 Junchen Wang
Junchen Wang Lifeng Zhang1,2†
Lifeng Zhang1,2† Liwang Qi
Liwang Qi- 1State Key Laboratory of Tree Genetics and Breeding, Research Institute of Forestry, Chinese Academy of Forestry, Beijing, China
- 2Key Laboratory of Tree Breeding and Cultivation, National Forestry and Grassland Administration, Research Institute of Forestry, Chinese Academy of Forestry, Beijing, China
Somatic embryogenesis (SE) techniques have been established for micropropagation or basic research related to plant development in many conifer species. The frequent occurrence of non-embryogenic callus (NEC) during SE has impose constraints on the application of somatic embryogenesis SE in Larix kaempferi (Lamb.) Carr, but the potential regulatory mechanisms are poorly understood. In this study, integrated transcriptomic and metabolomic analyses were performed in embryogenic callus (EC) and NEC originating from a single immature zygotic embryo to better decipher the key molecular and metabolic mechanisms required for embryogenic potential maintenance. The results showed that a total of 13,842 differentially expressed genes (DEGs) were found in EC and NEC, among which many were enriched in plant hormone signal transduction, starch and sucrose metabolism, phenylpropanoid biosynthesis, flavonoid biosynthesis, and the biosynthesis of amino acids pathways. Metabolite profiling showed that 441 differentially accumulated metabolites (DAMs) were identified in EC and NEC. Both EC and NEC had vigorous primary metabolic activities, while most secondary metabolites were upregulated in NEC. Many totipotency-related transcription factor (TF) genes such as BBMs, WUSs, and LEC1 showed higher expression levels in EC compared with NEC, which may result in the higher accumulation of indole 3-acetic acid (IAA) in EC. NEC was characterized by upregulation of genes and metabolites associated with stress responses, such as DEGs involved in jasmonic acid (JA) and ethylene (ETH) biosynthesis and signal transduction pathways, and DEGs and DAMs related to phenylpropanoid and flavonoid biosynthesis. We predicted and analyzed TFs that could target several key co-expressed structural DEGs including two C4H genes, two CcoAOMT genes and three HCT genes involved in phenylpropanoid and flavonoid biosynthesis. Based on the targeted relationship and the co-expression network, two ERFs (Lk23436 and Lk458687), one MYB (Lk34626) and one C2C2-dof (Lk37167) may play an important role in regulating phenolic acid and flavonoid biosynthesis by transcriptionally regulating the expression of these structural genes. This study shows an approach involving integrated transcriptomic and metabolic analyses to obtain insights into molecular events underlying embryogenic potential maintenance and the biosynthesis mechanisms of key metabolites involving TF regulation, which provides valuable information for the improvement of SE efficiency in L. kaempferi.
Introduction
Larix kaempferi (Lamb.) Carr is one of the most important fast-growing plantation species in China. It is widely distributed in alpine coniferous forests in northeastern and northern China, Inner Mongolia, and southwestern China (Zhang et al., 2010; Fan et al., 2020; Wu et al., 2021) and plays a substantial role in forest timber production as well as in ecological construction (Fan et al., 2020). However, L. kaempferi features a long-life span and complex genetic background, making it difficult to breed new lines with conventional methods especially by seed propagation (Zhang et al., 2014a; Zhang et al., 2021a). Therefore, to establish and develop efficient reproduction technologies is of great significance for multiplying true-to-type selected varieties with desired characteristics. Conifer somatic embryogenesis (SE) technology is powerful in the mass vegetative propagation of elite varieties or genotypes with desired traits, which has been applied in Larix species for more than three decades since it was first reported in Picea abies (Hakman et al., 1985) and L. decidua (Nagmani and Bonga, 1985). To date, SE technology has been regarded as an ideal system for basic research about gymnosperm growth and development and the related regulatory mechanisms (Zhang et al., 2021a). For example, stable gene transformation technology has been established in L. kaempferi SE system and has been utilized to have multiple genes functionally validated (Fan et al., 2020; Fan et al., 2021; Kang et al., 2021). However, micropropagation through SE technology has not been widely applied to plant regeneration in L. kaempferi due to several problems such as difficult embryonal-suspensor mass (ESM) induction, frequent abnormal embryo development, and challenges in vitro synchronization (Zhang et al., 2012b; Zhang et al., 2021a). Therefore, further knowledge is required to address these problems and realize the full potential of SE.
SE is a complex process of embryo development involving cell dedifferentiation and reprogramming to regenerate whole plants, which is regulated by a network of hundreds of genes (Mordhorst et al., 1997; Wang et al., 2020). Many studies focusing on plant SE were conducted in some short-lived angiosperms such as Cucumis sativus (Xue et al., 2022), Gossypium hirsutum (Guo et al., 2019; Kumar et al., 2021), and Fragaria vesca (Liu et al., 2022), and most in-depth knowledge of SE can be only obtained from Arabidopsis (Horstman et al., 2017; Karami et al., 2021; Li et al., 2022b; Paul et al., 2022). However, little is known from perennials including gymnosperm. Despite the promising achievements obtained in Picea abies and P. glauca (Klimaszewska et al., 2011; Varis et al., 2018; Nakamura et al., 2020), in most conifers including L. kaempferi, the whole process of SE can only be initiated from immature seeds, in practice immature zygotic embryos (Ávila et al., 2022). In the ESM induction of L. kaempferi, a cytological characteristic is that two distinct types of callus may be generated: embryogenic callus (EC) and non-embryogenic callus (NEC), during which the cells must dedifferentiate, activate their cell division cycle, and reorganize their physiological and metabolic states (Kumaravel et al., 2017; Wang et al., 2020). EC and NEC are different in their appearance and biochemical properties, but both of them can be maintained in medium with 2,4-Dichlorophenoxyacetic acid (2,4-D) and 6-Benzylaminopurine (6-BA). NEC are characterized by unorganized, dedifferentiated, continuously dividing cell masses (Kumar et al., 2021), whereas EC cells are in a state of vigorous proliferation and differentiation. These results furtherly reflect the distinctions in embryogenic potentials between EC and NEC. For L. kaempferi, EC consists of proembryogenic masses and have the ability to develop into rapidly proliferating early somatic embryos and eventually form a whole plant in growth medium, namely embryogenic potential (Bonga et al., 2009; Elhiti et al., 2013), while NEC have no embryogenic development ability. Nevertheless, limited knowledge is known about the molecular mechanisms underlying the formation of EC and NEC as well as the distinct embryogenic potentials between EC and NEC, which may be crucial for increasing the initiation rate of SE.
Besides the unexpected occurrence of NEC during ESM induction, EC may be frequently transformed to NEC during sub-culture process, which combinedly impose constraints on the stability of SE system in L. kaempferi (Zhang et al., 2010). Therefore, the maintenance of embryogenic state is crucial for the proper embryo development and global SE efficiency. However, the potential regulatory networks involved in these processes are poorly understood. In angiosperms, genes including SOMATIC EMBRYOGENESIS RECEPTOR KINASE (SERK) family members, BABY BOOM (BBM), WUSCHEL (WUS), and LEAFY COTYLEDON (LEC) are involved in the transition from NEC to EC (Gruel et al., 2018; Mendez-Hernandez et al., 2019) and the overexpression of these genes can increase embryo formation frequency (Lotan et al., 1998; Boutilier et al., 2002; Zuo et al., 2002; Lowe et al., 2016; Horstman et al., 2017; Li et al., 2022b), but the degree of the increase differed between monocot and dicot systems (Srinivasan et al., 2007; Zhang et al., 2014b; Florez et al., 2015; Kumar et al., 2021). Moreover, it has been reported that genes related to stress response such as abscisic aldehyde synthesis enzyme 2 (ABA2), abscisic acid insensitive 3 (ABI3), jasmonate ZIM-domain 1 (JAZ1), late embryogenesis abundant protein 1 (LEA1), and transcription factors like AUX/IAAs, NACs, WRKYs, MYBs, and ERFs were also involved in callus induction (Jin et al., 2014; Fehér, 2015; Guo et al., 2019; Salaün et al., 2021). In conifers, some studies have compared EC and NEC of several species including L. kaempferi (Zhang et al., 2010), P. balfouriana (Li et al., 2014), Pinus radiata (Bravo et al., 2017), Pseudotsuga menziesii (Gautier et al., 2019), P. abies (Nakamura et al., 2020), and Pinus pinaster (Ávila et al., 2022) from the perspectives of miRNA and their targeted gene expression, DNA methylation, histone modification, cytological or biochemical characteristics, and transcriptomic or proteomic profiling. Although a few putative gene markers for embryogenic cells such as BBM and WUS as mentioned above have been highlighted in these studies, whether these markers are universal for other plant species remains empirical.
The application of transcriptomics in diverse tree species have been very successful in providing a detailed gene expression profile of plant cells, which is important in understanding basic functions in tree biology (Ávila et al., 2022). In L. kaempferi, the availability of genetic and genomic resources has largely allowed its use in functional genomics approaches (Sun et al., 2022). Similar to transcriptomics or proteomics, metabolomics is an emerging omics technology aimed at identifying and quantifying endogenous molecule metabolites, the final products of cell biological regulation process (Lin et al., 2020). In recent years, “multi-omics” strategies have provided new insights in revealing underlying mechanisms in plant growth and development as well as in plant responses to external stresses (Xiao et al., 2021; Zhang et al., 2021c; Li et al., 2022a; Wang et al., 2022b). The combined analysis of transcriptome and metabolome data can be used to predict the gene function involved in the targeted metabolic pathway and simultaneously provide supporting information for gene mining (Fukushima et al., 2009). In conifers, the integrated analysis of the two omics were used in several species such as Pinus pinaster (Cañas et al., 2015), Pinus radiata (Escandón et al., 2017), P. abies (Dobrowolska et al., 2017; Blokhina et al., 2019), Pinus taeda (Mao et al., 2021), Pinus massoniana (Cai et al., 2021), and L. olgensis (Zhang et al., 2021b) to address the regulatory mechanisms underlying some problems in plant systems biology including tree responses to environmental changes or disease, the biosynthesis of targeted metabolites, and somatic embryo germination. Given the fact that SE induction is an initiation phase regulated by a complex network of numerous genes (Mordhorst et al., 1997; Wang et al., 2020), there must also be many metabolic activities functioning in this process, which can be reflected by the differences between EC and NEC. Moreover, the integration of transcriptomics and metabolomics may offer notable advantages to identify the biosynthetic mechanisms of key metabolic pathways underlying the formation of EC and NEC. However, no previous studies have compared EC and NEC using integrated transcriptomic and metabolic analysis.
The morphology of EC and NEC of L. kaempferi was addressed elsewhere (Zhang et al., 2010). In this work, transcriptomics and metabolomics were carried out on EC and NEC produced in the process of transdifferentiation. The aim of this study was to explore the gene expression and metabolic differences between EC and NEC and furtherly reveal the molecular mechanisms underlying key metabolites accumulation as well as embryogenic potential maintenance. To the best of our knowledge, this work is the first study in conifers trying to reveal the molecular basis of small-molecule metabolite biosynthesis during SE induction. Our results will provide new insights into conifer SE, with potential value to increase SE efficiency in L. kaempferi breeding program.
Materials and methods
Plant material
The embryonal-suspensor mass (ESM) was induced from L. kaempferi immature zygotic embryos on solid induction medium containing 450 mg/L glutamine, 500 mg/L casein hydrolysate, 1,000 mg/L inositol, 30 g/L sucrose, 3 g/L Gelrite, 10 µM 2,4-D, and 3.6 µM 6-BA in darkness at approximatively 23 °C (Zhang et al., 2014a; Zhang et al., 2021a). Two types of callus, EC and NEC, were found to emerge from a single immature zygotic embryo during this process (named cell line C6). Then, EC and NEC were sub-cultured every three weeks on solid proliferation medium supplemented with 450 mg/L glutamine, 500 mg/L casein hydrolysate, 1000 mg/L inositol, 30 g/L sucrose, 3 g/L Gelrite, 0.5 µM 2,4-D, and 0.18 µM 6-BA (Zhang et al., 2014a; Zhang et al., 2021a). After being sub-cultured for 15 days, the proliferating material of EC and NEC was collected (Figure 1), frozen in liquid nitrogen, and stored at -80 °C until use.

Figure 1 Embryogenic callus (EC) and non-embryogenic callus (NEC) in L. kaempferi. (A) EC. (B) NEC. Scale bar: 1mm.
RNA sequencing and functional annotation
RNA was extracted from three biological replicates of both EC and NEC. Samples were ground to fine powder. The total RNA was isolated using the RNAiso Plus and RNAiso-mate for Plant Tissue kits (Takara, Japan) according to the manufacturer’s instructions and was treated with DNase (Takara, Japan) to remove DNA. To ensure the accuracy of the sequencing data, the total RNA samples were prepared as follows. RNA degradation and contamination was monitored on 1% agarose gels and RNA purity was checked using the NanoPhotometer® spectrophotometer (IMPLEN, CA, USA). RNA concentration was quantified using Qubit® RNA Assay Kit in Qubit®2.0 Flurometer (Life Technologies, CA, USA). Finally, RNA integrity was assessed using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, CA, USA).
The cDNA library for each sample was sequenced on the Illumina sequencing platform by Metware Biotechnology Co., Ltd. (Wuhan, China). After that, many high-quality raw reads were selected, and clean reads were obtained by removing low-quality ones. All the clean reads were mapped separately to the L. kaempferi V1.0 genome using HISAT2 software. The basic local alignment search tool (BLAST) was used to annotate the functions of unigenes against protein databases, including Non-redundant Protein Database (NR), Gene ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), TrEMBL, Kyoto Encyclopedia of Genes and Genomes (KOG), Clusters of Orthologous Groups (COG), Protein Family Database (Pfam), and Non-redundant Protein Sequence Database (SwissProt) using BLAST with an e-value threshold of 1×10−5.
Identification of differentially expressed genes
To verify the transcription expression levels of all samples, fragments per kilobase of transcript per million mapped reads (FPKM) were calculated by featureCounts v1.6.2 to quantify the expression level of gene. Subsequently, differentially expressed genes (DEGs) between the two groups were filtered by DESeq2 software with |log2 fold change (FC)| ≥ 1 and false discovery rate (FDR) <0.01. The gene expression patterns were obtained through hierarchical clustering analysis. The GO and KEGG pathway enrichment analysis of DEGs was performed based on the hypergeometric test.
Quantitative real-time polymerase chain reaction (qRT-PCR) validation of RNA-seq data
The accuracy of RNA-seq data was verified using qRT-PCR. Total RNA was extracted from the calluses with RNAiso Plus and RNAiso-mate for Plant Tissue kits (TaKaRa, Dalian, China), after which 1 µg RNA was reverse transcribed using the PrimeScript™ RT Reagent Kit with gDNA Eraser (Perfect Real Time) (TaKaRa). The expression levels of selected genes were analyzed in a qRT-PCR assay conducted using a SYBR Premix Ex Taq Kit (TaKaRa) and the CFX96™ Real-Time
System. All primer sequences for these genes were shown in Supplementary Table 1. The specificity of the qRT-PCR primers was confirmed by separating the products on agarose gels and by sequencing (Supplementary Figure 1). The relative gene expression in each sample was calculated by the 2−△△ct method against LkEF1A1 (GenBank accession: JR153706) (Zhang et al., 2012a). SPSS 26.0 was used for ANOVA to test the significant differences of relative gene expression levels between EC and NEC and P < 0.05 was the threshold for significance.
Metabolomic analysis
Metabolomic analysis was conducted for three biological replicates of both EC and NEC. The freeze-dried calluses of L. kaempferi were homogenized in a ball mill. 100 mg powder was weighed and dissolved with 1.2 ml 70% methanol solution, vortexed 30 seconds every 30 minutes for 6 times in total, and placed in a refrigerator at 4°C overnight. After centrifugation at 12000 rpm for 10 min, the extracts were filtrated (0.22 μm pore size) for following ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) analysis by Metware Biotechnology Co., Ltd. (Wuhan, China).
Multiple reaction monitoring (MRM) was performed as previously described (Mao et al., 2021). Multivariate principal component analysis (PCA) and orthogonal partial least squares-discriminant analysis (OPLS-DA) were conducted using the base package and “MetaboAnalystR” in R. The multivariate analysis of variable importance in projection (VIP) in the OPLS-DA model was used to initially screen differentially accumulated metabolites (DAMs). The DAMs were identified based on VIP ≥ 1 and fabsolute log2FC (fold change) ≥ 1. K-means analysis and heatmap analysis based on hierarchical clustering were performed in R. Functional annotation and enrichment analysis of the DAMs were conducted based on the KEGG database (Du et al., 2021).
Correlation analysis of the transcriptome and metabolome data
Based on the transcriptome and metabolome data, Pearson’s correlation analysis was performed to explore the correlations between the DEGs and DAMs. The DEGs and DAMs were mapped to the KEGG pathway database to obtain their common pathway information. Only the detected correlations with a Pearson correlation coefficient (PCC)> 0.9 and P-value < 0.01 were selected. Subsequently, the related networks were visualized using the Cytoscape software.
Prediction of key transcription factors (TFs)
The DEGs highly related to key DAMs synthesis between EC and NEC were selected as target genes in this analysis. We used TBtools (Chen et al., 2020) to extract 2,000-bp sequences before the CDSs of the key genes as the potential promoter sequences based on the L. kaempferi genome data (Sun et al., 2022). The promoter sequences of these genes were uploaded to the online database PlantTFDB (http://planttfdb.gao-lab.org/) (Tian et al., 2020) and the potential TFs regulating target genes were predicted based on the Arabidopsis genome. BLAST program was employed to identify the best match between Arabidopsis and L. kaempferi genome. The significantly differentially expressed TFs between EC and NEC were selected as candidate TFs regulating the synthesis of key DAMs.
Results
Metabolomic profiling analysis
A widely targeted metabolomic analysis was performed to produce a metabolic profile by using UPLC-MS/MS system. An amount of 835 metabolites were identified and were divided into 14 categories, mainly containing lipids, flavonoids, phenolic acids, and amino acids and derivatives (Supplementary Tables 2, 3). Overall, primary metabolites showed a similar amount with secondary metabolites in all the detective metabolites, indicating that L. kaempferi callus possessed both vigorous primary and secondary metabolic activities. We used principal component analysis (PCA) to reveal the metabolite differences between EC and NEC. The results showed that PC1 and PC2 cumulatively explained 78.33% of the total variance of the samples (62.80% and 15.53% for PC1 and PC2, respectively), and all samples were grouped into two distinct clusters (Figure 2A). Moreover, the hierarchical clustering results also suggested the high repeatability of the biological replicates in each group and the significant differences in metabolites between EC and NEC (Figure 2B).
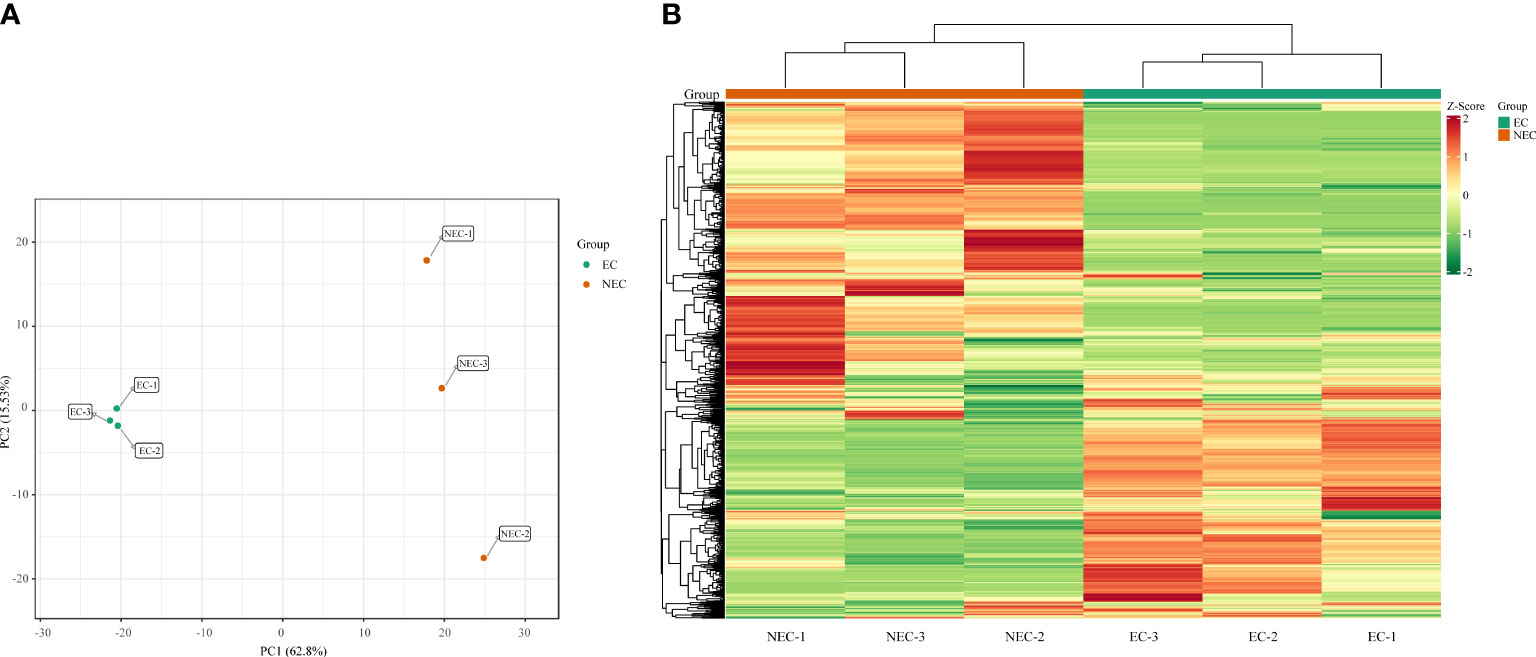
Figure 2 Principal component analysis (PCA) score plots and heat map of metabolite profile. (A) PCA score plots with each point representing an independent biological replicate; (B) Heatmap of metabolite abundance of EC and NEC according to hierarchical cluster analysis, where green, yellow, and red indicate low, intermediate, and high accumulation, respectively.
DAMs identification and enrichment analysis
The results of Orthogonal partial least squares-discriminant analysis (OPLS-DA) analysis and response permutation testing (RPT) were shown in Supplementary Figures 2A, B. DAMs between EC and NEC were screened and identified by using univariate and multivariate statistical analysis with threshold values of VIP ≥ 1 and fold change ≥ 2 or ≤ 0.5. Overall, 441 DAMs were obtained between the two groups, among which 177 metabolites were downregulated while 264 were upregulated in NEC compared to EC (Supplementary Figure 2C; Supplementary Table 4). Moreover, 30 and 79 metabolites were specifically accumulated in EC and NEC, respectively (Supplementary Table 4). The results of top ten metabolites with the largest fold change were shown in Supplementary Figure 2D, in which gallocatechin*, lysoPC 18:1*, p-coumaroylquinic acid-4’-O-glucuronide, nortrachelogenin 4-O-β-D-glucoside, Procyanidin C2*, catechin-catechin-catechin, and wogonin were the most differentially accumulated compounds between EC and NEC.
The DAMs obtained in this study contained a larger proportion of secondary metabolites than primary metabolites (Table 1). For primary metabolites including amino acids and derivatives, nucleotides and derivatives, lipids, and organic acids which accounted for relatively large proportions of all DAMs (175, 39.68%), 99 DAMs were downregulated while 76 were upregulated in NEC. For secondary metabolites including flavonoids, phenolic acids, lignans and coumarins, and alkaloids, 137 DAMs were upregulated while 63 were downregulated in NEC. There were 90 flavonoids and 82 phenolic acids differentially accumulated between EC and NEC, accounting for 20.41% and 18.59% of all the DAMs, respectively (39% in total). The 90 flavonoids were composed of 2 chalcones, 14 flavanols, 12 flavanones, 6 flavanonols, 24 flavones, 1 flavonoid carbonoside, and 31 flavonols, in which 70 DAMs were upregulated while 20 were downregulated in NEC. For phenolic acids, 55 DAMs were upregulated while 27 were downregulated in NEC (Supplementary Table 4).
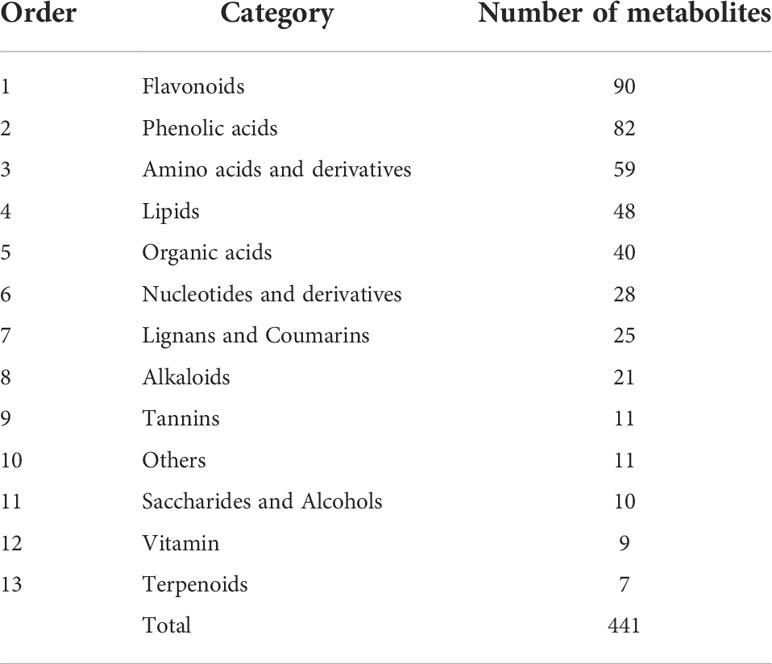
Table 1 Categories of the 441 DAMs between EC and NEC of L. kaempferi.
The results of KEGG enrichment of the DAMs showed that DAMs were mainly mapped onto “flavonoids biosynthesis”, “flavone and flavonol biosynthesis”, “arginine and proline metabolism”, “phenylalanine metabolism” and “phenylpropanoid biosynthesis” (Supplementary Figure 3).
Overview of RNA-seq and functional annotation
Six cDNA libraries from the EC and NEC of L. kaempferi were constructed and sequenced. A total of 297,123,278 raw reads and 276,606,454 clean reads were obtained. The average Q20 and Q30 values were 97.62 and 93.27%, respectively. The average of GC content was 44.76% (Table 2). Additionally, the overall mapped ratio exceeded 90% and the unique mapped ratio ranged from 90.36 to 91.88%. The overall expression patterns of all the samples demonstrated that the distribution of gene expression in all samples was similar, without extremely high/low expression in any sample (Supplementary Figure 4A). Moreover, the correlation between the samples showed the high repeatability of the three replicates within groups and the significant differences between EC and NEC (Supplementary Figure 4B). In general, the transcriptome sequencing quality and depth of the samples could well meet the necessary standard for the following analysis.
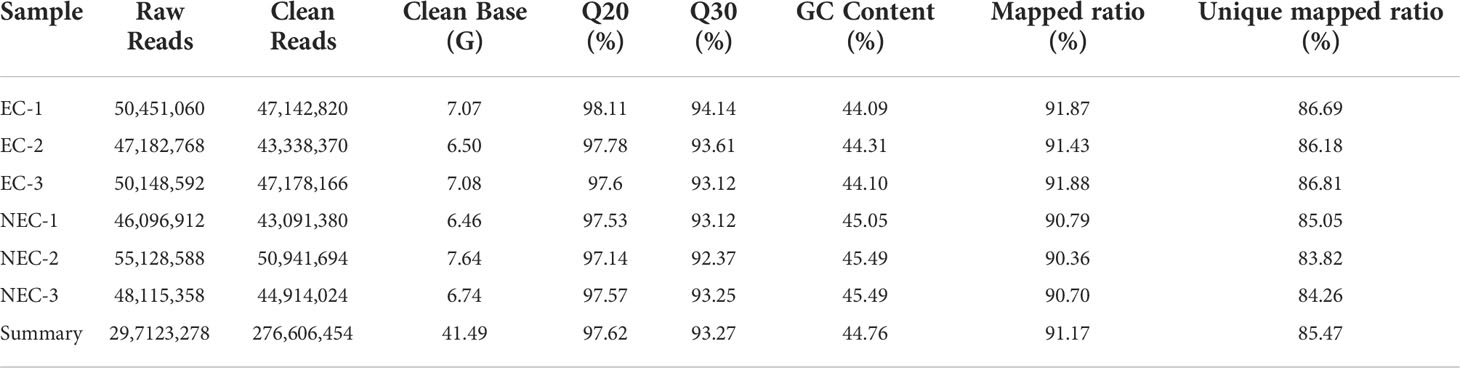
Table 2 Overview of transcriptome sequencing of complementary DNA from L. kaempferi callus.
DEGs identification and enrichment analysis
The FPKM method was applied to calculate the gene expression and DEGs were screened with adjusted P values < 0.05. A total of 13,842 significant DEGs were identified between EC and NEC (Supplementary Table 5). Among them, 5,798 genes were downregulated while 8,044 genes were upregulated in NEC compared to EC (Figure 3A). The expression patterns of EC and NEC libraries were compared, revealing that the colors of EC-1, EC-2 and EC-3 were similar and that these libraries were classified into the same cluster (Figure 3B).
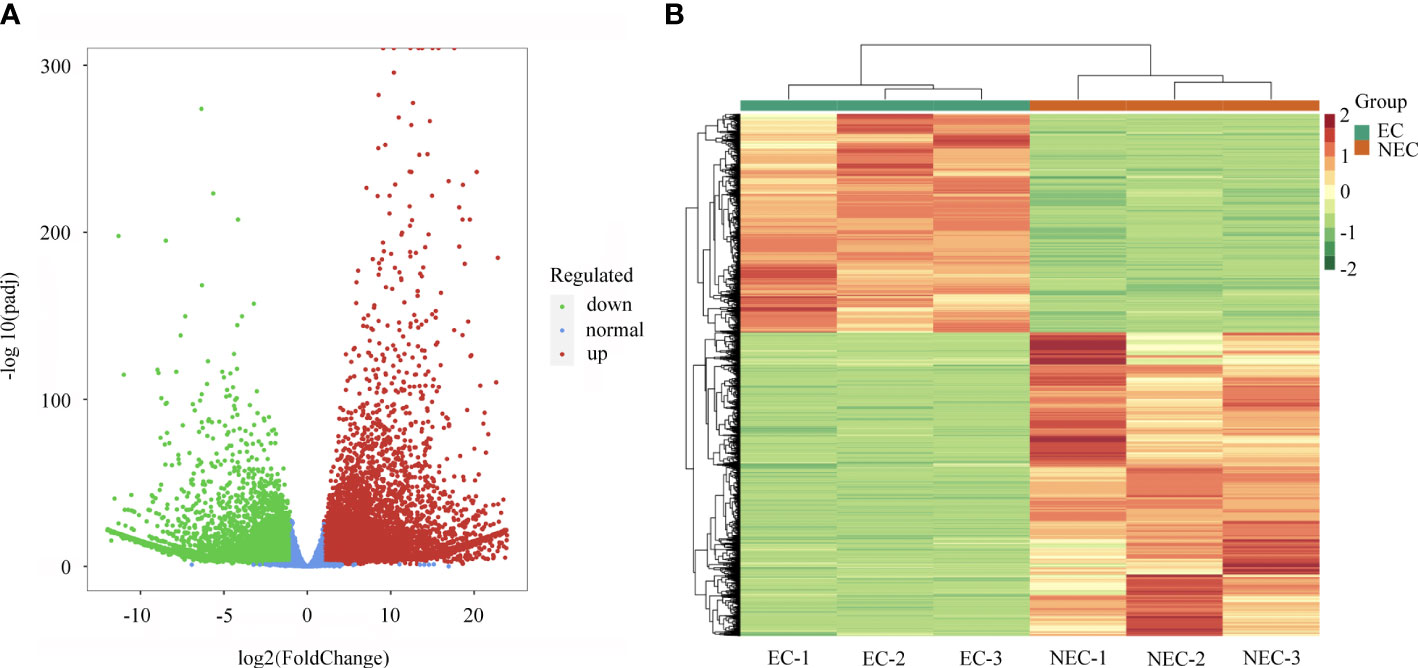
Figure 3 Analysis of differentially expressed genes (DEGs) between EC and NEC. (A) Volcano plot of DEGs between EC and NEC libraries, red and green dots represent the significantly upregulated and downregulated genes, respectively. (B) Heatmap of DEGs between EC and NEC libraries based on hierarchical clustering analysis.
To better understand the biological functions of DEGs, we functionally categorized the DEGs using GO and KEGG enrichment analysis. For GO analysis, the subcategory with the highest enrichment degree was “cellular anatomical entity” (7,576 genes), followed by “cellular process” (5,780 genes). “Cellular process” and “metabolic process” (4,791 genes) were the two subcategories with the highest enrichment degree in the biological process category. In the molecular function category, the “binding” (5,611 genes) and “catalytic activity” (5,468 genes) subcategories represented the two largest groups with the highest enrichment degrees (Supplementary Figure 5A). Similar to the functional categories, three GO-directed acyclic graphs were constructed. The number of significantly enriched GO terms obtained in the graphs of the biological process, cellular component, and molecular function category were 5, 8, and 6, respectively (Supplementary Figure 5B).
For KEGG analysis, the significant DEGs between EC and NEC libraries were assigned into five different KEGG terms in the order from top to down: metabolism, genetic information processing, environmental information processing, cellular processes, and organismal systems. A total of 4,598 DEGs were assigned into 134 KEGG pathways, in which the most DEGs were involved in “metabolic pathways” (1252 genes) and “biosynthesis of secondary metabolites” (1995 genes) (Supplementary Table 6; Supplementary Figure 6). The top 30 enriched KEGG pathways (P< 0.05) showed that the most highly enriched KEGG pathway was mainly related to the subcategory “plant hormone signal transduction” (336 genes), “amino sugar and nucleotide sugar metabolism” (145 genes), “Starch and sucrose metabolism” (327 genes), “MAPK signaling pathway” (340 genes), “phenylpropanoid biosynthesis” (196 genes), “pentose and glucuronate interconversions” (111genes), and “flavonoid biosynthesis” (111 genes) (Supplementary Figure 7). Additionally, “biosynthesis of amino acids” pathway was also representative in our results and 142 DEGs were enriched in it.
Analysis of differential gene expression in the phenylpropanoid and flavonoid biosynthesis pathway of L. kaempferi callus
To investigate the effects of differential gene expression on metabolic composition and content, we analyzed the expression profile of genes involved in the phenylpropanoid and flavonoid biosynthesis pathways in EC and NEC. According to the results of KEGG enrichment analysis of DEGs, there were 196 genes enriched in phenylpropanoid biosynthesis pathway, in which 107 genes were upregulated while 89 were downregulated in NEC. The 111 genes enriched in flavonoid biosynthesis pathway included 97 upregulated and 14 downregulated genes (Supplementary Table 7). Additionally, there were 16 co-regulated genes in the two pathways, among which ten genes were upregulated while six were downregulated. Overall, considerably more genes w ere upregulated in NEC than EC, which is consistent with the outcome of the secondary metabolome analysis (Supplementary Table 4).
The 16 co-regulated genes involved in both phenylpropanoid and flavonoid biosynthesis pathway were composed of three C4H genes (upregulated), six CcoAOMT genes (three upregulated and three downregulated), and seven HCT genes (four upregulated and three downregulated). Besides these genes, the remaining 97 upregulated genes in phenylpropanoid biosynthesis pathway included seven PAL genes, three 4CL genes, three COMT genes, four CAD genes, and five CCR genes. The 83 downregulated genes in phenylpropanoid biosynthesis pathway included one PAL gene, four 4CL genes, three COMT genes, nine CAD genes, and seven CCR genes. For flavonoid biosynthesis pathway, the remaining 87 upregulated genes included 22 CHS genes, four CHI genes, five F3H genes, five F3’H genes, seven F3’5’H genes, 12 DFR genes, six FLS genes, eight ANS genes, 14 ANR genes, and five LAR genes. The eight downregulated genes included one CHI gene, one F3H gene, two DFR genes, two FLS genes, and one LAR gene. Based on the differential gene expression profile, we reconstructed the phenylpropanoid and flavonoid biosynthesis pathways in L. kaempferi callus (Figure 4).
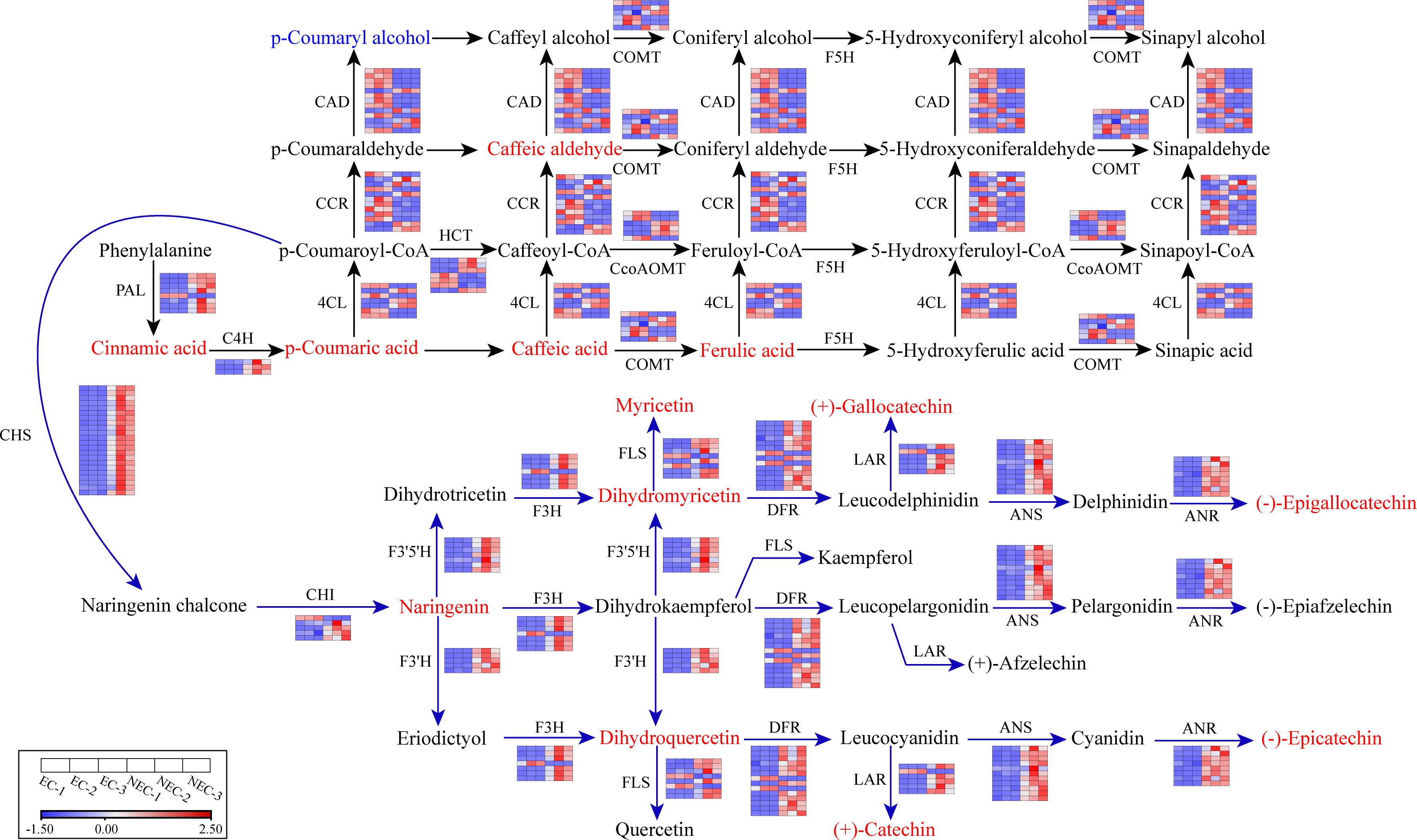
Figure 4 Part of phenylpropanoid and flavonoid metabolic subnetwork with genes and metabolites that constitute the process. Enzyme names and gene expression levels based on FPKM values are indicated at the side of each step. Red and blue blocks represent upregulation and downregulation of gene expression, respectively. Upregulated and downregulated metabolites were indicated in red and blue, respectively. Enzyme abbreviations: PAL, Phenylalanine ammonia-lyase; C4H, Cinnamate 4-hydroxylase; 4CL, 4-Coumarate-CoA ligase; CCR, Cinnamoyl-CoA reductase; HCT: Shikimate O-hydroxycinnamoyltransferase; CCoAOMT: Caffeoyl-CoA O-methyltransferase; COMT: Caffeic acid 3-O-methyltransferase; CHS, Chalcone synthase; CHI, Chalcone isomerase; F3H, Flavonoid 3-hydroxylase; F3′H, Flavonoid 3′ -hydroxylase; F3′5′H, Flavonoid 3’,5’-hydroxylase; FLS, Flavonol synthase; DFR, Dihydroflavonol 4-reductase; LAR, Leucoanthocyanidin reductase; ANS, Anthocyanidin synthase; ANR, Anthocyanidin reductase.
qRT-PCR validation of differential expression
To verify the accuracy of the transcriptome data, the expression levels of ten candidate genes related to phenolic acid and flavonoid biosynthesis were determined using qRT-PCR. The results showed that the expression levels of nine out of the ten candidate genes differed between EC and NEC, and showed expression patterns similar to those of the transcriptome data (Figure 5). Therefore, our RNA-seq and qRT-PCR analysis results showed high reliability and can be used for further analysis.
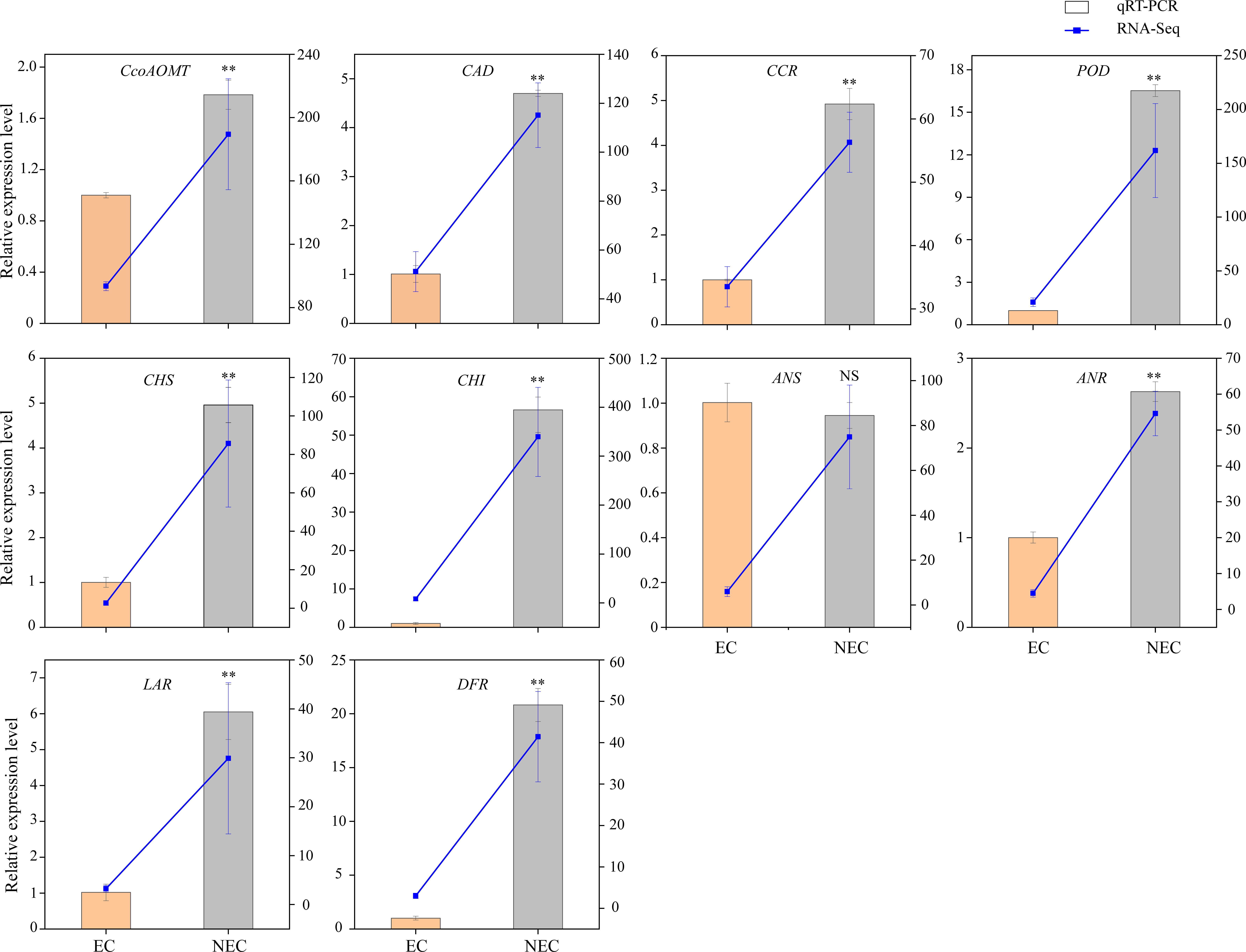
Figure 5 Comparison of expression profiles of ten selected genes in L. keampferi calluses as measured by quantitative real-time polymerase chain reaction (qRT-PCR) and RNA-seq. Columns represent gene expression determined by qRT-PCR (left y-axis), while lines represent gene expression determined by RNA-seq in FPKM values (right y-axis). The error bars represent the SD from three biological replicates. *, statistical significance at P < 0.05; **, statistical significance at P < 0.01; NS, not significant.
Correlation analysis between transcriptome and metabolome data
Firstly, we used KEGG enrichment analysis to screen out DAMs and DEGs that could be enriched in the same pathway (Figure 6; Supplementary Table 8). The results showed that the most DEGs and DAMs were enriched in “metabolic pathways” (1995 genes and 111 metabolites) and “biosynthesis of secondary metabolites” (1252 genes and 63 metabolites). Additionally, there are four KEGG pathways enriched with more DEGs and DAMs than other pathways– “biosynthesis of cofactors” (168 DEGs and 24 DAMs), “ABC transporters” (107 DEGs and 18 DAMs), “phenylpropanoid biosynthesis” (196 DEGs and 12 DAMs), and “flavonoid biosynthesis” (111 DEGs and 16 DAMs).
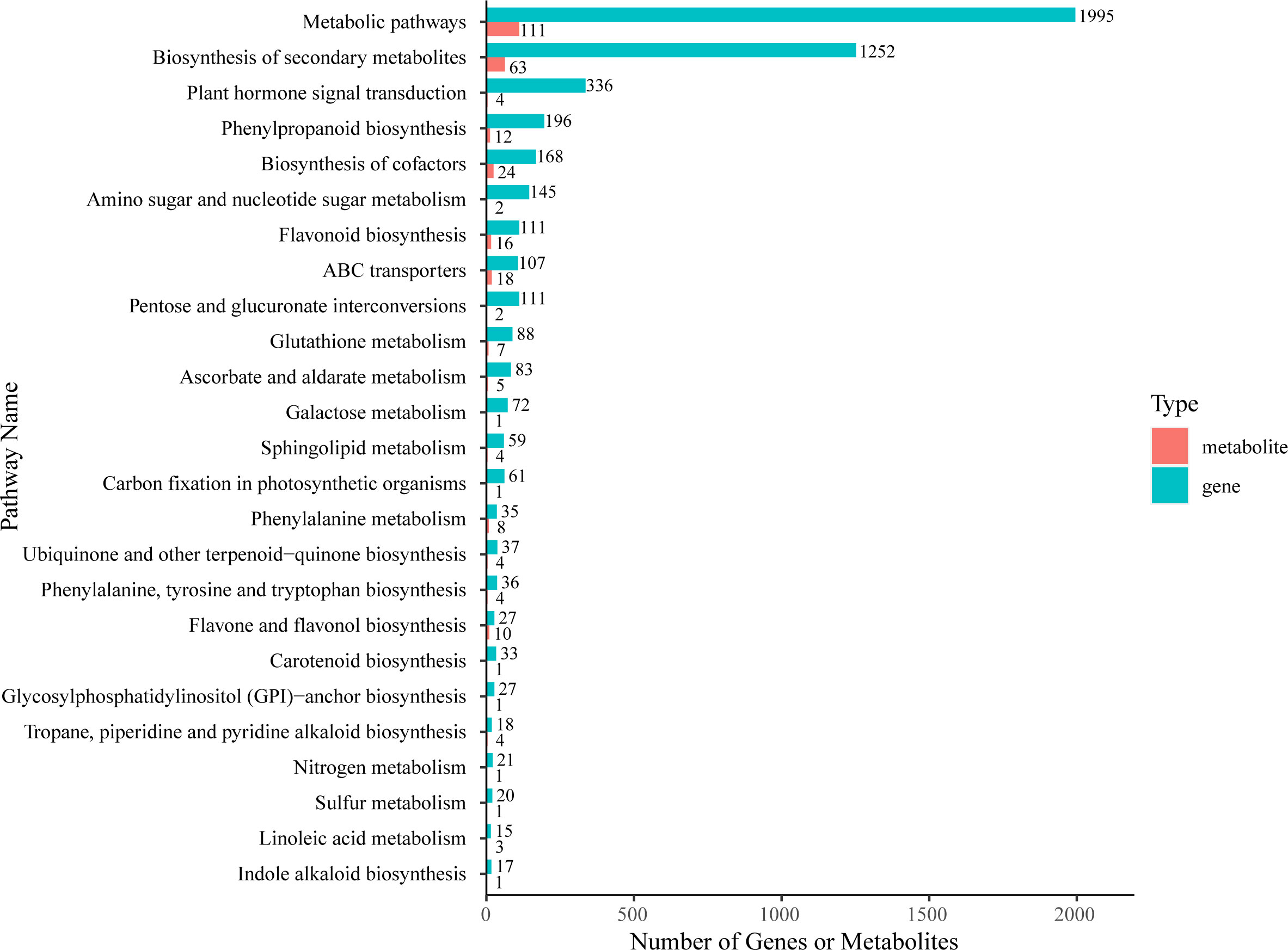
Figure 6 KEGG enrichment analysis of DEGs (green column) and DAMs (red column) that were enriched in the same pathway.
Pearson correlation analysis was performed to better understand the correlation relationship between DEGs and DAMs which were enriched in the same metabolic pathway. We selected 62 DEGs and 109 DEGs on the major branches in flavonoid biosynthesis and phenylpropanoid biosynthesis pathway, respectively, for this analysis.
The screening criteria were a P-value < 0.01 and a PCC-value > 0.9. In phenylpropanoid biosynthesis pathway, 53 DEGs were significantly correlated with 12 DAMs, resulting in 326 related pairs (159 positive and 167 negative pairs) (Supplementary Table 9). In flavonoid biosynthesis pathway, 94 DEGs were correlated with 16 DAMs and a total of 1137 related pairs (912 positive and 225 negative pairs) were found. Among them, it is ubiquitous that a single gene was regulated by multiple metabolites, or a single metabolite was regulated by multiple genes. For example, in phenylpropanoid biosynthesis pathway, the content of mws2213 (cinnamic acid) and pme1439 (p-Coumaric acid) was significantly correlated with the expression levels of 38 DEGs (18 positively correlated and 20 negatively correlated) and 30 DEGs (13 positively correlated and 17 negatively correlated), respectively. The expression level of the gene Lk17760 annotated as caffeoyl-CoA O-methyltransferase (CcoAOMT) was significantly positively correlated with the accumulation of HJN003 (1-O-sinapoyl-D-glucose), mws0906 (coniferin), mws2213 (Cinnamic acid), pmb0751 (trans-5-O-(p-Coumaroyl) shikimate), pme0021 (L-Phenylalanine), and pme2993 (scopoletin(7-Hydroxy-5-methoxycoumarin). Similarly, in flavonoid biosynthesis pathway, the accumulation of metabolites such as mws0044 (dihydroquercetin) and mws0054 (catechin*) were significantly correlated with the expression levels of 86 DEGs (76 positively correlated and 20 negatively correlated) and 78 DEGs (69 positively correlated and 9 negatively correlated), respectively. The expression level of Lk17613, which was annotated as naringenin 3-dioxygenase (F3H), showed significant positive correlation relationship with the accumulation with mws0032 (myricetin), mws0044 (dihydroquercetin), mws0049 (gallocatechin*), mws0054 (catechin*), mws0744 (dihydromyricetin), mws0914 (pinobanksin), mws1173 (garbanzol), and mws2118 (phlorizin) while it was negatively correlated with MWSHY0089 (sakuranetin) and pmb0751 (trans-5-O-(p-Coumaroyl) shikimate) (Supplementary Table 10).
Additionally, we found that the expression levels of the 14 co-regulated DEGs involved in both phenylpropanoid and flavonoid biosynthesis pathways were all significantly correlated with the accumulation of the DAMs enriched in the two pathways. For example, Lk17760 (annotated as caffeoyl-CoA O-methyltransferase) were significantly correlated with six metabolites (six positively correlated) in phenylpropanoid biosynthesis pathway and nine metabolites in flavonoid biosynthesis pathway (eight positively correlated and one negatively correlated). Therefore, these genes may play an important role in the accumulation of metabolites in the two pathways (Table 3).
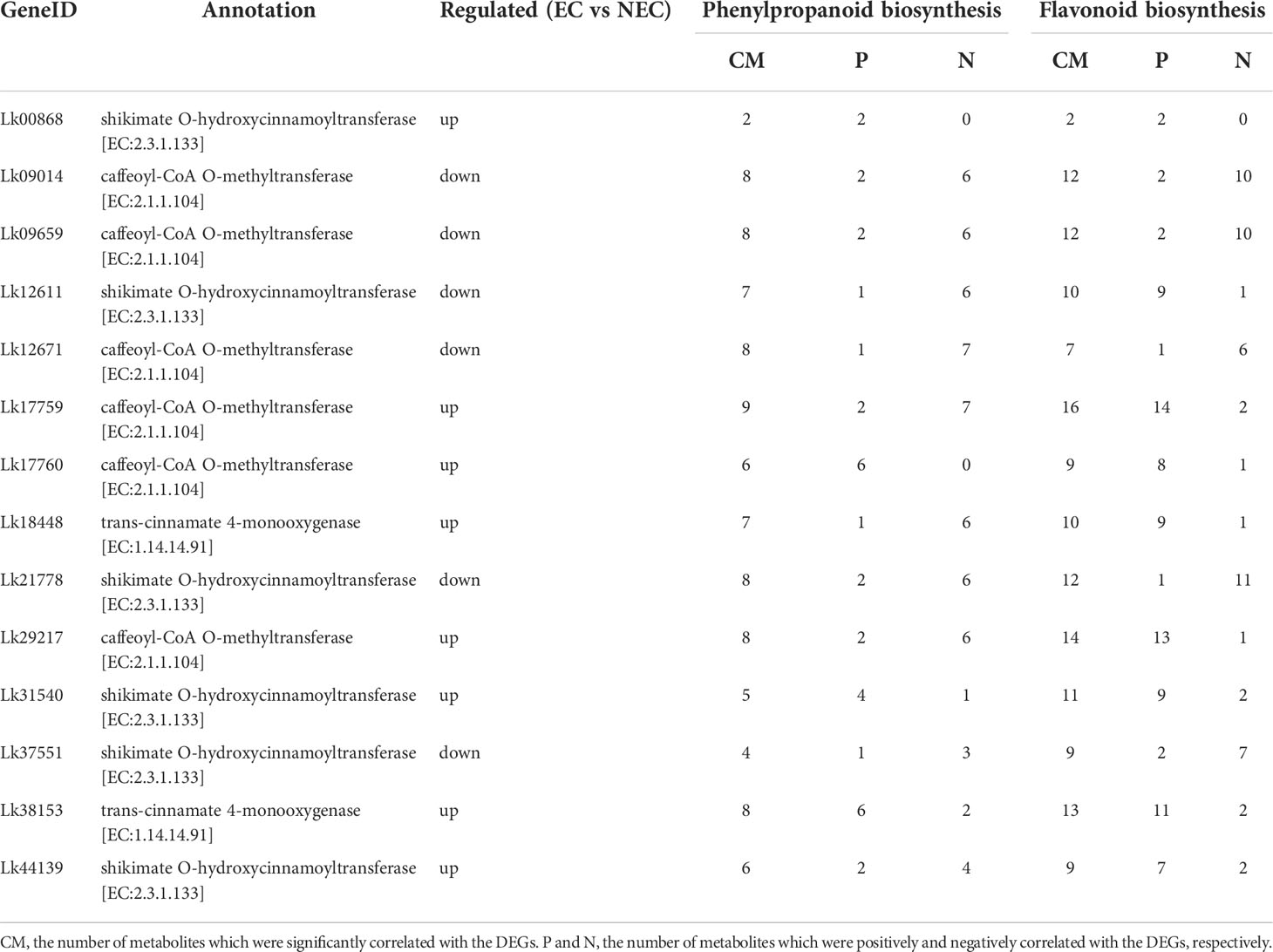
Table 3 Co-regulated DEGs and their relationship with DAMs in phenylpropanoid and flavonoid biosynthesis pathway.
Differential expression analysis of genes related to plant hormone signaling pathway
Based on the results of KEGG enrichment analysis of DEGs and DAMs above, we found that many DEGs and DAMs were enriched in “plant hormone biosynthesis” and “plant hormone signal transduction” pathways. 336 DEGs and four DAMs were enriched in “plant hormone signal transduction” pathway. In plant hormone biosynthesis, a total of 212 DEGs and 18 DAMs were involved in jasmonic acid (JA) (62 DEGs and three DAMs), ethylene (ETH) (59 DEGs and four DAMs), cytokinine (CTK) (38 DEGs and three DAMs), and IAA (53 DEGs and eight DAMs) biosynthesis.
In the JA pathway, the content of JA in NEC was found to be higher than that in EC (Supplementary Table 4). There were 41 genes related to JA signal transduction, in which 27 genes were upregulated while 14 were downregulated. The 27 upregulated genes in NEC included three genes encoding jasmonic acid-amido synthetase (JAR), six genes encoding jasmonate ZIM domain-containing protein (JAZ), three genes encoding coronatine-insensitive protein 1 (COI1), and 15 genes encoding transcription factor MYC2 (MYC2). The 14 downregulated genes contained one JAR gene, ten JAZ genes, one COI1 gene, and two MYC2 genes (Figure 7).
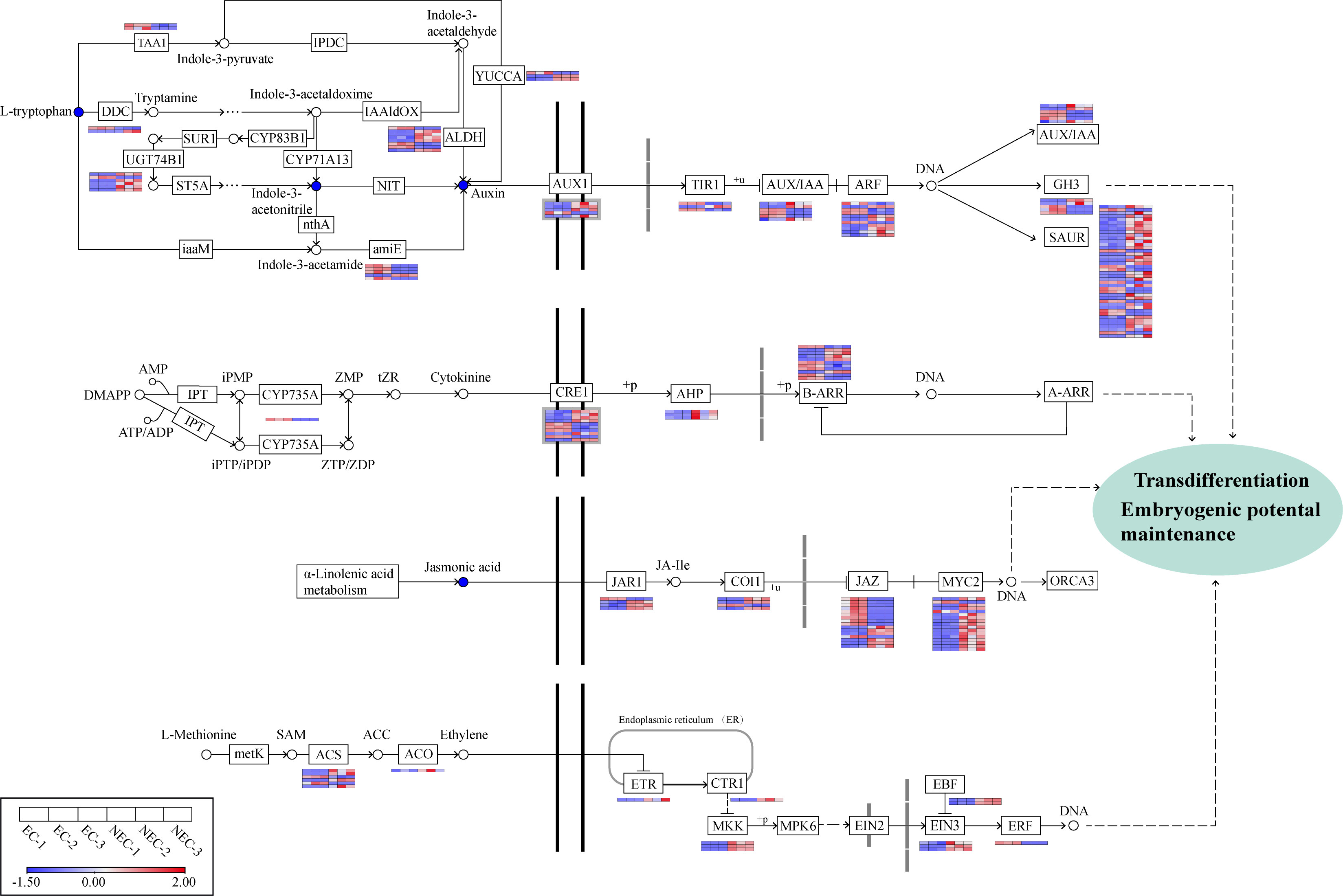
Figure 7 Analysis of DEGs involved in plant hormone synthesis and signal transduction pathways. Gene expression levels based on FPKM values are indicated at the side of each step. Red and blue blocks represent upregulation and downregulation of gene expression, respectively. Enzyme abbreviations involved in plant hormone synthesis: TAA1, L-tryptophan—pyruvate aminotransferase; DDC, L-tryptophan decarboxylase; UGT74B1, N-hydroxythioamide S-beta-glucosyltransferase; ALDH, aldehyde dehydrogenase; amiE, amidase; YUCCA, indole-3-pyruvate monooxygenase; CYP735A, cytokinin trans-hydroxylase; ACS, 1-aminocyclopropane-1-carboxylate synthase; ACO, aminocyclopropanecarboxylate oxidase.
L-Methionine to S-Adenosyl-L-methionine (SAM), SAM to 1-aminocyclopropane-1-carboxylic acid (ACC), and ACC to ETH are three key conversions in the ETH biosynthesis pathway (Zhang et al., 2022). In this study, four genes encoding 1-aminocyclopropane-1-carboxylate synthase (ACS) and one gene encoding aminocyclopropanecarboxylate oxidase (ACO) involved in the latter two catalytic processes were found to be upregulated in NEC while only two ACS genes were downregulated in NEC. 11 genes were enriched in the ETH signal transduction pathway, among which one gene encoding ethylene receptor (ETR), one gene encoding serine/threonine-protein kinase CTR1 (CTR1), three genes encoding mitogen-activated protein kinase kinase 4/5 (MKK4/5), three genes encoding ethylene-insensitive protein 3 (EIN3), and two genes encoding EIN3-binding F-box protein (EBF) were upregulated in NEC, whereas only one gene encoding ethylene-responsive transcription factor 1 (ERF1) was downregulated in NEC.
In the CTK biosynthesis pathway, one gene encoding the key enzyme cytokinin trans-hydroxylase (CYP735A) in CTK biosynthesis showed a higher expression in EC than NEC. Some genes related to CTK signal transduction such as six genes encoding cytokinin receptor 1 (CRE1), three genes encoding histidine-containing phosphotransfer protein (AHP), and five ARR-B gene family members were upregulated in NEC, whereas four CRE1 genes and six ARR-B genes were downregulated in NEC.
In our results, the content of three key metabolites in IAA biosynthesis pathway―L-tryptophan (mws0282), indole-3-acetonitrile (pmb0819), and indole 3-acetic acid (IAA) (pme1651) in EC was higher than that in NEC. Likewise, some genes involved in IAA biosynthesis and signal transduction pathways were differentially expressed between EC and NEC. Among the 53 genes related to IAA biosynthesis, three genes encoding aldehyde dehydrogenase (ALDH), four genes encoding amidase (amiE), one gene encoding aromatic-L-amino-acid/L-tryptophan decarboxylase (DDC), one gene encoding indole-3-pyruvate monooxygenase (YUC), one gene encoding N-hydroxythioamide S-beta-glucosyltransferase (UGT74B1), two genes encoding L-tryptophan—pyruvate aminotransferase (TAA1) were downregulated in NEC, whereas five ALDH genes, one amiE gene, one DDC gene, two YUC genes, and five UGT74B1 genes were upregulated in NEC. We screened out 72 DEGs associated with IAA signal transduction, including 48 upregulated and 24 downregulated genes in NEC, and the genes encoding SAUR family protein (SAUR) accounted for the largest proportion (42 genes/58.3%) of all these genes, among which 31 SAUR genes were upregulated while 11 were downregulated in NEC. Besides, the expression of one gene encoding auxin influx carrier (AUX1), two genes encoding transport inhibitor response 1 (TIR1), three genes encoding auxin-responsive protein IAA (AUX/IAA), four genes encoding auxin response factor (ARF), three genes encoding auxin responsive GH3 gene family (GH3) was downregulated in NEC, while four AUX1 genes, one TIR1 gene, three AUX/IAA genes, seven ARF genes, and two GH3 genes were upregulated in NEC (Figure 7).
Analysis of genes encoding TFs
TFs are of great importance in regulating various development-related processes as well as secondary metabolite production in plants. In this study, 3,066 genes were recognized as TF genes and belonged to 90 TF families. Among all the TF genes, 787 genes were differentially expressed between EC and NEC and can be furtherly classified into 73 TF families (Supplementary Table 11), among which 405 TF genes were upregulated while 382 were downregulated in NEC compared to EC. The TF families with the most genes which were differentially expressed between the two groups were ERF, bHLH, MYB, NAC, and LOB, with 64, 58, 46, 37, and 32 genes included, respectively (237 in total). Among these five major TF families, the number of all TF genes upregulated in NEC were greater than that were downregulated. Specifically, 134 TF genes were upregulated while 103 were downregulated in NEC, in which 37 ERFs, 32 bHLHs, 29 MYBs, 18 NACs, and 18 LOBs were found to be upregulated while 27 ERFs, 26 bHLHs, 17 MYBs, 19 NACs and 14 LOBs were downregulated in NEC. Besides, totipotency related TF genes showed contrasting expression levels between EC and NEC. Three BBM genes, 17 WUS genes, and one LEC1 gene were upregulated while only two WUS genes were downregulated in NEC compared to EC (Figure 8).
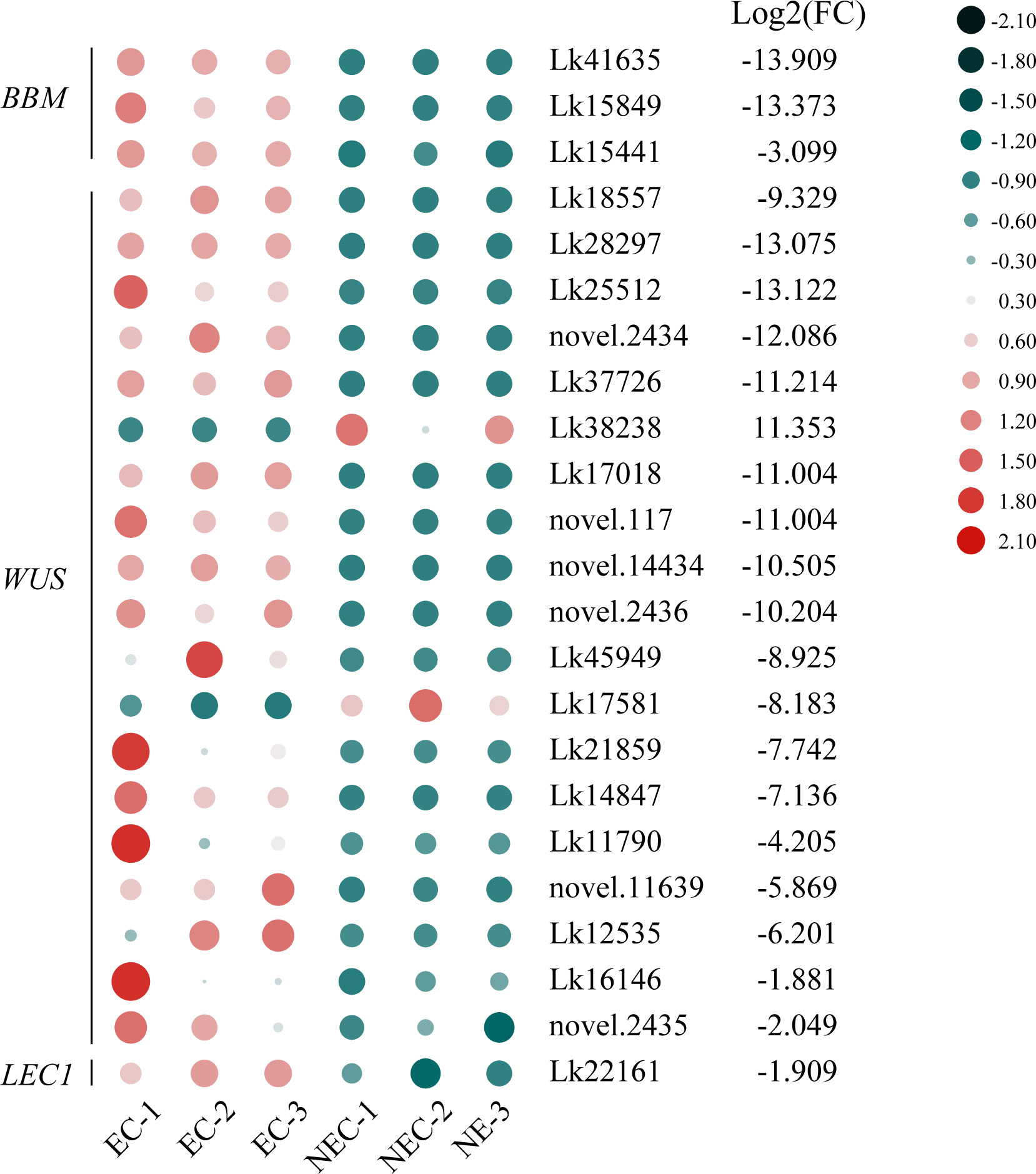
Figure 8 Expression patterns of totipotency-related transcription factor (TF) genes between EC and NEC in L. kaempferi. Gene expression levels were calculated based on FPKM values. Red and green dots represent upregulation and downregulation of gene expression, respectively.
Possible transcriptional regulatory network involved in phenolic acid and flavonoid biosynthesis
To more accurately identify the TFs that may play important roles in regulating the expression of functional genes involved in both phenolic acid and flavonoid biosynthesis, we used the PlantTFDB online database to predict the TFs that potentially regulated the co-regulated structural genes enriched in both phenylpropanoid and flavonoid biosynthesis pathways. The eight upregulated DEGs were selected for this analysis as the expression levels of these genes were more directly associated with the accumulation of the key metabolites (Table 3). Unfortunately, we did not find the TFs which may potentially regulate CcoAOMT (Lk29217), so we kept the remaining seven genes for further analysis. Based on the targeted relationship, a total of 34 differentially expressed TF genes between EC and NEC were identified as candidate regulatory genes (Table 4).
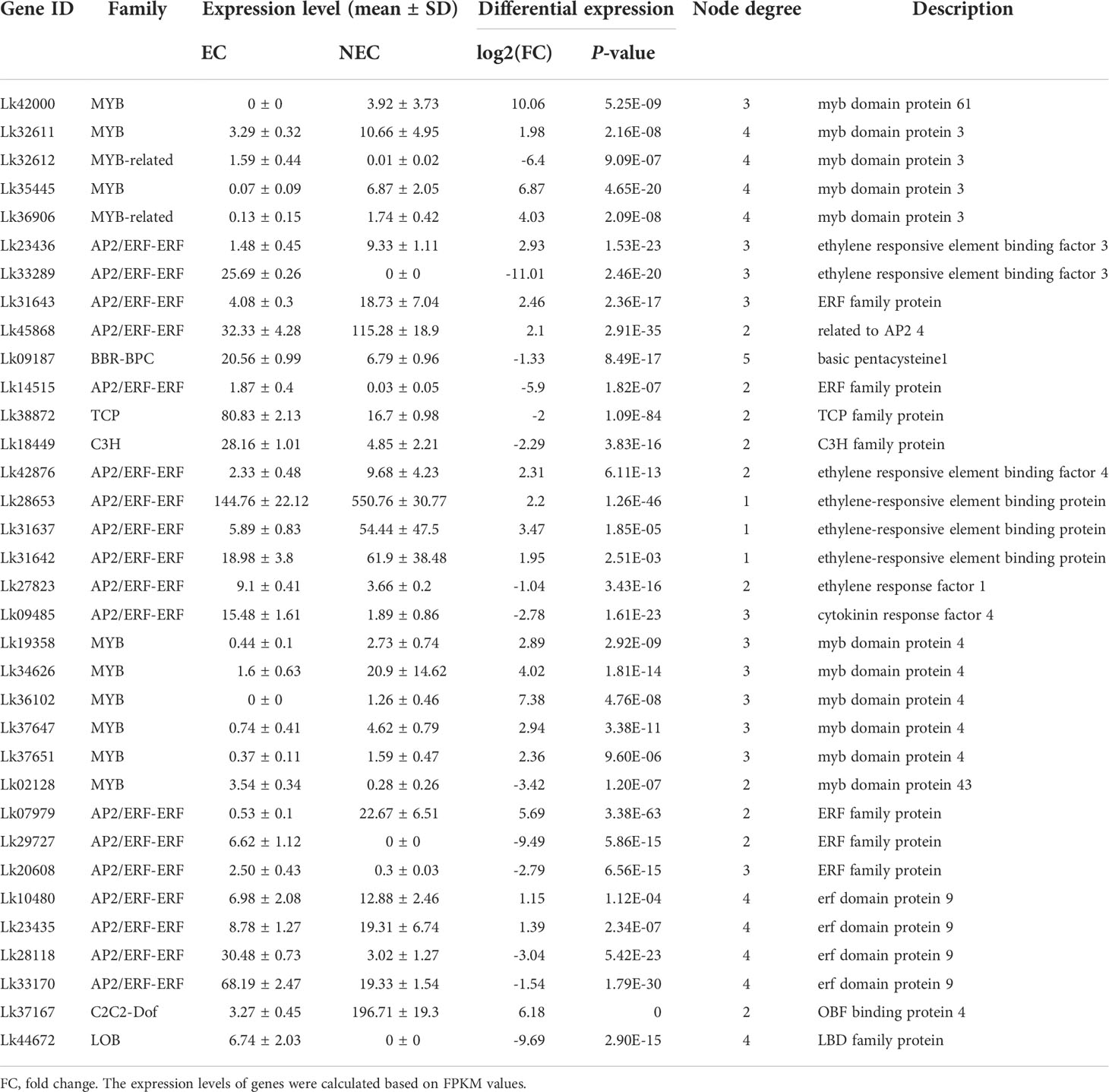
Table 4 The expression and annotation of candidate TFs regulating structural genes involved in flavonoid and phenolic acid biosynthesis.
The regulatory relationship between candidate TFs and the seven target genes was shown in Figure 9. The node degree of TFs was defined as the number of target genes one TF may regulate, which could represent the importance of TFs in the metabolic pathway (Xiao et al., 2021). The results showed that each TF had several target genes with a node degree ranging from 1 to 5 (Table 4; Figure 9). The node degree of BBR-BPC (Lk09187) was the highest among all the 34 TFs, with a value of five. Four ERFs including Lk10480, Lk23435, Lk28118, and Lk33170, LOB (Lk44672), two MYBs including Lk32611 and Lk35445, and two MYB-related TFs including Lk32612 and Lk36906 had the second highest node degree, with a value of four. The four ERFs all showed target relationship with one CcoAOMT gene (Lk17759), two C4H genes (Lk18448 and Lk38153), and one HCT gene (Lk44139). The two MYBs and two MYB-related TFs all showed target relationship with two CcoAOMT genes (Lk17759 and Lk17760), one C4H gene (Lk38153), and one HCT gene (Lk44139). Additionally, 11 TFs including five ERFs and six MYBs had a relatively high node degree, with a value of three. Thus, these TFs were considered as important TFs, which may be crucial in regulating the expression of key structural genes involved in the flavonoid and phenylpropanoid biosynthesis pathways and thus leading to the differential accumulation of flavonoids and phenolic acids between EC and NEC.
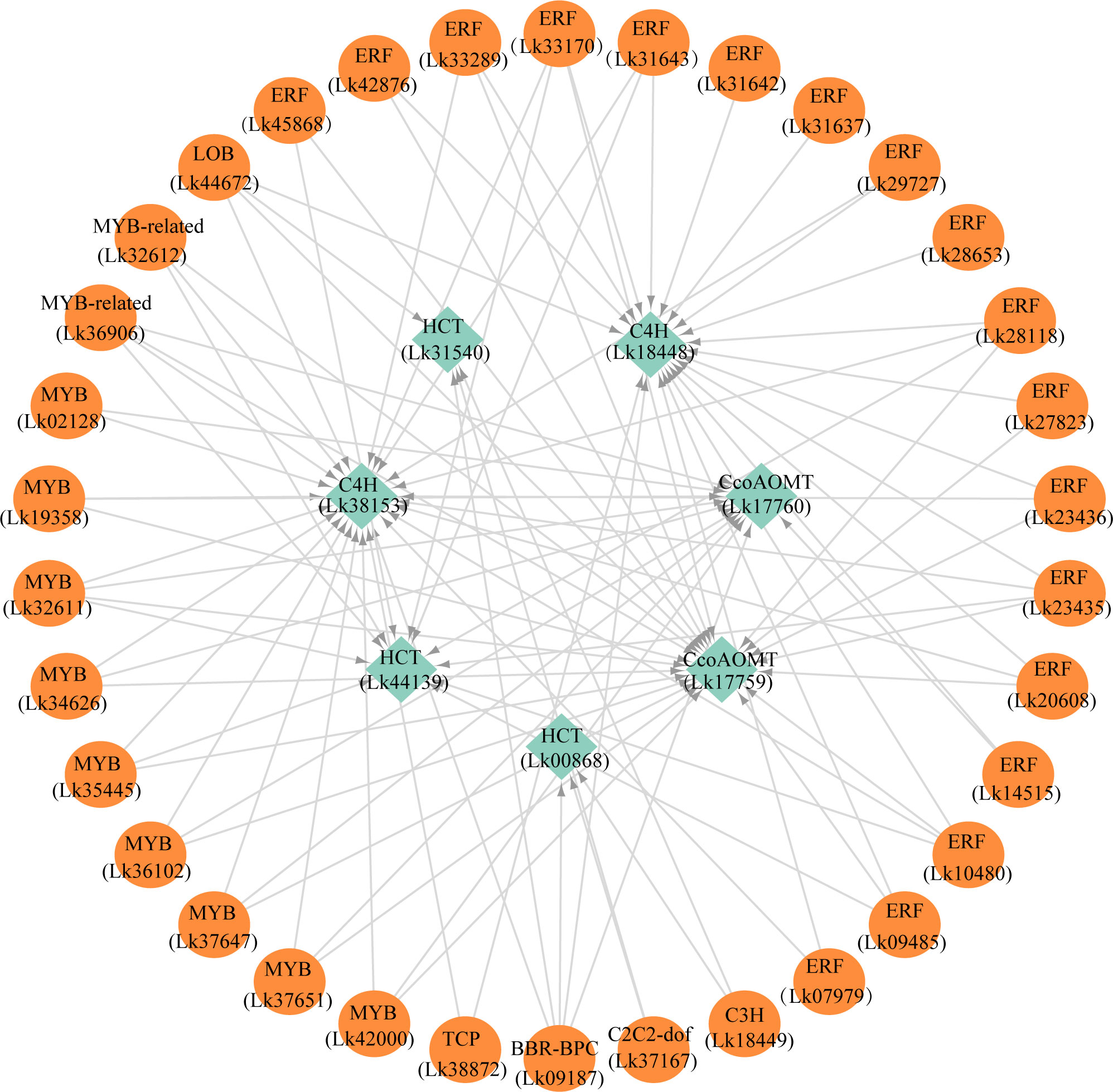
Figure 9 The potential regulatory network between TFs and key structural genes involved in phenylpropanoid and flavonoid biosynthesis pathways in L. kaempferi callus.
To better figure out the relationship between these candidate TF genes and the co-regulated structural genes, a Pearson correlation analysis was conducted based on the FPKM values of these genes (Figure 10). Although BBR-BPC and LOB had the highest node degrees among all TFs, their transcription levels all showed negative correlations with their target genes. For ERF members, the nine ERFs showed relatively high node degrees, but only Lk23436 was significantly positively correlated with all its target genes including two C4H genes (Lk18448 and Lk 38153) and one CcoAOMT gene (Lk17759). For MYB and MYB-related members, Lk34626 and Lk35445 were highly corelated with their target genes, whereas only Lk34626 showed high transcription level in NEC (FPKM= 20.90), which was approximately 13 times higher than that in EC. It’s worth mentioning that two candidate TFs (node degree= 2), C2C2-dof (Lk37167) and ERF (Lk45868), showed high transcription levels in NEC (FPKM= 196.71 and 115.28, respectively) and showed significantly positive correlations with their target genes. To sum up, we speculated that ERF (Lk23436 and Lk45868), MYB (Lk34626), and C2C2-dof (Lk37617) were the most important TFs in regulating phenolic acid and flavonoid biosynthesis.
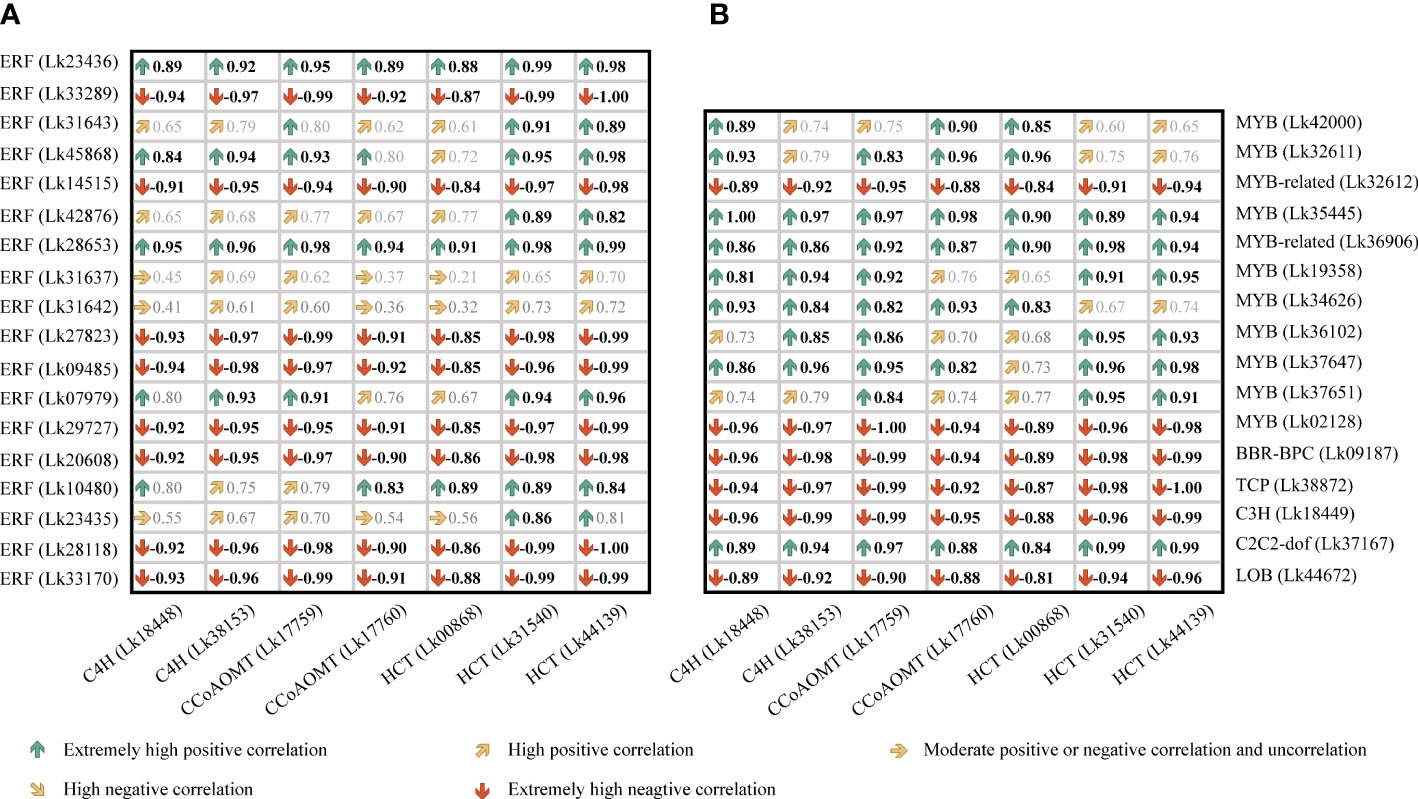
Figure 10 Correlation analysis of the expression levels between candidate TF genes and key structural genes involved in phenylpropanoid and flavonoid biosynthesis in L. kaempferi callus. (A) Correlation relationship between ERF genes and key structural genes. (B) Correlation relationship between other TF genes and key structural genes. Bold black indicates significant correlation at P< 0.05.
Regulation module governing the differential accumulation of phenolic acids and flavonoids between EC and NEC
According to the targeted relationship, the correlations between the TFs and the key structural genes based on the expression profile as well as the differential accumulation of metabolites in the phenylpropanoid and flavonoid biosynthesis pathways between groups, we preliminary proposed the transcriptional regulatory mechanisms underlying flavonoid and phenolic acid biosynthesis in L. kaempferi calluses (Figure 11). During SE induction, two types of callus including EC and NEC may be generated. They differed significantly in secondary metabolic activities and several TFs were involved in these metabolic processes. The transcription levels of ERF (Lk23436) and MYB (Lk34626) were significantly increased in NEC and they could bind C4H genes to activate their expression, contributing to the increase of p-Coumaric acid. The two substances including p-Coumaric acid and cinnamoyl-CoA were used as the substrates for further reactions. ERF (Lk45868) and C2C2-dof (Lk37617) bound HCT genes to promote the conversion of p-Coumaroyl-CoA to caffeoyl-CoA. Then, ERF (Lk23436) and MYB (Lk34626) furtherly enhanced the expression of CcoAOMT genes, leading to the conversion of caffeoyl-CoA to feruloyl-CoA. By integrating other structural genes such as 4CL, CCR, and CAD genes in phenylpropanoid biosynthesis pathway or CHS, F3H, and DFR genes in flavonoid biosynthesis pathway, the contents of phenolic acids and flavonoids were ultimately increased in NEC.
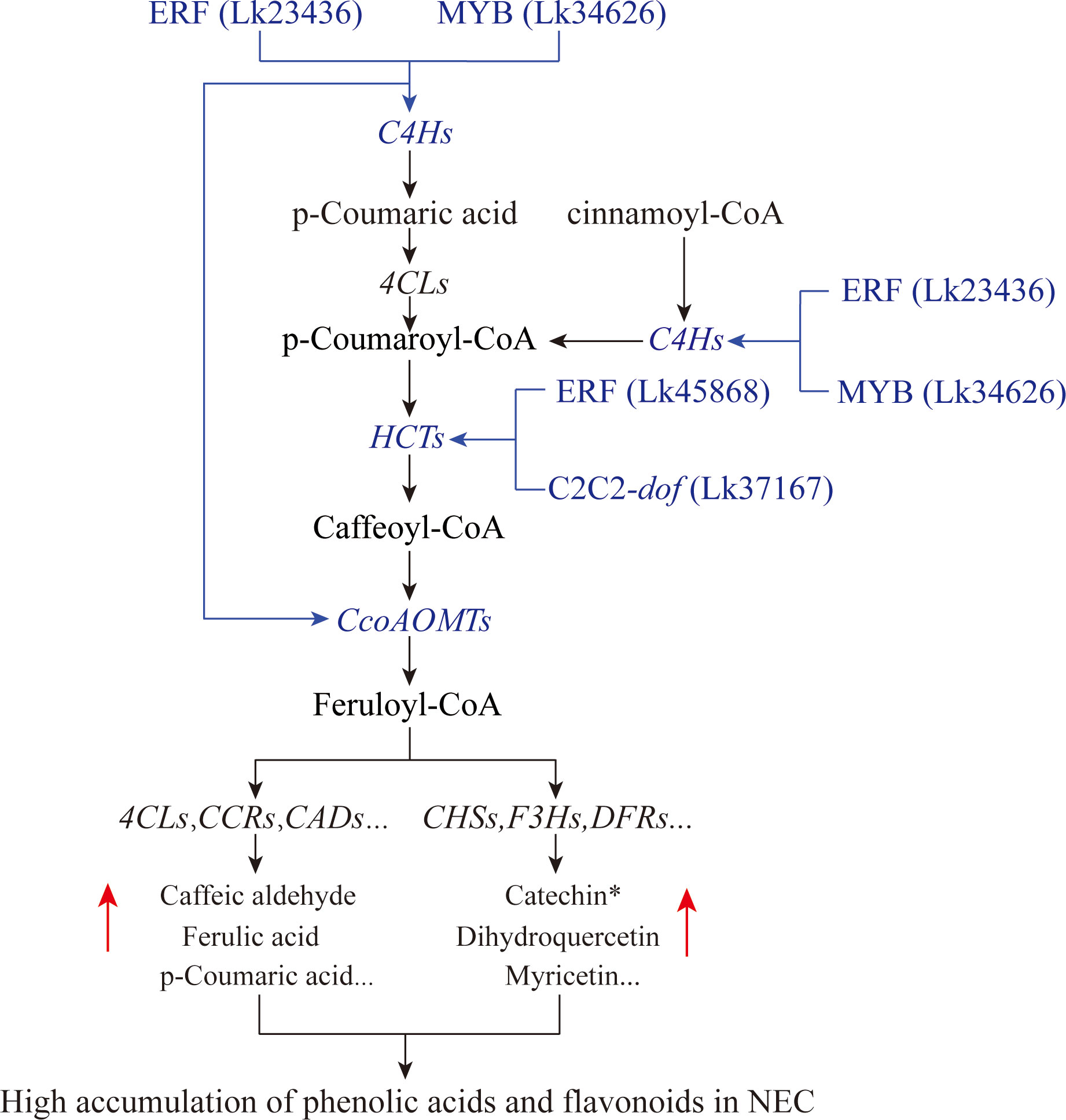
Figure 11 Potential transcriptional regulatory module governing the differential accumulation of flavonoids and phenolic acids between EC and NEC. Possible regulatory modules were highlighted in blue.
Discussion
As in many conifers, rooting of cuttings or seed propagation cannot satisfy the needs of mass reproduction of new lines with desired characteristics in L. kaempferi. SE is a flexible and efficient tool in providing enough uniform genetically improved material (Gautier et al., 2019), but the instability of EC induction has limited the application of SE in many plants. It has demonstrated that SE induction was a complex process regulated by a network of hundreds of genes (Mordhorst et al., 1997; Wang et al., 2020; Ávila et al., 2022). As metabolites are the end product of gene expression, we considered that many metabolic processes must be involved when EC and NEC were formed. However, metabolic changes between EC and NEC were rarely emphasized in conifers in previous studies. In this study, an integrated transcriptomic and metabolomic profiling was performed to identify genes/metabolic pathways that are differentially represented between groups and provides important insights into the molecular mechanisms underlying embryogenic potential maintenance and the differential accumulation of key metabolites between EC and NEC during SE induction in this species, thus leading to a better understanding of SE induction process that resulted in plant cell totipotency as well as the global association between gene expression and metabolite accumulation.
Essential role of totipotency-related TFs in EC
The obvious differences in color, texture, morphology or histology between EC and NEC of different species have been reported in several studies (Nagmani and Bonga, 1985; Maadon et al., 2016; Bravo et al., 2017; Gautier et al., 2019; Xue et al., 2022), but the most significant difference between EC and NEC is that EC cells have the potential to acquire totipotency, which can furtherly regenerate into a complete organism (Condic, 2014; Horstman et al., 2017; Li et al., 2022b). A number of plant TFs have been identified that can covert somatic cells into embryogenic, totipotent cells, in which BBM, WUS, and LEC1 have been highlighted in many studies (Su et al., 2009; Horstman et al., 2017; Su et al., 2021; Wang et al., 2022a). In this study, the expression levels of most of these TF genes were higher in EC than those in NEC (Figure 8), which may indicate the important roles of these TFs in cell dedifferentiation resulting in the totipotency fate determination of somatic plant cells during EC induction process in L. kaempferi. These results were consistent with the findings in Pinus radiata (Bravo et al., 2017). It’s thought that the initiation of SE requires an inductive signal that causes a somatic cell to change its fate and become totipotent (Su et al., 2021). In Arabidopsis, exogenous 2,4-D could evoke the increased transcript levels of totipotency-related TFs including BBM, WUS, LEC1, and LEC2 (Su et al., 2009; Wang et al., 2020). The changes of BBM and LEC2 may be directly induced by auxin response factor (ARF) TFs because of the presence of putative ARF-binding motifs in the promoter regions of BBM and LEC2 (Grzybkowska et al., 2020; Wang et al., 2020). In our results, several ARF genes also showed higher expression levels in EC (Figure 7). The expression of these genes may contribute to the high embryogenic potential of EC and endow part of EC cells with totipotency. Endogenous IAA levels are of primary importance for inducing SE (Fehér, 2015). During many types of SE, the inductive stimuli evoke endogenous auxin biosynthesis, leading to increased auxin levels and inducing totipotency in somatic cells (Braybrook and Harada, 2008). In Arabidopsis, BBM transcriptionally regulates LEC1 (Horstman et al., 2017) and YUC3 and YUC8 (Li et al., 2022b) and LEC1 transcriptionally regulates YUC10 (Junker et al., 2012). All these relationships act directly on endogenous IAA biosynthesis as the YUCs function as flavin-containing monooxygenases to catalyze indole-3-pyruvate acid (IPyA) to form IAA (Stepanova et al., 2011). Besides, enzymatic activity of tryptophan pyruvate aminotransferase (TAA1) converts tryptophan into the intermediate product indole-3-pyruvic acid (IPyA), which is also a key rate-limiting step of IAA biosynthesis. In this study, IAA was found to be highly accumulated in EC (Supplementary Table 3), which may be attributed to the higher expression level of one YUC gene (Lk07754) and one TAA1 gene (Lk22753) in EC. IAA participation in maintenance of multiplying cells has been demonstrated in SE of rubber tree (Hevea brasiliensis), and the higher accumulation of IAA in EC compared to NEC have been reported in Douglas-fir (Pseudotsuga menziesii) and Tamarillo (Solanum betaceum) (Gautier et al., 2019; Caeiro et al., 2022). Additionally, it has been reported that the increase in endogenous auxin biosynthesis play a role in determining developmental cell fate and in activating the proliferative activity of proembryo cells (Pérez-Pérez et al., 2019). These results may indicate that the maintenance of embryogenic potentials of EC is largely dependent on the high production of endogenous IAA. Consistent with the higher content of IAA in EC, we found that L-tryptophan, the precursor of IAA, also showed a higher accumulation in EC (Tables S3). To sum up, all these results may suggest a mechanism in the transdifferentiation during EC induction and the maintenance of embryogenic potentials of EC in L. kaempferi: on 2,4- D treated culture medium, the expression of cell-totipotency-related TF genes, BBM, were induced, probably by ARFs. BBM furtherly activated the expression of LEC1. The two TFs can in turn promote IAA biosynthesis by inducing YUCs expression, thereby forming a feed-forward loop to reinforce cell-fate maintenance and transition (Wang et al., 2022a). Additionally, WUS genes maintained high expression in EC, which may play a critical role in inducing somatic embryo on PGRs free medium.
Plant hormone signal transduction
In EC, the expression levels of many auxin-related genes were highly expressed in EC (Figure 7), indicating the important role of these auxin-responsive genes in L. kaempferi EC induction and embryogenic potential maintenance. We found that more auxin-related genes including AUX1s, AUX/IAAs, ARFs, GH3s, and SAURs showed higher expression levels in NEC than in EC, but the upregulation of these genes may be induced by the 2,4-D in the culture medium (Ji et al., 2015). In auxin-related responses, these TFs that control the target gene expression in response to auxin were indicated as playing a central role (Teale et al., 2006). In Arabidopsis, an expression analysis revealed that six ARFs were significantly upregulated, whereas five other ARF genes were downregulated in SE-induced explants (Wojcikowska and Gaj, 2017). Evaluation of SE efficiency from the ARF mutants further suggests that multiple ARFs redundantly contribute to SE in Arabidopsis (Wang et al., 2022a). Additionally, one possibility is that an unknown Aux/IAA-ARF complex binds to totipotency-related TF gene loci in somatic cells and the subsequent recruitment of the TOPLESS-histone deacetylase complex prevents totipotent gene expression (Wu et al., 2015). Therefore, the differential expression of totipotency-related genes and the differential accumulation of IAA between EC and NEC may be due to different ARF genes that are expressed in EC and NEC, which may be a possible reason leading to the differences in embryogenic potential between EC and NEC cells. However, the precise molecular mechanisms require further study.
Cytokinin plays key roles in the initiation and further development of embryogenic cultures (Cabrera-Ponce et al., 2018; Gautier et al., 2019). In this study, we observed that many genes related to zeatin biosynthesis and cytokinin signal transduction were differentially expressed between EC and NEC. In cucumber (Cucumis sativus L.), tRNA isopentenyltransferase genes (IPT) were found to be upregulated in EC compared to NEC and both IAA and CTK was promoted in EC, which led to the partial activation of related signal transduction pathway (Xue et al., 2022). However, the regulatory effects of genes involved in IAA and zeatin biosynthesis and signal transduction pathways on EC induction and maintenance are in a complex and comprehensive manner. Further research is needed in L. kaempferi SE.
It was of interest to note that it might be expected that, because 2,4- D and 6-BA were supplied in the medium, genes involved in auxin and cytokinin signal transduction pathways would be the only prominently featured hormone-related genes. However, this was not the case. We found that genes related to JA and ETH biosynthesis and signal transduction pathways were also differentially expressed between EC and NEC. Our results also showed that JA had a higher accumulation in NEC and many genes involved in JA biosynthesis and signal transduction were differentially expressed between EC and NEC, with more genes upregulated in NEC (Figure 7). For ETH related genes, most DEGs related to ETH biosynthesis and signal transduction showed higher expression levels in NEC (Figure 7). JA regulates diverse morphogenetic processes and defense responses in plants (Ruduś et al., 2009) and ETH is commonly involved in stress and development responses (Mantiri et al., 2008). Therefore, observed changes in JA and ETH content and the differential expression of related genes suggest that NEC may be under stress (Gautier et al., 2019).
Carbohydrate and amino acid metabolism
In primary metabolic aspects, “starch and sucrose metabolism” and “biosynthesis of amino acids” were the representative pathways involving a lot of DEGs and DAMs (Supplementary Table 12, 13). These results may indicate that both EC and NEC had vigorous primary metabolic activities. Starch and sucrose metabolism is closely related to cell division, tissue differentiation and organ formation (Eveland and Jackson, 2012; Xue et al., 2022) and may play a key role in determining embryogenesis (Baskaran et al., 2016). Previous studies have reported the differences in starch and sucrose content between EC and NEC. The higher accumulation of starch in cucumber EC may be the necessary prerequisite for further division of embryogenic cells (Xue et al., 2022). However, this seems not the case for Douglas-fir― starch content in NEC were significantly higher than that in EC, indicating that NEC was oriented toward energy storage in starch (Gautier et al., 2019). Although the metabolomic data provided limited information about saccharide metabolites, the large number of DEGs related to sugar metabolism may reflect the differences in carbohydrate and energy metabolism between EC and NEC. Additionally, many DEGs and DAMs between EC and NEC were related to the biosynthesis of amino acids (Supplementary Table 12, 13), which may indicate that the embryogenic potential may be affected by amino acid metabolism. It has been suggested that the content of free proline in callus may affect tissue differentiation, and the callus with higher content of free proline has stronger capacity of SE (Gerdakaneh et al., 2011; Xue et al., 2022). Phenylalanine and tryptophan are precursors for secondary metabolites and IAA, respectively. The higher accumulation of these amino acids may be due to the requirement of EC cells for frequent cell division and differentiation (Ng et al., 2016).
Molecular regulation of phenylpropanoid and flavonoid biosynthesis in L. kaempferi callus
In our results, flavonoids and phenolic acids accounted for 39% of all the DAMs (20.41% and 18.59%, respectively) (Table 1), and most of them were upregulated in NEC (Supplementary Table 4), which indicated that, compared to EC, more vigorous secondary metabolic activities may take place in NEC and thus led to the higher accumulation of flavonoids and phenolic acids in NEC. In contrast, primary metabolic activities were robust in both EC and NEC. In Douglas fir, 48 transcripts involved in the production of flavonoids were upregulated in NEC, and such increases in NEC were reflected by the tissue browning of NEC (Gautier et al., 2019). It was found that the secondary metabolites’ content, including anthocyanin, chlorophyll, total phenols, and flavonoids, induced in EC was lower than those in NEC in cotton embryogenesis (Zhang et al., 1992), and the higher accumulation of flavonoids in NEC was also detected in another report on cotton SE (Guo et al., 2019). It was also found that the active secondary metabolites may block the primary metabolism, resulting in delayed cell differentiation during SE (Sun et al., 2018). These findings suggest that the secondary metabolism levels in the NEC were higher, and the secondary metabolism affected cell division and differentiation by affecting the rate and intensity of the primary metabolism (Fan et al., 2022).
Flavonoids and phenolic acids are thought to contribute to the antioxidant potential in plants (Bajalan et al., 2016; Irakli et al., 2019). The biosynthesis of phenolic acids and flavonoids is conserved in plants, and the enzymes and genes involved in the two pathways have been well characterized in the last years (Zhou et al., 2019). Our results showed that most of the DEGs and DAMs were upregulated in NEC compared to EC, indicating that the biosynthesis of flavonoids and phenolic acids may play a critical role in L. kaempferi tissue transdifferentiation. The association analysis between metabolomics and transcriptomics is useful in identifying functional genes and elucidate the metabolic pathway of interest (Mao et al., 2021). The results of correlation analysis showed that the upregulation of multiple genes would ultimately result in the higher accumulation of several metabolites, which indicated that there is a complex regulatory mechanism between metabolite accumulation and gene expression in L. kaempferi calluses.
Many TFs have been proven to regulate phenylpropanoid or flavonoid biosynthesis in plants (Ma et al., 2018; Li et al., 2020a). For example, an ABA-responsive TF SmbZIP1 promotes phenolic acid biosynthesis by upregulation of expression of the biosynthetic gene C4H1 in Salvia miltiorrhiza (Deng et al., 2020). It has been reported that R2R3-MYB, bHLH, and WD40 repeat protein play an essential role in regulating biosynthesis (Gu et al., 2019). In this study, we found that the higher expression of seven co-regulated genes including two C4H genes, two CcoAOMT genes, and three HCT genes (Table 3) involved in both phenylpropanoid and flavonoid biosynthesis pathways could result in the higher accumulation of phenolic acids and flavonoids in NEC. The targeted relationship between the predicted TFs and the key structural genes may affect the biosynthesis of phenolic acids and flavonoids. It was found that MYB15 in Arabidopsis is required for the activation of phenylpropanoid biosynthesis genes such as PAL, C4H, 4CL, HCT, and CAD (Kim et al., 2020). It has also been reported that overexpression of FTMYBs in tartary buckwheat (Fagopyrum tataricum) resulted in higher accumulation of phenylpropanoid and anthocyanin accumulation in hairy roots and structural genes such as PAL, 4CL, C4H, CHI, and F3H were markedly upregulated in transgenic lines (Li et al., 2020b). Additionally, the regulatory effects of ERFs on phenylpropanoid or flavonoid biosynthesis have been also addressed in many studies. For example, PhERF6 could interact with EOBI to negatively regulate fragrance (volatile benzenoids/phenylpropanoid) biosynthesis in petunia (Petunia hybrida) flowers (Liu et al., 2017). In mulberry (Morus alba), the expression level of ERF5 gene showed high correlation with anthocyanin change pattern in the post-flowering stages and ERF5 could bind to the promotor regions of F3H and MYBA to transcriptionally activate their expression (Mo et al., 2022). The integrated transcriptome and metabolome analyses on two sugarcane (Saccharum spp.) varieties revealed that C2C2-dof may function as a hub gene in regulating phenylpropanoid biosynthesis, flavone and flavonol biosynthesis, and starch and sucrose metabolism (Yuan et al., 2022). These results supported our findings that there may be a potential targeting relationship between the four important TFs and several structural genes involved in phenolic acid and flavonoid biosynthesis, and their expression levels were significantly positively correlated with the target gene abundance (Figure 10 and 11). Therefore, one of our future tasks will focus on the validation of the functions of these TF genes and the target relationship between TFs and structural genes to furtherly reveal the relationship between second metabolite accumulation and embryogenic potential maintenance.
Conclusion
Tremendous differences in transcriptomic and metabolomic profiling were observed between EC and NEC of L. kaempferi. The high-frequency cell division and differentiation of EC cells may be supported by the feed-forward loop composed of high expression levels of ARF genes, totipotency-related TF genes, IAA biosynthesis genes, and resulted high IAA production in EC. Carbohydrate metabolism, amino acid metabolism, and plant hormone synthesis and signal transduction may also play an important role in the maintenance of embryogenic potential. NEC featured upregulation of stress response, with more genes or metabolites involved in JA and ETH biosynthesis and transduction, and phenylpropanoid and flavonoid biosynthesis upregulated. By analyzing the matching relationship between the TFs and the promoter sequences of key structural genes and the correlations between their transcription levels, we found that several TFs including ERF, MYB and C2C2-dof may be crucial in regulating phenolic acid and flavonoid biosynthesis. This work represents the first report providing integrated insights into transcriptional and metabolic events involved in EC induction and maintenance in conifer SE.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: NCBI, PRJNA884809.
Author contributions
JW performed the experiment, processed and analyzed the data, and wrote the first draft of the manuscript. LZ designed the experiment, prepared the plant material, analyzed the data and and carried out the manuscript revision. LQ and SZ supervised the work and carried out the manuscript revision. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the Research Fund of Key Laboratory of Tree Breeding and Cultivation of National Forestry and Grassland Administration (ZDRIF202101), the National Natural Science Foundation of China (32171811 and 31600544), and the National Transgenic Major Program of China (2018ZX08020-003).
Acknowledgments
We would like to thank all colleagues and friends who have contributed to this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1056930/full#supplementary-material
References
Ávila, C., Llebres, M. T., Castro-Rodriguez, V., Lobato-Fernandez, C., Reymond, I., Harvengt, L., et al. (2022). Identification of metabolic pathways differentially regulated in somatic and zygotic embryos of maritime pine. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.877960
Bajalan, I., Mohammadi, M., Alaei, M., Pirbalouti, A. G. (2016). Total phenolic and flavonoid contents and antioxidant activity of extracts from different populations of lavandin. Ind. Crop Prod. 87, 255–260. doi: 10.1016/j.indcrop.2016.04.059
Baskaran, P., Kumari, A., Naidoo, D., Van Staden, J. (2016). In vitro propagation and ultrastructural studies of somatic embryogenesis of Ledebouria ovatifolia. In Vitro Cell. Dev. -Pl. 52 (3), 283–292. doi: 10.1007/s11627-016-9762-9
Blokhina, O., Laitinen, T., Hatakeyama, Y., Delhomme, N., Paasela, T., Zhao, L., et al. (2019). Ray parenchymal cellsc ontribute to lignification of tracheids in developing xylem of Norway spruce. Plant Physiol. 181 (4), 1552–1572. doi: 10.1104/pp.19.00743
Bonga, J. M., Klimaszewska, K. K., von Aderkas, P. (2009). Recalcitrance in clonal propagation, in particular of conifers. Plant Cell Tiss. Org. 100 (3), 241–254. doi: 10.1007/s11240-009-9647-2
Boutilier, K., Offringa, R., Sharma, V. K., Kieft, H., Ouellet, T., Zhang, L., et al. (2002). Ectopic expression of BABY BOOM triggers a conversion from vegetative to embryonic growth. Plant Cell 14 (8), 1737–1749. doi: 10.1105/tpc.001941
Bravo, S., Bertín, A., Turner, A., Sepúlveda, F., Jopia, P., Parra, M. J., et al. (2017). Differences in DNA methylation, DNA structure and embryogenesis-related gene expression between embryogenic and non embryogenic lines of Pinus radiata d. don. Plant Cell Tiss. Org. 130 (3), 521–529. doi: 10.1007/s11240-017-1242-3
Braybrook, S. A., Harada, J. J. (2008). LECs go crazy in embryo development. Trends Plant Sci. 13 (12), 624–630. doi: 10.1016/j.tplants.2008.09.008
Cabrera-Ponce, J. L., Gonzalez-Gomez, I. A., Leon-Ramirez, C. G., Sanchez-Arreguin, J. A., Jofre, Y. G. A. E. (2018). Somatic embryogenesis in common bean Phaseolus vulgaris l. Methods Mol. Biol. 1815, 189–206. doi: 10.1007/978-1-4939-8594-4_12
Caeiro, A., Caeiro, S., Correia, S., Canhoto, J. (2022). Induction of somatic embryogenesis in tamarillo (Solanum betaceum cav.) involves increases in the endogenous auxin indole-3-acetic acid. Plants (Basel) 11 (10), 1347. doi: 10.3390/plants11101347
Cai, S., Jia, J., He, C., Zeng, L., Fang, Y., Qiu, G., et al. (2021). Multi-omics of pine wood nematode pathogenicity associated with culturable associated microbiota through an artificial assembly approach. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.798539
Cañas, R. A., Canales, J., Munoz-Hernandez, C., Granados, J. M., Avila, C., Garcia-Martin, M. L., et al. (2015). Understanding developmental and adaptive cues in pine through metabolite profiling and co-expression network analysis. J. Exp. Bot. 66 (11), 3113–3127. doi: 10.1093/jxb/erv118
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13 (8), 1194–1202. doi: 10.1016/j.molp.2020.06.009
Condic, M. L. (2014). Totipotency: What it is and what it is not. Stem Cells Dev. 23, 796–812. doi: 10.1089/scd.2013.0364
Deng, C., Shi, M., Fu, R., Zhang, Y., Wang, Q., Zhou, Y., et al. (2020). An ABA-responsive SmbZIP1 is involved in modulating biosynthesis of phenolic acids and tanshinones in Salvia miltiorrhiza. J. Exp. Bot. 71, 5948–5962. doi: 10.1093/jxb/eraa295/5863266
Dobrowolska, I., Businge, E., Abreu, I. N., Moritz, T., Egertsdotter, U. (2017). Metabolome and transcriptome profiling reveal new insights into somatic embryo germination in Norway spruce (Picea abies). Tree Physiol. 37 (12), 1752–1766. doi: 10.1093/treephys/tpx078
Du, Z., Lin, W., Yu, B., Zhu, J., Li, J. (2021). Integrated metabolomic and transcriptomic analysis of the flavonoid accumulation in the leaves of Cyclocarya paliurus at different altitudes. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.794137
Elhiti, M., Stasolla, C., Wang, A. (2013). Molecular regulation of plant somatic embryogenesis. In Vitro Cell. Dev. -Pl. 49 (6), 631–642. doi: 10.1007/s11627-013-9547-3
Escandón, M., Valledor, L., Pascual, J., Pinto, G., Canal, M. J., Meijón, M. (2017). System-wide analysis of short-term response to high temperature in Pinus radiata. J. Exp. Bot. 68 (13), 3629–3641. doi: 10.1093/jxb/erx198
Eveland, A. L., Jackson, D. P. (2012). Sugars, signalling, and plant development. J. Exp. Bot. 63 (9), 3367–3377. doi: 10.1093/jxb/err379
Fan, Y., Li, W., Li, Z., Dang, S., Han, S., Zhang, L., et al. (2021). Examination of the transcriptional response to LaMIR166a overexpression in Larix kaempferi (Lamb.) Carr. Biology-Basel 10 (7), 576. doi: 10.3390/biology10070576
Fan, Y., Li, Z., Zhang, L., Han, S., Qi, L. (2020). Metabolome and transcriptome association analysis reveals regulation of flavonoid biosynthesis by overexpression of LaMIR166a in Larix kaempferi (Lamb.) Carr. Forests 11 (12), 1367. doi: 10.3390/f11121367
Fan, Y., Tang, Z., Wei, J., Yu, X., Guo, H., Li, T., et al. (2022). Dynamic transcriptome analysis reveals complex regulatory pathway underlying induction and dose effect by different exogenous auxin IAA and 2,4-d during in vitro embryogenic redifferentiation in cotton. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.931105
Fehér, A. (2015). Somatic embryogenesis - stress-induced remodeling of plant cell fate. BBA-Gene Regul. Mech. 1849 (4), 385–402. doi: 10.1016/j.bbagrm.2014.07.005
Florez, S. L., Erwin, R. L., Maximova, S. N., Guiltinan, M. J., Curtis, W. R. (2015). Enhanced somatic embryogenesis in theobroma cacao using the homologous BABY BOOM transcription factor. BMC Plant Biol. 15, 121. doi: 10.1186/s12870-015-0479-4
Fukushima, A., Kusano, M., Redestig, H., Arita, M., Saito, K. (2009). Integrated omics approaches in plant systems biology. Curr. Opin. Chem. Biol. 13 (5-6), 532–538. doi: 10.1016/j.cbpa.2009.09.022
Gautier, F., Label, P., Eliasova, K., Leple, J. C., Motyka, V., Boizot, N., et al. (2019). Cytological, biochemical and molecular events of the embryogenic state in Douglas-fir (Pseudotsuga menziesii [Mirb.]). Front. Plant Sci. 10. doi: 10.3389/fpls.2019.00118
Gerdakaneh, M., Mozafari, A.-A., sioseh-mardah, A., Sarabi, B. (2011). Effects of different amino acids on somatic embryogenesis of strawberry (Fragaria × ananassa duch.). Acta Physiol. Plant 33 (5), 1847–1852. doi: 10.1007/s11738-011-0725-9
Gruel, J., Deichmann, J., Landrein, B., Hitchcock, T., Jonsson, H. (2018). The interaction of transcription factors controls the spatial layout of plant aerial stem cell niches. NPJ Syst. Biol. Appl. 4, 36. doi: 10.1038/s41540-018-0072-1
Grzybkowska, D., Nowak, K., Gaj, M. D. (2020). Hypermethylation of auxin-responsive motifs in the promoters of the transcription factor genes accompanies the somatic embryogenesis induction in arabidopsis. Int. J. Mol. Sci. 21 (18), 6849. doi: 10.3390/ijms21186849
Guo, H., Guo, H., Zhang, L., Tang, Z., Yu, X., Wu, J., et al. (2019). Metabolome and transcriptome association analysis reveals dynamic regulation of purine metabolism and flavonoid synthesis in transdifferentiation during somatic embryogenesis in cotton. Int. J. Mol. Sci. 20 (9), 2070. doi: 10.3390/ijms20092070
Gu, K.-D., Wang, C.-K., Hu, D.-G., Hao, Y.-J. (2019). How do anthocyanins paint our horticultural products? Sci. Hortic. -Amesterdam 249, 257–262. doi: 10.1016/j.scienta.2019.01.034
Hakman, I., FOWKE, L. C., Arnold, A. V., Eriksson, T. (1985). The development of somatic embryos in tissure cultures initiated from immature embryos of Picea abies (Norway spruce). Plant Sci. 38, 53–59. doi: 10.1016/0168-9452(85)90079-2
Horstman, A., Li, M., Heidmann, I., Weemen, M., Chen, B., Muino, J. M., et al. (2017). The BABY BOOM transcription factor activates the LEC1-ABI3-FUS3-LEC2 network to induce somatic embryogenesis. Plant Physiol. 175 (2), 848–857. doi: 10.1104/pp.17.00232
Irakli, M., Tsaliki, E., Kalivas, A., Kleisiaris, F., Sarrou, E., Cook, C. M. (2019). Effect omicronf genotype and growing year on the nutritional, phytochemical, and antioxidant properties of industrial hemp (Cannabis sativa l.) seeds. Antioxidants-Basel 8 (10), 491. doi: 10.3390/antiox8100491
Jin, F., Hu, L., Yuan, D., Xu, J., Gao, W., He, L., et al. (2014). Comparative transcriptome analysis between somatic embryos (SEs) and zygotic embryos in cotton: evidence for stress response functions in SE development. Plant Biotechnol. J. 12 (2), 161–173. doi: 10.1111/pbi.12123
Ji, X.-H., Zhang, R., Wang, N., Yang, L., Chen, X.-S. (2015). Transcriptome profiling reveals auxin suppressed anthocyanin biosynthesis in red-fleshed apple callus (Malus sieversii f. niedzwetzkyana). Plant Cell Tiss. Org. 123 (2), 389–404. doi: 10.1007/s11240-015-0843-y
Junker, A., Monke, G., Rutten, T., Keilwagen, J., Seifert, M., Thi, T. M., et al. (2012). Elongation-related functions of LEAFY COTYLEDON1 during the development of Arabidopsis thaliana. Plant J. 71 (3), 427–442. doi: 10.1111/j.1365-313X.2012.04999.x
Kang, Y., Li, W., Zhang, L., Qi, L. (2021). Over-expression of the cell-cycle gene LaCDKB1;2 promotes cell proliferation and the formation of normal cotyledonary embryos during Larix kaempferi somatic embryogenesis. Genes (Basel) 12 (9), 1435. doi: 10.3390/genes12091435
Karami, O., Rahimi, A., Mak, P., Horstman, A., Boutilier, K., Compier, M., et al. (2021). An Arabidopsis AT-hook motif nuclear protein mediates somatic embryogenesis and coinciding genome duplication. Nat. Commun. 12 (1), 2508. doi: 10.1038/s41467-021-22815-8
Kim, S. H., Lam, P. Y., Lee, M. H., Jeon, H. S., Tobimatsu, Y., Park, O. K. (2020). The arabidopsis R2R3 MYB transcription factor MYB15 is a key regulator of lignin biosynthesis in effector-triggered immunity. Front. Plant Sci. 11. doi: 10.3389/fpls.2020.583153
Klimaszewska, K., Overton, C., Stewart, D., Rutledge, R. G. (2011). Initiation of somatic embryos and regeneration of plants from primordial shoots of 10-year-old somatic white spruce and expression profiles of 11 genes followed during the tissue culture process. Planta 233 (3), 635–647. doi: 10.1007/s00425-010-1325-4
Kumaravel, M., Uma, S., Backiyarani, S., Saraswathi, M. S., Vaganan, M. M., Muthusamy, M., et al. (2017). Differential proteome analysis during early somatic embryogenesis in musa spp. AAA cv. grand naine. Plant Cell. Rep. 36 (1), 163–178. doi: 10.1007/s00299-016-2067-y
Kumar, S., Ruggles, A., Logan, S., Mazarakis, A., Tyson, T., Bates, M., et al. (2021). Comparative transcriptomics of non-embryogenic and embryogenic callus in semi-recalcitrant and non-recalcitrant upland cotton lines. Plants-Basel 10 (9), 1775. doi: 10.3390/plants10091775
Li, H., Li, Y., Yu, J., Wu, T., Zhang, J., Tian, J., et al. (2020a). MdMYB8 is associated with flavonol biosynthesis via the activation of the MdFLS promoter in the fruits of Malus crabapple. Hortic. Res. 7, 19. doi: 10.1038/s41438-020-0238-z
Li, D., Lu, X., Zhu, Y., Pan, J., Zhou, S., Zhang, X., et al. (2022a). The multi-omics basis of potato heterosis. J. Integr. Plant Biol. 64 (3), 671–687. doi: 10.1111/jipb.13211
Lin, W., Li, Y., Lu, Q., Lu, H., Li, J. (2020). Combined analysis of the metabolome and transcriptome identified candidate genes involved in phenolic acid biosynthesis in the leaves of Cyclocarya paliurus. Int. J. Mol. Sci. 21 (4), 1337. doi: 10.3390/ijms21041337
Li, X., Sathasivam, R., Park, N. I., Wu, Q., Park, S. U. (2020b). Enhancement of phenylpropanoid accumulation in tartary buckwheat hairy roots by overexpression of MYB transcription factors. Ind. Crop Prod. 156, 112887. doi: 10.1016/j.indcrop.2020.112887
Liu, D., Mu, Q., Li, X., Xu, S., Li, Y., Gu, T. (2022). The callus formation capacity of strawberry leaf explant is modulated by DNA methylation. Hortic. Res. 9, uhab073. doi: 10.1093/hr/uhab073
Liu, F., Xiao, Z., Yang, L., Chen, Q., Shao, L., Liu, J., et al. (2017). PhERF6, interacting with EOBI, negatively regulates fragrance biosynthesis in petunia flowers. New Phytol. 215 (4), 1490–1502. doi: 10.1111/nph.14675
Li, M., Wrobel-Marek, J., Heidmann, I., Horstman, A., Chen, B., Reis, R., et al. (2022b). Auxin biosynthesis maintains embryo identity and growth during BABY BOOM-induced somatic embryogenesis. Plant Physiol. 188 (2), 1095–1110. doi: 10.1093/plphys/kiab558
Li, Q., Zhang, S., Wang, J. (2014). Transcriptome analysis of callus from Picea balfouriana. BMC Genomics 15 (553), 1471–2164. doi: 10.1186/1471-2164-15-553
Lotan, T., Ohto, M., Yee, K. M., Lo, R., Yamagishi, K., Goldberg, R., et al. (1998). Arabidopsis LEAFY COTYLEDON1 is sufficient to induce embryo development in vegetative cells. Cell 93, 1195–1205. doi: 10.1016/S0092-8674(00)81463-4
Lowe, K., Wu, E., Wang, N., Hoerster, G., Hastings, C., Cho, M. J., et al. (2016). Morphogenic regulators Baby boom and Wuschel improve monocot transformation. Plant Cell 28 (9), 1998–2015. doi: 10.1105/tpc.16.00124
Maadon, S. N., Rohani, E. R., Ismail, I., Baharum, S. N., Normah, M. N. (2016). Somatic embryogenesis and metabolic differences between embryogenic and non-embryogenic structures in mangosteen. Plant Cell Tiss. Org. 127 (2), 443–459. doi: 10.1007/s11240-016-1068-4
Mantiri, F. R., Kurdyukov, S., Lohar, D. P., Sharopova, N., Saeed, N. A., Wang, X. D., et al. (2008). The transcription factor MtSERF1 of the ERF subfamily identified by transcriptional profiling is required for somatic embryogenesis induced by auxin plus cytokinin in Medicago truncatula. Plant Physiol. 146 (4), 1622–1636. doi: 10.1104/pp.107.110379
Mao, J., Huang, L., Chen, M., Zeng, W., Feng, Z., Huang, S., et al. (2021). Integrated analysis of the transcriptome and metabolome reveals genes involved in terpenoid and flavonoid biosynthesis in the loblolly pine (Pinus taeda l.). Front. Plant Sci. 12. doi: 10.3389/fpls.2021.729161
Ma, D., Reichelt, M., Yoshida, K., Gershenzon, J., Constabel, C. P. (2018). Two R2R3-MYB proteins are broad repressors of flavonoid and phenylpropanoid metabolism in poplar. Plant J. 96 (5), 949–965. doi: 10.1111/tpj.14081
Mendez-Hernandez, H. A., Ledezma-Rodriguez, M., Avilez-Montalvo, R. N., Juarez-Gomez, Y. L., Skeete, A., Avilez-Montalvo, J., et al. (2019). Signaling overview of plant somatic embryogenesis. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.00077
Mo, R., Guangming, H., Zhu, Z., Essemine, J., Dong, Z. D., Li, Y., et al. (2022). The ethylene response factor ERF5 regulates anthocyanin biosynthesis in ‘Zijin’ mulberry fruits by interacting with MYBA and F3H genes. Int. J. Mol. Sci. 23, 7615. doi: 10.3390/ijms23147615
Mordhorst, A. P., Toonen, M. A. J., De Vries, S. C., Meinke, D. (1997). Plant embryogenesis. Crit. Rev. Plant Sci. 16 (6), 535–576. doi: 10.1080/07352689709701959
Nagmani, R., Bonga, J. M. (1985). Embryogenesis in subcultured callus of Larix decidua. Can. J. For. Res. 15 (6), 1088–1091. doi: 10.1139/x85-177
Nakamura, M., Batista, R. A., Kohler, C., Hennig, L. (2020). Polycomb repressive complex 2-mediated histone modification H3K27me3 is associated with embryogenic potential in Norway spruce. J. Exp. Bot. 71 (20), 6366–6378. doi: 10.1093/jxb/eraa365
Ng, T. L., Karim, R., Tan, Y. S., Teh, H. F., Danial, A. D., Ho, L. S., et al. (2016). Amino acid and secondary metabolite production in embryogenic and non-embryogenic callus of fingerroot ginger (Boesenbergia rotunda). PloS One 11 (6), e0156714. doi: 10.1371/journal.pone.0156714
Paul, P., Joshi, S., Tian, R., Diogo Junior, R., Chakrabarti, M., Perry, S. E. (2022). The MADS-domain factor AGAMOUS-Like18 promotes somatic embryogenesis. Plant Physiol. 188 (3), 1617–1631. doi: 10.1093/plphys/kiab553
Pérez-Pérez, Y., El-Tantawy, A. A., Solís, M. T., Risueño, M. C., Testillano, P. S. (2019). Stress-induced microspore embryogenesis requires endogenous auxin synthesis and polar transport in barley. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.01200
Ruduś, I., Weiler, E. W., Kępczyńska, E. (2009). Do stress-related phytohormones, abscisic acid and jasmonic acid play a role in the regulation of Medicago sativa l. somatic embryogenesis? Plant Growth Regul. 59 (2), 159–169. doi: 10.1007/s10725-009-9399-3
Salaün, C., Lepiniec, L., Dubreucq, B. (2021). Genetic and molecular control of somatic embryogenesis. Plants-Basel 10 (7), 1467. doi: 10.3390/plants10071467
Srinivasan, C., Liu, Z., Heidmann, I., Supena, E. D., Fukuoka, H., Joosen, R., et al. (2007). Heterologous expression of the BABY BOOM AP2/ERF transcription factor enhances the regeneration capacity of tobacco (Nicotiana tabacum l.). Planta 225 (2), 341–351. doi: 10.1007/s00425-006-0358-1
Stepanova, A. N., Yun, J., Robles, L. M., Novak, O., He, W., Guo, H., et al. (2011). The arabidopsis YUCCA1 flavin monooxygenase functions in the indole-3-pyruvic acid branch of auxin biosynthesis. Plant Cell 23 (11), 3961–3973. doi: 10.1105/tpc.111.088047
Sun, R., Tian, R., Ma, D., Wang, S., Liu, C. (2018). Comparative transcriptome study provides insights into acquisition of embryogenic ability in upland cotton during somatic embryogenesis. J. Cotton Res. 1 (1), 9. doi: 10.1186/s42397-018-0010-1
Sun, C., Xie, Y. H., Li, Z., Liu, Y. J., Sun, X. M., Li, J. J., et al. (2022). The Larix kaempferi genome reveals new insights into wood properties. J. Integr. Plant Biol. 64 (7), 1364–1373. doi: 10.1111/jipb.13265
Su, Y. H., Tang, L. P., Zhao, X. Y., Zhang, X. S. (2021). Plant cell totipotency: Insights into cellular reprogramming. J. Integr. Plant Biol. 63 (1), 228–243. doi: 10.1111/jipb.12972
Su, Y. H., Zhao, X. Y., Liu, Y. B., Zhang, C. L., O'Neill, S. D., Zhang, X. S. (2009). Auxin-induced WUS expression is essential for embryonic stem cell renewal during somatic embryogenesis in arabidopsis. Plant J. 59 (3), 448–460. doi: 10.1111/j.1365-313X.2009.03880.x
Teale, W. D., Paponov, I. A., Palme, K. (2006). Auxin in action: signalling, transport and the control of plant growth and development. Nat. Rev. Mo.l Cell. Biol. 7 (11), 847–859. doi: 10.1038/nrm2020
Tian, F., Yang, D. C., Meng, Y. Q., Jin, J., Gao, G. (2020). PlantRegMap: charting functional regulatory maps in plants. Nucleic Acids Res. 48 (D1), D1104–D1113. doi: 10.1093/nar/gkz1020
Varis, S., Klimaszewska, K., Aronen, T. (2018). Somatic embryogenesis and plant regeneration from primordial shoot explants of Picea abies (L.) h. karst. somatic trees. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.01551
Wang, F. X., Shang, G. D., Wang, J. W. (2022a). Towards a hierarchical gene regulatory network underlying somatic embryogenesis. Trends Plant Sci. 27 (12), 1209–1217. doi: 10.1016/j.tplants.2022.06.002
Wang, F. X., Shang, G. D., Wu, L. Y., Xu, Z. G., Zhao, X. Y., Wang, J. W. (2020). Chromatin accessibility dynamics and a hierarchical transcriptional regulatory network structure for plant somatic embryogenesis. Dev. Cell 54 (6), 742–757. doi: 10.1016/j.devcel.2020.07.003
Wang, R., Shu, P., Zhang, C., Zhang, J., Chen, Y., Zhang, Y., et al. (2022b). Integrative analyses of metabolome and genome-wide transcriptome reveal the regulatory network governing flavor formation in kiwifruit (Actinidia chinensis). New Phytol. 233 (1), 373–389. doi: 10.1111/nph.17618
Wojcikowska, B., Gaj, M. D. (2017). Expression profiling of AUXIN RESPONSE FACTOR genes during somatic embryogenesis induction in arabidopsis. Plant Cell Rep. 36 (6), 843–858. doi: 10.1007/s00299-017-2114-3
Wu, C., Chen, D., Shen, J., Sun, X., Zhang, S. (2021). Estimating the distribution and productivity characters of Larix kaempferi in response to climate change. J. Environ. Manage. 280, 111633. doi: 10.1016/j.jenvman.2020.111633
Wu, M. F., Yamaguchi, N., Xiao, J., Bargmann, B., Estelle, M., Sang, Y., et al. (2015). Auxin-regulated chromatin switch directs acquisition of flower primordium founder fate. Elife 4, e09269. doi: 10.7554/eLife.09269
Xiao, Y., Ling, J., Yi, F., Ma, W., Lu, N., Zhu, T., et al. (2021). Transcriptomic, proteomic, and metabolic profiles of catalpa bungei tension wood reveal new insight into lignin biosynthesis involving transcription factor regulation. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.704262
Xue, W., Liu, N., Zhang, T., Li, J., Chen, P., Yang, Y., et al. (2022). Substance metabolism, IAA and CTK signaling pathways regulating the origin of embryogenic callus during dedifferentiation and redifferentiation of cucumber cotyledon nodes. Sci. Hortic. -Amesterdam 293, 110680. doi: 10.1016/j.scienta.2021.110680
Yuan, Z., Dong, F., Pang, Z., Fallah, N., Zhou, Y., Li, Z., et al. (2022). Integrated metabolomics and transcriptome analyses unveil pathways involved in sugar content and rind color of two sugarcane varieties. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.921536
Zhang, Y. Z., Clemens, A., Maximova, S. N., Guiltinan, M. J. (2014b). The Theobroma cacao B3 domain transcription factor TcLEC2 plays a duel role in control of embryo development and maturation. BMC Plant Biol. 14, 106. doi: 10.1186/1471-2229-14-106
Zhang, L.-f., Fan, Y.-R., Lan, Q., Qi, L.-W., Han, S.-Y. (2021a). Expression of the SPL-like gene LaSPL9 in Japanese larch (Larix leptolepis) is regulated by miR156 during somatic embryogenesis. Trees 35 (5), 1727–1737. doi: 10.1007/s00468-020-02081-9
Zhang, J., Liang, L., Xie, Y., Zhao, Z., Su, L., Tang, Y., et al. (2022). Transcriptome and metabolome analyses reveal molecular responses of two pepper (Capsicum annuum l.) cultivars to cold stress. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.819630
Zhang, L.-f., Li, W.-F., Xu, H.-Y., Qi, L.-W., Han, S.-Y. (2014a). Cloning and characterization of four differentially expressed cDNAs encoding NFYA homologs involved in responses to ABA during somatic embryogenesis in Japanese larch (Larix leptolepis). Plant Cell Tiss. Org. 117 (2), 293–304. doi: 10.1007/s11240-014-0440-5
Zhang, X., Sun, J., Liu, J. (1992). A comparative study on the biochemical metabolites between the embryogenic and non-embryogenic calli from the variety"Coker 201"of Gossypium hirsutum l. Acta Agron. Sin. 18, 175–182.
Zhang, L., Yan, S., Zhang, S., Yan, P., Wang, J., Zhang, H. (2021b). Glutathione, carbohydrate and other metabolites of Larix olgensis a. Henry reponse to polyethylene glycol-simulated drought stress. PloS One 16 (11), e0253780. doi: 10.1371/journal.pone.0253780
Zhang, L., Zhang, Z., Fang, S., Liu, Y., Shang, X. (2021c). Integrative analysis of metabolome and transcriptome reveals molecular regulatory mechanism of flavonoid biosynthesis in Cyclocarya paliurus under salt stress. Ind. Crop Prod. 170, 113823. doi: 10.1016/j.indcrop.2021.113823
Zhang, Y., Zhang, S., Han, S., Li, X., Qi, L. (2012b). Transcriptome profiling and in silico analysis of somatic embryos in Japanese larch (Larix leptolepis). Plant Cell Rep. 31 (9), 1637–1657. doi: 10.1007/s00299-012-1277-1
Zhang, J., Zhang, S., Han, S., Wu, T., Li, X., Li, W., et al. (2012a). Genome-wide identification of microRNAs in larch and stage-specific modulation of 11 conserved microRNAs and their targets during somatic embryogenesis. Planta 236 (2), 647–657. doi: 10.1007/s00425-012-1643-9
Zhang, S., Zhou, J., Han, S., Yang, W., Li, W., Wei, H., et al. (2010). Four abiotic stress-induced miRNA families differentially regulated in the embryogenic and non-embryogenic callus tissues of Larix leptolepis. Biochem. Biophys. Res. Commun. 398 (3), 355–360. doi: 10.1016/j.bbrc.2010.06.056
Zhou, S., Chen, J., Lai, Y., Yin, G., Chen, P., Pennerman, K. K., et al. (2019). Integrative analysis of metabolome and transcriptome reveals anthocyanins biosynthesis regulation in grass species Pennisetum purpureum. Ind. Crop Prod. 138, 111470. doi: 10.1016/j.indcrop.2019.111470
Keywords: embryogenic potential maintenance, embryogenic callus, non-embryogenic callus, transcriptomic, metabolomic, phenolic acids and flavonoids
Citation: Wang J, Zhang L, Qi L and Zhang S (2022) Integrated transcriptomic and metabolic analyses provide insights into the maintenance of embryogenic potential and the biosynthesis of phenolic acids and flavonoids involving transcription factors in Larix kaempferi (Lamb.) Carr.. Front. Plant Sci. 13:1056930. doi: 10.3389/fpls.2022.1056930
Received: 29 September 2022; Accepted: 01 November 2022;
Published: 17 November 2022.
Edited by:
Zhongxiong Lai, Fujian Agriculture and Forestry University, ChinaReviewed by:
Han Xu,Institut de la Recherche Interdiciplinaire de Toulouse (IRIT-ARI), France
Conglong Lian, Beijing Forestry University, Beijing, China
Copyright © 2022 Wang, Zhang, Qi and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shougong Zhang, bGFyY2hfcmlmQDE2My5jb20=
†These authors have contributed equally to this work and share first authorship