 Fuchuan Han
Fuchuan Han Yanshu Qu
Yanshu Qu Yicun Chen
Yicun Chen Li’an Xu
Li’an Xu Changwei Bi
Changwei Bi
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 16 November 2022
Sec. Plant Bioinformatics
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.1031769
Salix L. (willows) is one of the most taxonomically complex genera of flowering plants, including shrubs, tall trees, bushes, and prostrate plants. Despite the high species diversity, only five mitochondrial genomes (mitogenomes) have been released in this genus. Salix wilsonii is an important ornamental and economic willow tree in section Wilsonia of the genus Salix. In this study, the S. wilsonii mitogenome was assembled into a typical circular structure with a size of 711,456 bp using PacBio HiFi sequencing. A total of 58 genes were annotated in the S. wilsonii mitogenome, including 33 protein-coding genes (PCGs), 22 tRNAs, and 3 rRNAs. In the S. wilsonii mitogenome, four genes (mttB, nad3, nad4, and sdh4) were found to play important roles in its evolution through selection pressure analysis. Collinearity analysis of six Salix mitogenomes revealed high structural variability. To determine the evolutionary position of S. wilsonii, we conducted a phylogenetic analysis of the mitogenomes of S. wilsonii and 12 other species in the order Malpighiales. Results strongly supported the segregation of S. wilsonii and other five Salix species with 100% bootstrap support. The comparative analysis of the S. wilsonii mitogenome not only sheds light on the functional and structural features of S. wilsonii but also provides essential information for genetic studies of the genus Salix.
Mitochondria are semiautonomous organelles that originated from symbiotic bacteria within eukaryotic cells (Gupta and Golding, 1996; van Oven and Kayser, 2009). They have established a stable regulatory mechanism with the nuclear genome during long-term evolution. The nucleus plays a dominant role in the cell, while the growth of mitochondria is controlled by the nuclear genome and its genetic system. Plant mitochondria, the main sites of aerobic cellular respiratioFabaceae differ by approximatelyn, not only provide energy for complex intracellular biological activities but also are involved in lipid metabolism, cytoplasmic inheritance, and apoptosis (Green and Reed, 1998; Olson and Stenlid, 2001; McBride et al., 2006; Osiewacz et al., 2010). With the development of sequencing technologies, especially HiFi sequencing that balances length and high accuracy, increasing numbers of plant mitogenomes are being assembled and released. This development also drives progress in plant mitogenome research and provides important data for obtaining deeper insight into the structure, evolution, and function of mitogenomes (Palmer et al., 2000; Guo et al., 2016; Cole et al., 2018).
The mitogenome of plants is more complex in structure than that of other eukaryotes. In addition to a master circular molecule, it also includes linear conformations, branched structures, and numerous small circular molecules mediated by repeat sequences (Notsu et al., 2002; Kozik et al., 2019; Li et al., 2022). For example, the mitogenome of Populus simonii was assembled into three circular molecules, and all of them had protein-coding capability (Bi et al., 2022). Additionally, the sweet potato (Ipomoea batatas [L.] Lam) mitogenome was found to have a multicircular structure generated by repeat sequence-mediated recombination (Yang et al., 2022). The mitogenome of plants is much larger than that of other eukaryotes and differs even among related species (Kubo and Newton, 2008; Sloan et al., 2012). The largest terrestrial plant mitogenome is 11.3 Mb in size, while the smallest is approximately 66 kb (Sloan et al., 2012; Skippington et al., 2015). Additionally, the sizes of the mitogenomes of Medicago truncatula (271 kb) and Phaseolus vulgaris (588 kb) in the Fabaceae differ by approximately 300 kb (Bi et al., 2020). Studies have shown that foreign sequence insertions and numerous repeat sequences are the main causes of mitogenome expansion in plants (Richardson and Palmer, 2007; Li, 2009; Smith, 2011). Previous studies of Acer truncatum, Malus domestica, Cucumis sativus, and melon detected a significant number of sequences that are homologous to nuclear and chloroplast sequences (Alverson et al., 2011; Rodríguez-Moreno et al., 2011; Goremykin et al., 2012; Ma et al., 2022). Moreover, a study in Cucumis sativus revealed that numerous repeat sequences contribute significantly to the expansion of the mitogenome (Lilly and Havey, 2001; Alverson et al., 2011). In terms of genomic content among land plants, mitochondrial intron counts vary from only 4 in Viscum scurruloideum (Skippington et al., 2015) to >40 in hornworts (Anthoceros agrestis and Anthoceros angustus) (Gerke et al., 2020), while the number of protein-coding genes (PCGs) ranges from as few as 19 in Viscum scurruloideum to 41 in the liverworts Marchantia polymorpha (Oda et al., 1992) and Scapania ornithopodioides (Dong et al., 2019). The differences in gene numbers, intron counts, and intergenic contents of plant mitogenomes are probably caused by intracellular and horizontal transfer processes, which enable the integration of foreign DNA. However, to maintain functional stability, the number of genes in the mitogenome varies very little between species. These features suggest that the instability of plant mitogenomes has led to a significant lag in studies on them compared with the chloroplast and plastid genomes.
S. wilsonii, commonly known as Ziliu in China, is a deciduous tree that can grow up to 13 m tall. It belongs to the genus Salix, which represents one of the most taxonomically complex genera of flowering plants. It is frequently used as a horticultural plant due to its purple branches, and it is a fast-growing, economically valuable tree whose branches are frequently used for weaving (Kuzovkina et al., 2008; Wu et al., 2019). However, the assembly of plant mitogenomes has proven challenging because of numerous repeat sequences, intricate structure, and insertions of foreign sequences. In this work, PacBio HiFi reads were used to obtain a high-quality assembly of the S. wilsonii mitogenome. Comparative analysis of the S. wilsonii mitogenome is imperative for better elucidating the functional and structural features of S. wilsonii, which would facilitate evolutionary and genetic studies of Salix.
Materials were collected from fresh leaves of S. wilsonii on the campus of Nanjing Forestry University (32°04’41” N, 118°48’23” E). The fresh leaves were placed in a -80°C freezer for freezing and storage. DNA was extracted from the leaves. After the quality of the isolated DNA was checked, the libraries were constructed using the SMRTbell Express Template Preparation Kit 2.0 (Pacific Biosciences, CA, USA). A SMRTbell library was obtained after quality control testing. The library was sequenced on the PacBio Sequel II platform (Pacific Biosciences, CA, USA) (PacBio Sequel II System).
PacBio SMRT-Analysis software (https://www.pacb.com) was used for quality control of the raw polymerase reads. The obtained CCS reads were used to generate an assembly with Hifiasm v0.16 software (Cheng et al., 2021). All generated contigs were aligned to the reference mitogenomes of Salix suchowensis and other Salix species using BLASTn (Camacho et al., 2009), and finally a circular contig was found to be the S. wilsonii mitogenome sequence. The online tool GE-seq (https://chlorobox.mpimp-golm.mpg.de/geseq.html) (Tillich et al., 2017) was applied to annotate the S. wilsonii mitogenome, using the Salix purpurea and S. suchowensis mitogenomes as reference genomes. The threshold for protein, rRNA, tRNA, and DNA search identity was 85%. Subsequently, the annotation results were edited using Apollo to manually modify the GenBank file (Lewis et al., 2002). The circular map of the S. wilsonii mitogenome was visualized using the OrganellarGenomeDRAW program (Greiner et al., 2019).
Simple sequence repeats (SSRs) were identified using MISA v2.1 (Beier et al., 2017) (https://webblast.ipk-gatersleben.de/misa/). We identified units of 1-6 bp with the minimum number of repeats set to 8, 4, 4, 3, 3, and 3, respectively. The online version of Tandem Repeats Finder 4.09 (Benson, 1999) (https://tandem.bu.edu/trf/trf.html) was used to identify tandem sequence repeats based on default parameters. Dispersed repeats were searched by the online version of REPuter with the parameters minimal repeats and hamming Distance set to 30 and 3 bp, respectively (Kurtz et al., 2001) (http://bibiserv.techfak.uni-bielefeld.de/reputer/), and the results were verified by BLASTn (evalue < 1e-10, identity > 80) (Camacho et al., 2009).
We calculated nonsynonymous (Ka) and synonymous (Ks) substitution rates for 23 PCGs of S. wilsonii. Arabidopsis thaliana (NC_037304.1), Manihot esculenta (NC_045136.1), Passiflora edulis (NC_050950.1) and Populus tremula (NC_028096.1) were used as reference species in this study. Twenty-three PCGs of the reference species and S. wilsonii were compared using ParaAT 2.0 (default parameters) (Zhang et al., 2012). Subsequently, Ka/Ks values were calculated using KaKs_Calculator v.2.0 (Wang et al., 2010).
To study the collinearity between S. wilsonii and other members of the genus Salix, high-quality mitogenomes were downloaded from the NCBI (https://www.ncbi.nlm.nih.gov/genome) with the accession numbers shown in Table 1, which included Salix brachista, Salix cardiophylla, Salix paraflabellaris, Salix purpurea, and Salix suchowensis (Huang et al., 2019; Chen et al., 2020). Sequence alignment of S. wilsonii and the above species was performed using the subroutine nucmer of Mummer 3 (Marçais et al., 2018). The collinearity results were then filtered using the subroutine delta filter in Mummer 3 (identity>90, length>50). The collinearity results were statistically analyzed and visualized using ggplot2 (Wickham, 2016).
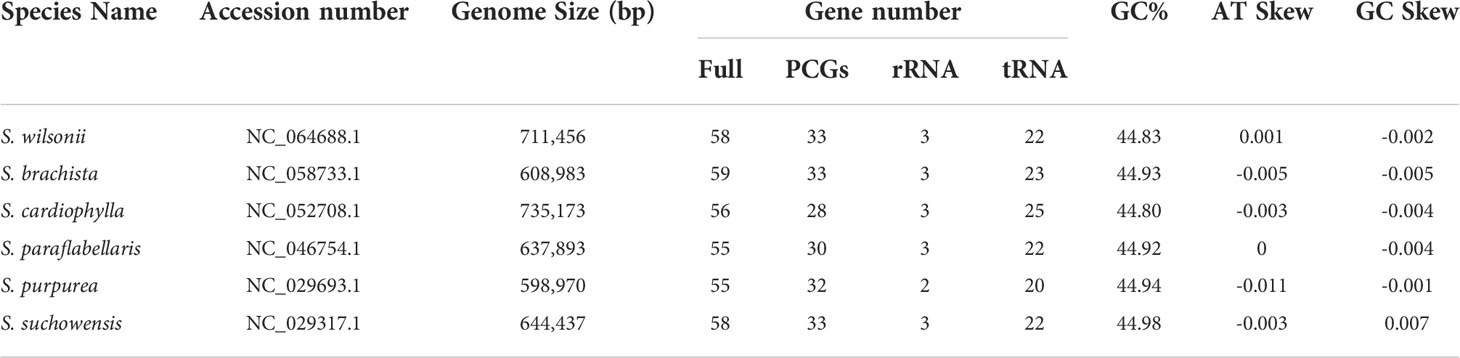
Table 1 Genomic features of six Salix mitogenomes.
To analyze the evolutionary position of S. wilsonii in the order Malpighiales, all released mitogenomes of Malpighiales in the NCBI (https://www.ncbi.nlm.nih.gov/genome) were selected for evolutionary analysis in this study. The species whose mitogenomes were selected included members of the Salicaceae (S. wilsonii, S. brachista, S. cardiophylla, S. paraflabellaris, S. purpurea, S. suchowensis, P. alba, P. davidiana, and P. tremula), Rhizophoraceae (Bruguiera sexangula; NC_056359.1), Euphorbiaceae (M. esculenta) and Passifloraceae (P. edulis). Additionally, A. thaliana was selected as an outgroup. The common PCGs among the 14 selected species were identified using Python scripts. The protein sequences were then subjected to multiple sequence alignment using MAFFT v7.475 (Katoh and Standley, 2013). Conserved regions of the aligned sequences were extracted using the Gblocks v0.91b default parameter (Castresana, 2000). The maximum likelihood (ML)-based evolutionary tree was constructed using IQ-TREE v2.1.2 with 1000 bootstraps, and the GTR+F+R2 model was selected according to Bayesian information criterion scores (Minh et al., 2020).
The HiFi reads were assembled into a typical circular structure 711,456 bp in size and submitted to NCBI under accession NC_064688.1 (Figure 1). In this study, the S. wilsonii mitogenome was annotated with a total of 58 genes, including 33 PCGs, 22 tRNA genes, and 3 rRNA genes (Table 1). The 33 PCGs consisted of 9 NADH dehydrogenase genes (nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, and nad9), 5 ATP synthase genes (atp1, atp4, atp6, atp8, and atp9), 4 cytochrome C biogenesis genes (ccmB, ccmC, ccmFc, and ccmFn), 3 cytochrome C oxidase genes (cox1, cox2, and cox3), a transport membrane protein (sdh4), a succinate dehydrogenase (mttB), a ubiquinol cytochrome c reductase (cob), a maturase (matR), 3 ribosomal protein (LSU) genes (rpl2, rpl10, and rpl16), and 5 ribosomal protein (SSU) genes (rps1, rps12, rps3, rps4, and rps7). The total length of the 33 PCGs was 30,298 bp, accounting for 4.26% of the S. wilsonii mitogenome; the total lengths of tRNA and rRNA genes accounted for 0.23% and 0.75%, respectively, while the total length of the intergenic region was close to 95% of the total length. Because the S. wilsonii mitogenome has no large repeat regions, all annotated PCGs and rRNA genes are single-copy genes, with only two tRNA genes having multiple copies (trnM-CAU and trnP-UGG). There were eight PCGs containing introns in the S. wilsonii mitogenome (nad1, nad2, nad4, nad5, rps3, nad7, rpl2, and ccmFc) (Table 2).
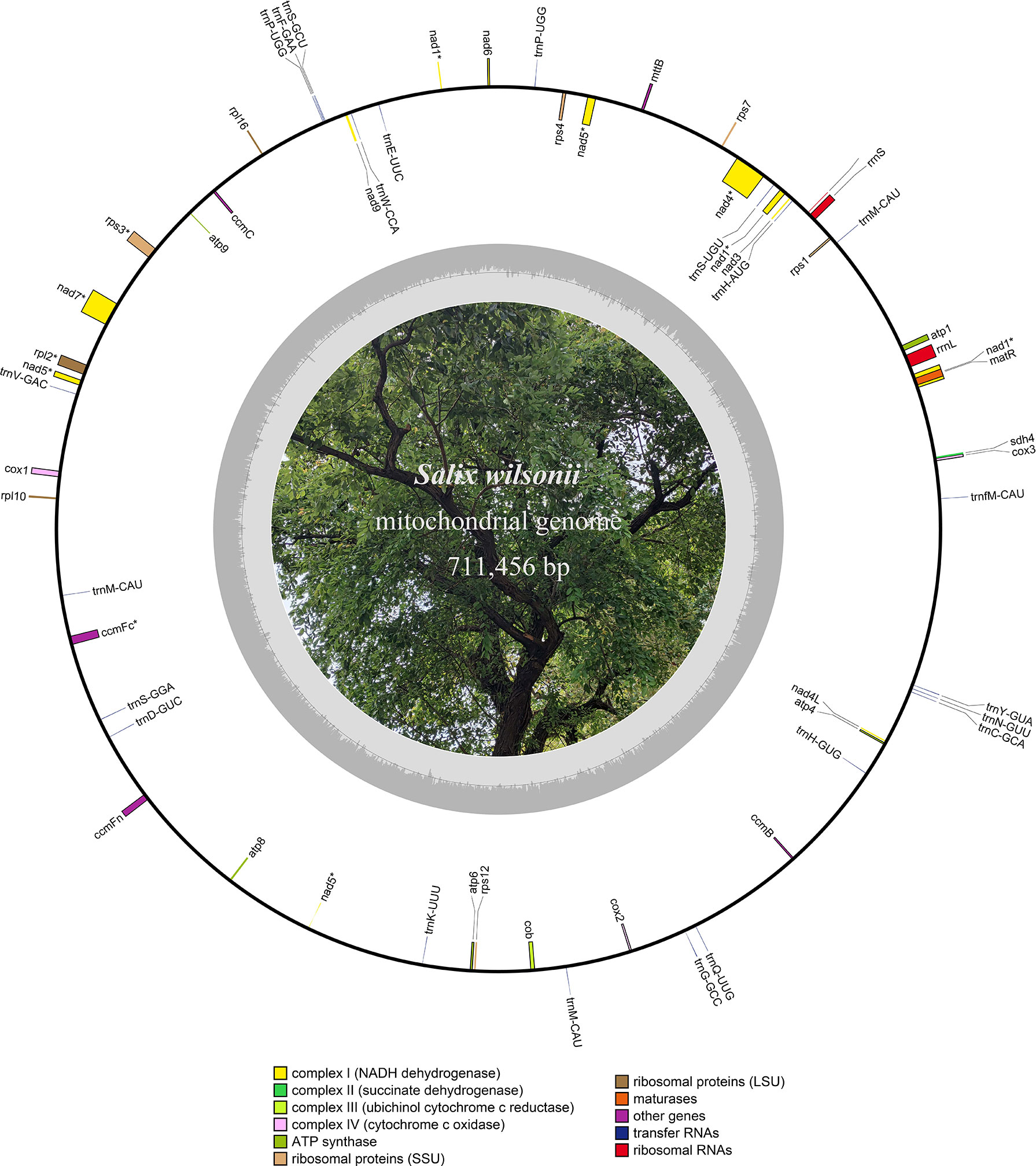
Figure 1 Circular map of the S. wilsonii mitogenome. The inner circle in gray represents the GC content of the chromosome.
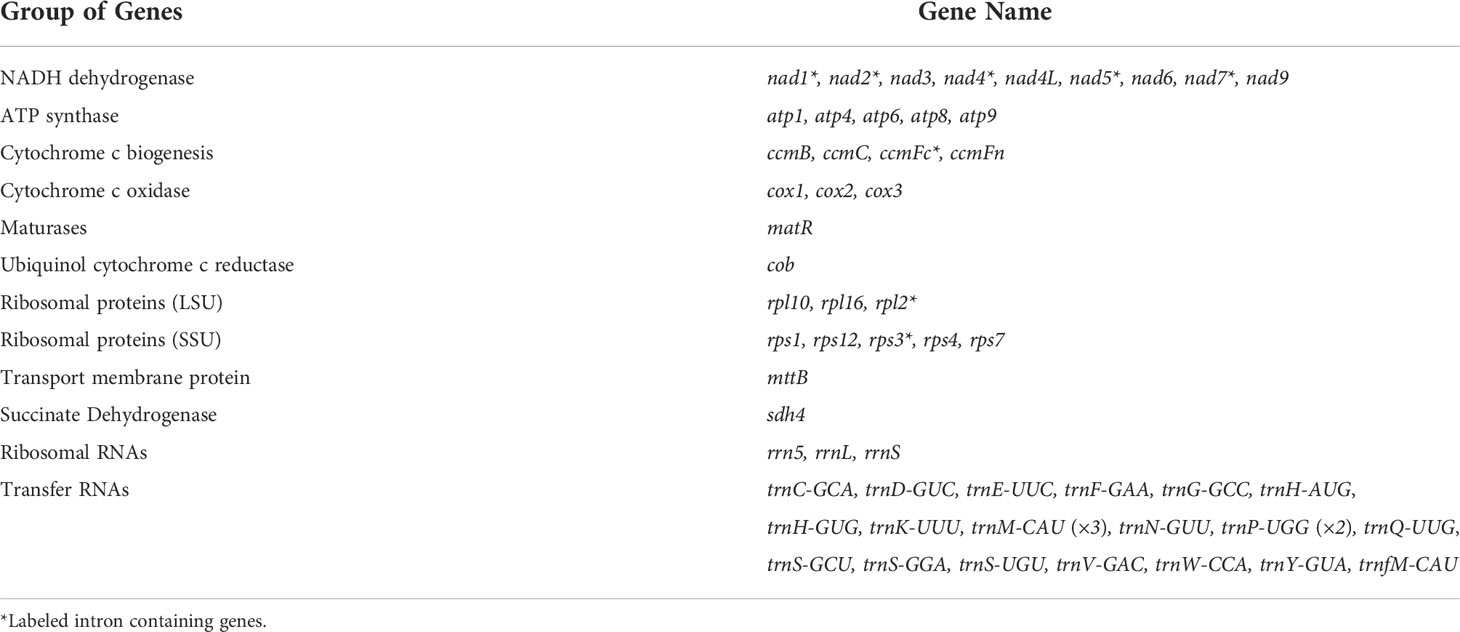
Table 2 Gene composition of the S. wilsonii mitogenome.
The GC content of S. wilsonii was 44.98% (A: 27.62%, C: 22.38%, G: 22.46%, and T: 27.55%), similar to that of other species in the genus Salix (Table 1). The positive AT skew in the mitogenome of S. wilsonii and the negative GC skew indicate a higher content of A and C bases. Thirty-one PCGs had ATG as the start codon, and the three stop codons used were TAA (54.55%), TAG (24.24%), and TGA (21.21%) (Supplementary Table 1). However, the start codons of mttB and rpl6 remain unclear and need to be verified experimentally.
SSRs are sequences usually consisting of 1-6 bp unit repeats and are widely distributed in plant mitogenomes (Powell et al., 1996). A total of 608 SSRs were found in the S. wilsonii mitogenome, with 259 mononucleotide repeats (42.6%) and 227 dinucleotide repeat (37.34%) being more abundant and 19 pentanucleotide (3.13%) and 4 hexanucleotide repeats (0.66%) being less abundant (Supplementary Table 2). The S. wilsonii mitogenome had the highest number of A/T repeats (225), accounting for 86.9% of all mononucleotide repeats, which was similar to findings for other Salix mitogenomes (Ye et al., 2017). Notably, AG/CT had the highest frequency of the dinucleotide repeats among all Salix mitogenomes (Figure 2A). Additionally, there was a fairly high frequency of AAAG/CTTT types among the tetranucleotide repeats (Figure 2A).
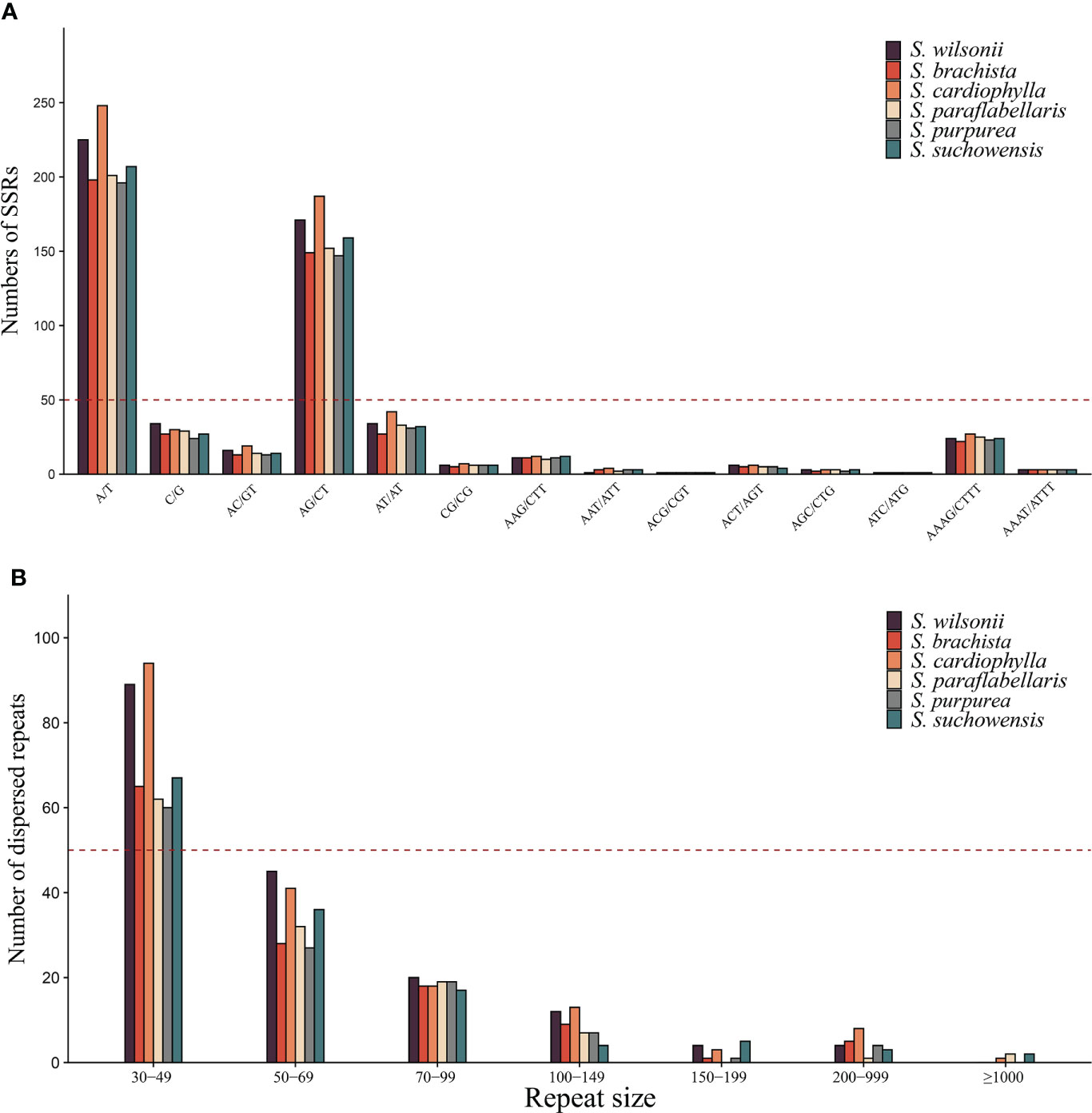
Figure 2 Repeat sequences of the genus Salix. (A) Identification frequencies of SSRs with different repeat types in the genus Salix. (B) The number of dispersed repeats of different lengths in the genus. Salix.
Tandem repeats are unstable in organisms, frequently mutate, are involved in the regulatory activities of the genome, and are closely related to genome recombination and rearrangement (Hannan, 2012). Tandem repeats were found to exist mainly in the intergenic regions of S. wilsonii, consisting of 10-30 bp (Supplementary Table S3). Most of them were present as two copies, except for the tandem repeat sequence that appears in the nad1 and trnS-UGU interval, with 4.2 copies.
Unlike tandem repeats, dispersed repeat sequences are distributed evenly throughout the mitogenome and promote or repress gene expression in the near-insertion site. Moreover, genes distributed in dispersed repeat sequences are more likely to display multiple copies, such as trnM-CAU and trnP-UGG. A total of 182 dispersed repeat sequences (21,658 bp) were found in the S. wilsonii mitogenome, accounting for 3% of the mitogenome. Eighty-nine dispersed repeat sequences were between 30 and 49 bp in length (48.9%), 45 dispersed repeat sequences were 50-69 bp in size (24.7%), and only two dispersed repeat sequences were >200 bp in size (Figure 2B). Dispersed repeat sequences varied considerably between species of the genus Salix, with only three species (S. cardiophylla, S. paraflabellaris, and S. suchowensis) having repeat sequences >1,000 bp.
The nucleotide substitution rates of mitochondrial PCGs are useful for inferring the direction and magnitude of natural selection acting on homologous PCGs during the evolution of diverged species (Li et al., 1985). A Ka/Ks ratio >1 implies positive or Darwinian selection (driving change), a ratio of exactly 1 indicates neutral selection, and a ratio of <1 implies purifying or stabilizing selection (acting against change). Mitochondria are important sites for cell metabolism, apoptosis, and energy production. To maintain mitochondrial function, deleterious mutations are removed by purifying selection during natural selection; therefore, the Ka value in most genes is smaller than the Ks value (Hurst, 2002; Shtolz and Mishmar, 2019). Twenty-four PCGs from the S. wilsonii mitogenome were compared with those from the mitogenomes of 4 other species, namely, A. thaliana, M. esculenta, P. edulis, and P. tremula (NC_ 028096.1), and the Ka/Ks ratio was calculated using the YN method in KaKs_Calculator v2.0. Most of the pairwise Ka/Ks ratios were <1 (Figure 3B), suggesting that most PCGs were under stabilizing selection during the evolution of S. wilsonii. These PCGs with Ka/Ks <1 may play important roles in stabilizing the normal function of mitochondria. In contrast, several genes were also found with Ka/Ks ratios >1 in most species, namely, atp4, ccmB, mttB, nad3, nad4, and sdh4, indicating that they had been under positive selection during evolution. In particular, the nad3 gene had an extremely high Ka/Ks ratio (S. wilsonii vs. M. esculenta: 2.63), indicating strong positive selection during the evolution of S. wilsonii and M. esculenta (Figure 3B). Additionally, the Ka and Ks values for most genes of P. tremula and S. wilsonii were close to 0, indicating a short time of differentiation between them (Figure 3A).
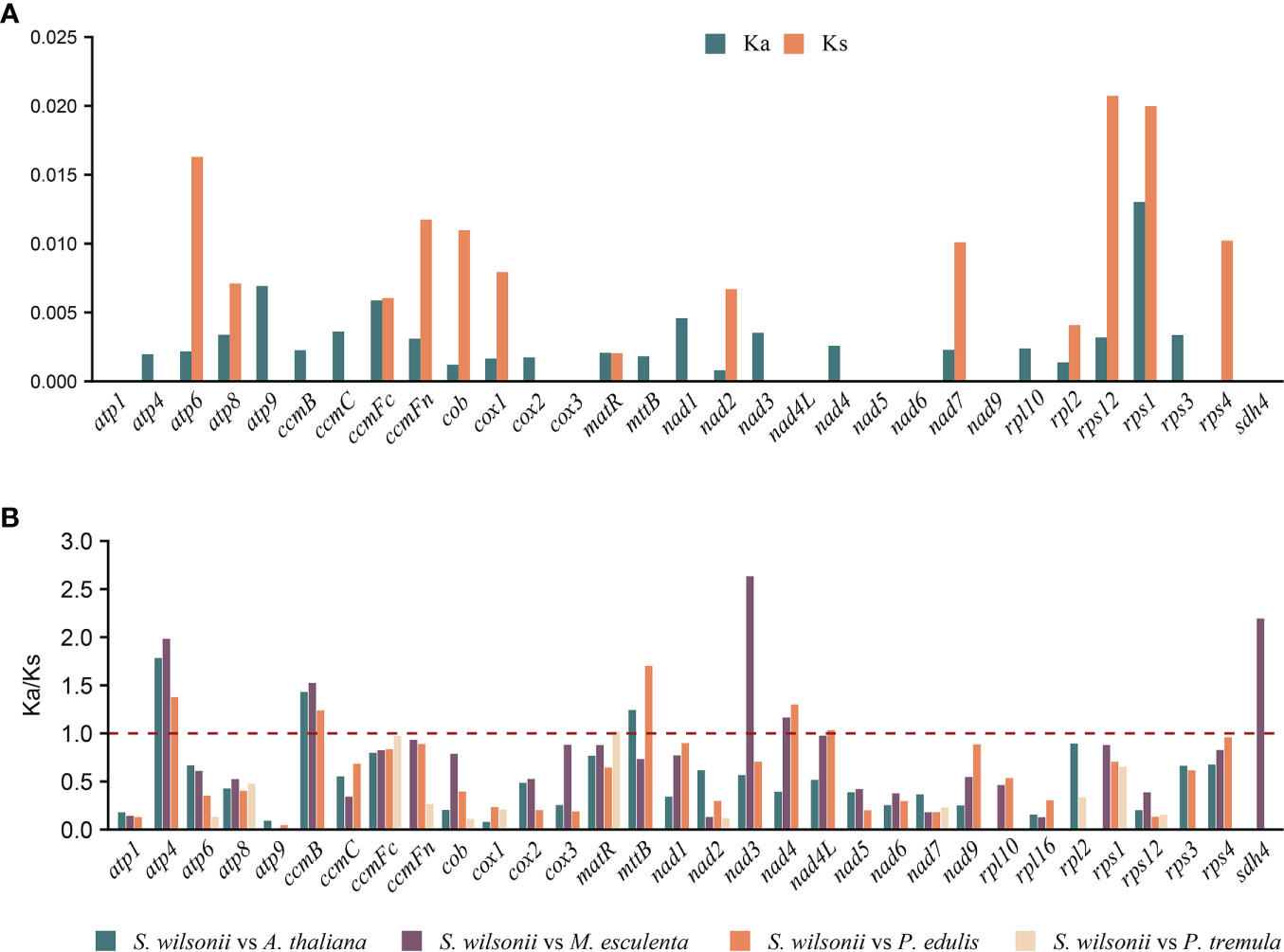
Figure 3 Nonsynonymous (Ka), synonymous (Ks), and Ka/Ks substitution values of S. wilsonii. (A) Values of Ka and Ks for P. tremula vs. S. wilsonii. (B) Ka/Ks values of 24 PCGs of S. wilsonii versus four other species.
Genome rearrangement resulting from repeat sequences is a major cause of the evolution of plant mitogenomes. The mitogenome of S. wilsonii was compared with those of five other species, namely, S. brachista, S. cardiophylla, S. paraflabellaris, S. purpurea, and S. suchowensis, using the nucmer program of MUMmer v3.23. Table 3 shows that there is strong collinearity between S. brachista and S. wilsonii. The 25 local colinear blocks (LCBs) between them make up 96.48% (587,518 bp) of the S. brachista mitogenome, and 99.83% (24,726 bp) of the S. brachista PCGs are in LCBs. Additionally, the mitogenomes of S. paraflabellaris (LCBs: 25, genome percentage: 92.97%, CDS percentage: 95.47%), S. purpurea (LCBs: 16, genome percentage: 90.33%, CDS percentage: 98.06%), and S. suchowensis (LCBs: 20, genome percentage: 94.27%, CDS percentage: 98.19%) also showed good collinearity relationships with the S. wilsonii mitogenome, with their LCBs accounting for more than 90% of their mitogenomes. Interestingly, there was poor collinearity between S. cardiophylla and S. wilsonii, but the protein-coding regions showed stronger collinearity, further suggesting that the PCGs are more stable. The dot plot indicates that there is frequent rearrangement in Salix mitogenomes (Figure 4), which may be related to numerous recombination events occurring in repeated sequences during evolution.
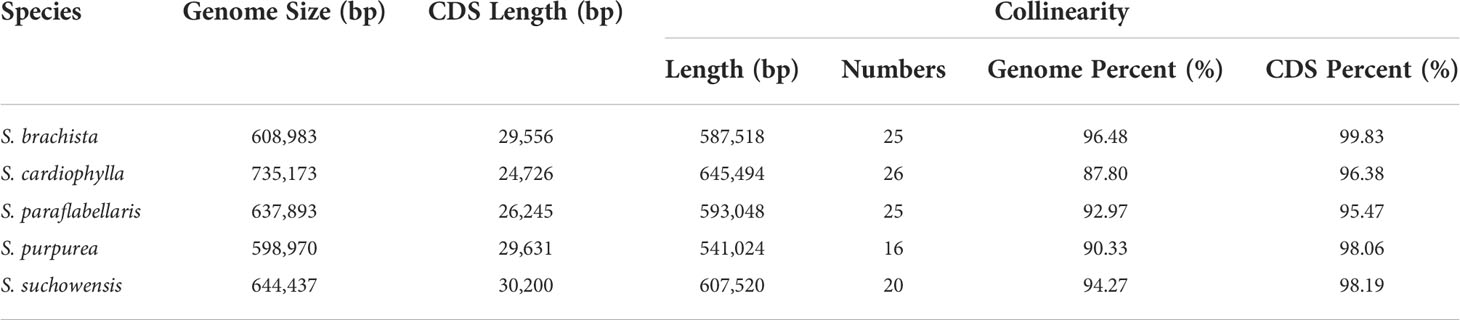
Table 3 Collinearity features between S. wilsonii and other five Salix mitogenomes.
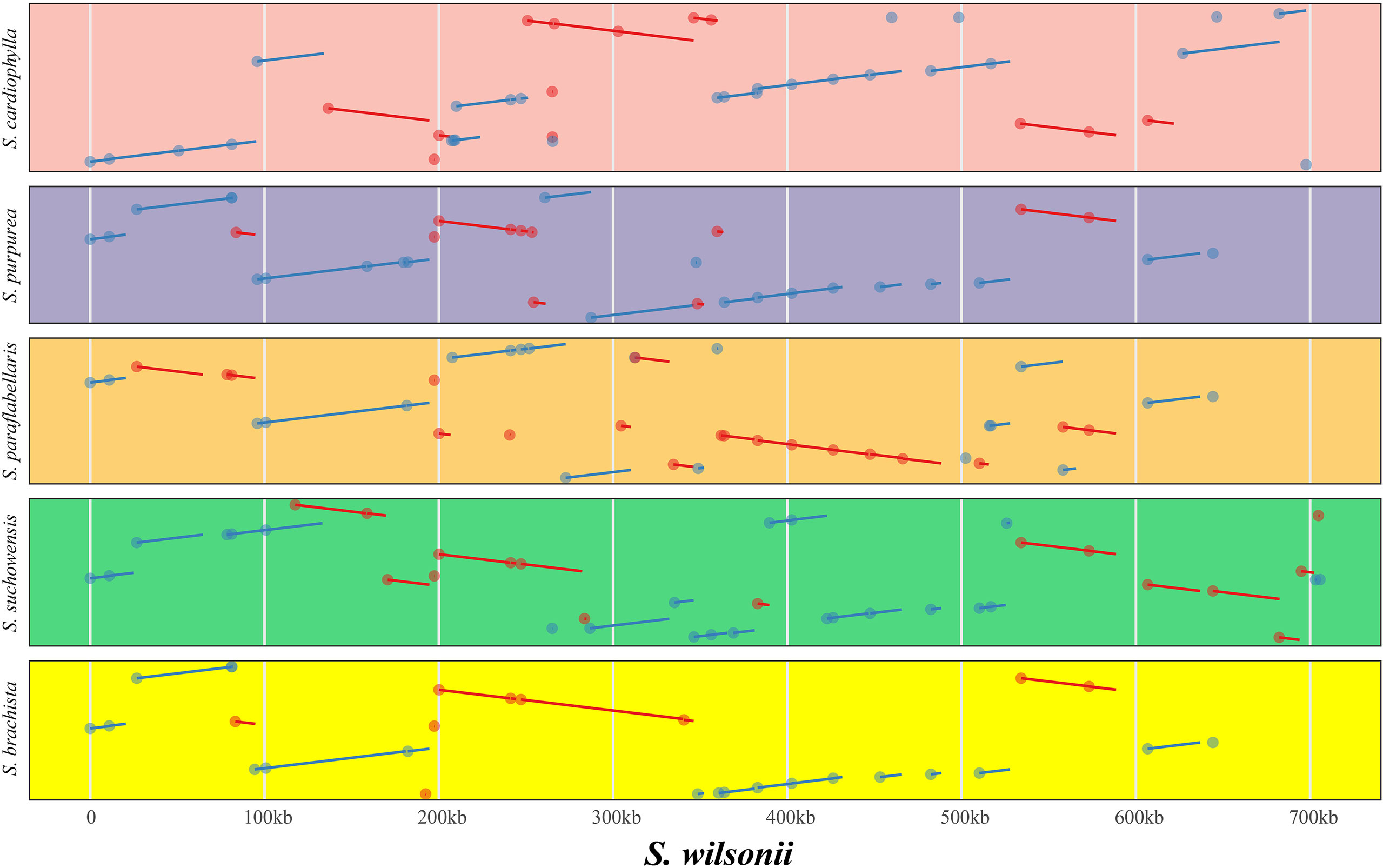
Figure 4 Covariance analysis of six different species in the genus Salix using S. wilsonii as the reference genome. The pink, purple, gold, green, and yellow represent the covariate regions of S. wilsonii vs. S. cardiophylla, S. wilsonii vs. S. purpurea, S. wilsonii vs. S. paraflabellaris, S.wilsonii vs. S. suchowensis, and S. wilsonii vs. S. brachista, respectively. The red and blue lines indicate forward and inverted covariance, respectively.
The phenomenon of horizontal sequence transfer occurs frequently between organelles and is an important cause of mitogenome expansion. In this study, multiple chloroplast-derived sequence transfer events in the S. wilsonii mitogenome were identified (Wu et al., 2019). Figure 5 shows numerous shared transfer sequences between the chloroplast genome and the mitogenome of S. wilsonii. A total of 43 DNA fragments with a total length of 23,368 bp were derived from the chloroplast genome, accounting for 3.28% of the S. wilsonii mitogenome (Supplementary Table 4), which was a very common proportion of the known angiosperm mitogenomes (Wang et al., 2019). A total of six sequences were longer than 1,500 bp in size, with the longest fragment measuring 2,800 bp. Among the chloroplast-derived sequences, four complete chloroplast PCGs (psaA, psbC, psbD, and psbH) and seven complete tRNA genes (trnp-UGG, trnD-GUC, trnM-CAU, trnN-GUU, trnV-GAC, trnV-UAC, and trnW-CCA) were observed. Additionally, our results also demonstrated that some PCGs, i.e., rrn16, psbB, clpP, psbA, rpoC2, rps7, rps12, and accD, migrated from the chloroplast genome to the mitogenome in S. wilsonii (Supplementary Table 4), and most of them lost their integrity during evolution.
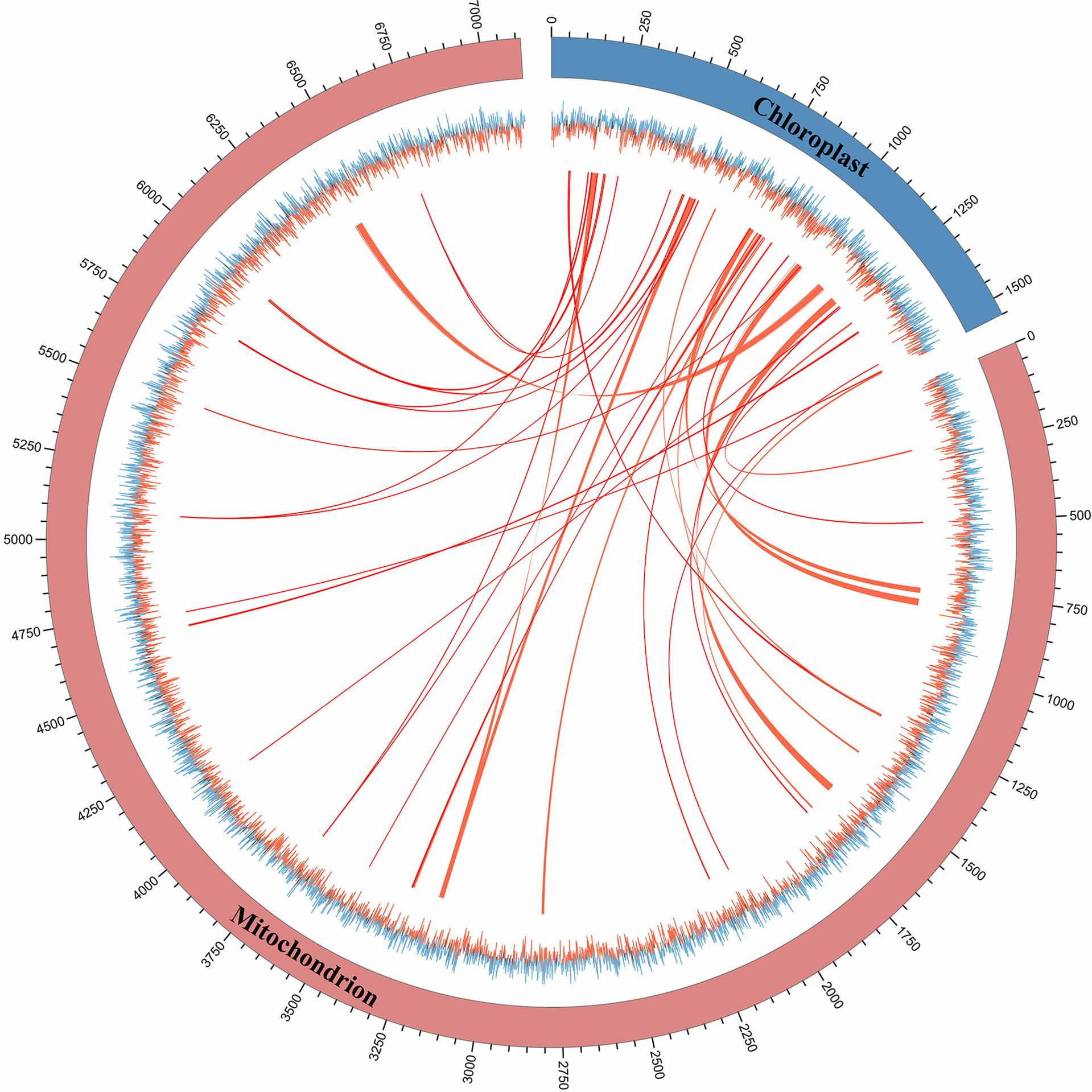
Figure 5 The shared transfer sequence of the mitogenome and chloroplast genome of S. wilsonii. The folded line represents the GC skew of the mitogenome and chloroplast genome. The linkage indicates the shared transfer sequence between the mitogenome and the chloroplast genome.
A phylogenetic analysis based on the conserved PCGs of S. wilsonii and 12 other species of Malpighiales, including 8 Salicaceae (5 Salix and 3 Populus), one Passifloraceae, one Rhizophoraceae, one Plantae, and one Magnoliophyta species, was performed (Figure 6). The phylogenetic tree strongly supported the segregation of S. wilsonii and five other Salix plants with 100% bootstrap support, as well as the segregation of Salix and Populus (100%) and the segregation of Passifloraceae and Salicaceae (100%). In addition, the Passifloraceae clustered with the Euphorbiaceae, which is consistent with traditional phylogenetic relationships (Group et al., 2016).
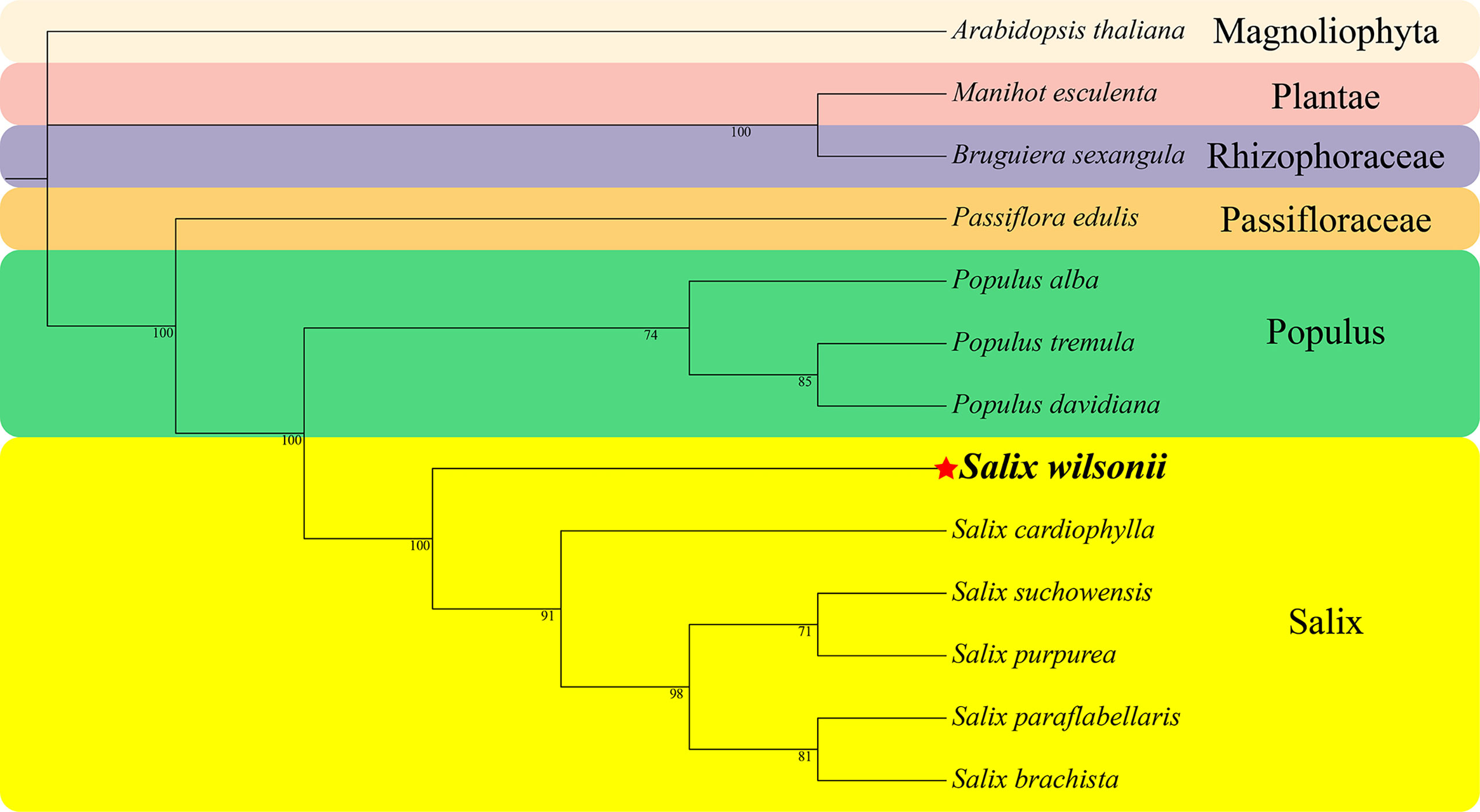
Figure 6 The phylogenetic relationships of Malpighiales. The number on each branch is the bootstrap support value. Colors indicate families of each species, where Populus and Salix of Salicaceae are indicated.
Due to the limitations of DNA sequencing technology, only 14 plant mitogenomes were released before 2005, and most of them were assembled by Sanger capillary sequencers (Jansen et al., 2005), such as those of Marchantia polymorpha (Oda et al., 1992), Nicotiana tabacum (Sugiyama et al., 2005), and some algae. Benefiting from the emergence of next-generation sequence (NGS) and TGS technology, as well as some brilliant assembly tools, an increasing number of mitogenomes of cash crops have been released, including those of Oryza sativa, Zea mays, Sorghum bicolor, and Cucumis sativus (Clifton et al., 2004; Tian et al., 2006; Alverson et al., 2011). However, limited by the short sequencing length of NGS technology and the high error rate of TGS technology, the de novo assembly of complex plant mitogenomes is challenging. With the rise of the PacBio HiFi sequencing method, which yields highly accurate long-read sequence datasets, it has rapidly become the ‘gold’ standard for the de novo assembly of genomes. Most published Salix mitogenomes were assembled by the next-generation sequencing data, which may result in incorrect assembly of some regions (Supplementary Table 5). In this research, the first de novo assembly of the S. wilsonii mitogenome was completed using PacBio HiFi sequence technology, providing a reference for its genetic study.
The plant mitogenome is a dynamically changing entity during evolution, showing great variation in structure and size among species (Bi et al., 2022). For this reason, studies of the plant mitogenome lag far behind those of the chloroplast genome. As of Aug. 2022, only 453 plant mitogenomes have been released in the NCBI Organelle Genome Database (https://www.ncbi.nlm.nih.gov/genome/), but over 7,400 chloroplast genomes have been released. Plant mitogenomes contain a large number of repeat sequences and foreign sequence insertions, resulting in gene loss and multiple copies, while repeat sequences mediate genomic rearrangements that also form multichromosomal structures. Studies have shown that 71.5% of the Malus domestica mtDNA sequence is highly similar to its nuclear DNA sequence and is the driving force of its mitogenome expansion (Goremykin et al., 2012). The melon (Cucumis melo L.) mitogenome is over 2.7 Mb in size, eight times larger than that of other cucurbits, and contains a large number of repeat sequences and a high content of nucleus-derived DNA, accounting for 42% and 47% of the total sequence (Rodríguez-Moreno et al., 2011), respectively. The high frequency of recombination mediated by three pairs of long repeats in the okra (Abelmoschus esculentus) mitogenome results in four molecules existing at the same time (Li et al., 2022). Acer truncatum and Glycine max contain numerous foreign sequences, and most genes with transferred sequences are tRNA genes (Chang et al., 2013; Ma et al., 2022).
The size of the mitogenome varies considerably in the genus Salix (Table 1), ranging from 735 kb (S. cardiophylla) to 599 kb (S. purpurea). Comparative analysis of the repeat sequences of the six Salix mitogenomes revealed that the mitogenomes of S. cardiophylla and S. wilsonii were larger and had more repeat sequences. This result further indicates the importance of repeat sequences for the expansion of plant mitogenomes. The numbers of genes and PCGs in the mitogenome were not positively correlate. In the genus Salix, the number of genes ranged from 55 to 59, and the number of PCGs ranged from 28 to 33 (Table 1). However, S. cardiophylla had the fewest PCGs, which contrasts with it having the largest mitogenome. Repeat sequence rearrangements can also result in gene loss and multiple copies (Bi et al., 2016; Liao et al., 2018; Cheng et al., 2021). However, the PCGs in the S. wilsonii mitogenome are all single-copy genes, and only the tRNA genes have multiple copies. There are no repeat sequences >300 bp in size in the S. wilsonii mitogenome, which may contribute to the stability of the genome structure and gene contents. In addition, five InDels were found in three mitochondrial PCGs (ccmFc, rps3, and rps7) in the genus Salix (Supplementary Table 6). The rps7 of S. wilsonii and S. brachista had 8 and 5 bp deletions forming a shift mutation, respectively (Figure 7), which may result in the loss of gene functions. In addition to repeat sequences, insertions of foreign sequences can also significantly affect the expansion of the mitogenome. Studies have shown that tRNA genes lost from mitochondria during evolution are generally compensated for by transferred sequences from chloroplasts (Woodson and Chory, 2008). Similar to the results for the A. truncatum mitogenome, the sequences in the S. wilsonii mitogenome transferred from the chloroplast genome were mostly tRNA genes. In conclusion, repeat and foreign sequences influence the expansion of the mitogenome and even affect some important functions.
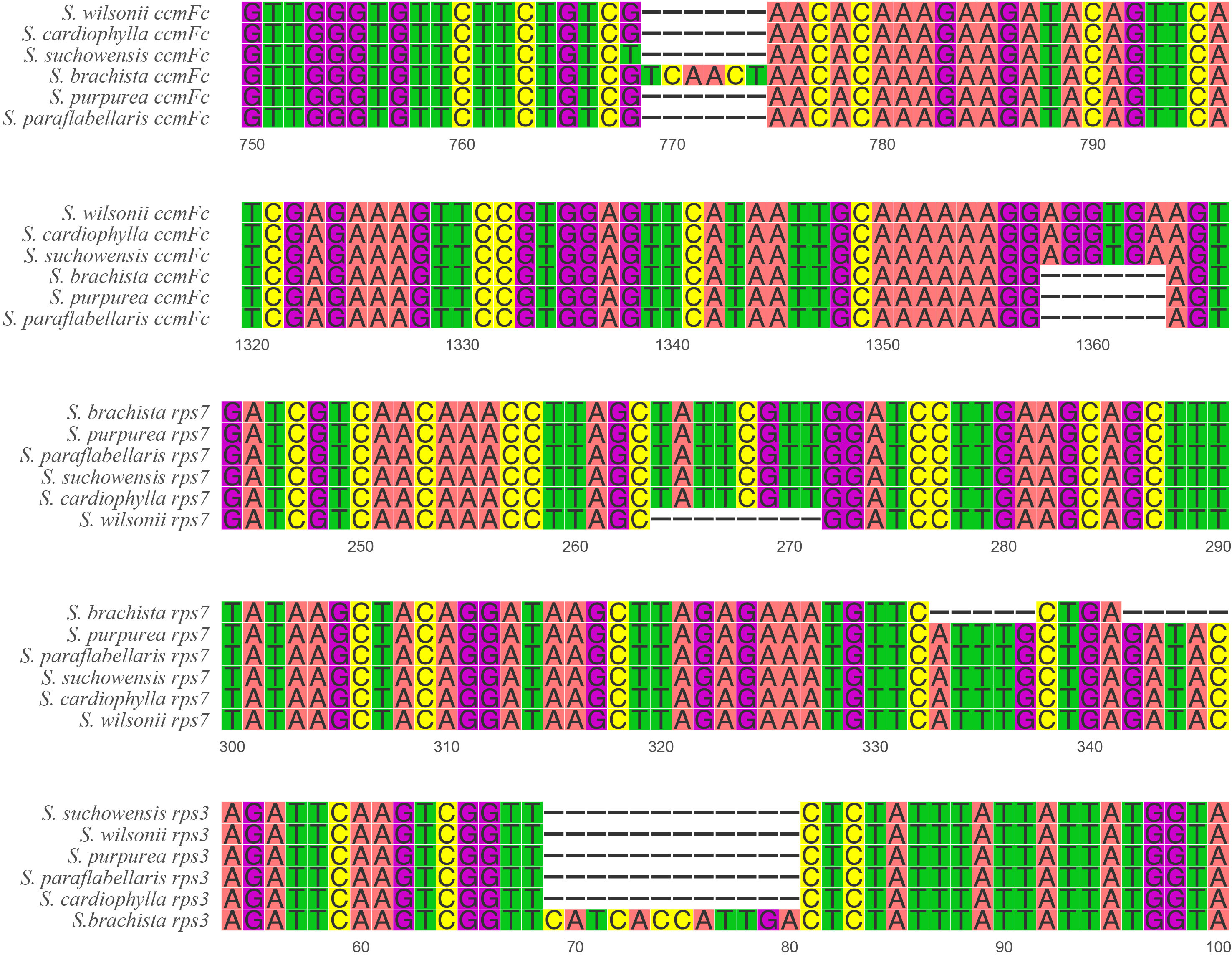
Figure 7 InDel distribution of PCGs in six Salix mitogenomes.
Repeated sequences mediate genomic rearrangements, causing significant variation in mitogenomes among species. Studies have proven that mitogenome structure is highly diverse in the genus Populus, with the mitogenomes of Populus simonii and Populus deltoides showing a multicircular structure (Bi et al., 2022; Qu et al., 2022) but those of Populus alba and Populus davidiana both having a single circular chromosome. The current findings indicate that all Salix mitogenomes have a single circular chromosome, but the collinearity within the genus Salix is not as strong as that with the genus Populus (Bi et al., 2022), suggesting rich species diversity in Salix. Notably, there was strong collinearity in the protein-coding regions within the genus Salix, a result similar to that in C. vulgaris (Turmel et al., 2003). The results further prove that the structure of the plant mitogenome is complicated and variable but that the sequences of its PCGs are highly conserved.
During evolution, most PCGs in mitochondria are relatively conserved, contributing to the maintenance of normal mitochondrial function. Analysis of selection pressure showed that atp4, ccmB, mttB, nad3, nad4, and sdh4 were subject to positive selection (Ka/Ks > 1) after ancestral differentiation (Figure 3). Additionally, the sdh4 gene is only present in S. wilsonii and M. esculenta among the five compared species, indicating that sdh4 is evolutionarily unstable and was frequently lost from mitogenome during evolution (Adams et al., 2001; Petersen et al., 2017).
The application of PacBio HiFi sequencing technology, which combines long reads and high accuracy, has rapidly changed previous sequencing strategies. In this study, the complete mitogenome of S. wilsonii was assembled and comparatively analyzed using PacBio HiFi sequencing. By comparative genomic analysis of the mitogenome of S. wilsonii, we determined the phylogenetic relationships of S. wilsonii. In addition, we inferred that a large number of repeat sequences and foreign sequences from the chloroplast genome are the main reasons for the expansion of the S. wilsonii mitogenome. The more conserved PCG region was further demonstrated by collinearity analysis, showing its contribution to the functional stability of mitochondria. In conclusion, this study of the mitogenome of S. wilsonii provides important information for evolutionary studies of S. wilsonii.
The original contributions presented in the study are publicly available. This data can be found here: NCBI, PRJNA880582.
FH: bioinformatic analyses, writing of original draft, writing of review, and editing. YQ: data curation, resources, and software. LX: validation and funding acquisition. YC: validation and formal analysis. CB: conceptualization, funding acquisition, supervision, writing of review and editing. All authors contributed to the article and approved the submitted version.
The work is supported by the Natural Science Foundation of Jiangsu Province (BK20220414) and the Natural Science Foundation of the Higher Education Institutions of Jiangsu Province. The work is also supported by the National Key Research and Development Program of China (2021YFD2201205) and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
The authors sincerely thank laboratory members for assistance with the study.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1031769/full#supplementary-material
Adams, K. L., Rosenblueth, M., Qiu, Y. L., Palmer, J. D. (2001). Multiple losses and transfers to the nucleus of two mitochondrial succinate dehydrogenase genes during angiosperm evolution. Genetics 158, 1289–1300. doi: 10.1093/genetics/158.3.1289
Alverson, A. J., Rice, D. W., Dickinson, S., Barry, K., Palmer, J. D. (2011). Origins and recombination of the bacterial-sized multichromosomal mitochondrial genome of cucumber(c)(w). Plant Cell 23, 2499–2513. doi: 10.1105/tpc.111.087189
Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. (2017). Misa-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–85. doi: 10.1093/bioinformatics/btx198
Benson, G. (1999). Tandem repeats finder: a program to analyze dna sequences. Nucleic Acids Res. 27, 573–80. doi: 10.1093/nar/27.2.573
Bi, C., Paterson, A. H., Wang, X., Xu, Y., Wu, D., Qu, Y. (2016). Analysis of the complete mitochondrial genome sequence of the diploid cotton gossypium raimondii by comparative genomics approaches. BioMed. Res. Int. 2016, 5040598. doi: 10.1155/2016/5040598
Bi, C., Qu, Y., Hou, J., Wu, K., Ye, N., Yin, T.. (2022). Deciphering the multi-chromosomal mitochondrial genome of populus simonii. Front. Plant Sci. 13, 914635. doi: 10.3389/fpls.2022.914635
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., Madden, T. L.. (2009). Blast+: architecture and applications. BMC Bioinf. 10, 421. doi: 10.1186/1471-2105-10-421
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–52. doi: 10.1093/oxfordjournals.molbev.a026334
Changwei, B., Na, L., Yiqing, X., Chunpeng, H., Zuhong, L. (2020). Characterization and analysis of the mitochondrial genome of common bean (phaseolus vulgaris) by comparative genomic approaches. Int. J. Mol. Sci. 21, 3778. doi: 10.3390/ijms21113778
Cheng, Y., He, X., Priyadarshani, S., Wang, Y., Ye, L., Shi, C., et al. (2021). Assembly and comparative analysis of the complete mitochondrial genome of suaeda glauca. BMC Genomics 22, 167. doi: 10.1186/s12864-021-07490-9
Chen, X., Zhang, L., Huang, Y., Zhao, F. (2020). Mitochondrial genome of;and its implications for infrageneric division of the genus of. Mitochondrial DNA Part B 5, 3485–3486. doi: 10.1080/23802359.2020.1827065
Chang, S., Wang, Y., Lu, J., Gai, J., Li, J., Chu, P., et al. (2013). The mitochondrial genome of soybean reveals complex genome structures and gene evolution at intercellular and phylogenetic levels. PLoS One 8, e56502. doi: 10.1371/journal.pone.0056502
Clifton, S. W., Minx, P., Fauron, C. M., Gibson, M., Allen, J. O., Sun, H., et al. (2004). Sequence and comparative analysis of the maize nb mitochondrial genome. Plant Physiol. 136, 3486–3503. doi: 10.1104/pp.104.044602
Cole, L. W., Guo, W., Mower, J. P., Palmer, J. D. (2018). High and variable rates of repeat-mediated mitochondrial genome rearrangement in a genus of plants. Mol. Biol. Evolution. 35, 2773–85. doi: 10.1093/molbev/msy176
Dong, S., Zhao, C., Zhang, S., Zhang, L., Wu, H., Liu, H., et al. (2019). Mitochondrial genomes of the early land plant lineage liverworts (marchantiophyta): conserved genome structure, and ongoing low frequency recombination. BMC Genomics 20, 953. doi: 10.1186/s12864-019-6365-y
Gerke, P., Szövényi, P., Neubauer, A., Lenz, H., Gutmann, B., McDowell, R., et al. (2020). Towards a plant model for enigmatic u-to-c rna editing: the organelle genomes, transcriptomes, editomes and candidate rna editing factors in the hornwort anthoceros agrestis. New Phytol. 225, 1974–92. doi: 10.1111/nph.16297
Goremykin, V. V., Lockhart, P. J., Viola, R., Velasco, R. (2012). The mitochondrial genome of malus domestica and the import-driven hypothesis of mitochondrial genome expansion in seed plants. Plant J. 71, 615–26. doi: 10.1111/j.1365-313X.2012.05014.x
Green, D. R., Reed, J. C. (1998). Mitochondria and apoptosis. Science 281, 1309–12. doi: 10.1126/science.281.5381.1309
Greiner, S., Lehwark, P., Bock, R. (2019). Organellargenomedraw (ogdraw) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64. doi: 10.1093/nar/gkz238
Group, A. P., Chase, M. W., Christenhusz, M.J., Fay, M. F., Byng, J., Judd, W., et al. (2016). An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: apg iv. Botanical J. Linn. Soc. 181, 1–20. doi: 10.1016/j.jep.2015.05.035
Guo, W., Grewe, F., Fan, W., Young, G. J., Knoop, V., Palmer, J. D., et al. (2016). Ginkgo and welwitschia mitogenomes reveal extreme contrasts in gymnosperm mitochondrial evolution. Mol. Biol. Evol. 33, 1448–60. doi: 10.1093/molbev/msw024
Gupta, R. S., Golding, B. G. (1996). The origin of the eukaryotic cell. Trends Biochem. Sci. 21, 166–71. doi: 10.1016/S0968-0004(96)20013-1
Hannan, A. J. (2012). Tandem repeat polymorphisms: mediators of genetic plasticity, modulators of biological diversity and dynamic sources of disease susceptibility. Adv. Exp. Med. Biol. 769, 1–9. doi: 10.1007/978-1-4614-5434-2_1
Huang, Y., Chen, X., Chen, K., Li, W., Shi, M., Chen, J. (2019). The complete mitochondrial genome of salix paraflabellaris , an endemic alpine plant of yunnan province of china. Mitochondrial DNA Part B 4, 1394–95 doi: 10.1080/23802359.2019.1598823
Hurst, L. D. (2002). The k a/ k s ratio: diagnosing the form of sequence evolution. Trends Genet. 18, 486. doi: 10.1016/s0168-9525(02)02722-1
Jansen, R. K., Raubeson, L. A., Boore, J. L., Depamphilis, C. W., Chumley, T. W., Haberle, R. C., et al. (2005). Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymology 395, 348–384. doi: 10.1016/S0076-6879(05)95020-9
Katoh, K., Standley, D. M. (2013). Mafft multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kozik, A., Rowan, B. A., Lavelle, D., Berke, L., Schranz, M. E., Michelmore, R. W., et al. (2019). The alternative reality of plant mitochondrial dna: one ring does not rule them all. PLoS Genet. 15, e1008373. doi: 10.1371/journal.pgen.1008373
Kubo, T., Newton, K. J. (2008). Angiosperm mitochondrial genomes and mutations. Mitochondrion 8, 5–14. doi: 10.1016/j.mito.2007.10.006
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R., et al. (2001). Reputer: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–42. doi: 10.1093/nar/29.22.4633
Kuzovkina, Y. A., Weih, M., Abalos, R. M., Charles, J., Hurst, S., McIvor, I., et al. (2008). Salix: botany and global horticulture. Hortic. Rev. 34, 447–89. doi: 10.1002/9780470380147.ch8
Lewis, S. E., Searle, S. M. J., Harris, N., Gibson, M., Iyer, V., Richter, J., et al. (2002). Apollo: a sequence annotation editor. Genome Biol. 3, RESEARCH0082. doi: 10.1186/gb-2002-3-12-research0082
Li, Z.-J., Li, H.-Q., Diao, X.-M. (2009). Methods for the identification of horizontal gene transfer (hgt) events and progress in related fields:methods for the identification of horizontal gene transfer (hgt) events and progress in related fields. Hereditas (Beijing) 30, 1108–14.
Li, J., Li, J., Ma, Y., Kou, L., Wei, J., Wang, W. (2022). The complete mitochondrial genome of okra (abelmoschus esculentus): using nanopore long reads to investigate gene transfer from chloroplast genomes and rearrangements of mitochondrial dna molecules. BMC Genomics 23, 481. doi: 10.1186/s12864-022-08706-2
Lilly, J. W., Havey, M. J. (2001). Small, repetitive dnas contribute significantly to the expanded mitochondrial genome of cucumber. Genetics 159, 317–328. doi: 10.1093/genetics/159.1.317
Li, W. H., Wu, C. I., Luo, C. C. (1985). A new method for estimating synonymous and nonsynonymous rates of nucleotide substitution considering the relative likelihood of nucleotide and codon changes. Mol. Biol. Evol. 2, 150–174. doi: 10.1093/oxfordjournals.molbev.a040343
Liao, X., Zhao, Y., Kong, X., Khan, A., Zhou, B., Liu, D., et al. (2018). Complete sequence of kenaf hibiscus cannabinus mitochondrial genome and comparative analysis with the mitochondrial genomes of other plants. Sci. Rep. 8, 1–13. doi: 10.1038/s41598-018-30297-w
Marçais, G., Delcher, A.L., Phillippy, A.M., Coston, R., Salzberg, S.L., and Zimin, A., et al. (2018). Mummer4: a fast and versatile genome alignment system. PLoS Comput. Biol. 14, e1005944. doi: 10.1371/journal.pcbi.1005944
Ma, Q., Wang, Y., Li, S., Wen, J., Zhu, L., Yan, K., et al. (2022). Assembly and comparative analysis of the first complete mitochondrial genome of acer truncatum bunge: a woody oil-tree species producing nervonic acid. BMC Plant Biol. 22, 29. doi: 10.1186/s12870-021-03416-5
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., von Haeseler, A., Lanfear, R. (2020). Iq-tree 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–34. doi: 10.1093/molbev/msaa015
McBride, H. M., Neuspiel, M., Wasiak, S. (2006). Mitochondria: more than just a powerhouse. Curr. Biol. 16, R551–R560. doi: 10.1016/j.cub.2006.06.054
Notsu, Y., Masood, S., Nishikawa, T., Kubo, N., Akiduki, G., Nakazono, M., et al. (2002). The complete sequence of the rice (oryza sativa l.) mitochondrial genome: frequent dna sequence acquisition and loss during the evolution of flowering plants. Mol. Genet. Genomics Mgg 268, 434–445. doi: 10.1007/s00438-002-0767-1
Oda, K., Yamato, K., Ohta, E., Nakamura, Y., Takemura, M., Nozato, N., et al. (1992). Gene organization deduced from the complete sequence of liverwort marchantia polymorpha mitochondrial dna: a primitive form of plant mitochondrial genome. J. Mol. Biol. 223, 1–7. doi: 10.1016/0022-2836(92)90708-r
Osiewacz, H. D., Brust, D., Hamann, A., Kunstmann, B., Luce, K., Müller-Ohldach, M., et al. (2010). Mitochondrial pathways governing stress resistance, life, and death in the fungal aging model podospora anserina. Ann. New York Acad. Sci. 1197, 54–66. doi: 10.1111/j.1749-6632.2010.05190.x
Olson, Å., Stenlid, J. (2001). Mitochondrial control of fungal hybrid virulence. Nature 411, 438. doi: 10.1038/35078147
Palmer, J. D., Adams, K. L., Cho, Y., Parkinson, C. L., Qiu, Y. L., Song, K., et al. (2000). Dynamic evolution of plant mitochondrial genomes: mobile genes and introns and highly variable mutation rates. Proc. Natl. Acad. Sci. United States America 97, 6960–6966. doi: 10.1073/pnas.97.13.6960
Petersen, G., Cuenca, A., Zervas, A., Ross, G. T., Graham, S. W., Barrett, C. F., et al. (2017). Mitochondrial genome evolution in alismatales: size reduction and extensive loss of ribosomal protein genes. PLoS One 12, e0177606. doi: 10.1371/journal.pone.0177606
Powell, W., Machray, G. C., Provan, J. (1996). Polymorphism revealed by simple sequence repeats. Trends Plant Sci. 1, 215–222. doi: 10.1016/1360-1385(96)86898-1
Qu, Y., Zhou, P., Tong, C., Bi, C., Xu, L. A. (2022). Assembly and analysis of the populus deltoides mitochondrial genome: the first report of a multicircular mitochondrial conformation for the genus populus. J. Forestry Res, 1–17. doi: 10.1007/s11676-022-01511-3
Richardson, A. O., Palmer, J. D. (2007). Horizontal gene transfer in plants. J. Exp. Bot. 58, 1–9. doi: 10.1093/jxb/erl148
Rodríguez-Moreno, L., González, V. M., Benjak, A., Martí, M. C., Puigdomènech, P., Aranda, M. A., et al. (2011). Determination of the melon chloroplast and mitochondrial genome sequences reveals that the largest reported mitochondrial genome in plants contains a significant amount of dna having a nuclear origin. BMC Genomics 12, 424. doi: 10.1186/1471-2164-12-424
Shtolz, N., Mishmar, D. (2019). The mitochondrial genome–on selective constraints and signatures at the organism, cell, and single mitochondrion levels. Front. Ecol. Evol. 7, 342. doi: 10.3389/fevo.2019.00342
Skippington, E., Barkman, T. J., Rice, D. W., Palmer, J. D. (2015). Miniaturized mitogenome of the parasitic plant viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc. Natl. Acad. Sci. United States America 112, E3515–E3524. doi: 10.1073/pnas.1504491112
Sloan, D. B., Alverson, A. J., Chuckalovcak, J. P., Wu, M., McCauley, D. E., Palmer, J. D., et al. (2012). Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 10, e1001241. doi: 10.1371/journal.pbio.1001241
Smith, D. R. (2011). Extending the limited transfer window hypothesis to inter-organelle dna migration. Genome Biol. Evol. 3, 743–748. doi: 10.1093/gbe/evr068
Sugiyama, Y., Watase, Y., Nagase, M., Makita, N., Yagura, S., Hirai, A., et al. (2005). The complete nucleotide sequence and multipartite organization of the tobacco mitochondrial genome: comparative analysis of mitochondrial genomes in higher plants. Mol. Genet. Genomics Mgg 272, 603–615. doi: 10.1007/s00438-004-1075-8
Tian, X., Zheng, J., Hu, S., Yu, J. (2006). The rice mitochondrial genomes and their variations. Plant Physiol. 140, 401–10. doi: 10.1104/pp.105.070060
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). Geseq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Turmel, M., Otis, C., Lemieux, C. (2003). The mitochondrial genome of chara vulgaris: insights into the mitochondrial dna architecture of the last common ancestor of green algae and land plants. Plant Cell 15, 1888–1903. doi: 10.1105/tpc.013169
van Oven, M., Kayser, M. (2009). Updated comprehensive phylogenetic tree of global human mitochondrial dna variation. Hum. Mutat. 30, E386–E394. doi: 10.1002/humu.20921
Wang, D., Zhang, Y., Zhang, Z., Zhu, J., Yu, J. (2010). Kaks_calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinf. 8, 77–80. doi: 10.1016/S1672-0229(10)60008-3
Wang, S., Li, D., Yao, X., Song, Q., Wang, Z., Zhang, Q., et al. (2019). Evolution and diversification of kiwifruit mitogenomes through extensive whole-genome rearrangement and mosaic loss of intergenic sequences in a highly variable region. Genome Biol. Evol. 11, 1192–1206. doi: 10.1093/gbe/evz063
Wickham, H. (2016). Package ‘ggplot2’: elegant graphics for data analysis (New York: Springer-Verlag) 10, 978–70.
Woodson, J. D., Chory, J. (2008). Coordination of gene expression between organellar and nuclear genomes. Nat. Rev. Genet. 9, 383–395. doi: 10.1038/nrg2348
Wu, D., Wang, Y., Zhang, L. (2019). The complete chloroplast genome sequence of an economic plant salix wilsonii. Mitochondrial DNA Part B 4, 3560–3562. doi: 10.1080/23802359.2019.1668311
Yang, Z., Ni, Y., Lin, Z., Yang, L., Chen, G., Nijiati, N., et al. (2022). De novo assembly of the complete mitochondrial genome of sweet potato (ipomoea batatas [l.] lam) revealed the existence of homologous conformations generated by the repeat-mediated recombination. BMC Plant Biol. 22, 1–12. doi: 10.1186/s12870-022-03665-y
Ye, N., Wang, X., Li, J., Bi, C., Xu, Y., Wu, D., et al. (2017). Assembly and comparative analysis of complete mitochondrial genome sequence of an economic plant salix suchowensis. Peerj 5, e3148. doi: 10.7717/peerj.3148
Keywords: Salix wilsonii, mitochondrial genome, comparative analysis, HiFi sequencing, assembly
Citation: Han F, Qu Y, Chen Y, Xu L and Bi C (2022) Assembly and comparative analysis of the complete mitochondrial genome of Salix wilsonii using PacBio HiFi sequencing. Front. Plant Sci. 13:1031769. doi: 10.3389/fpls.2022.1031769
Received: 03 September 2022; Accepted: 17 October 2022;
Published: 16 November 2022.
Edited by:
Jihong Hu, Northwest A&F University, ChinaReviewed by:
Sang-Ho Kang, National Institute of Agricultural Science, South KoreaCopyright © 2022 Han, Qu, Chen, Xu and Bi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Changwei Bi, YmljaHdlaUAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.