 Ling Qiao1,2†
Ling Qiao1,2† Hanlin Li1,2†Jie Wang2Jiajia Zhao2
Hanlin Li1,2†Jie Wang2Jiajia Zhao2 Xingwei Zheng2Bangbang Wu2Weijun Du1*Juanling Wang1*
Xingwei Zheng2Bangbang Wu2Weijun Du1*Juanling Wang1* Jun Zheng2*
Jun Zheng2*- 1College of Agronomy, State Key Laboratory of Sustainable Dryland Agriculture (in Preparation), Shanxi Agricultural University, Jinzhong, China
- 2Institute of Wheat Research, Shanxi Agricultural University, Linfen, China
Wheat founder parents have been important in the development of new wheat cultivars. Understanding the effects of specific genome regions on yield-related traits in founder variety derivatives can enable more efficient use of these genetic resources through molecular breeding. In this study, the genetic regions related to field grain number per spike (GNS) from the founder parent Linfen 5064 were analyzed using a doubled haploid (DH) population developed from a cross between Linfen 5064 and Nongda 3338. Quantitative trait loci (QTL) for five spike-related traits over nine experimental locations/years were identified, namely, total spikelet number per spike (TSS), base sterile spikelet number per spike (BSSS), top sterile spikelet number per spike (TSSS), fertile spikelet number per spike (FSS), and GNS. A total of 13 stable QTL explaining 3.91–19.51% of the phenotypic variation were found. The effect of six of these QTL, Qtss.saw-2B.1, Qtss.saw-2B.2, Qtss.saw-3B, Qfss.saw-2B.2, Qbsss.saw-5A.1, and Qgns.saw-1A, were verified by another DH population (Linfen 5064/Jinmai 47), which showed extreme significance (P < 0.05) in more than three environments. No homologs of reported grain number-related from grass species were found in the physical regions of Qtss.saw-2B.1 and Qtss.saw-3B, that indicating both of them are novel QTL, or possess novel-related genes. The positive alleles of Qtss.saw-2B.2 from Linfen 5064 have the larger effect on TSS (3.30%, 0.62) and have 66.89% in Chinese cultivars under long-term artificial selection. This study revealed three key regions for GNS in Linfen 5064 and provides insights into molecular marker-assisted breeding.
Introduction
Founder parents are not only successful cultivars that are cultivated in large areas but are also used extensively as parents in breeding programs. These valuable genetic resources are crucial to Chinese wheat breeding programs (Zhuang, 2003). Analyzing the genetic diversity of founder parents and the genetic basis of their widespread success can provide a foundation for more efficient use of these germplasm resources.
A Chinese wheat founder parent named Linfen 5064 is the pedigree of more than 80 high-quality strong gluten cultivars in China. Linfen 5064 has the strong-gluten trait, a high grain number per spike (GNS), and excellent agronomic traits (Qiao et al., 2018). Linfen 5064 and cultivars derived from it not only have high yields but have also been used as the main parents for improving wheat quality in Chinese breeding programs. The use of Linfen 5064 as the founder parent addressed three difficult points in the breeding for strong-gluten wheat (Qiao et al., 2018). The first difficultly is that quality is negatively correlated with GNS and thousand kernel weight (TKW). Chinese wheat cultivars with premium grain quality, such as Xinong 20, Fengdecun 5, Shiluan 02-1 and Jimai 20, usually have lower GNS and lower yields. The GNS of Linfen 5064 and cultivars and lines derived from it have higher yields than other high-quality cultivars. The second difficult point is that dwarfism is associated with late maturity. Linfen 5064 does not show this association as it matures early and is a semi-dwarf height of about 75 cm. Finally, Linfen 5064 overcomes the need to have the glutenin subunit combination 5 + 10 for good quality, since it lacks these subunits yet still has good quality. Therefore, the utilization of valuable traits of Linfen 5064, and the successful future breeding program of Wheat, it is essential to explore and analyze their genetic base.
In most wheat cultivars, a spike usually generates more than 10–20 spikelets, and each spikelet can differentiate into 9–10 florets (Cui et al., 2008). The differentiation of bract and floret primordia determines the number of spikelets and initial florets. During floret development, 60–80% of the initial florets either abort or otherwise lose fertility (Guo et al., 2015). The number of surviving florets which can eventually develop into grains determines the number of grains per spike (Zhang et al., 2021). GNS shows high heritability (Isham et al., 2021). Increasing GNS is an important way to increase grain yield. GNS can be divided into total spikelet number per spike (TSS), fertile spikelet number per spike (FSS), base sterile spikelet number per spike (BSSS), top sterile spikelet number per spike (TSSS), and grains per spikelet. The heritability of TSS was higher (Isham et al., 2021), but the number of grains per spikelet and spikelet propagation ability were greatly affected by the environment. The map-based cloning of common wheat genes lags that of other crops because of wheat's large genome size. Consequently, most studies focus on the quantitative trait loci (QTL) level of analysis, especially genes/QTL that control yield traits.
Hundreds of QTL for GNS have been found to be distributed across the 21 wheat chromosomes (Börner et al., 2002; Huang et al., 2006; Narasimhamoorthy et al., 2006; Li et al., 2007; Ma et al., 2007; Wu et al., 2012; Jia et al., 2013; Zhang et al., 2016; Cui et al., 2017; Guan et al., 2018; Keeble-Gagnere et al., 2018; Onyemaobi et al., 2018; Su et al., 2018; Deng et al., 2019; Fan et al., 2019; Liu et al., 2019; Yao et al., 2019). Some genes related to GNS had been reported, such as homology-based cloned genes TaTAR2.1-3A (Shao et al., 2017), TaCWI-4A (Jiang et al., 2015), TaMOC1-7A (Zhang et al., 2015), TaSnRK2.9-5A (Rehman et al., 2019), TaAPO-A1 (Muqaddasi et al., 2019), TaGW8-B1 (Yan et al., 2019), TaPHR3-A1 (Zheng et al., 2020), the Q gene (Chuck et al., 1998; Debernardi et al., 2017; Xie et al., 2018), and genes GNI-A1 (Sakuma et al., 2019) and WFZP identified via map-based cloning (Du et al., 2021). Genes for other traits of agronomic importance, such as flowering time (FT) and plant height (PH), can have significant effects on grain yield (Cuthbert et al., 2008; Zhou et al., 2017; Guan et al., 2018). Ppd-1 participates in the regulation of flower spike development in wheat, which affects the number of spikes and seed setting (Boden et al., 2015).
Although many QTL/genes associated with GNS have been reported in wheat, the major and stable QTL identified under multiple environments are still limited. In addition, the biparents used for mapping were mostly accessions aim at certain traits rather than founder cultivars, the use of QTL identified need long-term backcross process which is time-consuming and low efficiency. We especially used founder parent and core cultivars in breeding as biparents for mapping, the loci obtained and markers developed are easily used in breeding, also provide evidence on utilization of the derivatives. Two doubled haploid (DH) populations (Linfen 5064 × Nongda 3338 and Linfen 5064 × Jinmai 47) were analyzed for five GNS-related traits over the nine experimental locations/years to (1) identify and validate major, stable QTL for GNS that can be used for molecular marker-assisted breeding and (2) identify genetic regions associated with GNS of Linfen 5064, elucidate the genetic mechanism of GNS in the founder parent, and discover favorable allele variations.
Materials and Methods
Plant Materials
A total of two DH populations were used, 192 lines from the cross Linfen 5064 × Nongda 3338 (LN) and 194 lines from the cross Linfen 5064 × Jinmai 47 (LJ). Linfen 5064 is a Chinese wheat founder parent with strong gluten, a high GNS, and an excellent array of other characteristics (Qiao et al., 2018). Nongda 3338, developed by China Agricultural University, is a “core parental” breeding line for the North China Winter Wheat Breeding Program with high general combining ability and the dwarfing genes Rht-B1b and Rht-D1b (Kabir et al., 2015). Jinmai 47 has the advantages of drought tolerance, stable yield, and a high utilization rate of water and fertilizer (Song et al., 2017). The phenotypic difference between the two cultivars and Linfen 5064 was significant and there was obvious trait separation in the population. LN was used for QTL analysis and LJ was used to validate the effects of putative QTL identified in LN.
Field Evaluation
The two DH populations were planted as a single replication in three locations in 2018–2019, 2019–2020, and 2020–2021. Locations were in the Yaodu district in Shanxi province of China, at Linfen (36°08′N, 111°52′E, altitude 450 m) (19 YD, 20 YD, and 21 YD), Hancun (36°25′N, 111°67′E, altitude 450 m) (19 HC, 20 HC, and 21 HC), and Yuncheng (35°15′ N, 110°98′ W, altitude 369 m) (19 YC, 20YC, and 21 YC). The seed was sown in two 1.5 m rows per line spaced 0.3 m apart at 21 seeds per row. Field management practices were those commonly used in wheat production in the region.
Phenotypic Evaluation and Data Analysis
Ten days before harvest, data of five spike traits, TSS, BSSS, TSSS, FSS, and GNS, were collected by randomly choosing 10 plants in each line. FSS = TSS-BSSS-TSSS. The best linear unbiased prediction (BLUP) of target traits in different environments (Smith et al., 1998) and the broad-sense heritability (H2) were obtained using SAS (SAS Institute, Cary, NC, USA; https://www.sas.com). The SPSS18.0 software (SPSS, Chicago, Illinois, USA; http://en.wikipedia.org/wiki/SPSS) was used to perform Student's t-test (p < 0.05) and correlation analysis of phenotype values in different environments.
Genetic Map Construction and Linkage Analysis
The two DH and parental lines were genotyped with a 15 K single-nucleotide polymorphism (SNP) panel developed based on 20 resequencing datasets, 1,520 genotyping datasets collected globally from multiple platforms, and publicly released resequencing and exon capture data. These datasets were developed and optimized using GenoBait technology to finally yield 14,868 mSNP regions for use in this study.
The genetic map of LN was constructed using IciMapping 4.1 (Meng et al., 2015) and JoinMap 4.0. Markers were binned if the correlation coefficient between them was 1 using the BIN function in IciMapping 4.1 according to the method reported by Winfield et al. (2016). WinQTLCart version 2.5 (Wang et al., 2012) for composite interval mapping was used to detect QTL. The minimal logarithm of odds (LOD) score to accept the presence of a QTL was set at 2.5. QTL was considered major when more than 10% of the phenotypic variation was explained in at least one environment and it was detected in at least three environments, including the BLUP dataset. QTL either <1 cM apart or sharing common flanking markers were treated as a single locus.
Validation for the Major QTL Identified
Peak SNPs for stable QTL identified in the LN population were genotyped in the LJ population. The differences in spike-related traits between both groups in the LJ population were analyzed with a t-test in SAS V8.0.
Genes Identified in the Major QTL
Genes within the target region of major QTL were obtained using the genome browser (JBrowse) on the WheatOmics-bata website http://wheatomics.sdau.edu.cn/ (Ma et al., 2021). Functional annotation and enrichment analysis of genes in these regions were done using the gene ontology (GO) database and the R package cluster Profiler. Analysis of orthologs between wheat and rice used the Triticeae-Gene Tribe website (http://wheat.cau.edu.cn/TGT/). The expVIP public database (http://www.wheat-expression.com/) was used to search for the expression data of genes in 16 tissues and organs, perform log2 conversion processing, and analyze the expression patterns of genes.
The R software package LD heatmap of major QTL was used to draw the linkage disequilibrium heatmap according to the resequencing data in 145 landmark cultivars that were downloaded from https://wheat.cau.edu.cn/WheatUnion/ (Hao et al., 2020).
Results
Phenotypic Variation and Correlations of Five Traits in Nine Environments
Linfen 5064 had lower values for TSS and TSSS, and a higher value of GNS than Nongda 3338 (Table 1). The spike traits of the DH population showed continuous variation, suggesting multigene genetic control. The estimated H2 of five traits ranged from 0.78 to 0.92, indicating that these traits were significantly affected by genetic factors (Table 1). The Pearson correlation coefficients among different environments were significant (P < 0.05, Supplementary Table S1). Better among-environment correlations were observed for TSS than for FSS, TSSS, BSSS, and GNS.
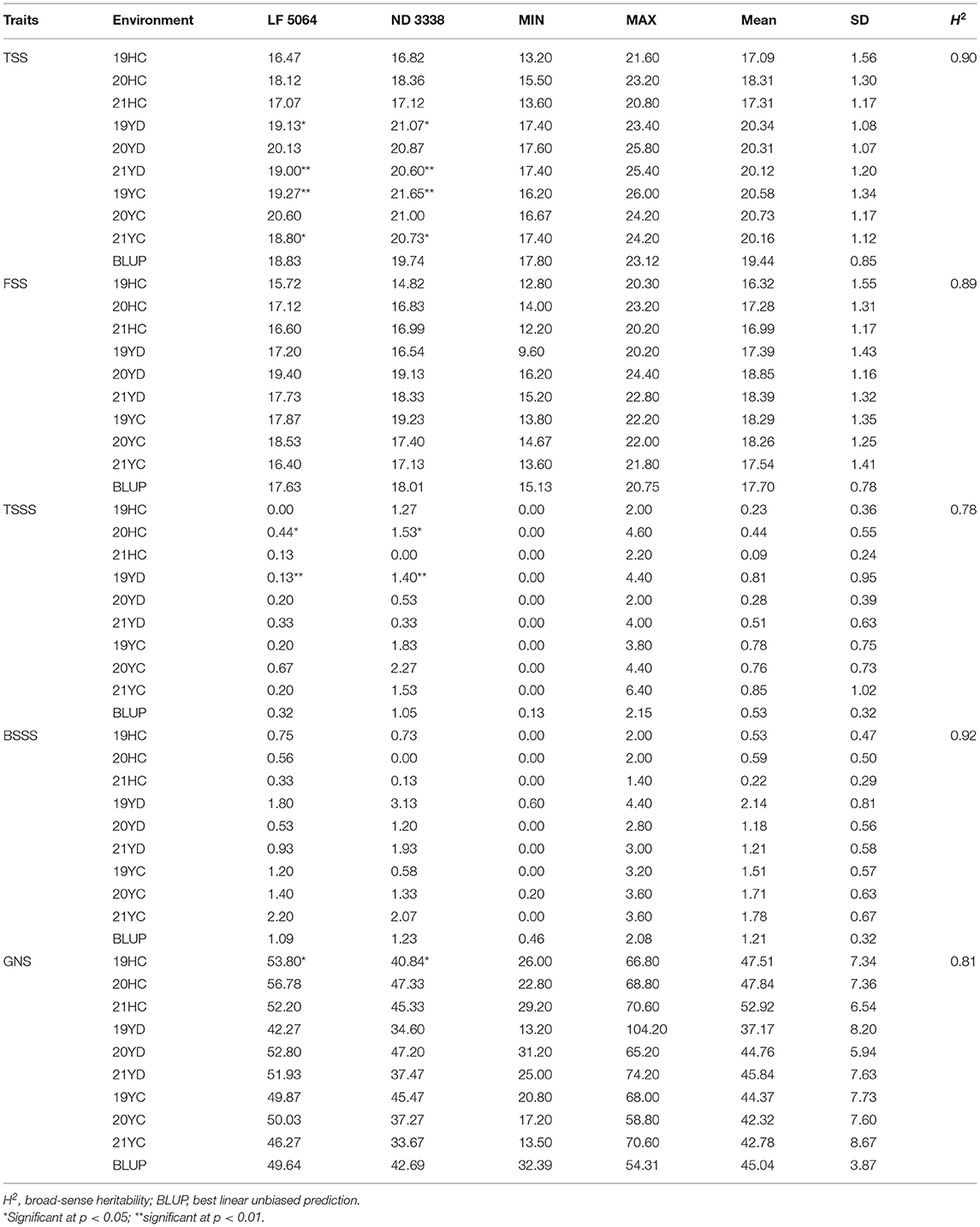
Table 1. Phenotypic variation and distribution of five spike-related traits in parents and the doubled haploid (Linfen 5064 × Nongda 3338) in nine field trials.
Phenotypic correlations among spike traits were evaluated using the BLUP dataset (Table 2). GNS significantly and positively correlated with FSS and TSS. GNS and FSS significantly and negatively correlated with BSSS and TSSS (p < 0.01, Table 2). The order of correlation coefficient with GNS were FSS (0.630) > TSSS (−0.437) > TSS (0.336) > BSSS (−0.162). These results showed that FSS and TSSS exerted great influence on GNS.
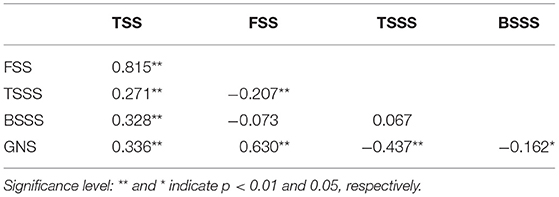
Table 2. Coefficients of pairwise Pearson correlations among five spike-related traits in the DH population Linfen 5064 × Nongda 3338.
Linkage Map Construction
In total, 841 SNP markers were used for constructing the LN genetic map. The map had 21 linkage groups, a total length of 3045.86 cM, and an average interval distance of 3.62 cM. The D genome had the lowest marker coverage, especially for chromosomes 5D and 6D. The maps of the A, B, and D genomes had, respectively, lengths of 1324.20, 1322.53, and 399.14 cM and densities of 3.99, 3.28, and 3.77 cM/marker (Supplementary Table S2).
QTL for Spikelet Number per Spike
A total of 64 QTL for TSS, FSS, TSSS, and BSSS were detected on 18 chromosomes (Supplementary Table S3) with 13 stable QTL identified (Table 3). QTL were found on all chromosomes except 1D, 6D, and 7D (Supplementary Table S3). The QTL explained 3.91–19.51% of the phenotypic variation in different environments. Linfen 5064 alleles contributed 30 of the 64 QTL, and Nongda 3338 contributed 34 alleles. Nine stable QTL, Qtss.saw-2B.1, Qtss.saw-2B.2, Qtss.saw-3B, Qtss.saw-4A.1, Qtss.saw-5A.1, Qtss.saw-5D, Qfss.saw-2B.2, Qbsss.saw-2B.2, and Qbsss.saw-5A.1 were detected in more than three environments and with BLUP values. Except for Qtss.saw-3B, Qtss.saw-5A.1, and Qbsss.saw-5A.1, the other six QTL explained more than 10% of the phenotypic variance and thus can be considered major stable QTL. The additive effect showed that the alleles of Qtss.saw-5A.1 and Qtss.saw-5D that increased TSS in grain were from Nongda 3338. The six stable QTL Qtss.saw-2B.1, Qtss.saw-2B.2, Qtss.saw-3B, Qtss.saw-4A.1, Qfss.saw-2B.2, and Qbsss.saw-2B.2 carried positive alleles from Linfen 5064. Qtss.saw-2B.2, Qfss.saw-2B.2, and Qbsss.saw-2B.2 were co-located in the 2B_54768734-2B_76515060 interval.
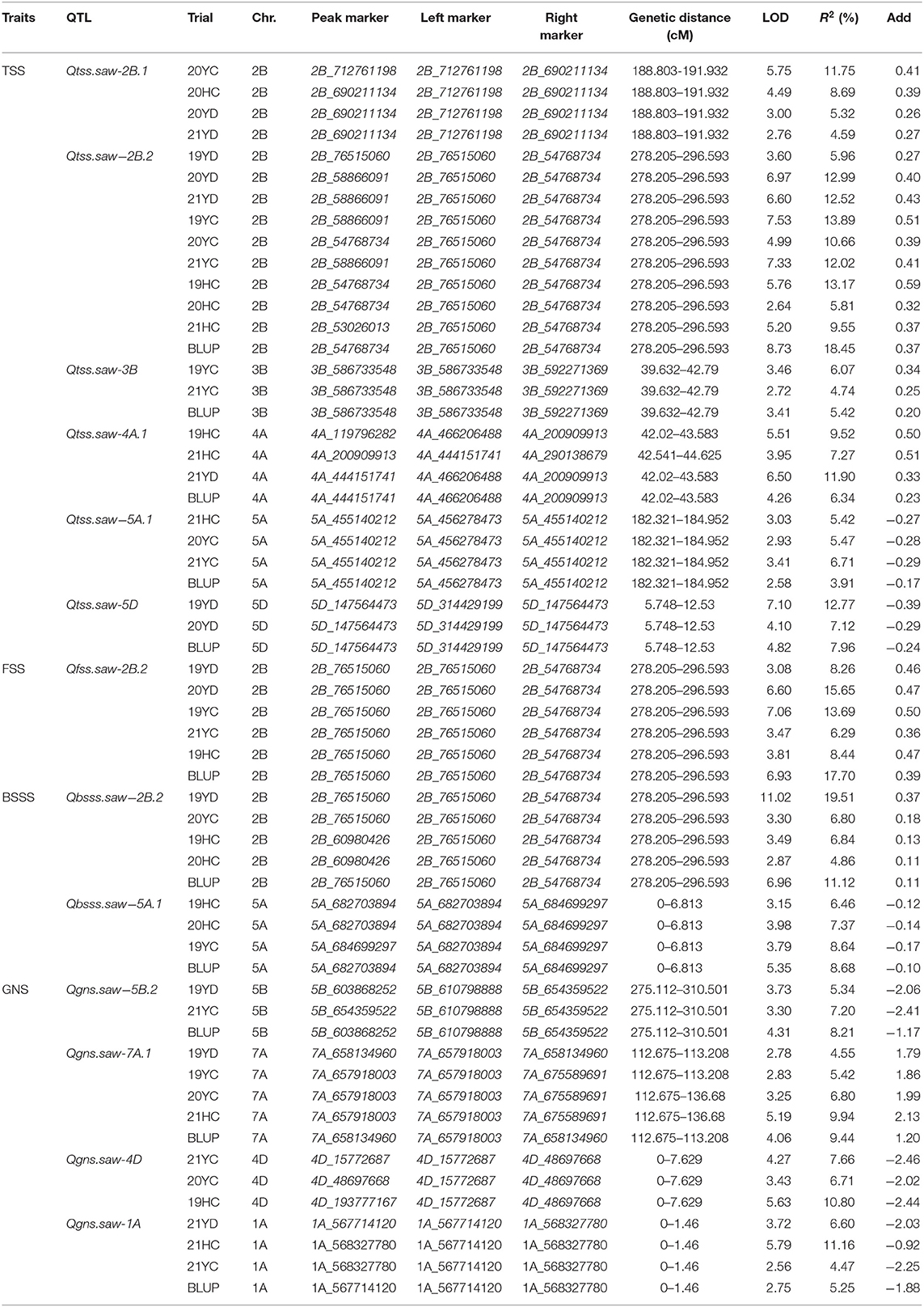
Table 3. Stable quantitative trait loci (QTL) detected for total spikelet number per spike (TSS), base sterile spikelet number per spike (BSSS), fertile spikelet number per spike (FSS), and grain number per spike (GNS) in the Linfen 5064 × Nongda 3338-derived doubled haploid population.
QTL for Grain Number per Spike
For GNS, 16 QTL were detected and these QTL explained 4.18–15.83% of the phenotypic variance (Supplementary Table S3). Four stable QTL, Qgns.saw-5B.2, Qgns.saw-7A.1, Qgns.saw-4D, and Qgns.saw-1A, explaining 4.47–11.16% of the phenotypic variance were identified in more than three environments and with BLUP values (Table 3). The additive effect of Qgns.saw-7A.1 was from Linfen 5064 indicating that Linfen 5064 contributed the allele for increased GNS. No stable QTL clusters for GNS and spikelet number per spike were detected on the same chromosome, indicating that the QTL of GNS were most likely independent of spikelet number per spike and therefore have great potential in wheat breeding.
QTL Validation
To further validate the stable QTL, the peak SNPs for each were used to evaluate their effects on corresponding traits in the LJ population. The peak markers for Qtss.saw-4A.1, Qtss.saw-5A.1, and Qgns.saw-4D were not polymorphic between the LJ parents, and thus could not be evaluated. The remaining 10 QTL were evaluated. The effect of Qtss.saw-5D, Qgns.saw-5B.2, Qgns.saw-7A.1, and Qbsss.saw-2B.2 did not differ significantly between the two groups in the LJ population (Figure 1). The effect of other six QTL, Qtss.saw-2B.1, Qtss.saw-2B.2, Qfss.saw-2B.2, Qtss.saw-3B, Qbsss.saw-5A.1, and Qgns.saw-1A, were highly significant (P < 0.05) in more than three environments. According to marker profiles of Qtss.saw-2B.1, Qtss.saw-2B.2, and Qtss.saw-3B, lines with homozygous alleles from Linfen 5064 had significantly higher (P < 0.05) values for TSS than those from Nongda 3338 and the difference ranged from 1.14 to 3.65%. The Qtss.saw-2B.2 lines homozygous for the Linfen 5064 alleles had significantly higher phenotypic values than those with the Jinmai 47 alleles irrespective of QTL region, with differences in TSS ranging from 1.29 to 3.21%. Lines with the positive allele from Qfss.saw-2B.2 had significantly greater FSS ranging from 0.88 to 3.38%, corresponding to 0.15–0.62 more spikelets than the lines with the alternate allele.
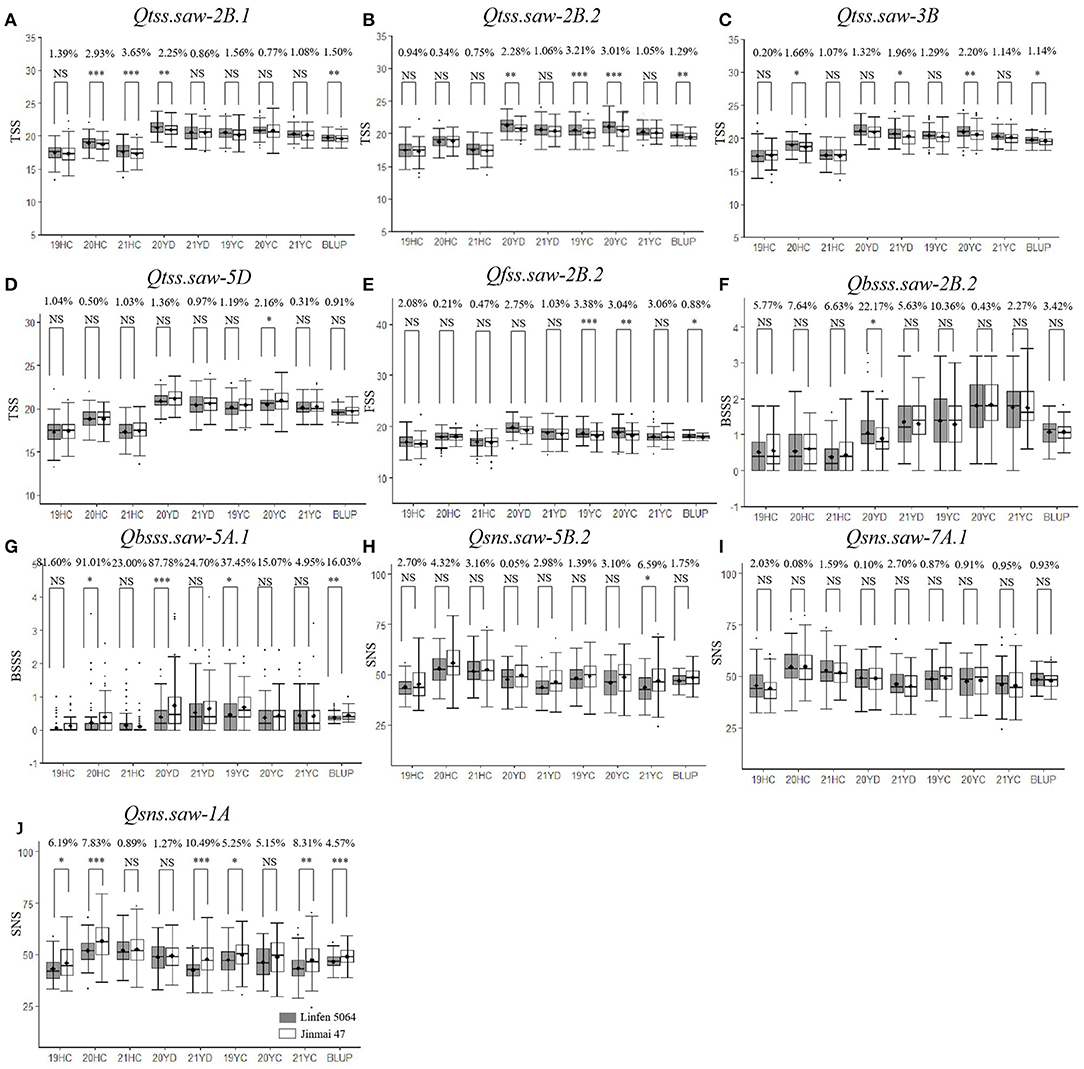
Figure 1. Validation of 10 stable quantitative trait loci (QTL) in LJ population. Effects of (A) Qtss.saw-2B.1, (B) Qtss.saw-2B.2, (C) Qtss.saw-3B, (D) Qtss.saw-5D, (E) on total spikelet number per spike (TSS) and effects of Qfss.saw-2B.2, (F) on fertile spikelet number per spike (FSS) and effects of Qbsss.saw-2B.2, (G) Qbsss.saw-5A.1, (H) on base sterile spikelet number per spike (BSSS) and effects of Qsns.saw-5B.2, (I) Qsns.saw-7A.1, (J) and Qsns.saw-1A on grain number per spike (GNS). *, **, *** and NS represent P < 0.05, P < 0.01, P < 0.001 and no significant difference,respectively.
Analyses of Additive Effects of the Major QTL
In the LN population, we detected six stable QTL for TSS (Qtss.saw-2B.1, Qtss.saw-2B.2, Qtss.saw-3B, Qtss.saw-4A.1, Qtss.saw-5A.1, and Qtss.saw-5D), two stable QTL for BSSS (Qbsss.saw-2B.2 and Qbsss.saw-5A.1), and four stable QTL for GNS (Qgns.saw-5B.2, Qgns.saw-7A.1, Qgns.saw-4D, and Qgns.saw-1A) (Table 3). The additive effects of these QTL on corresponding traits were analyzed based on linked markers. The average corresponding trait values increased as the number of positive alleles increased (Figures 2A–C). Lines with favorable alleles at all the six QTL regions had an average TSS increase of 2.25 vs. those possessing contrasting alleles (Supplementary Table S4, Figure 2A). Lines with both the positive alleles had significantly increased values for BSSS (Figure 2B). The combination of positive alleles from Qgns.saw-5B.2, Qgns.saw-7A.1, Qgns.saw-4D, and Qgns.saw-1A had the largest effect on GNS (Supplementary Table S4, Figure 2C).
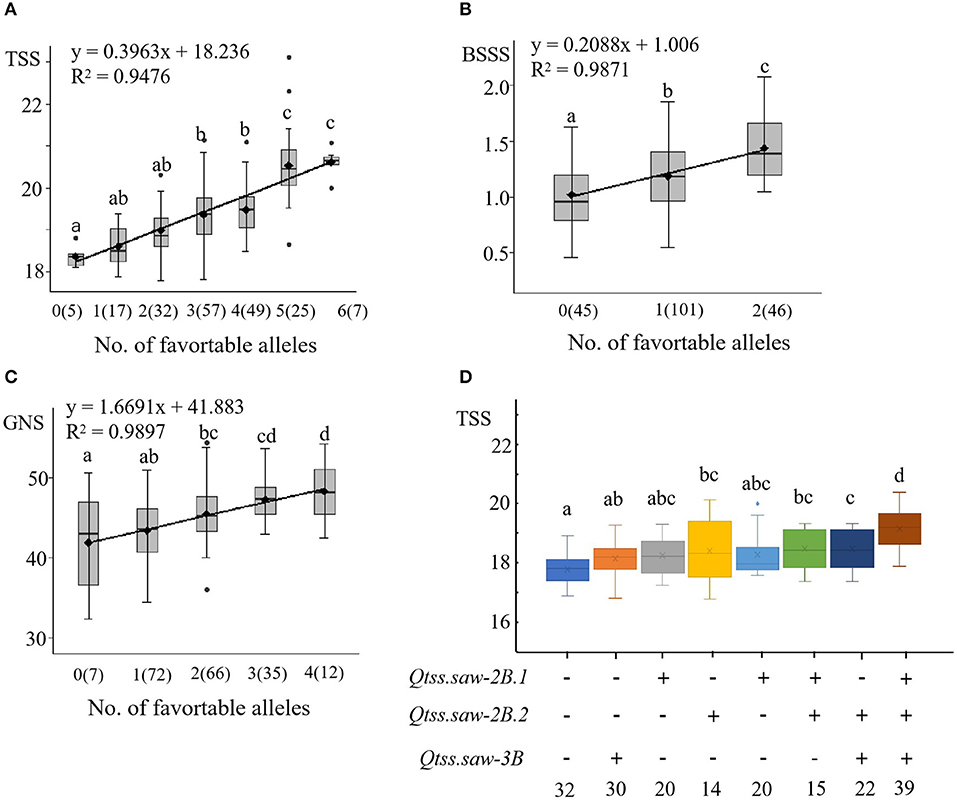
Figure 2. Linear regressions between the number of TSS, BSSS, and GNS (A–C) and additive effects of the QTL for TSS (D) in the LN population. The numbers of lines carrying the corresponding number of favorable alleles are shown in brackets. The letter above the bars indicated comparisons result at the significant level 0.05, respectively. Plus and minus represent lines with and without the positive alleles of the target QTL based on the flanking markers and the corresponding QTL.
Qtss.saw-2B.1, Qtss.saw-2B.2, and Qtss.saw-3B were validated in the LJ population, and the positive alleles of three QTL were derived from Linfen 5064, the additive effects on each corresponding trait were analyzed based on linked markers (Supplementary Table S5, Figure 2D). The combination of positive alleles from Qtss.saw-2B.1, Qtss.saw-2B.2, and Qtss.saw-3B (7.33%, 1.38) had the largest effect on TSS. Compared with lines lacking positive alleles for increased TSS, the positive allele from Qtss.saw-2B.2 significantly increased TSS by 3.30%, which was higher than that for the other single positive alleles of Qtss.saw-2B.1 (1.92%, 0.36) and Qtss.saw-3B (2.45%, 0.46). DH lines with both Qtss.saw-2B.1 and Qtss.saw-3B positive alleles significantly increased TSS (2.56%, 0.48) less than that of DH lines with single positive alleles of Qtss.saw-2B.2 (3.30%, 0.62). These results indicated that the positive allele of Qtss.saw-2B.2 from Linfen 5064 has a larger effect on TSS.
Distribution of Linfen 5064 Favorable Alleles Across Cultivars
The three stable QTL Qtss.saw-2B.1, Qtss.saw-2B.2, and Qtss.saw-3B were detected in more than three environments and were validated in the LJ population. The additive effects of these QTL were from Linfen 5064. Based on the resequencing of 145 wheat cultivars, linkage disequilibrium analysis was performed to assess variation sites within three target QTL regions (Figure 3). Qtss.saw-2B.1, Qtss.saw-2B.2, and Qtss.saw-3B had high recombination rates corresponding to recombination hotspot areas. Therefore, for three QTL the distribution of favorable alleles from Linfen 5064 was analyzed in 145 landmark cultivars (Table 4). The favorable alleles of Linfen 5064 for Qtss.saw-2B.2 had a lower proportion in the Chinese landraces (CL) (44%) and introduced modern cultivars (IMC) (45%), but a higher proportion in the modern Chinese cultivars (MCC) (77%). Therefore, the favorable alleles of Linfen 5064 at the Qtss.saw-2B.2 locus were selected because of their value in breeding new Chinese cultivars. Qtss.saw-2B.1 and Qtss.saw-3B with the positive Linfen 5064 alleles were less frequent in Chinese landmark cultivars (29.66 and 15.86%, respectively), indicating that Qtss.saw-3B landmark alleles tended to be replaced during breeding by the Linfen 5064 alleles.

Figure 3. Linkage disequilibrium heatmap of three target QTL regions (A) Qtss.saw-2B.1, (B) Qtss.saw-2B.2, and (C) Qtss.saw-3B.
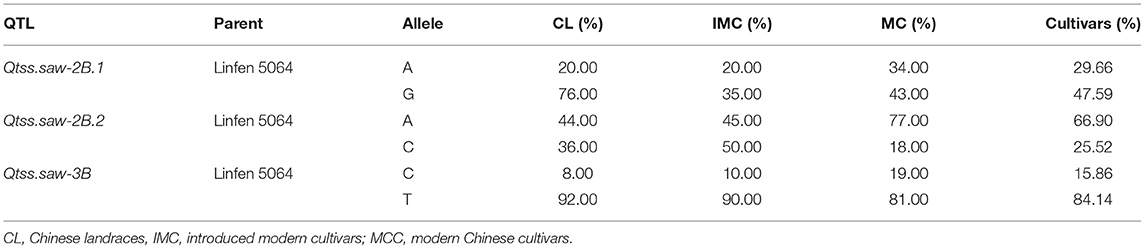
Table 4. The proportion of the Linfen 5064 favorable alleles detected in 145 cultivars Qtss.saw-2B.1, Qtss.saw-2B.2, and Qtss.saw-3B.
Genes Identified in the Major QTL
A series of orthologous GNS-related genes have been cloned in rice (Huang et al., 2009; Kyoko et al., 2009; Qiao et al., 2011; Gao et al., 2016) and wheat (Jiang et al., 2015; Zhang et al., 2015; Shao et al., 2017; Muqaddasi et al., 2019; Rehman et al., 2019), these genes always showed conserved functions across grass species (Valluru et al., 2014). Based on the result of local-blast browse through the IWGSC reference sequence, no homologs of the above genes were found in the physical regions of 690.21–712.76 Mb on 2BL and 586.73–592.27 Mb on 3BL in wheat. It indicated that there might be novel genes related to GNS among the two QTL, thus, these QTL were chosen for further analysis. Qtss.saw-2B.1 was in the interval 690.21–712.76 Mb on 2BL and where 260 genes have been found in the variety Chinese Spring (CS) (Supplementary Table S6). Gene annotation, expression pattern, and orthologous gene analysis indicate that three genes are likely involved in spike development (Supplementary Table S6, Supplementary Figure S1). The function of TraesCS2B02G500100, TraesCS2B02G500200, and TraesCS2B02G500300 are annotated as a series of molecular signals generated by the binding of the plant hormone abscisic acid to a receptor and ending with modulation of a cellular process. Qtss.saw-3B has 20 genes in CS and 13 common predicated genes between CS and rice (Supplementary Table S7). The genes were not preferentially expressed in spike and grain (Supplementary Figure S2).
Discussion
Linfen 5064 Possess Favorable Key Genomic Regions
Analyzing founder parents at the whole genome level and studying the genome regions of the founder parents of high value is important for wheat breeding, especially molecular marker-assisted breeding. As a founder parent, Linfen 5064 has greatly contributed to wheat breeding in China. The high-quality characteristics of Linfen 5064 are derived from the spring wheat SARICF74 introduced from the Centro Internacional de Mejoramiento de Maizy Trigo (CIMMYT). Linfen 5064 was selected from a cross of SARICF74 and Linfen 5694 for early maturity and good agronomic traits. In this study, two DH populations were constructed with Linfen 5064 as the female parent and with Nongda 3338 and Jinmai 47 as male parents. A total of 13 stable QTL were identified through the investigation of spike traits in three field locations over 3 years. Seven stable QTL carried positive alleles from Linfen 5064. For spikelet number per spike, the additive effect of Qtss.saw-2B.1, Qtss.saw-2B.2, Qtss.saw-3B, Qtss.saw-4A.1, Qfss.saw-2B.2, and Qbsss.saw-2B.2 were from Linfen 5064. And except for Qtss.saw-4A.1 and Qbsss.saw-2B.2, other QTL were validated in the LJ population. The QTL Qtss.saw-2B.2, Qfss.saw-2B.2, and Qbsss.saw-2B.2 were located in the same region. Therefore, Qtss.saw-2B.1, Qtss.saw-2B.2, and Qtss.saw-3B were the most important regions of Linfen 5064 controlling spikelet number per spike. Lines with the positive allele from Qtss.saw-2B.2 significantly increased TSS by 3.30%, which is higher than other single positive alleles of either Qtss.saw-2B.1 (1.92%) or Qtss.saw-3B (2.45%). The region Qtss.saw-2B.2 from Linfen 5064 had the larger effect on TSS and was present in 66.89% of Chinese landmark cultivars tested. For GNS, the only positive effect from a Linfen 5064 allele was from Qgns.saw-7A.1. The positive effects of Qgns.saw-5B.2, Qgns.saw-4D, and Qgns.saw-1A alleles were from Nongda 3338. The effect of Qgns.saw-1A was validated in the LJ population. These results indicate that this allele was unfavorable, but through breeding, improvement was made for the trait in Linfen 5064 presumably from contributions from other loci. This study examined the QTL of Linfen 5064 for GNS and analyzed the characteristics of genetic effects of related regions. These results further clarify the genetic contribution and intrinsic value of Linfen 5064 to GNS and provide a reference for future founder parent utilization and molecular breeding.
Qtss.saw-2B.1 and Qtss.saw-3B Are Novel Loci for Wheat Spike-Related Traits
To compare the intervals of the 13 QTL detected with those identified previously, we physically mapped these QTL on target chromosomes in CS. The QTL Qtss.saw-5D for TSS is physically located between 147.56 and 314.43 Mb on 5D (Table 3). It overlapped with a major QTL QSN.caas-5DL found in wheat by Li et al. (2018). Qgns.saw-7A.1 was located between 657.92 and 675.59 Mb on chromosome 7AL (Table 3). This region has QTL-rich clusters for wheat yield component traits. QSn-7A.2 (Fan et al., 2019), Qmt.tamu.7A.1 (Assanga et al., 2017), QTgw.cau-7A.4 (Guan et al., 2018), and Qkns.caas-7AL (Li et al., 2018) overlap with Qgns.saw-7A.1. Likewise, TaAPO-A1 is in this cluster, namely, Qkns.caas-7AL, QGne.nfcri-7A, and QGns.cau-7A.5 for Kernel number per spike, so it probably is the candidate gene of these QTL (Cao et al., 2020). TaAPO-A1 is orthologous to APO1, a rice gene that positively controls spikelet number on panicles (Muqaddasi et al., 2019). Qgns.saw-4D was located within 15.77–48.70 Mb on chromosome 4DS (Table 3). Comparative analysis revealed that this locus overlaps TB-D1 (Dixon et al., 2018), Rht-D1 (Peng et al., 1999), QTKW-4D-AN (Mohler et al., 2016), QGn.nau-4D (Jia et al., 2013), QTgw-4D, and QGns-4D (Liu et al., 2014), suggesting this region is a QTL-rich cluster for wheat yield component traits. Qtss.saw-2B.2, Qfss.saw-2B.2, and Qbsss.saw-2B.2 were co-located in the interval of 2B_54768734-2B_76515060 and physically mapped to 54.77–76.52 Mb on 2BS. This region has the Ppd-B1 gene which is a key component in the photoperiod regulatory flowering pathway (Beales et al., 2007; Nishida et al., 2013) and is associated with flag leaf size and grain yield (Kirby, 1992; Snape et al., 2001; Foulkes et al., 2004). No stable QTL have been reported previously overlapped with the other stable QTL from this study, Qtss.saw-2B.1, Qtss.saw-3B, Qtss.saw-5A, and Qgns.saw-5B.2. Both Qtss.saw-2B.1 and Qtss.saw-3B had significant effects on TSS and GNS that were detected in the validation population. Therefore, Qtss.saw-2B.1 and Qtss.saw-3B are likely novel loci for TSS. Therefore, spikelet development of wheat is a complex process, which is regulated by different types of genes. With the development of biotechnology, combining multiple technologies to analyze the development of GNS will help clarify the formation mechanism of GNS.
New Genes Were Identified in the Interval of the Stable QTL to Control Spike-Related Traits
Genes related to spike traits can be divided into two categories. The first category is flowering time (FT) genes which have significant effects on grain yield, namely, Vrn1, Vrn2/ZCCT1, Vrn3, and Ppd-D1 (Cuthbert et al., 2008; Zhou et al., 2017; Guan et al., 2018). Other genes were mainly involved in spike differentiation which influenced the number of grains per spike by regulating the rate and direction of differentiation. For example, aberrant panicle organization 1 (APO1) controls cell proliferation of the rice meristem, leading to the reduction of the primary and secondary branches of the panicle, thereby affecting panicle development (Kyoko et al., 2009). In addition, some genes can control panicle morphogenesis by regulating hormone and protein expression during rice growth (Huang et al., 2009; Qiao et al., 2011; Gao et al., 2016). BG1 regulates auxin transport and increases biomass, grain number per spike, and grain size to increase yield (Liu et al., 2015). In this study, we find three new genes for controlling spike-related traits. TraesCS2B02G500100, TraesCS2B02G500200, and TraesCS2B02G500300 and involved the phytohormone regulatory and ubiquitin proteasoma. In the next step, we will fine-mapping these QTL which will help explain the formation and development of GNS in wheat and develop linked molecular markers for use by breeders.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
JZhe, WD, JuW, and LQ designed the experiment and developed the original manuscript. LQ, HL, JZha, XZ, JiW, and BW performed the field experiments. LQ, HL, XZ, WD, and JZhe performed the phenotypic data analysis and the QTL detection. WD, JuW, and JZhe revised the manuscript. All authors approved the submitted version of the manuscript.
Funding
This study was supported by the State Key Laboratory of Integrative Sustainable Dryland Agriculture (in preparation), the Shanxi Agricultural University (No. 202105D121008-2-1), the Shanxi Scholarship Council of China (2020-159), the Shanxi Province Research Program (20210302124505), and the Agricultural Science Research of Shanxi Academy of Agricultural Sciences (YCX2020YQ47, YCX2020YQ34, and YZGC013).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to Professor Jian Gu, Food Crops Research Institute, Yunnan Academy of Agricultural Sciences, for providing help in developing DH populations. We also thank Professor Jianli Chen of the University of Idaho for critical advice on the design of these experiments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.808136/full#supplementary-material
References
Assanga, S. O., Fuentealba, M., Zhang, G. R., Tan, C., Dhakal, S., Rudd, J. C., et al. (2017). Mapping of quantitative trait loci for grain yield and its components in a US popular winter wheat TAM 111 using 90K SNPs. PLoS ONE 12:e189669. doi: 10.1371/journal.pone.0189669
Beales, J., Turner, A., Griffiths, S., Snape, J. W., and Laurie, D. A. (2007). A pseudo-response regulator is misexpressed in the photoperiod insensitive Ppd-D1a mutant of wheat (Triticum aestivum L.). Theor. Appl. Genet. 115, 721–733. doi: 10.1007/s00122-007-0603-4
Boden, S. A., Cavanagh, C., Cullis, B. R., Ramm, K., Greenwood, J. L., Finnegan, E. J., et al. (2015). Ppd-1 is a key regulator of inflorescence architecture and paired spikelet development in wheat. Nat Plants. 1:14016. doi: 10.1038/nplants.2014.16
Börner, A., Schumann, E., Fürste, A., Cöster, H., Leithold, B., Röder, M., et al. (2002). Mapping of quantitative trait locus determining agronomic important characters in hexaploid wheat (Triticum aestivum L.). Theor. Appl. Genet. 105, 921–936. doi: 10.1007/s00122-002-0994-1
Cao, S. H, Xu, D. A., Hanif, M., Xia, X. C., and He, Z. H. (2020). Genetic architecture underpinning yield component traits in wheat. Theor. Appl. Genet. 133, 1811–1823. doi: 10.1007/s00122-020-03562-8
Chuck, G., Meeley, R. B., and Hake, S. (1998). The control of maize spikelet meristem fate by the APETALA2-like gene indeterminate spikelet1. Gene Dev. 12, 1145–1154. doi: 10.1101/gad.12.8.1145
Cui, F., Zhang, N., Fan, X. L., Zhang, W., Chun, H. Z., Li, J. Y., et al. (2017). Utilization of a Wheat660K SNP array-derived high-density genetic map for high-resolution mapping of a major QTL for kernel number. Sci. Rep. 7:3788. doi: 10.1038/s41598-017-04028-6
Cui, J. M., Guo, T. C., Zhu, Y. j, Wang, C. Y., and Ma, X. M. (2008). Spike of Wheat. Beijing: Agriculture Press, 18–33.
Cuthbert, J. L., Somers, D. J., Brûlé-Babel, A. L., Brown, P. D., and Crow, G. H. (2008). Molecular mapping of quantitative trait loci for yield and yield components in spring wheat (Triticum aestivum L.). Theor. Appl. Genet. 117, 595–608. doi: 10.1007/s00122-008-0804-5
Debernardi, J. M., Lin, H., Chuck, G., Faris, J. D., and Dubcovsky, J. (2017). micro-RNA172 plays a crucial role in wheat spike morphogenesis and grain threshability. Development 144, 1966–1975. doi: 10.1242/dev.146399
Deng, M., Wu, F. K., Zhou, W. L., Li, J., Shi, H. R., Wang, Z. Q., et al. (2019). Mapping of QTL for total spikelet number per spike on chromosome 2D in wheat using a high-density genetic map. Genet. Mol. Biol. 42, 603–610. doi: 10.1590/1678-4685-gmb-2018-0122
Dixon, L. E., Greenwood, J. R., Bencivenga, S., Zhang, P., Cockram, J., Mellers, G., et al. (2018). TEOSINTE BRANCHED1 regulates inflorescence architecture and development in bread wheat (Triticum aestivum L.). Plant Cell. 30, 563–581. doi: 10.1105/tpc.17.00961
Du, D. J., Zhang, D. X., Yuan, J., Feng, M., Li, Z. J., Wang, Z. H., et al. (2021). FRIZZY PANICLE defines a regulatory hub for simultaneously controlling spikelet formation and awn elongation in bread wheat. New Phytol. 231, 814–833. doi: 10.1111/nph.17388
Fan, X. L., Cui, F., Ji, J., Zhang, W., Zhao, X. Q., Liu, J. J., et al. (2019). Dissection of pleiotropic QTL regions controlling wheat spike characteristics under different nitrogen treatments using traditional and conditional QTL mapping. Front. Plant Sci. 10:187. doi: 10.3389/fpls.2019.00187
Foulkes, M. J., Sylvester-Bradley, R., Worland, A. J., and Snape, J. W. (2004). Effects of a photoperiod-response gene Ppd-D1 on yield potential and drought resistance in UK winter wheat. Euphytica 135, 63–73. doi: 10.1023/B:EUPH.0000009542.06773.13
Gao, S. P., Fang, J., Xu, F., Wang, W., and Chu, C. C. (2016). Rice HOX12 regulates panicle exsertion by directly modulating the expression of ELONGATED UPPERMOST INTERNODE1, Plant Cell 28, 680–695. doi: 10.1105/tpc.15.01021
Guan, P. F., Lu, L. H., Jia, L. J., Kabir, M. R., Zhang, J. B., Lan, T. Y., et al. (2018). Global QTL analysis identifies genomic regions on Chromosomes 4A and 4B harboring stable loci for yield-related traits across different environments in wheat (Triticum aestivum L.). Front. Plant Sci. 9:529. doi: 10.3389/fpls.2018.00529
Guo, J., Zhang, Y., Shi, W. P., Zhang, B. Q., Xu, Y. H., et al. (2015). Association analysis of grain-setting rates in apical and basal spikelets in bread wheat (Triticum aestivum L.). Front. Plant Sci. 6:1029. doi: 10.3389/fpls.2015.01029
Hao, C. Y., Jiao, C. Z., Hou, J., Li, T., Liu, H. X., Wang, Y. Q., et al. (2020). Resequencing of 145 landmark cultivars reveals asymmetric sub-genome selection and strong founder genotype effects on wheat breeding in China. Mol. Plant. 13, 1733–1751. doi: 10.1016/j.molp.2020.09.001
Huang, X. Q., Cloutier, S., Lycar, L., Radovanovic, N., Humphreys, D. G., Noll, J. S., et al. (2006). Molecular detection of QTL for agronomic and quality traits in a doubled haploid population derived from two Canadian wheats (Triticum aestivum L.). Theor. Appl. Genet. 113, 753–766. doi: 10.1007/s00122-006-0346-7
Huang, X. Z., Qian, Q., Liu, Z. B., Sun, H. Y., He, S. Y., Luo, D., et al. (2009). Natural variation at the DEP1 locus enhances grain yield in rice. Nat. Genet. 41, 494–497. doi: 10.1038/ng.352
Isham, K., Wang, R., Zhao, W. D., Wheeler, J., Klassen, N., Akhunov, E., et al. (2021). QTL mapping for grain yield and three yield components in a population derived from two high-yielding spring wheat cultivars. Theor. Appl. Genet. 134, 2079–2095. doi: 10.1007/s00122-021-03806-1
Jia, H. Y., Wan, H. S., Yang, S. H., Zhang, Z. Z., Kong, Z. X., X, S. L., et al. (2013). Genetic dissection of yield-related traits in a recombinant inbred line population created using a key breeding parent in China's wheat breeding. Theor. Appl. Genet. 126, 2123–2139. doi: 10.1007/s00122-013-2123-8
Jiang, Y. M., Jiang, Q. Y., Hao, C. Y., Hou, J., Wang, L. F., Zhang, H. N., et al. (2015). A yield-associated gene TaCWI, in wheat: its function, selection and evolution in global breeding revealed by haplotype analysis. Theor. Appl. Genet. 128, 131–143. doi: 10.1007/s00122-014-2417-5
Kabir, M. R., Liu, G., Guan, P. F., Wang, F., Khan, A. A., Ni, Z. F., et al. (2015). Mapping QTL associated with root traits using two different populations in wheat (Triticum aestivum L.). Euphytica 206, 175–190. doi: 10.1007/s10681-015-1495-z
Keeble-Gagnere, G., Rigault, P., Tibbits, J., Pasam, R., Hayden, M., Forrest, K., et al. (2018). Optical and physical mapping with local finishing enables megabase-scale resolution of agronomically important regions in the wheat genome. Genome Biol. 19:112. doi: 10.1186/s13059-018-1475-4
Kirby, E. J. M. (1992). A field study of the number of main shoot leaves in wheat in relation to vernalization and photoperiod. J. Agric. Sci. 118, 271–278. doi: 10.1017/S0021859600070635
Kyoko, I. K., Naoko, Y., Tetsuo, O., Shigeru, I., Yasuo, N., Masahiko, M., et al. (2009). Expression Level of ABERRANT PANICLE ORGANIZATION1 determines rice inflorescence form through control of cell proliferation in the meristem. Plant Physiol. 150, 736–747. doi: 10.1104/pp.109.136739
Li, F., Wen, W., He, Z., Liu, J., Jin, H., Cao, S., et al. (2018). Genome-wide linkage mapping of yield-related traits in three Chinese bread wheat populations using high-density SNP markers. Theor. Appl. Genet. 131, 1903–1924. doi: 10.1007/s00122-018-3122-6
Li, S. S., Jia, J. Z., Wei, X. Y., Zhang, X. C., Li, L. Z., Chen, H. M., et al. (2007). A intervarietal genetic map and QTL analysis for yield traits in wheat. Mol. Breed. 20, 167–178. doi: 10.1007/s11032-007-9080-3
Liu, G., Jia, L. J., Lu, L. H., Qin, D. D., Zhang, J. P., Guan, P. F., et al. (2014). Mapping QTL of yield-related traits using RIL population derived from common wheat and Tibetan semi-wild wheat. Theor. Appl. Genet. 127, 2415–2432. doi: 10.1007/s00122-014-2387-7
Liu, J., Wu, B. H., Singh, R. P., and Velu, G. (2019). QTL mapping for micronutrients concentration and yield component traits in a hexaploid wheat mapping population. J. Cereal Sci. 88, 57–64. doi: 10.1016/j.jcs.2019.05.008
Liu, L. C., Tong, H. N., Xiao, Y. H., Che, R. H., Xu, F., Hu, B., et al. (2015). Activation of Big Grain1 significantly improves grain size by regulating auxin transport in rice. Proc. Natl. Acad. Sci. U.S.A. 112, 11102–11107. doi: 10.1073/pnas.1512748112
Ma, S. W., Wang, M., Wu, J. H., Guo, W. L., Chen, Y. M., Li, G. W., et al. (2021). WheatOmics: a platform combining multiple omics data to accelerate functional genomics studies in wheat. Mol. Plant. 14, 1965–1968. doi: 10.1016/j.molp.2021.10.006
Ma, Z. Q., Zhao, D. M., Zhang, C. Q., Zheng, Z. Z., Shu, L. X., Feng, L., et al. (2007). Molecular genetic analysis of five spike-related traits in wheat using RIL and immortalized F2 populations. Mol. Gen. Genomics 277, 31–42. doi: 10.1007/s00438-006-0166-0
Meng, L., Li, H. H., Zhang, L. Y., and Wang, J. K. (2015). QTL IciMapping: integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J. 3, 269–283. doi: 10.1016/j.cj.2015.01.001
Mohler, V., Albrecht, T., Castell, A., Diethelm, M., Schweizer, G., and Hartl, L. (2016). Considering causal genes in the genetic dissection of kernel traits in common wheat. J. Appl. Genet. 57, 467–476. doi: 10.1007/s13353-016-0349-2
Muqaddasi, Q. H., Brassac, J., Koppolu, R., Plieske, J., Ganal, M. W., and Röder, M. S. (2019). TaAPO-A1, an ortholog of rice ABERRANT PANICLE ORGANIZATION 1, is associated with total spikelet number per spike in elite hexaploid winter wheat varieties (Triticum aestivum L.). Sci. Rep. 9:13853. doi: 10.1038/s41598-019-50331-9
Narasimhamoorthy, B., Gill, B. S., Fritz, A. K., Nelson, J. C., and Broen-Guedira, G. L. (2006). Advanced backcross QTL analysis of a hard winter wheat × synthetic wheat population. Theor. Appl. Genet. 112, 787–796. doi: 10.1007/s00122-005-0159-0
Nishida, H., Yoshida, T., Kawakami, K., Fujita, M., Long, B., Akashi, Y., et al. (2013). Structural variation in the 5' upstream region of photoperiod-insensitive alleles Ppd-A1a and Ppd-B1a identified in hexaploid wheat (Triticum aestivum L.), and their effect on heading time. Mol. Breed. 31, 27–37. doi: 10.1007/s11032-012-9765-0
Onyemaobi, I., Ayalew, H., Liu, H., Siddique, K. H. M., and Yan, G. J. (2018). Identification and validation of a major chromosome region for high grain number per spike under meiotic stage water stress in wheat (Triticum aestivum L.). PLoS ONE 13:e194075. doi: 10.1371/journal.pone.0194075
Peng, J., Richards, D. E., Hartley, N. M., Murphy, G. P., Devos, K. M., Flintham, J. E., et al. (1999). 'Green revolution' genes encode mutant gibberellin response modulators. Nature 400, 256–261. doi: 10.1038/22307
Qiao, L., Liu, C., Zheng, X. W., Zhao, J. J., Shang, B. H., Ma, X. F., et al. (2018). Genetic analysis of haplotype-blocks from wheat founder parent Linfen 5064. Acta. Agron. Sin. 44, 931–937. doi: 10.3724/SP.J.1006.2018.00931
Qiao, Y. L., Piao, R., Shi, J., Lee, S. I., Jiang, W., Kim, B. K., et al. (2011). Fine mapping and candidate gene analysis of dense and erect panicle 3, DEP3 which confers high grain yield in rice (Oryza sativa L.). Theor Appl Genet. 122, 1439–1449. doi: 10.1007/s00122-011-1543-6
Rehman, S. U., Wang, J. Y., Chang, X. P., Zhang, X. Y., Mao, X. G., et al. (2019). A wheat protein kinase gene TaSnRK2.9-5A associated with yield contributing traits. Theor. Appl. Genet. 132, 907–919. doi: 10.1007/s00122-018-3247-7
Sakuma, S., Golan, G., Guo, Z. F., Ogawa, T., Tagiri, A., Sugimoto, K., et al. (2019). Unleashing floret fertility in wheat through the mutation of a homeobox gene. Proc. Natl. Acad. Sci. U.S.A. 116, 5182–5187. doi: 10.1073/pnas.1815465116
Shao, A., Ma, W. Y., Zhao, X. Q., Hu, M. Y., He, X., Teng, W., et al. (2017). The auxin biosynthetic TRYPTOPHAN AMINOTRANSFERASE RELATED TaTAR2.1-3A increases grain yield of wheat. Plant Physiol. 174, 2274–2288. doi: 10.1104/pp.17.00094
Smith, S. E., Kuehl, R. O., Ray, I. M., Hui, R., and Soleri, D. (1998). Evaluation of simple methods for estimating broad-sense heritability in stands of randomly planted genotypes. Crop Sci. 38, 1125–1129. doi: 10.2135/cropsci1998.0011183X003800050003x
Snape, J. W., Butterworth, K., Whitechurch, E., and Worland, A. J. (2001). Waiting for fine times: genetics of flowering time in wheat. Euphytica. 119, 185–190. doi: 10.1023/A:1017594422176
Song, Q. H., Liu, C. Y., Bachir, D. G., Chen, L., and Hu, Y. G. (2017). Drought resistance of new synthetic hexaploid wheat accessions evaluated by multiple traits and antioxidant enzyme activity. Field Crops Res. 210, 91–103. doi: 10.1016/j.fcr.2017.05.028
Su, Q. N., Zhang, X. L., Zhang, W., Zhang, N., Song, L. Q., Liu, L., et al. (2018). QTL detection for kernel size and weight in bread wheat (Triticum aestivum L.) using a high-density SNP and SSR-based linkage map. Front. Plant Sci. 9:1484. doi: 10.3389/fpls.2018.01484
Valluru, R., Reynolds, M. P., and Salse, J. (2014). Genetic and molecular bases of yield-associated traits: a translational biology approach between rice and wheat. Theor. Appl. Genet. 127, 1463–1489. doi: 10.1007/s00122-014-2332-9
Wang, S., Basten, C., and Zeng, Z. (2012). Windows QTL Cartographer 2.5. Raleigh: Department of Statistics, North Carolina State University.
Winfield, M. O., Allen, A. M., Burridge, A. J., Barker, G. L. A., Benbow, H. R., Wilkinson, P. A., et al. (2016). High-density SNP genotyping array for hexaploid wheat and its secondary and tertiary gene pool. Plant Biotechnol. J. 14, 1195–1206. doi: 10.1111/pbi.12485
Wu, X. S., Chang, X. P., and Jing, R. L. (2012). Genetic insight into yield-associated traits of wheat grown in multiple rain-fed environments. PLoS ONE 7:e31249. doi: 10.1371/journal.pone.0031249
Xie, Q., Li, N., Yang, Y., Lv, Y. L., Yao, H. N., Wei, R., et al. (2018). Pleiotropic effects of the wheat domestication gene Q on yield and grain morphology. Planta 247, 1089–1098. doi: 10.1007/s00425-018-2847-4
Yan, X. F., Zhao, L., Ren, Y., Dong, Z. Z., Cui, D. Q., and Chen, F. (2019). Genome-wide association study revealed that the TaGW8 gene was associated with kernel size in Chinese bread wheat. Sci. Rep. 9:2702. doi: 10.1038/s41598-019-38570-2
Yao, H. N., Xie, Q., Xue, S. L., Luo, J., Lu, J. K., Kong, Z. X., et al. (2019). HL2 on chromosome 7D of wheat (Triticum aestivum L.) regulates both head length and spikelet number. Theor. Appl. Genet. 132, 1789–1797. doi: 10.1007/s00122-019-03315-2
Zhang, B., Liu, X., Xu, W. N., Chang, J. Z., Li, A., Mao, X. G., et al. (2015). Novel function of a putative MOC1 ortholog associated with spikelet number per spike in common wheat. Sci. Rep. 5:12211. doi: 10.1038/srep12211
Zhang, H., Chen, J. S., Li, R. Y., Mao, X. G., Li, A., Wang, J. Y., et al. (2016). Conditional QTL mapping of three yield components in common wheat (Triticum aestivum L.). Crop J. 4, 220–228. doi: 10.1016/j.cj.2016.01.007
Zhang, Z., Li, J., Hu, N., Li, W., Qin, W., Li, J., et al. (2021). Spike growth affects spike fertility through the number of florets with green anthers before floret abortion in wheat. Field Crop. Res. 260:108007. doi: 10.1016/j.fcr.2020.108007
Zheng, X. W., Liu, C, Qiao, L., Zhao, J. J., Han, R., Wang, X. L., et al. (2020). The MYB transcription factor TaPHR3-A1 is involved in phosphate signaling and governs yield-related traits in bread wheat. J. Exp. Bot. 71, 5808–5822. doi: 10.1093/jxb/eraa355
Zhou, Y. P, Conway, B., Miller, D., Marshall, D., Cooper, A., Murphy, P., et al. (2017). Quantitative trait loci mapping for spike characteristics in hexaploid wheat. Plant Genome. 10, 1–15. doi: 10.3835/plantgenome2016.10.0101
Keywords: founder parent, Linfen 5064, wheat, grain number per spike, quantitative trait locus
Citation: Qiao L, Li H, Wang J, Zhao J, Zheng X, Wu B, Du W, Wang J and Zheng J (2022) Analysis of Genetic Regions Related to Field Grain Number per Spike From Chinese Wheat Founder Parent Linfen 5064. Front. Plant Sci. 12:808136. doi: 10.3389/fpls.2021.808136
Received: 03 November 2021; Accepted: 29 November 2021;
Published: 05 January 2022.
Edited by:
Xia Cui, Insititute of Vegetables and Flowers, Chinese Academy of Agricultural Sciences (CAAS), ChinaCopyright © 2022 Qiao, Li, Wang, Zhao, Zheng, Wu, Du, Wang and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weijun Du, ZHV3ZWlqdW42OEAxMjYuY29t; Juanling Wang, MTM5OTQyNjc1MDhAMTYzLmNvbQ==; Jun Zheng, c3hua3l6akAxMjYuY29t
†These authors have contributed equally to this work