 Liping Ren
Liping Ren Wenyang Wan1†
Wenyang Wan1†- 1Key Laboratory of Horticultural Plant Biology of Biological and Food Engineering School, Fuyang Normal University, Fuyang, China
- 2Horticultural Institute, Fuyang Academy of Agricultural Sciences, Fuyang, China
- 3State Key Laboratory of Tea Plant Biology and Utilization, International Joint Laboratory on Tea Chemistry and Health Effects, Anhui Agricultural University, Hefei, China
WRKY transcription factors (TFs), one of the largest TF families, serve critical roles in the regulation of secondary metabolite production. However, little is known about the expression pattern of WRKY genes during the germination and maturation processes of Toona sinensis buds. In the present study, the new assembly of the T. sinensis genome was used for the identification of 78 TsWRKY genes, including gene structures, phylogenetic features, chromosomal locations, conserved protein domains, cis-regulatory elements, synteny, and expression profiles. Gene duplication analysis revealed that gene tandem and segmental duplication events drove the expansion of the TsWRKYs family, with the latter playing a key role in the creation of new TsWRKY genes. The synteny and evolutionary constraint analyses of the WRKY proteins among T. sinensis and several distinct species provided more detailed evidence of gene evolution for TsWRKYs. Besides, the expression patterns and co-expression network analysis show TsWRKYs may multi-genes co-participate in regulating terpenoid biosynthesis. The findings revealed that TsWRKYs potentially play a regulatory role in secondary metabolite synthesis, forming the basis for further functional characterization of WRKY genes with the intention of improving T. sinensis.
1. Introduction
Toona sinensis (A. Juss) Roem, a deciduous native plant endemic to eastern and southeastern Asia and commonly known as Chinese toon, belongs to the Meliaceae family (Dong et al., 2013). In China, the tender buds of T. sinensis have been accepted widely as vegetables for its rich nutritional value and unique aroma (Zhai and Granvogl, 2019). The young leaves of T. sinensis are high in amino acids, vitamins, and other nutrients that are beneficial to human health (Ren et al., 2021). T. sinensis is often known as traditional Chinese medicine due to the use of its numerous tissues in the treatment of various of ailments. A recent study showed that terpenoids, phenylpropanoids, and flavonoids, known as bioactive substances derived from the extracts of T. sinensis leaves and bark have been identified to have anti-tumor, antioxidant, anti-inflammatory, antibacterial, antiviral, hepatoprotective, and hypoglycemic effects (Ji et al., 2021). T. sinensis in Taihe, Anhui has many varieties such as ‘Heiyouchun’, the most famous variety because of its taste, aroma, and nutritional value that was offered as a tribute as early as the Tang Dynasty (Yang et al., 2020a). In the early stage when the solar term of Grain Rain is coming, the ‘Heiyouchun’ shows the best quality, strong aroma, and good taste because its sprouts are thick, fat, and tender with the best oils, strong fragrances, and crunchiness (Sui et al., 2019).
WRKYs are plant-specific transcription factors (TFs) and have also been found in protozoans (Giardia lamblia) and amoeboid (Dictyostelium discoideum), indicating a long evolutionary history (Goyal et al., 2020; Mao et al., 2020). The DNA-binding domain of WRKY TFs is 60 amino acids long and has a highly conserved heptapeptide (WRKYGQK) signature motif on the N-terminus and a zinc finger-like motif on the C-terminus (Eulgem et al., 2000). This domain forms a four-stranded β-sheet whose stability is determined by a zinc-binding pocket at the end of the β-sheet, suggesting that the N-terminal conserved sequence can bind directly to DNA (Rushton et al., 2010; Rinerson et al., 2015). WRKYs are classified into three categories (Groups I–III) based on the number of conserved domains and the type of zinc finger structure. The first type (Group I) has a WRKY domain at the C-terminal and N-terminal, whereas the second type (Group II) also has a WRKY domain, and both types are C2H2 type zinc finger structures. The third type (Group III) is constituted of a single WRKY domain with the zinc finger structure of the C2HC type. Further, group II proteins could be classified into five primary subgroups (IIa+b, IIc, IId+e), depending on the evolutionary relationship of the WRKY domains (Eulgem et al., 2000; Agarwal et al., 2011).
Sweet Potato Factor 1 (SPF1), the first WRKY cDNA-encoding DNA-binding protein, was discovered in the 5’ upstream region of three genes associated with the synthesis of sporamin and amylase in sweet potato tuberous roots (Ipomoea batatas L.) (Ishiguro and Nakamura, 1994). Following that, WRKYs are found across the genome and in a multitude of crop species, including cotton (Gossypium hirsutum L.) (Dou et al., 2014), mouse-ear cress (Arabidopsis thaliana L.) (Wang et al., 2011), rice (Oryza sativa L.) (Ross et al., 2007), and sesame (Sesamum indicum L.) (Li et al., 2017), Banana (Musa acuminata) (Jia et al., 2022). Significant evidence shows that WRKYs are required for a variety of physiological processes, including embryogenesis (Yang et al., 2020b), seed dormancy and germination (Zhou et al., 2020), trichome initiation (Xie et al., 2021), root growth (Rosado et al., 2022), blooming time (Li et al., 2016), fruit ripening (Cheng et al., 2016), senescence (Fan et al., 2017), and metabolic activities (Schluttenhofer and Yuan, 2015). Additionally, WRKYs also act as both positive and negative regulators of plants’ responses to biotic and abiotic stresses (Song et al., 2014). It is worth noting that the regulatory activity of WRKYs is associated with several signaling pathways, including jasmonic acid, salicylic acid, and abscisic acid, all of which are related to abiotic stress responses (Chen et al., 2010; Dang et al., 2013; Yan et al., 2014).
Furthermore, it is improbable that the role of WRKYs will be confined to coordinated defensive reactions. The WRKYs regulate the biosynthetic genes involved in terpenoid synthesis by activating or inhibiting transcription, either alone or in combination with other TFs (Schluttenhofer and Yuan, 2015). GaWRKY1 from cotton has been demonstrated to bind specifically to the W-box in the CAD1-A promoter and control the activity of the cotton CAD1 gene, implicating a role in sesquiterpene biosynthesis regulation (Xu et al., 2004). When methyl jasmonate induces Medicago truncatula, several WRKY genes involved in the production of defensive chemicals (terpenoids and isoflavonoids) are upregulated (Naoumkina et al., 2008). In periwinkle(Catharanthus roseus), the CrWRKY1 gene is selectively expressed in roots, following JA and ethylene exposure, and it interacts with the DXS and SLS genes involved in steroid production, as well as with the regulators CrMYC2 and CrZCT (Suttipanta et al., 2011). These combined results provide insights on the terpene synthase mechanism of the WRKY gene family in plants. Terpenoids are versatile natural compounds that act as metabolic mediators, ecological communicators, and plant volatiles. As vegetables, terpenes, in addition to their major contribution to the taste of the plant, also have pharmacological effects: anti-cancer, anti-viral, and cholesterol-lowering. Recently, a study discovered 109 chemicals in T. sinensis tissues, including terpenoids, phenylpropanes, and flavonoids (Peng et al., 2019). Since the biosynthesis of terpene compounds is usually mediated by the terpene synthase (TPS) family, it is difficult to significantly increase the content of specific terpenoids through the regulation of a single enzyme gene. WRKY gene family regulate the secondary metabolism of various plants, especially the enzymes associated with terpenes biosynthesis. In particular, the WRKY gene family in T. sinensis has not been fully described, and the roles of the genes within the species remain unknown. Therefore, it is critical to identify and fully investigate the WRKY gene family related to terpene biosynthesis in T. sinensis.
As one of the best-known vegetables, few terpenoid-relative genes have been identified, and the molecular genetic basis of terpenoid biosynthesis pathways is still unveiled. This work found 78 members of the TsWRKYs genes family and determined their biochemical properties, phylogeny, gene structure, conserved motifs, gene promoters, chromosomal distribution, and evolution processes. In addition, TsWRKYs and terpenoid synthase gene expression patterns were analyzed across different young leaf sampling periods. Our study comprehensively revealed the information of the TsWRKYs, which is beneficial to promoting the discovery of its regulatory network and unique function in regulating the synthesis of volatile aromatic compounds.
2. Materials and methods
2.1. Identification of the WRKY genes family members in T. sinensis
The complete genome and proteome sequences of Arabidopsis were downloaded from the Arabidopsis Information Resource1. The T. sinensis data reported in this study are available under Accession No. CNP0000958 in the CNGB Nucleotide Sequence Archive2. The hidden Markov model (HMM) file of the WRKY domain (Accession Number PF03106) was downloaded from the Pfam database3 (Mistry et al., 2021), and HMMER3.0 was used for the identification of WRKY genes with an E-value setting of 1e-5. In addition, SMARAT4 and CCD5 were used to confirm all the potential TsWRKY genes (Chen et al., 2018; Lu et al., 2020; Letunic et al., 2021). The molecular weights (Mw), instability index (II), aliphatic index (AI), the Grand Average of Hydropathicity (GRAVY), and isoelectric points (pI) of the identified WRKY proteins were predicated on the Expasy website6 (Gasteiger et al., 2005). The subcellular locations were predicted using ProtComp – Version 9 from Softberry website 7.
2.2. Sequence analysis and Cis-regulatory element prediction of TsWRKYs genes
Multiple sequence alignments were created using ClustalW using default settings (Thompson et al., 2003), and then the WRKY proteins conserved domain sequences were modified in GeneDoc software (Nicholas, 1997). The distribution pattern of intron was analyzed by the Gene Structure Display Server (GSDS) (Hu et al., 2015)8. Conserved motif analysis of the identified T. sinensis WRKY proteins was carried out on the MEME online program (Bailey et al., 2015)9. The optimized parameters of MEME are as follows: the maximum number of motifs is 20, the motif width is between 8 and 50 aa, and the rest of the parameters are default. The promoters, which were extracted from 2000 bp upstream of the CDS region of TsWRKYs, were used for Cis-regulatory elements (CREs) prediction analysis by PlantCARE online software (Lescot et al., 2002)10.
2.3. Chromosomal distribution and gene duplication of TsWRKYs genes
The Circos (Krzywinski et al., 2009) was used to map the TsWRKY gene information based on T. sinensis genomic data. Tandem and segmental duplication of T. sinensis WRKY genes, as well as the synteny relationship between T. sinensis and six plant species genomes, were evaluated using MCScanX (Wang et al., 2012) and TBtools (Chen et al., 2020). The KaKs Calculator 2.0 (Wang et al., 2010) was used to estimate the non-synonymous (Ka) and synonymous (Ks) substitution of each duplicated WRKY gene. The sequence of WRKY proteins from Arabidopsis, tomato, citrus, maple, pineapple, and rice was downloaded from the NCBI11.
2.4. Phylogenetic analysis and classification of TsWRKYs genes
The conserved domains from the predicted WRKY proteins sequences were confirmed using multiple sequence alignments. The amino acid sequences of WRKY proteins in A. thaliana and T. sinensis were alignment by ClustalW. The phylogenetic trees were constructed using the Neighbor-Joining (NJ) method in MEGA 7.0 (Kumar et al., 2016) with the following pa-rameters: p-distance, pairwise deletion, and 1000 bootstrap replications. The neighbor-joining tree construction method of T. sinensis and other green line species refers to the research of Rinerson (Rinerson et al., 2015). The green line species including: Micromonas pusilla, Ostreococcus tauri, Ostreococcus lucimarinus, Dunaliella salina, Chlamydomonas reinhardtii, Gonium pectorale, Volvox carteri, Physcomitrella patens, Selaginella moellendorffii, Brachypodium distachyon, Oryza sativa, Glycine max, Arabidopsis thaliana, and T. sinensis.
2.5. Plant materials and gene expression analysis
The T. sinensis var. ‘Heiyouchun’ used in this study is universally recognized as the best variety because of its nutritional value, good taste, and unique aroma. ‘Heiyouchun’ was grown in the field at the Forestry Nursery of the Taihe County, Fuyang City, Anhui Province, China (118°48’8’’ N and 32°3’52’’ E). Young and healthy leaves with at least six branches and 5–10 cm in length were collected in four different sampling periods from March 30 to April 20, 2021. All collected samples were immediately frozen in liquid nitrogen and stored at −80°C.
Total RNA from samples was extracted using the RNA Extraction Kit 3.0 (Huayueyang Biotech, Beijing, China), with the RNase-free DNase I treatment to remove potential genomic DNA contamination. Qualified RNA was chosen as a template to produce the first-strand cDNA, as determined by gel electrophoresis and the A260/A280 ratio. Complementary cDNA was generated with SuperScript cDNA Synthesis Kit WX2050 (Huayueyang Biotech, Beijing, China). The specific TsWRKY gene primers were designed using Primer Premier 5, and TsActin gene served as the reference gene for normalization of the expression levels in different sampling periods. Supplementary Table S1 presents all the primer information. The qRT-PCR was performed with a 2×SYBR Green qPCR Mix (With ROX) (Sparkjade, Shandong, China), and amplification was performed using 96-well plates and CFX96 TouchTM RT-PCR system (Biorad, Los Angeles, CA, USA). Each reaction was performed in biological triplicates. The data from qRT-PCR amplification were analyzed using the 2−ΔΔCt method. A calculation of Bonferroni’s multiple comparisons test was performed using SPSS statistical software version 25. A mean fold change greater than 2 and a p value less than 0.05 were considered significant differences between the two groups.
3. Results
3.1. Identification of the WRKY proteins in T. sinensis
To thoroughly investigate the candidate WRKY genes in T. sinensis, 78 TsWRKY genes were finally identified, designated as TsWRKY1–TsWRKY78 based on the order of their HMM (Hidden Markov Model) search results, and were used for subsequent analysis. All extensive information TsWRKYs, including chromosomal location, subcellular localization prediction, protein length, molecular weight, GRAVY, instability index, and aliphatic index are provided on Table S2. Among the 78 TsWRKY proteins, TsWRKY14 and TsWRKY39 proteins were determined to be the smallest and the largest proteins, with 116 and 1161 amino acids (aa), respectively. The proteins’ mo-lecular weights varied from 13.5 to 125.8 kDa, and their pI values were from 4.94 to 9.73. According to the expected subcellular localization results, 67 and 11 TsWRKY proteins were found in the nucleus and extracellular areas, respectively. All of the TsWRKY proteins have a GRAVY of less than 0, which means that they are all hydrophilic proteins, and more information is shown in Table S2.
3.2. Multiple sequence alignment, phylogenetic analysis, and classification of TsWRKYs genes
Multiple sequence alignments of the WRKY domains, which cover about 60 amino acids, were used to analyze the evolutionary relationships of TsWRKY proteins. The WRKY domains of seven distinct Arabidopsis WRKY proteins (AtWRKY1, 18, 6, 8, 7, 14, and 30) were randomly chosen as representatives for further evaluation. Figure 1 shows the substantially conserved WRKY domain sequences. The majority of the proteins in this family (76 out of 78) share the conserved WRKY domain WRKYGQK, while TsWRKY33 and TsWRKY62 differ by an amino acid.
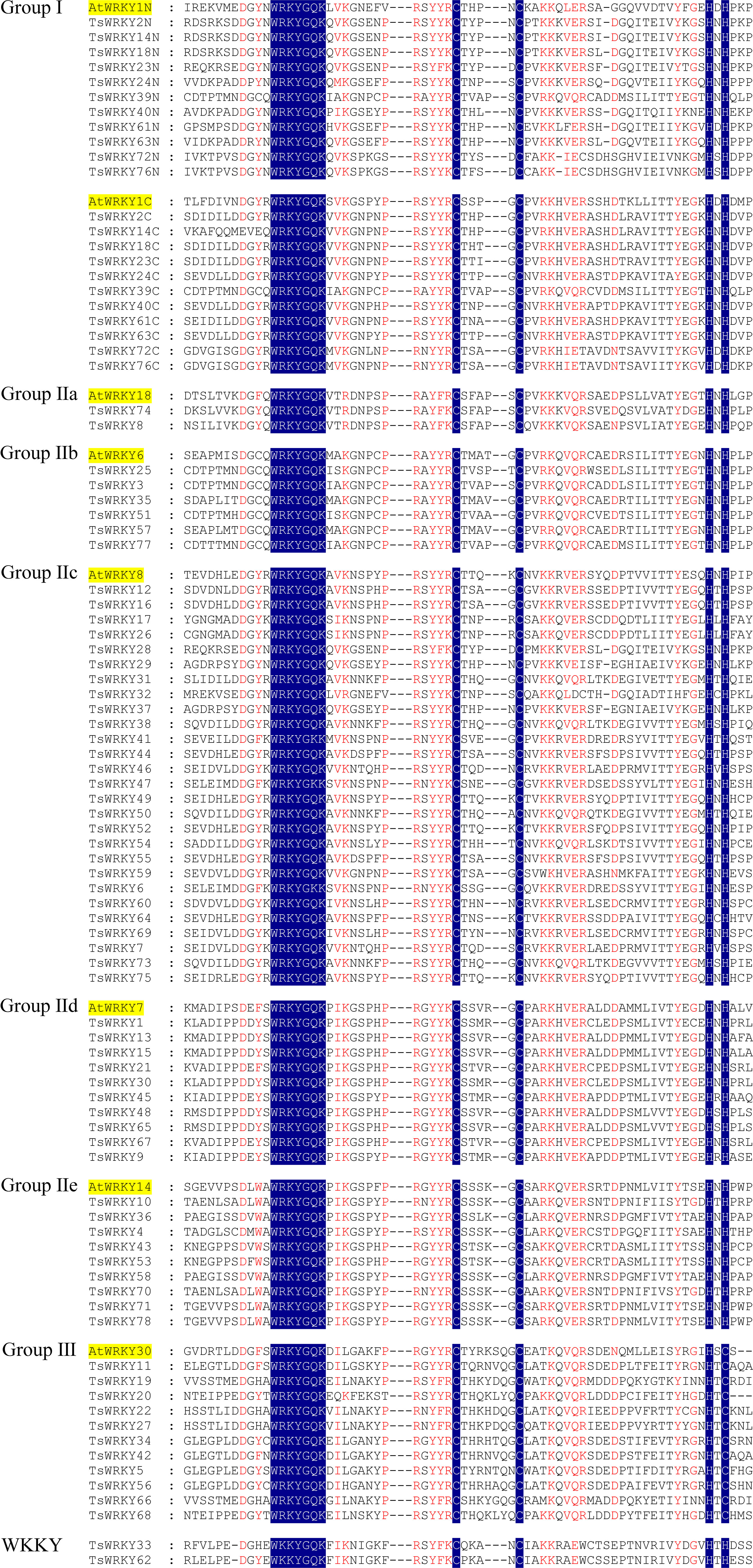
Figure 1 Multiple TsWRKY domain amino acid sequences were aligned with reference to AtWRKY. The letters “N” and “C” represent WRKY domains, which are found at the N and C terminal ends of amino acid sequences, respectively.
The phylogenetic analysis using the WRKY genes of Arabidopsis as a reference revealed that these TsWRKY genes are more precisely classified into groups I, II (a–e), and III (Eulgem et al., 2000; Zhang and Wang, 2005). Among the 78 members of the TsWRKY family, group II contains the most TsWRKY proteins (54), followed by group I (11) and group III (11). Additionally, each category may be subdivided into numerous subcategories. There are 11 TsWRKY proteins in group I that contain two WRKY conserved domains that are classified as N-terminal WRKYs (IN) or C-terminal WRKYs (IC) depending on their locations on the protein. Group II of the TsWRKY proteins can be divided into five subgroups, including two subgroups IIa, six subgroups IIb, 27 subgroups IIc, 10 subgroups IId, and nine subgroups IIe (Figure S1, Table S2). The 11 TsWRKY proteins in group III contain a zinc finger motif of the form C-X7-C-X23-H-X-C, which is identical to the AtWRKY30 in Arabidopsis subgroup III (Figure 1). We created a broader WRKY domain dataset for phylogenetic analysis to further understand the evolution of TsWRKYs familly. Figure 2 shows the neighbor-joining phylogenetic tree constructed by WRKY domains for 14 species. A scattered distribution of TsWRKYs was observed in groups I, II (a–e), and III. The TsWRKYs protein sequence branched away from algae, bryopsida, and pteridophyta but showed clustering with dicotyledons and monocotyledons within each subgroup. Interestingly, the TsWRKYs protein sequence branches tend to show close proximity between the two. For example, TsWRKY35-TsWRKY57 and TsWRKY25-TsWRKY51 pairs in group IIb, and TsWRKY13-TsWRKY15, TsWRKY21-TsWRKY67 and TsWRKY48-TsWRKY65 pairs in group IId (Figure 2; Figure S2). Group III WRKY family members could be significantly subdivided into eight clades, but all TsWRKYs proteins were found on Clade 1, 4, 6, and 8.
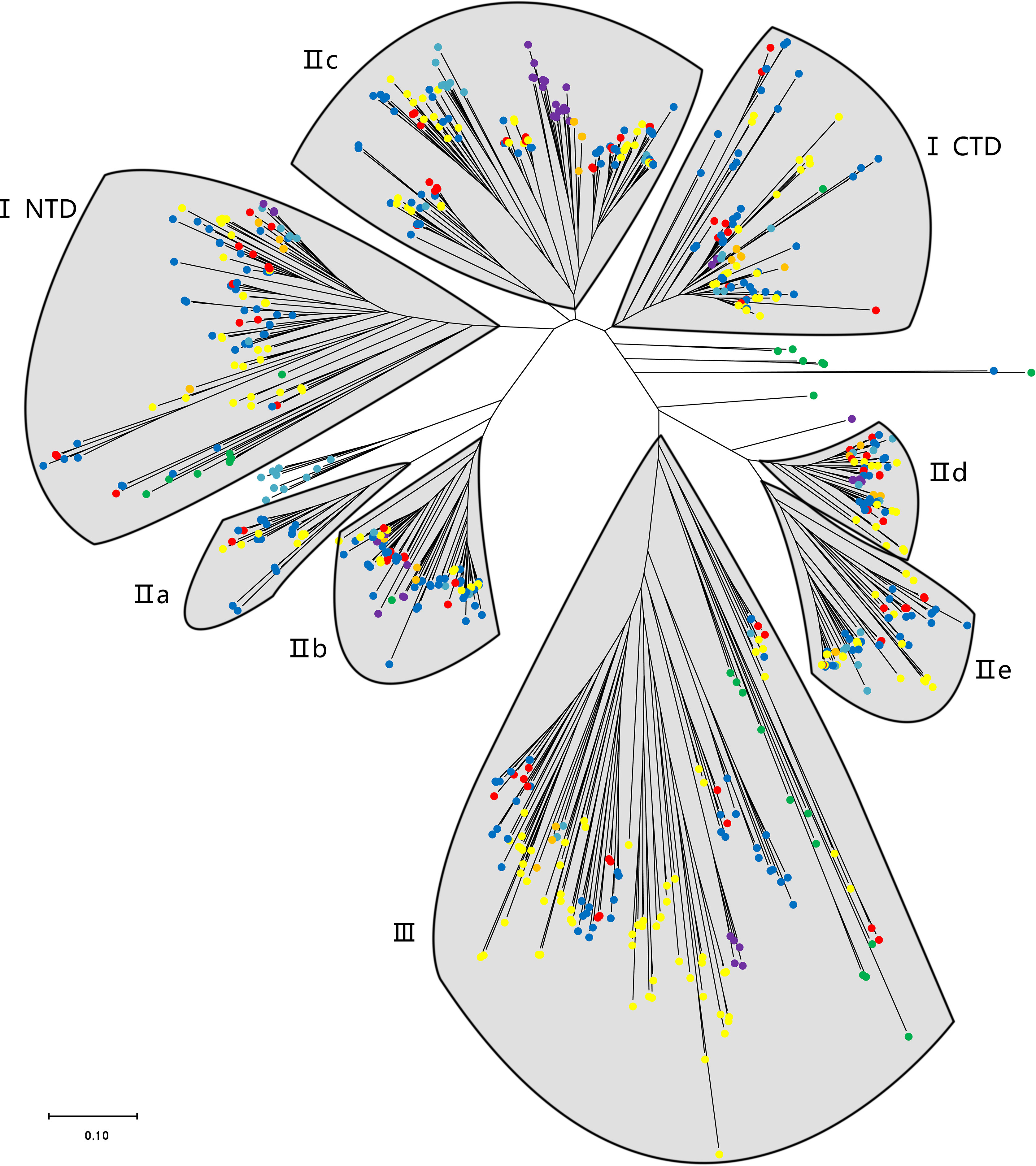
Figure 2 A phylogenetic tree of WRKY domains from T. sinensis and other species in the green lineage. The phylogenetic tree was derived from a MUSCLE alignment of WRKY domains from the following species: Micromonas pusilla, Ostreococcus tauri, Ostreococcus lucimarinus, Dunaliella salina, Chlamydomonas reinhardtii, Gonium pectorale, Volvox carteri, Physcomitrella patens, Selaginella moellendorffii, Brachypodium distachyon, Oryza sativa, Glycine max, Arabidopsis thaliana, and T. sinensis. The green, purple, orange, indigo, yellow, blue, and red dots represent unicellular green algae, bryopsida, pteridophyta, gymnosperms, monocotyledons, dicotyledons, and T. sinensis, respectively.
3.3. Gene structure and motif composition of TsWRKYs genes family
Figure 3B depicts the particular condition of the T. sinensis WRKY gene structures. The number of introns, with the exception of TsWRKY39 and TsWRKY59, ranges from 2 to 6, with an average of 3.59. The TsWRKY genes structure are composed of three exons and two introns in more than 60% (47 of the 78) of them. TsWRKY39, in particular, has the most exons and introns of all TsWRKYs, with 12 exons and 11 introns. Gene structures of genes in the same group, like IId and IIe, tend to be consistent in general.
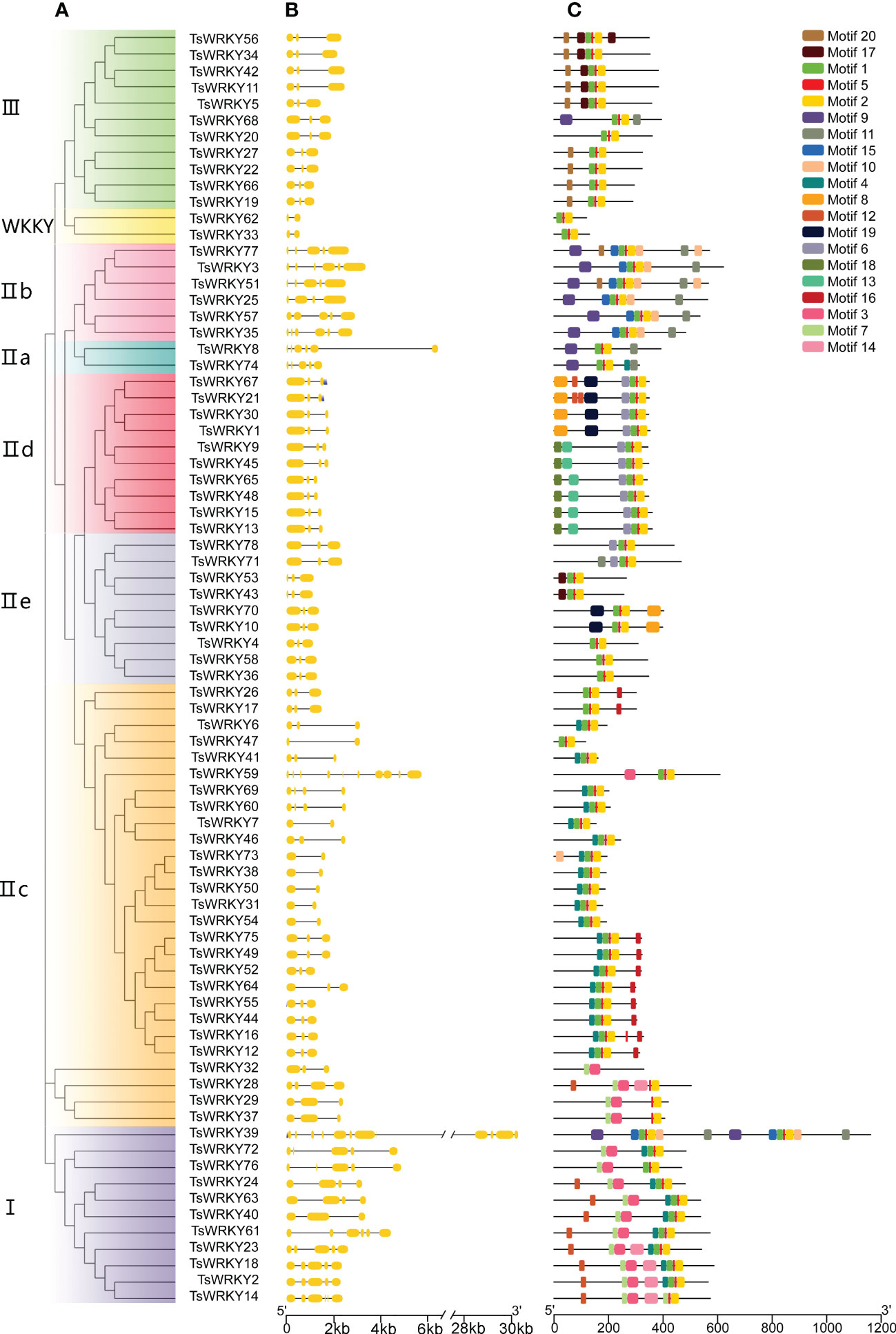
Figure 3 Phylogenetic tree, exon-intron distribution, and conserved protein motifs of TsWRKYs. (A) The TsWRKYs are divided into several groups, each with a distinct color. (B) Yellow rectangles and black lines represent exons and introns, respectively. (C) Each motif with conserved amino acid residues is represented in different color (motif 1 – 20).
The domain prediction results (Figure 3C) were validated by utilizing the MEME web server. By sequencing the TsWRKY proteins, 20 distinct motifs were discovered, comprising 8-50 amino acids. The majority of TsWRKYs contained motifs 1, 2, and 5, which corresponded to the DBD domain, while others contained motifs unique to each class. For instance, motif 8 is unique to group IId, while motif 10 is found only in groups IIa and IIb. Most importantly, each class had a distinct motif organization, and two genes that were tightly clustered on the tree usually exhibited identical motif patterns. Despite their heterogeneity in size and sequence, the projected WRKY domains and other conserved domains were cross-confirmed by the two combined approaches, implying that the group classifications are reliable.
3.4. Cis-regulatory element prediction of TsWRKYs genes
CREs are genomic sequence motifs located in the 5’ upstream region of genes that bind to motif-specific proteins and function as regulatory switches for downstream genes (Korkuć et al., 2013). As shown in Figure 4, the upstream 2000 bp regulatory regions of all TsWRKYs were extracted, several CREs were predicted using PlantCARE, and the 20 most common were visualized using TBtools software.
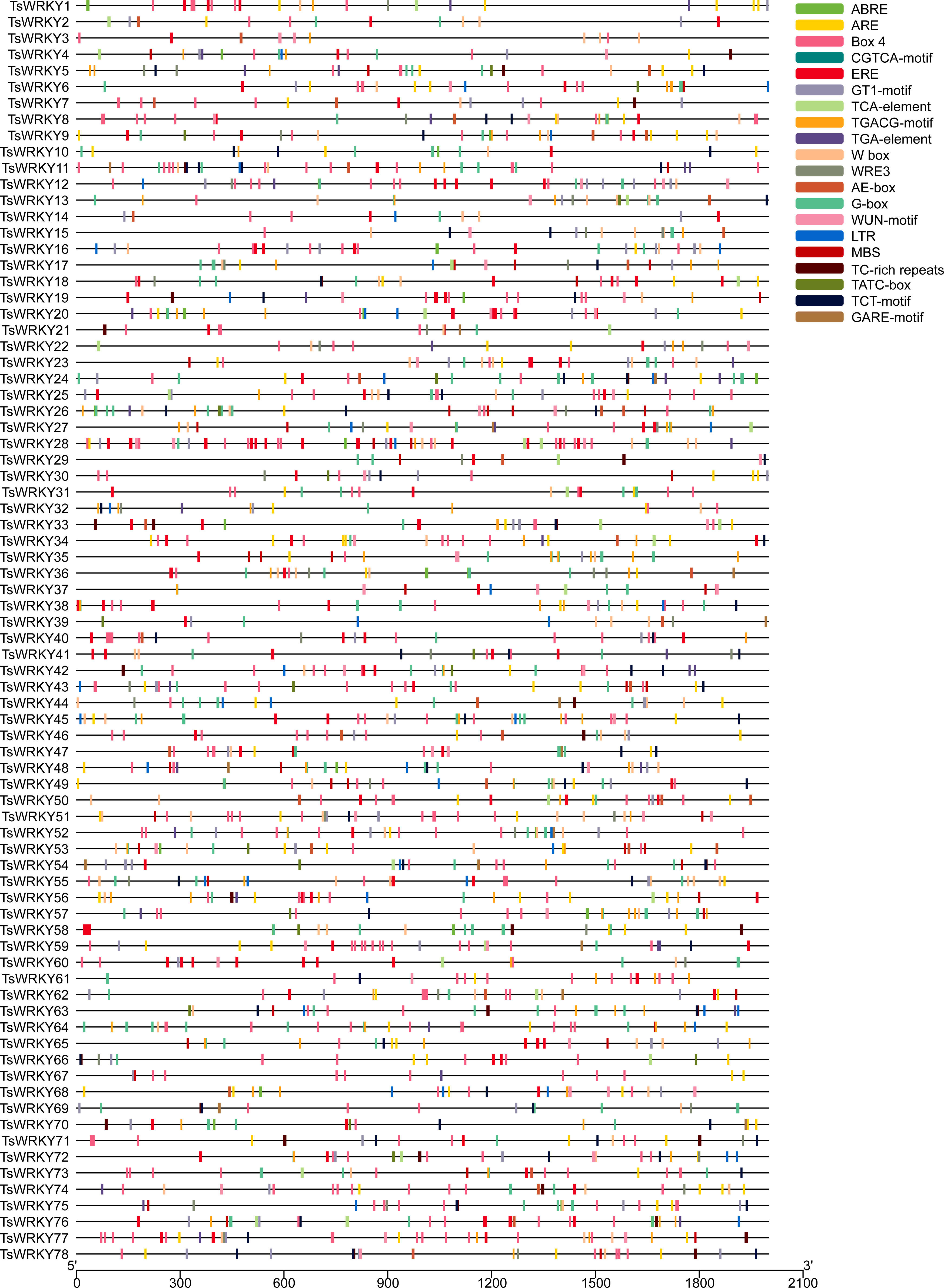
Figure 4 The prediction of CREs in the 2000 bp promoter upstream of the TsWRKY genes. The main CREs are showed in the upper right corner.
Our analysis revealed that T. sinensis contained many promoters’ core regulatory elements (CAAT-box, TATA-box), light responsive elements (Box 4, G-box, GT1-motif, AE-box, and TCT-motif), and W box elements. We observed a lot of abiotic stress responsive elements as well, such as wound-responsive elements (WUN-motif), drought-inducibility elements (MBS), dehydration, low-temp, salt stress responsive elements (DRE), low-temperature responsive elements (LTR and WRE3), and defense and stress responsive elements (TC-rich repeats). These are the hormone responsive elements: abscisic acid responsive elements (ABREs), methyl jasmonate (MeJA) responsive elements (CGTCA-motif and TGACG-motif), ethylene-responsive elements (EREs), auxin-responsive element (TGA-element), gibberellin-responsive elements (GARE-motif, P-box and TATC-box), and salicylic acid responsive element (TCA-element). Other CREs were also predicted, such as anaerobic responsive elements (AREs) and circadian control elements (circadian).
All TsWRKYs had at least one stress response-related CREs in this investigation. A total of 65 TsWRKY genes (83.3%) had one or more ABREs, which could be a sign that they have an ABA response when they are stressed. Additionally, more than 70% of TsWRKY genes have the EREs and AREs that have been speculated as having important promoter roles (Olive et al., 1991; Ohme-Takagi et al., 2000). TsWRKY27 and TsWRKY72 contained 16 out of 20 promoters in their promoter regions that surpass other TsWRKY genes. We also focused on CREs involved in wound, hypothermia, and drought responses, such as WUN-motif, LTR, MBS, and TC-rich repeats. The WRKY protein can be used efficiently in conjunction with W-box regions to activate or inhibit downstream target gene transcription (Jiang et al., 2017). It can form protein complexes with other active components, which improves transcription binding activity (Chi et al., 2013). Moreover, 62 TsWRKYs possessed one or even more W-boxes, implying that these WRKY genes are regulated by autoregulation or crossregulation (Rushton et al., 2010).
3.5. Chromosomal distribution and synteny analysis of TsWRKYs genes
The 78 TsWRKY genes were dispersed randomly throughout the 28 T. sinensis chromosomes (Figure 5). The bulk of the TsWRKYs were found at or at the ends of chromosomes. Of all TsWRKYs, 11 were identified on Chr24, scattered in several clusters, which is the largest number. On the contrary, there is only a single TsWRKY gene on Chr2, Chr4, Chr9, Chr10, Chr17, Chr20, Chr21, and Chr22. Tandem and segmental duplications both contribute to the generation of gene families throughout evolution (Cannon et al., 2004). Hence, we investigated the occurrences of TsWRKY genes duplication. On cross-referencing with Holub’s published research study, 20 TsWRKY genes (25.7%) were found to be tandem duplicated. Tandem duplication event is a chromosomal region within 200 kb, including multiple (two or more) members of a gene family (Holub, 2001). There were 10 distinct pairs of tandemly duplicated genes on Chr 1, 6, 11, 12, 13, 15, 16, 23, and 24. In addition to the tandem duplication events, 83 segmental duplication events involving 72 TsWRKY genes were discovered using the BLASTP and MCScanX approaches.
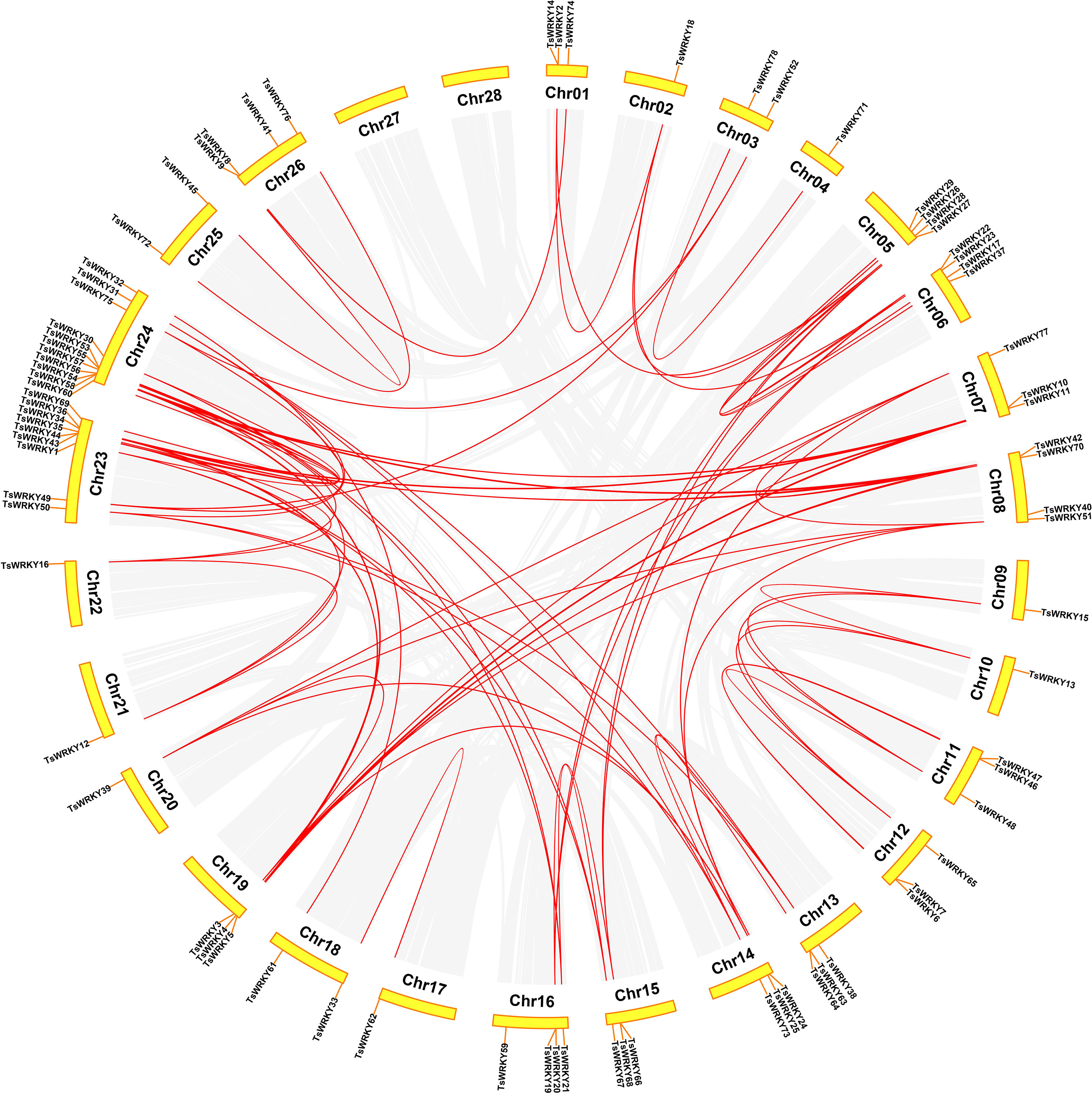
Figure 5 The WRKY genes segmental duplication in T. sinensis. The gray lines inside represent synteny blocks, whereas the red lines imply duplicated WRKY gene pairs that have been discovered.
Constructing syntenic graphs between T. sinensis and several typical species allows us to investigate the evolutionary clues for the T. sinensis WRKY gene family (Figure 6). The representative species consist of four dicots, Citrus clementina, Acer yangbiense, A. thaliana, and Lycopersicon esculentum, and two monocots, O. sativa and Ananas comosus. Syntenic links were found between 77 TsWRKY gene members and those in citrus (73), maple (70), tomato (70), Arabidopsis (63), pineapple (51), and rice (34). There were 136, 129, 113, 101, 73, and 45 orthologous pairings between the six species (tomato, citrus, maple, Arabidopsis, pineapple, and rice), respectively. In general, the TsWRKYs comprised more syntenic gene pairs in dicots than in monocots. C. clementina and A. yangbiense, well-known members of Sapindales, show greater synteny with T. sinensis, which belonged to Sapindales. Notably, in the interactive Venn map of WRKY genes across species (Figure 7A), 30 TsWRKY genes shared syntenic WRKY gene pairings with all six species, implying that these orthologous pairs existed prior to the ancestral split. Certain TsWRKY genes were shown to relate to 3, 4, or 5 collinear gene pairs (between T. sinensis and maple/citrus/tomato WRKY genes), indicating the possibility that these TsWRKY genes have significant roles in the evolution of the WRKY gene family. Syntenic gene pairings between T. sinensis and other species may be important for elucidating WRKY gene evolution. The Ka/Ks (non-synonymous substitution/synonymous substitution) ratios of the WRKY orthologous gene pairs of six species were computed to assess the evolutionary constraints operating on the T. sinensis WRKY gene family. Figure 7B shows that almost all TsWRKY orthologous gene pairs had Ka/Ks < 1. As a result, we speculated that the T. sinensis WRKY gene family may have been subjected to significant purifying selection forces throughout evolution (Hurst, 2002).
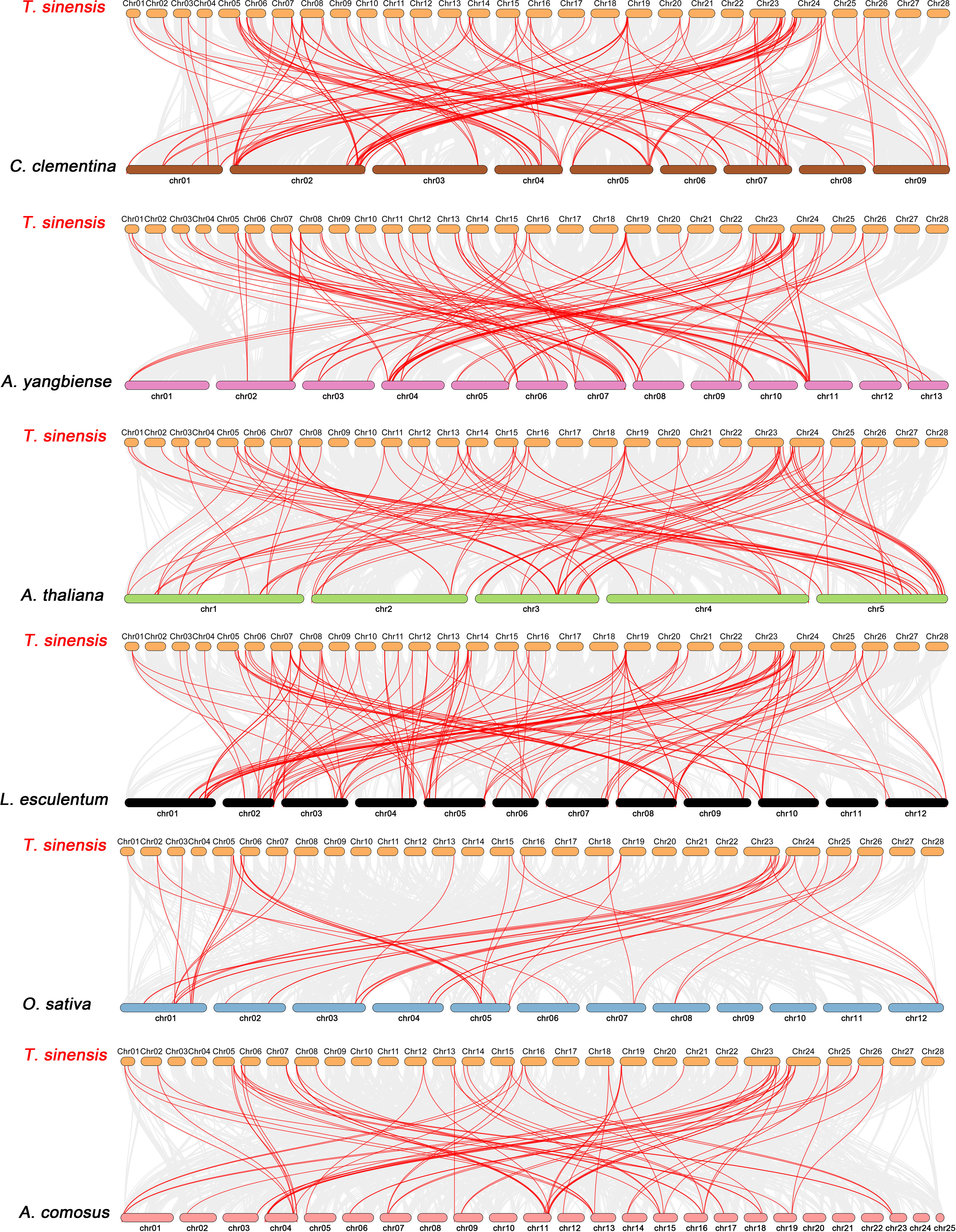
Figure 6 Synteny analysis of WRKY genes between T. sinensis and six representative plant species: Citrus clementina (C. clementina), Acer yangbiense (A. yangbiense), Arabidopsis thaliana (A. thaliana), Lycopersicon esculentum (L. esculentum), Oryza sativa (O. sativa), and Ananas comosus (A. comosus). Every horizontal bar indicates a different chromosome. The red curves represent the syntenic WRKY gene pairs, whereas the gray lines indicate the collinear blocks within T. sinensis and other plant genomes.
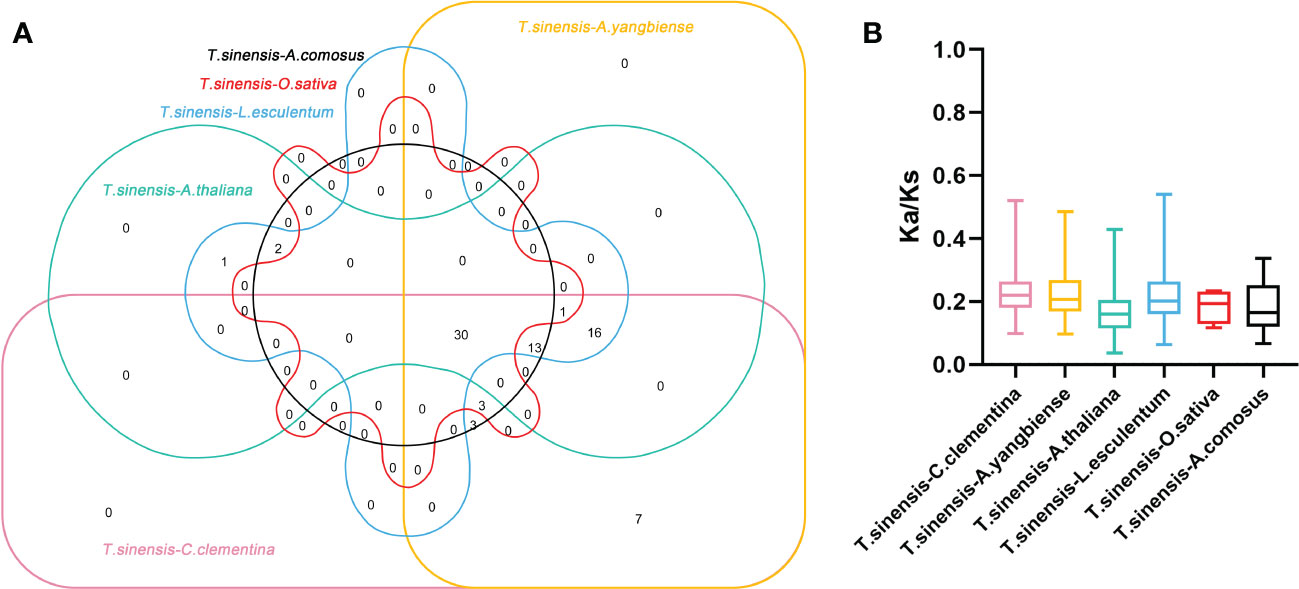
Figure 7 Non-redundant syntenic and evolution analysis of the WRKY gene families in a diversity of species. (A) The Venn diagram depicts syntenic WRKY genes found in multiple species. (B) The ratio of nonsynonymous to synonymous substitutions (Ka/Ks) of WRKY genes in T. sinensis and other six species: Citrus clementina (C. clementina), Acer yangbiense (A. yangbiense), Arabidopsis thaliana (A. thaliana), Lycopersicon esculentum (L. esculentum), Oryza sativa (O. sativa), and Ananas comosus (A. comosus).
3.6. Expression patterns of TsWRKYs genes and terpenoid synthases genes
Six main expression patterns of TsWRKY genes were observed (Figure 8). TsWRKY8 and 74 were upregulated at first and then downregulated, while TsWRKY12 and TsWRKY65 showed the opposite trend. Suppressed expression patterns were seen in 18 TsWRKY genes (7, 16, 17, 21, 26, 44, 46, 51, 55, 58, 59, 60, 61, 67, 69, 76, 77, and 78), while upregulated expressions were observed in 22 TsWRKY genes (5, 6, 18, 19, 22, 24, 32, 33, 35, 38, 39, 40, 41, 43, 45, 53, 57, 62, 63, 64, 66, and 68). Furthermore, the expression of nine TsWRKY genes (2, 13, 14, 15, 23, 28, 31, 34, and 56) were first upregulated, then downregulated, and then upregulated to a high level. The other TsWRKY genes (1, 3, 4, 9, 10, 11, 20, 25, 27, 29, 30, 36, 37, 42, 47, 48, 49, 50, 52, 54, 70, 71, 72, 73, 75, and 78) were not significantly altered (fold change ≥ 2) at different stages.
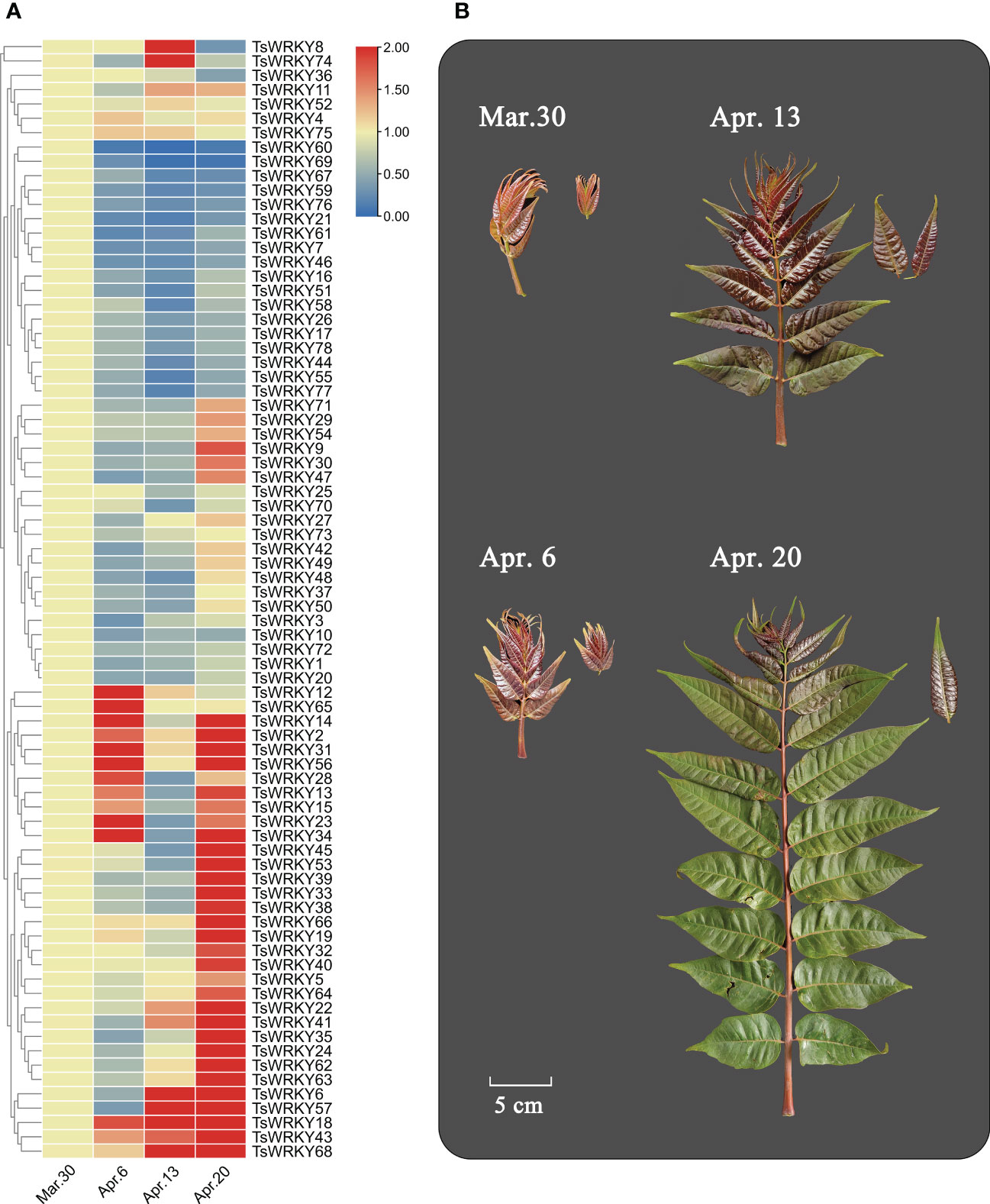
Figure 8 Differential transcription of TsWRKY genes in four different sampling periods (March 30, April 6, April 13, and April 20, 2021). (A) Blue and red indicate lower and higher transcript abundance, respectively, compared to initial data (March 30). (B) Samples of T. sinensis leaves from four periods.
We investigated the expression patterns of the genes involved in terpenoid biosynthesis in order to unravel the regulatory mechanism of terpenoid accumulation patterns in various developmental stages of T. sinensis. All of the key genes, except for TsFPPS, showed significant changes (fold change ≥ 2) in expression during development (Figure 9B). After April 6, the expressions of TsAACT, TsHMGS, TsHMGR, TsDXS, and TsDXR all changed distinctly. Notably, TsIDI, TsDXS, and TsDXR are more than five-fold changes in expression over four periods.
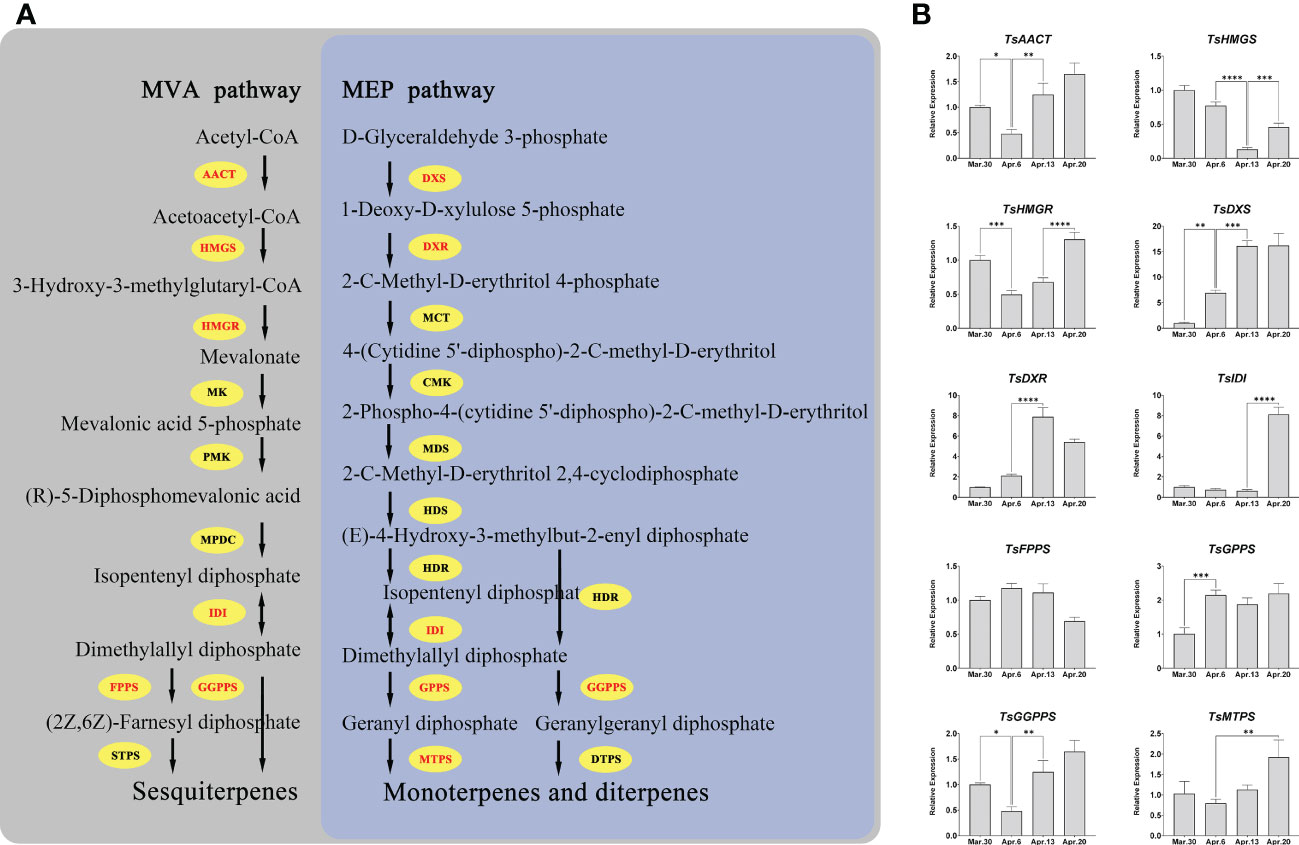
Figure 9 Pathways of terpenoid synthesis and the expression patterns of several key genes. (A) The main terpenoid biosynthesis pathway in plants; AACT, acetoacetyl-CoA thiolase; HMGS, 3-hydroxy-3-methylglutaryl-CoA synthase; HMGR, 3-hydroxy-3-methylglutaryl-CoA reductase; IDI, isopentenyl diphosphate isomerase; FPPS, farnesyl diphosphate synthase; DXS, 1-deoxyd-xylulose 5-phosphate synthase; DXR, 1-deoxy-D-xylulose 5-phosphate reductoisomerase; GPPS, geranyl diphosphate synthase; GGPPS, geranylgeranyl diphosphate synthases; PMK, phosphomevalonate kinase; MK, mevalonate kinase; MPDC, mevalonate diphosphate decarboxylase; MCT, 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase; CMK, 4-(cytidine 5’-diphospho)-2-C-methyl-D-erythritol kinase; MDS, 2-C-methyld-erythritol 2,4-cyclodiphosphate synthase; HDS, (E)-4-hydroxy-3-methylbut-2-enyl diphosphate synthase; HDR, (E)-4-hydroxy-3-methylbut-2-enyl diphosphate reductase; STPS, sesquiterpene synthase; MTPS, monoterpene synthase; DTPS, diterpene synthase. (B) *, **, ***, and **** mean a significant difference at P < 0.05, P < 0.01, P < 0.001, and P < 0.0001, respectively.
3.7. The co-expression network of TsWRKYs genes and terpenoid synthesis genes
The WRKYs usually control the expression of terpenoid synthesis genes by activating or repressing their promoters, thereby regulating the accumulation of terpenoids. We constructed a co-expression network of TsWRKYs with terpenoid synthesis genes (Table S3; Figure 10). The results showed that TsFPPS, TsIDI, TsMTPS, TsWRKY9, TsWRKY24, TsWRKY35, TsWRKY38, TsWRKY39, TsWRKY62, and TsWRKY64 are possible core members of the terpenoid synthesis co-expression network. TsFPPS, TsWRKY36, and TsWRKY75 were negatively correlated with other genes. By contrast, TsWRKY24, TsWRKY35, TsWRKY39, and TsWRKY40 are almost all positive correlated factors.
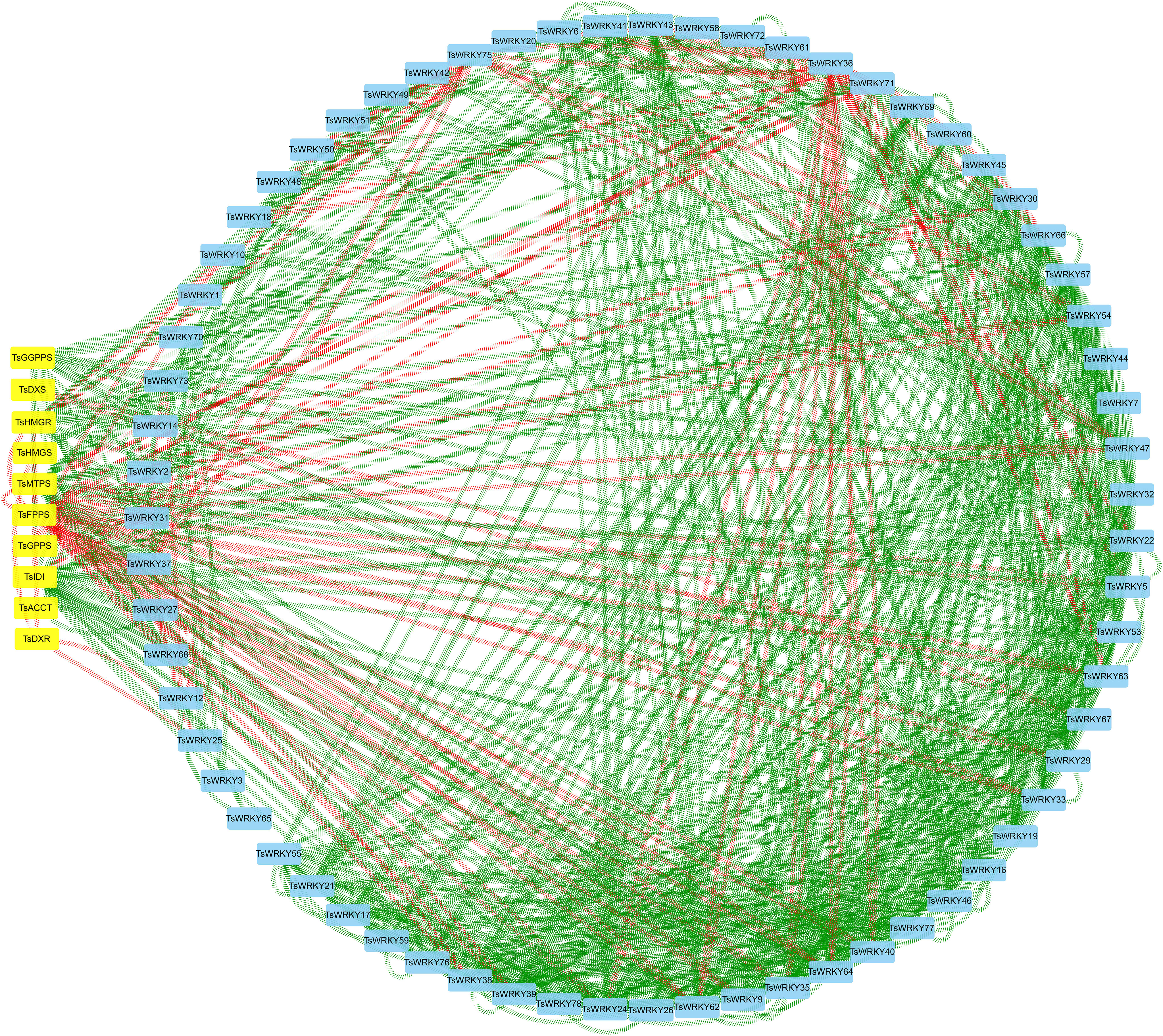
Figure 10 Correlation analysis of TsWRKYs and terpenoid synthesis genes (Pearson correlation coefficient > 0.95). The square nodes colored in blue are TsWRKYs, the square nodes colored in yellow are terpenoid synthesis genes, the green line represents positive correlation, and the red line represents negative correlation.
4. Discussion
Since the first WRKY proteins were identified, WRKY TFs have been well recognized in plants for their regulating functions in defense against abiotic and biotic stresses, growth and development, and secondary metabolism (Schluttenhofer and Yuan, 2015; Jiang et al., 2017; Abeysinghe et al., 2019). Terpenes are the signature volatile components of T. sinensis, and WRKYs are significantly involved in regulating the terpene pathway (Yang et al., 2012; Zhao et al., 2022). Neither the identification of the WRKY gene nor the regulation of terpenoids, the most iconic volatile substances in T. sinensis, has been reported. We describe WRKY TFs in T. sinensis and present the associated regulatory network of terpene biosynthesis.
4.1. Evolutionary characteristics of WRKY TFs in T. sinensis
Variation in the number of gene family members is a key mechanism for shaping adaptive natural variation during the evolution of species (Guo, 2013). We discovered 78 proper TsWRKY genes in this investigation. The results of the neighbour-joining phylogenetic tree of 14 species constructed with the WRKY protein sequence’s conserved domain indicate that they can be divided into three major groups (I–III) and five subgroups (IIa–IIe). WRKY TFs diverge early in the green lineage and TsWRKYs are more closely related to dicotyledons and monocotyledons. The number of presumed TsWRKYs are comparable to the count of WRKY genes in Fagopyrum tataricum (78) (He et al., 2019), and it is somewhat lower than those in S. lycopersicum L. (81) (Huang et al., 2012), but much lower than those in O. sativa L. ssp. indica (102) (Ross et al., 2007) and Glycine max (174) (Yang et al., 2017). These findings corroborated previous research that suggested herbaceous plants tend to have a larger number of WRKY genes than woody plants (Wu et al., 2016).
The variety of gene structures reflects the historical evidence of gene family evolution and serves as the foundation for phylogenetic categorization (Xiao et al., 2017). Wheat and tea plants have 0–5 introns and 0–11 introns, respectively, whereas TsWRKYs have 2–12 introns, suggesting that TsWRKYs have abundant gene structural variation (Ning et al., 2017; Wang et al., 2019). The intron-exon distribution pattern is comparable across members of the same subfamily, which is the basis for functional similarity among members of the same evolutionary group (Li et al., 2020a). For example, the number of introns in almost all members of group III is 2. Furthermore, TsWRKYs from group I contain many more introns than other groups, which implies that it is more likely that other groups came from group I (Chen et al., 2017a). Twenty conserved motifs were discovered in 78 TsWRKY proteins (Figure 3), with motifs 1 and 2 belonging to the WRKY conserved domains. Partiular motifs that only arose in one group, such as motif 10 in group IIa and group IIb proteins and motif 8 in subgroup IId, which has yet to be characterized for some roles, should be given additional attention.
According to a recent study, dicotyledons have experienced less evolutionary loss of the WRKY conserved domain than monocotyledons (Wei et al., 2012). This occurrence was validated in this investigation, with the majority of TsWRKYs having the conserved heptapeptid WRKYGQK motif, despite two TsWRKY genes having the variants WKKYGQK (TsWRKY33 and TsWRKY62, Group WKKY) (Figure 1). The WRKY proteins demonstrate a propensity for binding to W-box elements, and WRKYGQK motif changes may affect DNA-binding interactions with downstream genes, as previously discovered. As a result, additional exploration of the functional and binding properties of these two WRKY proteins is required (Chen et al., 2017a).
How did the number of members of the WRKY family expand from 1 in the unicellular green algae to 78 in the T. sinensis? The analysis of segmental and tandem duplications contributed to revealing the number and function of the TsWRKY gene family. Based on the chromosome distribution and synlinearity analysis results of the TsWRKYs, 83 segmental duplication events within 72 TsWRKY genes were observed, while tandem duplication events existed. We found that 20 out of the 78 genes (25.6%) in this family are tandem repeats in T. sinensis, suggesting that the abundance of tandem repeats may be a possible reason for the larger number of TsWRKYs. In addition, whole-genome duplication (WGD) events often lead to the growth of gene families, which is common in the evolution of angiosperms. Previous study revealed that T. sinensis has a large number of gene duplications and that WGD events happened approximately 7.8 and 71.5 million years ago (Mya) (Ji et al., 2021). We determined that, whereas some TsWRKY genes are the result of tandem duplication, segment duplication events are the driving force behind gene family evolution. The three basic evolutionary mechanisms are segmental duplication, tandem duplication, and transposition events like retroposition and replicative transposition. Individual gene duplication, chromosomal segment duplication, and even complete genome duplication supply the fresh materials required for gene generation (Yu et al., 2005). Gene duplication, which is associated with the generation of new gene functions, is one of the primary driving forces underlying genome evolution and also is essential to plant adaptive evolution (Moore and Purugganan, 2003; Kong et al., 2007). Tandem duplications produce highly diverse duplicates that have lineage-specific functions (Ezoe et al., 2020), and new research tea plants suggests that tandem duplication of genes plays an active part in flavor accumulation (Wang et al., 2021). Additionally, we speculated that the TsWRKY gene family may have been subjected to significant purifying selection forces throughout evolution because almost all TsWRKYs orthologous gene pairs had Ka/Ks<1. Divergence is a key feature of the evolution of paralogous homologous genes and DNA segments that make up fixed repeats. However, selection on copies of paralogous homologs would be relaxed in the case of full redundancy, which is when any number of functional copies of a gene give the same fitness. Therefore, this negative selection of TsWRKYs are associated with the post-fixation evolution of gene duplications.
4.2. Regulation of terpene biosynthesis by TsWRKYs genes in T. sinensis
The main volatile aromatic compounds of T. sinensis are terpenes (isopentene, monoterpenes, and sesquiterpenes), phenyl/phenylpropanes, and fatty acid derivatives. In T. sinensis, the terpenes have been demonstrated as the most important volatile compounds. The aroma of T. sinensis leaves is an important factor in determining its quality and an important criterion for measuring its economic value. During the germination and maturation process of T. sinensis tender buds, aromatic substances are gradually synthesized and accumulated. The results of real-time quantitative PCR revealed that 67.9 percent of TsWRKY had significantly varied transcript levels during the four stages of budding. Although little is known about the transcriptional regulatory network controlling terpenes synthesis, most of the identified regulators are WRKY TFs (Patra et al., 2013). It is reasonable to conclude that TsWRKYs expression patterns and the buildup of aromatic compounds are related. Therefore, we further analyzed the expression patterns of terpenoid synthase genes and detected that multiple genes of the MVA and MEP pathways are involved in regulating terpenoid accumulation. TsWRKYs showed a high correlation with the expression trends of 10 terpene synthesis genes. For example, TsFPPS, TsIDI, TsMTPS, TsWRKY9, TsWRKY24, and TsWRKY35 may coordinately regulate terpene synthesis.
WRKY genes are essential regulators of secondary metabolite production in plants (Luo et al., 2022), while their regulatory functions varied substantially due to distinct binding mechanisms (Li et al., 2020b). Evidence suggests that certain WRKYs, alone or in concert with other transcription factors, govern the biosynthesis of valuable natural products (Hsin et al., 2022). There have been several studies on the regulatory effects of WRKYs upon the activation or repression of genes involved in plant terpenoid production. Individual WRKY can be associated with a number of regulatory mechanisms, as SlWRKY73 transactivates the SlTPS3, SlTPS5, and SlTPS7 monoterpene synthase genes in tomato (S. lycopersicum) (Spyropoulou et al., 2014). Gossypol (sesquiterpene phytoalexins) in Gossypium arboretum (Xu et al., 2004), DP (diterpenoid phytoalexin) in rice (Akagi et al., 2014), Artemisinin (a type of sesquiterpene lactone) in Artemisia annua (Chen et al., 2017b), ginsenosides (a group of triterpene) in Panax quinquefolius (Sun et al., 2013), and tanshinone (one category of bioactive diterpenes) in Salvia miltiorrhiza are all regulated by WRKYs (Cao et al., 2018). From correlative analysis in sweet Osmanthus fragrans, it is speculated that the OfWRKY gene participates in aroma synthesis by regulating the synthesis of monoterpene volatiles, and that the expression of OfWRKYs are closely related to monoterpene synthesis (Ding et al., 2019). Heterologous expression of WRKY and MYC2 in Salvia sclarea causes coactivation of MEP-biosynthetic genes and accumulation of abietane diterpenes (Alfieri et al., 2018). The co-expression network between several key genes for terpene biosynthesis and TsWRKYs provides important insights into the terpene biosynthesis pathway in T. sinensis. It helps to further characterize the functions of candidate WRKY gene families in T. sinensis and provide new ideas for agronomic genetic improvement and quality variety breeding.
5. Conclusions
The 78 proper TsWRKYs were discovered in this investigation. Segment duplication events are determined to be the driving force behind the expansion of the TsWRKYs gene family. TsWRKYs proteins may have been subjected to significant purifying selection forces throughout evolution. Several TsWRKYs that may be involved in regulating terpenoid accumulation in the MVA and MEP pathways were identified. In summary, our study provides comprehensive information on TsWRKYs and could facilitate further research into the functions of TsWRKYs in regulating the synthesis of volatile aromatic compounds and improving the aroma of edible leaves based on an understanding of the regulatory network.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
LR, TG and XC conceived and designed the experiments. WW and DY performed the experiments. LR and TG analyzed the data. XD and ZM contributed reagents/materials/analysis tools. LR and WW wrote the paper. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant from the research funds for postdoctoral researchers in Anhui Province (2020B434), the Natural Science Key Foundations of the Anhui Bureau of Education (KJ2021A0677), the Key projects in teaching research of Anhui Provincial Department of Education (2021jyxm1086) and Fuyang Normal University’s major scientific and technological achievements incubator fund project (kjfh201703).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1063850/full#supplementary-material
Supplementary Figure 1 | A phylogenetic tree of WRKY domains from T. sinensis and A. thaliana.
Supplementary Figure 2 | A phylogenetic tree of WRKY domains from T. sinensis and other species in the green lineage.
Footnotes
- ^ http://www.arabidopsis.org/
- ^ https://db.cngb.org/search/project/CNP0000958/
- ^ http://pfam.xfam.org/
- ^ http://smart.embl-heidelberg.de/
- ^ https://www.ncbi.nlm.nih.gov/cdd/
- ^ http://web.expasy.org/protparam/
- ^ http://www.softberry.com/
- ^ http://gsds.gao-lab.org/
- ^ https://meme-suite.org/meme/tools/meme/
- ^ http://bioinformatics.psb.ugent.be/webtools/plantcare/html/
- ^ https://www.ncbi.nlm.nih.gov/
References
Abeysinghe, J. K., Lam, K. M., Ng, D. W. K. (2019). Differential regulation and interaction of homoeologous WRKY 18 and WRKY 40 in Arabidopsis allotetraploids and biotic stress responses. Plant J. 97 (2), 352–367. doi: 10.1111/tpj.14124
Agarwal, P., Reddy, M., Chikara, J. (2011). WRKY: its structure, evolutionary relationship, DNA-binding selectivity, role in stress tolerance and development of plants. Mol. Biol. Rep. 38 (6), 3883–3896. doi: 10.1007/s11033-010-0504-5
Akagi, A., Fukushima, S., Okada, K., Jiang, C.-J., Yoshida, R., Nakayama, A., et al. (2014). WRKY45-dependent priming of diterpenoid phytoalexin biosynthesis in rice and the role of cytokinin in triggering the reaction. Plant Mol. Biol. 86 (1), 171–183. doi: 10.1007/s11103-014-0221-x
Alfieri, M., Vaccaro, M. C., Cappetta, E., Ambrosone, A., De Tommasi, N., Leone, A. (2018). Coactivation of MEP-biosynthetic genes and accumulation of abietane diterpenes in Salvia sclarea by heterologous expression of WRKY and MYC2 transcription factors. Sci. Rep. 8 (1), 11009. doi: 10.1038/s41598-018-29389-4
Bailey, T. L., Johnson, J., Grant, C. E., Noble, W. S. (2015). The MEME suite. Nucleic Acids Res. 43 (W1), W39–W49. doi: 10.1093/nar/gkv416
Cannon, S. B., Mitra, A., Baumgarten, A., Young, N. D., May, G. (2004). The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 4 (1), 1–21. doi: 10.1186/1471-2229-4-10
Cao, W., Wang, Y., Shi, M., Hao, X., Zhao, W., Wang, Y., et al. (2018). Transcription factor SmWRKY1 positively promotes the biosynthesis of tanshinones in Salvia miltiorrhiza. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00554
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13 (8), 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chen, J., Gao, T., Wan, S., Zhang, Y., Yang, J., Yu, Y., et al. (2018). Genome-wide identification, classification and expression analysis of the HSP gene superfamily in tea plant (Camellia sinensis). Int. J. Mol. Sci. 19 (9), 2633. doi: 10.3390/ijms19092633
Cheng, Y., Jalal, A. G., Yu, J., Yao, Z., Ruan, M., Ye, Q., et al. (2016). Putative WRKYs associated with regulation of fruit ripening revealed by detailed expression analysis of the WRKY gene family in pepper. Sci. Rep. 6 (1), 1–11. doi: 10.1038/srep39000
Chen, F., Hu, Y., Vannozzi, A., Wu, K., Cai, H., Qin, Y., et al. (2017a). The WRKY transcription factor family in model plants and crops. Crit. Rev. Plant Sci. 36 (5-6), 311–335. doi: 10.1080/07352689.2018.1441103
Chen, H., Lai, Z., Shi, J., Xiao, Y., Chen, Z., Xu, X. (2010). Roles of Arabidopsis WRKY18, WRKY40 and WRKY60 transcription factors in plant responses to abscisic acid and abiotic stress. BMC Plant Biol. 10 (1), 281. doi: 10.1186/1471-2229-10-281
Chen, M., Yan, T., Shen, Q., Lu, X., Pan, Q., Huang, Y., et al. (2017b). Glandular trichome-specific WRKY 1 promotes artemisinin biosynthesis in Artemisia annua. New Phytol. 214 (1), 304–316. doi: 10.1111/nph.14373
Chi, Y., Yang, Y., Zhou, Y., Zhou, J., Fan, B., Yu, J. Q., et al. (2013). Protein–protein interactions in the regulation of WRKY transcription factors. Mol. Plant 6 (2), 287–300. doi: 10.1093/mp/sst026
Dang, F., Wang, Y., Yu, L., Eulgem, T., Lai, Y., Liu, Z., et al. (2013). CaWRKY40, a WRKY protein of pepper, plays an important role in the regulation of tolerance to heat stress and resistance to Ralstonia solanacearum infection. Plant Cell Environ. 36 (4), 757–774. doi: 10.1111/pce.12011
Ding, W., Ouyang, Q., Li, Y., Shi, T., Li, L., Yang, X., et al. (2019). Genome-wide investigation of WRKY transcription factors in sweet osmanthus and their potential regulation of aroma synthesis. Tree Physiol. 40 (4), 557–572. doi: 10.1093/treephys/tpz129
Dong, X., Zhu, Y., Bao, G., Hu, F., Qin, G. (2013). New limonoids and a dihydrobenzofuran norlignan from the roots of Toona sinensis. Molecules 18 (3), 2840–2850. doi: 10.3390/molecules18032840
Dou, L., Zhang, X., Pang, C., Song, M., Wei, H., Fan, S., et al. (2014). Genome-wide analysis of the WRKY gene family in cotton. Mol. Genet. Genomics 289 (6), 1103–1121. doi: 10.1007/s00438-014-0872-y
Eulgem, T., Rushton, P. J., Robatzek, S., Somssich, I. E. (2000). The WRKY superfamily of plant transcription factors. Trends Plant Sci. 5 (5), 199–206. doi: 10.1016/s1360-1385(00)01600-9
Ezoe, A., Shirai, K., Hanada, K. (2020). Degree of functional divergence in duplicates is associated with distinct roles in plant evolution. Mol. Biol. Evol. 38 (4), 1447–1459. doi: 10.1093/molbev/msaa302
Fan, Z., Tan, X., Shan, W., Kuang, J., Lu, W., Chen, J. (2017). BrWRKY65, a WRKY transcription factor, is involved in regulating three leaf senescence-associated genes in Chinese flowering cabbage. Int. J. Mol. Sci. 18 (6), 1228. doi: 10.3390/ijms18061228
Gasteiger, E., Hoogland, C., Gattiker, A., Wilkins, M. R., Appel, R. D., Bairoch, A. (2005). Protein identification and analysis tools on the ExPASy server. Proteomics Protoc. Handb., 571–607. doi: 10.1385/1-59259-890-0:571
Goyal, P., Manzoor, M. M., Vishwakarma, R. A., Sharma, D., Dhar, M. K., Gupta, S. (2020). A comprehensive transcriptome-wide identification and screening of WRKY gene family engaged in abiotic stress in Glycyrrhiza glabra. Sci. Rep. 10 (1), 1–18. doi: 10.1038/s41598-019-57232-x
Guo, Y. L. (2013). Gene family evolution in green plants with emphasis on the origination and evolution of Arabidopsis thaliana genes. Plant J. 73 (6), 941–951. doi: 10.1111/tpj.12089
He, X., Li, J., Chen, Y., Yang, J., Chen, X. (2019). Genome-wide analysis of the WRKY gene family and its response to abiotic stress in buckwheat (Fagopyrum tataricum). Open Life Sci. 14 (1), 80–96. doi: 10.1515/biol-2019-0010
Holub, E. B. (2001). The arms race is ancient history in Arabidopsis, the wildflower. Nat. Rev. Genet. 2 (7), 516–527. doi: 10.1038/35080508
Hsin, K., Hsieh, M., Lee, Y., Lin, K., Cheng, Y. (2022). Insight into the phylogeny and binding ability of WRKY transcription factors. Int. J. Mol. Sci. 23 (5), 2895. doi: 10.3390/ijms23052895
Huang, S., Gao, Y., Liu, J., Peng, X., Niu, X., Fei, Z., et al. (2012). Genome-wide analysis of WRKY transcription factors in Solanum lycopersicum. Mol. Genet. Genomics 287 (6), 495–513. doi: 10.1007/s00438-012-0696-6
Hu, B., Jin, J., Guo, A., Zhang, H., Luo, J., Gao, G. (2015). GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31 (8), 1296–1297. doi: 10.1093/bioinformatics/btu817
Hurst, L. D. (2002). The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet. 18 (9), 486–486. doi: 10.1016/s0168-9525(02)02722-1
Ishiguro, S., Nakamura, K. (1994). Characterization of a cDNA encoding a novel DNA-binding protein, SPF1, that recognizes SP8 sequences in the 5' upstream regions of genes coding for sporamin and β-amylase from sweet potato. Mol. Gen. Genet. 244 (6), 563–571. doi: 10.1007/BF00282746
Jiang, J., Ma, S., Ye, N., Jiang, M., Cao, J., Zhang, J. (2017). WRKY transcription factors in plant responses to stresses. J. Integr. Plant Biol. 59 (2), 86–101. doi: 10.1111/jipb.12513
Jia, C., Wang, Z., Wang, J., Miao, H., Zhang, J., Xu, B., et al. (2022). Genome-wide analysis of the banana WRKY transcription factor gene family closely related to fruit r ipening and stress. Plants 11 (5), 662. doi: 10.3390/plants11050662
Ji, Y. T., Xiu, Z., Chen, C. H., Wang, Y., Yang, J. X., Sui, J. J., et al. (2021). Long read sequencing of Toona sinensis (A. juss) roem: A chromosome-level reference genome for the family Meliaceae. Mol. Ecol. Resour. 21 (4), 1243–1255. doi: 10.1111/1755-0998.13318
Kong, H., Landherr, L. L., Frohlich, M. W., Leebens-Mack, J., Ma, H., DePamphilis, CW (2007). Patterns of gene duplication in the plant SKP1 gene family in angiosperms: evidence for multiple mechanisms of rapid gene birth. Plant J. 50 (5), 873–885. doi: 10.1111/j.1365-313X.2007.03097.x
Korkuć, P., Schippers, J. H. M., Walther, D. (2013). Characterization and identification of cis-regulatory elements in Arabidopsis based on single-nucleotide polymorphism information. Plant Physiol. 164 (1), 181–200. doi: 10.1104/pp.113.229716
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19 (9), 1639–1645. doi: 10.1101/gr.092759.109
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33 (7), 1870–1874. doi: 10.1093/molbev/msw054
Lescot, M., Déhais, P., Thijs, G., Marchal, K., Moreau, Y., Van de Peer, Y., et al. (2002). PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30 (1), 325–327. doi: 10.1093/nar/30.1.325
Letunic, I., Khedkar, S., Bork, P. (2021). SMART: recent updates, new developments and status in 2020. Nucleic Acids Res. 49 (D1), D458–D460. doi: 10.1093/nar/gkaa937
Li, X., He, L., An, X., Yu, K., Meng, N., Duan, C., et al. (2020b). VviWRKY40, a WRKY transcription factor, regulates glycosylated monoterpenoid production by VviGT14 in grape berry. Genes 11 (5), 485. doi: 10.3390/genes11050485
Li, D., Liu, P., Yu, J., Wang, L., Dossa, K., Zhang, Y., et al. (2017). Genome-wide analysis of WRKY gene family in the sesame genome and identification of the WRKY genes involved in responses to abiotic stresses. BMC Plant Biol. 17 (1), 1–19. doi: 10.1186/s12870-017-1099-y
Li, W., Wang, H., Yu, D. (2016). Arabidopsis WRKY transcription factors WRKY12 and WRKY13 oppositely regulate flowering under short-day conditions. Mol. Plant 9 (11), 1492–1503. doi: 10.1016/j.molp.2016.08.003
Li, H., Yue, Y., Ding, W., Chen, G., Li, L., Li, Y., et al. (2020a). Genome-wide identification, classification, and expression profiling reveals R2R3-MYB transcription factors related to monoterpenoid biosynthesis in Osmanthus fragrans. Genes 11 (4), 353. doi: 10.3390/genes11040353
Luo, Y., Huang, X., Song, X., Wen, B., Xie, N., Wang, K., et al. (2022). Identification of a WRKY transcriptional activator from Camellia sinensis that regulates methylated EGCG biosynthesis. Horticult Res. 2022 (9), uhac024. doi: 10.1093/hr/uhac024
Lu, S., Wang, J., Chitsaz, F., Derbyshire, M. K., Geer, R. C., Gonzales, N. R., et al. (2020). CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 48 (D1), D265–D268. doi: 10.1093/nar/gkz991
Mao, P., Jin, X., Bao, Q., Mei, C., Zhou, Q., Min, X., et al. (2020). WRKY transcription factors in Medicago sativa l.: genome-wide identification and expression analysis under abiotic stress. DNA Cell Biol. 39 (12), 2212–2225. doi: 10.1089/dna.2020.5726
Mistry, J., Chuguransky, S., Williams, L., Qureshi, M., Salazar, G. A., Sonnhammer, E. L., et al. (2021). Pfam: The protein families database in 2021. Nucleic Acids Res. 49 (D1), D412–D419. doi: 10.1093/nar/gkaa913
Moore, R. C., Purugganan, M. D. (2003). The early stages of duplicate gene evolution. Proc. Natl. Acad. Sci. 100 (26), 15682–15687. doi: 10.1073/pnas.2535513100
Naoumkina, M. A., He, X. Z., Dixon, R. A. (2008). Elicitor-induced transcription factors for metabolic reprogramming of secondary metabolism in Medicago truncatula. BMC Plant Biol. 8 (1), 132. doi: 10.1186/1471-2229-8-132
Nicholas, K. B. (1997). GeneDoc: analysis and visualization of genetic variation. Embnew. News 4, 14.
Ning, P., Liu, C., Kang, J., Lv, J. (2017). Genome-wide analysis of WRKY transcription factors in wheat (Triticum aestivum l.) and differential expression under water deficit condition. PeerJ 5, e3232. doi: 10.7717/peerj.3232
Ohme-Takagi, M., Suzuki, K., Shinshi, H. (2000). Regulation of ethylene-induced transcription of defense genes. Plant Cell Physiol. 41 (11), 1187–1192. doi: 10.1093/pcp/pcd057
Olive, M. R., Walker, J. C., Singh, K., Ellis, J. G., Llewellyn, D., Peacock, W. J., et al. (1991). The anaerobic responsive element. Plant Mol. Biol. 212 (1), 673–684. doi: 10.1007/978-1-4615-3304-7_67
Patra, B., Schluttenhofer, C., Wu, Y., Pattanaik, S., Yuan, L. (2013). Transcriptional regulation of secondary metabolite biosynthesis in plants. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 1829 (11), 1236–1247. doi: 10.1016/j.bbagrm.2013.09.006
Peng, W., Liu, Y., Hu, M., Zhang, M., Yang, J., Liang, F., et al. (2019). Toona sinensis: a comprehensive review on its traditional usages, phytochemisty, pharmacology and toxicology. Rev. Bras. Farmacognosia 29 (1), 111–124. doi: 10.1016/j.bjp.2018.07.009
Ren, L., Wan, W., Xie, Y., Chen, B., Wang, G., Yin, D., et al. (2021). Identification of volatile aroma compounds of Toona sinensis (a. juss) roem buds and investigation of genes expression profiles confering aroma production. Pak. J. Bot. 53 (4), 1459–1464. doi: 10.30848/Pjb2021-4(32
Rinerson, C. I., Rabara, R. C., Tripathi, P., Shen, Q. J., Rushton, P. J. (2015). The evolution of WRKY transcription factors. BMC Plant Biol. 15 (1), 1–18. doi: 10.1186/s12870-015-0456-y
Rosado, D., Ackermann, A., Spassibojko, O., Rossi, M., Pedmale, U. V. (2022). WRKY transcription factors and ethylene signaling modify root growth during the shade-avoidance response. Plant Physiol. 188 (2), 1294–1311. doi: 10.1093/plphys/kiab493
Ross, C. A., Liu, Y., Shen, Q. J. (2007). The WRKY gene family in rice (Oryza sativa). J. Integr. Plant Biol. 49 (6), 827–842. doi: 10.1111/j.1744-7909.2007.00504.x
Rushton, P. J., Somssich, I. E., Ringler, P., Shen, Q. J. (2010). WRKY transcription factors. Trends Plant Sci. 15 (5), 247–258. doi: 10.1016/j.tplants.2010.02.006
Schluttenhofer, C., Yuan, L. (2015). Regulation of specialized metabolism by WRKY transcription factors. Plant Physiol. 167 (2), 295–306. doi: 10.1104/pp.114.251769
Song, A., Li, P., Jiang, J., Chen, S., Li, H., Zeng, J., et al. (2014). Phylogenetic and transcription analysis of chrysanthemum WRKY transcription factors. Int. J. Mol. Sci. 15 (8), 14442–14455. doi: 10.3390/ijms150814442
Spyropoulou, E. A., Haring, M. A., Schuurink, R. C. (2014). RNA Sequencing on Solanum lycopersicum trichomes identifies transcription factors that activate terpene synthase promoters. BMC Genomics 15 (1), 1–16. doi: 10.1186/1471-2164-15-402
Sui, J., Qu, C., Yang, J., Zhang, W., Ji, Y. (2019). Transcriptome changes in the phenylpropanoid pathway in senescing leaves of Toona sinensis. Acta Physiologiae Plantarum 41 (7), 1–15. doi: 10.1007/s11738-019-2915-9
Sun, Y., Niu, Y., Xu, J., Li, Y., Luo, H., Zhu, Y., et al. (2013). Discovery of WRKY transcription factors through transcriptome analysis and characterization of a novel methyl jasmonate-inducible PqWRKY1 gene from Panax quinquefolius. Plant Cell Tissue Organ Culture 114 (2), 269–277. doi: 10.1007/s11240-013-0323-1
Suttipanta, N., Pattanaik, S., Kulshrestha, M., Patra, B., Singh, S. K., Yuan, L. (2011). The transcription factor CrWRKY1 positively regulates the terpenoid indole alkaloid biosynthesis in Catharanthus roseus. Plant Physiol. 157 (4), 2081–2093. doi: 10.1104/pp.111.181834
Thompson, J. D., Gibson, T. J., Higgins, D. G. (2003). Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinf. 1), 2.3. doi: 10.1002/0471250953.bi0203s00
Wang, Y., Chen, F., Ma, Y., Zhang, T., Sun, P., Lan, M., et al. (2021). An ancient whole-genome duplication event and its contribution to flavor compounds in the tea plant (Camellia sinensis). Horticult Res. 2021 (8), 176–188. doi: 10.1038/s41438-021-00613-z
Wang, Y., Tang, H., DeBarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40 (7), e49–e49. doi: 10.1093/nar/gkr1293
Wang, Q., Wang, M., Zhang, X., Hao, B., Kaushik, S., Pan, Y. (2011). WRKY gene family evolution in Arabidopsis thaliana. Genetica 139 (8), 973–983. doi: 10.1007/s10709-011-9599-4
Wang, P., Yue, C., Chen, D., Zheng, Y., Zhang, Q., Yang, J., et al. (2019). Genome-wide identification of WRKY family genes and their response to abiotic stresses in tea plant (Camellia sinensis). Genes Genomics 41 (1), 17–33. doi: 10.1007/s13258-018-0734-9
Wang, D., Zhang, Y., Zhang, Z., Zhu, J., Yu, J. (2010). KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinf. 8 (1), 77–80. doi: 10.1016/S1672-0229(10)60008-3
Wei, K., Chen, J., Chen, Y., Wu, L., Xie, D. (2012). Molecular phylogenetic and expression analysis of the complete WRKY transcription factor family in maize. DNA Res. 19 (2), 153–164. doi: 10.1093/dnares/dsr048
Wu, Z., Li, X., Liu, Z., Li, H., Wang, Y., Zhuang, J. (2016). Transcriptome-wide identification of Camellia sinensis WRKY transcription factors in response to temperature stress. Mol. Genet. Genomics 291 (1), 255–269. doi: 10.1007/s00438-015-1107-6
Xiao, Y., Zhou, L., Lei, X., Cao, H., Wang, Y., Dou, Y., et al. (2017). Genome-wide identification of WRKY genes and their expression profiles under different abiotic stresses in Elaeis guineensis. PloS One 12 (12), e0189224. doi: 10.1371/journal.pone.0189224
Xie, L., Yan, T., Li, L., Chen, M., Ma, Y., Hao, X., et al. (2021). The WRKY transcription factor AaGSW2 promotes glandular trichome initiation in Artemisia annua. J. Exp. Bot. 72 (5), 1691–1701. doi: 10.1093/jxb/eraa523
Xu, Y. H., Wang, J. W., Wang, S., Wang, J., Chen, X. Y. (2004). Characterization of GaWRKY1, a cotton transcription factor that regulates the sesquiterpene synthase gene (+)-δ-cadinene synthase-a. Plant Physiol. 135 (1), 507–515. doi: 10.1104/pp.104.038612
Yang, C., Fang, X., Wu, X., Mao, Y., Wang, L., Chen, X. (2012). Transcriptional regulation of plant secondary metabolism. J. Integr. Plant Biol. 54 (10), 703–712. doi: 10.1111/j.1744-7909.2012.01161.x
Yang, Y., Ma, Y., Yang, S., Yue, X., Peng, W. (2020a). Chemical components analysis of Toona sinensis bark and wood by pyrolisis-gas chromatography-mass spectrometry. Asia-Pacific J. Chem. Eng. 15, e2487. doi: 10.1002/apj.2487
Yang, Y., Wang, N., Zhao, S. (2020b). Functional characterization of a WRKY family gene involved in somatic embryogenesis in Panax ginseng. Protoplasma 257 (2), 449–458. doi: 10.1007/s00709-019-01455-2
Yang, Y., Zhou, Y., Chi, Y., Fan, B., Chen, Z. (2017). Characterization of soybean WRKY gene family and identification of soybean WRKY genes that promote resistance to soybean cyst nematode. Sci. Rep. 7 (1), 1–13. doi: 10.1038/s41598-017-18235-8
Yan, H., Jia, H., Chen, X., Hao, L., An, H., Guo, X. (2014). The cotton WRKY transcription factor GhWRKY17 functions in drought and salt stress in transgenic Nicotiana benthamiana through ABA signaling and the modulation of reactive oxygen species production. Plant Cell Physiol. 55 (12), 2060–2076. doi: 10.1093/pcp/pcu133
Yu, J., Wang, J., Lin, W., Li, S., Li, H., Zhou, J., et al. (2005). The genomes of Oryza sativa: a history of duplications. PloS Biol. 3 (2), e38. doi: 10.1371/journal.pbio.0030038
Zhai, X., Granvogl, M. (2019). Characterization of the key aroma compounds in two differently dried Toona sinensis (A. juss.) roem. by means of the molecular sensory science concept. J. Agric. Food Chem. 67 (35), 9885–9894. doi: 10.1021/acs.jafc.8b06656
Zhang, Y., Wang, L. (2005). The WRKY transcription factor superfamily: its origin in eukaryotes and expansion in plants. BMC Evolutionary Biol. 5 (1), 1–12. doi: 10.1186/1471-2148-5-1
Zhao, Q., Zhong, X., Zhu, S., Wang, K., Tan, G., Meng, P., et al. (2022). Research advances in Toona sinensis, a traditional chinese medicinal plant and popular vegetable in China. Diversity 14 (7), 572. doi: 10.3390/d14070572
Keywords: Toona sinensis, WRKY transcription factors, gene evolution, terpene synthesis, genome-wide analysis
Citation: Ren L, Wan W, Yin D, Deng X, Ma Z, Gao T and Cao X (2023) Genome-wide analysis of WRKY transcription factor genes in Toona sinensis: An insight into evolutionary characteristics and terpene synthesis. Front. Plant Sci. 13:1063850. doi: 10.3389/fpls.2022.1063850
Received: 07 October 2022; Accepted: 13 December 2022;
Published: 20 January 2023.
Edited by:
Shengqing Shi, Chinese Academy of Forestry, ChinaReviewed by:
Lanting Zeng, Chinese Academy of Sciences (CAS), ChinaMohammed Ali Abd Elhammed Abd Allah, Desert Research Center, Egypt
Copyright © 2023 Ren, Wan, Yin, Deng, Ma, Gao and Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ting Gao, Z2FvdGluZzIwMTgwMkAxNjMuY29t; Xiaohan Cao, Y2FveGlhb2hhbkAxNjMuY29t
†These authors have contributed equally to this work and share first authorship