGEN, China) and used to construct long-read libraries for the ONT platform.1 Libraries were prepared following the ONT’1D Genomic DNA by Ligation (Kit 9 chemistry)-PromethION’ protocol and sequenced using the PromethION protocol. In addition, high-quality DNA was broken into random fragments, and an Illumina paired-end library was constructed with an insert size of 350 bp and sequenced using the Illumina HiSeq X Ten platform.
For Hi-C sequencing, leaves were fixed with 1% formaldehyde solution in MS buffer (10 mM potassium phosphate, pH 7.0; 50 mM NaCl; 0.1 M sucrose) at room temperature for 30 min in a vacuum. After fixation, the leaves were incubated at room temperature for 5 min under a vacuum in MC buffer with 0.15M glycine. Approximately 2 g of fixed tissue was homogenized with liquid nitrogen, resuspended in nuclei isolation buffer, and filtered with a 40-nm cell strainer. The procedures for enriching nuclei from flow through and subsequent denaturation were performed according to a 3C protocol. The chromatin extraction procedures were similar to those described previously. In brief, chromatin was digested for 16 h with 400 U HindIII restriction enzyme (NEB) at 37°C. DNA ends were labeled with biotin and incubated at 37°C for 45 min, and the enzyme was inactivated with 20% SDS solution. DNA ligation was performed by the addition of T4 DNA ligase (NEB) and incubation at 16°C for 4–6 h. After ligation, proteinase K was added for reverse cross-linking during overnight incubation at 65°C. DNA fragments were purified and dissolved in 86 μL of water, and unligated ends were then removed. Purified DNA was fragmented to a size of 300–500 bp, and DNA ends were repaired. Finally, DNA fragments labeled with biotin were separated on Dynabeads M-280 Streptavidin (Life Technologies). Hi-C libraries were assessed for quality and sequenced on an Illumina HiSeq X Ten sequencer.
De novo assembly of all Nanopore long reads was performed using wtdbg2 v2.5 (Ruan and Li, 2020). Because Nanopore reads contain systematic errors in homopolymeric regions, we polished the consensus assembly three times using the Nanopore reads as input to Racon v1.3.1 (Vaser et al., 2017) and then three additional times using Illumina reads as input to Pilon v1.22 (Walker et al., 2014).
The Hi-C data were mapped to the original scaffold genome using BWA v0.7.7 (Li, 2009) and only reads with unique alignment positions were extracted to construct a chromosome-scale assembly using LACHESIS v201701 (Burton et al., 2013).
We used both CEGMA (Core Eukaryotic Gene Mapping Approach) (Parra et al., 2007; Supplementary Table 4) and BUSCO (Benchmarking Universal Single-Copy Orthologs) (Simão et al., 2015; Supplementary Table 5) to evaluate the completeness of the assembly.
TEs were identified in the genome assembly at both the DNA and protein levels. We used RepeatModeler, RepeatScout (Tarailo-Graovac and Chen, 2009), Piler (Edgar and Myers, 2005), and LTR_FINDER (Xu and Wang, 2007) to develop a de novo TE library. RepeatMasker (Tarailo-Graovac and Chen, 2009) was used for DNA-level identification with Repbase and the de novo TE library. Tandem repeats were identified using Tandem Repeats Finder (Benson, 1999). At the protein level, RepeatProteinMask (Tarailo-Graovac and Chen, 2009) was used to conduct WU-BLASTX searches against the TE protein database. Overlapping TEs that belonged to the same type of repeat were integrated together.
We used homology-based, de novo, and transcriptome-based approaches to predict protein-coding genes in the H. mutabilis genome. For homolog-based prediction, sequences of homologous proteins from six plants (A. thaliana, C. capsularis, D. zibethinus, G. raimondii, H. umbratica, H. syriacus, and T. cacao) were downloaded from Ensembl, NCBI, or JGI. Protein sequences were aligned to the genome using TBLASTN with an E-value cutoff of 1 × 10–5. The blast hits were concatenated using solar (Yu et al., 2007). For each blast hit, GeneWise v2.4.1 (Birney and Clamp, 2004) was used to predict the exact gene structure in the corresponding genomic regions. The five ab initio gene prediction programs AUGUSTUS v3.0.2 (Stanke and Morgenstern, 2005), Genescan v1.0 (Aggarwal and Ramaswamy, 2002), GeneID (Parra et al., 2000), GlimmerHMM v3.0.2 (Majoros et al., 2004), and SNAP (Korf, 2004) were used for de novo protein prediction. To further optimize the genome annotation, RNA-seq data from floral, leaf, and stem tissues were aligned to the H. mutabilis genome using TopHat v2.0.13 (Trapnell et al., 2009) to identify exon regions and splice junctions. The alignment results were then used as input for Cufflinks v2.1.1 (Trapnell et al., 2010) in order to assemble transcripts into gene models. Trinity (Grabherr et al., 2011) was used with default parameters to assemble the RNA-seq data, and PASA (Haas et al., 2003) was used to improve the gene structures. A weighted and non-redundant gene set was generated by EVidenceModeler (EVM) (Haas et al., 2008), which merged all gene models predicted using the three approaches above. PASA adjusted the gene models generated by EVM based on information from the transcriptome assembly.
The functional annotation of protein-coding genes was evaluated by BLASTP (E-value ≤ 1 × 10-5) against two integrated protein sequence databases, Swiss-Prot (Bairoch and Apweiler, 2000) and the NCBI non-redundant (NR) database. Protein domains were annotated using InterProScan v4.8 to search InterPro v32.0 (Mulder and Apweiler, 2007), which includes the Pfam, PRINTS, PROSITE, ProDom, and SMART databases. Gene Ontology (GO) (Ashburner et al., 2000) terms for each gene were obtained from the corresponding InterPro descriptions. Putative pathway assignments for each gene were obtained by blasting against the KEGG (Kanehisa and Goto, 2006) database with an E-value cutoff of 1 × 10–5.
tRNA genes were predicted by tRNAscan-SE (Lowe and Eddy, 1997), and miRNA and snRNA fragments were identified using Infernal (Nawrocki et al., 2009) with the Rfam (Griffiths-Jones et al., 2005) database. rRNA genes were identified using BLASTN (E-value ≤ 1 × 10–10) against the plant rRNA database.
First, nucleotide and protein data from nine species (A. trichopoda, A. thaliana, B. ceiba, C. capsularis, D. zibethinus, G. raimondii, H. syriacus, R. chinensis, and T. cacao) were downloaded from Ensembl, NCBI, and JGI. The longest transcript was selected from the alternatively spliced transcripts of each gene, and genes with ≤ 50 amino acids were removed. Nucleotide and protein data from H. mutabilis and the other nine angiosperms were clustered into orthologous groups using BLASTP and OrthoMCL v2.0.9, and an MCL inflation of 1.5 was used as the cluster granularity setting (Li et al., 2003). A phylogenetic tree was constructed using shared single-copy orthologs. Protein sequences of the orthologs were aligned using MUSCLE (Edgar, 2004), and the protein alignments were transformed to CDS alignments. We then concatenated the CDS alignments into a “supermatrix” from which the phylogenetic tree was constructed using the maximum likelihood (ML) TREE algorithm in RAxML v8.1.13 (Stamatakis, 2006) with the best-scoring protein substitution model (GTRGAMMA) and 1,000 bootstrap replicates. The MCMCtree program in the PAML package (Yang, 1997) was used to estimate divergence times among the ten species. Three fossil calibration points were used for restraining the age of the nodes: 23–48 Mya (Million years ago) for the MRCA of T. cacao–G. raimondii, 65–107 Mya for the MRCA of G. raimondii–A. thaliana (Wang et al., 2012), and 103–109 Mya for the MRCA of Malvales–Rosales (Wikström et al., 2001). CAFE was used to identify expansions and contractions within orthologous gene families by comparing cluster size differences between the ancestor and other species (De Bie et al., 2006). To estimate the synonymous substitutions per synonymous site (Ks), all paralogous gene pairs were analyzed with the ML method in PAML (Yang, 1997). MCscan (Tang et al., 2008) was used to analyze genome collinearity in H. mutabilis.
To identify NBS-encoding genes, representative genes from each plant genome were screened using a raw Hidden Markov Model (HMM3.0) (Marchin et al., 2005) to search for the Pfam NBS family PF00931 domain with an E-value cut-off of 1.0. All putative NBS protein sequences were analyzed and manually curated based on a TBLASTN search against known R gene sequences in GenBank. To further identify TIR homologs and sequences that encoded CC and LRR motifs, candidate NBS-LRR protein sequences were characterized using SMART (Schultz et al., 1998), the Pfam database (Finn et al., 2013), and the COILS program (Lupas et al., 1991) with a threshold of 0.9 to specifically detect the CC domain.
For analysis of flowering gene(s), petals were manually collected from three color cultivars at the same time (2–3 p.m.) and at three stages of color development in ‘Drunk girl’ (white at the 9 a.m., blended white and pink at the 12 a.m., and fully pink at 6 p.m.) and these samples were immediately frozen in liquid nitrogen and then used for RNA sequencing, and total RNA was extracted using an RNAprep Pure Plant Kit (TIANGEN, China). The quality and quantity of the RNA samples were evaluated using a NanoPhotometer (Implen, CA, United States), a Qubit 3.0 Fluorometer (Thermo Fisher Scientific, United States), and an Agilent 2,100 Bioanalyzer (Agilent Technologies, United States). All RNA samples with integrity values greater than 7.0 were used for cDNA library construction and sequencing. The cDNA libraries were prepared using the NEB Next Ultra RNA Library Prep Kit (E7350L, NEB, United States), and 150-bp paired-end sequencing was performed on the Illumina NovaSeq 6000 platform (Illumina, CA, United States).
We assembled the H. mutabilis genome using a combination of Illumina HiSeq X Ten and Oxford Nanopore PromethION sequencing. We generated 315.22 Gb (104-fold coverage) of raw 150-bp paired-end Illumina reads and 469.91 Gb (155-fold coverage) of raw Nanopore reads. The genome size was estimated to be 3032.98 Mb based on the 17-mer depth distribution (Supplementary Table 1 and Supplementary Figure 1). Nanopore long reads were assembled into contigs and scaffolds using wtdbg2 v2.5,13 resulting in a final assembly of 2.68 Gb with 5,464 contigs and a contig N50 of 2.22 Mb (Supplementary Table 2). Its GC percentage was 35.36%, similar to that of the H. syriacus genome (34.04%). In total, 363.5 Gb of clean reads were obtained from Hi-C sequencing (over 121-fold coverage). We used these data to construct chromosome-scale scaffolds, resulting in a total of 5,598 contigs, with a scaffold N50 of 54.70 Mb and a total length of 2,676,237,573 bp (Supplementary Table 10 and Figure 3A).
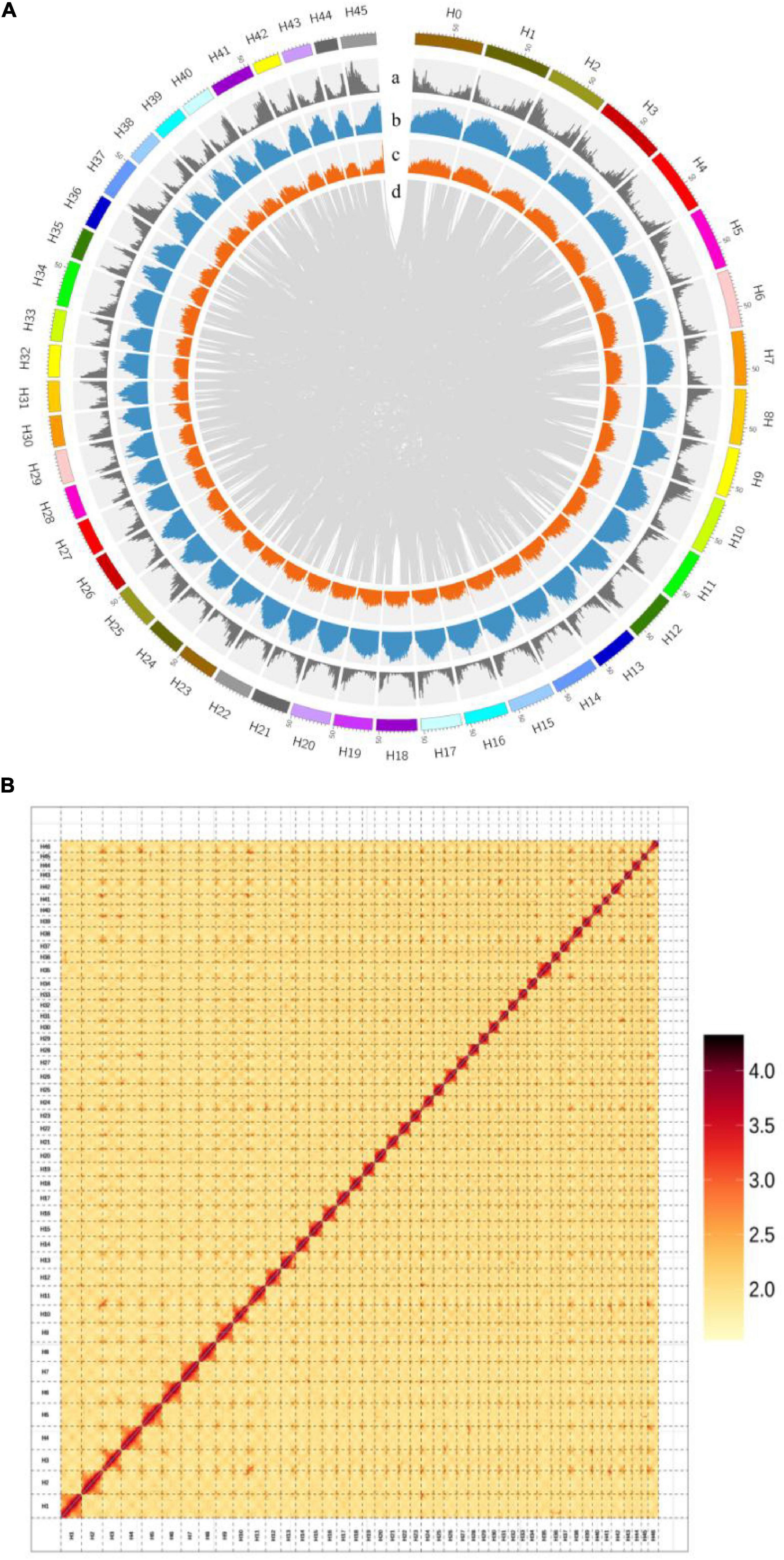
Figure 3. (A) Chromosomal features of H. mutabilis. (a) Gene density; (b) Repeat density; (c) AT content; (d) Syntonic blocks. (B) Hi-C map of the H. mutabilis genome showing genome-wide all-by-all interactions. The map shows a high resolution of individual chromosomes that were scaffolded and assembled independently. Color intensity indicates the frequency of Hi-C interaction links from low (yellow) to high (red).
Next, The clustering of contig by hierarchical clustering of the Hi-C data was performed. Hi-C linkage was used as a criterion to measure the degree of tightness of the association between different contigs by standardizing the digestion sites of DpnII on the genome sketch. The contigs were assembled into 46 pseudo-chromosomes using LACHESIS package tools. The Illumina paired-end reads were mapped to the assembled genome to assess assembly accuracy, resulting in a 98.81% mapping rate (Supplementary Table 3 and Figure 3B). The genome assembly captured 96.77% of the core eukaryotic genes from CEGMA18 (Supplementary Table 4) and 92.6% of the Embryophyta OrthoDB gene set in BUSCO19 (Supplementary Table 5), indicating a high level of completeness.
We identified 1.56 Gb of non-redundant repetitive elements, representing approximately 55.85% of the H. mutabilis genome assembly. Because long terminal repeat retrotransposons (LTR-RTs) typically make a significant contribution to large genome size (Zhao and Ma, 2013), we estimated LTR-RT insertion time in H. mutabilis. We identified a round of LTR-RT burst approximately 2.5 million years ago (Mya), especially for the Ty3/Gypsy-del and Ty1/Copia-Retrofit families (Supplementary Table 6). The transposable elements (TEs) were primarily long terminal repeats (LTRs), which accounted for approximately 53% of the genome (Supplementary Figure 2).
We used de novo and homology-based gene prediction approaches and combined their results to annotate 118,222 protein-coding genes in the H. mutabilis genome. The average transcript length was 2,466.97 bp, with an average of 4.53 exons per gene and an average exon length of 218.86 bp. Compared with other model plants and Malvaceae species, the H. mutabilis genome contained a larger number of genes: H. syriacus (82,827 genes), Arabidopsis thaliana (26,869), Theobroma cacao (29,144), G. raimondii (35,526), Corchorus capsularis (29,356), Herrania umbratica (29,262), and Durio zibethinus (63,819) (Supplementary Table 7).
In addition to RNA-coding genes, we also identified 827 mature microRNAs (miRNAs), 3,604 transfer RNAs (tRNAs), 3,423 ribosomal RNAs (rRNAs), and 9,370 small nuclear RNAs (snRNAs) in the H. mutabilis genome (Supplementary Table 11).
The probable functions of the predicted genes were assessed by searching against public databases, including Swiss-Prot, NR, InterPro, and KEGG of 118,222 predicted genes in the H. mutabilis genome, 113,821 (96.3%) were assigned potential functions as a result of these database searches (Supplementary Table 8).
Although morphological investigations have placed H. mutabilis in the Malvaceae family, there is still no phylogenomic analysis of its evolutionary position within the family based on whole-genome data. Here, we compared the H. mutabilis genome with the genome sequences of nine other angiosperm plants (H. syriacus, A. thaliana, Rosa chinensis, Bombax ceiba, Amborella trichopoda, G. raimondii, T. cacao, and C. capsularis). Orthologous protein groups were identified within the genomes, yielding a total of 30,208 gene families and 198 single-copy orthologs across ten species. There were 2,781 gene families specific to H. mutabilis, and 10,593 gene families were shared among all species investigated (Figure 4). We detected 4,558 gene families expansion when H. mutabilis and H. syriacus have diverged (Figure 5C).
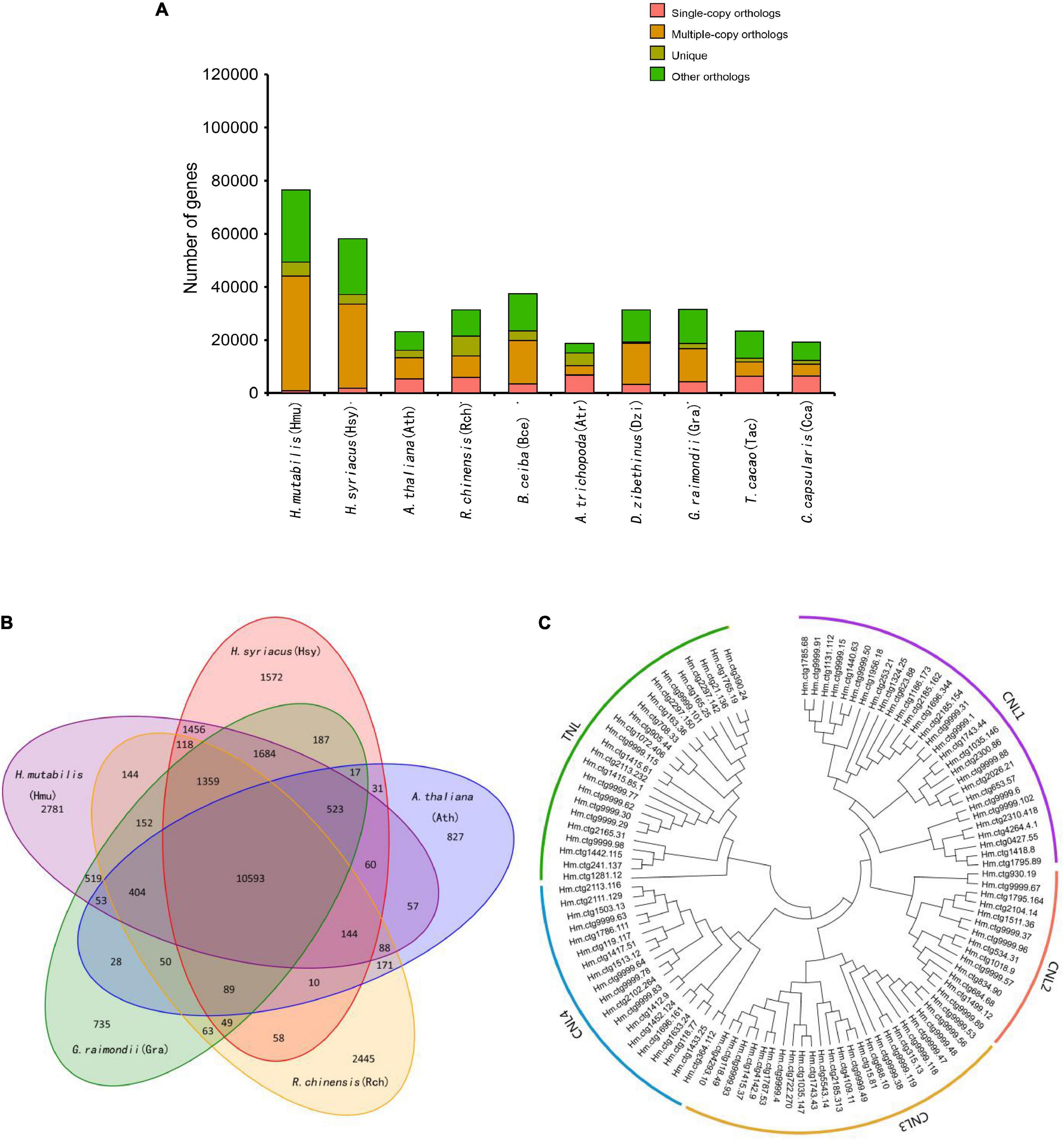
Figure 4. Comparative genomics and evolution of gene numbers. (A) The number of genes in cluster of ten plant species, showing a high gene number in H. mutabilis compared with the model plant A. thaliana and other angiosperm species. The number of multiple-copy paralogs is high in H. mutabilis. (B) Venn diagram showing the numbers of shared gene families among H. mutabilis (Hmu), A. thaliana (Ath), H. syriacus (Hsy), R. chinensis (Rch), and G. raimondii (Gra). (C) Phylogenetic relationships of NBS genes in H. mutabilis.
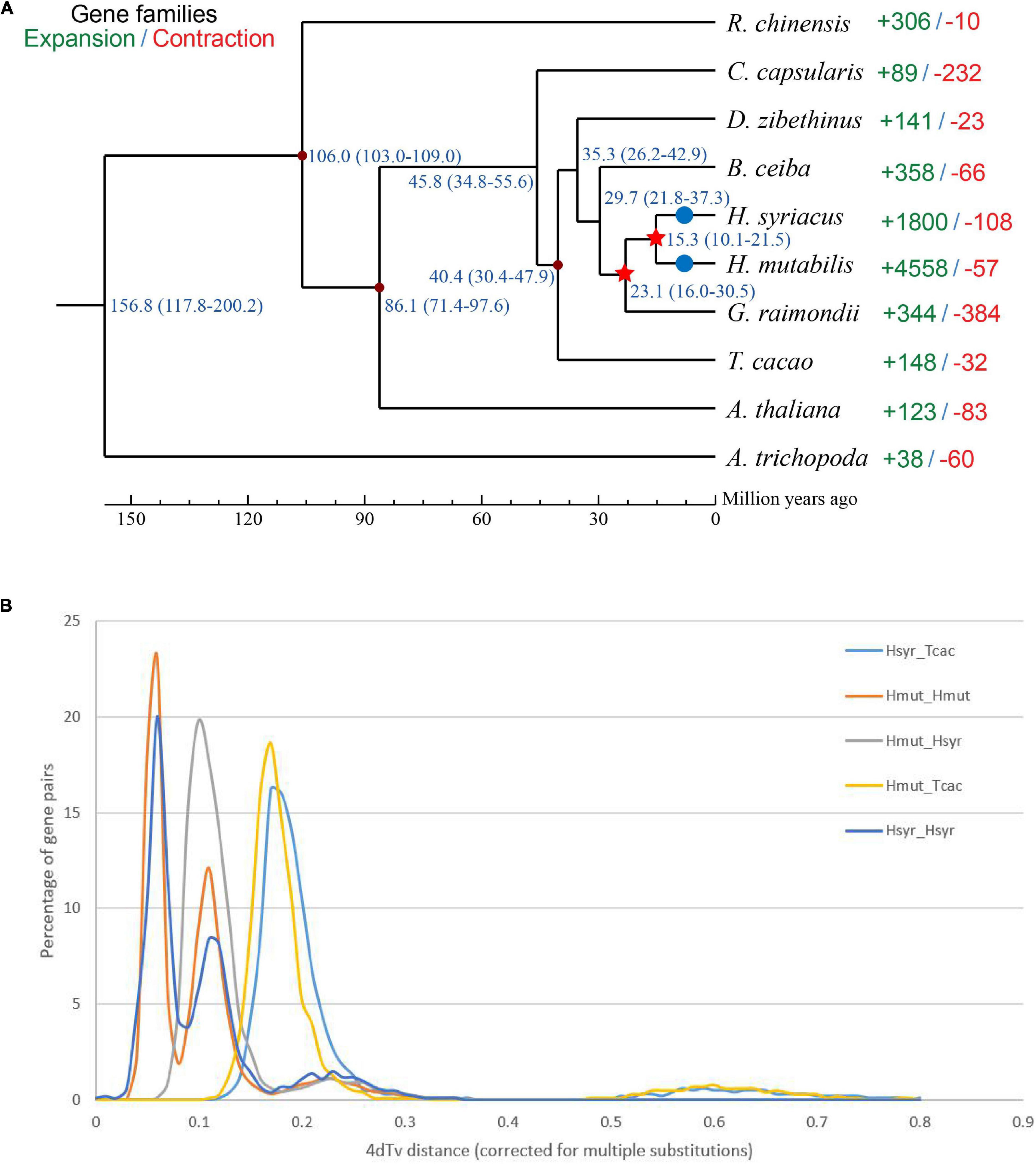
Figure 5. (A) Phylogenetic tree showing the close relationship between H. mutabilis and H. syriacus. Numbers outside the parentheses represent the divergence times of species (Mya), and numbers inside the parentheses indicate the confidence intervals. The red points indicated calibration points, and the red stars indicate WGD events and blue circles indicate diploidization events. (B) Distribution of 4Dtv distances.
We constructed a phylogenetic tree based on single-copy genes using PAML and estimated the divergence times among the 10 species. The Malvaceae family appeared to have diverged from a Tiliaceae–Malvaceae most recent common ancestor (MRCA) approximately 45.8 (34.8–55.6) Mya, and the Hibiscus-Gossypium divergence was estimated at 23.1 (16.0–30.5) Mya. H. mutabilis was most closely related to H. syriacus, with an estimated divergence time of approximately 15.3 (10.1–21.5) Mya (Figure 5A). In addition to the paleohexaploidization event shared by the eudicots, we observed three additional whole-genome duplication (WGD) events in H. mutabilis. Hibiscus and Gossypium shared a WGD event (13.3–20.0 Mya), H. mutabilis and H. syriacus shared a WGD event (10.76–21.51 Mya), and H. mutabilis and H. syriacus each underwent a WGD event (4.61–9.00 Mya) (Figure 5B).
Genetic and molecular mechanisms of floral development are highly conserved among different plant species (Schiessl et al., 2014) and include four major flowering pathways that have been well characterized in A. thaliana. H. mutabilis is similar to H. syriacus, a short-day flowering plant with a long bloom period that produces more than 30 blossoms per day. Like those of H. syriacus, the flowers of H. mutabilis open daily and last for less than 48 h. Because flowering time is frequently dependent on gene copy number (Grover et al., 2015), we investigated the copy numbers of genes involved in the four major flowering pathways in A. thaliana, T. cacao, G. raimondii, A. trichopoda, H. syriacus, and H. mutabilis. Copy numbers of most flowering-related genes were higher in H. mutabilis than in other plants, including T. cacao, G. raimondii, A. trichopoda, and H. syriacus. In particular, the copy number of the plant-specific nuclear protein GIGANTEA (GI) was three to four times greater in H. mutabilis than in A. trichopoda, T. cacao, or G. raimondii (Table 1).
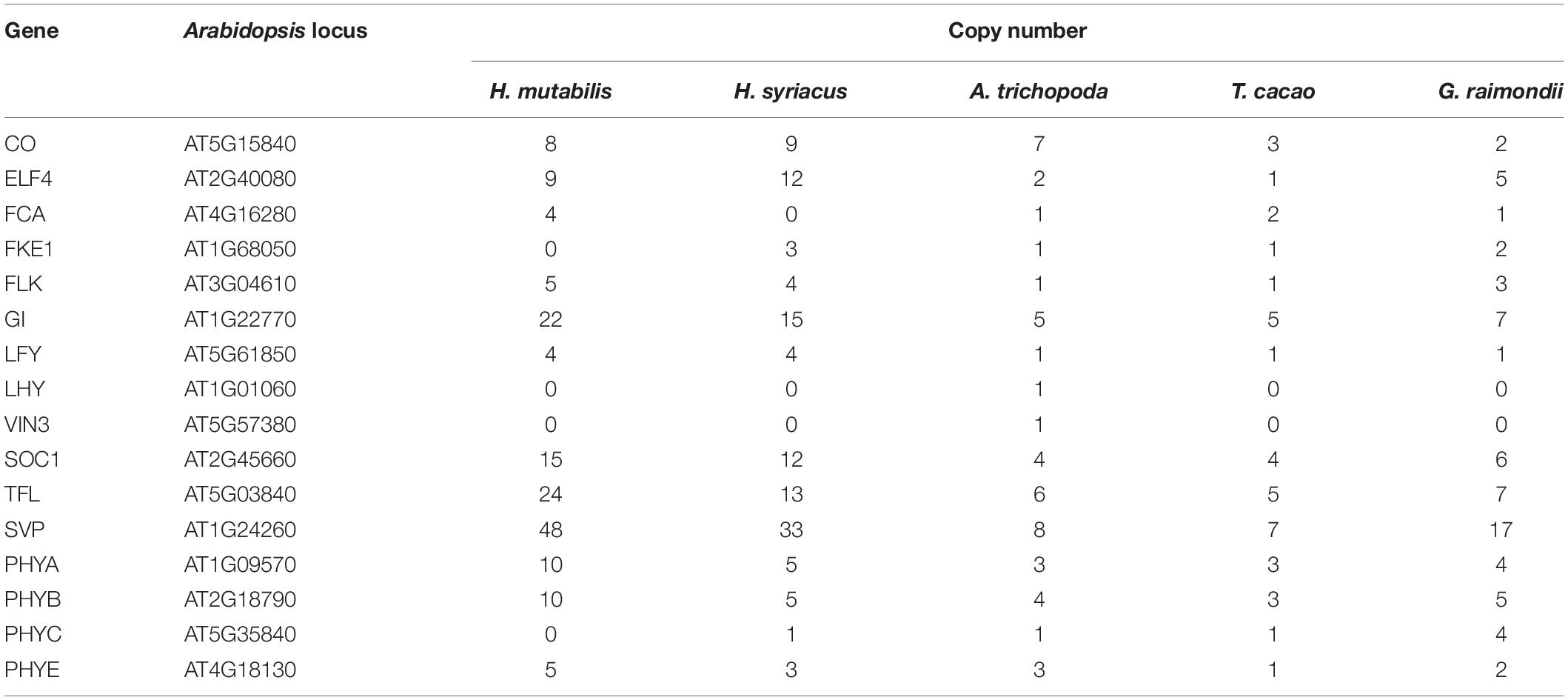
Table 1. Copy numbers of genes encoding flowering time regulators in five plant species.
Nucleotide-binding site (NBS) and carboxy-terminal LRR domains are found in the majority of R proteins (DeYoung and Innes, 2006; Takken et al., 2006). Based on resistance domain analyses in the H. mutabilis genome, a total of 490 NBS-containing resistance genes were identified and classified into six groups: CC-NBS-LRR, CC-NBS, TIR-NBS-LRR, TIR-NBS, NBS-LRR, and NBS. In total, their gene numbers were approximately three times greater in H. mutabilis than in A. thaliana (170). This trend was particularly striking for the NBS genes, whose numbers were much higher in H. mutabilis (192 genes) than in H. syriacus (54), T. cacao (9), G. raimondii (4), and A. thaliana (3). Although H. mutabilis had the highest number of NBS-containing resistance genes among the five angiosperms (Table 2), its number of NBS-containing genes as a percentage of total genes was the lowest. All six NBS-containing groups existed in each plant genome, but their distributions differed among species.
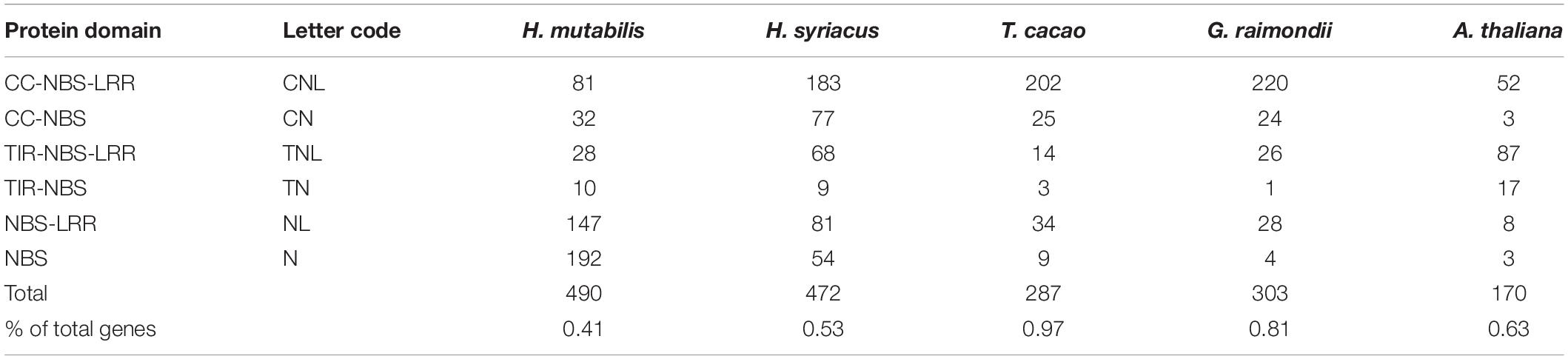
Table 2. Numbers and classifications of genes encoding NBS-containing resistance proteins in five plant species.
Global gene expression patterns were quantified in three stages of the floral color transition of H. mutabilis ‘Drunk girl’: white (Stage 1), blended white and pink (Stage 2), and fully pink (Stage 3). A total of 9,492 genes were up-regulated from Stage 1 to Stage 2, and more than 15,000 genes were up-regulated from Stage 1 to Stage 3. A total of 8,481 genes were down-regulated from Stage 1 to Stage 2, and 15,839 genes were down-regulated from Stage 1 to Stage 3. In particular, we analyzed expression changes in anthocyanin-related genes at the three flower stages and present the results in a heatmap (Figure 6A). A number of key anthocyanin biosynthesis-related genes, such as Hmchs, HmchI, and Hmans, increased in expression from Stage 1 to Stage 3, consistent with the pattern of floral color development.
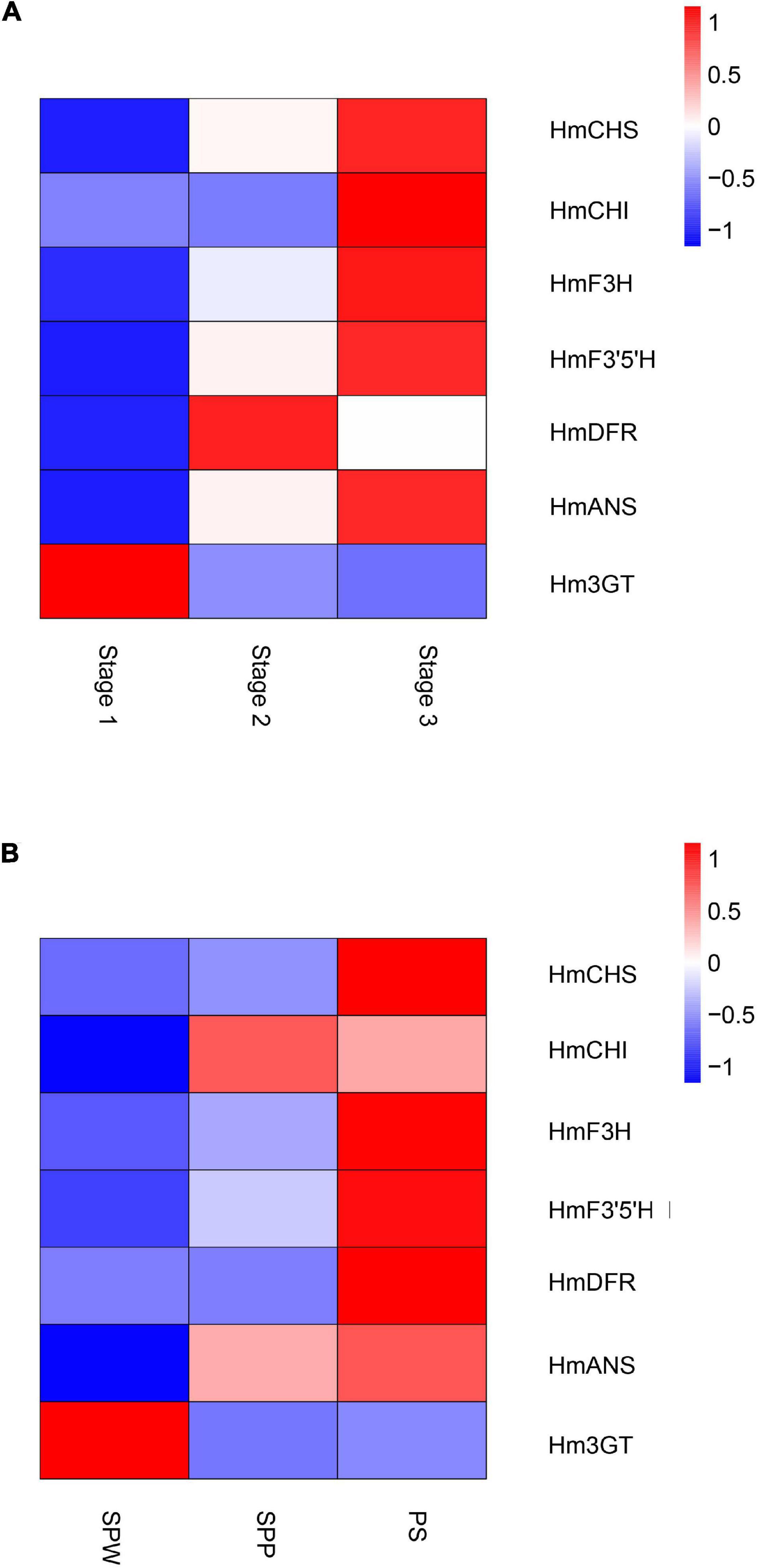
Figure 6. Heatmaps show the expression of key genes in the anthocyanin biosynthesis pathway of H. mutabilis. (A) Gene expression changes in flowers of H. mutabilis ‘Drunk girl’ as their color transitions from white to fully pink. (B) Gene expression in flowers of three different color cultivars: ‘single-petal white’ (SPW), ‘single-petal pink’ (SPP), and ‘Purple silk’ (PS).
Key anthocyanin biosynthesis-related genes also differed in expression among different color cultivars of H. mutabilis, including ‘single-petal white,’ ‘single-petal pink,’ and ‘Purple silk.’ The highest expression levels were generally found in ‘Purple silk,’ which is a deep purple color form (Figure 6B).
Completeness and continuity are important indicators of genome assembly quality. In this study, we took advantage of the longer read lengths offered by ONT sequencing that have proven advantageous in the assembly of other plant genomes such as Solanum pennellii (Schmidt et al., 2017) and Chrysanthemum nankingense (Song et al., 2018). Here, we report the first genome data for H. mutabilis and estimate its genome size to be 2.68 Gb, far larger than that of H. syriacus. Our H. mutabilis genome assembled using Nanopore reads had a contig N50 of 2.02 Mb. We then used Hi-C data to cluster the contigs into forty-six chromosomes with a final scaffold N50 of 54.70 Mb. The genome contained complete copies of 92.6% of the BUSCO orthologs examined. This genome sequence will contribute to our understanding of the biosynthesis of natural products such as anthocyanins, although additional research is needed to directly link specific genes to individual traits. Nonetheless, our high-quality, annotated genome sequence provides insights into determinate flowering time and flower color in H. mutabilis.
Compared with the H. syriacus genome, the H. mutabilis genome was larger and contained more protein-coding genes. H. mutabilis and H. syriacus share an MRCA approximately 15.3 (10.1–21.5) Mya, and investigation of WGD timing in the H. mutabilis genome showed that two WGDs occurred after H. mutabilis–H. syriacus divergence and H. mutabilis speciation. WGD events and tandem duplications are the most important determinants of genome size variation in angiosperms (Piegu et al., 2006; El Baidouri and Panaud, 2013). This recent WGD event not only caused genome expansion in H. mutabilis, but may also have contributed to the morphological and physiological diversity of H. mutabilis. We inferred that gene losses, which had different frequencies in H. mutabilis and H. syriacus, made the H. syriacus genome smaller than that of H. mutabilis. H. mutabilis has a long bloom period and high blossom turnover. The copy numbers of most flowering-related genes, such as GI, CONSTANS (CO), and SUPPRESSOR OF OVEREXPRESSION OF CONSTANS1 (SOC1) were higher in H. mutabilis than in T. cacao, A. thaliana, and H. syriacus. These results show that H. mutabilis preserved many copies of flowering-related genes during the transition from a polyploid to a diploid genome.
The dynamic color change from white to pink in H. mutabilis ‘Drunk girl’ flowers is reported to be caused by variations in anthocyanin contents (Liu et al., 2008). Flavonoids are the major molecules involved in plant pigmentation (Lai et al., 2014) and include anthocyanins, flavan-3-ols (catechins and proanthocyanidins), flavanonols, flavonols, flavones, and phenolic acid (Lou et al., 2014). To date, regulation of the flavonoid pathway has been shown to occur primarily at the transcriptional level (Mol et al., 1998). Different species have distinct regulated genes, and these appear to be among the most important candidate genes for flower color determination (Casimiro-Soriguer et al., 2016; Jiao et al., 2020). To investigate the expression of anthocyanin-related genes over the course of flower development and in different color forms, we combined the high-quality genome sequence generated here with RNA-seq data from H. mutabilis The expression levels of anthocyanin biosynthetic genes such as Hmchs, HmchI, and Hmans were correlated and increased as flowers transitioned from white to pink. The pink flower color in cotton rose is related to the synthesis of cyanidin-based pigments (Chen et al., 2014), and our results indicate that low CHS, CHI, and ANS expression may inhibit cyanidin production in white flowers. Thus, combined genomic and transcriptomic analysis of H. mutabilis flowers indicated that structural genes had important roles in anthocyanin biosynthesis during the transition from white to pink flower coloration. In maize, an MYB-related protein and a bHLH containing protein interact to activate genes in the anthocyanin biosynthetic pathway (Schwinn et al., 2006). However, the functions of transcription factors, including MYBs, bHLHs, and WD40s, are unknown in H. mutabilis. The investigation of these TFs in cotton rose will be a subject for further research.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, PRJNA717149.
YY and XLL: conceptualization. XG, MZ, and XDL: formal analysis. FL and ZZ: project administration. JM and XZ: resources. YY and XS: writing—original draft. XLL: writing—review and editing. All authors have read and agreed to the published version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.818206/full#supplementary-material
Aggarwal, G., and Ramaswamy, R. (2002). Ab initio gene identification: prokaryote genome annotation with GeneScan and GLIMMER. J. Biosci. 27, 7–14. doi: 10.1007/BF02703679
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Bairoch, A., and Apweiler, R. (2000). The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48. doi: 10.1093/nar/28.1.45
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Birney, E., and Clamp, M. (2004). GeneWise and GenomeWise. Genome Res. 14, 988–995. doi: 10.1101/gr.1865504
Burton, J. N., Adey, A., Patwardhan, R. P., Qiu, R., Kitzman, J. O., and Shendure, J. (2013). Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125. doi: 10.1038/nbt.2727
Casimiro-Soriguer, I., Narbona, E., Buide, M. L., Del Valle, J. C., and Whittall, J. B. (2016). Transcriptome and biochemical analysis of a flower color polymorphism in Silene littorea (Caryophyllaceae). Front. Plant Sci. 7:204. doi: 10.3389/fpls.2016.00204
Chen, Y., Mao, Y., Liu, H., Yu, F., Li, S., and Yin, T. (2014). Transcriptome analysis of differentially expressed genes relevant to variegation in peach flowers. PLoS One 9:e90842. doi: 10.1371/journal.pone.0090842
De Bie, T., Cristianini, N., Demuth, J. P., and Hahn, M. W. (2006). CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271. doi: 10.1093/bioinformatics/btl097
DeYoung, B. J., and Innes, R. W. (2006). Plant NBS-LRR proteins in pathogen sensing and host defense. Nat. Immunol. 7, 1243–1249. doi: 10.1038/ni1410
Durbin, M., Lundy, K., Morrell, P., Torres-Martinez, C., and Clegg, M. (2004). Genes that determine flower color: the role of regulatory changes in the evolution of phenotypic adaptations. Mol. Phylogenet. Evol. 29, 507–518. doi: 10.1016/S1055-7903(03)00196-9
Edgar, R., and Myers, E. (2005). PILER: identification and classification of genomic repeats. Bioinformatics 21, 152–158. doi: 10.1093/bioinformatics/bti1003
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
El Baidouri, M., and Panaud, O. (2013). Comparative genomic paleontology across plant kingdom reveals the dynamics of TE-Driven genome evolution. Genome Biol. Evol. 5, 954–965. doi: 10.1093/gbe/evt025
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2013). Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230. doi: 10.1093/nar/gkt1223
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Griffiths-Jones, S., Moxon, S., Marshall, M., Khanna, A., Eddy, S. R., and Bateman, A. (2005). Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, 121–124. doi: 10.1093/nar/gki081
Grover, C. E., Gallagher, J. P., and Wendel, J. F. (2015). Candidate gene identification of flowering time genes in cotton. Plant Genome 8:eplantgenome2014.2012.0098. doi: 10.3835/plantgenome2014.12.0098
Haas, B. J., Delcher, A. L., Mount, S. M., Wortman, J. R., Smith, R. K. Jr., Hannick, L. I., et al. (2003). Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666. doi: 10.1093/nar/gkg770
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9:R7. doi: 10.1186/gb-2008-9-1-r7
Jiao, F., Zhao, L., Wu, X., Song, Z., and Li, Y. (2020). Metabolome and transcriptome analyses of the molecular mechanisms of flower color mutation in tobacco. BMC Genom. 21:611. doi: 10.1186/s12864-020-07028-5
Kanehisa, M., and Goto, S. (2006). KEGG: kyoto encyclopedia of genes and genomes. Artif. Intell. 28, 27–30. doi: 10.1093/nar/28.1.27
Kim, Y. M., Kim, S., Koo, N., Shin, A. Y., Yeom, S. I., Seo, E., et al. (2016). Genome analysis of Hibiscus syriacus provides insights of polyploidization and indeterminate flowering in woody plants. DNA Res. 24, 71–80. doi: 10.1093/dnares/dsw049
Koes, R., Quattrocchio, F., and Mol, J. (1994). The flavonoid biosynthetic pathway in plants: Function and evolution. Bioessays 16, 123–132. doi: 10.1002/bies.950160209
Lai, B., Li, X. J., Hu, B., Qin, Y. H., Huang, X. M., Wang, H. C., et al. (2014). LcMYB1 is a key determinant of differential anthocyanin accumulation among genotypes, tissues, developmental phases and ABA and light stimuli in Litchi chinensis. PLoS One 9:e86293. doi: 10.1371/journal.pone.0086293
Li, H. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, L., Stoeckert, C. J. Jr., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Li, X., Tang, D., Du, H., and Shi, Y. (2018). Transcriptome sequencing and biochemical analysis of perianths and coronas reveal flower color formation in Narcissus pseudonarcissus. Int. J. Mol. Sci. 19:4006. doi: 10.3390/ijms19124006
Li, Y. P., Zhang, X. L., Wu, W. T., Miao, S. X., and Chang, J. L. (2015). Chromosome and karyotype analysis of Hibiscus mutabilis f. mutabilis. Front. Life Sci. 8, 300–304. doi: 10.1080/21553769.2015.1041166
Liu, D., Mei, Q., Wan, X., Que, H., Li, L., and Wan, D. (2015). Determination of rutin and isoquercetin contents in Hibiscus mutabilis folium in different collection periods by HPLC. J. Chromatogr. Sci. 53, 1680–1684. doi: 10.1093/chromsci/bmv071
Liu, J. Q., Jin, H. Q., Yuan, H. Y., and Lu, X. P. (2008). Mechanism analysis of variety corolla from Hibiscus mutabilis L. Northern Hortic. 11, 113–116.
Lou, Q., Liu, Y., Qi, Y., Jiao, S., Tian, F., Jiang, L., et al. (2014). Transcriptome sequencing and metabolite analysis reveals the role of delphinidin metabolism in flower colour in grape hyacinth. J. Exp. Bot. 65, 3157–3164. doi: 10.1093/jxb/eru168
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Lupas, A., Van Dyke, M., and Stock, J. (1991). Predicting coiled coils from protein sequences. Science 252, 1162–1164. doi: 10.1126/science.252.5009.1162
Majoros, W., Pertea, M., and Salzberg, S. (2004). TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879. doi: 10.1093/bioinformatics/bth315
Marchin, M., Kelly, P. T., and Fang, J. (2005). Tracker: continuous HMMER and BLAST searching. Bioinformatics 21, 388–389. doi: 10.1093/bioinformatics/bti012
Meer, I. M., Stuitje, A., and Mol, J. N. M. (1993). “Regulation of general phenylpropanoid and flavonoid gene expression,” in Control of Plant Gene Expression, ed. D. P. S. Verma (Boca Raton, FL: CRC Press), 125–155.
Mol, J., Grotewold, E., and Koes, R. (1998). How genes paint flowers and seeds. Trends Plant Sci. 3, 212–217. doi: 10.1016/S1360-1385(98)01242-4
Moustafa, E., and Wong, E. (1967). Purification and properties of chalconeflavone isomerase from soya bean seed. Phytochemistry 6, 625–632. doi: 10.1016/S0031-9422(00)86001-X
Mulder, N., and Apweiler, R. (2007). InterPro and InterProScan: tools for protein sequence classification and comparison. Methods Mol. Biol. 396, 59–70. doi: 10.1007/978-1-59745-515-2_5
Nawrocki, E. P., Kolbe, D. L., and Eddy, S. R. (2009). Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337. doi: 10.1093/bioinformatics/btp157
Ohmiya, A., Kishimoto, S., Aida, R., Yoshioka, S., and Sumitomo, K. (2006). Carotenoid cleavage dioxygenase (CmCCD4a) contributes to white color formation in chrysanthemum petals. Plant Physiol. 142, 1193–1201. doi: 10.1104/pp.106.087130
Parra, G., Blanco, E., and Guigó, R. (2000). GeneId in Drosophila. Genome Res. 10, 511–515. doi: 10.1101/gr.10.4.511
Parra, G., Bradnam, K., and Korf, I. (2007). CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067. doi: 10.1093/bioinformatics/btm071
Piegu, B., Guyot, R., Picault, N., Roulin, A., Sanyal, A., Kim, H., et al. (2006). Doubling genome size without polyploidization: dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Res. 16, 1262–1269. doi: 10.1101/gr.5290206
Ruan, J., and Li, H. (2020). Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17, 155–158. doi: 10.1038/s41592-019-0669-3
Schiessl, S., Samans, B., Hüttel, B., Reinhard, R., and Snowdon, R. J. (2014). Capturing sequence variation among flowering-time regulatory gene homologs in the allopolyploid crop species Brassica napus. Front. Plant Sci. 5:404. doi: 10.3389/fpls.2014.00404
Schmidt, M. H., Vogel, A., Denton, A. K., Istace, B., Wormit, A., van de Geest, H., et al. (2017). De novo assembly of a new Solanum pennellii accession using nanopore sequencing. Plant Cell 29, 2336–2348. doi: 10.1105/tpc.17.00521
Schultz, J., Milpetz, F., Bork, P., and Ponting, C. P. (1998). SMART, a simple modular architecture research tool: identification of signaling domains. Proc. Natl. Acad. Sci. U.S.A. 95, 5857–5864. doi: 10.1073/pnas.95.11.5857
Schwinn, K., Venail, J., Shang, Y., Mackay, S., Alm, V., Butelli, E., et al. (2006). A small family of MYB-regulatory genes controls floral pigmentation intensity and patterning in the genus Antirrhinum. Plant Cell 18, 831–851. doi: 10.1105/tpc.105.039255
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Song, C., Liu, Y., Song, A., Dong, G., Zhao, H., Sun, W., et al. (2018). The Chrysanthemum nankingense genome provides insights into the evolution and diversification of chrysanthemum flowers and medicinal traits. Mol. Plant 11, 1482–1491. doi: 10.1016/j.molp.2018.10.003
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Stanke, M., and Morgenstern, B. (2005). AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33, W465–W467. doi: 10.1093/nar/gki458
Takken, F. L., Albrecht, M., and Tameling, W. I. (2006). Resistance proteins: molecular switches of plant defence. Curr. Opin. Plant Biol. 9, 383–390. doi: 10.1016/j.pbi.2006.05.009
Tang, H., Bowers, J. E., Wang, X., Ming, R., Alam, M., and Paterson, A. H. (2008). Synteny and collinearity in plant genomes. Science 320, 486–488. doi: 10.1126/science.1153917
Tarailo-Graovac, M., and Chen, N. (2009). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinf. 25, 4.10.1–4.10.14. doi: 10.1002/0471250953.bi0410s25
Trapnell, C., Pachter, L., and Salzberg, S. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. doi: 10.1093/bioinformatics/btp120
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. doi: 10.1038/nbt.1621
Vaser, R., Sovic, I., Nagarajan, N., and Sikic, M. (2017). Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746. doi: 10.1101/gr.214270.116
Walker, B., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Wang, K., Wang, Z., Li, F., Ye, W., Wang, J., Song, G., et al. (2012). The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 44, 1098–1103. doi: 10.1038/ng.2371
Wang, M., Tu, L., Yuan, D., Zhu, D., Shen, C., Li, J., et al. (2019). Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 51, 224–229. doi: 10.1038/s41588-018-0282-x
Wikström, N., Savolainen, V., and Chase, M. W. (2001). Evolution of the angiosperms: calibrating the family tree. Proc. Biol. Sci. 268, 2211–2220. doi: 10.1098/rspb.2001.1782
Xu, Z., and Wang, H. (2007). LTR-FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268. doi: 10.1093/nar/gkm286
Yang, T. J., Kim, J. S., Lim, K. B., Kwon, S. J., Kim, J. A., Jin, M., et al. (2005). The Korea Brassica genome project: a glimpse of the Brassica genome based on comparative genome analysis with Arabidopsis. Comp. Funct. Genom. 6, 138–146. doi: 10.1002/cfg.465
Yang, Z. (1997). PAML: a program package for phylogenetic analysis by maximum likelihood. Bioinformatics 13, 555–556. doi: 10.1093/bioinformatics/13.5.555
Yu, X. J., Zheng, H. K., Wang, J., Wang, W., and Su, B. (2007). Detecting lineage-specific adaptive evolution of brain-expressed genes in human using rhesus macaque as outgroup. Genomics 88, 745–751. doi: 10.1016/j.ygeno.2006.05.008
Zhang, B., Liu, C., Wang, Y., Yao, X., Wang, F., Wu, J., et al. (2015). Disruption of a CAROTENOID CLEAVAGE DIOXYGENASE 4 gene converts flower colour from white to yellow in Brassica species. New Phytol. 206, 1513–1526. doi: 10.1111/nph.13335
Zhang, L., Xu, Y., Zhang, X., Ma, X., Zhang, L., Liao, Z., et al. (2020). The genome of kenaf (Hibiscus cannabinus L.) provides insights into bast fiber and leaf shape biogenesis. Plant Biotechnol. J. 18, 1796–1809. doi: 10.1111/pbi.13341
Keywords: Hibiscus mutabilis, genome, Hi-C, phylogenetic affiliation, floral regulators
Citation: Yang Y, Liu X, Shi X, Ma J, Zeng X, Zhu Z, Li F, Zhou M, Guo X and Liu X (2022) A High-Quality, Chromosome-Level Genome Provides Insights Into Determinate Flowering Time and Color of Cotton Rose (Hibiscus mutabilis). Front. Plant Sci. 13:818206. doi: 10.3389/fpls.2022.818206
Received: 19 November 2021; Accepted: 20 January 2022;
Published: 14 February 2022.
Edited by:
Surya Saha, Boyce Thompson Institute, United StatesReviewed by:
Kun_Li Xiang, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences (CAAS), ChinaCopyright © 2022 Yang, Liu, Shi, Ma, Zeng, Zhu, Li, Zhou, Guo and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoli Liu, NjczOTExMzkwQHFxLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.