 Qiang Han1‡
Qiang Han1‡ Yu-Hung Hung2,3‡
Yu-Hung Hung2,3‡ Changqing Zhang2,3
Changqing Zhang2,3 Arthur Bartels1
Arthur Bartels1 Matthew Rea1†
Matthew Rea1† Hanwen Yang1
Hanwen Yang1 Christine Park1
Christine Park1 Xiang-Qian Zhang2,3,4Robert L. Fischer5*
Xiang-Qian Zhang2,3,4Robert L. Fischer5* Wenyan Xiao1*
Wenyan Xiao1* Tzung-Fu Hsieh2,3*
Tzung-Fu Hsieh2,3*- 1Department of Biology, Saint Louis University, St. Louis, MO, United States
- 2Department of Plant and Microbial Biology, North Carolina State University, Raleigh, NC, United States
- 3Plants for Human Health Institute, North Carolina State University, Kannapolis, NC, United States
- 4College of Food Science and Engineering, Foshan University, Foshan, China
- 5Department of Plant and Microbial Biology, University of California, Berkeley, Berkeley, CA, United States
The Arabidopsis DEMETER (DME) DNA glycosylase demethylates the central cell genome prior to fertilization. This epigenetic reconfiguration of the female gamete companion cell establishes gene imprinting in the endosperm and is essential for seed viability. DME demethylates small and genic-flanking transposons as well as intergenic and heterochromatin sequences, but how DME is recruited to these loci remains unknown. H1.2 was identified as a DME-interacting protein in a yeast two-hybrid screen, and maternal genome H1 loss affects DNA methylation and expression of selected imprinted genes in the endosperm. Yet, the extent to which H1 influences DME demethylation and gene imprinting in the Arabidopsis endosperm has not been investigated. Here, we showed that without the maternal linker histones, DME-mediated demethylation is facilitated, particularly in the heterochromatin regions, indicating that H1-bound heterochromatins are barriers for DME demethylation. Loss of H1 in the maternal genome has a very limited effect on gene transcription or gene imprinting regulation in the endosperm; however, it variably influences euchromatin TE methylation and causes a slight hypermethylation and a reduced expression in selected imprinted genes. We conclude that loss of maternal H1 indirectly influences DME-mediated demethylation and endosperm DNA methylation landscape but does not appear to affect endosperm gene transcription and overall imprinting regulation.
Introduction
DNA methylation regulates important processes in eukaryotic genomes including gene transcription, transposon silencing and genomic imprinting (Law and Jacobsen, 2010). In plants, de novo methylation is guided by small interfering RNAs in a process known as the RNA-directed DNA methylation (RdDM) (Matzke and Mosher, 2014). Once DNA methylation is established, it is maintained upon replication by distinct DNA methyltransferases to preserve cell identity and genome integrity. Plant DNA methylation is found in CG, CHG, and CHH sequence contexts (H is A, C, or T) and is mainly targeted to transposons or repetitive sequences of the genome. DNA methylation also needs to be dynamically reconfigured during development to enable transition to a new cell fate and transcriptional state. Such epigenetic reconfiguration plays a prominent role in animal reproduction and is required for reproductive success in flowering plant (Ono and Kinoshita, 2021). Removal of DNA methylation in plant is catalyzed by the DNA glycosylase DEMETER (DME), repressor of silencing1 (ROS1), DEMETER LIKE2 (DML2), and DML3 in Arabidopsis (Agius et al., 2006; Gehring et al., 2006) through a Base Excision Repair (BER) pathway. Whereas ROS1, DML2 and DML3 are more widely expressed and function to counteract the spread of RdDM methylation into nearby coding genes, DME demethylation during reproduction establishes gene imprinting in the endosperm and is essential for seed viability (Choi et al., 2002; Gehring et al., 2006; Gent et al., 2022; Xu et al., 2022).
During Arabidopsis reproduction, the central cell genome is extensively demethylated at about nine thousand loci prior to fertilization. This epigenetic reconfiguration differentiates the imprints of parental genomes and establishes the parent-of-origin specific expression of many imprinted genes, including two essential components of the PRC2 complex (i.e., MEDEA and FIS2) crucial for seed development (Choi et al., 2002; Jullien et al., 2006; Huh et al., 2008). In pollen, DME also demethylates the vegetative cell genome to reinforce DNA methylation of the sperm and to ensure a robust pollen germination in certain ecotypes (Schoft et al., 2011; Ibarra et al., 2012). Genomic regions hypermethylated in dme mutant reside primarily in gene-flanking small euchromatic transposons as well as in many intergenic and heterochromatin sequences. Such loci that satisfy differential methylation criteria were generally referred to as DME target loci, even though a direct physical DME localization remains to be demonstrated. (Ibarra et al., 2012). How DME is recruited to distinct genomic regions with different chromatin states remains elusive, although the Facilitates Chromatin Transactions (FACT) histone chaperone is required for demethylation of heterochromatin and certain imprinted loci in the central cell (Ikeda et al., 2011; Frost et al., 2018). FACT complex is known to play a pivotal role in almost all chromatin-related processes, including transcription, replication, and DNA repair (Hondele and Ladurner, 2013). This is because nucleosomes are barriers to these processes that require access to the nucleosomal DNA, and the FACT complex is needed to facilitate destabilizing and disassembling nucleosomes. Interestingly, the FACT complex is only required for DME demethylation in central cell but not in vegetative nuclei (Frost et al., 2018), highlighting a difference in their chromatin conformation. There are three canonical histone H1 variants in Arabidopsis, the ubiquitous H1.1 and H1.2 variants and the stress-inducible H1.3 (Gantt and Lenvik, 1991; Ascenzi and Gantt, 1999a; Wierzbicki and Jerzmanowski, 2005; Kotlinski et al., 2017). The vegetative cell has a decondensed nuclei and highly dispersed heterochromatin (Schoft et al., 2009); the two canonical linker histone H1.1 and H1.2 are specifically absent in the vegetative cell, which might contribute to the differential requirement of FACT for DME function (Hsieh et al., 2016; He et al., 2019).
H1 linker histones are conserved eukaryotic nuclear proteins required to maintain higher order chromatin structure and DNA methylation patterns in plants and animals (Fan et al., 2005; Wierzbicki and Jerzmanowski, 2005; Zemach et al., 2013). H1 is found more enriched in heterochromatin than euchromatin in Arabidopsis and is thought to participate in heterochromatin condensation in plant cells (Ascenzi and Gantt, 1999b; Rutowicz et al., 2015; Choi et al., 2020). H1 loss causes dispersion of heterochromatin, nucleosome reorganization, and de-repression of H1-bound genes (Rutowicz et al., 2019; Choi et al., 2020). It has been suggested that H1-enriched chromatins are less accessible to DNA modifying enzymes, which is supported by the observations that gain in heterochromatin DNA methylation due to H1 loss likely results from increased access by DNA methyltransferases to the less condensed heterochromatin (Zemach et al., 2013; Lyons and Zilberman, 2017). However, euchromatic TEs exhibit a loss in DNA methylation in h1 double mutant (h1.1-1,h1.2-1), for reasons not fully understood (Zemach et al., 2013). Although earlier studies have shown that reducing the level of histone H1 affected DNA methylation patterns of some imprinted genes in mammals and plants (Fan et al., 2005; Wierzbicki and Jerzmanowski, 2005; Rea et al., 2012), how H1 takes part in these processes has not been elucidated.
H1.2 interacts with DME in a yeast two-hybrid screen and an in vitro pull-down assay and its isoforms H1.1 and H1.3 were also shown to interact with DME (Rea et al., 2012). In the endosperm derived from a cross between female h1 triple mutant (h1.1, h1.2-1, h1.3) and wild-type pollen, the expression of selected DME-regulated imprinted genes (MEA, FIS2, FWA) was reduced, accompanied with an increase in the methylation of their maternal alleles, suggesting that H1 might play a role in mediating DME demethylation. However, loss of H1 differentially influences DNA methylation in heterochromatic and euchromatic TEs, raising the possibility that these effects might be epistatic to DME action. Here we showed that both H1.1 and H1.2 promoters are active in the central cell, with H1.1 being more ubiquitously expressed in ovule while H1.2 is preferentially expressed in the central cell. Our methylome analysis showed that H1 loss in the maternal genome resulted in endosperm heterochromatin hypomethylation, suggesting that the central cell chromatin is less condensed and more accessible by DME in the absence of H1. Maternal H1 loss differentially affected methylation pattern of canonical DME DMRs but did not substantially affect parent-of-origin specific expression of a list of reported imprinted genes. Our results revealed that loss of H1 indirectly affects DME-mediated DNA demethylation in the central cell and endosperm genome methylation but does not alter overall gene transcription or imprinting regulation in the endosperm.
Materials and methods
Plant materials and growth conditions
Arabidopsis plants were grown on soil in growth chambers under 16h, 23°C day/8h, 22°C night growth condition. h1 mutant T-DNA insertional lines were obtained from ABRC and used to generate h1 higher order mutants. h1.1-1 (SALK_128430C) has a T-DNA insertion in the first exon, at 133bp downstream of the start codon. h1.2-1 (Salk_002142) has a T-DNA insertion in the promoter region and the h1.3-1 (SALK_025209) has a T-DNA insertion also in the promoter region, 62bp upstream of the transcription start. H1.2 expression was still detectable in the h1.2-1 allele. We therefore searched for another stronger h1.2 T-DNA line and found CS438975 (we named as h1.2-2), which has a T-DNA insertion in the first exon, at 115 bp downstream of the ATG. Each h1 single mutant was backcrossed to wild type for 4-6 time before analysis and used for generating higher order mutants. Expression of H1.2 was undetectable in the h1.2-2 mutant allele. This h1.2-2 allele has since been used in multiple studies (Zemach et al., 2013; Lyons and Zilberman, 2017; He et al., 2019; Choi et al., 2020; Choi et al., 2021; Bourguet et al., 2022).
GUS reporter constructs
The promoter region of histone H1.1 was PCR amplified with primers H1.1promoter_HindIII_fwd (5’-CCCAAGCTTAAGATGTTTTAGATTGATTT-3’) and H1.1promoter_BamHI_rev (5’-CGCGGATCCCGTCTTCTGAACTTAAGATC-3’). The promoter region of histone H1.2 was PCR amplified with primers H1.2promoter_SalI_fwd2 (5’-ACGCGTCGACGGTTAGATTTTGAATTGGAA-3’) and H1.2promoter_XbaI_rev2 (5’-TGCTCTAGACTTCTTCTCTCTCAGAAACT-3’). The promoter region of histone H1.3 was PCR amplified with primers H1.3promoter_HindIII_fwd (5’-CCCAAGCTTAGAGTTTTAGCTTAGTTTTA-3’) and H1.3promoter_BamHI_rev (5’-CGCGGATCCTAGAGGATTAGTGAAAGTGT-3’). The amplified H1 promoter regions were fused with reporter gene GUS (β-glucuronidase) in the binary vector pBI101, respectively. The constructs were confirmed by sequencing and transformed into Arabidopsis Col-0 by Agrobacterium infiltration. After screening for transgenic plants, stable transgenic T2 or later plants were used for GUS staining analysis. Tissue samples from young seedlings, different stages of floral buds and seeds were harvested and put immediately into the GUS staining buffer (10 mM sodium phosphate buffer, pH 7.2, 0.5% Triton X-100, and 1 mg/mL 5-bromo-4-chloro-3-indolyl- β-glucuronic acid) on ice. Samples in the staining buffer were vacuum infiltrated on ice for 30 minutes. Samples were incubated at 37°C with rotation for 18 hours or less. Tissues were destained using 30%, 50%, 70% ethanol in sequence for 30 minutes each time with rotation. Samples were imbibed in seed clearing solution or diH2O on slides for imaging.
GFP reporter constructs
The H1.1 promoter region (the 1296-bp genomic DNA of the H1.1 promoter sequence starting from the end of 3’UTR of the upstream gene to the end of 5’UTR of H1.1) was PCR amplified with primers H1.1pro_SalI_fwd (5’- ACG CGT CGA CAA GAT GTT TTA GAT TGA TTT -3”) and H1.1pro_SalI_rev (5’- ACG CGT CGA CCG TCT TCT GAA CTT AAG ATC-3’). The H1.2 promoter region (the 2036-bp genomic DNA including 1607-bp upstream of transcription start site of H1.2 plus 429-bp 5’UTR of H1.2) was PCR amplified with primers His1.2 5’ SalI (5’- ACGCGTCGACTGGTTCGAGTATTTTA-3’) and His1.2 3’ XbalI (5’- GCTCTAGACTTCTTCTCTCTCAGAAA-3’). The H1.3 promoter region (the 1279-bp genomic DNA of the H1.3 promoter sequence starting from the end of 3’UTR of the upstream gene to the end of 5’UTR of H1.3) was PCR amplified with primers H1.3proSalIfwd (5’- ACG CGT CGA CAG AGT TTT AGC TTA GTT TTA AAA ATC -3’) and H1.3proSalIrev (5’- ACG CGT CGA CTA GAG GAT TAG TGA AAG TGT-3’). The amplified H1 promoter regions were cloned into vector pBI-EGFP. These transcriptional fusion reporter constructs were confirmed by sequencing and transformed into Arabidopsis Col-0 by Agrobacterium infiltration. After screening for transgenic plants, stable transgenic T2 or later plants were used for GFP expression analysis. Flowers before and after fertilization were harvested. Ovules at flower stage 12-13 and seeds were dissected out and imbibed in diH2O on slides for imaging using confocal microscopy. GFP was excited by the 488 nm laser line and was detected in a range between 505 nm to 535 nm. Approximately 40 stable transgenic lines were obtained and screened, 4-6 GUS reporter lines and 3-5 GFP reporter lines for each promoter construct were examined and representative expression pattern were reported in Figures 1, S1–S3.
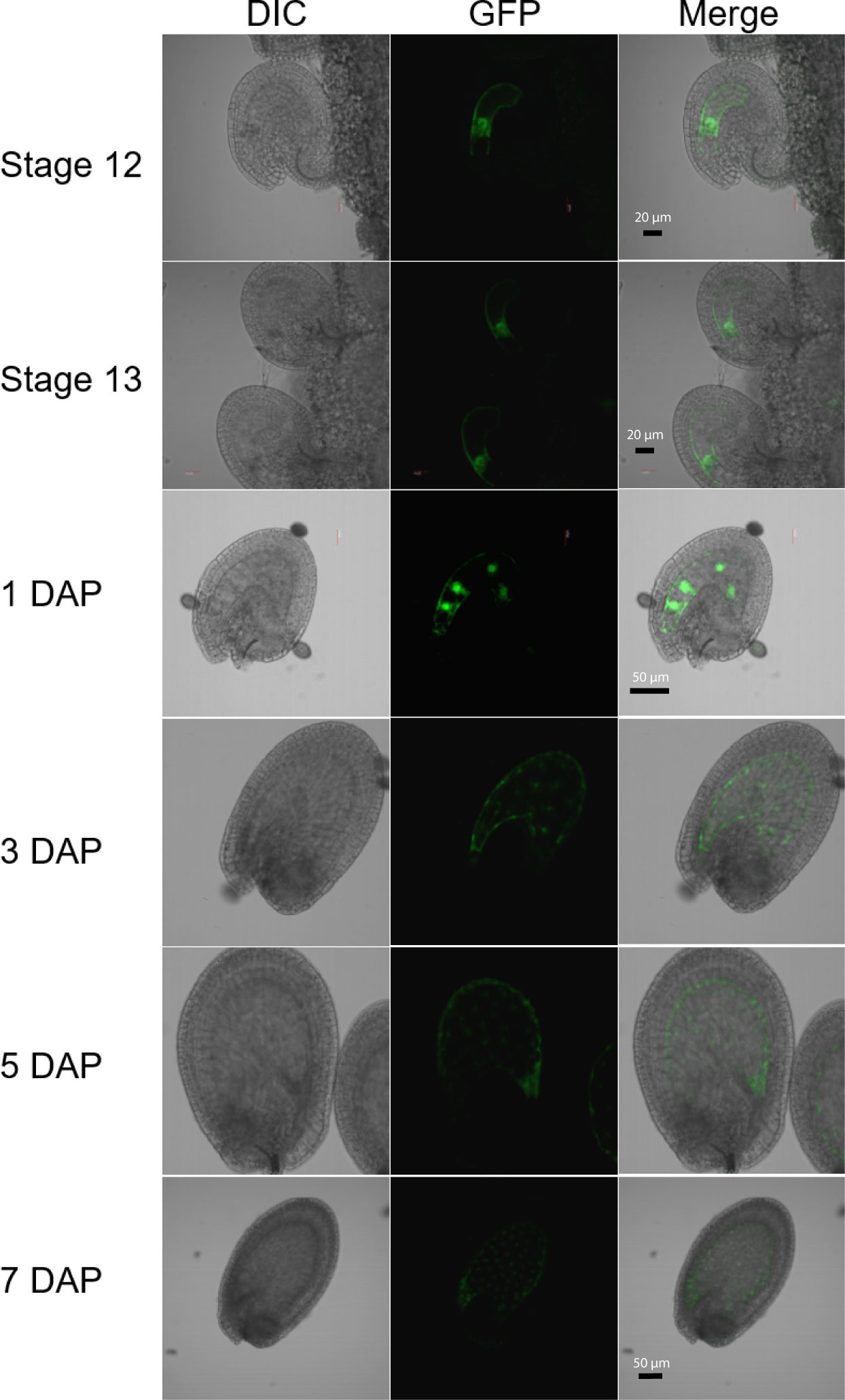
Figure 1 Expression of the H1.2 promoter::GFP transgene in Arabidopsis ovules and young developing seeds. Ovules and seeds with expression of H1.2 promoter:GFP transgene were photographed using confocal fluorescence microscope. Ovules at flower stage 12 and stage 13 and seeds at 1, 3, 5, and 7 DAP were hand-dissected for imaging. The GFP signal is shown in green. DAP, Days After Pollination; DIC, Differential Interference Contrast. Scale bars: State 12 and 13, bar = 20 um; 1-5 DAP, bar = 50 um; 7 DAP, bar = 50 um.
The pFWA::GFP construct was a gift from Dr. Tetsu Kinoshita (Yokohama City University) and was the same construct used in (Ikeda et al., 2011). The MEDEA promoter-GFP construct contains a 4.238 bp of MEA promoter amplified with pMEAF (5’-AGACGGACGTCCTGACGCTAACGTCCTGTCAAACCCGTCCCGTAA-3’) and pMEAR (5’-TCTGCCTTCGCCATTAACCACTCGCCTCTTCTTTTTTTCTC -3’), an Arabidopsis H2B (AT5G22880) fused, nuclear-localized GFP amplified with meaHTB2F (5’-AGTGGTTAATGGCGAAGGCAGATAAGAAACCA-3’) and GFPmeaR (5’-GCTGCTTCTCCTCAGATCAAAAATTACTTGTACAGCTCGTCCATGC-3’) from pBIn1GFP plasmid (Zhang et al., 2008), a 2.1 kb MEDEA 3’-end sequence amplified with 3meaF (5’-TTTTTGATCTGAGGAGAAGCAGCAATTCAAGCA-3’) and 3meaR (5’-ACTCTAGGGACTAGTCCCGGGTTTCATATTCTTGATTCGCCAAATCAGTG-3’). The 3 fragments were concatenated using the Gibson assembly method. The backbone plasmid is a binary plasmid vector, pFGAMh (a hygromycin resistant version of pFGC5941) described before (Zhang et al., 2019). Both constructs were transformed into wild type and the h1 triple homozygous mutant Arabidopsis plants, respectively. The transgenic plants were analyzed for GFP expression in ovules and seeds. For pMEA::GFP, we obtained and analyzed 8 independent lines in wild type and 7 independent lines in h1 triple mutant. For pFWA::GFP, 12 and 7 independent lines were obtained and analyzed in wild-type Col-0 and in h1 triple mutant, respectively. Representative images are presented in Figures 2, S5.
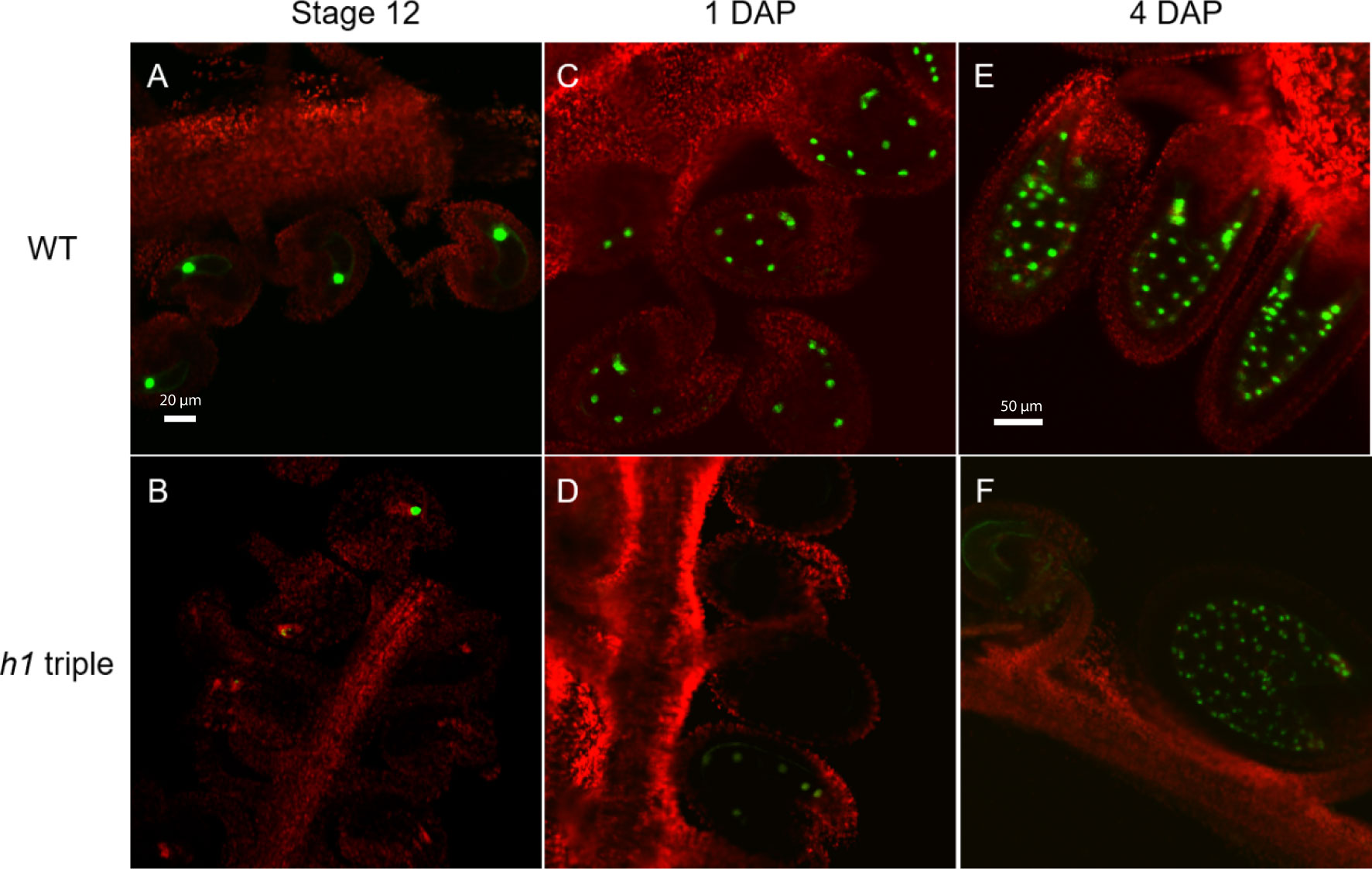
Figure 2 Expression of the H1.2 promoter::GFP transgene is reduced in the central cell of h1 hybrid endosperm. Fluorescence images of pMEA::GFP expression signals in wild type (A, C, E) and in h1 triple mutant endosperm (B, D, F). The GFP and chlorophyll fluorescence signals were pseudo-colored as green and red, respectively. Fluorescence micrographs of ovules at flower stage 12 (A, B), 24-hr after pollination (C, D), and 96-hr after pollination (E, F).
Purification of endosperm nuclei through fluorescence activated cell sorting (FASC)
Embryo and endosperm from 7 - 8 DAP seeds were dissected under a dissecting microscope. For DNA extraction, embryo and endosperm were collected in a tube with 140 ul nuclei extraction buffer and protease inhibitor as described (Zheng and Gehring, 2019), and ground with a pestle on ice. Each sample was ground for 2 minutes, then 800ul nuclei staining buffer was added. Tubes containing nuclei staining buffer were wrapped with aluminum foil to avoid light. The nuclei in extraction buffer and staining buffer were filtered twice with CellTrics 30 um filter on ice, and kept on ice until FASC. The nuclei were sorted at the Flow Cytometry Research Core Facility at Doisy Research Center, Saint Louis University, using a BD FACSAriaI IIu equipped with a 407 nm violet laser using the following parameters: Nozzle size: 100 microns; Sheath pressure: 30 psi; Droplet frequency: 28,100 drops/sec.; Precision mode: Purity. Nuclei were gated based on signals from the DAPI channel.
For RNA extraction, embryos and endosperms were dissected and put into 1.5 ml Eppendorf tubes floated on liquid nitrogen. After collection, embryos and endosperms are kept in -80°C. Nuclei extraction buffer and nuclei staining buffer are added before cell sorting. After sorting, purified embryo and endosperm nuclei were collected with 1.5 ml Eppendorf tube in 50 ml TRIzol™ Reagent (Invitrogen), Zymo RNA Lysis buffer or other RNA lysis buffer which were used for RNA extraction immediately after sorting.
Whole-genome bisulfite sequencing and DNA methylome analysis
Crosses were performed between h1 triple mutant h1.1-1 h1.2-2 h1.3-1 (female) and wild type Ler (male). Embryos and endosperms were collected 7 - 8 days after crossing and desired nuclei fractions purified through FACS. Genomic DNA was isolated from FACS-purified nuclei using CTAB method as described (Ibarra et al., 2012). Approximately 2-5 ng of purified genomic DNA was spiked with 1% (w/w) of unmethylated cl857 Sam7 Lambda DNA (Promega, Madison, WI) and sheared to about 400bp using Covaris M220 (Covaris Inc., Woburn, Massachusetts) under the following settings: target BP, 350; peak incident power, 75 W; duty factor, 10%; cycles per burst, 200; treatment time, 60 second; sample volume 50μl. The sheared DNA was cleaned up and recovered by 1.2x AMPure XP beads followed by one round of sodium bisulfite conversion using the EZ DNA Methylation-Lightning Kit (Zymo Research Corporation, Irvine, CA) as outlined in the manufacturer’s instruction with 80 min of conversion time. Bisulfite sequencing libraries were constructed using the ACCEL-NGS Methyl-Seq DNA library kit (Product Code 30024, Swift Biosciences, Ann Arbor, MI) per manufacturer’s instruction. The PCR enriched libraries were purified twice with 0.8x (v/v) AMPure XP beads to remove adaptor dimers. High throughput sequencing was performed by Novogene Corporation (Davis, CA.). Sequencing reads from three individual transgenic lines were used in the analysis (Table S1). Sequenced reads were mapped to the TAIR10 (whole genome) and TAIR8 (allele-specific) reference genomes and DNA methylation analyses were performed as previously described (Ibarra et al., 2012).
Fractional CG methylation in 50-bp windows across the genome was compared between embryo and endosperm from h1/H1 seeds as well as wild-type embryo, endosperm, and dme-2 endosperm (GSE38935). Windows with a fractional CG methylation difference of at least 0.3 (Fisher’s exact test p-value < 0.001) were merged to generate larger differentially methylated regions (DMRs) if they occurred within 300 bp. Merged DMRs were retained for further analysis if the fractional CG methylation difference across the merged DMR > 0.3 (Fisher’s exact test p-value < 10-6), and if the DMR is at least 100-bp long. The merged DMR list is in the Supplemental Dataset S1. Distribution of DMRs along genes and whole genome methylation average metaplots of genes and TEs were plotted as described previously (Ibarra et al., 2012; Zhang et al., 2019). DNA methylation kernel density plots compare fractional methylation within 50-bp windows. We used windows with at least 10 informative sequenced cytosines and fractional methylation of at least 0.5 in at least one of the samples being compared as described before (Ibarra et al., 2012; Zhang et al., 2019).
RNA sequencing
Embryos and endosperms were collected 8 days after cross and total RNA was extracted using RNAeasy kit (Qiagen) plus on-column DNase I digestion. Three independent sets of total RNAs from embryo and endosperm of h1 Col x Ler and wild-type Col x Ler were isolated. Illumina cDNA libraries were constructed with the Ovation RNA-seq System V2 (NuGen Technologies) per manufacturer’s instruction and as described (Hsieh et al., 2011). Adapter trimming was performed using the fastp program (Chen et al., 2018). For allele-specific transcriptome analysis, adapter-trimmed sequencing reads were mapped to the TAIR8 Col-0 and Ler cDNA scaffold using custom scripts as before (Hsieh et al., 2011). Individual reads were assigned to Col-0 or Ler ecotype based on the SNPs detected between the two ecotypes. Each gene received Col-0 and Ler scores, which represented the number of reads aligned to the corresponding ecotype. These numbers were used to calculate the maternal transcript proportion for each gene. For endosperm and embryo transcriptomes, adapter-trimmed sequencing reads were aligned to TAIR10 for transcript assignation and quantification with Salmon (v.1.9.0) (Patro et al., 2017). DESeq2 was used to identify differentially expressed genes (Love et al., 2014).
Results
Histone H1.2 promoter is active in the central cell and developing endosperm
Although DME is preferentially active in the central cell, it is also expressed in the pollen vegetative cell (VC) and demethylates the VC genome during Arabidopsis reproduction (Schoft et al., 2011; Ibarra et al., 2012). However, H1.1 and H1.2 are present in sperm but undetectable in vegetative nuclei and H1.3 expression is not detected in pollen (Hsieh et al., 2016; He et al., 2019). This indicates that H1 is not required for DME demethylation in VC. To examine whether histone H1 is expressed in central cell, we fused the three H1 genes’ promoter regions with the GFP reporter gene respectively and transferred these transcriptional GFP reporter constructs into Arabidopsis Col-0. Although both H1.1 and H1.2 were reported to be widely expressed (Rutowicz et al., 2015), we were surprised to see that the H1.2 promoter was preferentially active in the central cell of stage 12 flowers but undetectable during earlier gametogenesis (Figure 1). After fertilization, H1.2 expression is observed in the nuclear cytoplasmic domain (NCD) of early endosperm, but the expression level decreases as endosperm develops (Figure 1). H1.2 expression was not visible in embryo although very weak GFP signals sometimes can be seen in the septum, funiculus, and integuments (Figure S1). The H1.1 promoter-GFP construct was more ubiquitously expressed in ovules and seeds including the integument, central cell, embryo, and endosperm (Figure S2). The H1.3 promoter-GFP was generally not expressed but GFP signals were visible around incision areas and in the septum. These signals are likely artifactually induced during dissecting of ovules because H1.3 expression was stress inducible (Rutowicz et al., 2015) and H1.3 promoter-GUS did not show similar expression pattern (Figure S3).
The same set of H1 promoters driving the GUS reporter genes confirmed what was reported that both H1.1 and H1.2 are widely expressed, with H1.1 being more abundantly expressed than H1.2 whereas H1.3 is very lowly expressed in the tissues assessed (Figure S3). Gamete expression of H1.2 has been reported by Song et al. with an average expression of 135.86 (TPM normalized) in the central cell which is about 15-fold higher than the average expression of 9.26 in the egg cell (Song et al., 2020). After fertilization, however, the transcripts of H1.1 and H1.2, but not those of H1.3, can be detected from pre-globular embryo proper through mature embryo, with H1.1 being expressed at 2x - 4x more abundantly than H1.2, according to the Gene Networks in Seed Development database (Figure S4) (Belmonte et al., 2013).
Central cell-preferred expression of H1.2 in the mature ovule was unexpected. We wondered whether H1.2 expression in the central cell is regulated by DME. To test this, we crossed the homozygous pH1.2:GFP transgenic plants with DME/dme-2 heterozygotes and obtained the F1 plants that were heterozygous for DME/dme-2 and hemizygous for the pH1.2:GFP transgene. In the F2 generation, we examined the pH1.2:GFP expression in the DME/dme-2 mutant and their segregating wild-type siblings. We observed that 48% of the female gametophytes with ovules showed strong GFP signal in the DME/DME pH1.2:GFP/- (87 ovules expressed GFP out of 183 total ovules, 87:96, 1:1, χ2 = 0.44, P > 0.50). and 47% of expression in the DME/dme-2 pH1.2:GFP/- plants (83 ovules expressed GFP out of 176 total ovules, 83:93, 1:1, χ2 = 0.56, P > 0.46). This result indicates that H1.2 expression in the central cell is not regulated by DME. This is consistent with H1.2 not being an imprinted gene as we reported earlier (Rea et al., 2012).
Loss of histone H1 affects the expression of MEA and FWA before and after fertilization
We previously showed that in the endosperm derived from h1 x Ler cross (referred to as h1/H1 cross thereinafter), the expression of MEA and FWA was reduced (Rea et al., 2012). To gain a better understanding on how loss of H1 affects MEA and FWA expression, we used MEA and FWA promoter::GFP constructs and transferred them into Arabidopsis wild type Col-0 and the h1 triple mutant. Transgenic plants homozygous for pMEA::GFP and pFWA::GFP were used to examine MEA and FWA gene expression. In wild-type plants, MEA is specifically expressed in the central cell nucleus of stage 12 flower and in the endosperm after fertilization (Figure 2). In h1 mutant, fluorescent intensity of the pMEA::GFP transgene was lower than that in the wild type and the number of GFP positive ovules and seeds was significantly reduced. We detected 67.1% of h1 ovules (94 out of 140) expressing the pMEA::GFP transgene compared with 85.3% (262 out of 307) in wild-type at stage 12 flowers (Table 1). As the seeds develop, the number of h1 mutant seeds expressing the pMEA::GFP transgene increased to 79.1% compared to 91.5% in WT at 1 day after pollination (DAP) and reached 94.4% at 4 DAP which was comparable with what was seen in wild-type (95.2%). The lower ratio of GFP-positive h1 ovules suggests that the h1 mutant ovules mature slightly slower than wild type ovules for unknown reason. However, the GFP fluorescent intensity was noticeably lower in the h1 mutant than in wild type using the same parameter settings under the fluorescence microscope. The pFWA::GFP transgene also exhibited a similar expression pattern as the pMEA::FP transgene. In the h1 triple mutant seeds at 1 DAP, the GFP fluorescent intensity was much lower than that in wild type and the percentage of pFWA::GFP expression seeds (84.5%) was less than wild type (94.6%) (Figure S5, Table 1). As the h1 triple mutant seeds developed, nearly all seeds expressed the pFWA : GFP transgene, but the GFP intensity remains lower than in wild type. In summary, h1 mutations reduce MEA and FWA gene expression in the central cell and endosperm.
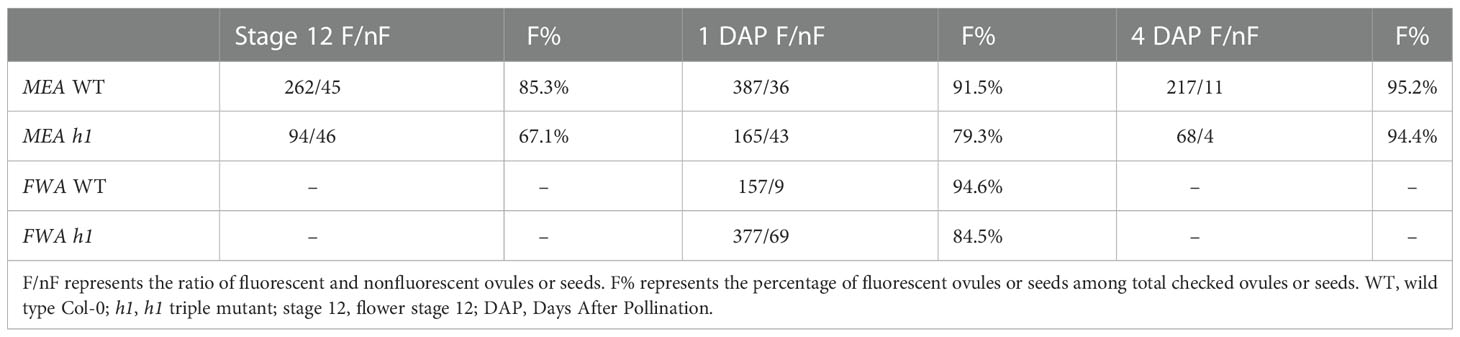
Table 1 The effect of the h1 mutation on the expression of MEA and FWA in the central cell and endosperm.
Maternal H1 loss influences DME-mediated DNA demethylation in the central cell and endosperm genome methylation
The reduction in MEA and FWA expression and the increase in DNA methylation in their maternal alleles in h1/H1 endosperm (Rea et al., 2012) suggested that H1 might play a role in assisting DME demethylation in the central cell. If H1 is necessary for DME demethylation, the majority of canonical DME DMRs would exhibit a lack of DNA demethylation (or hypermethylation) in the endosperm. Alternatively, the reduced MEA and FWA expression seen in h1/H1 endosperm could be due to a locus-specific effect caused by maternal h1 mutations that makes it less favorable for DME demethylation. In this scenario, we would expect to see the majority of DME DMRs are properly demethylated, accompanied by specific loci exhibiting an increase or reduction in DNA methylation. Since loss of DME results in a striking seed abortion phenotype (Choi et al., 2002), the largely normal h1 mutant seeds (Rea et al., 2012) suggested that DME demethylation should be largely intact in these seeds, at least in the DME-regulated Polycomb Repressive Complex 2 (PRC2)-encoding genes (i.e., MEA and FIS2) critical for seed viability (Huh et al., 2008). To investigate the effect of h1 mutations on DME demethylation, we carried out genome-wide bisulfite sequencing of the h1/H1 endosperm derived from crosses between h1 triple mutant or wild-type female (Col-0) and wild-type Ler as the male parent. To avoid contamination of seed coat tissues, we prepared crude nuclei from the hand dissected embryo and endosperm tissues and used fluorescence activated cell sorting (FACS) to isolate pure embryo (2C and 4C) and endosperm (3C and 6C) nuclei for BS-seq library construction and sequencing (Figure S6). We used the Swift Bioscience’s Adapase™ technique (see Materials and Methods) that was successfully used for methylation profiling from ultra-low amounts of input genomic DNA (Luo et al., 2017; Luo et al., 2018). Methylomes from three biological replicates of h1/H1 embryo and endosperm were generated. We used the Col-Ler SNPs to sort and assign uniquely mapped reads to their respective parents of origin. The maternal:paternal read ratios for endosperm and embryo libraries tightly followed the expected ratios (2:1 for endosperm and 1:1 for embryo), indicating sample purity of FAC sorted nuclei with little maternal seedcoat contamination (Table S1). Pearson correlation coefficients were highly concordant between bio-reps (Table S2) and reads from the same tissue/genotype were pooled for subsequent analysis. We first looked at the methylation profiles of selected DME target genes in genome browser. Their methylation profiles are clearly lower in the h1/H1 endosperm than that in the h1/H1 embryo. However, the methylation levels of FWA and MEA (and to a more variable degree in FIS2) promoters were higher in h1/H1 endosperm (Figure S7, upper panel), which is consistent with what we reported before that demethylation of the maternal alleles in these loci were less effective in the h1/H1 endosperm (Rea et al., 2012). By contrast, YUC10, SDC, and DRB2 exhibited a lower DNA methylation profiles in the h1/H1 endosperm (Figure S7, lower panel), suggesting that maternal h1 mutations variably influenced the methylation of certain imprinted genes.
The two polymorphic parental strains (Col x Ler) allowed assessment of methylation difference between the two parental genomes. In wild-type endosperm, kernel density plot of fractional DNA methylation difference between the paternal Ler and the maternal Col genome exhibited a center peak around zero and a small population of 50-bp windows toward the positive tail-end, representing localized demethylated regions hypomethylated on the maternal genome by DME (Ibarra et al., 2012) (Figure S8A, blue trace). The fractional methylation difference between paternal (wild-type Ler) and maternal (h1 Col) genomes also displayed a zero-centered peak and a more profound positive end bump, suggesting a more extended demethylation on the maternal h1 genome (Figure S8A, red trace). However, the availability of SNPs between the Col-0 and Ler genomes (~400,000 SNPs) (Hsieh et al., 2011; Ibarra et al., 2012) limits assessment of only a small fraction of the genome, we instead focused subsequent analysis at the genome-wide scale.
Wild-type endosperm and embryo methylome comparison is one accepted proxy for deducing DME actions in the central cell because DME is not active in the egg cell (Hsieh et al., 2009). Thus, comparing the differentially methylated regions between wild-type Col-0 x Ler embryo and endosperm (referred to as H1/H1-DMRs) would inform DME activity in the wild-type central cell. Likewise, comparing the DMRs of embryo and endosperm derived from h1 mutant x Ler (referred to as h1/H1-DMRs) would provide insights into how loss of H1 affects DME demethylation. Since the canonical DME DMRs (dme vs wt endosperm or dme-hyper DMRs) are well-defined (Ibarra et al., 2012), we separated the genome into loci within or outside dme-hyper DMRs and used kernel density estimate to compare the embryo-endosperm difference between Col x Ler and h1 mutant x Ler seeds. For canonical DME DMRs, embryo and endosperm CG methylation difference in wild-type (H1/H1) (Figure 3A, blue trace) and h1/H1 seeds (green trace) nearly overlap, indicating that the DME action is largely intact without maternal H1. For regions outside canonical DME DMRs, the embryo and endosperm difference are reduced (Figure 3A, orange trace, peak shifted toward zero) in h1/H1 compared with H1/H1 seeds (Figure 3A, red trace, peak is slightly toward the positive side). Since CG methylation is higher in embryo than in endosperm (Hsieh et al., 2009; Ibarra et al., 2012), this reduction indicates a slight overall CG hypermethylation outside of DME DMRs in the h1/H1 endosperm. These features suggest that there is no large-scale change in DNA methylation profile in h1/H1 seeds.
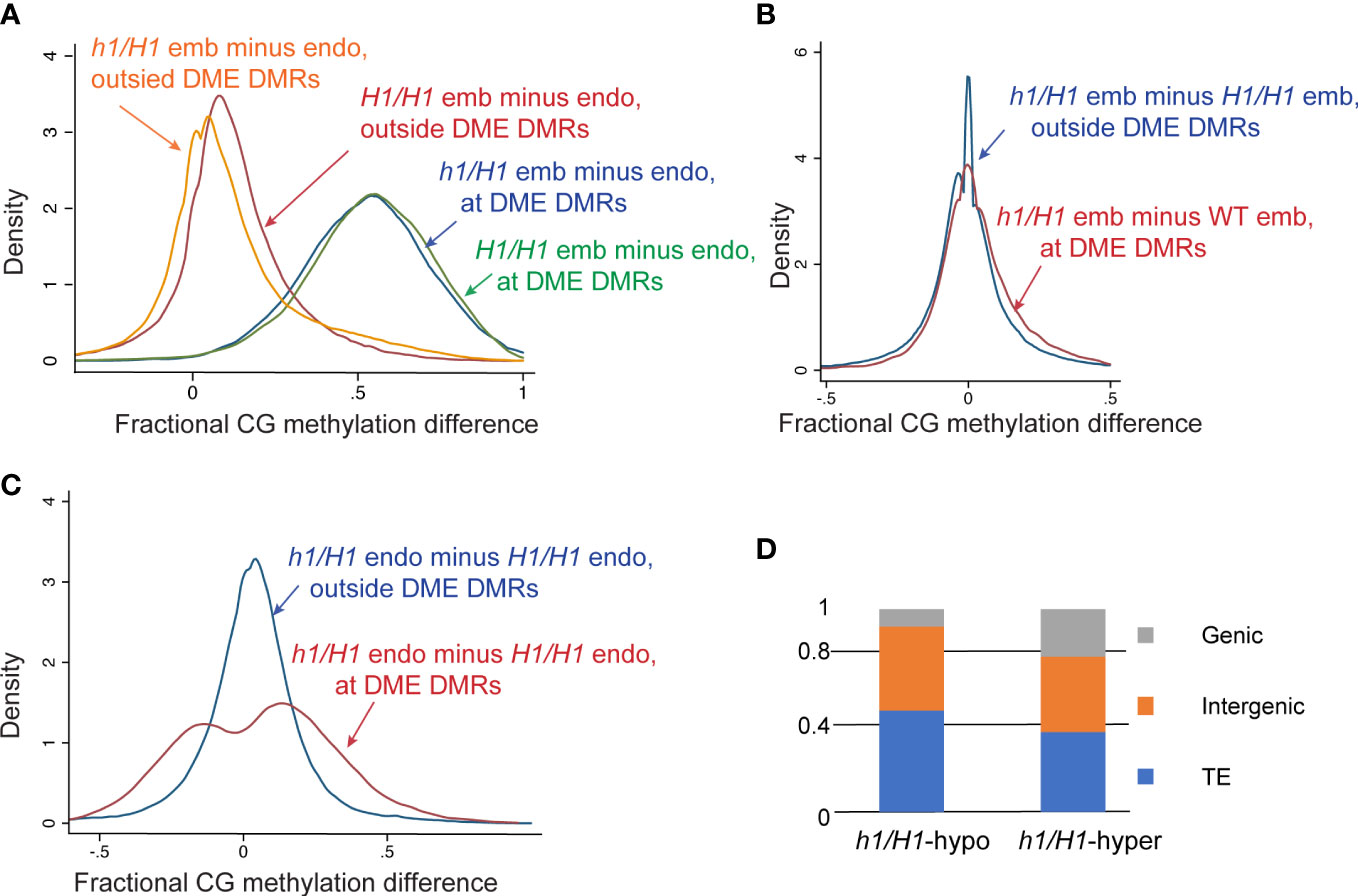
Figure 3 Methylome analysis of embryo and endosperm derived from the maternal wild type or h1 mutant crossed with Ler pollen. (A) Kernel density plots of CG methylation difference between embryo and endosperm (embryo minus endosperm) in wild type H1/H1 (blue and magenta traces) or mutant h1/H1 endosperm (green and orange traces). Methylated loci were grouped into canonical DME DMRs (50-bp windows, N=36281, blue and green traces) and outside DMR loci (N=338297, magenta and orange traces). (B) Kernel density plots of CG methylation difference between H1/H1 and h1/H1 embryo in DME DMRs (magenta) and non-DMR loci (blue). (C) Kernel density plots of CG methylation difference between H1/H1 and h1/H1 endosperm in DME DMRs (magenta) and non-DMR loci (blue). (D) Percent distribution of the three different genomic features in canonical DME DMRs that loose (h1/H1-hypo) or gain (h1/H1-hyper) in h1/H1 endosperm compared with wild type.
We next compared the difference between h1/H1 and H1/H1 methylomes. DNA methylation was largely unchanged between h1/H1 and H1/H1 embryos with the peak of fractional methylation difference sharply centered at zero (Figure S8B, blue trace) flanked by two minor shoulder peaks, indicating that localized minor hyper and hypo methylations exist in the h1/H1 mutant embryo. This is also evident in whole genome methylation metaplots showing near identical CG methylation profiles between h1/H1 and H1/H1 embryos (Figure S9). By contrast, CG DNA methylation of the h1/H1 endosperm is higher compared to H1/H1 endosperm, with the fractional methylation difference peak shifted toward the right side and flanked by broader shoulders (Figure S8B, red trace). This suggests that a slight CG hypermethylation occurs in the h1/H1 endosperm. Whole genome CG metaplots revealed this hypermethylation occurs in coding sequences and to a higher degree in longer TEs (Figure S9). This is consistent with earlier reports that heterochromatin TEs gain DNA methylation in h1 mutant and support the hypothesis that H1-rich heterochromatin impedes DNA methyltransferase access to the DNA templates (Zemach et al., 2013; Lyons and Zilberman, 2017). Interestingly, this heterochromatin TE hypermethylation is not observed in the h1/H1 embryo as little change in DNA methylation is observed between h1/H1 and H1/H1 embryos (Figures 3B, S9).
To gain a better insight into how H1 loss affects DME demethylation, we plotted the fractional methylation difference between h1/H1 and H1/H1 seeds within and outside of the canonical DME DMRs. Whereas very little difference between these two groups of loci was observed in embryo (Figure 3B), a more widespread difference can be seen within the canonical DME DMRs in the endosperm (Figure 3C, red trace), indicating that the loss of H1 differentially affected demethylation at the canonical DME loci. DME DMRs more demethylated in h1/H1 endosperm (Figure 3C, h1/H1-hypo loci, fractional methylation difference <0, left peak of red trace) are enriched for TEs (50.2% versus 39.5%) but depleted for genic sequence (8.2% versus 23.6%) (Figure 3D) relative to DME DMRs hypermethylated in the h1/H1 endosperm (Figure 3C, h1/H1-hyper loci, difference >0, right peak of red trace). This suggest that without H1, certain heterochromatin loci are more demethylated, which is consistent with the model that H1-bound nucleosomes restrict accessibility of chromatin modifying enzymes (Zemach et al., 2013; Lyons and Zilberman, 2017). Why some DME loci (Figure 3C, h1/H1-hyper sites) are less demethylated in the h1/H1 endosperm is unknown, but they are enriched for chromatin states 3 and 7, hallmark features for intragenic sequences (Figure S10) (Sequeira-Mendes et al., 2014). For loci outside the canonical DME DMRs, there is also a slight difference between h1/H1 and H1/H1 endosperm (Figure 3C, blue trace with peak shifted toward positive side and a broader shoulder compared with embryo plot). Thus, maternal h1 mutant specifically affects the methylation pattern of h1/H1 endosperm but not embryo, implicating the presence of an altered DME demethylation activity in the h1 mutant central cell.
We next compared the differentially methylated regions between wild-type embryo versus endosperm (embryo hyper-DMRs in the presence of H1 or H1/H1-DMRs, n=8207), and between h1/H1 embryo versus endosperm (embryo hyper-DMRs in the absence of H1 or h1/H1-DMRs, n=11552) (Materials and Methods) using previously established criteria (Supplementary Dataset 1, DMR lists) (Ibarra et al., 2012). The h1/H1-DMRs cover over 6 million bases, more than twice longer than the H1/H1-DMRs (Figure 4A). About 56% of the h1/H1-DMRs overlap with H1/H1-DMRs (Figure 4B). On average, the h1/H1-DMRs are significantly longer in size (Figure 4C, t=29.1221, p=0, Welch’s t-test). These observations suggest that without H1, DME demethylation is less constrained. Natural depletion of H1 in the Arabidopsis vegetative cell (VC) is associated with heterochromatin de-condensation and ectopic H1 expression in VC impedes DME from accessing heterochromatic transposons (He et al., 2019). Consistent with this model, the h1/H1-unique DMRs are highly enriched for the heterochromatin states (State 8 & 9, Figure 4D) (Sequeira-Mendes et al., 2014). This is also supported by the DMR distribution plot across the genome showing h1/H1-DMRs are more abundant in the pericentromeric and genic regions (Figures 4E, F).
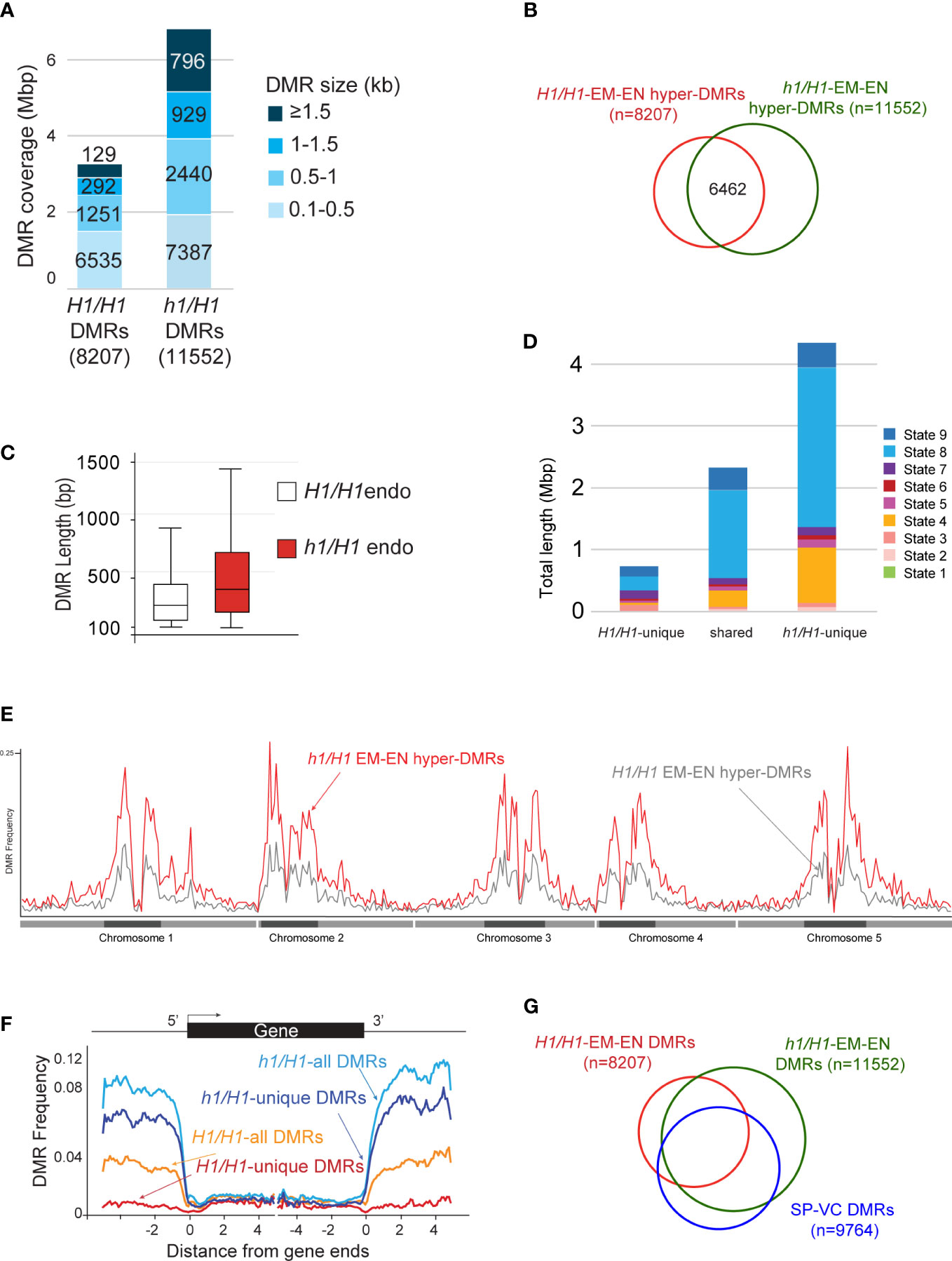
Figure 4 Analysis of the hyper DMRs of embryo vs endosperm derived from maternal wild type or h1 mutant crossed with Ler pollen. (A) Wild-type H1/H1 and mutant h1/H1 DMRs grouped by size, with the cumulative total length they cover shown. (B) Venn diagram depicting overlaps between H1/H1 and h1/H1 DMRs. (C) Boxplot showing the length distribution of H1/H1 and h1/H1 DMRs. (D) Chromatin state distribution, and the total length they covered, within H1/H1-unique, shared, and h1/H1-unique DMRs. States 1 to 7 correspond to euchromatin, and states 8 and 9 correspond to AT- and GC-rich heterochromatin, respectively. (E) Distribution frequency of DMRs along the 5 chromosomes. Dark blocks represent centromere and peri-centromeric regions of each chromosome. (F) Distribution frequency of DMRs with respect to coding genes. Genes were aligned at the 5’- or the 3’-end, and the proportion of genes with DMRs in each 100-bp interval is plotted. DMR distribution is shown with respect to all H1/H1-DMRs (orange trace), H1/H1-unique DMRs (red trace), all h1/H1-DMRs (light blue trace), and h1/H1-unique DMRs (dark blue trace). (G) Venn diagram showing overlaps between H1/H1, h1/H1, and sperm vs vegetative cell DMRs.
DME is also active in the vegetative cell where it demethylates a slightly larger number of loci (referred to as SP-VC DMRs, n=9764) than in the central cell (canonical DMRs, dme mutant versus wild-type endosperm, n=8672) (Ibarra et al., 2012). About 56% of the DME DMRs in the female gametophyte (canonical DME DMRs) and 50% of H1/H1-DMRs (EM vs EN, derived from Col-0 x Ler seeds) overlap with the SP-VC DMRs (Ibarra et al., 2012). One factor attributed to this moderate overlap between the male and female gametophytes is the H1-depleted, heterochromatin-decondensed vegetative nuclei that is thought to facilitate DME access to the heterochromatin targets (He et al., 2019). Notably, greater than 80% (and up to 86%) of known DME DMRs (i.e., the SP-VC DMRs, canonical DME DMRs, and WT-DMRs) overlap with the h1/H1-DMRs (Figure 4G; Table S3). These observations strongly support the model that lack of H1 facilitates DME access to the heterochromatin regions, which was the primary attribute for the increased number of h1/H1-DMRs.
Maternal h1 mutations do not alter gene transcription and imprinting regulation in Arabidopsis endosperm
To assess how loss of H1 influences endosperm gene transcription and imprinting regulation, we manually dissected and collected endosperm from 7-DAP F1 seeds derived from Col-0 x Ler (H1/H1) and h1 (Col-0) x Ler (h1/H1) crosses. Since endosperm RNA-seq using manually dissected materials is prone to seed coat contamination (Schon and Nodine, 2017), we first tried to isolate nuclei RNAs from FACS-purified 3C and 6C endosperm nuclei. However, due to the low input amount of dissected endosperm and instability of nascent transcripts in the nuclei, we were unable to purify any detectable amounts of nuclei RNAs in our experimental setting using multiple RNA-isolation methods. We therefore proceeded with regular RNA-seq analysis using manually dissected endosperm (Hsieh et al., 2011). We used the tissue enrichment test tool (Schon and Nodine, 2017) to evaluate the extend of seedcoat contamination in our endosperm and embryo RNA-seq datasets and found that indeed a modest degree of seedcoat enrichment can be detected, with a general seed coat (GSC) enrichment score of 5 to 10 among endosperm samples and 1.8 to 3 among embryo datasets (Figure S11), which is consistent with, but slightly lower than, all the manually dissected endosperm datasets assessed by an earlier study (Schon and Nodine, 2017). Pearson correlation coefficients between biological replicates showed that they were highly concordant (Table S4). Principal component analysis showed that samples were primarily separated by tissue type, with the PC1 capturing 82% of variation while genotype difference does not differentiate tissue samples (Figure S12). This is also reflected in the low numbers of up- and down-regulated DEGs between h1/H1 and H1/H1 endosperm or embryo (fold change >2, adjusted p value < 0.05, 31 up and 20 down in endosperm, 5 up and 13 down in embryo, Supplemental Dataset 2). No GO term enrichment was detected, likely due to the small numbers of DEGs identified. RNA-seq data showed that the expression of FIS2 and FWA were reduced in the h1-wt endosperm (with a log2 fold change of -0.41 for FIS2 and -1.23 for FWA), which is consistent with what we had observed before (Rea et al., 2012). The expression of MEA varied between replicates but overall unchanged between wild-type and h1 endosperm (log2 fold change 0.2). All three genes were expressed at low levels. Since histone H1 genes are not imprinted in the endosperm (Rea et al., 2012), loss of H1 function in the gamete and central cell is likely compensated by the functional paternal copies upon fertilization. By contrast, a complete loss of H1 in the homozygous h1 triple mutant caused a significant mis-regulation of > 900 genes in leaves and seedlings (Choi et al., 2020) and 701 gene in Arabidopsis seedlings (Rutowicz et al., 2019). Maternal depletion of H1 also did not cause TE mis-regulation (3 up- and 2 down-regulated in endosperm, 2 up- and 4 down-regulated in embryo), in contrast to the reported 18 TEs significantly dysregulated (Choi et al., 2020) and the 1.5% of TEs been mis-regulated (Rutowicz et al., 2019) in h1 plants.
We used the Col-Ler SNPs to distinguish and sum the number of reads derived from their respective parents as we did before (Hsieh et al., 2011). To minimize mis-interpretation of possible seedcoat contamination that could skew the maternal ratio of expressed genes, we focused our analysis only on a published list of high confident imprinted gens (148 MEGs and 81 PEGs) (Schon and Nodine, 2017) and assessed any difference between H1/H1 and h1/H1 endosperm data that were generated by the same procedure. Among them, 80 MEGs and 42 PEGs have sufficient read numbers for proper comparison among our datasets. Overall, the maternal ratios of MEGs and PEGs are similar between H1/H1 and h1/H1 endosperm, with the MEGs showing a small increase in the maternal ratio (student’s t-test, P < 0.01) but not the PEGs (P > 0.05) (Figure 5A). Scatter plot of maternal transcript proportions among analyzed imprinted genes showed that they are highly correlated between H1/H1 and h1/H1 endosperm (Pearson correlation r = 0.91, n=122, Figure 5B) indicating that loss of maternal H1 activity does not affect overall gene imprinting regulation. Genome-wide the correlation of maternal transcript proportion between H1/H1 and h1/H1 endosperm is also high (r = 0.83, n=9054, Figure S13), consistent with the low DEGs detected between them. In summary, our results show that maternal genome h1 mutations have a very limited effect on endosperm gene transcription or imprinted regulation.
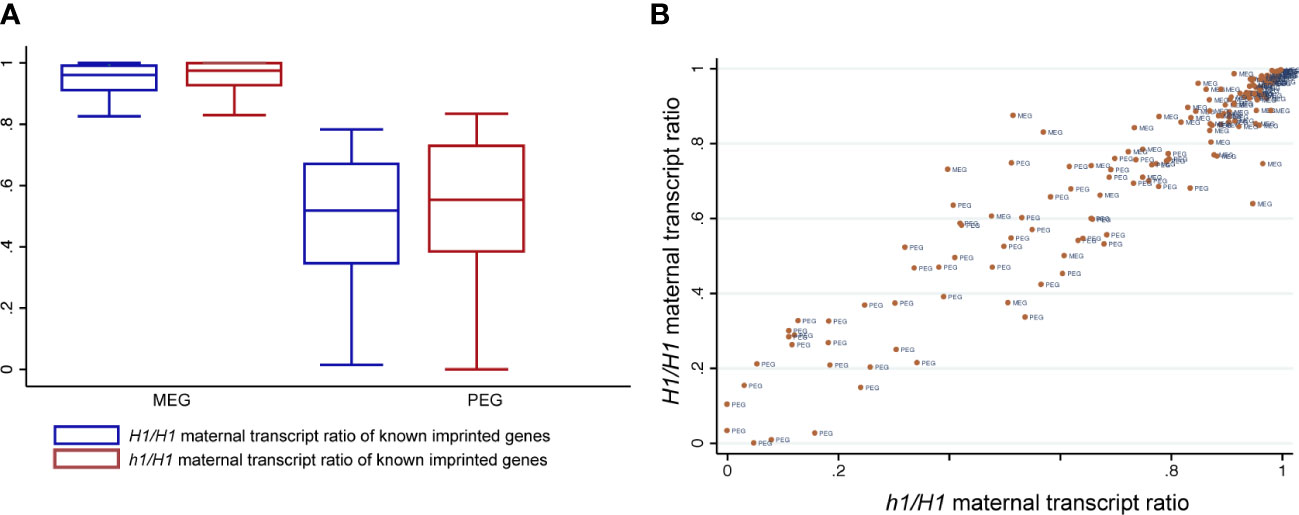
Figure 5 Known imprinted genes and their status of imprinted expression in H1/H1 and h1/H1 endosperm. (A) Boxplot showing the distribution of maternal transcript proportions of selected MEGs and PEGs in H1/H1and h1/H1 endosperm. (B) Scatterplot showing the correlation of maternal transcript proportion of each imprinted gene between H1/H1and h1/H1 endosperm.
Discussion
DME, a multi-domain novel glycosylase that initiates active DNA demethylation process by removing the methylated cytosine bases in the central cell, regulates gene imprinting and is essential for seed development in Arabidopsis (Choi et al., 2002; Gehring et al., 2006; Ibarra et al., 2012). In pollen, DME also demethylates the VC genome at thousands of loci to reinforce gamete TE silencing and to ensure robust pollen germination in certain accessions (Schoft et al., 2011; Ibarra et al., 2012). DME preferentially influences small and genic-flanking transposons whose demethylation affects the transcription of nearby coding genes, it also demethylates many intergenic and heterochromatin sequences. How DME is specifically recruited to these loci remain elusive, due to its ephemeral expression in the gamete companion cells not accessible by common experimental methods. Using a yeast two-hybrid screen, Rea et al. identified linker histone H1.2 as a candidate DME-interacting protein and demonstrated that they did physically interact in an in vitro pull-down assay (Rea et al., 2012). Furthermore, in the female h1 triple mutant (h1.1, h1.2-1, h1.3) x wild-type Ler endosperm, the expression of selected DME-regulated imprinted genes (MEA, FIS2, FWA) were reduced and the methylation of their maternal alleles were increased, suggesting that DME demethylation might be impaired due to H1 loss on the maternal genome. However, H1 loss differentially influences DNA methylation in heterochromatic and euchromatic TEs prior to DME action (Zemach et al., 2013; Rutowicz et al., 2015), making it difficult to delineate if H1 directly or indirectly involves in DME demethylation in the central cell. Furthermore, DME and its more ubiquitous paralog ROS1 are likely recruited by and make contact with specialized local chromatin environments (Qian et al., 2012), as demonstrated by a recent study showing that ROS1 binds to all 4 histone proteins in vitro (Parrilla-Doblas et al., 2022). Since demethylation takes place in chromatin environment where DNA wraps around nucleosomes connected by H1 linker histones, the interaction between H1 and DME might simply reflect the obligated physical contact between them.
H1.2 and DME are co-expressed in the central cell
In Arabidopsis, the reproductive phase is initiated late in adult plant with the specification of meiocyte precursors known as the spore mother cells (SMCs) that transition from vegetative to reproductive cell fate. This transition is accompanied by a temporary eviction of H1.1 and H1.2 in the megaspore mother cells, the female SMCs (She et al., 2013; She and Baroux, 2015; Ingouff et al., 2017). During male gametogenesis, both H1 variants are absent in late microspore stage, remain absent in the VC nuclei, but are present in the sperm nuclei (Hsieh et al., 2016; He et al., 2019). The absence of H1 in the VCs where DME is active indicates that H1 is not a requirement for DME demethylation, at least not in the VCs.
Although we found that the H1.1 and H1.2 promoters are active in the central cell where DME acts (Figures 1, S1, S2), it remains to be independently validated whether the H1 proteins are indeed present in the central cell. However, the fact that the FACT complex is specifically required for DME demethylation in the central cell heterochromatin but not in the VC (Frost et al., 2018) suggests that CC heterochromatin is more compact than in VC, and the presence or absence of H1 proteins could contribute to this difference. Detailed study is required to elucidate the expression dynamics of H1 proteins during female gametogenesis and throughout seed development.
H1 mutations did not cause a dme-like seed abortion phenotype
If H1 is required for normal DME function in the central cell, loss of H1 would be expected to induce certain degree of seed abortion similar to what’s seen in the dme mutant. We previously observed 16.7% seed abortion in self-pollinated F3 triple mutant h1.1-1 h1.2-1 h1.3-1 plants (Rea et al., 2012). However, the h1.2-1 allele has residual H1.2 expression due to T-DNA insertion in the promoter region. This promoted us to find another stronger h1.2-2 allele (see Materials and Methods). During the course of this study, many reports have used this h1.2-2 allele for their studies (Zemach et al., 2013; Lyons and Zilberman, 2017; He et al., 2019; Choi et al., 2020). Each of the h1 single mutants was backcrossed to WT for 4-6 time before analysis or used for generating the h1 triple mutant. We examined seed phenotype of h1 single, double, and triple mutants (h1.1-1 h1.2-2 h1.3-1) using different batches of plants. Seed abortion rates of h1 single mutants h1.1-1, h1.2-2, and h1.3-1 were 1.80%, 1.61%, and 1.26%, respectively, which were slightly higher than that of wild type Col-0 (0.62%) (Table S5). The h1 double mutants h1.1-1 h1.2-2 and h1.2-2 h1.3-1 clearly have a cumulative effect on seed abortion, which reach 6.71% and 2.80% respectively. The h1 triple mutant (h1.1-1 h1.2-2 h1.3-1) has the highest seed abortion rate (7.35%) (Table S5), which shows statistically significant difference from any other h1 single and double mutants except h1.1-1 h1.2-2. The statistical analysis showed that seed abortion between the single mutant h1.1-1 and h1.2-2 and the h1.1h1.2 double mutant was not significant (Mann–Whitney U test), but we cannot completely rule out the potential synergistic interaction between h1.1 and h1.2 on seed abortion. This result suggests that both H1.1 and H1.2 play a role in seed development. Interestingly, we also observed a large variation of seed abortion rate in histone h1 mutants. In the h1 triple mutant, the spectrum of seed abortion rate in individual silique varied widely from 0% to > 40%, and with more than 61% (113 out of 184) of examined siliques carrying aborted seeds (Table S6). This large variation of seed abortion can reflect subtle epigenetic influence of H1 on seed development as it has now been shown that H1 affects flowering time, stomata and lateral root formation, and stress response (Rutowicz et al., 2019). This can also suggest that epigenetic effect of H1 on seed development might be easily influenced by environment or subtle plant growth conditions. Since each h1 single mutant was backcrossed to wild-type for 4-6 times, we assumed the seed abortion phenotype was caused by the T-DNA insertion. However, we cannot unequivocally ascribe the seed phenotype to the h1 knockout without the proof of complementation results. In summary, H1 loss likely has a subtle influence on seed development but h1 mutants do not exhibit a dme-like prominent seed abortion phenotype, and H1 is not a strict requirement for DME function in the central cell.
Maternal h1 triple mutant affects chromatin architecture and influences DME demethylation
H1 binds to the nucleosome and the linker DNA between two adjacent nucleosomes and is required for higher-order chromatin organization; yet, despite its ubiquitous presence in the nucleus and important nuclear functions, loss of H1 has very limited effects on gene transcription and TE silencing (Rutowicz et al., 2019). H1-bound nucleosomes are thought to be natural barriers to the enzymes that modify DNA, such as DNA methyltransferases (Zemach et al., 2013; Lyons and Zilberman, 2017), DNA repair enzymes, and DNA demethylases (Frost et al., 2018; He et al., 2019). Consequently, loss of H1 would facilitate access of DNA methyltransferases to the less condensed heterochromatin and resulted in a gain in heterochromatic TEs methylation as reported in (Zemach et al., 2013). By contrast, euchromatin TEs exhibited a loss in DNA methylation in h1 homozygous mutant, for reasons currently not fully understood (Zemach et al., 2013).
Our h1 x Ler endosperm methylome data supports this general model of H1 function. Our results suggest that loss of H1 in the central cell (or in the maternal genome) allowed easier access of DME, particularly to the heterochromatic regions. This is reflected in a 2x increased in the number of EMB-ENDO DMRs in h1/H1 compared with H1/H1 seeds (Figures 4A, B), and the gained (h1/H1-unique) DMRs are enriched for heterochromatin states (Figure 4D) and in the pericentromeric regions (Figure 4E). Since there was little change between h1/H1 and H1/H1 embryo methylome, we can assume the difference in endosperm methylome was caused by DME action in the h1 central cell. Our data also suggests that H1-bound nucleosomes impede DME demethylation process and the removal of H1 resulted in a significant increase in DMR length (Figure 4C). This notion was supported by our earlier studies that the FACT complex is specifically needed in the H1-enriched heterochromatin target loci (Frost et al., 2018; Zhang et al., 2019). Within the canonical DME DMRs (dme vs wild-type endosperm), we did not observe any significant difference between h1/H1 embryo and endosperm (Figure 3A, blue and green traces); but a small degree of difference is visible outside DME canonical loci with a slight hypomethylation in h1/H1 endosperm (Figure 3A), consisting with gaining more DMRs in h1/H1 due to a more extensive demethylation in the h1 CC genomes. A direct comparison between h1/H1 and H1/H1 endosperm revealed that loss of H1 differentially affects DME canonical DMRs (Figure 3C, red trace). Canonical loci hypermethylated in h1/H1 endosperm (Figure 3C, positive side of the red trace) are enriched for genic coding sequences whereas loci hypomethylated (red trace, negative side) are more enriched for intergenic and heterochromatin sequences (Figure 3D). Taken together, our methylome data showed the effect of H1 loss on DME demethylation most likely is indirect.
H1 loss in the maternal genome has a very limited influence on gene transcription and imprinting regulation
Our attempt to isolate RNAs from FACS-purified endosperm nuclei was unsuccessful upon multiple trials with different RNA extraction protocols. This prevented us from performing a more detailed analysis on how loss of maternal H1 influences gene imprinting regulation. Due to the presence of maternal seedcoat tissues, revealed by tissue enrichment test (Figure S11), we instead limited our allele-specific expression analysis on a list of previously reported high-confident imprinted genes (Schon and Nodine, 2017) and qualitatively assessed whether maternal H1 loss induced a significant distortion on their allele-specific expression. Very few differentially expressed genes or TEs were identified between h1/H1 and H1/H1 in endosperm and in embryo, suggesting that the wild-type H1 alleles from pollen can quickly compensate for the loss of maternal copies upon fertilization. We used the ratio of maternal over total transcripts as a measurement of parental bias and did not identify any deviation between h1/H1 and H1/H1 among known imprinted genes. Overall, the maternal-bias score for each imprinted gene were highly correlated between h1/H1 and H1/H1, with selected individual gene showing some variation visible on the scatter plot (Figure 5B). These observations showed that even with a greater degree of maternal genome demethylation in h1/H1, the influences on endosperm gene transcription or imprinting regulation was very limited.
We reported earlier the expression of MEA, FIS2, and FWA were reduced and their maternal copies were hypermethylated in h1/H1 endosperm (Rea et al., 2012). Here we showed that the flanking sequences of these three genes are indeed hypermethylated in the h1/H1 endosperm relative to wild type and except for MEA whose expression was variable in our RNA-seq datasets, the expression of FIS2 and FWA was reduced in the h1/H1 endosperm. This was independently supported by the promoter reporter constructs showing MEA and FWA expression were reduced in h1 endosperm compared with wild type (Figures 2, S5). We believe this is due to the differential effect on DME demethylation in the absence of H1 (Figure 3C). However, we also observed a variable methylation change in other selected imprinted genes (Figure S7). The reason why maternal H1 loss caused hypermethylation in some genes but not others remains to be investigated.
Data availability statement
The high throughput sequencing datasets reported in this study are deposited in the NCBI GEO under accession number GSE217279.
Author contributions
RF, WX, and T-FH conceived the project; QH, Y-HH, CZ, AB, MR, HY, and CP performed the experiments; X-QZ, QH, Y-HH, WX, T-FH analyzed the data; RF, WX, and T-FH wrote the article with contributions of all the authors. All authors contributed to the article and approved the submitted version.
Funding
This work is supported by the NIFA Hatch Project 02413 (to T-FH), NSF Grant MCB-1715115 (to T-FH and WX), and NIH Grant R01-GM69415 (to RF).
Acknowledgments
We thank the Arabidopsis Biological Resource Center at Ohio State University for providing Arabidopsis mutant seed stocks, Dr. Grant Kolar and Caroline Murphy at Research Microscopy and Histology Core at SLU for providing assistance with confocal microscopy, Joy Eslick at Flow Cytometry Research Core Facility at SLU for helping with nuclei sorting.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1070397/full#supplementary-material
References
Agius, F., Kapoor, A., Zhu, J. K. (2006). Role of the arabidopsis DNA glycosylase/lyase ROS1 in active DNA demethylation. Proc. Natl. Acad. Sci. U.S.A. 103, 1796–1801. doi: 10.1073/pnas.0603563103
Ascenzi, R., Gantt, J. S. (1999a). Molecular genetic analysis of the drought-inducible linker histone variant in arabidopsis thaliana. Plant Mol. Biol. 41, 159–169. doi: 10.1023/a:1006302330879
Ascenzi, R., Gantt, J. S. (1999b). Subnuclear distribution of the entire complement of linker histone variants in arabidopsis thaliana. Chromosoma 108, 345–355. doi: 10.1007/s004120050386
Belmonte, M. F., Kirkbride, R. C., Stone, S. L., Pelletier, J. M., Bui, A. Q., Yeung, E. C., et al. (2013). Comprehensive developmental profiles of gene activity in regions and subregions of the arabidopsis seed. Proc. Natl. Acad. Sci. U.S.A. 110, E435–E444. doi: 10.1073/pnas.1222061110
Bourguet, P., Yelagandula, R., To, T. K., Osakabe, A., Alishe, A., Lu, R. J.-H., et al. (2022). The histone variant H2A.W cooperates with chromatin modifications and linker histone H1 to maintain transcriptional silencing of transposons in arabidopsis. bioRxiv. doi: 10.1101/2022.05.31.493688
Chen, S., Zhou, Y., Chen, Y., Gu, J. (2018). Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Choi, Y., Gehring, M., Johnson, L., Hannon, M., Harada, J. J., Goldberg, R. B., et al. (2002). DEMETER, a DNA glycosylase domain protein, is required for endosperm gene imprinting and seed viability in Arabidopsis. Cell 110, 33–42. doi: 10.1016/S0092-8674(02)00807-3
Choi, J., Lyons, D. B., Kim, M. Y., Moore, J. D., Zilberman, D. (2020). DNA Methylation and histone H1 jointly repress transposable elements and aberrant intragenic transcripts. Mol. Cell 77, 310–323.e317. doi: 10.1016/j.molcel.2019.10.011
Choi, J., Lyons, D. B., Zilberman, D. (2021). Histone H1 prevents non-CG methylation-mediated small RNA biogenesis in arabidopsis heterochromatin. Elife 10, e72676. doi: 10.7554/eLife.72676.sa2
Fan, Y., Nikitina, T., Zhao, J., Fleury, T. J., Bhattacharyya, R., Bouhassira, E. E., et al. (2005). Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell 123, 1199–1212. doi: 10.1016/j.cell.2005.10.028
Frost, J. M., Kim, M. Y., Park, G. T., Hsieh, P. H., Nakamura, M., Lin, S. J. H., et al. (2018). FACT complex is required for DNA demethylation at heterochromatin during reproduction in arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 115, E4720–E4729. doi: 10.1073/pnas.1713333115
Gantt, J. S., Lenvik, T. R. (1991). Arabidopsis thaliana H1 histones. analysis of two members of a small gene family. Eur. J. Biochem. 202, 1029–1039. doi: 10.1111/j.1432-1033.1991.tb16466.x
Gehring, M., Huh, J. H., Hsieh, T. F., Penterman, J., Choi, Y., Harada, J. J., et al. (2006). DEMETER DNA glycosylase establishes MEDEA polycomb gene self-imprinting by allele-specific demethylation. Cell 124, 495–506. doi: 10.1016/j.cell.2005.12.034
Gent, J. I., Higgins, K. M., Swentowsky, K. W., Fu, F. F., Zeng, Y., Kim, D. W., et al. (2022). The maize gene maternal derepression of r1 encodes a DNA glycosylase that demethylates DNA and reduces siRNA expression in the endosperm. Plant Cell 10, 3685–3701. doi: 10.1093/plcell/koac199
He, S., Vickers, M., Zhang, J., Feng, X. (2019). Natural depletion of histone H1 in sex cells causes DNA demethylation, heterochromatin decondensation and transposon activation. Elife 8, e42530. doi: 10.7554/eLife.42530.035
Hondele, M., Ladurner, A. G. (2013). Catch me if you can: How the histone chaperone FACT capitalizes on nucleosome breathing. Nucleus 4, 443–449. doi: 10.4161/nucl.27235
Hsieh, P. H., He, S., Buttress, T., Gao, H., Couchman, M., Fischer, R. L., et al. (2016). Arabidopsis male sexual lineage exhibits more robust maintenance of CG methylation than somatic tissues. Proc. Natl. Acad. Sci. U.S.A. 113, 15132–15137. doi: 10.1073/pnas.1619074114
Hsieh, T. F., Ibarra, C. A., Silva, P., Zemach, A., Eshed-Williams, L., Fischer, R. L., et al. (2009). Genome-wide demethylation of arabidopsis endosperm. Science 324, 1451–1454. doi: 10.1126/science.1172417
Hsieh, T. F., Shin, J., Uzawa, R., Silva, P., Cohen, S., Bauer, M. J., et al. (2011). Regulation of imprinted gene expression in arabidopsis endosperm. Proc. Natl. Acad. Sci. U.S.A. 108, 1755–1762. doi: 10.1073/pnas.1019273108
Huh, J. H., Bauer, M. J., Hsieh, T.-F., Fischer, R. L. (2008). Cellular programming of plant gene imprinting. Cell 132, 735–744. doi: 10.1016/j.cell.2008.02.018
Ibarra, C. A., Feng, X., Schoft, V. K., Hsieh, T. F., Uzawa, R., Rodrigues, J. A., et al. (2012). Active DNA demethylation in plant companion cells reinforces transposon methylation in gametes. Science 337, 1360–1364. doi: 10.1126/science.1224839
Ikeda, Y., Kinoshita, Y., Susaki, D., Iwano, M., Takayama, S., Higashiyama, T., et al. (2011). HMG domain containing SSRP1 is required for DNA demethylation and genomic imprinting in arabidopsis. Dev. Cell 21, 589–596. doi: 10.1016/j.devcel.2011.08.013
Ingouff, M., Selles, B., Michaud, C., Vu, T. M., Berger, F., Schorn, A. J., et al. (2017). Live-cell analysis of DNA methylation during sexual reproduction in arabidopsis reveals context and sex-specific dynamics controlled by noncanonical RdDM. Genes Dev. 31, 72–83. doi: 10.1101/gad.289397.116
Jullien, P. E., Kinoshita, T., Ohad, N., Berger, F. (2006). Maintenance of DNA methylation during the arabidopsis life cycle is essential for parental imprinting. Plant Cell 18, 1360–1372. doi: 10.1105/tpc.106.041178
Kotlinski, M., Knizewski, L., Muszewska, A., Rutowicz, K., Lirski, M., Schmidt, A., et al. (2017). Phylogeny-based systematization of arabidopsis proteins with histone H1 globular domain. Plant Physiol. 174, 27–34. doi: 10.1104/pp.16.00214
Law, J. A., Jacobsen, S. E. (2010). Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 11, 204–220. doi: 10.1038/nrg2719
Love, M. I., Huber, W., Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi: 10.1186/s13059-014-0550-8
Luo, C., Keown, C. L., Kurihara, L., Zhou, J., He, Y., Li, J., et al. (2017). Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science 357, 600–604. doi: 10.1126/science.aan3351
Luo, C., Rivkin, A., Zhou, J., Sandoval, J. P., Kurihara, L., Lucero, J., et al. (2018). Robust single-cell DNA methylome profiling with snmC-seq2. Nat. Commun. 9, 3824. doi: 10.1038/s41467-018-06355-2
Lyons, D. B., Zilberman, D. (2017). DDM1 and lsh remodelers allow methylation of DNA wrapped in nucleosomes. Elife 6, e30674. doi: 10.7554/eLife.30674.028
Matzke, M. A., Mosher, R. A. (2014). RNA-Directed DNA methylation: An epigenetic pathway of increasing complexity. Nat. Rev. Genet. 15, 394–408. doi: 10.1038/nrg3683
Ono, A., Kinoshita, T. (2021). Epigenetics and plant reproduction: Multiple steps for responsibly handling succession. Curr. Opin. Plant Biol. 61, 102032. doi: 10.1016/j.pbi.2021.102032
Parrilla-Doblas, J. T., Morales-Ruiz, T., Ariza, R. R., Martinez-Macias, M. I., Roldan-Arjona, T. (2022). The c-terminal domain of arabidopsis ROS1 DNA demethylase interacts with histone H3 and is required for DNA binding and catalytic activity. DNA Repair (Amst). 115, 103341. doi: 10.1016/j.dnarep.2022.103341
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A., Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. doi: 10.1038/nmeth.4197
Qian, W., Miki, D., Zhang, H., Liu, Y., Zhang, X., Tang, K., et al. (2012). A histone acetyltransferase regulates active DNA demethylation in arabidopsis. Science 336, 1445–1448. doi: 10.1126/science.1219416
Rea, M., Zheng, W., Chen, M., Braud, C., Bhangu, D., Rognan, T. N., et al. (2012). Histone H1 affects gene imprinting and DNA methylation in arabidopsis. Plant J. 71, 776–786. doi: 10.1111/j.1365-313X.2012.05028.x
Rutowicz, K., Lirski, M., Mermaz, B., Teano, G., Schubert, J., Mestiri, I., et al. (2019). Linker histones are fine-scale chromatin architects modulating developmental decisions in arabidopsis. Genome Biol. 20, 157. doi: 10.1186/s13059-019-1767-3
Rutowicz, K., Puzio, M., Halibart-Puzio, J., Lirski, M., Kotlinski, M., Kroten, M. A., et al. (2015). A specialized histone H1 variant is required for adaptive responses to complex abiotic stress and related DNA methylation in arabidopsis. Plant Physiol. 169, 2080–2101. doi: 10.1104/pp.15.00493
Schoft, V. K., Chumak, N., Choi, Y., Hannon, M., Garcia-Aguilar, M., Machlicova, A., et al. (2011). Function of the DEMETER DNA glycosylase in the arabidopsis thaliana male gametophyte. Proc. Natl. Acad. Sci. U.S.A. 108, 8042–8047. doi: 10.1073/pnas.1105117108
Schoft, V. K., Chumak, N., Mosiolek, M., Slusarz, L., Komnenovic, V., Brownfield, L., et al. (2009). Induction of RNA-directed DNA methylation upon decondensation of constitutive heterochromatin. EMBO Rep. 10, 1015–1021. doi: 10.1038/embor.2009.152
Schon, M. A., Nodine, M. D. (2017). Widespread contamination of arabidopsis embryo and endosperm transcriptome data sets. Plant Cell 29, 608–617. doi: 10.1105/tpc.16.00845
Sequeira-Mendes, J., Araguez, I., Peiro, R., Mendez-Giraldez, R., Zhang, X., Jacobsen, S. E., et al. (2014). The functional topography of the arabidopsis genome is organized in a reduced number of linear motifs of chromatin states. Plant Cell 26, 2351–2366. doi: 10.1105/tpc.114.124578
She, W., Baroux, C. (2015). Chromatin dynamics in pollen mother cells underpin a common scenario at the somatic-to-reproductive fate transition of both the male and female lineages in arabidopsis. Front. Plant Sci. 6, 294. doi: 10.3389/fpls.2015.00294
She, W., Grimanelli, D., Rutowicz, K., Whitehead, M. W., Puzio, M., Kotlinski, M., et al. (2013). Chromatin reprogramming during the somatic-to-reproductive cell fate transition in plants. Development 140, 4008–4019. doi: 10.1242/dev.095034
Song, Q., Ando, A., Jiang, N., Ikeda, Y., Chen, Z. J. (2020). Single-cell RNA-seq analysis reveals ploidy-dependent and cell-specific transcriptome changes in arabidopsis female gametophytes. Genome Biol. 21, 178. doi: 10.1186/s13059-020-02094-0
Wierzbicki, A. T., Jerzmanowski, A. (2005). Suppression of histone H1 genes in arabidopsis results in heritable developmental defects and stochastic changes in DNA methylation. Genetics 169, 997–1008. doi: 10.1534/genetics.104.031997
Xu, Q., Wu, L., Luo, Z., Zhang, M., Lai, J., Li, L., et al. (2022). DNA Demethylation affects imprinted gene expression in maize endosperm. Genome Biol. 23, 77. doi: 10.1186/s13059-022-02641-x
Zemach, A., Kim, M. Y., Hsieh, P. H., Coleman-Derr, D., Eshed-Williams, L., Thao, K., et al. (2013). The arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 153, 193–205. doi: 10.1016/j.cell.2013.02.033
Zhang, C., Barthelson, R. A., Lambert, G. M., Galbraith, D. W. (2008). Global characterization of cell-specific gene expression through fluorescence-activated sorting of nuclei. Plant Physiol. 147, 30–40. doi: 10.1104/pp.107.115246
Zhang, C., Hung, Y. H., Rim, H. J., Zhang, D., Frost, J. M., Shin, H., et al. (2019). The catalytic core of DEMETER guides active DNA demethylation in arabidopsis. Proc. Natl. Acad. Sci. U. S. A. 116, 17563–17571. doi: 10.1073/pnas.1907290116
Keywords: epigenetics, DNA demethylation, gene imprinting, DNA glycosylase, linker histone H1
Citation: Han Q, Hung Y-H, Zhang C, Bartels A, Rea M, Yang H, Park C, Zhang X-Q, Fischer RL, Xiao W and Hsieh T-F (2022) Loss of linker histone H1 in the maternal genome influences DEMETER-mediated demethylation and affects the endosperm DNA methylation landscape. Front. Plant Sci. 13:1070397. doi: 10.3389/fpls.2022.1070397
Received: 14 October 2022; Accepted: 01 December 2022;
Published: 22 December 2022.
Edited by:
Nobutoshi Yamaguchi, Nara Institute of Science and Technology (NAIST), JapanReviewed by:
Alexandre Berr, UPR2357 Institut de biologie moléculaire des plantes (IBMP), FranceCélia Baroux, University of Zurich, Switzerland
Copyright © 2022 Han, Hung, Zhang, Bartels, Rea, Yang, Park, Zhang, Fischer, Xiao and Hsieh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert L. Fischer, cmZpc2NoZXJAYmVya2VsZXkuZWR1; Wenyan Xiao, d2VueWFuLnhpYW9Ac2x1LmVkdQ==; Tzung-Fu Hsieh, dGhzaWVoM0BuY3N1LmVkdQ==
†Present address: Matthew Rea, Department of Epigenetics, Van Andel Institute, Grand Rapids, MI, United States
‡These authors have contributed equally to this work