 Richard G. J. Hodel
Richard G. J. Hodel Elizabeth A. Zimmer
Elizabeth A. Zimmer Bin-Bin Liu1,2
Bin-Bin Liu1,2 Jun Wen
Jun Wen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 25 January 2022
Sec. Plant Systematics and Evolution
Volume 12 - 2021 | https://doi.org/10.3389/fpls.2021.820997
This article is part of the Research TopicPhylogenomic Discordance in Plant SystematicsView all 15 articles
Plant biologists have debated the evolutionary origin of the apple tribe (Maleae; Rosaceae) for over a century. The “wide-hybridization hypothesis” posits that the pome-bearing members of Maleae (base chromosome number x = 17) resulted from a hybridization and/or allopolyploid event between progenitors of other tribes in the subfamily Amygdaloideae with x = 8 and x = 9, respectively. An alternative “spiraeoid hypothesis” proposed that the x = 17 of Maleae arose via the genome doubling of x = 9 ancestors to x = 18, and subsequent aneuploidy resulting in x = 17. We use publicly available genomic data—448 nuclear genes and complete plastomes—from 27 species representing all major tribes within the Amygdaloideae to investigate evolutionary relationships within the subfamily containing the apple tribe. Specifically, we use network analyses and multi-labeled trees to test the competing wide-hybridization and spiraeoid hypotheses. Hybridization occurred between an ancestor of the tribe Spiraeeae (x = 9) and an ancestor of the clade Sorbarieae (x = 9) + Exochordeae (x = 8) + Kerrieae (x = 9), giving rise to the clade Gillenieae (x = 9) + Maleae (x = 17). The ancestor of the Maleae + Gillenieae arose via hybridization between distantly related tribes in the Amygdaloideae (i.e., supporting the wide hybridization hypothesis). However, some evidence supports an aspect of the spiraeoid hypothesis—the ancestors involved in the hybridization event were likely both x = 9, so genome doubling was followed by aneuploidy to result in x = 17 observed in Maleae. By synthesizing existing genomic data with novel analyses, we resolve the nearly century-old mystery regarding the origin of the apple tribe. Our results also indicate that nuclear gene tree-species tree conflict and/or cytonuclear conflict are pervasive at several other nodes in subfamily Amygdaloideae of Rosaceae.
Throughout the Rosaceae, there is pervasive conflict between phylogenetic relationships inferred using the nuclear vs. chloroplast genomes. Among major lineages of the Rosaceae, variation in chromosome number is prevalent, and there have been frequent whole genome duplications in the family. Many lineages of the Rosaceae contain economically important species; the Maleae, with over 1,000 species, includes commercially important fruit crops, such as apples and pears, as well as many ornamentals. In addition to apples and pears, the subfamily Amygdaloideae contains many other important species such as cherries, almonds, peaches, apricots, and plums. The branching order among the three subfamilies of the Rosaceae—Amygdaloideae, Dryadoideae, and Rosoideae—is uncertain. Nuclear data indicate that the Dryadoideae are sister to the Amygdaloideae + Rosoideae (Xiang et al., 2017), whereas phylogenetic relationships reconstructed using plastome data have still not conclusively resolved the branching order. Recent analyses inferred that the Rosoideae are sister to Amygdaloideae + Dryadoideae when using whole plastome data, or that the Amygdaloideae are sister to the Dryadoideae + Rosoideae when using whole plastomes with most ambiguous sites removed (Zhang et al., 2017). In the Amygdaloideae, the relationships between many tribes conflict when the nuclear and chloroplast topologies are compared (Figure 1; Xiang et al., 2017; Zhang et al., 2017). Furthermore, within the Rosaceae, many relationships between tribes were inconsistent between the nuclear and chloroplast genomes, such as the placement of all tribes within the Rosoideae except for Ulmarieae (Xiang et al., 2017; Zhang et al., 2017). Cytonuclear conflict also exists within the Rosaceae at shallower systematic scales (e.g., within the tribe Maleae; Liu et al., 2019, 2020a,b, 2021).
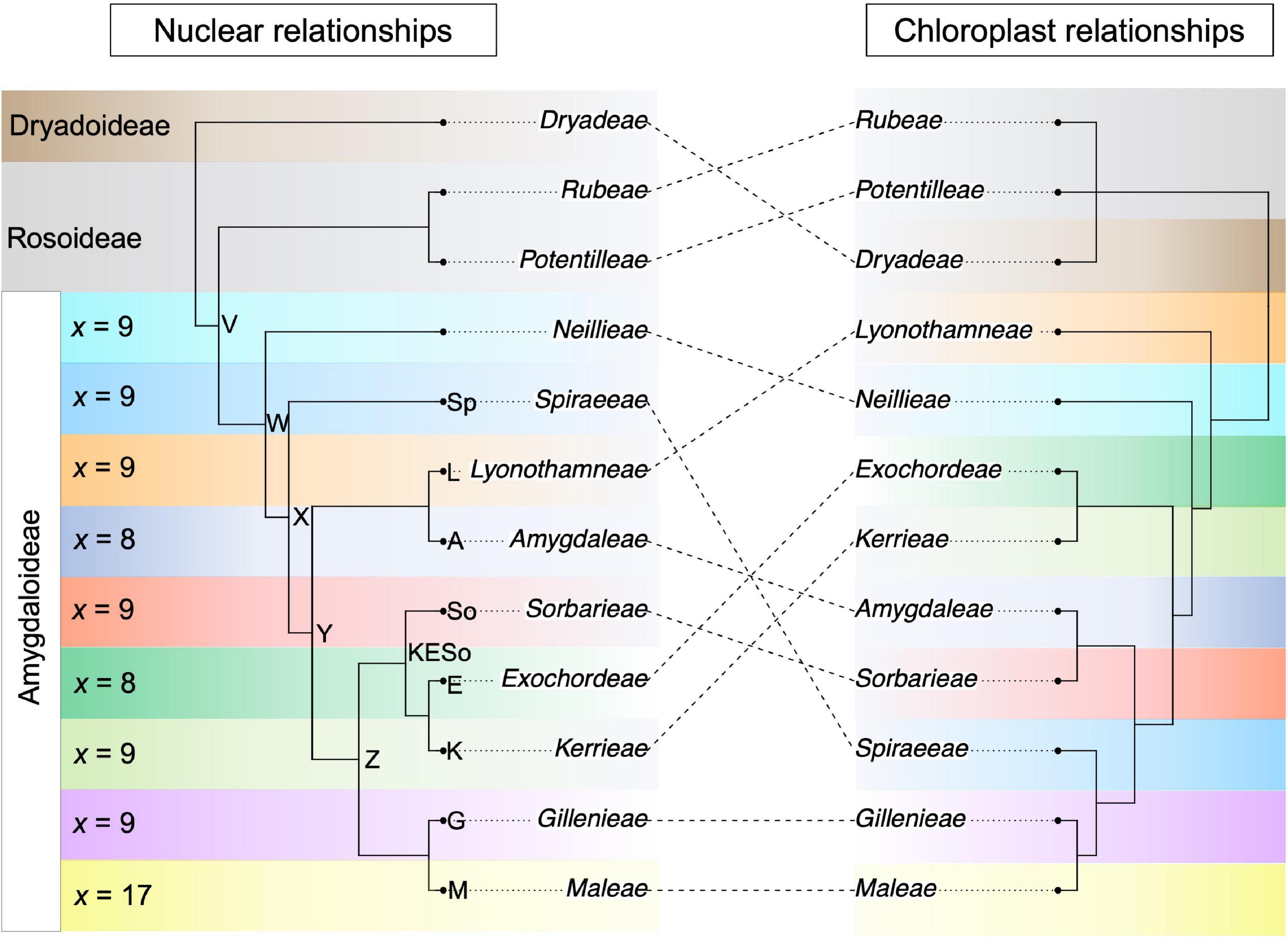
Figure 1. Nuclear- and chloroplast-inferred phylogenetic relationships among tribes within the Amygdaloideae, rooted using the other two subfamilies within the Rosaceae, the Dryadoideae and Rosoideae. Conflict between the nuclear and chloroplast topologies is shown using dotted lines. The tribal and subfamilial relationships are based on topologies from Xiang et al.(2017; nuclear) and Zhang et al.(2017; chloroplast). For each tribe in the Amygdaloideae, the base chromosome number is indicated to the left of the nuclear phylogeny. Note that one genus in the Maleae is not x = 17 but rather is x = 15 (Vauquelinia). Select nodes in the nuclear phylogeny are labeled with letters or abbreviations to facilitate reference to these nodes in the text.
For nearly a century, plant biologists have debated the evolutionary origin of the apple tribe Maleae (Rosaceae; formerly Maloideae). Species in the tribe Maleae are characterized by a base chromosome number of x = 17 (except for x = 15 in Vauquelinia Corrêa ex Bonpl.)—distinct from other tribes in the Rosaceae, which typically are x = 7, 8, or 9 (Evans and Campbell, 2002). Within the Amygdaloideae, the subfamily containing the Maleae, all tribes except the Maleae are x = 8 or 9 (Robertson et al., 1991). Because the base chromosome number of Maleae was approximately double that of all its close relatives, early researchers investigated hypotheses of a polyploid origin of the pome-bearing members of the apple subtribe Malinae, which includes all Maleae except for three early diverging dry fruit lineages including genera Kageneckia Ruiz and Pav. (x = 17), Lindleya Kunth (x = 17) and Vauquelinia (x = 15) (Nebel, 1929; Campbell et al., 1995). Darlington and Moffett (1930) proposed hypotheses of autopolyploidy, which were quickly refuted by Sax (1931, 1932, 1933) after observing predominantly univalents in triploids during meiosis, as opposed to multivalents. Sax proposed an explanation of allopolyploidy occurring between x = 8 and x = 9 progenitors from the subfamily Spiraeoideae (now as part of Amygdaloideae; Potter et al., 2007). The “wide-hybridization hypothesis” formulated in the 1930s posits that the Malinae (base chromosome number x = 17) resulted from an ancient hybridization event between progenitors from other tribes in the subfamily Amygdaloideae that have x = 8 and x = 9, respectively. The “wide-hybridization” hypothesis was favored by Stebbins (1950) and was further supported by studies using isozymes decades later (Chevreau et al., 1985; Weeden and Lamb, 1987).
An alternative “spiraeoid hypothesis” proposed that the 17 (or in rare cases 15) chromosomes found in Maleae arose via the genome doubling of an x = 9 spiraeoid ancestor to x = 18, and subsequent aneuploidy resulting in x = 17 (Goldblatt, 1976; Evans and Campbell, 2002). This hypothesis is referred to as the “spiraeoid” hypothesis because the participants in allopolyploidy were considered a member of spiraeoid taxa (Goldblatt, 1976), in particular, the ancestor of the tribe Gillenieae (Evans and Campbell, 2002), which was traditionally placed in the formerly recognized subfamily Spiraeoideae (also see Gladkova, 1972). A genetic investigation of the origin of the apple tribe using one nuclear gene (Evans and Campbell, 2002) favored the spiraeoid hypothesis while rejecting the wide-hybridization hypothesis. Their study inferred that an ancestor of the tribe Gillenieae (x = 9), which is sister to the Maleae, experienced genome doubling and subsequent aneuploidy. Other molecular analyses of the Rosaceae did not explicitly test hypotheses explaining the origin of the apple tribe (e.g., Potter et al., 2002, 2007). To date, the two competing hypotheses have not been tested using genomic data. Recent phylogenomic studies identified pervasive cytonuclear conflict throughout the Amygdaloideae, which contains the Maleae, suggesting that ancient hybridization and/or allopolyploidization may have impacted the diversification of this group.
The time is ripe to re-evaluate these hypotheses using analyses that consider phylogenomic data from both nuclear and chloroplast genomes, and methodologies that explicitly incorporate discordance and/or reticulation into phylogenies. As researchers obtain more DNA sequence data from both the nuclear and chloroplast genomes, it is becoming increasingly clear that cytonuclear conflict is prevalent in many plant lineages (Huang et al., 2014; Bruun-Lund et al., 2017; Lee-Yaw et al., 2018; Hodel et al., 2021; Liu et al., 2021; Wang et al., 2021; Xu et al., 2021). Here, we limit our focus to studying and resolving cytonuclear conflict within the Amygdaloideae. Our objectives in this paper are to: (1) Test the competing wide hybridization and spiraeoid hypotheses, and investigate the role of genome doubling in the origin of the apple tribe using genomic data from the nuclear and chloroplast genomes, and (2) Characterize cytonuclear conflict within the Amygdaloideae, a clade with pervasive reticulate evolution, and identify explanations for the observed conflict. Specifically, we integrate data from Xiang et al. (2017)—hundreds of nuclear genes—and plastomes from Zhang et al. (2017), supplemented by chloroplast sequence data from NCBI, to investigate pervasive cytonuclear conflict within the Amygdaloideae that may provide insights into the evolutionary origin of the apple tribe.
Subfamily Amygdaloideae contains approximately 1,500 species organized into nine tribes (Figure 1). Some tribes, such as Maleae and Amygdaleae are represented by hundreds of species, whereas others such as Gillenieae and Lyonothamneae each contain a single genus. We selected representatives from each tribe, as well as species from the other Rosaceae subfamilies Dryadoideae and Rosoideae, with the goal of obtaining a representative sampling of Amygdaloideae tribes while limiting the number of taxa included so that certain analyses (i.e., phylogenetic networks) would be computationally feasible. First, we downloaded the 148-taxa alignments of 882 nuclear genes from Xiang et al. (2017) from TreeBASE (study ID = 19726). Briefly, Xiang et al. (2017) isolated RNA from young leaf, floral bud, or fruit tissue, performed transcriptome sequencing, and identified putative low copy candidate orthologous genes to use in phylogenentic analyses. The publicly available alignments consisted of consensus sequences from the candidate orthologous genes. As the authors of Xiang et al. (2017) note, a large proportion of these 882 nuclear genes are suspected hidden paralogs, and they used several paralog filtering steps. Xiang et al. (2017) primarily used smaller filtered subsets of genes (571, 444, 256, and 113 genes) in phylogenomic analyses. In the present study, phylogenies were first constructed using all 148 taxa to identify putative paralogous gene trees. We inferred each of 882 gene trees from the sequence alignments using RAxML v8.2.11 (Stamatakis, 2014) with the GTRGAMMA model of evolution, 20 independent ML searches, and 100 bootstrap replicates. For consistency with Xiang et al. (2017), we screened all gene trees using TreSpEx (Struck, 2014) with the a priori paralogy screening function with a bootstrap threshold of 95—with two masking filters—first using established ordinal relationships, and then using subfamilial relationships. The ordinal and subfamilial filters were used in Xiang et al. (2017) to remove suspected hidden paralogs, so we used this strategy for consistency. Our TreSpEx paralog trimming left 448 putative orthologs out of 882. When we investigated including paralogs in our analyses (i.e., using all 882 genes), our species tree topology did not match the dominant topology presented in Xiang et al. (2017). Therefore, we proceeded using our 448 gene set, which did match the dominant topology from Xiang et al. (2017). We trimmed taxa from the 448-gene alignments using the “pxrmt” command in phyx (phylogenetic tools for unix; Brown et al., 2017) to reduce the data matrix down to 27 species. We included at least one species from each of the nine tribes in the Amygdaloideae and two species each from the Rosoideae and Dryadoideae, as well as one outgroup species, Ziziphus jujuba Mill. (Rhamnaceae). The trimming of taxa was done to facilitate downstream analyses (i.e., network analyses implemented in SNaQ) that become computationally intractable when larger numbers of taxa (i.e., > 30) are included. Whenever possible, we selected species represented by both nuclear data (from Xiang et al., 2017) and complete plastomes (from Zhang et al., 2017).
For the species not represented by plastome data in Zhang et al. (2017), we downloaded complete plastomes from NCBI for all species except Physocarpus opulifolius (L.) Maxim. (Table 1), which was represented in the nuclear data from Xiang et al. (2017) but not in the plastome data from Zhang et al. (2017). For P. opulifolius, we downloaded RNA-Seq reads from NCBI (accession number ERR2040427; Table 1) and used FastPlast1 to de novo assemble reads into contigs. The contigs were mapped to a reference plastome [Malus domestica (Suckow) Borkh., accession number: MK434916.1; Table 1] to complete the assembly. Because this species was the only taxon without a complete plastome sequence, we included two additional Physocarpus (Cambess.) Raf. plastomes from Zhang et al. (2017), labeled by the authors as Physocarpus sp. A and Physocarpus sp. B, in our preliminary chloroplast phylogenetic analyses to verify that the phylogenetic position of our newly assembled plastome was as expected. MAFFT (Katoh and Standley, 2013) was used to align the plastomes with settings “--maxiterate 5000 --localpair --adjustdirectionaccurately.” Resulting alignments were trimmed using TrimAl with the “-automated1” heuristic. The “pxclsq” command in phyx was separately used to filter the alignment based on either 20, 30, 40, 50, or 60% column occupancy required. We compared the phylogenetic trees resulting from all alignments, and after determining there was no change in topology, we used the TrimAl-trimmed tree in subsequent plastome phylogenetic analyses.
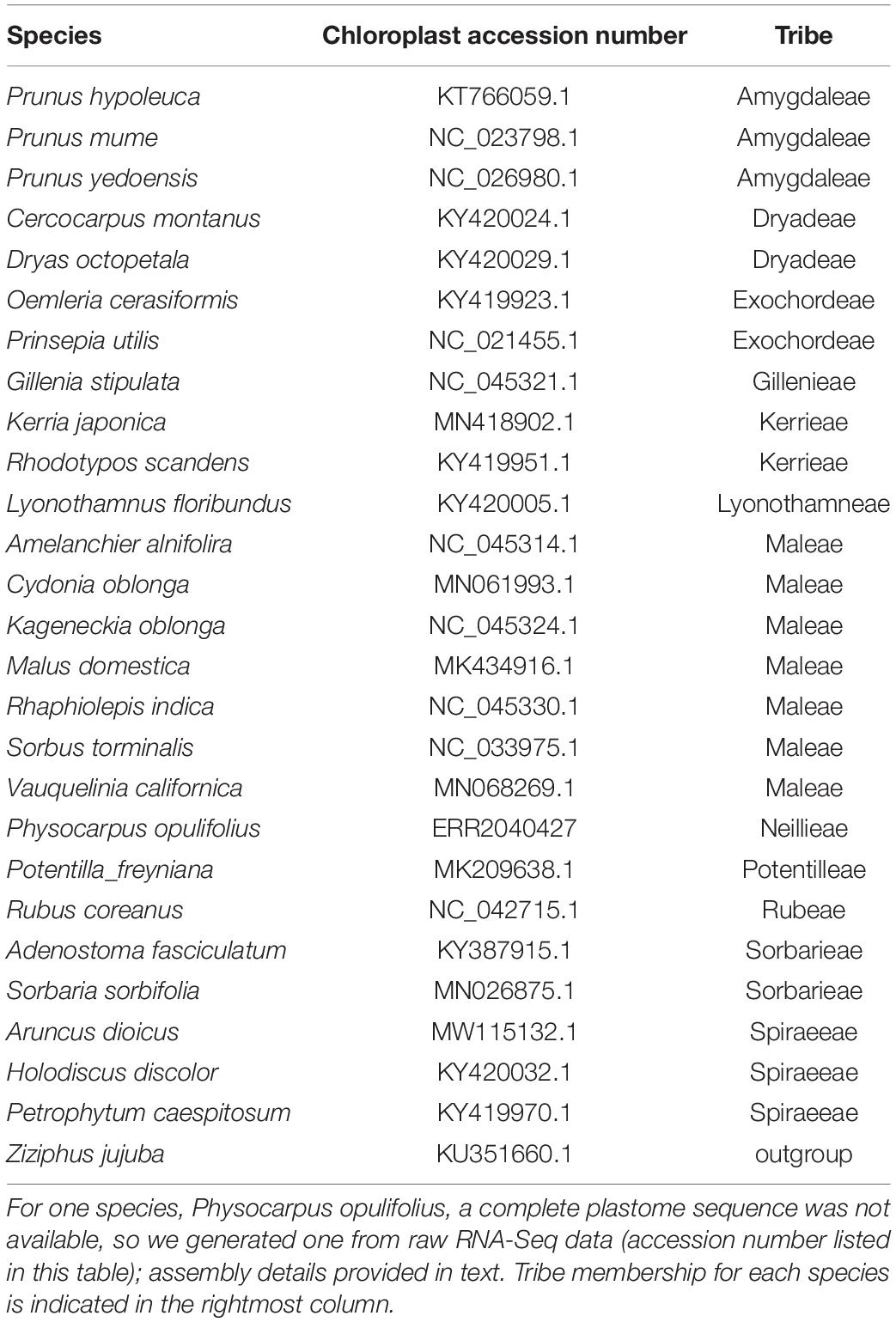
Table 1. For all 27 focal species used in our study, the NCBI accession number of the plastome sequence is listed.
We assessed the phylogenetic relationships among the 27 species using RAxML and ASTRAL to ensure that the nuclear topology reflected the relationships from Xiang et al. (2017). The ML analysis was conducted in RAxML using a concatenated supermatrix of the 448 orthologues, with the GTRGAMMA model of evolution, 20 independent ML searches, and 100 bootstrap replicates. Both unpartitioned and partitioned (-q) analyses were used. The coalescent analyses were conducted in ASTRAL (Mirarab et al., 2014), a tree estimation program consistent with the coalescent, and using quartet support values to measure confidence in species relationships. The quartet support scores indicate the percentage of quartets in gene trees that are concordant with a given branch and therefore can show the amount of gene tree conflict associated with a branch. Quartet scores provide more information about uncertainty at key nodes than bootstrap scores, which can be inappropriately inflated in some phylogenomic datasets (Roycroft et al., 2020). We also used RAxML to ensure that the chloroplast relationships from Zhang et al. (2017) were recapitulated, using the GTRGAMMA model of evolution, 20 independent ML searches, and 100 bootstrap replicates. Phylogenetic trees were visualized and manipulated using IcyTree (Vaughan, 2017) and Interactive Tree of Life (Letunic and Bork, 2021). The “cophylo” function in the R package phytools (Revell, 2012) was used to visualize concordance between the nuclear and plastome phylogenies. Unless otherwise noted, all software analyses were run on the Smithsonian Institution High Performance Cluster (SI/HPC, “Hydra”).2
To assess if a reticulate tree (i.e., a phylogenetic network) better represented the nuclear gene tree data than a purely bifurcating tree, we used the program SNaQ, which is implemented in PhyloNetworks (Solís-Lemus et al., 2017). The phylogenomic network method SNaQ, which uses a pseudolikelihood method, explicitly accommodates hybridization by representing certain nodes as having received genetic material from two parental lineages with inheritance probabilities γ and 1-γ. The RAxML-inferred gene trees for all 448 orthologues were used as input and summarized using quartet concordance factors (i.e., the proportion of gene trees with a given quartet; Larget et al., 2010). In SNaQ, networks are optimized based on the branch lengths and inheritance probabilities in phylogenetic network space as measured by a pseudodeviance score. The pseudodeviance score represents a multiple of the network’s log-likelihood score up to a constant where the network perfectly fits the data. Lower pseudo-deviance scores always indicate a better fit, but as hmax increases, the pseudodeviance score always improves (Solís-Lemus and Ané, 2016). Accordingly, the rate of change in the pseudodeviance score between hmax values can be used to assess the optimal hmax (Baudry et al., 2011). We constructed networks using hmax values ranging from 0 to 5. For the initial optimization (hmax = 0), the ASTRAL tree was used as a starting network with no hybridization edges, and for subsequent hmax values, the optimal network estimated by the preceding lower hmax value was used as the starting topology. We ran 10 independent searches for each hmax value and the optimal number of hybridization edges was assessed by plotting hmax against the log-likelihood score (i.e., network score) of the optimal network for each hmax value.
The program phyparts (Smith et al., 2015) was used to assess gene tree conflict in the nuclear dataset. This program compares rooted gene trees with the rooted species tree to identify topologically concordant, discordant, and uninformative gene trees for each species tree node. Because rooted gene trees were necessary, fewer gene trees (440 out of 448) were available for this analysis due to the absence of the outgroup in some gene trees. We used a gene tree bootstrap support cutoff of 50% (-s 50); below this threshold gene trees were considered to be uninformative for a given node. A phyparts analysis using no bootstrap support cutoff was also run for comparison. The results of each phyparts analysis were visualized as piecharts on the phylogeny using the phypartspiecharts.py jupyter notebook (by Matt Johnson).3 Nodes of interest, as identified by network analysis and the above conflict analysis, were further investigated using the “alternative relationship test” implemented in phyckle (Smith et al., 2020). The alternative relationship test takes as input two or more user specified bipartitions, which are used as a constraint when running RAxML to infer every gene tree from the sequence matrices. Log-likelihood scores are calculated for each gene tree and then compared to determine which topology (i.e., between the user-inputted bipartitions) is optimal for every gene tree. The number of gene trees and/or the summed difference of log-likelihood scores between the gene trees can then used to determine support for one bipartition vs. others.
The software package GRAMPA (Thomas et al., 2017) was used to identify the parental lineages involved in a hybridization event leading to an allopolyploid lineage. GRAMPA makes use of multiply-labeled (MUL) trees, which are topologies in which selected species can appear twice, a common way of representing polyploid relationships when constrained by a bifurcating phylogeny. The algorithm implemented in GRAMPA uses least common ancestor reconciliation of gene trees and species trees (Goodman et al., 1979; Page, 1994) to place polyploidy events on a phylogeny. Branches of the species tree with disproportionately high numbers of gene duplications can be used to identify polyploidy events. The use of MUL-trees enables accurate inferences of allopolyploidy vs. autopolyploidy, because all subgenomes involved in allopolyploidy can be represented as descendants of different parental linages. Under scenarios of allopolyploidy, we would expect the homoeologs that result from an allopolyploidy event to be sister to different diploid taxa (Thomas et al., 2017). Using hypotheses from the literature, and guided by the SNaQ results, we tested the following hypotheses of allopolypoidy. We considered either the Maleae (i.e., node M; Figure 1) or the Gillenieae + Maleae (node G) as possible clades that were a result of allopolyploidization (“-h1” inputs). We investigated the following nodes as potential secondary parental branches (“-h2” inputs): nodes labeled A, L, Sp, S, K, E, KESo, W, X, Y, Z (Figure 1). If the wide hybridization hypothesis is supported, we would expect a node further removed from the Gillenieae + Maleae clade to be selected as the secondary parental branch (e.g., Sp). Conversely, if the spiraeoid-origin hypothesis is supported, we would expect the “-h2” node to be adjacent to a branch representing an ancestor of Gillenieae (e.g., Z).
To reconcile any differences between the phylogenetic network and MUL-tree results, we used one additional approach to test for histories of hybridization in the Amygdaloideae. The program Hybrid Detector (HyDe) uses phylogenetic invariants under a coalescent model with hybridization to infer probability of hybridization of three ingroup taxa relative to an outgroup taxon (Blischak et al., 2018). In this framework, the parameter γ represents the probability that gene trees with a hybrid population sister to parent X would arise under the parental population trees, whereas 1-γ would be the probability of a hybrid population being sister to parent Y. Based on the SNaQ results and GRAMPA results, we tested several sets of taxa for histories of hybridization in HyDe. Using the SNaQ results as a guide, we tested the hybrid status of three ingroups (Maleae + Gillenieae, Spiraeeae, Sorbarieae) relative to an outgroup (Neillieae), and based on the GRAMPA results, we tested for hybridization using three ingroups (Maleae + Gillenieae, Spiraeeae, Kerrieae + Exochordeae + Sorbarieae) and the same outgroup (Neillieae). This outgroup was chosen because it was sister to all other Amygdaloideae tribes when using nuclear data (Figure 1).
Our phylogenetic analyses recovered all subfamilies and tribes as monophyletic (Figure 2). In the nuclear phylogeny, the Dryadoideae (represented by Cercocarpus montanus Raf. and Dryas octopetala L.) and Rosoideae (Potentilla freyniana Bornm. and Rubus coreanus Miq.) were successively sister to the Amygdaloideae (Figure 2). Within the Amygdaloideae, the Neillieae (Physocarpus opulifolius) and Spiraeeae [Aruncus dioicus (Walter) Fernald, Holodiscus discolor (Pursh) Maxim., and Petrophytum caespitosum (Nutt.) Rydb.] were successively sister to a clade containing the remaining seven tribes (Figure 2). The Amygdaleae [Prunus hypoleuca (Koehne) J.Wen, Prunus mume Siebold & Zucc., and Prunus × yedoensis Matsum.] and Lyonothamneae (Lyonothamnus floribundus Gray) then form a clade sister to the remaining five tribes. The Exochordeae (Prinsepia utilis Royle. and Oemleria cerasiformis (Torr. & Gray ex Hook. & Arn.) J.W.Landon), Kerrieae [Kerria japonica (L.) DC. and Rhodotypos scandens (Thunb.) Makino], and Sorbarieae [Adenostoma fasciculatum Hook. & Arn. and Sorbaria sorbifolia (L.) A.Braun] formed a clade that is sister to the clade comprised of Gillenieae [Gillenia stipulata (Muhl. ex Willd.) Nutt.] and Maleae [Cydonia oblonga Mill., Sorbus torminalis (L.) Crantz, Malus domestica, Rhaphiolepis indica (L.) Lindl. ex Ker Gawl., Amelanchier alnifolia (Nutt.) Nutt., Vauquelinia californica (Torr.) Sarg., and Kageneckia oblonga Ruiz & Pav.]. In the chloroplast phylogeny, the Dryadoideae and Rosoideae were sister to the Amygdaloideae (Figure 2). The Lyonothamneae and Neillieae were successively sister to the seven remaining tribes. Then, the Exochordeae and Kerrieae formed a clade sister to the remaining five tribes. The Amygdaleae and Sorbarieae made up a clade sister to the Spiraeeae, Gillenieae, and Maleae. Within this final clade, the Spiraeeae were sister to Maleae + Gillenieae (Figure 2).

Figure 2. Nuclear- and chloroplast-inferred phylogenetic relationships for all 27 focal species. Tribes/subfamilies are color-coded using the same scheme as Figure 1, and tribes in the Amygdaloideae are labeled. Disagreement between the chloroplast and nuclear phylogenies is indicated by dotted lines between the phylogenies. Note that in the bottom half of the phylogenies, even though the dotted lines do not intersect, there are key differences in the branching order of clades between the two phylogenies. In the nuclear phylogeny, node support was assessed using ASTRAL quartet support scores and nodes are labeled if the quartet support was greater than 50 (hollow triangles), greater than 70 (solid diamonds), or greater than 90 (asterisks). Any unlabeled nodes in the nuclear phylogeny had quartet support scores < 50. In the chloroplast phylogeny, support was measured via bootstrapping in RAxML and nodes with bootstrap scores less than 70% are labeled (inverted hollow triangles) as are nodes with less than 95% but greater than 70% (solid squares). All unlabeled nodes in the chloroplast phylogeny have 100% bootstrap support.
The phylogeny constructed using nuclear data recapitulated results from Xiang et al. (2017) with our reduced-taxa dataset (Figure 2 and Supplementary Figure 1). In the nuclear datasets, there were several topological differences between the nuclear coalescent and concatenation trees (Figure 2 and Supplementary Figure 1)—differences that also existed among different datasets in Xiang et al. (2017). When comparing our nuclear phylogenies, the key difference was the placement of the Amygdaleae + Lyonothamneae clade, which was sister to the Kerrieae + Exochordeae + Sorbarieae in the concatenation trees, but in the coalescent tree was sister to these three tribes as well as the Maleae + Gillenieae (Figure 2 and Supplementary Figure 1). Within the Maleae, there were also discrepancies between the coalescent tree and concatenation trees, and between the unpartitioned and partitioned concatenation trees (Figure 2 and Supplementary Figure 1). In the coalescent topology, Rhaphiolepis indica and Malus domestica were respectively successively sister to Cydonia oblonga and Sorbus torminalis (Figure 2). However, in the unpartitioned ML phylogeny, Malus was sister to Cydonia whereas Sorbus was sister to Rhaphiolepis Lindl (Supplementary Figure 1). Meanwhile, in the partitioned ML tree, Malus domestica was sister to Rhaphiolepis indica and Cydonia oblonga was sister to Sorbus torminalis (Supplementary Figure 1). Hereafter, we use our ASTRAL topology as the nuclear topology for clarity because it matches the predominant topology presented in Xiang et al. (2017).
Our reduced-taxa plastome phylogeny matched the ML whole plastome tree topology, as opposed to the ambiguous-sites-removed tree, from Zhang et al. (2017) (Figure 2). For simplicity, we use this plastome tree in subsequent comparisons with the nuclear phylogeny, because the primary topological difference between plastome trees from Zhang et al. (2017) involved the branching order of subfamilies, not the relationships among Amygdaloideae tribes, which is our focus. As expected, there were numerous differences between our plastome and nuclear phylogenies (Figure 2) throughout the tree, including major relationships between subfamilies. In the plastome phylogeny, the Dryadoideae were sister to the Rosoideae, whereas in all nuclear trees, the Dryadoideae were sister to the Rosoideae + Amygdaloideae. There were also many differences in intertribal relationships, including virtually every tribe except the Gillenieae + Maleae (Figure 2). The different alignment strategies that we used for the plastome sequence alignment did not influence the inferred topology of the chloroplast phylogeny, but there was variation in the bootstrap percentages at certain nodes between the different alignments (Supplementary Figure 2).
The SNaQ network analysis inferred that one hybridization event was optimal (Figure 3 and Supplementary Figure 3). The hybridization edge indicated that the clade Gillenieae + Maleae was 57.2% sister to the Sorbarieae [represented by Adenostoma Hook. & Arn. and Sorbaria (Ser.) A.Braun], and 42.8% sister to the Spiraeeae [represented by Aruncus L., Holodiscus (K.Koch) Maxim., and Petrophytum (Nutt. ex Torr. & A.Gray) Rydb.; Figure 3]. The position of the Sorbarieae (57.2% sister to Gillenieae + Maleae) contrasted with both the nuclear topology (Sorbarieae sister to Exochordeae + Kerrieae) and the plastome topology (Sorbarieae sister to Amygdaleae) (Figures 2, 3). Notably, the position of the Spiraeeae as 42.8% sister to the Gillenieae + Maleae was congruent with the plastome topology, where Spiraeeae was sister to Gillenieae + Maleae. Essentially, the major hybridization edge was similar to the nuclear topology, while the minor hybridization edge was consistent with the plastome topology (Figures 2, 3). The other networks with hmax = 2–5 all included a hybridization edge similar to the hmax = 1 network (Supplementary Figure 4). As hmax increased, the network score always improved, although the very small changes in network score as hmax increases from 1 to 5 indicated that hmax = 1 was indeed the optimal network. Nevertheless, the hybrid edges in other networks can still provide valuable insights. The SNaQ network with hmax = 2 showed that the second hybridization edge was between Prunus hypoleuca of the Maddenia group (formerly in the genus Maddenia Hook. f & Thomson; Wen and Shi, 2012) and the lineage ancestral to Lyonothamneae + Amygdaleae (Supplementary Figure 4). This hybridization edge indicated that Prunus hypoleuca is 87.9% sister to the other Prunus L. species, and 12.1% sister to the ancestor of Lyonothamneae + Amygdaleae.
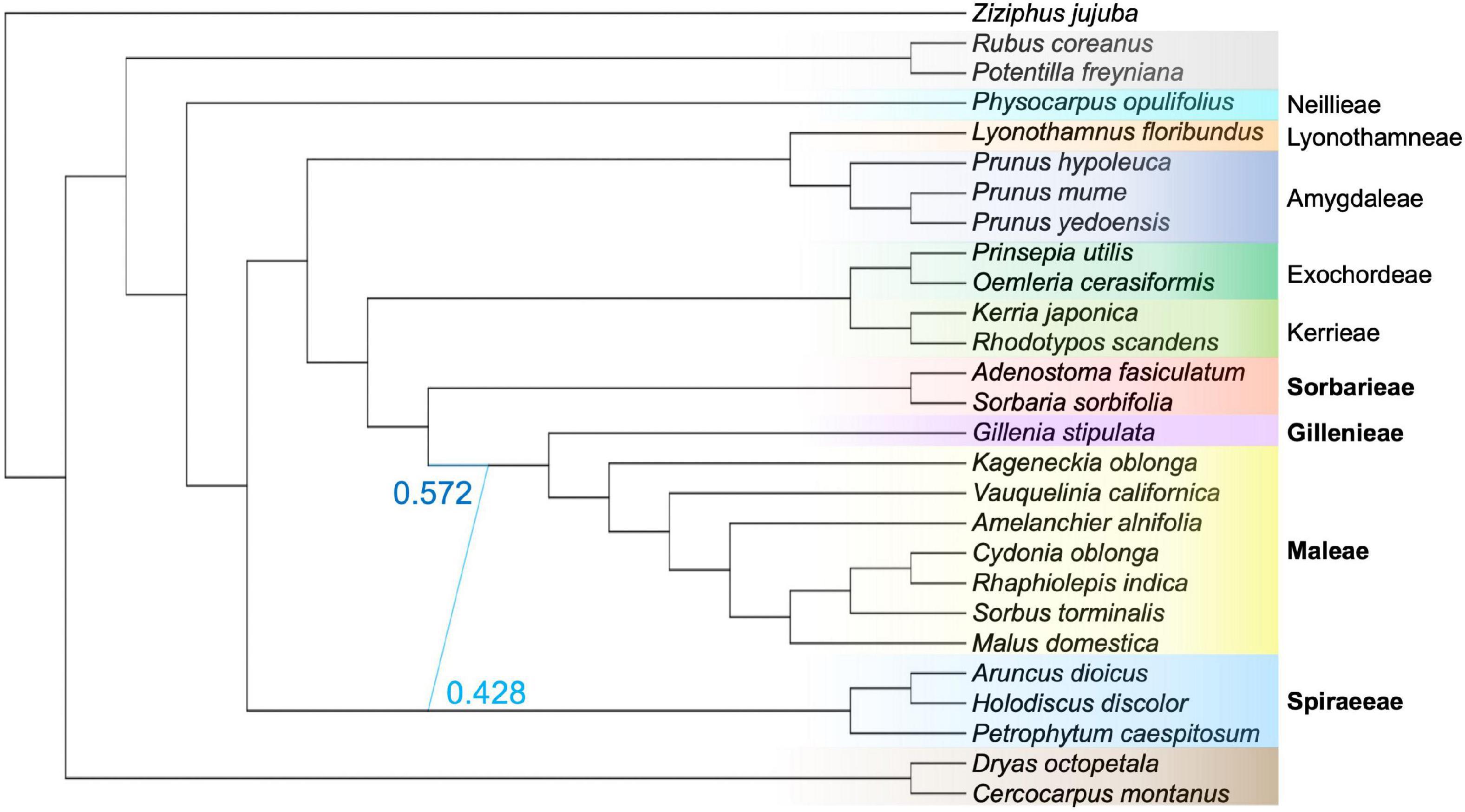
Figure 3. The optimal SNaQ network using 27 species with hmax = 1. The SNaQ analysis inferred that the lineage Gillenia + Maleae is 57.2% sister to an ancestor of Sorbarieae (represented here by Adenostoma fasiculatum and Sorbaria sorbifolia), and 42.8% sister to an ancestor of Spiraeeae (represented here by Aruncus dioicus, Holodiscus discolor, Petrophytum caespitosum). The color coding shows tribe/subfamily membership and is identical to the scheme in Figures 1, 2. Tribes in the Amygdaloideae are labeled to the right, and bold lettering indicates tribes involved in the hybridization edge.
The phyparts analysis indicated a wide range of gene tree conflict relative to the species tree, from virtually no conflict (e.g., the node defining the Spiraeeae; node Sp1 in Figure 4), to pervasive conflict where nearly 10 times more genes were discordant with the species tree topology than were concordant (e.g., node Z—the node defining Gillenieae + Maleae as sister to Exochordeae + Kerrieae + Sorbarieae; Figure 4). The nodes with a greater proportion of gene trees in conflict with the species tree than congruent with the species tree generally reflected nodes which disagree between the nuclear and chloroplast phylogenies, even though the data used for this analysis were nuclear gene trees and the nuclear species tree. The nodes with high conflict included deep nodes such as those displaying uncertainty regarding subfamilial relationships (node V; Figure 4) and the one reflecting the uncertainty of the position of the Spiraeeae tribe relative to the other tribes of the Amygdaloideae (node X; Figure 4). Moreover, the sister relationship between Amygdaleae + Lyonothamneae and a clade comprised of five other tribes (Sorbarieae, Kerrieae, Exochordeae, Gillenieae, and Maleae) showed high gene tree/species tree conflict (node Y; Figure 4). One other relatively deep node, representing the clade Kerrieae + Exochordeae + Sorbarieae (node KESo; Figure 4) exhibited high gene tree/species tree conflict, with over twice as many gene trees discordant as concordant. There were also several nodes with high degrees of discord within the Maleae, but investigating these shallower relationships is beyond the scope of this study, and we focused our taxon sampling with the goal of investigating deeper relationships in the tree as opposed to investigating documented discordance within the Maleae. When no bootstrap cutoff was used to consider whether gene trees were informative for a given node, the results were qualitatively similar (Supplementary Figure 5), so we focused on reporting the proportions of gene trees using the 50% bootstrap threshold (Figure 4). Based on the results of the SNaQ analysis, we used the phyckle “alternative relationship test” to further investigate support for the placement of the Spiraeeae using nuclear genes. We found that over 45% of nuclear genes (203 out of 448) support the chloroplast topology over the nuclear topology regarding the placement of Spiraeeae (Table 2). Moreover, the sum of log-likelihood differences across all genes indicated greater gene tree support for the chloroplast topology than the nuclear topology (Table 2).
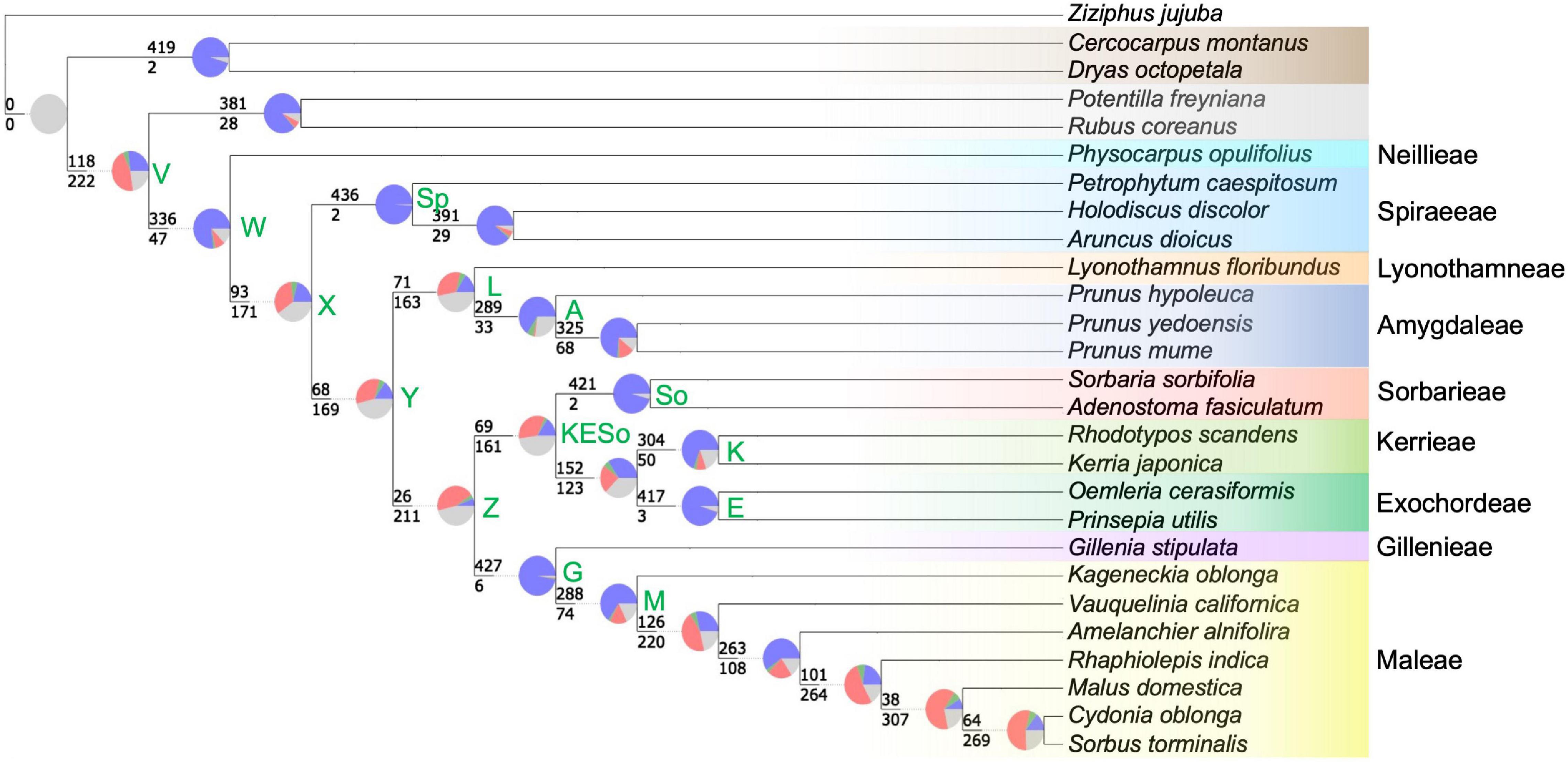
Figure 4. The phyparts tree indicating conflict at each node of the nuclear phylogeny using the 448-gene alignment and requiring gene trees to have 50% bootstrap support to be considered informative at a given node. Because phyparts uses rooted gene trees, only 440 genes were included in this analysis. At each node, the pie charts indicate the proportion of homologs supporting the clade defined by the node is shown in blue, the proportion supporting the primary alternative for that clade are green, the proportion supporting all other alternatives for the clade are red, and the proportion of homologs with less than 50% bootstrap support are shown in gray. Along each branch, the top number shows the number of genes concordant with the species tree at the associated node, and the bottom number represents the number of genes discordant with the species tree for the clade of interest. The color-coding of species names indicates tribe/subfamily membership and is consistent with Figures 1–3. As in Figure 1, select nodes are labeled with letters or abbreviations to enable easy reference in the text. Amygdaloideae tribes are labeled to the right of the species names.

Table 2. The results of the phyckle analysis investigating gene tree support for alternative topologies regarding the phylogenetic placement of the tribe Spiraeeae.
The GRAMPA analysis revealed that an allopolyploid event likely occurred in the clade that resulted in Gillenieae + Maleae. The most parsimonious tree (score = 14,733) was a MUL-tree with multiple tips of all taxa within the Gillenieae + Maleae, with one clade sister to Exochordeae + Kerrieae + Sorbarieae, and one clade sister to the Spiraeeae (Figure 5). This MUL-tree was more parsimonious than the singly labeled tree (score = 14,777), which is considered evidence of allopolyploidy. The result that the Spiraeeae are one parental participant in an allopolyploidy event was consistent with the SNaQ network results. One difference between the most parsimonious GRAMPA MUL-tree and the optimal SNaQ network was that the GRAMPA tree shows Exochordeae + Kerrieae + Sorbarieae as sister to the Gillenieae + Maleae, whereas in the SNaQ network, an ancestor of the Sorbarieae was one half of the hybridization edge (Figures 3, 5).
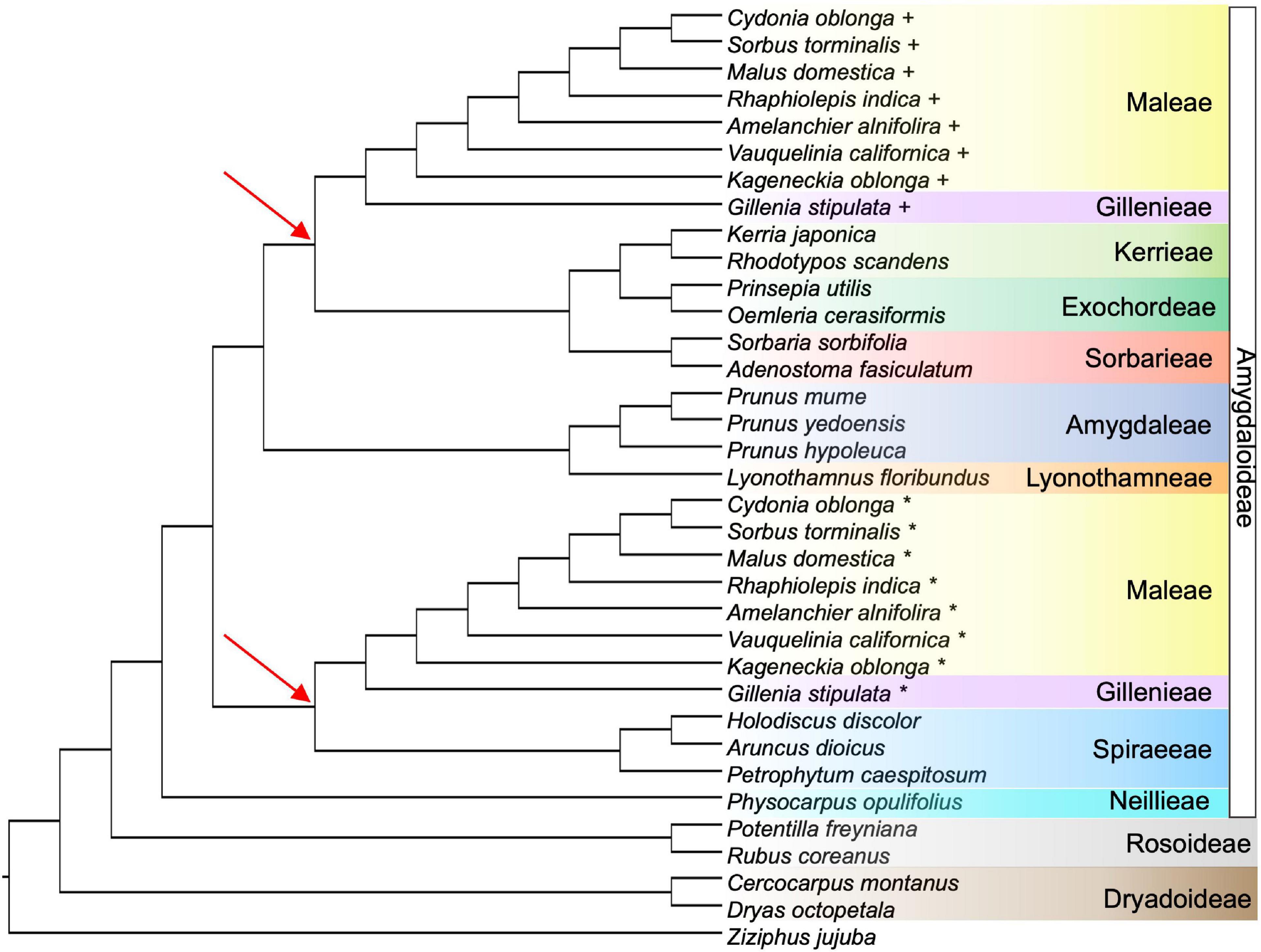
Figure 5. The most parsimonious tree from the GRAMPA analysis, which is a multi-labeled (MUL) tree indicating two tips for all species in Maleae + Gillenieae. For each species with multiple labels, the first tip is indicated by a plus sign and the second tip is shown using an asterisk. One Gillenieae + Maleae clade is sister to Kerrieae + Exochordeae + Sorbarieae, and the other Gillenieae + Maleae clade is sister to Spiraeeae. The red arrows highlight the nodes defining different lineages sister to the multi-labeled taxa in Maleae + Gillenieae. The color-coding of species shows tribe/subfamily and is consistent with all previous figures.
Hybrid Detector analyses confirmed aspects of both the SNaQ and GRAMPA results (Table 3). While using the Neillieae as an outgroup, the HyDe analysis inferred that the clade Maleae + Gillenieae was a hybrid with parents Spiraeeae and Sorbarieae, confirming the phylogenetic network result, and rejecting the possibility that either parental lineage (i.e., Spiraeeae or Sorbarieae) could be the hybrid lineage in this case (Table 3). The γ-value from the test that showed Maleae + Gillenieae as a hybrid lineage was 0.262 (Table 3). A similar analysis to test the relationship found using GRAMPA recovered support for Maleae + Gillenieae as a hybrid lineage with parents Spiraeeae and Kerrieae + Exochordeae + Sorbarieae (Table 3). Here the γ-value when Maleae + Gillenieae were a hybrid lineage was 0.526 (Table 3).

Table 3. The two hybridization hypotheses tested using Hybrid Detector.
The important role of hybridization and genome doubling in generating plant diversity is becoming apparent (Soltis and Soltis, 2009; Folk et al., 2018). However, there are few well-supported examples of large, successful groups such as Maleae originating via wide hybridization and/or allopolyploidy from the ancestor of a small lineage (i.e., Gillenia Moench) (Evans and Campbell, 2002). Based on several complementary analyses—the comparison of nuclear and chloroplast phylogenies, phylogenetic network analyses, and allopolyploidy analyses using MUL-trees—we test the competing wide hybridization and spiraeoid hypotheses, and investigate the role of genome doubling, to explain the origin of the apple tribe. Here, we present multiple lines of evidence indicating that an ancestor of the Spiraeeae was likely the maternal participant in an ancient hybridization event and an ancestor of the clade Sorbarieae + Exochordeae + Kerrieae was likely the paternal participant, although there was some minor variation in analyses regarding the identity of the paternal parent (Figures 3, 5). This hybridization event likely explains the origin of the clade Gillenieae + Maleae (Figure 3). Our results indicate that aspects of both existing hypotheses explaining the origin of the apple tribe are correct, but also aspects of each were incorrect. Our results also indicate that nuclear gene tree-species tree conflict and/or cytonuclear conflict are pervasive at several nodes in the Amygdaloideae. This suggests that beyond the hybrid origin of the apple clade, other lineages in the Amygdaloideae have reticulate evolutionary histories characterized by hybridization and/or allopolyploidy. Below, we discuss the details of our results and their implications on the origin of the apple tribe, as well as the possible explanations for high conflict nodes elsewhere in the subfamily Amygdaloideae.
Our results suggest the ancestor of the Maleae + Gillenieae originated via hybridization between distantly related tribes in the Amygdaloideae (i.e., the wide hybridization hypothesis, which states that the Maleae are the result of an ancient hybridization event between progenitors from other tribes in the subfamily Amygdaloideae) (Figure 3). Specifically, there was a hybridization event between an ancestor of the tribe Spiraeeae (x = 9) and an ancestor of Sorbarieae (x = 9) + Exochordeae (x = 8) + Kerrieae (x = 9), which gave rise to the clade comprised of Gillenieae (x = 9) + Maleae (x = 17) (Figure 3). This result is largely congruent with the wide hybridization hypothesis, except that we found that the clade Gillenieae + Maleae was the result of a wide hybridization event, as opposed to just the Maleae (Figure 3). Our results also partially support the spiraeoid hypothesis (i.e., the 17 chromosomes found in Maleae arose via the genome doubling of an x = 9 ancestor to x = 18, and subsequent aneuploidy resulting in x = 17), specifically regarding the role of whole genome duplication in the origin of the Maleae (Figure 5). The ancestors involved in the hybridization event leading to Gillenieae + Maleae had base chromosome numbers of x = 8 or 9, so there may have been genome doubling, possibly followed by aneuploidy if two x = 9 taxa were involved, to result in the x = 17 observed in the Maleae (Figure 5). Regardless of the ancestral chromosome number (x = 8 vs. x = 9), the genome doubling aspect of the spiraeoid hypothesis is supported by our results. However, given that our network analysis found that the clade Gillenieae + Maleae was the result of a hybridization event, and the base chromosome number of Gillenieae is x = 9, a genome doubling event preceding Gillenieae + Maleae can readily explain the x = 17 observed in the Maleae but not x = 9 in Gillenieae (Figure 3). The Gillenieae lineage may have undergone diploidization following an allopolyploidy event whereas the Maleae did not. Interpretation of the GRAMPA analysis favors this explanation because the most parsimonious allopolyploidy event precedes Gillenieae + Maleae, as opposed to only Maleae. Alternatively, perhaps there was a second genome doubling event of Maleae after an initial hybridization event leading to Gillenieae + Maleae. We favor the latter explanation, which is consistent with our SNaQ analysis and with results from Xiang et al. (2017), who noted nodes in the Maleae with evidence of whole genome duplications (WGDs) after the divergence of the Maleae from the ancestor of Gillenieae + Maleae (see Xiang et al., 2017; Figure 5). The annotated genome assembly of another Gillenieae species, Gillenia trifoliata, revealed that many syntenic blocks in Gillenia trifoliata mapped to two locations in Malus domestica, as would be expected with a history of genome doubling (Ireland et al., 2021). Moreover, the same syntenic blocks correspond to single orthologous regions in other Rosaceae species [Rubus occidentalis (raspberry) of Rosoideae and Prunus persica (peach)] of Amygdaloideae, suggesting that it is unlikely that the Gillenieae underwent a WGD and subsequent diploidization—a simpler explanation is that the WGD occurred after an initial hybridization leading to Gillenieae + Maleae.
Across the plant tree of life, diversification via genome duplication is relatively common. It is becoming increasingly clear that following WGD events, the genomes of organisms are particularly malleable and that genomic rearrangements may spur key functional innovations. Genome evolution associated with WGDs has often been studied in crop species, many of which are polyploid. For example, controlled crosses of early generation allopolyploid wheat revealed that aneuploidy is common following WGDs (Zhang et al., 2013). However, there are examples of variation in genome size and organization after WGD events in non-model systems. In the neopolyploid Tragopogon L., massive chromosomal variation followed an allopolyploidy event (Chester et al., 2012), including aneuploidy in 69% of cases. Within a single genus of ca. 250 species (clover; genus Trifolium L.), there have been many deviations from the ancestral chromosome state (2n = 16), including at least 22 instances of polyploidy and 19 occurrences of aneuploidy (Ellison et al., 2006). The diversification of the Gillenieae-Maleae clade may represent another example of lineages that diversified following chromosomal rearragnements via allopolyploidy and aneuploidy.
Many previous studies have hypothesized an allopolyploid origin of the apple tribe (Sax, 1931, 1932, 1933; Stebbins, 1950; Chevreau et al., 1985; Weeden and Lamb, 1987; Robertson et al., 1991; Evans and Campbell, 2002; Vamosi and Dickinson, 2006; Potter et al., 2007). The wide-hybridization hypothesis, favored until 2002, considered many lineages as possible participants in hybridization and/or allopolyploidy, but strong evidence for any particular lineage was lacking. The spiraeoid hypothesis was supported by one duplicated nuclear gene (GBSSI-1 and GBSSI-2; Evans and Campbell, 2002), inferring that both parental participants in allopolyploidy were ancestors of the Gillenieae lineage. Our network analyses (Figure 3) indicate that a hybridization event between an ancestor of the Spiraeeae and Sorbarieae leading to Gillenieae + Maleae, whereas allopolyploid analyses (Figure 5) indicate that an ancestor of the Spiraeeae and a common ancestor of Sorbarieae + Exochordeae + Kerrieae were likely the parental participants in allopolyploidy. Both of these scenarios were confirmed as possible hybridization events using separate analyses (i.e., HyDe; Table 3). Given the low support for the KESo and Z nodes (quartet support scores = 50.44 and 39.70, respectively, and high degrees of gene tree conflict; Figure 4), perhaps the topological uncertainty in the nuclear phylogeny is causing the discrepancy between the SNaQ and GRAMPA analyses (Figures 3, 5). When considering the bifurcating nuclear and plastome topologies (Figure 2), and considering the proportions of nuclear gene trees that support the nuclear vs. chloroplast topologies (Table 2), it becomes evident that the Spiraeeae ancestor was most likely the maternal donor to a hybridization or allopolyploid event because Spiraeeae is sister to Gillenieae + Maleae in the plastome tree, and that the ancestor of Sorbarieae + Exochordeae + Kerrieae was the paternal participant because this relationship is more similar to the nuclear tree than the plastome tree.
There are multiple nodes with pervasive conflict, both among the subfamilies of the Rosaceae and within the Amygdaloideae. These include nodes V (Rosoideae—Amygdaloideae sister), Y (Lyonothamneae + Amygdaleae sister to clade defined by node Z), L (Lyonothamneae sister to Amygdaleae), Z (Kerrieae + Exochordeae + Sorbarieae sister to Gillenieae +Maleae), and KESo (Sorbarieae sister to Kerrieae + Maleae) (Figure 4). Xiang et al. (2017) produced six distinct Rosaceae phylogenies based on data filtering (between 113 and 882 genes included) and tree-inference method (concatenation with ML inference in RAxML vs. a coalescent species tree approach implemented in ASTRAL). They defined nodes as highly supported (100% bootstrap support in all trees), moderately supported (90% bootstrap support in at least five trees, and 85% support in all six trees), poorly supported (80% bootstrap support in three or more trees and 40% support in all six trees), and unresolved (not meeting the above criteria). Multiple nodes with pervasive conflict according to our phyparts analysis were considered highly supported (Y,V,L) or moderately supported (KESo) in Xiang et al. (2017). Only one key node with high gene tree conflict (Z) was considered poorly supported in Xiang et al. (2017), and no nodes with pervasive conflict identified in our analyses were listed as unresolved. None of the above nodes were consistent with the plastome tree, either from Zhang et al. (2017) or from our analyses. Clearly, there is substantial conflict among nuclear gene trees within the Amygdaloideae, in addition to the documented cytonuclear discord. Histories of reticulate evolution appear common in this group, beyond the allopolyploid origin of the apple tribe.
We cannot be certain of the cause of conflict in many of the nodes in the Amygdaloideae. Potential biological explanations for gene tree discord may include incomplete lineage sorting (ILS) or hybridization. Processes such as ILS may also lead to gene tree-species tree conflict in the absence of hybridization. However, there is strong evidence for the hybrid origin of the Gillenieae + Maleae. SNaQ is robust to ILS in that it can incorporate uncertainty in user-estimated gene trees and handle gene tree discordance caused by ILS (Solís-Lemus and Ané, 2016). The comparison of pseudodeviance network scores in SNaQ between the ASTRAL tree, which accommodates ILS, and the hmax = 1 network, which can accommodate ILS and hybrid edges, clearly favors the hmax = 1 network. The phyckle analysis of the node defining the position of the Spiraeeae (i.e., node X, Figure 4 and Table 2) on its own can identify the proportion of nuclear genes that support species tree or alternative relationships, but does not explicitly identify sources of conflict. However, the nearly equal distribution of nuclear genes that support the nuclear topology and the chloroplast topology, when considered alongside the other analyses (e.g., SNaQ, GRAMPA, and HyDe), add evidence that a history of hybridization via allopolyploidy shaped evolutionary histories of the sampled genes. While we do not have specific expectations for the proportion of gene trees that may conflict with the species tree solely due to ILS, that so many gene trees support the alternative chloroplast topology, as opposed to a distribution of different topologies induced by ILS, provides more evidence for an instance of hybridization. That over 45% of nuclear genes (203 out of 448) support the chloroplast topology over the nuclear topology with regard to the placement of Spiraeeae is another piece of evidence that the maternal participant in allopolyploidy leading to the apple tribe was an ancestor of the Spiraeeae. The large number of nuclear genes that favor the chloroplast topology may in part explain past uncertainty in phylogenetic studies investigating the Rosaceae or its subfamilies.
The tribe Lyonothamneae is represented by a monotypic genus, Lyonothamnus A.Gray. The position of this tribe varies greatly between the plastome phylogeny (Lyonothamneae sister to all other tribes in the Amygdaloideae) and nuclear phylogeny (Lyonothamneae sister to Amygdaleae). Furthermore, there is substantial nuclear gene tree conflict at this node (L; Figure 4). The SNaQ network with hmax = 2 showed that the second hybridization edge was between Prunus hypoleuca of the Maddenia group and the lineage ancestral to Lyonothamneae + Amygdaleae (Supplementary Figure 4). Essentially, this hybridization edge means that Prunus hypoleuca of the Maddenia group (Wen and Shi, 2012) is 87.9% sister to the other Prunus species, and 12.1% sister to the ancestor of Lyonothamneae + Amygdaleae. The interpretation of this hybridization edge is less straightforward than the hmax = 1 edge. However, there is evidence from previous studies that a WGD occurred near the base of the Amygdaleae (Xiang et al., 2017), and other studies have hypothesized that ancient hybridization and/or allopolyploidy were involved in the diversification of Prunus (Chin et al., 2014; Zhao et al., 2016, 2018; Hodel et al., 2021), the sole accepted genus in the Amygdaleae. Future studies with denser taxon-sampling in the Amygdaleae and hundreds of nuclear loci combined with chloroplast data are needed to investigate the evolutionary history of the Lyonothamneae + Amygdaleae.
Although assessing discordance within the Maleae is not a focus of this paper, we note that in the hmax = 4 and hmax = 5 SNaQ networks (Supplementary Figure 4), there are hybridization edges that indicate possible hybridization within the Maleae. The hmax = 4 hybrid edge shows Kageneckia 96.2% sister to all other Maleae but also 3.8% sister to Maleae + Gillenieae. In the hmax = 5 network, the hybrid edge indicates the ancestor of subtribe Malinae (pome-bearing Maleae, i.e., Maleae excluding Kageneckia and Vauquelinia) is 97.7% sister to Vauquelinia and also 2.3% sister to Maleae + Gillenieae. Taken in isolation, these hybrid edges mean little, especially given the discrepancy between the γ values of the major and minor hybridization edges. However, when considered in concert with previously documented discord within the Maleae, the conflict documented via phyparts at multiple nodes in the Maleae (Figure 4), as well as evidence of genome doubling at multiple nodes within the Maleae (Xiang et al., 2017), the hmax = 4 and hmax = 5 SNaQ results point to hybridization, especially introgression, as a possible mechanism explaining phylogenomic discord within the Maleae. Further targeted investigations are needed to address discordance within the Maleae.
The well-documented discordance between chloroplast and nuclear phylogenies in the Amygdaloideae could also be explained by chloroplast capture. This phenomenon occurs when native cytoplasm is replaced by foreign cytoplasm via hybridization followed by repeated backcrossing (Rieseberg and Soltis, 1991). In closely related species that are sexually compatible, chloroplast capture can be pervasive and lead to cytonuclear discordance. In the Amygdaloideae, there have been several instances of chloroplast capture documented. In the Amygdaleae tribe, cytonuclear discord was attributed to chloroplast capture in several Prunus species, including North American plums (Rohrer et al., 2008) and East Asian cherries (Cho et al., 2014). Within the Maleae, there is also evidence of chloroplast capture as a mechanism causing cytonuclear discord. Strong discordance between nuclear and plastid phylogenies regarding the placement of the Maleae genera Malacomeles (Decne.) Decne. and Peraphyllum Nutt. supports ancient chloroplast capture events in SW North America (Liu et al., 2020a). Because chloroplast capture involves hybridization followed by recurrent backcrossing, it occurs more frequently at shallower systematic scales among sexually compatible species. Accordingly, chloroplast capture could explain the SNaQ hybridization edges detected within the Maleae and Amygdaleae at higher values of hmax (Supplementary Figure 4). Additionally, although we did not detect cytonuclear discord within the Maleae in our sampling, histories consistent with chloroplast capture may explain the pervasive gene tree conflict at nodes within the Maleae (Figure 4).
Previous molecular studies of the Rosaceae typically used either nuclear (e.g., Evans and Campbell, 2002) or chloroplast data (Potter et al., 2002). The single-nuclear gene GBSSI phylogeny by Evans and Campbell (2002) could not resolve the position of the Spiraeeae, and the branching order of the Spiraeeae, Sorbarieae, Exochordeae, Amygdaleae, and Dryadeae was a polytomy. Potter et al. (2002) used two chloroplast genes and recovered a phylogeny that placed the Spiraeeae + Sorbarieae sister to the Gillenieae + Maleae + Amygdaleae. However, there was poor bootstrap support (i.e., < 75%) for all these relationships except Gillenieae + Maleae. Potter et al.’s (2002) chloroplast phylogeny also found that Lyonothamneae were sister to all other Amygdaloid tribes with 100% bootstrap support. One study that used data from both nuclear and chloroplast genomes is Potter et al. (2007), with six nuclear and four chloroplast loci to create a consensus phylogeny, inferred that the Spiraeeae were sister to the Gillenieae + Maleae (i.e., the dominant chloroplast topology from Zhang et al. (2017) and the present study), albeit with low support (44% bootstrap and 57% Bayesian clade credibility). Potter et al. (2007) excluded two nuclear loci from their analyses due to results “inconsistent in some ways with the majority of other data.” Two of the anomalous results caused by the two excluded nuclear genes they report are inconsistent placement of the Spiraeeae and the lack of a sister relationship between Lyonothamneae and the rest of the Amygdaloideae (referred to as Spiraeoideae in Potter et al., 2007).
Subsequent results, from Xiang et al. (2017) and Zhang et al. (2017) and the present study, contextualize and explain the results from earlier molecular studies. The position of Lyonothamneae is clearly quite different in the chloroplast and nuclear genomes, and this is reflected throughout the literature; studies with only chloroplast data repeatedly find Lyonothamneae sister to the rest of Amygdaloideae, typically with strong support. In contrast, this relationship is never found in studies using only nuclear data. The position of the Spiraeeae has been variable in studies from the literature, but it is now becoming clear that much of the uncertainty with regard to its placement is due to a history of WGDs in the Amygdaloideae. Specifically, in this paper we characterize one instance of allopolyploidy, in which an ancestor of the Spiraeeae was likely the maternal participant in allopolyploidization. Given the distinct chloroplast and nuclear topologies regarding the placement of Spiraeeae, and the fact that nearly half of nuclear genes sampled in this study favor the chloroplast topology, it is unsurprising that earlier molecular studies using fewer than 10 markers were unable to confidently resolve the position of Spiraeeae. Although we used a set of complementary analyses to resolve the origin of the apple tribe, there is clearly more phylogenetic uncertainty due to reticulate evolutionary histories in the Amygdaloideae. The different positions of the Lyonothamneae in the nuclear and chloroplast phylogenies, coupled with the SNaQ network results and previous evidence of WGD events leading to and within the Amygdaleae, indicate that future targeted efforts should be focused on resolving the evolutionary history of the Lyonothamneae and Amygdaleae.
Over the past several decades, systematists have embraced the need for incorporating genealogical information from nuclear genes to obtain robust estimates of phylogeny. Chloroplast data were favored for many years due to their high copy number which translated to easy generation of homologous loci for many individuals and/or species (Thode et al., 2020; Wang et al., 2020; Welker et al., 2020). Those studies fell out of favor due to the limited information regarding ancestry given their typical uniparental inheritance. It is now becoming clear that reticulation is prevalent at many phylogenetic scales due to hybridization or other processes (Lee-Yaw et al., 2018). In cases of pervasive reticulation, nuclear AND chloroplast data are now necessary complements to one another if researchers hope to resolve reticulate complexes. Our study highlights how synthesizing results from existing studies cannot only reconcile differences from two recent studies, but also answer century old questions that have been continually debated in the literature. Our study also highlights the need to revisit and reconsider phylogenetic relationships, even when they have been found to be highly supported using metrics such as bootstrapping. In a number of recent studies (e.g., Soltis et al., 2004; Prasanna et al., 2020; Walker et al., 2021), careful analyses of conflict have revealed that we should not be overly confident in apparently resolved relationships. In conclusion, our results from multiple lines of evidence confirmed the hybrid origin of the Maleae + Gillenieae clade and supported the polyploidy-aneuploidy-origin aspect of the hypothesis of Maleae (x = 17 or 15) originating from the tribe Gillenieae (x = 9) as proposed by Evans and Campbell (2002). Future research may provide a complete picture of the role of hybridization in the early diversification of Maleae, especially regarding the formation of the chromosome number of 15 in Vauquelinia and the evolutionary mechanisms leading from dry fruits (capsules) to fleshy fruits (pomes).
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
RH, EZ, and JW conceptualized the project. RH and B-BL obtained and analyzed the data. RH led the writing of the manuscript. All authors edited drafts and approved the final version of the manuscript.
This work was supported by a Smithsonian Institution Peter Buck Fellowship to RH.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Greg Stull, two reviewers, and the associate editor for many helpful comments on the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.820997/full#supplementary-material
Baudry, J.-P., Maugis, C., and Michel, B. (2011). Slope heuristics: overview and implementation. Stat. Comput. 22, 455–470. doi: 10.1007/s11222-011-9236-1
Blischak, P. D., Chifman, J., Wolfe, A. D., and Kubatko, L. S. (2018). HyDe: a python package for genome-scale hybridization detection. Syst. Biol. 67, 821–829. doi: 10.1093/sysbio/syy023
Brown, J. W., Walker, J. F., and Smith, S. A. (2017). Phyx: phylogenetic tools for unix. Bioinformatics 33, 1886–1888. doi: 10.1093/bioinformatics/btx063
Bruun-Lund, S., Clement, W. L., Kjellberg, F., and Rønsted, N. (2017). First plastid phylogenomic study reveals potential cyto-nuclear discordance in the evolutionary history of Ficus L. (Moraceae). Mol. Phylogenet. Evol. 109, 93–104. doi: 10.1016/j.ympev.2016.12.031
Campbell, C. S., Donoghue, M. J., Baldwin, B. G., and Wojciechowski, M. F. (1995). Phylogenetic relationships in Maloideae (Rosaceae): evidence from sequences of the internal transcribed spacers of nuclear ribosomal DNA and its congruence with morphology. Am. J. Bot. 82, 903–918. doi: 10.1002/j.1537-2197.1995.tb15707.x
Chester, M., Gallagher, J. P., Symonds, V. V., Da Silva, A. V. C., Mavrodiev, E. V., Leitch, A. R., et al. (2012). Extensive chromosomal variation in a recently formed natural allopolyploid species, Tragopogon miscellus (Asteraceae). Proc. Natl. Acad. Sci. U.S.A. 109, 1176–1181. doi: 10.1073/pnas.1112041109
Chevreau, E., Lespinasse, Y., and Gallet, M. (1985). Inheritance of pollen enzymes and polyploid origin of apple (Malus x domestica Borkh.). Theor. Appl. Genet. 71, 268–277. doi: 10.1007/BF00252066
Chin, S.-W., Shaw, J., Haberle, R., Wen, J., and Potter, D. (2014). Diversification of almonds, peaches, plums and cherries – molecular systematics and biogeographic history of Prunus (Rosaceae). Mol. Phylogenet. Evol. 76, 34–48. doi: 10.1016/j.ympev.2014.02.024
Cho, M. S., Kim, C. S., Kim, S. H., Kim, T. O., Heo, K. I., Jun, J., et al. (2014). Molecular and morphological data reveal hybrid origin of wild Prunus yedoensis (Rosaceae) from Jeju Island, Korea: implications for the origin of the flowering cherry. Am. J. Bot. 101, 1976–1986. doi: 10.3732/ajb.1400318
Darlington, C. D., and Moffett, A. A. (1930). Primary and secondary chromosome balance in Pyrus. J. Genet. 22, 129–151. doi: 10.1007/bf02983843
Ellison, N. W., Liston, A., Steiner, J. J., Williams, W. M., and Taylor, N. L. (2006). Molecular phylogenetics of the clover genus (Trifolium-Leguminosae). Mol. Phylogenet. Evol. 39, 688–705. doi: 10.1016/j.ympev.2006.01.004
Evans, R. C., and Campbell, C. S. (2002). The origin of the apple subfamily (Maloideae; Rosaceae) is clarified by DNA sequence data from duplicated GBSSI genes. Am. J. Bot. 89, 1478–1484. doi: 10.3732/ajb.89.9.1478
Folk, R. A., Soltis, P. S., Soltis, D. E., and Guralnick, R. (2018). New prospects in the detection and comparative analysis of hybridization in the tree of life. Am. J. Bot. 105, 364–375. doi: 10.1002/ajb2.1018
Goldblatt, P. (1976). Cytotaxonomic studies in the tribe Quillajeae (Rosaceae). Ann. Miss. Bot. Gard. 63, 200–206. doi: 10.2307/2395226
Goodman, M., Czelusniak, J., William Moore, G., Romero-Herrera, A. E., and Matsuda, G. (1979). Fitting the gene lineage into its species lineage, a parsimony strategy illustrated by cladograms constructed from globin sequences. Syst. Biol. 28, 132–163. doi: 10.2307/2412519
Hodel, R. G. J., Zimmer, E., and Wen, J. (2021). A phylogenomic approach resolves the backbone of Prunus (Rosaceae) and identifies signals of hybridization and allopolyploidy. Mol. Phylogenet. Evol. 160:107118. doi: 10.1016/j.ympev.2021.107118
Huang, D. I, Hefer, C. A., Kolosova, N., Douglas, C. J., and Cronk, Q. C. B. (2014). Whole plastome sequencing reveals deep plastid divergence and cytonuclear discordance between closely related balsam poplars, Populus balsamifera and P. trichocarpa (Salicaceae). New Phytol. 204, 693–703. doi: 10.1111/nph.12956
Ireland, H. S., Wu, C., Deng, C. H., Hilario, E., Saei, A., Erasmuson, S., et al. (2021). The Gillenia trifoliata genome reveals dynamics correlated with growth and reproduction in Rosaceae. Hortic. Res. 8:233. doi: 10.1038/s41438-021-00662-4
Katoh, K., and Standley, D. M. (2013). Mafft multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Larget, B. R., Kotha, S. K., Dewey, C. N., and Ané, C. (2010). Bucky: gene tree/species tree reconciliation with Bayesian concordance analysis. Bioinformatics 26, 2910–2911. doi: 10.1093/bioinformatics/btq539
Lee-Yaw, J. A., Grassa, C. J., Joly, S., Andrew, R. L., and Rieseberg, L. H. (2018). An evaluation of alternative explanations for widespread cytonuclear discordance in annual sunflowers (Helianthus). New Phytol. 221, 515–526. doi: 10.1111/nph.15386
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Liu, B.-B., Campbell, C. S., Hong, D. Y., and Wen, J. (2020a). Phylogenetic relationships and chloroplast capture in the Amelanchier-Malacomeles-Peraphyllum clade (Maleae, Rosaceae): evidence from chloroplast genome and nuclear ribosomal DNA data using genome skimming. Mol. Phylogenet. Evol. 147:106784. doi: 10.1016/j.ympev.2020.106784
Liu, B.-B., Hong, D.-Y., Zhou, S. L., Xu, C., Dong, W. P., Johnson, G., et al. (2019). Phylogenomic analyses of the Photinia complex support the recognition of a new genus Phippsiomeles and the resurrection of a redefined Stranvaesia in Maleae (Rosaceae). J. Syst. Evol. 57, 678–694. doi: 10.1111/jse.12542
Liu, B.-B., Liu, G.-N., Hong, D.-Y., and Wen, J. (2020b). Eriobotrya belongs to Rhaphiolepis (Maleae, Rosaceae): evidence from chloroplast genome and nuclear ribosomal DNA data. Front. Plant Sci. 10:1731. doi: 10.3389/fpls.2019.01731
Liu, B.-B., Ren, C., Kwak, M., Hodel, R. G. J., Xu, C., He, J., et al. (2021). Phylogenomic analyses in the apple genus Malus s.l. reveal widespread hybridization and allopolyploidy driving the diversifications, with insights into the complex biogeographic history in the Northern Hemisphere. bioRxiv [Preprint] doi: 10.1101/2021.10.12.464085
Mirarab, S., Reaz, R., and Bayzid, M. S. (2014) ASTRAL: genome-scale coalescent-based species tree estimation. Bioinformatics 30, i541–i548.
Page, R. D. M. (1994). Maps between trees and cladistic analysis of historical associations among genes, organisms, and areas. Syst. Biol. 43, 58–77. doi: 10.1093/sysbio/43.1.58
Potter, D., Eriksson, T., Evans, R. C., Oh, S., Smedmark, J. E. E., Morgan, D. R., et al. (2007). Phylogeny and classification of Rosaceae. Plant Syst. Evol. 266, 5–43.
Potter, D., Gao, F., Bortiri, P. E., Oh, S.-H., and Baggett, S. (2002). Phylogenetic relationships in Rosaceae inferred from chloroplast matK and trnL-trnF nucleotide sequence data. Plant Syst. Evol. 231, 77–89.
Prasanna, A. N., Gerber, D., Kijpornyongpan, T., Aime, M. C., Doyle, V. P., and Nagy, L. G. (2020). Model choice, missing data, and taxon sampling impact phylogenomic inference of deep basidiomycota relationships. Syst. Biol. 69, 17–37. doi: 10.1093/sysbio/syz029
Revell, L. J. (2012). phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 3, 217–223. doi: 10.1111/j.2041-210x.2011.00169.x
Rieseberg, L. H., and Soltis, D. E. (1991). Phylogenetic consequences of cytoplasmic gene flow in plants. Evol. Trends Plants 5, 65–84.
Robertson, K. R., Phipps, J. B., Rohrer, J. R., and Smith, P. G. (1991). A synopsis of genera in Maloideae (Rosaceae). Syst. Bot. 16:376. doi: 10.2307/2419287
Rohrer, J. R., O’brien, M. A., and Anderson, J. A. (2008). Phylogenetic analysis of North American plums (Prunus sect. Prunocerasus: Rosaceae) based on nuclear Leafy and s6pdh sequences. J. Bot. Res. Inst. Tx 2, 401–414.
Roycroft, E. J., Moussalli, A., and Rowe, K. C. (2020). Phylogenomics uncovers confidence and conflict in the rapid radiation of Australo-Papuan rodents. Syst. Biol. 69, 431–444. doi: 10.1093/sysbio/syz044
Sax, K. (1933). The origin of the Pomoideae. Proc. Am. Soc. Hortic. Sci. 30, 147–150. doi: 10.1046/j.1365-2672.1997.00377.x
Smith, S. A., Moore, M. J., Brown, J. W., and Yang, Y. (2015). Analysis of phylogenomic datasets reveals conflict, concordance, and gene duplications with examples from animals and plants. BMC Evol. Biol. 15:150. doi: 10.1186/s12862-015-0423-0
Smith, S. A., Walker-Hale, N., Walker, J. F., and Brown, J. W. (2020). Phylogenetic conflicts, combinability, and deep phylogenomics in plants. Syst. Biol. 69, 579–592. doi: 10.1093/sysbio/syz078
Solís-Lemus, C., and Ané, C. (2016). Inferring phylogenetic networks with maximum pseudolikelihood under incomplete lineage sorting. PLoS Genet. 12:e1005896. doi: 10.1371/journal.pgen.1005896
Solís-Lemus, C., Bastide, P., and Ané, C. (2017). PhyloNetworks: a package for phylogenetic networks. Mol. Biol. Evol. 34, 3292–3298. doi: 10.1093/molbev/msx235
Soltis, D. E., Albert, V. A., Savolainen, V., Hilu, K., Qiu, Y. L., Chase, M. W., et al. (2004). Genome-scale data, angiosperm relationships, and “ending incongruence”: a cautionary tale in phylogenetics. Trends Plant Sci. 9, 477–483. doi: 10.1016/j.tplants.2004.08.008
Soltis, P. S., and Soltis, D. E. (2009). The role of hybridization in plant speciation. Annu. Rev. Plant Biol. 60, 561–588. doi: 10.1146/annurev.arplant.043008.092039
Stamatakis, A. (2014). Raxml version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Struck, T. H. (2014). Trespex-detection of misleading signal in phylogenetic reconstructions based on tree information. Evol. Bioinformat. 10, 51–67. doi: 10.4137/EBO.S14239
Thode, V. A., Lohmann, L. G., and Sanmartín, I. (2020). Evaluating character partitioning and molecular models in plastid phylogenomics at low taxonomic levels: a case study using Amphilophium (Bignonieae, Bignoniaceae). J. Syst. Evol. 58, 1071–1089.
Thomas, G. W. C., Ather, S. H., and Hahn, M. W. (2017). Gene-tree reconciliation with MUL-trees to resolve polyploidy events. Syst. Biol. 66, 1007–1018. doi: 10.1093/sysbio/syx044
Vamosi, J. C., and Dickinson, T. A. (2006). Polyploidy and diversification: a phylogenetic investigation in Rosaceae. Int. J. Plant Sci. 167, 349–358.
Vaughan, T. G. (2017). IcyTree: rapid browser-based visualization for phylogenetic trees and networks. Bioinformatics 33, 2392–2394. doi: 10.1093/bioinformatics/btx155
Walker, J. F., Smith, S. A., Hodel, R. G. J., and Moyroud, E. (2021). Concordance-based approaches for the inference of relationships and molecular rates with phylogenomic data sets. Syst. Biol. 7:syab052. doi: 10.1093/sysbio/syab052
Wang, H. X., Morales-Briones, D. F., Moore, M. J., Wen, J., and Wang, H. F. (2021). A phylogenomic perspective on gene tree conflict and character evolution in Caprifoliaceae using target enrichment data, with Zabelioideae recognized as a new subfamily. J. Syst. Evol. 59, 897–914.
Wang, Y.-B., Liu, B.-B., Nie, Z.-L., Chen, H.-F., Chen, F.-J., Figlar, R. B., et al. (2020). Major clades and a revised classification of Magnolia and Magnoliaceae based on whole plastid genome sequences via genome skimming. J. Syst. Evol. 58, 673–695. doi: 10.1111/jse.12588
Weeden, N., and Lamb, R. (1987). Genetics and linkage analysis of 19 isozyme loci in apple. J. Am. Soc. Hortic. Sci. 112, 865–872.
Welker, C. A. D., Mckain, M. R., Estep, M. C., Pasquet, R. S., Chipabika, G., Pallangyo, B., et al. (2020). Phylogenomics enables biogeographic analysis and a new subtribal classification of the Andropogoneae (Poaceae—Panicoideae). J. Syst. Evol. 58:10031030.
Wen, J., and Shi, W. (2012). Revision of the Maddenia clade of Prunus (Rosaceae). PhytoKeys 11, 39–59. doi: 10.3897/phytokeys.11.2825
Xiang, Y., Huang, C. H., Hu, Y., Wen, J., Li, S., Yi, T., et al. (2017). Evolution of Rosaceae fruit types based on nuclear phylogeny in the context of geological times and genome duplication. Mol. Biol. Evol. 34, 262–281.
Xu, L.-L., Yu, R.-M., Lin, X.-R., Zhang, B.-W., Li, N., Lin, K., et al. (2021). Different rates of pollen and seed gene flow cause branch-length and geographic cytonuclear discordance within Asian butternuts. New Phytol. 232, 388–403. doi: 10.1111/nph.17564
Zhang, H., Bian, Y., Gou, X., Zhu, B., Xu, C., Qi, B., et al. (2013). Persistent whole-chromosome aneuploidy is generally associated with nascent allohexaploid wheat. Proc. Natl. Acad. Sci. U.S. A. 110, 3447–3452. doi: 10.1073/pnas.1300153110
Zhang, S. D., Jin, J. J., Chen, S. Y., Chase, M. W., Soltis, D. E., Li, H. T., et al. (2017). Diversification of Rosaceae since the late Cretaceous based on plastid phylogenomics. New Phytol. 214, 1355–1367. doi: 10.1111/nph.14461
Zhao, L., Jiang, X.-W., Zuo, Y., Liu, X.-L., Chin, S.-W., Haberle, R., et al. (2016). Multiple events of allopolyploidy in the evolution of the racemose lineages in Prunus (Rosaceae) based on integrated evidence from nuclear and plastid data. PLoS One 11:e0157123. doi: 10.1371/journal.pone.0157123
Keywords: allopolyploidy, ancient hybridization, cytonuclear conflict, genome doubling, phylogenetic networks, phylogenomics, reticulate evolution
Citation: Hodel RGJ, Zimmer EA, Liu B-B and Wen J (2022) Synthesis of Nuclear and Chloroplast Data Combined With Network Analyses Supports the Polyploid Origin of the Apple Tribe and the Hybrid Origin of the Maleae—Gillenieae Clade. Front. Plant Sci. 12:820997. doi: 10.3389/fpls.2021.820997
Received: 23 November 2021; Accepted: 20 December 2021;
Published: 25 January 2022.
Edited by:
Susann Wicke, Humboldt University of Berlin, GermanyReviewed by:
Natascha D. Wagner, University of Göttingen, GermanyCopyright © 2022 Hodel, Zimmer, Liu and Wen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard G. J. Hodel, cmljaGllaG9kZWxAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.