
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 03 November 2022
Sec. Plant Bioinformatics
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.1004359
This article is part of the Research Topic Crop Population Genomics and Pan-omics for Gene Discovery and Breeding View all 6 articles
 Javaria Tabassum1,2†
Javaria Tabassum1,2† Qasim Raza3,4†
Qasim Raza3,4† Awais Riaz4,5†
Awais Riaz4,5† Shakeel Ahmad1,6
Shakeel Ahmad1,6 Muhammad Abdul Rehman Rashid7
Muhammad Abdul Rehman Rashid7 Muhammad Arshad Javed2
Muhammad Arshad Javed2 Zulfiqar Ali8
Zulfiqar Ali8 Fengyu Kang1
Fengyu Kang1 Iqrar Ahmad Khan3,9
Iqrar Ahmad Khan3,9 Rana Muhammad Atif3,8*
Rana Muhammad Atif3,8* Ju Luo1*
Ju Luo1*DNA-binding with one finger (Dof) transcription factors have been demonstrated to regulate various stresses and developmental processes in plants. Their identification and comparative evolutionary analyses in cultivated and wild species of genus oryza were yet to be explored. In this context, we report a comprehensive genomics atlas of DNA-binding with one finger (Dof) family genes in 13 diverse rice genomes (five cultivated and eight rice wild-relatives) through a genome-wide scanning approach. A galore of 238 Dof genes, identified across the genus Oryza, are categorized into seven distinct subgroups by comparative phylogenetic analysis with the model plant Arabidopsis. Conserved motifs and gene structure analyses unveiled the prevalence of species- and subgroups-specific structural and functional diversity that is expediating with the evolutionary period. Our results indicate that Dof genes might have undergone strong purifying selections and segmental duplications to expand their gene family members in corresponding Oryza genomes. We speculate that miR2927 potentially targets the Dof domain to regulate gene expression under different climatic conditions, which are supported by in-silico and wet-lab experiments-based expression profiles. In a nutshell, we report several superior haplotypes significantly associated with early flowering in a treasure trove of 3,010 sequenced rice accessions and have validated these haplotypes with two years of field evaluation-based flowering data of a representative subpanel. Finally, we have provided some insights on the resolution of Oryza species phylogeny discordance and divergence highlighting the mosaic evolutionary history of the genus Oryza. Overall, this study reports a complete genomic landscape of the Dof family in cultivated and wild Oryza species that could greatly facilitate in fast-track development of early maturing and climate-resilient rice cultivars through modern haplotype-led breeding.
Transcription factors (TFs) are regulatory proteins that bind to specific nucleotide sequences in the promoter region of target genes and regulate their expressions both at transcriptional and post-transcriptional levels (Riechmann et al., 2000). This regulation affects various downstream fundamental processes crucial for optimal growth and development. Therefore, identification as well as functional characterization of TFs are indispensable for understanding and exploiting their regulatory functions.
Among several known TF families, members of the DNA-binding with one finger (Dof) subfamily are plant-specific proteins belonging to the zinc finger multigene superfamily (Noguero et al., 2013). Dof proteins are distributed throughout the plant kingdom; from simple unicellular to complex multicellular organisms (Moreno-Risueno et al., 2007). N-terminal of these proteins contains a single copy of 52 amino acids long highly conserved core Dof domain, whereas C-terminal contains a less conserved transcriptional activation domain. The Dof domain forms a C2-C2 zinc finger motif that binds to the AAAG cis-regulatory element in the promoter region of target genes (Yanagisawa, 2016). Earlier evidence suggests that besides DNA binding activity, Dof proteins also participate in protein-protein interactions and physically interact with other transcription factor families such as bZIP, MYB, WRKY, and ZFP (Yanagisawa, 2002; Isabel-LaMoneda et al., 2003; Zou et al., 2008; Wei et al., 2016). Dof TFs play multifarious roles in plants including cell signaling (Riechmann et al., 2000), hormonal signaling (Moreno-Risueno et al., 2007), differentiation of guard cells (Negi et al., 2013), fatty acid metabolism (Ibáñez-Salazar et al., 2014), flower abscission (Wei et al., 2010), formation of interfascicular cambium (Guo et al., 2009), photoperiodic flowering (Fornara et al., 2009), nitrogen assimilation (Wang et al., 2013), regulation of flowering time (Liu et al., 2020), seed and seed development (Ruta et al., 2020), and tolerance against abiotic and biotic stresses (Bhullar et al., 2007; Liu et al., 2019). Keeping in view their diversified roles in biological and physiological processes, it is imperative to explore these proteins in the genomes of important crop species and gain insights into their evolutionary patterns.
Rice (Oryza sativa L.) is one of the oldest and most consumed cereal crops. More than 50% of the world’s population solely depends upon it to fulfill their daily calorie requirements (Fukagawa and Ziska, 2019; Raza et al., 2020). The Oryza genus contains at least 22 diploids (2n = 2X = 24) and 5 tetraploids (2n = 4X = 48) species, which originated from a common wild grass ancestor about 130 million years ago and spread to different parts of the world (Sangeetha et al., 2020). Among all known rice species, only two diploid species Oryza sativa (Asian rice) and Oryza glaberrima (African rice) are cultivated around the world. O. sativa is cultivated on more than 90% of the total world area under rice cultivation and the majority of it is produced and consumed in Asian countries (Fukagawa and Ziska, 2019). Rice also serves as a model cereal crop due to its small genome size, ease of genetic transformation, and genomic synteny with other important grass species (Jackson, 2016). The draft genome sequences of two subspecies of O. sativa (indica and japonica) were completed in 2002 (Goff et al., 2002; Yu et al., 2002) and high-quality genome annotation of japonica rice was available in 2004 (Matsumoto et al., 2005), which was later improved in 2013 (Kawahara et al., 2013) and serves as a foundation of cereal genomics (Matsumoto et al., 2016). The genomes of O. glaberrima (Wang et al., 2014) and several other cultivated and wild rice species have also been sequenced (Stein et al., 2018). Comparative genomic studies among sequenced rice genomes would help investigations on evolution and genomic diversity among Oryza species and could provide deeper insights into crop improvement under rapidly changing global environments (Raza et al., 2020).
Genome-wide analysis of Dof family TFs has been reported in several plant species such as Arabidopsis and rice (Lijavetzky et al., 2003), sorghum (Kushwaha et al., 2011), cotton (Chattha et al., 2020) and olive (Mariyam et al., 2021). However, a comparative genome-wide analysis of Dof family TFs among cultivated and wild rice relatives is missing so far. With the advent of next-generation sequencing technologies, many cultivated rice cultivars and wild relatives have been sequenced in the recent past (Stein et al., 2018; Wang et al., 2018). The availability of this genomic data provides an opportunity to perform comparative genomic studies and gain new insights into the genomic diversity and evolutionary patterns of Oryza species. So far, only a single comparative study of Dof family transcription factors has been reported in cultivated and ancestral cotton species (Chattha et al., 2020). Here, in this study, we performed comprehensive whole-genome analyses and identified Dof family TFs from 10 cultivated and wild rice species including O. sativa (indica and japonica), O. nivara, O. rufipogon, O. glaberrima, O. barthii, O. glumipatula, O. meridionalis, O. punctata, and O. brachyantha. Based upon comparative phylogenetic analyses, the rice Dof TFs were classified into different subgroups. Gene structures, conserved motifs, and gene duplication analyses were also performed to understand the evolutionary patterns. Furthermore, promoter analysis, prediction of miRNA target sites and in-silico, as well as wet-lab experiments-based expression analyses in different tissues and conditions, were also investigated. Moreover, superior haplotypes for earliness breeding were also identified along with resolving the complex evolutionary phylogenetic relationships among closely related species. In conclusion, this study provides a complete genomic landscape of Dof family TFs in ten Oryza genomes that could be further exploited for wider cultivation and crop improvement by humans.
The complete genome assemblies along with annotation files of four Oryza sativa indica genotypes i.e. 93-11, Minghui 63 (MH63), Zhenshan 97 (ZS97), and Shuhui498 (R498) were retrieved (Supplementary Table S1) for the identification of Dof encoding genes across these genomes. Moreover, genome assemblies of nine other Oryza species including O. sativa japonica, O. barthii, O. brachyantha, O. glaberrima, O. glumipatula, O. meridionalis, O. nivara, O. punctata, and O. rufipogon were were also downloaded (Supplementary Table S1) for further comparative analysis of Dof genes. These downloaded genome sequences were thoroughly examined for the identification of Dof family members in all given rice species using CLC Sequence Viewer version 7 (CLC Bio, QIAGEN A/S, Aarhus, Denmark). The presence of the conserved Dof domain was further validated through a conserved domain search tool (accessed online at https://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml) (Marchler-Bauer et al., 2015). The molecular weight and isoelectric points of all the identified Dof genes were computed using Expasy Compute pI/Mw web tool (https://web.expasy.org/compute_pi/) (Gasteiger et al., 2003). The renaming of identified genes was done according to the Oryza species-specific chromosomal location information as adapted for other plant species (Yanagisawa, 2002; Chattha et al., 2020 Supplementary Table S3). For instance, OsInDof1.5 refers to the fifth Dof gene present on chr. 1 of O. sativa indica. (Supplementary Table S4).
The protein sequences of all the identified Dofs from four genomes of indica rice were aligned using Clustal Omega (Sievers et al., 2014; accessed at https://www.ebi.ac.uk/Tools/msa/clustalo/) using default parameters. The resultant multiple sequence alignment was used for constructing a phylogenetic tree using the neighbor-joining method employing molecular evolutionary genetics analysis software (MEGA-X) (Kumar et al., 2018). The similarities and differences among the Dofs from each of the four genomes were visualized using a Venn diagram through a webtool (accessed at https://bioinformatics.psb.ugent.be/webtools/Venn/).
The Dof protein sequences of Arabidopsis (Yanagisawa, 2002) and ten Oryza species were subjected to multiple sequence alignment using MAFFT (v7) (accessible at https://mafft.cbrc.jp/alignment/server/) (Katoh et al., 2018) by choosing L-INS-i algorithm with default parameters. The maximum likelihood (ML) comparative phylogenetic tree was inferred with IQTree (Nguyen et al., 2015) by choosing JTTDCMut+F+I+G4 best fit substitution model (Kalyaanamoorthy et al., 2017) according to the Bayesian information criterion. The consistency of the ML tree was validated by setting an Ultrafast bootstrap value of 1000 (Hoang et al., 2018). The final phylogenetic tree was visualized with MEGA-X (Kumar et al., 2016). The Dof genes were classified into different groups by adopting the classification scheme of Yanagisawa (2002). Paralogous and orthologous genes were assigned based on the results of comparative phylogenetic analysis.
The GFF3 files of all Oryza species were subjected to TBtools for displaying the gene structures (Chen et al., 2020). For conserved motifs analysis, peptide sequences of all Oryza species Dof genes were uploaded in MEME v5.4.1 (Bailey et al., 2009) with the following parameters: motif distribution, zero or one occurrence per sequence; the number of motifs, 10; motifs width, 6–200, while all other parameters were kept default. The identified motifs were mapped onto the peptide lengths of all Dof genes with TBtools (Chen et al., 2020). The detailed information of identified motifs in all Oryza species is given in Supplementary Table S5.
Coding sequences of all identified Dof genes were blasted against each other using Sequence Demarcation Tool V1.2 (Muhire et al., 2014) for computing the sequence similarities. Gene pairs with approximate≥ 90% similarity (E value <1e-10) were considered as duplicated genes and retained for further analysis. Furthermore, the CDS sequences of duplicated gene pairs were aligned with ClustalW in MEGA7 and subjected to Synonymous Non-synonymous Analysis Program V2.1.1 (www.hiv.lanl.gov/content/sequence/SNAP/SNAP.html) to compute the substitution rates. The ratio of non-synonymous to synonymous substitutions was also calculated to assume which type of codon selection operated on duplicated gene pairs during their evolution. Moreover, the approximate divergence time between duplicated gene pairs was also computed by using the formulae T=Ks/2r × 10-6 assuming a substitution rate (r) of 6.5 × 10-9 substitutions/synonymous site/year (El Baidouri et al., 2017). Finally, a circular diagram showing the links between duplicated gene pairs among ten Oryza genomes was created with shiny Circos (Yu et al., 2018). The detailed information of all duplicated gene pairs is given in Supplementary Table S6.
The 2 kb upstream sequences from the transcription start site of all Dof genes were retrieved from the Regulatory Sequence Analysis Tool homepage (http://rsat.eead.csic.es/plants/index.php) (Nguyen et al., 2018) by searching through corresponding gene IDs. To prevent overlap with neighboring genes, the sequences were clipped when a predicted gene is located within the promoter region. Cis-acting regulatory elements (CAREs) were identified by uploading the promoter sequences in PlantCARE database with default parameters (Lescot et al., 2002). The CAREs without any putative function/role were discarded, as well as those involved in core transcription initiation function in eukaryotes such as TATA and CAAT/CAT boxes. The identified CAREs were mapped onto the promoter regions of all Oryza species Dof genes with TBtools (Chen et al., 2020). The detailed results of promoter analysis are given in Supplementary Table S7 and summarized in Supplementary Table S8.
miRNAs potentially targeting the identified Dof genes in two cultivated rice genomes (O. sativa indica and japonica) were predicted by uploading their transcript sequences on the web-based psRNA Target Server (https://www.zhaolab.org/psRNATarget/) (Dai et al., 2018). Stringent selection criteria with a penalty score of ≤2.0 were adopted for the identification of targets using the already reported rice miRNA database available at the above-mentioned web server. Only frequently occurring miRNAs were considered for creating the final miRNA target site prediction figure.
The normalized (Log2 fold change) expression profiles in root and shoot tissues of O. sativa japonica in response to six plant hormones including abscisic acid, auxin, brassinosteroid, cytokinin gibberellin, and jasmonic acid were retrieved from the RiceXPro database (http://ricexpro.dna.afrc.go.jp) (Sato et al., 2013). These data were collected from 7-days old rice seedlings individually treated with different plant hormones and incubated for different periods. Details of expression analysis and normalization were described by Sato et al. (2013). Similarly, O. sativa indica expression data of low light-sensitive (IR8) and tolerant (Swarnaprabha) rice genotypes were mined from Sekhar et al. (2019). Briefly, both rice genotypes were exposed to low light treatments for one day (T1), three days (T3), and five days (T5) along with control treatments during the active tillering stage (40 days after germination) before the final expression analysis. Finally, the heatmaps of retrieved expression data were generated with TBtools (Chen et al., 2020).
An indica rice cultivar “9311” with sequenced genome data was used for gene expression studies under high temperature, low temperature, and low light stress conditions. Seedlings were grown for three weeks under controlled laboratory conditions at 27 ± 2°C with a relative humidity of 60% under a photoperiod of 14h/10h (day/night) before the application of stress treatments. High temperature (40 ± 1°C), low temperature (15 ± 1°C), and low light intensity (20% of control; 60 µmolm-2s-1) were administered for one week during 14h daylight periods only. Leaf and stem tissues were harvested from control and treated seedlings and stored at -80°C for subsequent RNA isolation.
Total RNA was isolated and reverse transcribed using RNA-prep pure plant kit (TIANGEN) and HiScript II QRT SuperMix (Vazyme), respectively (Chun et al., 2020). A quantitative real-time polymerase chain reaction (qRT-PCR) for 14 OsInDofs was performed using SYBR green mixture on an ABI 7500 RT-PCR detection system. Rice Ubiquitin was used as internal control and for normalization of expression data as described previously (Chun et al., 2020). The forward and reverse primer sequences of all genes are listed in Supplementary Table S10.
The 3K rice genomes (RG) variation data comprising 3,010 rice accessions belonging to indica, japonica, aus/boro, aromatic basmati/sadri, and admixture groups was mined through the rice functional genomics & breeding (RFGB v2.0) database (Sun et al., 2017; Wang et al., 2020). In the 3K RG panel, several haplotypes have been predicted using single nucleotide polymorphisms (SNPs) based on non-synonymous substitutions in the coding region of protein-encoding genes. By choosing O. sativa japonica Dof genes having orthologs in all other studied Oryza genomes, we selected only those haplotypes which were significantly associated with days to heading phenotypic trait (Wang et al., 2020) (Supplementary Table S11).
A sub-panel of 22 rice accessions belonging to all major and minor groups of the 3K RG panel was selected for field evaluation (Supplementary Table S12). Thirty-days old seedlings of sub-panel accessions were transplanted in the puddled field at a distance of 22.5 × 22.5 cm with a fertilizer dose of 116:87:62 (NPK) during cropping seasons 2020 and 2021 at the experimental area of Rice Research Institute, Kala Shah Kaku, Pakistan. Standard agronomic and plant protection practices were followed during both growing seasons. Two rows of each accession (32 plants) were considered as two independent replications and days to heading data (from sowing date) were recorded from five randomly selected guarded plants from each row. Averaged data of both replications and seasons were considered for haplotype validation.
To gain insight into the evolution and expansion of Dof TF-encoding genes in O. sativa indica, the genomes of four important indica varieties (93-11, MH63, ZS97, and R498) were thoroughly scanned. The systematic analysis revealed that ZS97 genome harbored the highest number of non-redundant Dof genes i.e 28 genes. On the contrary, only 23 Dof genes were identified MH63 genome. Both 93-11 and R498 genomes contained 27 Dof genes. Further comparative analysis revealed the presence of orthologs of 19 common Dof genes in all the four indica genomes (Figure 1A; Supplementary Table S2). The ZS97 genome does not contain any Dof gene on chromosome no. 8 while remaining all three genomes showed the presence of a single locus on chr. 8 (Figure 1B). Contrarily, the 93-11 genome did not contain any gene on chr. 6 as compared to the other three genomes. An extra Dof gene on chr. 10 of 93-11 genome seems to have resulted from segmental duplication. Overall, the gene duplication might have played role in the expansion of Dof family members in ZS97 and 93-11 genomes. Copy number variation of some Dof genes on chr. 5, 7 10 and 12 have been observed in these indica rice genomes. For further comparative analysis, the Dof genes identified from 93-11 genome were used as representative of O. sativa indica group.
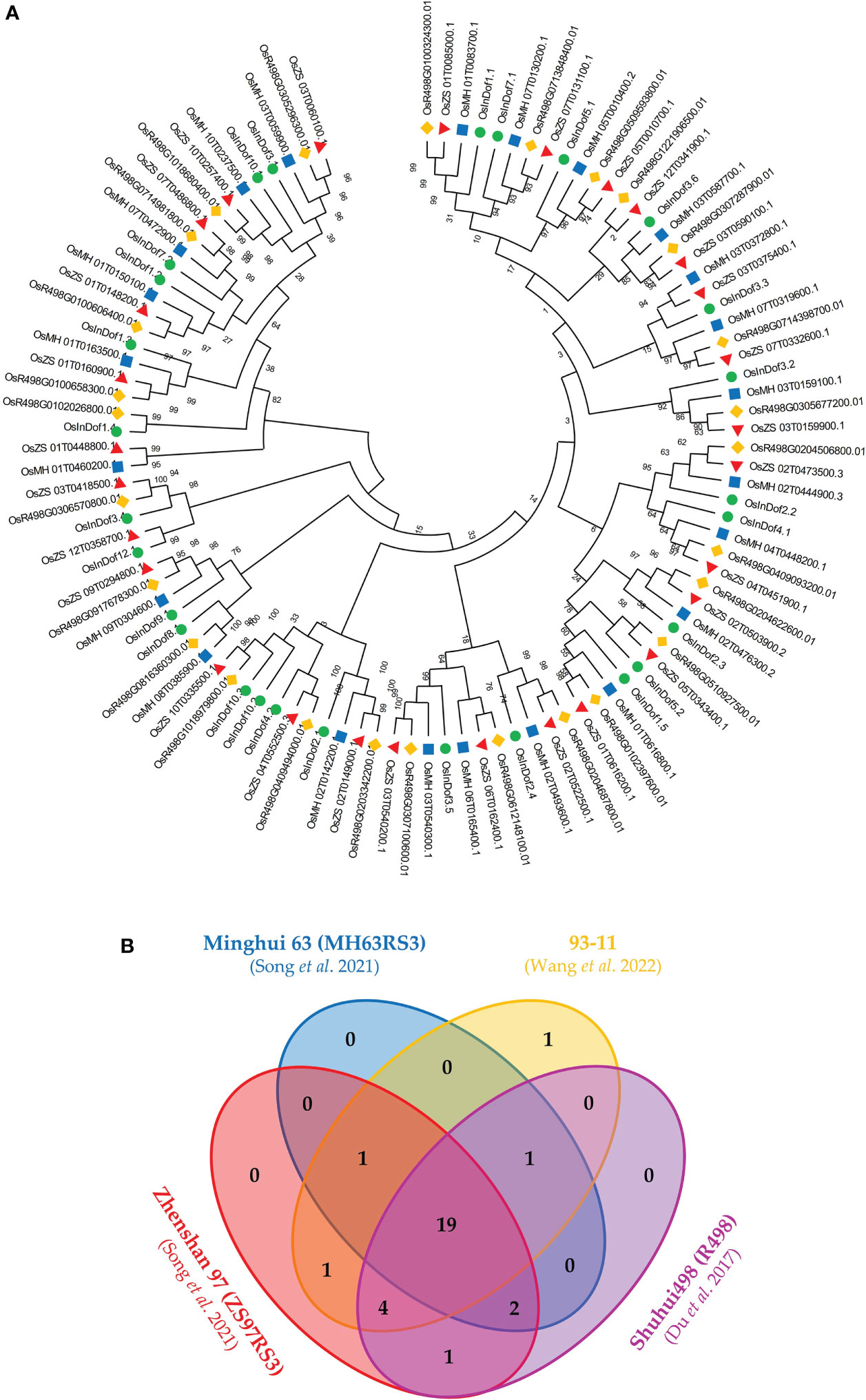
Figure 1 Comparison of Dof proteins among four elite genotypes (93-11, MH63, ZS97, and R498) of O. sativa indica. (A) The comparative phylogenetic tree of Dofs from four elite genotypes of indica rice. The evolutionary history was inferred using the Neighbor-Joining method (Saitou and Nei, 1987). The evolutionary distances were computed using the Poisson correction method (Zuckerkandl and Pauling, 1965) and are in the units of the number of amino acid substitutions per site. This analysis involved 105 protein sequences. All positions containing gaps and missing data were eliminated (complete deletion option). There was a total of 51 positions in the final dataset. Evolutionary analyses were conducted in MEGA X (Kumar et al., 2018). (B) Comparison of presence of Dof protein among four indica rice elite genotypes through Venn diagram.
Through comprehensive genome-wide scanning of whole genomes by using the Dof domain amino acids sequence as a query, a total of 250 Dof domain encoding genes were identified in 10 rice genomes. After excluding incomplete Dof domain-containing and non-annotated genes, 238 non-redundant and full-length protein encoding Dof genes were considered for further analyses (Supplementary Table S3).
The O. nivara genome assembly contained the highest (30) number of Dof genes, followed by O. glumipatula (28), O. sativa indica (27), O. rufipogon (27), O. sativa japonica (25), O. meridionalis (25), O. punctata (25), O. glaberrima (23) and O. barthii (19) (Figure 2A). Whereas the O. brachyantha genome harbored the least number (9) of Dof genes. Furthermore, the Dof groups II, V, and VI members were overrepresented, whereas groups III and VII members were less represented in all these genomes. Interestingly, O. brachyantha and O. barthii genomes lack Dof members of at least two (III and V) and one (VI) subgroups, respectively (Figure 2A), which could have been lost during the speciation events in rice crop evolution.
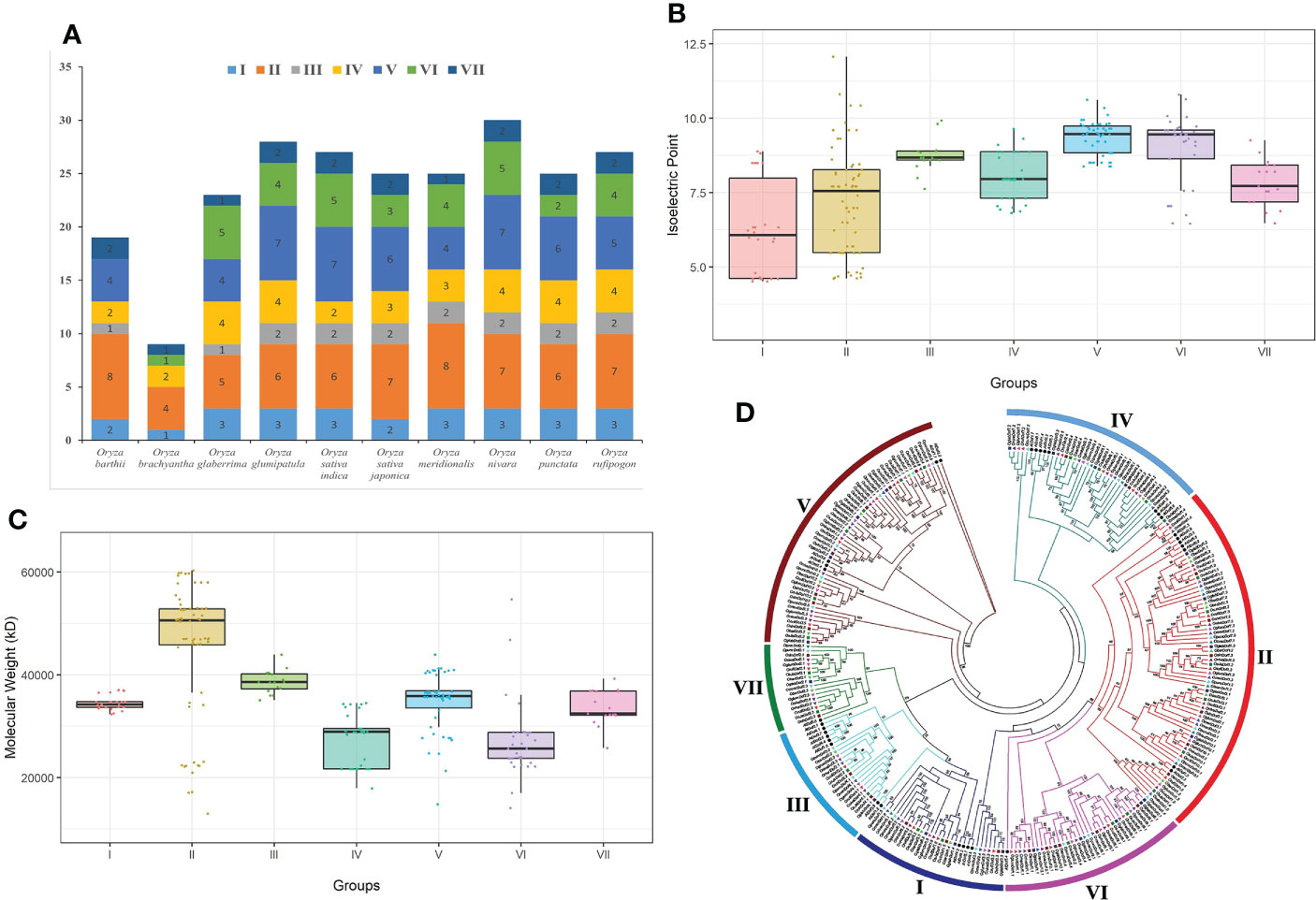
Figure 2 Classification and distribution of Dof genes among ten Oryza genomes. (A) Distribution of Dof genes in ten Oryza genomes. Each bar represents an individual genome with values on top of bars indicating the total number of Dof genes in that genome. (B) Distribution of iso-electric points and (C) molecular weights of Dof proteins belonging to different subgroups, respectively. (D) Comparative maximum likelihood phylogenetic tree of Arabidopsis and Oryza species Dof genes inferred with IQTree software (Nguyen et al., 2015) after MAFFT alignment (Katoh et al., 2018) of amino acid sequences. Dof subgroups are indicated with different colors. Arabidopsis Dof genes are labeled with solid black circles, whereas Oryza species Dof genes are labeled with species-specific triangles of different colors. Bootstrap values (≥50%) as calculated from 1,000 replicates are displayed at tree nodes.
A comparative maximum likelihood (ML) phylogenetic tree was inferred with IQTree using Arabidopsis and 10 rice genomes Dof genes to explore the evolutionary relationships among identified genes (Figure 2D). The classification of Dof genes in the resultant phylogenetic tree was carried out folowing classification scheme proposed by Yanagisawa (2002). All rice Dof genes were classified into seven different groups. Group II contained the highest number of genes (64; ~27%), followed by group V (50; ~21%), group VI (33; ~14%), group IV (32; ~13%), and the group I (26; ~11%), whereas groups VII (17; ~7%) and III (16; ~7%) harbored the least number of Dof genes. In general, the genes of 10 rice genomes showed more sequence homology among them as compared with Arabidopsis, as Arabidopsis Dof genes were clustered into separate clades showing distant orthologous relationships with rice genes (Figure 2D). This classification of rice Dof genes into seven different groups might be due to group-specific conserved amino acid motifs among the protein sequences of specific group members. Collectively, these results suggest that different Dof genes might function within different conditions, which is supported by genetic differences in their isoelectric points (Figure 2B) and molecular weights (Figure 2C).
The amino acid sequences of all Dof genes were explored to predict conserved motifs among them. All identified genes contained at least one Dof domain in their protein sequences (Figure 3 and Supplementary Figures S1A–S1H, Supplementary Table S5). Members of group II contained the highest number of motifs (2–10), whereas members of groups I and VII contained the least number of motifs (1–3). All other groups contained a variable number of motifs (1–8). In general, members of the same subgroups shared conserved motifs, whereas substantial differences were observed among members of different groups supporting the evolutionary phylogenetic relationships.
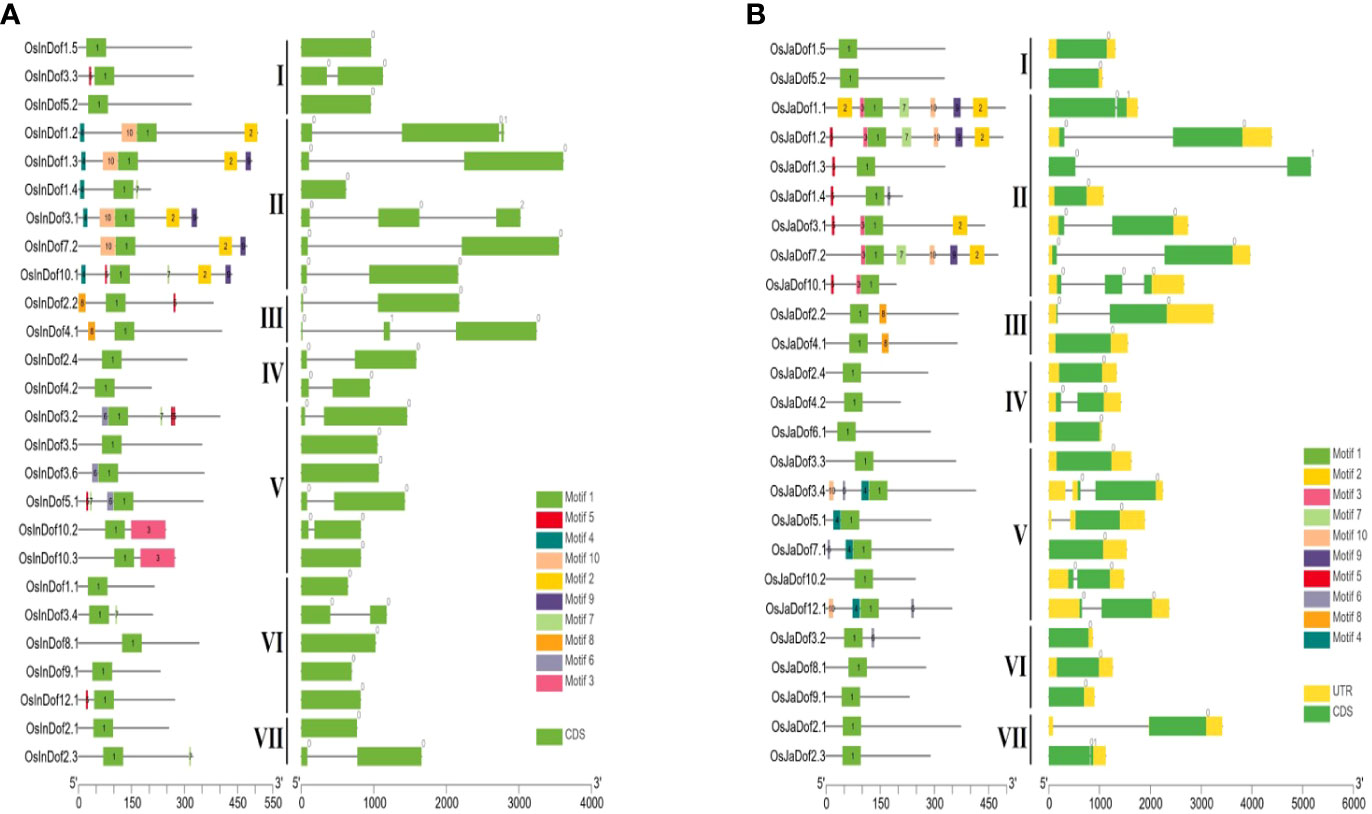
Figure 3 Conserved motifs and gene structures of Dof genes in (A) indica and (B) japonica rice. Conserved motifs were identified by uploading amino acid sequences to the MEME online tool (Bailey et al., 2009) and displayed on protein lengths with TBtools (Chen et al., 2020). Similarly, gene structures were drawn on genomic lengths by using respective GFF3 files of both rice types. Sub-groups are indicated with Roman numbers (I−VII) in the middle of both figures.
Similarly, gene structures were drawn to explore structural differences among different Dof subgroups and genes. A highly significant number of genes (~92%) are either intronless or one intron, with only < 6% and < 2% genes containing two and three introns, respectively (Figure 3; Supplementary Figures S1A–S1H; Supplementary Table S3). Whereas, only two genes harbored four introns in their structures. Collectively, these results suggest potential structural and functional conservation among members of the same subgroups but diversification between members of different subgroups.
Duplicated Dof gene pairs were identified by comparing the coding sequences and those pairs showing ≥90% sequence similarity were considered as duplicated. A total of 136 duplicated gene pairs corresponded to 30 unique Dof genes of ten Oryza genomes (Figure 4; Supplementary Table S6). Interestingly, all these duplicated gene pairs were sub-group specific, only identified in subgroups II and VII, and exclusively located on chromosomes 2, 3, and 7. We also identified at least three duplicated gene pairs among closely related Oryza genomes with 100% identical coding sequences. The duplicated gene pairs among all Oryza genomes ranged from 2 to 5 with the highest duplicated pairs found between O. barthii and O. meridionalis (Supplementary Table S6).
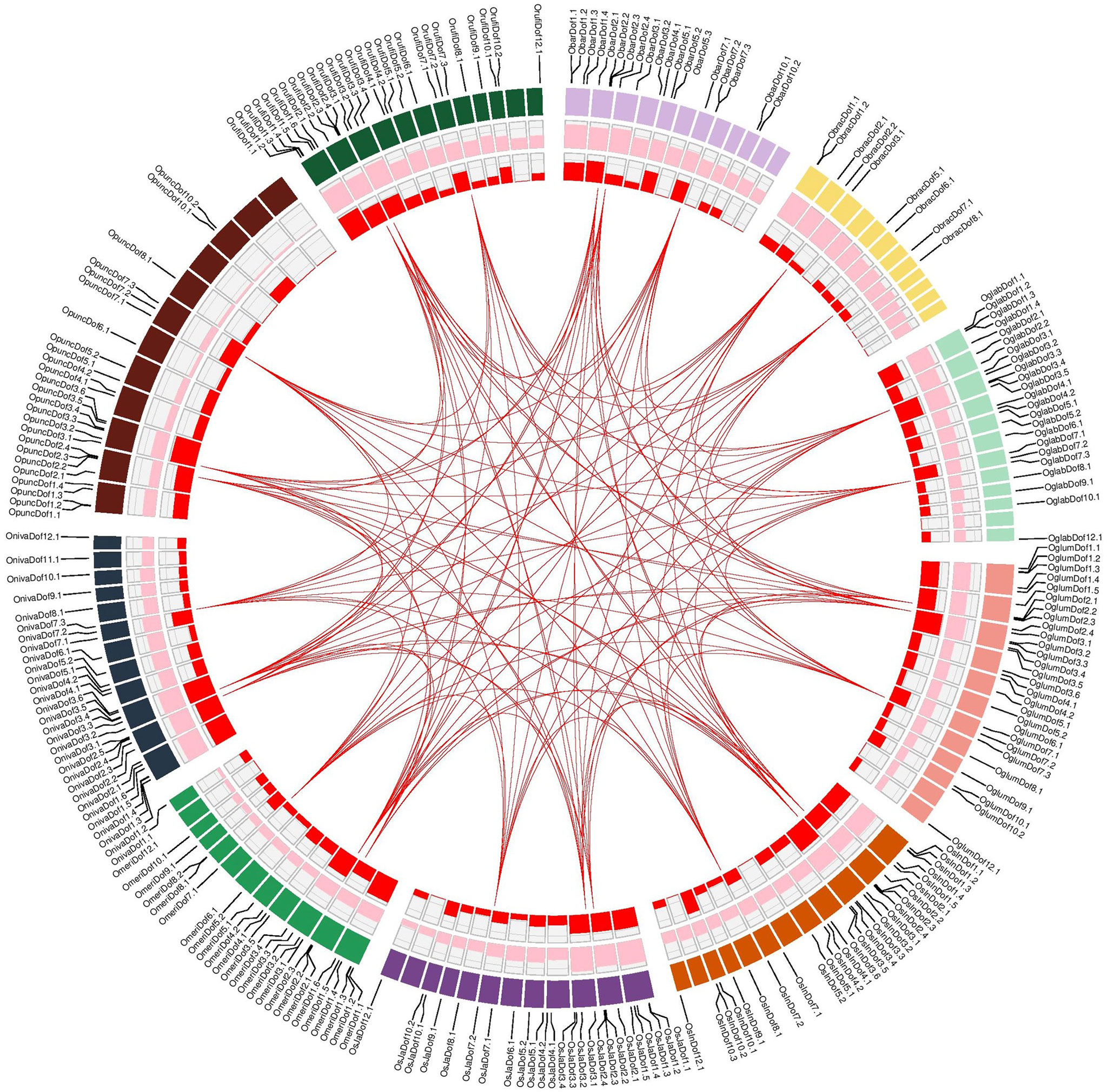
Figure 4 Genomic landscape of Dof genes among ten Oryza genomes. Circular diagram from outside to inside are gene names and locations on individual species-specific colored chromosomal bands, density of high confidence protein-encoding genes (count/Mb; min = 65, max = 133), density of Dof genes (count/Mb; min = 0.0, max = 0.17) and links indicating duplicated genes among ten rice species. Detailed information about duplicated gene pairs is provided in Table S6.
Furthermore, we also computed substitution rates among duplicated gene pairs to investigate different evolutionary patterns. In general, Dof genes were under strong purifying selections as nearly 85% of the duplicated pairs had a Ka/Ks ratio < 1, whereas only 12% of pairs were under positive selections (Ka/Ks > 1) (Supplementary Table S6). Moreover, all these duplicated genes were estimated to be diverged between 0.23 to 24.50 million years ago (MYA). Collectively, these results suggest that purifying selections are operating on codons to expand the Dof family members in Oryza genomes.
Cis-acting regulatory elements (CAREs) are found in the promoter region of a gene which binds to the transcription factors and regulates the gene expression. A variety of CAREs were predicted in the promoters of Dof genes of all Oryza genomes using the PlantCARE database (Supplementary Table S7). All these CAREs corresponded to diverse biological functions including abiotic and biotic stresses, plant hormones, and growth and development-related processes. Interestingly, the promoters of all identified Dof genes were extremely enriched with light, abscisic acid, methyl jasmonate, and anaerobic induction responsive CAREs (Supplementary Figures S2A–S2J; Supplementary Table S8). These results indicate that Dof family members can regulate diverse biological and physiological processes with highly significant regulatory roles in light responsiveness and hormone signaling.
Various miRNAs post-transcriptionally target several transcription factor families in different crops to regulate the gene expression patterns. Here, we identified several kinds of miRNAs which can potentially target the transcripts of identified Dof genes of the two most common Oryza sativa subspecies (indica and japonica) (Supplementary Table S9). Among identified miRNAs, three types (osa-miR2925, osa-miR2927 and osa-miR5075) were most frequent as the transcripts of 19 indica (~70%) and 17 japonica (68%) Dof genes contained the putative binding sites of these miRNAs (Figure 5). Surprisingly, osa-miR2927 was observed to be potentially targeting the Dof domain sequence of nearly 48% of both O. sativa sub-species genes. Likewise, osa-miR2925 potentially targets the Dof domain in a few gene transcripts and predominately the downstream regions of the Dof domain in all genes, whereas osa-miR5057 only targets the upstream or downstream regions of the Dof domain in all genes of both sub-species (Figure 5). Collectively, these results highlight that Dof genes are frequent targets of several miRNAs with significant roles of osa-miR2927, osa-miR2925, and osa-miR5075 in post-transcriptional regulation of Dof genes expressions.
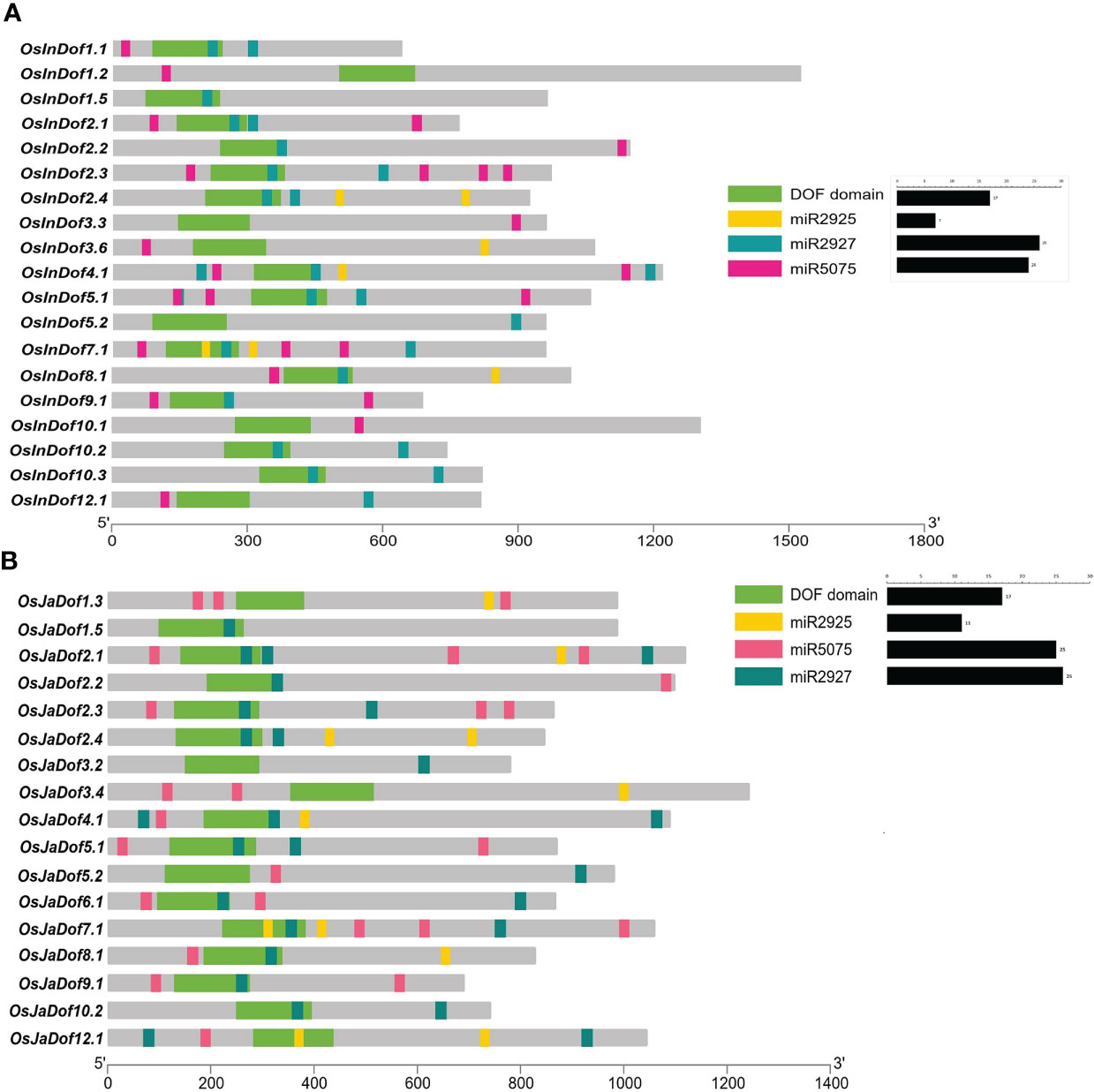
Figure 5 miRNA’s potentially targeting (A) indica and (B) japonica Dof genes. Previously reported information related to rice miRNAs and their potential targets was retrieved from the psRNA Target Server (Dai et al., 2018) and target sites were mapped onto the transcripts of Dof genes with TBtools (Chen et al., 2020).
Comprehensive expression profiling could provide clues toward the understanding of gene functions. Here, we examined the global gene expression patterns of O. sativa japonica Dof genes in root and shoot tissues in response to six plant hormones (Figure 6A). In general, the majority of the genes (~68%) were deregulated after plant hormone treatments. As expected, nearly all OsJaDofs were highly deregulated in roots and shoots of jasmonic acid and abscisic acid-treated seedlings. Interestingly, these genes were significantly downregulated in response to jasmonic acid but up-regulated in response to abscisic acid treatments in root tissues (Figure 6A), however, unevenly deregulated in shoot tissues. Similarly, all these genes are inconsistently and marginally deregulated in response to auxin, brassinosteroid, cytokinin, and gibberellin treatments in root and shoot tissues. Notably, OsJaDof10.1 significantly up-regulated in roots and shoots of abscisic acid-treated seedlings. Likewise, OsJaDof1.1 and OsJaDof2.1 significantly down-regulated in roots and shoots of jasmonic acid-treated seedlings. Overall, these results indicate the vital roles of Dof genes in phytohormones perception, signal transduction, and downstream effects to stimulate the rice defense response mechanisms against different stresses.
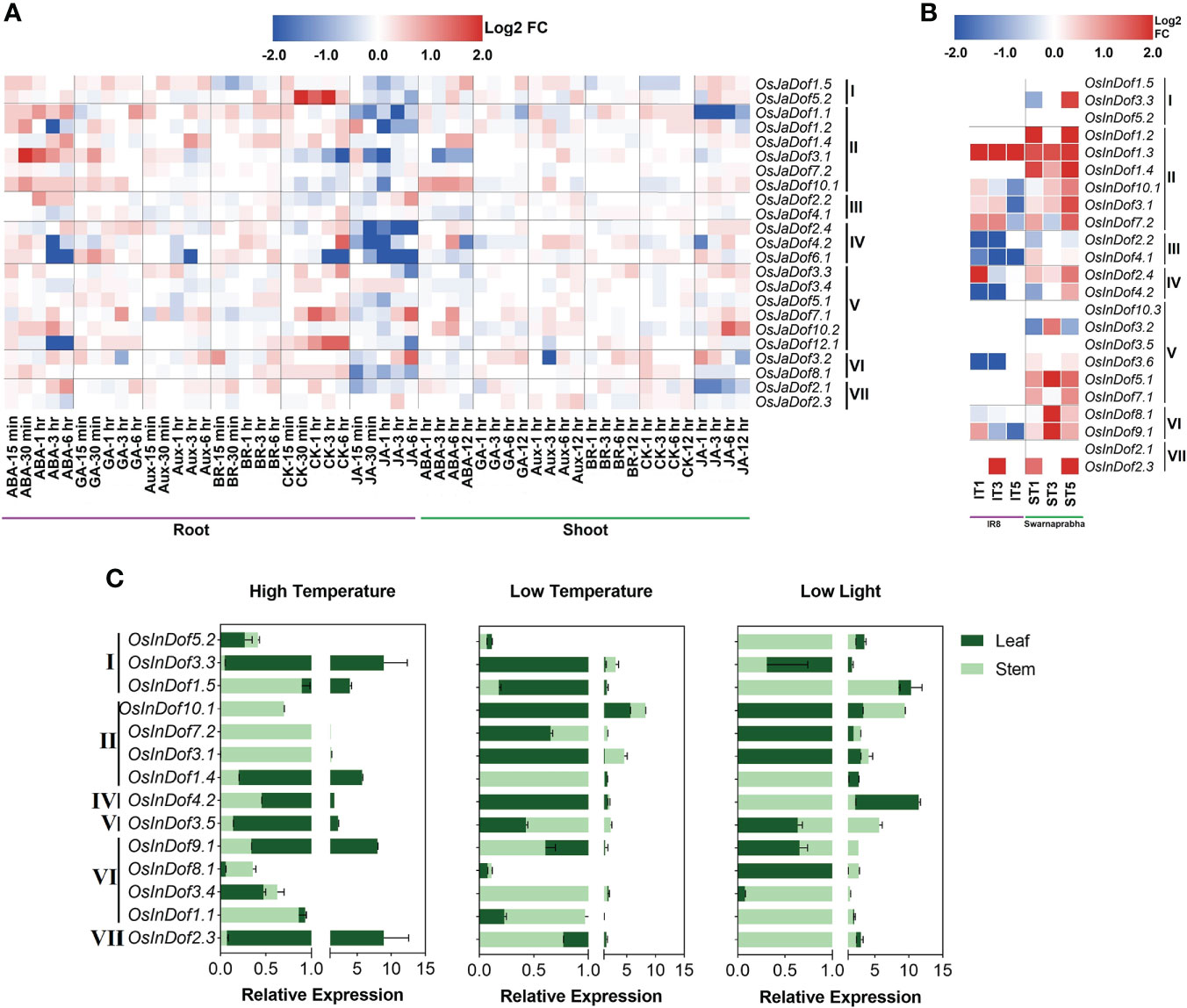
Figure 6 Expression profiles of Dof genes under stressful conditions. (A) RNA-seq-based normalized (Log2 fold change) expression patterns of O. sativa japonica Dof genes in phytohormones treated roots and shoots. Hormone treatments are listed in columns and Dof genes in rows. Roman numbers on the left of the gene names indicate different Dof sub-groups. Abbreviations; ABA (abscisic acid), GA (gibberellic acid), Aux (auxin), BR (brassinosteroid), CK (cytokinin), JA (jasmonic acid). (B) RNA-seq-based normalized (Log2 fold change) expression patterns of O. sativa indica Dof genes in response to low light treatments in susceptible (IR8) and tolerant (Swarnaprabha) rice genotypes. Abbreviations: IT1 (IR8 treated with low light for one day), IT3 (IR8 treated with low light for three days), IT5 (IR8 treated with low light for five days), ST1 (Swarnaprabha treated with low light for one day), ST3 (Swarnaprabha treated with low light for three days) and ST5 (Swarnaprabha treated with low light for five days). (C) Relative expression profiles of O. sativa indica Dof genes after one week of high temperature (40 ± 1°C), low temperature (15 ± 1°C), and low light (20% of control; 60 µmolm-2s-1) treatments during 14h daylight periods in 93-11 indica rice seedlings.
Similarly, the expression patterns of O. sativa indica Dofs were investigated in two contrasting rice genotypes for low light responsiveness (Figure 6B). Consistent with O. sativa japonica Dofs expression patterns under hormone stress, the majority of the OsInDofs (>66%) were deregulated after exposure to low light treatments. Interestingly, all these genes were up-regulated in low light tolerant rice (Swarnaprabha), with exception of a few genes which marginally down-regulated. Contrarily, only 44% of all OsInDofs were deregulated in low light-sensitive rice (IR8) and a majority of these (63%) were downregulated after exposure to at least one low light treatment (Figure 6B). Collectively, these results suggest that accelerated expressions of OsInDofs are detrimental for maintaining optimum photosynthetic processes under low light conditions.
Furthermore, in-silico expression profiles were validated by investigating qRT-PCR-based relative expression patterns under low light, high-temperature, and low-temperature treatments (Figure 6C; Supplementary Table S10). After one week of low light treatment (20% of control; 60 µmolm-2s-1), the majority of the investigated genes were significantly expressed in both leaf and stem tissues. Notably, nearly all genes were strongly expressed in stems, whereas genes of at least four Dof subgroups (I, II, IV, and VII) were also highly expressed in leaves as compared with control. As expected, the majority of these strongly expressed genes also showed higher relative expression profiles in low light tolerant rice (Figure 6B). Similarly, nearly 50% of genes were significantly expressed in leaves after exposure to one week of high temperature (40 ± 1°C) treatment during 14h daylight period (Figure 6C). Likewise, 11 investigated genes out of 14 were also highly expressed in at least one tested tissue after exposure to one week of low light (15 ± 1°C) treatment. Notably, at least five genes (OsInDof1.4, OsInDof1.5, OsInDof2.3, OsInDof3.3, OsInDof4.2) showed significantly higher relative expressions in leaves and two genes (OsInDof3.1, OsInDof7.2) showed significantly higher relative expressions in stems under all three stress treatments (Figure 6C). Together, these wet-lab-based expression profiles strongly support the in-silico results and suggest multifarious roles of Dof genes in regulating the growth and development and tolerance to abiotic stresses.
Dof genes have been recently reported to be strongly associated with heading date in rice (Huang et al., 2020). We took advantage of previously reported 3,010 rice genomes variation and phenotypic data (Wang et al., 2020), and mined haplotype blocks significantly associated with days to heading (DTH) trait in coding regions of four orthologous Dof genes harbored by all ten rice genomes (Figure 7; Supplementary Table S11). Furthermore, we also validated the identified haplotypes with field experiments data across two consecutive growing seasons (Supplementary Table S12). In total, we identified 26 haplotypes (six for OsJaDof1.1, five for OsJaDof3.1 and OsJaDof5.2 each, and ten for OsJaDof7.2) with significant variation in their days to heading data. For OsJaDof1.1, haplotypes 4 and 2 (Hap4, Hap2) were found to be superior as rice accession harboring these haplotypes have taken 91 and 93 days on average for flowering starting from germination, respectively (Figure 7A). Similarly, Hap4 and Hap5 of OsJaDof3.1 took 89 and 92 days for flowering, respectively (Figure 7B). Likewise, Hap9 and Hap1 of OsJaDof5.2 took 82 and 92 days for flowering, respectively (Figure 7C). Whereas Hap2 of OsJaDof7.2 was found to be superior to all others, however, we could not validate it with field evaluation-based experiments as none of the sub-panel accessions harbored this haplotype, although Hap3 was also found to be superior and validated with field evaluation (Figure 7D). Overall, our results indicate that superior haplotypes for early flowering are contributed by wild relatives of rice and will be helpful to broaden the genetic diversity of cultivated rice. Moreover, these alleles can also be used to develop varying photoperiod-specific rice varieties.
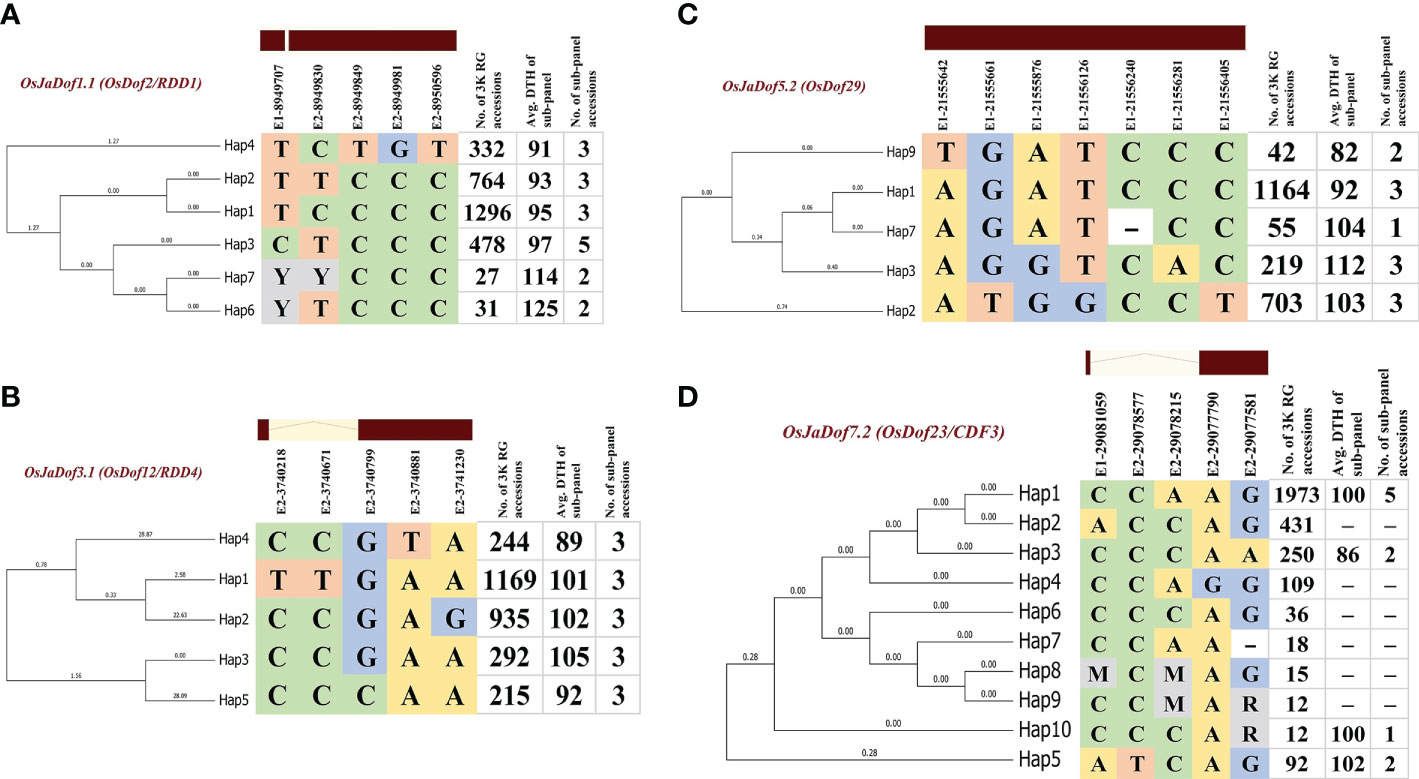
Figure 7 Superior haplotypes of four orthologous genes across genus Oryza including (A) OsJaDof1.1, (B) OsJaDof3.1 (C) OsJaDof5.2, and (D) OsJaDof7.2 significantly associated with days to heading data from field evaluation. The genomic positions of non-synonymous substitutions in the coding region of genes are indicated above the haplotype blocks with E1 and E2 indicating exon 1 and exon 2, respectively followed by the genomic position of SNP-based polymorphism.
The exact evolutionary phylogenetic relationships among Oryzae species remained elusive, particularly due to phylogeny discordance among published reports (Degnan and Rosenberg, 2009; Zwickl et al., 2014). In this study, we inferred a single-species phylogeny using five single-copy orthologs from all ten rice genomes (Figure 8A). We noted two independent origins of Asian cultivated japonica and indica rice, whereas African cultivated O. glaberrima rice shared a closer evolutionary phylogenetic relationship with South American O. glumipatula rice, rather than the O. sativa complex. The real-time method-based time tree indicated a more recent divergence of two African rice genomes (~0.03 Mya), followed by O. sativa vg. japonica divergence from O. rufipogon (~0.29 Mya) and O. sativa vg. indica divergence from O. nivara (~0.50 Mya). As expected, the eight AA genomes (O. sativa indica and japonica, O. nivara, O. rufipogon, O. glaberrima, O. barthii, O glumipatula, and O. meridionalis) were found to be recently diverged from the BB genome (O. punctata), as compared with FF genome (O. brachyantha).
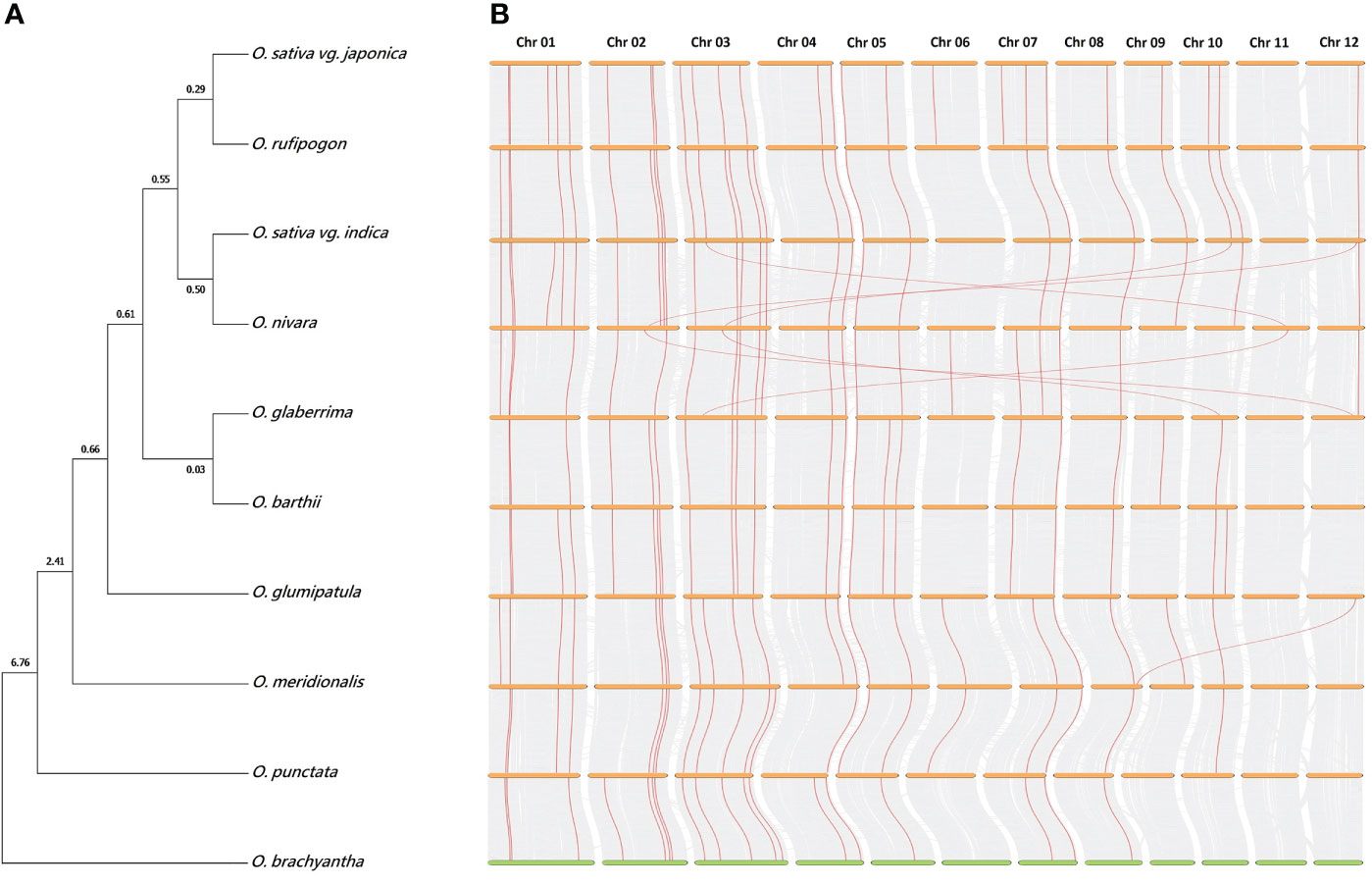
Figure 8 Evolutionary phylogenetic relationships and chromosomal synteny analysis among Oryza genomes. (A) Species phylogeny was inferred after MAFFT v7 multiple sequence alignment (Katoh et al., 2018) and neighbor-joining real-time tree creation with MEGA7 (Kumar et al., 2016) using coding sequences of five orthologous Dof genes present across all ten Oryza genomes. O. brachyantha was considered as an outgroup while estimating evolutionary divergence time in million years (above tree nodes). (B) Chromosomal synteny analysis among adjacent species was carried out with TBtools (Chen et al., 2020) by using GFF3 and peptide sequence files of ten Oryza genomes. Gene links indicate orthologous Dof genes and their chromosomal locations among different genomes.
Furthermore, we also investigated genetic conservation and synteny among Oryza genomes. Notably, dual synteny plots highlighted >90% synteny among all ten genomes (Figure 8B), as nearly all ancestral wild species Dof genes had orthologs in descendant cultivated species. We noted a higher turnover of Dof genes among AA genomes, particularly among cultivated species complex, and few translocations between O. sativa vg. indica and O. nivara and between O. nivara and O. glaberrima Dof genes. Collectively, these results reveal higher genetic conservation across the Oryza genomes.
Dof genes belong to plant kingdom-specific transcription factor family that play important roles during the growth and development of nearly all plant species. In the recent past, significant progress was made towards comprehensive genome-wide identification and functional characterization of Dof family genes in rice which could facilitate marker-assisted development of rice cultivars. For example, two more recent genome-wide studies independently identified a total of 30 Dofs in the O. sativa japonica genome and reported their phylogenetic relationships, gene structures, conserved motifs, gene duplications, and expression patterns in different tissue under normal and cold stress conditions (Khan et al., 2021; Liu et al., 2021). Our results demonstrated the presence of copy number variation of Dof genes on some chromosomes of among indica rice genomes. The copy number variation of Dof genes in wheat has been shown to increase the stem solidness (Nilsen et al., 2020) which can ultimately be exploited for the development of wheat stem sawfly resistance.
Rice OsDof12/RDD4 has been shown to be a genetic regulator of plant architecture, as its overexpression could lead to shortened plant height, erect leaves, narrow leaf blades, and smaller panicles (Wu et al., 2015). RDD1 and RDD4 promote efficient uptake and accumulation of several nutrients such as NH4+, Na+, SO42‾, Cl‾, PO43‾, and sucrose under low-nutrient conditions and contribute to increased grain productivity (Iwamoto and Tagiri, 2016; Iwamoto et al., 2021). OsDof4 regulates photoperiodic flowering under short and long days (Wu et al., 2017b). OsDOF18 affects nitrogen use efficiency in roots by inducing ammonium transporters and mediating the ammonium transport and nitrogen distribution (Wu et al., 2017a). OsDOF11 modulates sugar distribution in different plant organs during the pathogenic invasion by accelerating the expression of SUT and SWEET genes (Wu et al., 2018). OsDOF24 delays leaf senescence by deactivating jasmonate biosynthetic pathways (Shim et al., 2019). OsDOF15 positively regulates primary root elongation under salt stress by restricting ethylene biosynthesis in developing roots (Qin et al., 2019). Moreover, minor effects of 11 Dofs contribute diverse functions in promoting and suppressing heading dates under contrasting photoperiodic conditions (Huang et al., 2020). These recent advancements, as well as the results presented in this study, could contribute significantly toward the genomics-assisted development of tailor-made rice cultivars.
Wild crop relatives constitute an important genetic reservoir for improvement due to their tolerance to different stresses and wider adaptability (Jena, 2010; Atwell et al., 2014). Harnessing superior traits for crop improvement hold enormous promises, as revealed by the introgression of bacterial resistance from wild rice to cultivated rice (Song et al., 1995). To precisely harness the genetic potential of wild relatives, a clear understanding of genomic conservation and diversification is crucial. Taking advantage of recently sequenced wild relatives of cultivated rice (Stein et al., 2018), we comprehensively identified 238 full-length protein-encoding Dof genes through genome-wide scans in ten Oryza genomes (Figures 2A, D; Supplementary Table S3). Previously, Dof genes were only identified in three cultivated (O. sativa indica and japonica, O. glaberrima) and one wild relative of rice (O. brachyantha) (Lijavetzky et al., 2003; Jacquemin et al., 2014; Khan et al., 2021; Liu et al., 2021), ignoring other important wild rice species. Furthermore, variable number of Dof phylogenetic subgroups were reported in those studies, thus lacking consistency in phylogenetic classification. Based on evolutionary phylogenetic relationships, Arabidopsis Dof genes were classified into seven subgroups (I–VII) (Yanagisawa, 2002), which serve as a reference for Dof phylogenetic classification in later studies. The Oryza Dof genes identified in this study were also clustered into seven distinct subgroups showing distant orthologous relationships with Arabidopsis genes in each subgroup (Figure 2A). Interestingly, two subgroups (II and V) were overrepresented in all investigated genomes, with O. brachyantha harboring the least gene count and entirely lacking subgroup V, as well as subgroup III members (Figure 2D). The reason could be the shortest genome size and extended evolutionary history, as O. brachyantha diverged >15 million years ago, whereas all other Oryza genomes diverged ≤ 6.76 million years ago (Stein et al., 2018). Conversely to previous reports on genome-wide identification of Dof genes in O. sativa japonica (Lijavetzky et al., 2003; Khan et al., 2021; Liu et al., 2021), only 25 non-redundant OsJaDofs containing complete Dof domain were identified in this study (Figures 2A, D; Supplementary Table S3), highlighting those previous studies also included incomplete Dof domain-containing genes. Overall, our results demonstrate an abundance of wider genetic and genomic resources in domesticated and wild rice relatives for further crop improvement.
Dof genes were reported to be occurred in ancient days, even before the diversification of angiosperms (Yanagisawa, 2002). Because of an extended evolutionary history, different Dof subgroups might come to play diversified roles. This could be largely due to amino acid substitutions in genomic regions outside the conserved Dof domain. Therefore, it is not unexpected that different Dof subgroups show distinct and/or overlapping functions (Yanagisawa, 2004). In this study, we also observed wider genetic diversity in iso-electric points and molecular weights of different Dof subgroups (Figures 2B, C), which was supported by conserved motifs and gene structure analyses (Figure 3 and Supplementary Figures S1A–S1H). Members of different subgroups demonstrated a prevalence of structural diversification between them, however, conservation among members of the same subgroup. Moreover, subgroup-specific gene duplications among Dofs of ten Oryza genomes (Figure 4) and gene expression profiles in response to phytohormones and low light treatments (Figures 6A, B), strongly reinforced the current results. Thus, the current catalog of Oryza Dof genes might disclose both the subgroup-specific and species-specific structural and functional diversity that is expediating with the passage of the evolutionary period.
Dofs are plant-specific transcription factors that bind to the specific CAREs present in their promoter regions and regulate diverse biological, physiological, and molecular functions both at transcriptional and post-transcriptional levels (Yanagisawa, 2004). Previous reports have shed light on their multifarious roles in regulating the growth and development, as well as resistance or tolerance to biotic and abiotic stresses throughout the plant kingdom as briefly reviewed in previous reports (Gupta et al., 2015; Waqas et al., 2020; Zhuo et al., 2020). In this study, a variety of CAREs corresponding to diverse potential functions, especially tolerance to abiotic and hormone stresses, were identified in promoters of Dof genes of all Oryza species (Supplementary Tables S5, S6). Furthermore, the majority of the genes of the two most important cultivated Oryza species (O. sativa indica and japonica) were significantly deregulated in response to low light and phytohormone treatments (Figures 6A, B), strongly backing the promoter analysis results. Recent reports have also revealed their regulatory roles in cold, heat, and shade tolerance (Sekhar et al., 2019; He et al., 2020; Liu et al., 2021) confirming their diverse roles in rice growth and development and wider adaptability in hostile environments. Collectively, these results strongly support the notion about Dofs act as dominant transcriptional regulators of plant life cycle and wider adaptability. However, further deeper insights at gene regulatory networks and systems biology levels are required to confirm this hypothesis.
The genetic variation and phenotypic agronomic data of the 3K RG panel is a treasure trove for molecular rice breeders for exploiting haplotype diversity (Sun et al., 2017; Wang et al., 2020). In this study, we identified significant haplotypes associated with days to heading field data across the 3K RG panel and validated these haplotypes with a two-year field evaluation of a representative sub-panel (Figure 7; Supplementary Tables S9, S10). The superior haplotypes predominately originated from wild rice accessions and flowered significantly earlier than all other identified haplotypes. These findings are backed by the significant contribution of Dof genes to the missing heritability of heading dates in rice (Huang et al., 2020). Introgression of these superior haplotypes through modern haplotype-led breeding could assist in designing early maturing rice cultivars under fluctuating climatic conditions. However, multi-location field data-based confirmatory studies involving a larger number of 3K RG representative accessions need to be carried out before successfully harnessing the potential of these superior haplotypes in applied research programs. Systems-level deeper insights into the economically important traits, underlying genes, and associated epigenetic factors could greatly facilitate the development of customizable rice ideotypes. Once realized, this strategy would accelerate the way toward designer rice plants.
Species diversity among important crop plants is of paramount importance for their broader cultivation followed by adaptation and holds promise for further crop improvement. Here, we resolved controversial areas of Oryza species phylogeny by untangling the complex evolutionary phylogenetic relationships among closely related species (Degnan and Rosenberg, 2009; Zwickl et al., 2014) (Figure 8A). Our results highlighted rapid species diversification rate of approximately 0.50 million years per new species among AA genomes, which is at par with many other rapidly evolving taxa (Madriñán et al., 2013; Zhu et al., 2014). For example, we found recent independent origins of two widely cultivated subspecies indica and japonica, whereas another cultivated species O. glaberrima revealed more recent divergence from wild O. barthii rice and higher evolutionary relatedness with South American wild rice O. glumipatula (Stein et al., 2018). However, despite rapid species diversification, footprints of higher gene flow are still detectable, as nearly all wild Dof genes had orthologs in recently evolved cultivated species (Figure 8B). This rapid diversification coupled with higher genetic conservation and gene flow among wild and cultivated species facilitates wider adaptability under changing climatic conditions and provides special opportunities for broader cultivation and further crop improvement.
In this study, we reported a complete catalog of full-length Dof family genes in ten cultivated and wild Oryza genomes through genome-wide scanning. A total of 238 Dof genes were classified into seven distinct phylogenetic subgroups sharing structural and functional diversity. Our results revealed exclusive localization of duplicated gene pairs on 2nd, 3rd, and 7th chromosomes and Dof domain sequences potentially containing target sites for miR2927 across the Oryza genomes. Transcriptional profiles in different tissues under phytohormones and low light treatments strongly support that Dof genes act as dominant transcriptional regulators during rice growth and development and wider adaptation under changing environments. Furthermore, superior haplotypes significantly associated with early flowering across the 3K RG panel were reported and validated with two years of field experiments based on the morphometric evaluation that could be exploited through haplotype-led breeding for fast-track development of early flowering rice cultivars. Finally, we resolved Oryza species’ phylogeny discordance and observed higher genetic conservation and gene flow among Oryzae species which provide better opportunities for wider adaptability and further crop improvement.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
RA and JL conceived the idea. JT, QR, AR, JL and RA designed the experiments. JT, QR, AR, SA and MR retrieved and curated the data. JT, QR, AR, SA, MR and FK performed analyses. JT, QR and AR performed formal analyses and drafted the manuscript. MJ, ZA, IK, JL and RA reviewed and edited the manuscript. IK, JL and RA provided resources, supervised the experiments, and acquired funding for research. All authors have read and approved the final manuscript.
This work was supported by Agricultural Sciences and Technologies Innovation Program of the Chinese Academy of Agricultural Sciences (CAAS); and Higher Education Commission of Pakistan (HEC) through their grant of Precision Agriculture and Analytics Lab (PAAL) under National Centre in Big Data and Cloud Computing (NCBC).
The authors are thankful to the Rice Research Institute, Kala Shah Kaku, Sheikhupura, Pakistan for providing institutional support for carrying out field experimentation. The infrastructural support from Centre for Advanced Studies in Agriculture and Food Security (CAS-AFS), at University of Agriculture Faisalabad, Pakistan is also grateful acknowledged.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1004359/full#supplementary-material
Atwell, B. J., Wang, H., Scafaro, A. P. (2014). Could abiotic stress tolerance in wild relatives of rice be used to improve oryza sativa? Plant Sci. 215–216, 48–58. doi: 10.1016/J.PLANTSCI.2013.10.007
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME suite: Tools for motif discovery and searching. Nucleic Acids Res 37, W202–W208. doi: 10.1093/nar/gkp335
Bhullar, S., Datta, S., Advani, S., Chakravarthy, S., Gautam, T., Pental, D., et al. (2007). Functional analysis of cauliflower mosaic virus 35S promoter: re-evaluation of the role of subdomains B5, B4 and B2 in promoter activity. Plant Biotechnol. J. 5, 696–708. doi: 10.1111/J.1467-7652.2007.00274.X
Chattha, W. S., Atif, R. M., Iqbal, M., Shafqat, W., Farooq, M. A., Shakeel, A. (2020). Genome-wide identification and evolution of dof transcription factor family in cultivated and ancestral cotton species. Genomics 112, 4155–4170. doi: 10.1016/J.YGENO.2020.07.006
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chun, Y., Fang, J., Zafar, S. A., Shang, J., Zhao, J., Yuan, S., et al. (2020). MINI SEED 2 (MIS2) encodes a receptor-like kinase that controls grain size and shape in rice. Rice 13, 1–17. doi: 10.1186/S12284-020-0368-9/FIGURES/9
Dai, X., Zhuang, Z., Zhao, P. X. (2018). psRNATarget: A plant small RNA target analysis server, (2017 Release). Nucleic Acids Res. 46, W49–W54. doi: 10.1093/NAR/GKY316
Degnan, J. H., Rosenberg, N. A. (2009). Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 24, 332–340. doi: 10.1016/J.TREE.2009.01.009
El Baidouri, M., Murat, F., Veyssiere, M., Molinier, M., Flores, R., Burlot, L., et al. (2017). Reconciling the evolutionary origin of bread wheat (Triticum aestivum). New Phytol 213, 1477–1486. doi: 10.1111/nph.14113
Fornara, F., Panigrahi, K. C. S., Gissot, L., Sauerbrunn, N., Rühl, M., Jarillo, J. A., et al. (2009). Arabidopsis DOF transcription factors act redundantly to reduce CONSTANS expression and are essential for a photoperiodic flowering response. Dev. Cell 17, 75–86. doi: 10.1016/J.DEVCEL.2009.06.015
Fukagawa, N. K., Ziska, L. H. (2019). Rice: Importance for global nutrition. J. Nutr. Sci. Vitaminol. 65, S2–S3. doi: 10.3177/JNSV.65.S2
Gasteiger, E., Gattiker, A., Hoogland, C., Ivanyi, I., Appel, R. D., Bairoch, A. (2003). ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res 31, 3784–3788. doi: 10.1093/nar/gkg563
Goff, S. A., Ricke, D., Lan, T. H., Presting, G., Wang, R., Dunn, M., et al. (2002). A draft sequence of the rice genome (Oryza sativa l. ssp. japonica). Sci. (1979) 296, 92–100. doi: 10.1126/SCIENCE.1068275/SUPPL_FILE/TMRI-AGREEMENT-MAR02.PDF
Guo, Y., Qin, G., Gu, H., Qu, L. J. (2009). Dof5.6/HCA2, a dof transcription factor gene, regulates interfascicular cambium formation and vascular tissue development in arabidopsis. Plant Cell 21, 3518–3534. doi: 10.1105/TPC.108.064139
Gupta, S., Malviya, N., Kushwaha, H., Nasim, J., Bisht, N. C., Singh, V. K., et al. (2015). Insights into structural and functional diversity of dof (DNA binding with one finger) transcription factor. Planta 241, 549–562. doi: 10.1007/S00425-014-2239-3/3
He, C., Zhang, H. Y., Zhang, Y. X., Fu, P., You, L. L., Xiao, W. B., et al. (2020). Cytosine methylations in the promoter regions of genes involved in the cellular oxidation equilibrium pathways affect rice heat tolerance. BMC Genomics 21, 1–16. doi: 10.1186/S12864-020-06975-3/FIGURES/6
Hoang, D. T., Chernomor, O., Von Haeseler, A., Minh, B. Q., Vinh, L. S. (2018). UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522. doi: 10.1093/molbev/msx281
Huang, Y., Han, Z., Cheng, N., Luo, M., Bai, X., Xing, Y. (2020). Minor effects of 11 dof family genes contribute to the missing heritability of heading date in rice (Oryza sativa l.). Front. Plant Sci. 10 1739. doi: 10.3389/FPLS.2019.01739/BIBTEX
Ibáñez-Salazar, A., Rosales-Mendoza, S., Rocha-Uribe, A., Ramírez-Alonso, J. I., Lara-Hernández, I., Hernández-Torres, A., et al. (2014). Over-expression of dof-type transcription factor increases lipid production in chlamydomonas reinhardtii. J. Biotechnol. 184, 27–38. doi: 10.1016/J.JBIOTEC.2014.05.003
Isabel-LaMoneda, I., Diaz, I., Martinez, M., Mena, M., Carbonero, P. (2003). SAD: a new DOF protein from barley that activates transcription of a cathepsin b-like thiol protease gene in the aleurone of germinating seeds. Plant J. 33, 329–340. doi: 10.1046/J.1365-313X.2003.01628.X
Iwamoto, M., Tagiri, A. (2016). MicroRNA-targeted transcription factor gene RDD1 promotes nutrient ion uptake and accumulation in rice. Plant J. 85, 466–477. doi: 10.1111/TPJ.13117
Iwamoto, M., Tsuchida-Mayama, T., Ichikawa, H. (2021). The transcription factor gene RDD4 contributes to the control of nutrient ion accumulation in rice. Physiologia Plantarum 172, 2059–2069. doi: 10.1111/PPL.13434
Jackson, S. A. (2016). Rice: The first crop genome. Rice 9, 1–3. doi: 10.1186/S12284-016-0087-4/FIGURES/1
Jacquemin, J., Ammiraju, J. S. S., Haberer, G., Billheimer, D. D., Yu, Y., Liu, L. C., et al. (2014). Fifteen million years of evolution in the oryza genus shows extensive gene family expansion. Mol. Plant 7, 642–656. doi: 10.1093/MP/SST149
Jena, K. K. (2010). The species of the genus oryza and transfer of useful genes from wild species into cultivated rice, o. sativa. Breed. Sci. 60, 518–523. doi: 10.1270/JSBBS.60.518
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., Von Haeseler, A., Jermiin, L. S. (2017). ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Katoh, K., Rozewicki, J., Yamada, K. D. (2018). MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Briefings Bioinf 20, 1160–1166. doi: 10.1093/bib/bbx108
Kawahara, Y., de la Bastide, M., Hamilton, J. P., Kanamori, H., Mccombie, W. R., Ouyang, S., et al. (2013). Improvement of the oryza sativa nipponbare reference genome using next generation sequence and optical map data. Rice 6, 1–10. doi: 10.1186/1939-8433-6-4
Khan, I., Khan, S., Zhang, Y., Zhou, J. (2021). Genome-wide analysis and functional characterization of the dof transcription factor family in rice (Oryza sativa l.). Planta 253, 1–14. doi: 10.1007/S00425-021-03627-Y/FIGURES/7
Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/MOLBEV/MSW054
Kushwaha, H., Gupta, S., Singh, V. K., Rastogi, S., Yadav, D. (2011). Genome wide identification of dof transcription factor gene family in sorghum and its comparative phylogenetic analysis with rice and arabidopsis. Mol. Biol. Rep. 38, 5037–5053. doi: 10.1007/S11033-010-0650-9/TABLES/6
Lescot, M., Déhais, P., Thijs, G., Marchal, K., Moreau, Y., Van De Peer, Y., et al. (2002). PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327. doi: 10.1093/NAR/30.1.325
Lijavetzky, D., Carbonero, P., Vicente-Carbajosa, J. (2003). Genome-wide comparative phylogenetic analysis of the rice and arabidopsis dof gene families. BMC Evolutionary Biol. 3, 1–11. doi: 10.1186/1471-2148-3-17/TABLES/3
Liu, J., Cheng, Z., Xie, L., Li, X., Gao, J. (2019). Multifaceted role of PheDof12-1 in the regulation of flowering time and abiotic stress responses in moso bamboo (Phyllostachys edulis). Int. J. Mol. Sci. 20, 424. doi: 10.3390/IJMS20020424
Liu, X., Liu, Z., Hao, Z., Chen, G., Qi, K., Zhang, H., et al. (2020). Characterization of dof family in pyrus bretschneideri and role of PbDof9.2 in flowering time regulation. Genomics 112, 712–720. doi: 10.1016/J.YGENO.2019.05.005
Liu, J., Meng, Q., Xiang, H., Shi, F., Ma, L., Li, Y., et al. (2021). Genome-wide analysis of dof transcription factors and their response to cold stress in rice (Oryza sativa l.). BMC Genomics 22, 1–12. doi: 10.1186/S12864-021-08104-0/FIGURES/6
Madriñán, S., Cortés, A. J., Richardson, J. E. (2013). Páramo is the world’s fastest evolving and coolest biodiversity hotspot. Front. Genet. 4. doi: 10.3389/FGENE.2013.00192/ABSTRACT
Marchler-Bauer, A., Derbyshire, M. K., Gonzales, N. R., Lu, S., Chitsaz, F., Geer, L. Y., et al. (2015). CDD: NCBI’s conserved domain database. Nucleic Acids Res 43, D222–D226. doi: 10.1093/nar/gku1221
Mariyam, Shafiq, M., Haseeb, M., Atif, R. M., Naqvi, S. A. A. A. A., Ali, N., et al. (2021). Genome-wide identification and characterization of a plant-specific dof transcription factor gene family in olive (Olea europaea) and its comparison with arabidopsis. Horticulture Environment Biotechnol. 62 (6), 949–968. doi: 10.1007/s13580-021-00366-7
Matsumoto, T., Wu, J., Itoh, T., Numa, H., Antonio, B., Sasaki, T. (2016). The nipponbare genome and the next-generation of rice genomics research in Japan. Rice 9, 1–11. doi: 10.1186/S12284-016-0107-4/FIGURES/4
Matsumoto, T., Wu, J., Kanamori, H., Katayose, Y., Fujisawa, M., Namiki, N., et al. (2005). The map-based sequence of the rice genome. Nature 7052 436, 793–800. doi: 10.1038/nature03895
Moreno-Risueno, MÁ., Martínez, M., Vicente-Carbajosa, J., Carbonero, P. (2007). The family of DOF transcription factors: From green unicellular algae to vascular plants. Mol. Genet. Genomics 277, 379–390. doi: 10.1007/S00438-006-0186-9/FIGURES/5
Muhire, B. M., Varsani, A., Martin, D. P. (2014). SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS One 9, e108277. doi: 10.1371/journal.pone.0108277
Negi, J., Moriwaki, K., Konishi, M., Yokoyama, R., Nakano, T., Kusumi, K., et al. (2013). A dof transcription factor, SCAP1, is essential for the development of functional stomata in arabidopsis. Curr. Biol. 23, 479–484. doi: 10.1016/J.CUB.2013.02.001/ATTACHMENT/B579DAAF-2454-42A4-884A-450DD6FC26ED/MMC3.XLSX
Nguyen, N. T. T., Contreras-Moreira, B., Castro-Mondragon, J. A., Santana-Garcia, W., Ossio, R., Robles-Espinoza, C. D., et al. (2018). RSAT 2018: regulatory sequence analysis tools 20th anniversary. Nucleic Acids Res. 46, W209–W214. doi: 10.1093/NAR/GKY317
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nilsen, K. T., Walkowiak, S., Xiang, D., Gao, P., Quilichini, T. D., Willick, I. R., et al. (2020). Copy number variation of TdDof controls solid-stemmed architecture in wheat. Proc. Natl. Acad. Sci. 117 (46), 28708–28718. doi: 10.1073/pnas.2009418117
Noguero, M., Atif, R. M., Ochatt, S., Thompson, R. D. (2013). The role of the DNA-binding one zinc finger (DOF) transcription factor family in plants. Plant Sci. 209, 32–45. doi: 10.1016/j.plantsci.2013.03.016
Qin, H., Wang, J., Chen, X., Wang, F., Peng, P., Zhou, Y., et al. (2019). Rice OsDOF15 contributes to ethylene-inhibited primary root elongation under salt stress. New Phytol. 223, 798–813. doi: 10.1111/NPH.15824
Raza, Q., Riaz, A., Bashir, K., Sabar, M. (2020). Reproductive tissues-specific meta-QTLs and candidate genes for development of heat-tolerant rice cultivars. Plant Mol. Biol 104, 97–112. doi: 10.1007/s11103-020-01027-6
Riechmann, J. L., Heard, J., Martin, G., Reuber, L., Jiang, C. Z., Keddie, J., et al. (2000). Arabidopsis transcription factors: Genome-wide comparative analysis among eukaryotes. Sci. (1979) 290, 2105–2110. doi: 10.1126/science.290.5499.2105
Ruta, V., Longo, C., Lepri, A., De Angelis, V., Occhigrossi, S., Costantino, P., et al. (2020). The DOF transcription factors in seed and seedling development. Plants 9, 218. doi: 10.3390/PLANTS9020218
Saitou, N., Nei, M. (1987). The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Sangeetha, J., Thangadurai, D., Fayeun, L. S., Akinwale, J. A., Habeeb, J., Maxim, S. S., et al. (2020). Origin and evolution of rice as domesticated food crop. Rice Res. Qual. Improvement: Genomics Genet. Eng., 1–14. doi: 10.1007/978-981-15-4120-9_1
Sato, Y., Takehisa, H., Kamatsuki, K., Minami, H., Namiki, N., Ikawa, H., et al. (2013). RiceXPro version 3.0: expanding the informatics resource for rice transcriptome. Nucleic Acids Res. 41, D1206–D1213. doi: 10.1093/NAR/GKS1125
Sekhar, S., Panda, D., Kumar, J., Mohanty, N., Biswal, M., Baig, M. J., et al. (2019). Comparative transcriptome profiling of low light tolerant and sensitive rice varieties induced by low light stress at active tillering stage. Sci. Rep. 1 9, 1–14. doi: 10.1038/s41598-019-42170-5
Sievers, F., Higgins, D. G. (2014). Clustal omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol. 1079, 105–16. doi: 10.1007/978-1-62703-646-7_6
Shim, Y., Kang, K., An, G., Paek, N. C. (2019). Rice DNA-binding one zinc finger 24 (OsDOF24) delays leaf senescence in a jasmonate-mediated pathway. Plant Cell Physiol. 60, 2065–2076. doi: 10.1093/pcp/pcz105
Song, W. Y., Wang, G. L., Chen, L. L., Kim, H. S., Pi, L. Y., Holsten, T., et al. (1995). A receptor kinase-like protein encoded by the rice disease resistance gene, Xa21. Sci. (1979) 270, 1804. doi: 10.1126/SCIENCE.270.5243.1804
Stein, J. C., Yu, Y., Copetti, D., Zwickl, D. J., Zhang, L., Zhang, C., et al. (2018). Genomes of 13 domesticated and wild rice relatives highlight genetic conservation, turnover and innovation across the genus oryza. Nat. Genet. 2 50, 285–296. doi: 10.1038/s41588-018-0040-0
Sun, C., Hu, Z., Zheng, T., Lu, K., Zhao, Y., Wang, W., et al. (2017). RPAN: rice pan-genome browser for ∼3000 rice genomes. Nucleic Acids Res. 45, 597–605. doi: 10.1093/NAR/GKW958
Wang, Y., Fu, B., Pan, L., Chen, L., Fu, X., Li, K. (2013). Overexpression of arabidopsis Dof1, GS1 and GS2 enhanced nitrogen assimilation in transgenic tobacco grown under low-nitrogen conditions. Plant Mol. Biol. Rep. 31, 886–900. doi: 10.1007/S11105-013-0561-8/FIGURES/8
Wang, W., Mauleon, R., Hu, Z., Chebotarov, D., Tai, S., Wu, Z., et al. (2018). Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 7703 557, 43–49. doi: 10.1038/s41586-018-0063-9
Wang, M., Yu, Y., Haberer, G., Marri, P. R., Fan, C., Goicoechea, J. L., et al. (2014). The genome sequence of African rice (Oryza glaberrima) and evidence for independent domestication. Nat. Genet. 9 46, 982–988. doi: 10.1038/ng.3044
Wang, C. C., Yu, H., Huang, J., Wang, W. S., Faruquee, M., Zhang, F., et al. (2020). Towards a deeper haplotype mining of complex traits in rice with RFGB v2.0. Plant Biotechnol. J. 18, 14–16. doi: 10.1111/PBI.13215
Waqas, M., Shahid, L., Shoukat, K., Aslam, U., Azeem, F., Atif, R. M. (2020). Role of DNA-binding with one finger (Dof) transcription factors for abiotic stress tolerance in plants. Transcription Factors Abiotic Stress Tolerance Plants 1–14, 1–14. doi: 10.1016/B978-0-12-819334-1.00001-0
Wei, C. Q., Chien, C. W., Ai, L. F., Zhao, J., Zhang, Z., Li, K. H., et al. (2016). The arabidopsis b-BOX protein BZS1/BBX20 interacts with HY5 and mediates strigolactone regulation of photomorphogenesis. J. Genet. Genomics 43, 555. doi: 10.1016/J.JGG.2016.05.007
Wei, P. C., Tan, F., Gao, X. Q., Zhang, X. Q., Wang, G. Q., Xu, H., et al. (2010). Overexpression of AtDOF4.7, an arabidopsis DOF family transcription factor, induces floral organ abscission deficiency in arabidopsis. Plant Physiol. 153, 1031–1045. doi: 10.1104/PP.110.153247
Wu, Y., Lee, S. K., Yoo, Y., Wei, J., Kwon, S. Y., Lee, S. W., et al. (2018). Rice transcription factor OsDOF11 modulates sugar transport by promoting expression of sucrose transporter and SWEET genes. Mol. Plant 11, 833–845. doi: 10.1016/J.MOLP.2018.04.002
Wu, Q., Li, D., Li, D., Liu, X., Zhao, X., Li, X., et al. (2015). Overexpression of OsDof12 affects plant architecture in rice (Oryza sativa l.). Front. Plant Sci. 6. doi: 10.3389/FPLS.2015.00833/BIBTEX
Wu, Q., Liu, X., Yin, D., Yuan, H., Xie, Q., Zhao, X., et al. (2017a). Constitutive expression of OsDof4, encoding a C2-C2 zinc finger transcription factor, confesses its distinct flowering effects under long- and short-day photoperiods in rice (Oryza sativa l.). BMC Plant Biol. 17, 1–13. doi: 10.1186/S12870-017-1109-0/FIGURES/6
Wu, Y., Yang, W., Wei, J., Yoon, H., An, G. (2017b). Transcription factor OsDOF18 controls ammonium uptake by inducing ammonium transporters in rice roots. Moleucles Cells 40, 178–185. doi: 10.14348/MOLCELLS.2017.2261
Yanagisawa, S. (2002). The dof family of plant transcription factors. Trends Plant Sci. 7, 555–560. doi: 10.1016/S1360-1385(02)02362-2
Yanagisawa, S. (2004). Dof domain proteins: Plant-specific transcription factors associated with diverse phenomena unique to plants. Plant Cell Physiol. 45, 386–391. doi: 10.1093/PCP/PCH055
Yanagisawa, S. (2016). Structure, function, and evolution of the dof transcription factor family. Plant Transcription Factors: Evolutionary Struct. Funct. Aspects, 183–197. doi: 10.1016/B978-0-12-800854-6.00012-9
Yu, J., Hu, S., Wang, J., Wong, G. K. S., Li, S., Liu, B., et al. (2002). A draft sequence of the rice genome (Oryza sativa l. ssp. indica). Sci. (1979) 296, 79–92. doi: 10.1126/SCIENCE.1068037/SUPPL_FILE/WEBSUPPL_EXINDAT_PARSE.XLS
Yu, Y., Ouyang, Y., Yao, W. (2018). ShinyCircos: An R/Shiny application for interactive creation of circos plot. Bioinformatics 34, 1229–1231. doi: 10.1093/bioinformatics/btx763
Zhuo, M., Sakuraba, Y., Yanagisawa, S. (2020). A jasmonate-activated MYC2–Dof2.1–MYC2 transcriptional loop promotes leaf senescence in arabidopsis. Plant Cell 32, 242–262. doi: 10.1105/TPC.19.00297
Zhu, T., Xu, P. Z., Liu, J. P., Peng, S., Mo, X. C., Gao, L. Z. (2014). Phylogenetic relationships and genome divergence among the AA- genome species of the genus oryza as revealed by 53 nuclear genes and 16 intergenic regions. Mol. Phylogenet. Evol. 70, 348–361. doi: 10.1016/J.YMPEV.2013.10.008
Zou, X., Neuman, D., Shen, Q. J. (2008). Interactions of two transcriptional repressors and two transcriptional activators in modulating gibberellin signaling in aleurone cells. Plant Physiol. 148, 176–186. doi: 10.1104/PP.108.123653
Zuckerkandl, E., Pauling, L. (1965). “Evolutionary divergence and convergence in proteins,” in Evolving genes and proteins. Eds. Bryson, V., Vogel, H. J. (Academic Press, New York: Institute of Microbiology of Rutgers), 97–166. Edited in.
Keywords: rice, DNA-binding with one finger, gene evolution, phylogenetics, comparative genomics
Citation: Tabassum J, Raza Q, Riaz A, Ahmad S, Rashid MAR, Javed MA, Ali Z, Kang F, Khan IA, Atif RM and Luo J (2022) Exploration of the genomic atlas of Dof transcription factor family across genus Oryza provides novel insights on rice breeding in changing climate. Front. Plant Sci. 13:1004359. doi: 10.3389/fpls.2022.1004359
Received: 27 July 2022; Accepted: 05 September 2022;
Published: 03 November 2022.
Edited by:
Bo Song, Agricultural Genomics Institute at Shenzhen (CAAS), ChinaCopyright © 2022 Tabassum, Raza, Riaz, Ahmad, Rashid, Javed, Ali, Kang, Khan, Atif and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ju Luo, bHVvanUwMUBjYWFzLmNu; Rana Muhammad Atif, ZHJhdGlmQHVhZi5lZHUucGs=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.