 Yushuai Wang
Yushuai Wang Aimei Dai
Aimei Dai Tian Tang
Tian Tang- State Key Laboratory of Biocontrol and Guangdong Key Laboratory of Plant Resources, School of Life Sciences, Sun Yat-sen University, Guangzhou, China
Transposable elements (TEs) are an important source of genetic diversity and can be co-opted for the regulation of host genes. However, to what extent the pervasive TE colonization of plant genomes has contributed to stress adaptation remains controversial. Plants inhabiting harsh environments in nature provide a unique opportunity to answer this question. We compared TE compositions and their evolutionary dynamics in the genomes of two mangrove species: the pioneer Sonneratia alba and its less salt-tolerant relative S. caseolaris. Age distribution, strength of purifying selection and the removal rate of LTR (long terminal repeat) retrotransposons were estimated. Phylogenetic analysis of LTR retrotransposons and their distribution in the genome of S. alba were surveyed. Small RNA sequencing and whole-genome bisulfite sequencing was conducted using leaves of S. alba. Expression pattern of LTR retrotransposons and their nearby genes were examined using RNA-seq data of S. alba under different salt treatments. S. alba possesses more TEs than S. caseolaris. Particularly, many more young Gypsy LTR retrotransposons have accumulated in S. alba than in S. caseolaris despite an increase in purifying selection against TE insertions. The top two most abundant Gypsy families in S. alba preferentially insert in gene-poor regions. They are under relaxed epigenetic repression, probably due to the presence of CHROMO domains in their 3′-ends. Although a considerable number of TEs in S. alba showed differential expression under salt stress, only four copies were significantly correlated with their nearby genes in expression levels. One such TE-gene pair involves Abscisic acid 8'-hydroxylase 3 functioning in abscisic acid catabolism. This study sheds light on the evolutionary dynamics and potential function of TEs in an extremophile. Our results suggest that the conclusion on co-option of TEs should be cautious even though activation of TEs by stress might be prevalent.
Introduction
Transposable elements (TEs) are genetically diverse mobile sequences that can move to new sites in genomes by a “copy-and-paste” mechanism via an RNA intermediate (Class I, retrotransposons) or a “cut-and-paste” mechanism (Class II, transposons) (Wicker et al., 2007; Bourque et al., 2018). TE insertions often disrupt gene function or regulation and induce ectopic/non-allelic recombination events (Chuong et al., 2017). As a result, their activities are repressed by host epigenetic silencing and small interfering RNAs (siRNAs) (Fultz et al., 2015). Bursts of TE abundance are further subject to mutational decay and are eliminated by genome purging mechanisms in the long run. In many higher eukaryotes TEs constitute more than half of the DNA (Fedoroff, 2012) and play a critical role in the evolution of genome size and organization (Lisch, 2013). It is thus important to dissect the evolutionary dynamics of TEs in the genome.
While TEs are often deleterious or effectively neutral, there is growing evidence that TE activity can help the host adapt to environmental challenges (Casacuberta and Gonzalez, 2013; Horvath et al., 2017). High copy number and mutagenicity make TEs a major source of genetic innovation that can be activated by environmental cues (Capy et al., 2000; Grandbastien, 2015; Rey et al., 2016). This may accelerate mutation rates and generate variability upon which natural selection could act. Indeed, recent findings show that TEs can facilitate rapid adaptation to novel environments in species with limited genetic variation, such as Capsella rubella the annual inbreeding forb (Niu et al., 2019) and Cardiocondyla obscurior the invasive inbreeding ant (Schrader et al., 2014). Meanwhile, some TEs are known to carry stress-responsive regulatory elements. These elements, once activated by stress, may have a large role in remodeling regulatory networks and reshaping transcriptomes (Naito et al., 2009; Cavrak et al., 2014; Grandbastien, 2015; Chuong et al., 2017). Despite many reports that individual TE integration events result in changes in gene expression or even phenotypes under stress conditions (Butelli et al., 2012; Lisch, 2013), there is empirical evidence that regulatory function of TEs is not always strong (de Souza et al., 2013; Simonti et al., 2017).
Mangroves are a group of phylogenetically diverse woody species that invaded and adapted to the extreme and extremely fluctuating environments in the tropical intertidal zone (Tomlinson, 1986). These species have undergone multiple population bottlenecks during their evolutionary history and typically harbor extremely low standing genetic variation (Zhou et al., 2011; Guo et al., 2018). A recent study reported massive and convergent TE load reduction in mangroves, when compared to non-mangrove species (Lyu et al., 2018). However, we found a continuous host-TE battle masked by the TE load reduction in Rhizophora apiculata, the tall-stilted mangrove, suggesting TE activities may enable mangroves to maintain genetic diversity (Wang et al., 2018). It still remains unclear how often TE activities are significant to the evolution of mangrove genomes and to what extent TEs may have influenced gene expression during mangrove stress adaptation.
In this study, we surveyed the evolutionary dynamics and potential regulatory functions of TEs in Sonneratia alba Sm., the pioneer mangrove (Tomlinson, 1986), in comparison with its relative S. caseolaris (L.) Engl. Although both are mangroves, S. alba is more salt-tolerant than S. caseolaris and often grows on the seaward side of mangrove forests with maximal growth in 5–50% sea water (Ball and Pidsley, 1995). In contrast, S.caseolaris is common in the inner parts of mangrove forests (Bunt et al., 1982) and in some instances has also been found growing in fully fresh water (Rahim and Bakar, 2018). Here we report recent amplification of Gypsy LTR (long terminal repeat) retrotransposons in S. alba. Nevertheless, this burst of Gypsy retrotransposons has only a weak effect on salt stress gene expression. Our results suggest that the conclusion on co-option of TEs should be cautious even though activation of TEs by stress might be prevalent.
Materials and Methods
Annotation and Classification of Transposable Elements
Whole genome sequences of Sonneratia alba, Sonneratia caseolaris, and Eucalyptus grandis were retrieved from previous studies (Myburg et al., 2014; Lyu et al., 2018). LTR retrotransposons were de novo mined, classified and named using work flows described previously (Wicker et al., 2007; Wang et al., 2018). Candidate LTR retrotransposons that possessed complete Gag-Pol protein sequences were considered as intact elements. The annotation of long interspersed nuclear elements (LINE), short interspersed nuclear elements (SINE), and DNA transposons (MULE, hAT, PIF, CACTA, and TcMar) was carried out as follows. First, sequences of the most conserved coding domain of each TE family were retrieved form the NCBI database (Coordinators, 2018) and used as queries against each genome using the TARGeT pipeline (Han et al., 2009) with the e-value cutoff of 0.001. Then, the identified homologs together with their 5 kb up- and downstream sequences were aligned within individual TE families using MAFFT (Katoh et al., 2002) and resolved into lineages using phylogenetic analyses as implemented in SiLiX (Miele et al., 2011). The terminals of each putative element were determined manually according to sequence similarity with its closely related elements. Miniature inverted-repeat transposable elements (MITEs) and Helitron copies were identified using MITE_HUNTER (Han and Wessler, 2010) and HelitronScanner (Xiong et al., 2014) with default parameters, respectively. Finally, all de novo identified TEs were added to RepBase (v.23.06) (Bao et al., 2015) for each genome separately and used to annotate TE contents in each species using RepeatMasker (v.4.0.0) (Smit et al., 2013-2015).
Dating LTR Retrotransposon Elements
Age of each LTR retrotransposon element was estimated based on sequence divergence between their LTR pair (SanMiguel et al., 1998). The pairs from individual LTR retrotransposons were aligned using MUSCLE (v.3.8.31) with default parameters (Edgar, 2004). Sequence divergence k between each pair of LTRs was estimated using Kimura's two-parameter model (Kimura, 1980). The age (T) of each LTR retrotransposons was estimated using the formula T = k/2r, where r is the substitution rate of 1.3 × 10−8 substitutions per site per year (Ma and Bennetzen, 2004).
Distribution of TEs Across Genomic Regions
The insertion frequency of TEs categories in intergenic regions, introns, and coding regions in S. alba and S. caseolaris were estimated according to the procedure described before (Wright et al., 2003). Briefly, the genome sequences and annotated gene positions of S. alba and S. caseolaris were obtained from Lyu et al. (2018). For each species, each scaffold (>100 kb) was submitted to a BLAST search against our TE database to determine the positions of each TE superfamily along the entire set of scaffolds. TE positions were then verified manually to eliminate all redundancies and record the presence of nested insertions. To estimate gene density, sequences were scanned using 100 kb sliding windows with 50 kb steps across each scaffold. Gene density of each window was calculated as the fraction of the total length occupied by coding sequences.
Phylogenetic Analyses of LTR Retrotransposons
Multiple alignments of full-length sequences of intact Copia or Gypsy retrotransposons were conducted using MAFFT (Katoh et al., 2002). Maximum likelihood phylogenetic trees were inferred with FastTree (Price et al., 2009) using a generalized time-reversible (GTR) model of nucleotide substitution. The generated trees were visualized with Figtree (v.1.4.2) (Rambaut, 2012). Resampling of estimated log-likelihoods (RELL)-like local support values was performed using FastTree (Price et al., 2009) with default parameters.
Identification and Sequence Analysis of the Chromodomain in LTR Retrotransposons
Chromodomains residing in LTR retrotransposons were identified by tBLASTx (NCBI) searches against the nr database (NCBI) with default parameters (Boratyn et al., 2012). Multiple alignments of the chromodomain amino acid sequences were performed using MAFFT (Katoh et al., 2002). Tertiary structures of the chromodomains peptides were predicted using the Phyre2 server with default parameters (Kelley et al., 2015). Known chromodomain amino acid sequences were retrieved from the NCBI database (Coordinators, 2018), including DmHP1 (GenBank accession number, M57574) from Drosophila melanogaster, MAGGY (L35053) and Pyret (AB062507) from Magnaporthe grisea, Cft1 (AF051915) from Cladosporium fulvum, PpatensLTR1 (XM_001752430) from Physcomitrella patens, Tf2-12 (RVW30894) from Vitis vinifera, sr35 (AC068924), rn8 (AK068625), and RIRE3 (AC119148) from Oryza sativa, Tekay (AF050455) from Zea mays, RetroSor2 (AF061282) from Sorghum bicolor, and Tma (AF147263) from Arabidopsis thaliana.
Epigenome Sequencing and Analyses
Young leaves of S. alba were collected from Qinlan Harbor, Hainan, China. Genomic DNA and total RNA were extracted separately using the modified CTAB protocol (Yang et al., 2008). Bisulfite (BS-seq) and small RNA sequencing were conducted with two biological replicates each according to previously published procedures (Wen et al., 2016; Wang et al., 2018). Briefly, bisulfite (BS-seq) sequencing libraries were prepare with the Zymo EZ DNA Methylation-Gold Kit (Zymo Research, Orange, CA, USA) and EpiGnome™ Kit (Epicenter, Madison, WI, USA) following manufacturer's recommendations. Sequencing was performed on an Illumina HiSeq 2500 platform (Illumina, San Diego, CA, USA) in paired-end mode to generate 150 bp reads with Q30 quality control. Small RNA libraries were prepared using the standard protocols of the NEBNext® Multiplex Small RNA Library Prep Set for Illumina® (Set 2) (NEB, Ipswich, MA, USA) and were sequenced using an Illumina HiSeq 2500 platform (Illumina, San Diego, CA, USA).
Bisulfite sequencing reads were filtered for quality using Trimmomatic (v.0.32) (Bolger et al., 2014) and mapped to the S. alba genome using Bismark (v.0.16.3) (Krueger and Andrews, 2011) with default parameters. Only uniquely mapping reads were retained. Methylation levels of each LTR retrotransposon copy were calculated as the proportion of Cs among Cs and Ts [#C/(#C+#T)] by methylation context (CG, CHG and CHH) using custom Perl scripts. Only sites covered by more than three reads were used for the calculation. Results were visualized using R (v.3.1.3) (https://www.R-project.org).
Small RNA sequencing reads were quality controlled following procedures published previously (Wen et al., 2016) while structural non-coding RNAs, such as ribosomal RNA (rRNA), transfer RNA (tRNA), small nuclear RNA (snRNA), small nucleolar RNA (snoRNA), and known microRNAs (miRNAs) were removed. The remaining reads were considered as putative siRNAs and aligned to S. alba genomes using Bowtie (Langmead et al., 2009) with no mismatches allowed. SiRNA density of a particular LTR retrotransposon element was calculated as read counts per kilo base pair per element. Multiply-mapping reads were weighted by the number of their mapping locations. All statistical calculations were performed in R (v.3.1.3).
Expression Analyses of TEs and TE-Flanking Genes Under Salt Treatments
Transcriptome data from S. alba under the 0, 250, and 500 mM NaCl treatments were obtained from Feng et al. (2020) (NCBI Sequence Read Archive database accession No. PRJNA615770). Sequencing adapters and low quality reads were filtered using Trimmomatic (v.0.32) (Bolger et al., 2014). Clean reads were mapped to coding genes or intact LTR retrotransposons separately using Bowtie (Langmead et al., 2009) allowing no more than two mismatches. We calculated the expression level of each LTR retrotransposon element as reads per kilobase of transcript. Expression levels of individual genes were analyzed using HTSeq (v.0.6.1) (Anders et al., 2015) with the parameter -s set to “no” and normalized to Reads Per Kilobase per Million mapped reads (RPKM). Only genes located within 3 kb up- or downstream of the insertion of an intact or truncated LTR retrotransposon were considered in our analysis. The expression differences of TEs or genes across salt treatments were identified using DESeq2 (Love et al., 2014) requiring Benjamini-Hochberg multiple testing corrected (Benjamini and Hochberg, 1995). FDR < 0.05 and ≥2-fold change.
Identification of cis-Acting Regulatory Elements of LTR Retrotransposons
The CREs within LTRs of the consensus sequence of each LTR retrotransposon family or within the whole sequence of each truncated LTR retrotransposon were identified using PlantCARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/search_CARE.html) (Lescot et al., 2002). Gypsy and Copia families that have more than ten copies were considered in this analysis.
MiRNA Target Prediction
Plant mature miRNAs and their precursors were retrieved from miRBase (Release 22) (Kozomara et al., 2019). The identification of the known and novel miRNAs was performed by miRDeep2 software package (Friedlander et al., 2012) with the default parameters. The expression level of each identified miRNAs was normalized to Reads Per Million mapped reads (RPM). MiRNAs that with a RPM > 1 in both biological replicates were considered expressed and retained for further analysis. The prediction of the putative target genes for the known and novel miRNAs was done with the psRNATarget (Dai et al., 2018) using an expectation score of 2.5 as the cutoff in the prediction. All LTR retrotransposons including the intact and truncated elements and solo-LTRs were used in these analyses.
Results
TEs Are More Abundant in S. alba Than in S. caseolaris
S. alba and S. caseolaris diverged about ~11.6 million years (Myr) ago (Yang et al., 2016). The whole genome sequences of S. caseolaris (~208Mb, 1C = 259 Mb) and S. alba (~207Mb, 1C = 284 Mb) have recently been de novo assembled (Lyu et al., 2018). We used the whole genome of Eucalyptus grandis (Myburg et al., 2014) as the outgroup following a previous study (Xu et al., 2017) and studied the evolutionary dynamics of TEs in S. alba in comparison with S. caseolaris. We annotated TEs in each species considering both TE structure and sequence homology and clustered them into families using the same procedure (see Methods).
Consistent with the report of TE load reduction in mangroves (Lyu et al., 2018), we found TEs only accounted for 5.3% (11,091,467 bp) and 11.0% (22,805,675 bp) of the S. caseolaris and S. alba genomes, respectively, far less than the proportion of TEs in the E. grandis genome (27.7%, 191,425,687 bp, Figure 1A). In all three species, LTR retrotransposons are the dominant TE components (Figure 1A). Nevertheless, Gypsy retrotransposons are more abundant than Copia retrotransposons in the two Sonneratia species while the opposite pattern is observed in the outgroup E. grandis (Figure 1A). The expansion of Gypsy retrotransposons is more prominent in S. alba (12.7 Mb or 6.1%) than in S. caseolaris (3.3 Mb or 1.6%, Figure 1B). We identified 947 intact LTR retrotransposon copies comprising 180 Copia and 767 Gypsy elements in S. alba and 125 intact LTR retrotransposon copies consisting of 51 Copia and 74 Gypsy elements in S. caseolaris (Supplementary Table 1). Although mangroves have experienced an overall reduction in TE load (Lyu et al., 2018), more Gypsy retrotransposons have accumulated in S. alba than in S. caseolaris.
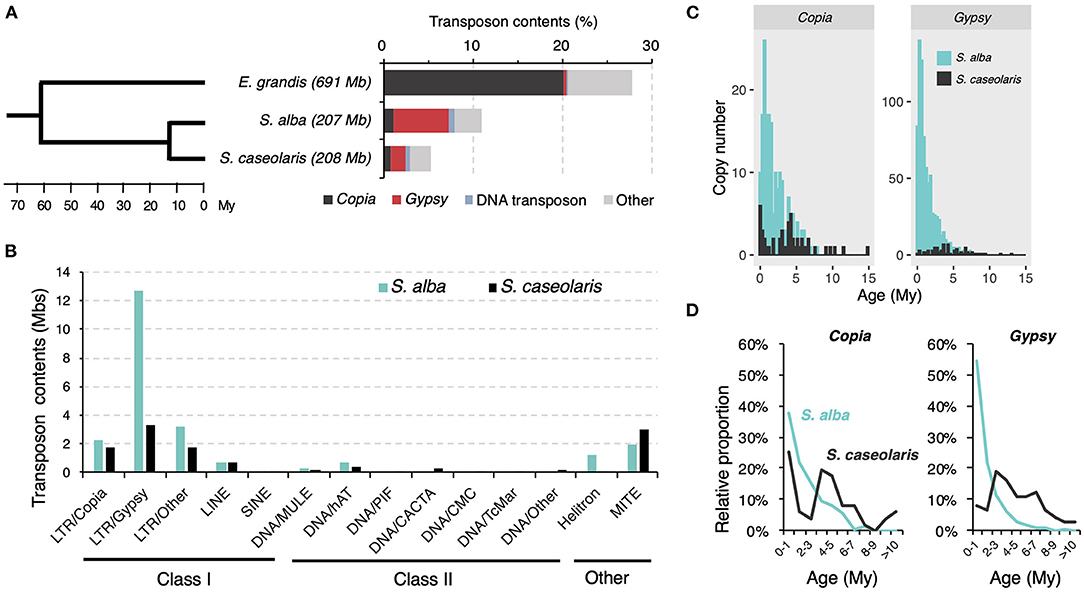
Figure 1. TE composition in Sonneratia alba and S. caseolaris. (A) Bar plot shows the proportions of transposon groups in S. alba, S. caseolaris, and Eucalyptus grandis (the outgroup). Tree topology is adopted from Lyu et al. (2018). (B) Histogram shows the distribution of transposon contents across families between S. alba and S. caseolaris. (C) Histogram shows the copy number of Copia and Gypsy elements in S. alba and S. caseolaris grouped by age. (D) Line graph shows the relative proportion of total Copia and Gypsy elements in different age bins.
Gypsy Retrotransposons Experienced Recent Bursts in S. alba
To explore the dynamics of TE accumulation, we first estimated ages of the intact Gypsy and Copia retrotransposons by comparing LTR sequences flanking each element and assuming a molecular clock of 1.3 × 108 (Ma and Bennetzen, 2004). We found these retrotransposons were much younger in S. alba (Copia: 1.47 Myr old; Gypsy: 0.87 Myr old) than in S. caseolaris (Copia: 3.85 Myr old and Gypsy: 3.99 Myr old). These differences are highly significant (Mann-Whitney U-test, both P < 0.001, Figure 1C). The recent burst of TE activity is most striking for Gypsy elements in S. alba. Young (< 1 Myr old) Gypsy elements account for 54.4% of the intact elements in S. alba but only 8.1% in S. caseolaris (Figure 1D). By contrast, the proportions of young Copia elements are more similar between S. alba (37.9%) and S. caseolaris (25.5%, Figure 1D). Therefore, there are more recent originations of the Gypsy element in S. alba than in S. caseolaris.
We then inferred the intensity of purifying selection against deleterious TE insertions by comparing TE distribution in relation to coding regions. We found TE insertions are strongly under-represented in coding regions compared to introns and intergenic regions (Table 1), consistent with the prediction of the deleterious insertion model (Finnegan, 1992). The abundance of all TE categories regardless of their locations are significantly lower in S. alba than in S. caseolaris (Table 1), suggesting TEs are under stronger purifying selection in the former species. However, the abundance of Gypsy elements in introns and intergenic regions was comparable between S. alba and S. caseolaris (Mann-Whitney U-test, all P > 0.05), suggesting that the insertions of Gypsy into introns and intergenic regions have experienced relaxation of purifying selection in S. alba. It is also possible that purifying selection against Gypsy insertions into coding regions is even stronger than that for other TE families in S. alba; otherwise, we should see more Gypsy insertions in the coding regions of that species.
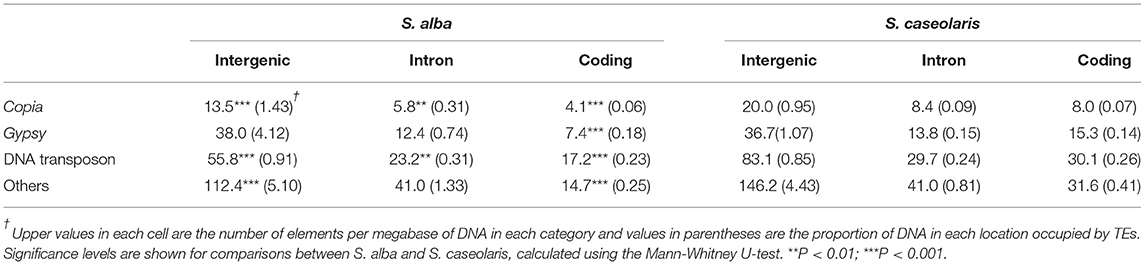
Table 1. Location of TE insertions in relation to coding regions in S. alba and S. caseolaris.
Finally, we analyzed the removal rate of LTR elements by estimating the ratio of solo-LTRs to intact elements (S:I). Unequal intra-element homologous recombination resulting in solo-LTRs is known to be one of the major mechanisms eliminating LTR retrotransposons (Devos et al., 2002). As shown in Table 2, the S:I ratio is the lowest for Gypsy elements in S. alba (0.34), followed by Copia in S. alba (1.79) and then the Copia and Gypsy elements in S. caseolaris (2.45 and 3.72). Taken together, these results suggest that despite strong purifying selection, high proliferation activity and low elimination rate jointly contributed to the rapid accumulation of Gypsy elements in the S. alba genome.
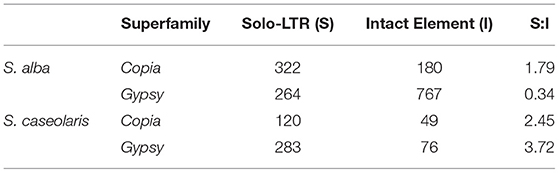
Table 2. Comparison of the ratios of solo-LTRs to intact elements (S:I) for Copia and Gypsy superfamilies.
Recent Bursts of Two Gypsy Families in S. alba Are Associated With Integration Site Preference
To compare the evolutionary histories of Copia and Gypsy retrotransposons in the two Sonneratia species, we conducted phylogenetic analyses using full length sequences of intact elements for the Copia and Gypsy superfamilies separately. The maximum-likelihood phylogenetic trees grouped LTR retrotransposon copies of S. alba and S. caseolaris into 26 Copia and 23 Gypsy families, half of which (13 out of 26 for Copia and 11 out of 23 for Gypsy) were shared by both genomes (Supplementary Figure 1). Remarkably, we found that two Gypsy families (RLG_1 and RLG_8) are ~10 times more abundant in S. alba (RLG_1: 226 copies and RLG_8: 273 copies) than in S. caseolaris (RLG_1: 20 and RLG_8: 22, Supplementary Figure 1). These two Gypsy families account for 64.6% of the young copies (<1 Myr) and 65.1% of the total intact Gypsy elements in S. alba (Figure 2A). Furthermore, three RLG_1 elements and four RLG_8 elements have identical 5′ and 3′ LTR sequences and perfect 5-bp Target Site Duplications (TSDs), suggesting they are newly originated copies. Therefore, recent bursts of RLG_1 and RLG_8 in S. alba are the major contributors to the observed expansion of Gypsy elements.
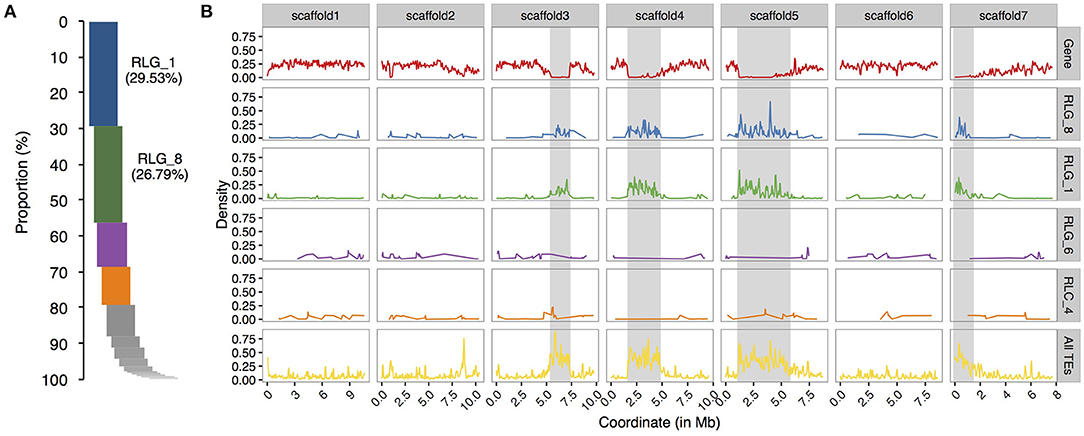
Figure 2. Relative proportion and chromosome distribution of LTR retrotransposon families in S. alba. (A) Proportions of intact copy numbers from different LTR retrotransposon families ranked in a descending order. Note that RLG_1 and RLG_8 are the top two abundant LTR retrotransposons in S. alba. (B) Chromosome distribution of coding genes (the upper row), the top four abundant LTR retrotransposon families (RLG_1, RLG_8, and RLG_4 of Gypsy and RLC_6 of Copia), and all TEs along the seven largest scaffolds of the S. alba genome. The putative heterochromatin regions (gene density < 0.05) are indicated with the gray shadow. Note that the distributions of RLG_1 and RLG_8 elements track the genomic locations of putative heterochromatic regions but no such pattern is observed for RLG_4 and RLC_6.
Plant LTR retrotransposons are preferentially associated with large heterochromatic regions that flank functional centromeres (Kumar and Bennetzen, 1999). We wondered whether such integration site preference, leading to loose epigenetic repression, might have contributed to the expansion of RLG_1 and RLG_8 families. We scanned scaffolds of the S. alba genome using 100 kb sliding windows with a step-size of 50 kb and estimated gene density in each window (see Methods). As expected, about half of the RLG_1 copies (50.9%) and 2/5 of the RLG_8 copies (40.7%) are located in gene-poor regions (gene density < 0.05) in S. alba. In contrast, the proportion is only 8.1 and 12.5% for the third and fourth most abundant LTR retrotransposon families (Figure 2B).
Further analysis of the structure of RLG_1 and RLG_8 detected a chromodomain (CHD) in the 3′ end of both families. CHD was first identified in the heterochromatin protein-1 (HP1) of Drosophila melanogaster (Paro and Hogness, 1991) and considered to be involved in chromatin remodeling and the formation of heterochromatin (Paro and Hogness, 1991). Sequence analyses of CHD proteins from diverse organisms classified CHD of the RLG_1 and RLG_8 families into Group I chromodomains (Figure 3A). Both of them possess a highly conservative protein secondary structure (Figure 3B). CHD in Gypsy elements, also named chromoviruses, has been experimentally demonstrated to promote the integration of newly emerging retroelements into heterochromatic regions (Gao et al., 2008).
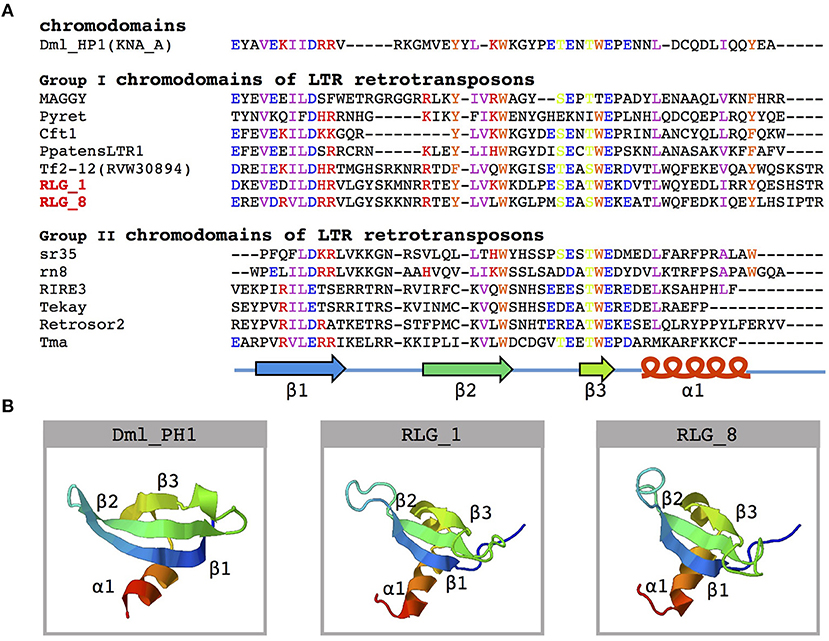
Figure 3. Amino acid sequence alignment and structure prediction of the chromodomain. (A) Multiple alignments of amino acid sequences of known chromodomains and chromo-like motifs from the RLG_1 and RLG_8 families. A schematic representation of β-strands (arrows) and α-helix (cylinder) is on the bottom. The GenBank accession numbers for the sequences used are: DmHP1(KNA_A)-M57574 from Drosophila melanogaster; MAGGY-L35053 and Pyret-AB062507 from Magnaporthe grisea; Cft1-AF051915 from Cladosporium fulvum; PpatensLTR1-XM_001752430 from Physcomitrella patens; Tf2-12-RVW30894 from Vitis vinifera; sr35-AC068924, rn8-AK068625 and RIRE3-AC119148 from Oryza sativa; Tekay-AF050455 from Zea mays; RetroSor2-AF061282 from Sorghum bicolor; Tma-AF147263 from Arabidopsis thaliana. (B) Predicted three-dimensional structure of chromodomains from the DmHP1 protein (M57574) and the RLG_1 and RLG_8 families.
Expanded Gypsy Families Are Under Relaxed Epigenetic Repression
If CHD indeed promotes the insertions of RLG_1 and RLG_8 into heterochromatin regions, we would expect that epigenetic repression of these elements is relaxed in the host genome. In plants, CHH methylation mediated by 24-nt siRNAs is responsible for the establishment of de novo methylation against the emergence of new TE copies (Law and Jacobsen, 2010). We thus estimated correlations between gene density, CHH methylation level, and 24-nt siRNA abundance for the RLG_1 and RLG_8 copies in S. alba. Gene density of the window in which each RLG_1 and RLG_8 copy resides was used in this analysis.
For both RLG_1 and RLG_8, copies residing in the gene-poor regions (gene density < 0.05) exhibited a significantly lower level of CHH methylation (RLG_1: median, hereafter, 10.6% and RLG_8: 15.9%) than the rest (RLG_1: 44.0% and RLG_8: 37.7%, Mann-Whitney U-test, all P < 0.001, Figure 4A). The 24-nt siRNA targeting pattern is in accordance with the difference in methylation levels, i.e., a lower level of 24-nt siRNA abundance on TEs residing in gene-poor regions than the other TEs (4.0 vs. 7.5 reads per element per kilobase for RLG_1 and 2.9 vs. 5.8 for RLG_8, Mann-Whitney U-test, all P <0.001; Figure 4A). These results support the idea that members of RLG_1 and RLG_8 have experienced relaxed epigenetic repression associated with their insertion preference. Gene-poor regions might represent a “safe haven” for these elements to coexist with host cells.
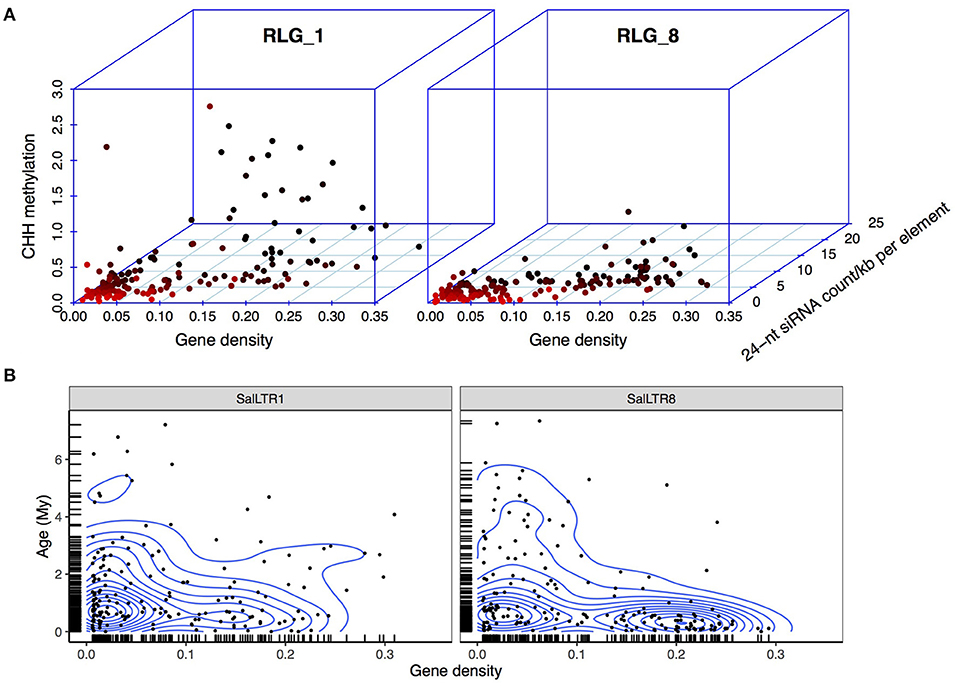
Figure 4. Epigenetic repression and insertional locations of RLG_1 and RLG_8 in S. alba. (A) Correlations between CHH methylation level, 24-nt siRNA abundance of the RLG_1 and RLG_8 elements, and gene density of the 100 Kb region in which each retroelement is located in the S. alba genome. Each dot represents an intact RLG_1 or RLG_8 element and is colored in red to black gradient according to gene density of the genomic region in which the element is located. (B) Age distribution of the RLG_1 or RLG_8 elements along genomic regions with different gene density. Contour lines represent estimated two dimensional kernel density.
Weak Effect of TE Expansion on Salt Stress Gene Expression in S. alba
Despite the overall preference for integration into gene-poor regions, there is still a considerable fraction of young copies (< 1 Myr) of RLG_1 (21.8%) and RLG_8 (45.2%) inserted into genomic regions with gene density >0.15 (Figure 4B). It is possible that recently amplified TEs can affect nearby gene expression by carrying repressive epigenetic markers or creating new regulatory elements (Naito et al., 2009; Chuong et al., 2017). Recently, Feng et al. (2020) reported a comparative transcriptome study of S. alba treated with 0, 25 0, and 500 mM NaCl. They used the 250 mM NaCl condition as a control and 0 and 500 mM NaCl as stresses with low or high salt (Feng et al., 2020). Using these data, we asked whether intact LTR retrotransposons in S. alba can be activated by salt stress and how TE expansion might affect gene expression under stress conditions.
Many more of the 767 and 180 intact Gypsy and Copia retrotransposons were expressed in roots (Gypsy: 420 and Copia: 15) than in leaves (Gypsy: 169 and Copia: 11) (Figure 5A). This is coincident with the fact that roots are directly exposed to the saline environment. An intact retrotransposon was considered to be expressed if it had more than 5 reads in both biological replicates under at least one of the low, medium, or high salinity conditions. There was more upregulation (log2 fold change > 1) than downregulation (log2 fold change >−1) for these retroelements in all comparisons, most prominently for the comparison between 500 and 250 mM NaCl in roots (Figure 5A). However, only four retrotransposons were significantly mis-regulated due to large variation among replicates.
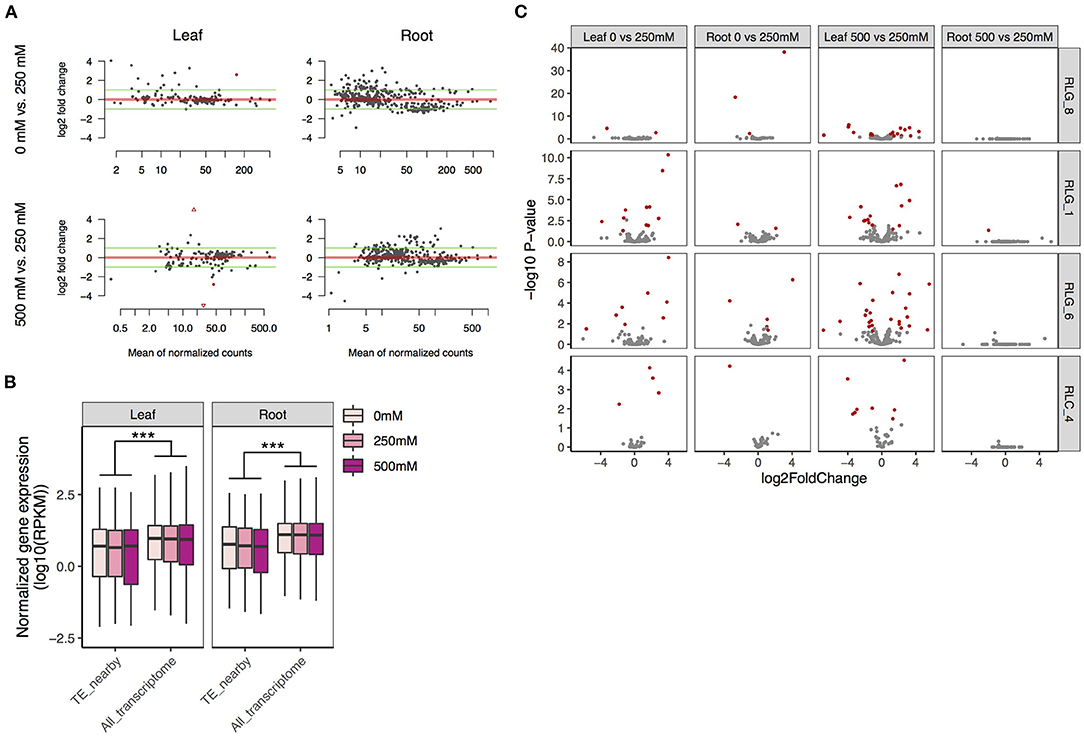
Figure 5. Expression patterns of TEs and their flanking genes under salt treatments in S. alba. (A) Fold changes of the expression levels of intact Gypsy and Copia retrotransposons across salt treatments (the upper: 0 mM vs. 250 mM NaCl, the lower: 250 mM vs. 500 mM NaCl). (B) Expression levels of genes neighboring copies of the four most abundant LTR retrotransposon families in comparison with the whole transcriptome. (C) Fold expression change of genes next to copies of the four most abundant LTR retrotransposon families across salt treatments (the upper: 0 mM vs. 250 mM NaCl, the lower: 500 mM vs 250 mM NaCl). Genes with significant mis-expression (FDR < 0.05 and ≥2-fold change) between treatments are indicated in red. Significance was determined by the Mann-Whitney U test: ***P < 0.001.
We further checked changes of expression in genes with intact TEs in introns, within 3 kb upstream of the transcription start site, or downstream of the transcription termination site for the top four most abundant LTR retrotransposon families in S. alba. Gene expression level was calculated as reads per kb per million reads (RPKM). For all the four families, genes with neighboring TEs were expressed at a significantly lower level than the whole transcriptome under all conditions (Mann-Whitney U-test, all P < 0.001 in both leaves and roots; Figure 5B), supporting the view that TEs carry repressive epigenetic marks that may reduce flanking gene expression (Naito et al., 2009; Chuong et al., 2017). While a few TE-adjacent genes were significantly mis-regulated (FDR < 0.05 and fold change ≥2) by salt (Figure 5C, Supplementary Table 2), this set of loci was not enriched in mis-expressed genes relative to the whole transcriptome in any of our comparisons (Fisher exact test, all P > 0.05, Supplementary Table 3). We also estimated correlations between TE expression levels and their nearby genes in leaves or roots across the three salt conditions (Supplementary Table 4). Only two significantly correlated pairs were identified in leaves: a RLC_4 copy and its 1307-bp upstream gene of unknown function (Pearson's correlation, r = 0.99, adjusted P < 0.05), and a RLG_8 copy and its 505-bp downstream gene Abscisic acid 8'-hydroxylase 3 (Pearson's correlation, r = 0.99, adjusted P < 0.05). Abscisic acid 8'-hydroxylase catalyzes the first step in the oxidative degradation of (+)-ABA (Krochko et al., 1998). Among the four ABA 8'-hydroxylase genes in Arabidopsis, CYP707A3 supports the drought tolerance of plants via maintenance of ABA levels (Umezawa et al., 2006).
Besides intact LTR retrotransposons, truncated elements and solo-LTRs may also affect expression of nearby genes. We then examined expression changes of all the truncated retrotransposons (3,279) and solo-LTRs (586) as well as expression changes of their nearby genes in S. alba as described above. Similar with the intact elements, the truncated LTR retrotransposons were expressed more in roots (615) than in leaves (355), and exhibited more upregulation (log2 fold change > 1) than downregulation (log2 fold change > −1) upon high salinity treatment (Supplementary Figure 2). In contrast, solo-LTRs were rarely expressed (Supplementary Figure 2). A total of 24 truncated retrotransposons and one solo-LTR were significantly mis-expressed in roots or leaves between treatments. Two TE-gene pairs were found to have significant expression correlation across conditions of different salt treatments. One pair was identified in leaves, involving a truncated RLG_6 copy (RLG_6_T1) and its 1,306-bp downstream gene thylakoid soluble phosphoprotein of 9 kDa (TSP9) (Pearson's correlation, r = 0.99, adjusted P < 0.01). TSP9, is a plant-specific protein in the photosynthetic thylakoid membrane that regulates light harvest in Arabidopsis (Carlberg et al., 2003; Fristedt et al., 2009). The other pair was identified in roots, involving another truncated RLG_6 copy (RLG_6_T2) and its 705-bp upstream gene glutamate-tRNA ligase, also known as glutamyl-tRNA synthetase (Pearson's correlation, r = −0.99, adjusted P < 0.05).
Taken together, we identified four TE-gene pairs with significant expression correlation in S. alba that involve both intact and truncated LTR retrotransposons. Among the S. caseolaris orthologs of these four genes, only TSP9 had a truncate RLG_6 copy inserted in its downstream region. However, none of the RLG_6 copies was expressed in leaves of S. caceolaris under normal condition (Wang et al., 2021). Orthologs of the other three genes in S. caseolaris had no insertion of RLC_4, RLG_8 or RLG_6 (BLASTn, all e-value > 0.05) in their flanking 10-kb up- or downstream region.
Potential cis- and Trans-Acting Roles of TEs on Gene Expression in S. alba
It is very well-known that LTRs contain CREs that can confer stress-inducible expression pattern on TEs and their flanking genes (Cavrak et al., 2014). To infer the potential cis-acting role of LTR retrotransposons, we predicted CREs in LTRs for each family using PlantCARE (Lescot et al., 2002). Consensus sequences of Gypsy and Copia families that have more than ten copies were used in this analysis. We identified abundant CREs responsive to various biotic and abiotic stresses in LTRs, whereas none of these CREs is responsive to salt stress (Figure 6). RLG_1 and RLG_8 contained CREs responsive to anoxic, drought, low temperature, pathogen and fungal elicitor, which are not more abundant than CREs residing in LTRs of other families (Figure 6). Interestingly, the LTRs of RLG_8 harboring three ABA-responsive elements (ABREs) involved in ABA and drought responsiveness (Figure 6) is coincident with a RLG_8 copy positively co-expressed with Abscisic acid 8'-hydroxylase 3 the gene nearby (see above). ABREs and MYB binding sites (MBSs) involved in drought inducibility were also found in the other three retrotransposons that showed significant expression correlation with their flanking genes (Figure 6). These results suggest that cis-regulatory role of LTR retrotransposons in S. alba, although rare, might occur via cross talk between drought and salt stress signaling pathways.
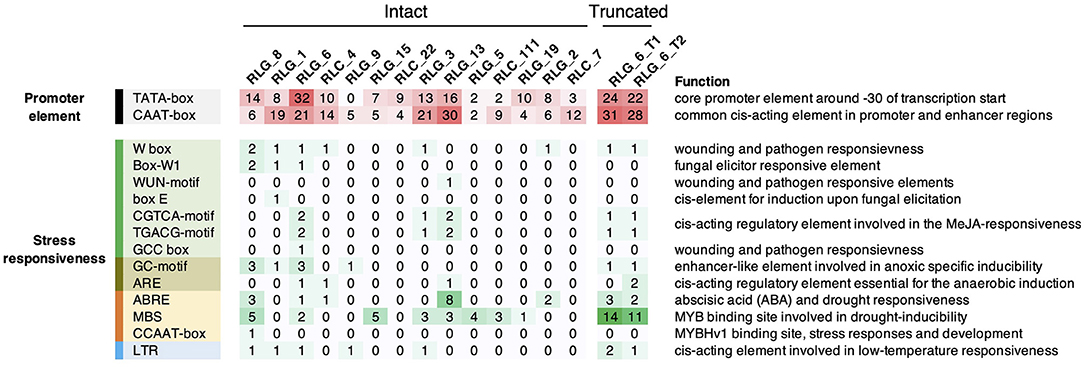
Figure 6. Summary of cis-acting regulatory elements (CREs) in the LTRs of Gypsy and Copia families. Only families with more than ten copies in S. alba were shown. Also shown are CREs in the full length of the two truncated elements (RLG_6_T1 and RLG_6_T2) which significantly correlated with their nearby genes in expression levels. Color depths and numbers indicate the abundance of specific CREs.
Besides cis-acting regulation, TEs can also function in trans, either resembling microRNA (miRNA) genes (Li et al., 2011) or acting as miRNA sponges competing with target transcripts of the same miRNA (Cho and Paszkowski, 2017; Cho, 2018; Palazzo and Koonin, 2020). To test these possibilities in S. alba, we annotated known and novel miRNAs using small RNA sequencing data described above with miRDeep2 (Friedlander et al., 2012) and predicted miRNA targets using psRNATarget (Dai et al., 2018). An expectation score of 2.5 was used as the cutoff in the target prediction. All LTR retrotransposons including the intact and truncated elements and solo-LTRs were used in these analyses. We identified 105 known and 14 novel miRNAs expressed (Reads per million mapped reads, RPM > 1) in both biological replicates; none was derived from TEs (Supplementary Table 5). We then used the criteria described by Cho and Paszkowski (2017) to identify TE-gene pairs with potential miRNA competition. Among the 65 LTR retrotransposons predicted to contain potential miRNA binding sites, only 21 were expressed in roots or leaves of S. alba under either condition of salt treatments (Supplementary Table 6). Requiring co-expression of both TEs and genes, we identified six TE-gene pairs with sequence matching in sense orientation and miRNA-binding sites within the matching regions. Nevertheless, none of them showed significantly expression correlation across different salt treatment conditions (Pearson correlation, all P > 0.05, Supplementary Table 7). Therefore, trans-acting role of LTR retrotransposons is negligible in S. alba.
Discussion
In her seminal studies of maize, McClintock proposed that TEs are “controlling elements” that may reprogram genome responses to environmental challenges (McClintock, 1984). Although still controversial, this idea has been supported by increasing empirical evidence (Naito et al., 2009; Cavrak et al., 2014; Horvath et al., 2017). TEs can be activated by stress, generate genetic variation, and modulate gene expression (Feschotte, 2008; Henaff et al., 2014). While induced TE activity has long been thought to benefit the host, it is puzzling that mangroves have experienced convergent reduction of TE loads despite being adapted to extreme environments with limited standing genetic variation (Guo et al., 2018; Lyu et al., 2018). How often TE activities are regulated by environmental cues and how these activities may contribute to a significant adaptive genome response in natural environments remain intriguing questions.
In asking whether divergent habitats promote differential TE accumulation, we found TEs that were accumulating more in S. alba than in S. caseolaris despite the overall tendency of unloading the TE burden in mangroves (Figures 1A,B). This result, together with the fact that S. alba faces higher salinity than S. caseolaris, is consistent with the view that TEs tend to be active in plants growing in challenging habitats. Besides high salinity, other stresses such as anoxia, nutrient deprivation and fungal pathogens are also associated with the frequently submerged environments of the S. alba habitat. The large fraction of young copies in S. alba (Figures 1C,D) suggests that TE originations are more frequent in S. alba than S. caseolaris. As young elements do not have enough time to degenerate, genome purging mechanisms should have little effect, if any, on their copy number. Although we do not have direct evidence that increased mobility in S. alba is triggered by environmental stress, transcriptome analyses suggest that more TEs are constitutively expressed in roots than leaves (Figure 5A), supporting this idea. Interestingly, purifying selection against TE insertions was even stronger in S. alba than in S. caseolaris (Table 1). The high origination rate and strong purifying selection might have led to fast turnover of TEs in S. alba, echoing a previous report of constant TE-host conflict masked by unloading of the TE burden in R. apiculata (Wang et al., 2018). Difference in unequal homologous recombination, as inferred by the S:I ratio (Table 2) may further contribute to the differential TE accumulation between the two Sonneratia species.
The recent burst of TE abundance is particularly prominent for the Gypsy retrotransposons in S. alba. We found that the two most abundant Gypsy families in S. alba (RLG_1 and RLG_8) mainly accumulate in gene-poor regions. This is likely due to a balance between TE integration site preference and post-integration selection processes (Sultana et al., 2017). The presence of the CHD domain in RLG_1 and RLG_8 likely drive heterochromatic integration site preference. However, the same integration site preference should be maintained in S. caseolaris if the CHD domain is sufficient to locate TE insertions to the same genomic region. Therefore, integration site preference alone cannot explain the difference in TE originations between species. Alternatively, if the two families differed in integration site preference between species, e.g., because of divergence of host proteins involved in TE tethering, the amplified copies should be much older than our observation (< 1 Myr), given that S. alba and S. caseolaris have diverged for ~11.6 Myr. We propose that the differential accumulation of RLG_1 and RLG_8 between species was probably triggered by environmental stress. It is possible that severe environments reactivated TEs more in S. alba than in S. caseolaris and then heterochromatin provided a “safe haven” for the accidentally inserted copies, allowing them to proliferate rapidly with relaxed epigenetic repression. The synergy between these mechanisms may constitute a self-reinforcing and self-perpetuating cycle of TE origination leading to long-term maintenance of genetic diversity. This hypothesis needs to be rigorously tested using data on stress-activated retrotransposition. We do see more up- than down-regulation of RLG_1 and RLG_8 during salt stress (Supplementary Figure 3), suggesting stress can activate TE transcription. The presence of CREs responsive to anoxia and drought but not to salt in the LTRs of RLG_1 and RLG_8 suggests that the cross-talk between different stress signaling pathways might play a significant role in the potential TE activation in S. alba.
Although stress-induced TE activity in S. alba is plausible, our transcriptome analyses suggest that TE expansion into regulatory sequences is rare. This is evident in the observation that TEs are mainly expressed in roots whereas the vast majority of differentially expressed TE-adjacent genes were found in leaves (Figure 5). Comparative transcriptome analyses between S. alba and S. caceolaris also suggest that TE expansion in S. alba influences between-species expression divergence mainly via carrying repressive epigenetic markers rather than creating new regulatory elements (Supplementary Material). There are only four potential cases of TE co-option in S. alba (Supplementary Table 4). In one case, unlike constitutive expression of CYP707A3 in Arabidopsis, Abscisic acid 8'-hydroxylase 3 in S. alba and the RLG_8 copy upstream of it exhibited salt-induced expression in leaves only under 500 mM NaCl, leading to a significant positive correlation in their expression levels (Supplementary Table 4). This result together with the presence of ABREs in the LTRs of RLG_8 suggests that the RLG_8 copy might have been integrated into a stress-responsive gene regulatory network involving ABA catabolism. In Arabidopsis, cyp707a3 mutant plants exhibit enhanced drought tolerance (Umezawa et al., 2006). Our result might also suggest that the strong salt tolerance of S. alba is achieved via reduced ABA degradation. We were unable to make any biological inferences in the other case of the RLC_4 copy, since the gene next to it has no known function. The rest two cases both involved truncated RLG_6 copies that possess tens of MBSs involved in drought-inducibility (Figure 6). The positive expression correlation of one RLG_6 copy with TSP9 in leaves and the negative expression correlation of another RLG_6 copy with glutamate-tRNA ligase in roots suggest that TEs might mediate the cross-talk between photosynthesis and nitrogen metabolism during the adaptation of S. alba to mangrove swamps.
It should be noted that a retrotransposon is potentially responsive to diverse stress conditions. As we only examined one stress condition (salinity), it is possible that co-option of TEs under other stress conditions was missed in this study. Nevertheless, it has been shown that enhancers often evolve from ancient TE sequences (Lynch et al., 2015; Villar et al., 2015). The evolutionary age of a TE was found to correlate positively with its likelihood of contributing to a regulatory element in human and mouse (Simonti et al., 2017). As the vast majority of retrotransposons in S. alba are relatively young, they may not have had enough time to get integrated into gene regulatory networks unless strong selection favors maintenance of the TE insertions, such as the Hopscotch insertion increasing tb1 expression in maize (Studer et al., 2011). Considering the small effective population size of S. alba (Zhou et al., 2007, 2011), natural selection may not be efficient enough to drive rapid fixation of TE insertions even though some TEs are capable of modulating expression of nearby genes.
Conclusion
In summary, our results together with a previous study (Wang et al., 2018) suggest that expansion of Gypsy retrotransposons, probably triggered by stress, is prevalent in mangroves despite the overall tendency of unloading the TE burden. The extent of TE proliferation across families is influenced by integration site preference and is subject to epigenetic repression and genome purging mechanisms. Although genetic variation generated by TE activities may be beneficial to host adaption, TE exaptation in S. alba is rare at least in terms of salt stress. Further experiments are needed to test the hypothesis of rapid transcriptional regulatory rewiring mediated by TE mobilization even though this hypothesis is appealing for extremophiles in which stress-induced reactivation of TEs is thought to be frequent.
Data Availability Statement
The datasets presented in this study can be found in GenBank repository, BioProject PRJNA636918.
Author Contributions
TT and YW planned, designed the research, and wrote the manuscript. YW and AD analyzed the data. All authors read and approved the submission of the final manuscript.
Funding
This study was funded by the National Science Foundation of China (31770246 and 31970245), the National Key Research and Development Program of China (2017YFC0506101), the Science and Technology Program of Guangzhou (201707020035), the Fundamental Research Funds for the Central Universities, Sun Yat-sen University (2021qntd26), Guangdong Basic and Applied Basic Research Foundation (2021A1515010808), the program of Guangdong Key Laboratory of Plant Resources (2019PlantKF10), the China Postdoctoral Science Foundation (2019TQ0391), and the Chang Hungta Science Foundation of Sun Yat-sen University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.830079/full#supplementary-material
References
Anders, S., Pyl, P. T., and Huber, W. (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. doi: 10.1093/bioinformatics/btu638
Ball, M. C., and Pidsley, S. M. (1995). Growth responses to salinity in relation to distribution of two mangrove species, Sonneratia alba and S. lanceolata, in Northern Australia. Funct. Ecol. 9:77. doi: 10.2307/2390093
Bao, W., Kojima, K. K., and Kohany, O. (2015). Repbase update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6:11. doi: 10.1186/s13100-015-0041-9
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Boratyn, G. M., Schaffer, A. A., Agarwala, R., Altschul, S. F., Lipman, D. J., and Madden, T. L. (2012). Domain enhanced lookup time accelerated BLAST. Biol. Direct 7:12. doi: 10.1186/1745-6150-7-12
Bourque, G., Burns, K. H., Gehring, M., Gorbunova, V., Seluanov, A., Hammell, M., et al. (2018). Ten things you should know about transposable elements. Genome Biol. 19:199. doi: 10.1186/s13059-018-1577-z
Bunt, J. S., Williams, W. T., and Clay, H. J. (1982). River water salinity and the distribution of mangroves species along several rivers in north Queensland. Aust. J. Bot. 30, 401–412. doi: 10.1071/BT9820401
Butelli, E., Licciardello, C., Zhang, Y., Liu, J., Mackay, S., Bailey, P., et al. (2012). Retrotransposons control fruit-specific, cold-dependent accumulation of anthocyanins in blood oranges. Plant Cell 24, 1242–1255. doi: 10.1105/tpc.111.095232
Capy, P., Gasperi, G., Biemont, C., and Bazin, C. (2000). Stress and transposable elements: co-evolution or useful parasites? Heredity 85 (Pt. 2), 101–106. doi: 10.1046/j.1365-2540.2000.00751.x
Carlberg, I., Hansson, M., Kieselbach, T., Schroder, W. P., Andersson, B., and Vener, A. V. (2003). A novel plant protein undergoing light-induced phosphorylation and release from the photosynthetic thylakoid membranes. Proc. Natl. Acad. Sci. U.S.A. 100, 757–762. doi: 10.1073/pnas.0235452100
Casacuberta, E., and Gonzalez, J. (2013). The impact of transposable elements in environmental adaptation. Mol. Ecol. 22, 1503–1517. doi: 10.1111/mec.12170
Cavrak, V. V., Lettner, N., Jamge, S., Kosarewicz, A., Bayer, L. M., and Mittelsten Scheid, O. (2014). How a retrotransposon exploits the plant's heat stress response for its activation. PLoS Genet. 10:e1004115. doi: 10.1371/journal.pgen.1004115
Cho, J. (2018). Transposon-Derived Non-coding RNAs and Their Function in Plants. Front. Plant Sci. 9:600. doi: 10.3389/fpls.2018.00600
Cho, J., and Paszkowski, J. (2017). Regulation of rice root development by a retrotransposon acting as a microRNA sponge. Elife 6:e54. doi: 10.7554/eLife.30038.054
Chuong, E. B., Elde, N. C., and Feschotte, C. (2017). Regulatory activities of transposable elements: from conflicts to benefits. Nat. Rev. Genet. 18, 71–86. doi: 10.1038/nrg.2016.139
Coordinators, N. R. (2018). Database resources of the national center for biotechnology information. Nucleic Acids Res. 46, D8–D13. doi: 10.1093/nar/gkv1290
Dai, X., Zhuang, Z., and Zhao, P. X. (2018). psRNATarget: a plant small RNA target analysis server (2017 release). Nucleic Acids Res. 46, W49–W54. doi: 10.1093/nar/gky316
de Souza, F. S., Franchini, L. F., and Rubinstein, M. (2013). Exaptation of transposable elements into novel cis-regulatory elements: is the evidence always strong? Mol. Biol. Evol. 30, 1239–1251. doi: 10.1093/molbev/mst045
Devos, K. M., Brown, J. K., and Bennetzen, J. L. (2002). Genome size reduction through illegitimate recombination counteracts genome expansion in Arabidopsis. Genome Res. 12, 1075–1079. doi: 10.1101/gr.132102
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Fedoroff, N. V. (2012). Presidential address. Transposable elements, epigenetics, and genome evolution. Science 338, 758–767. doi: 10.1126/science.338.6108.758
Feng, X., Xu, S., Li, J., Yang, Y., Chen, Q., Lyu, H., et al. (2020). Molecular adaptation to salinity fluctuation in tropical intertidal environments of a mangrove tree Sonneratia alba. BMC Plant Biol. 20:178. doi: 10.1186/s12870-020-02395-3
Feschotte, C. (2008). Transposable elements and the evolution of regulatory networks. Nat. Rev. Genet. 9, 397–405. doi: 10.1038/nrg2337
Finnegan, D. J. (1992). Transposable Elements. The Genome of Drosophila Melanogaster. New York, NY: Academic Press, 1096–1107. doi: 10.1016/B978-0-12-450990-0.50010-1
Friedlander, M. R., Mackowiak, S. D., Li, N., Chen, W., and Rajewsky, N. (2012). miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 40, 37–52. doi: 10.1093/nar/gkr688
Fristedt, R., Carlberg, I., Zygadlo, A., Piippo, M., Nurmi, M., Aro, E. M., et al. (2009). Intrinsically unstructured phosphoprotein TSP9 regulates light harvesting in Arabidopsis thaliana. Biochemistry 48, 499–509. doi: 10.1021/bi8016334
Fultz, D., Choudury, S. G., and Slotkin, R. K. (2015). Silencing of active transposable elements in plants. Curr. Opin. Plant Biol. 27, 67–76. doi: 10.1016/j.pbi.2015.05.027
Gao, X., Hou, Y., Ebina, H., Levin, H. L., and Voytas, D. F. (2008). Chromodomains direct integration of retrotransposons to heterochromatin. Genome Res. 18, 359–369. doi: 10.1101/gr.7146408
Grandbastien, M. A. (2015). LTR retrotransposons, handy hitchhikers of plant regulation and stress response. Biochim. Biophys. Acta 1849, 403–416. doi: 10.1016/j.bbagrm.2014.07.017
Guo, Z., Li, X., He, Z., Yang, Y., Wang, W., Zhong, C., et al. (2018). Extremely low genetic diversity across mangrove taxa reflects past sea level changes and hints at poor future responses. Glob. Chang. Biol. 24, 1741–1748. doi: 10.1111/gcb.13968
Han, Y., Burnette, J. M. 3rd, and Wessler, S. R. (2009). TARGeT: a web-based pipeline for retrieving and characterizing gene and transposable element families from genomic sequences. Nucleic Acids Res. 37:e78. doi: 10.1093/nar/gkp295
Han, Y., and Wessler, S. R. (2010). MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 38:e199. doi: 10.1093/nar/gkq862
Henaff, E., Vives, C., Desvoyes, B., Chaurasia, A., Payet, J., Gutierrez, C., et al. (2014). Extensive amplification of the E2F transcription factor binding sites by transposons during evolution of Brassica species. Plant J. 77, 852–862. doi: 10.1111/tpj.12434
Horvath, V., Merenciano, M., and Gonzalez, J. (2017). Revisiting the relationship between transposable elements and the eukaryotic stress response. Trends Genet. 33, 832–841. doi: 10.1016/j.tig.2017.08.007
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. doi: 10.1038/nprot.2015.053
Kimura, M. (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120. doi: 10.1007/BF01731581
Kozomara, A., Birgaoanu, M., and Griffiths-Jones, S. (2019). miRBase: from microRNA sequences to function. Nucleic Acids Res. 47, D155–D162. doi: 10.1093/nar/gky1141
Krochko, J. E., Abrams, G. D., Loewen, M. K., Abrams, S. R., and Cutler, A. J. (1998). (+)-Abscisic acid 8'-hydroxylase is a cytochrome P450 monooxygenase. Plant Physiol. 118, 849–860. doi: 10.1104/pp.118.3.849
Krueger, F., and Andrews, S. R. (2011). Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572. doi: 10.1093/bioinformatics/btr167
Kumar, A., and Bennetzen, J. L. (1999). Plant retrotransposons. Annu. Rev. Genet. 33, 479–532. doi: 10.1146/annurev.genet.33.1.479
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. doi: 10.1186/gb-2009-10-3-r25
Law, J. A., and Jacobsen, S. E. (2010). Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 11, 204–220. doi: 10.1038/nrg2719
Lescot, M., Dehais, P., Thijs, G., Marchal, K., Moreau, Y., Van de Peer, Y., et al. (2002). PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327. doi: 10.1093/nar/30.1.325
Li, Y., Li, C., Xia, J., and Jin, Y. (2011). Domestication of transposable elements into MicroRNA genes in plants. PLoS ONE 6:e19212. doi: 10.1371/journal.pone.0019212
Lisch, D. (2013). How important are transposons for plant evolution? Nat. Rev. Genet. 14, 49–61. doi: 10.1038/nrg3374
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Lynch, V. J., Nnamani, M. C., Kapusta, A., Brayer, K., Plaza, S. L., Mazur, E. C., et al. (2015). Ancient transposable elements transformed the uterine regulatory landscape and transcriptome during the evolution of mammalian pregnancy. Cell Rep. 10, 551–561. doi: 10.1016/j.celrep.2014.12.052
Lyu, H., He, Z., Wu, C. I., and Shi, S. (2018). Convergent adaptive evolution in marginal environments: unloading transposable elements as a common strategy among mangrove genomes. New Phytol. 217, 428–438. doi: 10.1111/nph.14784
Ma, J., and Bennetzen, J. L. (2004). Rapid recent growth and divergence of rice nuclear genomes. Proc. Natl. Acad. Sci. U.S.A. 101, 12404–12410. doi: 10.1073/pnas.0403715101
McClintock, B. (1984). The significance of responses of the genome to challenge. Science 226, 792–801. doi: 10.1126/science.15739260
Miele, V., Penel, S., and Duret, L. (2011). Ultra-fast sequence clustering from similarity networks with SiLiX. BMC Bioinformatics 12:116. doi: 10.1186/1471-2105-12-116
Myburg, A. A., Grattapaglia, D., Tuskan, G. A., Hellsten, U., Hayes, R. D., Grimwood, J., et al. (2014). The genome of Eucalyptus grandis. Nature 510, 356–362. doi: 10.1038/nature13308
Naito, K., Zhang, F., Tsukiyama, T., Saito, H., Hancock, C. N., Richardson, A. O., et al. (2009). Unexpected consequences of a sudden and massive transposon amplification on rice gene expression. Nature 461, 1130–1134. doi: 10.1038/nature08479
Niu, X. M., Xu, Y. C., Li, Z. W., Bian, Y. T., Hou, X. H., Chen, J. F., et al. (2019). Transposable elements drive rapid phenotypic variation in Capsella rubella. Proc. Natl. Acad. Sci. U.S.A. 116, 6908–6913. doi: 10.1073/pnas.1811498116
Palazzo, A. F., and Koonin, E. V. (2020). Functional long non-coding RNAs evolve from junk transcripts. Cell 183, 1151–1161. doi: 10.1016/j.cell.2020.09.047
Paro, R., and Hogness, D. S. (1991). The Polycomb protein shares a homologous domain with a heterochromatin-associated protein of Drosophila. Proc. Natl. Acad. Sci. U.S.A. 88, 263–267. doi: 10.1073/pnas.88.1.263
Price, M. N., Dehal, P. S., and Arkin, A. P. (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. doi: 10.1093/molbev/msp077
Rahim, A. C., and Bakar, M. F. A. (2018). Pidada—Sonneratia Caseolaris. USA: Academic Press. doi: 10.1016/B978-0-12-803138-4.00043-5
Rey, O., Danchin, E., Mirouze, M., Loot, C., and Blanchet, S. (2016). Adaptation to global change: a transposable element-epigenetics perspective. Trends Ecol. Evol. 31, 514–526. doi: 10.1016/j.tree.2016.03.013
SanMiguel, P., Gaut, B. S., Tikhonov, A., Nakajima, Y., and Bennetzen, J. L. (1998). The paleontology of intergene retrotransposons of maize. Nat. Genet. 20, 43–45. doi: 10.1038/1695
Schrader, L., Kim, J. W., Ence, D., Zimin, A., Klein, A., Wyschetzki, K., et al. (2014). Transposable element islands facilitate adaptation to novel environments in an invasive species. Nat. Commun. 5:5495. doi: 10.1038/ncomms6495
Simonti, C. N., Pavlicev, M., and Capra, J. A. (2017). Transposable element exaptation into regulatory regions is rare, influenced by evolutionary age, and subject to pleiotropic constraints. Mol. Biol. Evol. 34, 2856–2869. doi: 10.1093/molbev/msx219
Smit, A., Hubley, R., and Green, P. (2013-2015). RepeatMasker Open-4.0. Available online at: http://www.repeatmasker.org.
Studer, A., Zhao, Q., Ross-Ibarra, J., and Doebley, J. (2011). Identification of a functional transposon insertion in the maize domestication gene tb1. Nat. Genet. 43, 1160–1163. doi: 10.1038/ng.942
Sultana, T., Zamborlini, A., Cristofari, G., and Lesage, P. (2017). Integration site selection by retroviruses and transposable elements in eukaryotes. Nat. Rev. Genet. 18, 292–308. doi: 10.1038/nrg.2017.7
Umezawa, T., Okamoto, M., Kushiro, T., Nambara, E., Oono, Y., Seki, M., et al. (2006). CYP707A3, a major ABA 8'-hydroxylase involved in dehydration and rehydration response in Arabidopsis thaliana. Plant J. 46, 171–182. doi: 10.1111/j.1365-313X.2006.02683.x
Villar, D., Berthelot, C., Aldridge, S., Rayner, T. F., Lukk, M., Pignatelli, M., et al. (2015). Enhancer evolution across 20 mammalian species. Cell 160, 554–566. doi: 10.1016/j.cell.2015.01.006
Wang, Y., Dai, A., Chen, Y., and Tang, T. (2021). Gene body methylation confers transcription robustness in mangroves during long-term stress adaptation. Front. Plant Sci. 12:2104. doi: 10.3389/fpls.2021.733846
Wang, Y., Liang, W., and Tang, T. (2018). Constant conflict between Gypsy LTR retrotransposons and CHH methylation within a stress-adapted mangrove genome. New Phytol. 220, 922–935. doi: 10.1111/nph.15209
Wen, M., Lin, X., Xie, M., Wang, Y., Shen, X., Liufu, Z., et al. (2016). Small RNA transcriptomes of mangroves evolve adaptively in extreme environments. Sci. Rep. 6:27551. doi: 10.1038/srep27551
Wicker, T., Sabot, F., Hua-Van, A., Bennetzen, J. L., Capy, P., Chalhoub, B., et al. (2007). A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 8, 973–982. doi: 10.1038/nrg2165
Wright, S. I., Agrawal, N., and Bureau, T. E. (2003). Effects of recombination rate and gene density on transposable element distributions in Arabidopsis thaliana. Genome Res. 13, 1897–1903. doi: 10.1101/gr.1281503
Xiong, W., He, L., Lai, J., Dooner, H. K., and Du, C. (2014). HelitronScanner uncovers a large overlooked cache of Helitron transposons in many plant genomes. Proc. Natl. Acad. Sci. U.S.A. 111, 10263–10268. doi: 10.1073/pnas.1410068111
Xu, S., He, Z., Guo, Z., Zhang, Z., Wyckoff, G. J., Greenberg, A., et al. (2017). Genome-Wide convergence during evolution of mangroves from woody plants. Mol. Biol. Evol. 34, 1008–1015. doi: 10.1093/molbev/msw277
Yang, G., Zhou, R., Tang, T., and Shi, S. (2008). Simple and efficient isolation of high-quality total RNA from Hibiscus tiliaceus, a mangrove associate and its relatives. Prep. Biochem. Biotechnol. 38, 257–264. doi: 10.1080/10826060802164991
Yang, Y., Duke, N. C., Peng, F., Li, J., Yang, S., Zhong, C., et al. (2016). Ancient geographical barriers drive differentiation among sonneratia caseolaris populations and recent divergence from S. lanceolata. Front. Plant Sci. 7:1618. doi: 10.3389/fpls.2016.01618
Zhou, R., Ling, S., Zhao, W., Osada, N., Chen, S., Zhang, M., et al. (2011). Population genetics in nonmodel organisms: II. natural selection in marginal habitats revealed by deep sequencing on dual platforms. Mol.Biol. Evol. 28, 2833–2842. doi: 10.1093/molbev/msr102
Keywords: transposable elements (TEs), genome evolution, TE co-option, stress adaptation, mangroves
Citation: Wang Y, Dai A and Tang T (2022) Weak Effect of Gypsy Retrotransposon Bursts on Sonneratia alba Salt Stress Gene Expression. Front. Plant Sci. 12:830079. doi: 10.3389/fpls.2021.830079
Received: 06 December 2021; Accepted: 27 December 2021;
Published: 17 January 2022.
Edited by:
Ruslan Kalendar, University of Helsinki, FinlandReviewed by:
Carlos M. Vicient, Centre for Research in Agricultural Genomics, Spanish National Research Council (CSIC), SpainMatej Lexa, Masaryk University, Czechia
Copyright © 2022 Wang, Dai and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tian Tang, bHNzdHRAbWFpbC5zeXN1LmVkdS5jbg==