 Yanhong Song
Yanhong Song Chaochao Li
Chaochao Li Lifeng Liu1
Lifeng Liu1 Houcheng Zhou
Houcheng Zhou- 1Zhengzhou Fruit Research Institute, Chinese Academy of Agricultural Sciences, Zhengzhou, China
- 2Key Laboratory of Horticultural Plant Biology (Ministry of Education), Huazhong Agricultural University, Wuhan, China
The genus Fragaria consists of a rich diversity of ploidy levels with diploid (2x), tetraploid (4x), pentaploid (5x), hexaploidy (6x), octoploid (8x) and decaploid (10x) species. Only a few studies have explored the origin of diploid and octoploid strawberry, and little is known about the roles of tetraploidy and hexaploidy during the evolution of octoploid strawberry. The chloroplast genome is usually a stable circular genome and is widely used in investigating the evolution and matrilineal identification. Here, we assembled the chloroplast genomes of F. x ananassa cv. ‘Benihoppe’ (8x) using Illumina and HiFi data seperately. The genome alignment results showed that more InDels were located in the chloroplast genomes based on the PacBio HiFi data than Illumina data. We obtain highly accurate chloroplast genomes assembled through GetOrganelle using Illumina reads. We assembled 200 chloroplast genomes including 198 Fragaria (21 species) and 2 Potentilla samples. Analyses of sequence variation, phylogenetic and PCA analyses showed that Fragaria was divided into five groups. F. iinumae, F. nilgerrensis and all octoploid accessions formed Group A, C and E separately. Species native to western China were clustered into Group B. Group D consisted of F. virdis, F. orientalis, F. moschata, and F. vesca. STRUCTURE and haplotype network confirmed that the diploid F. vesca subsp. bracteata was the last maternal donator of octoploid strawberry. The dN/dS ratio estimated for the protein-coding genes revealed that genes involved in ATP synthase and photosystem function were under positive selection. These findings demonstrate the phylogeny of totally 21 Fragaria species and the origin of octoploid species. F. vesca was the last female donator of octoploid, which confirms the hypothesis that the hexaploid species F. moschata may be an evolutionary intermediate between the diploids and wild octoploid species.
1 Introduction
Chloroplasts are organelles with seni-autonomous genetic systems, playing a vital role in energy converters for higher plants (Neuhaus and Emes, 2000). Compared with the nuclear genome, the plant chloroplast genomes have relative conservation in structure composition and gene type (Wu et al., 2011; Bock and Knoop, 2012; Dong et al., 2012; Dong et al., 2013; Liang et al., 2020). The chloroplast genomes are ideal for studying plant phylogenetic analysis and species identification due to their simple structure, lack of recombination, and uniparental inheritance characteristics (Clegg et al., 1994; Ahmed et al., 2013; Mower and Vickrey, 2018). The first chloroplast genome (Nicotiana tabacum) was sequenced in 1986 (Shinozaki et al., 1986). As the sequencing technology develops, there has been a sharp increase in the number of chloroplast genomes from cereals, fruits, vegetables, and other flowering plants (Maier et al., 1995; Schmitz-Linneweber et al., 2001; Wu et al., 2010; Ruhfel et al., 2014; Yan et al., 2022). Comparative chloroplast genomes of Gossypium, Atractylodes, Musa, Medicago, Citrus, and other species were conducted to reveal genetic variation, phylogenetic relationship, and plastome evolution (Xu et al., 2013; Carbonell-Caballero et al., 2015; Zhang et al., 2020; Wang et al., 2021; Brock et al., 2022; Jiao et al., 2022; Li et al., 2022; Song et al., 2022; Yisilam et al., 2022). Combined with the nuclear genomes, chloroplast genomes were used to explore the haplotype development and phylogenetics relationship of Japanese apricot from different geographical locations (Huang et al., 2022).
Genome assembly toolkits and sequencing reads are two keys to accurate genomes. Next-generation sequencing (NGS) methods became an effective approach to producing chloroplast genome sequences after Sanger sequencing. However, short-read produce large amounts of DNA fragments ranging from 50-400 bp, which makes it challenging to assemble accurate genomes, especially for repeat-rich samples. The long-reads of third-generation sequencing (TGS), such as Oxford Nanopore Technologies (ONT) ultra-long reads and Pacific Biosciences (PacBio) highly accurate long read (HiFi), delivers even up to 200 kb long reads (Istace et al., 2017). Circular consensus sequencing (CCS) reads are long reads with a low error rate, meaning they allow the assembly of repeated regions. CCS reads were used to accurately assemble and detect SNPs of chloroplast genomes (Li et al., 2014). Although there is no systematic comparison between the HiFi CCS and short-read for assembling chloroplast genomes, the quality expectation for such small but important genomes is as high as complete and accurate in the community. Specifically, whether and how HiFi reads could be used to generate high-quality chloroplast genomes is untested. GetOrganelle is an efficient and accurate toolkit for de novo assembly of organelle genomes (Freudenthal et al., 2020; Jin et al., 2020; Odago et al., 2021; Ruang-Areerate et al., 2021; Singh et al., 2021; Drown et al., 2022; Liu et al., 2022; Zhao et al., 2022). It can assemble better plastomes using low coverage WGS data compared with NOVOplasty. Thus, we chose GetOrganelle to obtain accurate chloroplast genomes as a reference to correct genome sequences based on CCS reads.
The genus Fragaria includes ~25 identified species and comprises natural ploidy levels consisting of diploids (2n =14), tetraploids (4n =28), pentaploids (5n =35), hexaploids (6n =42), octoploids (8n =56) and decaploids (10n =70) (Staudt, 1962; Hummer et al., 2009; Staudt, 2009). Previous genome studies have focused on diploids and octoploids (Shulaev et al., 2011; Hardigan et al., 2019; Edger et al., 2019; Feng et al.,2021; Qiao et al., 2021), the origin and evolution of tetraploid and hexaploid strawberry remain unknown. China has been a distribution center of Fragaria resources for fourteen of twenty-five species spread in northeastern, northwestern, and southwestern China (Deng and Lei, 2005; Lei et al., 2017). The tetraploid strawberry includes five species: F. orientalis, F. moupinensis, F. corymbosa, F. gracilis, and F. tibetica. Except for F. orientalis, other species are native to China. Fragaria moschata (musk strawberry or hautbois strawberry) is native to Central Europe but has been replaced by F.x ananassa at the end of the 19th century (Darrow, 1966). The tetraploid and hexaploid strawberry are dioecious (individuals are either females or males) and unique characteristics in fruit aroma and resistance. F. moupinensis has strong adaptability and disease resistance (Guo et al., 2018). Fragaria moschata resists diseases like bacterial angular leaf spot disease (Maas et al., 1995) and powdery mildew (Kantor, 1984). They are cultivated commercially for their intense aroma and flavor (Kantor, 1984).
The cultivated strawberry (Fragaria x ananassa) is a young species formed less than 300 years ago through a spontaneous hybridization between the allo-octoploid species Fragaria virginiana and Fragaria chiloensis (Darrow, 1966; Given et al., 1988; Staudt, 2009). It spread globally from France for its fruit flavor and juicy flesh. It is an allo-octoploid species (2n =8× = 56), composed of 56 chromosomes organized in four diploid progenitor species. Previous phylogenetic studies reported that octoploid genomes consisted of four or five diploid progenitors (Fedorova, 1946; Tennessen et al., 2014; Kamneva et al., 2017; Yang and Davis, 2017; Edger et al., 2019; Liston et al., 2020; Feng et al., 2021). F. vesca and F. iinumae as two of the diploid progenitor species have been identified, the other species are still controversial. Based on a near-complete chromosome-scale assembly for octoploid strawberry ‘Camorosa’, phylogenetic analyses provided genome-wide support for the two unknown progenitors: F. virids and F. nipponica (Edger et al., 2019). According to the geographical distribution, tetraploid and hexaploid species may be involved in the evolution of octoploid strawberry (Edger et al., 2019). However, Liston et al., found no support for F. virids, F. nipponica and F. moschata as ancestors (Liston et al., 2020). Research also drawn a conclusion that F. viridis was not the diploid progenitor (Feng et al.,2021) using sppIDer (Langdon et al., 2018). In summary, the cytoplasm donor of wild strawberry remains unknown.
For the present study, we assembled the chloroplast genomes of F. x ananassa cv. ‘Benihoppe’ based on Illumina and CCS reads. Next, we sequenced 33 samples, including tetraploid species (F. orientalis, F. moupinensis, F. corymbose, F. gracilis, F. tibetica) and hexaploidy species (F. moschata) with Illumina HiSeq X Ten platform. We collected a total of 200 illumina data including NCBI database source, consisting of twenty-one Fragaria species and Potentilla. With the GetOrganelle toolkit, we obtained 165 complete circular chloroplast genomes successfully. Our main objects were to (1) compare the chloroplast genomes assembled with long- and short-read data; (2) conduct population genomic analyses of chloroplast genomes of Fragaria genus; (3) shed new insights on the population constructure and evolutionary history of strawberry. These results provided new insights into Fragaria species cluster and the origin of octoploid strawberry.
2 Materials and methods
2.1 Plant material and DNA sequencing
We conducted a comparison of chloroplast genomes of F. x ananassa cv. ‘Benihoppe’ assembled with long- and short-read data. In order to perform the test, we extracted DNA of the young leaves of ‘Benihoppe’ for the construction of CCS libraries and Illumina short-read libraries and sequenced them on the PacBio Sequel and Illumina HiSeq X Ten platform reapectively. A total of 10 Gb of HiFi reads and 20 Gb of Illumina reads were generated for the assembly of chloroplast genomes.
A total of 200 Illumina sequences were examined in this study. Of these, Illumina paired-end sequences of 167 accessions were downloaded from the NCBI Sequence Read Archive database (https://www.ncbi.nlm.nih.gov/sra) (Table S2). The rest of 33 Fragaria accessions, including F. orientalis, F. moupinensis, F. corymbose, F. gracilis, F. tibetica and F. moschata, were newly sequenced. The Illumina sequence are available in the NCBI SRA (BioProject ID: PRJNA913463). The fresh and young leaves of Fragaria accessions were collected from Zhengzhou Fruit Research Institute, CAAS. Extraction of the whole genomic DNA from fresh leaves of these species was performed with a modified Cetyltrimethylammonium bromide (CTAB) method. A 150 bp of paired-end libraries were constructed and PE150 sequencing was performed on the Illumina HiSeq X Ten platform.
2.2 Chloroplast genome assembly and annotation
We used Fastp software (v0.20.1) to filter the low-quality reads of 200 next-generation sequencing data, and then assembled by GetOrganelle (v 1.7.6) pipeline (Jin et al., 2020) with the optimized parameters “-fast -k 65,105,127 -w 0.68 -t 10 -f embplant_pt”. We obtained Illumina short-reads and PacBio HiFi sequences for Fragaria x ananassa cv. ‘Benihoppe’ to compare and correct the chloroplast genomes. About 10 Gb CCS clean reads were used to assemble the ‘Benihoppe’ genome contigs with the default parameters with Canu (v2.2, Koren et al., 2017) and hifiasm (v 0.15.5-r 350, Cheng et al., 2021). The chloroplast genome contigs based on HiFi reads were selected from the Blast using the ‘Benihoppe’ chloroplast genome based on short-reads as a reference. Three different hifiasm_contigs and canu_contigs could cover the chloroplast genome separately. Comparison of pair-wise alignment of ‘Benihoppe’ chloroplast genomes was performed by the mVISTA (http://genome.lbl.gov/vista/mvista/submit.shtml) with the Shuffle-LAGAN mode. Sequences of ‘Benihoppe’ chloroplast genome based on short-reads were used as a reference.
We obtained 165 complete chloroplast genome sequences. All circular chloroplast genomes were annotated with the PGA (Qu et al., 2019) and GeSeq (Tillich et al., 2017) (https://chlorobox.mpimp-golm.mpg.de/geseq.html), by using five GenBank-formatted file of F. x ananassa (MZ851773, KY358226), F. vesca (JF345175), F. orientalis (NC_035501) and F. moschata (MW537852) as the database. By comparing the annotation results and removing the incorrect annotations, we obtained 85 protein-coding genes (PCGs), 37 tRNAs and 8 rRNAs.
2.3 Mapping, variant calling and annotation
Clean reads of 200 next-generation sequencing data were then separately mapped to the ‘Benihoppe’ chloroplast genome using Burrows-Wheeler Aligner (BWA) software (v0.7.17-r1188) (Li and Durbin, 2009). Alignment files were converted SAM (Sequence Alignment Map) files into sorted BAM (binary version of SAM) files with SAMtools (v1.11). Then, the removal of duplicates was performed using Picard Tools (v2.27.4). Finally, the Variant Call Format (VCF) was obtained with Deepvariant (rc1.0.0). The GVCF files from 200 accessions were consolidated into a single VCF file using GLnexus (v1.2.7). The VCF file was used to annotate mutation sites using the software snpEff (v5.1) (Cingolani et al., 2012).
2.4 Phylogeny and population structure analyses
We constructed phylogenetic trees using a maximum likelihood-based method. Vcftools (v0.1.16) was used to extract sequence variation data from VCF files. The phylogenetic tree was conducted by IQ-TREE (v2.1.2) (Nguyen et al., 2015) with the ‘GTR + I+G’ model and 1000 bootstrap replicates based on sequence variation. Based on the evolution relationship and ploidy level within 198 samples, we defined six subgroups in diploid species and three subgroups in tetraploid species for further analysis (F. iinumae as 2x-1, F. nubicola and F. nipponica as 2x-2, daltoniana and F. chinensis as 2x-3, F. nilgerrensis as 2x-4, F. virdis as 2x-5, F. vesca, F. mandschurica and F.bucharica as 2x-6; F. moupinensis and F. tibetica as 4x-1, F. corymbose and F. gracilis as 4x-2, F. orientalis as 4x-3).
To analyze the population structure of chloroplast genomes in Fragaria, we conducted the principal component analysis (PCA) for 198 Fragaria accessions with filter SNPs using Plink (v1.9) pipeline (Purcell et al., 2007). Then, we conducted the ADMIXTURE (v1.3.0) (Alexander et al., 2009) to estimate the genetic ancestry of 198 Fragaria samples. The K=2 to 12 hypothetical ancestral populations were formed, and k=5 and k=11 is shown in Figure 1.
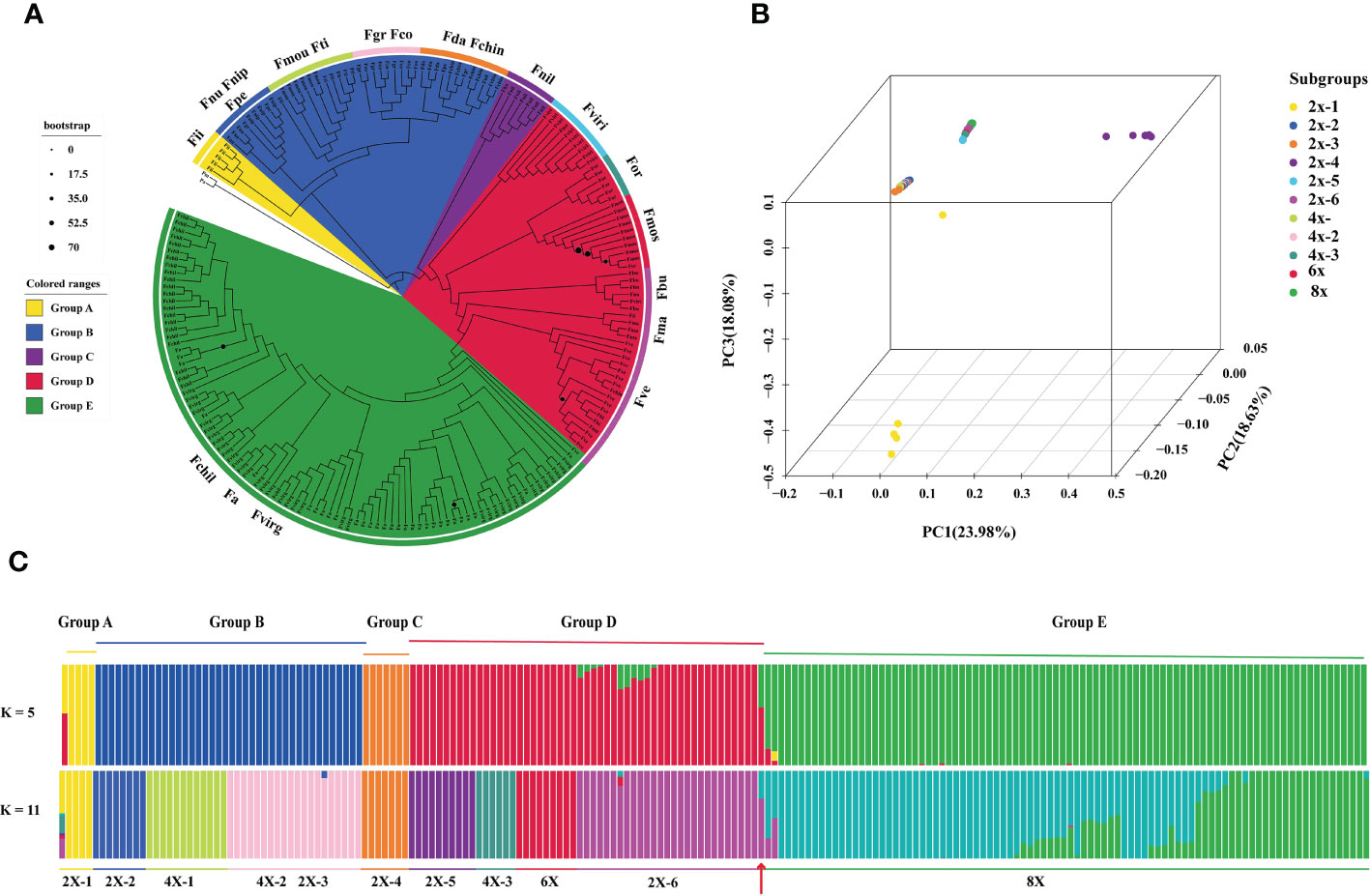
Figure 1 Analysis of Fragaria population genetic structure. (A) Phylogenetic tree based on sequence divergence of chloroplast genomes, with the colors of each branch region indicating the different groups (group A, B, C, D, E and outgroup). The colored circle outside the specie names represented the eleven subgroups: 2x-1, Fii, F iinumae; 2x-2, Fnu, F nubicola, Fnip, F nipponica, Fpe, F pentaphylla; 2x-3, Fda, F daltoniana, Fchin, F chinensis;2x-4, Fnil, F nilgerrensis; 2x-5, Fviri, F viridis; 2x-6, Fbu, F bringhurstii; Fma, F mandschurica; Fve, F vesca; 4x-1, Fmou, F moupinensis; Fti, F tibetica; 4x-2, Fgr, F gracilis; Fco, F corymbosa; 4x-3, F orientalis; 6x, Fmos, F moschata;8x, Fvirg, F virginiana; Fchil, F chiloensis; Fa, F.x ananassa. The blue circle sizes are shown as the percentage of bootstrap support less than 70%; (B) Principal component analysis (PCA) plot of the 198 Fragaria accessions, PC1, PC2 and PC3 explained 23.98%, 18.64% and 18.08% proportion of variance; (C) Population stratification analyses of Fragaria species. ADMIXTURE plots for representative Fragaria accessions and the outgroup for K= 5 and K= 11. The order of Fragaria species was in line with the phylogenetic tree. The red arrow indicates the diploid F vesca subsp. bracteate.
2.5 Genetic differentiation and population gene selection and haplotype
For the assessment of genetic differentiation and sequence divergence of the Fragaria population, we performed a sliding windows analysis to compute the F-statistics (Fst) and nucleotide diversity (π) based on the sequence variation as recommended by genomics_general (https://github.com/simonhmartin/genomics_general). The nucleotide diversity (π) should only be computed within species. We clustered the subgroups by their extremely close relationship using a phylogenetic tree. We calculated π over multiple species in a subgroup for its minor sequence divergence.
To study the molecular evolution of twenty-two Fragaria species, the patterns of synonymous (dS), nonsynonymous (dN) nucleotide substitutions and the ratio of nonsynonymous to synonymous rates (dN/dS) were calculated in PAML (v4.10.5) using the CODEML option with codon frequencies estimated using the F3 × 4 model, after removing the duplicated gene and the stop codon of the gene. We conducted a haplotype network for all Fragaria species using POPART (v1.7.1) (Leigh & Bryant, 2015) to calculate the gene flow diversity of haplotypes.
3 Results
3.1 De novo chloroplast genome assembly based on short- and long-read data
GetOrganelle was used to accurately assemble the chloroplast genomes of Fragaria x ananassa cv. ‘Benihoppe’. With the rapid development of high-throughput sequencing technologies, it is feasible to assemble complete chloroplast genomes using the low-coverage whole-genome sequencing data. Following the decreased HiFi sequencing costs in recent years, we want to know if HiFi reads could be used to generate high-precision chloroplast genomes. First, the Illumina data of ‘Benihoppe’ ranging from 1G to 10G was used to test the lowest necessary sequencing coverage to assemble complete chloroplast genomes with GetOrganelle. The results showed that 2G reads were enough to get the circular chloroplast genomes, and a further increase in the sequencing data did not improve the genomes further (Wang et al., 2018; Jin et al., 2020).
Furthermore, we conducted chloroplast genome assembly using HiFi data of the same cultivated strawberry ‘Benihoppe’. About 10 Gb of CCS clean reads were used to assemble the primary contigs with default parameters of Hifiasm (v 0.15.5-r 350) and Canu (v 2.2). Taking the chloroplast genome assembled by short-read as a reference, three contigs can overlap the whole genome (Figure S1). Visualized alignment of the three versions of chloroplast genomes (v1_Illumina, v2_Hifiasm_contig, and v3_Canu_contig) sequences using mVISTA (Frazer et al., 2004). Each horizontal row represents the pairwise sequence alignment identity percent. Compared with two versions of long-read genomes, the genome based on Canu-contigs has more SNP and minor InDels (especially the 34-46 kb region). Seven and four obvious InDels were found in Hiasm-contigs and Canu-contigs. Verification through PCR amplification and Sanger sequencing (primers are shown in Table S1) was used to verify the differences between the three versions. Chloroplast genomes with short-read data have much higher accuracy in InDels except for those located around 66kb (aligned sequences were shown in Figures 2, S2–7). We manually corrected the sequences of chloroplast genomes assembled using Illumina reads by GetOgranelle, and taked the genome as a reference for further analysis.
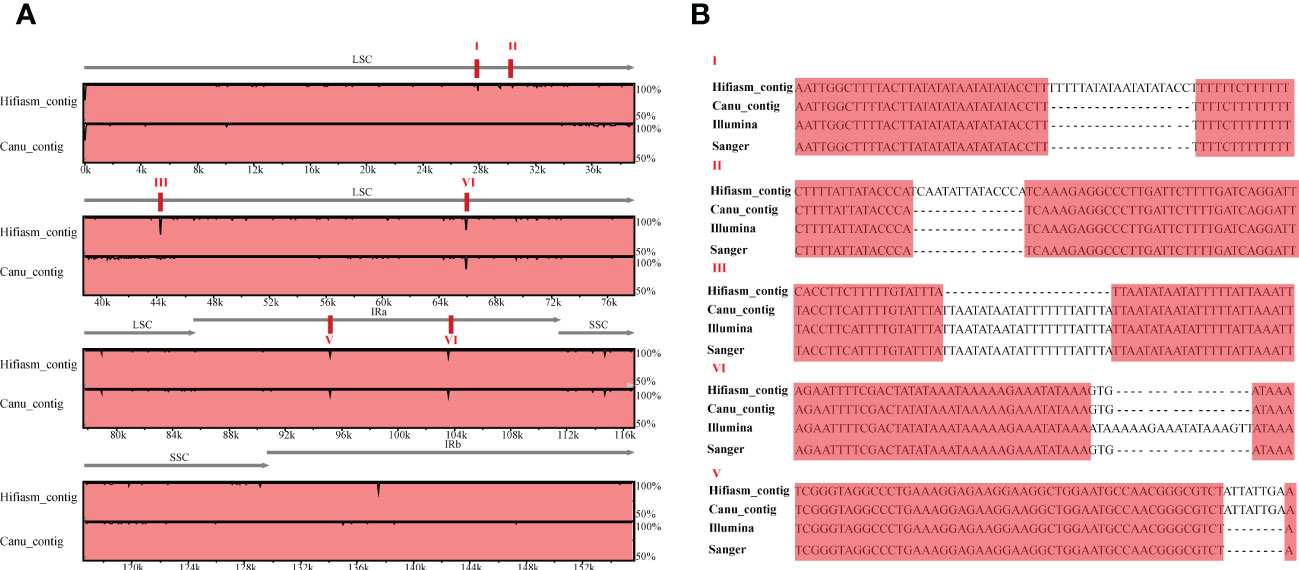
Figure 2 Visualized alignment and identity percent among the Fragaria chloroplast genomes based on three assembly methods relying on short- and long-read sequencing. (A)The figure was generated using mVISTA. The visible “peaks and valleys” graph shows the pairwise sequence alignement identity with the Benihoppe chloroplast genome assembled using Illumina data. The top and bottom percentages are displayed to the right of every row. The red boxes indicate six positions of identified InDels; (B) The verification of PCR amplification and Sanger sequence of the obvious InDels in A.
By comparing the annotation resulted by PGA and Geseq, and removing the incorrect annotations, eighty-five protein-coding genes (PCGs), 37 transfer RNA (tRNA) genes, and 8 ribosomal RNA (rRNA) genes were predicted in the F. x ananassa cv. ‘Benihoppe’.
3.2 Phylogenetic analyses and structure of strawberry species
In order to apply the assembly methods to the genetic study of strawberry population, we assembled 165 complete circular chloroplast genomes from the 200 samples using GetOrganelle (Figure 3; Table 1). The average length of the plastid genomes is 155,644 bp. The genome sizes among complete genomes ranged from 155,493 bp for F. viridis to 155,809 bp for F. hayatae. The GC contents were 37.18% (F. iinumae) - 37.29% (F. viridis).
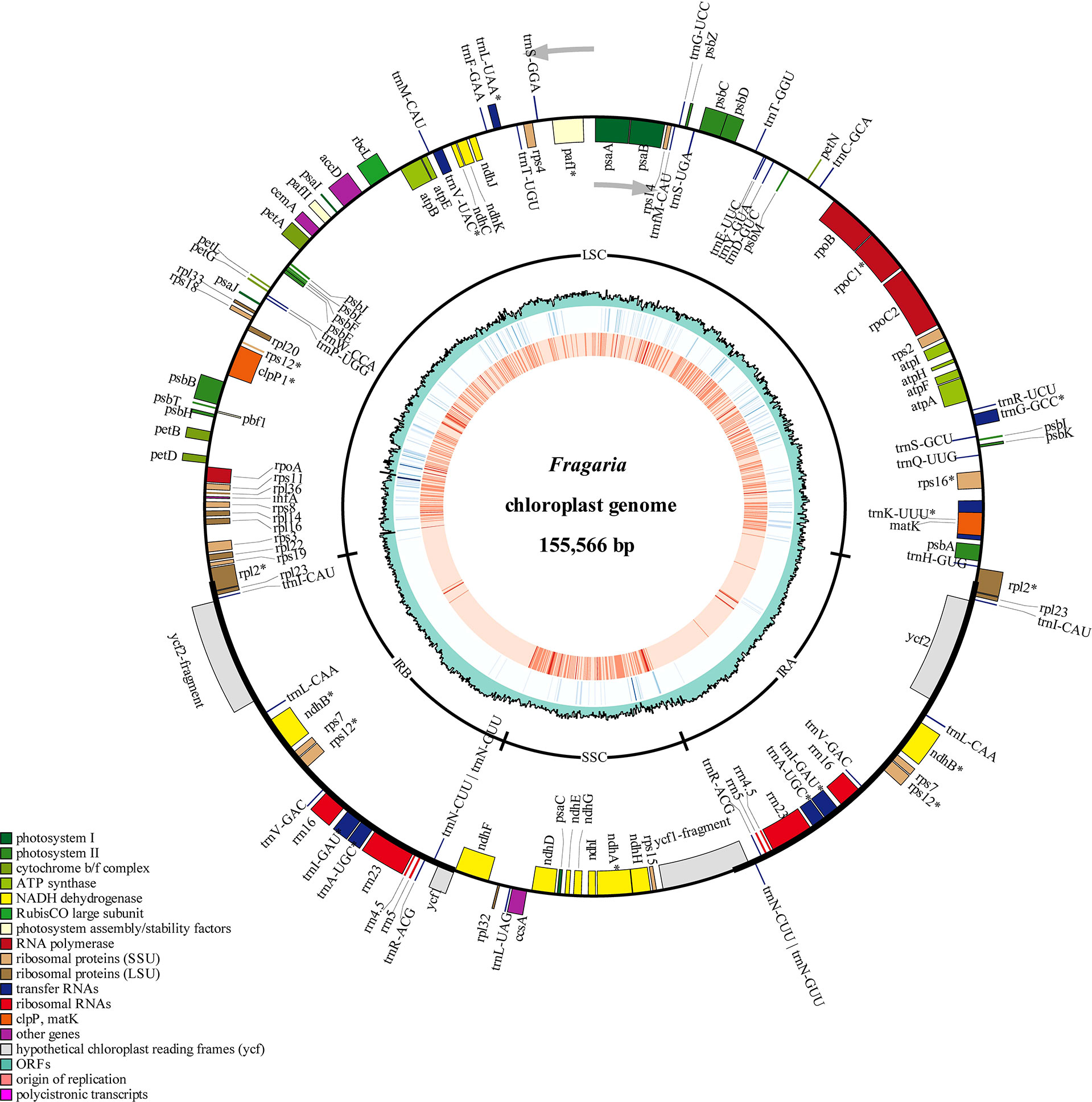
Figure 3 Chloroplast genome map of 198 Fragaria accessions. The gene position, quadripartite structure, GC content, density of InDels and SNP distribution were shown from the outer to inner rings. The outermost rectangles represented annotate genes belonging to different functional groups. Gene blocks on the outside and inside the circle indicated the clockwise and anticlockwise transcribed genes, respectively.
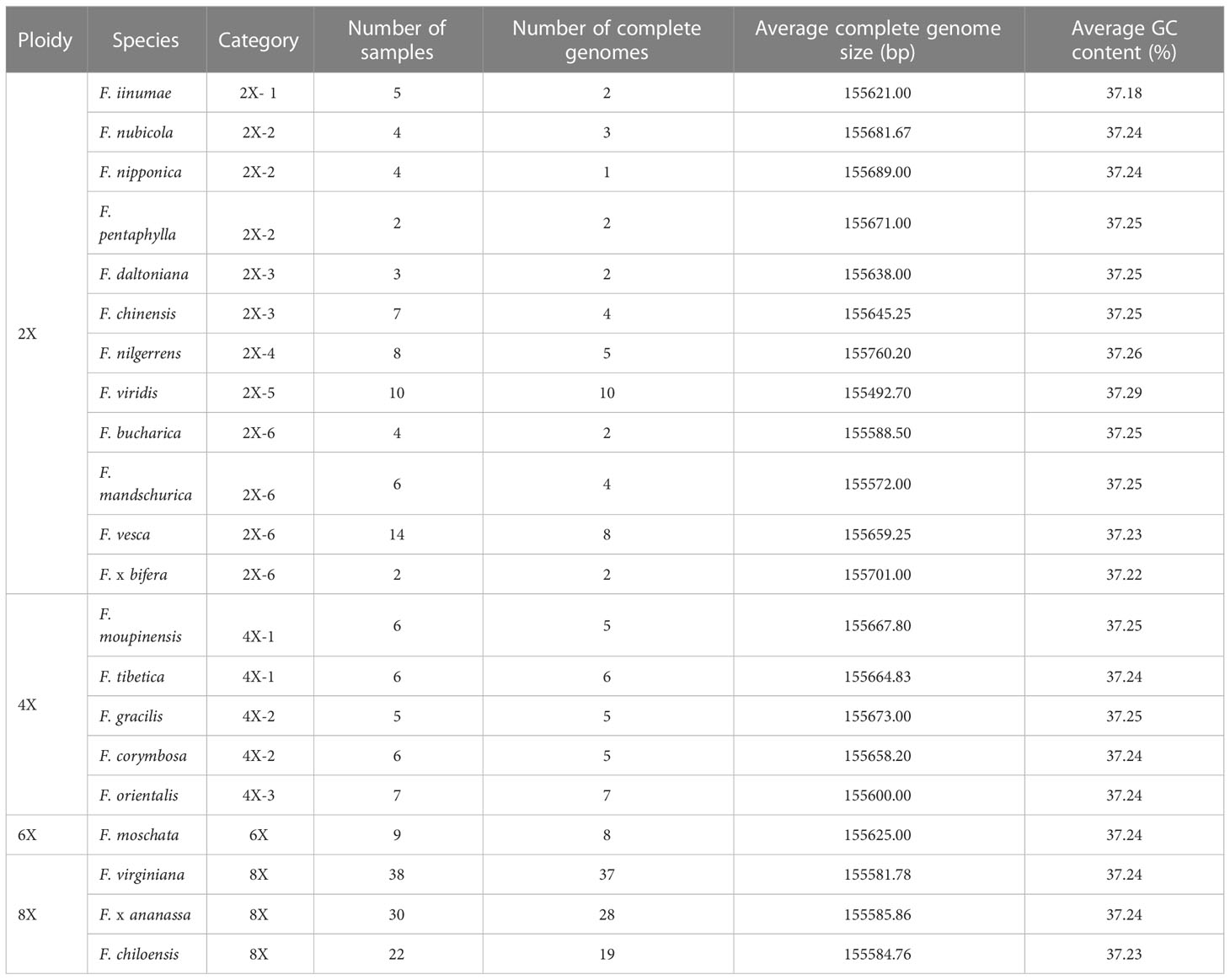
Table 1 Chloroplast genome features of 22 Fragaria species (21 wild species and 1 cultivated strawberry).
A comparison of the chloroplast genomes within Fragaria species showed that the sequence is highly conserved. Taking the chloroplast genome of ‘Benihoppe’ assembled with short-read data as reference. Among the 200 accessions, 4,551 single nucleotide polymorphisms (SNPs) and 621 small insertions and deletions (InDels) were identified. To further explore the roles of tetraploid and hexaploid species in polyploid formation, the phylogenetic trees were inferred using sequence variation of 198 accessions, with the genus Potentilla as the outgroups (Figure 1A). All Fragaria species could be divided into five groups. F. iinumae, the oldest extant species, and F. nilgerrensis formed a single Group A and Group C, separately. Group B included F. nubicola, F. nipponica, F. moupinensis (4x), F. tibetica (4x), F. gracilis (4x), F. corymbosa (4x), F. daltoniana (2x), and F. chinensis (2x). Group B contained four tetraploid species, in agreement with previous phylogenetic analyses using chloroplast sequences (Potter et al., 2009; Njuguna et al., 2013). In previous studies, F. pentaphylla (2x) and F. nubicola (2x) were supposed to be the progenitors of F. moupinensis (4x) and F. tibetica (4x) by target capturing sequence (Kamneva et al., 2017). F. corymbosa (4x) might be originated from F. chinensis (2x) for geographical distribution and similarity in morphological traits (Staudt, 2009). From our phylogenetic tree, tetraploid F. corymbosa (4x), F. gracilis (4x), and diploid F. chinensis (2x) are in the same clade, and F. chinensis (2x) might not be the female donator. The diploid species F. virdis (2x) was sister to the tetraploid species F. orientalis (4x-3) and hexaploid species F. moschata in group D. Meanwhile, F. vesca subsp. bracteata was the latest diploid donator to F x ananassa.
We conducted principal components analysis (PCA) to visualize the relationships between the Fragaria samples (Figure 1B). The Fragaria species formed four groups: group A (2x-1), group B and group C (2x-4), forming three distinct groups in accord with the clustering results of phylogenetic trees. Group D and E clustered together. Further, we applied the ADMIXTRUE analysis to all the samples based on sequence divergence. With K=5, Fragaria species groups were in line with the taxa of the phylogenetic tree. Haplotype analyses showed specific proof that k=5 could be distinguished from 198 accessions of 21 species (Figure 4). F. x ananassa had a mixture of F. vesca subsp. bracteate, suggesting recent introgression between these two species (Figure 1C). When K=11, it was notable that F. x ananassa originated from natural hybridization between F. chiloensis and F. virginiana.
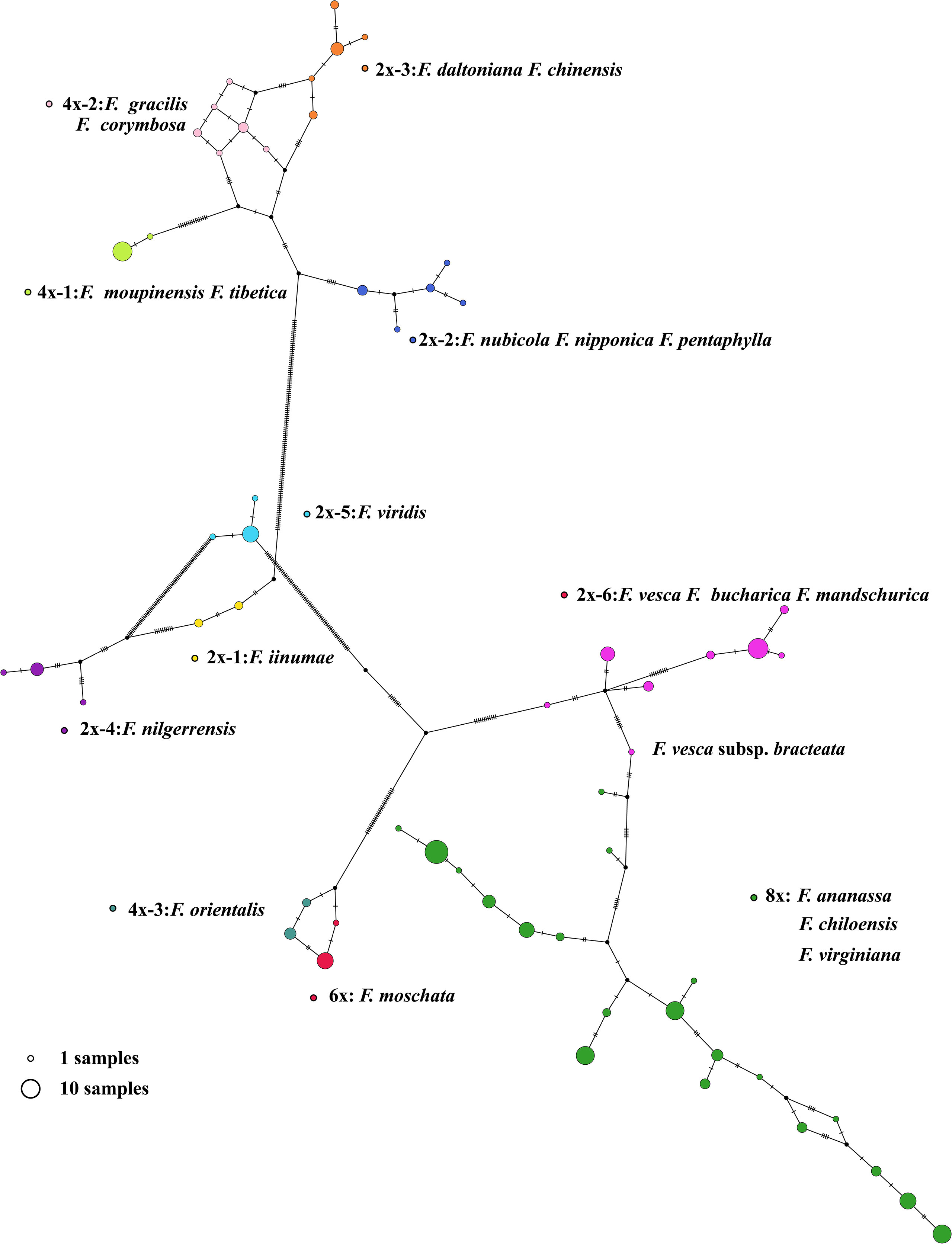
Figure 4 The chloroplast haplotypes network of Fragaria species. The size of the circle represents the number of haplotypes. Dots represent putative haplotypes. Mutations are represented by perpendicular dashes. The red arrow indicates the diploid F. vesca subsp. bracteate.
3.3 Genetic diversity within Fragaria species
To further analyze the relationship among species, we defined F. moupinensis (4x), F. tibetica (4x), as 4x-1, and F. corymbose (4x), F. gracilis (4x) as 4x-2. All Fragaria species were divided into 11 subgroups in Table 1. We calculated differentiation values (Fst) across all pairwise taxa comparisons. Lower Fst values were found between taxa in the same groups. For example, the overall lowest Fst was observed between 4x-1/2 and 2x-2/3. Among tetraploid species, 4x-3 shows a high Fst value compared to 4x-1 and 4x-2. Results are consistent with PCA analysis (Figure 1B). However, overall lowest Fst was obtained between 8x and other species (Figure 5A).
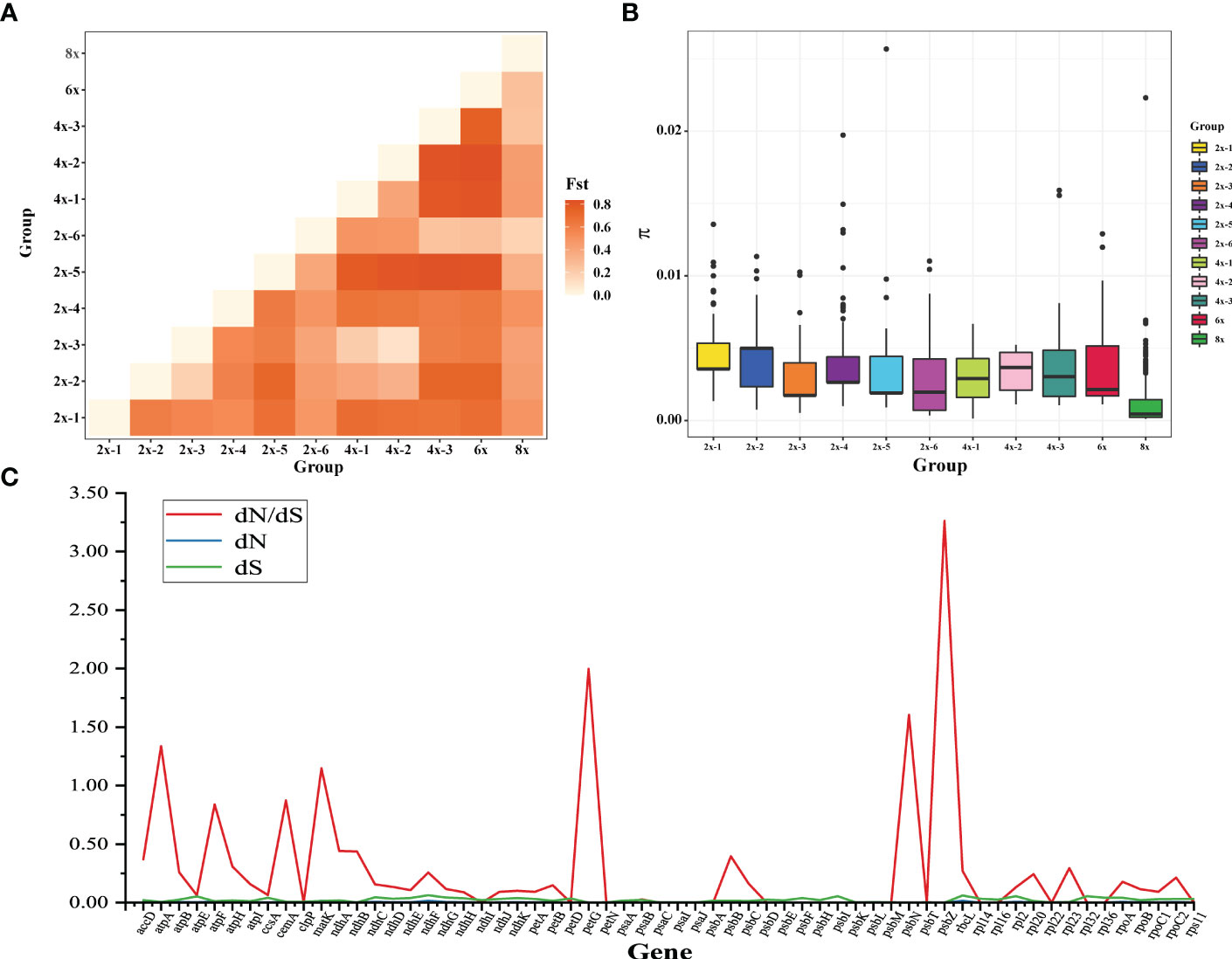
Figure 5 Analysis of Fragaria population genetic structure. (A) the pairwise Fst values between species of different ploidy levels; (B) Nucleotide diversity of chloroplast genome sequences of Fragaria species; (C) The estimations of the ratio of nonsynonymous to synonymous rates (dN/dS) of plastid protein-coding genes (PCGs).
The nucleotide diversity (π) was used to assess the level of sequence divergence in the chloroplast genomes of Fragaria species. The value of π showed that the lowest nucleotide diversity was found in octoploid accessions, even though 8x has more accessions than others in our study (Figure 5B). F. virdis (2x-5) underwent more mutation to 4x-3, 6x, and 2x-6. We couldn’t infer their ancestors from the absence of samples (extinct or uncollected progenitors), although they share the same clade in the phylogenetic tree. We also calculated the dN/dS ratio of protein-coding genes in the Fragaria chloroplast genomes (Figure 5C). The average dN/dS ratio of the 72 common protein-coding genes studied in the genomes was 0.27. The protein accD, matK, petG, psbN, and psbZ were under positive selection due to dn/ds ratio above 1. These genes involve in ATP synthase and photosystem function.
4 Discussion
4.1 Assembly of chloroplast genomes of Fragaria species
Strawberry genus, Fragaria L. includes ~25 identified species and comprises natural ploidy levels ranging from diploid (2n =14) to decaploid (10n =70), making it a research model for studying ploidy variations. Previously, the chloroplast genomes of 25 accessions representing 21 Fragaria species were assembled using genomic DNA and PCR pool sequencing, with 49% - 99% completeness (Njuguna et al., 2013). Twenty-seven (Li et al., 2021) and ten (Sun et al., 2021) Fragaria species were sequenced and obtained a chloroplast genome size of 155,479~155,832bp and 155,459~155,705 bp, respectively. In this study, we assemble the chloroplast genomes of F. x ananassa cv. ‘Benihoppe’ using short- and long-read data. Previous studies have assembled chloroplast genomes using PacBio and ONT data (Ferrarini et al., 2013; Wu et al., 2014; Redwan et al., 2015). After error correction, PacBio sequence data has an advantage on generating complete genome assemblies. A circular consensus sequencing (CCS) strategy was applied to assemble accurate genomes (Li et al., 2014).
Nevertheless, there is still a dearth of knowledge about the best approach to obtain accurate genomes. Here, we compared the genomes between short- and long-read data. The alignment results showed that chloroplast genomes based on hifiasm_contigs detected more InDels than canu_contigs (Figure 2). We used PCR to amplify target DNA segments, including InDels, to check which assembly was the most accurate. We found that the chloroplast genome of ‘Benihoppe’ using GetOrganelle with Illumina data was the most highly accurate genome assembly. To conclude, we suggest using short read Illumina data for chloroplast genome studies.
In our study, 165 new complete circular chloroplast genomes were obtained from 200 samples within 21 Fragaria species were obtained using the GetOrganelle toolkit, with an average of 155,644 bp in length (Figure 3). The chloroplast genomes of plants have highly conserved structures, with a quadripartite structure including two copies of an IR region and large and small single-copy (LSC and SSC) regions. Although several studies have revealed variation and evolution of the whole chloroplast genomes, to the best of our current knowledge, the accession number we assembled represents a more comprehensive analysis to date. As the sequencing technology and toolkits develop, it is possible to obtain complete and accurate chloroplast genomes. Based on the Fragaria population here, the results bring light to the origin and diversity of the genus. For example, haplotype analysis showed that F. vesca subsp. bracteata is the direct maternal source of octoploid strawberry (Figure 4).
4.2 Diversity and phylogenetic studies
Earlier phylogenetic analyses relied on the DNA sequence of partial chloroplast genomes or several genes (Jansen et al., 2007). Even though the use of DNA fragments enhanced the analysis, there are still uncertainties in information in the taxa relationship. Complete chloroplast genome sequences are valuable for phylogeny group classification and evolution of plant species. Based on pollen morphology and distribution, A Eurasian-American Fragaria group included six diploid, one tetraploid, one hexaploidy, and all octoploid species (Staudt, 2009). The classification is consistent with previous phylogenetic studies (Njuguna et al., 2013; Li et al., 2021; Sun et al., 2021). F. nilgerrensis species contains two subspecies nilgerrensis and hayatae. In our phylogenetic analysis, F. nilgerrensis forms an independent group C. The PCA results also showed that F. nilgerrensis separates from the rest Fragaria species (Figure 1B). However, it is uncertain that F. nilgerrensis was placed as a sister to F. chinensis or F. virdis in the previous chloroplast genomes and nuclear genomes analysis (Potter et al., 2009; Rousseau-Gueutin et al., 2009; Qiao et al., 2021). Chloroplast capture resulting from hybridization may explain the discordance between trees based on chloroplast DNA and nuclear genes (Soltis and Kuzoff, 1995). Chloroplast haplotype analysis showed that F. nilgerrensis was closely associated with F. iinumae. F. nilgerrensis is a widely distributed diploid strawberry native to southwest China and provides valuable genetic variations for breeding. This species is very different from other species and is easy to identify. The decaploid strawberry cultivar ‘Tokun’ origined from the hybridization between F. nilgerrensis and F. x ananassa (Noguchi, 2011). The evolutionary relationships of F. nubicola, F. pentaphylla, F. moupinensis, F. tibetica, F. corymbosa, F. gracilis, F. chinensis, and F. daltoniana have never been revealed (Rousseau-Gueutin et al., 2009; Li et al., 2021). These species, clustered into group B, are limited in distribution in Western China (Lei et al., 2017). In our results, F. nubicola and F. pentaphylla are sisters to the F. moupinensis and F. tibetica with 100% bootstrap support (Figure 3). Taking into account the overlapping geographical distribution in Southwest China and similar morphological characteristics of F. pentaphylla, F. moupinensis, and F. tibetica, F. pentaphylla may be a common female parent of tetraploid species F. moupinensis and F. tibetica (Rousseau-Gueutin et al., 2009; Kamneva et al., 2017; Li et al., 2021).
In our study, 4x-2 (F. corymbose and F. gracilis) and 2x-3 (F. chinensis and F. daltoniana) are sister species, and these species are distributed in Northwest China. Moreover, the morphological characteristics of F. corymbose and F. gracilis are similar in runners, petioles and calyx. Our phylogenetic analysis also supported that F. corymbosa and F. gracilis may share the same ancestor (Rousseau-Gueutin et al., 2009). Staudt et al. (2009) pointed out F. chinensis may be one of the ancestors of F. corymbose. Combined with the results of the phylogenetic tree and haplotype network, F. nubicola and F. pentaphylla may be the ancestors of F. moupinensis, F. tibetica, F. corymbosa, F. gracilis for sharing the overlapping geographical distribution. More samples need further research to explore the origin and evolution of these species.
F. virdis belonged to group D and was a sister to F. orientalis, F. moschata, F. bucharica, F. mandschurica, and F. vesca in this clade. Nevertheless, it is difficult to conclude that F. virdis is the ancestor of the rest of the species in group D, for the lack of adequate within-species diversity samples. This can partially explaine why antecedent research did not support the F. virdis as one of the subgenomes of octoploid strawberry. The tetraploid species F. orientalis and hexaploid species F. moschata share the same female ancestor (Figure 4). Regarding the diploid species in 2x-6, F. mandshurica is related to F. vesca, which may occur gene introgression from F. mandshurica to F. vesca (Hummer et al., 2013). Haplotype network shows that F. vesca subsp. bracteate haplotype was the latest female donor to octoploid strawberry, which means hexaploid species F. moschata may contribute to the octoploid event.
4.3 Effects of the geographical distribution of wild species on Fragaria evolution
Wild Fragaria species are valuable resources for cultivated strawberry breeding improvement. The nucleotide diversity (π) of Fragaria chloroplast genomes shows low diversity of 8x accessions (Figure 5B). The overall lower Fst was obtained between 8x and other species (Figure 5A). These results suggested that wild species have significantly contributed to cultivated strawberry. Middle or East Asia was regarded as a center of diversity from which native diploid and tetraploid species spread (Staudt, 1999). According to the phylogenetic tree, species clustered into groups A, B, and C are native to Asia, especially China. There is little known about tetraploid species in group B. It is more likely that these species are limited to Asia, and excluded from the formation of octoploid. Haplotype network also supports this speculation (Figure 4).
Interspecific hybridization between Fragaria species with lower ploidy levels was used to develop gene introgression into octoploid cultivars (Bors and Sullivan, 2005). The distribution of species in group D and E are from Asia to Europe. F. virids is distributed in Asia and Europe, and partially overlaps with the hexaploid F. moschata natived to Europe (Edgar et al., 2018). F. orientalis is distributed from Asia to Eastern Siberia, and haplotypes in F. orientalis and F. moschata were closer relationships (Figure 4). F. vesca is the most widely distributed diploid. and F. vesca subsp. bracteata is native to North America (Staudt, 1999), which coincides with F. chiloensis and F. virginiana geographical distribution. Consequently, species in group D are most likely to have contributed to the formation of octoploid strawberry.
Conclusion
In this study, we conducted a comparison of the chloroplast genomes assembled with short- and long-read data of F. ananassa cultivated species ‘Benihoppe’. we concluded that the chloroplast genome assemblies based on Illumina data were more accurate than CCS reads. We assembled 200 chloroplast genomes including 21 Fragaria species and outgroups. Based on sequence diversity, the phylogenetic tree and PCA analysis showed that Fragaria species could be divided into five groups. The F. nilgerrensis species form a single clade, in line with its unique morphological observation. Furthermore, we support that F. vesca subsp. bracteata was the last maternal donor to octoploid strawberry, which speculated that F. moschata may involve in the origin of octoploid strawberry.
Data availability statement
The data presented in the study are deposited in the NCBI SRA (BioProject ID: PRJNA913463).
Author contributions
YS and CL performed the research and analyzed the data. These authors contributed equally to this work and share the first authorship. HZ designed the chloroplast genomes research. LL, PH, GL, XZ and HZ contributed to the collection and conservation of wild resources. YS wrote the manuscript. All authors contributed to the article and approved the submitted version. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the National Key Research and Development Program (2019YFD1000203), the Agricultural Science and Technology Innovation Program (CAAS-ASTIP-2021-ZFRI) and the Major Science and Technology Projects of Henan Province (221100110400).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1065218/full#supplementary-material
Supplementary Figure 1 | Schematic diagram of sequence coverage of the Fragaria ananassa cv. Benihoppe chloroplast genome. The red line at the top of the schematic represents the ‘Benihoppe’ chloroplast genomes using Illumina data. The orange and bold blue lines indicate the contigs produced by Hifiasm and Canu software, respectively.
Supplementary Figure 2–7 | The sequence alignment of PCR amplification and Sanger sequence of the obvious InDels I-VI in, respectively.
References
Ahmed, I., Matthews, P. J., Biggs, P. J., Naeem, M., Mclenachan, P. A., Lockhart, P. J. (2013). Identification of chloroplast genome loci suitable for high-resolution phylogeographic studies of colocasia esculenta (L.) schott (Araceae) and closely related taxa. Mol. Ecol. Resour. 13, 929–937. doi: 10.1111/1755-0998.12128
Alexander, D. H., Novembre, J., Lange, K. (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. doi: 10.1101/gr.094052.109
Bors, R. H., Sullivan, J. A. (2005). Interspecific hybridization of fragaria vesca subspecies with f. nilgerrensis, f. nubicola, f. pentaphylla and f. viridis. J. Am. Soc Hortic. Sci. 130, 418–423. doi: 10.21273/JASHS.130.3.418
Brock, J. R., Mandáková, T., Mckain, M., Lysak, M. A., Olsen, K. M. (2022). Chloroplast phylogenomics in camelina (Brassicaceae) reveals multiple origins of polyploid species and the maternal lineage of c. sativa. Hortic. Res. 9, uhab050. doi: 10.1093/hr/uhab050
Carbonell-Caballero, J., Alonso, R., Ibanez, V., Terol, J., Talon, M., Dopazo, J. (2015). A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus citrus. Mol. Biol. Evol. 32, 929–937. doi: 10.1093/molbev/msv082
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H., Li, H. (2021). Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175. doi: 10.1038/s41592-020-01056-5
Cingolani, P., Platts, A., Wang Le, L., Coon, M., Nguyen, T., Wang, L., et al (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92.
Clegg, M. T., Gaut, B. S., Learn, G. H., Jr., Morton, B. R. (1994). Rates and patterns of chloroplast DNA evolution. Proc. Natl. Acad. Sci. U. S. A. 91, 6795–6801. doi: 10.1073/pnas.91.15.6795
Darrow, G. M. (1966). The strawberry history, breeding and physiology (New York: Holt, Rinehart and Winston).
Deng, M. Q., Lei, J. J. (2005). China Fruit records–volume strawberry (Beijing: China Forest Press), 20–103.
Dong, W., Liu, J., Yu, J., Wang, L., Zhou, S. (2012). Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS One 7, e35071. doi: 10.1371/journal.pone.0035071
Dong, W., Xu, C., Cheng, T., Lin, K., Zhou, S. (2013). Sequencing angiosperm plastid genomes made easy: a complete set of universal primers and a case study on the phylogeny of saxifragales. Genome Biol. Evol. 5, 989–997. doi: 10.1093/gbe/evt063
Drown, M. K., Deliberto, A. N., Flack, N., Doyle, M., Westover, A. G., Proefrock, J. C., et al. (2022). Sequencing bait: Nuclear and mitogenome assembly of an abundant coastal tropical and subtropical fish, atherinomorus stipes. Genome Biol. Evol. 14, evac111. doi: 10.1093/gbe/evac111
Edger, P. P., Poorten, T. J., Vanburen, R., Hardigan, M. A., Colle, M., Mckain, M. R. (2019). Origin and evolution of the octoploid strawberry genome. Nat. Genet. 51, 541–547. doi: 10.1038/s41588-019-0356-4
Fedorova, N. J. (1946). Crossability and phylogenetic relations in the main European species of fragaria. Compil. Natl. Acad. Sci. USSR. 52, 545–547.
Feng, C., Wang, J., Harris, A. J., Folta, K. M., Zhao, M., Kang, M. (2021). Tracing the diploid ancestry of the cultivated octoploid strawberry. Mol. Biol. Evol. 38, 478–485. doi: 10.1093/molbev/msaa238
Ferrarini, M., Moretto, M., Ward, J. A., Šurbanovski, N., Stevanović, V., Giongo, L., et al. (2013). An evaluation of the PacBio RS platform for sequencing and de novo assembly of a chloroplast genome. BMC Genomics 14, 670. doi: 10.1186/1471-2164-14-670
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic. Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Freudenthal, J. A., Pfaff, S., Terhoeven, N., Korte, A., Ankenbrand, M. J., Förster, F. (2020). A systematic comparison of chloroplast genome assembly tools. Genome Biol. 21, 254. doi: 10.1186/s13059-020-02153-6
Given, N. K., Venis, M. A., Gierson, D. (1988). Hormonal regulation of ripening in the strawberry, a non-climacteric fruit. Planta 174, 402–406. doi: 10.1007/BF00959527
Guo, R., Xue, L., Luo, G., Zhang, T., Lei, J. (2018). Investigation and taxonomy of wild fragaria resources in Tibet, China. Genet. Resour. Crop Evol. 65, 405–415. doi: 10.1007/s10722-017-0541-1
Hardigan, M. A., Feldmann, M. J., Lorant, A., Bird, K. A., Famula, R., Acharya, C., et al. (2019). Genome synteny has been conserved among the octoploid progenitors of cultivated strawberry over millions of years of evolution. Front. Plant Sci. 10, 1789. doi: 10.3389/fpls.2019.01789
Huang, X., Ni, Z., Shi, T., Tao, R., Yang, Q., Luo, C., et al. (2022). Novel insights into the dissemination route of Japanese apricot (Prunus mume sieb. et zucc.) based on genomics. Plant J. 110, 1182–1197. doi: 10.1111/tpj.15731
Hummer, K., Ballington, J., Finn, C., Davis, T. (2013). Asian Germplasm Influences on American Berry Crops. HortScience 48, 1090–1094
Hummer, K.E., Nathewet, P., Yanagi, T. (2009). Decaploidy in Fragaria iturupensis (Rosaceae). Am. J. Bot. 96, 713–716.
Istace, B., Friedrich, A., D'agata, L., Faye, S., Payen, E., Beluche, O., et al. (2017). de novo assembly and population genomic survey of natural yeast isolates with the Oxford nanopore MinION sequencer. GigaScience 6, 1–13. doi: 10.1093/gigascience/giw018
Jansen, R. K., Cai, Z., Raubeson, L. A., Daniell, H., Depamphilis, C. W., Leebens-Mack, J., et al. (2007). Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. U. S. A. 104, 19369–19374. doi: 10.1073/pnas.0709121104
Jiao, Y. X., He, X. F., Song, R., Wang, X. M., Zhang, H., Aili, R., et al. (2022). Recent structural variations in the medicago chloroplast genomes and their horizontal transfer into nuclear chromosomes. J. Syst. Evol. doi: 10.1111/jse.12900
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., Depamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241. doi: 10.1186/s13059-020-02154-5
Kamneva, O. K., Syring, J., Liston, A., Rosenberg, N. A. (2017). Evaluating allopolyploid origins in strawberry (Fragaria) using haplotypes generated from target capture sequencing. BMC Evol. Biol. 17, 180. doi: 10.1186/s12862-017-1019-7
Kantor, T. S. (1984). Results of breeding and genetic work aimed at creating economically valuable cultivars from incongruent crossing of fragaria × ananas. × fragaria moschata. Soviet Genet. 19, 1621–1635.
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Langdon, Q. K., Peris, D., Kyle, B., Hittinger, C. T. (2018). sppIDer: A species identification tool to investigate hybrid genomes with high-throughput sequencing. Mol. Biol. Evol. 35, 2835–2849. doi: 10.1093/molbev/msy166
Leigh, J. W., Bryant, D. (2015). Popart: full-feature software for haplotype network construction. Methods Ecol. Evol. 6, 1110–1116. doi: 10.1111/2041-210X.12410
Lei, J. J., Xue, L., Guo, R. X., Dai, H. P. (2017). The fragaria species native to China and their geographical distribution. Acta Hortic. 1156, 37–46. doi: 10.17660/ActaHortic.2017.1156.5
Liang, H., Zhang, Y., Deng, J., Gao, G., Ding, C., Zhang, L., et al. (2020). The complete chloroplast genome sequences of 14 curcuma species: Insights into genome evolution and phylogenetic relationships within zingiberales. Front. Genet. 11, 802. doi: 10.3389/fgene.2020.00802
Li, C., Cai, C., Tao, Y., Sun, Z., Jiang, M., Chen, L., et al. (2021). Variation and evolution of the whole chloroplast genomes of fragaria spp. (Rosaceae). Front. Plant Sci. 12, 754209. doi: 10.3389/fpls.2021.754209
Li, S., Duan, W., Zhao, J., Jing, Y., Feng, M., Kuang, B., et al. (2022). Comparative analysis of chloroplast genome in saccharum spp. and related members of 'Saccharum complex'. Int. J. Mol. Sci. 23 7661. doi: 10.3390/ijms23147661
Li, H., Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, Q., Li, Y., Song, J., Xu, H., Xu, J., Zhu, Y., et al. (2014). High-accuracy de novo assembly and SNP detection of chloroplast genomes using a SMRT circular consensus sequencing strategy. New Phytol. 204, 1041–1049. doi: 10.1111/nph.12966
Liston, A., Wei, N., Tennessen, J. A., Li, J., Dong, M., Ashman, T.-L. (2020). Revisiting the origin of octoploid strawberry. Nat. Genet. 52, 2–4. doi: 10.1038/s41588-019-0543-3
Liu, Y., Zhu, X., Wu, M., Xu, X., Dai, Z., Gou, G. (2022). The complete chloroplast genome of critically endangered chimonobambusa hirtinoda (Poaceae: Chimonobambusa) and phylogenetic analysis. Sci. Rep. 12, 9649. doi: 10.1038/s41598-022-13204-2
Maas, J. L., Pooler, M. R., Galletta, G. J. (1995). Bacterial angular leafspot disease strawberry: Present status and prospects for control. Adv. Strawberry Res. 14, 18–24.
Maier, R. M., Neckermann, K., Igloi, G. L., Kössel, H. (1995). Complete sequence of the maize chloroplast genome: Gene content, hotspots of divergence and fine tuning of genetic information by transcript editing. J. Mol. Biol. 251, 614–628. doi: 10.1006/jmbi.1995.0460
Mower, J. P., Vickrey, T. L. (2018). “Chapter nine - structural diversity among plastid genomes of land plants,” in Advances in botanical research. Eds. Chaw, S.-M., Jansen, R. K. (Academic Press), 263–292.
Neuhaus, H. E., Emes, M. J. (2000). Nonphotosynthetic metabolism in plastids. Annu. Rev. Plant Physiol. Plant Mol. Biol. 51, 111–140. doi: 10.1146/annurev.arplant.51.1.111
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Njuguna, W., Liston, A., Cronn, R., Ashman, T. L., Bassil, N. (2013). Insights into phylogeny, sex function and age of fragaria based on whole chloroplast genome sequencing. Mol. Phyl. Evol. 66, 17–29. doi: 10.1016/j.ympev.2012.08.026
Noguchi, Y. (2011). “Tokun” : A new decaploid interspecific hybrid strawberry having the aroma of the wild strawberry. J. Japan Assoc. Odor Environ. 42, 122–128. doi: 10.2171/jao.42.122
Odago, W. O., Waswa, E. N., Nanjala, C., Mutinda, E. S., Wanga, V. O., Mkala, E. M., et al. (2021). Analysis of the complete plastomes of 31 species of hoya group: Insights into their comparative genomics and phylogenetic relationships. Front. Plant Sci. 12, 814833. doi: 10.3389/fpls.2021.814833
Potter, D., Luby, J., Harrison, R. (2009). Phylogenetic relationships among species of fragaria (Rosaceae) inferred from non-coding nuclear and chloroplast DNA sequences. Syst. Bot. 25, 337–348. doi: 10.2307/2666646
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Qiao, Q., Edger, P. P., Xue, L., Qiong, L., Lu, J., Zhang, Y., et al. (2021). Evolutionary history and pan-genome dynamics of strawberry (Fragaria spp.). Proc. Natl. Acad. Sci. U. S. A. 118, e2105431118. doi: 10.1073/pnas.2105431118
Qu, X. J., Moore, M. J., Li, D. Z., Yi, T. S. (2019). PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 15, 50. doi: 10.1186/s13007-019-0435-7
Redwan, R. M., Saidin, A., Kumar, S. V. (2015). Complete chloroplast genome sequence of MD-2 pineapple and its comparative analysis among nine other plants from the subclass commelinidae. BMC Plant Biol. 15, 196. doi: 10.1186/s12870-015-0587-1
Rousseau-Gueutin, M., Gaston, A., Aïnouche, A., Aïnouche, M. L., Olbricht, K., Staudt, G., et al. (2009). Tracking the evolutionary history of polyploidy in fragaria l. (strawberry): New insights from phylogenetic analyses of low-copy nuclear genes. Mol. Phyl. Evol. 51, 515–530. doi: 10.1016/j.ympev.2008.12.024
Ruang-Areerate, P., Kongkachana, W., Naktang, C., Sonthirod, C., Narong, N., Jomchai, N., et al. (2021). Complete chloroplast genome sequences of five bruguiera species (Rhizophoraceae): comparative analysis and phylogenetic relationships. Peer J. 9, e12268. doi: 10.7717/peerj.12268
Ruhfel, B. R., Gitzendanner, M. A., Soltis, P. S., Soltis, D. E., Burleigh, J. G. (2014). From algae to angiosperms–inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes. BMC Evol. Biol. 14, 23. doi: 10.1186/1471-2148-14-23
Schmitz-Linneweber, C., Maier, R. M., Alcaraz, J.-P., Cottet, A., Herrmann, R. G., Mache, R. (2001). The plastid chromosome of spinach (Spinacia oleracea): complete nucleotide sequence and gene organization. Plant Mol. Biol. 45, 307–315. doi: 10.1023/A:1006478403810
Shinozaki, K., Ohme, M., Tanaka, M., Wakasugi, T., Hayashida, N., Matsubayashi, T., et al. (1986). The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression. EMBO J. 5, 2043–2049. doi: 10.1002/j.1460-2075.1986.tb04464.x
Shulaev, V., Sargent, D. J., Crowhurst, R. N., Mockler, T. C., Folkerts, O., Delcher, A. L., et al. (2011). The genome of woodland strawberry (Fragaria vesca). Nat. Genet. 43, 109–116. doi: 10.1038/ng.740
Singh, N., Patil, P., Sowjanya, P. R., Parashuram, S., Natarajan, P., Babu, K., et al. (2021). Chloroplast genome sequencing, comparative analysis, and discovery of unique cytoplasmic variants in pomegranate (Punica granatum l.). Front. Genet. 12, 704075. doi: 10.3389/fgene.2021.704075
Soltis, D. E., Kuzoff, R. K. (1995). Discordance between nuclear and chloroplast phylogenies in the heuchera group (Saxifragaceae). Evolution 49, 727–742. doi: 10.2307/2410326
Song, W., Ji, C., Chen, Z., Cai, H., Wu, X., Shi, C., et al. (2022). Comparative analysis the complete chloroplast genomes of nine musa species: Genomic features, comparative analysis, and phylogenetic implications. Front. Plant Sci. 13, 832884. doi: 10.3389/fpls.2022.832884
Staudt, G. (1962). Taxonomic studies in the genus fragaria typification of fragaria species known at the time of Linnaeus. Botany 40, 869–886.
Staudt, G. (1999). Systematics and geographic distribution of the American strawberry species: Taxonomic studies in the genus fragaria (Rosaceae:Potentilleae) (Berkeley, CA: University of California Press).
Sun, J., Sun, R., Liu, H., Chang, L., Li, S., Zhao, M., et al. (2021). Complete chloroplast genome sequencing of ten wild fragaria species in China provides evidence for phylogenetic evolution of fragaria. Genomics 113, 1170–1179. doi: 10.1016/j.ygeno.2021.01.027
Tennessen, J. A., Govindarajulu, R., Ashman, T.-L., Liston, A. (2014). Evolutionary origins and dynamics of octoploid strawberry subgenomes revealed by dense targeted capture linkage maps. Genome Biol. Evol. 6, 3295–3313. doi: 10.1093/gbe/evu261
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq– versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Wang, W., Schalamun, M., Morales-Suarez, A., Kainer, D., Schwessinger, B., Lanfear, R. (2018). Assembly of chloroplast genomes with long- and short-read data: a comparison of approaches using eucalyptus pauciflora as a test case. BMC Genom. 19, 977. doi: 10.1186/s12864-018-5348-8
Wang, Y., Wang, S., Liu, Y., Yuan, Q., Sun, J., Guo, L. (2021). Chloroplast genome variation and phylogenetic relationships of atractylodes species. BMC Genom. 22, 103. doi: 10.1186/s12864-021-07394-8
Wu, F. H., Chan, M. T., Liao, D. C., Hsu, C. T., Lee, Y. W., Daniell, H., et al. (2010). Complete chloroplast genome of oncidium Gower ramsey and evaluation of molecular markers for identification and breeding in oncidiinae. BMC Plant Biol. 10, 68. doi: 10.1186/1471-2229-10-68
Wu, Z., Gui, S., Quan, Z., Pan, L., Wang, S., Ke, W., et al. (2014). A precise chloroplast genome of nelumbo nucifera (Nelumbonaceae) evaluated with Sanger, illumina MiSeq, and PacBio RS II sequencing platforms: insight into the plastid evolution of basal eudicots. BMC Plant Biol. 14, 289. doi: 10.1186/s12870-014-0289-0
Wu, C. S., Wang, Y. N., Hsu, C. Y., Lin, C. P., Chaw, S. M. (2011). Loss of different inverted repeat copies from the chloroplast genomes of pinaceae and cupressophytes and influence of heterotachy on the evaluation of gymnosperm phylogeny. Genome Biol. Evol. 3, 1284–1295. doi: 10.1093/gbe/evr095
Xu, Q., Xiong, G., Li, P., He, F., Huang, Y., Wang, K., et al. (2013). Correction: Analysis of complete nucleotide sequences of 12 gossypium chloroplast genomes: Origin and evolution of allotetraploids. PloS One 8, e37128. doi: 10.1371/annotation/47563c17-536c-465d-9b93-cd35a78f6e66
Yang, Y., Davis, T. M. (2017). A new perspective on polyploid fragaria (Strawberry) genome composition based on Large-scale, multi-locus phylogenetic analysis. Genome Biol. Evol. 9, 3433–3448. doi: 10.1093/gbe/evx214
Yan, L. J., Zhu, Z. G., Wang, P., Fu, C. N., Guan, X. J., Kear, P., et al. (2022). Comparative analysis of 343 plastid genomes of solanum section petota : insights into potato diversity, phylogeny and species discrimination. J. Syst. Evol. doi: 10.1111/jse.12898
Yisilam, G., Wang, C. X., Xia, M. Q., Comes, H. P., Li, P., Li, J., et al. (2022). Phylogeography and population genetics analyses reveal evolutionary history of the desert resource plant lycium ruthenicum (Solanaceae). Front. Plant Sci. 13, 915526. doi: 10.3389/fpls.2022.915526
Zhang, X., Sun, Y., Landis, J. B., Lv, Z., Shen, J., Zhang, H., et al. (2020). Plastome phylogenomic study of gentianeae (Gentianaceae): widespread gene tree discordance and its association with evolutionary rate heterogeneity of plastid genes. BMC Plant Biol. 20, 340. doi: 10.1186/s12870-020-02518-w
Keywords: Fragaria, chloroplast genome assembly, population structure, diversity, evolutionary
Citation: Song Y, Li C, Liu L, Hu P, Li G, Zhao X and Zhou H (2023) The population genomic analyses of chloroplast genomes shed new insights on the complicated ploidy and evolutionary history in Fragaria. Front. Plant Sci. 13:1065218. doi: 10.3389/fpls.2022.1065218
Received: 15 November 2022; Accepted: 30 December 2022;
Published: 15 February 2023.
Edited by:
JianJun Jin, Columbia University, United StatesReviewed by:
Tao Zhou, Xian Jiaotong University, ChinaHuasheng Peng, China Academy of Chinese Medical Sciences, China
Copyright © 2023 Song, Li, Liu, Hu, Li, Zhao and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Houcheng Zhou, emhvdWhvdWNoZW5nQGNhYXMuY24=
†These authors have contributed equally to this work and share first authorship