 Qi-Qing Cheng
Qi-Qing Cheng Yue Ouyang
Yue Ouyang Zi-Yu Tang
Zi-Yu Tang Chi-Chou Lao1
Chi-Chou Lao1 Chun-Song Cheng
Chun-Song Cheng Hua Zhou
Hua Zhou
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Plant Sci. , 23 December 2021
Sec. Plant Biotechnology
Volume 12 - 2021 | https://doi.org/10.3389/fpls.2021.791219
With the development of sequencing technology, the research on medicinal plants is no longer limited to the aspects of chemistry, pharmacology, and pharmacodynamics, but reveals them from the genetic level. As the price of next-generation sequencing technology becomes affordable, and the long-read sequencing technology is established, the medicinal plant genomes with large sizes have been sequenced and assembled more easily. Although the review of plant genomes has been reported several times, there is no review giving a systematic and comprehensive introduction about the development and application of medicinal plant genomes that have been reported until now. Here, we provide a historical perspective on the current situation of genomes in medicinal plant biology, highlight the use of the rapidly developing sequencing technologies, and conduct a comprehensive summary on how the genomes apply to solve the practical problems in medicinal plants, like genomics-assisted herb breeding, evolution history revelation, herbal synthetic biology study, and geoherbal research, which are important for effective utilization, rational use and sustainable protection of medicinal plants.
Medicinal plants, in the simple definition, are plants that can be used for medicinal purposes; in the detailed definition, are plants that have been verified and used for a long time as traditional medicines, have been found to have medicinal value in modern research, or contain medicinal ingredients in them. And they can provide the essential resources for human life, such as drugs, nourishment, condiments, and medicinal oil. They also uncovered and promoted the evolution of nature, animals, and humans. The foundation of all life is the genetic code. Therefore, access to the primary DNA sequence and how genes are encoded within the genome has become a basic resource in biology (Hamilton and Robin Buell, 2012). The genomics study of medicinal plants is to elucidate their molecular mechanism to prevent human diseases, by utilizing the genetic information and regulatory network of the species and the omics technologies, accordingly, to reveal their effect on the human body from the level of the genome. Now the process of genome sequencing in plants lags behind that in microorganisms and animals. Due to the lack of genomic information, there is a lack of communication between medicinal plants and modern life sciences, and the new frontier life science technology is hardly be applied to their research. Over the years, the works of research on medicinal plant medicines mainly focus on chemistry and pharmacology, the studies to uncover the biological nature of medicinal plants need to be strengthened.
Regarding plant genome sequencing methods and strategies, radical changes have taken place in the past 5 years, and medicinal plant genome sequencing is no exception. Previous reviews summarized the status of sequenced plant genomes before 2012 (Hamilton and Robin Buell, 2012), the status of sequenced angiosperm genomes before 2018 (Chen et al., 2018), and the impact of third generation genomic technologies on plant genome assembly (Jiao and Schneeberger, 2017). In addition, there were also Chinese reviews that proposed and introduced the Herb Genome Program (Chen et al., 2010) and 1,000 genome projects of medicinal plants (Chen et al., 2019). As sequencing cost reduces drastically and long-read sequencing technology develops quickly in recent years, it is certain that the genome continues to be improved, while more and more large and complicated medicinal plant genomes are reported. The future of revealing the secret of medicinal plant biology is bright. However, there is still not a review covering the medicinal plant genomes that have been released so far and introducing the development of sequencing strategies and applications.
In this manuscript, we conducted a systematic review of medicinal plants genome research. Moreover, the genome situation, sequencing technology development, and application of medicinal plant genomes are discussed. This review provides a historical perspective on the current situation of genomes in medicinal plant biology and highlights the use of rapidly developing sequencing technologies in plant biology. Challenges in genomics for medicinal plants are improved to some extent by long-read sequencing technologies regarding the current limitations. Multiple omics methods are integrated to make better use of medicinal plant genome data and to solve practical problems meeting in the breeding and medical fields. We also conduct a comprehensive summary of the application of medicinal plant genomes, to promote the studies of important questions in plant biology, like genomics-assisted herb breeding, herbal synthetic biology, and geoherbal research, which are significant for securing the future of medicinal plants and their active compounds.
The systematic literature search was performed by the following PRISMA guidelines (Moher et al., 2009). Firstly, it was performed through electronic databases, including PubMed (National Library of Medicine, United States), EMBASE (Elsevier, Netherlands), and Web of Science (Clarivate Analytics, United States) databases published until June 4, 2021. Studies were selected using the term “medicinal plant genome.” Additionally, we also searched the studies from the plaBiPD (Forschungszentrum Jülich GmbH, Germany) database and identified the medicinal plants from all the plants which have reported genomes. About the medicinal plant genomes, a total of 5,064 articles were identified initially by retrieving the electronic database comprehensively. Among these, 1,678 articles were from PubMed, 1,982 articles were from EMBASE, and 1,404 articles were from Web of Science, 173 articles were from the plaBiPD database, 831 articles were excluded for duplicates. A total of 4,189 articles were excluded by scanning the titles and the abstracts, and the exclusion reasons included irrelevant articles, not studies, and so on. Fifty-nine articles were excluded by reading the full-text manuscripts, with the exclusion reasons of reviews, not for medicinal plants and not for whole-genome sequencing and no mention of medicinal related content. Finally, a total of 158 articles were included in this meta-analysis. A flowchart of articles search and selection is shown in Figure 1. According to our statistical result, there were at least 161 reference genomes reported in 158 articles belonging to 126 medicinal plants published. We counted the number of journals that have published medicinal plant genomes, there were a totally of 40 journals, and the corresponding journal name and article number are provided in Supplementary Table 1. Since 2010, articles about medicinal plant genomes have appeared in journals almost every year. Since 2017, the number of medicinal plant genome articles has increased significantly.
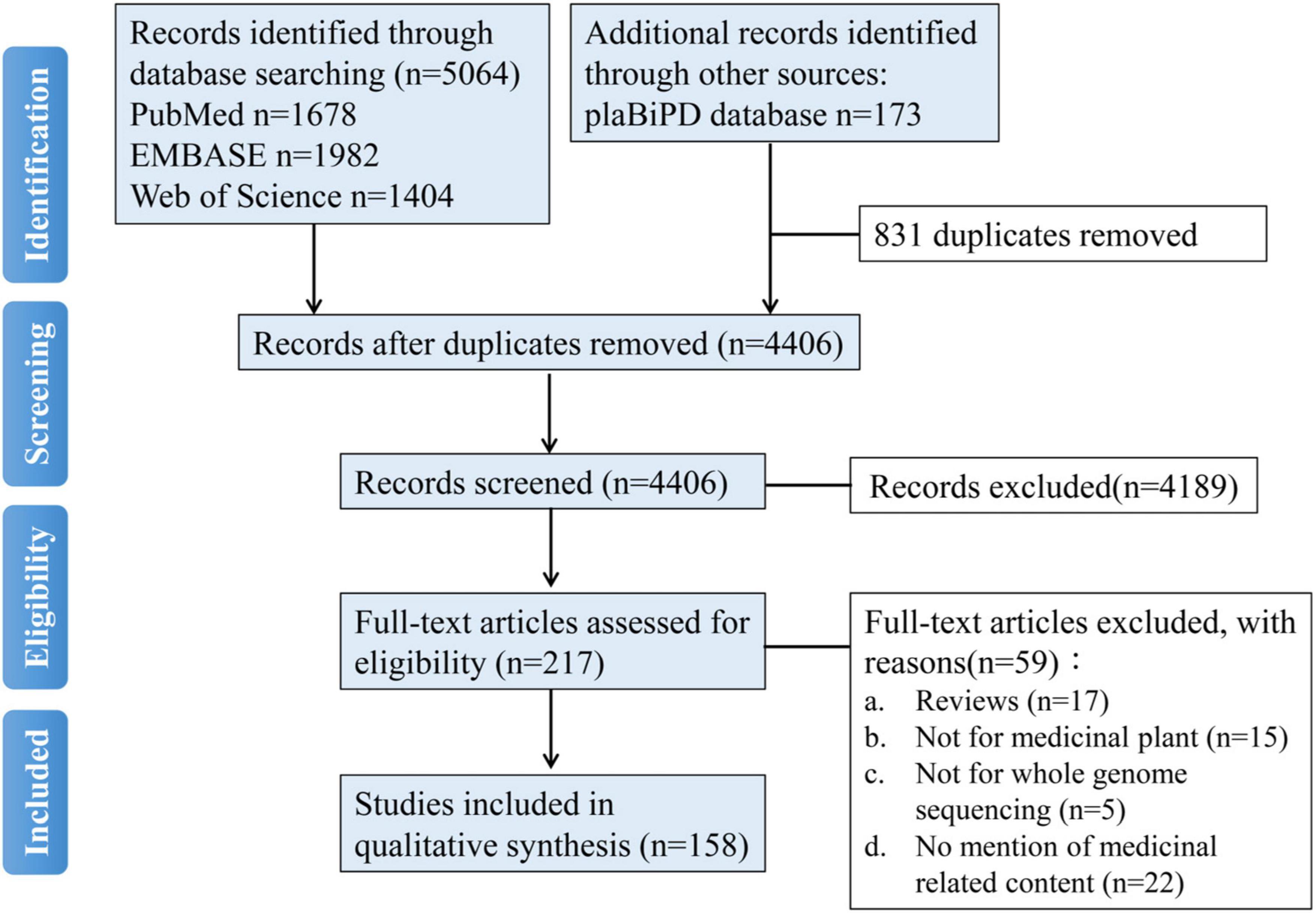
Figure 1. A flowchart of literature search and selection for a systematic review about medicinal plant genomes.
The medicinal plant genomes are more complex than animal genomes, so the process of sequencing the medicinal plant genomes has been hindered, and it entered a period of rapid development from 2016. This may be due to the decline in sequencing price and the development of long-read sequencing technologies. The status of medicinal plant genomes articles reported each year is shown in Figure 2A. In 2020, the number of published medicinal plant genomes has reached up to 53. In 2021, 33 medicinal plant genome articles have been published until June 4th, and the total article number is inferred to be more than 60. As more and more medicinal plant genomes have been revealed, several plants have been sequenced twice or multiple times for genomes. Among these repeatedly sequenced medicinal plant genomes, some are because of sequencing at the same time, some are due to improved level and quality, and some are genomes of different varieties from the same species. Among those 53 medicinal plant genomes reported in 2020, 18 genomes were reported repeatedly, accounting for 34%. This tells us that sequencing technology is continuously developing and progressing, bringing us to a completer and more accurate genome. Take Panax notoginseng (Chinese name: Sanqi) as an example, five versions of its genomes have been reported, the first two versions published in 2017 were sequenced by the next-generation sequencing (NGS) technology of Illumina platform (Chen et al., 2017; Zhang D. et al., 2017), and the recent three versions published in 2020 and 2021 were sequenced by the third-generation sequencing technologies of Pacific Biosciences (PacBio) and Oxford Nanopore (ONT) (Fan G. et al., 2020; Jiang et al., 2021; Yang et al., 2021b). The latest two versions of the genome were assembled to the chromosome level, the length of the assembled sequences was hundreds of times longer than the first two versions, and the accuracy and credibility of annotation have also been greatly improved. The statistical results of detailed information about medicinal plant genomes were shown in Table 1.
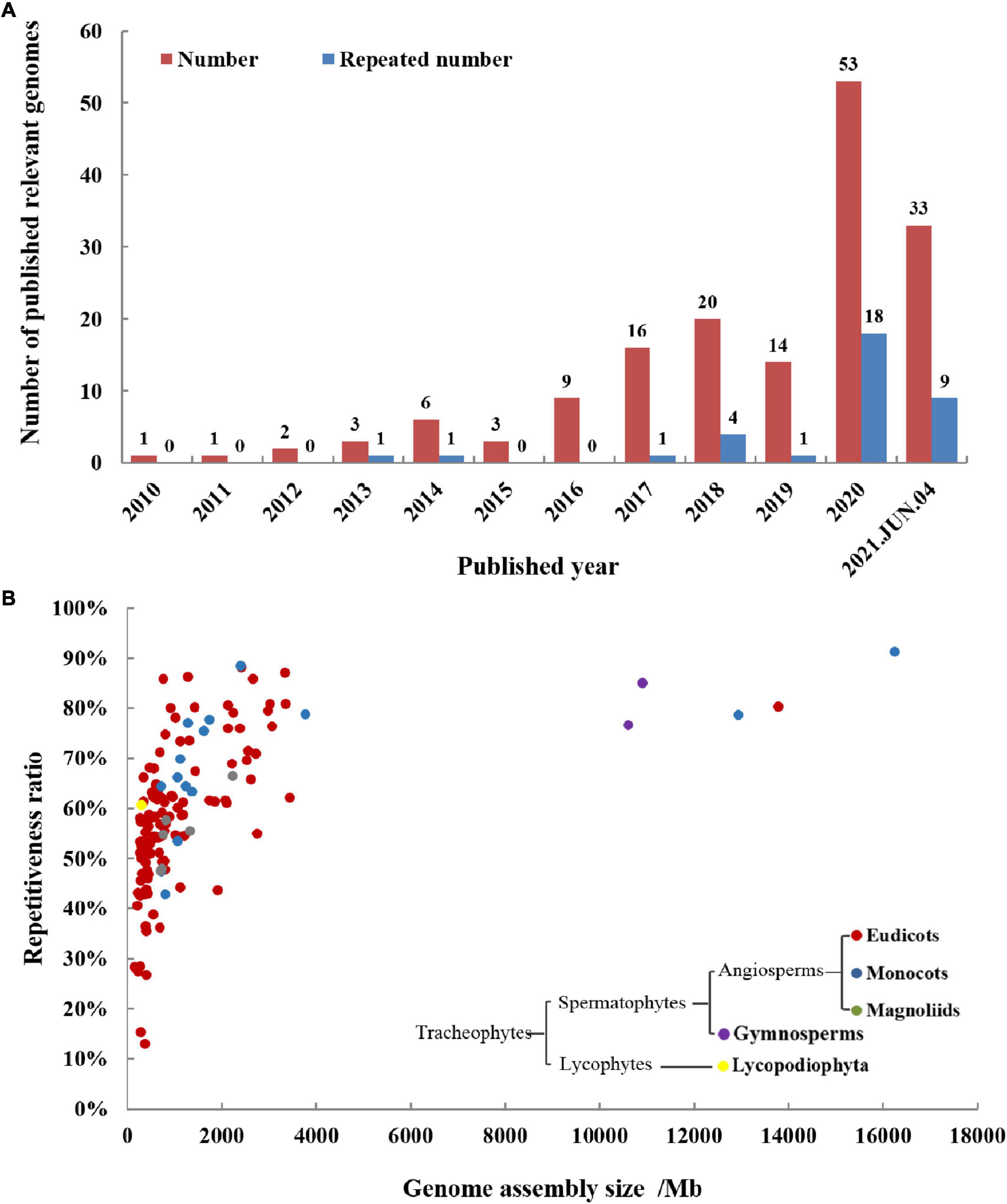
Figure 2. Publication history (A) and general information (B) of medicinal plant genomes. (A) The total number and repeated sequencing number of medicinal plant genomes are increasing year by year, proving that it has received more and more scientific research attention. (B) The figure shows published medicinal genome assemblies analyzed for genome-wide repeat levels and genome size. The repetitiveness of most medicinal plant genomes is generally high and correlated to genome size. The sequenced medicinal plants are divided into five groups based on phylogeny, including lycopodiophyta, gymnosperms, eudicots, monocots, and magnoliids, and eudicot accounts for the majority of them.
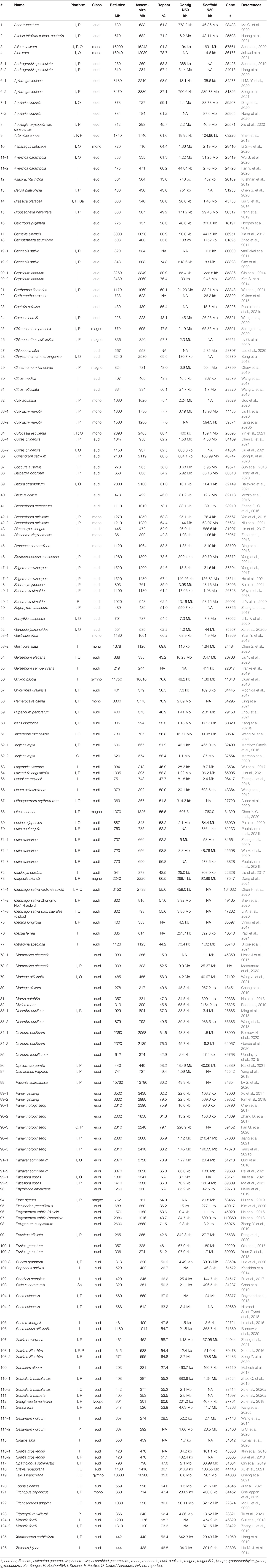
Table 1. The statistical results of medicinal plant genome published journals.
With the widespread application of NGS technology, genome sequencing of medicinal plants has become more feasible due to the greatly reduced cost and time required to complete the project. According to the whole genome sequence, the basic information of biology and biomedical functions can be well understood.
We have made statistics on the medicinal plant genome articles over the years, and have a basic understanding of the general characteristics of the reported medicinal plant genomes. The comparison of size and repetitiveness ratio of these published medicinal plant genomes and their evolution relationship is shown in Figure 2B. Among them, the genomes of five medicinal plants are much larger than other medicinal plants, they are Allium sativum, Paeonia suffruticosa, Aloe vera, Taxus wallichiana, and Ginkgo biloba. In the plants whose genomes have been sequenced, there are 123 angiosperms (including 12 monocots, 105 eudicots, and 6 magnoliids), two gymnosperms, and one lycopodiophyta plant. The simplified phylogeny of the major clades of sequenced medicinal plants is also shown in Figure 2B. Angiosperms account for the vast majority of sequenced medicinal plants, and eudicots make up the majority of angiosperms. Genome size has a positive correlation with the ratio of repetitive elements, when the genome size is larger, the proportion of repetitive elements also tends to be correspondingly larger. Most of the genome size is concentrated within 4 Gb, and the repetitiveness ratio sequences are concentrated between 30 and 90%.
It has been said that plant genome reports were formulaic and lack biology significance, their descriptions mainly included the assembly, protein-coding genes, repeats, evolution analysis, some aspects of biology, usually with a focus on transcription factors and active compounds biosynthesis pathway (Michael and Jackson, 2013). According to these published medicinal plant genomes, most of them have not yet been used to solve specific application problems, such as discovering new medicinal mechanisms, cultivating new resistant varieties, explaining evolutionary events, and so on. But the assembly of the genomes provides us with the guarantee of the database. Once we need the support of genetic information, the genome is the solid foundation and reference.
Medicinal plants are the main sources of medicine, and their records for medicinal usage can be traced back to almost 5,000 years ago in China, India, and Egypt (Moss and Yuan, 2006; Jamshidi-Kia et al., 2018). They are also the precious resource libraries for many chemical drugs, currently, more than one-third of clinical medications are derived from plant extracts or their derivatives (Chen and Song, 2016). The sequencing and demystification of the genome can give us a better understanding of the biosynthesis and regulation of bioactive compounds. Artemisinin-derived plant named Artemisia annua is one of the most famous medicinal plants, while the discovery of artemisinin has won the 2015 Nobel Prize in Physiology or Medicine (Su and Miller, 2015). A semi-synthetic system has been used to improve the production of artemisinin greatly (Paddon et al., 2013). Further revealing the genome of A. annua provides a comprehensive understanding of artemisinin biosynthesis and leads to improvement in artemisinin production. Before A. annua genome revelation, studies manipulating artemisinin biosynthesis focused on either upstream (Nafis et al., 2011) or downstream (Yuan et al., 2015) genes on the artemisinin biosynthesis pathway. Then the combined study and analysis of A. annua genomic and associated transcriptomic data proposed other efficient strategies to increase the production of artemisinin, one was to simultaneously enhance the expression of enzyme genes in different steps in the biosynthesis pathway including the upstream (HMGR), midstream (FPS) and downstream (DBR2), and the other was to overexpress the expression of transcription factors like AaMYB2 that could regulate the expression of ADS, CYP71AV1, DBR2, and ALDH1 in artemisinin biosynthesis pathway, which could significantly improve artemisinin and dihydroartemisinic acid content, providing a new insight for increasing the supply of artemisinin from plant sources (Shen et al., 2018).
In addition to improving the content of active compounds, it is also necessary to ensure the agronomic traits and enhance the resistance ability to stresses of medicinal plants. Genome sequencing can help identify the genes associated with agronomic and disease resistance traits, and can target control of the genes to cultivate new varieties of medicinal plants with highly effective ingredients, excellent agronomic features, and high resistance abilities. P. notoginseng, a well-known medicinal plant, is susceptible to a wide range of pathogens, so its cultivation faces several challenges (Ou et al., 2011). The sequencing of the P. notoginseng chromosome level genome, combining a genome-wide association study on 240 cultivated individuals, successfully identified 63 genes associated with dry root weight (included genes encoding cysteine/histidine-rich C1 domain proteins), 168 genes associated with stem thickness (included APC6, WRKY71, and RWA3, etc.) and 33 genes associated with disease resistance trait (included genes encoding LRR receptor-like serine/threonine-protein kinases) (Fan G. et al., 2020). These valuable resources of P. notoginseng can provide new opportunities to harness the full potential of its economic and medicinal values.
Moreover, some medicinal plants also play an important role in evolution, and the discovery of their genomes can help to understand the evolutionary relationship of plants. Ginkgo biloba is a living fossil without living relatives, which represents one of the four extant gymnosperm lineages (cycads, ginkgo, conifers, and gnetophytes). Its genome showed that LTR-RT insertions and two whole-genome duplications (WGD) events in evolution history contribute to the large genome size and long introns. In angiosperms, chromosomal breakages and fusions, as well as uneven gene loss, might occur to prevent a continuous growth in genome size (Schnable et al., 2009), and this mechanism for removing transposable elements (TEs) might lack and lead to enormous genome size in gymnosperms like ginkgo. The outstanding defense ability of ginkgo resulted from the remarkable duplication of resistance genes and enrichment of relevant pathways. The ginkgo genome sheds light on sequencing large plant genomes and helps to know the genetic and evolutionary process of land plants in natural evolution (Guan et al., 2016).
The quality of genome assembly directly affects the quality of the whole genome. Contig N50 and scaffold N50 are the primary indicators for evaluating genome assembly results. Generally, the longer the contig N50 and scaffold N50 are, the better the assembly result is. As shown in Table 1, in 2017 and before, most of the reported medicinal plant genomes used the NGS technologies, such as Illumina and Roche/454, and the length of contig N50 ranged from a few kilobases to dozens of kilobases. In 2018, half of the published genomes used a combination of next- and third-generation sequencing technologies, such as Illumina + PacBio and Illumina + Oxford Nanopore. In 2019 and beyond, the sequencing strategy of combining next- and third-generation has been applied to the majority of the reported genomes. It can be seen from Figure 3 that the length of contig N50 became long since 2018, and then increased year by year. By 2020, the length has been greatly improved, the length of contig N50 was generally increased to the range between a few hundred kilobases and several megabases. The length of contig N50 was similar in the medicinal plant genomes published in 2020 and 2021. And the longest length was as long as 21.23 Mb (Cheng et al., 2021). It shows that the popularization and application of third-generation sequencing have brought convenience to scientific research, and at the same time have greatly improved the quality and integrity of the genome.
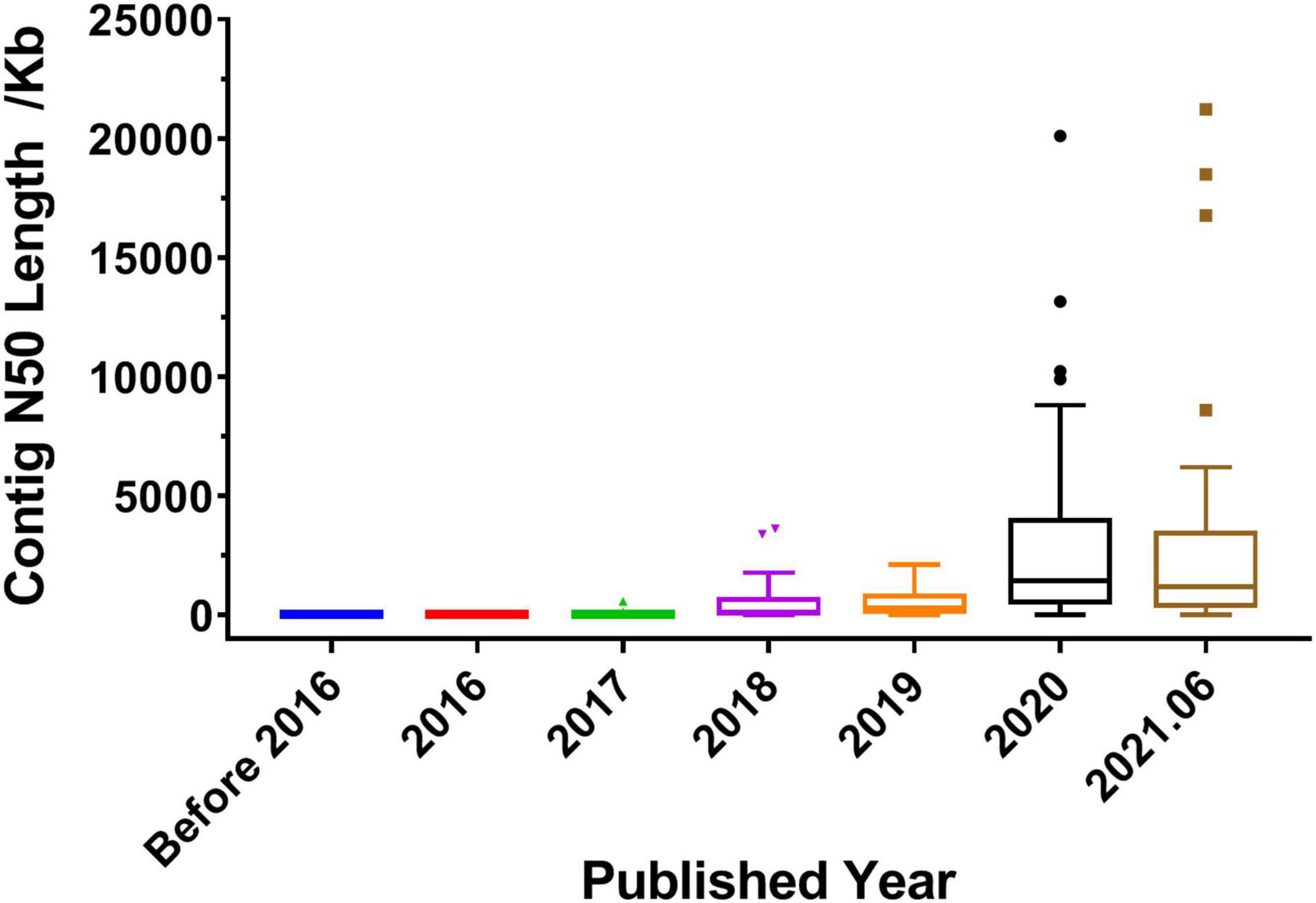
Figure 3. Distribution of contig N50 length in published medicinal plant genomes. Before 2016 represents the published years before 2016, and 2021.06 represents the time between 2021 January and June.
The development process of sequencing strategy on medicinal plant genomes has experienced three stages, germination stage, development stage, and expansion stage (Figure 4).

Figure 4. The development process of medicinal plant genome sequencing.
The start of genomics is from the early 1990s, and automated sequencing methods using dideoxy chain termination with fluorescent molecules developed, which is known as Sanger sequencing. The effectiveness of the Sanger platform for large eukaryotic genomes was first reported in 2000 for Drosophila melanogaster, ushering in a new era of genomics (Adams et al., 2000). This method was also applied in plant biology, like sequencing ESTs in Arabidopsis thaliana (Newman et al., 1994), and then sequencing the whole genome of various plants, like Oryza sativa (Yu et al., 2002), Populus trichocarpa (Tuskan et al., 2006), Carica papaya (Ming et al., 2008) and Brachypodium distachyon (International Brachypodium Initiative, 2010). However, there are still gaps and errors in the assembly of these genomes, so they are not completely “finished,” because the process of “finishing” needs inspection and experimental resolution of inconsistencies, and it’s a time-consuming, tough, and expensive work (Hamilton and Robin Buell, 2012). In the germination stage of the development process about medicinal plant genome sequencing, considering this and cost, the Sanger sequencing method is only used to sequence the genome of major economic crops that are also regarded as medicinal plants, like Ricinus communis, to provide references and templates for subsequent research.
After 2011, the NGS technology develops rapidly and occupies the position of the mainstream sequencing platform, becoming the preferred technology for sequencing the medicinal plant genomes. The widely and mainly used NGS platforms are Roche 454 platform and Illumina platform.
Roche 454 platform is the first commercially successful NGS system. This sequencing method uses a high-throughput pyrosequencing technology (Margulies et al., 2005). This platform utilizes emulsion PCR to detect the pyrophosphate released during nucleotide incorporation. In 2005, the read length of Roche 454 was only 100–150 bp with 20 Mb output data per run (Mardis, 2008). In 2008, the 454 GS FLX Titanium system appeared, with a reading length up to 700 bp and 0.7 G output data per run within 24 h. In late 2009, Roche simplified the library preparation and data processing and improved the output to 14 G per run (Liu et al., 2012). In 2012, the platform upgraded to the FLX+ and could generate 1 million reads, with a reading length up to 1,000 bp.
Illumina platform is a high-throughput technology of sequencing by synthesis using reversible dye terminators developed by Solexa and then purchased by Illumina in 2008 (Bentley et al., 2008). The mechanism of the Illumina platform is bridge PCR, which is different from the Roche/454 platform. The library DNA with fixed adaptors is denatured to single strands and linked on the flow cell, followed by bridge amplification to synthesize clusters of clonal DNA fragments. The library splices into single strands by linearization enzyme (Mardis, 2008), and then four kinds of fluorescently labeled nucleotides which have been modified with a terminator complement the template one base at a time, the signal is captured, then the terminator and fluorescent dye are cleaved, and a new round of synthesis repeats until coming up to the desired read length. In late 2011, the paired-end mode of the Hi-Seq2000 Illumina platform could generate more than 250 million reads total sequences of one lane.
Because the throughput of Hi-Seq 2000 is higher, the price is lower, and the application range is wider than Roche/454, the application of the Illumina platform in the medicinal plant genome sequencing occupies the mainstream position. The Illumina platform is widely applied for expression profiling, de novo sequencing, and re-sequencing in plant sequencing, like Thellungiella parvula (Dassanayake et al., 2011) and Arabidopsis thaliana (Cao et al., 2011). As more and more medicinal plant genomes have been reported, the medicinal plant genome sequencing has begun to enter the development stage, many large size medicinal plant genomes were successfully sequenced. However, another difficulty of plant genomes is the high repetition in the genome, so it is difficult to accurately assemble them by the NGS technologies.
The development of third-generation sequencing has overcome this problem. The most widely applied long-read sequencing platform is Single-Molecule Real-Time (SMRT) sequencing of Pacific Biosciences company. SMRT sequencing is run on cells, which have tiny wells called zero-mode waveguides (ZMWs). In each ZMWs, a DNA polymerase/template complex gets immobilized, and synthesizes a new DNA strand (Jiao and Schneeberger, 2017). Each incorporation generates a light pulse that can be recognized for differently labeled nucleotides (Eid et al., 2009). PacBio systems can sequence reads with an average size of about 20 kb and a maximum length of over 60 kb (Kim K. E. et al., 2014; Vanburen et al., 2015). Although the sequencing error rate of raw reads is up to 15%, self-correction by adequate coverage sequencing data (Chin et al., 2013) or correction with NGS data (Bashir et al., 2012; Koren et al., 2012) enables genome assemblies with the accuracy of over 99.999% simply by running bioinformatics analysis software (Chin et al., 2016). Besides the PacBio SMRT platform, there is also another long-read sequencing platform introduced by ONT Technologies, which provided access to their first sequencing system in 2014 (Quick et al., 2014; Deamer et al., 2016). Single DNA molecules are run through nanopores, and individual nucleotides create characteristic disruptions in them, which reveal the sequence of the nucleotides. The reads length and sequencing accuracy are similar with PacBio reads, and the longest reads can reach up to 200 kb. First, whole-genome assemblies using ONT data have reached N50 values of multiple hundred kb for fungal genomes, and bacterial genomes could be fully assembled with a nucleotide accuracy of over 99% (Goodwin et al., 2015; Loman et al., 2015).
The emergence of third-generation sequencing technology has made a great leap in sequencing read length and brought medicinal plant genome sequencing into a stage of rapid development. The strategy used in this stage is a combination of second- and third-generation sequencing technologies, which can ensure long read length, high throughput, and reasonable sequencing price at the same time. Medicinal plant genomes are large and have high-ratio repetitive elements, the frequently-used strategy is combining high coverage Illumina and low coverage PacBio SMRT or ONT data. Because third-generation sequencing can provide long-read sequences to increase the assembly accuracy and genome draft quality, but the price is relatively high, so Illumina platform is used to guarantee enough sequencing data. And this can make it possible to assemble large and complex medicinal plant genomes to the chromosome level. After these years of sequencing development, the medicinal plants not only can obtain draft genome relevant information and dig out target protein-coding genes, but also recognize the chromosome-level of the genome to discover the evolution, gene cluster’s function, repetitive elements effect, and so on.
We found that not only does the number of medicinal plant genomes sequenced continue to increase, but the number of medicinal plant genomes sequenced repeatedly is also increasing. Why? First of all, because the genomes of many medicinal plants have not been revealed yet, many teams are performing de novo sequencing of the genomes at the same time, and accordingly publish them at the same time. Then, with the continuous development of gene sequencing technology, we can obtain longer sequencing read lengths, so as to assemble more complete and accurate high-level genomes. Genomes assembled to the chromosome level are the current trend. The information that the genome gives us is no longer a contig or scaffold, but the chromosome and the position of a gene on the chromosome.
There are 25 medicinal plants with two reported genomes, three medicinal plants with three reported genomes, and one plant with five reported genomes. Representative medicinal plants include Momordica charantia (bitter gourd), Salvia miltiorrhiza (Danshen), Punica granatum (pomegranate), Panax notoginseng (Sanqi), Panax ginseng (Asian ginseng), etc. Bitter gourd and danshen have two reported versions of the genome. Bitter gourd completed the de novo assembly of the genome draft in 2017, as well as basic annotation and evolutionary analysis (Urasaki et al., 2017). In 2020, using PacBio long-read sequencing technology, the Momordica charantia genome was assembled to the chromosome level, and further investigate the genomic changes under domestication (Matsumura et al., 2020). The genome of Salvia miltiorrhiza was also assembled to eight chromosomes, the assembled genome size increased from 538 to 594.75 Mb, and the proportion of repetitive elements also increased from 54.44 to 64.84% (Xu et al., 2016; Song Z. et al., 2020). Punica granatum (pomegranate), which is a popular and nutritious fruit with medicinal properties, has three published genome versions (Qin et al., 2017; Yuan Z. et al., 2018; Luo et al., 2020). The third version of the genome is assembled to the chromosome level, and it is a high-quality genome map of the soft-seed pomegranate, which helps to clarify the genetic divergence between soft- and hard-seeded varieties and provides insights into the genetic diversity and population structure of pomegranates (Luo et al., 2020). Panax notoginseng (Sanqi) is a well-known TCM whose genome research is sought after by scientists, and a total of five versions have been reported. The three recent versions are assembled to the chromosome level (Fan G. et al., 2020; Jiang et al., 2021; Yang et al., 2021b), which are more complete than the previously available genome assemblies (Chen et al., 2017; Zhang D. et al., 2017), further reveal the biosynthesis pathways of ginsenosides and dencichine, as well as provide a resource for further exploration of the saponin biosynthesis, cultivation, and breeding of P. notoginseng. Panax ginseng (Asian ginseng), reputed as the king of medicinal herbs, belongs to the same genus Panax, which also has two versions of reported genomes (Xu et al., 2017; Kim et al., 2018). Both of these two genomes provide a comprehensive understanding for functional and evolutionary analysis as well as ginsenoside biosynthesis. Additionally, Kim et al. (2018) identified fatty acid desaturases that can increase freezing tolerance and chlorophyll a/b binding protein genes which enable efficient photosynthesis under low light. However, the read length of both genomes is not long enough by the current standards, and there is still space for further improvement in the integrity and accuracy of the ginseng genome.
The genes related to medicinal plant growth and development, disease resistance, important genetic traits, and germplasm characters which are the important functional genes in medicinal plants, taking advantage of genome annotation information, discovering good genes, using genetic engineering methods to break the reproductive isolation, and cultivating the new species with excellent agronomic characters and high content of active ingredients, so that it can lay the foundation for the large-amount extraction of active ingredients and extensive clinical application. By combing transcriptome and resequencing of individual species within or between species, the large-scale molecular markers can be identified rapidly and accurately, and genetic linkage study of molecular markers and qualified characters can also be accelerated, the phenotypes of medicinal plants and the relationship of physical characteristics and genotypes are discovered quickly so that efficiency of breeding are improved obviously.
The study of Scutellaria baicalensis (Huangqin) genome sequencing revealed that a specialized metabolic pathway for the synthesis of 40-deoxyflavonebioactives evolved in the genus Scutellaria and found that the gene encoding a specific cinnamate coenzyme A ligase likely obtained its new function following recent mutations and that four genes encoding enzymes in the 40-deoxyflavone pathway are present as tandem repeats in the genome of Huangqin. Further analysis discovered that gene duplications, segmental duplication, gene amplification, and point mutations coupled to gene neo- and subfunctionalizations were involved in the evolution of 40-deoxyflavone synthesis in Scutellaria. These results not only provide significant insight into the evolution of specific flavone biosynthetic pathways in the mint family Lamiaceae but also facilitate the development of tools for enhancing bioactive productivity by molecular breeding in plants (Zhao Q. et al., 2019).
Whole-genome sequencing cannot only elucidate the biosynthesis pathways of natural products but also give insight into their evolution. The evolution will bring the whole genome change, like WGD and whole-genome triplication (WGT), to adapt to the environment alteration and explain the characters of plants. We summarized the WGD and WGT events of some representative species reported in the medicinal plant genome articles, and these situations are shown in Figure 5. These WGD and WGT events are summarized and introduced into three types of plants, which are eudicots, monocots, and magnoliids.

Figure 5. The whole genome duplication (WGD) and whole genome triplication (WGT) events in representative medicinal plants.
In the eudicots part, we select five representative branches to demonstrate the situation. The representative medicinal plants of Araliaceae and Apiaceae are clustered together. P. ginseng, P. notoginseng, and E. senticosus belong to Araliaceae, and P. notoginseng is diploid, while P. ginseng and E. senticosus are tetraploid. Two rounds of WGD were discovered in these Araliaceae plants, the first round occurred around 29.6 Mya, P. ginseng, and E. senticosus both had the second round of WGD, which were found almost 2.2 Mya in P. ginseng and 13 Mya in E. senticosus, respectively. Additionally, these recent WGDs were discovered to contribute to the ability of P. ginseng to overwinter and E. senticosus to adapt to cold environment, enabling them to live and spread broadly through the cold area (Kim et al., 2018; Jiang et al., 2021; Yang et al., 2021a). These two rounds of WGD occurred in the family Araliaceae after divergence with the Apiaceae, which may be one of the reasons why its genome was bigger than other medicinal plants. In the D. carota and A. graveolens that belonged to Apiaceae, one shared WGD occurred in about 43 Mya, and one recent WGD only existed in A. graveolens in approximately 1.9 Mya, and this duplication contributed to the expansion of terpene synthase gene families (Song X. et al., 2020). The second branch in the eudicots part includes six plants belonging to Lamiales, one shared WGD (almost 60.7 Mya) was identified in S. baicalensis, S. barbata, S. miltiorrhiza, and S. indicum, which might be responsible for chromosomal expansion and rearrangement (Xu et al., 2020a), and two rounds of WGD were found in S. splendens and L. angustifolia, which could result in the gene families expansion related to terpenoid biosynthesis (Li et al., 2021). In P. cuspidatum, it experienced current lineage-specific WGD at 6.6 Mya after the divergence with F. tataricum from the ancestor, and it shared the ancient and common WGD with F. tataricum at 65 Mya (Zhang Y. et al., 2019), after this WGD, the genome of F. tataricum experienced dramatic chromosomal rearrangements, resulting in very fragmented intra-genome collinear blocks (Zhang L. et al., 2017). There is also a WGT event identified and reported in the medicinal plant genome articles. T. wilfordii was found to have a WGT event in approximately 21 Mya, which enabled it to cope better with and adapt to the markedly changed environment, and the duplication of the triptolide biosynthesis genes were almost generated by this WGT event, suggesting this WGT event was important to the evolution of triptolide biosynthesis (Tu et al., 2020).
In the monocots part, A. sativum and H. citrina are the representatives. A. sativum has undergone two rounds of WGD, suggesting WGD can be the important driving force of the proliferation of TEs and genome expansion in garlic (Sun et al., 2020). Otherwise, H. citrina experienced a recent WGD event at about 15.73 Mya, which was the main factor resulting in multiple copies of the orthologous genes (Qing et al., 2021). In the magnoliids part, C. salicifolius and P. nigrum are the representatives. Two rounds of ancient WGD were inferred in the C. salicifolious genome, one was shared by Calycanthaceae at ∼87 Mya after its divergence with Lauraceae, and the other was dating back to approximately 142 Mya in the ancestry of Magnoliales and Laurales (Lv Q. et al., 2020). Meanwhile, the P. nigrum genome was speculated to have a WGD event at ∼17.9 Mya, which brought genetic changes that were responsible for the particular biosynthesis of piperine (Hu et al., 2019).
Domestication is a complex evolutionary process, which is one of the most important technological innovations in human history, humans use plants to change their morphology and physiology traits, distinguishing them from wild ancestors, and ultimately giving rise to the current human cultures (Diamond, 2002; Hancock, 2005). Some of the domesticated plants are medicinal plants. The timing and geographical origins of domesticated traits, as well as the genes that lead to changes in traits, can be sent to find clues from genomic information (Purugganan and Fuller, 2009).
Coix is a widely cultivated grass crop with high nutritional and medicinal value, which has been domesticated as early as the Neolithic era. However, its genetic research and breeding were hampered by the lack of a sequenced genome. Two chromosome-level genomes of coix have been reported simultaneously, which belong to elite cultivar Beijing (Liu H. et al., 2020) and wild relative Coix aquatica Daheishan (Guo et al., 2020), respectively. They both find that hull thickness is an important domestication trait between the wild relatives and cultivars, and selection of papery hull from the stony hull in wild progenitors was a key step in coix domestication. Combining resequencing analysis and comparative analysis, several domesticated loci or genes (like loci in the ∼2 to 150 kb region upstream of ub3) and two major quantitative trait loci associated with hull thickness and color (Ccph1 and Ccph2), were discovered to be the potential identification loci for domestication. These findings will greatly facilitate and benefit the molecular breeding of coix and provide a powerful reference for the domestication and evolution of medicinal plants.
The active components of medicinal plants with complex and diverse structures are the material basis for their medicinal effect, and it’s also an important source of new drug discovery. However, many medicinal plant materials often face a series of problems in the process of development and utilization, for example, the growth of many medicinal materials is greatly affected by environmental factors; some rare herbs grow slowly and are difficult to grow by artificial cultivation; most of the active ingredients are low in content, complex in chemical structure and difficult in chemical synthesis; traditional methods of natural extraction or artificial chemical synthesis cannot meet the needs of scientific research and new drug development. Synthetic biology will be an effective way to resolve these problems.
As high-throughput sequencing technology for genome and transcriptome studies have developed rapidly, using bioinformatics method and functional genomics approach to screen and identify enzyme-coding genes on specific secondary biosynthesis pathway from a large number of the original species of medicinal plants, which will greatly accelerate the analysis process of secondary biosynthesis pathway and lay a solid foundation for herbal synthetic biology research of medicinal plants.
Tripterygium wilfordii genome is one of the typical examples. Because of the extremely low yield of triptolide extracted from T. wilfordii, its original plant cannot be grown on a large scale, and the current chemical synthesis route is limited to a yield of less than 1.64%. A more promising method to obtain more triptolide could be metabolic engineering, which can be realized via a synthetic biology strategy. However, it required elucidation of the triptolide biosynthesis pathway. Therefore, the sequencing of the T. wilfordii genome was completed, and cytochrome P450 TwCYP728B70 involved in triptolide biosynthesis was identified, accordingly, the triptolide content in the CYP728B70 overexpression line increased obviously (Tu et al., 2020). It’s important to make full use of genomic resources to reveal the biosynthesis pathways of active compounds in medicinal plants and use candidate genes in these pathways for the heterologous bioproduction under synthetic biology strategy.
Geoherbs, controlled by genetic factors and affected by environmental conditions, are representative of high-quality medicinal materials. The utilization of sequencing technology and data can provide useful tools to elucidate the molecular mechanism of geoherbs. For the same medicinal plants in different areas, epigenomic studies of medicinal plants can be carried out to clarify the genetic variation of different production areas, especially the modification effect of different environments on the epigenome of medicinal material, including DNA methylation modification, small RNA sequencing analysis, chromatin immunoprecipitation analysis, and so on. In addition, microorganisms in soil are also important factors in the growth environment of geoherbs. Metagenomic analysis of soil microbial community can be sequenced to provide the basis for revealing the interaction between soil microorganisms and the growth of medicinal plants.
Recently, 545 genomes of ginkgo trees sampled from 51 populations across the world were sequenced to identify three refugia in China and detect multiple cycles of population expansion and reduction along with glacial admixture between relict populations in the southwestern and southern refugia, and multiple anthropogenic introductions of ginkgo were proved to occur from eastern China into different continents. This study provides insight into the evolutionary history of ginkgo and helps to provide protection and utilization way for its valuable genomic resources (Zhao Y. P. et al., 2019).
Because of the rapid development and progress of sequencing technology, more and more biosynthesis pathways of active ingredients from medicinal plants have been revealed. The early-stage was based on the mining from transcriptome data, and the later stage was based on the combined mining from genome and transcriptome data. Although transcriptome sequencing has so far occupied a major position in the research of biosynthesis pathways of medicinal ingredients, genome data can provide more important information, for example, it can reveal the evolution process of biosynthesis pathway genes, thereby efficiently synthesizing secondary metabolites with medicinal activity. In the opium poppy genome, a great discovery about a gene cluster including 15 genes was reported. Meanwhile, in its evolution process, the events like gene duplication, rearrangement, and fusion, could lead to the aggregation and co-expression of genes in the two metabolic pathways of noscapine and morphinan, so that it resulted in the formation of this supergene cluster, which could synergistically synthesize the medicinal ingredients in opium poppy (Guo et al., 2018). Therefore, the opium poppy genome helps to decipher the mystery of the synthesis of secondary metabolites. It is not only beneficial to the development of molecular plant breeding tools and cultivating new varieties, but also has great guiding significance for the selective improvement of the production of alkaloids with different efficacy in future artificial synthesis.
It also provides new ideas for the application of medicinal plant genomes. Through the evolution process, gene duplications and neofunctionalization can generate gene clusters, which may relate to specialized metabolites, and this phenomenon has already been observed in several model plants, like A. thaliana, Zea mays, and Solanum lycopersicum (Bharadwaj et al., 2021). In medicinal plants, we can refer to the research strategy of the opium poppy (Guo et al., 2018), which can help us understand the formation process of gene clusters related to medicinal active ingredients and improve their biosynthesis efficiency.
The continuous emergence of high-quality genomes has made the application of comparative genomics analysis more and more extensive and in-depth, and it is also a powerful tool for researchers to dig out biological problems and explain biological phenomena (Nobrega and Pennacchio, 2004). Comparative genomics, based on genome mapping and sequencing technology, are generally referred to as comparative analysis of the structural and functional gene regions of the genomes among multiple species or multiple individuals (populations) from one species. Specifically, it is to compare the similarities and differences in the structural characteristics, study the contraction and expansion of gene families, discover the differentiation time and evolution relationship, analyze the generation and evolution of new genes, etc.
One representative example of comparative genomics among different medicinal plant genome species can be Scutellaria baicalensis and Scutellaria barbata. The comparative genomic analysis of them showed the recent LTR may result in chromosomal rearrangement and expansion, and tandem duplication of paralogs after their speciation might contribute to the divergent evolution of flavonoid biosynthesis gene families, which provided a significant foundation for the evolution and chemodiversity studies in the Lamiaceae (Xu et al., 2020a).
Moreover, a representative of comparative genomics among different populations in the same species can be Forsythia suspense. Genome-wide comparative analysis was then conducted for the 15 natural populations across its current distribution range. The results revealed that candidate genes associated with local adaptation were functionally correlated with heterogeneous environmental factors, and supported the hypothesis that adaptive differentiation should be highly obvious in the genes of signal crosstalk between different environmental variables, which gave insights into the fundamental genetic mechanisms of the local adaptation to climatic gradients in plant species (Li L.-F. et al., 2020).
The use of medicinal plants has a long history and diverse application methods. Related works of research mainly focus on the discovery of chemical basis and the analysis of pharmacodynamic effect, but the understanding of medicinal plant genetic resources is relatively weak. Therefore, the research on the genome of medicinal plants should make use of the latest technologies and achievements of genomics, and integrate the studies of structural genome, functional genome, transcriptome, proteome, epigenome, metagenome, synthetic biology, metabolome, bioinformatics, and other relevant databases. Therefore, the essence of medicinal plants can be revealed, the relationship among genetic resources, chemical quality, and drug efficacy can be recognized.
We are most concerned about the medicinal value of medicinal plants. The medicinal value is not only reflected in the content of their medicinal ingredients, but also the stability of the quality of their medicinal materials. Now medicinal plant genomes can be annotated to obtain protein-coding genes, especially biosynthesis genes of active ingredients, analyze their evolutionary history and domestication process, and discover genes that respond to environmental stresses to help improve their resistance and ability. However, the powerful ability of the medicinal plant group has not yet been manifested, and its ability to solve the difficulties in practical applications remains to be developed. How to use the information of the medicinal plant genome to transform and obtain excellent medicinal plant varieties has not yet been realized. Determining suitable model medicinal plants is of great significance to the research on the practical application of medicinal plant genomes. The determination of appropriate model medicinal plants is of great significance to the study of the genomics of medicinal plants. From the perspective of general biological characteristics, it usually should have the traits of a short age cycle, many offspring, and stable phenotype. As for genetic resources, the genome should be relatively small, easy to sequence, and genetic transformation is relatively easy. As for medicinal characteristics, it should be suitable for secondary metabolite biosynthesis and production research. Therefore, the establishment and improvement of a suitable model medicinal plant platform will greatly enhance the application value of medicinal plant genomes.
The assembly of plant genomes is a challenging problem because of their high repetitiveness due to TEs, extreme genome sizes, and polyploid nature. With the development and emergence of long-read sequencing (Eid et al., 2009; Deamer et al., 2016) and long-range scaffolding methods such as optical mapping (Schwartz et al., 1993), chromosome conformation capture (Burton et al., 2013), and DNA dilution-based technologies (Amini et al., 2014; Zheng et al., 2016), the medicinal plant genome sequencing overcomes weaknesses of short-read assemblies and becomes possible to assemble to the chromosome-level (Jiao and Schneeberger, 2017). Although there have been medicinal plants that enable the assembly of entire chromosomes, most medicinal plants just still obtained long scaffolds or super-scaffolds. And now we have got a large amount of sequencing data from medicinal plants, how to effectively explore and apply them to dig deeper information is still facing problems and challenges.
Moreover, thanks to the advancement and development of sequencing technology and bioinformatics algorithms, at least one hundred medicinal plant genomes have been obtained. How to use them thoroughly and effectively has attracted the attention of many institutions and researchers. In recent years, several databases of medicinal plant genomes have already been built, such as the Herbal Medicine Omics Database1 (Wang X. et al., 2018), 1K Medicinal Plant Genome Database,2 and Database of 10,000 Medicinal Plants.3 These databases summarize the medicinal plant genomes that have been reported at this stage or aim to build a biological big data platform for medicinal plants, linking the omics data, active ingredients, disease information, and other information to promote their modernization. All of the above indicate that the medicinal plant genome has entered the stage of big data association research from the stage of exploring the unknown. Moreover, because of the limitations of previous technologies and methods, the disclosed medicinal plant genome information is limited. If the obtained medicinal plant genome information is aggregated and shared through the database, this should be a huge treasure to be unearthed, which will prompt the research efficiency of medicinal plants.
Thanks to the invention of the long-read sequencing technology, the research on medicinal plant genomes has developed rapidly and is no longer limited by their huge genome size and high repetitive sequences. The number of genomes reported in the past 2 years has increased significantly, and the quality of genomes has also been greatly improved, most of which have been assembled to the chromosome level. Correspondingly, the sequencing strategy they adopted has also been continuously updated, making them more and more widely used, answering and solving many problems in scientific researches and practical applications, including herb breeding assistance, evolutionary history revealing, domestication process understanding, herb synthetic biology study, geoherbal research and comparative genome analysis, these are of great significance to the effective use and sustainable protection of medicinal plants, which can improve their research efficiency and promote their modern development.
Q-QC planned the manuscript outline, wrote the draft, and created the figures and tables. YO, Z-YT, C-CL, Y-YZ, and C-SC proofread the manuscript. HZ supervised the study and revised the manuscript. All authors contributed to the article and approved the submitted version.
This project was funded by the Science and Technology Development Fund, Macao SAR (Project Nos. 0001/2020/AKP, 0061/2019/AGJ, 0027/2017/AMJ, and 062/2017/A2) and the National Key Research and Development Program of China (Project No. 2017YFE0119900).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.791219/full#supplementary-material
Adams, M. D., Celniker, S. E., Holt, R. A., Evans, C. A., Gocayne, J. D., Amanatides, P. G., et al. (2000). The genome sequence of Drosophila melanogaster. Science 287, 2185–2195. doi: 10.1126/science.287.5461.2185
Amini, S., Pushkarev, D., Christiansen, L., Kostem, E., Royce, T., Turk, C., et al. (2014). Haplotype-resolved whole-genome sequencing by contiguity-preserving transposition and combinatorial indexing. Nat. Genet. 46, 1343–1349. doi: 10.1038/ng.3119
Auber, R. P., Suttiyut, T., McCoy, R. M., Ghaste, M., Crook, J. W., Pendleton, A. L., et al. (2020). Hybrid de novo genome assembly of red gromwell (Lithospermum erythrorhizon) reveals evolutionary insight into shikonin biosynthesis. Hortic. Res. 7:82. doi: 10.1038/s41438-020-0301-9
Bashir, A., Klammer, A. A., Robins, W. P., Chin, C., Webster, D., Paxinos, E., et al. (2012). A hybrid approach for the automated finishing of bacterial genomes. Nat. Biotechnol. 30, 701–707. doi: 10.1038/nbt.2288
Bentley, D. R., Balasubramanian, S., Swerdlow, H. P., Smith, G. P., Milton, J., Brown, C. G., et al. (2008). Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456, 53–59. doi: 10.1038/nature07517
Bharadwaj, R., Kumar, S. R., Sharma, A., and Sathishkumar, R. (2021). Plant metabolic gene clusters: evolution, organization, and their applications in synthetic biology. Front. Plant Sci. 12:697318. doi: 10.3389/fpls.2021.697318
Bornowski, N., Hamilton, J. P., Liao, P., Wood, J. C., Dudareva, N., and Buell, C. R. (2020). Genome sequencing of four culinary herbs reveals terpenoid genes underlying chemodiversity in the Nepetoideae. DNA Res. 27, 1–12. doi: 10.1093/dnares/dsaa016
Brose, J., Lau, K. H., Dang, T. T. T., Hamilton, J. P., do Vale Martins, L., Hamberger, B., et al. (2021). The Mitragyna speciosa (Kratom) Genome: a resource for data-mining potent pharmaceuticals that impact human health. G3 (Bethesda) 11:jkab058. doi: 10.1093/g3journal/jkab058
Burton, J. N., Adey, A., Patwardhan, R. P., Qiu, R., Kitzman, J. O., and Shendure, J. (2013). Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125. doi: 10.1038/nbt.2727
Cao, J., Schneeberger, K., Ossowski, S., Günther, T., Bender, S., Fitz, J., et al. (2011). Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat. Genet. 43, 956–963. doi: 10.1038/ng.911
Chan, A. P., Crabtree, J., Zhao, Q., Lorenzi, H., Orvis, J., Puiu, D., et al. (2010). Draft genome sequence of the oilseed species Ricinus communis. Nat. Biotechnol. 28, 951–956. doi: 10.1038/nbt.1674
Chang, Y., Liu, H., Liu, M., Liao, X., Sahu, S. K., Fu, Y., et al. (2019). The draft genomes of five agriculturally important African orphan crops. Gigascience 8, 1–16. doi: 10.1093/gigascience/giy152
Chaw, S.-M., Liu, Y.-C., Wu, Y.-W., Wang, H.-Y., Lin, C.-Y. I., Wu, C.-S., et al. (2019). Stout camphor tree genome fills gaps in understanding of flowering plant genome evolution. Nat. Plants 5, 63–73. doi: 10.1038/s41477-018-0337-0
Chellappan, B. V., Shidhi, P. R., Vijayan, S., Rajan, V. S., Sasi, A., Nair, A. S., et al. (2019). High quality draft genome of arogyapacha (Trichopus zeylanicus), an important medicinal plant endemic to Western Ghats of India. G3 9, 2395–2404. doi: 10.1534/g3.119.400164
Chen, D., Pan, Y., Wang, Y., Cui, Y.-Z., Zhang, Y.-J., Mo, R., et al. (2021). The chromosome-level reference genome of Coptis chinensis provides insights into genomic evolution and berberine biosynthesis. Hortic. Res. 8:121. doi: 10.1038/s41438-021-00559-2
Chen, F., Dong, W., Zhang, J., Guo, X., Chen, J., Wang, Z., et al. (2018). The sequenced angiosperm genomes and genome databases. Front. Plant Sci. 9:418. doi: 10.3389/fpls.2018.00418
Chen, H., Zeng, Y., Yang, Y., Huang, L., Tang, B., Zhang, H., et al. (2020). Allele-aware chromosome-level genome assembly and efficient transgene-free genome editing for the autotetraploid cultivated alfalfa. Nat. Commun. 11:2494. doi: 10.1038/s41467-020-16338-x
Chen, S. L., and Song, J.-Y. (2016). [Herbgenomics]. Zhongguo Zhong Yao Za Zhi 41, 3881–3889. doi: 10.4268/cjcmm20162101
Chen, S. L., Sun, Y. Z., Xu, J., Luo, H. M., Sun, C., He, L., et al. (2010). Strategies of the study on Herb genome program. Yaoxue Xuebao 45, 807–812.
Chen, S. L., Wu, W.-G., Wang, C.-X., Xiang, L., Shi, Y.-H., Zhang, D., et al. (2019). [Molecular genetics research of medicinal plants]. Zhongguo Zhong Yao Za Zhi 44, 2421–2432. doi: 10.19540/j.cnki.cjcmm.20190514.102
Chen, S., Wang, X., Wang, Y., Zhang, G., Song, W., Dong, X., et al. (2020). Improved de novo assembly of the achlorophyllous orchid Gastrodia elata. Front. Genet. 11:580568. doi: 10.3389/fgene.2020.580568
Chen, S., Wang, Y., Yu, L., Zheng, T., Wang, S., Yue, Z., et al. (2021). Genome sequence and evolution of Betula platyphylla. Hortic. Res. 8:37. doi: 10.1038/s41438-021-00481-7
Chen, W., Kui, L., Zhang, G., Zhu, S., Zhang, J., Wang, X., et al. (2017). Whole-Genome sequencing and analysis of the Chinese herbal plant Panax notoginseng. Mol. Plant 10, 899–902. doi: 10.1016/j.molp.2017.02.010
Chen, Y.-C., Li, Z., Zhao, Y.-X., Gao, M., Wang, J.-Y., Liu, K.-W., et al. (2020). The Litsea genome and the evolution of the laurel family. Nat. Commun. 11:1675. doi: 10.1038/s41467-020-15493-5
Cheng, J., Wang, X., Liu, X., Zhu, X., Li, Z., Chu, H., et al. (2021). Chromosome-level genome of Himalayan yew provides insights into the origin and evolution of the paclitaxel biosynthetic pathway. Mol. Plant 14, 1199–1209. doi: 10.1016/j.molp.2021.04.015
Chin, C. S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. doi: 10.1038/nmeth.2474
Chin, C.-S., Peluso, P., Sedlazeck, F. J., Nattestad, M., Concepcion, G. T., Clum, A., et al. (2016). Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050–1054. doi: 10.1038/nmeth.4035
Cui, P., Lin, Q., Fang, D., Zhang, L., Li, R., Cheng, J., et al. (2018). Tung Tree (Vernicia fordii, Hemsl.) Genome and transcriptome sequencing reveals co-ordinate up-regulation of fatty acid β-oxidation and triacylglycerol biosynthesis pathways during Eleostearic acid accumulation in seeds. Plant Cell Physiol. 59, 1990–2003. doi: 10.1093/pcp/pcy117
Dassanayake, M., Oh, D.-H., Haas, J. S., Hernandez, A., Hong, H., Ali, S., et al. (2011). The genome of the extremophile crucifer Thellungiella parvula. Nat. Genet. 43, 913–918. doi: 10.1038/ng.889
Deamer, D., Akeson, M., and Branton, D. (2016). Three decades of nanopore sequencing. Nat. Biotechnol. 34, 518–524. doi: 10.1038/nbt.3423
Diamond, J. (2002). Evolution, consequences and future of plant and animal domestication. Nature 418, 700–707. doi: 10.1038/nature01019
Ding, X., Mei, W., Huang, S., Wang, H., Zhu, J., Hu, W., et al. (2018). Genome survey sequencing for the characterization of genetic background of Dracaena cambodiana and its defense response during dragon’s blood formation. PLoS One 13:e0209258. doi: 10.1371/journal.pone.0209258
Ding, X., Mei, W., Lin, Q., Wang, H., Wang, J., Peng, S., et al. (2020). Genome sequence of the agarwood tree Aquilaria sinensis (Lour.) Spreng: the first chromosome-level draft genome in the Thymelaeceae family. Gigascience 9:giaa013. doi: 10.1093/gigascience/giaa013
Dong, S., Liu, M., Liu, Y., Chen, F., Yang, T., Chen, L., et al. (2021). The genome of Magnolia biondii Pamp. provides insights into the evolution of Magnoliales and biosynthesis of terpenoids. Hortic. Res. 8, 38. doi: 10.1038/s41438-021-00471-9
Eid, J., Fehr, A., Gray, J., Luong, K., Lyle, J., Otto, G., et al. (2009). Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138. doi: 10.1126/science.1162986
Fan, G., Liu, X., Sun, S., Shi, C., Du, X., Han, K., et al. (2020). The chromosome level genome and genome-wide association study for the agronomic traits of Panax notoginseng. iScience 23, 101538. doi: 10.1016/j.isci.2020.101538
Fan, Y., Sahu, S. K., Yang, T., Mu, W., Wei, J., Cheng, L., et al. (2020). Dissecting the genome of star fruit (Averrhoa carambola L.). Hortic. Res. 7:94. doi: 10.1038/s41438-020-0306-4
Franke, J., Kim, J., Hamilton, J. P., Zhao, D., Pham, G. M., Wiegert-Rininger, K., et al. (2019). Gene discovery in gelsemium highlights conserved gene clusters in monoterpene indole alkaloid biosynthesis. ChemBioChem 20, 83–87. doi: 10.1002/cbic.201800592
Fu, Y., Li, L., Hao, S., Guan, R., Fan, G., Shi, C., et al. (2017). Draft genome sequence of the Tibetan medicinal herb Rhodiola crenulata. Gigascience 6, 1–5. doi: 10.1093/gigascience/gix033
Gao, S., Wang, B., Xie, S., Xu, X., Zhang, J., Pei, L., et al. (2020). A high-quality reference genome of wild Cannabis sativa. Hortic. Res. 7:73. doi: 10.1038/s41438-020-0295-3
Gonda, I., Faigenboim, A., Adler, C., Milavski, R., Karp, M.-J., Shachter, A., et al. (2020). The genome sequence of tetraploid sweet basil, Ocimum basilicum L., provides tools for advanced genome editing and molecular breeding. DNA Res. 27, 1–10. doi: 10.1093/dnares/dsaa027
Goodwin, S., Gurtowski, J., Ethe-Sayers, S., Deshpande, P., Schatz, M. C., and McCombie, W. R. (2015). Oxford Nanopore sequencing, hybrid error correction, and de novo assembly of a eukaryotic genome. Genome Res. 25, 1750–1756. doi: 10.1101/gr.191395.115
Guan, R., Zhao, Y., Zhang, H., Fan, G., Liu, X., Zhou, W., et al. (2016). Draft genome of the living fossil Ginkgo biloba. Gigascience 5:49. doi: 10.1186/s13742-016-0154-1
Guo, C., Wang, Y., Yang, A., He, J., Xiao, C., Lv, S., et al. (2020). The coix genome provides insights into Panicoideae evolution and papery hull domestication. Mol. Plant 13, 309–320. doi: 10.1016/j.molp.2019.11.008
Guo, L., Winzer, T., Yang, X., Li, Y., Ning, Z., He, Z., et al. (2018). The opium poppy genome and morphinan production. Science 362, 343–347. doi: 10.1126/science.aat4096
Hamilton, J. P., and Robin Buell, C. (2012). Advances in plant genome sequencing. Plant J. 70, 177–190. doi: 10.1111/j.1365-313X.2012.04894.x
Hancock, J. F. (2005). Contributions of domesticated plant studies to our understanding of plant evolution. Ann. Bot. 96, 953–963. doi: 10.1093/aob/mci259
He, N., Zhang, C., Qi, X., Zhao, S., Tao, Y., Yang, G., et al. (2013). Draft genome sequence of the mulberry tree Morus notabilis. Nat. Commun. 4:2445. doi: 10.1038/ncomms3445
He, S., Dong, X., Zhang, G., Fan, W., Duan, S., Shi, H., et al. (2021). High quality genome of Erigeron breviscapus provides a reference for herbal plants in Asteraceae. Mol. Ecol. Resour. 21, 153–169. doi: 10.1111/1755-0998.13257
He, Y., Peng, F., Deng, C., Xiong, L., Huang, Z., Zhang, R., et al. (2018). Building an octaploid genome and transcriptome of the medicinal plant Pogostemon cablin from Lamiales. Sci. Data 5:180274. doi: 10.1038/sdata.2018.274
He, Y., Xiao, H., Deng, C., Xiong, L., Nie, H., and Peng, C. (2016). Survey of the genome of Pogostemon cablin provides insights into its evolutionary history and sesquiterpenoid biosynthesis. Sci. Rep. 6:26405. doi: 10.1038/srep26405
Hibrand Saint-Oyant, L., Ruttink, T., Hamama, L., Kirov, I., Lakhwani, D., Zhou, N. N., et al. (2018). A high-quality genome sequence of Rosa chinensis to elucidate ornamental traits. Nat. Plants 4, 473–484. doi: 10.1038/s41477-018-0166-1
Hong, Z., Li, J., Liu, X., Lian, J., Zhang, N., Yang, Z., et al. (2020). The chromosome-level draft genome of Dalbergia odorifera. Gigascience 9, 1–8. doi: 10.1093/gigascience/giaa084
Hoopes, G. M., Hamilton, J. P., Kim, J., Zhao, D., Wiegert-Rininger, K., Crisovan, E., et al. (2018). Genome assembly and annotation of the medicinal plant Calotropis gigantea, a producer of anticancer and antimalarial cardenolides. G3 Genes Genomes Genetics 8, 385–391. doi: 10.1534/g3.117.300331
Hu, L., Xu, Z., Wang, M., Fan, R., Yuan, D., Wu, B., et al. (2019). The chromosome-scale reference genome of black pepper provides insight into piperine biosynthesis. Nat. Commun. 10:4702. doi: 10.1038/s41467-019-12607-6
Huang, H., Liang, J., Tan, Q., Ou, L., Li, X., Zhong, C., et al. (2021). Insights into triterpene synthesis and unsaturated fatty-acid accumulation provided by chromosomal-level genome analysis of Akebia trifoliata subsp. australis. Hortic. Res. 8:33. doi: 10.1038/s41438-020-00458-y
International Brachypodium Initiative (2010). Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 463, 763–768. doi: 10.1038/nature08747
Iorizzo, M., Ellison, S., Senalik, D., Zeng, P., Satapoomin, P., Huang, J., et al. (2016). A high-quality carrot genome assembly provides new insights into carotenoid accumulation and asterid genome evolution. Nat. Genet. 48, 657–666. doi: 10.1038/ng.3565
Itkin, M., Davidovich-Rikanati, R., Cohen, S., Portnoy, V., Doron-Faigenboim, A., Oren, E., et al. (2016). The biosynthetic pathway of the nonsugar, high-intensity sweetener mogroside V from Siraitia grosvenorii. Proc. Natl. Acad. Sci. U.S.A. 113, E7619–E7628. doi: 10.1073/pnas.1604828113
Jaiswal, S. K., Mahajan, S., Chakraborty, A., Kumar, S., and Sharma, V. K. (2021). The genome sequence of Aloe vera reveals adaptive evolution of drought tolerance mechanisms. iScience 24:102079. doi: 10.1016/j.isci.2021.102079
Jamshidi-Kia, F., Lorigooini, Z., and Amini-Khoei, H. (2018). Medicinal plants: past history and future perspective. J. Herbmed Pharmacol. 7, 1–7. doi: 10.15171/jhp.2018.01
Ji, Y., Xiu, Z., Chen, C., Wang, Y., Yang, J., Sui, J., et al. (2021). Long read sequencing of Toona sinensis (A. Juss) Roem: a chromosome-level reference genome for the family Meliaceae. Mol. Ecol. Resour. 21, 1243–1255. doi: 10.1111/1755-0998.13318
Jiang, Z., Tu, L., Yang, W., Zhang, Y., Hu, T., Ma, B., et al. (2021). The chromosome-level reference genome assembly for Panax notoginseng and insights into ginsenoside biosynthesis. Plant Commun. 2:100113. doi: 10.1016/j.xplc.2020.100113
Jiao, W.-B., and Schneeberger, K. (2017). The impact of third generation genomic technologies on plant genome assembly. Curr. Opin. Plant Biol. 36, 64–70. doi: 10.1016/j.pbi.2017.02.002
Kang, S.-H., Kim, B., Choi, B.-S., Lee, H. O., Kim, N.-H., Lee, S. J., et al. (2020b). Genome assembly and annotation of soft-shelled adlay (Coix lacryma-jobi Variety ma-yuen), a cereal and medicinal crop in the poaceae family. Front. Plant Sci. 11:630. doi: 10.3389/fpls.2020.00630
Kang, M., Wu, H., Yang, Q., Huang, L., Hu, Q., Ma, T., et al. (2020a). A chromosome-scale genome assembly of Isatis indigotica, an important medicinal plant used in traditional Chinese medicine. Hortic. Res. 7:18. doi: 10.1038/s41438-020-0240-5
Kang, S.-H., Pandey, R. P., Lee, C.-M., Sim, J.-S., Jeong, J.-T., Choi, B.-S., et al. (2020c). Genome-enabled discovery of anthraquinone biosynthesis in Senna tora. Nat. Commun. 11:5875. doi: 10.1038/s41467-020-19681-1
Kellner, F., Kim, J., Clavijo, B. J., Hamilton, J. P., Childs, K. L., Vaillancourt, B., et al. (2015). Genome-guided investigation of plant natural product biosynthesis. Plant J. 82, 680–692. doi: 10.1111/tpj.12827
Kim, J., Kang, S.-H., Park, S.-G., Yang, T.-J., Lee, Y., Kim, O. T., et al. (2020). Whole-genome, transcriptome, and methylome analyses provide insights into the evolution of platycoside biosynthesis in Platycodon grandiflorus, a medicinal plant. Hortic. Res. 7:112. doi: 10.1038/s41438-020-0329-x
Kim, K. E., Peluso, P., Babayan, P., Yeadon, P. J., Yu, C., Fisher, W. W., et al. (2014). Long-read, whole-genome shotgun sequence data for five model organisms. Sci. Data 1, 1–10. doi: 10.1038/sdata.2014.45
Kim, N.-H., Jayakodi, M., Lee, S.-C., Choi, B.-S., Jang, W., Lee, J., et al. (2018). Genome and evolution of the shade-requiring medicinal herb Panax ginseng. Plant Biotechnol. J. 16, 1904–1917. doi: 10.1111/pbi.12926
Kim, S., Park, M., Yeom, S.-I., Kim, Y.-M., Lee, J. M., Lee, H.-A., et al. (2014). Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat. Genet. 46, 270–278. doi: 10.1038/ng.2877
Kitashiba, H., Li, F., Hirakawa, H., Kawanabe, T., Zou, Z., Hasegawa, Y., et al. (2014). Draft Sequences of the Radish (Raphanus sativus L.) Genome. DNA Res. 21, 481–490. doi: 10.1093/dnares/dsu014
Koren, S., Schatz, M. C., Walenz, B. P., Martin, J., Howard, J. T., Ganapathy, G., et al. (2012). Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 30, 693–700. doi: 10.1038/nbt.2280
Krishnan, N. M., Pattnaik, S., Jain, P., Gaur, P., Choudhary, R., Vaidyanathan, S., et al. (2012). A draft of the genome and four transcriptomes of a medicinal and pesticidal angiosperm Azadirachta indica. BMC Genomics 13:464. doi: 10.1186/1471-2164-13-464
Kumari, P., Singh, K. P., and Rai, P. K. (2020). Draft genome of multiple resistance donor plant Sinapis alba: an insight into SSRs, annotations and phylogenetics. PLoS One 15:e0231002. doi: 10.1371/journal.pone.0231002
Lau, K. H., Bhat, W. W., Hamilton, J. P., Wood, J. C., Vaillancourt, B., Wiegert-Rininger, K., et al. (2020). Genome assembly of Chiococca alba uncovers key enzymes involved in the biosynthesis of unusual terpenoids. DNA Res. 27, 1–12. doi: 10.1093/dnares/dsaa013
Li, A., Liu, A., Du, X., Chen, J.-Y., Yin, M., Hu, H.-Y., et al. (2020). A chromosome-scale genome assembly of a diploid alfalfa, the progenitor of autotetraploid alfalfa. Hortic. Res. 7:194. doi: 10.1038/s41438-020-00417-7
Li, C., Li, X., Liu, H., Wang, X., Li, W., Chen, M.-S., et al. (2020). Chromatin architectures are associated with response to dark treatment in the oil crop Sesamum indicum, based on a high-quality genome assembly. Plant Cell Physiol. 61, 978–987. doi: 10.1093/pcp/pcaa026
Li, J., Wang, Y., Dong, Y., Zhang, W., Wang, D., Bai, H., et al. (2021). The chromosome-based lavender genome provides new insights into Lamiaceae evolution and terpenoid biosynthesis. Hortic. Res. 8:53. doi: 10.1038/s41438-021-00490-6
Li, L.-F., Cushman, S. A., He, Y.-X., and Li, Y. (2020). Genome sequencing and population genomics modeling provide insights into the local adaptation of weeping forsythia. Hortic. Res. 7:130. doi: 10.1038/s41438-020-00352-7
Li, M.-Y., Feng, K., Hou, X.-L., Jiang, Q., Xu, Z.-S., Wang, G.-L., et al. (2020). The genome sequence of celery (Apium graveolens L.), an important leaf vegetable crop rich in apigenin in the Apiaceae family. Hortic. Res. 7:9. doi: 10.1038/s41438-019-0235-2
Li, S.-F., Wang, J., Dong, R., Zhu, H.-W., Lan, L.-N., Zhang, Y.-L., et al. (2020). Chromosome-level genome assembly, annotation and evolutionary analysis of the ornamental plant Asparagus setaceus. Hortic. Res. 7:48. doi: 10.1038/s41438-020-0271-y
Li, Y., Wei, H., Yang, J., Du, K., Li, J., Zhang, Y., et al. (2020). High-quality de novo assembly of the Eucommia ulmoides haploid genome provides new insights into evolution and rubber biosynthesis. Hortic. Res. 7:183. doi: 10.1038/s41438-020-00406-w
Liang, Q., Li, H., Li, S., Yuan, F., Sun, J., Duan, Q., et al. (2019). The genome assembly and annotation of yellowhorn (Xanthoceras sorbifolium Bunge). Gigascience 8, 1–15. doi: 10.1093/gigascience/giz071
Liang, Y., Chen, S., Wei, K., Yang, Z., Duan, S., Du, Y., et al. (2020). Chromosome level genome assembly of Andrographis paniculata. Front. Genet. 11:701. doi: 10.3389/fgene.2020.00701
Lin, Y., Min, J., Lai, R., Wu, Z., Chen, Y., Yu, L., et al. (2017). Genome-wide sequencing of longan (Dimocarpus longan Lour.) provides insights into molecular basis of its polyphenol-rich characteristics. Gigascience 6, 1–14. doi: 10.1093/gigascience/gix023
Liu, H., Shi, J., Cai, Z., Huang, Y., Lv, M., Du, H., et al. (2020). Evolution and domestication footprints uncovered from the genomes of Coix. Mol. Plant 13, 295–308. doi: 10.1016/j.molp.2019.11.009
Liu, L., Li, Y., Li, S., Hu, N., He, Y., Pong, R., et al. (2012). Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012:251364. doi: 10.1155/2012/251364
Liu, M. J., Zhao, J., Cai, Q.-L., Liu, G.-C., Wang, J.-R., Zhao, Z.-H., et al. (2014). The complex jujube genome provides insights into fruit tree biology. Nat. Commun. 5:5315. doi: 10.1038/ncomms6315
Liu, S., Liu, Y., Yang, X., Tong, C., Edwards, D., Parkin, I. A. P., et al. (2014). The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 5:3930. doi: 10.1038/ncomms4930
Liu, X., Liu, Y., Huang, P., Ma, Y., Qing, Z., Tang, Q., et al. (2017). The genome of medicinal plant Macleaya cordata provides new insights into benzylisoquinoline alkaloids metabolism. Mol. Plant 10, 975–989. doi: 10.1016/j.molp.2017.05.007
Liu, Y., Tang, Q., Cheng, P., Zhu, M., Zhang, H., Liu, J., et al. (2020). Whole-genome sequencing and analysis of the Chinese herbal plant Gelsemium elegans. Acta Pharm. Sin. B 10, 374–382. doi: 10.1016/j.apsb.2019.08.004
Liu, Y., Wang, B., Shu, S., Li, Z., Song, C., Liu, D., et al. (2021). Analysis of the Coptis chinensis genome reveals the diversification of protoberberine-type alkaloids. Nat. Commun. 12:3276. doi: 10.1038/s41467-021-23611-0
Loman, N. J., Quick, J., and Simpson, J. T. (2015). A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods 12, 733–735. doi: 10.1038/nmeth.3444
Lu, M., An, H., and Li, L. (2016). Genome survey sequencing for the characterization of the genetic background of Rosa roxburghii tratt and leaf ascorbate metabolism genes. PLoS One 11:e0147530. doi: 10.1371/journal.pone.0147530
Luo, X., Li, H., Wu, Z., Yao, W., Zhao, P., Cao, D., et al. (2020). The pomegranate (Punica granatum L.) draft genome dissects genetic divergence between soft- and hard-seeded cultivars. Plant Biotechnol. J. 18, 955–968. doi: 10.1111/pbi.13260
Lv, Q., Qiu, J., Liu, J., Li, Z., Zhang, W., Wang, Q., et al. (2020). The Chimonanthus salicifolius genome provides insight into magnoliid evolution and flavonoid biosynthesis. Plant J. 103, 1910–1923. doi: 10.1111/tpj.14874
Lv, S., Cheng, S., Wang, Z., Li, S., Jin, X., Lan, L., et al. (2020). Draft genome of the famous ornamental plant Paeonia suffruticosa. Ecol. Evol. 10, 4518–4530. doi: 10.1002/ece3.5965
Ma, D., Dong, S., Zhang, S., Wei, X., Xie, Q., Ding, Q., et al. (2021). Chromosome-level reference genome assembly provides insights into aroma biosynthesis in passion fruit (Passiflora edulis). Mol. Ecol. Resour. 21, 955–968. doi: 10.1111/1755-0998.13310
Ma, L., Wang, Q., Mu, J., Fu, A., Wen, C., Zhao, X., et al. (2020). The genome and transcriptome analysis of snake gourd provide insights into its evolution and fruit development and ripening. Hortic. Res. 7:199. doi: 10.1038/s41438-020-00423-9
Ma, Q., Sun, T., Li, S., Wen, J., Zhu, L., Yin, T., et al. (2020). The Acer truncatum genome provides insights into nervonic acid biosynthesis. Plant J. 104, 662–678. doi: 10.1111/tpj.14954
Mahesh, H. B., Subba, P., Advani, J., Shirke, M. D., Loganathan, R. M., Chandana, S. L., et al. (2018). Multi-Omics driven assembly and annotation of the sandalwood (Santalum album) Genome. Plant Physiol. 176, 2772–2788. doi: 10.1104/pp.17.01764
Mardis, E. R. (2008). The impact of next-generation sequencing technology on genetics. Trends Genet. 24, 133–141. doi: 10.1016/j.tig.2007.12.007
Margulies, M., Egholm, M., Altman, W. E., Attiya, S., Bader, J. S., Bemben, L. A., et al. (2005). Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380. doi: 10.1038/nature03959
Marrano, A., Britton, M., Zaini, P. A., Zimin, A. V., Workman, R. E., Puiu, D., et al. (2020). High-quality chromosome-scale assembly of the walnut (Juglans regia L.) reference genome. Gigascience 9, 1–16. doi: 10.1093/gigascience/giaa050
Martínez-García, P. J., Crepeau, M. W., Puiu, D., Gonzalez-Ibeas, D., Whalen, J., Stevens, K. A., et al. (2016). The walnut (Juglans regia) genome sequence reveals diversity in genes coding for the biosynthesis of non-structural polyphenols. Plant J. 87, 507–532. doi: 10.1111/tpj.13207
Matsumura, H., Hsiao, M.-C., Lin, Y.-P., Toyoda, A., Taniai, N., Tarora, K., et al. (2020). Long-read bitter gourd (Momordica charantia) genome and the genomic architecture of nonclassic domestication. Proc. Natl. Acad. Sci. U.S.A. 117, 14543–14551. doi: 10.1073/pnas.1921016117
Michael, T. P., and Jackson, S. (2013). The first 50 plant genomes. Plant Genome 6, 1–7. doi: 10.3835/plantgenome2013.03.0001in
Ming, R., Hou, S., Feng, Y., Yu, Q., Dionne-Laporte, A., Saw, J. H., et al. (2008). The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 452, 991–996. doi: 10.1038/nature06856
Ming, R., VanBuren, R., Liu, Y., Yang, M., Han, Y., Li, L.-T., et al. (2013). Genome of the long-living sacred lotus (Nelumbo nucifera Gaertn.). Genome Biol. 14:R41. doi: 10.1186/gb-2013-14-5-r41
Mochida, K., Sakurai, T., Seki, H., Yoshida, T., Takahagi, K., Sawai, S., et al. (2017). Draft genome assembly and annotation of Glycyrrhiza uralensis, a medicinal legume. Plant J. 89, 181–194. doi: 10.1111/tpj.13385
Moher, D., Liberati, A., Tetzlaff, J., and Altman, D. G. (2009). Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 6:e1000097. doi: 10.1371/journal.pmed.1000097
Moss, J., and Yuan, C. S. (2006). Herbal medicines and perioperative care. Anesthesiology 105, 441–442. doi: 10.1097/00000542-200609000-00002
Nafis, T., Akmal, M., Ram, M., Alam, P., Ahlawat, S., Mohd, A., et al. (2011). Enhancement of artemisinin content by constitutive expression of the HMG-CoA reductase gene in high-yielding strain of Artemisia annua L. Plant Biotechnol. Rep. 5, 53–60. doi: 10.1007/s11816-010-0156-x
Neller, K. C. M., Diaz, C. A., Platts, A. E., and Hudak, K. A. (2019). De novo assembly of the pokeweed genome provides insight into pokeweed antiviral protein (PAP) gene expression. Front. Plant Sci. 10:1002. doi: 10.3389/fpls.2019.01002
Newman, T., deBruijn, F. J., Green, P., Keegstra, K., Kende, H., McIntosh, L., et al. (1994). Genes galore: a summary of methods for accessing results from large-scale partial sequencing of anonymous Arabidopsis cDNA clones. Plant Physiol. 106, 1241–1255. doi: 10.1104/pp.106.4.1241
Niu, Z., Zhu, F., Fan, Y., Li, C., Zhang, B., Zhu, S., et al. (2021). The chromosome-level reference genome assembly for Dendrobium officinale and its utility of functional genomics research and molecular breeding study. Acta Pharm. Sin. B 11, 2080–2092. doi: 10.1016/j.apsb.2021.01.019
Nobrega, M. A., and Pennacchio, L. A. (2004). Comparative genomic analysis as a tool for biological discovery. J. Physiol. 554, 31–39. doi: 10.1113/jphysiol.2003.050948
Nong, W., Law, S. T. S., Wong, A. Y. P., Baril, T., Swale, T., Chu, L. M., et al. (2020). Chromosomal-level reference genome of the incense tree Aquilaria sinensis. Mol. Ecol. Resour. 20, 971–979. doi: 10.1111/1755-0998.13154
Ou, X., Jin, H., Guo, L., Yang, Y., Cui, X., Xiao, Y., et al. (2011). [Status and prospective on nutritional physiology and fertilization of Panax notoginseng]. Zhongguo Zhong Yao Za Zhi 36, 2620–2624. doi: 10.4268/cjcmm20111904
Paddon, C. J., Westfall, P. J., Pitera, D. J., Benjamin, K., Fisher, K., McPhee, D., et al. (2013). High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 496, 528–532. doi: 10.1038/nature12051
Patil, A. B., Shinde, S. S., Raghavendra, S., Satish, B. N., Kushalappa, C. G., and Vijay, N. (2021). The genome sequence of Mesua ferrea and comparative demographic histories of forest trees. Gene 769, 145214. doi: 10.1016/j.gene.2020.145214
Pei, L., Wang, B., Ye, J., Hu, X., Fu, L., Li, K., et al. (2021). Genome and transcriptome of Papaver somniferum Chinese landrace CHM indicates that massive genome expansion contributes to high benzylisoquinoline alkaloid biosynthesis. Hortic. Res. 8:5. doi: 10.1038/s41438-020-00435-5
Peng, X., Liu, H., Chen, P., Tang, F., Hu, Y., Wang, F., et al. (2019). A chromosome-scale genome assembly of paper mulberry (Broussonetia papyrifera) provides new insights into its forage and papermaking usage. Mol. Plant 12, 661–677. doi: 10.1016/j.molp.2019.01.021
Peng, Z., Bredeson, J. V., Wu, G. A., Shu, S., Rawat, N., Du, D., et al. (2020). A chromosome-scale reference genome of trifoliate orange (Poncirus trifoliata) provides insights into disease resistance, cold tolerance and genome evolution in Citrus. Plant J. 104, 1215–1232. doi: 10.1111/tpj.14993
Pootakham, W., Naktang, C., Kongkachana, W., Sonthirod, C., Yoocha, T., Sangsrakru, D., et al. (2021a). De novo chromosome-level assembly of the Centella asiatica genome. Genomics 113, 2221–2228. doi: 10.1016/j.ygeno.2021.05.019
Pootakham, W., Sonthirod, C., Naktang, C., Nawae, W., Yoocha, T., Kongkachana, W., et al. (2021b). De novo assemblies of Luffa acutangula and Luffa cylindrica genomes reveal an expansion associated with substantial accumulation of transposable elements. Mol. Ecol. Resour. 21, 212–225. doi: 10.1111/1755-0998.13240
Pu, X., Li, Z., Tian, Y., Gao, R., Hao, L., Hu, Y., et al. (2020). The honeysuckle genome provides insight into the molecular mechanism of carotenoid metabolism underlying dynamic flower coloration. New Phytol. 227, 930–943. doi: 10.1111/nph.16552
Purugganan, M. D., and Fuller, D. Q. (2009). The nature of selection during plant domestication. Nature 457, 843–848. doi: 10.1038/nature07895
Qin, C., Yu, C., Shen, Y., Fang, X., Chen, L., Min, J., et al. (2014). Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization. Proc. Natl. Acad. Sci. U.S.A. 111, 5135–5140. doi: 10.1073/pnas.1400975111
Qin, G., Xu, C., Ming, R., Tang, H., Guyot, R., Kramer, E. M., et al. (2017). The pomegranate (Punica granatum L.) genome and the genomics of punicalagin biosynthesis. Plant J. 91, 1108–1128. doi: 10.1111/tpj.13625
Qin, S., Wu, L., Wei, K., Liang, Y., Song, Z., Zhou, X., et al. (2019). A draft genome for Spatholobus suberectus. Sci. Data 6:113. doi: 10.1038/s41597-019-0110-x
Qing, Z., Liu, J., Yi, X., Liu, X., Hu, G., Lao, J., et al. (2021). The chromosome-level Hemerocallis citrina Borani genome provides new insights into the rutin biosynthesis and the lack of colchicine. Hortic. Res. 8:89. doi: 10.1038/s41438-021-00539-6
Quick, J., Quinlan, A. R., and Loman, N. J. (2014). A reference bacterial genome dataset generated on the MinIONTM portable single-molecule nanopore sequencer. Gigascience 3, 1–6. doi: 10.1186/2047-217X-3-22
Rai, A., Hirakawa, H., Nakabayashi, R., Kikuchi, S., Hayashi, K., Rai, M., et al. (2021). Chromosome-level genome assembly of Ophiorrhiza pumila reveals the evolution of camptothecin biosynthesis. Nat. Commun. 12:405. doi: 10.1038/s41467-020-20508-2
Rajewski, A., Carter-House, D., Stajich, J., and Litt, A. (2021). Datura genome reveals duplications of psychoactive alkaloid biosynthetic genes and high mutation rate following tissue culture. BMC Genomics 22:201. doi: 10.1186/s12864-021-07489-2
Raymond, O., Gouzy, J., Just, J., Badouin, H., Verdenaud, M., Lemainque, A., et al. (2018). The Rosa genome provides new insights into the domestication of modern roses. Nat. Genet. 50, 772–777. doi: 10.1038/s41588-018-0110-3
Ren, H., Yu, H., Zhang, S., Liang, S., Zheng, X., Zhang, S., et al. (2019). Genome sequencing provides insights into the evolution and antioxidant activity of Chinese bayberry. BMC Genomics 20:458. doi: 10.1186/s12864-019-5818-7
Schnable, P. S., Ware, D., Fulton, R. S., Stein, J. C., Wei, F., Pasternak, S., et al. (2009). The B73 maize genome: complexity, diversity, and dynamics. Science 326, 1112–1115. doi: 10.1126/science.1178534
Schwartz, D. C., Li, X., Hernandez, L. I., Ramnarain, S. P., Huff, E. J., and Wang, Y. K. (1993). Ordered restriction maps of Saccharomyces cerevisiae chromosomes constructed by optical mapping. Science 262, 110–114. doi: 10.1126/science.8211116
Shang, J., Tian, J., Cheng, H., Yan, Q., Li, L., Jamal, A., et al. (2020). The chromosome-level wintersweet (Chimonanthus praecox) genome provides insights into floral scent biosynthesis and flowering in winter. Genome Biol. 21:200. doi: 10.1186/s13059-020-02088-y
Shen, C., Du, H., Chen, Z., Lu, H., Zhu, F., Chen, H., et al. (2020). The chromosome-level genome sequence of the Autotetraploid alfalfa and Resequencing of core Germplasms provide genomic resources for alfalfa research. Mol. Plant 13, 1250–1261. doi: 10.1016/j.molp.2020.07.003
Shen, Q., Zhang, L., Liao, Z., Wang, S., Yan, T., Shi, P., et al. (2018). The genome of Artemisia annua provides insight into the evolution of Asteraceae family and Artemisinin biosynthesis. Mol. Plant 11, 776–788. doi: 10.1016/j.molp.2018.03.015
Song, C., Liu, Y., Song, A., Dong, G., Zhao, H., Sun, W., et al. (2018). The Chrysanthemum nankingense genome provides insights into the evolution and diversification of Chrysanthemum flowers and medicinal traits. Mol. Plant 11, 1482–1491. doi: 10.1016/j.molp.2018.10.003
Song, X., Sun, P., Yuan, J., Gong, K., Li, N., Meng, F., et al. (2021). The celery genome sequence reveals sequential paleo-polyploidizations, karyotype evolution and resistance gene reduction in apiales. Plant Biotechnol. J. 19, 731–744. doi: 10.1111/pbi.13499
Song, X., Wang, J., Li, N., Yu, J., Meng, F., Wei, C., et al. (2020). Deciphering the high-quality genome sequence of coriander that causes controversial feelings. Plant Biotechnol. J. 18, 1444–1456. doi: 10.1111/pbi.13310
Song, Z., Lin, C., Xing, P., Fen, Y., Jin, H., Zhou, C., et al. (2020). A high-quality reference genome sequence of Salvia miltiorrhiza provides insights into tanshinone synthesis in its red rhizomes. Plant Genome 13:e20041. doi: 10.1002/tpg2.20041
Su, W., Jing, Y., Lin, S., Yue, Z., Yang, X., Xu, J., et al. (2021). Polyploidy underlies co-option and diversification of biosynthetic triterpene pathways in the apple tribe. Proc. Natl. Acad. Sci. U.S.A. 118, e2101767118. doi: 10.1073/pnas.2101767118
Su, X. Z., and Miller, L. H. (2015). The discovery of artemisinin and the Nobel Prize in Physiology or Medicine. Sci. China Life Sci. 58, 1175–1179. doi: 10.1007/s11427-015-4948-7
Sun, G., Xu, Y., Liu, H., Sun, T., Zhang, J., Hettenhausen, C., et al. (2018). Large-scale gene losses underlie the genome evolution of parasitic plant Cuscuta australis. Nat. Commun. 9:2683. doi: 10.1038/s41467-018-04721-8
Sun, W., Leng, L., Yin, Q., Xu, M., Huang, M., Xu, Z., et al. (2019). The genome of the medicinal plant Andrographis paniculata provides insight into the biosynthesis of the bioactive diterpenoid neoandrographolide. Plant J. 97, 841–857. doi: 10.1111/tpj.14162
Sun, X., Zhu, S., Li, N., Cheng, Y., Zhao, J., Qiao, X., et al. (2020). A chromosome-level genome assembly of garlic (Allium sativum) provides insights into genome evolution and allicin biosynthesis. Mol. Plant 13, 1328–1339. doi: 10.1016/j.molp.2020.07.019
Tu, L., Su, P., Zhang, Z., Gao, L., Wang, J., Hu, T., et al. (2020). Genome of Tripterygium wilfordii and identification of cytochrome P450 involved in triptolide biosynthesis. Nat. Commun. 11:971. doi: 10.1038/s41467-020-14776-1
Tuskan, G. A., Difazio, S., Jansson, S., Bohlmann, J., Grigoriev, I., Hellsten, U., et al. (2006). The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313, 1596–1604. doi: 10.1126/science.1128691
Upadhyay, A. K., Chacko, A. R., Gandhimathi, A., Ghosh, P., Harini, K., Joseph, A. P., et al. (2015). Genome sequencing of herb Tulsi (Ocimum tenuiflorum) unravels key genes behind its strong medicinal properties. BMC Plant Biol. 15:212. doi: 10.1186/s12870-015-0562-x
Urasaki, N., Takagi, H., Natsume, S., Uemura, A., Taniai, N., Miyagi, N., et al. (2017). Draft genome sequence of bitter gourd (Momordica charantia), a vegetable and medicinal plant in tropical and subtropical regions. DNA Res. 24, 51–58. doi: 10.1093/dnares/dsw047
vanBakel, H., Stout, J. M., Cote, A. G., Tallon, C. M., Sharpe, A. G., Hughes, T. R., et al. (2011). The draft genome and transcriptome of Cannabis sativa. Genome Biol. 12:R102. doi: 10.1186/gb-2011-12-10-r102
Vanburen, R., Bryant, D., Edger, P. P., Tang, H., Burgess, D., Challabathula, D., et al. (2015). Single-molecule sequencing of the desiccation-tolerant grass Oropetium thomaeum. Nature 527, 508–511. doi: 10.1038/nature15714
Vining, K. J., Johnson, S. R., Ahkami, A., Lange, I., Parrish, A. N., Trapp, S. C., et al. (2017). Draft genome sequence of Mentha longifolia and development of resources for mint cultivar improvement. Mol. Plant 10, 323–339. doi: 10.1016/j.molp.2016.10.018
Wang, J., Xu, S., Mei, Y., Cai, S., Gu, Y., Sun, M., et al. (2021). A high-quality genome assembly of Morinda officinalis, a famous native southern herb in the Lingnan region of southern China. Hortic. Res. 8:135. doi: 10.1038/s41438-021-00551-w
Wang, L., He, F., Huang, Y., He, J., Yang, S., Zeng, J., et al. (2018). Genome of wild mandarin and domestication history of mandarin. Mol. Plant 11, 1024–1037. doi: 10.1016/j.molp.2018.06.001
Wang, L., Yu, S., Tong, C., Zhao, Y., Liu, Y., Song, C., et al. (2014). Genome sequencing of the high oil crop sesame provides insight into oil biosynthesis. Genome Biol. 15:R39. doi: 10.1186/gb-2014-15-2-r39
Wang, M., Zhang, L., and Wang, Z. (2021). Chromosomal-Level reference genome of the neotropical tree Jacaranda mimosifolia D. Don. Genome Biol. Evol. 13, 2–7. doi: 10.1093/gbe/evab094
Wang, P., Yi, S., Mu, X., Zhang, J., and Du, J. (2020). Chromosome-Level genome assembly of Cerasus humilis using PacBio and Hi-C technologies. Front. Genet. 11:956. doi: 10.3389/fgene.2020.00956
Wang, X., Xu, Y., Zhang, S., Cao, L., Huang, Y., Cheng, J., et al. (2017). Genomic analyses of primitive, wild and cultivated citrus provide insights into asexual reproduction. Nat. Genet. 49, 765–772. doi: 10.1038/ng.3839
Wang, X., Zhang, J., He, S., Gao, Y., Ma, X., Gao, Y., et al. (2018). HMOD: an omics database for herbal medicine plants. Mol. Plant 11, 757–759. doi: 10.1016/j.molp.2018.03.002
Wang, Y., Fan, G., Liu, Y., Sun, F., Shi, C., Liu, X., et al. (2013). The sacred lotus genome provides insights into the evolution of flowering plants. Plant J. 76, 557–567. doi: 10.1111/tpj.12313
Wang, Z., Hobson, N., Galindo, L., Zhu, S., Shi, D., McDill, J., et al. (2012). The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. Plant J. 72, 461–473. doi: 10.1111/j.1365-313X.2012.05093.x
Wu, H., Zhao, G., Gong, H., Li, J., Luo, C., He, X., et al. (2020). A high-quality sponge gourd (Luffa cylindrica) genome. Hortic. Res. 7:128. doi: 10.1038/s41438-020-00350-9
Wu, S., Shamimuzzaman, M., Sun, H., Salse, J., Sui, X., Wilder, A., et al. (2017). The bottle gourd genome provides insights into Cucurbitaceae evolution and facilitates mapping of a Papaya ring-spot virus resistance locus. Plant J. 92, 963–975. doi: 10.1111/tpj.13722
Wu, S., Sun, W., Xu, Z., Zhai, J., Li, X., Li, C., et al. (2020). The genome sequence of star fruit (Averrhoa carambola). Hortic. Res. 7:95. doi: 10.1038/s41438-020-0307-3
Wu, Z., Liu, H., Zhan, W., Yu, Z., Qin, E., Liu, S., et al. (2021). The chromosome-scale reference genome of safflower (Carthamus tinctorius) provides insights into linoleic acid and flavonoid biosynthesis. Plant Biotechnol. J. 19, 1725–1742. doi: 10.1111/pbi.13586
Wuyun, T., Wang, L., Liu, H., Wang, X., Zhang, L., Bennetzen, J. L., et al. (2018). The hardy rubber tree genome provides insights into the evolution of polyisoprene biosynthesis. Mol. Plant 11, 429–442. doi: 10.1016/j.molp.2017.11.014
Xia, E. H., Zhang, H.-B., Sheng, J., Li, K., Zhang, Q.-J., Kim, C., et al. (2017). The tea tree genome provides insights into tea flavor and independent evolution of caffeine biosynthesis. Mol. Plant 10, 866–877. doi: 10.1016/j.molp.2017.04.002
Xia, M., Han, X., He, H., Yu, R., Zhen, G., Jia, X., et al. (2018). Improved de novo genome assembly and analysis of the Chinese cucurbit Siraitia grosvenorii, also known as monk fruit or luo-han-guo. Gigascience 7, 1–9. doi: 10.1093/gigascience/giy067
Xia, Z., Huang, D., Zhang, S., Wang, W., Ma, F., Wu, B., et al. (2021). Chromosome-scale genome assembly provides insights into the evolution and flavor synthesis of passion fruit (Passiflora edulis Sims). Hortic. Res. 8:14. doi: 10.1038/s41438-020-00455-1
Xie, J., Zhao, H., Li, K., Zhang, R., Jiang, Y., Wang, M., et al. (2020). A chromosome-scale reference genome of Aquilegia oxysepala var. kansuensis. Hortic. Res. 7:113. doi: 10.1038/s41438-020-0328-y
Xu, H., Song, J., Luo, H., Zhang, Y., Li, Q., Zhu, Y., et al. (2016). Analysis of the genome sequence of the medicinal plant Salvia miltiorrhiza. Mol. Plant 9, 949–952. doi: 10.1016/j.molp.2016.03.010
Xu, J., Chu, Y., Liao, B., Xiao, S., Yin, Q., Bai, R., et al. (2017). Panax ginseng genome examination for ginsenoside biosynthesis. Gigascience 6, 1–15. doi: 10.1093/gigascience/gix093
Xu, X., Yuan, H., Yu, X., Huang, S., Sun, Y., Zhang, T., et al. (2021). The chromosome-level Stevia genome provides insights into steviol glycoside biosynthesis. Hortic. Res. 8:129. doi: 10.1038/s41438-021-00565-4
Xu, Z., Pu, X., Gao, R., Demurtas, O. C., Fleck, S. J., Richter, M., et al. (2020b). Tandem gene duplications drive divergent evolution of caffeine and crocin biosynthetic pathways in plants. BMC Biol. 18:63. doi: 10.1186/s12915-020-00795-3
Xu, Z., Gao, R., Pu, X., Xu, R., Wang, J., Zheng, S., et al. (2020a). Comparative genome analysis of Scutellaria baicalensis and Scutellaria barbata reveals the evolution of active flavonoid biosynthesis. Genomics Proteomics Bioinformatics 18, 230–240. doi: 10.1016/j.gpb.2020.06.002
Xu, Z., Xin, T., Bartels, D., Li, Y., Gu, W., Yao, H., et al. (2018). Genome analysis of the ancient tracheophyte Selaginella tamariscina reveals evolutionary features relevant to the acquisition of desiccation tolerance. Mol. Plant 11, 983–994. doi: 10.1016/j.molp.2018.05.003
Yan, L., Wang, X., Liu, H., Tian, Y., Lian, J., Yang, R., et al. (2015). The genome of Dendrobium officinale illuminates the biology of the important traditional Chinese orchid herb. Mol. Plant 8, 922–934. doi: 10.1016/j.molp.2014.12.011
Yang, J., Zhang, G., Zhang, J., Liu, H., Chen, W., Wang, X., et al. (2017). Hybrid de novo genome assembly of the Chinese herbal fleabane Erigeron breviscapus. Gigascience 6, 1–7. doi: 10.1093/gigascience/gix028
Yang, X., Yue, Y., Li, H., Ding, W., Chen, G., Shi, T., et al. (2018). The chromosome-level quality genome provides insights into the evolution of the biosynthesis genes for aroma compounds of Osmanthus fragrans. Hortic. Res. 5:72. doi: 10.1038/s41438-018-0108-0
Yang, Z., Chen, S., Wang, S., Hu, Y., Zhang, G., Dong, Y., et al. (2021a). Chromosomal-scale genome assembly of Eleutherococcus senticosus provides insights into chromosome evolution in Araliaceae. Mol. Ecol. Resour. 21, 2204–2220. doi: 10.1111/1755-0998.13403
Yang, Z., Liu, G., Zhang, G., Yan, J., Dong, Y., Lu, Y., et al. (2021b). The chromosome-scale high-quality genome assembly of Panax notoginseng provides insight into dencichine biosynthesis. Plant Biotechnol. J. 19, 869–871. doi: 10.1111/pbi.13558
Yin, J., Jiang, L., Wang, L., Han, X., Guo, W., Li, C., et al. (2021). A high-quality genome of taro (Colocasia esculenta (L.) Schott), one of the world’s oldest crops. Mol. Ecol. Resour. 21, 68–77. doi: 10.1111/1755-0998.13239
Yu, J., Hu, S., Wang, J., Wong, G. K.-S., Li, S., Liu, B., et al. (2002). A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 296, 79–92. doi: 10.1126/science.1068037
Yuan, Y., Jin, X., Liu, J., Zhao, X., Zhou, J., Wang, X., et al. (2018). The Gastrodia elata genome provides insights into plant adaptation to heterotrophy. Nat. Commun. 9:1615. doi: 10.1038/s41467-018-03423-5
Yuan, Y., Liu, W., Zhang, Q., Xiang, L., Liu, X., Chen, M., et al. (2015). Overexpression of artemisinic aldehyde Δ11 (13) reductase gene-enhanced artemisinin and its relative metabolite biosynthesis in transgenic Artemisia annua L. Biotechnol. Appl. Biochem. 62, 17–23. doi: 10.1002/bab.1234
Yuan, Z., Fang, Y., Zhang, T., Fei, Z., Han, F., Liu, C., et al. (2018). The pomegranate (Punica granatum L.) genome provides insights into fruit quality and ovule developmental biology. Plant Biotechnol. J. 16, 1363–1374. doi: 10.1111/pbi.12875
Zhang, D., Li, W., Xia, E., Zhang, Q., Liu, Y., Zhang, Y., et al. (2017). The medicinal herb panax Notoginseng genome provides insights into Ginsenoside biosynthesis and genome evolution. Mol. Plant 10, 903–907. doi: 10.1016/j.molp.2017.02.011
Zhang, G. Q., Xu, Q., Bian, C., Tsai, W.-C., Yeh, C.-M., Liu, K.-W., et al. (2016). The Dendrobium catenatum Lindl. genome sequence provides insights into polysaccharide synthase, floral development and adaptive evolution. Sci. Rep. 6:19029. doi: 10.1038/srep19029
Zhang, J., Tian, Y., Yan, L., Zhang, G., Wang, X., Zeng, Y., et al. (2016). Genome of plant Maca (Lepidium meyenii) illuminates genomic basis for high-altitude adaptation in the central andes. Mol. Plant 9, 1066–1077. doi: 10.1016/j.molp.2016.04.016
Zhang, L., Li, X., Ma, B., Gao, Q., Du, H., Han, Y., et al. (2017). The tartary buckwheat genome provides insights into rutin biosynthesis and abiotic stress tolerance. Mol. Plant 10, 1224–1237. doi: 10.1016/j.molp.2017.08.013
Zhang, L., Liu, M., Long, H., Dong, W., Pasha, A., Esteban, E., et al. (2019). Tung tree (Vernicia fordii) genome provides a resource for understanding genome evolution and improved oil production. Genomics Proteomics Bioinformatics 17, 558–575. doi: 10.1016/j.gpb.2019.03.006
Zhang, T., Ren, X., Zhang, Z., Ming, Y., Yang, Z., Hu, J., et al. (2020). Long-read sequencing and de novo assembly of the Luffa cylindrica (L.) Roem. genome. Mol. Ecol. Resour. 20, 511–519. doi: 10.1111/1755-0998.13129
Zhang, Y., Zheng, L., Zheng, Y., Zhou, C., Huang, P., Xiao, X., et al. (2019). Assembly and annotation of a draft genome of the medicinal plant Polygonum cuspidatum. Front. Plant Sci. 10:1274. doi: 10.3389/fpls.2019.01274
Zhao, D., Hamilton, J. P., Pham, G. M., Crisovan, E., Wiegert-Rininger, K., Vaillancourt, B., et al. (2017). De novo genome assembly of Camptotheca acuminata, a natural source of the anti-cancer compound camptothecin. Gigascience 6, 1–7. doi: 10.1093/gigascience/gix065
Zhao, Q., Yang, J., Cui, M.-Y., Liu, J., Fang, Y., Yan, M., et al. (2019). The reference genome sequence of Scutellaria baicalensis provides insights into the evolution of Wogonin biosynthesis. Mol. Plant 12, 935–950. doi: 10.1016/j.molp.2019.04.002
Zhao, Y. P., Fan, G., Yin, P. P., Sun, S., Li, N., Hong, X., et al. (2019). Resequencing 545 ginkgo genomes across the world reveals the evolutionary history of the living fossil. Nat. Commun 10:4201. doi: 10.1038/s41467-019-12133-5
Zheng, G. X. Y., Lau, B. T., Schnall-Levin, M., Jarosz, M., Bell, J. M., Hindson, C. M., et al. (2016). Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat. Biotechnol. 34, 303–311. doi: 10.1038/nbt.3432
Zheng, X., Chen, D., Chen, B., Liang, L., Huang, Z., Fan, W., et al. (2021). Insights into salvianolic acid B biosynthesis from chromosome-scale assembly of the Salvia bowleyana genome. J. Integr. Plant Biol. 63, 1309–1323. doi: 10.1111/jipb.13085
Zhou, W., Li, B., Li, L., Ma, W., Liu, Y., Feng, S., et al. (2018). Genome survey sequencing of Dioscorea zingiberensis. Genome 61, 567–574. doi: 10.1139/gen-2018-0011
Keywords: medicinal plant, genome, sequencing, long-read sequencing technology, application
Citation: Cheng Q-Q, Ouyang Y, Tang Z-Y, Lao C-C, Zhang Y-Y, Cheng C-S and Zhou H (2021) Review on the Development and Applications of Medicinal Plant Genomes. Front. Plant Sci. 12:791219. doi: 10.3389/fpls.2021.791219
Received: 08 October 2021; Accepted: 23 November 2021;
Published: 23 December 2021.
Edited by:
Qi Chen, Kunming University of Science and Technology, ChinaReviewed by:
Wei Gao, Capital Medical University, ChinaCopyright © 2021 Cheng, Ouyang, Tang, Lao, Zhang, Cheng and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hua Zhou, aHpob3VAbXVzdC5lZHUubW8=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.