
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 30 November 2021
Sec. Plant Metabolism and Chemodiversity
Volume 12 - 2021 | https://doi.org/10.3389/fpls.2021.780970
 Chanchan Liu1,2†
Chanchan Liu1,2† Qiyu Gao3†
Qiyu Gao3† Zhuo Shang3Jian Liu1Siwei Zhou4Jingjie Dang1Licheng Liu1Iris Lange5
Zhuo Shang3Jian Liu1Siwei Zhou4Jingjie Dang1Licheng Liu1Iris Lange5 Narayanan Srividya5
Narayanan Srividya5 B. Markus Lange5Qinan Wu1,2*
B. Markus Lange5Qinan Wu1,2* Wei Lin1,2,3,6*
Wei Lin1,2,3,6*
Monoterpenoids are the main components of plant essential oils and the active components of some traditional Chinese medicinal herbs like Mentha haplocalyx Briq., Nepeta tenuifolia Briq., Perilla frutescens (L.) Britt and Pogostemin cablin (Blanco) Benth. Pulegone reductase is the key enzyme in the biosynthesis of menthol and is required for the stereoselective reduction of the Δ2,8 double bond of pulegone to produce the major intermediate menthone, thus determining the stereochemistry of menthol. However, the structural basis and mechanism underlying the stereoselectivity of pulegone reductase remain poorly understood. In this study, we characterized a novel (−)-pulegone reductase from Nepeta tenuifolia (NtPR), which can catalyze (−)-pulegone to (+)-menthone and (−)-isomenthone through our RNA-seq, bioinformatic analysis in combination with in vitro enzyme activity assay, and determined the structure of (+)-pulegone reductase from M. piperita (MpPR) by using X-ray crystallography, molecular modeling and docking, site-directed mutagenesis, molecular dynamics simulations, and biochemical analysis. We identified and validated the critical residues in the crystal structure of MpPR involved in the binding of the substrate pulegone. We also further identified that residues Leu56, Val282, and Val284 determine the stereoselectivity of the substrate pulegone, and mainly contributes to the product stereoselectivity. This work not only provides a starting point for the understanding of stereoselectivity of pulegone reductases, but also offers a basis for the engineering of menthone/menthol biosynthetic enzymes to achieve high-titer, industrial-scale production of enantiomerically pure products.
Terpenoids are a large and structurally diverse group of natural products widely distributed in plants, microorganisms, and insects. More than 55,000 terpenoids have been identified so far, and the majority of them display diverse biological activities (Christianson, 2008). For example, some plant terpenoids function as chemical defense agents against predation and pathogens (Hijaz et al., 2016; Mahizan et al., 2019), and some play regulatory roles in the interactions with other plants and environment (Abbas et al., 2017). Among them, monoterpenoids are a type of terpenoids composed of two isoprene units and are widely distributed in plants with great therapeutic potential, such as traditional Chinese medicinal herbs like Mentha haplocalyx Briq (Duan et al., 2015), Nepeta tenuifolia Briq (Liu et al., 2018), Perilla frutescens (L.) Britt (Zhou et al., 2021), and Pogostemin cablin (Blanco) Benth (Wojtunik-Kulesza et al., 2019). As one of the representative monoterpenoids, menthol and its biosynthetic pathway as well as related biosynthetic enzymes have been identified (Gao et al., 2020). Briefly, the C10 menthol skeleton is formed by the condensation of isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP) (Gao et al., 2020), which are obtained from the 2C-methyl-D-erythritol-4-phosphate (MEP) pathway or the mevalonic acid (MVA) pathway in plant cells, followed by cyclization catalyzed by (−)-limonene synthase (LS). Subsequently, the C10 skeleton is further modified by a series of tailoring enzymes (e.g., monooxygenase, dehydrogenase, reductase, and isomerase) to generate (−)-menthol and the diastereomer (+)-isomenthol (Supplementary Figure 1A).
(−)-Menthone, the second most abundant monoterpenoid in peppermint essential oil, is a critical intermediate in menthol biosynthesis (Lange, 2015; Chen et al., 2019). In Mentha piperita, (−)-menthone is directly synthesized from (+)-pulegone through the reduction of C2–C8 alkene double bond catalyzed by (+)-pulegone reductase (EC number 1.3.1.81). Pulegone reductase is a double bond reductase (DBR) which belongs to the NADPH-dependent, medium-chain dehydrogenase/reductase (MDR) superfamily. Some DBRs in the MDR superfamily proteins have become important biotechnological tools for asymmetric synthesis due to their high stereoselectivity in the alkene double bond reduction (Currin et al., 2018). It has been reported that the reduction reaction proceeds through stereoselective transfer of a hydride from NADPH to the carbon of the substrate (Mcconkey et al., 2000; Ringer et al., 2003; Zebec et al., 2016; Paramasivan and Mutturi, 2017; Cheallaigh et al., 2018; Currin et al., 2018; Liu et al., 2018; Bergman et al., 2019; Wojtunik-Kulesza et al., 2019). In recent years, genes and enzymes accounting for double bond reduction in plant secondary metabolites have been identified through biochemical and structural studies. For instance, Arabidopsis thaliana L. homolog DBR (AtDBR, UniProt ID: Q39172) reduces the C2-C8 double bond of p-coumaryl- and coniferyl-aldehydes (Youn et al., 2006). The Nicotiana tabacum L. DBR (NtDBR, UniProt ID: Q9SLN8) has been shown to be active toward a variety of α,β-unsaturated alkenes (Mansell et al., 2013), such as (−)-cinnamaldehyde and 1-nitrocyclohexene. In addition, Rubus idaeus L. raspberry DBR (RiDBR, UniProt ID: G1FCG0) catalyzes the reduction of 4-hydroxybenzalacetone and 3-methoxy-4-hydroxybenzalacetone to raspberry ketone and zingerone, respectively (Simon et al., 2017). A DBR from Malus domestica L. (MdDBR, UniProt ID: A0A5N5GUE7) has been isolated and characterized recently (Caliandro et al., 2021) and suggested to be involved in the biosynthesis of polyphenolic compounds (e.g., dihydrochalcones) that are beneficial in human diet. The structures of several plant derived DBRs (e.g., AtDBR, NtDBR, and RiDBR) have been reported. However, stereoselectivity of the DBRs for substrates and products remains poorly understood.
In this report, we first performed functional characterization of pulegone reductases from Nepeta tenuifolia (NtPR) and Mentha piperita (MpPR, UniProt ID: Q6WAU0), from which we observed the two DBRs possess selectivity for both substrates and products in different diastereomeric forms. Phylogenetic analysis revealed both NtPR and MpPR belong to the MDR superfamily, but they are present in two different clades. To investigate the structural basis and underlying mechanism of the stereoselectivity, we performed molecular docking, molecular dynamic simulation, site-directed mutagenesis analysis, and biochemical assays based on the crystal structure of MpPR. We uncovered the key residues in the pulegone binding pockets of NtPR and MpPR, and illustrated that the hydrophobic interactions and potential steric hindrances may mainly contribute to their stereoselectivity toward substrates and products. This study not only deepens our understanding on the substrate and product selectivity of alkene reductases, but also provides a structural and mechanistic basis for the engineering and directed evolution of pulegone reductases, enabling the production of enantiomerically pure end-product in industry.
The (+)-pulegone reductase gene from Mentha piperita and (−)-pulegone reductase gene from Nepeta tenuifolia were cloned into the pET28a vector under control of the bacteriophage T7 gene promoters using NheI and HindIII, respectively. The (−)-pulegone reductase gene from Agastache rugose was cloned into the pET28a vector under control of the bacteriophage T7 gene promoters using BamHI and SacI. Each resulting plasmid was transformed into E. coli strain BL21(DE3) (Invitrogen). A single colony of the resulting transformants was used to inoculate 50 mL of LB broth containing 50 mg/mL kanamycin, and the culture was incubated at 37°C for 16 h with shaking. An aliquot (10 mL) was used to inoculate 1 L of LB broth containing 50 mg/mL kanamycin, followed by the incubation with agitation at 37°C, 180 rpm until the OD600 reached 0.8–1.0. Subsequently, the culture was supplemented with isopropyl-b-D-thiogalactoside to the final concentration of 0.5 mM for protein expression, and then the culture was incubated for 16 h at 20°C. Cells were harvested by centrifugation (5,000 × g; 15 min at 4°C), re-suspended in buffer A (10 mM Tris-HCl, pH 8.0, 200 mM NaCl, and 5% glycerol), and lysed using an EmulsiFlex-C5 cell disruptor (Avestin). The lysate was centrifuged (20,000 × g; 30 min at 4°C) and the cell debris was removed. The supernatant was loaded onto a 5 mL column of Ni2+-NTA-agarose (Qiagen) pre-equilibrated with buffer A. The column was washed with 10 × 5 mL buffer A containing 25 mM imidazole followed by elution with 30 mL buffer A containing 200 mM imidazole. The eluate was concentrated to ∼10 mg/mL and further purified by gel filtration chromatography on a HiLoad 16/60 Superdex 200 prep grade column (GE Healthcare) in 10 mM Tris-HCl, pH 8.0, 50 mM NaCl, 5 mM MgCl2, and 1 mM DTT. The peak fractions were collected and concentrated to the final concentration of 20 mg/mL in the same buffer using 30 kDa MWCO Amicon Ultra-15 centrifugal ultrafilters (EMD Millipore), and stored in aliquots at –80°C. The yields of the proteins were ∼20 mg/mL, and purities were ∼95%. Site-directed mutations were prepared using one step PCR method, and the mutated proteins were expressed and purified by following the same protocol as the wild-type proteins. High and low molecular weight (mass) calibration kits (GE Health-care) were used to calibrate the molecular mass of wild type MpPR.
The reduction reaction (0.4 mL) catalyzed by pulegone reductase was performed in buffer B (50 mM KH2PO4, 10% sorbitol, 1 mM DTT, pH 7.5), containing 20 μM substrate [(+)-pulegone (CAS No: 89-82-7) or (−)-pulegone (CAS No: 3391-90-0), Sigma], 10 mM NADPH tetrasodium salt hydrate (CAS No:2646-71-1, Sigma), 6 mM glucose-6-phosphate, 20 U glucose-6-phosphate dehydrogenase (Solarbio), and 30 μM MpPR or 36 μM NtPR. 0.2 mL of n-hexane was added on the top of the reaction solution. Reaction was carried out at 31°C for 1 h (MpPR) and 16 h (NtPR) with slowly stirring. The reaction was terminated by placing the reaction vial at –20°C for 2 h. The upper organic phase was transferred into a new 2 mL glass vial containing a conical glass insert and immediately analyzed by GC-MS and chiral GC. The negative control containing inactive enzyme was generated by heating the reconstitution mix for 15 min at 95°C.
GC-MS analysis was performed by Agilent 7890B/7000C, equipped with a HP 5MS capillary column (30 m × 0.25 mm; film thickness 0.25 μm). The programmed temperatures of the column were set as follows: 85°C for 4 min, 85–130°C at 5°C/min, 130°C for 2 min, 130–140°C at 5°C/min, 140°C for 3 min. Ion source temperature was set at 230°C. Electron ionization (EI) mass spectra were acquired over the mass range 50–500 Da at the energy of 70 eV. Chiral GC analysis was carried out using Agilent 8860, equipped with a CYCLODEX-B capillary column (30 m × 0.32 mm; film thickness 0.25 μM). The programmed temperatures of the column were set as follows: 80–95°C at 2°C/min, 95–110°C at 0.5°C/min. Injector temperature and carrier gas (Nitrogen) detector temperature were set at 250°C.Nitrogen was used as the carrier gas at a flow rate of 1 mL/min with an injection volume of 1 μL, no split.
The reduction reaction was initiated in a 400 μL solution containing 50 mM KH2PO4, 10% sorbitol, 1 mM DTT, 10 mM NADPH tetrasodium salt hydrate, 6 mM glucose-6-phosphate, 20 U glucose-6-phosphate dehydrogenase (Solarbio), together with the wild-type and mutated MpPR, ArPR, and NtPR (Supplementary Table 1). Other conditions are the same as the in vitro enzyme catalysis assay. Quantitative analysis was performed by the comparison of the peak areas of products to the standards of known concentrations obtained from chiral GC analysis (Agilent 8860 equipped with a HP-5 capillary column; the column condition is the same as described above). All biotransformation reactions were performed in at least duplicates, and the results are averages of the data. The yields of menthone and iso-menthone at each concentration were calculated by the comparison of the peak areas of products to the standards of known concentrations. The kinetic parameters Km and kcat were calculated using Equation 1.
where V0 is the initial velocity, [E] is the enzyme concentration, [S] is the substrate concentration, Vmax is the maximum velocity; Km is the Michaelis constant and kcat is calculated from Vmax/[E].
Robotic crystallization trials were performed for MpPR and NtPR as well as co-crystallization with NADPH tetrasodium salt hydrate and (+)- or (−)-pulegone using a Griffin liquid handling system (Art Robbins Instruments), commercial screening solutions (Emerald Biosystems, Hampton Research, and Qiagen), and the sitting-drop vapor-diffusion technique (drop: 0.2 μL protein plus 0.2 μL screening solution; reservoir: 60 μL screening solution; 20°C). 1,200 conditions were screened. Under several conditions, MpPR crystals appeared within 2 weeks. Conditions were optimized using the hanging-drop vapor-diffusion technique at 20°C. The optimized crystallization condition for MpPR was as follows: 1 M ammonium sulfate, 0.1 M Bis-Tris (pH 5.5), 1% W/V PEG 3,350 at 20°C. Crystals were transferred to reservoir solution containing 20% (v/v) glycerol and flash-cooled with liquid nitrogen. Diffraction data were collected from cryo-cooled crystals at SSRF BL17U. Data were processed using HKL2000 (Otwinowski and Minor, 1997) and CCP4i (Winn et al., 2011). The resolution cut-off criteria were: (i) I/σ > = 2.0, (ii) CC1/2 (highest resolution shell) > 0.5.
The structure of MpPR was solved by molecular replacement with MOLREP (Adams et al., 2010; Vagin and Teplyakov, 2010) using the structure of native raspberry ketone synthase from Rubus idaeus (PDB ID 6EOW) as a starting model. The molecular replacement solution was good, and an automatic model building was performed with Phenix (Adams et al., 2010). Additional model building was done manually with Coot (Emsley and Cowtan, 2004) and refined with Phenix. The final model of MpPR was refined to 2.7 Å resolution. The final model for MpPR was refined to Rwork and Rfree of 0.27/0.29 (Table 1).
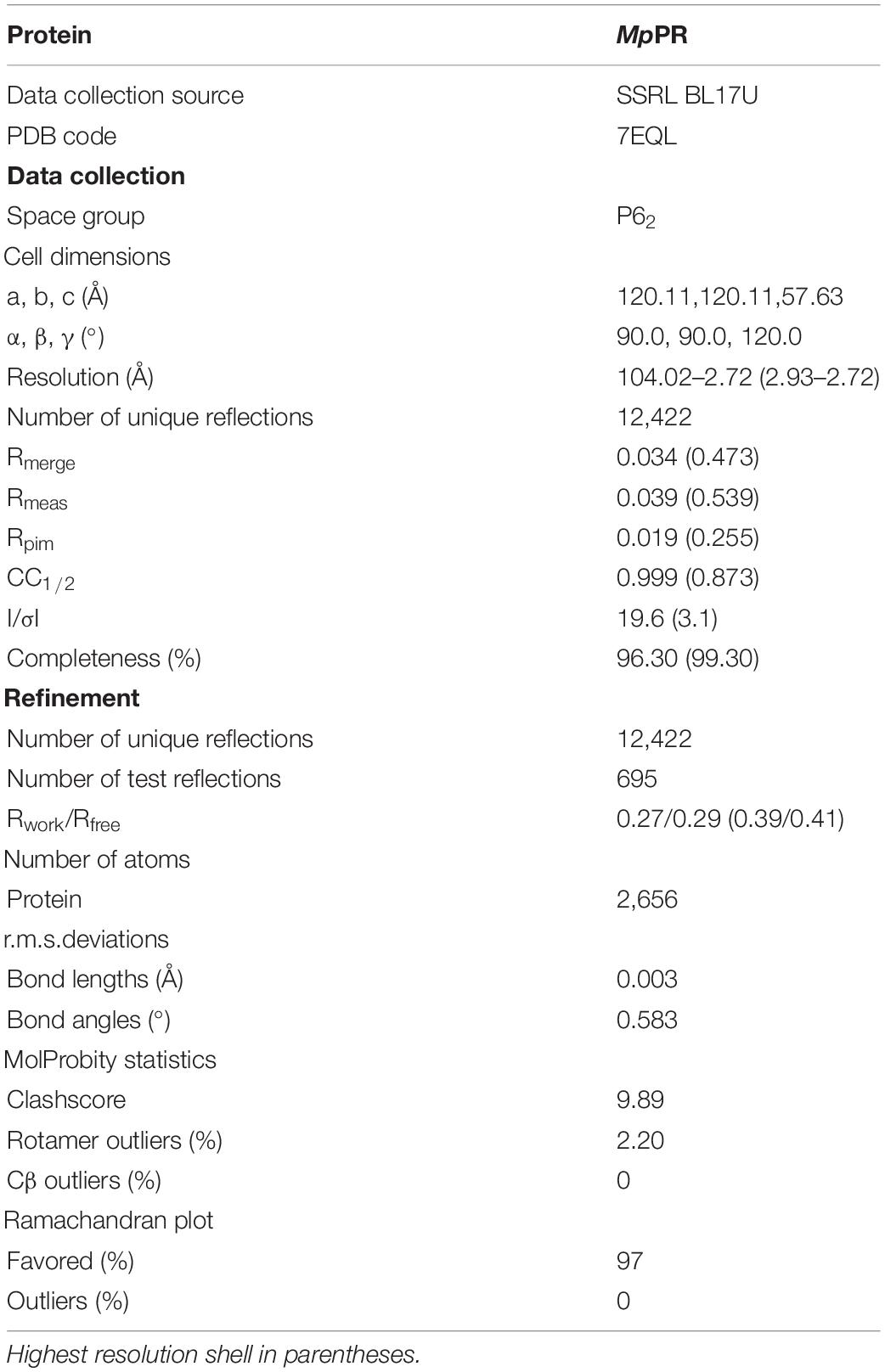
Table 1. Structure data collection and refinement statistics.
All molecular docking studies were performed using Autodock4.2 package (Morris et al., 2009). Briefly, crystal structure of MpPR enzyme was docked with (+)-pulegone. The molecule was added with non-polar hydrogens and assigned partial atomic charges using AutoDockTools (ADT) (Morris et al., 2009). The coordinates of NADP(H) and (+)-pulegone in MpPR structure was generated based on the coordinates of hydroxybenzalacetone from the crystal structure of RiDBR (PDB ID: 6EOW) and the coordinates of p-coumaryl aldehyde of the crystal of AtDBR (PDB ID: 2J3J) in combination with CORINA Classic online service. A grid box with 40 × 40 × 40 grid points and 0.2 Å grid spacing centered roughly at the pulegone binding position was used as the searching space. 100 runs of Larmarckian Genetic Algorithm were performed to search the protein-ligand interactions. The results were clustered and ranked. Result analyses and figure rendering were performed using PyMOL. The structure of (−)-pulegone reductase from N. tenuifolia was modeled by the online artificial intelligence tfold2 program.1
The crystal structure of MpPR was deposited into Protein Data Bank under accession number 7EQL. UniProt IDs: Q6WAU0 for MpPR; Q39172 for AtDBR; Q9SLN8 for NtDBR; G1FCG0 for RiDBR; A0A5N5GUE7 for MdDBR. National Center Bioinfomratic Center (NCBI) accession number: MZ504956 for NtPR; MZ504957 for ArPR.
Pulegone reductase from Mentha piperita (MpPR) that catalyzes the reduction of the C2–C8 double bond of (+)-pulegone to (−)-menthone using NADP(H) as a co-factor has been established for decades (Lange, 2015). Besides M. piperita, many other medicinal herbs, such as Nepeta tenuifolia and Agastache rugosa are also producers of essential oils, in which menthone and menthol are the major components (Chen et al., 2019). Intriguingly, it has been reported that N. tenuifolia and A. rugosa produce (+)-menthone and (+)-menthol as the major metabolites, in contrast to M. piperita that predominantly biosynthesizes (−)-menthone and (−)-menthol (Chen et al., 2019).
To investigate the underlying reasons for the substrate selectivity between M. piperita and N. tenuifolia or A. rugosa, we first compared the evolutionary relatedness of these three plants by performing molecular phylogenetic analysis based on a cascade of 28S-18S-5.8S rDNA sequences from their genomes. The results clearly showed that M. piperita, N. tenuifolia, A. rugose, Sesamum indicum, and Amborella trichopoda were clustered into a big group. More importantly, M. piperita, N. tenuifolia, and A. rugose were further classified into an individual subcluster, indicating that the three plants may be evolutionarily related (Supplementary Figure 2). Next, we analyzed menthone biosynthesis pathway in the genus M. piperita in which was well studied before. Limonene synthases, isopiperitenone reductase, and pulegone reductase have been proven to be able to catalyze the committed step in the biosynthesis of menthone until now (Supplementary Figure 1B; Ringer et al., 2003, 2005). Moreover, the structure-function relationships underlying the formation of limonene enantiomers in limonene synthases and candidate active site residues with critical roles in catalyzing reactions that involve accommodating reaction intermediates of opposite enantiomeric series have been identified (Hyatt et al., 2007; Srividya et al., 2020). However, there is no information concerning the biosynthesis mechanism of limonene downstream product enantiomers and relevant biosynthesis enzymes like isopiperitenone reductase or pulegone reductase so far. Therefore, we performed RNA-seq analysis on N. tenuifolia that produces (−)-pulegone and (+)-menthone in order to locate the candidate genes of (−)-pulegone reductase (Liu et al., 2021). Based on our N. tenuifolia RNA-seq data, three candidate genes of pulegone reductase (cluster-16657.51589, cluster-16657.19187, cluster-16657.38628) sharing high identities (∼65, ∼75, and ∼64%, respectively) with MpPR were proposed to be NtPR (Supplementary Figure 3). We then selected cluster-16657.51589 as our target gene in view of its highest expression level in leaves and the leaves contain the highest ratio of monoterpenes (e.g., pulegone, menthone) among all the tissues of N. tenuifolia (Supplementary Figure 4). Subsequently, we performed sequence alignment analysis of NtPR (cluster-16657.51589) and MpPR together with another four DBRs (AtDBR, NtDBR, MdDBR, and RiDBR) in the MDR superfamily. The alignment revealed that most residues in the proposed substrate binding pocket across all aligned MDR superfamily proteins are conserved (Supplementary Figure 5, labeled with green triangles). Moreover, NtPR and MpPR showed high sequence similarity with the major exception that six amino acid residues (Leu56, Tyr78, Phe281, Val282, Val284, and Tyr287 numbered as in MpPR, Ser59, Asp81, Tyr283, Leu284, Tyr286, and Arg289 numbered as in NtPR) in the proposed pulegone binding pocket are different (Supplementary Figure 5), suggesting these amino acids might contribute to substrate or product specificity. Thus, we cloned NtPR and MpPR genes and expressed the respective pulegone reductase in Escherichia coli BL21(DE3) strain for biochemical and structural studies (Supplementary Figure 6).
To experimentally validate the substrate specificity of NtPR and MpPR, we firstly carried out in vitro enzyme activity assays, where the pulegone reductase was fed with (+)- and (−)-pulegone, respectively. The products were analyzed and quantified using gas chromatography-mass spectrometry (GC-MS). The results clearly showed that NtPR has a higher preference for (−)-pulegone to generate (−)-isomenthone (57%) and (+)-menthone (43%), confirmed by chiral GC and the fragments generated from electron impact mass spectrometry (EIMS) by comparison with standards (Figures 1A–D). In contrast, MpPR is more inclined to convert (+)-pulegone to the major (−)-menthone (69%) and the minor (+)-isomenthone (31%) as reported previously (Figures 1A–D; Lange, 2015). Next, we plotted the Michaelis–Menten curve and calculated the enzyme kinetic parameters for NtPR after feeding with (+)- or (−)-pulegones. Apparently, NtPR has lower catalytic activities on (+)- or (−)-pulegone [Kcat 0.39 10–4s–1 for (+)-pulegone or 0.53 10–4s–1 for (−)-pulegone] compared to that of MpPR [Kcat 863.66 10–4s–1for (+)-pulegone and 1595.06 10–4s–1 for (−)-pulegone]. NtPR displayed higher binding affinity toward (−)-pulegone (Km 57.18 μM) than that for (+)-pulegone (Km = 163.30 μM), whereas the Vmax for both (+)- and (−)-pulegones appears similar (Figure 1B), confirming that (−)-pulegone is the more favorable substrate that NtPR can bind rather than (+)-pulegone. In comparison with NtPR, MpPR displayed higher binding affinity toward (+)-pulegone (Km 3.00 μM) than that for (−)-pulegone (Km 8.63 μM) (Figure 1B). To further investigate the substrate selectivity of MpPR and NtPR, we fed the pulegone reductases with other alkene double bond-containing substrates, including (+)-menthone, (−)-menthone, (+)-limonene, (−)-limonene, (+)-menthofuran, (−)-(1R,4S)-p-mentha-2,8-dien-1-ol, carveol, (−)-perilllc alcohol, and (−)-carvone. The results revealed that MpPR and NtPR can adopt (+)- and (−)-pulegones as substrate exclusively, and they do not show any activity toward other substrates (Supplementary Figure 7), suggesting MpPR and NtPR are the DBRs with high substrate specificity.
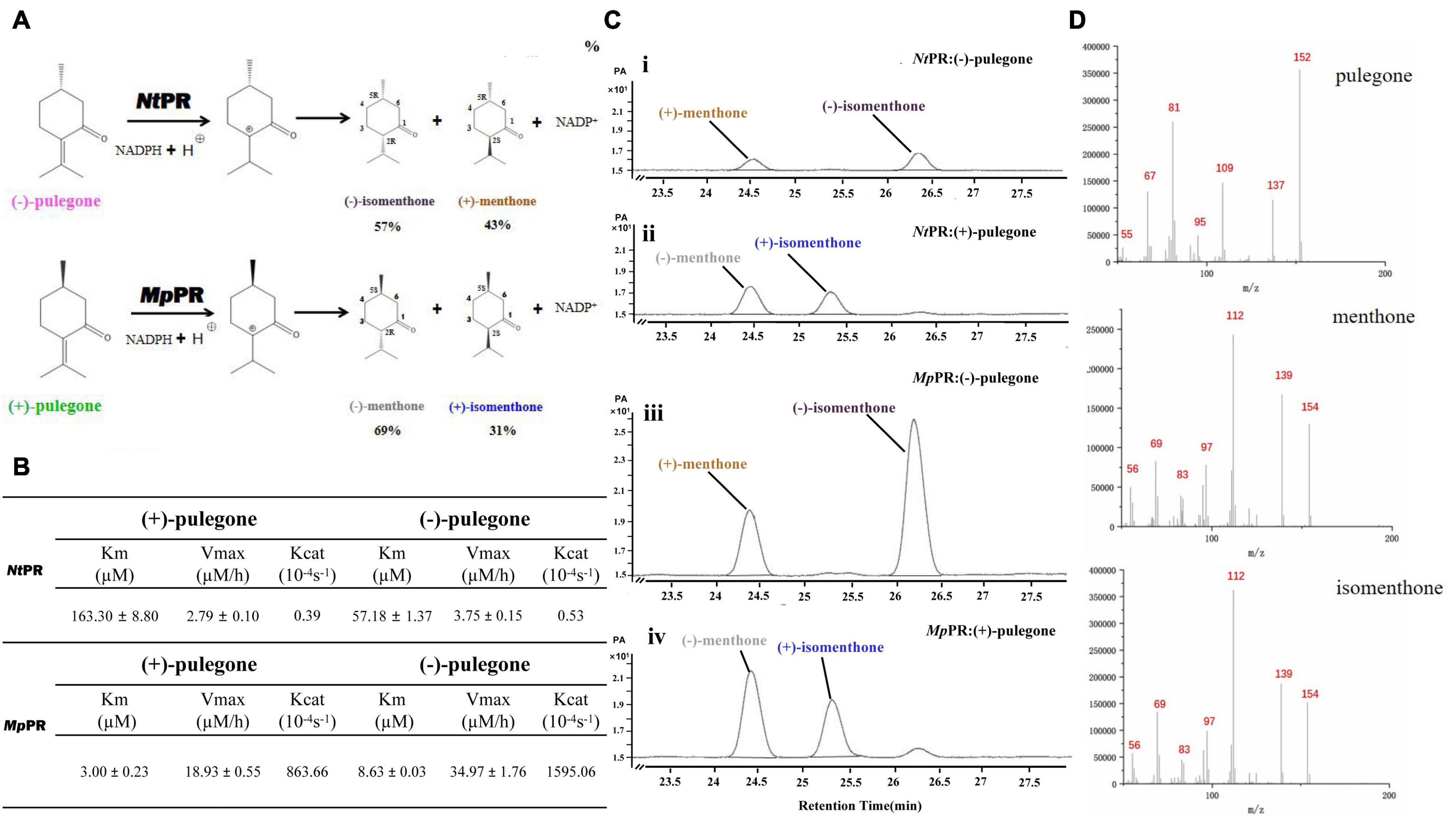
Figure 1. Functional characterization of (–)-pulegone reductase from N. tenuifolia and (+)-pulegone reductase from M. piperita. (A) Reduction of pulegone to menthone/isomenthone catalyzed by pulegone reductases from N. tenuifolia (NtPR) and M. piperita (MpPR). (B) Kinetic parameters of NtPR or MpPR that reduces (+)-pulegone or (–)-pulegone as the substrate, respectively. (C,D) NtPR- and MpPR-catalyzed in vitro conversion of pulegone to menthone/isomenthone monitored by chiral GC and GC/MS. Reactions were performed as described in Materials and Methods. Each reaction contained 20 μM substrate, and 30 μM MpPR or 36 μM NtPR. Left panel, GC chromatograms of methanolic extracts of the reactions: (i) (–)-pulegone + NtPR + NADPH, (ii) (+)-pulegone + NtPR + NADPH, (iii) (–)-pulegone + MpPR + NADPH, and (iv) (+)-pulegone + MpPR + NADPH. The horizontal axis represents retention time and the vertical axis represents relative abundance; right panel, GC/MS spectra of pulegone and menthone (SIM mode, m/z).
Using NtPR sequence as a query, we further investigated the evolutionary relationship of NtPR, MpPR, and other DBRs in the MDR superfamily. 67 DBRs belonging to the MDR superfamily from plants that have complete genome data in KEGG database were chosen for the maximum-likelihood phylogenetic analysis. As expected, the phylogenetic tree revealed that NtPR forms a unique clade, which suggests NtPR may be evolutionarily distinct from other known MDR reductases (Figure 2 and Supplementary Figure 8). In contrast, the clade where MpPR locates has more homologous members. The results inspired us to perform in-depth structural analysis of NtPR and MpPR toward the better understanding of substrate and product stereoselectivity.
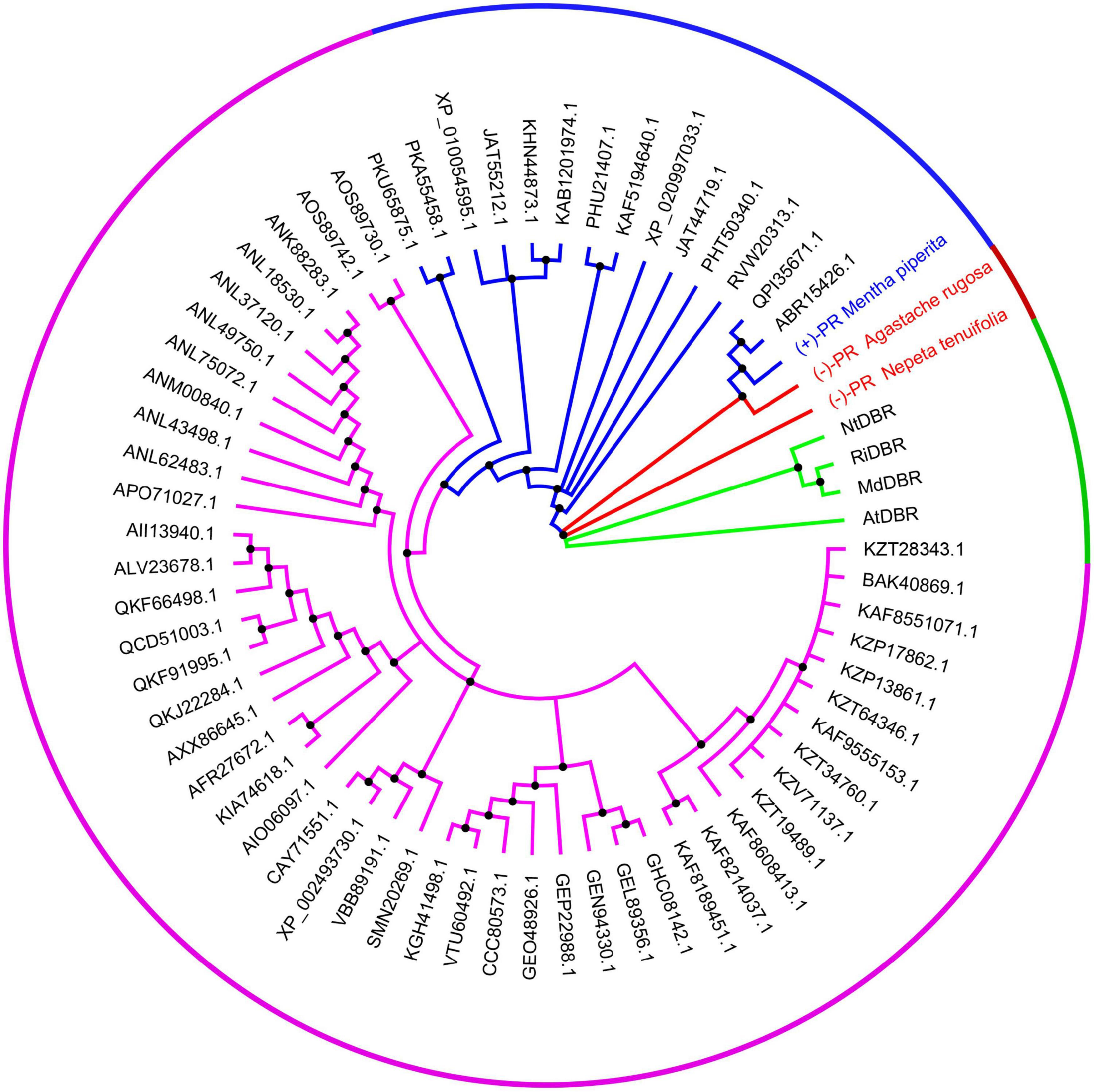
Figure 2. Phylogenetic analysis of alkene reductases from the MDR superfamily. The clades of alkene reductases from the MDR superfamily in which new identified pulegone reductase N. tenuifolia (–)-pulegone reductase (NtPR), A. rugose (–)-pulegone reductase (ArPR), and previous identified pulegone reductases including M. piperita (+)-pulegone reductase (MpPR) located are highlighted in red and blue, respectively. The neighboring clade mainly bearing the double bond reductases, such as MdDBR, AtDBR, NtDBR, and RiDBR from the MDR superfamily, the structures and functions of which have been previously investigated, is highlighted in green. The rest clade including other unrelated alkene reductases belonging to the MDR superfamily but the structures and functions of which have not been previously well studied is highlighted in purple. The alkene reductases IDs from the MDR superfamily mentioned above are referring to the National Center for Biotechnology Information (NCBI). All source species for the alkene reductases from the MDR superfamily used in the phylogenetic analysis are listed in Supplementary Table 3.
To elucidate the molecular basis underlying the differences in substrate and product specificities, we attempted to obtain co-crystals of MpPR and NtPR with NADP(H) and (±)-pulegone. Finally, the crystal structure of MpPR was obtained at 2.7 Å resolution (Figure 3A), although the attempts to acquire crystal of NtPR and co-crystals of the enzyme with either NADP+, (+)-pulegone or both NADP+ and (+)-pulegone failed, due to the hydrophobic and volatile nature of pulegone. Statistics of data collection and model refinement for MpPR crystal are summarized in Table 1. Alternatively, the structure of NtPR was in silico modeled using the tfold2 program (Figure 3B), an artificial intelligence-based online structural modeling tool. By structure superimposition analysis, the overall structures of both MpPR and NtPR are similar to the known MDR superfamily enzymes RiDBR, NtDBR, AtDBR, and MdDBR (the RMSDs are 0.99, 0.85, 1.07, and 1.04 Å, Figures 3B–F), which form a different clade in the phylogenetic tree (in green, Figure 2).
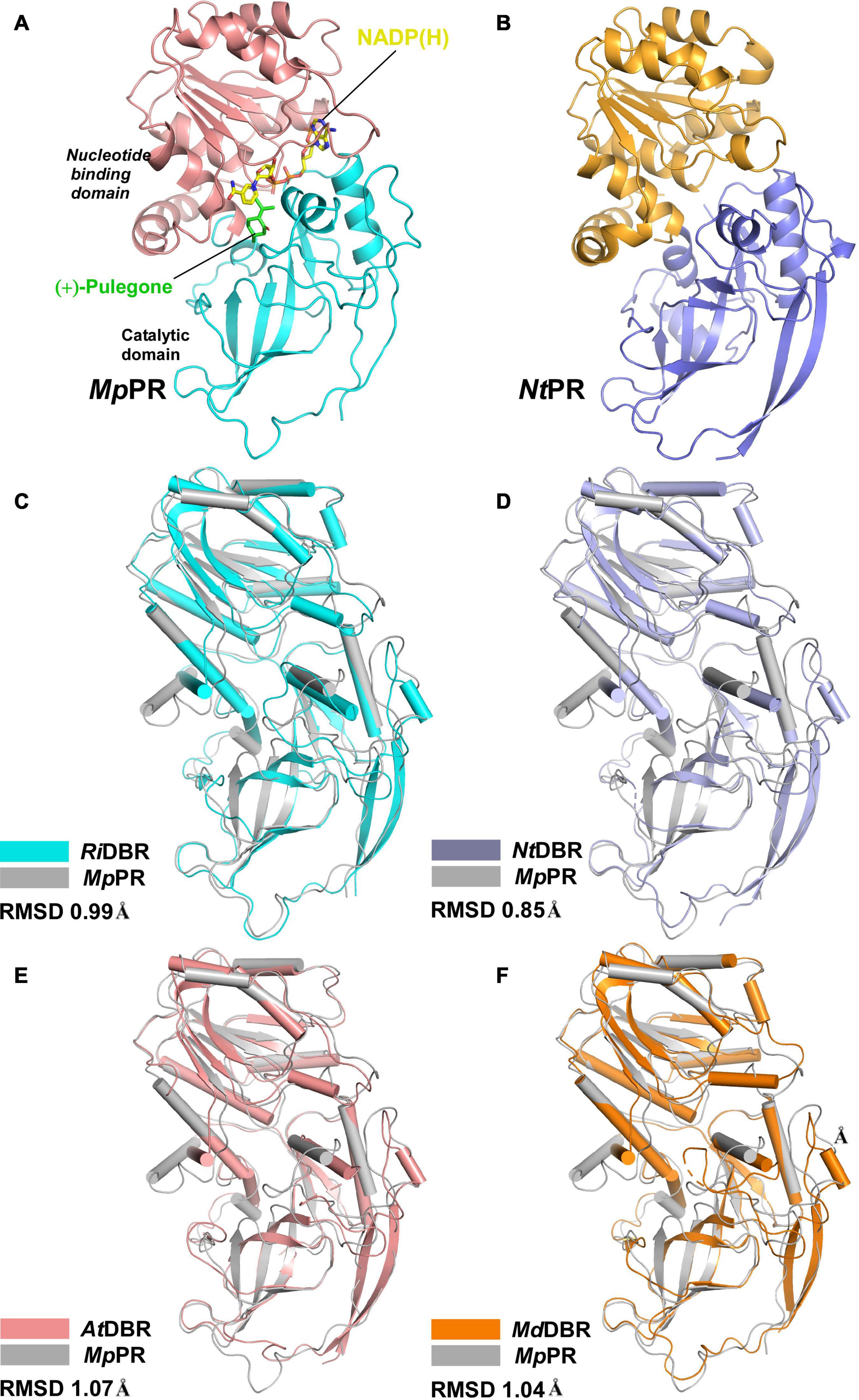
Figure 3. Overall structures of pulegone reductases from Mentha piperita and Nepeta tenuifolia. (A) Overall structure of (+)-pulegone reductase from M. piperita (MpPR) bound with NADP(H). Residues 1–134 and 310–341 are colored cyan, and residues 135–309 are colored salmon. The NADP(H) and modeled (+)-pulegone are shown as yellow and green stick models, respectively. (B) Modeled structure of pulegone reductase from N. tenuifolia (NtPR) using the tfold2 program. Residues 1–134 and 310–344 are colored blue, and residues 135–309 are colored orange. (C) Structure superimposition of MpPR and RiDBR, (D) structure superimposition of MpPR and NtDBR, (E) structure superimposition of MpPR and AtDBR. (F) Structure superimposition of MpPR and MdDBR. the RMSDs of RiDBR, NtDBR, AtDBR, and MdDBR with MpPR are shown.
The crystal structure of MpPR revealed an asymmetric unit containing only one monomer of apo-MpPR. The PDBePISA server calculations and gel filtration of the protein suggested that the biologically relevant form of the enzyme is a homodimer as reported previously for other MDR superfamily members (Supplementary Figure 9). The structure consists of two typical conserved N-terminal and C-terminal domains that are connected by a short loop. The N-terminal catalytic domain (residues 1–134 and 310–349) includes three a-helices and nine b-sheets forming a twisted partial b-barrel-like structure, while the C-terminal nucleotide coenzyme-binding Rossmann-fold domain (residues 135–309) features seven a-helices and six b-sheets, forming a typical six-stranded, parallel b-sheet sandwiched by three helices on each side (Figure 3A). The two domains are separated by a cleft containing a deep pocket that accommodates the cofactor and forms the active site (Figure 3A).
We did not observe the unambiguous electronic density for NADP(H) in the map of co-crystal of MpPR in complex with NADP(H), but two extra density peaks accounting for the phosphate group of NADP(H) were present, probably due to the crystal packing. Sequence alignment of MpPR with other DBRs (e.g., AtDBR and NtDBR) in the MDR superfamily showed that most residues predicted to interact with NADP(H) are conserved and located at the nucleotide coenzyme-binding domain of MpPR (Supplementary Figure 5, shown as purple rectangle). In order to locate the relative position of NADP(H) in MpPR, we superimposed the phosphate group of NADP(H) in the crystal structure of MpPR with that in AtDBR (PDB ID 2J3J and 2J3K). The superimposed model exhibited that the residues Asn51, Ser164, Lys189, Tyr205, and Asn331 in MpPR may form hydrogen bonds with the phosphate groups and the ribose rings of NADP(H) (Figure 4A). A number of hydrophobic residues consisting of Pro52, Tyr53, Met135, Ala162, Val165, Ala184, Cys251, Met253, Val254, Phe281, Val282, and Val283 were found surrounding the NADP(H) backbone to further stabilize the NADP(H) molecule (Figure 4A).
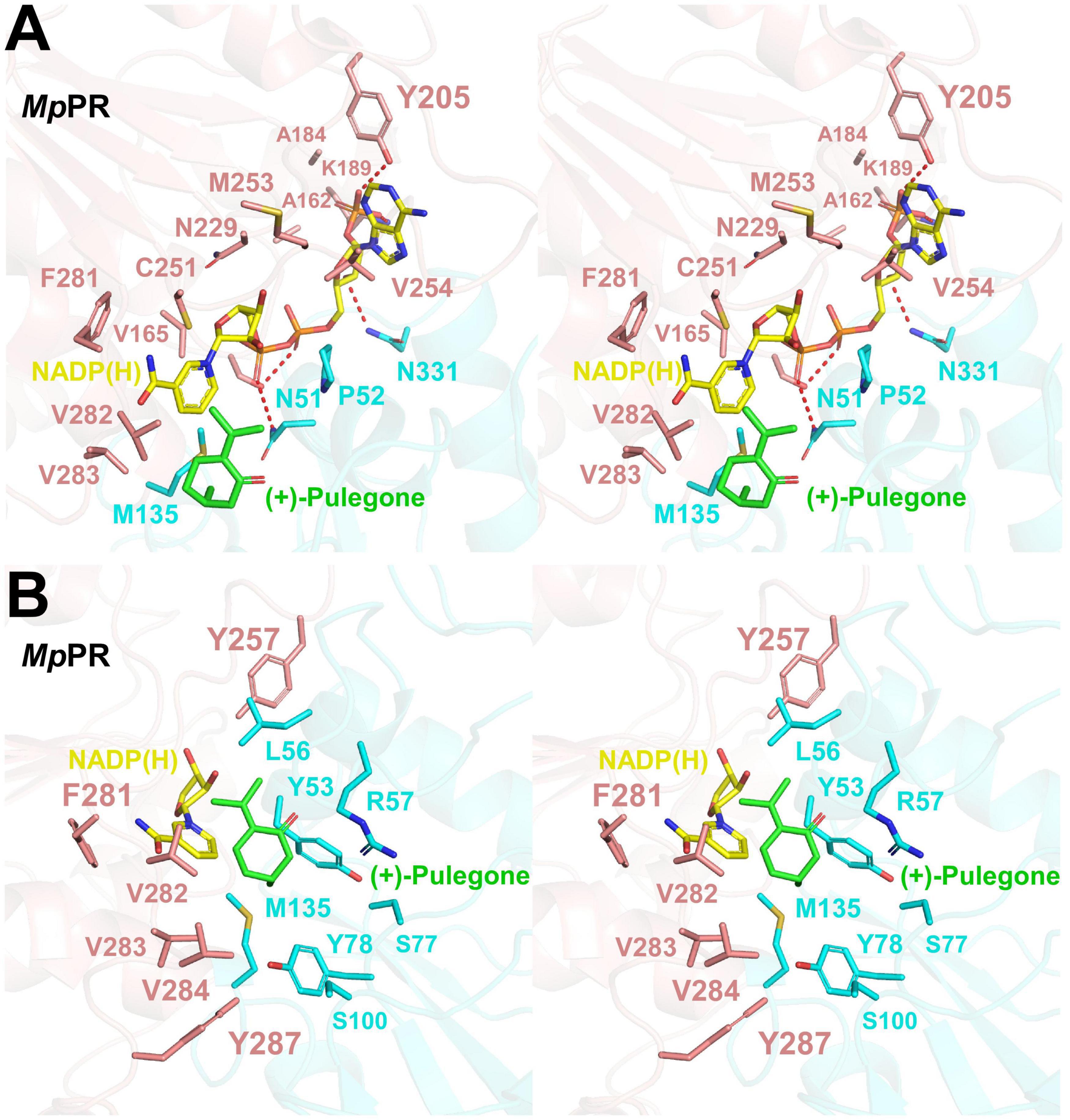
Figure 4. Structural modeling analysis of the NADP(H) and (+)-pulegone binding site of pulegone reductases from Mentha piperita (MpPR). (A) Zoom-in stereo view shows the key residues of MpPR involved in the interaction with NADP(H). Color codes are the same as those in Figure 3. Red dashed lines indicate hydrogen bonds. (B) Zoom-in stereo view shows the key residues of MpPR involved in the interaction with (+)-pulegone. Color codes are the same as those in Figure 3.
Due to the lack of co-crystal structure of MpPR with the substrate, we docked (+)-pulegone into the MpPR structure based on the ternary complex structure of its homolog AtDBR (PDB ID 2J3J and 2J3K). In contrast to the predicted NADP(H) binding pocket, residues Tyr53, Leu65, Tyr78, Met135, Tyr257, Phe281, Val282, Val283, Val284, and Tyr287 form a hydrophobic network stabilizing (+)-pulegone mainly via hydrophobic and van der Waals interactions (Figure 4B). In addition, residues Ser77 and Ser100 also contribute to the binding of (+)-pulegone by van der Waals interaction. Residues Tyr53 and Arg57 may interact with the ketone moiety of (+)-pulegone through polar interactions to further stabilize the binding of (+)-pulegone (Figure 4B). All these residues form a feasible (+)-pulegone binding pocket in the structure of MpPR.
To validate the predicted residues in pulegone binding pocket, we generated 25 single mutants by mutating 13 most relevant residues in the binding pockets of MpPR and assessed the effect of point mutation on enzyme activity and the yield of final product (Supplementary Figure 6). In vitro enzyme catalysis assays showed that the wild-type MpPR can convert (+)-pulegone to the products (−)-menthone and (+)-isomenthone with the ratio of 2:1 (Figure 5A and Supplementary Figure 10). The yield of the products decreased when the MpPR mutants L56A, R57A, M135A, Y257A, F281A, V282A, V283A, V284A, and S77G were used in the reactions. Based on the docking model of MpPR with NADP(H) and (+)-pulegone, the mutation R57A may disrupt the potential weak hydrogen bonding interaction with the ketone group of (+)-pulegone, thus reducing the enzyme activity slightly. The key residues Leu56, Val282, and Val284 in the binding pocket was proposed to interact with (+)-pulegone through hydrophobic effect. The hypothesis was evidenced by the mutation of Leu56, Val282, and Val284 to the non-polar residues with larger hydrophobic side chain (Leu56 to Ile and Val, Val282 to Leu, and Val284 to Phe, Leu, and Tyr) led to a remarkable increase in the yield of products when compared with the wild-type enzyme (Figure 5). Site-directed mutagenesis analysis validated the amino acid residues Leu56, Arg57, Ser77, Tyr257, Phe281, Val282, Val283, and Val284 in MpPR binding pocket are critical to substrate binding and catalysis, consistent with the interactions revealed from the docking model of MpPR in complex with NADP(H) and (+)-pulegone.
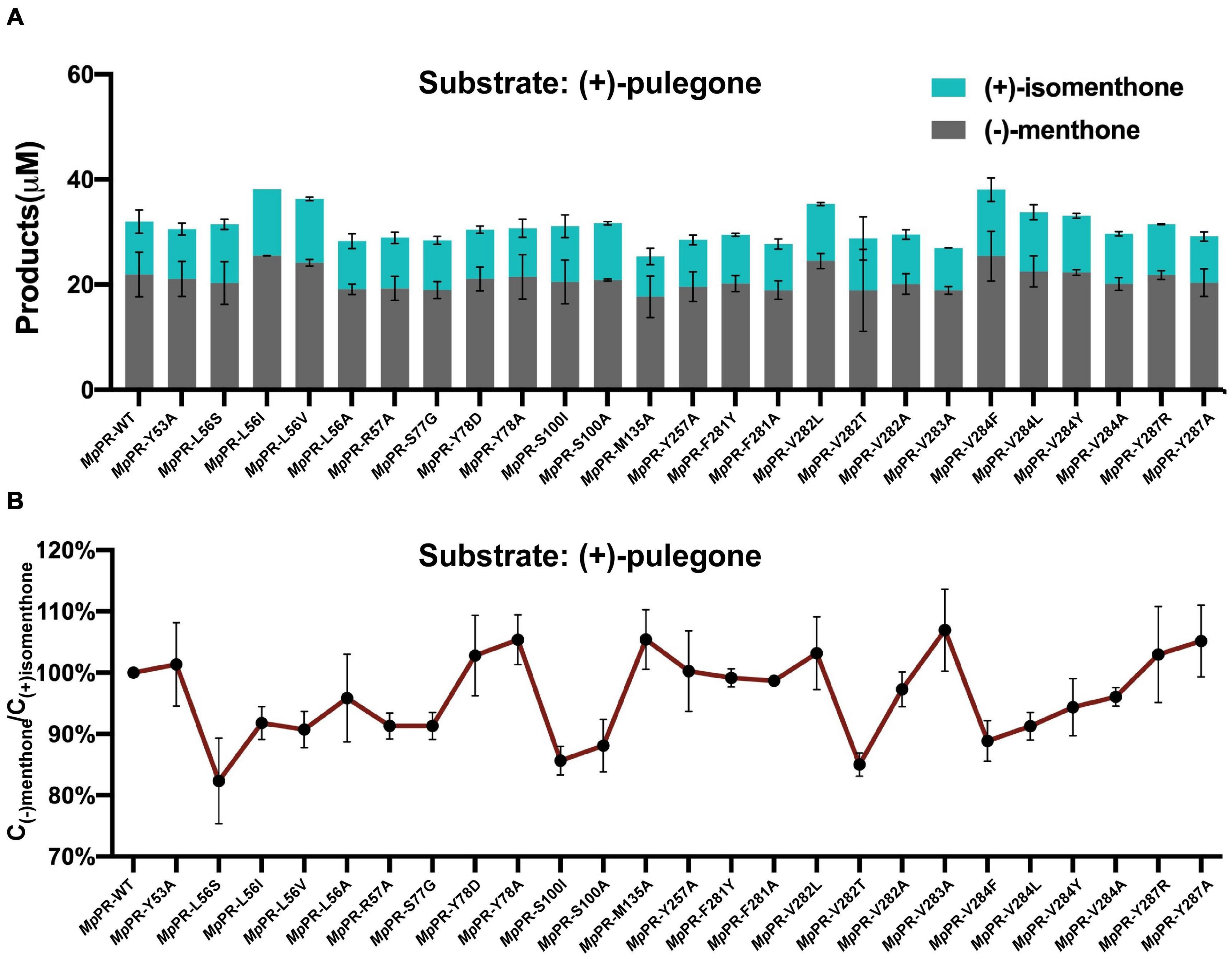
Figure 5. Site-directed mutagenesis analysis of the (+)-pulegone binding site of pulegone reductases from Mentha piperita (MpPR). (A) Quantification of the reduction products (–)-menthone and (+)-isomenthone converted from the substrate (+)-pulegone by wild-type and mutated MpPR. Reactions (0.4 mL) were performed in buffer (50 mM KH2PO4, 10% sorbitol, 1 mM DTT, pH 7.5) containing 20 μM substrate,10 mM NADPH tetrasodium salt hydrate, 6 mM glucose-6-phosphate, 20 U glucose-6-phosphate dehydrogenase and 30 μM MpPR. 0.2 mL of n-hexane was added on the top of the reaction solution. Reaction was carried out at 31°C for 1 h. The concentration of products in n-hexane was determined by gas chromatography. Wild-type MpPR and its mutants react under the same conditions. (B) The percentage ratio of the products (–)-menthone and (+)-isomenthone converted from (A). Wild type’s ratio of (–)-menthone and (+)-isomenthone is normalized to 100%. Data are presented as mean ± S.D. of triplicate experiments.
Once the putative residues (i.e., Leu56, Arg57, Ser77, Tyr257, Phe281, Val282, Val283, and Val284) in the (+)-pulegone binding pocket of MpPR were validated, we set out to explore the underlying mechanism on why MpPR has preference to (+)-pulegone rather than (−)-pulegone as observed in our GC-MS analysis and enzyme kinetic study (Figure 1). Firstly, (+)- and (−)-pulegones were docked into the crystal structure of MpPR with NADP(H), respectively, and we observed that the angle of C-5 methyl group (5-Me) of pulegone in the binding pocket likely determines the substrate selectivity of MpPR. Specifically, 5-Me of (+)-pulegone perfectly interacts with the hydrophobic network constituted by Tyr53, Leu56, Tyr78, M135, Val282, Val283, Val284, and Tyr287, stabilizing the binding between MpPR and (+)-pulegone. In contrast, 5-Me of (−)-pulegone does not totally fit into the hydrophobic network and may destabilize the binding with MpPR (Figures 6A,B). To validate the hypothesis, we calculated the forces and energies generated during the binding between pulegone and MpPR using molecular dynamic (MD) simulations. The free energy of binding (ΔGbinde) calculated for (+)-pulegone to MpPR (–98.95 kcal/mol) is significantly lower than that of (−)-pulegone (–89.17 kcal/mol), indicating a tendency to adopt (+)-pulegone as the substrate by MpPR (Figure 6C). Furthermore, the non-polar interaction (ΔGGAd) might contribute to the free energy of binding to the most extent with the values of –29.97 kcal/mol for (+)-pulegone and –43.12 kcal/mol for (−)-pulegone (Figure 6C). The MD simulations also predicted and compared the contribution of each amino acid residue involved in the non-polar interaction with (+)- and (−)-pulegones. MD simulations results showed that the residues Tyr78, Met135, Val282, Val283, Val284, Tyr287, and particularly Tyr53 constitute much stronger hydrophobic interactions with (+)-pulegone compared with those for (−)-pulegone, suggesting these residues are highly likely involved in stereoselectivity of the substrate (Figure 6D). The calculated results are highly consistent with our hypothesis that the interaction between these residues and 5-Me of (+)-pulegone stabilize their binding and thus led to the substrate specificity.
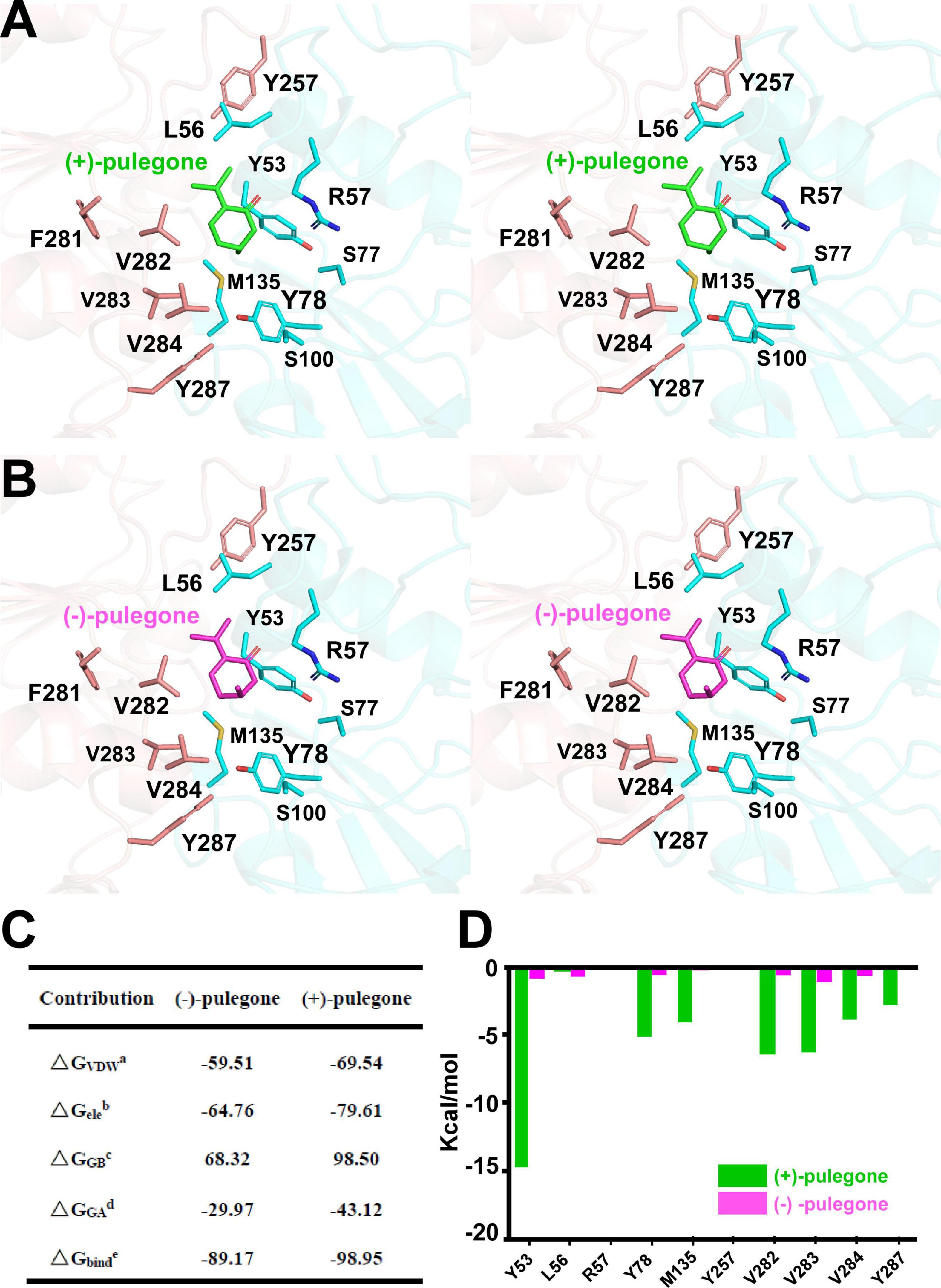
Figure 6. Structural docking results and predicted binding parameters of Mentha piperita (+)-pulegone reductase (MpPR) bound with the substrate (+)-pulegone or (–)-pulegone by MD simulations. (A) Stereo view shows the key residues of MpPR in the interaction with docked (+)-pulegone. (B) Stereo view shows the key residues of MpPR in the interaction with docked (–)-pulegone. (C) Calculated intermolecular forces and binding free energies between MpPR and (±)-pulegone by MD simulations. ΔGVDWa, Contribution to the free energy of binding from the van der Waals interaction; ΔGeleb, Contribution to the free energy of binding from the electrostatic interaction; ΔGGBc, Contribution to the free energy of binding from the polar interaction; ΔGGAd, Contribution to the free energy of binding from the non-polar interaction; ΔGbinde, Free energy of binding. (D) Strengths of hydrophobic interactions contributed by residues located at the potential pulegone binding pocket of MpPR predicted by MD simulations.
To further experimentally investigate the importance of these predicted residues involved in substrate stereoselectivity, we mutated the hydrophobic residues Leu56, Val282, and Val284 in the binding pocket of (+)-pulegone in MpPR to the corresponding residues Ser59, Leu285, and Tyr287 in NtPR. The enzyme kinetic measurements of the wild-type MpPR revealed that its binding affinity to (+)-pulegone (Km ∼3.00 μM) is about threefold higher than that to (−)-pulegone (Km ∼8.63 μM). The mutations L56S, V282L, and V284Y led to 8–100-fold decrease in the binding affinity to both (+)- and (−)-pulegone (Figures 7A–E). Importantly, the mutation V282L and V284Y almost diminished the substrate stereoselectivity of MpPR as evidenced by the very similar Km values for both (+)- and (−)-pulegone, possibly attributed to the larger side chains of the mutants that strengthen the hydrophobic interaction with (−)-pulegone (Figures 7A–E). In contrast, the mutation L56S did not display a significant effect on the substrate selectivity, suggesting Leu56 is not involved in the substrate specificity.
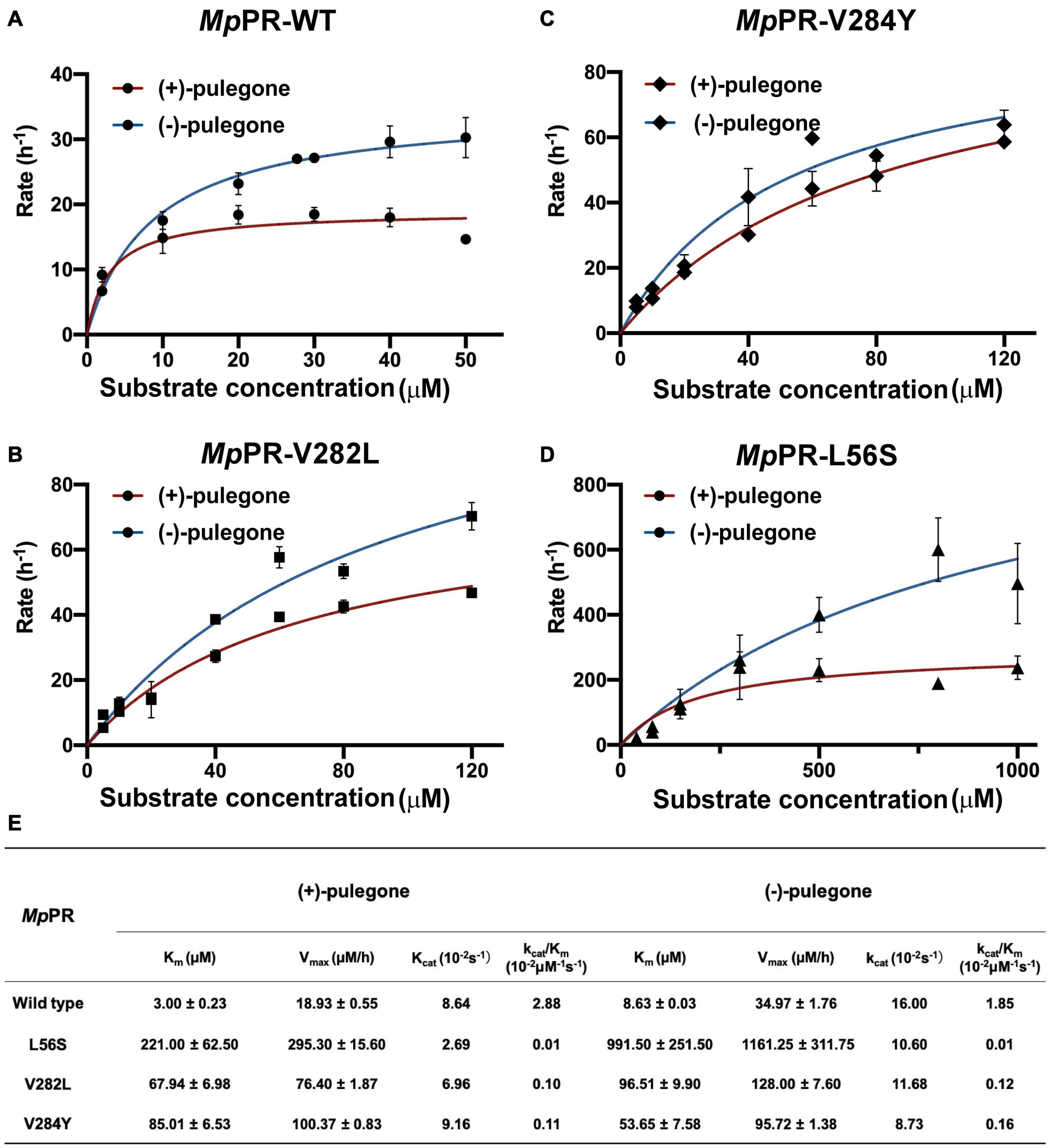
Figure 7. Measurement of kinetic parameters for the wild-type Mentha piperita (+)-pulegone reductase (MpPR) and its mutants. (A–D) Michaelis Menten plot of for wild-type, V282L, V284Y, and L56S MpPR using (+)-pulegone or (–)-pulegone as the substrate. Reactions were performed as described in Materials and Methods. Supplementary Table 1 provides enzyme concentration and substrate concentration for each reaction. (E) Summary of enzyme kinetic parameters of the wild-type MpPR and its mutants. Values are means ± S.D. and error bars indicate the S.D. for three biological replicates.
In vitro enzyme catalysis assays coupled with GC-MS analysis showed that the wild-type MpPR can convert (+)-pulegone to the products (−)-menthone and (+)-isomenthone with the ratio of 2:1. This observation encouraged us to explore the potential mechanism on the product stereoselectivity of MpPR. Given that the only difference between the structures of (−)-menthone and (+)-isomenthone lies in the angle of the 2-isopropyl group (2-iPr), we therefore proposed that the product stereoselectivity may be attributed to the stereochemistry of 2-iPr, which could result in different interaction strengths and binding affinities to MpPR. To test this hypothesis, we first docked (−)-menthone and (+)-isomenthone into the crystal structure of MpPR, respectively. The docking model showed that the 2-iPr of (−)-menthone is well positioned in the binding pocket of MpPR and stabilized by Tyr53, Leu56, Val282, Val283, and Val284, while the 2-iPr of (+)-isomenthone crashes the main chain of residue Tyr53 in the pocket, thus requiring higher energies to generate and stabilize with MpPR (Figures 8A,B). Subsequently, the binding affinities of (−)-menthone and (+)-isomenthone to MpPR were calculated using MD simulations. The enzyme kinetic parameters supported our deduction from the structural analysis as the free binding energy (ΔGbinde) for (−)-menthone (–119.43 kcal/mol) is higher than that for (+)-isomenthone (–112.75 kcal/mol) (Figure 8C). Moreover, the MD simulations also predicted the potential contributions of the surrounding residues to the non-polar hydrophobic interactions with (−)-menthone or (+)-isomenthone. The residues Tyr53, Leu56, Tyr78, Met135, Val282, Val283, and Val284 have major contributions to the binding of the products (Figure 8D). Particularly, Tyr53, M135, and V284 are inclined to stabilize (−)-menthone rather than (+)-isomenthone as revealed by the lower ΔGGAd values, whereas Tyr78, Val282, and Val283 have larger contribution to the stabilization of (+)-isomenthone through hydrophobic interactions (Figure 8D).
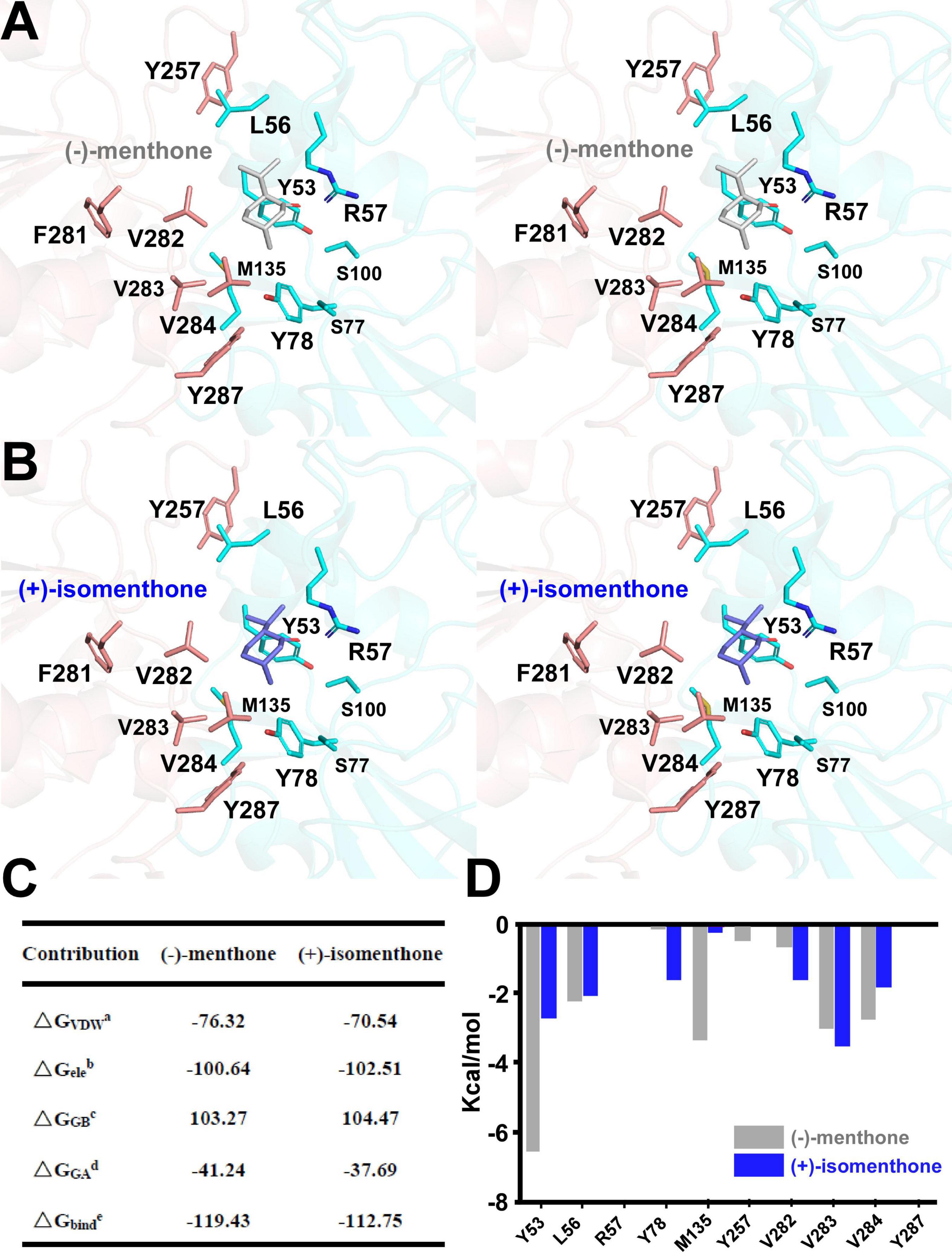
Figure 8. Structural docking results and predicted binding parameters of Mentha piperita (+)-pulegone reductase (MpPR) with the products (–)-menthone or (+)-isomenthone by MD simulations. (A) Stereo view shows the key residues of MpPR interacting with the product (–)-menthone. (B) Stereo view shows the key residues of MpPR interacting with the product (+)-isomenthone. (C) Calculated intermolecular forces and free binding energies between MpPR and (–)-menthone or (+)-isomenthone by MD simulations. The parameters are the same as in Figure 5C. (D) Strengths of hydrophobic interactions contributed by the binding pocket residues of MpPR predicted by MD simulations.
Next, we performed an in-depth analysis of the site-directed mutagenesis data to evaluate the impact of these residues on the stereoselectivity of products (−)-menthone and (+)-isomenthone. The ratio of the products (−)-menthone and (+)-isomenthone was calculated for each mutant and normalized based on those for the wild-type MpPR. The results revealed that the mutants L56S, L56I, L56V, S100I, S100A, V282T, V284F, V284L, V284Y, and V284A significantly reduced the ratio of (−)-menthone to (+)-isomenthone, but have little impact on the total yield of the products (Figures 5A,B), suggesting that the residues Leu56, Ser100, Val282, and Val284 may play critical roles in the stereoselectivity of the products (−)-menthone and (+)-isomenthone, which was consistent with our docking and MD simulation results described above.
In the enzyme catalysis assay and GC-MS analysis, we found that MpPR can also adopt (−)-pulegone as its substrate but with low affinity to generate (+)-menthone (∼46%) and the (−)-isomenthone (∼54%) (Figure 1C, Supplementary Figure 10, and Supplementary Table 1). To investigate the stereoselectivity of MpPR for products (+)-menthone and (−)-isomenthone, we performed docking study and MD simulations. The docking analysis show that the product either (+)-menthone or (−)-isomenthone can well fit the proposed product binding pocket and does not have big crash with the surrounding residues (Supplementary Figures 11A,B), indicating (+)-menthone or (−)-isomenthone may be all favored by MpPR. The MD simulations results supported the hypothesis because the free binding energy of product (+)-menthone (–141.26 kcal/mol) and (−)-isomenthone (–116.74 kcal/mol) is both relatively low (Supplementary Figure 11C), which is consistent with our GC-MS data. Furthermore, the residues Y53, L56, V282, and V284 in the binding pocket of MpPR are predicted to mainly contribute to the non-polar hydrophobic interactions with (+)-menthone and (−)-isomenthone in the MD simulations (Supplementary Figure 11D). To experimentally validate these residues responsible for product stabilization, we performed mutagenesis analysis and assayed the abilities of mutants to convert (−)-pulegone to (+)-menthone and (−)-isomenthone. Compared with the wild-type enzyme, the mutation L56S, Y78D, and V282L decreased the ratio of products (+)-menthone and (−)-isomenthone, like the results obtained using the native substrate (+)-pulegone (Supplementary Figure 12). In contrast, the mutation L56I, L56V, R57A, Y78A, M135A, V282A, and V284A significantly improved the percentage yield of (+)-menthone, suggesting the residues Leu56, Arg57, Tyr78, Met135, Val282, and V284 might be critical to the stereoselectivity of products (+)-menthone and (−)-isomenthone (Supplementary Figure 12B).
Monoterpenes are constituents of essential oils and oleoresins used commercially in the flavor, fragrance, and medicine industries. Currently, monoterpenes are mainly extracted from plants using chemical methods, which is costly and environmentally unfriendly. Moreover, the yield of monoterpenes isolated from natural or engineered plants remain unsatisfactory and do not meet the large need in industry. Importantly, most of the medicinal plants grow slowly, and the planting area is limited due to the climate and other environmental conditions (Wang et al., 2015). Although chemical synthesis is another option, multiple chiral centers in most monoterpene structures pose a big challenge to total chemical synthesis and the cost to obtain enantiomerically pure monoterpenes is high. Thus, looking for alternative sustainable supply of industrially important monoterpenes is of great importance. The development of synthetic biology and metabolic engineering tools provides great opportunities for heterologous biosynthesis of monoterpenoids in microbial cells (Oswald et al., 2007; Herrero et al., 2008; Fischer et al., 2011; Lange, 2015; Zebec et al., 2016; Rajput et al., 2018; Bergman et al., 2019; Gao et al., 2020). Industrial-scale production of certain terpenoids using engineered microorganisms have been achieved, such as artemisinin acid and ginsenosides (Paddon et al., 2013; Wang et al., 2015; Wei et al., 2015; Talman et al., 2019). To achieve this goal, elucidation of monoterpenoid biosynthetic pathways, especially the key biosynthetic enzymes, is extremely essential the reconstruction of monoterpenoid pathways in microbial chassis cells. Considerable attention has been paid on the mechanism of the cyclization reactions in monoterpenoid biosynthesis, however, many fascinating puzzles and stereochemical anomalies remain unclear (Finefield et al., 2012).
(−)-Menthone is the second most abundant monoterpene [15–20% (v/v)] of peppermint essential oil and the substrate of (−)-menthol, which also represents anti-inflammatory, antiviral, and anti-bacterial activity (Shalayel et al., 2016). Natural (−)-menthone is crystallized from dementholized peppermint or cornmint essential oil, the source of which is limited to plants. Commercial (−)-menthone is always sold as a mixture with up to 29% (v/v) (+)-isomenthone (Lange, 2015). (+)-Menthone is the opposite chirality compound, occupying 20–30% (v/v) in N. tenuifolia essential oil, and display similar activities as (−)-menthone (Chen et al., 2017; Liu et al., 2018). The major source of commercial (+)-menthone is medicinal plants (Marumoto et al., 2017), and the yield is often varied due to the source and status of the plants. Therefore, achieving heterologous production of menthone in microbial host is a promising way for reproducible and high-titer production of enantiomerically pure menthone in industry.
Pulegone reductases catalyze the formation of menthone and determine the substrate specificity and product stereoselectivity. However, the structure of pulegone reductase and the underlying mechanism for substrate specificity and product stereoselectivity remain largely unknown. The implementation of a synthetic biology approach to the production of menthone was also hampered by the absence of the mechanism studies for pulegone reductase. In our study, we characterized a novel (−)-pulegone reductase from Nepeta tenuifolia (NtPR), which displays opposite substrate specificity and product stereoselectivity to those for (+)-pulegone reductase isolated from Mentha piperita (MpPR). Comparative analysis of the structures, key amino acids residues in the pulegone binding pocket, and enzyme kinetics for NtPR and MpPR using the combined bioinformatics, structure biology and biochemistry approaches led us to better understand the underlying mechanism of the substrate and product stereoselectivity. Through site-directed mutagenesis, we obtained several MpPR mutants that improve the percentage of (−)-menthone to 70% and of (+)-menthone to 68%. The recombinant pulegone reductase could be further engineered to increase the production and percentage of menthone. Therefore, increasing the ratio/yield of major product menthone in the enzymatic reaction is critical to heterologous biosynthesis of menthone in microbial hosts.
Recently, molecular reaction mechanisms have been proposed for several DBRs, such as AtDBR (Youn et al., 2006) and RiDBR, in which the conjugated double bond of the substrate is in equilibrium with an α,β-conjugated enolate intermediate. In this condition, a hydride transfer occurs from the C-4 of the nicotine amide of NADP(H) (catalytic carbon) to the β carbon of the enolate intermediate, with a subsequent protonation of its α carbon. In AtDBR, this process is facilitated by the stabilization of the propenal transition state by a π-π interaction between Tyr53 and the phenolic ring of the substrate p-coumaryl aldehyde (Youn et al., 2006). In RiDBR ternary structure, a π-π stacking between the substrate hydroxybenzalacetone and nicotinamide aromatic rings is observed, with a hydride transfer distance of 3.06 Å to the alkene double bond (Simon et al., 2017). For MpPR and NtPR (Mansell et al., 2013), a π-π stacking may be formed between the double bond from the substrate pulegone and nicotinamide aromatic rings to facilitate hydride transfer (Figure 4).
Our extensive structural and biochemical analyses on MpPR and NtPR in this study provide a novel insight into the solution to meet the growing demand for high-quality natural (±)-menthone products in the future. Recently, we discovered another novel pulegone reductase from Agastache rugosa (ArPR) through genomic analysis of A. rugosa, a medicinal plant belonging to the genus of Labiatae that produces therapeutic volatile oils. ArPR is evolutionarily related to NtPR and showed a higher preference for (−)-pulegone over (+)-pulegone (Supplementary Figure 13), suggesting (−)-pulegone reductase may be widely spread in medicinal plants of genera Nepeta and Labiatae. Pulegone reductase discovered in our study provides a valuable reservoir of genetic resources for biocatalytic applications, and an example to explore the stereoselectivity of monoterpenoid biosynthetic enzymes in plants. The crystal structures of MpPR, NtPR, PpPR, or ArPR in complex with its substrate and products are anticipated in the future for the comprehensive understanding of the stereoselectivity of pulegone reductases.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: http://www.wwpdb.org/, 7EQL.
CL, QW, IL, NS, BL, and WL designed experiments. QG and CL performed the bulk of the experiments. QG, CL, IL, and NS contributed to gene identification and cloning, protein expression, purification, and crystallization. JL, LL, QG, NS, and CL contributed to enzymatic assay experiments and oil distillation. SZ contributed to molecular phylogenetic analysis. JL contributed to molecular dynamic simulations. CL, ZS, QW, and WL analyzed the data and wrote the manuscript. QW, BL, and WL conceived the project. All authors contributed to the article and approved the submitted version.
This work was financially supported by the National Natural Science Foundation of China (81903526, 81991523, 82072240, 81903756, 81973435, and 81473313), Jiangsu Province of China (BK20190798 to WL), the Open Project of State Key Laboratory of Natural Medicines (No. SKLNMKF202004 to WL), the Open Project of Chinese Materia Medica First-Class Discipline of Nanjing University of Chinese Medicine (No. 2020YLXK008 to WL), the Open Project of Natural Science Foundation of Nanjing University of Chinese Medicine (No. NZY81903756 to CL), the Fok Ying Tung Education Foundation, and Jiangsu Specially Appointed Professor Talent Program to WL, a grant from the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences (Grant No. DE–SC0001553) to BL.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the staff at BL19U1/BL17U1-SSRF for technical assistance in X-ray diffraction data collection and staff at the experiment center for science and technology of Nanjing University of Chinese Medicine for experimental supports.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.780970/full#supplementary-material
Abbas, F., Ke, Y., Yu, R., Yue, Y., Amanullah, S., Jahangir, M. M., et al. (2017). Volatile terpenoids: multiple functions, biosynthesis, modulation and manipulation by genetic engineering. Planta 246, 803–816. doi: 10.1007/s00425-017-2749-x
Adams, P. D., Afonine, P. V., Bunkoczi, G., Chen, V. B., Davis, I. W., Echols, N., et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221. doi: 10.1107/S0907444909052925
Bergman, M. E., Davis, B., and Phillips, M. A. (2019). Medically Useful Plant Terpenoids: Biosynthesis, Occurrence, and Mechanism of Action. Molecules 2019:24.
Caliandro, R., Polsinelli, I., Demitri, N., Musiani, F., Martens, S., and Benini, S. (2021). The structural and functional characterization of Malus domestica double bond reductase MdDBR provides insights towards the identification of its substrates. Int. J. Biol. Macromol. 171, 89–99. doi: 10.1016/j.ijbiomac.2020.12.190
Cheallaigh, A. N., Mansell, D. J., Toogood, H. S., Tait, S., Lygidakis, A., Scrutton, N. S., et al. (2018). Chemoenzymatic Synthesis of the Intermediates in the Peppermint Monoterpenoid Biosynthetic Pathway. J. Nat. Prod. 81, 1546–1552. doi: 10.1021/acs.jnatprod.7b01026
Chen, C-J., Zi-Ying, Z., Su-Su, D., Wei, W., Fu-Rng, X., Yong-Xian, C., et al. (2019). Efficacy and mechanism of Mentha haplocalyx and Schizonepeta tenuifolia essential oils on the inhibition of Panax notoginseng pathogens. Industrial. Crops Products 2019:145.
Chen, S. G., Cheng, M. L., Chen, K. H., Horng, J. T., Liu, C. C., Wang, S. M., et al. (2017). Antiviral activities of Schizonepeta tenuifolia Briq. against enterovirus 71 in vitro and in vivo. Sci. Rep. 7:935. doi: 10.1038/s41598-017-01110-x
Christianson, D. W. (2008). Unearthing the roots of the terpenome. Curr. Opin. Chem. Biol. 12, 141–150. doi: 10.1016/j.cbpa.2007.12.008
Currin, A., Dunstan, M. S., Johannissen, L. O., Hollywood, K. A., Vinaixa, M., Jervis, A. J., et al. (2018). Engineering the “Missing Link” in Biosynthetic (-)-Menthol Production: Bacterial Isopulegone Isomerase. ACS Catal. 8, 2012–2020. doi: 10.1021/acscatal.7b04115
Duan, S. L., Zeng, W. X., and Sun, L. L. (2015). Chemical constituents of volatile oils in drug pair of menthae haplocalycis herba and schizonepetae spica by GC-MS. Chin. J. Exp. Tradit. Med. Form. 21, 50–54. doi: 10.13422/j.cnki.syfjx.2015110050
Emsley, P., and Cowtan, K. (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132.
Finefield, J. M., Sherman, D. H., Kreitman, M., and Williams, R. M. (2012). Enantiomeric natural products: occurrence and biogenesis. Angew. Chem. Int. Ed. Engl. 51, 4802–4836.
Fischer, M. J., Meyer, S., Claudel, P., Bergdoll, M., and Karst, F. (2011). Metabolic engineering of monoterpene synthesis in yeast. Biotechnol. Bioeng. 108, 1883–1892.
Gao, Q., Wang, L., Zhang, M., Wei, Y., and Lin, W. (2020). Recent Advances on Feasible Strategies for Monoterpenoid Production in Saccharomyces cerevisiae. Front. Bioeng. Biotechnol. 8:609800. doi: 10.3389/fbioe.2020.609800
Herrero, O., Ramon, D., and Orejas, M. (2008). Engineering the Saccharomyces cerevisiae isoprenoid pathway for de novo production of aromatic monoterpenes in wine. Metab Eng 10, 78–86. doi: 10.1016/j.ymben.2007.11.001
Hijaz, F., Nehela, Y., and Killiny, N. (2016). Possible role of plant volatiles in tolerance against huanglongbing in citrus. Plant Signal Behav. 11:e1138193. doi: 10.1080/15592324.2016.1138193
Hyatt, D. C., Youn, B., Zhao, Y., Santhamma, B., Coates, R. M., Croteau, R. B., et al. (2007). Structure of limonene synthase, a simple model for terpenoid cyclase catalysis. Proc. Natl. Acad. Sci. U S A 104, 5360–5365. doi: 10.1073/pnas.0700915104
Lange, B. M. (2015). Biosynthesis and Biotechnology of High-Value p-Menthane Monoterpenes, Including Menthol, Carvone, and Limonene. Adv. Biochem. Eng. Biotechnol. 148, 319–353. doi: 10.1007/10_2014_289
Liu, C., Srividya, N., Parrish, A. N., Yue, W., Shan, M., Wu, Q., et al. (2018). Morphology of glandular trichomes of Japanese catnip (Schizonepeta tenuifolia Briquet) and developmental dynamics of their secretory activity. Phytochemistry 150, 23–30. doi: 10.1016/j.phytochem.2018.02.018
Liu, L., Yin, M., Lin, G., Wang, Q., Zhou, P., Dai, S., et al. (2021). Integrating RNA-seq with functional expression to analyze the regulation and characterization of genes involved in monoterpenoid biosynthesis in Nepeta tenuifolia Briq [Plant Physiol. Biochem. 167 (October 2021) 31-41]. Plant Physiol. Biochem. 167:911. doi: 10.1016/j.plaphy.2021.09.020
Mahizan, N. A., Yang, S. K., Moo, C. L., Song, A. A., Chong, C. M., Chong, C. W., et al. (2019). Terpene Derivatives as a Potential Agent against Antimicrobial Resistance (AMR) Pathogens. Molecules 2019:24. doi: 10.3390/molecules24142631
Mansell, D. J., Toogood, H. S., Waller, J., Hughes, J. M., Levy, C. W., Gardiner, J. M., et al. (2013). Biocatalytic Asymmetric Alkene Reduction: Crystal Structure and Characterization of a Double Bond Reductase from Nicotiana tabacum. ACS Catal. 3, 370–379. doi: 10.1021/cs300709m
Marumoto, S., Okuno, Y., Hagiwara, Y., and Miyazawa, M. (2017). Biotransformation of (-)-(1R,4S)-Menthone and (+)-(1S,4R)-Menthone by the Common Cutworm Spodoptera litura Larvae. J. Oleo Sci. 66, 883–888. doi: 10.5650/jos.ess17005
Mcconkey, M. E., Gershenzon, J., and Croteau, R. B. (2000). Developmental regulation of monoterpene biosynthesis in the glandular trichomes of peppermint. Plant Physiol. 122, 215–224. doi: 10.1104/pp.122.1.215
Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., et al. (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791. doi: 10.1002/jcc.21256
Oswald, M., Fischer, M., Dirninger, N., and Karst, F. (2007). Monoterpenoid biosynthesis in Saccharomyces cerevisiae. FEMS Yeast Res. 7, 413–421.
Otwinowski, Z., and Minor, W. (1997). Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326.
Paddon, C. J., Westfall, P. J., Pitera, D. J., Benjamin, K., Fisher, K., Mcphee, D., et al. (2013). High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 496, 528–532. doi: 10.1038/nature12051
Paramasivan, K., and Mutturi, S. (2017). Progress in terpene synthesis strategies through engineering of Saccharomyces cerevisiae. Crit. Rev. Biotechnol. 37, 974–989.
Rajput, J. D., Bagul, S. D., Pete, U. D., Zade, C. M., Padhye, S. B., and Bendre, R. S. (2018). Perspectives on medicinal properties of natural phenolic monoterpenoids and their hybrids. Mol. Divers 22, 225–245. doi: 10.1007/s11030-017-9787-y
Ringer, K. L., Davis, E. M., and Croteau, R. (2005). Monoterpene metabolism. Cloning, expression, and characterization of (-)-isopiperitenol/(-)-carveol dehydrogenase of peppermint and spearmint. Plant Physiol. 137, 863–872. doi: 10.1104/pp.104.053298
Ringer, K. L., Mcconkey, M. E., Davis, E. M., Rushing, G. W., and Croteau, R. (2003). Monoterpene double-bond reductases of the (-)-menthol biosynthetic pathway: isolation and characterization of cDNAs encoding (-)-isopiperitenone reductase and (+)-pulegone reductase of peppermint. Arch Biochem. Biophys. 418, 80–92. doi: 10.1016/s0003-9861(03)00390-4
Shalayel, M. H. F., Asaad, A. M., and Qureshi, M. A. (2016). Anti-bacterial activity of peppermint (Mentha piperita) extracts against some emerging multi-drug resistant human bacterial pathogens. J. Herb. Med. 2016, 27–30.
Simon, J. M., David, B., Yonek, B., Karen, M. P., and Paul, S. F. (2017). A cell-free synthetic biochemistry platform for raspberry ketone production. bioRxiv doi: 10.1101/202341
Srividya, N., Lange, I., and Lange, B. M. (2020). Determinants of Enantiospecificity in Limonene Synthases. Biochemistry 59, 1661–1664. doi: 10.1021/acs.biochem.0c00206
Talman, A. M., Clain, J., Duval, R., Menard, R., and Ariey, F. (2019). Artemisinin Bioactivity and Resistance in Malaria Parasites. Trends Parasitol. 35, 953–963.
Vagin, A., and Teplyakov, A. (2010). Molecular replacement with MOLREP. Acta Crystallogr. D. Biol. Crystallogr. 66, 22–25.
Wang, P., Wei, Y., Fan, Y., Liu, Q., Wei, W., Yang, C., et al. (2015). Production of bioactive ginsenosides Rh2 and Rg3 by metabolically engineered yeasts. Metab. Eng. 29, 97–105.
Wei, W., Wang, P., Wei, Y., Liu, Q., Yang, C., Zhao, G., et al. (2015). Characterization of Panax ginseng UDP-Glycosyltransferases Catalyzing Protopanaxatriol and Biosyntheses of Bioactive Ginsenosides F1 and Rh1 in Metabolically Engineered Yeasts. Mole. Plant 8, 1412–1424. doi: 10.1016/j.molp.2015.05.010
Winn, M. D., Ballard, C. C., Cowtan, K. D., Dodson, E. J., Emsley, P., Evans, P. R., et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. D. Biol. Crystallogr. 67, 235–242. doi: 10.1107/S0907444910045749
Wojtunik-Kulesza, K. A., Kasprzak, K., Oniszczuk, T., and Oniszczuk, A. (2019). Natural Monoterpenes: Much More than Only a Scent. Chem. Biodivers 16:e1900434. doi: 10.1002/cbdv.201900434
Youn, B., Kim, S. J., Moinuddin, S. G., Lee, C., Bedgar, D. L., Harper, A. R., et al. (2006). Mechanistic and structural studies of apoform, binary, and ternary complexes of the Arabidopsis alkenal double bond reductase At5g16970. J. Biol. Chem. 281, 40076–40088. doi: 10.1074/jbc.M605900200
Zebec, Z., Wilkes, J., Jervis, A. J., Scrutton, N. S., Takano, E., and Breitling, R. (2016). Towards synthesis of monoterpenes and derivatives using synthetic biology. Curr. Opin. Chem. Biol. 34, 37–43.
Keywords: pulegone reductase, stereoselectivity, molecular dynamics simulations, menthol biosynthesis, Mentha piperita
Citation: Liu C, Gao Q, Shang Z, Liu J, Zhou S, Dang J, Liu L, Lange I, Srividya N, Lange BM, Wu Q and Lin W (2021) Functional Characterization and Structural Insights Into Stereoselectivity of Pulegone Reductase in Menthol Biosynthesis. Front. Plant Sci. 12:780970. doi: 10.3389/fpls.2021.780970
Received: 22 September 2021; Accepted: 09 November 2021;
Published: 30 November 2021.
Edited by:
Supaart Sirikantaramas, Chulalongkorn University, ThailandReviewed by:
Dinesh A. Nagegowda, Central Institute of Medicinal and Aromatic Plants, Council of Scientific and Industrial Research (CSIR), IndiaCopyright © 2021 Liu, Gao, Shang, Liu, Zhou, Dang, Liu, Lange, Srividya, Lange, Wu and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qinan Wu, d3VxbkBuanVjbS5lZHUuY24=; Wei Lin, d2VpbGluQG5qdWNtLmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.