 Wyclif Ochieng Odago1,2,3
Wyclif Ochieng Odago1,2,3 Emmanuel Nyongesa Waswa1,2,3
Emmanuel Nyongesa Waswa1,2,3 Consolata Nanjala1,2,3
Consolata Nanjala1,2,3 Elizabeth Syowai Mutinda1,2,3
Elizabeth Syowai Mutinda1,2,3 Vincent Okelo Wanga1,2,3
Vincent Okelo Wanga1,2,3 Elijah Mbandi Mkala1,2,3
Elijah Mbandi Mkala1,2,3 Millicent Akinyi Oulo1,2,3
Millicent Akinyi Oulo1,2,3 Yan Wang1,2,3
Yan Wang1,2,3 Cai-Fei Zhang1,2*
Cai-Fei Zhang1,2* Guang-Wan Hu1,2,3*
Guang-Wan Hu1,2,3* Qing-Feng Wang1,2,3
Qing-Feng Wang1,2,3- 1CAS Key Laboratory of Plant Germplasm Enhancement and Specialty Agriculture, Wuhan Botanical Garden, Chinese Academy of Sciences, Wuhan, China
- 2Sino-Africa Joint Research Center, Chinese Academy of Sciences, Wuhan, China
- 3University of Chinese Academy of Sciences, Beijing, China
Hoya is a genus in Apocynaceae-Asclepiadoideae, known for its showy wax flowers, making it a popular ornamental plant. However, phylogenetic relationships among most Hoya species are not yet fully resolved. In this study, we sequenced 31 plastomes of Hoya group species using genome skimming data and carried out multiple analyses to understand genome variation to resolve the phylogenetic positions of some newly sequenced Chinese endemic species. We also screened possible hotspots, trnT-trnL-trnF, psba-trnH, and trnG-UCC, ndhF, ycf1, matK, rps16, and accD genes that could be used as molecular markers for DNA barcoding and species identification. Using maximum likelihood (ML) and Bayesian Inference (BI), a species phylogeny was constructed. The newly assembled plastomes genomes showed the quasi-tripartite structure characteristic for Hoya and Dischidia with a reduced small single copy (SSC) and extremely enlarged inverted repeats (IR). The lengths ranged from 175,404 bp in Hoya lacunosa to 179,069 bp in H. ariadna. The large single copy (LSC) regions ranged from 80,795 bp (Hoya liangii) to 92,072 bp (Hoya_sp2_ZCF6006). The massively expanded IR regions were relatively conserved in length, with the small single-copy region reduced to a single gene, ndhF. We identified 235 long dispersed repeats (LDRs) and ten highly divergent hotspots in the 31 Hoya plastomes, which can be used as DNA barcodes for species identification. The phylogeny supports Clemensiella as a distinct genus. Hoya ignorata is resolved as a relative to Clade VI species. This study discloses the advantages of using Plastome genome data to study phylogenetic relationships.
Introduction
Hoya is the second largest genus in Apocynaceae-Asclepiadoideae with at least 300 species, after Ceropegia L. (ca. 357 species) (Bruyns et al., 2017). The genus consists of sub-shrub lianas composed of epiphytic climbers that grow in dense tropical forests of South, Southeast and East Asia, and Australasia (Lamb et al., 2016). The flowers have rotated corollas, staminal coronas with revolute margins, pollinia with pellucid margins, and elongated, slender, and fusiform seeds with hairs attached at their terminal part (Omlor, 1998). Due to their showy flowers, ease of growing, and popularity as ornamental plants, they have been grown in botanical gardens around the world. However, overexploitation from the wild may exist.
Dischidia is a genus closely related to the genus Hoya found in South East Asia. It consists of approximately 80 species (Livshultz et al., 2005). The best-known species is Dischidia major, which has pitcher leaves used as nesting sites by arboreal ants. Unlike Hoya, Dischidia is poorly known and has not been studied closely. Some species have oil-rich structures in the seeds that are attractive to ants and may facilitate seed dispersal (Rintz, 1980).
The first broadly sampled Hoya phylogeny used a few chloroplast loci (matK gene; trnH-psbA, psbA-atpB, trnT-trnL, and trnL-trnF intergenic spacers) and have been heavily informed by nuclear (ITS and 5′ ETS) (Wanntorp et al., 2006, 2014). They erected six main intrageneric lineages (Clade I–VI) comprising many species with mostly congruent plastid and nuclear affinities. Most interclade relationships remained ambiguous even in the study by Rodda et al. (2020a) with more samples. Using first phylogenomic data from complete plastomes (Rodda and Niissalo, 2021) further resolved the interclade and some intra-clade relationships and re-defined Hoya s.str. as comprising only Clade III–VI (Rodda and Niissalo, 2021). Phylogenetic analyses of 42 Hoya plastomes using exons larger than 90 bp erected a new clade Y, which was not captured by Rodda et al. (2020b) demonstrating that any new data may return further insights.
Nevertheless, all the taxonomic and phylogenetic conclusions are inferred from unreliable and dynamic morphological features or DNA fragments with limited polymorphic information loci, which may inevitably bias the phylogenetic reference (Philippe et al., 2011). Additionally, future studies on Hoya will pay more attention to population genetics and the true biogeographic origin. All these studies rely on high-resolution molecular markers and robust phylogeny, but the limited and low-resolution DNA markers heavily inhibited the comprehensive evaluation of Hoya resources. Therefore, it is imperative to develop efficient molecular markers to resolve the current problems.
The plastome, in addition to the nuclear and mitochondrial genomes, is one of the genetic systems that help to understand genetically inherited traits as it exhibits multiple evolutionary histories in angiosperms. Generally, phylogenetic inferences using nuclear genomes are unrealistic for their costly situation and lack enough genomic data (Wang et al., 2014; Olsen et al., 2016). On the other hand, mitochondrial genomes are unsuitable for phylogenetic analysis due to their slow evolutionary rate (Palmer and Herbon, 1988). Plastomes have independent evolutionary routes and are characterized by uniparental inheritance, moderate nucleotide substitutions, haploid status, and no homologous recombination compared to mitochondrion genomes (Shaw et al., 2005; Hansen et al., 2007). In parallel to that, these features of plastomes make them particularly suitable for phylogenetic and biogeographic studies of plants (Huang et al., 2014; Walker et al., 2014; Attigala et al., 2016). With the pileup of angiosperm plastomes, comparative genomics and phylogenomics of closely related plastomes are very useful for grasping the genome evolution regarding structure variations, nucleotide substitutions, and gene losses (Barrett et al., 2016; Raman and Park, 2016; Hu et al., 2017).
The first sequenced complete plastomes of Hoya were as follows: Hoya carnosa (H. carnosa) reported by Wei et al. (2020), Hoya liangii (H. liangii) and Hoya pottsii (H. pottsii) reported by Tan et al. (2018). Additionally, Rodda and Niissalo (2021) reported 20 newly sequenced plastomes of species in the Hoya group. While plastomes usually contain ≈110–130 protein-coding genes, ≈30 transfer RNAs (tRNAs) genes, and ≈4 ribosomal RNAs (rRNA) (Guisinger et al., 2010) organized in the LSC and small single copy (SSC), separated by the IR regions (including all rRNA gene), the Hoya plastomes have lost almost the entire SSC region due to the expansion of the two inverted repeats (IRs) (Wei et al., 2020; Rodda and Niissalo, 2021). Currently, the cp genomes of Hoya are rare and far much less for the clades Clemensiella and Eriostemma.
We sequenced complete plastomes of 31 species of the Hoya group (Dischidia australis, D. griffithii, D. nummularia, D. ruscifolia, Hoya angustifolia (pottsii), H. ariadna, H. caudata, H. chinghungensis, H. commutata, H. dimorpha, H. griffithii, H. kerrii, H. lacunosa, H. lanceolata subs. Bella, H. liangii, H. longifolia, H. meliflua subs. fraterna, H. ovalifolia, H. pandurata, H. pottsii, H. pubicalyx, H. radicalis, H. rigida, H. sylvatica, Hoya sp. 11 ZCF6107, Hoya sp. 2 ZCF6006, Hoya sp. 3 ZCF6076, Hoya sp. 4 ZCF6004, Hoya sp. 8 ZCF6076, H. thomsonii, and H. volubilis) then, conducted comparative genomics and phylogenomics analyses by integrating previously published cp genomes from Tan et al. (2018); Wei et al. (2020), and Rodda and Niissalo (2021). We aim to compare and characterize the cp genomes among selected species of Hoya, identify and select molecular markers suitable for population genetics, reconstruct the species relationships of the six extant clades of the Hoya group. This study provides useful genomic information for molecular evolutionary and phylogenetic studies of the Hoya group and genetic resources for breeding and improving the species.
Materials and Methods
Sampling, DNA Extraction, and Sequencing
A total of 31 Hoya group species (Supplementary Table 1) were collected from the orchards from Xishuangbanna Tropical Garden and South China Botanical Garden, CAS, and their voucher specimens were deposited at the herbarium of Wuhan Botanical Garden, CAS (HIB). Total genomic DNA was extracted from silica-dried leaves using a modified cetyl trimethylammonium bromide (CTAB) protocol (Li J. et al., 2013), and quality was assessed by agarose gel electrophoresis. Total DNA was sent to Novogene Company (Beijing, China)1 for short insert (350 bp) library construction and next-generation sequencing. Pair end reads of 2 × 150 bp for all tested species were generated on an Illumina Hiseq 4,000 genome analyzer platform. Original reads were filtered using the FASTX-Toolkit2 to acquire high-quality data by deleting adaptors and low-quality reads.
Chloroplast Genome Assembly, Annotation, and Comparison
Filtered high-quality reads were assembled into complete plastomes using GetOrganelle v1.7.5 (Jin et al., 2020) with the following settings: word size set to (w -6), number of rounds to 10 (R -10). Finally, the complete paths were viewed in Bandage 0.8.1 (Wick et al., 2015).
The resulting complete circular plastomes were annotated by Plastid Genome Annotator (PGA) (Qu et al., 2019) and GeSeq (Tillich et al., 2017). The resulting sequences were manually checked in Geneious 8.0.4 (Kearse et al., 2012) using reference plastid genome H. carnosa (Wei et al., 2020) to avoid annotation errors. In addition, tRNAs were further verified using the tRNAScan-SE search server (Schattner et al., 2005). All newly assembled chloroplast genomes were deposited in GenBank (accession numbers are shown in Supplementary File 1). The circular genome map with structural features was generated using OGDRAW (Greiner et al., 2019).
Genome Comparison
Out of 31 sequenced plastomes, 20 species were chosen to represent each clade for genome comparison using H. carnosa as the reference sequence. The plastomes were grouped into nine representatives for each clade except Dischidia clade then aligned in progressive mauve (Darling et al., 2010) implemented in mauve v.2.4.0 (Darling et al., 2004) to detect any form of rearrangement in Hoya. The IR expansion and contraction among 20 chloroplast genomes were visualized by the online program IRscope (Amiryousefi et al., 2018).
Characterization of Repetitive Sequences
Simple sequence repeats (SSRs) across the 31 plastomes were extracted using the online web tool MISA3 (Beier et al., 2017) with the following parameters: ten repetitions for mononucleotide motifs, eight for dinucleotide motifs, and three for Penta and hexanucleotide motifs. Identification of the long dispersed repeats (LDRs): forward (F), palindromic (P), reverse (R), and complement (C) repeats analysis was done using the REPuter program4, with a minimum repeat size of 30 bp and a Hamming distance of 3 (Kurtz et al., 2001). Nucleotide diversity (Pi) was calculated by sliding window analysis conducted in DnaSP v.6.11.01 (Librado and Rozas, 2009), using a window length of 600 bp and a step size of 200 bp.
Phylogenetic Analysis
The phylogenetic tree was constructed based on 72 protein-coding genes of 45,624 characters common among all the 55 Hoya group species and the two outgroup species; Jasminanthes maingayi and Marsdenia flavescens (Supplementary File 1). The nucleotide sequences were aligned using MAFFT v.7.2.2 (Katoh and Standley, 2013). Each alignment sequence was first trimmed using TrimAI v.1.2 (Capella-Gutiérrez et al., 2009) with default settings to reduce poorly aligned regions. The resulting trimmed alignments were then filtered with Gblocks (Talavera and Castresana, 2007) to clean the sequences from poorly aligned positions and too divergent regions. The final alignment for all datasets was concatenated in Phylo Suite v.1.2.1 (Zhang et al., 2020). Using Bayesian Information Criterion (BIC), the best-fit models for the phylogenetic analysis were GTR, GTR + G, and GTR + I + G, under settings (R cluster) for the concatenated alignment as implemented in ModelFinder. This study employed two different phylogenetic algorithms/optimality criteria: maximum likelihood (ML) and Bayesian Inference (BI). The ML tree was constructed using IQ-tree (Nguyen et al., 2015) implemented in Phylosuite with the best-fit models determined by ModelFinder and 1,000 replicates for ultrafast bootstrapping (Minh et al., 2013). BIs were performed by MrBayes v.3.2.7 (Ronquist et al., 2012) under the GTR + G model with four chains and two parallel runs. The Monte Carlo Markov chains (MCMCs) were run for 10 million generations and sampled at a frequency of every 1,000 generations. The first 25% of the trees were discarded as burn-in, and the remaining trees were used to build a majority-rule consensus tree and establish posterior probability values for each branch. The stationarity was considered to be met since the average SD of split frequencies remained below 0.115631. The final phylogenetic results were visualized with FigTree v.1.4.4 (Rambaut, 2018).
Detection of Selection Pressure
We applied the site model method implemented in CodeML (Gao et al., 2019) to detect positively selected sites in Hoya group species. Our selection analysis was based on 72 protein coding-region sequences after all stop codons were removed. The positive selection models (M2a and M8) and their respective null models (M1a and M7) implemented in the site model were used to conduct the adaptive evolution analysis. Likelihood ratio tests (LRTs) were performed two times to compare the difference in the log-likelihoods between the nested codon-based models (Yang, 1998). The Bayes Empirical Bayes (BEB) method was used to identify the most likely codons under positive selection (Yang et al., 2005).
Results
Characterization of the Chloroplast Genomes
Approximately 173.84 GB of paired-end quality reads were obtained from Illumina sequencing for the 31 Hoya group species from China (Table 1). All plastomes showed the quasi-tripartite structure characteristic for Dischidia and Hoya with a strongly reduced SSC and significantly enlarged IR regions (Figures 1A,B). Their lengths varied in sizes ranging from 175,404 bp in Hoya lacunosa to 179,069 bp in H. ariadna, mainly because of length variation in LSC (80,795 bp in H. liangii to 92,072 bp in Hoya sp2 ZCF6006). The SSC region only includes a single gene (ndhF), ranging from 2,265 in Hoya pandurata to 2,306 in Hoya caudata. Slight variation characterizes the sizes of their IR regions and overall guanine-cytosine (GC) contents (Table 1).
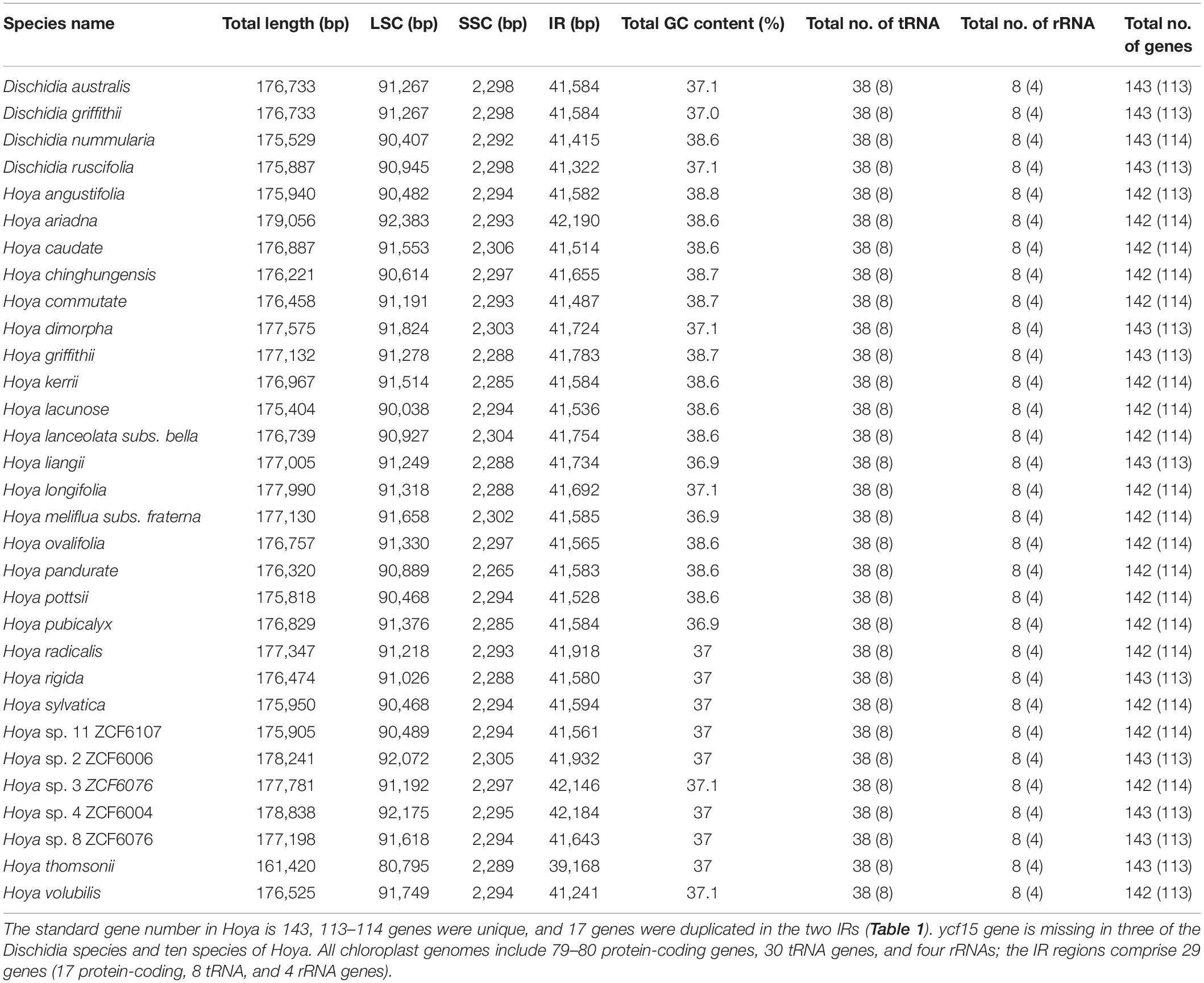
Table 1. Summary of 31 Hoya plastome features that were assembled and annotated.
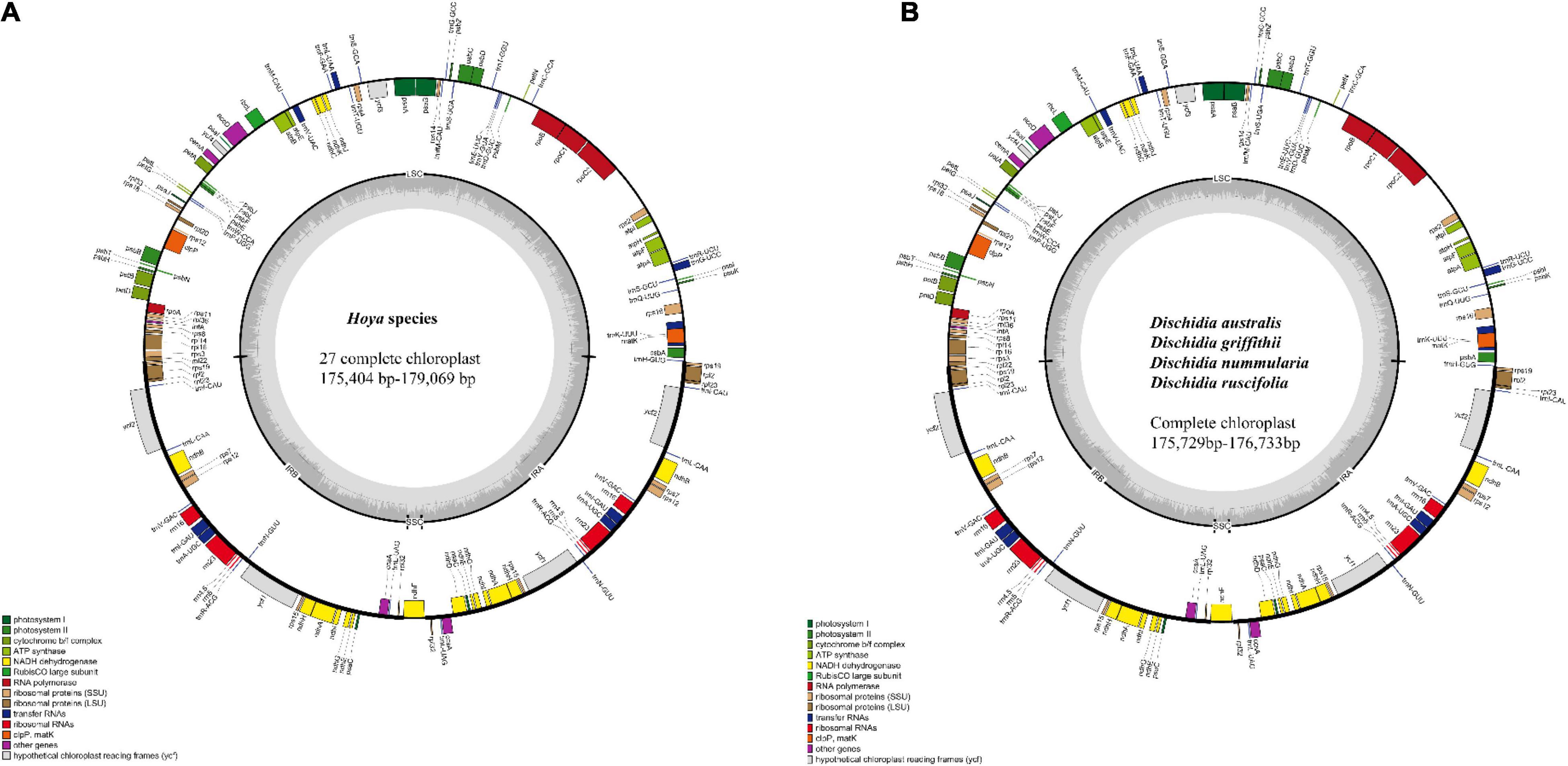
Figure 1. The genome maps of (A) 27 Hoya species; (B) 4 Dischidia species. The genes inside the circle are transcribed in the clockwise direction and those outside in the anticlockwise direction. The different colors represent the genes of different functional groups. The thick lines denote the extent of IRa and IRb, which separates the chloroplast genome into LSC and SSC. LSC: large single copy; SSC: small single copy. Plastome structure variation.
Plastome Structure Variation
All the Hoya plastomes showed the same order and orientation of syntenic blocks (Figure 2), indicating that Hoya plastomes are highly conserved and collinear. Nevertheless, a few local changes representing variable regions were detected, with several evident inversions mainly located in SC regions, especially within the nucleotide of 125,000–145,000 bp.

Figure 2. Rearrangements in 7 Hoya plastomes using the mauve multiple alignment algorithm. Different colors represent different collinear blocks. The lines linking the collinear blocks represent homology between different genomes. The scale above each genome indicates nucleotide positions, and the white regions represent elements specific to a genome. IR contraction and expansion.
Inverted Repeats Contraction and Expansion
The LSC/IRb boundary was consistently located downstream of the rpl22 gene within the 3′ part of the rpl22 gene (Figure 6). The IRb/SSC SSC/IRa junctions were located 35–40 bp upstream and downstream of the ndhF gene (Figure 6). The IRa/LSC junction fell within the rps19-trnH (GUG) spacer. IR contraction and expansion in the Hoya plastomes ultimately lead to the length variations of the four structural segments and whole-genome sequences.
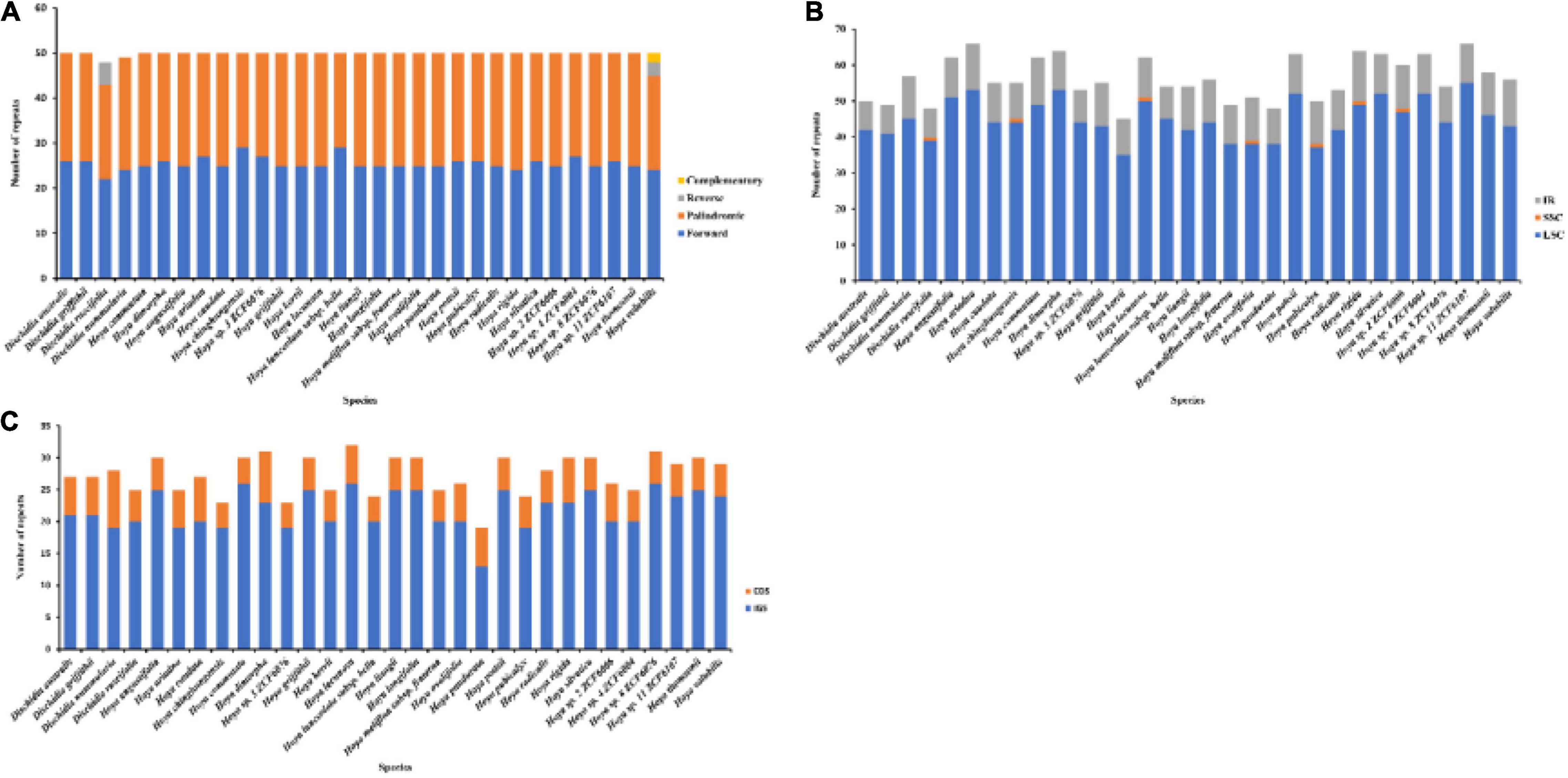
Figure 3. Analyses of repeat sequences and SSRs in 31 Hoya group plastomes. (A) Frequency of the four repeat types. (B) Frequency of SSRs in LSC, SSC, and IR. (C) Frequency of SSRs in IGS and CDS. LSC, large single copy; SSC, small single copy; SSR, simple sequence repeats.
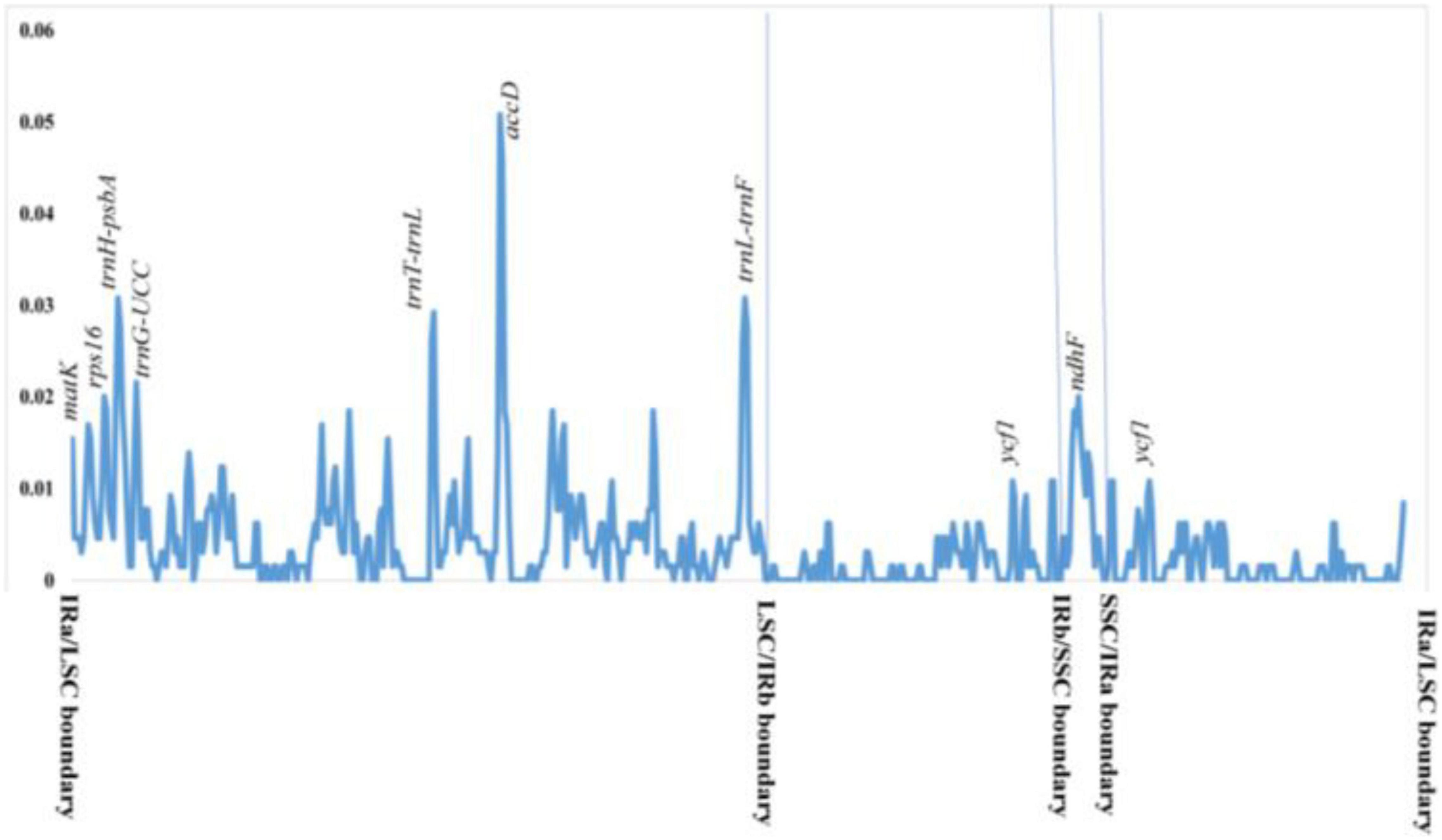
Figure 4. Nucleotide diversity (Pi) in 31 Hoya group complete plastomes. The values represent different diversity for different genes and regions.
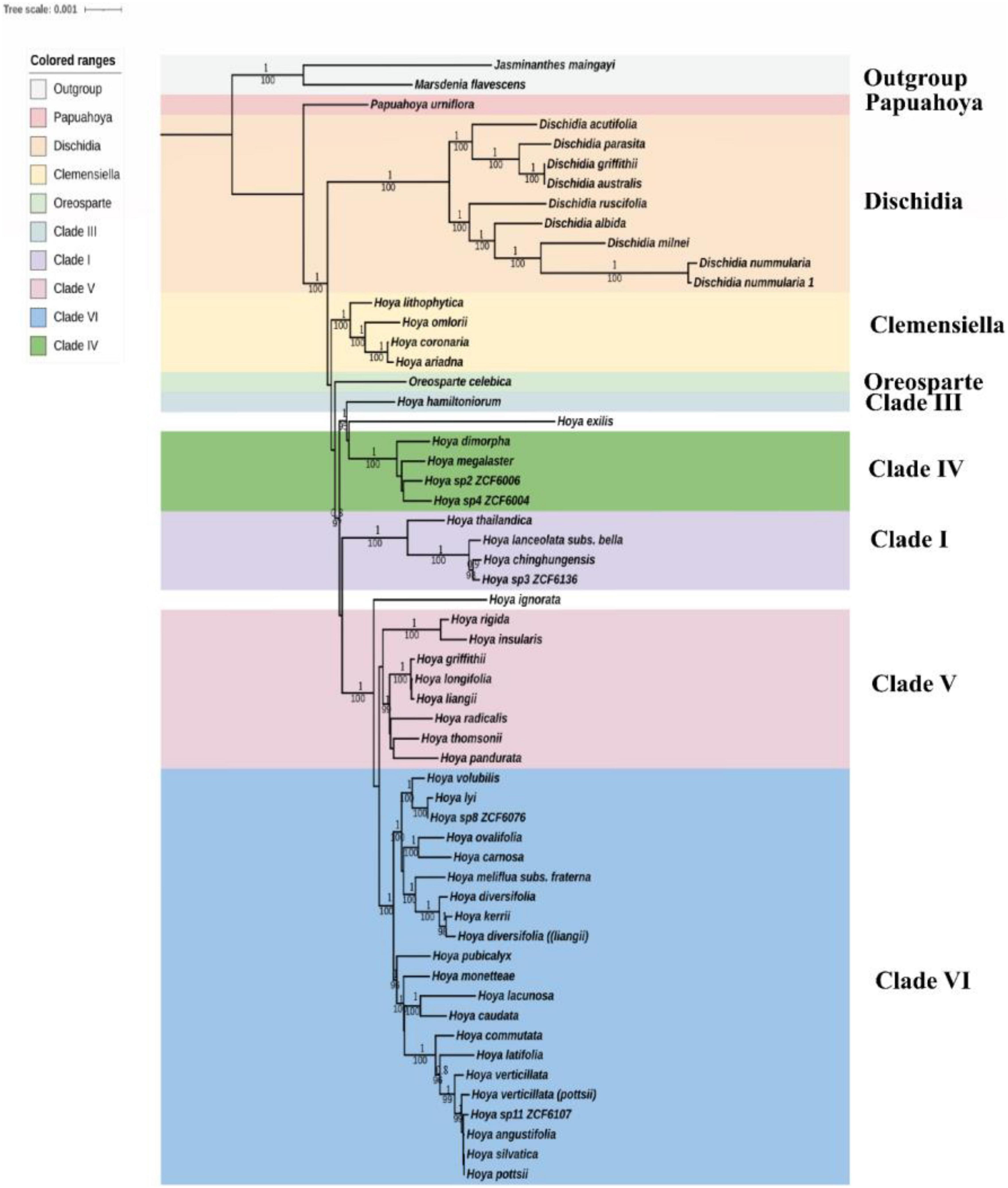
Figure 5. Phylogenetic tree of Hoya inferred from 57 species using ML. CDS tree reconstruction is rooted with Marsdenia and Jasminanthes as outgroups. Bootstrap support values and Bayesian inferred posterior probabilities are given below and above the branches, respectively. Different colors represent different clades following Wanntorp et al. (2014) and Rodda and Niissalo (2021).

Figure 6. Junction sites comparison of LSC, SSC, and IR for 20 Hoya group plastomes. JLB, junction line between LSC and IRb; JSB, junction line between IRb and SSC; JSA, junction line between SSC and IRa; JLA, junction line between IRa and LSC; LSC, large single copy; SSC, small single copy.
Repeat Analysis
We identified a total number of 235 SSR motifs in our set of 31 Hoya group plastomes (four Dischidia, 27 Hoya). Mono-nucleotide repeats were more prevalent and most of them belonged to the A/T type (163 repeats) followed by AAG/CTT (60 repeats). Penta and hexanucleotides were rare (5 repeats each); the rarest were A/G (2 repeats). Additionally, four types of LDRs (forward, palindromic, reverse, and complement), each with a motif length longer than 30 bp, were detected (Figure 3 and Supplementary Table 1). Finally, a total of 1,446 repeats, 709 forward (F) and 725 palindromic (P), were detected across all chloroplast genomes with eight reverse (R) and four complementary (C) in the plastomes of Dischidia ruscifolia and Hoya volubilis, respectively.
Nucleotide diversity (Pi) was calculated by sliding window analysis to observe sequence divergence and determine highly divergent hotspots. Both single-copy regions were identified as having greater sequence divergence than the IR region (Figure 4). With a Pi-value cut-off point of 0.02, eight highly variable gene regions were identified: intergenic spacers (trnT-trnL-trnF, psba-trnH, and trnG-UCC), ndhF, ycf1, matK, rps16, and accD genes. Six of the highly variable regions were located in the LSC, while one was in the SSC and IR regions.
The Synonymous and Non-synonymous Substitution Rate Analysis
Using Jasminanthes maingayi as the outgroup, we computed and compared the dN/dS of H. ariadna, H. caudata, H. chinghungensis, H. pubicalyx, H. longifolia, H. thomsonii, and H. rigida. According to the statistical neutrality test, 10 genes in the seven plastomes of selected Hoya species were under positive selection. The genes were majorly involved in adenosine triphosphate (ATP) synthesis (atpA, atpB, atpE, and petL), RNA processing (matK), NADH dehydrogenase (ndhA, ndhB, and ndhD), and other genes (accD and clpP) (Table 2). According to the M8 model, atpA harbored four sites under positive selection, followed by atpB, which had three sites. However, the LRT indicated that the models (M2a and M8) were significantly better than the control models (M1a and M7), proving the presence of codons under positive selection. Further analysis from BEB scores indicated an intense positive selection pressure on 15 codons (Table 2).
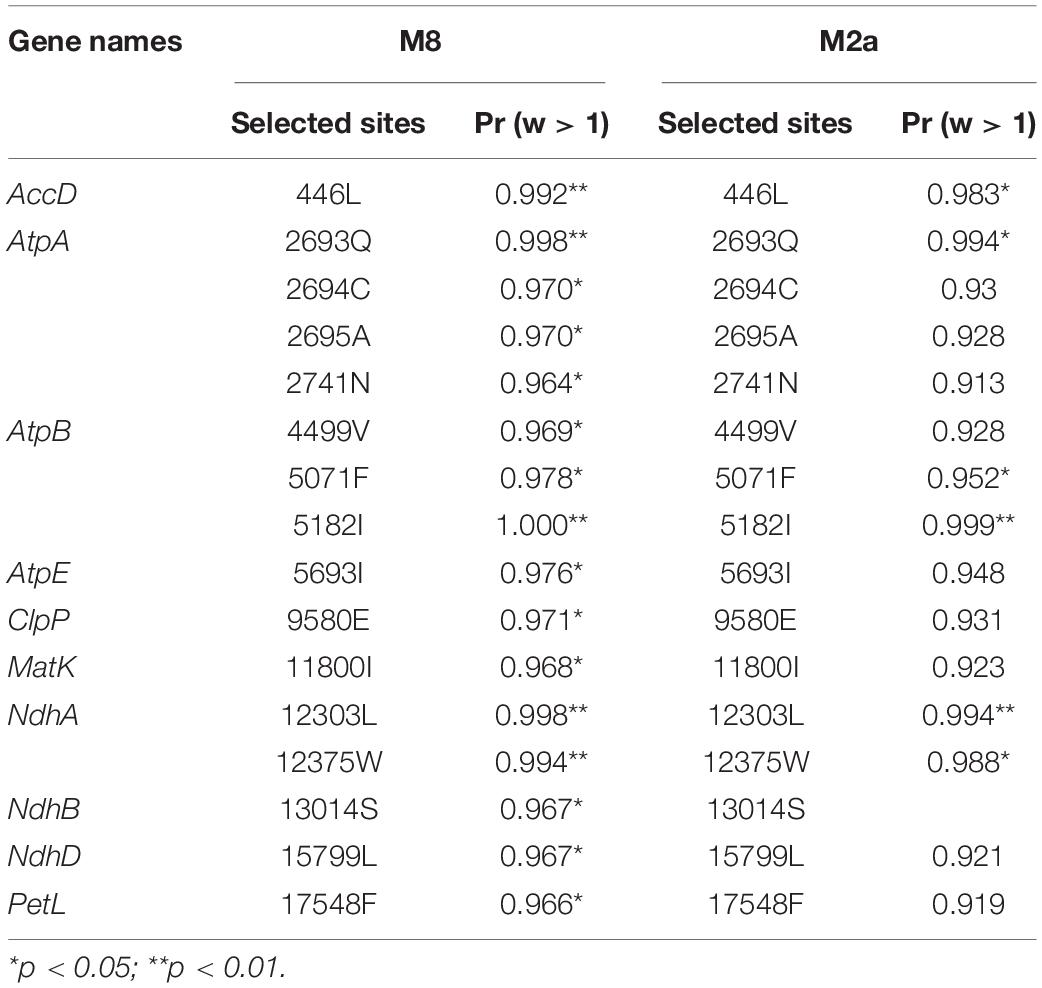
Table 2. Positive selected sites detected in the cp genomes of Hoya.
Phylogenetic Analysis
The phylogenetic tree was constructed based on 72 protein-coding genes common to all currently available Hoya group species (Supplementary File 1), i.e., two more representatives of Marsdenieae as outgroups to root the trees (Figure 5). The multiple sequence alignment comprised of protein-coding sequences with 45,624 characters and 2,332 variable sites. To compare the clades, we referred to Wanntorp et al. (2014), Rodda et al. (2020a), and Rodda and Niissalo (2021). The 55 in-group taxa formed nine distinct clades (Supplementary Figure 1), and both ML and BI yielded similar topologies (Figure 5). Dischidia is the second early diverged clade after Papuahoya, and it comprises nine species. Dischidia griffithii and D. australis form a clade with D. parasita, a species native to the Philippines. Hoya ariadna (a species belonging to the section Eriostemma) formed a stable sister relationship with species from clade II/Clemensiella [100% (Bootstrap support), 1 (Bayesian posterior probability)]. Within clade IV, there are two unidentified species (Hoya sp. 2 ZCF6006 and Hoya sp. 4 ZCF6004) that form a well-supported relationship to Hoya megalaster from section Physostelma (100% BS, 1 BPP). Clade V comprises eight montane subtropical species whose relationships are well supported (99% BS, 1 BPP). Clade VI is the most recently diverged and widespread in Hoya sensu lato. Furthermore, unidentified species (Hoya sp. 8 ZCF6076 and Hoya sp. 11 ZCF6107) are strongly supported to belong to clade VI (99–100 BS, 0.8–1 BPP), a lineage morphologically characterized by flowers with dark-colored nectar. Hoya sp. 3 ZCF6076 from Yunnan, China and H. chinghungensis are part of Clade I (BS = 100, PP = 1) and are closely related to H. lanceolata subsp. bella. Overall, the emended taxon set recovers the same intrageneric, interclade relationships as found by Rodda and Niissalo (2021).
Discussion
Chloroplast Genome Variation
The overall plastome sequences in the 31 Hoya group examined were highly conserved, and they did not exhibit the standard quadripartite structure similar to the other angiosperm plastomes. Their SSC was massively reduced with only one gene (ndhF) present and their IR regions much more prominent than other higher plants. This could be due to the complexity arising in assembling Hoya group plastomes, as reported by Crook (2017). Furthermore, the ndhF gene has been reported to be notoriously difficult to assemble, as they move between the IRb and SSC, sometimes straddling both regions (Davis and Soreng, 2010). Consequently, our data were manually adjusted using Geneious predictions to correct the poorly assembled plastomes. The exact number and contents of the genes were predicted in this study, suggesting that the evolution of the gene sequences was consistent across the 31 species. As a result, Hoya group species’ plastomes contain a total of 113–114 unique genes, such as 79–80 protein-coding genes, 30 tRNAs, and four rRNAs. The expansion and contraction of the IR is the main reason for variation in genomic size; rearrangements, such as inversion of genes and SSC, are common in plastome genomes (Liu et al., 2018). Additionally, it has been reported to have occurred in Marsdenieae and other Apocynaceae plastomes (Straub et al., 2011). Comparably, the newly sequenced Hoya (Figure 2) had similar rearrangements.
Plastome Structure Variation
The sequence divergence of IR regions was lower compared to LSC and SSC, with the accD gene being the most divergent (Figure 4). This was caused by IR having very few protein-coding genes, short IGS, and mostly tRNAs and rRNA, which are more conserved than exons and introns of the protein-coding genes. Similarly, Straub et al. (2011) reported this on Asclepias syriaca but Rodda and Niissalo (2021) did not mention it in the recent Hoya plastomes. Intergenic regions, especially the rpl32-trnL, have been used for phylogenetic and evolutionary studies at the species level (Dong et al., 2012; Zecca et al., 2012; Jara-Arancio et al., 2018) due to its high nucleotide diversity, making it a mutationally active region. On the contrary, we discovered that the mutationally active plastome region in Hoya is the 3′ region of ndhF locus, accD, and matK genes, and the intergenic spacers (trnT-trnL, trnH-psbA, and ycf1) are the lowest (Figure 4). Similar to most land plants, the ycf1 is the largest open reading frame (ORFs). It is located at the boundary of the IR and SSC, its diversity and length make it a better candidate for phylogenetic studies than other genes (Neubig et al., 2009). While still being one of the most variable regions, in the Hoya group, the ycf1 is outcompeted by the only remaining SSC gene, the ndhF gene.
Simple Sequence Repeats
Both LDRs and SSRs are useful genetic markers due to their abundance in chloroplast genomes, high degree of polymorphism, and co-dominance (Ivanovych and Volkov, 2018). In a previous study, Straub et al. (2011) focused only on the repeat sequences based on the nuclear genome. Similar to most angiosperms, sequence repeats for A/T were more prevalent than those of G/C in the Hoya group plastomes. This may represent bias in the base composition, which is potentially affected by the tendency of the genome to change to A-T rather than to G-C (Li X. et al., 2013). Notably, these microsatellites are likely to have originated from multiple paralogous loci of the Hoya group plastomes, as is the case with all the microsatellites. This is the ultimate proof of the utility of these markers for identifying intraspecific variation. There was variation in the distribution of LDRs and SSRs in non-coding intergeneric spacers (IGS) vs. coding region (CDS), with repeats being concentrated in IGS, in line with previous studies (e.g., Shen et al., 2018; Meng et al., 2019)
Positive Selection
Testing synonymous and non-synonymous nucleotide substitution is vital in gene evolution studies (Drouin et al., 2008). Accordingly, the ratio ω = dN/dS has become a standard measure of selective pressure. Our study is the first to report on the selection pressures acting on protein-coding genes of Hoya s.l. From our findings, instances of multiple positive selections in different genes are involved in various functions, such as ATP synthesis (atpA, atpB, atpE, and petL), RNA processing (matK), NADH dehydrogenase (ndhA, ndhB, and ndhD), and other gene functions (accD and clpP). A total of 15 codons were detected to be under positive selection with high confidence levels (posterior probability > 0.95; Table 2).
Phylogeny
Our plastome matrix of 72 protein-coding genes and 57 species based on ML and Bayesian analyses (Figure 5) represented the most extensive sampling of the protein-coding genes to date and was mainly in congruence with the previous results (Wanntorp et al., 2006, 2014; Rodda et al., 2020a; Rodda and Niissalo, 2021). The seven major clades established by Wanntorp et al. (2014) are well supported in our study (BS = 98–100, PP = 1.00). This reveals strong support for the relationships of these enigmatic species (H. griffithii, H. Kerrii H. meliflua subs. fraterna, H. ovalifolia, and H. thomsonii) sampled for the first time. These species were ambiguous taxa in the previous study done by Wanntorp et al. (2014) and their relationships could not be resolved using plastid loci trnT-trnL, trnH-psbA, and nuclear datasets (ITS and ITS). Moreover, Rodda and Niissalo (2021) did not resolve the relationships possibly due to sampling problems. Our analysis further confirms previous studies (Rodda et al., 2020a; Rodda and Niissalo, 2021), which recognized the monophyletic genera Dischidia and Oreosparte (BS = 100; PP = 1.0), and placed Clade II (Clemensiella) outside Hoya s.str. Dischidia remains the sister clade to Hoya s.l., i.e., Oreosparte. Phylogenetic relationships within the Hoya group on a large scale have been ambiguous mainly due to poor infrageneric resolution since there is no infrageneric system established up to date (Rodda et al., 2020b). For example, Hoya caudata is resolved as the sister of H. lacunosa (BS = 100, PP = 1.0), yet taxonomically and morphologically distinct. Hoya caudata, a widespread species distributed in S. Asia, Malesia, and Australasia, belongs to section Peltostemma, while H. lacunosa from S.W China, an oddball in Wanntorp et al. (2014) with conflicting nuclear and plastid affinities, is sect. Otostemma. Species of Hoya sect. Otostemma has revolute lobes, rotate corolla with boat-shaped corona segments. Their anthers are incumbent on the stigma, with the apex simple, acute, and pollinia attached at the base, close together, and linearly compressed (Kloppenburg, 2001). However, the Hoya sect. Peltostemma is distinguished through the inclined corona scales and long extended anther appendages. In addition, the stigma head is hollow on the point and slow to open. Clade II (sect. Clemensiella) on the other hand comprising H. omlorii, H. coronaria, H. ariadna, and H. lithophytica is sharply different from the rest of the Hoya group clades. Their branches are fleshy, their retinaculum is rather large, and the pollinia are more club shaped and moreover do not have a keel on the outer edge. Furthermore, Rodda and Niissalo (2021) suggested that it should be treated as a sub-genus of Hoya, despite its phylogenetic placement outside Hoya s.str. Our findings, such as the enigmatic species Hoya ariadna, confirm Rodda and Niissalo (2021) topology, hence, recognizing Clemensiella as a distinct genus, but the support of the critical branch remains ambiguous (BS = 76, PP = 0.7). In addition, the number of species belonging to Clemensiella is still small but with a wider search into the Malay Peninsula and the Sunda Islands, more species could be added. Species, such as H. purpurea Blume and H. neoguineensis Engler from New Guinea, H. guppyi Oliv., and H. affinis Hemsl. from the Solomon Islands, belong to this section/clade and they are particularly found in the forest edges and along streams (Kloppenburg, 2001; Wanntorp et al., 2014).
Conclusion
Through the use of next-generation sequencing (NGS), 31 new plastomes of the Hoya group species were assembled and analyzed and used to complement existing data sets. The gene content, gene order, and GC contents were conserved in Hoya s.l. genomes, which share a unique chloroplast quasi-tripartite genome structure with Dischidia. Highly divergent regions (ndhF, ycf1, rpl22, matK, trnT-trnL, and trnL-trnF) and repeats (-mono, -tri, -Penta, and -hexanucleotides) that could potentially serve as molecular markers for phylogenetics were identified. All the phylogenetic analyses using the species of the Hoya group strongly supported the relationships among the species within the genus. The results and data presented in this study provide insights into the evolutionary relationships and biogeographic history of the Hoya group species. More detailed taxon sampling will further contribute to our understanding of phylogenetic dynamics in the Hoya group lineages.
Data Availability Statement
The data presented in this study can be found in the GenBank repository. The accession number can be found in the Supplementary Material.
Author Contributions
WO, C-FZ, G-WH, and Q-FW participated in the design of the study and carried out the experiments. YW, C-FZ, and G-WH collected the materials. WO, EW, CN, ESM, VW, EMM, and MO contributed to data analysis and draft manuscript writing. WO, C-FZ, G-WH, and VW revised the draft manuscript. All authors read and approved the final version of the manuscript.
Funding
The study was funded by the Biological Resources Program, Chinese Academy of Sciences (KFJ-BRP-017-10), China National Plant Specimen Resource Center (E0117G1001), International Partnership Program of Chinese Academy of Sciences (151853KYSB20190027), and Sino-Africa Joint Research Center, CAS (SAJC202101).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the Department of Gardening and Horticulture, Xishuangbannan Tropical Botanical Garden, Chinese Academy of Sciences (CAS), and South China Botanical Garden, CAS for providing samples; Haibo Mo, Jingfeng Zhang, and Hui Jiang for their kind help.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.814833/full#supplementary-material
Footnotes
- ^ http://www.novogene.com
- ^ http://hannonlab.cshl.edu/fastx_toolkit
- ^ http://pgrc.ipk-gatersleben.de/misa/
- ^ https://bibiserv.cebitec.uni-bielefeld.de/reputer
References
Amiryousefi, A., Hyvönen, J., and Poczai, P. (2018). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220
Attigala, L., Wysocki, W. P., Duvall, M. R., and Clark, L. G. (2016). Phylogenetic estimation and morphological evolution of Arundinarieae (Bambusoideae: Poaceae) based on plastome phylogenomic analysis. Mol. Phylogenet. Evol. 101, 111–121. doi: 10.1016/j.ympev.2016.05.008
Barrett, C. F., Baker, W. J., Comer, J. R., Conran, J. G., Lahmeyer, S. C., Leebens-Mack, J. H., et al. (2016). Plastid genomes reveal support for deep phylogenetic relationships and extensive rate variation among palms and other commelinid monocots. New Phytol. 209, 855–870. doi: 10.1111/nph.13617
Beier, S., Thiel, T., Münch, T., Scholz, U., and Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Bruyns, P. V., Klak, C., and Hanáèek, P. (2017). A revised, phylogenetically-based concept of Ceropegia (Apocynaceae). S. Afr. J. Bot. 112, 399–436. doi: 10.1016/j.sajb.2017.06.021
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Crook, T. (2017). Plastome Assembly in the Wax Plants (Hoya) and Near Relatives (Marsdenieae, Apocynaceae). Philadelphia: University of Pennsylvania.
Darling, A. C. E., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
Davis, J. I., and Soreng, R. J. (2010). Migration of endpoints of two genes relative to boundaries between regions of the plastid genome in the grass family (Poaceae). Am. J. Bot. 97, 874–892. doi: 10.3732/ajb.0900228
Dong, W., Liu, J., Yu, J., Wang, L., and Zhou, S. (2012). Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS One 7:e35071. doi: 10.1371/journal.pone.0035071
Drouin, G., Daoud, H., and Xia, J. (2008). Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol. Phylogene. Evol. 49, 827–831. doi: 10.1016/j.ympev.2008.09.009
Gao, F., Chen, C., Arab, D. A., Du, Z., He, Y., and Ho, S. Y. W. (2019). EasyCodeML: a visual tool for analysis of selection using CodeML. Ecol. Evol. 9, 3891–3898. doi: 10.1002/ece3.5015
Greiner, S., Lehwark, P., and Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3. 1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64. doi: 10.1093/nar/gkz238
Guisinger, M. M., Chumley, T. W., Kuehl, J. V., Boore, J. L., and Jansen, R. K. (2010). Implications of the plastid genome sequence of Typha (Typhaceae. J. Mol. Evol. 70, 149–166. doi: 10.1007/s00239-009-9317-3
Hansen, A. K., Escobar, L. K., Gilbert, L. E., and Jansen, R. K. (2007). Paternal, maternal, and biparental inheritance of the chloroplast genome in Passiflora (Passifloraceae): implications for phylogenetic studies. Am. J. Bot. 94, 42–46. doi: 10.3732/ajb.94.1.42
Hu, Y., Woeste, K. E., and Zhao, P. (2017). Completion of the chloroplast genomes of five Chinese Juglans and their contribution to chloroplast phylogeny. Front. Plant Sci. 7:1955. doi: 10.3389/fpls.2016.01955
Huang, H., Shi, C., Liu, Y., Mao, S.-Y., and Gao, L.-Z. (2014). Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol. Biol. 14:151. doi: 10.1186/1471-2148-14-151
Ivanovych, Y., and Volkov, R. (2018). Genetic relatedness of sweet cherry (Prunus avium L.) cultivars from Ukraine determined by microsatellite markers. J. Hortic. Sci. Biotechnol. 93, 64–72.
Jara-Arancio, P., Vidal, P. M., and Arroyo, M. T. K. (2018). Phylogenetic reconstruction of the genus Triptilion (Asteraceae, Nassauvieae) based on nuclear and chloroplast DNA sequences. J. Syst. Evol. 56, 120–128. doi: 10.1111/jse.12294
Jin, J.-J., Yu, W.-B., Yang, J.-B., Song, Y., DePamphilis, C. W., Yi, T.-S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 1–31. doi: 10.1186/s13059-020-02154-5
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kloppenburg, D. (2001). Hoya Sections : a Complete Study With Modifications and Addition. Fresno, CA: D. Kloppenburg.
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Lamb, A., Rodda, M., Gokulsing, L., Bosuang, S., and Rahayu, S. (2016). A Guide to Hoyas of Borneo. Borneo: Natural History Publications.
Li, J., Wang, S., Yu, J., Wang, L., and Zhou, S. (2013). A modified CTAB protocol for plant DNA extraction. Chinese Bull. Bot. 48:72. doi: 10.3724/sp.j.1259.2013.00072
Li, X., Gao, H., Wang, Y., Song, J., Henry, R., Wu, H., et al. (2013). Complete chloroplast genome sequence of Magnolia grandiflora and comparative analysis with related species. Sci. China Life Sci. 56, 189–198. doi: 10.1007/s11427-012-4430-8
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Liu, H., Ding, C., He, J., Cheng, J., Pei, L., and Xie, L. (2018). Complete chloroplast genomes of Archiclematis, Naravelia and Clematis (Ranunculaceae), and their phylogenetic implications. Phytotaxa. 343, 214–226. doi: 10.11646/phytotaxa.343.3.2
Livshultz, T., Tran, T. B., Bounphanmy, S., and Schott, D. (2005). Dischidia (Apocynaceae, Asclepiadoideae) in Laos and Vietnam. Blumea J. Plant Taxon. Plant Geogr. 50, 113–134. doi: 10.3767/000651905X623300
Meng, D., Xiaomei, Z., Wenzhen, K., and Xu, Z. (2019). Detecting useful genetic markers and reconstructing the phylogeny of an important medicinal resource plant, Artemisia selengensis, based on chloroplast genomics. PLoS One 14:e0211340. doi: 10.1371/journal.pone.0211340
Minh, B. Q., Nguyen, M. A. T., and von Haeseler, A. (2013). Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195. doi: 10.1093/molbev/mst024
Neubig, K. M., Whitten, W. M., Carlsward, B. S., Blanco, M. A., Endara, L., Williams, N. H., et al. (2009). Phylogenetic utility of ycf 1 in orchids: a plastid gene more variable than mat K. Plant Syst. Evol. 277, 75–84. doi: 10.1007/s00606-008-0105-0
Nguyen, L.-T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Olsen, J. L., Rouzé, P., Verhelst, B., Lin, Y.-C., Bayer, T., Collen, J., et al. (2016). The genome of the seagrass Zostera marina reveals angiosperm adaptation to the sea. Nature 530, 331–335. doi: 10.1038/nature16548
Omlor, R. (1998). Generische Revision der’Marsdenieae’(‘Asclepiadaceae’). Herzogenrath: Shaker Verlaga.
Palmer, J. D., and Herbon, L. A. (1988). Plant mitochondrial DNA evolved rapidly in structure, but slowly in sequence. J. Mol. Evol. 28, 87–97. doi: 10.1007/bf02143500
Philippe, H., Brinkmann, H., Lavrov, D. V., Littlewood, D. T. J., Manuel, M., Wörheide, G., et al. (2011). Resolving difficult phylogenetic questions: why more sequences are not enough. PLoS Biol. 9:e1000602. doi: 10.1371/journal.pbio.1000602
Qu, X.-J., Moore, M. J., Li, D.-Z., and Yi, T.-S. (2019). PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 15, 1–12. doi: 10.1186/s13007-019-0435-7
Raman, G., and Park, S. (2016). The complete chloroplast genome sequence of Ampelopsis: gene organization, comparative analysis, and phylogenetic relationships to other angiosperms. Front. Plant Sci. 7:341. doi: 10.3389/fpls.2016.00341
Rintz, R. E. (1980). The peninsular Malayan species of Dischidia (Asclepiadaceae). Blumea Biodivers. Evol. Biogeogr. Plants 26, 81–126.
Rodda, M., and Niissalo, M. A. (2021). Plastome evolution and organisation in the Hoya group (Apocynaceae). Sci. Rep. 11, 1–13. doi: 10.1038/s41598-021-93890-6
Rodda, M., Simonsson, N., Ercole, E., Khew, G., Niissalo, M., Rahayu, S., et al. (2020a). Phylogenetic studies in the Hoya group (Apocynaceae, Marsdenieae): the position of Anatropanthus and Oreosparte. Willdenowia 50:119. doi: 10.3372/wi.50.50112
Rodda, M., Simonsson, N., Ercole, E., Khew, G., Niissalo, M., Rahayu, S., et al. (2020b). Phylogenetic studies in the Hoya group (Apocynaceae, Marsdenieae): the position of Anatropanthus and Oreosparte. Willdenowia 50, 119–138.
Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi: 10.1093/nar/gki366
Shaw, J., Lickey, E. B., Beck, J. T., Farmer, S. B., Liu, W., Miller, J., et al. (2005). The tortoise and the hare II: relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 92, 142–166. doi: 10.3732/ajb.92.1.142
Shen, X., Guo, S., Yin, Y., Zhang, J., Yin, X., Liang, C., et al. (2018). Complete chloroplast genome sequence and phylogenetic analysis of Aster tataricus. Molecules 23:2426. doi: 10.3390/molecules23102426
Straub, S. C. K., Fishbein, M., Livshultz, T., Foster, Z., Parks, M., Weitemier, K., et al. (2011). Building a model: developing genomic resources for common milkweed (Asclepias syriaca) with low coverage genome sequencing. BMC Genom. 12:211. doi: 10.1186/1471-2164-12-211
Talavera, G., and Castresana, J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Sys. Biol. 56, 564–577. doi: 10.1080/10635150701472164
Tan, X.-H., Wang, J.-H., Zhao, K.-K., Zhu, Z.-X., and Wang, H.-F. (2018). Complete plastome sequence of Hoya pottsii Traill and Hoya liangii Tsiang (Apocynaceae). Mitochondrial DNA Part B 3, 1176–1177. doi: 10.1080/23802359.2018.1524720
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Walker, J. F., Zanis, M. J., and Emery, N. C. (2014). Comparative analysis of complete chloroplast genome sequence and inversion variation in Lasthenia burkei (Madieae, Asteraceae). Am. J. Bot. 101, 722–729. doi: 10.3732/ajb.1400049
Wang, W., Haberer, G., Gundlach, H., Gläßer, C., Nussbaumer, T., Luo, M. C., et al. (2014). The Spirodela polyrhiza genome reveals insights into its neotenous reduction fast growth and aquatic lifestyle. Nat. Comm. 5, 1–13. doi: 10.1038/ncomms4311
Wanntorp, L., Grudinski, M., Forster, P. I., Muellner-Riehl, A. N., and Grimm, G. W. (2014). Wax plants (Hoya, Apocynaceae) evolution: epiphytism drives successful radiation. Taxon 63, 89–102. doi: 10.12705/631.3
Wanntorp, L., Kocyan, A., and Renner, S. S. (2006). Wax plants disentangled: a phylogeny of Hoya (Marsdenieae, Apocynaceae) inferred from nuclear and chloroplast DNA sequences. Mol. Phylogenet. Evol. 39, 722–733. doi: 10.1016/j.ympev.2006.01.022
Wei, X.-F., Zeng, S.-J., Zhang, G.-Q., Tang, G.-D., and Huang, J.-X. (2020). Complete plastome sequence of Hoya carnosa (L. f.) R. Br.(Apocynaceae). Mitochondrial DNA Part B 5, 522–523. doi: 10.1080/23802359.2019.1710596
Wick, R. R., Schultz, M. B., Zobel, J., and Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Yang, Z. (1998). Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol. Biol. Evol. 15, 568–573. doi: 10.1093/oxfordjournals.molbev.a025957
Yang, Z., Wong, W. S. W., and Nielsen, R. (2005). Bayes empirical Bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 22, 1107–1118. doi: 10.1093/molbev/msi097
Zecca, G., Abbott, J. R., Sun, W.-B., Spada, A., Sala, F., and Grassi, F. (2012). The timing and the mode of evolution of wild grapes (Vitis). Mol. Phylogenet. Evol. 62, 736–747. doi: 10.1016/j.ympev.2011.11.015
Keywords: chloroplast, Hoya, Dischidia, phylogeny, barcoding, genomics
Citation: Odago WO, Waswa EN, Nanjala C, Mutinda ES, Wanga VO, Mkala EM, Oulo MA, Wang Y, Zhang C-F, Hu G-W and Wang Q-F (2022) Analysis of the Complete Plastomes of 31 Species of Hoya Group: Insights Into Their Comparative Genomics and Phylogenetic Relationships. Front. Plant Sci. 12:814833. doi: 10.3389/fpls.2021.814833
Received: 14 November 2021; Accepted: 24 December 2021;
Published: 08 February 2022.
Edited by:
Robert Philipp Wagensommer, University of Bari Aldo Moro, ItalyCopyright © 2022 Odago, Waswa, Nanjala, Mutinda, Wanga, Mkala, Oulo, Wang, Zhang, Hu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cai-Fei Zhang, emhhbmdjZkB3YmdjYXMuY24=; Guang-Wan Hu, Z3Vhbmd3YW5odUB3YmdjYXMuY24=