 Jing-Na Ru1,2†Ze-Hao Hou2†Lei Zheng2Qi Zhao2Feng-Zhi Wang3
Jing-Na Ru1,2†Ze-Hao Hou2†Lei Zheng2Qi Zhao2Feng-Zhi Wang3 Jun Chen2Yong-Bin Zhou2
Jun Chen2Yong-Bin Zhou2 Ming Chen2
Ming Chen2 You-Zhi Ma2Ya-Jun Xi1*
You-Zhi Ma2Ya-Jun Xi1* Zhao-Shi Xu2*
Zhao-Shi Xu2*- 1State Key Laboratory of Crop Stress Biology for Arid Areas, College of Agronomy, Northwest A&F University, Yangling, China
- 2Institute of Crop Science, Chinese Academy of Agricultural Sciences/National Key Facility for Crop Gene Resources and Genetic Improvement, Key Laboratory of Biology and Genetic Improvement of Triticeae Crops, Ministry of Agriculture, Beijing, China
- 3Hebei Key Laboratory of Crop Salt-Alkali Stress Tolerance Evaluation and Genetic Improvement/Cangzhou Academy of Agriculture and Forestry Sciences, Cangzhou, China
DEAD-box RNA helicases constitute the largest subfamily of RNA helicase superfamily 2 (SF2), and play crucial roles in plant growth, development, and abiotic stress responses. Wheat is one of the most important cereal crops in worldwide, and abiotic stresses greatly restrict its production. So far, the DEAD-box RNA helicase family has yet to be characterized in wheat. Here, we performed a comprehensive genome-wide analysis of the DEAD-box RNA helicase family in wheat, including phylogenetic relationships, chromosomal distribution, duplication events, and protein motifs. A total of 141 TaDEAD-box genes were identified and found to be unevenly distributed across all 21 chromosomes. Whole genome/segmental duplication was identified as the likely main driving factor for expansion of the TaDEAD-box family. Expression patterns of the 141 TaDEAD-box genes were compared across different tissues and under abiotic stresses to identify genes to be important in growth or stress responses. TaDEAD-box57-3B was significantly up-regulated under multiple abiotic stresses, and was therefore selected for further analysis. TaDEAD-box57-3B was localized to the cytoplasm and plasma membrane. Ectopic expression of TaDEAD-box57-3B in Arabidopsis improved tolerance to drought and salt stress as measured by germination rates, root lengths, fresh weights, and survival rates. Transgenic lines also showed higher levels of proline and chlorophyll and lower levels of malonaldehyde (MDA) than WT plants in response to drought or salt stress. In response to cold stress, the transgenic lines showed significantly better growth and higher survival rates than WT plants. These results indicate that TaDEAD-box57-3B may increase tolerance to drought, salt, and cold stress in transgenic plants through regulating the degree of membrane lipid peroxidation. This study provides new insights for understanding evolution and function in the TaDEAD-box gene family.
Introduction
RNA helicases unwind double-stranded RNA in an ATP-dependent manner through hydrolysis (Lorsch, 2002; Gong et al., 2005; Cordin et al., 2006; Linder, 2006; Chen et al., 2014). The DEAD-box RNA helicases constitute the largest subfamily of RNA helicase superfamily 2 (SF2), and are ubiquitous in both prokaryotes and eukaryotes (Mingam et al., 2004; Rocak and Linder, 2004). Within the SF2 superfamily, DEAD-box, DEAH-box, and DExD/H-box families are distinguished by variations in motif II; DEAD-box RNA helicases are named after the highly conserved residues Asp-Glu-Ala-Asp (D-E-A-D) comprising motif II (Caruthers and McKay, 2002; Cordin et al., 2006; Linder and Owttrim, 2009). DEAD-box RNA helicases contain at least nine conserved motifs in the helicase core domain, which are involved in ATPase and helicase activities (Tanner et al., 2003; Banroques et al., 2011; Linder and Jankowsky, 2011; Putnam and Jankowsky, 2013). In addition to the conserved motifs, the N- and C-terminal extensions in the DEAD-box family vary in length and composition, and these play roles in substrate binding specificity, helicase activity, subcellular localization, and interaction with components (Cordin et al., 2006; Fairman-Williams et al., 2010; Banroques et al., 2011; Byrd and Raney, 2012).
DEAD-box RNA helicases have been reported to function in all aspects of RNA metabolism (Bowers et al., 2006; Cordin et al., 2006; Linder, 2006; Linder and Fuller-Pace, 2013; Putnam and Jankowsky, 2013; Russell et al., 2014). For example, DEAD-box RNA helicase AtRH57 may play a role in the formation of small ribosomal subunits (Hsu et al., 2014). DEAD-box RNA helicase RCF1 maintains proper splicing of pre-mRNAs (Guan et al., 2013). DEAD-box protein UAP56 interacts with the mRNA export factors ALY2 and MOS11, which are involved in exporting mRNA from the plant cell nucleus (Kammel et al., 2013). UPF2 eliminates aberrant mRNAs that are derived from various sources, which is necessary for the nonsense-mediated mRNA decay process (Shi et al., 2012). RH50 is involved in efficient translation of plastid proteins by comigrating with ribosomal particles, indicating that RH50 may act as a 23S-4.5S rRNA maturation factor, functionally overlapping with the plastid signaling factor GUN1 (Paieri et al., 2018). DEAD-box RNA helicases disrupt misfolded RNA structures, thus acting as chaperones to promote correct RNA folding through unwinding activity or RNA-protein association/dissociation (Lorsch, 2002; Gong et al., 2005; Kant et al., 2007).
DEAD-box RNA helicases have also been reported to participate in plant growth and development (Kant et al., 2007; Shi et al., 2012; Bush et al., 2015, 2016; Huang et al., 2016a,b; Chen D. et al., 2020). ISE2 is involved in posttranscriptional gene silencing and cell fate determination, affecting plasmodesmata structure and function through the regulation of RNA metabolism and subsequent gene expression (Kobayashi et al., 2007). AtRH7/PRH75-knockout mutants show auxin-related developmental defect phenotypes that have also been observed in several ribosomal protein mutants, indicating that AtRH7 may participate in rRNA biogenesis and plant development (Huang et al., 2016a). eIF4A interacts with cyclin-dependent kinase A (CDKA) in proliferating cells and is modulated by phosphorylation, thereby affecting translation. Loss of eIF4A1 function results in reduced plant growth and dramatically decreased fertility (Bush et al., 2015, 2016). An rh29 mutant was shown to have nonfunctional gametophytes and defective pollen tube growth in the pistil, implying that gametophyte-specific RH29 may play important roles in functional maturation of male and female gametophytes (Chen D. et al., 2020). Rice OsRH2 and OsRH34 were identified as core components of the exon junction complex (EJC) and are involved in regulating plant height and development of pollen and seeds in rice (Huang et al., 2016b).
Increasing evidence has revealed that DEAD-box RNA helicases play crucial roles in plant abiotic stress responses, including drought, salt, temperature, and oxidative stress. A los4-1 mutation has been shown to reduce cold regulation of C-repeat binding factors (CBFs) and their target genes, and mutants exhibit a chilling-sensitive phenotype. This indicates that CRYOPHYTE/LOS4 may play an important role in mRNA export and stress responses (Gong et al., 2005). strs1 and strs2 mutants have increased tolerance to salt, osmotic, and heat stresses, whereas STRS-overexpressing lines display less stress tolerance. STRS proteins are involved in epigenetic silencing of gene expression through re-localization to suppress the stress responses in Arabidopsis (Kant et al., 2007; Khan et al., 2014). AtRH9 and AtRH25 affect seed germination under salt stress, and different nucleic acid binding properties lead to differences in cold tolerance (Kim et al., 2008). Rice OsPDH45 mediates salinity stress tolerance by controlling the antioxidant system and protecting the photosynthetic machinery (Gill et al., 2013). The rice nucleolar DEAD-box RNA helicase TOGR1 is regulated by temperature and circadian rhythm, and is associated with the small subunit (SSU) to facilitate effective pre-rRNA processing; it is therefore involved in regulating rice thermotolerant growth as a key pre-rRNA chaperone for rRNA homeostasis (Wang D. et al., 2016). Additionally, OsTOGR1 may be a positive factor for regulation of heat stress tolerance in Chinese cabbage (Yarra and Xue, 2020). tcd33 displays an albino phenotype and severe defects of chloroplast structure under 20°C condition; however, overexpression of TCD33 confers cold tolerance by regulating rice chloroplast development and the expression of cold-responsive genes (Wang X. M. et al., 2020).
Genome-wide analyses of the RNA Helicase gene family have been undertaken in Arabidopsis, rice, tomato, maize, cotton, and sweet potato (Umate et al., 2010; Xu et al., 2013a,b; Wan et al., 2020). However, few reports exist describing identification and functional analysis of DEAD-box genes in wheat. Wheat is one of the most important cereal crops worldwide, serving as a key staple crop for humans (Oono et al., 2013; Marcussen et al., 2014). Severe abiotic stresses limit wheat production (Zhan et al., 2017). Because wheat now has a fully annotated reference genome, studies related to gene function are now more feasible than they were previously (Appels et al., 2018). Identifying stress resistance genes and exploring their functions are important goals towards improving stress responses through wheat molecular breeding. In this study, we revealed that TaDEAD-box57-3B improved diverse abiotic stresses resistance in transgenic Arabidopsis through regulating the degree of membrane lipid peroxidation. This study provides a basis for better understanding the TaDEAD-box gene family and the function of TaDEAD-box57-3B in response to abiotic stresses.
Materials and Methods
Identification of DEAD-box Genes in Wheat
The identification of wheat DEAD-box gene family was performed according to the previous methods described by Yu et al. (2019) with some revisions. Firstly, to identify the TaDEAD-box genes in wheat, protein sequences of all DEAD-box genes in Arabidopsis and rice were downloaded from TAIR - Home Page (TAIR)1 and Rice Genome Annotation Project (RGAP) databases2 with National Center for Biotechnology Information (NCBI) as a complementary database3. Wheat reference sequences were downloaded from the Ensembl Plants database4 to build a local protein database. The protein sequences of all Arabidopsis and rice were used as queries to perform local BLASTp searches (with e-value < 1e-10) against local wheat database. Secondly, the candidate sequences were used to build a wheat-specific HMM profile through hmmbuild program. Then the hmmsearch program was used to obtain all TaDEAD-box candidate members. All candidate proteins were submitted to NCBI Batch CD-search5 and Pfam databases6 for examining the presence of DEAD-box domain. We identified all candidate sequences with the conserved D-E-A-D motif. After validation, all candidate TaDEAD-box genes were identified in the wheat genome (Supplementary Table 1). The molecular weight (MW) and isoelectric point (pI) values were predicted by online tool ExPASy7. Subcellular localizations were predicted through online tools BUSCA8.
Multiple Sequence Alignment and Phylogenetic Analysis
Multiple sequence alignment of amino acid sequences was performed using ClustalX, and then optimum model was used to generate the interspecific phylogenetic tree (Arabidopsis, rice, wheat) by MEGA X based on the maximum-likelihood (ML) method with 1000 bootstrap replicates to determine the statistical reliability of the phylogenetic trees. In addition, the phylogenetic tree of TaDEAD-box family was built by MEGA X based on neighbor-joining (NJ) method with 1000 bootstrap replicates to identify the consistency of phylogenetic tree.
Chromosomal Location and Gene Duplication
The wheat annotated reference genome sequences were downloaded from Ensembl Plants database. All TaDEAD-box genes were mapped against the 21 wheat chromosomes using positional information by TBtools software (Chen C. et al., 2020). Homoeologous genes were identified through phylogeny tree and Ensembl Plants database (Schilling et al., 2020). Segmental and tandem duplication events were recognized according to the previous study (Wang M. et al., 2016; Fan et al., 2019). Related synteny blocks and duplicated gene pairs were calculated using MCScanX (e-value ≤ 1e-10) and visualized using Circos software (Krzywinski et al., 2009). The nonsynonymous substitution rate (Ka), synonymous substitution rate (Ks) and the Ka/Ks ratio between duplicated gene pairs were calculated by TBtools software. Ka/Ks ratio > 1 represents positive selection, Ka/Ks ratio = 1 represents neutrality, and Ka/Ks ratio of < 1 represents purifying selection.
Gene Structure and Conserved Motifs
The coding sequences and genome sequences of TaDEAD-box genes were obtained from the Ensembl Plants database, and the exon/intron structure was visualized by TBtools software (Chen et al., 2018). The online MEME program9 with 15 motifs and motif width of 6-50 amino acids was applied to identify the conserved motifs of all members (Bailey et al., 2009). The results were visualized and rearranged by TBtools software (Chen C. et al., 2020).
Cis-Elements Analysis
To analysis the possible biological functions and transcriptional regulation of 141 TaDEAD-box genes. The 2.0 kb region sequences upstream from start codons were downloaded from Ensembl Plants database and then submitted to PlantCARE database for cis-elements analysis10 (Lescot et al., 2002).
Expression Pattern Analysis by Transcriptomic Data
Transcriptomic data (choulet_URGI) used to analyze the expression patterns of TaDEAD-box genes in different tissues and stages were downloaded from Wheat Expression browser11 database (Borrill et al., 2016; Ramirez-Gonzalez et al., 2018). Besides, transcriptomic data (SRP045409, SRP043554) were also extracted to study the expression level of TaDEAD-box genes under abiotic stresses (Li et al., 2015; Liu et al., 2015). The expression patterns of TaDEAD-box genes were estimated using (log2TPM+1) (transcripts per million) values through pheatmap package of R project13. The TPM values of TaDEAD-box genes in different tissues and under abiotic stresses (cold, drought, high-temperature) were shown in Supplementary Table 7.
Plant Materials and Stress Treatments
The wheat cultivar Xiaobaimai grown in Hoagland’s liquid medium was used for gene expression patterns analysis. The wild type Arabidopsis cultivar Columbia-0 was used as background for transformation and subsequent phenotypic assays. All the plants were grown in a greenhouse with 60% relative humidity, 22°C, and a photoperiod of 16-h light/8-h dark. Seven-day-old wheat seedlings were used for dehydration (250 mM Mannitol), salt (200 mM NaCl), heat (42°C), and cold (4°C) stress treatments. Seedlings were transferred into solutions containing 250 mM Mannitol or 200 mM NaCl for dehydration or salt treatment. Seedlings were placed in 42°C or 4°C incubator for heat or cold treatment. The samples were collected at 0, 0.5, 1, 2, 4, 8, 12, 24 h after treatments, and then immediately frozen in liquid nitrogen and stored at -80°C for RNA extraction.
RNA Isolation and Quantitative Real-Time PCR
Total RNA was extracted from wheat leaves using RNA plant extraction kit (Zhuangmeng, Beijing, China), and Transcript® All-in-One First-Strand cDNA Synthesis SuperMix (TransGen Biotech, Beijing, China) was used for reverse transcription following the manufacturer’s instruction. Quantitative Real-Time PCR (qRT-PCR) was performed on an Applied Biosystems 7500 Real-Time PCR System with Super Real PreMix Plus (SYBR Green) (TIANGEN, China). The wheat β-actin (GenBank: MF405765.1) was used as an internal control for normalization of the template cDNA. Each experiment was performed with three biological replicates. The quantitative results analysis was performed using the 2–ΔΔCT method (Udvardi et al., 2008). The primers used for qRT-PCR are listed in Supplementary Table 8.
Subcellular Localization
The full-length coding sequence (CDS) of TaDEAD-box57-3B was amplified from wheat cultivar Xiaobaimai and Jinhe991, and no allelic variations were observed. The CDS excluding the termination codons of TaDEAD-box57-3B was amplified and cloned into p16318hGFP vector under control of the CaMV35S promoter. For transient expression assays, 7-day-old wheat seedlings were used for the isolation of wheat protoplasts as previously described (Liu P. et al., 2013). The p16318hGFP-TaDEAD-box57-3B and control plasmids were transformed into wheat protoplasts mediated by polyethylene glycol 4000 (PEG4000) as described by Liu P. et al. (2013). The transfected protoplasts were incubated at 22°C for 18 h in darkness, after which GFP signals were observed with 488 nm and 543 nm illumination by a confocal laser scanning microscopy (LSM700; CarlZeiss). The primers are listed in Supplementary Table 8.
Generation of Transgenic Arabidopsis
The CDS excluding the termination codons of TaDEAD-box57-3B was cloned into plant expression vector pCAMBIA1302 under the control of CaMV35S promoter. After sequencing, the correct plasmid pCAMBIA1302-TaDEAD-box57-3B was transformed into Arabidopsis by Agrobacterium tumefaciens-mediated floral dip method (Clough and Bent, 1998). The transformed seeds were surface sterilized with sodium hypochlorite and selected on 1/2 MS medium containing 50 ug/ml hygromycin and then transferred to soil (rich soil: vermiculite = 1:1). T3 generation plants were plants were cultured at 22°C and 60% relative humidity with a photoperiod of 16-h light/8-h dark. Three homozygous T3 lines were selected for the following phenotypic analysis.
The Abiotic Stress Treatments of Transgenic Arabidopsis
For the germination assay, T3 generation seeds of transgenic lines and WT were grown on Murashige and Skoog (MS) and MS medium supplemented with 300 mM, 400 mM mannitol, 100mM, and 125 mM NaCl. The seeds were kept at 4°C for 3 d to break dormancy, and then transferred to a tissue culture room with a photoperiod of 16-h light/8-h dark at 22°C (60% relative humidity; 100 μmol m–2 s–1 light intensity). We defined the seed germinated when radicles had emerged from the seed coat. Seed germination was followed for the next 6 days, and the germination rate was calculated as a percentage. For the root growth assay, 5-day-old seedlings were transferred to MS medium with or without 200 mM, 300 mM mannitol, 100mM, and 125 mM NaCl for another 7 days, and then the total root lengths and fresh wights were measured. All experiments contained three independent replicates.
For tolerance tests, the disinfected seeds were planted in MS medium for 7 days, and then transplanted to the soil (1:1 mixture of rich soil and vermiculite) at 22°C with a photoperiod of 16-h light/8-h dark. The drought phenotype analysis was performed as described (Liu W. X. et al., 2013). For drought treatment, 21-day-old seedlings were treated by withdrawing irrigation until there were significant wilting differences between WT and transgenic plants. The survival rates were measured after re-watering for 7 days. For salt treatment, 7-day-old seedlings were transferred to the soil at 22°C with a photoperiod of 16-h light/8-h dark, and then 21-day-old seedlings were irrigated with 200 mM NaCl until significant differences were observed between WT and transgenic plants. The survival rates were recorded and all the assays were repeated three times.
For cold treatment, disinfected seeds were planted in MS medium for 5 days, and then were placed at 4°C for 1 d followed by −8°C for 1 h. After freezing treatment, the plants were transferred to 4°C for 12 h and then resumed normal grown at 22°C for additional 3 days. The survival rates were recorded and all the tests contained three independent replicates.
Measurements of Chlorophyll, Proline, and Malonaldehyde (MDA) Contents
To better understand the function of TaDEAD-box57-3B under abiotic stresses, the physiological indicators (chlorophyll, proline, MDA contents) were measured according to the manuals of the corresponding kit (CPL-1-G, PRO-1-Y, MDA-1-Y, Cominbio, Suzhou, China). After drought treatment for 2 weeks and salt treatment for 1 week, the leaves were mixed and collected for physiological indicator measurement. Absorbance values were measured with a Varioskan LUX Multimode Microplate Reader (Thermo Fisher Scientific). All of the experiments were repeated three times.
Statistical Analysis
All the experiments contained three independent replicates. Values are means ± standard deviations (SDs) of three replicates, and asterisks (∗ or ∗∗) represent the significant differences at p < 0.05 or p < 0.01, respectively (ANOVA test).
Results
Identification of TaDEAD-box Genes in Wheat
Based on conserved domains and motifs, a total of 141 TaDEAD-box candidate genes were identified in the wheat genome (Triticum aestivum IWGSC). For convenience, these candidate genes were renamed based on their chromosomal locations, from TaDEAD-box1-1A to TaDEAD-box141-7D. Detailed information and physicochemical properties of these genes are summarized in Supplementary Table 1. The amino acid sequences ranged from 354 to 1347 in length, with an average of 647. Physicochemical properties varied greatly in the TaDEAD-box family. The molecular weight (MW) values ranged from 39.04 kDa to 148.50 kDa, with an average of 71.55 kDa, and isoelectric point (pI) values ranged from 4.92 to 10.17, with 76.6% of members (108/141) exhibiting alkaline pI values.
Phylogenetic and Homologous Analysis of TaDEAD-box Members
To analyze evolutionary relationships among the 141 TaDEAD-box proteins, phylogenetic analysis was conducted using an additional 57 DEAD-box proteins from Arabidopsis and 47 from rice, for a total of 245 genes (Figure 1). A separate phylogenetic tree was also built using only the TaDEAD-box proteins for comparison. Based on amino acid sequence similarities and the phylogenetic analysis from Arabidopsis, DEAD-box members were divided into nine groups (I-IX). Group I harbored the most DEAD-box members of the three species, implying that genes in Group I were frequently retained during evolution.
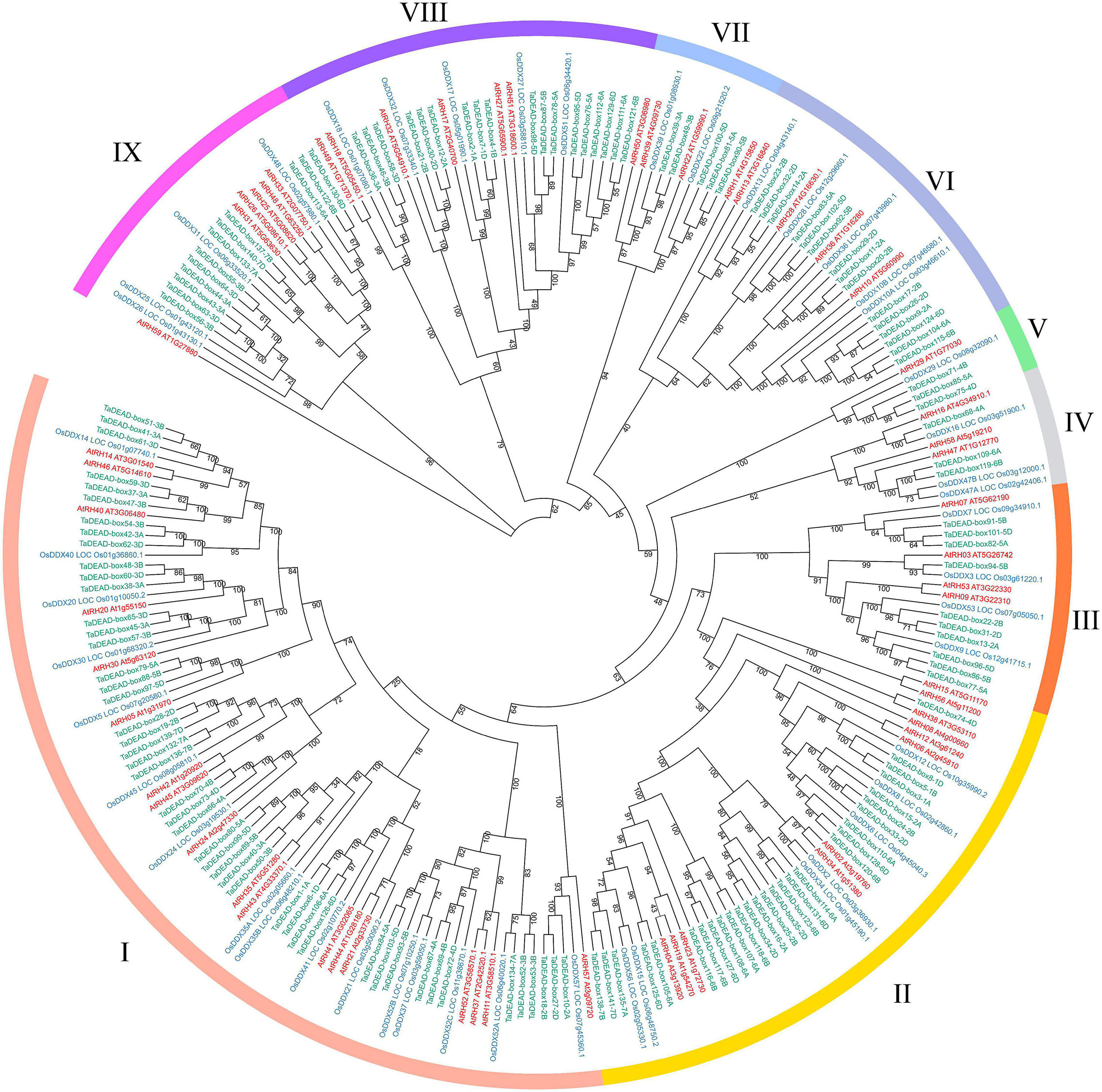
Figure 1. Phylogenetic analysis of DEAD-box proteins in wheat, rice and Arabidopsis. A total of 245 proteins were used to construct the maximum-likelihood (ML) phylogenetic tree by MEGA X with 1,000 bootstrap replicates. The DEAD-box proteins were divided into nine groups (I-IX), which were marked with different colors. The members of three species were, respectively, marked with different colors: red for Arabidopsis, blue for rice, and green for wheat.
Because there are three homoeologous subgenomes in common wheat (A, B, and D), every wheat gene may be part of a three-homoeolog triad (Yu et al., 2019). We identified homoeologous genes through phylogenetic tree construction and BLAST searches. The results revealed that the 141 TaDEAD-box genes represented 141 homoeologs of 55 genes: 38 genes had all three homoeologs, 10 genes had two homoeologs, and seven genes had only one homoeolog (Table 1). Prior research showed that 35.8% of genes in the wheat genome have three homoeologous genes (Appels et al., 2018; Xu et al., 2021), meaning that TaDEAD-box homoeologs are retained at a higher rate; 69.1% of TaDEAD-box genes vs. 35.8% of genes on average are present in triads, 18.2% vs. 13.2% are present in pairs, and 12.7% vs. 37.1% are present as a single gene. Consequently, the high homoeolog retention rate may explain why the number of DEAD-box genes in wheat is much higher than it is in Arabidopsis and rice.

Table 1. Distribution pattern of TaDEAD-box homoeologs.
Chromosomal Location and Duplication Events of TaDEAD-box Genes
The 141 TaDEAD-box genes were unevenly distributed across all 21 wheat chromosomes (Figure 2). A total of 9, 27, 30, 10, 28, 28, and 10 genes were present on chromosomes 1 through 7, respectively, and the numbers of TaDEAD-box genes per chromosome ranged from 2 to 12 (Supplementary Figure 1). Chromosome 1B contained the fewest TaDEAD-box genes (∼1.4%), whereas chromosome 2B contained the most (∼8.5%), followed by 6A (∼7.8%). These results suggested that duplication of TaDEAD-box genes may have occurred more often on wheat chromosomes 2, 3, 5, and 6. TaDEAD-box genes clustered together in one region of the chromosome may belong to the same group. For example, TaDEAD-box107-6A/TaDEAD-box108-6A and TaDEAD-box117-6B/TaDEAD-box118-6B, which belonged to Group II, were tightly linked on chromosome 6. TaDEAD-box55-3B/TaDEAD-box56-3B and TaDEAD-box63-3D/TaDEAD-box64-3D belonged to group IX and were tightly linked on chromosome 3 (Figure 2). Chromosome translocation and inversion analysis showed that one triad (TaDEAD-box70-4B/TaDEAD-box73-4D/TaDEAD-box66-4A) was involved in inversion between the long and short arms of chromosome 4A, and another triad (TaDEAD-box85-5A/TaDEAD-box71-4B/TaDEAD-box75-4D) was involved in translocation between the long arms of chromosomes 4A and 5A (Supplementary Table 2).
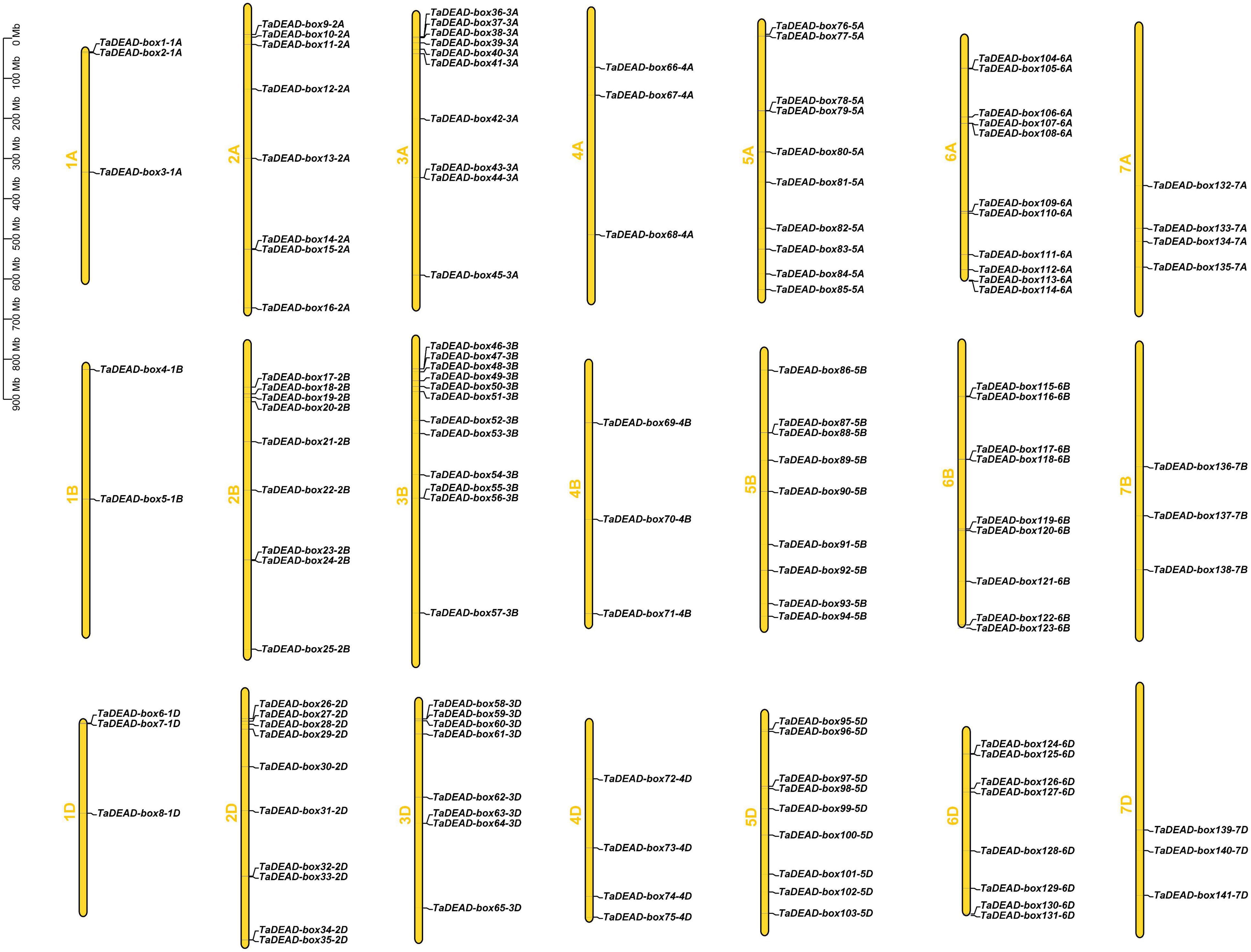
Figure 2. Chromosomal distribution of the 141 TaDEAD-box genes. The 141 TaDEAD-box genes were unevenly distributed on all 21 wheat chromosomes. The chromosome numbers are denoted left of each chromosome, and the bar locates on the left side shows the size of chromosome in megabases (MB).
To elucidate the expansion mechanism within this gene family, we conducted collinearity analysis of TaDEAD-box genes within the wheat genome. Using the definitions of Wang M. et al. (2016) and Fan et al. (2019), we evaluated tandem and segmental duplication events of TaDEAD-box genes. There were 128 genes located within syntenic blocks, forming 138 pairs of duplicated genes (Figure 3 and Supplementary Table 3). Approximately 79.3% (107/135) of the duplicated TaDEAD-box genes were clustered on chromosomes 2, 3, 5, and 6, and Group I contained the most duplicated genes, consistent with the results above. Specifically, ∼4.3% (6/141) of TaDEAD-box genes that were located adjacent to one another on the same chromosome were derived from tandem duplication events, forming three tandem duplicated pairs: TaDEAD-box55-3B/TaDEAD-box56-3B, TaDEAD-box63-3D/TaDEAD-box64-3D, and TaDEAD-box117-6B/TaDEAD-box118-6B. In addition, we further observed that ∼88.7% (125/141) of TaDEAD-box genes were derived from whole genome duplication (WGD)/segmental duplication, such as TaDEAD-box135-7A/TaDEAD-box138-7. It thus appears that WGD/segmental duplication is the main driving factor behind expansion of TaDEAD-box genes in the wheat genome.
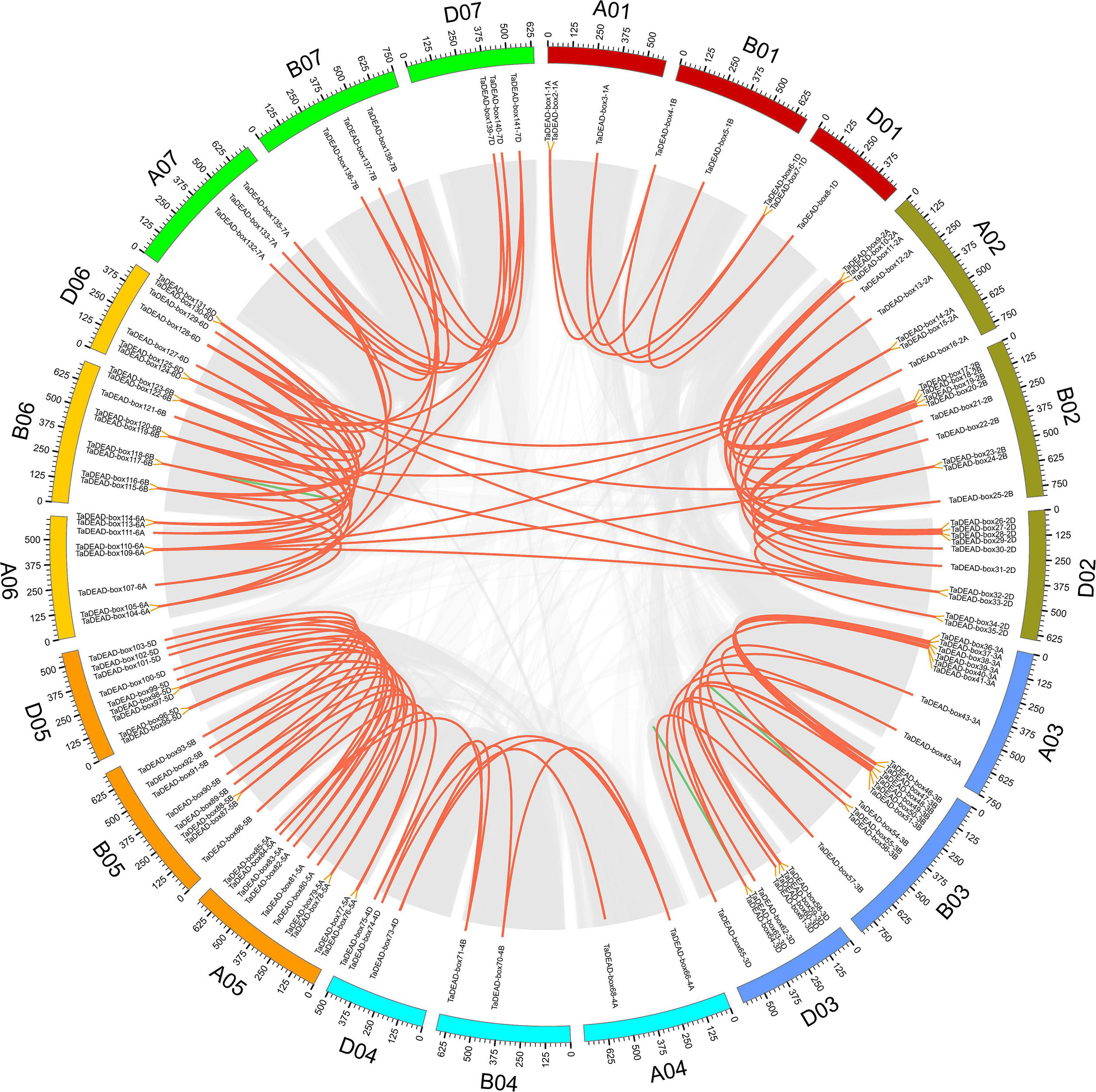
Figure 3. Distribution and duplication events of TaDEAD-box genes across wheat genome. All TaDEAD-box genes were mapped to 21 wheat chromosomes in a circle using Circos, and WGD/segmental and tandem duplications were mapped to their respective locations. Gray regions indicate all synteny blocks within the wheat genome, red and green lines, respectively, represent WGD/segmental and tandem duplications. The chromosome numbers are marked inside of the circle.
To investigate the evolutionary forces acting on the TaDEAD-box genes, the Ka/Ks ratios were calculated for duplicate gene pairs. The Ka/Ks ratios of all except one duplicate pair were less than 1 and ranged from 0 (TaDEAD-box135-7A/TaDEAD-box138-7B) to 0.62 (TaDEAD-box117-6B/TaDEAD-box127-6D) (Figure 4). The Ka/Ks ratios of the three TaDEAD-box tandem duplicated gene pairs ranged between 0.22 and 0.54 (Supplementary Table 3). These results revealed that most duplicated TaDEAD-box genes were under purifying selection.
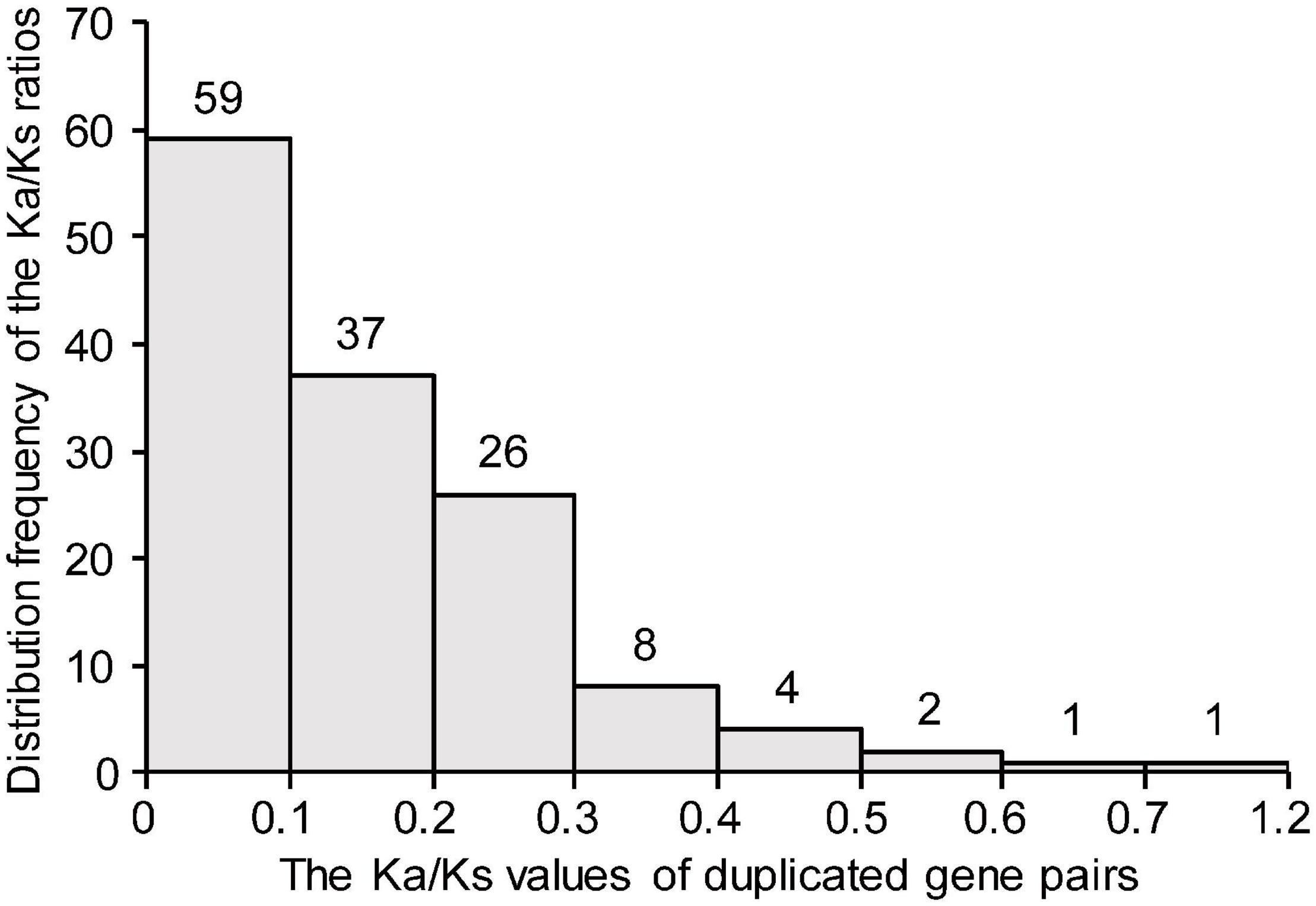
Figure 4. Histogram of distribution frequency of pairwise Ka/Ks ratios for duplicated TaDEAD-box genes. Ka, nonsynonymous substitution rate; Ks, synonymous substitution rate.
Gene Structure and Conserved Motif Analysis of TaDEAD-box Members
Exon-intron structures and conserved motifs were compared between the 141 TaDEAD-box members (Figure 5). Differences in exon-intron structure are associated with the evolution and function among members of the gene family. Except for five genes that contained no intron, TaDEAD-box genes had between 1 and 15 introns (Figure 5 and Supplementary Table 4). TaDEAD-box32-2D harbored the most exons and introns (16 and 15, respectively). Members of Groups I and II had the most introns, followed by Group VIII and VII. Members of Group I ranged from 1-13 and had an average of six introns, except for one gene with no intron. Members of Group II had between three and nine introns, with 75% (21/28) having six or more. There was significant variation among members of Group VII; three genes had no intron, three genes contained five to six introns, and the others contained more than 11. In Group VIII, almost 88.9% (16/18) genes contained 7 or more introns, except for one gene contained two introns and the other with no intron (Figure 5). Genes in the same branch were homeologs and had similar gene structure, suggesting that exon-intron structure was highly correlated with phylogenetic relationship. However, a few exceptions existed among homologous genes; for example, TaDEAD-box130-6D, TaDEAD-box113-6A, and TaDEAD-box122-6B had differences in exon and intron numbers.
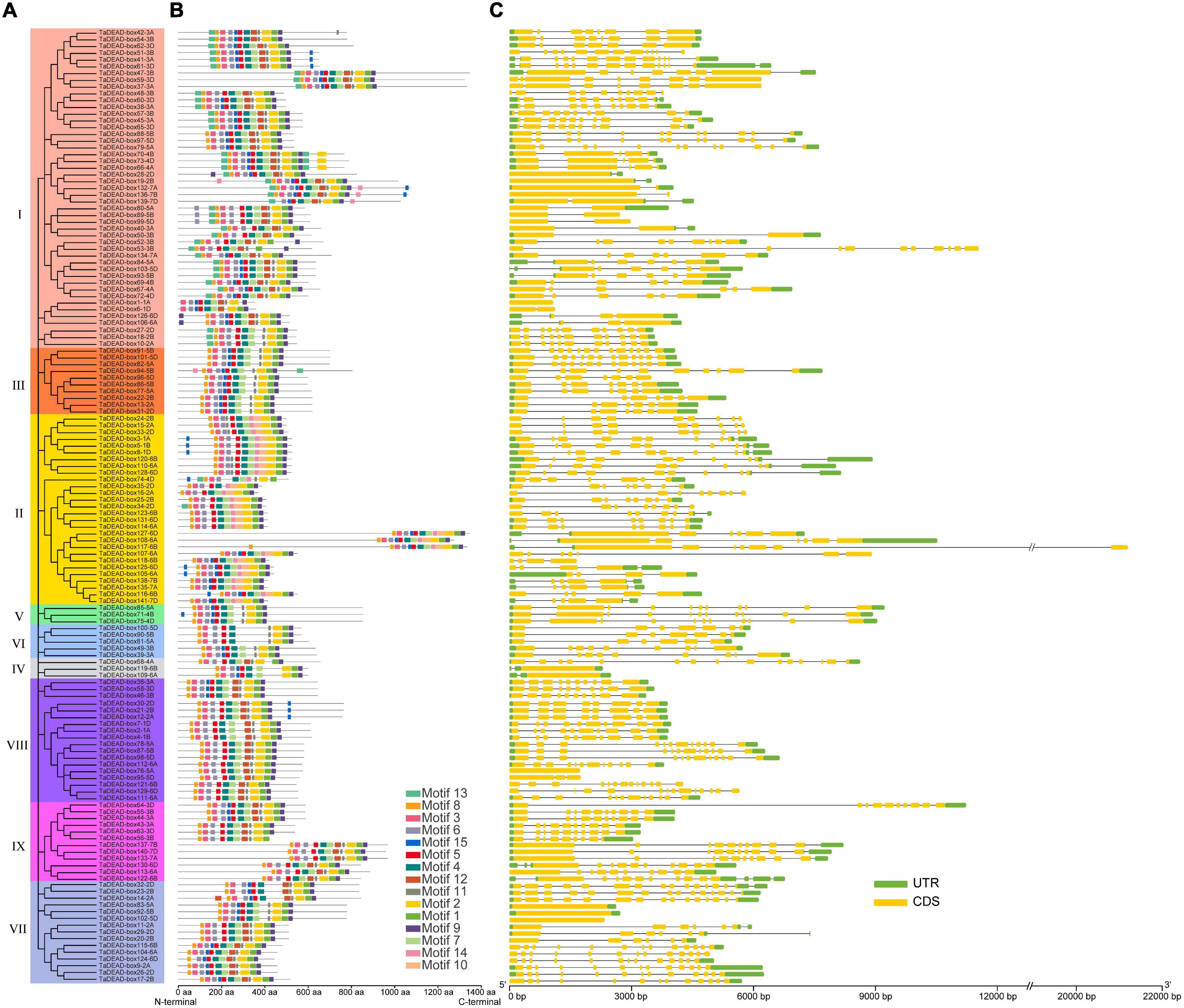
Figure 5. Phylogenetic tree, motif distribution and gene structure analysis of 141 TaDEAD-box members. (A) Phylogenetic tree of DEAD-box members in wheat. A total of 141 TaDEAD-box proteins were used to construct the neighbor-joining (NJ) phylogenetic tree by MEGA X with 1,000 bootstrap replicates. The different groups are marked with different colors. (B) Motif distribution of 141 TaDEAD-box proteins. The different colored boxes represent 15 motifs, and the box lengths represent motif lengths. (C) The exon-intron structure analysis of 141 TaDEAD-box genes. Exon and introns lengths are displayed proportionally, yellow boxes represent exons, black lines represent introns, and green boxes represent 5′ and 3′ UTR.
Fifteen conserved motifs with 6-50 amino acids each were identified using MEME (Figure 5 and Supplementary Table 5). Motifs 1, 2, 3, 4, 5, 6, 8, 7, and 9 were present in almost all proteins, and motif 4 contained the highly conserved sequence residues Asp-Glu-Ala-Asp (D-E-A-D) (Figure 5). In addition, motif 11 was present in all members, with the exception of Group II, where less than 22% members contained motif 11. However, some motifs were specific to individual groups; for example, motif 10 was only found in Group II, and motifs 13 and 14 were only found in Groups I, II, and III. Group-specific motifs such as these may account for functional diversity of individual groups. Interestingly, the identified motifs were concentrated close to the C-terminal in TaDEAD-box108-6A/TaDEAD-box117-6B/TaDEAD-box 127-6D (Group II) and TaDEAD-box133-7A/TaDEAD-box137-7B/TaDEAD-box140-7D (Group IX). These results indicate that TaDEAD-box proteins in the same subclade often had similar motif composition, which was consistent with their phylogenetic relationship. However, a few exceptions were found in homologous proteins. For example, motif 3 was found in TaDEAD-box115-6B and TaDEAD-box104-6A, but not in TaDEAD-box124-6D. Within the same branch, the proteins contained different motifs, and the genes encoding them also differed in the arrangement of introns and exons (e.g., TaDEAD-box1-1A/TaDEAD-box6-1D, TaDEAD-box56-3B/TaDEAD-box63-3D, and TaDEAD-box136-7B/TaDEAD-box139-7D. These differences may be due to the evolution of the TaDEAD-box members in wheat. Overall, our phylogenetic analysis revealed that phylogeny and function were associated with the diversity of exon-intron structure and structural motif distribution.
Promoter Cis-Element Analysis of TaDEAD-box Genes
Gene expression is often regulated by cis-elements in the promoter region. We found 3,178 potential cis-elements in the promoter regions of the 141 TaDEAD-box genes (Supplementary Table 6). These were divided into four broad categories: light-responsive elements (44.0%), hormone-responsive elements (40.1%), environmental stress-responsive elements (8.5%), and plant growth-related elements (7.4%) (Supplementary Figure 2). Among the 269 environmental stress-responsive elements, the proportion of low temperature-responsive elements was highest (45.4%), followed by drought response (40.9%), and wound-responsive elements accounted for the lowest proportion (1.1%). Among the 1275 hormone-responsive elements, methyl jasmonate (MeJA)-responsive elements accounted for the highest proportion (46.0%), with a lower proportion of abscisic acid-responsive elements (33.0%). These results show that TaDEAD-box genes may be involved in plant abiotic stress responses, especially low temperature and drought stress (Supplementary Figure 2). In addition, among the plant growth-related elements, meristem expression elements were the most abundant, followed by metabolism regulation elements, suggesting involvement of TaDEAD-box genes in plant growth and development.
Expression Patterns Analysis of TaDEAD-box Genes
In order to study the expression patterns of TaDEAD-box genes in different tissues and developmental stages, transcriptomic data (choulet_URGI) were analyzed and a tissue-specific expression clustering heatmap generated (Figure 6A and Supplementary Table 7). Some genes displayed low transcription levels, including TaDEAD-box80-5A, 89-5B, 99-5D, 112-6A, 111-6A, 104-6A, and 19-2B, which clustered together, and TaDEAD-box6-1D, which was not expressed in the examined tissues and developmental stages. Some genes exhibited tissue-specific expression, for instance, TaDEAD-box44-3A, 55-3B, 90-5B, 100-5D, 109-6B, 81-5A, 39-3A and 49-3B, which clustered together and were highly expressed in the leaf. However, some genes were constitutively expressed in wheat: TaDEAD-box135-7A, 138-7B, 141-7D, 57-3B, 65-3D, 82-5A, 91-5B, 101-5D, 116-6B, 105-6A, 125-6D, 123-6B, 114-6A, 131-6D, 93-5B, 84-5A, and 103-5D clustered together and displayed high transcription levels in most tissues throughout plant development (Figure 6A). In general, TaDEAD-box genes exhibited a great deal of tissue-specific expression, indicating that they may be involved in adaptation to different physiological processes.
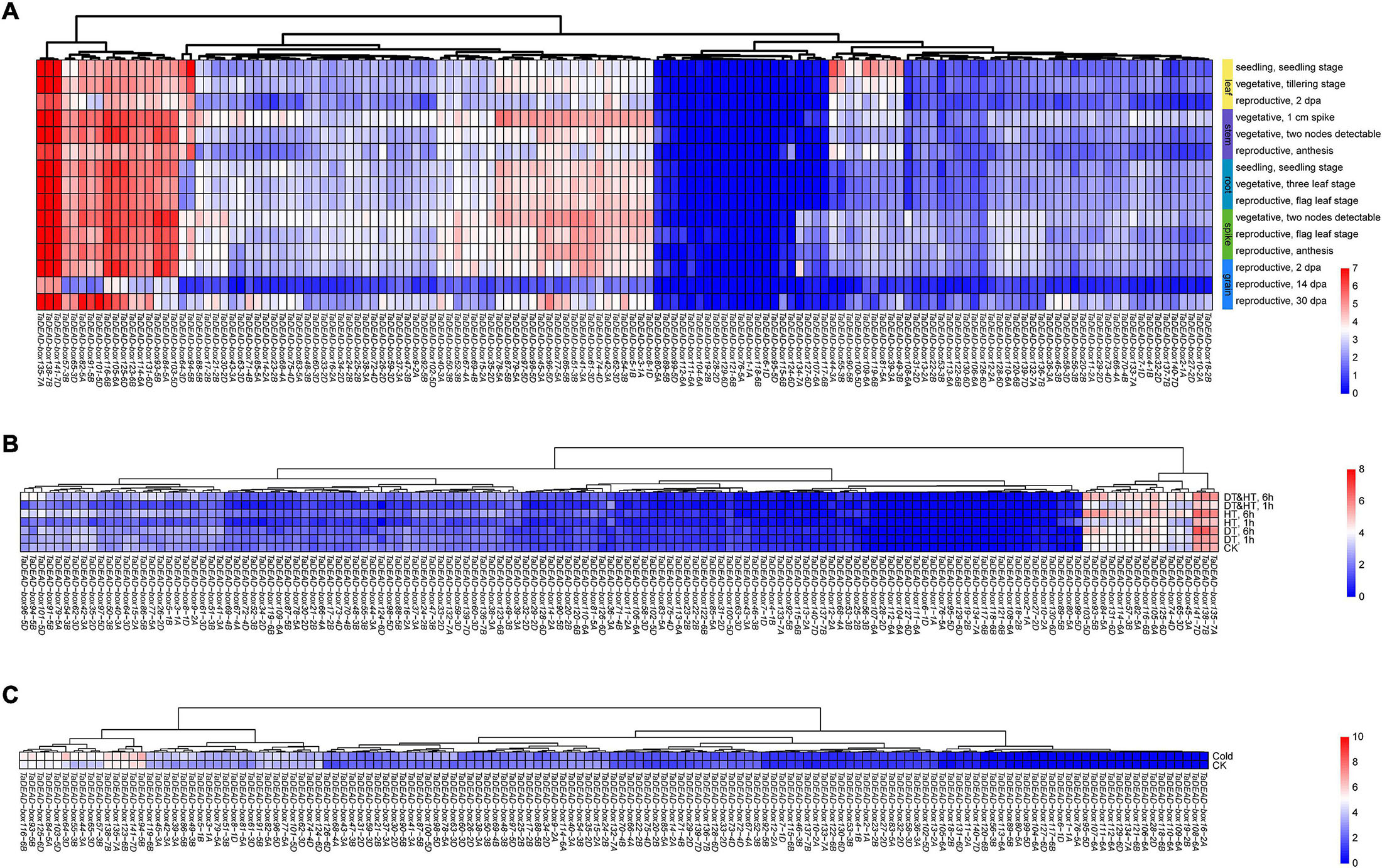
Figure 6. Hierarchical clustering of expression patterns of 141 TaDEAD-box genes using transcriptome data. The expression pattern heatmaps of TaDEAD-box genes were generated through pheatmap package of R project13. The expression levels are indicated by the different color bars, red: higher expression, blue: lower expression. (A) Expression patterns of 141 TaDEAD-box genes in different tissues and development stages. Columns represent 141 TaDEAD-box genes, and rows represent different developmental tissues/stages, including leaf, stem, root, spike, and grain. (B) Expression patterns of 141 TaDEAD-box genes under drought and high-temperature treatments. Columns represent 141 TaDEAD-box genes, and rows represent control and stress treatments. (C) Expression patterns of 141 TaDEAD-box genes under cold treatment. Columns represent 141 TaDEAD-box genes, and rows represent control and cold treatment. CK, control check; DT, drought treatment; HT, high-temperature treatment.
In order to analyze the expression patterns of TaDEAD-box genes under abiotic stresses, transcriptomic data (SRP045409, SRP043554) were analyzed with an abiotic stress expression clustering heatmap (Figures 6B,C and Supplementary Table 7). A large number of genes were expressed at low levels under drought, high temperature, and cold treatments, including TaDEAD-box117-6B, 127-6D, 118-6B, and 19-2B, which clustered together. TaDEAD-box1-1A, 6-1D, 76-5A, and 95-5D were not detectable at all either before or after stress treatments. However, 16 genes were differentially expressed under drought and high-temperature treatments (TaDEAD-box135-7A, 138-7B, 141-7D, 45-3A, 65-3D, 74-4D, 125-6D, 105-6A, 116-6B, 82-5A, 57-3B,114-6A, 131-6D, 84-5A, 93-5B, 103-5D) (Figure 6B), and a further 15 genes were differentially expressed under cold treatment (TaDEAD-box94-5B, 141-7D, 123-6B, 135-7A, 138-7B, 57-3B, 65-3D, 44-3A, 55-3B, 64-3D, 103-5D, 84-5A, 125-6D, 93-5B, 116-6B) (Figure 6C). We selected 10 genes for further analysis that responded to abiotic stresses and were highly expressed throughout growth and development: TaDEAD-box141-7D, 135-7A, 138-7B, 57-3B, 65-3D, 125-6D, 103-5D, 93-5B, 84-5A, and 116-6B.
TaDEAD-box Genes Were Involved in Abiotic Stress Responses
Expression of the 10 selected genes was studied at a finer scale with qRT-PCR (Figure 7), and the primers are listed in Supplementary Table 8. Results showed differing responses to abiotic stresses. Under dehydration treatment, TaDEAD-box57-3B, TaDEAD-box84-5A and TaDEAD-box116-6B were significantly up-regulated (> 5-fold) and peaked at 24 h, 8 h, and 24 h, respectively (Figure 7A). Aside from TaDEAD-box103-5D, the remaining six genes showed a slight response to dehydration treatment (Figure 7A). After salt treatment, TaDEAD-box57-3B and TaDEAD-box93-5B were up-regulated (> 5-fold), whereas TaDEAD-box116-6B, TaDEAD-box125-6D, and TaDEAD-box135-7A were down-regulated (Figure 7B). Under cold stress, five genes were significantly up-regulated (> 30-fold); remarkably, TaDEAD-box57-3B, TaDEAD-box65-3D, and TaDEAD-box116-6B were up-regulated more than 80-fold, suggesting that these genes may play an important role in cold response (Figure 7C). However, TaDEAD-box135-7A, TaDEAD-box103-5D, and TaDEAD-box141-7D were down-regulated after cold treatment (Figure 7C). Following heat treatment, five genes were up-regulated (> 5-fold); TaDEAD-box125-6D showed the highest expression levels (> 60-fold) after heat treatment for 24 h (Figure 7D). TaDEAD-box57-3B was significantly up-regulated under all tested treatments and was therefore selected for further analysis.
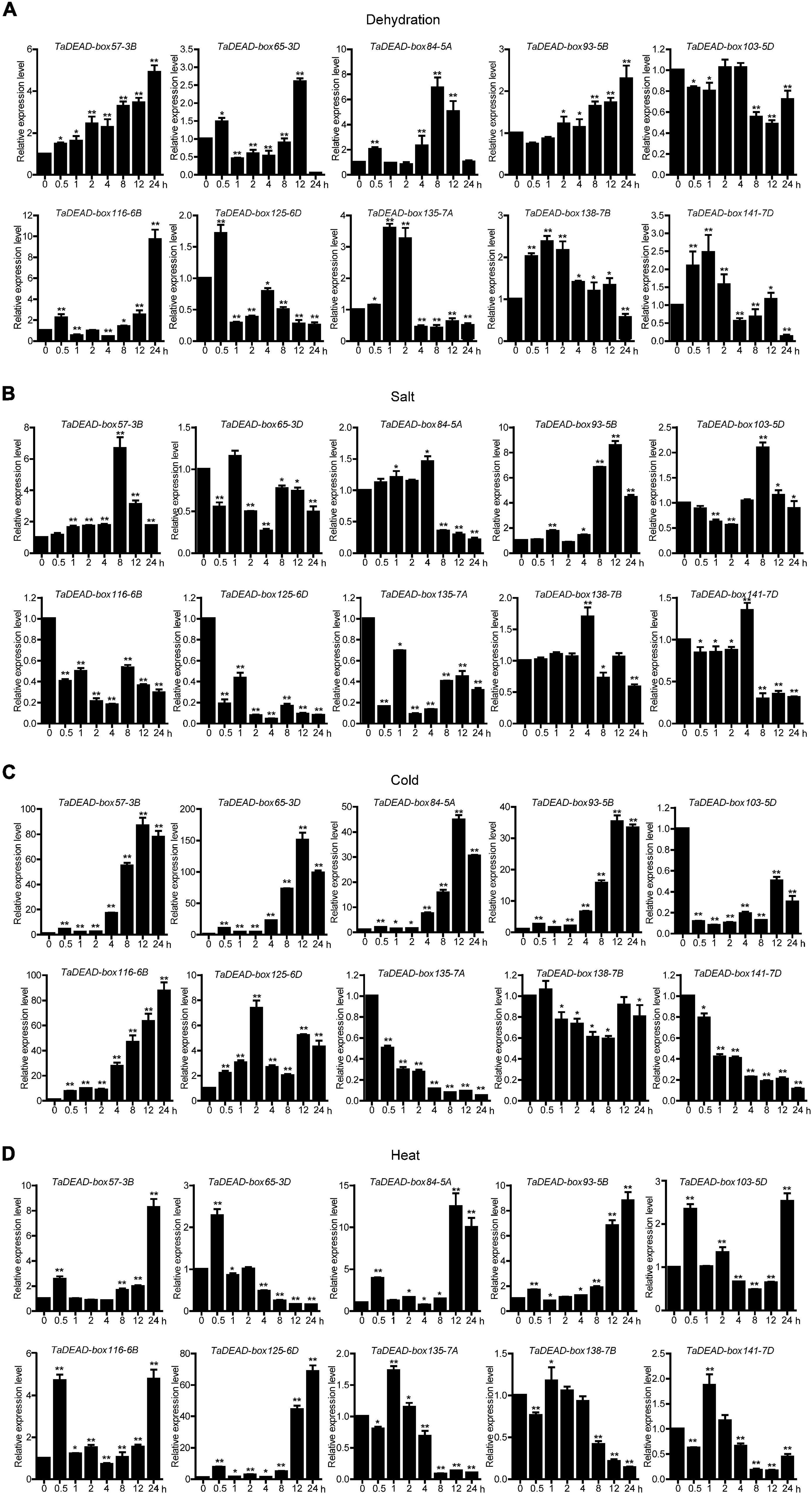
Figure 7. Quantitative Real-Time PCR (qRT-PCR) analysis of 10 TaDEAD-box genes under dehydration (A), salt (B), cold (C) and heat (D) treatments. Seven-day-old wheat seedlings were used for dehydration (250 mM Mannitol), salt (200 mM NaCl), heat (42°C), and cold (4°C) stress treatments. The wheat β-actin was used as an internal control. The data represent means ± SD of three biological replications. ANOVA test was used to analyze significant differences (*p < 0.05, **p < 0.01).
TaDEAD-box57-3B Was Located in Cytoplasm and Plasma Membrane
Subcellular localizations of the 141 TaDEAD-box family members were predicted through using BUSCA (Supplementary Table 1). For transient expression assays, the p16318hGFP control and recombinant plasmids were transformed into wheat protoplasts mediated by PEG4000. GFP expression in protoplasts was observed using a confocal laser scanning microscope. Relative to the control where GFP expression was dispersed throughout the cell, TaDEAD-box57-3B was localized in the cytoplasm and plasma membrane (Figure 8).
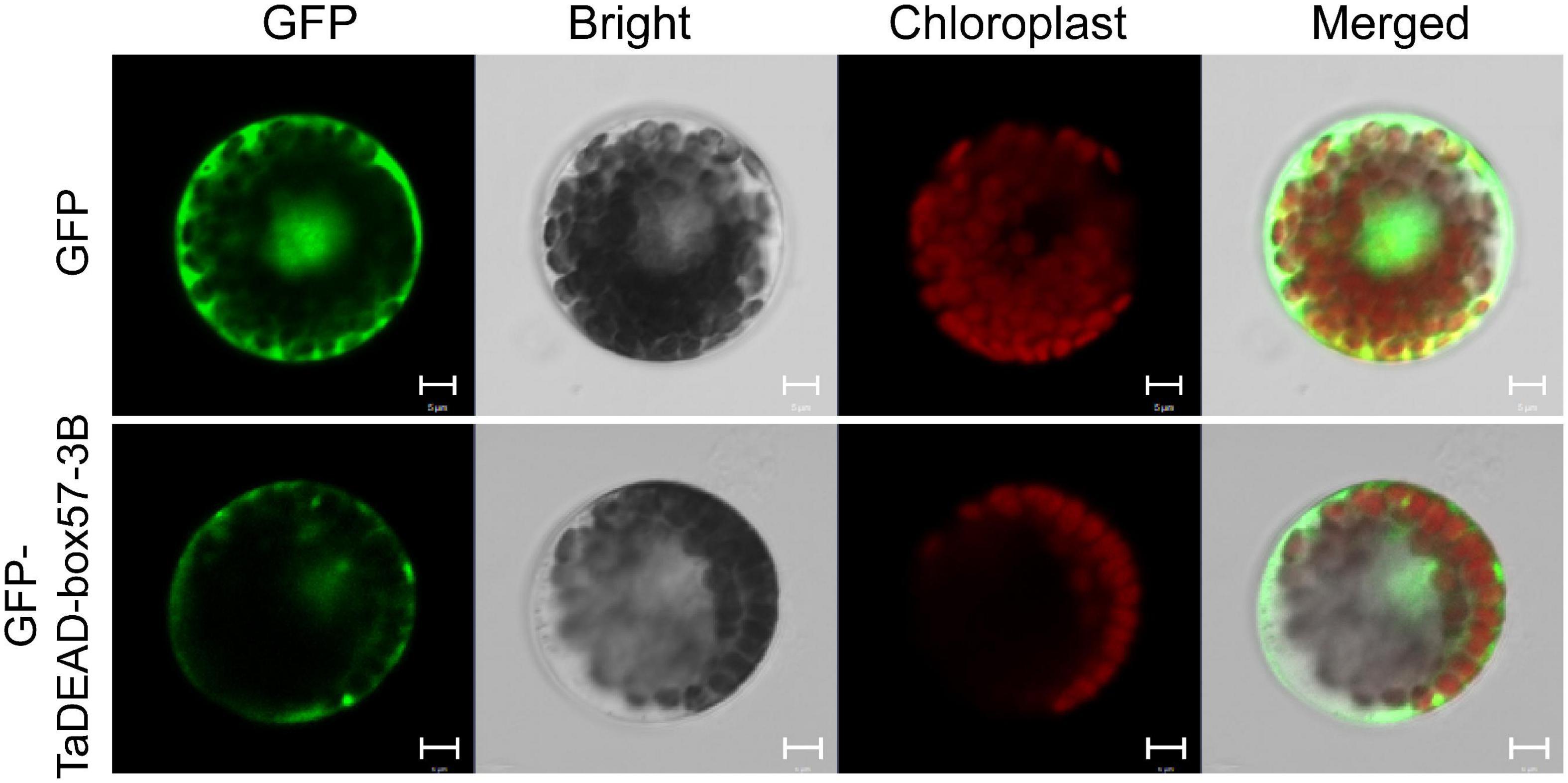
Figure 8. Subcellular localization of TaDEAD-box57-3B. The p16318hGFP control vector and recombinant constructs were transiently expressed in wheat protoplasts. The green indicates GFP signals, and the red indicates chloroplast autofluorescence. Results were observed after transformation for 18 h with confocal microscopy. Scale bars = 5 μm.
Ectopic Expression of TaDEAD-box57-3B Enhanced Drought Tolerance in Arabidopsis
TaDEAD-box57-3B under the control of CaMV35S was transformed into Arabidopsis, and three homozygous T3 lines were selected for phenotypic analysis. First, transgenic and wild-type (WT) seeds were used for germination assays. After surface sterilization, transgenic and WT seeds were grown on MS medium as a control or on MS supplemented with mannitol (300 mM or 400 mM). In the control medium, there was no significant difference in germination rate between WT and transgenic Arabidopsis. Germination of both WT and transgenic lines was inhibited in the 300 mM mannitol treatment, although the transgenic lines showed higher germination rates than the WT (Figures 9A,B). Germination rates of WT and transgenic lines were also inhibited to varying degrees in the 400 mM mannitol treatment; for example, the germination rates of the transgenic lines on the sixth day were approximately 80%, significantly higher than the rate of the WT (∼45%) (Figures 9A,B).
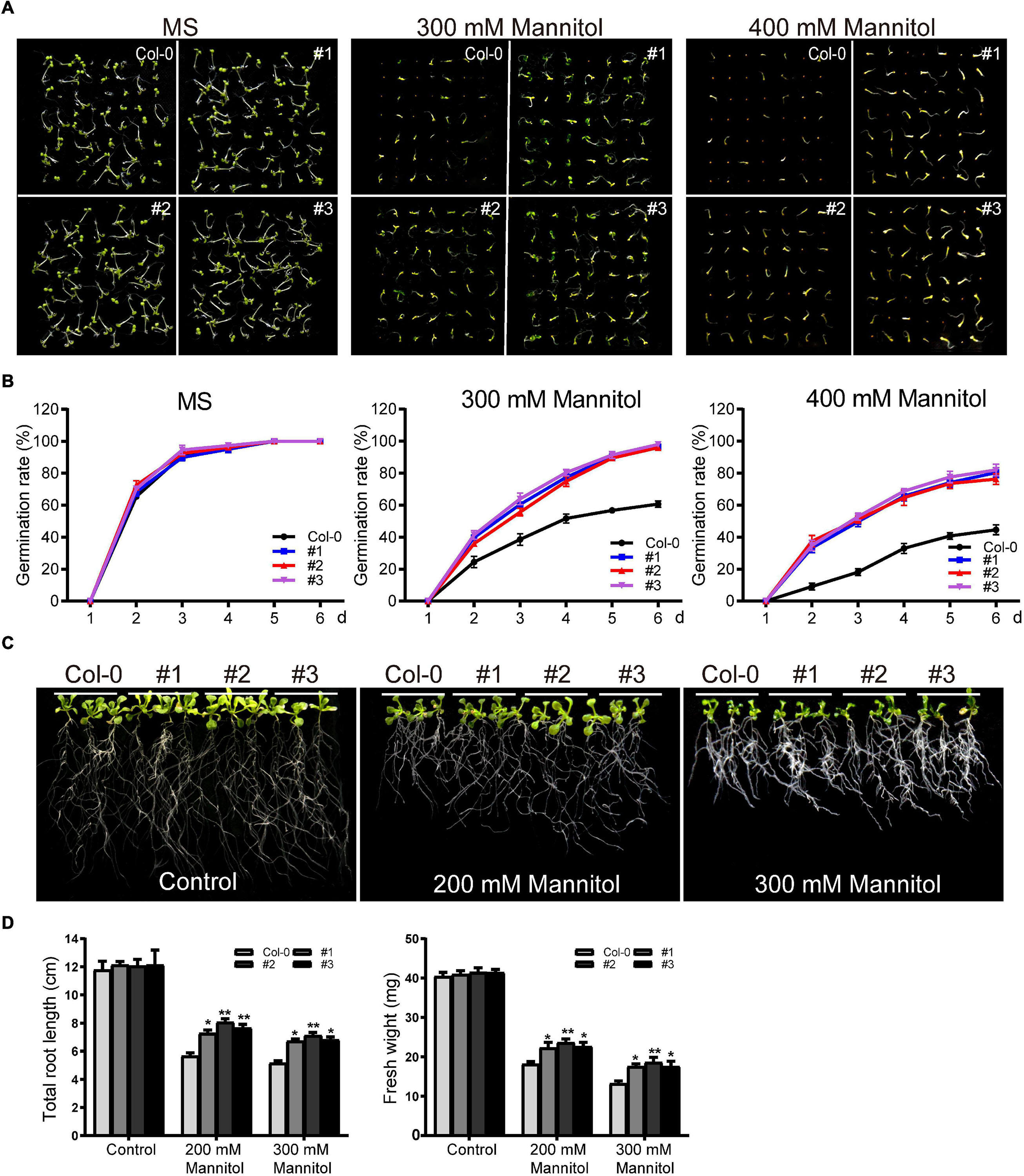
Figure 9. Phenotype analysis of TaDEAD-box57-3B transgenic plants under dehydration treatment. (A) Seed germination assays of WT and TaDEAD-box57-3B transgenic lines. Seeds were grown on MS and MS medium supplemented with 300 mM and 400 mM mannitol. (B) The germination rates of WT and TaDEAD-box57-3B transgenic lines. The germination rate was calculated for the next 6 days. (C) Root length assays of WT and TaDEAD-box57-3B transgenic lines. Five-day-old seedlings were transferred to MS medium with or without 200 mM, 300 mM mannitol. (D) The total root lengths and fresh weights of WT and TaDEAD-box57-3B transgenic lines. All the data represent the means ± SDs of three independent biological replicates and asterisks (*or **) represent the significant differences at p < 0.05 or p < 0.01(ANOVA test).
To explore the function of TaDEAD-box57-3B on seedlings, root growth assays and drought resistance tests in soil were performed. For root growth assays, 5-day-old seedlings were transferred to MS medium containing 200 mM or 300 mM mannitol, and the total root length and fresh weight were calculated. There was no significant difference in root length or fresh weight between WT and transgenic lines in the control medium. In the 200 mM mannitol treatment, growth was inhibited for both WT and transgenic plants; however, transgenic lines showed less growth inhibition than the WT, with longer roots and higher fresh weights (Figures 9C,D). In 300 mM mannitol, growth was more severely inhibited for WT and transgenic lines; however, consistent with the results of 200 mM mannitol treatment, the effect was decreased in the transgenic plants compared to the WT for both total root lengths and fresh weights (Figures 9C,D).
To measure drought resistance in soil, 21-day-old seedlings were exposed to drought conditions by withdrawing irrigation until there was a significant difference between WT and transgenic plants, after which the survival rates were counted. Before drought treatment, there was no significant difference in morphology between WT and transgenic lines. After drought treatment, the transgenic plants exhibited significantly increased drought tolerance, whereas WT plants were severely wilted and comparatively sensitive to drought treatment, with ∼48.1% survival (Figures 10A,B).
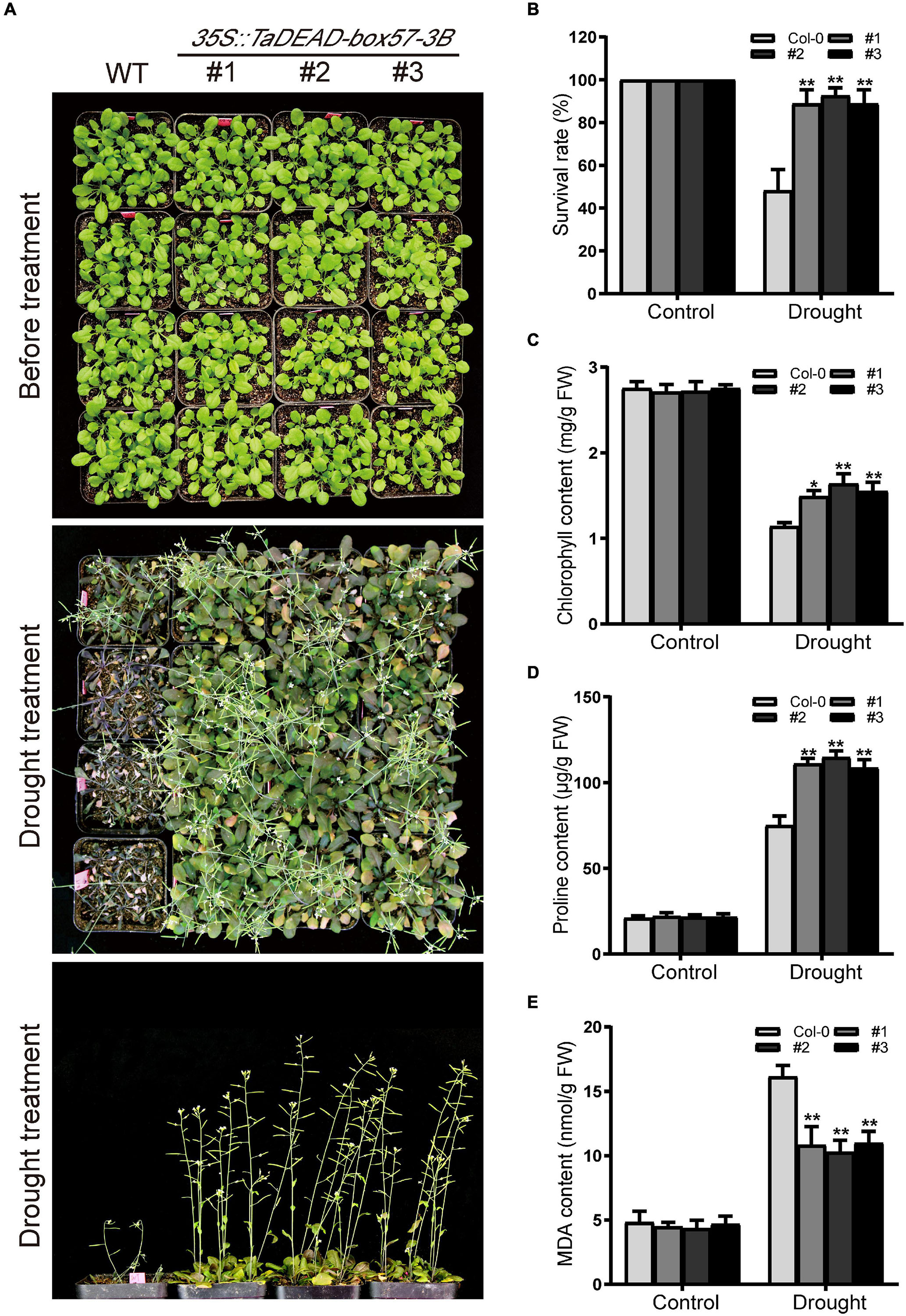
Figure 10. Ectopic expression of TaDEAD-box57-3B enhanced drought tolerance in transgenic Arabidopsis. (A) Phenotypes of 21-day-old WT and TaDEAD-box57-3B transgenic plants under drought stress. (B) The survival rates of WT and transgenic plants under drought condition was monitored 7 days after rewatering. (C-E) The physiological indicators of WT and transgenic plants under normal and stress treatments, including chlorophyll (C), proline (D), and MDA (E) contents. All the data represent the means ± SDs of three independent biological replicates and asterisks (*or **) represent the significant differences at p < 0.05 or p < 0.01(ANOVA test).
To explain the mechanism of enhanced drought tolerance, stress-related physiological indicators including levels of chlorophyll, proline, and MDA were measured under control and drought conditions. After drought treatment, levels of proline and MDA were increased and chlorophyll contents were decreased in the WT, whereas the transgenic lines showed significantly lower MDA contents but higher proline and chlorophyll content. There were no significant differences between the lines under standard conditions (Figures 10C–E). Collectively, these findings indicate that ectopic expression of TaDEAD-box57-3B enhanced drought tolerance in transgenic Arabidopsis.
Ectopic Expression of TaDEAD-box57-3B Increased Salt Resistance in Arabidopsis
For germination assays, WT and transgenic seeds were grown on standard MS medium or MS supplemented with NaCl (100 mM or 125 mM) after surface sterilization. There was no significant difference in germination rates between WT and transgenic plants in the control group. In the salt-treated plants, germination was inhibited in both the WT and transgenic lines; consistent with our drought treatment results, the transgenic lines showed higher germination rates (Figures 11A,B). Germination was inhibited to varying degrees for WT and transgenic plants grown in 125 mM NaCl, with the germination rates of transgenic lines at six days ∼89.7%, significantly higher than the rate of the WT (∼74.5%) (Figures 11A,B).
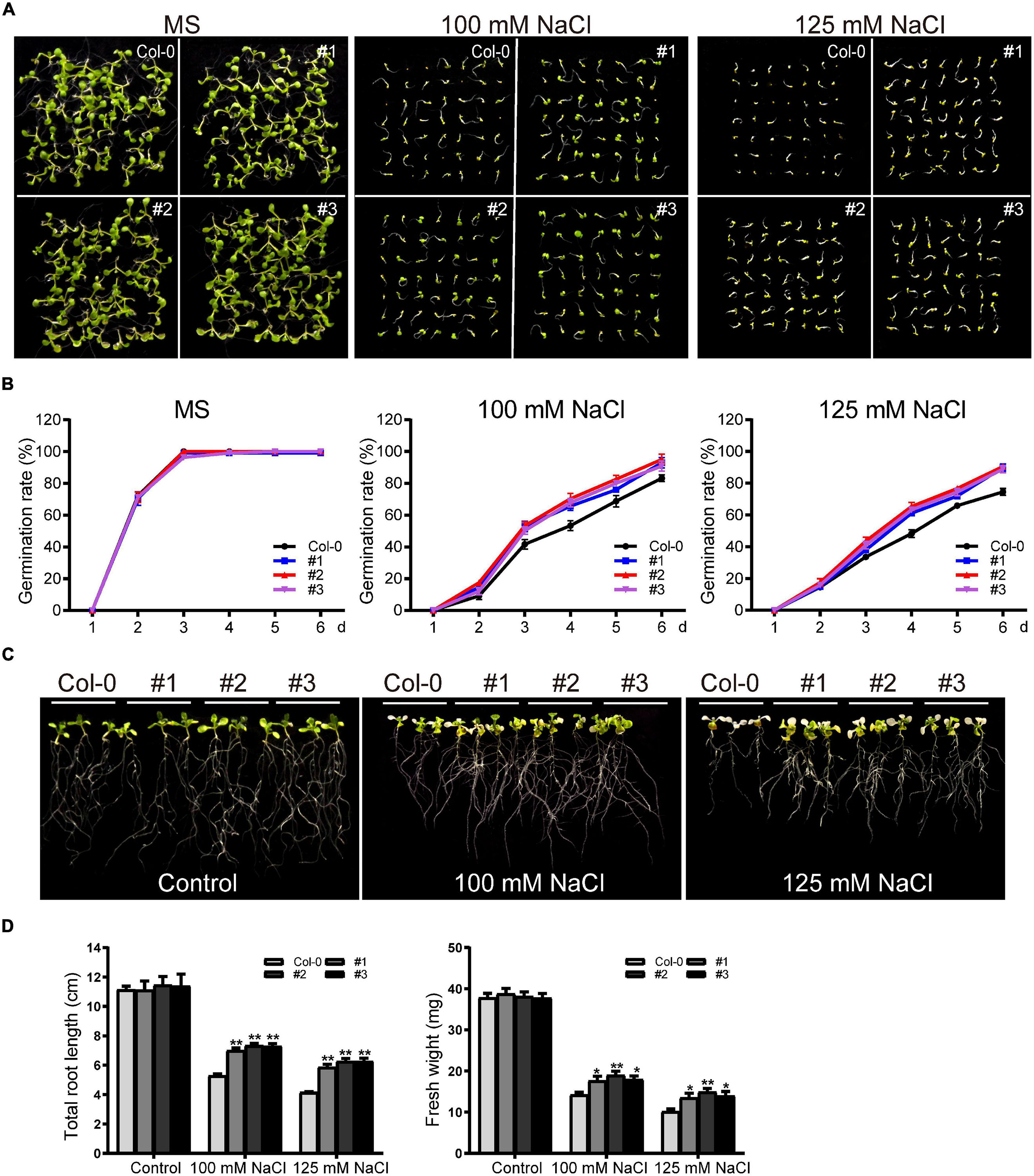
Figure 11. Phenotype analysis of TaDEAD-box57-3B transgenic plants under salt treatment. (A) Seed germination assays of WT and TaDEAD-box57-3B transgenic lines. Seeds were grown on MS and MS medium supplemented with 100 mM and 125 mM NaCl. (B) The germination rates of WT and TaDEAD-box57-3B transgenic lines. The germination rate was calculated for the next 6 days. (C) Root length assays of WT and TaDEAD-box57-3B transgenic lines. Five-day-old seedlings were transferred to MS and MS medium supplemented with 100 mM and 125 mM NaCl. (D) The total root lengths and fresh weights of WT and TaDEAD-box57-3B transgenic lines. The data are shown as the means ± SD obtained from three biological replicates. ANOVA test was used to analyze significant differences (*p < 0.05, **p < 0.01).
We next performed root growth assays and drought resistance tests in soil to identify the role of TaDEAD-box57-3B in salt stress response. After 5-day-old seedlings were transferred to MS medium supplemented with 100 mM and 125 mM NaCl for another 7 days, we measured the total root lengths and fresh weights. No significant difference was observed between WT and transgenic lines under normal conditions. However, in those treated with 100 mM NaCl, growth was inhibited in both WT and transgenic plants. Again, the transgenic lines showed significantly better growth than WT, with longer roots and higher fresh weights (Figures 11C,D). Treatment with 125 mM NaCl caused poor growth in the WT and transgenic lines; plants were bleached and had shorter roots and lower fresh weights than those grown in the control medium, but all of these effects were less severe in the transgenic lines compared to the WT (Figures 11C,D).
In order to determine the level of salt resistance in soil, 21-day-old plants were irrigated with 200 mM NaCl solution and the survival rates were counted. There was no obvious difference in morphology between the WT and transgenic lines before salt treatment (Figure 12A), and after treatment, transgenic plants exhibited increased salt resistance compared to the WT (Figure 12B). Later, the physiological indicators (chlorophyll, proline, and MDA contents) were measured under normal and salt treatment. In the control treatment, there was no obvious difference in physiological indicators. After salt treatment, the chlorophyll contents were decreased in both WT and transgenic plants, but transgenic lines looked greener with higher chlorophyll contents than the WT (Figure 12C). Proline and MDA contents were increased in all genotypes under salt treatment, but the transgenic lines showed significantly lower MDA contents and higher proline levels compared to the WT (Figures 12D,E). These results demonstrated that ectopic expression of TaDEAD-box57-3B in Arabidopsis contributed to a stronger capacity to resist salt stress.
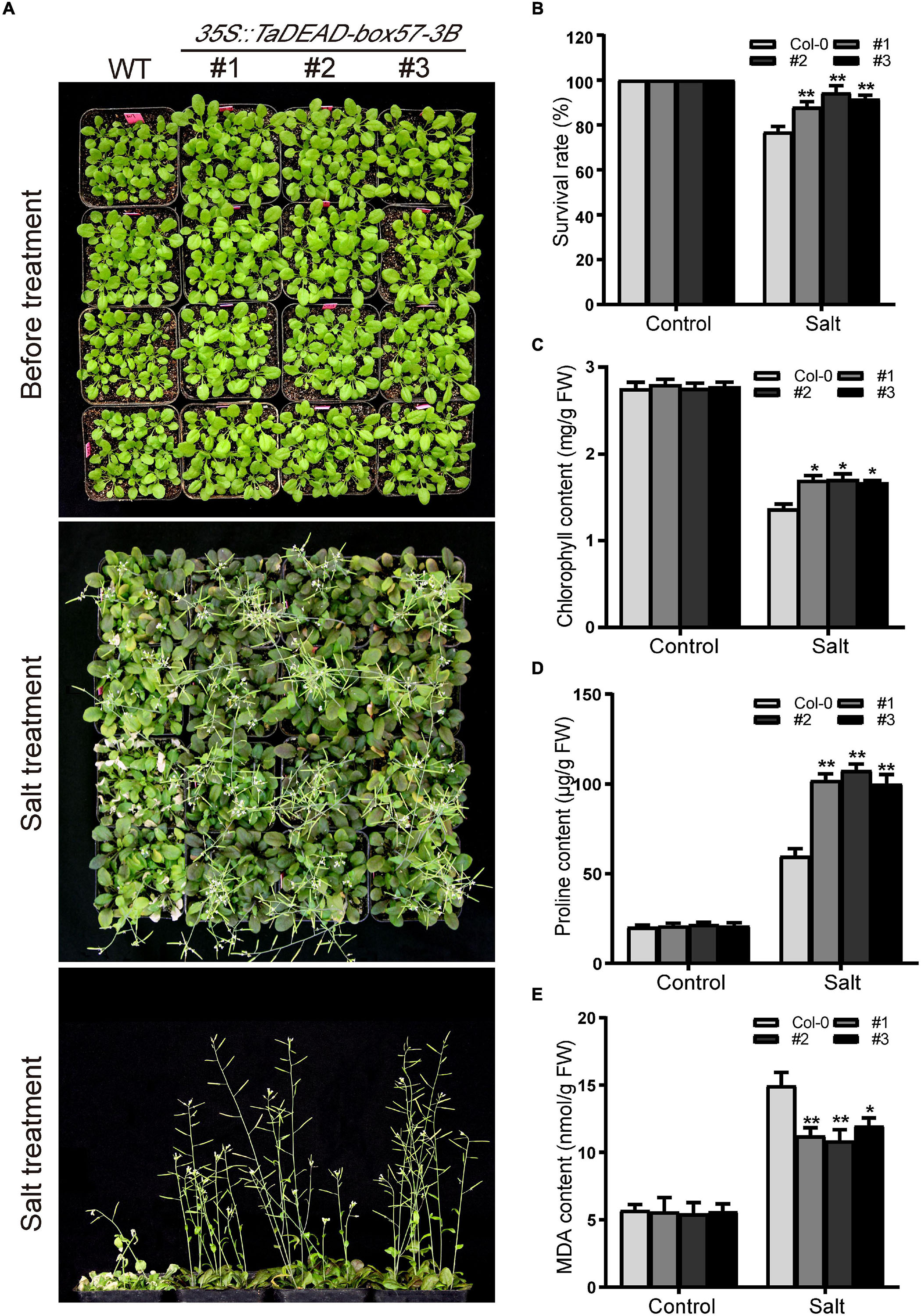
Figure 12. Ectopic expression of TaDEAD-box57-3B increased salt resistance in transgenic Arabidopsis. (A) Phenotypes of 21-day-old WT and TaDEAD-box57-3B transgenic plants under salt stress. (B) The survival rates of WT and transgenic plants under salt treatment. (C–E) The physiological indicators of WT and transgenic plants under normal and stress treatments, including chlorophyll (C), proline (D), and MDA (E) contents. The data are shown as the means ± SD obtained from three biological replicates. ANOVA test was used to analyze significant differences (*p < 0.05, **p < 0.01).
Ectopic Expression of TaDEAD-box57-3B Increased Cold Tolerance in Arabidopsis
TaDEAD-box57-3B was up-regulated more than 80-fold in cold treatment compared to standard growth conditions, suggesting that it may play an important role in cold response. Using the three TaDEAD-box57-3B overexpressing lines, we observed the phenotypes of the WT and transgenic lines under cold stress. There was no significant difference between WT and transgenic plants under normal conditions (Figure 13). However, in response to cold treatment, the transgenic lines showed significantly improved growth compared to the WT in terms of survival rates (> 50% vs. < 30%, respectively). These results suggest that ectopic expression of TaDEAD-box57-3B may improve cold tolerance in transgenic Arabidopsis.
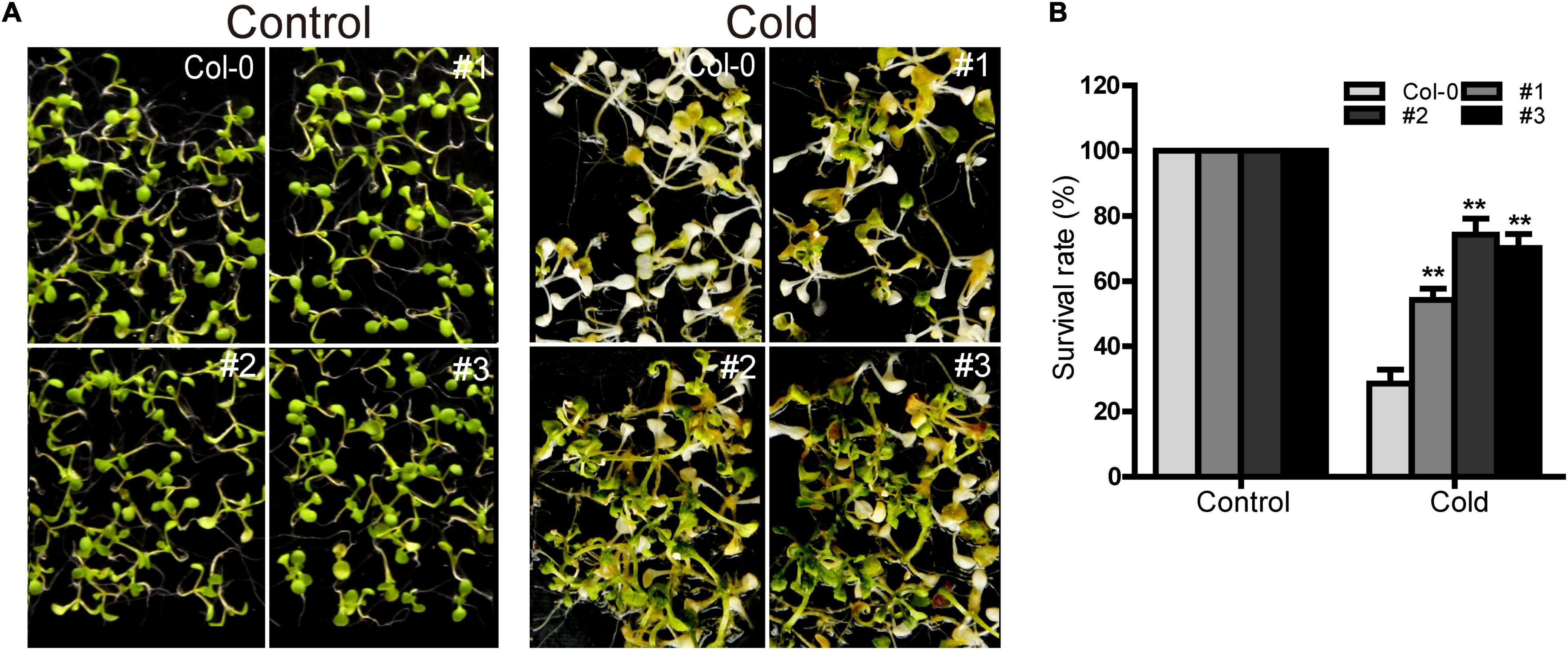
Figure 13. Ectopic expression of TaDEAD-box57-3B increased cold tolerance in transgenic Arabidopsis. (A) Phenotypes of WT and TaDEAD-box57-3B transgenic plants under cold stress. (B) Survival rates of WT and TaDEAD-box57-3B transgenic lines under cold stress. Five-day-old seedlings were placed at 4°C for 1 d, –8°C for 1 h, 4°C for 12 h and then resumed at 22°C. The data are shown as the means ± SD obtained from three biological replicates. ANOVA test was used to analyze significant differences (**p < 0.01).
Discussion
DEAD-box RNA helicases play an important role in RNA metabolism (Bowers et al., 2006; Cordin et al., 2006; Linder, 2006; Linder and Fuller-Pace, 2013; Putnam and Jankowsky, 2013), and are widely involved in plant growth, development, and stress responses (Guan et al., 2013; Khan et al., 2014; Bush et al., 2016; Liu et al., 2016; Paieri et al., 2018; Chen D. et al., 2020; Lu et al., 2020). However, knowledge of DEAD-box RNA helicases has so far been limited in wheat. Here, we conducted a comprehensive analysis of the DEAD-box gene family in wheat, and characterized the function of an abiotic stress-responsive family member, TaDEAD-box57-3B. This work provides a foundation for better understanding the roles of TaDEAD-box members in plant growth, development, and stress responses.
TaDEAD-box family members are more numerous in wheat than in Arabidopsis, rice, tomato, and maize by nearly 2.47-fold, 3-fold, 3.36-fold, and 2.47-fold, respectively. This may be due to a higher homoeolog retention rate in wheat. DEAD-box genes are unevenly distributed across the chromosomes; for example, chromosome 1 contains the most DEAD-box genes in Arabidopsis and rice, and only one DEAD-box gene was located in particular chromosome among these species (Xu et al., 2013a,b; Wan et al., 2020). In wheat, the 141 TaDEAD-box genes were also unevenly distributed on all 21 chromosomes (Figure 2). Whole genome duplication (WGD) is a large-scale process of gene multiplication at the chromosomal level that can generate abundant duplicated genes, playing a vital role in the expansion of some gene families (Schmutz et al., 2010; Lee et al., 2013; Su et al., 2020; Xu et al., 2021). For example, the significant expansion of TaSCPL genes in the wheat genome is attributed to tandem duplication (10.5%) and WGD/segmental duplication (64.8%). Duplicate MADS-box genes may be derived from tandem or segmental duplication, which would produce many homologs on different chromosomes (Duan et al., 2015; Schilling et al., 2020; Zhao et al., 2021). Expansion of ANK genes in the soybean genome is also attributed to tandem and segmental duplication events (35.8% and 46.0%, respectively) (Zhao et al., 2020). Consistent with these prior findings, ∼4.3% and 88.7% of TaDEAD-box genes were determined to be derived from tandem and WGD/segmental duplication events, respectively. This suggests that WGD/segmental duplication is the main driving factor for expansion of DEAD-box genes in the wheat genome (Figure 3). Gene family expansion is hypothesized to contribute to the ability of a species to adapt to various environments, thus enabling their wide distribution (Theissen et al., 2016; Zhao et al., 2021).
Overall, the diversity of gene structure and structural motifs was strongly associated with the evolutionary relationships and functions among members of the gene family. Genes in the same branch of the phylogenetic tree constructed from DEAD-box genes were generally homoeologs with similar gene structure; however, there were a few exceptions (Figure 5). In fact, structural divergences commonly exist in gene families and can generate homologous genes with different functions, although the mechanism is still unclear (Xu et al., 2012; Zhao et al., 2021). Nine motifs were present in almost all of the TaDEAD-box proteins, with motif 4 containing the highly conserved D-E-A-D sequence. Some motifs were restricted to specific groups within the gene family, e.g., motif 10 was specific to Group II. As expected, proteins in the same branch of the phylogenetic tree generally exhibited similar motif distribution (Figure 5). However, some proteins within the same branch contained different motifs, which may be due to differences in the arrangement of introns and exons.
Cis-elements in the promoter region are highly related to the regulation of gene expression. There were several stress response-related cis-elements in 10 TaDEAD-box genes identified as having different responses to abiotic stressors, indicating that these genes may be involved in plant abiotic stress responses. These results provide a foundation for further functional identification of TaDEAD-box genes in wheat. Increasing evidence has indicated that DEAD-box RNA helicases play important roles in plant abiotic stress responses (Khan et al., 2014; Bush et al., 2016; Huang et al., 2016a; Wang D. et al., 2016; Chen D. et al., 2020; Lu et al., 2020). Some abiotic stress-related DEAD-box RNA helicases have been characterized; for example, RCF1 is essential for pre-mRNA splicing and cold response in plants (Guan et al., 2013). AtRH7 interacts with AtCSP3, which acts as an RNA chaperone involved in cold adaptation, affecting rRNA biogenesis and playing an important role in plant growth under cold stress (Liu et al., 2016). Rice OsBIRH1 increases oxidative stress tolerance in transgenic plants by elevating expression levels of the oxidative defense genes AtApx1, AtApx2, and AtFSD1 (Li et al., 2008). Rice OsRH58 is induced by salt, drought, and heat stress, and can act as an RNA chaperone to improve salt or drought stress tolerance by increasing chloroplast mRNA translation (Nawaz and Kang, 2019). Rice OsRH42, which is located in the splicing speckles, interacts with U2 small nuclear RNA, thus playing a crucial role in accurate pre-mRNA splicing and cold adaptation in rice (Lu et al., 2020).
Transcriptomic data showed that TaDEAD-box57-3B was significantly up-regulated under diverse abiotic stresses, and it was therefore selected for further experimental analysis. Under drought or salt stress, TaDEAD-box57-3B transgenic Arabidopsis plants showed higher germination rates, root lengths, fresh weights and survival rates; moreover, transgenic lines had significantly better growth and higher survival rates than the WT after cold treatment. Chlorophyll content was measured as a proxy for the degree of cellular injury, and MDA content was quantified as a measure of membrane lipid peroxidation (Gunes et al., 2011; Hong et al., 2016; Wang T. T. et al., 2020; Chen et al., 2021). Proline, a common osmoprotectant, is a regulator of osmotic pressure to maintain plant homeostasis in responses to adverse conditions, and generally increases in response to abiotic stress (Szekely et al., 2008; Su et al., 2020; Wang et al., 2021; Zhao et al., 2021). These three components (chlorophyll, MDA, and proline content) can be used as physiological indicators to measure plant stress resistance. Drought and salt stress can cause oxidative and hyperosmotic damage (Zhu, 2016; Gong et al., 2020; Chen et al., 2021; Xu et al., 2021). We therefore measured these stress-related physiological indicators under normal (control) growth conditions and drought or salt treatment. Transgenic lines showed significantly lower MDA, higher proline, and higher chlorophyll contents than WT plants after drought or salt treatment, whereas there were no obvious differences in the control treatment. The higher accumulation of proline and chlorophyll and the lower MDA content may contribute to the tolerance of TaDEAD-box57-3B transgenic plants to drought and salt stresses. Collectively, our results indicate that TaDEAD-box57-3B plays a positive role in drought, salt, and cold responses. It is still unclear that whether TaDEAD-box57-3B affects chlorophyll, proline and MDA contents through RNA metabolism, therefore, further studies are necessary to clarify the possible role of TaDEAD-box57-3B in RNA metabolism under stresses.
To ensure sustainable production of wheat, crop varieties with enhanced stress tolerance should be cultivated. TaDEAD-box57-3B is a potential candidate gene for improving abiotic stress resistance, although further studies are required to identify the function and stress response mechanism in wheat. Discovering novel stress-responsive genes, as we have done here, and studying the mechanisms of action provides a theoretical basis and technical support for crop molecular breeding.
Conclusion
We performed a comprehensive genome-wide analysis of the DEAD-box RNA helicase family in wheat, including analyses of phylogeny, chromosomal distribution, duplication events, gene structure, and protein motifs. A total of 141 TaDEAD-box genes were identified and expression patterns compared using transcriptomic data. Expression levels of 10 genes were confirmed by qRT-PCR; among them, TaDEAD-box57-3B was significantly up-regulated under diverse abiotic stresses, and further analysis indicated it was involved in tolerance to drought, salt, and cold treatments through regulating the degree of membrane lipid peroxidation. This study provides new insights for understanding the evolution and function of the TaDEAD-box gene family.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
Z-SX coordinated the project, conceived and designed the experiments, and edited the manuscript. J-NR performed the experiments and wrote the first draft. Z-HH, LZ, QZ, F-ZW, JC, Y-BZ, and MC conducted data analysis. Y-JX and Y-ZM contributed with valuable discussions. All authors reviewed and approved the final manuscript.
Funding
This research was financially supported by the National Natural Science Foundation of China (31871624), the Central Public-interest Scientific Institution Basal Research Fund (S2021ZD02), and the Agricultural Science and Technology Innovation Program (CAAS-ZDRW202109 and CAAS-ZDRW202002).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to Yong-Wei Liu from Institute of Genetics and Physiology, Hebei Academy of Agriculture and Forestry Sciences/Plant Genetic Engineering Center for providing the wheat seeds.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.797276/full#supplementary-material
Supplementary Figure 1 | The distribution frequency of TaDEAD-box genes per chromosome.
Supplementary Figure 2 | Cis-elements distribution of 141 TaDEAD-box genes. (A) Distribution of cis-elements in different biological processes. (B) Hormone responsive cis-elements. (C) Stress related cis-elements. (D) Plant growth related cis-elements.
Supplementary Table 1 | Detailed information of 141 wheat DEAD-box genes identified in wheat genome.
Supplementary Table 2 | Chromosome inversion and translocation events in TaDEAD-box homoeologs.
Supplementary Table 3 | Ka, Ks, and Ka/Ks values of duplicated TaDEAD-box genes.
Supplementary Table 4 | Exon and intron numbers of 141 TaDEAD-box genes.
Supplementary Table 5 | List of conserved motifs inTaDEAD-box proteins.
Supplementary Table 6 | Cis-elements in the promoter region of 141 TaDEAD-box genes.
Supplementary Table 7 | The TPM values of 141 TaDEAD-box genes from transcriptome data.
Supplementary Table 8 | The primers used in this study.
Footnotes
- ^ https://www.arabidopsis.org/
- ^ http://rice.uga.edu/
- ^ https://www.ncbi.nlm.nih.gov/
- ^ http://plants.ensembl.org/index.html
- ^ https://www.ncbi.nlm.nih.gov/Structure
- ^ http://pfam.xfam.org/
- ^ https://web.expasy.org/compute_pi/
- ^ http://busca.biocomp.unibo.it/
- ^ http://meme-suite.org/tools/meme
- ^ http://bioinformatics.psb.ugent.be/webtools/plantcare/html/
- ^ http://www.wheat-expression.com
References
Appels, R., Eversole, K., Feuillet, C., Keller, B., and Rogers, J. (2018). Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361:eaar7191.
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. doi: 10.1093/nar/gkp335
Banroques, J., Cordin, O., Doere, M., Linder, P., and Tanner, N. K. (2011). Analyses of the functional regions of DEAD-box RNA “helicases” with deletion and chimera constructs tested in vivo and in vitro. J. Mol. Biol. 413, 451–472. doi: 10.1016/j.jmb.2011.08.032
Borrill, P., Ramirez-Gonzalez, R., and Uauy, C. (2016). expVIP: a Customizable RNA-seq Data Analysis and Visualization Platform. Plant Physiol. 170, 2172–2186. doi: 10.1104/pp.15.01667
Bowers, H. A., Maroney, P. A., Fairman, M. E., Kastner, B., Luhrmann, R., Nilsen, T. W., et al. (2006). Discriminatory RNP remodeling by the DEAD-box protein DED1. RNA 12, 903–912. doi: 10.1261/rna.2323406
Bush, M. S., Crowe, N., Zheng, T., and Doonan, J. H. (2015). The RNA helicase, eIF4A-1, is required for ovule development and cell size homeostasis in Arabidopsis. Plant J. 84, 989–1004. doi: 10.1111/tpj.13062
Bush, M. S., Pierrat, O., Nibau, C., Mikitova, V., Zheng, T., Corke, F. M., et al. (2016). eIF4A RNA Helicase Associates with Cyclin-Dependent Protein Kinase A in Proliferating Cells and Is Modulated by Phosphorylation. Plant Physiol. 172, 128–140. doi: 10.1104/pp.16.00435
Byrd, A. K., and Raney, K. D. (2012). Superfamily 2 helicases. Front. Biosci. 17, 2070–2088. doi: 10.2741/4038
Caruthers, J. M., and McKay, D. B. (2002). Helicase structure and mechanism. Curr. Opin. Struct. Biol. 12, 123–133. doi: 10.1016/s0959-440x(02)00298-1
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chen, D., Wang, Y., Zhang, W., Li, N., Dai, B., Xie, F., et al. (2020). Gametophyte-specific DEAD-box RNA helicase 29 is required for functional maturation of male and female gametophytes in Arabidopsis. J. Exp. Bot. 71, 4083–4092. doi: 10.1093/jxb/eraa190
Chen, C., Xia, R., Chen, H., and He, Y. (2018). TBtools, a Toolkit for Biologists integrating various biological data handling tools with a user-friendly interface. bioRxiv[Preprint]. doi: 10.1101/289660
Chen, J., Zhang, Y., Liu, J., Xia, M., Wang, W., and Shen, F. (2014). Genome-wide analysis of the RNA helicase gene family in Gossypium raimondii. Int. J. Mol. Sci. 15, 4635–4656. doi: 10.3390/ijms15034635
Chen, Z. F., Ru, J. N., Sun, G. Z., Du, Y., Chen, J., Zhou, Y. B., et al. (2021). Genomic-Wide Analysis of the PLC Family and Detection of GmPI-PLC7 Responses to Drought and Salt Stresses in Soybean. Front. Plant Sci. 12:631470. doi: 10.3389/fpls.2021.631470
Clough, S. J., and Bent, A. F. (1998). Floral dip: a simplified method for Agrobacterium -mediated transformation of Arabidopsis thaliana. Plant J. 16, 735–743. doi: 10.1046/j.1365-313x.1998.00343.x
Cordin, O., Banroques, J., Tanner, N. K., and Linder, P. (2006). The DEAD-box protein family of RNA helicases. Gene 367, 17–37. doi: 10.1016/j.gene.2005.10.019
Duan, W., Song, X., Liu, T., Huang, Z., Ren, J., Hou, X., et al. (2015). Genome-wide analysis of the MADS-box gene family in Brassica rapa (Chinese cabbage). Mol. Genet. Genomics 290, 239–255. doi: 10.1007/s00438-014-0912-7
Fairman-Williams, M. E., Guenther, U. P., and Jankowsky, E. (2010). SF1 and SF2 helicases: family matters. Curr. Opin. Struct. Biol. 20, 313–324. doi: 10.1016/j.sbi.2010.03.011
Fan, K., Yuan, S., Chen, J., Chen, Y., Li, Z., Lin, W., et al. (2019). Molecular evolution and lineage-specific expansion of the PP2C family in Zea mays. Planta 250, 1521–1538. doi: 10.1007/s00425-019-03243-x
Gill, S. S., Tajrishi, M., Madan, M., and Tuteja, N. (2013). A DESD-box helicase functions in salinity stress tolerance by improving photosynthesis and antioxidant machinery in rice (Oryza sativa L. cv. PB1). Plant Mol. Biol. 82, 1–22. doi: 10.1007/s11103-013-0031-6
Gong, Z., Dong, C. H., Lee, H., Zhu, J., Xiong, L., Gong, D., et al. (2005). A DEAD box RNA helicase is essential for mRNA export and important for development and stress responses in Arabidopsis. Plant Cell. 17, 256–267. doi: 10.1105/tpc.104.027557
Gong, Z., Xiong, L., Shi, H., Yang, S., Herrera-Estrella, L. R., Xu, G., et al. (2020). Plant abiotic stress response and nutrient use efficiency. Sci. China Life Sci. 63, 635–674. doi: 10.1007/s11427-020-1683-x
Guan, Q., Wu, J., Zhang, Y., Jiang, C., Liu, R., Chai, C., et al. (2013). A DEAD box RNA helicase is critical for pre-mRNA splicing, cold-responsive gene regulation, and cold tolerance in Arabidopsis. Plant Cell 25, 342–356. doi: 10.1105/tpc.112.108340
Gunes, A., Inal, A., Adak, M. S., Bagci, E. G., Cicek, N., and Eraslan, F. (2011). Effect of drought stress implemented at pre- or post-anthesis stage on some physiological parameters as screening criteria in chickpea cultivars. Russ. J. Plant Physiol. 55, 59–67. doi: 10.1134/s102144370801007x
Hong, Y., Zhang, H., Huang, L., Li, D., and Song, F. (2016). Overexpression of a Stress-Responsive NAC Transcription Factor Gene ONAC022 Improves Drought and Salt Tolerance in Rice. Front. Plant Sci. 7:4. doi: 10.3389/fpls.2016.00004
Hsu, Y. F., Chen, Y. C., Hsiao, Y. C., Wang, B. J., Lin, S. Y., Cheng, W. H., et al. (2014). AtRH57, a DEAD-box RNA helicase, is involved in feedback inhibition of glucose-mediated abscisic acid accumulation during seedling development and additively affects pre-ribosomal RNA processing with high glucose. Plant J. 77, 119–135. doi: 10.1111/tpj.12371
Huang, C. K., Shen, Y. L., Huang, L. F., Wu, S. J., Yeh, C. H., and Lu, C. A. (2016a). The DEAD-Box RNA Helicase AtRH7/PRH75 Participates in Pre-rRNA Processing, Plant Development and Cold Tolerance in Arabidopsis. Plant Cell Physiol. 57, 174–191. doi: 10.1093/pcp/pcv188
Huang, C. K., Sie, Y. S., Chen, Y. F., Huang, T. S., and Lu, C. A. (2016b). Two highly similar DEAD box proteins, OsRH2 and OsRH34, homologous to eukaryotic initiation factor 4AIII, play roles of the exon junction complex in regulating growth and development in rice. BMC Plant Biol. 16:84. doi: 10.1186/s12870-016-0769-5
Kammel, C., Thomaier, M., Sorensen, B. B., Schubert, T., Langst, G., Grasser, M., et al. (2013). Arabidopsis DEAD-box RNA helicase UAP56 interacts with both RNA and DNA as well as with mRNA export factors. PLoS One 8:e60644. doi: 10.1371/journal.pone.0060644
Kant, P., Kant, S., Gordon, M., Shaked, R., and Barak, S. (2007). STRESS RESPONSE SUPPRESSOR1 and STRESS RESPONSE SUPPRESSOR2, two DEAD-box RNA helicases that attenuate Arabidopsis responses to multiple abiotic stresses. Plant Physiol. 145, 814–830. doi: 10.1104/pp.107.099895
Khan, A., Garbelli, A., Grossi, S., Florentin, A., Batelli, G., Acuna, T., et al. (2014). The Arabidopsis STRESS RESPONSE SUPPRESSOR DEAD-box RNA helicases are nucleolar- and chromocenter-localized proteins that undergo stress-mediated relocalization and are involved in epigenetic gene silencing. Plant J. 79, 28–43. doi: 10.1111/tpj.12533
Kim, J. S., Kim, K. A., Oh, T. R., Park, C. M., and Kang, H. (2008). Functional characterization of DEAD-box RNA helicases in Arabidopsis thaliana under abiotic stress conditions. Plant Cell Physiol. 49, 1563–1571. doi: 10.1093/pcp/pcn125
Kobayashi, K., Otegui, M. S., Krishnakumar, S., Mindrinos, M., and Zambryski, P. (2007). INCREASED SIZE EXCLUSION LIMIT 2 encodes a putative DEVH box RNA helicase involved in plasmodesmata function during Arabidopsis embryogenesis. Plant Cell 19, 1885–1897. doi: 10.1105/tpc.106.045666
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Lee, T. H., Tang, H., Wang, X., and Paterson, A. H. (2013). PGDD: a database of gene and genome duplication in plants. Nucleic Acids Res. 41, D1152–D1158. doi: 10.1093/nar/gks1104
Lescot, M., Dehais, P., Thijs, G., Marchal, K., Moreau, Y., Van de Peer, Y., et al. (2002). PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327. doi: 10.1093/nar/30.1.325
Li, D., Liu, H., Zhang, H., Wang, X., and Song, F. (2008). OsBIRH1, a DEAD-box RNA helicase with functions in modulating defence responses against pathogen infection and oxidative stress. J. Exp. Bot. 59, 2133–2146. doi: 10.1093/jxb/ern072
Li, Q., Zheng, Q., Shen, W., Cram, D., Fowler, D. B., Wei, Y., et al. (2015). Understanding the biochemical basis of temperature-induced lipid pathway adjustments in plants. Plant Cell. 27, 86–103. doi: 10.1105/tpc.114.134338
Linder, P. (2006). Dead-box proteins: a family affair–active and passive players in RNP-remodeling. Nucleic Acids Res. 34, 4168–4180. doi: 10.1093/nar/gkl468
Linder, P., and Fuller-Pace, F. V. (2013). Looking back on the birth of DEAD-box RNA helicases. Biochim. Biophys. Acta 1829, 750–755. doi: 10.1016/j.bbagrm.2013.03.007
Linder, P., and Jankowsky, E. (2011). From unwinding to clamping - the DEAD box RNA helicase family. Nat. Rev. Mol. Cell Biol. 12, 505–516. doi: 10.1038/nrm3154
Linder, P., and Owttrim, G. W. (2009). Plant RNA helicases: linking aberrant and silencing RNA. Trends Plant Sci. 14, 344–352. doi: 10.1016/j.tplants.2009.03.007
Liu, P., Xu, Z. S., Pan-Pan, L., Hu, D., Chen, M., Li, L. C., et al. (2013). A wheat PI4K gene whose product possesses threonine autophophorylation activity confers tolerance to drought and salt in Arabidopsis. J. Exp. Bot. 64, 2915–2927. doi: 10.1093/jxb/ert133
Liu, W. X., Zhang, F. C., Zhang, W. Z., Song, L. F., Wu, W. H., and Chen, Y. F. (2013). Arabidopsis Di19 functions as a transcription factor and modulates PR1, PR2, and PR5 expression in response to drought stress. Mol. Plant 6, 1487–1502. doi: 10.1093/mp/sst031
Liu, Y., Tabata, D., and Imai, R. (2016). A Cold-Inducible DEAD-Box RNA Helicase from Arabidopsis thaliana Regulates Plant Growth and Development under Low Temperature. PLoS One 11:e0154040. doi: 10.1371/journal.pone.0154040
Liu, Z., Xin, M., Qin, J., Peng, H., Ni, Z., Yao, Y., et al. (2015). Temporal transcriptome profiling reveals expression partitioning of homeologous genes contributing to heat and drought acclimation in wheat (Triticum aestivum L.). BMC Plant Biol. 15:152. doi: 10.1186/s12870-015-0511-8
Lorsch, J. R. (2002). RNA chaperones exist and DEAD box proteins get a life. Cell 109, 797–800. doi: 10.1016/s0092-8674(02)00804-8
Lu, C. A., Huang, C. K., Huang, W. S., Huang, T. S., Liu, H. Y., and Chen, Y. F. (2020). DEAD-Box RNA Helicase 42 Plays a Critical Role in Pre-mRNA Splicing under Cold Stress. Plant Physiol. 182, 255–271. doi: 10.1104/pp.19.00832
Marcussen, T., Sandve, S. R., Heier, L., Spannagl, M., Pfeifer, M., Jakobsen, K. S., et al. (2014). Ancient hybridizations among the ancestral genomes of bread wheat. Science 345:1250092. doi: 10.1126/science.1250092
Mingam, A., Toffano-Nioche, C., Brunaud, V., Boudet, N., Kreis, M., and Lecharny, A. (2004). DEAD-box RNA helicases in Arabidopsis thaliana: establishing a link between quantitative expression, gene structure and evolution of a family of genes. Plant Biotechnol. J. 2, 401–415. doi: 10.1111/j.1467-7652.2004.00084.x
Nawaz, G., and Kang, H. (2019). Rice OsRH58, a chloroplast DEAD-box RNA helicase, improves salt or drought stress tolerance in Arabidopsis by affecting chloroplast translation. BMC Plant Biol. 19:17. doi: 10.1186/s12870-018-1623-8
Oono, Y., Kobayashi, K., Kawahara, Y., Yazawa, T., Handa, H., Itoh, T., et al. (2013). Characterisation of the wheat (Triticum aestivum L.) transcriptome by de novo assembly for the discovery of phosphate starvation-responsive genes: gene expression in Pi-stressed wheat. BMC Genomics 14:77. doi: 10.1186/1471-2164-14-77
Paieri, F., Tadini, L., Manavski, N., Kleine, T., Ferrari, R., Morandini, P., et al. (2018). The DEAD-box RNA Helicase RH50 Is a 23S-4.5S rRNA Maturation Factor that Functionally Overlaps with the Plastid Signaling Factor GUN1. Plant Physiol. 176, 634–648. doi: 10.1104/pp.17.01545
Putnam, A. A., and Jankowsky, E. (2013). DEAD-box helicases as integrators of RNA, nucleotide and protein binding. Biochim. Biophys. Acta 1829, 884–893. doi: 10.1016/j.bbagrm.2013.02.002
Ramirez-Gonzalez, R. H., Borrill, P., Lang, D., Harrington, S. A., Brinton, J., Venturini, L., et al. (2018). The transcriptional landscape of polyploid wheat. Science 361:6403. doi: 10.1126/science.aar6089
Rocak, S., and Linder, P. (2004). DEAD-box proteins: the driving forces behind RNA metabolism. Nat. Rev. Mol. Cell Biol. 5, 232–241. doi: 10.1038/nrm1335
Russell, R., Jarmoskaite, I., and Lambowitz, A. M. (2014). Toward a molecular understanding of RNA remodeling by DEAD-box proteins. RNA Biol. 10, 44–55. doi: 10.4161/rna.22210
Schilling, S., Kennedy, A., Pan, S., Jermiin, L. S., and Melzer, R. (2020). Genome-wide analysis of MIKC-type MADS-box genes in wheat: pervasive duplications, functional conservation and putative neofunctionalization. New Phytol. 225, 511–529. doi: 10.1111/nph.16122
Schmutz, J., Cannon, S. B., Schlueter, J., Ma, J., Mitros, T., Nelson, W., et al. (2010). Genome sequence of the palaeopolyploid soybean. Nature 463, 178–183. doi: 10.1038/nature08670
Shi, C., Baldwin, I. T., and Wu, J. (2012). Arabidopsis plants having defects in nonsense-mediated mRNA decay factors UPF1, UPF2, and UPF3 show photoperiod-dependent phenotypes in development and stress responses. J. Integr. Plant Biol. 54, 99–114. doi: 10.1111/j.1744-7909.2012.01093.x
Su, H. G., Zhang, X. H., Wang, T. T., Wei, W. L., Wang, Y. X., Chen, J., et al. (2020). Genome-Wide Identification, Evolution, and Expression of GDSL-Type Esterase/Lipase Gene Family in Soybean. Front. Plant Sci. 11:726. doi: 10.3389/fpls.2020.00726
Szekely, G., Abraham, E., Cseplo, A., Rigo, G., Zsigmond, L., Csiszar, J., et al. (2008). Duplicated P5CS genes of Arabidopsis play distinct roles in stress regulation and developmental control of proline biosynthesis. Plant J. 53, 11–28. doi: 10.1111/j.1365-313X.2007.03318.x
Tanner, N. K., Cordin, O., Banroques, J., Doère, M., and Linder, P. (2003). The Q motif: a newly identified motif in DEAD box helicases may regulate ATP binding and hydrolysis. Mol. Cell. 11, 127–138. doi: 10.1016/s1097-2765(03)00006-6
Theissen, G., Melzer, R., and Rumpler, F. (2016). MADS-domain transcription factors and the floral quartet model of flower development: linking plant development and evolution. Development 143, 3259–3271. doi: 10.1242/dev.134080
Udvardi, M. K., Czechowski, T., and Scheible, W. R. (2008). Eleven golden rules of quantitative RT-PCR. Plant Cell. 20, 1736–1737. doi: 10.1105/tpc.108.061143
Umate, P., Tuteja, R., and Tuteja, N. (2010). Genome-wide analysis of helicase gene family from rice and Arabidopsis: a comparison with yeast and human. Plant Mol. Biol. 73, 449–465. doi: 10.1007/s11103-010-9632-5
Wan, R., Liu, J., Yang, Z., Zhu, P., Cao, Q., and Xu, T. (2020). Genome-wide identification, characterisation and expression profile analysis of DEAD-box family genes in sweet potato wild ancestor Ipomoea trifida under abiotic stresses. Genes Genom. 42, 325–335. doi: 10.1007/s13258-019-00910-x
Wang, D., Qin, B., Li, X., Tang, D., Zhang, Y., Cheng, Z., et al. (2016). Nucleolar DEAD-Box RNA Helicase TOGR1 Regulates Thermotolerant Growth as a Pre-rRNA Chaperone in Rice. PLoS Genet 12:e1005844. doi: 10.1371/journal.pgen.1005844
Wang, M., Yue, H., Feng, K., Deng, P., Song, W., and Nie, X. (2016). Genome-wide identification, phylogeny and expressional profiles of mitogen activated protein kinase kinase kinase (MAPKKK) gene family in bread wheat (Triticum aestivum L.). BMC Genom. 17:668. doi: 10.1186/s12864-016-2993-7
Wang, T. T., Yu, T. F., Fu, J. D., Su, H. G., Chen, J., Zhou, Y. B., et al. (2020). Genome-Wide Analysis of the GRAS Gene Family and Functional Identification of GmGRAS37 in Drought and Salt Tolerance. Front. Plant Sci. 11:604690. doi: 10.3389/fpls.2020.604690
Wang, X. M., Kong, R. R., Zhang, T., Gao, Y. Y., Xu, J. L., Lee, G., et al. (2020). A DEAD-box RNA helicase TCD33 that confers chloroplast development in rice at seedling stage under cold stress. J. Plant Physiol. 248:153138. doi: 10.1016/j.jplph.2020.153138
Wang, Z. Q., Yu, T. F., Sun, G. Z., Zheng, J. C., Chen, J., Zhou, Y. B., et al. (2021). Genome-Wide Analysis of the Catharanthus roseus RLK1-Like in Soybean and GmCrRLK1L20 Responds to Drought and Salt Stresses. Front. Plant Sci. 12:614909. doi: 10.3389/fpls.2021.614909
Xu, G., Guo, C., Shan, H., and Kong, H. (2012). Divergence of duplicate genes in exon-intron structure. Proc. Natl. Acad. Sci. U.S.A. 109, 1187–1192. doi: 10.1073/pnas.1109047109
Xu, R., Zhang, S., Huang, J., and Zheng, C. (2013a). Genome-wide comparative in silico analysis of the RNA helicase gene family in Zea mays and Glycine max: a comparison with Arabidopsis and Oryza sativa. PLoS One 8:e78982. doi: 10.1371/journal.pone.0078982
Xu, R., Zhang, S., Lu, L., Cao, H., and Zheng, C. (2013b). A genome-wide analysis of the RNA helicase gene family in Solanum lycopersicum. Gene 513, 128–140. doi: 10.1016/j.gene.2012.10.053
Xu, X., Zhang, L., Zhao, W., Fu, L., Han, Y., Wang, K., et al. (2021). Genome-wide analysis of the serine carboxypeptidase-like protein family in Triticum aestivum reveals TaSCPL184-6D is involved in abiotic stress response. BMC Genomics 22:350. doi: 10.1186/s12864-021-07647-6
Yarra, R., and Xue, Y. (2020). Ectopic expression of nucleolar DEAD-Box RNA helicase OsTOGR1 confers improved heat stress tolerance in transgenic Chinese cabbage. Plant Cell Rep. 39, 1803–1814. doi: 10.1007/s00299-020-02608-x
Yu, X., Han, J., Wang, E., Xiao, J., Hu, R., Yang, G., et al. (2019). Genome-Wide Identification and Homoeologous Expression Analysis of PP2C Genes in Wheat (Triticum aestivum L.). Front. Genet. 10:561. doi: 10.3389/fgene.2019.00561
Zhan, H., Yue, H., Zhao, X., Wang, M., Song, W., and Nie, X. (2017). Genome-Wide Identification and Analysis of MAPK and MAPKK Gene Families in Bread Wheat (Triticum aestivum L.). Genes 8:284. doi: 10.3390/genes8100284
Zhao, J. Y., Lu, Z. W., Sun, Y., Fang, Z. W., Chen, J., Zhou, Y. B., et al. (2020). The Ankyrin-Repeat Gene GmANK114 Confers Drought and Salt Tolerance in Arabidopsis and Soybean. Front. Plant Sci. 11:584167. doi: 10.3389/fpls.2020.584167
Zhao, W., Zhang, L. L., Xu, Z. S., Fu, L., Pang, H. X., Ma, Y. Z., et al. (2021). Genome-Wide Analysis of MADS-Box Genes in Foxtail Millet (Setaria italica L.) and Functional Assessment of the Role of SiMADS51 in the Drought Stress Response. Front. Plant Sci. 12:659474. doi: 10.3389/fpls.2021.659474
Keywords: DEAD-box, genome-wide analysis, expression profile, abiotic stress response, regulation mechanism, wheat
Citation: Ru J-N, Hou Z-H, Zheng L, Zhao Q, Wang F-Z, Chen J, Zhou Y-B, Chen M, Ma Y-Z, Xi Y-J and Xu Z-S (2021) Genome-Wide Analysis of DEAD-box RNA Helicase Family in Wheat (Triticum aestivum) and Functional Identification of TaDEAD-box57 in Abiotic Stress Responses. Front. Plant Sci. 12:797276. doi: 10.3389/fpls.2021.797276
Received: 18 October 2021; Accepted: 01 November 2021;
Published: 09 December 2021.
Edited by:
Ramamurthy Mahalingam, Cereal Crops Research Unit, Agricultural Research Service, United States Department of Agriculture, United StatesReviewed by:
Hunseung Kang, Chonnam National University, South KoreaChristell Van Der Vyver, Stellenbosch University, South Africa
Copyright © 2021 Ru, Hou, Zheng, Zhao, Wang, Chen, Zhou, Chen, Ma, Xi and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ya-Jun Xi, eGl5YWp1bjIwMDJAMTI2LmNvbQ==; Zhao-Shi Xu, eHV6aGFvc2hpQGNhYXMuY24=
†These authors have contributed equally to this work