 Rui Xiang
Rui Xiang Bilal Ahmad
Bilal Ahmad Chen Liang
Chen Liang Xiaoxin Shi1
Xiaoxin Shi1 Guoqiang Du
Guoqiang Du Li Wang
Li Wang- 1College of Horticulture, Hebei Agricultural University, Baoding, China
- 2Department of Horticulture, Muhammad Nawaz Sharif (MNS)-University of Agriculture Multan, Multan, Pakistan
- 3Shijiazhuang Fruit Research Institute, Hebei Academy of Agricultural and Forestry Sciences, Shijiazhuang, China
RNA-directed DNA methylation (RdDM) is an important epigenetic pathway in plants and mediates transcriptional silencing by siRNAs. Different gene families have role in the regulation of the RdDM pathway and there is a lack of information about these gene families in the grapes (Vitis vinifera L.). Here, we mentioned the genome-wide identification, bioinformatics analysis, evolutionary history, and expression profiling of VvRdDM pathway genes against various stresses, hormonal treatments as well as in different organs. Sixty VvRdDM genes belonging to fourteen different families were identified. All the genes were unevenly distributed and chromosome 4 contained the highest number of genes (7). Most of the genes showed similar exon-intron and motif distribution patterns within the same subfamilies. Out of 14 families, only members of 4 families underwent duplication events during the evolutionary process and 50% of members of the AGO family are the result of duplication events. Based on Ka/Ks ratio all duplicated gene pairs have a negative mode of selection. VvRdDM pathway genes showed differential spatiotemporal expression patterns against different hormone and stress treatments. Further, with multiple transcriptome analysis, some VvRdDM genes showed a broad spectrum of high expression in different organs at various stages, and VvRdDM genes also displayed different expression in seeded and seedless cultivars during different phases of seed development. This proposed that VvRdDM genes may play multiple roles in grape growth and development, especially in seed development. qRT-PCR analysis of selected genes further verified the critical roles of RdDM genes in multiple biological processes, especially in seed development/ovule abortion i.e., VvIDN2a, VvDRD1a, VvRDR1a, and VvRDR6. Our study provides detailed information about VvRdDM genes in perspective of gene structure and evolution, as well as expression pattern against different stress, hormones and in different plants parts. It provides new candidate gene resources for further functional characterization and molecular breeding of grapes.
Introduction
Epigenetics investigates heritable changes in gene function that occur without a change in DNA sequence (Holliday, 2006). Most of the epigenetic changes (Histone modification, DNA methylation, chromatin remodeling, and chemical modification) occur due to microRNAs (miRNAs). The epigenetic mechanisms are tightly connected to cellular development and differentiation (Barter et al., 2012). DNA methylation is a conserved epigenetic silencing process that is involved in a variety of biological functions, including the regulation of gene expression, prevention of the spread of transposons, and defense against these organisms. Additionally, changes in DNA methylation can result in abnormal development (Zhang et al., 2018). Mostly DNA methylation in plants occurs in the CG, CHG, and CHH sequences (where “H” can be either an A, C, or T), and it is especially prevalent over heterochromatic transposable elements (TEs) and repetitions, where it is crucial for transcriptional gene silencing (TGS) (Zhang et al., 2018). However, many plant species have DNA methylation over gene bodies but in this case precise role is still unknown (Bewick and Schmitz, 2017).
In plants, DNA methylation usually happens by RNA-directed DNA methylation (RdDM). RdDM is a biological mechanism in which DNA methylation is controlled by non-coding RNA molecules. RdDM-induced DNA methylation is usually linked to transcriptional suppression of the genomic regions targeted by the pathway (Erdmann and Picard, 2020). DNA methylation can change gene expression through direct and indirect mechanisms. Further, when it occurs in promoter region of a gene it inhibits transcription (Wierzbicki et al., 2008; Zhang et al., 2018). The key players of RdDM pathway are Pol IV (RNA Polymerase 4), RDR2 (RNA-dependent RNA polymerase 2), DCL3 (Dicer-like 3), and AGO4 (Argonaute 4) (Dinh et al., 2013). According to the canonical RdDM pathway, which is best explained in Arabidopsis, generation of 24-nt small interfering RNAs (siRNAs) depends on Pol IV. Pol IV transcribes single strand RNA (ssRNA), which is converted to double strand RNA (dsRNA) by RDR2. Then dsRNA is diced by protein DCL3 resulting in siRNAs (siRNAs 24nt). These siRNAs are loaded in AGO4 protein and AGO4 interacts with RISC (RNA-induced silencing complex) and methylates DNA (Matzke and Mosher, 2014). Apart from these proteins, some other proteins also play roles in RdDM pathways for example, SHH1 (SAWADEE HOMEO-DOMAIN HOMOLOG 1), CLSY (CLASSY), NRPD (NUCLEAR RNA POLYMERASE D), DRM (DOMAINS REARRANGED METHYLASE), RDM (RNA- DIRECTED DNA METHYLATION), RRP6L (RRP6-like), DRD (DEFECTIVE IN RNA-DIRECTED DNA METHYLATION), DDM (DECREASE IN DNA METHYLATION), IDN (INVOLVED IN DE NOVO), HEN (HUA ENHANCER). Moreover, SHH, NRPD, NRPE (DNA-DIRECIED RNA polymerase V subunit 1), DRM, RDM1, RDM2, RDM3, RMD4, and RDR have SAWADEE, RNA_pol Rpb2, RNA_pol Rpb1, DNA methylase, RdDM_RDM1, RNA_pol Rpb4, Spt5-NGN, lwr1, and RdRP conserved domain, respectively (Wassenegger and Krczal, 2006; Ream et al., 2009; He et al., 2009a; He et al., 2009b; Henderson et al., 2010; Hirtreiter et al., 2010; Zhang et al., 2013; Sasaki et al., 2014). CLSY, DRD, and DDM have two conserved domains; Helicase_C and SNF2-rel_dom (Smith et al., 2007; Hu et al., 2013). RRP6L also has two domains (DNA_pol_A_exo1 and HRDC) (Lange et al., 2008). IDNs (zf-XS, XS and XH), HEN (Hen1_Lam_C, Methyltransf_31, and dsRBD2), and AGO (n-side PAZs, C-side Piwi, and ArgoMid) each has three conserved domains (Ausin et al., 2009; Huang et al., 2009; Zhao et al., 2014). Further, DCL has six conserved domains (DEAD, Helicase-C, Dicer_dimer, PAZ, Ribonuclease_3, and dsrm) (Margis et al., 2006). In most of the crops, AGO, DCL, and RDR gene families have been identified and characterized.
DNA methylation performs diverse functions in plant growth and development including stress tolerance (Zhang et al., 2018). RdDM-related genes have been well studied in various crops for their critical roles in stress tolerance. For example, RNA Pol V (NRPE), RDR1/2/6, DCL2/4, CaDCLs, and RDR1 have been reported for their roles against different stresses in different crops i.e., Arabidopsis and pepper (Garcia-Ruiz et al., 2010; López et al., 2011; Qin et al., 2018). Further, SlAGO4 showed a negative role in salt and drought stress tolerance in tomato (Huang et al., 2016). DNA methylation may have potential roles in flowering, fruit setting, seed development, and fruit ripening. In Arabidopsis low levels of methylation caused delay in flowering (Jones and Sung, 2014). AtRRP6L1 and AtRRP6L2 regulated flowering in Arabidopsis via repressing the expression of FLC (Flowering Locus C) (Shin and Chekanova, 2014). On the other hand, high levels of DNA methylation are important for orange fruit development and ripening (Huang et al., 2019). In the abscission zone of the citrus sinensis fruit, key players of the RdDM pathway are down-regulated (Sabbione et al., 2019). Further, low DNA methylation levels in apples resulted in reduced fruit size (Daccord et al., 2017). In tomato a change in hypermethylation in CNR (COLORLESS NON-RIPENING) indicated abnormal fruit ripening (Manning et al., 2006). Cheng et al. (2018) has also reported the involvement of RdDM pathway in strawberry fruit ripening. During the ripening process in apples, DNA methylation of the MdMYB10 promoter affected gene expression and fruit color (El-Sharkawy et al., 2015).
The role of RdDM in seed development and germination has been studied (Xiao et al., 2006; Zhong et al., 2020). Furthermore, the data suggested that DNA methylation can play a critical role in seed dormancy (Kawakatsu et al., 2017). The association of DNA methylation with seed development has been reported in Arabidopsis thaliana and Brassica rapa (Grover et al., 2018). For example, the involvement of AtHEN1 in ovule development (Wei et al., 2020). Various phases of tissue have different degrees of DNA methylation. For example, the shoot apical meristem of early peach seedlings exhibited a higher amount of DNA methylation than adults (Bitonti et al., 2002). Most probably, the diverse DNA methylation levels in different tissues at different growth stages are due to different DNA methylation pathways (Bartels et al., 2018) and gene expression levels.
Grapevine (Vitis vinifera L.) is among the top fruit crops grown all over the world. The grapes are consumed in different ways including fresh fruit (table), wine, juices, and raisins (dried) (This et al., 2006). RdDM pathway genes have been identified in different plant species including Arabidopsis (Kurihara et al., 2008), Brassica rapa (Cao et al., 2016), Soybean (An et al., 2017), Citrus (Mosharaf et al., 2020), Apple (Li et al., 2019), Rice (Ahmad et al., 2019), Maize (Qian et al., 2011), Tomato (Bai et al., 2012), and Ginkgo (Gao et al., 2020). However, in most of the species scientists have reported only three RdDM (DCL, AGO, and RDR) gene families. There is a lack of information about RdDM genes in grapevine. The involvement of RdDM genes in different mechanisms of plant growth, development, and response to different stresses (biotic and abiotic) and antiviral defense, justifies the need of detailed bioinformatics studies of RdDM genes in grapevine. Here, we elaborated comprehensive genome-wide identification of RdDM grapevine genes, including phylogenetic analysis, chromosomal positions, intron-exon distribution, motif analysis, evolutionary history, selection pressure, and cis-elements analysis. Various transcriptome analyses under biotic stress, abiotic stress, hormone treatments, and at different stages of growth were performed. The expression of selected genes was further investigated during progressive phases of seed development in seedless and seeded cultivars. Moreover, the integration of all these studies can provide inklings about VvRdDM pathway genes function and their involvement in multiple biological processes. Our study provides basic information about RdDM grapevine genes and will provide new candidate genes for functional studies.
Materials and methods
Identification of RNA-directed DNA methylation genes in grapes
To identify RdDM pathway genes in grapes, the grape genome was retrieved from Grape Genome CRIBI Biotechnology (http://genomes.cribi.unipd.it/) and Grape Genome Database (http://www.genoscope.cn.fr). Protein sequences involved in the RdDM pathway of the Arabidopsis were downloaded from the TAIR database (http://www.arabidopsis.org/) and were used as a query in Pfam database (http://pfam.xfam.org/search#tabview=tab1) to obtained the Hidden Markov Model (HMM) profile of the RdDM conserved domain. SPDE software was used with the hmmer search function (HMMER 3.0) to obtain candidate genes (Xu et al., 2021). Arabidopsis RdDM pathway protein sequences were used as a query in National Center for Biotechnology Information (NCBI; http://www.ncbi.nlm.nih.gov/), Grape Genome Database, and Grape Genome CRIBI Biotechnology using BLAST-P program (e-value threshold of 1e−10) (Altschul et al., 1997) to search for sequences of VvRdDM homologous genes. The genes obtained from HMM and BLAST-P were combined. To further determine the reliability of RdDM pathways genes in grape, search results of the protein sequences were checked with SMART (http://smart.embl-heidelberg.de), Pfam database and proteins with incomplete domains were removed.
Cis-acting regulatory elements prediction
2 Kb upstream region (Xi et al., 2017) of the VvRdDM genes was retrieved from the latest Vitis vinifera genome assembly and annotations were downloaded from Phytozomev12 (http://www.phytozome.net) using the SPDE. PlantCARE database was used for conserved cis-elements prediction (Lescot et al., 2002).
Phylogenetic analysis and nomenclature
The full length amino acid sequences of RdDM proteins belonging to A. thaliana, Oryza sativa, Solanum lycopersicum, and V. vinifera were downloaded from TAIR and JGI Data Portal (https://data.jgi.doe.gov/). Multiple alignment of RdDM protein sequences was performed using ClustalW program with default settings. MEGA7.0 software was used for phylogenetic tree construction (Kumar et al., 2018). The phylogenetic trees were generated with the following parameters; Neighbor-Joining (NJ) method, p-distance, complete deletion, and 1000 bootstrap replicates (Ahmad et al., 2020). The candidate grapevine genes were named based on phylogenetic relationships and sequence homologies with corresponding Arabidopsis homologs (Grimplet et al., 2014).
Exon-intron distribution and conserved motifs analysis
The conserved motif distribution patterns of VvRdDM genes were assessed using Expectation Maximization for Motif Elicitation (MEME) online software (version 5.4.1) (https://meme-suite.org/meme/tools/meme) (Bailey and Elkan, 1994) with the following parameters: default settings and maximum number of motifs were designated to identify 20 motifs. SMART program and Pfam database were used to annotate the MEME motifs. TBtools were used to visualize the results of conserved motif analysis (Chen et al., 2020). The full genomic sequences and respective coding sequences of RdDM grape genes were obtained from Grape Genome Database and online Gene Structure Display Server 2.0 (http://gsds.gao-lab.org/index.php) was used for exon-intron analysis.
Evolutionary history analysis and estimation of selection pressure
Tandem duplication was determined using the Houlb-described criterion (Holub, 2001). The Plant Genome Duplication Database (http://chibba.agtec.uga.edu/duplication/) was used to retrieve syntenic pairs between Arabidopsis and grapes and among grape genes (Lee et al., 2012). The Circos diagrams were created using TBtools (Chen et al., 2020). The synonymous substitution rate (Ks) and non-synonymous substitution rate (Ka) of duplicated genes were calculated using an online software (http://services.cbu.uib.no/tools/kaks). For estimating selection pressure, the Ka/Ks ratio was calculated (Siltberg and Liberles, 2002).
Plant materials
In this experiment, 2 seeded (‘Zuijinxiang’ and ‘Kyoho’) and 2 seedless (‘Crimson seedless’ and ‘Flame seedless’) cultivars were used. All the plants were grown in grapevine germplasm orchard of Hebei Agricultural University, Baoding, Hebei, China (38°51′N, 115°29′E) under field conditions, where the average annual temperature is 13.4°C, (Below - 4.3°C in winter and above 26.4°C in summer), the average annual sunshine hours are 2511, and the mean annual precipitation is 498.9 mm. Seed samples were taken from berries using tweezers at 4 different growth stages, 20, 30, 40, and 50 days after full bloom (DAF). All the samples were quickly dipped in liquid nitrogen and preserved at -80°C for further studies. Three biological replicates were obtained at each time point.
Expression analysis of RdDM pathway genes in grapes
Expression analysis of the VvRdDM pathway genes were carried out using transcriptome data of NCBI SRA database (https://www.ncbi.nlm.nih.gov/sra/?term=), the corresponding accession IDs are as followings: for biotic stress treatment: downy mildew infection (SRP013835), botrytis cinerea infection (SRP120480), powdery mildew infection (SRP253455); For abiotic stress treatment: cold (SRP202053), heat (SRP091989); For hormone treatment: gibberellic acid (GA3) (SRP045605), methyl jasmonate (MeJA) and salicylic acid (SA) (SRP378285), abscisic acid (ABA) (SRP098802); For organ development at different stages: seeds (SRP081137), buds (SRP159132), inflorescences (SRP045605), berries (SRP265116). Then the raw transcriptomic data was checked using FastQC (v0.11.9) software, and low-quality reads and adapters were removed using Trimmomatic v0.39. All clean reads were mapped to the V. vinifera reference genome (PN40024.v4) by Hisat2 software. Gene expression quantification [Transcripts Per kilobase of exon model per Million mapped reads (TPM)] were estimated using StringTie (v2.2.1) (Pertea et al., 2016). R package DeSeq2 (Love et al., 2014) with the principle adjusted p-value (padj) < 0.05 and the |log2 fold change (log2 FC) | ≥ 1 was used for the identification of significantly differently expressed genes (DEGs). Heat maps were generated using TBtools.
RNA extraction and qRT-expression profiling
The OmniPlant RNA Kit (Dnase I, Comwin Biotech, Beijing, China) was utilized for the extraction of total RNA. For checking the quality and OD value, Agarose gel electrophoresis and nano drop spectrophotometer (Thermo Fisher Scientific, Yokohama, Japan) were used, respectively. Reverse transcription was carried out after gDNA was eliminated using the Prime ScriptRTase (Trans Gen Biotech, Beijing, China). gDNA Eraser (contains Dnase) eliminated genome DNA. Additionally, a qPCR assay on crude RNA was carried out to determine the level of gDNA contamination. Then cDNA was diluted to 200 ng/μl by using distilled water. Primer Premier 7 software was used for designing specific primers (Supplementary Table S1). Each reaction was carried out in 20 μl volume comprising of 1 μl cDNA,1 μl of each primer (1.0 μM), 10 μl of 2 X Fast Super EvaGreen® qPCR Mastermix (US Everbright Inc., Suzhou, China), and 7 μl sterile distilled H2O. Every reaction was performed three times. With three biological and three technical replicates, the quantitative real time-PCR (qRT-PCR) was carried out using the SYBR Green (Applied Biosystem) in the LightCycler®96 System (Applied Biosystems, Roche, Shanghai, China). We used the grape ACTIN gene (GenBank Accession number NC 012010) as an internal reference, and relative gene expression was computed using the 2 −ΔΔCT method (Livak and Schmittgen, 2001).
Statistical analysis
SPSS Statistics 22.0 software was used for data analysis. One way analysis of variance (ANOVA) on ranks followed by tukey post-hoc analysis was used to evaluate the overall significance of the data (Tukey’s test; p < 0.05) (Wang et al., 2020). The graphs were created using sigma Plot 14.
Results
VvRdDM genes identification
There were 60 RdDM genes found in grapes. The obtained genes were structurally analyzed and all genes were divided into different gene families according to conserved domains. There were 4, 2, 3, 2, 1, 9, 2, 10, 1, 3, 1, 4, 5, and 13 genes in SHH, CLSY, NRPD, NRPE, DRM, RDM, RRP6L, DRD, DDM, IDN, HEN, DCL, RDR, AGO sub-family, respectively. The protein prediction lengths of SHH, RDM, DRD, DCL, RDR, and AGO sub-families ranged from 248- 697, 174-1034, 729-2266, 1340-1689, 919-1128, and 879-1039 amino acids, respectively. The complete information about VvRdDM genes including gene locus ID, chromosomal position, coding sequence length, and protein sequence length are shown in Table 1. All the genes were named as per earlier criteria i.e., based on protein sequence similarity with Arabidopsis RdDM genes.
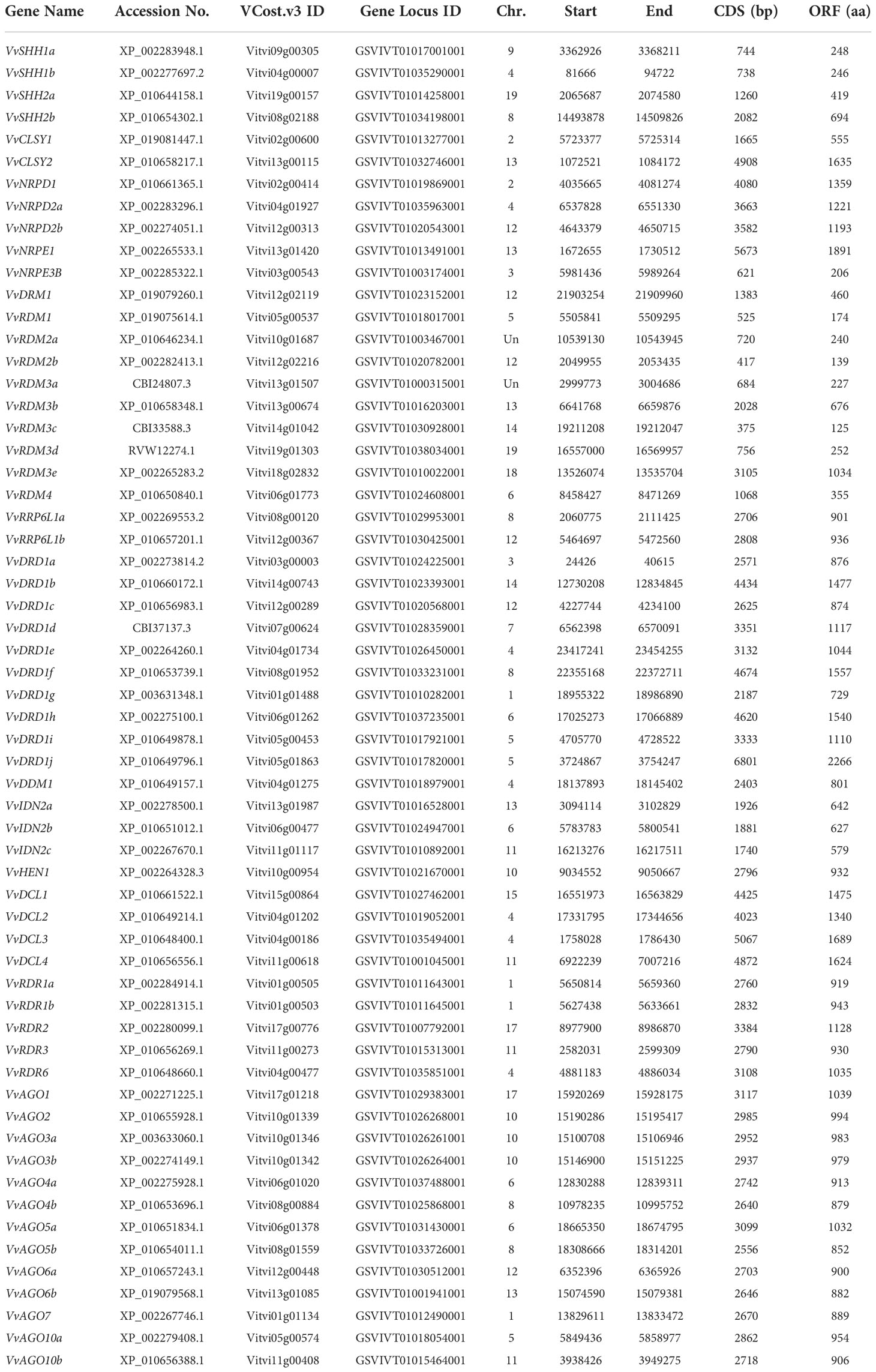
Table 1 List of VvRdDM genes.
Phylogenetic and structural analysis
To study the evolutionary relationship of the grape RdDM genes. The phylogenetic trees were constructed among Arabidopsis, grape, tomato, and rice RdDM subfamilies i.e., AGO, DCL, and RDR. According to Figure 1A, AGO proteins can be divided into 4 subgroups including AGO4, ZIPPY/AGO7, AGO1, and MEL/AGO5, and the protein affinities among dicots are closer. Further, AGO4 subgroup was more conserved with respect to proteins number. Except tomato all other have same number of proteins. The same trend of distribution in different subgroups was noted in all families of RdDM genes of grapes except RDM and DRD. Four VvDCLs proteins were divided in four subgroups: DCL1, DCL2, DCL3, and DCL4 (Figure 1D). Likewise, VvRDRs protein were distributed in RDR1, RDR2, RDR6, and RDR3/4/5 (Figure 1B). The VvRDMs and VvDRDs proteins of grapes were grouped into three sub-classes: RDM3, RDM2/4, RDM1 (Figure 1C) and I, II, III (Figure 1E), respectively. The more differences between grapes and Arabidopsis RDM and DRD protein numbers indicate that the RDM and DRD have observed more changes during the evolutionary process.
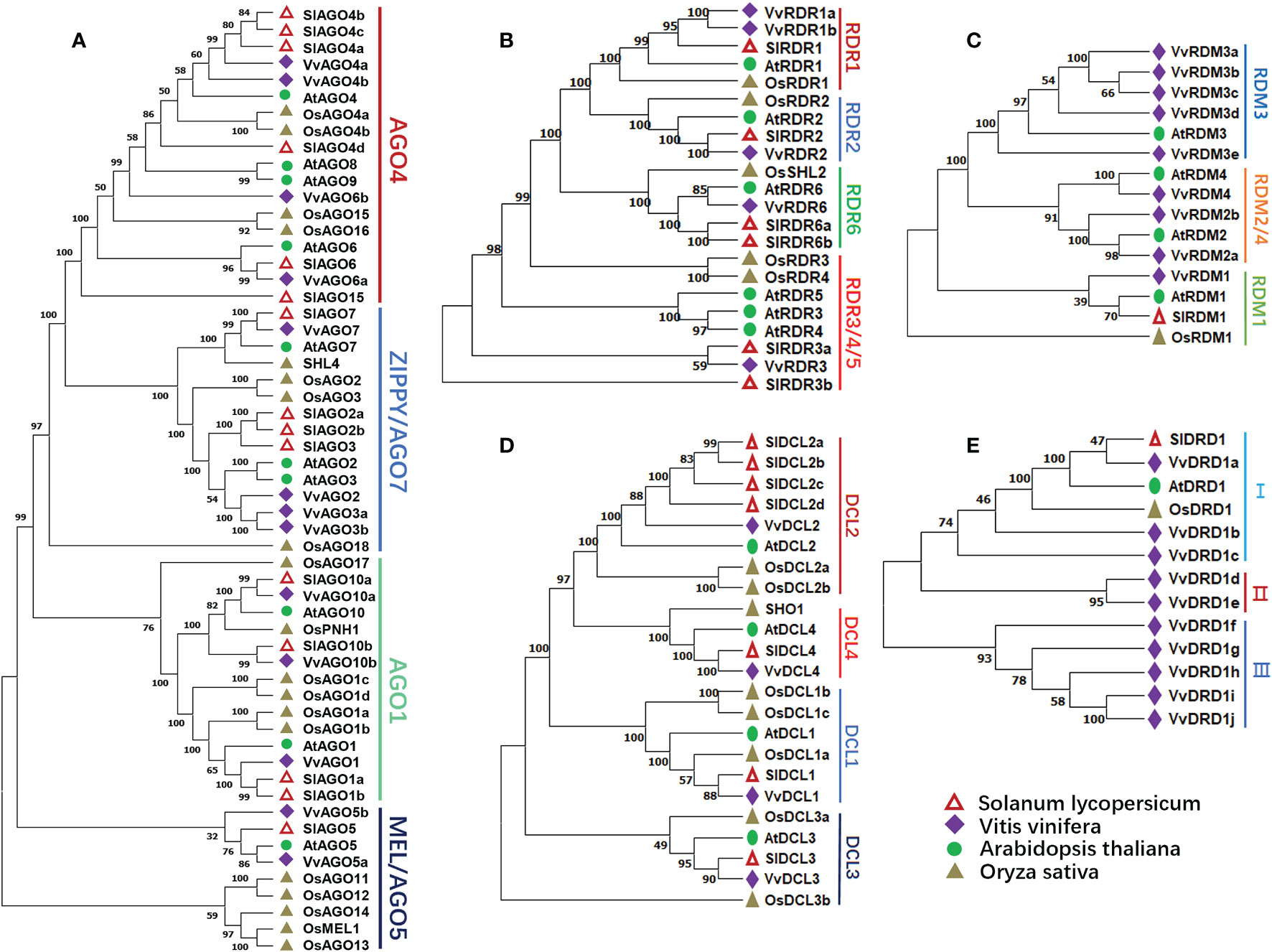
Figure 1 Phylogenetic analysis of AGO, RDR, RDM, DCL, and DRD proteins from grape, Arabidopsis, tomato, and rice. Hollow triangle, diamonds, circles, and triangle represent tomato, grapes, Arabidopsis, rice proteins, respectively. Numbers near the branches denote bootstrap values. (A) Phylogenetic tree of AGO proteins. AGO4, AGO7, AGO1, and AGO5 represent different subgroups. (B) Phylogenetic tree of RDR proteins. RDR1, 2, 3, 4, and 5 represent different subgroups. (C) Phylogenetic tree of RDM proteins. RDM1, 2, 3, and 4 represent different subgroups. (D) Phylogenetic tree of DCL proteins. DCL1, 2, 3, and 4 represent different subgroups. (E) Phylogenetic tree of DRD proteins. I, II, and III represent different subgroups.
For the better understanding of gene evolutionary process, the exon-intron structure of the grape RdDM genes was studied. The number of exons ranged from 2 (VvRDM1) to 32 (VvDRD1j) (Figure 2). As the AGO subfamily is concerned, there were almost the same number of exons within the same subgroup except AGO7 subgroup. This indicates that AGO7 is less conserved as compared to other groups. In addition, the DRD, RDM, RDR, SHH, DCL, NRPD, and IDN sub-family contained 8-32, 4-22, 4-18, 6-10, 21-25, 10-25, and 6-7 exons, respectively. Except, DCL and IDN sub-families, there were differences in the exon-intron distribution in the subfamilies and even within same subgroups, reflecting the diversity emerged during evolution. However, four duplicated gene pairs showed conserved (VvAGO10a-VvAGO10b, VvIDN2a-VvIDN2b, VvAGO5a-VvAGO5b, and VvSHH1a-VvSHH1b) exon-intron number.

Figure 2 Phylogenetic tree and exon-intron distribution of VvRdDM genes. Purple boxes and black lines denote exons and intron, respectively.
Most of the subfamilies including AGO, DRD, RDR, SHH, DCL, IDN, RRP6L, and CLSY displayed conserved motif distribution patterns (Figure 3). Further, DRD, DCL, CLSY, and DDM subfamilies have a common structural domain (Helicase_C), but with different position and number. However, the members of RDM sub-family (supergene family) have different domains and protein sequences. Four duplicated gene pairs having same exon-intron distribution (as mentioned above) also showed similar motif distribution patterns.
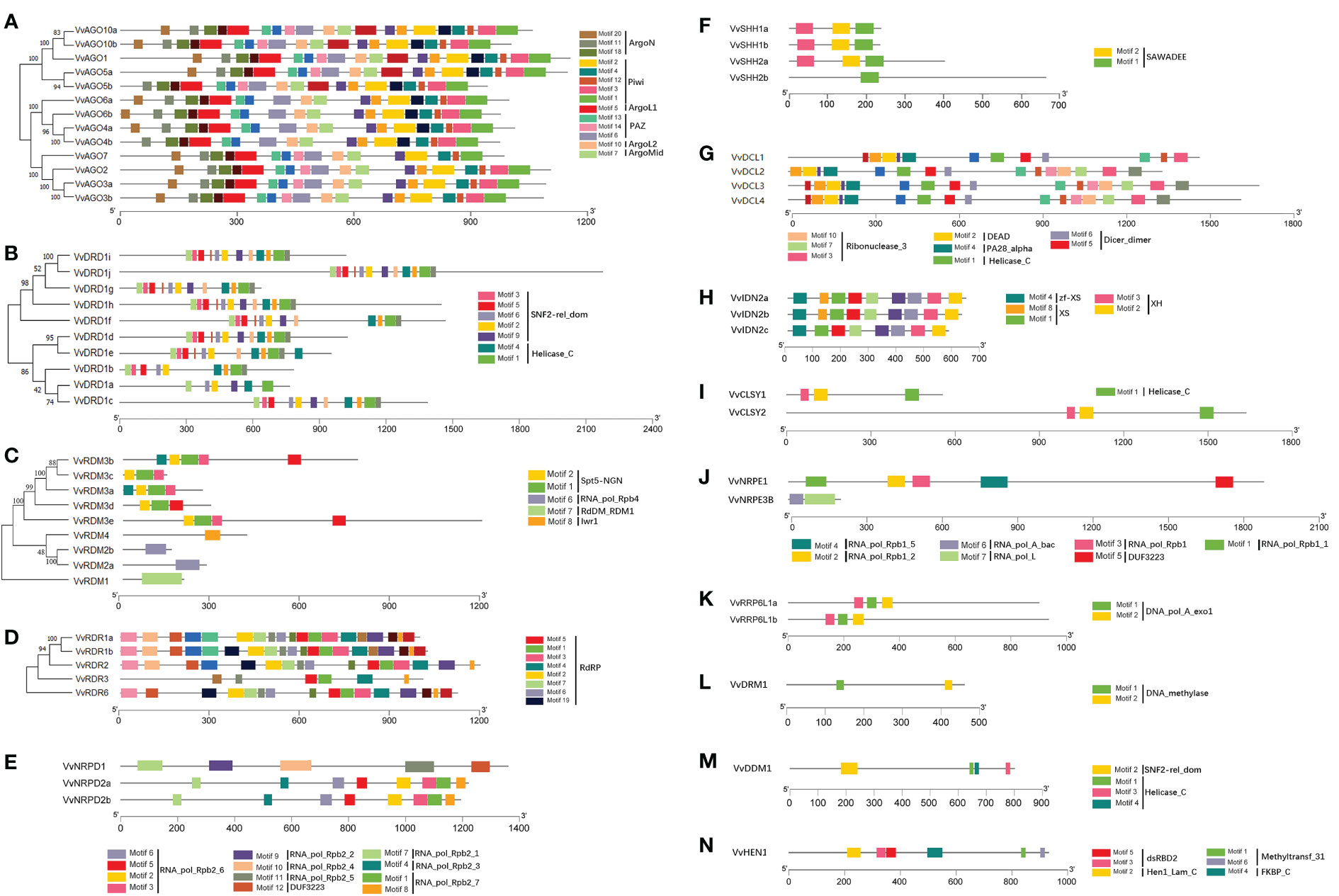
Figure 3 Conserved motifs analysis of VvRdDM genes. Different color boxes represent different motifs The letters (A–N) represent the different subfamilies.
Promoter analysis of VvRdDM genes
The distribution patterns of different types of cis-regulatory elements may provide inklings about gene expressions and functions. A total of 18 cis-elements were identified and can be divided into 4 functional types; (i) Hormone responsive (MeJA, ABA, Auxin, SA, GA), (ii) Light responsive, (iii) Stress responsive (Defense and stress, Anaerobic, Drought, Cold), (iv) Participate in plant development (Endosperm, Meristem, Seed specific, Circadian control).
Except four (VvNRPD1, VvNRPD2a, VvDRD1i, VvAGO3a), all other genes have hormone responsive elements including MeJA responsive element (CGTCA-motif, TGACG-motif), the ABA responsive element (ABRE), the Auxin responsive element (TGA-element, AuxRR-core), the SA responsive element (TCA-element) and GA responsive element (P-box,TATC-box) (Figure 4A). However, the ABRE were more abundant accounting up to 31% share in hormone responsive elements (Figure 4B).
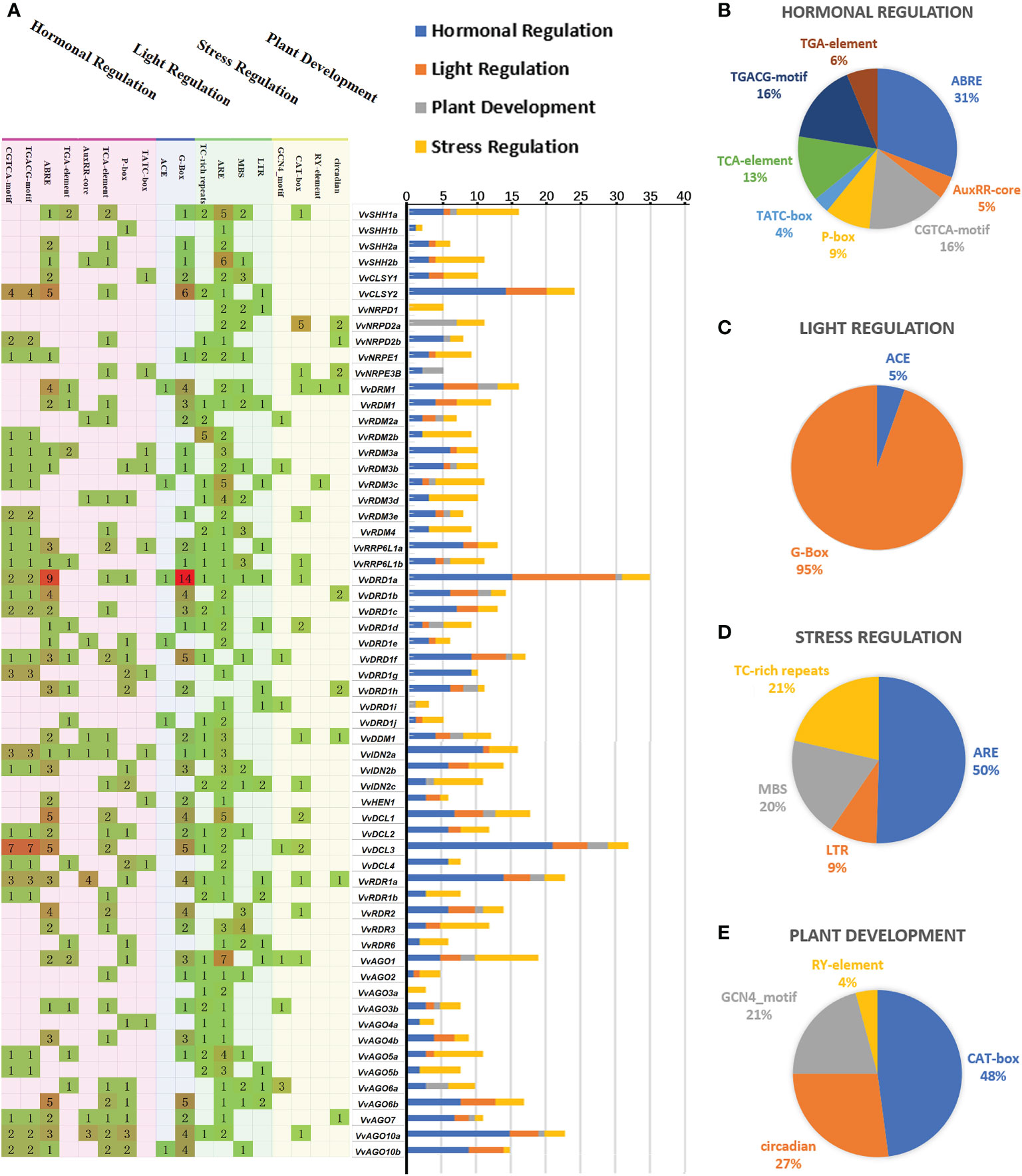
Figure 4 Cis-acting element analysis of VvRdDM genes. (A) The numbers and type of cis-elements in the VvRdDM promoter regions. The cis-elements belonging to different categorizes are denoted with different colors. (B) Relative percentage of hormonal regulation cis-elements. (C) Relative percentage of light regulating cis-elements. (D) Relative percentage of stress regulating cis-elements. (E) Relative percentage of plant development related.
Stress response cis-elements were present in most of the genes i.e., ARE (involvement in anaerobic induction), LTR (Low temperature-responsive elements), and TC-rich repeats (defense and stress response). Further, the presence of plant growth and development (CAT-box, circadian, GCN4-motif, and RY-elements) related elements suggests the involvement of VvRdDM genes in grapevine growth and development.
Expansion patterns and chromosomal arrangements of VvRdDM genes
VvRdDM genes were mapped on grape chromosomes by utilizing the available grape genome annotation information. All genes were unequally present on 19 chromosomes and there was no gene on chromosome 16. Chromosome 4 contained the maximum number of genes (7) (Figure 5). Further, total 8 genes undergone segmental duplication in the form of 4 pairs (VvAGO10b-VvAGO10a, VvIDN2a-VvIDN2b, VvAGO5a-VvAGO5b, and VvSHH1a-VvSHH1b) and two events of tandem duplication (VvRDR1a-VvRDR1b and VvAGO2-VvAGO3a-VvAGO3b) were observed among five genes. Interesting, AGO family genes have undergone both duplication events and more than 50% genes (7 out of 13) are the result of duplication events. These results suggest that segmental and tandem duplication both have a role in the expansion of some VvRdDM sub-families. Further, Ka/Ks ratio was calculated for the estimation of selection pressure. According to the results all the duplicated genes have purifying mode of selection i.e., Ka/KS < 1 (Supplementary Table S2).
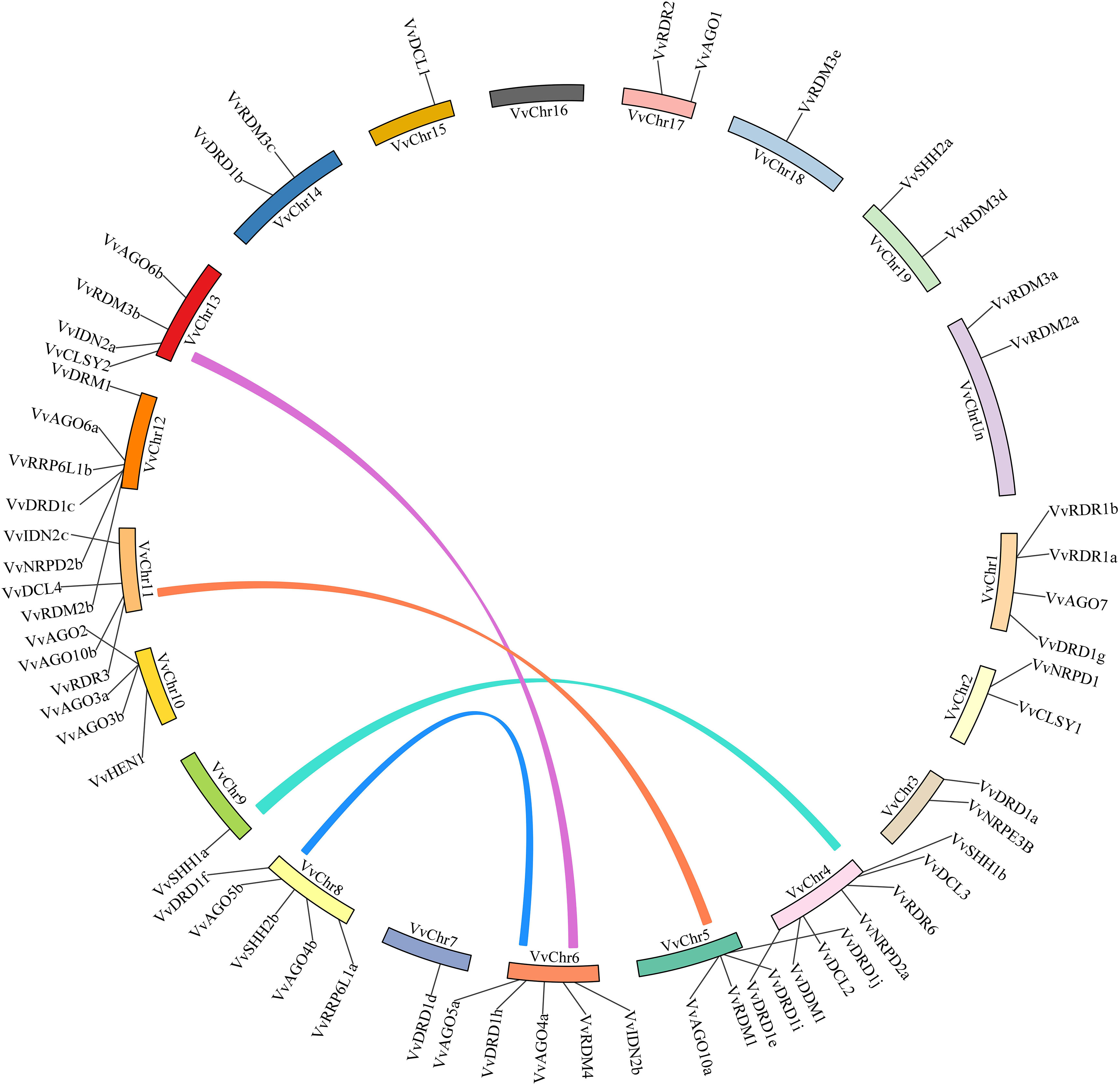
Figure 5 Chromosomal distribution and Synteny analysis. The corresponding genes on two chromosomes were regarded as segmental duplications and colored bars connecting two chromosomal regions indicate syntenic regions. Chr: chromosomes.
Evolutionary relationships between arabidopsis and grapes
To get further information about evolution of VvRdDM genes, the syntenic relationships between grape and Arabidopsis RdDM genes were found. There were 19 syntenic blocks among 17 VvRdDM and 19 AtRdDM genes (Figure 6; Supplementary Table S3). In each syntenic pair, both members belonged to the same subgroup. These syntenic relationships among grape and Arabidopsis suggest that these homologous genes have common ancestors before the speciation. Interestingly, VvCLSY2 paired with three different Arabidopsis genes (VvCLSY2-AT5G05410/AT3G11020/AT2G40340). Further, AT5G59390 also paired with more than one grapevine genes (AT5G59390-VvIDN2a/VvIDN2b). These results suggest that VvCLSY2 and AT5G59390 have gained more changes during the evolutionary process.
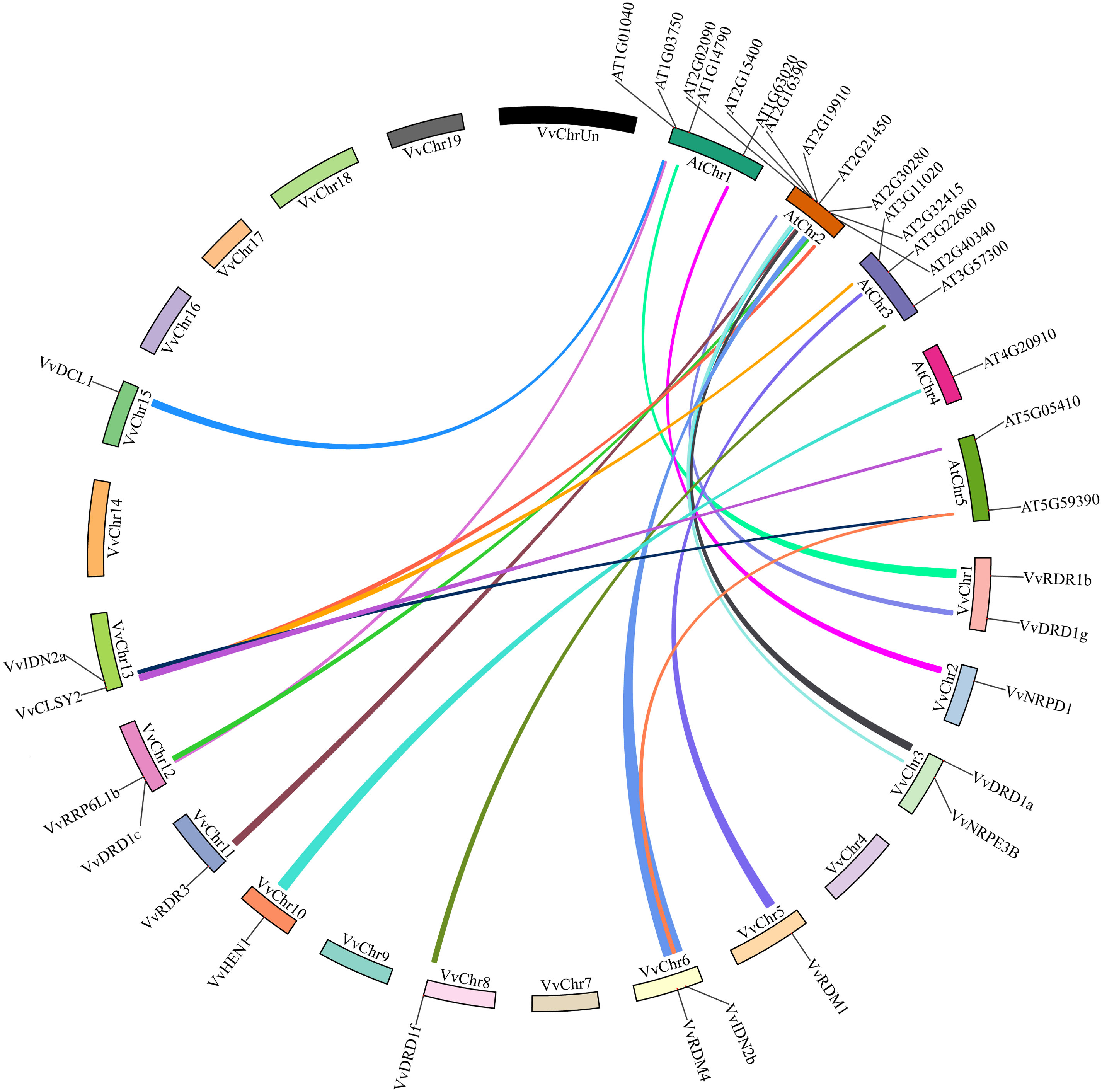
Figure 6 Synteny analysis of VvRdDM genes between grapes and Arabidopsis. Relative positions were estimated according to the grape and Arabidopsis chromosomes; colored lines denote syntenic regions.
Transcriptomic analysis of VvRdDM genes under biotic stress, abiotic stress and hormone treatment
To comprehensively explore potential functions of VvRdDM genes, expression patterns were investigated with a variety of transcriptome data. VvRdDM genes showed significantly differential expression against botrytis cinerea and downy mildew infection. For example, following B. cinerea infection, five genes (VvRDR6, VvAGO4a, VvDCL3, VvRDM3b, VvNRPD1) showed significant down-regulation and one gene (VvDRM1) displayed significant up-regulation expression at berry ripened stage. However, some of the VvRdDM genes (VvRDR1a, VvAGO1, VvAGO4a, VvAGO5a, etc) showed time specific expression under botrytis cinerea treatment i.e., higher expression level at berry hard green stage than ripened stage. Further, expressions of VvDCL2, VvAGO3a, and VvAGO3b were significantly down-regulated with downy mildew infection. However, none of the VvRdDM genes showed significantly different expression after infection with powdery mildew (Figure 7). After heat stress (35°C, 40°C, 45°C) treatments the number of significantly expressed genes was highest (13) at the 40°C. Interestingly, VvAGO3a showed a significant up-regulation against all temperature treatments.
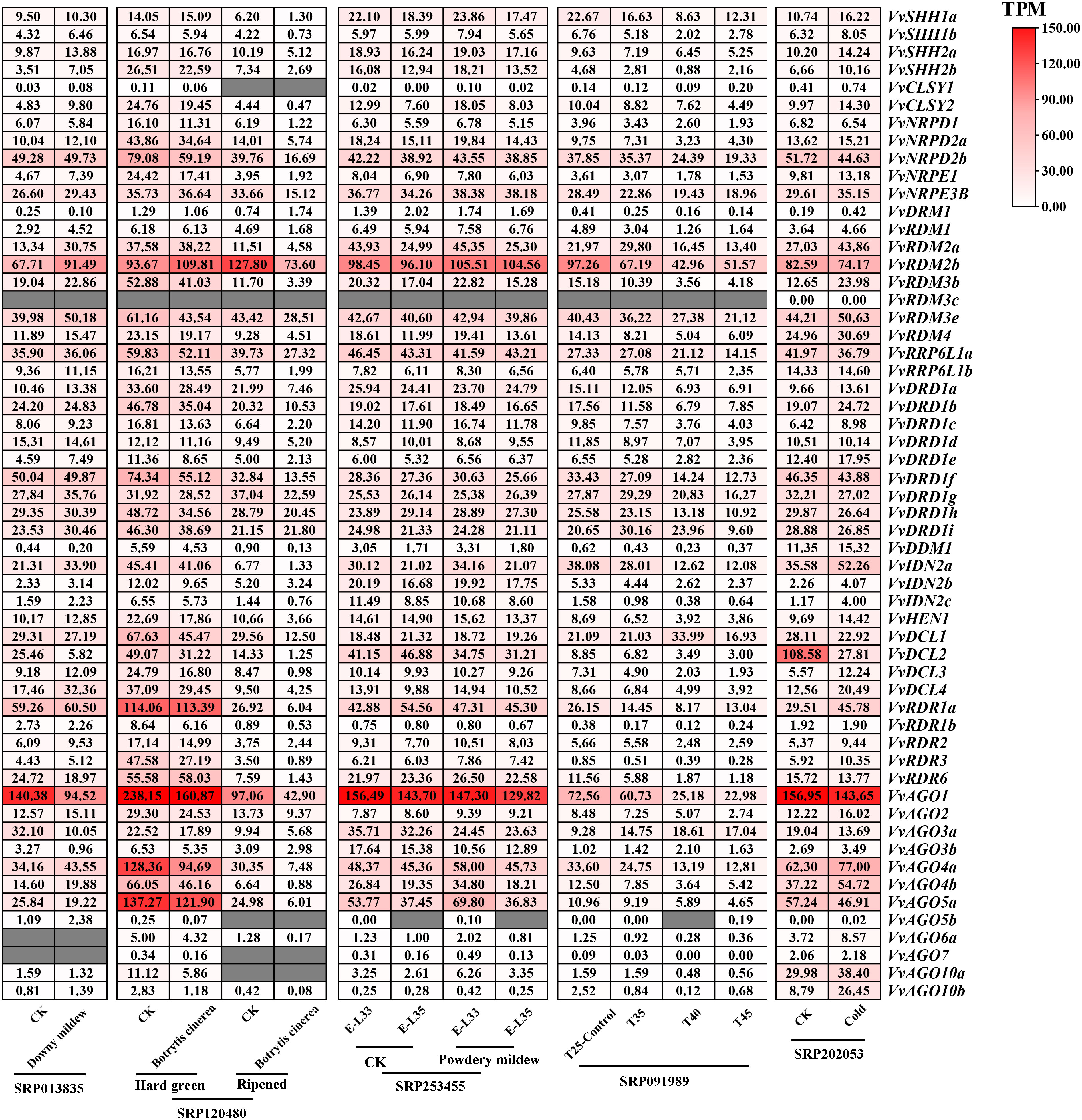
Figure 7 Transcriptomic expression of VvRdDM genes following biotic and abiotic stresses. Transcriptome data were downloaded from the NCBI SRA datasets (SRP013835, SRP120480, SRP253455, SRP091989 and SRP202053). For transcriptomic analysis plant samples following different stress were collected; For downy mildew infected leaves; Botrytis cinerea infected fruits at 2 stages (hard green and ripened stage); Powdery mildew treated fruits at 2 growth stages (EL33 and EL35). For high temperature stress, plants grown at 25°C were taken as control, and 35°C, 40°C, and 45°C were the treatments. Low temperature treatment is 4°C, plants grown at 25°C were taken as control. The white and red color scale denotes low and high expression levels, respectively. No gene expression is denoted with grey color. Numbers denote TPM of VvRdDMs.
Moreover, following cold treatment at 4°C, VvIDN2c and VvAGO10b were significantly up-regulated, and it is worth noting that the significantly down-regulated gene VvDCL2 also had a high change in expression (108.58 to 27.81). VvRdDM genes showed better response to heat stress (Figure 7) i.e., VvNRPD2b, VvRDM2b, VvIDN2a, VvAGO1, and VvAGO4a showed decrease in expression with rise in temperature.
Under hormones treatment (Figure 8), most VvRdDM genes showed significantly different expression at 24 h post treatment. For example, VvDDM1 and VvAGO6a were significantly up-regulated and VvDCL2 was significantly down-regulated after GA3 treatment. Following MeJA treatment VvRDR3 and VvAGO7 were significantly up-regulated and VvRDR1b was significantly down-regulated. After ABA treatment some of the genes were significantly down-regulated i.e., VvRDR1b, VvRDR1a, VvDCL2 and VvAGO5a.
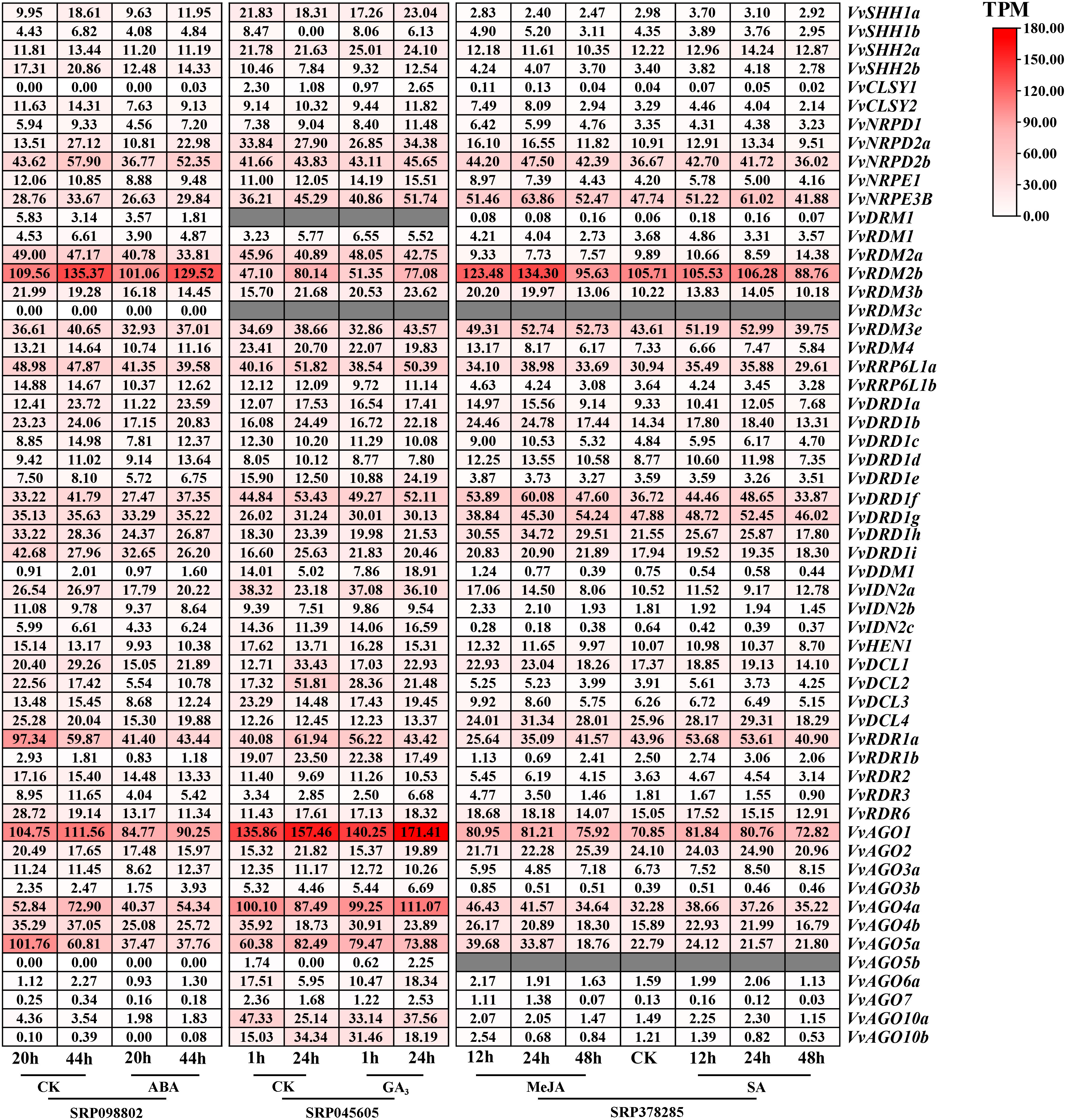
Figure 8 Transcriptomic expression of VvRdDM genes following different hormone treatments. Transcriptome data were retrieved from the NCBI SRA datasets (SRP09880, SRP045605 and SRP378285). The white and red color scale denotes low and high expression levels, respectively. No gene expression is denoted with grey color. Numbers denote TPM of VvRdDMs.
Expression profiling of VvRdDM genes in various organs and developmental stage
Transcriptomic study of RdDM grapevine genes was undertaken to acquire some hints regarding the probable roles of RdDM grapevine genes in different organs (buds, inflorescences, berries, seeds). The results showed (Figure 9) that the VvNRPD2b, VvNRPE3B, VvRDM2b, VvRDM3e, VvRRP6L1a, VvDRD1f, VvDRD1g, VvAGO1, and VvAGO4a gene showed a broad spectrum of high expression in different organs at various stages, especially VvRDM2b and VvAGO1. During the process of bud development, VvRdDM genes (VvNRPD2b, VvRDM2b, VvRRP6L1a, VvDCL1, etc.) initially showed an increase in expression followed by a subsequent decrease. During April most of the genes showed a significant decrease in expression. However, some of the genes like VvRDM2a, VvRDR1a, and VvAGO4a showed a trend of initial decrease followed by increase in expression. Further, some genes (VvRDM2b, VvDCL2, VvAGO5a, etc.) also showed significant changes at the berry ripening stage, and this trend (change of expression) was also linked with the growth and development of grape organs.
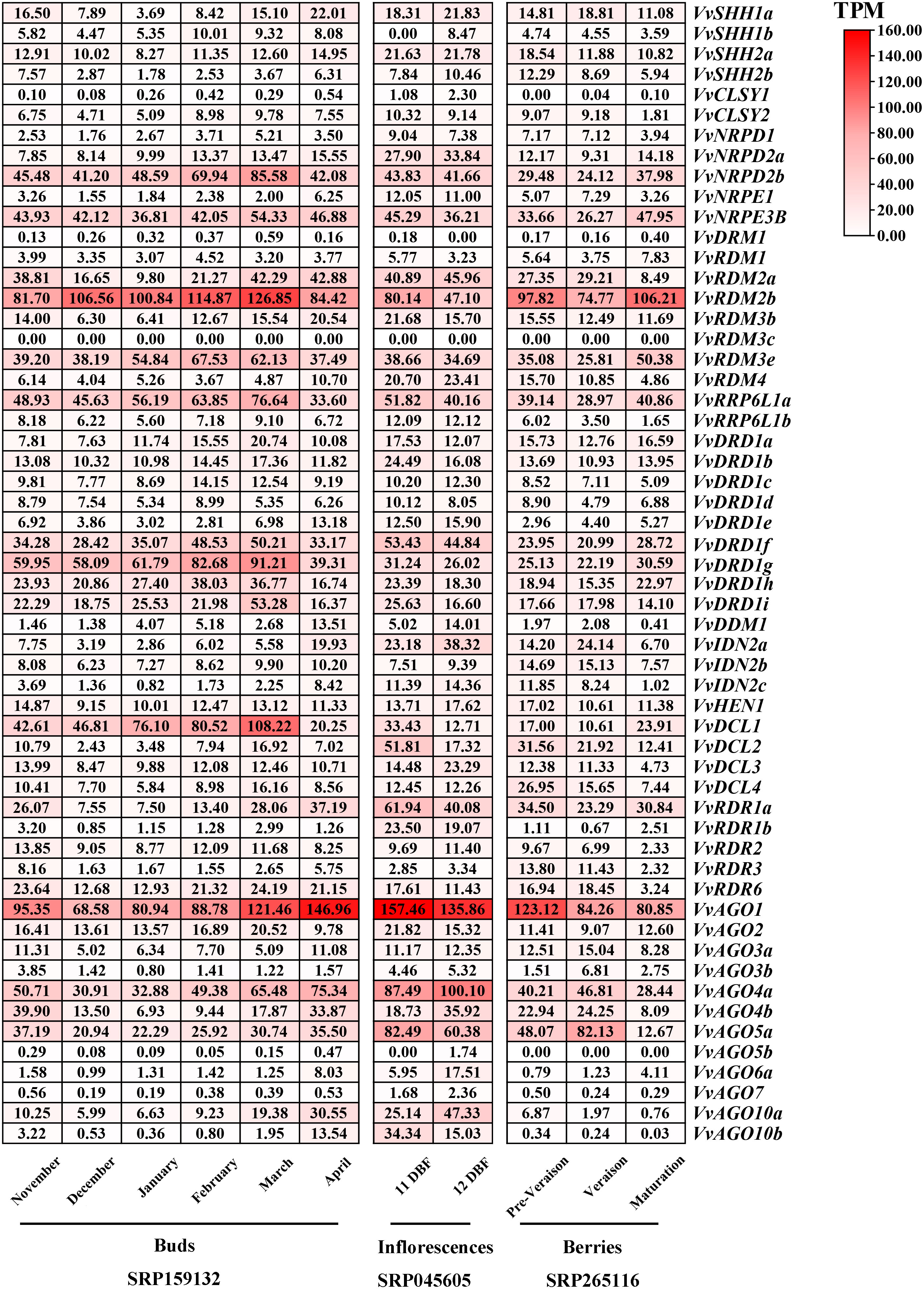
Figure 9 Transcriptomic expression of VvRdDM genes in different organs at various periods of time. The bud samples were collected at every month from November to April. The inflorescence samples were taken at 11 and 12 days before flowering (DBF). The fruit samples were collected at three developmental stages (pre-veraison, veraison, maturation). Transcriptome data were downloaded from the NCBI SRA datasets (SRP159132, SRP045605 and SRP265116). The white and red color scale denotes low and high expression levels, respectively. No gene expression is denoted with grey color. Numbers denote TPM of VvRdDMs.
The transcriptomic expressions of genes during seed development, are presented in the form of a ratio between seedless and seeded (Seedless/Seeded) cultivars at specific time points (initial stage, stage with the highest weight, and stage with the lowest weight during seedless progeny seed development) (Wang et al., 2016) (Figure 10A). The expression of VvRDR1a, VvRDR1b, VvRDR3, VvRDR6, and VvRRP6L1b was significantly higher in seedless progenies than in seeded progenies. Surprisingly, no gene showed significantly higher expression during all stages of seed development in seeded cultivar as compared to seedless. However, VvAGO5b, VvAGO10a, VvCLSY1, and VvDRD1a displayed significantly higher expression in seeded progeny only at one or at two times points. Overall, transcriptomic analysis suggests that some of the VvRdDM genes might have some roles in seed development or ovule abortion.
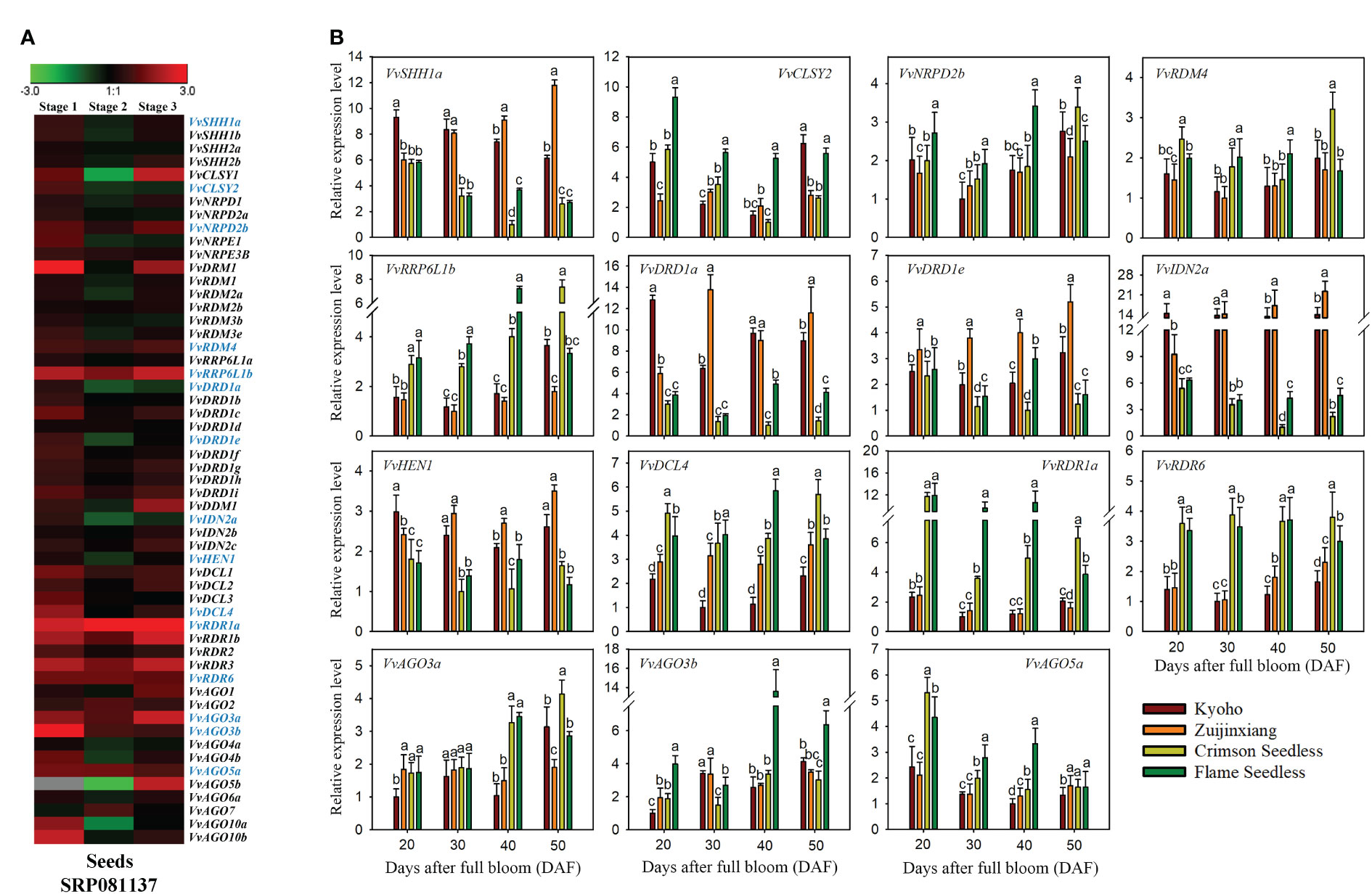
Figure 10 Transcriptomic and expression analysis of grapevine VvRdDM genes during seed development. (A) Heat map of grapevine VvRdDM genes in seeded and seedless progenies during seed development, transcriptome data were downloaded from the NCBI SRA datasets (SRP081137). Data were normalized by log2 the ratio of TPM (seedless/seeded) of each gene during the all three stages. The red and green boxes indicate the relatively high and low expression levels of each gene, respectively. (B) qRT-PCR analysis of VvRdDM genes in four cultivars during seed development. Different cultivars are denoted with different colors. Different letters denote statistically significant differences (ANOVA with a Tukey post-hoc analysis, 5% level). The mean ± S.D. of three biological replicates are mentioned.
Based on transcriptomic analysis, 15 (VvSHH1a, VvCLSY2, VvNRDP2b, VvRDM4, VvRRP6L1b, VvDRD1a, VvDRD1e, VvIDN2a, VvHEN1, VvDCL4, VvRDR1a, VvRDR6, VvAGO3a, VvAGO3b, and VvAGO5a) genes were selected for further qRT-PCR analysis. The qRT-PCR analysis of selected genes was performed to check whether the trend of expression (differential) is common in different cultivars of grapes or it is variety specific. As shown in Figure 10B, VvSHH1a, VvDRD1a, and VvIDN2a, were significantly highly expressed during different stages of seed development in seeded progenies. Further all three genes showed relatively higher change ratio between seedless and seeded cultivars at 40 and 50 DAF as compared to earlier stages. Based on the findings, we postulate that these genes might have a significant role in seed development. On other hand, few genes were highly expressed in specific stages of seed development in seedless cultivars, for example, VvRRP6L1b, VvDCL4, VvRDR1a, and VvRDR6. However, VvRDR1a and VvRDR6 showed significant higher expression during all phases of seed development in seedless cultivars than seeded ones. These findings imply that these genes may contribute to ovule abortion. The outcomes of qRT-PCR and transcriptome were generally in agreement.
Discussion
In plants, RdDM pathway genes control the epigenetic states via De novo methylation and play important roles in multiple biological processes (Matzke and Mosher, 2014; Zhao and Chen, 2014; Matzke et al., 2015). Due to recent advancements in whole-genome mapping and next-generation sequencing, plant breeders are paying more attention in epigenetic research and its use in breeding programmes. The RdDM pathway genes have been characterized in many plants i.e., Arabidopsis, Citrus, Tomato, etc (Kurihara et al., 2008; Qian et al., 2011; Bai et al., 2012; Cao et al., 2016; An et al., 2017; Ahmad et al., 2019; Li et al., 2019; Gao et al., 2020; Mosharaf et al., 2020). However, there have been limited reports for RdDM pathway genes in grapes. Previously, Zhao et al. (2014) identified only three important RdDM pathway-related gene families in grapes i.e., AGO, DCL, and RDR. Here by using the latest high-quality grape reference genome data, we identified 60 RdDM grapevine genes belonging to 14 different subfamilies. Moreover, integrated bioinformatics, transcriptomics, and expression profiling to identify their potential functional roles.
Proteins sequences of one monocot (rice) and three dicots (Arabidopsis, grapes, and tomato) species were used to study the phylogenetic relationships of different RdDM families. Based on phylogenetic studies, VvAGO, VvDCL, and VvRDR proteins were more closely related with SlAGO, SlDCL, and SlRDR proteins, respectively. However, VvRDM proteins were closely related to AtRDM proteins. These results suggest that AGO, DCL, and RDR protein families of grapes and tomato have similarities in evolution and ancestral history. Likewise, RDM proteins of grapes and Arabidopsis might have common ancestors. The pattern of exon-intron distribution within a gene family can provide inklings about evolutionary history. Some of the VvRdDM genes families (AGO, DCL, and IDN) showed similar exon-intron numbers within the same sub-families, proposing that these subfamilies are functionally conserved. Other families showed differences in exon-intron number and distribution patterns even within the subfamilies. These results suggest that during the evolutionary process these subfamilies have either gained or lost exon/introns. Changes in exon number during the evolutionary process play important functions in the evolution of new gene families (Xu et al., 2012). Further, the newly evolved genes are initially redundant and gain new functions with the passage of time (Dias et al., 2003). Like intron-exon arrangement, most of the families (DRD, DCL, CLSY, and DDM) showed similar motif distribution patterns within the subfamilies suggesting these subfamilies are conserved during the evolutionary process. More interestingly, DRD, DCL, CLSY, and DDM all have a common domain (Helicase_C), which consists of ATP-dependent proteins which participate in epigenetic mechanisms via chromatin remodeling by using the energy provided by ATP (Flaus et al., 2006). This indicates this domain has some key roles in the RdDM pathways. However, RDM members showed differences in motif arrangements.
Gene duplications and exon-intron patterns have played a key role in the expansion of gene families during the evolutionary process (Cannon et al., 2004). In case of duplicated gene pairs previous trend was observed i.e., members of four duplicated gene pairs (VvAGO10a-VvAGO10b, VvIDN2a-VvIDN2b, VvAGO5a-VvAGO5b, and VvSHH1a-VvSHH1b) showed a similar trend of exon-intron and motif distribution patterns. Moreover, some duplicated gene pairs (VvAGO10a-VvAGO10b, VvAGO3a-VvAGO3b) showed similar expression patterns under high temperature treatment (Figure 11). Only thirteen genes (8 segmental and 5 tandem) belonging to 4 families (AGO, IDN, SHH, and RDR) are the result of duplication events. All duplicated gene pairs have a purifying mode of selection. These results suggest that these four families are diversified and all others are conserved. Further, the AGO family is more diversified because 50% of members of this family have undergone duplication events. Overall, in the expansion of VvRdDM gene families, we noticed the previous trend that segmental duplication occurs more often than tandem duplication (Cannon et al., 2004). Synteny analysis can provide inklings about the gene functions. We identified 19 syntenic pairs between grape and Arabidopsis RdDM genes. These findings propose that these homologous genes have common ancestors before the speciation. However, relying solely on syntenic linkages to explain evolutionary relationships is difficult. Interestingly, two genes VvCLSY2 and AT5G59390 paired with more than one gene, implying that these genes have gained more changes during the evolutionary process. Based on the stated functions of orthologous genes in one species (Arabidopsis) functions can be predicted in other species (grapes).
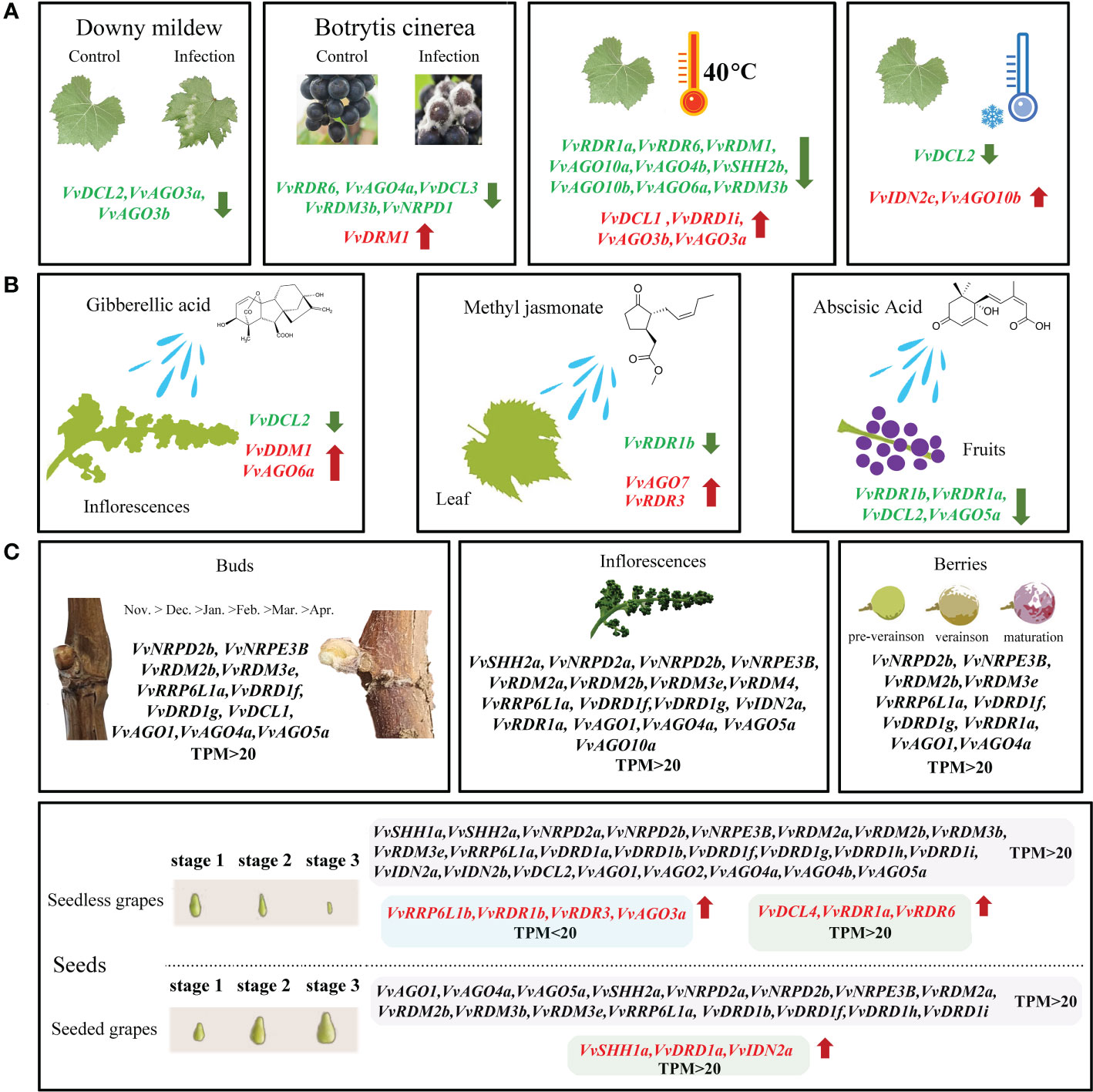
Figure 11 Transcriptomic and expression analysis of grapevine VvRdDM genes. The green and red font represents significant down and up-regulation, respectively. (A) The differential expression of VvRdDM genes following different biotic (downy mildew, botrytis cinerea), and abiotic (heat, cold) treatments. (B) The differential expression of VvRdDM genes under hormone treatments (GA3, MeJA, ABA). (C) The expression of VvRdDM genes in different organs: buds, inflorescence, seeds, and berries. The genes having TPM>20 are listed. In the seeds, the red font indicates that the expression was significantly up-regulated during the seed development in the seedless cultivars as compared with the seeded cultivars and vice versa.
According to promoter analysis, most of the VvRdDM genes have hormone-responsive cis-elements. However, ABA response element (ABRE) was a more abundant hormone-responsive cis-element, and genes VvDCL2, VvRDR1a (contained ABRE elements) significantly responded ABA treatment, moreover, the DEGs after MeJA treatment (VvRDR1b, VvAGO7-CGTCA-motif, TGACG-motif) and GA3 treatment (VvDCL2, VvAGO6a, VvAGO7-P-box) also contained related elements, suggesting the involvement of VvRdDM genes in hormone signaling pathways. Previously, Mosharaf et al. (2020) has also reported the presence of more hormone-responsive cis-elements in CsRdDM genes. Apart from hormone-responsive elements, stress and growth-related elements were also identified, signifying the role of RdDM genes in disease resistance and grapevine growth signaling pathways. These findings predict that regulation of RdDM genes is a complex process and rationalizes the need for further research.
Transcriptome data from SRA can provide some information for further research. In this study, previously published transcriptome data was used to evaluate the response and expression of VvRdDM genes against different treatments. Following different stress treatments or in different organs, the AGO family showed high expression or significant expression change as compared to control, suggesting it’s involvement in multiple biological processes (Figure 11). Previously, AGO family members have displayed diverse functions in different species. For example, in A. thaliana AGO mutant showed enhanced susceptibility to fungi (Ellendorff et al., 2009) and in another study AtAGO4 mutants were associated with resistance to bacterial pathogens (Le et al., 2014). SlAGO4A has been reported for its role in salt and drought stress tolerance in tomato (Huang et al., 2016) and OsAGO17 regulated seed size in rice (Zhong et al., 2020). AGO proteins are core components of the RNA-induced silencing complex (RISC) (Hutvagner and Simard, 2008), and this may be the potential explanation to its diverse roles. VvRDM2b and VvAGO1 (Figure 7–9) gene showed a broad spectrum of high expression in different organs at various stages even under different treatment. VvRdDM genes not only showed diverse expression but also showed spatio-temporal expression patterns. For example, VvRDR1a, VvIDN2a, and VvNRPD2b, were significantly more expressed at the hard green stage than at the ripened stage. VvAGO5a was significantly differentially expressed at all stages of fruit ripening. Most of the VvRdDM genes showed significant down-regulation following different treatments (Figure 11). Surprisingly, VvDCL2 displayed same expression trend under the conditions of downy mildew, low temperature, GA3 and ABA treatment. Further, the expression level of VvDCL2 was significantly down-regulated following low temperature treatment (108 to 27) suggesting some role in the adaptation of grape to low temperatures. Our results are also supported by the earlier findings i.e., the activity of DCL2 in A. thaliana was related with temperature (Zhang et al., 2012). Prupe DCL under drought treatment was differentially expressed in peach (Belal et al., 2022). These findings suggest, that the functions of DCL2 are diverse, and further research is needed for exploring the exact functions of VvDCL2. The VvRdDM genes showed differential expressions in different organs, indicating their involvement in the growth and development process. Our results are in line with the previous findings, as AtRRP6L1 and AtRRP6L2 (homologous genes of VvRRP6L1a) regulated flowering (Shin and Chekanova, 2014), DDM1 (homolog VvDDM1) mutant effected seed development process (Xiao et al., 2006) in A. thaliana. Silencing of FvAGO4 (homolog VvAGO4a) during fruit development resulted in early ripening of strawberry fruit (Cheng et al., 2018). Based on these findings, we speculate that VvRdDM genes can play critical role in the growth and development of grapes.
Expression patterns of fifteen genes during seed development were compared among four different (2 seeded and 2 seedless) cultivars of grapes. Interestingly, three genes (VvSHH1a, VvDRD1a, and VvIDN2a) showed higher expression in seeded cultivars especially at 40 and 50 DAF, signifying their key roles in grapevine seed development pathways. Previous results support our findings; for example, in Hypericum perforatum L, a gene with high similarity to AtIDN2 demonstrated differential expression in the ovule of apomictic H. perforatum. Furthermore, in A. thaliana, silencing the IDN2 gene led to decreased seed set and changes in seed size (Basso et al., 2019). However, in seedless cultivars, VvRDR1a and VvRDR6 showed significantly higher expression throughout all phases of seed formation. Similar trend of higher expression was noted during transcriptomic analysis. Relying on the findings, we can predict that these two genes might have roles in ovule abortion in grapes. Our results are in concordant with the previous findings of Grover et al. (2018) who noticed the abortion of brassica rapa seeds due to maternal alterations in the Pol IV-dependent small RNA pathway. They reported that RdDM pathway genes have critical roles in brassica rapa seed development. These findings rationalize the need for a future study about the functional characterization of these (VvRDR1a and VvRDR6) genes.
Conclusion
A total of 60 RdDM genes in grapevine were identified, and comprehensive bioinformatic and expression analysis were performed. VvRdDM proteins were evaluated in terms of chromosomal positions, exon-intron distribution, phylogenetic, and evolutionary history. Expression analysis including transcriptomic analysis of VvRdDM genes following different stresses and hormonal treatments and qRT-PCR analysis of selected VvRdDM genes during different stages of seed development suggested their roles in multiple biological processes. VvRDR1a and VvRDR6 gene were proposed as candidate gene resources for further functional characterization. Our study provides new resources for grape molecular breeding and inklings about the role of epigenetics in grapevine growth and development.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
LW and RX designed the research. LW and GD supervised the experiments. RX performed most of the experiments. BA, LY and CL provided technical assistance. RX and XS analyzed the data. BA, RX and LW wrote the article with contributions of all the authors. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Natural Science Youth Foundation of Hebei, China (C2021204146), the Scientific Research Program of Hebei Educational Commission (QN2020232), the Strat-up Funds of Hebei Agricultural University to LW (YJ201806), the Earmarked Fund for Hebei Agriculture Research System (13000022P00E90410004M), and the Earmarked Fund for Sci-Tech Innovation in Modern Breeding Industry (21326310D).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1089392/full#supplementary-material
References
Ahmad, F., Farman, K., Waseem, M., Rana, R. M., Nawaz, M. A., Rehman, H. M., et al. (2019). Genome-wide identification, classification, expression profiling and DNA methylation (5mC) analysis of stress-responsive ZFP transcription factors in rice (Oryza sativa l.). Gene. 718, 144018. doi: 10.1016/j.gene.2019.144018
Ahmad, B., Yao, J., Zhang, S., Li, X., Zhang, X., Yadav, V., et al. (2020). Genome-wide characterization and expression profiling of GASA genes during different stages of seed development in grapevine (Vitis vinifera l.) predict their involvement in seed development. Int. J. Mol. Sci. 21 (3), 1088. doi: 10.3390/ijms21031088
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25 (17), 3389–3402. doi: 10.1093/nar/25.17.3389
An, Y. C., Goettel, W., Han, Q., Bartels, A., Liu, Z., Xiao, W. (2017). Dynamic changes of genome-wide DNA methylation during soybean seed development. Sci. Rep. 7 (1), 12263. doi: 10.1038/s41598-017-12510-4
Ausin, I., Mockler, T. C., Chory, J., Jacobsen, S. E. (2009). IDN1 and IDN2 are required for de novo DNA methylation in Arabidopsis thaliana. Nat. Struct. Mol. Biol. 16 (12), 1325–1327. doi: 10.1038/nsmb.1690
Bailey, T. L., Elkan, C. (1994). Fitting a mixture model by expectation maximization to discover motifs in bipolymers. Proc. Int. Conf Intell. Syst. Mol. Biol. 2, 28–36.
Bai, M., Yang, G. S., Chen, W. T., Mao, Z. C., Kang, H. X., Chen, G. H., et al. (2012). Genome-wide identification of dicer-like, argonaute and RNA-dependent RNA polymerase gene families and their expression analyses in response to viral infection and abiotic stresses in Solanum lycopersicum. Gene. 501 (1), 52–62. doi: 10.1016/j.gene.2012.02.009
Bartels, A., Han, Q., Nair, P., Stacey, L., Gaynier, H., Mosley, M., et al. (2018). Dynamic DNA methylation in plant growth and development. Int. J. Mol. Sci. 19 (7), 2144. doi: 10.3390/ijms19072144
Barter, M., Bui, C., Young, D. (2012). Epigenetic mechanisms in cartilage and osteoarthritis: DNA methylation, histone modifications and microRNAs. Osteoarthritis Cartilage. 20 (5), 339–349. doi: 10.1016/j.joca.2011.12.012
Basso, A., Barcaccia, G., Galla, G. (2019). Annotation and expression of IDN2-like and FDM-like genes in sexual and aposporous Hypericum perforatum l. accessions. Plants. 8 (6), 158. doi: 10.3390/plants8060158
Belal, M. A., Ezzat, M., Zhang, Y., Xu, Z., Cao, Y., Han, Y. (2022). Integrative analysis of the DICER-like (DCL) genes from peach (Prunus persica): A critical role in response to drought stress. Front. Ecol. Evol. 10. doi: 10.3389/fevo.2022.923166
Bewick, A. J., Schmitz, R. J. (2017). Gene body DNA methylation in plants. Curr. Opin. Plant Biol. 36, 103–110. doi: 10.1016/j.pbi.2016.12.007
Bitonti, M. B., Cozza, R., Chiappetta, A., Giannino, D., Castiglione, M. R., Dewitte, W., et al. (2002). Distinct nuclear organization, DNA methylation pattern and cytokinin distribution mark juvenile, juvenile-like and adult vegetative apical meristems in peach (Prunus persica (L.) batsch). J. Exp. Botany. 53 (371), 1047–1054. doi: 10.1093/jexbot/53.371.1047
Cannon, S. B., Mitra, A., Baumgarten, A., Young, N. D., May, G. (2004). The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 4 (1), 1–21. doi: 10.1186/1471-2229-4-10
Cao, J. Y., Xu, Y. P., Li, W., Li, S. S., Rahman, H., Cai, X. Z. (2016). Genome-wide identification of dicer-like, argonaute, and RNA-dependent RNA polymerase gene families in brassica species and functional analyses of their arabidopsis homologs in resistance to Sclerotinia sclerotiorum. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.01614
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13 (8), 1194–1202. doi: 10.1016/j.molp.2020.06.009
Cheng, J., Niu, Q., Zhang, B., Chen, K., Yang, R., Zhu, J.-K., et al. (2018). Downregulation of RdDM during strawberry fruit ripening. Genome Biol. 19 (1), 212. doi: 10.1186/s13059-018-1587-x
Daccord, N., Celton, J.-M., Linsmith, G., Becker, C., Choisne, N., Schijlen, E., et al. (2017). High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 49 (7), 1099–1106. doi: 10.1038/ng.3886
Dias, A. P., Braun, E. L., McMullen, M. D., Grotewold, E. (2003). Recently duplicated maize R2R3 myb genes provide evidence for distinct mechanisms of evolutionary divergence after duplication. Plant Physiol. 131 (2), 610–620. doi: 10.1104/pp.012047
Dinh, T. T., O’Leary, M., Won, S. Y., Li, S., Arroyo, L., Liu, X., et al. (2013). Generation of a luciferase-based reporter for CHH and CG DNA methylation in Arabidopsis thaliana. Silence. 4 (1), 1–11. doi: 10.1186/1758-907X-4-1
Ellendorff, U., Fradin, E. F., de Jonge, R., Thomma, B. P. (2009). RNA Silencing is required for Arabidopsis defence against Verticillium wilt disease. J. Exp. Bot. 60 (2), 591–602. doi: 10.1093/jxb/ern306
El-Sharkawy, I., Liang, D., Xu, K. (2015). Transcriptome analysis of an apple (Malus× domestica) yellow fruit somatic mutation identifies a gene network module highly associated with anthocyanin and epigenetic regulation. J. Exp. Botany. 66 (22), 7359–7376. doi: 10.1093/jxb/erv433
Erdmann, R. M., Picard, C. L. (2020). RNA-Directed DNA methylation. PloS Genet. 16 (10), e1009034. doi: 10.1371/journal.pgen.1009034
Flaus, A., Martin, D. M., Barton, G. J., Owen-Hughes, T. (2006). Identification of multiple distinct Snf2 subfamilies with conserved structural motifs. Nucleic Acids Res. 34 (10), 2887–2905. doi: 10.1093/nar/gkl295
Gao, C., Deng, M., Yang, X., Yu, W., Cai, J., Shi, Y., et al. (2020). Genome-wide identification and coexpression network analysis of DNA methylation pathway genes and their differentiated functions in Ginkgo biloba l. Forests. 11 (10), 1076. doi: 10.3390/f11101076
Garcia-Ruiz, H., Takeda, A., Chapman, E. J., Sullivan, C. M., Fahlgren, N., Brempelis, K. J., et al. (2010). Arabidopsis RNA-dependent RNA polymerases and dicer-like proteins in antiviral defense and small interfering RNA biogenesis during Turnip mosaic virus infection. Plant Cell. 22 (2), 481–496. doi: 10.1105/tpc.109.073056
Grimplet, J., Adam-Blondon, A.-F., Bert, P.-F., Bitz, O., Cantu, D., Davies, C., et al. (2014). The grapevine gene nomenclature system. BMC Genomics 15 (1), 1–14. doi: 10.1186/1471-2164-15-1077
Grover, J. W., Kendall, T., Baten, A., Burgess, D., Freeling, M., King, G. J., et al. (2018). Maternal components of RNA-directed DNA methylation are required for seed development in Brassica rapa. Plant J. 94 (4), 575–582. doi: 10.1111/tpj.13910
He, X.-J., Hsu, Y.-F., Pontes, O., Zhu, J., Lu, J., Bressan, R. A., et al. (2009a). NRPD4, a protein related to the RPB4 subunit of RNA polymerase II, is a component of RNA polymerases IV and V and is required for RNA-directed DNA methylation. Genes Dev. 23 (3), 318–330. doi: 10.1101/gad.1765209
He, X.-J., Hsu, Y.-F., Zhu, S., Liu, H.-L., Pontes, O., Zhu, J., et al. (2009b). A conserved transcriptional regulator is required for RNA-directed DNA methylation and plant development. Genes Dev. 23 (23), 2717–2722. doi: 10.1101/gad.1851809
Henderson, I. R., Deleris, A., Wong, W., Zhong, X., Chin, H. G., Horwitz, G. A., et al. (2010). The de novo cytosine methyltransferase DRM2 requires intact UBA domains and a catalytically mutated paralog DRM3 during RNA–directed DNA methylation in Arabidopsis thaliana. PloS Genet. 6 (10), e1001182. doi: 10.1371/journal.pgen.1001182
Hirtreiter, A., Damsma, G. E., Cheung, A. C. M., Klose, D., Grohmann, D., Vojnic, E., et al. (2010). Spt4/5 stimulates transcription elongation through the RNA polymerase clamp coiled-coil motif. Nucleic Acids Res. 38 (12), 4040–4051. doi: 10.1093/nar/gkq135
Holliday, R. (2006). Epigenetics: a historical overview. Epigenetics. 1 (2), 76–80. doi: 10.4161/epi.1.2.2762
Holub, E. B. (2001). The arms race is ancient history in Arabidopsis, the wildflower. Nat. Rev. Genet. 2 (7), 516–527. doi: 10.1038/35080508
Huang, Y., Ji, L., Huang, Q., Vassylyev, D. G., Chen, X., Ma, J.-B. (2009). Structural insights into mechanisms of the small RNA methyltransferase HEN1. Nature. 461 (7265), 823–827. doi: 10.1038/nature08433
Huang, H., Liu, R., Niu, Q., Tang, K., Zhang, B., Zhang, H., et al. (2019). Global increase in DNA methylation during orange fruit development and ripening. Proc. Natl. Acad. Sci. 116 (4), 1430–1436. doi: 10.1073/pnas.1815441116
Huang, W., Xian, Z., Hu, G., Li, Z. (2016). SlAGO4A, a core factor of RNA-directed DNA methylation (RdDM) pathway, plays an important role under salt and drought stress in tomato. Mol. Breeding. 36 (3), 1–3. doi: 10.1007/s11032-016-0439-1
Hutvagner, G., Simard, M. J. (2008). Argonaute proteins: key players in RNA silencing. Nat. Rev. Mol. Cell Biol. 9 (1), 22–32. doi: 10.1038/nrm2321
Hu, Y., Zhu, N., Wang, X., Yi, Q., Zhu, D., Lai, Y., et al. (2013). Analysis of rice Snf2 family proteins and their potential roles in epigenetic regulation. Plant Physiol. Biochem. 70, 33–42. doi: 10.1016/j.plaphy.2013.05.001
Jones, A. L., Sung, S. (2014). Mechanisms underlying epigenetic regulation in Arabidopsis thaliana. Integr. Comp. Biol. 54 (1), 61–67. doi: 10.1093/icb/icu030
Kawakatsu, T., Nery, J. R., Castanon, R., Ecker, J. R. (2017). Dynamic DNA methylation reconfiguration during seed development and germination. Genome Biol. 18 (1), 1–12. doi: 10.1186/s13059-017-1251-x
Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evolution. 35 (6), 1547. doi: 10.1093/molbev/msy096
Kurihara, Y., Matsui, A., Kawashima, M., Kaminuma, E., Ishida, J., Morosawa, T., et al. (2008). Identification of the candidate genes regulated by RNA-directed DNA methylation in Arzabidopsis. Biochem. Biophys. Res. Commun. 376 (3), 553–557. doi: 10.1016/j.bbrc.2008.09.046
Lange, H., Holec, S., Cognat, V., Pieuchot, L., Le Ret, M., Canaday, J., et al. (2008). Degradation of a polyadenylated rRNA maturation by-product involves one of the three RRP6-like proteins in Arabidopsis thaliana. Mol. Cell. Biol. 28 (9), 3038–3044. doi: 10.1128/MCB.02064-07
Lee, T.-H., Tang, H., Wang, X., Paterson, A. H. (2012). PGDD: a database of gene and genome duplication in plants. Nucleic Acids Res. 41 (D1), D1152–D1158. doi: 10.1093/nar/gks1104
Le, T. N., Schumann, U., Smith, N. A., Tiwari, S., Au, P. C., Zhu, Q. H., et al. (2014). DNA Demethylases target promoter transposable elements to positively regulate stress responsive genes in Arabidopsis. Genome Biol. 15 (9), 458. doi: 10.1186/s13059-014-0458-3
Lescot, M., Déhais, P., Thijs, G., Marchal, K., Moreau, Y., Van de Peer, Y., et al. (2002). PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30 (1), 325–327. doi: 10.1093/nar/30.1.325
Li, W.-F., Ning, G.-X., Mao, J., Guo, Z.-G., Zhou, Q., Chen, B.-H. (2019). Whole-genome DNA methylation patterns and complex associations with gene expression associated with anthocyanin biosynthesis in apple fruit skin. Planta. 250 (6), 1833–1847. doi: 10.1007/s00425-019-03266-4
Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2– ΔΔCT method. Methods. 25 (4), 402–408. doi: 10.1006/meth.2001.1262
López, A., Ramírez, V., García-Andrade, J., Flors, V., Vera, P. (2011). The RNA silencing enzyme RNA polymerase v is required for plant immunity. PloS Genet. 7 (12), e1002434. doi: 10.1371/journal.pgen.1002434
Love, M. I., Huber, W., Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15 (12), 550. doi: 10.1186/s13059-014-0550-8
Manning, K., Tör, M., Poole, M., Hong, Y., Thompson, A. J., King, G. J., et al. (2006). A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 38 (8), 948–952. doi: 10.1038/ng1841
Margis, R., Fusaro, A. F., Smith, N. A., Curtin, S. J., Watson, J. M., Finnegan, E. J., et al. (2006). The evolution and diversification of dicers in plants. FEBS Letters. 580 (10), 2442–2450. doi: 10.1016/j.febslet.2006.03.072
Matzke, M. A., Kanno, T., Matzke, A. J. (2015). RNA-Directed DNA methylation: The evolution of a complex epigenetic pathway in flowering plants. Annu. Rev. Plant Biol. 66, 243–267. doi: 10.1146/annurev-arplant-043014-114633
Matzke, M. A., Mosher, R. A. (2014). RNA-Directed DNA methylation: an epigenetic pathway of increasing complexity. Nat. Rev. Genet. 15 (6), 394–408. doi: 10.1038/nrg3683
Mosharaf, M. P., Rahman, H., Ahsan, M. A., Akond, Z., Ahmed, F. F., Islam, M. M., et al. (2020). In silico identification and characterization of AGO, DCL and RDR gene families and their associated regulatory elements in sweet orange (Citrus sinensis l.). PloS One 15 (12), e0228233. doi: 10.1371/journal.pone.0228233
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T., Salzberg, S. L. (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and ballgown. Nat. Protoc. 11 (9), 1650–1667. doi: 10.1038/nprot.2016.095
Qian, Y., Cheng, Y., Cheng, X., Jiang, H., Zhu, S., Cheng, B. (2011). Identification and characterization of dicer-like, argonaute and RNA-dependent RNA polymerase gene families in maize. Plant Cell Rep. 30 (7), 1347–1363. doi: 10.1007/s00299-011-1046-6
Qin, L., Mo, N., Muhammad, T., Liang, Y. (2018). Genome-wide analysis of DCL, AGO, and RDR gene families in pepper (Capsicum annuum l.). Int. J. Mol. Sci. 19 (4), 1038. doi: 10.3390/ijms19041038
Ream, T. S., Haag, J. R., Wierzbicki, A. T., Nicora, C. D., Norbeck, A. D., Zhu, J.-K., et al. (2009). Subunit compositions of the RNA-silencing enzymes pol IV and pol V reveal their origins as specialized forms of RNA polymerase II. Mol. Cell. 33 (2), 192–203. doi: 10.1016/j.molcel.2008.12.015
Sabbione, A., Daurelio, L., Vegetti, A., Talon, M., Tadeo, F., Dotto, M. (2019). Genome-wide analysis of AGO, DCL and RDR gene families reveals RNA-directed DNA methylation is involved in fruit abscission in Citrus sinensis. BMC Plant Biol. 19 (1), 401. doi: 10.1186/s12870-019-1998-1
Sasaki, T., Lorković, Z. J., Liang, S.-C., Matzke, A. J., Matzke, M. (2014). The ability to form homodimers is essential for RDM1 to function in RNA-directed DNA methylation. PloS One 9 (2), e88190. doi: 10.1371/journal.pone.0088190
Shin, J. H., Chekanova, J. A. (2014). Arabidopsis RRP6L1 and RRP6L2 function in FLOWERING LOCUS c silencing via regulation of antisense RNA synthesis. PloS Genet. 10 (9), e1004612. doi: 10.1371/journal.pgen.1004612
Siltberg, J., Liberles, D. (2002). A simple covarion-based approach to analyse nucleotide substitution rates. J. Evolutionary Biol. 15 (4), 588–594. doi: 10.1046/j.1420-9101.2002.00416.x
Smith, L. M., Pontes, O., Searle, I., Yelina, N., Yousafzai, F. K., Herr, A. J., et al. (2007). An SNF2 protein associated with nuclear RNA silencing and the spread of a silencing signal between cells in Arabidopsis. Plant Cell. 19 (5), 1507–1521. doi: 10.1105/tpc.107.051540
This, P., Lacombe, T., Thomas, M. R. (2006). Historical origins and genetic diversity of wine grapes. Trends Genet. 22 (9), 511–519. doi: 10.1016/j.tig.2006.07.008
Wang, L., Ahmad, B., Liang, C., Shi, X., Sun, R., Zhang, S., et al. (2020). Bioinformatics and expression analysis of histone modification genes in grapevinepredict their involvement in seed development, powdery mildew resistance, andhormonal signaling. BMC Plant Biology 20 (1), 1–20. doi: 10.1186/s12870-020-02618-7
Wang, L., Hu, X., Jiao, C., Li, Z., Fei, Z., Yan, X., et al. (2016). Transcriptomeanalyses of seed development in grape hybrids reveals a possible mechanisminfluencing seed size. BMC Genomics 17 (1), 1–15. doi: 10.1186/s12864-016-3193-1
Wassenegger, M., Krczal, G. (2006). Nomenclature and functions of RNA-directed RNA polymerases. Trends Plant Science. 11 (3), 142–151. doi: 10.1016/j.tplants.2006.01.003
Wei, S. J., Chai, S., Zhu, R. M., Duan, C. Y., Zhang, Y., Li, S. (2020). HUA ENHANCER1 mediates ovule development. Front. Plant Sci. 11. doi: 10.3389/fpls.2020.00397
Wierzbicki, A. T., Haag, J. R., Pikaard, C. S. (2008). Noncoding transcription by RNA polymerase pol IVb/Pol V mediates transcriptional silencing of overlapping and adjacent genes. Cell. 135 (4), 635–648. doi: 10.1016/j.cell.2008.09.035
Xiao, W., Brown, R. C., Lemmon, B. E., Harada, J. J., Goldberg, R. B., Fischer, R. L. (2006). Regulation of seed size by hypomethylation of maternal and paternal genomes. Plant Physiol. 142 (3), 1160–1168. doi: 10.1104/pp.106.088849
Xi, Y., Liu, J., Dong, C., Cheng, Z.-M. (2017). The CBL and CIPK gene family in grapevine (Vitis vinifera): genome-wide analysis and expression profiles in response to various abiotic stresses. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.00978
Xu, G., Guo, C., Shan, H., Kong, H. (2012). Divergence of duplicate genes in exon–intron structure. Proc. Natl. Acad. Sci. 109 (4), 1187–1192. doi: 10.1073/pnas.1109047109
Xu, D., Lu, Z., Jin, K., Qiu, W., Qiao, G., Han, X., et al. (2021). SPDE: A multi-functional software for sequence processing and data extraction. Bioinformatics. 37(20), 3686–3687. doi: 10.1093/bioinformatics/btab235
Zhang, H., Lang, Z., Zhu, J. K. (2018). Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 19 (8), 489–506. doi: 10.1038/s41580-018-0016-z
Zhang, H., Ma, Z.-Y., Zeng, L., Tanaka, K., Zhang, C.-J., Ma, J., et al. (2013). DTF1 is a core component of RNA-directed DNA methylation and may assist in the recruitment of pol IV. Proc. Natl. Acad. Sci. 110 (20), 8290–8295. doi: 10.1073/pnas.1300585110
Zhang, X., Zhang, X., Singh, J., Li, D., Qu, F. (2012). Temperature-dependent survival of Turnip crinkle virus-infected arabidopsis plants relies on an RNA silencing-based defense that requires dcl2, AGO2, and HEN1. J. Virol. 86 (12), 6847–6854. doi: 10.1128/jvi.00497-12
Zhao, Y., Chen, X. (2014). Non-coding RNAs and DNA methylation in plants. Natl. Sci. Review. 1 (2), 219–229. doi: 10.1093/nsr/nwu003
Zhao, H., Zhao, K., Wang, J., Chen, X., Chen, Z., Cai, R., et al. (2014). Comprehensive analysis of dicer-like, argonaute, and RNA-dependent RNA polymerase gene families in grapevine (Vitis vinifera). J. Plant Growth Regulation. 34 (1), 108–121. doi: 10.1007/s00344-014-9448-7
Keywords: RdDM, seed development, grapes, DNA methylation, expression analysis
Citation: Xiang R, Ahmad B, Liang C, Shi X, Yang L, Du G and Wang L (2022) Systematic genome-wide and expression analysis of RNA-directed DNA methylation pathway genes in grapes predicts their involvement in multiple biological processes. Front. Plant Sci. 13:1089392. doi: 10.3389/fpls.2022.1089392
Received: 04 November 2022; Accepted: 25 November 2022;
Published: 09 December 2022.
Edited by:
Chenxia Cheng, Qingdao Agricultural University, ChinaReviewed by:
Xiangpeng Leng, Qingdao Agricultural University, ChinaSongtao Jiu, Shanghai Jiao Tong University, China
Copyright © 2022 Xiang, Ahmad, Liang, Shi, Yang, Du and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Wang, dml2aUBoZWJhdS5lZHUuY24=; Guoqiang Du, Z2R1QGhlYmF1LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship