 Ambika Baldev Gaikwad1*
Ambika Baldev Gaikwad1* Tanvi Kaila2
Tanvi Kaila2 Avantika Maurya1
Avantika Maurya1 Ratna Kumari1
Ratna Kumari1 Parimalan Rangan1
Parimalan Rangan1 Dhammaprakash Pandhari Wankhede1
Dhammaprakash Pandhari Wankhede1 K. V. Bhat1
K. V. Bhat1- 1Division of Genomic Resources, Indian Council of Agricultural Research (ICAR)-National Bureau of Plant Genetic Resources, New Delhi, India
- 2Indian Council of Agricultural Research (ICAR)-National Institute for Plant Biotechnology, New Delhi, India
Piper nigrum, also known as black pepper, is an economically and ecologically important crop of the genus Piper. It has been titled as the king of spices due to its wide consumption throughout the world. In the present investigation, the chloroplast genome of P. nigrum has been assembled from a whole genome sequence by integrating the short and long reads generated through Illumina and PacBio platforms, respectively. The chloroplast genome was observed to be 161,522 bp in size, having a quadripartite structure with a large single copy (LSC) region of 89,153 bp and a small single copy (SSC) region of 18,255 bp separated by a copy of inverted repeats (IRs), each 27,057 bp in length. Taking into consideration all the duplicated genes, a total of 131 genes were observed, which included 81 protein-coding genes, 37 tRNAs, 4 rRNAs, and 1 pseudogene. Individually, the LSC region consisted of 83 genes, the SSC region had 13 genes, and 18 genes were present in each IR region. Additionally, 216 SSRs were detected and 11 of these were validated through amplification in 12 species of Piper. The features of the chloroplast genome have been compared with those of the genus Piper. Our results provide useful insights into evolutionary and molecular studies of black pepper which will contribute to its further genetic improvement and breeding.
Introduction
Chloroplasts are essential intracellular organelles having an independent genome with several genes responsible for the process of photosynthesis. Most higher plants harbor a single, circular, double-stranded chloroplast DNA with a quadripartite structure that includes a long single copy (LSC) region, a small single copy (SSC) region, and two copies of large inverted repeat (IRa and IRb) regions (Bock, 2007). Chloroplast genomes are highly conserved in terms of their size, gene content, and structure with a few exceptions (Dong et al., 2013; Daniell et al., 2016). According to the Organelle Genome Resource-NCBI (www.ncbi.nlm.nih.gov/genome/browse#!/organelles/), 4,557 chloroplast genomes of land plants are reported until December 2020.
Magnoliidae is one of the largest and early-diverging Angiosperm clade representing four orders—Canellales, Piperales, Laurales, and Magnoliales—that comprise around 10,000 species in 20 families (Massoni et al., 2015; Chase et al., 2016; Sinn et al., 2018). So far, 384 chloroplast genomes belonging to Magnoliidae have been submitted to NCBI-Organelle Genome Resource of which 48 belong to Piperales and only 15 to the genus Piper. Piperaceae is considered to be the most diverse group of magnoliids with around more than 4,300 species. The genus Piper possesses more than 2,000 species (Divya et al., 2015) including the economically important crop black pepper (Piper nigrum).
Black pepper has been titled as the “king of spices” due to its wide consumption throughout the world for inducing a pungent flavor in culinary dishes. The Western Ghats region of India is known to be the center of origin and diversity for this species with around 30 countries growing it commercially, mainly India, Indonesia, Malaysia, China, Thailand, and Brazil (Kubitzki and Rohwer, 1993; Ravindran, 2000). The medicinal value of secondary metabolites produced by P. nigrum reveals its role in digestive disorders, hepatoprotection, antithyroidism, anti-inflammation, antidepressant, immunomodulator, antispasmodic, antioxidant, and many more (Ahmad et al., 2012).
In the present investigation, we report on the complete sequence of the P. nigrum chloroplast genome. Detailed characterization of the black pepper chloroplast including size, gene content, structure repeats, and GC content and comparative genomics analysis of the chloroplast genome provide insights into functional genomics (Li et al., 2019). Moreover, phylogenetic analysis of 58 genes and 37 taxa consisting of 34 angiosperms and 3 gymnosperms provides insight into the phylogenetic position of P. nigrum in accordance with other magnoliids, monocots, and eudicots.
Material and methods
DNA extraction and next-generation sequencing
Leaves of black pepper landrace ‘Thottumuriyan’ were used to extract high-quality genomic DNA that was assessed for its quality and quantity using Qubit. Two paired-end libraries were prepared from genomic DNA using TruSeq DNA PCR-Free LT sample Prep Kit (Illumina, San Diego, USA) as per standard protocol and quantified using Bioanalyzer (Agilent Technologies, USA) and qPCR (Kapa Biosystems, Wilmington, MA, USA) sequenced on HiSeq1000 and MiSeq (Illumina, San Diego, CA, USA) using 2 × 100 and 2 × 150 bases paired-end chemistry, respectively. Pacific Biosciences (PacBio) SMRT bell sequencing libraries were prepared for long read generation and were sequenced on PacBio Sequel. A total of 82.74- and 14.65-GB data were generated through Illumina sequencing and PacBio, respectively.
Chloroplast genome assembly and annotation
The FastQC software (Andrews, 2010) was used to check the quality of the sequenced reads, and then the Trimmomatic software (Bolger et al., 2014) was used to trim and filter the low-quality reads. Filtered data were mapped to the reference chloroplast genome using CLC Genomics workbench 9.0. Mapped reads of PacBio sequence data were extracted and assembled using the Organelle PBA software (Soorni et al., 2017) which was polished through the Pilon software (Walker et al., 2014) using Illumina sequence data.
For the preliminary annotation of the chloroplast genome, the D online program GeSeq (Tillich et al., 2017) offered by MPI-MP Chlorobox was used and the output was curated. The tRNA genes were identified by the ARAGORN v1.2.38 tRNA annotator (Laslett and Canback, 2004), which was available at the GeSeq server. The OGDraw (Greiner et al., 2019) online server of Chlorobox was used to construct a gene map of the chloroplast genome with default settings and checked manually. The black pepper chloroplast genome sequence was assembled to 161,522 bp and submitted to NCBI, GenBank accession No. MK883818 (Supplementary File 1).
Repeat structure and comparative genome analysis
Codon usage and GC content were analyzed using CodonW and Molecular Evolutionary Genetics Analysis (MEGA 7.0) (Kumar et al., 2016), respectively. To investigate the size and location of repeat sequences comprising forward, reverse, palindromic, and complement repeats, REPuter online server was used with a minimal size of 30 bp, >90% identity, and hamming distance of 3 between two repeat copies. Also, Tandem Repeats Finder (Benson, 1999) was used to extract the tandem repeats present in the chloroplast genome of P. nigrum and the other reported species of Piper. Simple sequence repeats (SSRs) were detected using the MISA software (Beier et al., 2017) with parameters of eight, four, four, three, three, and three repeat units for mono-, di-, tri-, tetra-, penta-, and hexanucleotide SSRs, respectively. mVISTA (Mayor et al., 2000) was used to perform whole genome sequence alignment of P. nigrum along with other members of Piperaceae comprising Piper kadsura, Piper laetispicum, Piper cenocladum, and Piper auritum.
Validation of SSRs
A set of 11 primer pairs were synthesized using the software Primer3 (Untergasser et al., 2012) and validated in 21 accessions of Piper representing 12 different species (Piper nigrum, Piper longum, Piper arboreum, Piper argyrophyllum, Piper attenuatum, Piper betel, Piper chaba, Piper hymenophyllum, Piper trichostachyon, Piper wallichi, Piper columbrinum, and Piper sarmentosum). Genomic DNA was isolated from the leaf using the cTAB DNA extraction method. The PCR reaction consisted of 1× PCR buffer, 2.5 mM of MgCl2, 1 µM of primer, 0.2 mM of each dNTP, 1 U of Taq DNA polymerase (NEB), and 15 ng of template DNA in a total volume of 20 µl and cycled at 95°C for 5 min followed by 35 cycles of denaturation at 95°C for 1 min, annealing at 55°C for 1 min and extension at 72°C for 1 min followed by a final extension at 72°C for 10 min. The amplification products were resolved on a QIAxcel multicapillary system using QIAxcel High Resolution Kit 1200 (QIAGEN, No. 929002, New Delhi,Qiagen India Pvt. Ltd.) 50-800 bp v2.0 QX DNA size marker (QIAGEN, No. 929561, New Delhi,Qiagen India Pvt. Ltd.) and 15 bp/1,000 bp Qx alignment marker (QIAGEN No. 929521, New Delhi,Qiagen India Pvt. Ltd.) with the high-resolution run method OM700. The allelic sizes of each sample were calculated in the form of gel profiles and peaks using the QIAxcel ScreenGel software (QIAGEN, v1.5).
Synonymous substitution
The numbers of synonymous substitutions per synonymous sites (Ks) and non-synonymous substitutions per non-synonymous sites (Ka) and functional polymorphism within magnoliids were calculated using the DnaSP software (Rozas et al., 2017).
Phylogenetic analysis
Phylogenetic analysis was performed with 58 genes common to all 37 complete chloroplast genomes downloaded from NCBI (Supplementary File 2). The 37 complete chloroplast genomes used in the study are those of species representing 31 orders of the phylum Tracheophyta. These include 3 orders from the gymnosperms; those of Amborella, Austrobaileyales, and Chloranthales; 4 orders of the magnoliids; the Alismatid, Lilioid, and Commelinid monocots; and 17 orders of the eudicots. Firstly, each gene from all 37 taxa was aligned individually using ClustalW 2.0 (Thompson et al., 1994). On an online server, gBlocks was used to create conserved blocks with default settings, eliminating the poorly aligned regions. Maximum likelihood analysis was performed with PhyML using the subtree pruning and regrafting (SPR) topological moves with Smart Model Selection (SMS) of the substitution model based on Akaike information criterion (AIC) and 100 bootstrapping.
Results and discussion
Features of the Piper nigrum chloroplast genome
With the increased use of next-generation sequencing, the number of reported angiosperm chloroplast genomes has increased. Nonetheless, only 15 chloroplast genomes belonging to the genus Piper have been sequenced. Recently, the complete chloroplast genome sequences of Piper hancei (Zhang et al., 2021) and P. sarmentosum (Geng et al., 2022) have been reported. A comparative analysis of eight Piper species with P. nigrum has been also reported (Li et al., 2022). In the present investigation, the chloroplast genome of P. nigrum was sequenced and analyzed. The complete assembled chloroplast genome of P. nigrum is a circular model with the length of 161,522 bp, having a typical quadripartite structure comprising an LSC region of 89,153 bp and an SSC region of 18,255 bp separated by a copy of inverted repeats (IRs) each 27,057 bp in length (Figure 1). Piper nigrum differs slightly from other reported Piper species in terms of the total length of the chloroplast genome. It is smaller than P. laetispicum (161,721 bp), but larger than P. kadsura (161,486 bp), P. cenocladum (160,624 bp), P. auritum (160,624 bp), and P. hancei (161,476 bp) in terms of length.
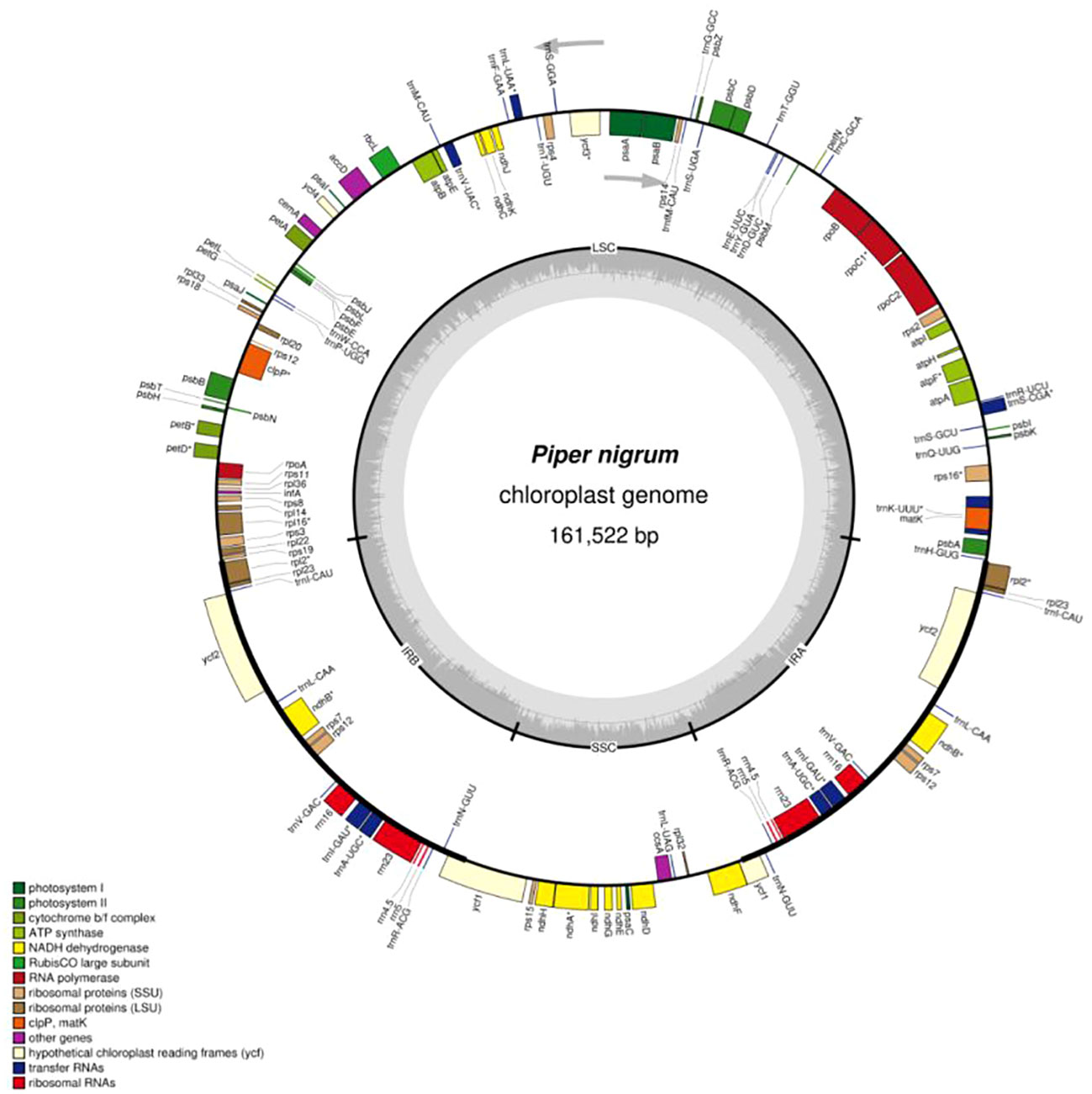
Figure 1 Circular map of the Piper nigrum chloroplast genome.
The P. nigrum chloroplast genome was observed to have 131 genes, which include 81 protein-coding genes, 37 tRNAs, 4 rRNAs, and 1 pseudogene (Table 1). On considering each region individually, it was observed that the LSC consists of 83 genes, the SSC consists of 13 genes, and 18 genes were present in the IR region. The duplication of tRNA genes in the LSC region has been observed in various angiosperms (Tsudzuki et al., 1994; Kaila et al., 2016; Kaila et al., 2017). However, as with other reported Piperaceae chloroplast genomes (Cai et al., 2006), the tRNA gene was not observed to be duplicated in P. nigrum. The gene rps12 was observed to be trans-spliced with one end present in the LSC region and another duplicated end present in the IR region. Trans-splicing of the rps12 gene has also been reported in other chloroplast genomes like Pinus taeda, P. kadsura, and Fagus crenata (Lee et al., 2016; Asaf et al., 2018; Worth et al., 2019). In consonance with the previous studies, the trnK-UUU gene was observed to be harboring the largest intron (2,548 bp) which includes the matK gene.
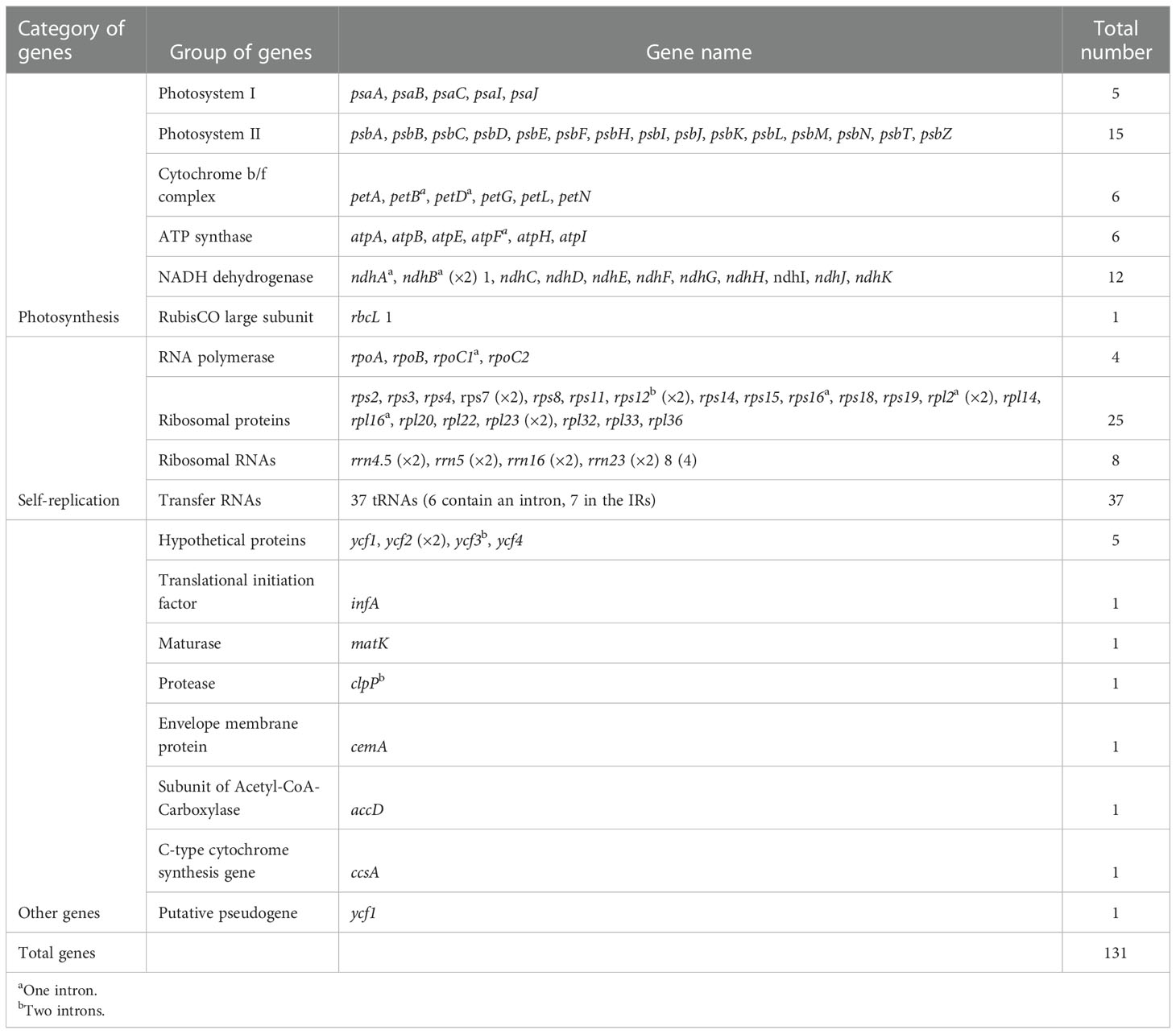
Table 1 List of genes in the chloroplast genome of Piper nigrum.
Pseudogenes are similar to functional genes except that they do not produce functional proteins. One reason for this may be a disrupted open reading frame due to the presence of an internal stop codon(s). In P. nigrum, the ycf1 gene was identified to be the only pseudogene, similar to P. kadsura (Lee et al., 2016) and P. laetispicum (Wang et al., 2018), while Cinnamomum camphora and Persea americana are reported to have three (rpl23, ycf1, and ycf2) and two (ycf1 and ycf2) pseudogenes, respectively (Li et al., 2019; Liu et al., 2019).
The average AT content of the P. nigrum chloroplast genome was observed to be 61.7% which is similar to other reported Piper species (Cai et al., 2006; Lee et al., 2016; Wang et al., 2018; Li et al., 2022). The single copy regions LSC (63.2%) and SSC (67.9%) possess higher AT content than the repeat regions (57.1%) (Table 2). The lower AT content of the IR region can be associated with the presence of rRNA genes, which possesses a decreased number of AT nucleotides, thereby contributing to genome stabilization and sequence complexity. Also, the AT content for the CDS region (protein-coding region) was observed to be 60.9% (Table 2). Coding regions have higher GC content and therefore lower AT content than non-coding regions. While if we consider the different regions of the chloroplast genome, the IR regions have the lowest AT content and the SSC regions have the highest AT content. This is due to the presence of NADH genes present in the SSC region that are reported to have the lowest GC content as compared with any class of genes (Cai et al., 2006).
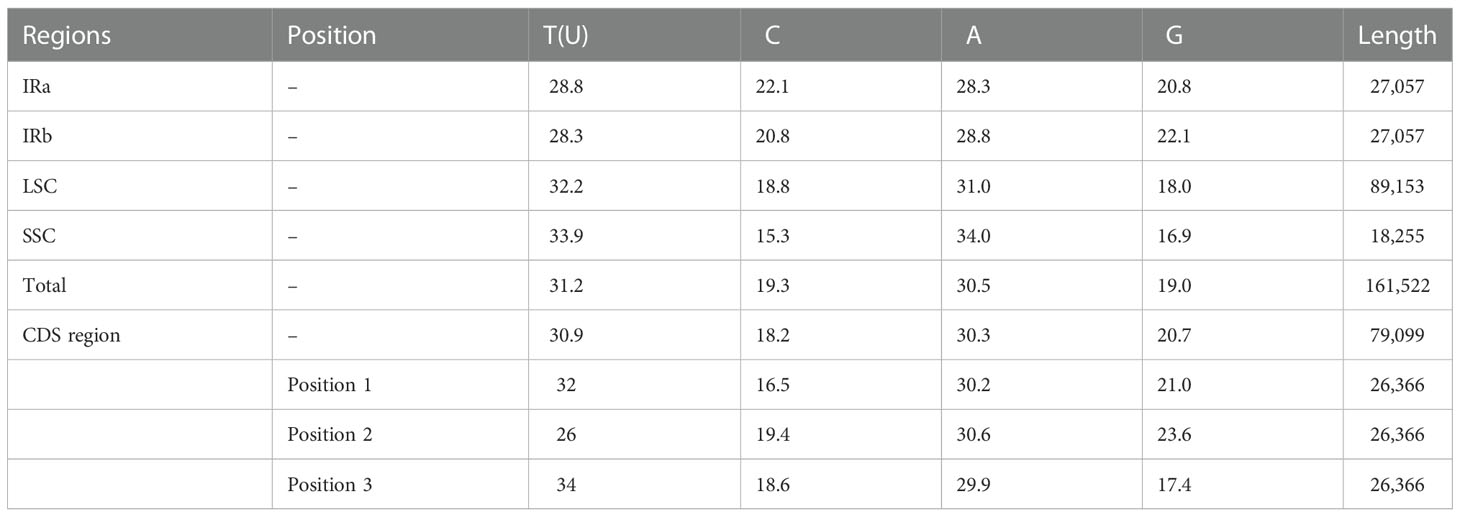
Table 2 Nucleotide composition in different regions of the chloroplast genome of Piper nigrum.
The chloroplast genome of P. nigrum consists of 18 intron-containing genes, viz., ycf3, rpoC1, atpF, rps16, rpl2, ndhB, ndhA, rpl16, petB, petD, clpP, rps12, and 6 tRNA genes (Table 3). Three genes, viz., ycf3, rps12, and clpP, consist of two introns, while the rest have one intron. Similar observations have been made for other plants belonging to the Magnoliid clade like P. cenocladum and Drimys granadensis (Cai et al., 2006) which harbor 18 intron-containing genes with three genes spanning 2 introns. The recently reported chloroplast genome of P. hancei also harbors 18 intron-containing genes, 16 of which contain one intron and 2 genes (clpP and ycf3) possess two introns (Zhang et al., 2021). In contrast, a recent analysis of eight Piper chloroplast genomes reveal 14 intron-containing genes of which 10 protein-coding genes and 2 tRNA genes had a single intron and 2 genes had two introns (Li et al., 2022). Different from these results, the chloroplast genome of P. sarmentosum was reported to have 21 intron-containing genes, consisting of 8 tRNA genes and 13 protein-coding genes of which ycf3 and clpP possess two introns each and the others contain only one intron (Geng et al., 2022). Similarly, C. camphora (Li et al., 2019) which also belongs to the magnoliids consists of 17 intron-containing genes which include 3 genes having two introns. In the case of the P. nigrum chloroplast genome, the protein-coding region accounts for 49.6% of the genome, while the tRNA and rRNA regions comprise 1.7% and 5.5% of the whole genome, respectively (Table 4). Therefore, the remaining genome consists of introns, intergenic regions, and pseudogenes. Within the Piperaceae, no change is observed in the number of protein-coding genes, rRNA, and GC percentage. Except for P. auritum which is reported to have 36 tRNAs and no pseudogene (NC_034697, unpublished), P. cenocladum (Cai et al., 2006) that contains no pseudogene, and P. hancei (Zhang et al., 2021) that consists of 36 tRNAs, the rest of the Piper species contain 37 tRNAs and 1 pseudogene (ycf1), respectively.
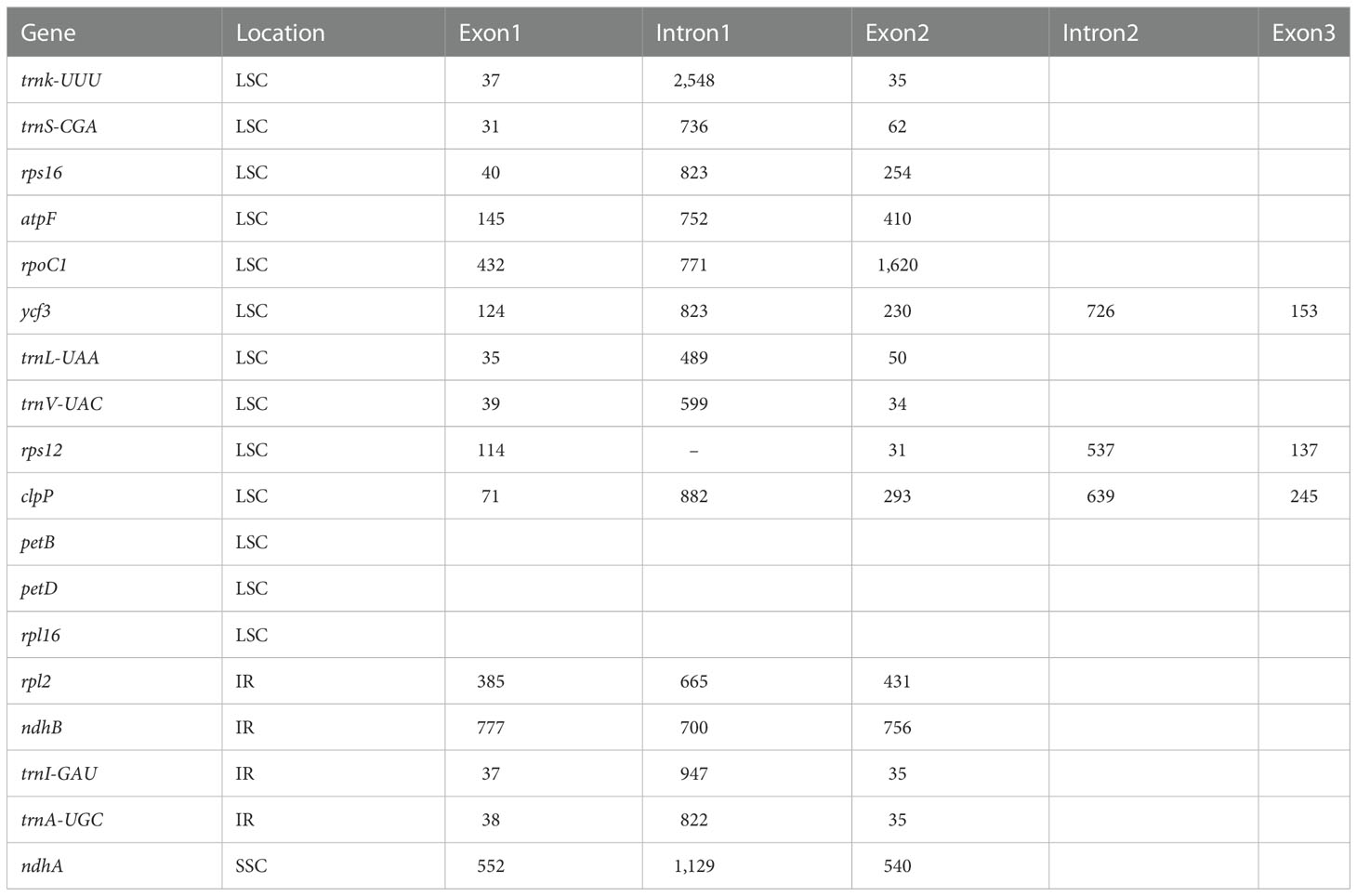
Table 3 Length of exons and introns in chloroplast genes of Piper nigrum.

Table 4 Gene content comparison of Piper nigrum chloroplast with reported Piper species and other magnoliids.
Considering the other members of the Magnoliid clade, Persea was found to have 81 protein-coding genes, whereas Cinnamomum and Calycanthus of Laurales (Chen et al., 2018) and Liriodendron of Magnoliales (Park et al., 2019) consist of 84 protein-coding genes. Cinnamomum and Persea comprise 36 tRNA, whereas Calycanthus and Liriodendron have 37 tRNAs. Drimys of the order Canellales consists of 85 protein-coding genes and 44 tRNAs which is the maximum number of tRNAs present within magnoliids (Table 4). In comparison to other magnoliids, ycf1 is present as a pseudogene at the SSC/IRa junction in Piper species, whereas in Drimys, Calycanthus, and Liriodendron, the 3' fragment of ycf1 is present at the IRb/SSC junction and a complete copy of ycf1 is present at the SSC/IRa junction.
Codon usage
Codon usage for the P. nigrum chloroplast genome was calculated with the CodonW software. It was observed that 61 codons encoded for 20 amino acids and 3 stop codons. A total of 26,366 codons represented 85 protein-coding genes, 79,099 nucleotides in length. Among the amino acids, serine was the most (2,687 codons) and tryptophan was the least (550 codons) encoded amino acids (Figure 2). Codon usage bias is a phenomenon in which certain codons are preferred repeatedly over the others. This usage varies for different genomes and genes. Biasness was also observed in the presentation of a nucleotide at the third codon position. The AT content varies according to codon position with the highest at the third codon position, followed by the first and second codon positions (Morton, 2003). The above observation is also supported by relative synonymous codon usage (RSCU) values. It was found that 40.7% of codons ended with A and T, whereas 23.4% of codons ended with C and G. It is known that organellar proteins are primarily encoded by codons having A or U at the third codon position. Similar results have been reported for various plastid genomes in the past (Cai et al., 2006; Asaf et al., 2018).

Figure 2 Codon usage of the CDS region of the chloroplast genome of Piper nigrum.
Gene content
The chloroplast genomes have evolved over a period of time. On comparing the algal and embryophyte plastomes, it is shown that a number of genes (tufA, ftsH, odpB, and rpl5) have been lost or transferred to the nucleus and a few genes, namely, matK, ycf1, and ycf2, have been gained by the abovementioned plastomes (Turmel et al., 2006). There also exist differences in the gene content among the angiosperms as genes like infA, accD, and rpl22 have been reported to be lost in legumes (Doyle et al., 1995), and on the other hand, accD, ycf1, and ycf2 have been lost in Poaceae (Guisinger et al., 2010). Complete loss or pseudogenization of ndh genes has been shown in heterotrophic plants in the past. Nonetheless, events showing ndh gene loss have also been observed in Pinaceae, gnetophytes, and autotrophic orchids (Wakasugi et al., 1994; Braukmann et al., 2009; Kim et al., 2015).
Among the Mesangiospermae, magnoliids are the third largest group after the eudicots and monocots. Except for a few differences in gene content, the magnoliids are identical to tobacco and many unarranged angiosperms in terms of gene order. The ACRS gene, which has been reported in a number of plastid genomes as the ycf68 gene, was identified in Calycanthus based on its similarity to a mitochondrial ACR sensitivity toxin gene of Citrus jambhiri (Ohtani et al., 2002). Due to the presence of internal stop codons, it has been reported as a pseudogene in a number of plastomes (Curci et al., 2015; Yan et al., 2015). However, it was not annotated in the P. nigrum chloroplast genome and hence observed to be absent from the plastid genome. Similar observations were made for D. granatenis and P. cenocladum (Cai et al., 2006), belonging to the magnoliid clade. Likewise, another gene, namely, ycf15, was not annotated in the P. nigrum plastid genome and hence was found to be absent. The ycf15 has also been reported to be a non-functional protein-coding gene (Shi et al., 2013) and hence a pseudogene due to the presence of internal stop codons (Yan et al., 2015; Kaila et al., 2017). It is found to be present in the Calycanthus plastome but absent in the D. granatenis and P. cenocladum plastid genomes (Cai et al., 2006). If we consider the phenomenon of nuclear substitution, then one such gene which is well-studied is rps16. The rps16 gene has been found to be present as a pseudogene in some plastid genomes (Roy et al., 2010; Kaila et al., 2016) or has been completely lost from the plastid genome due to non-functional protein sequence or incorrect splicing of intron due to mutation at the 5' or 3' splicing site. Like in certain legumes, the loss of rps16 from the chloroplast genome and its replacement by nuclear copy have been reported (Ueda et al., 2008). However, in P. nigrum, its copy has been retained in the chloroplast genome similar also to P. cenocladum, P. laetispicum, and P. auritum. As mentioned above, the chloroplast genome of Piper is similar to tobacco, and it has been shown that rps16 is indispensable for tobacco plastome function (Fleischmann et al., 2011). A better regulation of plastome rps16 gene in certain conditions or the non-presence of chloroplast-targeting sequence in nuclear-encoded peptide has been proposed as the reasons behind the retention of plastid rps16 gene in some plants (Keller et al., 2017). Some differences in gene content may also lead to the expansion of the IR region, which is a common phenomenon occurring in the plastid genome. One such example is duplication and inclusion of the trnH gene in the IR region of the D. granatenis chloroplast genome, hence leading to the expansion of the IR region (Cai et al., 2006). Such duplication of the trnH gene has not been observed in any other plant belonging to magnoliids or basal angiosperms. Similarly, such duplication was not observed in the P. nigrum plastid genome.
Repeat structures
Repeat structure analysis through the REPuter software (Kurtz et al., 2001) revealed that P. nigrum comprises 23 palindromic, 18 forward, 2 complement, and 1 reverse repeat along with 55 tandem repeats that were predicted using the Tandem Repeats Finder. The length of the largest forward and palindromic repeat was 52 bp each. Among the tandem repeats, 64% of the repeats were present in the intergenic region, 28% in the coding region, and 8% in the intronic region. Palindromic repeats were observed to be present in majority within all the reported species of Piper (Figure 3). Reverse and complement repeats were absent in P. laetispicum, whereas no complement repeat was present in P. auritum. Piper auritum had a minimum number of tandem repeats (Wang et al., 2018; NC_034697). Likewise, palindromic and forward repeats were the most abundant repeat types in other plant types like P. americana (Liu et al., 2019) and Aristolochia contorta (Zhou et al., 2017). These repeats have been reported to be responsible for chloroplast genome rearrangement, gene duplication, and gene expression (Do et al., 2014; Vieira et al., 2014). It is due to their role in genome rearrangement that they have proved to be helpful in phylogenetic studies (Cavalier-Smith, 2002). Improper recombination and slipped strand mispairing of these repeat units result in plastome rearrangement and sequence variation among different plastid genomes. These repeats are also responsible for substitutions and indels in the chloroplast genome (Yi et al., 2013). They have also proved to be an informational source for developing markers and therefore play an important role in population and phylogenetic studies (Nie et al., 2012).
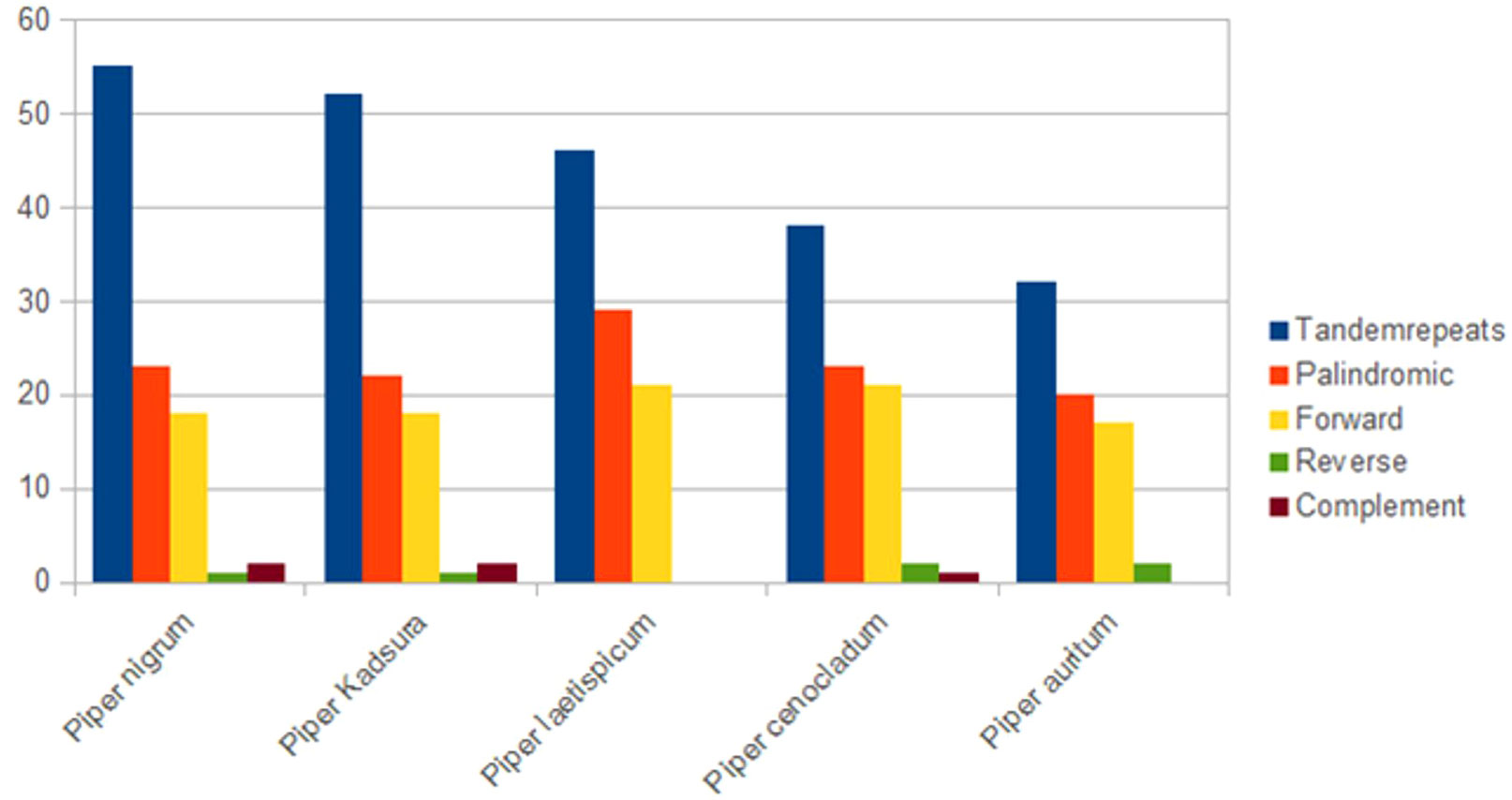
Figure 3 Repeat sequences in five Piper species plastid genomes calculated using REPuter and Tandem Repeats Finder.
Simple sequence repeats
A group of tandem repeats comprising DNA sequences of 1-6 nucleotides long are called microsatellites or SSRs. They are present in large numbers in both coding and non-coding regions. They are highly reproducible, abundant in number, polymorphic, and uniparentally inherited. Variation in SSRs at a specific locus contributes to its use as a molecular marker (Olmstead and Palmer, 1994). Therefore, they are used in evolutionary studies, phylogenetic relationships, and plant population genetics. A total of 216 SSRs were found using the MISA perl script [http://pgrc.ipk-gatersleben.de/misa/] (Beier et al., 2017). The number of SSRs extracted was similar to those reported in other Piper species like P. kadsura (215 SSRs), P. laetispicum (216 SSRs), P. cenocladum (198 SSRs), and P. auritum (189 SSRs). Among the abovementioned 216 SSRs, 65% were present in the LSC region, 13% in the SSC region, and 11% in the IR regions. It was also observed that 32% of the SSRs were present in the coding region and 13% in the non-coding region, and the maximum SSRs (53%) were present in the intergenic region. Thus, the maximum number of SSRs is present in the non-coding region than in the coding region. Similar observations have also been made for P. taeda (Asaf et al., 2018) and Cyamopsis tetragonoloba (Kaila et al., 2017). If we look individually at the coding region, then it is observed that the ycf1 gene had a maximum number of repeats. Previous studies have also reported the ycf1 gene as the most variable locus, and similar observations have been made for Glycine species, Cynara cardunculus, and Camellia species (Huang et al., 2014; Curci et al., 2015; Ozyigit et al., 2015). Among the 216 SSRs found, the most abundant were mononucleotide repeats (147), followed by dinucleotide (49), trinucleotide (6), tetranucleotide (9), pentanucleotide (3), and hexanucleotide repeats (2). On comparison, it was found that all the repeat types were present in different Piper species except pentanucleotides and hexanucleotides, which were absent in P. cenocladum and P. auritum (Figure 4). In P. nigrum, the A/T repeats were found to be in majority with 138 in number out of 147 mononucleotide repeats present. The AT/AT was the most common among the dinucleotide repeats and five out of six trinucleotide repeats were AAT/ATT (Figure 5). Such A/T richness of mononucleotides has been observed earlier by Wheeler et al. (2014). This biasness in base composition and the presence of polyadenine (A) or polythymine (T) rich repeats have been reported earlier in species like C. tetragonoloba (Kaila et al., 2017), P. taeda (Asaf et al., 2018), P. bournei (Li et al., 2017), and Aristolochia debilis (Zhou et al., 2017).
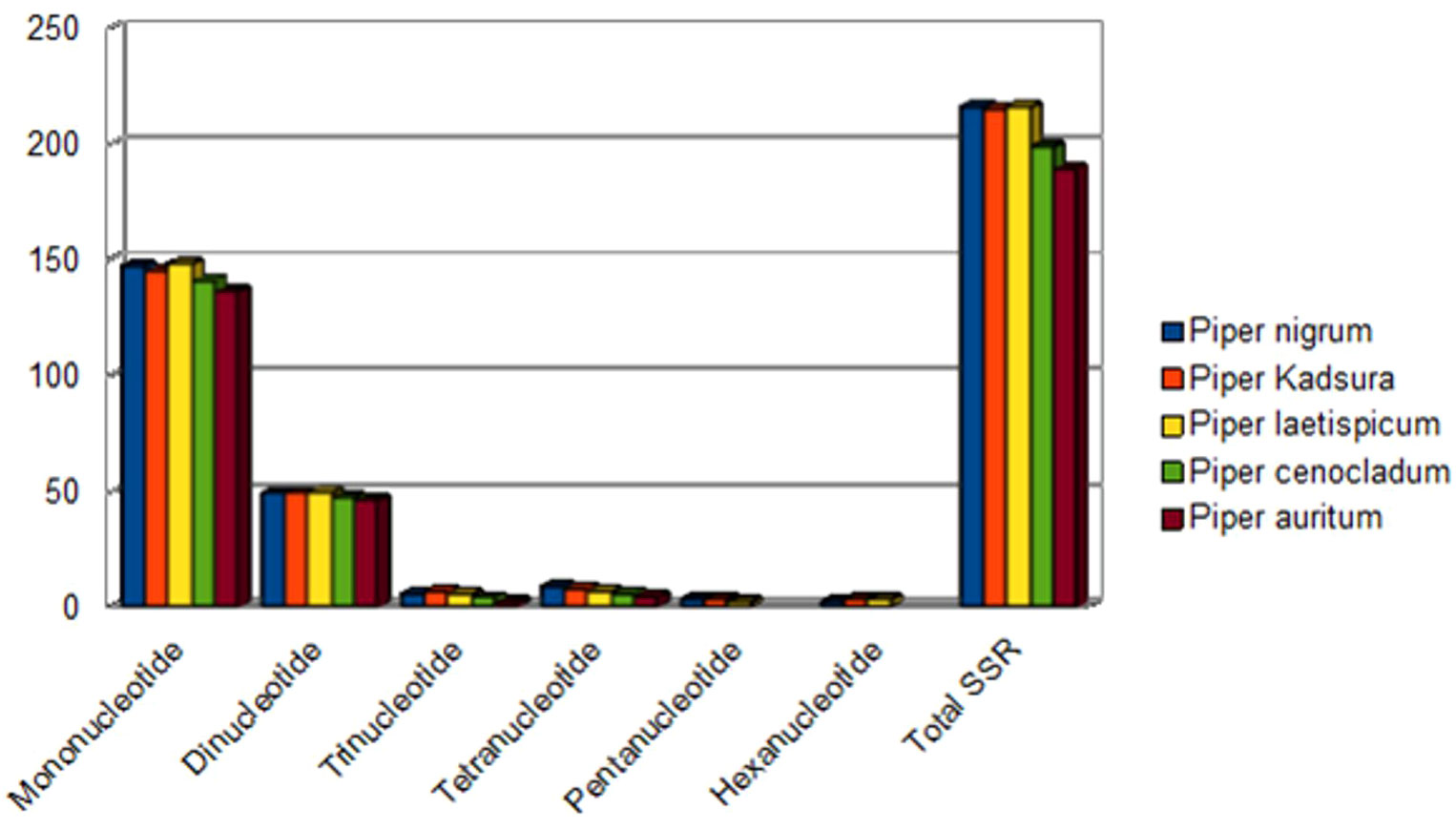
Figure 4 Simple sequence repeats (SSRs) in plastid genomes of five Piper species.
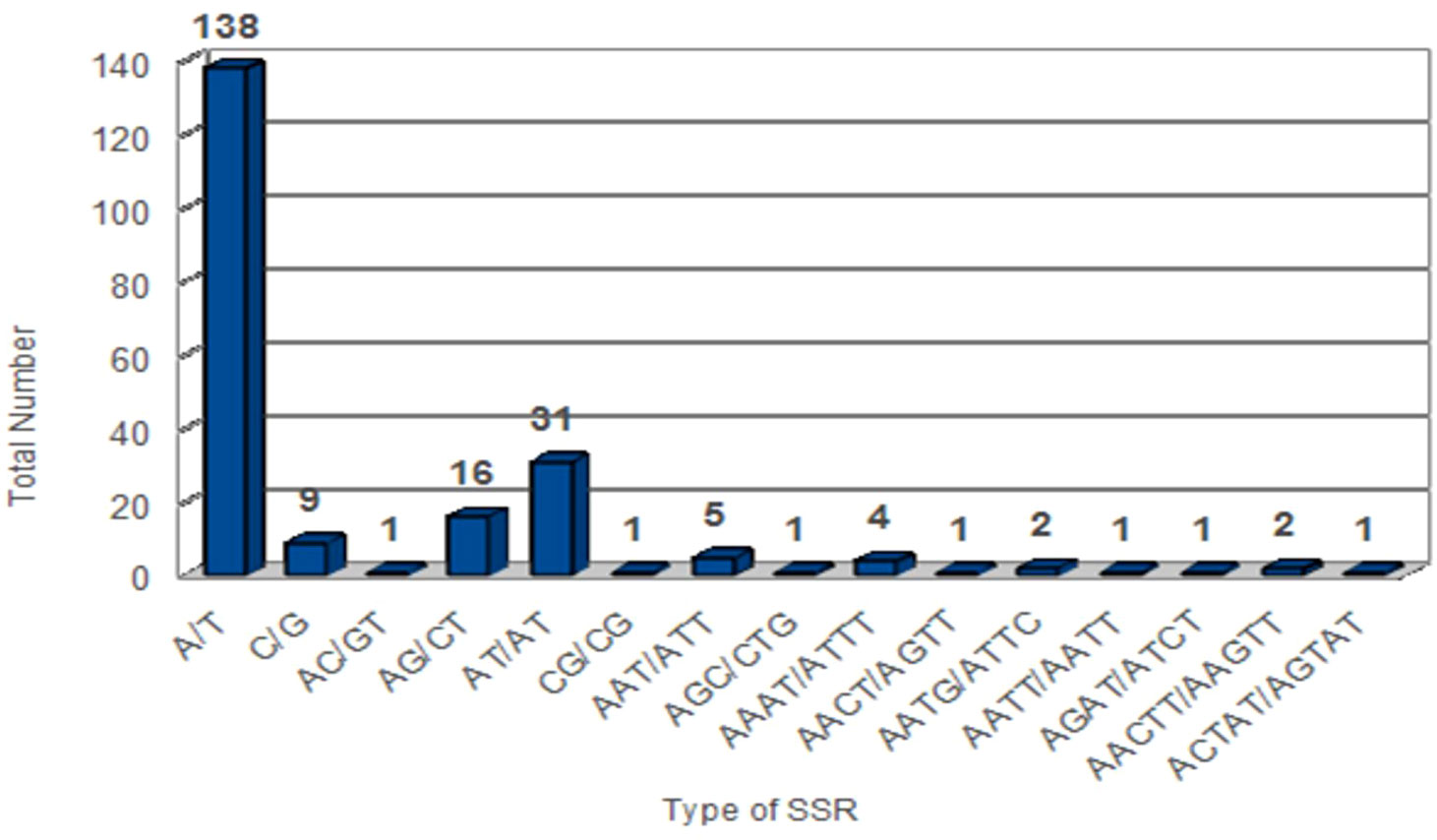
Figure 5 Distribution of SSRs based on their repeat type in Piper nigrum..
Wet lab validation at 11 SSR loci across 12 Piper species (including P. nigrum) resulted in the amplification of 60 alleles (Figure 6). The 11 loci included di-, tri-, tetra-, and pentanucleotide repeat core motifs (Table 5). The number of alleles detected ranged from 4 to 11, with the maximum number of alleles detected at the locus at BPC8 with a pentanucleotide core repeat. Amplification at this locus indicates the presence of pentanucleotide repeats in P. longum, P. arboreuum, P. argyrophyllum, P. attenuatum, P. betel, P. chaba, P. trichostachyon, P. sarmentosum, P. columbrinum, P. wallichi, and P. hymenophyllum. The high degree of polymorphism detected across the 12 Piper species allows for the identification of hypervariable regions informative for DNA barcoding and is a source for further evolutionary and phylogenetic studies in Piper species.
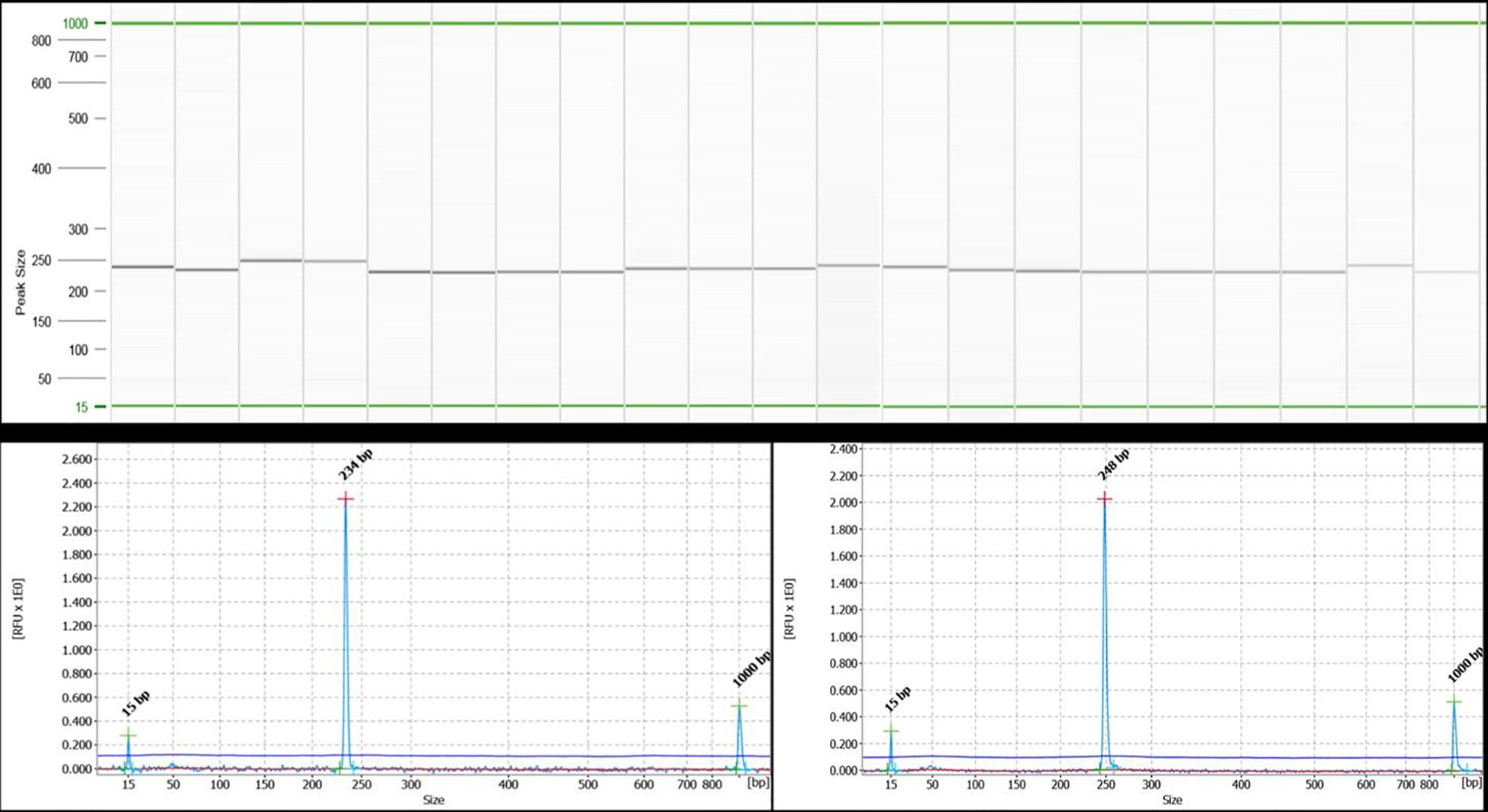
Figure 6 QIAxcel gel image of PCR amplification using the black pepper chloroplast SSR marker BPC5 in 21 accessions of 12 different species of black pepper as captured on the QIAxcel ScreenGel software. Lanes 1 and 2: Piper longum; lanes 3 and 4: Piper arboreum; lanes 5 and 6: Piper argyrophylum; lane 7: Piper attenuatum; lanes 8–10: Piper betel; lanes 11 and 12: Piper chaba; lane 13: Piper hymenophyllum; lanes 14 and 15: Piper trichostachyon; lane 16: Piper wallichi; lanes 17–19: Piper nigrum; lane 20: Piper columbrinum; lane 21: Piper sarmentosum. The lane marked as “M” is DNA molecular weight standard 50-800 bp v2.0 Qx DNA size marker. A representative electropherogram showing allele size of 234 bp for lane 2 and 248 bp for lane 3 has been shown below the gel image.
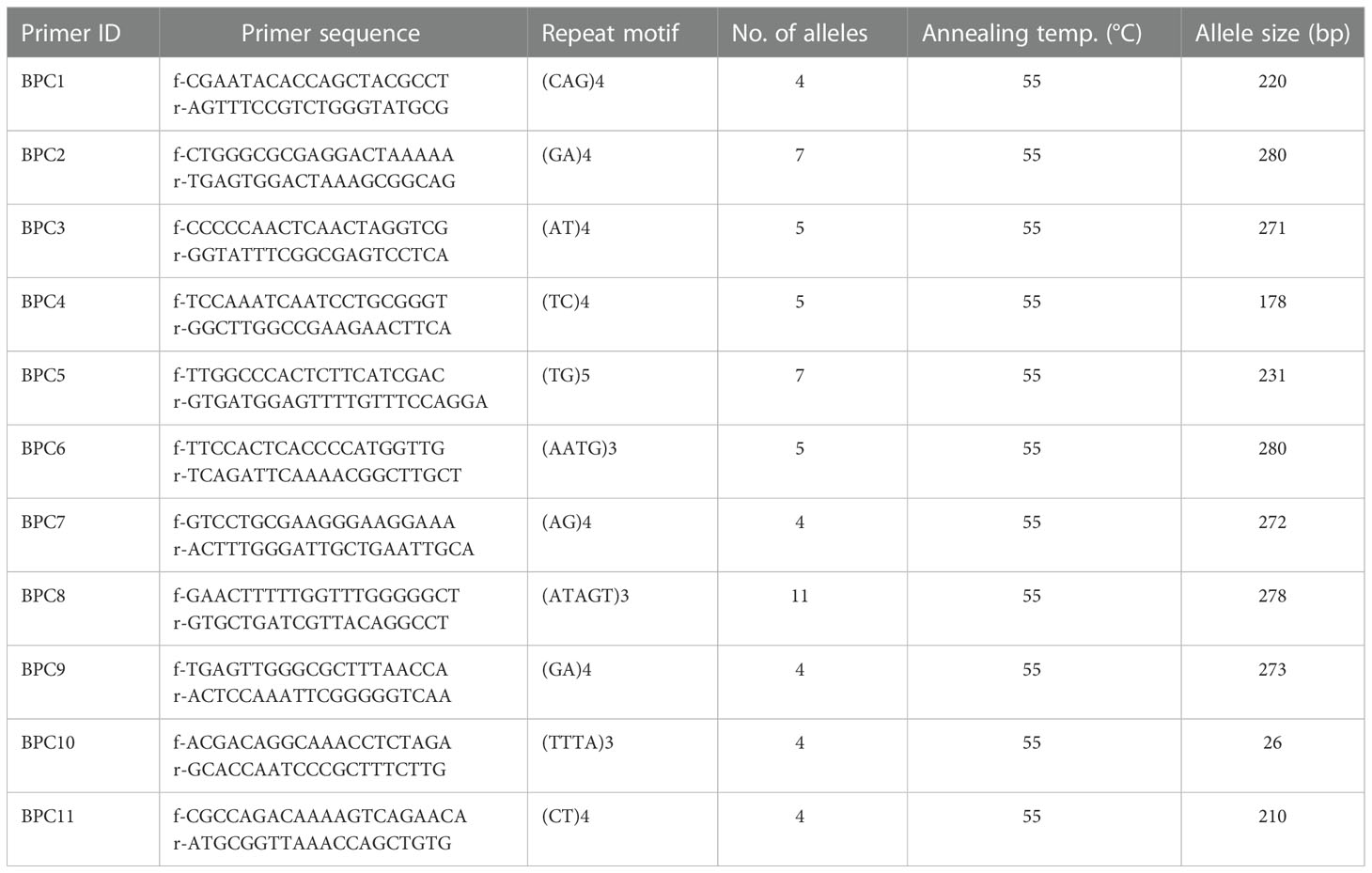
Table 5 Details of 11 chloroplast SSR primers validated in 12 Piper species (21 accessions).
Comparison of IR boundaries
The IR regions provide stability to the chloroplast genome by exhibiting intramolecular recombination between the two IR copies (Palmer et al., 1987; Palmer, 1991). The expansion and contraction of IRs lead to the conversion of single-copy genes to duplicate copies and vice versa, respectively, thereby leading to variation in the plastome size among the angiosperms (Sabir et al., 2014). The chloroplast genomes of all Piper species are highly conserved, but expansion and contraction at the IR/SSC boundary regions leads to variation in the size of the genomes (Goulding et al., 1996). Here, we have compared the IR/SSC boundaries of P. nigrum with four other Piper species, namely, P. kadsura, P. laetispicum, P. cenocladum, and P. auritum. It was observed that 10 completely duplicated genes were present in the IR region of the P. nigrum chloroplast genome. In Figure 7, it can be seen that an intergenic region of 1,106 bp is present between ndhF and ycf1 genes at the SSC/IRa junction in P. cenocladum, whereas ndhF and ycf1 genes are overlapping at the SSC/Ira junction in other Piper species. The size of an overlapping fragment of the ndhF gene varies in different Piper species, hence contributing to fluctuating IR boundaries among different plastomes. The ycf1 gene (5,474 bp) is present at the IRb/SSC junction in P. nigrum, P. kadsura, and P. laetispicum, whereas it is absent in P. cenocladum and P. auritum. A fragment of the ycf1 gene of different sizes is present as a pseudogene at the SSC/IRa junction in all the species. The IR region of P. nigrum (27,057 bp) was observed to be larger than P. kadusra (24,657 bp), P. laetispicum (26,133 bp), and P. cenocladum (27,039 bp).

Figure 7 Contraction and expansion at junctions of four regions (LSC, SSC, IRa, IRb) of the plastid genome of five Piper species.
Gene order
The plastomes of five Piper species, viz., P. laetispicum (Wang et al., 2018), P. auritum (NC_034697), P. cenocladum (Cai et al., 2006), P. nigrum, and P. kadsura (Lee et al., 2016) were compared with the help of the Mauve software (Darling et al., 2004). In the comparison of different Piper species, no change was observed in the gene order within Piperaceae except for the absence of the ycf1 gene at the IRb/SSC junction in P. cenocladum and P. auritum (Figure 8). On analysis, it was found that the sequence of the ycf1 gene was present at the IRb/SSC junction in both the P. cenocladum and P. auritum plastid genomes, but it is not being annotated. Therefore, it can be inferred that a significant level of synteny can be seen between the plastid genome of different Piper species. One difference that can be accounted for is the inclusion of the ycf1 gene in the IR region, which varies among different plastid genomes and therefore contributes to variation in IR size and ultimately to the chloroplast genome size.

Figure 8 Gene order comparison of the chloroplast genome within Piperaceae.
Phylogenetic analysis
Chloroplast genomes have proved to be useful in evolutionary, phylogenetic, and molecular systematic studies. They are an important resource for exploring evolutionary histories within and among different species. Various studies have compared intergenic spacers, protein-coding genes, and complete chloroplast genome sequences and have, therefore, contributed to our understanding of evolutionary relationships among different angiosperms and gymnosperms. In the present investigation, the phylogenetic data set included 58 protein-coding genes (38,262 bp after the removal of poorly aligned regions) from 37 taxa, comprising 34 angiosperms and 3 gymnosperms. Maximum likelihood analysis resulted in a single tree with higher bootstrap values (Figure 9). Twenty-eight out of 35 nodes showed 100% bootstrap value. The 100% bootstrap value strongly supports the monophyly of magnoliids. Magnoliids with 100% support value form two distinct clades: one with Canellales and Piperales and the other Magnoliales and Laurales. Piperales with four species of Piper got further divided into two clades with 100% bootstrap value. Piper nigrum is positioned with P. kadsura in one clade, and P. auritum and P. cenocladum were observed to be more similar. Our findings were in agreement with the results reported by Wang et al. (2018), where P. nigrum and P. kadsura were placed in one clade.
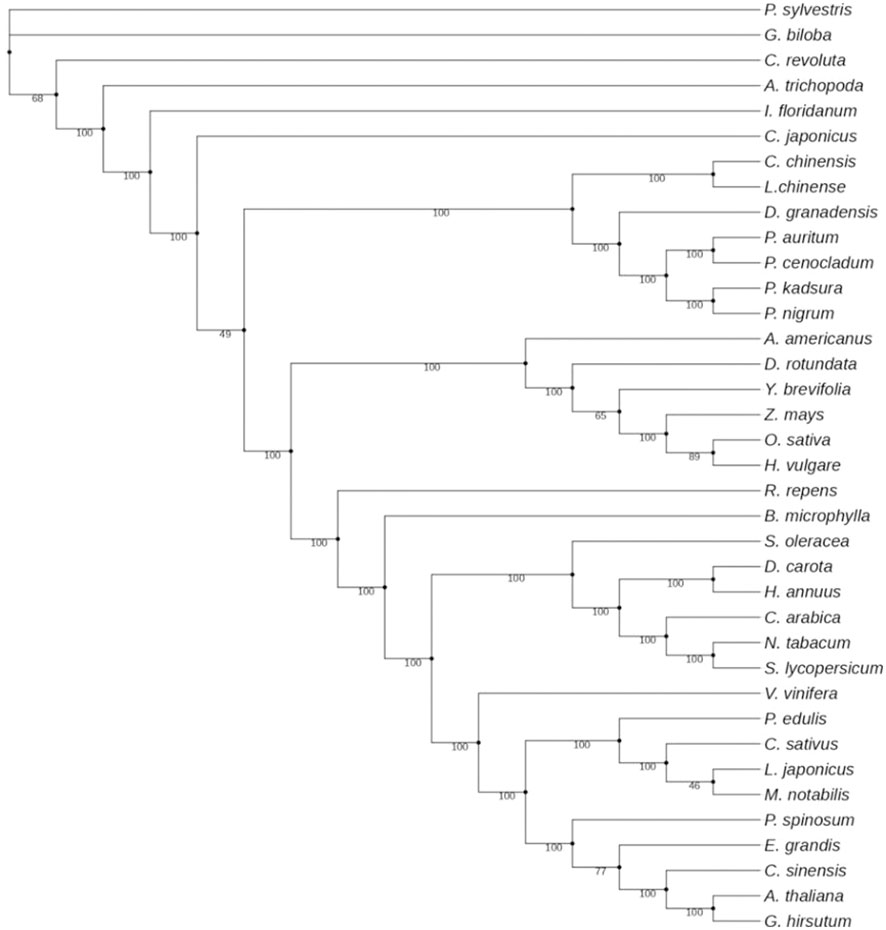
Figure 9 Phylogenetic tree of 37 different species based on 58 protein-coding genes.
Conclusion
In the present investigation, the chloroplast genome of P. nigrum was sequenced by integrating two platforms: Illumina and PacBio. The sequenced chloroplast genome was assembled using the Organelle PBA software and the Pilon software. The chloroplast genome having 161,522 bp length showed a typical quadripartite structure with 81 protein-coding genes, 37 tRNAs, 4 rRNAs, and 1 pseudogene. Codon usage analysis revealed that serine was the most coded amino acid, and 61 codons were present for coding 20 amino acids. The gene rps16, which has been observed to be lost from some of the plastid genomes, is present in the P. nigrum chloroplast genome similar also to other Piper species. Repeat analysis was also done, and there were 23 palindromic, 18 forward, 2 complement, and 1 reverse repeat along with 55 tandem repeats that were predicted. Of the 216 SSRs identified, the majority were present in the intergenic region. Phylogenetic analysis done with 58 protein-coding genes for 37 taxa, comprising 34 angiosperms and 3 gymnosperms, revealed the presence of P. nigrum and P. kadsura in one clade. This study has laid the foundation for future evolutionary and molecular studies on Piperales species.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
Conceptualization and funding acquisition: AG and KB. Investigation and data analysis: AG, TK, AM, and RK. Manuscript preparation: AG, TK, PR and DW. All authors contributed to the article and approved the submitted version.
Funding
This work is funded by the grant received under the project Indian Council of Agricultural Research-Consortium Research Platform on Genomics (project number 1007341).
Acknowledgments
The authors acknowledge the financial support from the ICAR-Consortium Research Platform on Genomics and the facilities provided by the ICAR-National Bureau of Plant Genetics Resources, New Delhi.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1095781/full#supplementary-material
References
Ahmad, N., Fazal, H., Abbasi, B. H., Farooq, S., Ali, M., Khan, M. A. (2012). Biological role of Piper nigrum (Black pepper): A review. Asian Pac. J. Trop. BioMed., 5 1945–1953. doi: 10.1016/S2221-1691(12)60524-3
Andrews, S. (2010) FastQC: a quality control tool for high throughput sequence data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastq.
Asaf, S., Khan, A. L., Khan, M. A., Shahzad, R., Kang, S. M. L., Harrasi, A. A., et al. (2018). Complete chloroplast genome sequence and comparative analysis of loblolly pine (Pinus taeda l.) with related species. PloS One 13 (3), e0192966. doi: 10.1371/journal.pone.0192966
Beier, S., Thiel, T., Mu¨ nch, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33 (16), 2583–2585. doi: 10.1093/bioinformatics/btx198
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27 (2), 573–580. doi: 10.1093/nar/27.2.573
Bock, R. (2007). “Structure, function, and inheritance of plastid genomes,” in Cell and molecular biology of plastids. Ed. Bock, R. (Berlin, Heidelberg: Springer), 29–63.
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi: 10.1093/bioinformatics/btu170
Braukmann, T. W. A., Kuzmina, M., Stefanovic, S. (2009). Loss of all plastid ndh genes in gnetales and conifers: extent and evolutionary significance for the seed plant phylogeny. Curr. Genet. 55 (3), 323–337. doi: 10.1007/s00294-009-0249-7
Cai, Z., Penaflor, C., Kuehl, J. V., Leebens-Mack, J., Carlson, J. E., dePamphilis, C. W., et al. (2006). ). complete plastid genome sequences of Drimys, liriodendron, and Piper: implications for the phylogenetic relationships of magnoliids. BMC Evol. Biol. 6, 77. doi: 10.1186/1471-2148-6-77
Cavalier-Smith, T. (2002). Chloroplast evolution: Secondary symbiogenesis and multiple losses. Curr. Biol. 12 (2), R62–R64. doi: 10.1016/S0960-9822(01)00675-3
Chase, M. W., Christenhusz, M. J. M., Fay, M. F., Byng, J. W., Judd, W. S., Soltis, D. E., et al. (2016). An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc 181 (1), 1–20. doi: 10.1111/boj.12385
Chen, X., Yang, J., Zhang, H., Bai, R., Zhang, X., Bai, G., et al. (2018). The complete chloroplast genome of Calycanthus chinensis, an endangered species endemic to china. conserv. Genet. Resour. 11, 55–58. doi: 10.1007/s12686-017-0966-z
Curci, P. L., De Paola, D., Danzi, D., Vendramin, G. G., Sonnante, G. (2015). Complete chloroplast genome of the multifunctional crop globe artichoke and comparison with other asteraceae. PloS One 10, e120589. doi: 10.1371/journal.pone.0120589
Daniell, H., Lin, C. S., Yu, M., Chang, W. J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17, 134. doi: 10.1186/s13059-016-1004-2
Darling, A. C. E., Mau, B., Blattner, F. R., Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Divya, K. G., Nair, M. C., Shaji, P. K., Nair, P. K. K. (2015). Pollen morphology of pepper cultivars and their wild allies from southern Western ghats, kerala, India. Int. J. Adv. Res. 3 (3), 344–353.
Do, H. D. K., Kim, J. S., Kim, J. H. (2014). A trnI CAU triplication event in the complete chloroplast genome of Paris verticillata m. bieb. (Melanthiaceae, liliales). Genome Biol. Evol. 6 (7), 1699–1706. doi: 10.1093/gbe/evu138
Dong, W., Xu, C., Cheng, T., Lin, K., Zhou, S. (2013). Sequencing angiosperm plastid genomes made easy: A complete set of universal primers and a case study on the phylogeny of saxifragales. Genome Biol. Evol. 5, 989–997. doi: 10.1093/gbe/evt063
Doyle, J. J., Doyle, J. L., Palmer, J. D. (1995). Multiple independent losses of 2 genes and one intron from legume chloroplast genomes. Syst. Bot. 20 (3), 272–294. doi: 10.2307/2419496
Fleischmann, T. T., Scharff, L. B., Alkatib, S., Hasdorf, S., Schottler, M. A., Bock, R. (2011). Nonessential plastid-encoded ribosomal proteins in tobacco: A developmental role for plastid translation and implications for reductive genome evolution. Plant Cell. 23, 3137–3155. doi: 10.1105/tpc.111.088906
Geng, X., Zhu, Y., Ren, Z., Chen, R., Liu, Q. (2022). The complete chloroplast genome of Piper sarmentosum roxburg 1820 (Piperaceae). Mitochondrial DNA Part B. 7 (5), 854–855. doi: 10.1080/23802359.2022.2074805
Goulding, S. E., Wolfe, K. H., Olmstead, R. G., Morden, C. W. (1996). Ebb and flow of the chloroplast inverted repeat. Mol. Genet. Genom. 252, 195–206. doi: 10.1007/bf02173220
Greiner, S., Lehwark, P., Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic. Acids Res. 47 (W1), W59–W64. doi: 10.1093/nar/gkz238
Guisinger, M., Chumley, T., Kuehl, J., Boore, J., Jansen, R. (2010). Implications of the plastid genome sequence of typha (Typhaceae, poales) for understanding genome evolution in poaceae. J. Mol. Evol. 70 (2), 149–166. doi: 10.1007/s00239-009-9317-3
Huang, H., Shi, C., Liu, Y., Mao, S., Gao, L. (2014). Thirteen camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol. Biol. 14, 1–17. doi: 10.1186/1471-2148-14-151
Kaila, T., Chaduvla, P. K., Rawal, H. C., Saxena, S., Tyagi, A., Mithra, S., et al. (2017). Chloroplast genome sequence of clusterbean (Cyamopsis tetragonoloba l.): Genome structure and comparative analysis. Genes 8 (9), 212. doi: 10.3390/genes8090212
Kaila, T., Chaduvla, P. K., Saxena, S., Bahadur, K., Gahukar, S. J., Chaudhury, A., et al. (2016). Chloroplast genome sequence of pigeonpea (Cajanus cajan (L.) millspaugh) and Cajanus scarabaeoides (L.) thouars: Genome organization and comparison with other legumes. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.01847
Keller, J., Rousseau-Gueutin, M., Martin, G. E., Morice, J., Boutte, J., Coissac, E., et al. (2017). The evolutionary fate of the chloroplast and nuclear rps16 genes as revealed through the sequencing and comparative analyses of four novel legume chloroplast genomes from lupinus. DNA Res. 24 (4), 343–358. doi: 10.1093/dnares/dsx006
Kim, H. T., Kim, J. S., Moore, M. J., Neubig, K. M., Williams, N. H., Whitten, W. M., et al. (2015). Seven new complete plastome sequences reveal rampant independent loss of the ndh gene family across orchids and associated instability of the inverted Repeat/Small single-copy region boundaries. PloS One 10 (11), e0142215. doi: 10.1371/journal.pone.0142215
Kubitzki, K., Rohwer, J. G. (1993). “Betrich,” in Flowering plants, dicotyledons magnolid, hamamelid and caryophyliid families (Berlin, Germany: Springer Verlag).
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29 (22), 4633–4642. doi: 10.1093/nar/29.22.4633
Laslett, D., Canback, B. (2004). ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32 (1), 11–16. doi: 10.1093/nar/gkh152
Lee, J. H., Choi, I. S., Choi, B. H., Yang, S., Choi, G. (2016). The complete plastid genome of Piper kadsura (Piperaceae), an East Asian woody vine. mitochondrial DNA a: DNA mapp. Seq. Anal. 27 (5), 3555–3556. doi: 10.3109/19401736.2015.1074216
Li, J., Fan, R., Xu, J., Hu, L., Su, F., Hao, C. (2022). Comparative analysis of the chloroplast genomes of eight Piper species and insights into the utilization of structural variation in phylogenetic analysis. Front. Genet. 13, 925252. doi: 10.3389/fgene.2022.925252
Li, P., Jia, G., Xin, G., Cai, X. (2019). The complete chloroplast genome of Cinnamomum camphora (L.) presl., a unique economic plant to China. Mitochondrial DNA Part B. 4 (2), 2511–2512. doi: 10.1080/23802359.2019.1640083
Li, Y., Xu, W., Zou, W., Jiang, D., Liu, X. (2017). Complete chloroplast genome sequences of two endangered phoebe (Lauraceae) species. Bot. Stud. 58 (1), 37. doi: 10.1186/s40529-017-0192-8
Liu, J., Gong, L. D., Qi, L., Zheng, C., Niu, Y. F., Shi, C. (2019). Complete plastid genome of Persea americana mill. (Ebenales) and phylogenetic analysis. Mitochondrial DNA Part B. 4 (1), 2033–2034. doi: 10.1080/23802359.2019.1617073
Massoni, J., Couvreur, T. L., Sauquet, H. (2015). Five major shifts of diversification through the long evolutionary history of magnoliidae (angiosperms). BMC Evol. Biol. 15, 49. doi: 10.1186/s12862-015-0320-6
Mayor, C., Brudno, M., Schwartz, J. R., Poliakov, A., Rubin, E. M., Frazer, K. A., et al. (2000). VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 16, 1046. doi: 10.1093/bioinformatics/16.11.1046
Morton, B. R. (2003). The role of context-dependent mutations in generating compositional and codon usage bias in grass chloroplast DNA. Mol. Evol. 56, 616–629. doi: 10.1007/s00239-002-2430-1
Nie, X., Lv, S., Zhang, Y., Du, X., Wang, L., Biradar, S. S., et al. (2012). Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PloS One 7, e36869. doi: 10.1371/journal.pone.0036869
Ohtani, K., Yamamoto, H., Akimitsu, K. (2002). Sensitivity to Alternaria alternata toxin in citrus because of altered mitochondrial RNA processing. Proc. Natl. Acad. Sci. U.S.A. 99, 2439–2444. doi: 10.1073/pnas.042448499
Olmstead, R. G., Palmer, J. D. (1994). Chloroplast DNA systematics: a review of methods and data analysis. Am. J. Bot. 8, 1205–1224. doi: 10.2307/2445483
Ozyigit, I. I., Dogan, I., Filiz, E. (2015). In silico analysis of simple sequence repeats (SSRs) in chloroplast genomes of glycine species. Plant Omics. 8, 24–29.
Palmer, J. D. (1991). “Plastid chromosomes: Structure and evolution,” in Molecular biology of plastids. Ed. Bogorad, L. (San Diego, CA, USA: Academic Press), 5–53.
Palmer, J. D., Osorio, B., Aldrich, J., Thompson, W. F. (1987). Chloroplast DNA evolution among legumes: Loss of a large inverted repeat occurred prior to other sequence rearrangements. Curr. Genet. 11, 275–286. doi: 10.1007/BF00355401
Park, J., Kim, Y., Kwon, W., Xi, H., Kwon, M. (2019). The complete chloroplast genome of tulip tree, Liriodendron tulifipera l. (Magnoliaceae): investigation of intra-species chloroplast variations. Mitochondrial DNA Part B. 4 (2), 2523–2524. doi: 10.1080/23802359.2019.1598822
Roy, S., Ueda, M., Kadowaki, K., Tsutsumi, N. (2010). Different status of the gene for ribosomal protein S16 in the chloroplast genome during evolution of the genus Arabidopsis and closely related species. Genes Genet. Syst. 85, 319–326. doi: 10.1266/ggs.85.319
Rozas, U., Ferrer-Mata, A., Sánchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of Large data sets. Mol.Biol. Evol. 34, 12, 3299–3302. doi: 10.1093/molbev/msx248
Sabir, J., Schwarz, E., Ellison, N., Zhang, J., Baeshen, N. A., Mutwakil, M., et al. (2014). Evolutionary and biotechnology implications of plastid genome variation in the inverted-repeat-lacking clade of legumes. Plant Biotechnol. J. 12, 743–754. doi: 10.1111/pbi.12179
Shi, C., Liu, Y., Huang, H., Xia, E. H., Zhang, H. B., Gao, L. Z. (2013). Contradiction between plastid gene transcription and function due to complex post transcriptional splicing: an exemplary study of ycf15 function and evolution in angiosperms. PloS One 8, e59620. doi: 10.1371/journal.pone.0059620
Sinn, B. T., Sedmak, D. D., Kelly, L. M., Freudenstein, J. V. (2018). Total duplication of the small single copy region in the angiosperm plastome: Rearrangement and inverted repeat instability in asarum. Am. J. Bot. 105 (1), 71–84. doi: 10.1002/ajb2.1001
Soorni, A., Haak, D., Zaitlin, D., Bombarely, A. (2017). Organelle_PBA, a pipeline for assembling chloroplast and mitochondrial genomes from PacBio DNA sequencing data. BMC Genomics 18 (1), 49. doi: 10.1186/s12864-016-3412-9
Thompson, J. D., Higgins, D. G., Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 (22), 4673–4680. doi: 10.1093/nar/22.22.4673
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45 (W1), W6–W11. doi: 10.1093/nar/gkx391
Tsudzuki, J., Ito, S., Tsudzuki, T., Wakasugi, T., Sugiura, M. (1994). A new gene encoding tRNAPro (GGG) is present in the chloroplast genome of black pine: A compilation of 32 tRNA genes from black pine chloroplasts. Curr. Genet. 26, 153–158. doi: 10.1007/BF00313804
Turmel, M., Otis, C., Lemieux, C. (2006). The chloroplast genome sequence of Chara vulgaris sheds new light into the closest green algal relatives of land plants. Mol. Biol. Evol. 23 (6), 1324–1338. doi: 10.1093/molbev/msk018
Ueda, M., Nishikawa, T., Fujimoto, M., Takanashi, H., Arimura, S. I., Tsutsumi, N., et al. (2008). Substitution of the gene for chloroplast RPS16 was assisted by generation of a dual targeting signal. Mol. Biol. Evol. 25, 1566–1575. doi: 10.1093/molbev/msn102
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3–new capabilities and interfaces. Nucleic Acids Res. 40 (15), e115. doi: 10.1093/nar/gks596
Vieira, L., Faoro, H., Rogalski, M., Fraga, H., Cardoso, R. L. A., de Souza, E. M., et al. (2014). The complete chloroplast genome sequence of Podocarpus lambertii: genome structure, evolutionary aspects, gene content and SSR detection. PloS One 9 (3), e90618. doi: 10.1371/journal.pone.0090618
Wakasugi, T., Tsudzuki, J., Ito, S., Nakashima, K., Tsudzuki, T., Sugiura, M. (1994). Loss of all ndh genes as determined by sequencing the entire chloroplast genome of the black pine pinus thunbergii. Proc. Natl. Acad. Sci. U.S.A. 91 (21), 9794–9798. doi: 10.1073/pnas.91.21.9794
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One 9 (11), e112963. doi: 10.1371/journal.pone.0112963
Wang, M. T., Wang, J. H., Zhao, K. K., Zhu, Z. X., Wang, H. F. (2018). Complete plastome sequence of Piper laetispicum (Piperaceae): An endemic plant species in south China. Mitochondrial DNA Part B. 3 (2), 1035–1036. doi: 10.1080/23802359.2018.1511850
Wheeler, G. L., Dorman, H. E., Buchanan, A., Challagundla, L., Wallace, L. E. (2014). A review of the prevalence, utility, and caveats of using chloroplast simple sequence repeats for studies of plant biology. Appl. Plant Sci. 2, 1400059. doi: 10.3732/apps.1400059
Worth, J. R. P., Liu, L., Wei, F. J., Tomaru, N. (2019). The complete chloroplast genome of Fagus crenata (subgenus Fagus) and comparison with F. engleriana (subgenus Engleriana). PeerJ 7, e7026. doi: 10.7717/peerj.7026
Yan, L., Lai, X., Li, X., Wei, C., Tan, X., Zhang, Y. (2015). Analyses of the complete genome and gene expression of chloroplast of sweet potato [Ipomoea batata]. PloS One 10, e124083. doi: 10.1371/journal.pone.0124083
Yi, X., Gao, L., Wang, B., Su, Y. J., Wang, T. (2013). The complete chloroplast genome sequence of Cephalotaxus oliveri (Cephalotaxaceae): Evolutionary comparison of cephalotaxus chloroplast DNAs and insights into the loss of inverted repeat copies in gymnosperms. Genome Biol. Evol. 5 (4), 688–698. doi: 10.1093/gbe/evt042
Zhang, L., Hu, X., Liu, M. (2021). Complete chloroplast genome sequences of the medicinal plant Piper hancei. Mitochondrial DNA Part B. 6 (9), 2775–2776. doi: 10.1080/23802359.2021.1967217
Keywords: black pepper, chloroplast genome, gene content, gene order, phylogeny
Citation: Gaikwad AB, Kaila T, Maurya A, Kumari R, Rangan P, Wankhede DP and Bhat KV (2023) The chloroplast genome of black pepper (Piper nigrum L.) and its comparative analysis with related Piper species. Front. Plant Sci. 13:1095781. doi: 10.3389/fpls.2022.1095781
Received: 11 November 2022; Accepted: 15 December 2022;
Published: 12 January 2023.
Edited by:
Pranav Pankaj Sahu, Czech Academy of Sciences, CzechiaReviewed by:
Mehanathan Muthamilarasan, University of Hyderabad, IndiaSantosh Kumar Gupta, National Institute of Plant Genome Research (NIPGR), India
Copyright © 2023 Gaikwad, Kaila, Maurya, Kumari, Rangan, Wankhede and Bhat. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ambika Baldev Gaikwad, YW1iaWthLmdhaWt3YWRAaWNhci5nb3YuaW4=; YW1iaWthYmdAZ21haWwuY29t